
Home » Posts tagged 'HYPERTENTION'
Tag Archives: HYPERTENTION
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TAK 272, For Hypertension, Takeda’s Next Sartan
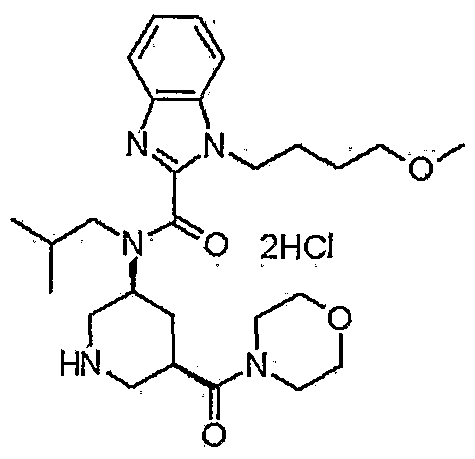
TAK 272
C27 H41 N5 O4 . Cl H, 536.106
CAS.1202269-24-6. MonoHCl
1202265-90-4 DIHCL
Base cas…1202265-63-1
Metanesulfonate…1202266-34-9
Takeda Pharmaceutical Company Limited, INNOVATOR
see……….http://www.allfordrugs.com/2015/10/21/tak-272-for-hypertension-takedas-next-sartan/
1-(4-methoxybutyl)-N-(2-methylpropyl)-N-[(3S,5R)-5-(morpholin-4-ylcarbonyl)-piperidin-3-yl]-1H-benzimidazole-2-carboxamide
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
N-Isobutyl-1-(4-methoxybutyl)-N-[5(R)-(morpholin-4-ylcarbonyl)piperidin-3(S)-yl]-1H-benzimidazole-2-carboxamide hydrochloride
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidine-3 – yl] -1H- benzimidazole-2-carboxamide hydrochloride,
![]()
The compound is used as renin inhibitor for treating diabetic nephropathy and hypertension
Takeda’s TAK-272, was reported to be in phase II in October 2015), an oral renin inhibitor, for treating diabetic nephropathy and hypertension
- 01 Apr 2015Takeda completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 18 Feb 2015Takeda plans a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 13 Feb 2015Takeda plans a phase I pharmacokinetics trial in Renal or Hepatic impairment patients in Japan (NCT02367872)

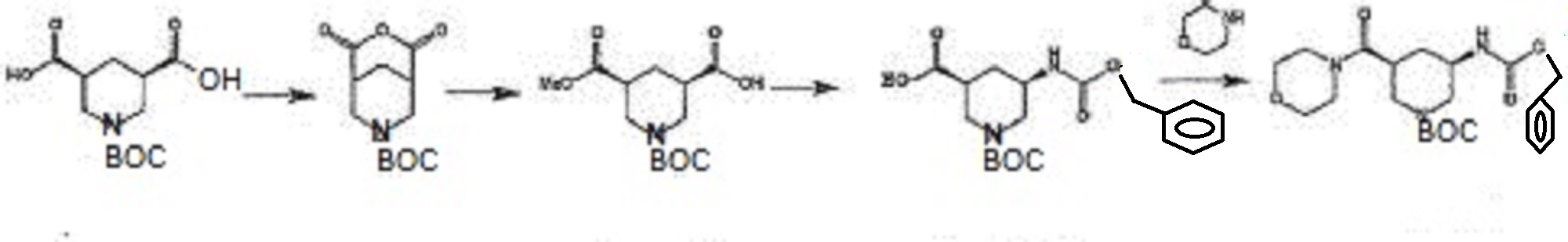
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.
(Wherein each symbol is as described in Patent Document 2.)
Patent literature
Patent Document 2: International Publication No. 2007/077005
WO2009154300
https://www.google.co.in/patents/WO2009154300A2?cl=en
INTERMEDIATES FOR CONSTRUCTION

USE THIS ONE




Reference Example 31 tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate and 1- (4-methoxybutyl) -N-
(2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4- ylcarbonyl)piperidin-3-yl]-lH-benzimidazole-2-carboxamide
tert-Butyl (3S, 5R) -3-{ [ ( {2- [ (4- methoxybutyl) amino] phenyl}amino) (oxo) acetyl] (2- methylpropyl) amino} -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.11 g) was dissolved in acetic acid (50 ml), and the mixture was stirred at 😯0C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure, the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give tert- butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl } (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (5.85 g) , and a fraction eluted with ethyl acetate-methanol (85:15) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2- methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin- 3-yl] -lH-benzimidazole-2-carboxamide (580 mg) . [0424] tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl ) piperidine-1-carboxylate 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3. 46 ( 6H, m) , 3.49-3. 91 (1OH, m) , 3. 95-4 . 47 (5H, m) , 7 . 18-7 . 51 (3H, m) , 7. 56-7 . 84 ( IH, m) .
MS (ESI+, m/e) 600 (M+l )
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl)piperidin-3-yl] -lH-benzimidazole-2-carboxamide BASE
1H-NMR (CDCl3) δ 0.64-0.74 (2H, m) , 0.95-1.07 (4H, m) , 1.43-
1.74 (3H, m) , 1.84-2.41 (4H, m) , 2.48-2.67 (IH, m) , 2.67-3.01
(3H, m), 3.03-3.44 (8H, m) , 3.47-3.78 (9H, m) , 4.06-4.46 (3H, m) , 7.28-7.47 (3H, m) , 7.62-7.81 (IH, m) . MS (ESI+, m/e) 500 (M+l)
Example 10
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-
4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
tert-Butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5-
(morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, and the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide (4.40 g) . The obtained 1- (4-methoxybutyl) -N- (2-methylpropyl) – N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -IH- benzimidazole-2-carboxamide (2.20 g) was dissolved in ethyl acetate (20 ml) , 4M hydrogen chloride-ethyl acetate (5 ml) and methanol (20 ml) were added, and the mixture was stirred at room temperature for 5 min. The reaction mixture was concentrated under reduced pressure to give the object product (2.52 g).
dihydrochloride
1H-NMR (DMSO-d6) δ 0.63-0.76 (2H, m) , 0.85-1.00 (4H, m) , 1.40-
1.60 (2H, m) , 1.68-1.89 (2H, m) , 1.93-2.17 (2H, m) , 2.20-2.44
(2H, m) , 2.81-3.81 (2OH, m) , 4.19-4.39 (3H, m) , 7.23-7.46 (2H, m) , 7.57-7.81 (2H, m) , 8.38-9.77 (2H, m) .
MS (ESI+, m/e) 500 (M+l)
Example 252
1- ( 4-methoxybutyl ) -N- ( 2-methylpropyl ) -N- [ ( 3S 1. 5R) -5- (morpholin- 4-ylcarbonyl ) piperidin-3-yl ] -lH-benzimidazole-2-carboxamide methanesulfonate

l-(4-Methoxybutyl) -N- (2-methylpropyl) -N- [ (3S,5R)-5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide (208 mg) was dissolved in ethyl acetate (2 ml) , a solution of methanesulfonic acid (40 μl) in ethyl acetate (1 ml) was added at 75°C, hexane (1 ml) was added, and the mixture was heated under reflux and stood at room temperature overnight. The precipitated crystals were collected by filtration, and dried at 7O0C for 3 hr to give the object product (158 mg) . MS (ESI+, m/e) 500 (M+l) melting point : 144.40C
EXTRAS IF REQD .………….
Example 32
methyl (3R, 5S)-5-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] piperidine-3-carboxylate dihydrochloride [0675]

MS (ESI+, m/e) 445 (M+l)
Example 33
(3R, 5S) -5- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yljcarbonyl} (2-methylpropyl) amino] piperidine-3-carboxylic acid dihydrochloride

MS (ESI+, m/e) 431 (M+l)
Reference Example 29
{ [ ( 3S , 5R) -1- (tert-butoxycarbonyl ) -5- (morpholin-4- ylcarbonyl ) piperidin-3~yl ] ( 2-itιethylpropyl ) amino } (oxo ) acetic acid
To a solution of tert-butyl (3S,5R)~3-{ [ethoxy (oxo) acetyl] (2-methylpropyl) amino}-5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate (10.3 g) in ethanol (40 ml) was added 2M aqueous sodium hydroxide solution (22 ml) , and the mixture was stirred at room temperature for 6 hr. The reaction mixture was adjusted to pH 7 with IM hydrochloric acid, and extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure to give the object product (10.3 g) .
1H-NMR (CDCl3) δ 0.78-0.99 (6H, m) , 1.37-1.52 (9H, m) , 1.79- 2.16 (3H, m) , 2.38-3.86 (14H, m) , 3.93-4.43 (2H, m) . MS (ESI+, m/e) 442 (M+l)
Reference Example 28
tert-butyl (3S, 5R) -3-{ [ethoxy (oxo) acetyl] (2- methylpropyl ) amino } -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate
To a solution of tert-butyl (3S, 5R) -3- [ (2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.24 g) and diisopropylethylamine (10.5 ml) in DMA (100 ml) was added dropwise ethyl chloroglyoxylate (3.4 ml) at 0°C. The reaction mixture was stirred at room temperature for 15 hr, and the reaction mixture was concentrated. An aqueous sodium bicarbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (10.3 g) . 1H-NMR (CDCl3) δ 0.84-1.00 (6H, m) , 1.37 (3H, q) , 1.42-1.53 (9H, m) , 1.80-2.19 (3H, m) , 2.26-2.42 (IH, m) , 2.59-2.96 (IH, in) , 2.97-3.30 (3H, m) , 3.37-3.92 (9H, m) , 4.01-4.26 (2H, m) , 4.26- 4.40 (2H, m) . MS (ESI4-, m/e) 470 (M+l) “
Reference Example 22 tert-butyl (3S, 5R) -3- [ (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate
[0369] tert-Butyl (3S,5R)-3-{ [ (benzyloxy) carbonyl] aminoJ-5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (58 g) and palladium (II) hydroxide-carbon (5 g) were suspended in methanol (400 ml) and the mixture was stirred under a hydrogen atmosphere (1 atom) at room temperature for 16 hr. The palladium catalyst was filtered off, and the filtrate was concentrated under reduced pressure. The obtained residue and acetic acid (8.8 ml) were dissolved in methanol (400 ml), 2- methylpropanal (14.0 ml) was added, and the mixture was stirred at room temperature for 1 hr. Sodium triacetoxyborohydride (40.4 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the concentrate was basified with 3.5M aqueous potassium carbonate solution, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:5) – ethyl acetate-hexane (1:1) was concentrated under reduced pressure to give the object product (33.3 g) .
1H-NMR (CDCl3) δ: 0.90 (6H, d) , 1.46 (9H, s) , 1.54 (IH, d) , 1.69 (IH, dt), 1.96-2.12 (2H, m) , 2.23-2.37 (IH, m) , 2.47 (3H, d) , 2.66 (IH, d) , 3.61 (IH, br s) , 3.55 (2H, d) , 3.69 (5H, ddd) , 4.01-4.46 (2H, m) .
Example 6 1-tert-butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate [0318]

(3S, 5R) -1- (tert-Butoxycarbonyl) -5-(methoxycarbonyl)piperidine-3-carboxylic acid (2.83 g) was suspended in toluene (36 ml), diphenylphosphoryl azide (2.60 ml) and triethylamine (1.70 ml) were added, and the mixture was stirred at 100°C for 1 hr. The reaction mixture was cooled to room temperature, benzyl alcohol (1.53 ml) and triethylamine (7.00 ml) were added and the mixture was stirred at 80°C for 3 hr. The reaction mixture was concentrated, the residue was dissolved in ethyl acetate, and the solution was washed with water, 0.5M hydrochloric acid, saturated aqueous sodium hydrogen carbonate and saturated brine in this order, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:3 – 3:1) was concentrated under reduced pressure. The obtained residue was dissolved in methanol (60 ml), 10% palladium carbon (50% in water) (150 mg) was added and the mixture was stirred under a hydrogen pressurization (5 atom) at ambient temperature and normal pressure for 5 hr. The catalyst was filtered off, and the filtrate was concentrated under reduced pressure to give the object product (1.83 g) as an oil.
1H-NMR (CDCl3) δ 1.22-1.43 (4H, m) , 1.46 (9H, s), 2.27-2.79 (4H, m) , 3.70 (3H, s) , 4.13 (2H, br s) [0320] In the same manner as in the method shown in Reference Example 6, the following compound (Reference Example 7) was obtained.
Reference Example 8
1-tert-butyl 3-methyl (3R, 5S) -5- [ (2- methylpropyl) amino] piperidine-1, 3-dicarboxylate [0325]

1-tert-Butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate (1.83 g) , isobutyraldehyde (0.78 ml) and acetic acid (0.49 ml) were dissolved in methanol (50 ml), and the mixture was stirred at room temperature for 30 min. Sodium triacetoxyborohydride (3.80 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 7 hr. The reaction mixture was concentrated under reduced pressure, the concentrate was basified with aqueous sodium bicarbonate, and extracted with ethyl acetate. The extract was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:1) – ethyl acetate 100% – ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (1.42 g) as an oil.
1H-NMR (CDCl3) δ 0.90 (6H, d) , 1.22-1.38 (3H, m) , 1.46 (9H, s) , 1.69 (IH, dt), 2.23-2.39 (2H, m) , 2.44-2.59 (IH, m) , 2.47 (2H, d) , 2.74 (IH, br s) , 3.69 (3H, s) , 4.18-4.34 (2H, m)
Reference Example 27
N- (4-methoxybutyl) benzene-1, 2-diamine

To a solution of phenylenediamine (10.8 g) and 4- methoxybutyl methanesulfonate (9.11 g) in acetonitrile (100 ml) was added potassium carbonate (20.7 g) , and the mixture was stirred heated under reflux for 15 hr. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (35:65) was concentrated under reduced pressure to give the object product (5.44 g) . 1H-NMR (CDCl3) δ 1.67-1.82 (4H, m) , 3.13 (2H, t) , 3.24-3.39 (6H, m) , 3 . 38 -3 . 50 ( 2H, m) , 6 . 62 – 6 . 74 ( 3H, m) , 6 . 81 ( IH, in) . MS ( ESI+ , m/e ) 195 (M+l )
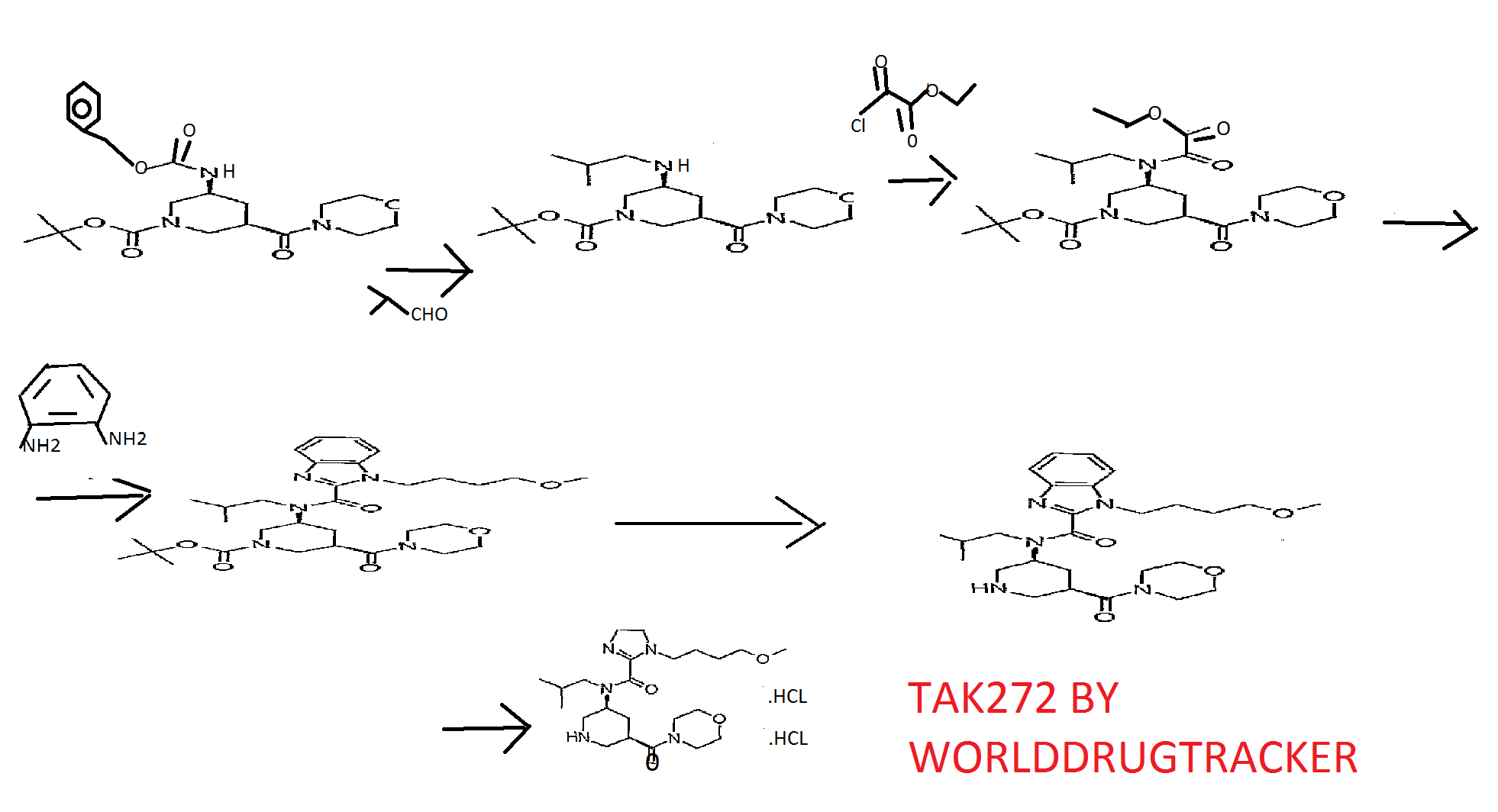
Reference Example 146 tert-butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl]carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate

A solution of tert-butyl (3S, 5R) -3- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (200 mg) , 4-itιethoxybutyl methanesulfonate (107 mg) and cesium carbonate (254 mg) in N,N-dimethylacetamide (5 ml) was stirred at 60°C for 15 hr. After cooling to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate (10 ml*2) . The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (5:95 – 3:7) was concentrated under reduced pressure to give the object product (190 mg) . 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3.46 (6H, m) , 3.49-3.91 (1OH, m) , 3.95-4.47 (5H, m) , 7.18-7.51 (3H, m) , 7.56-7.84 (IH, m) . MS (ESI+, m/e) 600 (M+l)
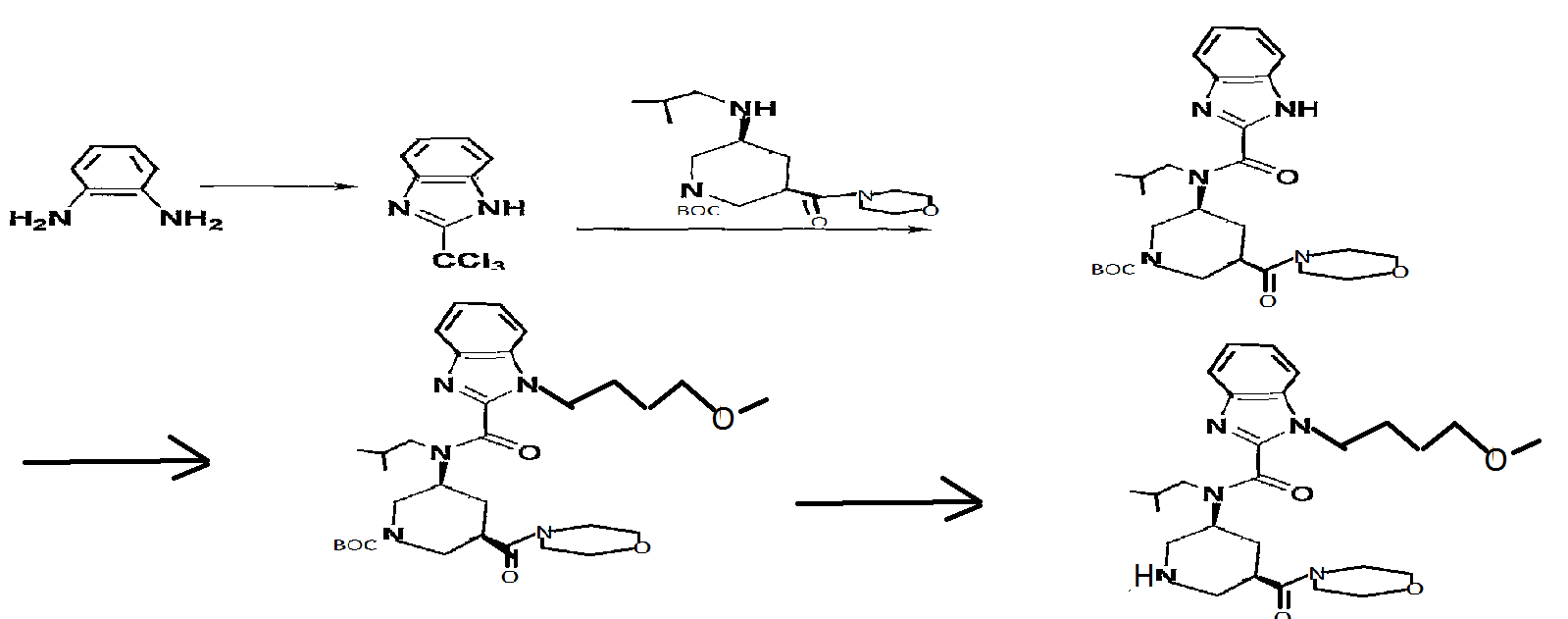
ALTERNATE METHOD IN THIS PATENT



Reference Example 61
2- (trichloromethyl) -lH-benzimidazole

O-Phenylenediamine (25 g) was dissolved in acetic acid (750 ml), and methyl 2, 2, 2-trichloroacetimidate (28.5 ml) was added dropwise over 15 min. After stirring at room temperature for 1 hr, the reaction mixture was concentrated to about 150 ml, and poured into water (1500 ml) . The precipitated crystals were collected by filtration, washed with water (1000 ml) and suspended in toluene (500 ml) . The solvent was evaporated under reduced pressure. The residue was again suspended in toluene (500 ml) and the solvent was evaporated under reduced pressure. The residue was dried under reduced pressure to give the object product (51.8 g) . 1H-NMR (CDCl3) δ 7.31-7.45 (2H, m) , 7.49-7.55 (IH, m) , 7.89 (IH, d) , 9 . 74 ( IH, br s )
Reference Example 64
1-tert-butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate

2- (Trichloromethyl) -lH-benzimidazole (19 g) and 1-tert- butyl 3-methyl (3R, 5S) -5- [ (2-methylpropyl) amino] piperidine- 1,3-dicarboxylate (25 g) were dissolved in THF (1200 ml), sodium hydrogen carbonate (67 g) and water (600 ml) were added, and the mixture was stirred at room temperature for 1 hr and at 5O0C for 1 hr. After evaporation of the solvent, the residue was extracted 3 times with ethyl acetate (700 ml) . The extract was washed successively with 10%-aqueous citric acid solution (500 ml) and brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure.
The residue was dissolved in ethyl acetate (1000 ml), subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (30.6 g) .
1H-NMR (CDCl3) δ 0.78-1.09 (6 H, m) , 1.17-1.55 (9 H, m) , 1.77-2.95 (5 H, m) , 3.11-3.79 (6 H, m) , 3.99-4.73 (4 H, m) , 7.24- 7.41 (2 H, m) , 7.45-7.59 (1 H, m) , 7.72-7.88 (1 H, m) , 10.66-10.98 (1 H, m)MS (ESI+, m/e) 459 (M+l)
Reference Example 69
1-tert-butyl 3-methyl (3R, 5S) -5- [ { [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] piperidine-1 , 3-dicarboxylate

1-tert-Butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate (30 g) and 4-methoxybutyl methanesulfonate (12.5 g) were dissolved in DMA (600 ml), cesium carbonate (32 g) was added, and the mixture was stirred at 70°C for 12 hr. The reaction mixture was poured into ice water (1000 ml), and the mixture was extracted twice with ethyl acetate (1000 ml) . The extract was washed with brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:4 – 1:1) was concentrated under reduced pressure to give the object product (28.7 g) .
1H-NMR (CDCl3) δ 0.76 (4H, d) , 1.01 (2H, d) , 1.30-1.52 (9H, m) , 1.58-2.07 (4H, m) , 2.10-2.93 (4H, m) , 3.27-3.75 (12H, m) , 4.06-4.57 (5H, m) , 7.26-7.48 (3H, m) , 7.79 (IH, d) MS (ESI+, m/e) 545 (M+l)
Example 71
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide

tert-Butyl (3S, 5R) -3- [{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, the residue was diluted with aqueous sodium bicarbonate,…and, the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (4.40 g) . MS (ESI+, m/e) 500 (M+l)
Example 101
1- (5-methoxypentyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide dihydrochloride

[1144] tert-Butyl (3S, 5R) -3- [ { [1- (5-methoxypentyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (123 mg) was dissolved in 4M hydrogen chloride-ethyl acetate (5 ml) , and the mixture was stirred at room temperature for 3 hr. The reaction mixture was concentrated, and the residue was subjected to reversed-phase preparative HPLC and the eluted fraction was concentrated under reduced pressure. The residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. 4M Hydrogen chloride-ethyl acetate (1 ml) was added and the mixture was stirred for 5 min. The solvent was evaporated under reduced pressure to give the object product (76 mg) . MS (ESI+, m/e) 514 (M+l)
PATENT
WO2013122260
http://www.google.co.in/patents/WO2013122260A1?cl=en
PATENT
WO 2011158880
http://www.google.co.in/patents/WO2011158880A1?cl=en
Reference Example 1
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -1H- benzimidazole -2 – carboxamide hydrochloride (A-type crystal)
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) was suspended dissolved piperidine-1-carboxylate The (300g) in 3N- hydrochloric acid water (1200mL) and Ethyl acetate (60mL), and stirred over 3 h at 25 ~ 35 ℃. After completion of the reaction, it was added ethyl acetate (2400mL) in the same temperature. After the addition, it was added 25% aqueous ammonia (600mL) with cooling. After the addition stirring and extracted the organic layer of 5% aqueous ammonia (600mL) was added and stirred. After stirring, the resulting organic layer it was concentrated until the solvent no longer distilled off. After concentrated, dissolved with ethyl acetate (1500mL), and transferred to solution to the crystallizer vessel, and washed with ethyl acetate (750mL). After washing, it was raised in stirring under 45 ~ 55 ℃. After raising the temperature, at the same temperature 4N- hydrogen chloride – it was dropped ethyl acetate (131.3mL). After dropping, it was to dissolve the precipitate at the same temperature. After dissolution confirmation, it was added heptane (750mL) at 40 ~ 50 ℃, after the addition, then cooled to 25 ~ 35 ℃. After cooling, the addition of A-type crystals of the seed crystals (300mg) which was obtained according to the method described in Example 265 of WO2009 / 154300, and stirred for 30 minutes or more. After stirring, the temperature was raised to 40 ~ 45 ℃, it was dropped heptane (1500mL). After the completion of the dropping, it was stirred at the same temperature. Then gradually cooled to 5 ℃ below, followed by stirring at the same temperature for 1 hour. After stirring, ethyl acetate and filtered crystals – heptane: washed with (1 1,600mL), to obtain a wet crystal. The obtained wet crystals dried under reduced pressure at 50 ℃, 1- (4- methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-yl carbonyl) piperidin-3-yl] -1H- obtained a crystalline powder of benzimidazole-2-carboxamide hydrochloride (A-type crystal, 198.82g, 74.1% yield). FINAL PRODUCT
TERT BUTYL DERIVATIVE, N-1
Reference Example 4
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate 1)
o- nitro aniline (50.0g, 0.362mol), tetrabutylammonium bromide (58.3g, 0.181mol), potassium bromide (43.1g, 0.362mol) in toluene (500mL ) and it was added. At a temperature of 20 ~ 30 ℃ 1- chloro-4-methoxy-butane (66.6g, 0.543mol) and, I was added to 50w / v% sodium hydroxide solution (145mL, 1.81mol). The reaction was heated to a temperature 85 ~ 95 ℃, and stirred for 6 hours. After cooling to a temperature 20 ~ 30 ℃, the reaction mixture water (250mL), 1N- aqueous hydrochloric acid (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), it was washed successively with water (250mL). After concentration under reduced pressure the organic layer to Contents (250mL), was added toluene (100mL), was obtained
N- (4- methoxy-butyl) -2-nitroaniline in toluene (350mL, 100% yield).
1 H-NMR (300MHz, CDCl 3) δ 1.64-1.89 (m, 4H), 3.25-3.39 (m, 2H), 3.35 (s, 3H), 3.44 (t, J = 6.1 Hz, 2H), 6.63 ( ddd, J = 8.5, 6.9, 1.2 Hz, 1H), 6.86 (dd, J = 8.5, 1.2 Hz, 1H), 7.43 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 8.07 (br s, 1H ), 8.17 (dd, J = 8.5, 1.5 Hz, 1H).
2) N- (4-methoxy-butyl) -2-10 percent in nitroaniline of toluene solution (350mL) Pd / C (K-type, 50% water-containing product) (10.0g) and toluene (100mL) it was added. Hydrogen pressure of 0.1MPa, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. A stream of nitrogen, the catalyst was filtered, I was washed with toluene (100mL). After the water in the filtrate was separated off and adding magnesium sulfate (25.0g) at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered over magnesium sulfate, washed with toluene (100mL), was obtained N- (4- methoxybutyl) -o- toluene solution of phenylenediamine (100% yield).
1 H NMR (500 MHz, CDCl 3) δ1.67-1.78 (m, 4H), 3.12-3.14 (m, 2H), 3.32 (br, 3H), 3.35 (s, 3H), 3.41-3.47 (m, 2H), 6.63-6.69 (m, 2H), 6.69-6.74 (m, 1H), 6.82 (td, J = 7.57, 1.58 Hz, 1H).
3) N- (4- methoxy-butyl) -o- After the toluene solution of phenylenediamine cooled to a temperature 0 ~ 10 ℃, acetic acid (65.2g, 1.09mol) and 2,2,2 trichloroacetimide acid methyl ( 70.3g, 0.398mol) and I were added. After stirring for 30 minutes at a temperature 0 ~ 10 ℃, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. The reaction was 5w / v% saline (250mL), 2N- aqueous hydrochloric acid / 5w / v% sodium chloride solution: a mixture of (1 1) (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), 5w / v It was washed successively with% saline solution (250mL). A stream of nitrogen, was added magnesium sulfate (25.0g) to the organic layer at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered magnesium sulfate, and washed with toluene (100mL). The filtrate was concentrated under reduced pressure and the amount of contents (150mL). Stir the concentrated solution at a temperature 20 ~ 30 ℃, was allowed to precipitate crystals, was added dropwise heptane (750mL). The crystals bleeding is heated to a temperature 40 ~ 50 ℃, after stirring for 30 min, cooled to a temperature 0 ~ 10 ℃, and the mixture was stirred at the same temperature for 2 hours.The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,150 mL). And dried under reduced pressure at 40 ℃, it was obtained 1- (4-methoxy-butyl) -2-fine brown crystals of trichloromethyl -1H- benzimidazole (96.5g, 82.9% yield from o- nitroaniline).
1 H-NMR (300MHz, CDCl 3) δ: 1.68-1.85 (m, 2H), 1.99-2.17 (m, 2H), 3.37 (s, 3H), 3.48 (t, J = 6.1 Hz, 2H), 4.50 -4.65 (m, 2H), 7.27-7.49 (m, 4H), 7.82-7.93 (m, 1H).
. Anal Calcd for C 13 H 15 Cl 3 N 2 O:. C, 48.55; H, 4.70; N, 8.71; Cl, 33.07 Found: C, 48.30; H, 4.61; N, 8.74; Cl, 33.30.
4) pyridine-3,5-dicarboxylic acid (110g, 0.66mol), it was dropped methanol (660 mL) mixture of concentrated sulfuric acid at a temperature of 50 ℃ or less of (226.0g, 2.30mol). Thereafter, the mixture was stirred and heated to a temperature 55 ~ 65 ℃ 7 hours. The reaction was the temperature 40 ~ 50 ℃, was added water (220mL). And further dropping temperature 40-50 5% aqueous ammonia at ℃ (about 1.10L) was adjusted to pH8.0 ~ 8.5. After stirring at a temperature 40 ~ 50 ℃ 30 minutes and stirred for 1 hour and cooled to a temperature 0 ~ 10 ℃. Was collected by filtration precipitated crystals, methanol – water (1: 3,165mL), and washed successively with water (440mL). To obtain a white crystalline powder pyridine-3,5-dicarboxylic acid dimethyl and dried under reduced pressure at 50 ℃ (105.0g, 82.0% yield).
1 H-NMR (300 MHz, CDCl 3) δ 4.00 (s, 6H), 8.87 (s, 1H), 9.37 (s, 2H).
. Anal Calcd for C 9 H 9 NO 4:. C, 55.39; H, 4.65; N, 7.18; O, 32.79 Found: C, 55.42; H, 4.65; N, 7.16.
5) 1 L autoclave pyridine-3,5-dicarboxylic acid dimethyl (100g, 0.51mol) and was charged with dimethylacetamide (400mL), temperature 30 ℃ below with trifluoroacetic acid (59.2mL, after dropping the 0.77mol), 10% Pd-C (PE-type) the (20.0g) it was added. Hydrogen pressure of 0.5 ~ 0.7MPa, it was stirred for 12 hours at a temperature of 55 ~ 65 ℃. The catalyst was filtered off, it was washed with dimethylacetamide (50mL × 2). Triethylamine and the combined filtrates at a temperature 20 ~ 30 ℃ (77.8g, 0.77mol) was added dropwise, and adjusted to pH9.0 ~ 10.0. Temperature 30 ~ 40 ℃ by di -tert- butyl (134g, 0.614mol) was added dropwise and stirred at the same temperature for 2 hours. After the reaction mixture as a 20 ~ 30 ℃, it was added ethyl acetate (600mL), washed with water (900mL). The aqueous layer it was re-extracted with ethyl acetate (400mL). The combined organic layers 5w / v% citric acid -10w / v% sodium chloride solution (600mL), 3% aqueous sodium bicarbonate (600mL), and washed successively with water (600mL). Contents The organic layer (200mL) until it was concentrated under reduced pressure, methanol (250mL) was added to the concentrated solution, and then concentrated under reduced pressure until Contents (200mL). The addition of methanol (250mL) again concentrate, After concentration under reduced pressure until Contents (200mL), was added methanol (2.40L). The solution in water (18.5g, 1.03mol), cesium carbonate (417g, 1.28mol) was added and stirred for about 24 hours at a temperature 55 ~ 65 ℃. The reaction solution was the temperature 20 ~ 30 ℃, concentrated to Contents (700mL), it was added tetrahydrofuran (500mL). The solution temperature at 15 ~ 35 ℃ 2N- hydrochloric acid solution (1.28L, 2.56mol) was added dropwise and adjusted to pH3.0 ~ 3.5, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Extracted with ethyl acetate (750mL × 2), and the organic layer was washed with 10w / v% aqueous sodium chloride solution (500mL × 3). Contents The organic layer (300mL) until it was concentrated under reduced pressure, to obtain a weight content by adding ethyl acetate (650mL).Heating the concentrate to a temperature of 55 ~ 65 ℃, it was added dropwise heptane (500mL). It cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. The precipitated crystals were collected by filtration, ethyl acetate – heptane: was washed with (1 1,120mL). Dried under reduced pressure at 50 ℃ 1- (tert- butoxycarbonyl) to give a white crystalline powder of piperidine-3,5-dicarboxylic acid (113.3g, 80.9% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.40 (s, 9H), 1.44-1.61 (m, 1H), 2.21-2.26 (m, 1H), 2.31-2.41 (m, 2H), 4.10- 4.12 (m, 2H).
. Anal Calcd for C 12 H 19 NO 6:. C, 52.74; H, 7.01; N, 5.13; O, 35.13 Found: C, 52.96; H, 6.99; N, 5.39.
6) Under a nitrogen stream, 1- (tert- butoxycarbonyl) piperidine-3,5-dicarboxylic acid (5.00g, 18.3mmol) was suspended in tetrahydrofuran (10.0mL), trifluoroacetic acid anhydride at a temperature 20 ~ 30 ℃ It was dropping things (3.80mL, 27.5mmol). After the completion of the dropping, it was stirred for 1 hour at a temperature of 20 ~ 30 ℃. It was added dropwise heptane (20.0mL) at a temperature 20 ~ 30 ℃ the reaction solution, and stirred for 3 hours then cooled to a temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with heptane (3.00mL). Dried under reduced pressure at 40 ℃ 2,4- dioxo-3-oxa-7-azabicyclo [3,3,1] white crystalline powder of nonane-7-carboxylic acid tert- butyl was obtained (4.03g, yield 86.1%).
1 H-NMR (300 MHz, CDCl 3) δ 1.43 (s, 9H), 1.93-1.99 (m, 1H), 2.40-2.46 (m, 1H), 3.06-3.11 (m, 4H), 4.50-4.54 ( m, 2H).
. Anal Calcd for C 12 H 17 NO 5:. C, 56.46; H, 6.71; N, 5.49; O, 31.34 Found: C, 56.51; H, 6.63; N, 5.69.
7) Under a nitrogen stream, quinidine (69.9g, 0.215mol) and was charged with tetrahydrofuran (200mL), and cooled to a temperature -5 ~ 5 ℃. At the same temperature 2,4-dioxo-3-oxa-7-azabicyclo [3,3,1] nonane-7-carboxylic acid tert- butyl (50.0g, 0.196mol) was added and washed with tetrahydrofuran (50.0mL) crowded. Temperature -5 ~ 5 methanol at ℃ (9.41g, 0.29 4mol) was added dropwise, and the mixture was stirred for 2 hours at a temperature -5 ~ 5 ℃. Ethyl acetate (350mL) to the reaction mixture, was by adding minute solution 20w / v% citric acid aqueous solution (250mL). The aqueous layer it was re-extracted with ethyl acetate (125mL × 2). The organic layers were combined 20w / v% aqueous solution of citric acid (250mL), I was washed successively with water (250mL × 2). The organic layer it was concentrated under reduced pressure. To the residue ethanol (100mL) was added ethyl acetate (450mL) was heated to a temperature 60 ~ 70 ℃, (R) – was added phenethylamine (23.7g, 0.196mol). Temperature 50-60 for one hour at ℃, 1 hour at a temperature of 20 ~ 30 ℃, it was stirred for 1 hour at a temperature of -5 ~ 5 ℃. The precipitated crystals were collected by filtration, ethanol – ethyl acetate: and washed with (2 9,100mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidin-3 to give a white crystalline powder of the carboxylic acid (1R) -1- phenylethylamine salt It was (55.7g, 69.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.42 (s, 9H), 1.43-1.51 (m, 3H), 2.06-2.14 (m, 1H), 2.21-2.26 (m, 1H), 2.39- 2.44 (m, 1H), 2.52-2.53 (m, 1H), 2.57 (br s, 2H), 3.64 (s, 3H), 4.12 (br s, 2H), 4.19-4.26 (m, 1H), 7.30- 7.40 (m, 3H), 7.45-7.48 (m, 2H).
. Anal Calcd for C 21 H 32 N 2 O 6:. C, 61.75; H, 7.90; N, 6.86; O, 23.50 Found: C, 61.54; H, 7.77; N, 6.86.
8) (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (1R) -1- phenylethylamine salt (20.0g, 49.0mmol), methanol (20mL) and it was charged with water (80mL). Temperature 20-30 citric acid at ℃ (11.3g, 58.8mmol) was added dropwise a solution prepared by dissolving in water (20.0mL), and the mixture was stirred 1.5 hours at the same temperature. The precipitated crystals were collected by filtration and washed with water (60mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- give a white crystalline powder (methoxycarbonyl) piperidine-3-carboxylic acid (13.5g, 96.1% yield ).
1 H-NMR (300 MHz, CDCl 3) δ 1.40 (s, 9H), 1.46-1.59 (m, 1H), 2.22-2.27 (m, 1H), 2.37-2.45 (m, 2H), 2.63-2.73 ( m, 2H), 3.63 (s, 3H), 4.14 (br s, 2H), 12.51 (br s, 1H).
. Anal Calcd for C 13 H 21 NO 6:. C, 54.35; H, 7.37; N, 4.88; O, 33.41 Found: C, 54.14; H, 7.28; N, 4.85.
9) Under a nitrogen stream, (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 104mmol), triethylamine (31.7g, 313mmol) and toluene ( It was charged with 180mL). Diphenylphosphorylazide at a temperature of 15 ~ 35 ℃ (28.7g, 313mmol) I was dropped a toluene (30.0mL) solution. After stirring at a temperature 30 ± 5 ℃ 30 minutes, and the mixture was stirred and heated to a temperature 65 ~ 75 ℃ 30 minutes. Temperature 60 ~ 70 ℃ in the benzyl alcohol (12.4g, 115mmol) it was dropped. To a temperature 80 ~ 90 ℃ was stirred and heated for 3 hours. The reaction mixture was cooled to a temperature 20 ~ 30 ℃, sodium nitrite (7.20g, 104mmol) and after stirring was added a solution prepared by dissolving in water (150mL) 1 hour, the aqueous layer was separated. The organic layer 5w / v% aqueous sodium bicarbonate solution (150mL), 20w / v% aqueous citric acid solution (150mL), washed successively with 5w / v% aqueous sodium chloride solution (150mL), the organic layer was concentrated under reduced pressure. The residue methanol (60.0mL) was added and concentrated under reduced pressure to. The more we went once in the same manner.To the residue was added methanol and the content amount of the (90.0g). Temperature 15 ~ 35 ℃ 2N- aqueous sodium hydroxide (62.6mL, 125mmol) was added and stirred for 1 hour at a temperature 30 ± 5 ℃. Temperature 20 ~ 30 ℃ in methanol (120mL), was added to 20w / v% aqueous citric acid solution (300mL), it was a pH3.0 ~ 3.5. After stirring for 30 minutes at a temperature 50 ~ 60 ℃, cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. It was stirred for 1 hour at the temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with water (90.0mL). And dried under reduced pressure at 50 ℃ (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) to yield a white crystalline powder piperidine-3-carboxylic acid (35.0 g, 88.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 2.11 (d, J = 12.4 Hz, 1H), 2.40-2.48 (m, 4H), 2.62 (br s, 1H), 4.08 (t, J = 14.4 Hz, 2H), 5.04 (s, 2H), 7.31-7.41 (m, 5H), 12.53 (br s, 1H).
. Anal Calcd for C 19 H 26 N 2 O 6:. C, 60.30; H, 6.93; N, 7.40; O, 25.37 Found: C, 60.03; H, 6.99; N, 7.41.
10) Under a nitrogen stream, (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 79.3mmol), morpholine (7.60 g, 87.2mmol), 1- hydroxybenzotriazole monohydrate (2.43g, it was charged with 15.9mmol) and dimethylacetamide (90.0mL). Hydrochloride 1-ethyl at a temperature 20 ~ 30 ℃ -3- (3- dimethylaminopropyl) carbodiimide (16.7g, 87.1mmol) after addition and stirred for 1 hour at a temperature 45 ~ 55 ℃. Temperature 45 ~ 55 ℃ with tetrahydrofuran (90.0mL), sequentially dropwise addition of water (210mL), and stirred for 1 hour. After stirring for 1 hour and cooled to a temperature 20 ~ 30 ℃, were collected by filtration the precipitated crystals, tetrahydrofuran – water: washing with (1 3,120mL). And dried under reduced pressure at 50 ℃ tert- butyl piperidine -1- (3S, 5R) -3 – a white crystalline powder of {[(benzyloxy) carbonyl] amino} -5 (morpholin-4-yl-carbonyl) carboxylate It was obtained (32.7g, 92.3% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 1.49-1.57 (m, 1H), 1.87 (d, J = 12.3 Hz, 1H), 2.43 (br s, 1H), 2.63-2.71 (m, 1H), 2.79-2.83 (m, 1H), 3.37-3.54 (m, 9H), 3.89 (d, J = 11.5 Hz, 1H), 4.06 (br s, 1H), 5.03 (s , 2H), 7.30-7.38 (m, 5H).
. Anal Calcd for C 23 H 33 N 3 O 6:. C, 61.73; H, 7.43; N, 9.39; O, 21.45 Found: C, 61.59; H, 7.50; N, 9.43.
11) tert- Butyl piperidin -1- (3S, 5R) -3 – {[(benzyloxy) carbonyl] amino} -5- (morpholin-4-ylcarbonyl) carboxylate (30.0g, 67.0mmol), isobutyraldehyde (7.25g, 101mmol), it was charged with 10% Pd-C (PE type) (1.50g) and methanol (240mL).Hydrogen pressure of 0.2 ~ 0.3MPa, it was stirred for 4 hours at a temperature of 20 ~ 30 ℃. The catalyst is filtered off and washed with methanol (60.0mL). The filtrate was concentrated under reduced pressure, ethyl acetate was added (60.0mL), and concentrated under reduced pressure again. The residue ethyl acetate was added, followed by the amount of contents (360mL). Temperature 45-55 succinate by heating to ℃ (7.90g, 67.0mmol) was added. After stirring for 1 hour at a temperature 45 ~ 55 ℃, cooled to a temperature 20 ~ 30 ℃, and stirred for 1 hour. The precipitated crystals were collected by filtration, and washed with ethyl acetate (90.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [(2- methyl-propyl) amino] -5- (morpholin-4-yl-carbonyl) piperidine – 1-carboxylate white crystals of alert succinate got sex powder (30.2g, 92.5% yield).
1 H-NMR (300 MHz, D 2 O) δ 1.02 (s, 3H), 1.04 (s, 3H), 1.47 (s, 9H), 1.97-2.09 (m, 2H), 2.26-2.30 (m, 1H ), 2.55 (s, 4H), 2.99 (d, J = 7.0 Hz, 2H), 3.23 (br s, 1H), 3.39-3.45 (m, 2H), 3.53-3.80 (m, 10H), 3.82-3.93 (br s, 1H).
. Anal Calcd for C 23 H 41 N 3 O 8:. C, 56.66; H, 8.48; N, 8.62; O, 26.25 Found: C, 56.48; H, 8.46; N, 8.39.
12) tert- Butyl (3S, 5R) -3 – [(2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine – 1 – carboxylate succinate (30.3g, 62.2mmol), acetonitrile (60.0mL) and, it was charged with water (40.0mL). Then after stirring was added potassium carbonate (34.4g, 0.249mmol) 10 minutes, 1- (4-methoxybutyl) -2-trichloromethyl -1H- benzimidazole (20.0g, 62.2mmol) was added. After stirring for 2 hours at a temperature of 70 ~ 80 ℃, it was added dimethyl sulfoxide (15.0mL), and the mixture was stirred for 6 hours at a temperature 70 ~ 80 ℃. After cooling the reaction mixture to a temperature 20 ~ 30 ℃, water (120mL), it was separated and by adding toluene (240mL). The organic layer 10w / v% sodium chloride solution (100mL), 10w / v% aqueous solution of citric acid (100mL), it was washed sequentially with 10w / v% sodium chloride solution (100mL). The organic layer of activated carbon Shirasagi A a (1.0g) was added, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Activated carbon was filtered, washed with toluene (40.0mL), and concentrated under reduced pressure of the filtrate to 110 mL. By heating to a temperature 35 ~ 45 ℃ was added dropwise heptane (280mL). At a temperature 35 ~ 45 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5 – and the mixture was stirred for 1 hour at (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was added to the same temperature the crystals (10mg) of the acrylate. Heptane (140mL) was stirred and added dropwise to 30 minutes at a temperature 35 ~ 45 ℃. It was cooled to a temperature 20 ~ 30 ℃ and stirred for 2 hours. The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,40.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] – 5- (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was obtained a pale yellowish crystalline powder of alert (27.7g, 74.2% yield).
1 H-NMR (300 MHz, CDCl 3) δ 0.68-0.80 (m, 3H), 0.96-1.08 (m, 3H), 1.31 (br s, 5H), 1.49 (s, 4H), 1.61-1.71 (m , 2H), 1.71 (br s, 0.5H), 1.92-2.05 (m, 3H), 2.05-2.24 (m, 2H), 2.45 (br s, 1H), 2.60 (br s, 1H), 2.72-2.96 (m, 2H), 3.26-3.35 (m, 3H), 3.35-3.47 (m, 2H), 3.47-3.73 (m, 10H), 4.02-4.26 (m, 2H), 4.26-4.34 (m, 1H) , 4.34-4.47 (m, 0.5H), 7.25-7.29 (m, 1H), 7.29-7.41 (m, 1H), 7.41-7.53 (m, 1H), 7.64 (br s, 0.5H), 7.79 (d , J = 8.2 Hz, 0.5H).
. Anal Calcd for C 32 H 49 N 5 O 6:. C, 64.08; H, 8.23; N, 11.68; O, 16.01 Found: C, 63.82; H, 8.12; N, 11.64.
PATENT
TAKEDA PHARMACEUTICAL COMPANY LIMITED [JP/JP]; 1-1, Doshomachi 4-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP)
Provided is a method for producing a synthetic intermediate of a heterocyclic compound having a renin inhibitory activity and effective as a prophylactic or therapeutic drug against diabetic renal disease, hypertension, and the like. A method for producing a compound represented by formula (III-1a), (III-1b), (III-1c), and/or (III-1d) [where the symbols in the formulas are as defined in the description], or a salt thereof, said method characterized in that a compound represented by formula (Ia) or (Ib) [where the symbols in the formulas are as defined in the description] or a salt thereof is reacted with a compound represented by formula (II) [where the symbols in the formula are as defined in the description] or a salt thereof in the presence of an aluminum compound and a chiral amine compound.
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.[Formula 3]

(Wherein each symbol is as described in Patent Document 2.)
Prior art documents
Patent literaturePatent Document 1: Patent No. 4,800,445 Patent
reaction vessel (1R, 3S) – was added to cyclohexane-1,3-dicarboxylic acid (10g) and THF (20mL), 5 It was cooled to ℃. It was added dropwise trifluoroacetic anhydride (8.19mL), and the mixture was stirred for about 1 hour. The reaction mixture was allowed to warm to room temperature, heptane (20mL) was added, up to 5 ℃ was cooled and stirred for about 30 minutes. The precipitate was filtered off, washed with heptane to give the title compound. Yield (6.7g)
reactor in THF (240ml), (3S, 5R) -1- (tert – butoxycarbonyl) -5- (morpholine-4-carbonyl) piperidine-3-carboxylic acid (20.0g), triethylamine (12.2mL) and diphenylphosphoryl azide (15.1mL) They were charged and allowed to react for 1 hour at 60 ℃, cooled to 25 ℃. After cooling the THF (60ml) and sodium trimethyl silanolate (19.7g) to charged 0 ℃ separately reaction vessel, was added dropwise to this was allowed to react before the reaction solution over about 1 hour, 0 at 0 ℃. 5 hours it was allowed to react. 0 slowly added dropwise acetic acid (40mL) at ℃, After stirring for 10 minutes, was added ethanol (60ml) and isobutyraldehyde (5.3mL) at 25 ℃, and stirred for 10 minutes. Then added sodium borohydride (1.88g), and the mixture was stirred for 30 minutes, and further addition of sodium borohydride (1.88g) at 25 ℃, and the mixture was stirred for 30 minutes. After completion of the reaction, water (100mL) was added and stirred for 10 minutes at room temperature. The organic layer was concentrated, then added dropwise slowly toluene (140ml) and 5N aqueous sodium hydroxide solution (120ml), the layers were separated. After washing and addition of aqueous 1N sodium hydroxide (100ml) the organic layer was washed 1N aqueous sodium hydroxide (100ml) was added again organic layer. The aqueous layers were combined and extracted by addition of toluene (100ml). The organic layers were combined, washed with 10w / v% aqueous sodium chloride solution (100ml), and the organic layer was concentrated. It was added ethanol (100ml), after it was concentrated under reduced pressure until about 60ml, warmed to 60 ℃ by the addition of ethyl acetate (40ml). Was added succinic acid (6.9g), After stirring for 30 minutes, it was added dropwise ethyl acetate (200ml) at 60 ℃, and stirred for 30 minutes. After stirring for 1 hour at room temperature, and the mixture was stirred for 1 hour at 0 ℃. The crystals were collected by filtration and washed with a mixture of ethyl acetate / n-heptane (6/1) (60mL). The obtained crystals at an external temperature of 50 ℃ to constant weight and then dried under reduced pressure to give the title compound as almost white crystals. Yield (22.8g)
the reaction vessel in chlorobenzene (7.5mL) and quinine (0.70g ) is added and stirred, it was added dropwise DIBAL1.0M hexane solution (2.16mL). The reaction mixture was cooled to -40 ℃, tert – butyl 2,4-dioxo-3-oxa-7-azabicyclo [3.3.1] was added nonane-7-carboxylic acid ester (0.50g), about 1 hour stirring. Was added chlorobenzene to another reaction vessel (2.5mL) and morpholine (0.17mL), the resulting solution was cooled to -40 ℃ was added dropwise to the previous reaction solution. After completion of the reaction, the mixture was separated with ethyl acetate and 10w / w% aqueous citric acid solution, and the resulting aqueous layer was re-extracted with ethyl acetate. The organic layers were combined, washed with 10w / w% saline, and concentrated to give the title compound. 1 H NMR (500 MHz, DMSO-D 6 ) delta ppm 1.41 (s, 9 H), 1.47 – 1.72 (M, 1 H), 1.89 – 2.10 (M, 1 H), 2.36 – 2.49 (M, 1 H ), 2.55 – 2.83 (m, 3 H), 3.40 – 3.50 (m, 2 H), 3.51 -.. 3.57 (m, 4 H), 3.59 (br s, 2 H), 3.83 – 4.04 (m, 1 H), 4.05 – 4.29 (m, 1 H), 12.52 (s, 1 H) optical purity of 94.3% EE <HPLC analytical conditions> column: CHIRALPAK IC (Co., Ltd. Daicel) column temperature: constant around 15 ℃ Temperature Mobile phase: A solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 70: 30 B solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 50 : 50 gradient program
| WO2010150840A1 | 24 Jun 2010 | 29 Dec 2010 | Dainippon Sumitomo Pharma Co., Ltd. | N-substituted-cyclic amino derivative |
| WO2011158880A1 | 15 Jun 2011 | 22 Dec 2011 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| WO2012062687A1 * | 7 Nov 2011 | 18 May 2012 | F. Hoffmann-La Roche Ag | Triazole derivatives and their use for neurological disorders |
| WO2013122260A1 | 14 Feb 2013 | 22 Aug 2013 | Takeda Pharmaceutical Company Limited | Tablet |
| CN103221402B * | 7 Nov 2011 | 17 Jun 2015 | 霍夫曼-拉罗奇有限公司 | 三唑衍生物及其用于神经障碍的用途 |
| US8329691 | 14 Oct 2008 | 11 Dec 2012 | Takeda Pharmaceutical Company Limited | Amide compounds and use of the same |
| US8389511 | 19 Dec 2008 | 5 Mar 2013 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic heterocyclic derivative |
| US8658639 | 24 Jun 2010 | 25 Feb 2014 | Dainippon Sumitomo Pharma Co., Ltd | N-substituted-cyclic amino derivative |
| US8742097 | 2 Nov 2011 | 3 Jun 2014 | Hoffmann-La Roche Inc. | Triazole compounds I |
| US9018374 | 15 Jun 2011 | 28 Apr 2015 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| US9090601 | 28 Jan 2010 | 28 Jul 2015 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
///////////TAK 272, Hypertension
BMS-248360, A NEW SARTAN ON HORIZON

2-[4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-[(3,3-dimethyl-2-oxopyrrolidin-1-yl)methyl]phenyl]-N-(3,4-dimethyl-1,2-oxazol-5-yl)benzenesulfonamide
4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide,
4′- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 ‘-biphenyl] -2-sulfonamide
4‘-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N–(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide
BMS-248360
PRECLINICAL …..treating hypertension
Bristol Myers Squibb Co, INNOVATOR
Hypertension remains one of the largest unmet medical needs in the 21st century, especially when one considers that hypertension is the portent of future debilitating cardiovascular disease. While many drugs are available for treating the disease, approximately one-third of the hypertensive population is still not adequately treated. Of the more recent avenues explored for treating hypertension, disruption of the effects of either angiotensin II (AII) or endothelin-1 (ET-1) has shown promise. These endogenous vasoactive peptides are among the most potent vasoconstrictors and cell proliferative factors identified to date. AII is the effector molecule of the renin−angiotensin system (RAS), and a large number of AII receptor (AT1) antagonists, including irbesartan , have been developed for treating hypertension


SYNTHESIS
picked from…….http://www.drugfuture.com/synth/syndata.aspx?ID=324487

EP 1094816; JP 2002519380; US 2002143024; WO 0001389
The intermediate biphenyl aldehyde (XI) is prepared by two related methods. 4-Bromo-3-methylbenzonitrile (I) is oxidized to aldehyde (II) via radical bromination with N-bromosuccinimide/benzoyl peroxide, followed by treatment with trimethylamine N-oxide. Suzuki coupling of aryl bromide (II) with the pinacol boronate (III) affords biphenyl (IV). After protection of the aldehyde moiety of (IV) as the corresponding ethylene ketal (V), its cyano group is reduced to aldehyde (VI) employing DIBAL in THF. Subsequent reduction of (VI) with NaBH4 leads to alcohol (VII), which is further converted into the benzyl bromide (VIII) by means of CBr4/PPh3. Bromide (VIII) is condensed with the spiro imidazolone (IX) in the presence of NaH, to produce (X). Then acidic hydrolysis of the ethylene ketal and SEM groups of (X) gives rise to the intermediate aldehyde (XI)
NEXT

Alternatively, reduction of 4-bromo-3-formylbenzonitrile ethylene ketal (XII) by means of DIBAL leads to aldehyde (XIII), which is further reduced to alcohol (XIV) with NaBH4. After bromination of (XIV) with CBr4/PPh3, the resultant benzyl bromide (XV) is condensed with the spiro imidazolone (IX), yielding (XVI). Then, acidic ketal hydrolysis in (XVI) furnishes aldehyde (XVII). Suzuki coupling between aryl bromide (XVII) and boronic acid (XVIII) gives biphenyl (XIX). The SEM group of (XIX) is then removed under acidic conditions to provide (XI)

Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC

Coupling of 2-bromobenzenesulfonyl chloride (I) with 5-amino-3,4-dimethylisoxazole (II) affords sulfonamide (III), which is further protected as the N-methoxyethoxymethyl derivative (IV) employing MEM-chloride in DMF. Lithiation of bromosulfonamide (IV), followed by treatment with trimethyl borate and acidic work up leads to the boronic acid intermediate (V). This is then subjected to Suzuki coupling with 4-bromo-3-methylbenzaldehyde (VI) to yield the biphenyl adduct (VII). After reduction of aldehyde (VII) to the benzylic alcohol (VIII) with NaBH4, reaction with methanesulfonyl chloride and diisopropylethylamine gives rise to the mesylate (IX) (1-3).

Mesylate (IX) is condensed with ethyl 2-propyl-4-ethylimidazole-5-carboxylate (X) yielding (XI). Simultaneous ester group hydrolysis and MEM group deprotection under acidic conditions gives rise to the imidazolecarboxylic acid (XII). This is finally coupled with methylamine via activation with CDI to produce the desired N-methyl carboxamide (1-3).
Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC
PAPER
 BMS 248360
BMS 248360The ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
15 as a white solid (40 mg, 31%):
mp 104−110 °C;
1H NMR (CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H), 1.08 (s, 3H), 1.14 (s, 3H), 1.36 (m, 2H), 1.61 (m, 2H), 1.75−2.06 (m, 13H), 2.17 (s, 3H), 2.39 (m, 2H), 4.18 (m, 2H), 4.71 (m, 2H), 7.02−7.93 (m, 7H);
13CNMR (CDCl3 ) δ 7.82, 11.91, 14.79, 23.36, 25.50, 25.61, 27.11, 28.81, 29.88, 35.33, 38.42, 41.48, 44.59, 46.24, 46.47, 109.29, 125.15, 125.76, 129.68, 130.58, 131.76, 133.20, 134.07, 137.15, 138.27, 139.11, 139.57, 155.81, 162.68, 162.91, 181.25, 187.83.
Anal. (C36H45N5O5S) C, H, N, S.
……………………………
PATENT
US 2002143024
http://www.google.com/patents/US20020143024
 Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.
Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.

N-(3,4-Dimethyl-5-iso-xazolyl)-2′-formyl-4′-(hydroxy-methyl)-N-[[2-(tri-methylsilyl)ethoxy]- methyl][1,1′- biphenyl]-2- sulfonamide
Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
[0414]

Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
A. 4′-Cyano-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130° C. for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: Rf=0.57, silica gel, 1:2 hexane/EtOAc.
B. 2′-(1,3-Dioxolan-2-yl)-4′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0° C. was added DIBAL-H (1M in CH2Cl2, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCl solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: Rf=0.45, silica gel, 2:3 hexane/EtOAc.
C. 2′-(1,3-Dioxolan-2-yl)-4′-hydroxymethyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4′-Bromomethyl-2′-(1,3-dioxolan-2-yl)-N-(3,4′-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: Rf=0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
3D (750 mg, 1.3 mmol) was treated with 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2Cl2/MeOH to afford 3E as a gum (830 mg, 93%): Rf=0.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2Cl2 /MeOH to afford the title compound as a gum (480 mg, 72%): Rf=0.16, silica gel, 100:5 CH2Cl2/MeOH.
Example 4 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2′-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl][1,1′-biphenyl]-2-sulfonamide

To 3F (110 mg, 0.20 mmol) in CH2Cl2 (4 mL) was added 4-amino-2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3 Å molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2Cl2 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2Cl2, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104-110° C. Analysis calculated for C36H45N5O5S.0.8 H2O: Calc’d: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide (Alternative Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)]-1,3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at −78° C. The solution was allowed to warm to 0° C. After 1 hour, a solution of Rochelle’s salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hydroxymethyl)phenyl)]-1,3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0° C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification.
C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-1,3-dioxolane
Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0° C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane).
D. 2-(1,3-Dioxolan-2-yl)-4-[(2-n-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]bromobenzene
[0436] Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol, 2.5 eq) was added in portions over 15 minutes to a mixture of 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0° C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and 1N hydrochloric acid (30 mL) was heated at 65° C. for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5-isoxazolyl)[(2-methoxyethoxy)methyl]amino]sulfonyl]phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G=3F) in 73% yield: Rf=0.2 (silica gel using CH2Cl2/MeOH [100:5]).
PATENT
EP 1237888; WO 0144239
Example 3 4′-r(2-Butyl-4-oxo-1.3-diazaspiror4.41non-l-en-3-yl)methvn-2′-formyl-N-
(3, 4-dimethyl-5-isoxazolyl)-[ 1,1 ‘-biphenyl] -2-sulfonamide

A. 4′-Cvano-2>-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1.1 ‘-biphenyl] -2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130°C for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: R^0.57, silica gel, 1:2 hexane EtOAc.
B. 2,-(1.3-Dioxolan-2-yl)-4′-formyl-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1 , l’-biphenyl] -2-sulfonamide To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0°C was added DIBAL- H (IM in CH2C12, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCI solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: R^O.45, silica gel, 2:3 hexane/EtOAc. C. 2′-(1.3-Dioxolan-2-yl)-4′-hvdroxymethyl-N-(3.4-dimethyl-5- isoxazolyl)-N-(2-methoxyethoxymethyl) [1.1 ‘-biphenyl] -2- sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4l-Bromomethyl-2,-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)- N-(2-methoxyethoxymethyl) [1 , l’-biphenyl] -2-sulfonamide 3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: R^0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn- 2,-(1.3- dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [ 1. l’-biphenyll -2-sulfonamide 3D (750 mg, 1.3 mmol) was treated with 2-re-butyl-l,3- diazaspiro[4.4]non-l-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2CL/MeOH to afford 3E as a gum (830 mg, 93%): R^O.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-r(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,– formyl-N-(3.4-dimethyl-5-isoxazolyl)-[l.l’-biphenyl1-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2C12 /MeOH to afford the title compound as a gum (480 mg, 72%): R^O.16, silica gel, 100:5 CH.Cl MeOH.
Example 4
4′- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 ‘-biphenyl] -2-sulfonamide
To 3F (110 mg, 0.20 mmol) in CH2C12 (4 mL) was added 4-amino- 2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3A molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2C12 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2C12, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104- 110°C. Analysis calculated for C36H45N5O5S • 0.8 H2O: Calc’d: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,-formyl-N-
(3,4-dimethyl-5-isoxazolyl)-[l,l’-biphenyl]-2-sulfonamide (Alternative
Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)1-1.3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-l,3- dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at -78 °C. The solution was allowed to warm to 0 °C. After 1 hour, a solution of Rochelle’s salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hvdroxymethyl)phenyl)l-1.3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0 °C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification. C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-l,3-dioxolane Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0 °C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-l,3-dioxolane).
D. 2-( 1 ,3-Dioxolan-2-yl)-4- [ (2-re-butyl-4-oxo- 1 ,3-diazaspiro [4.4] non- 1- en-3-yl)methyl] bromobenzene Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol,
2.5 eq) was added in portions over 15 minutes to a mixture of 2-rc-butyl- l,3-diazaspiro[4.4]non-l-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0°C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2- formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and IN hydrochloric acid (30 mL) was heated at 65°C for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-.(2-Butyl-4-oxo-1.3-diazaspiro■4.41non-l-en-3-yl)methyl1-2,– formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) f 1.1 ‘-biphenyl] -2-sulfonamide Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5- isoxazolyl) [(2-methoxyethoxy)methyl] amino] sulfonyl] phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4’-[ 2-Butyl-4-oxo-1.3-diazaspiro[4■41non-l-en-3-yl)methvn-2,– formyl-N-(3 ,4-dimethyl-5-isoxazolyl)- fi .1 ‘-biphenyl] -2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G = 3F) in 73% yield: R^0.2 (silica gel using CH2ClJ eOH [100:5]).
| Patent | Submitted | Granted |
|---|---|---|
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6638937] | 2002-10-03 | 2003-10-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6835741] | 2004-06-03 | 2004-12-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6852745] | 2004-07-01 | 2005-02-08 |
///////////BMS-248360, Preclinical, SARTAN, BMS, HYPERTENTION
CCCCC1=NC2(CCCC2)C(=O)N1CC3=CC(=C(C=C3)C4=CC=CC=C4S(=O)(=O)NC5=C(C(=NO5)C)C)CN6CCC(C6=O)(C)C
Allisartan isoproxil

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
![]()
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.

2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension

CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.

Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.

As regards to the solid physical properties of the compound of formula (I), the patent document of CN101024643A discloses that it is a white solid, and its melting point is 134.5-136° C. However, CN101024643A dose not disclose the crystalline structure of the compound of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.

CN200710094021.4 discloses another method for preparing the compounds of formula I, the following route B, the starting material by nucleophilic substitution, oxidation, an ester, a tetrazole ring to obtain a compound of formula I, the first step of the method nucleophilic substitution easy to generate an imidazole ring -3 para isomer impurities difficult to remove; the last step into the ring to use sodium azide, operating dangerous.

CN201210020174.5 disclosed a series of anti-hypertensive compound and preparation method, the following line C, the temperature control in the first step of its preparation O ~ 5 ° C, a mixed solution of acetone and water, with a 5% aqueous solution of sodium hypochlorite oxidation, yield 70%, the second step use of potassium permanganate, manganese dioxide will produce the same, and a yield of only 40%, the first two steps total yield of 28%, is very low, and the post-treatment methods are by column separation, the first two steps are used are organic and inorganic mixed solvent is not conducive to recovery, not suitable for scale-up.






Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)

To a 20L reactor 9800ml of methanol, stirring was started, the rotational speed is added at 200r / min 1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20 ° C with stirring 3h, the reaction mixture was filtered to give a pale yellow clear filtrate. The filtrate was concentrated under reduced pressure to move 20L flask, vacuum degree of 0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

850ml of absolute ethanol was added to the 3L reaction vessel was charged with crude Alicante medoxomil (800.lg, 1.45mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1300ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration, the purified Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

950ml of absolute ethanol was added to the 5L reaction vessel was charged with crude Alicante medoxomil (549.9g, 1.72mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1200ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = cake was washed with a mixture of 1/3, and dried under reduced pressure after filtration to obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
Example 122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)

To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn. The solution was stirred at room temperature for 20 minutes. Then 0.562 g of 1-chloromethyl isopropyl carbonate was added and the mixture was reacted at 45-50° C. for 16 hours. After the reaction was completed, the mixture solution was filtered, and 30 ml of water was added into the filtrate. The resulting mixture was extracted with 30 ml of ethyl acetate twice. The organic phase was dried and concentrated to give 1.724 g of oil, which was directly used in the next reaction without purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.

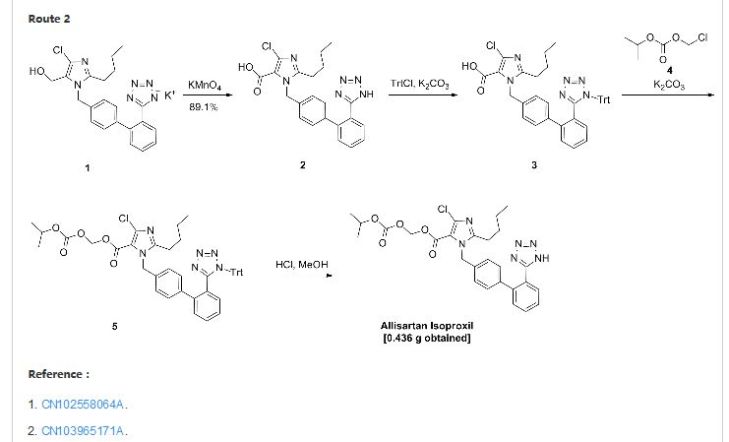



| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
……………….
update……………..
Example 1
Weigh 25g 2- butyl-4-chloro-1- [2 ‘- (1-trityl–1H- tetrazol-5-yl) -1,1’-biphenyl – methyl] – imidazole 5-carboxylic acid, 1 – [(isopropoxy) – carbonyloxy] -, methyl ester, was added to a 500ml three-necked flask, methanol was added 200ml, refluxed for 9h, methanol was distilled off under reduced pressure to give crude Alicante medoxomil .
To the residue (i.e., medoxomil crude Alicante) were added 33ml of isopropanol and 66ml of n-heptane, heated to 76 ℃ stirred for 2h. After cooling to 60 ℃ stirring for 1h, and then the system was slowly cooled to 0 ℃, stirring was continued for 3h. Filtered, the filter cake was washed with n-heptane. At 40 ℃ 8 hours and dried in vacuo to give 15.3g Alicante medoxomil (purity 99.3%) as a XRD spectrum as shown in Figure, the main peak of the diffraction peaks as shown in the following table, the DSC spectrum shown in figure II . Compared with the published crystal, the crystal obtained by the absence of significant electrostatic phenomena.
Shanghai , CHINA







