
Home » Posts tagged 'hepatitis C'
Tag Archives: hepatitis C
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves Mavyret (glecaprevir and pibrentasvir) for Hepatitis C


The U.S. Food and Drug Administration today approved Mavyret (glecaprevir and pibrentasvir) to treat adults with chronic hepatitis C virus (HCV) genotypes 1-6 without cirrhosis (liver disease) or with mild cirrhosis, including patients with moderate to severe kidney disease and those who are on dialysis. Mavyret is also approved for adult patients with HCV genotype 1 infection who have been previously treated with a regimen either containing an NS5A inhibitor or an NS3/4A protease inhibitor but not both.
Mavyret is the first treatment of eight weeks duration approved for all HCV genotypes 1-6 in adult patients without cirrhosis who have not been previously treated. Standard treatment length was previously 12 weeks or more.
“This approval provides a shorter treatment duration for many patients, and also a treatment option for certain patients with genotype 1 infection, the most common HCV genotype in the United States, who were not successfully treated with other direct-acting antiviral treatments in the past,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. According to the Centers for Disease Control and Prevention, an estimated 2.7 to 3.9 million people in the United States have chronic HCV. Some patients who suffer from chronic HCV infection over many years may have jaundice (yellowish eyes or skin) and complications, such as bleeding, fluid accumulation in the abdomen, infections, liver cancer and death.
There are at least six distinct HCV genotypes, or strains, which are genetically distinct groups of the virus. Knowing the strain of the virus can help inform treatment recommendations. Approximately 75 percent of Americans with HCV have genotype 1; 20-25 percent have genotypes 2 or 3; and a small number of patients are infected with genotypes 4, 5 or 6.
The safety and efficacy of Mavyret were evaluated during clinical trials enrolling approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. Results of the trials demonstrated that 92-100 percent of patients who received Mavyret for eight, 12 or 16 weeks duration had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients’ infection had been cured.
Treatment duration with Mavyret differs depending on treatment history, viral genotype, and cirrhosis status.
The most common adverse reactions in patients taking Mavyret were headache, fatigue and nausea.
Mavyret is not recommended in patients with moderate cirrhosis and contraindicated in patients with severe cirrhosis. It is also contraindicated in patients taking the drugs atazanavir and rifampin.
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected adult patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. HBV reactivation in patients treated with direct-acting antiviral medicines can result in serious liver problems or death in some patients. Health care professionals should screen all patients for evidence of current or prior HBV infection before starting treatment with Mavyret.
The FDA granted this application Priority Review and Breakthrough Therapydesignations.
The FDA granted approval of Mavyret to AbbVie Inc.
 |
|
| Clinical data | |
|---|---|
| Trade names | Maviret (combination with pibrentasvir) |
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| Synonyms | ABT-493 |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C38H46F4N6O9S |
| Molar mass | 838.87 g·mol−1 |
Glecaprevir (INN,[1] codenamed ABT-493) is a hepatitis C virus (HCV) nonstructural (NS) protein 3/4A protease inhibitor that was identified jointly by AbbVie and Enanta Pharmaceuticals. It is being developed as a treatment of chronic hepatitis C infection in co-formulation with an HCV NS5A inhibitor pibrentasvir. Together they demonstrated potent antiviral activity against major HCV genotypes and high barriers to resistance in vitro.[2]
On December 19, 2016, AbbVie submitted New Drug Application to U.S. Food and Drug Administration for glecaprevir/pibrentasvir (trade name Maviret) regimen for the treatment of all major genotypes (1–6) of chronic hepatitis C.[3]
References
- Jump up^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 76” (PDF). World Health Organization. p. 503. Retrieved 25 February 2017.
- Jump up^ Lawitz, EJ; O’Riordan, WD; Asatryan, A; Freilich, BL; Box, TD; Overcash, JS; Lovell, S; Ng, TI; Liu, W; Campbell, A; Lin, CW; Yao, B; Kort, J (28 December 2015). “Potent Antiviral Activities of the Direct-Acting Antivirals ABT-493 and ABT-530 with Three-Day Monotherapy for Hepatitis C Virus Genotype 1 Infection”. Antimicrobial Agents and Chemotherapy. 60 (3): 1546–55. PMC 4775945
 . PMID 26711747. doi:10.1128/AAC.02264-15.
. PMID 26711747. doi:10.1128/AAC.02264-15. - Jump up^ “AbbVie Submits New Drug Application to U.S. FDA for its Investigational Regimen of Glecaprevir/Pibrentasvir (G/P) for the Treatment of All Major Genotypes of Chronic Hepatitis C”. AbbVie Inc. North Chicago, Illinois, U.S.A. December 19, 2016. Retrieved 25 February 2017.
 |
|
| Identifiers | |
|---|---|
| Synonyms | ABT-530 |
| CAS Number | |
| Chemical and physical data | |
| Formula | C57H65F5N10O8 |
| Molar mass | 1,113.20 g·mol−1 |
Pibrentasvir is an antiviral agent.[1] In the United States, it is approved for use with glecaprevir as the combination drug glecaprevir/pibrentasvir (Mavyret) for the treatment of hepatitis C.[2]
References
- Jump up^ Ng, Teresa I.; Krishnan, Preethi; Pilot-Matias, Tami; Kati, Warren; Schnell, Gretja; Beyer, Jill; Reisch, Thomas; Lu, Liangjun; Dekhtyar, Tatyana; Irvin, Michelle; Tripathi, Rakesh; Maring, Clarence; Randolph, John T.; Wagner, Rolf; Collins, Christine (2017). “In Vitro Antiviral Activity and Resistance Profile of the Next-Generation Hepatitis C Virus NS5A Inhibitor Pibrentasvir”. Antimicrobial Agents and Chemotherapy. 61 (5): e02558–16. PMID 28193664. doi:10.1128/AAC.02558-16.
- Jump up^ Linda A. Johnson (August 3, 2017). “FDA OKs new drug to treat all forms of hepatitis C”. Fox Business.
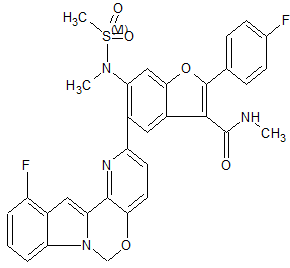
MK 8876
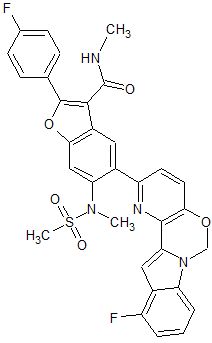

MK 8876
CAS 1426960-33-9
2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-(N-methylmethanesulfonamido)-1-benzofuran-3-carboxamide
| 2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-[methyl(methylsulfonyl)amino]-3-benzofurancarboxamide |
| Molecular Formula | C32H24F2N4O5S | |
| Molecular Weight | 614.62 |
- Originator Merck & Co
- Class Antivirals
- Phase I Hepatitis C
Most Recent Events
- 11 Oct 2013 Phase-I clinical trials in Hepatitis C in Germany (PO)
- 11 Oct 2013 Phase-I clinical trials in Hepatitis C in Moldova (PO)
- 23 Aug 2013 Preclinical trials in Hepatitis C in USA (PO)
DATA
2-(4-Fluorophenyl)-5-(11-fluoro-6H-pyrido[2′,3′:5,6][1,3]oxazino[3,4-a]indol-2-yl)-N-methyl-6-(N-methylmethanesulfonamido)-1-benzofuran-3-carboxamide
MK-8876 off-white solid
1H NMR (500 MHz, DMSO-d6) δ 8.56 (q, J = 4.7 Hz, 1H), 8.06–8.01 (m, 2H), 8.05 (s, 1H), 7.86 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.46–7.40 (m, 2H), 7.29–7.22 (m, 1H), 7.11 (s, 1H), 6.94 (dd, J = 10.6, 7.9 Hz, 1H), 6.27 (s, 2H), 3.31 (s, 3H), 2.96 (s, 3H), 2.85 (d, J = 4.7 Hz, 3H);
13C NMR (125.7 MHz, DMSO-d6) δ 162.86, 162.82 (d, JC–F = 248.5 Hz), 155.74 (d, JC–F = 246.1 Hz), 153.80, 152.43, 152.28, 147.20, 137.08, 137.00 (d, JC–F = 10.8 Hz), 136.36, 136.20, 132.37, 129.50 (d, JC–F = 8.6 Hz), 127.17, 125.45 (d, JC–F = 3.1 Hz), 125.08, 125.02, 123.70 (d, JC–F = 7.7 Hz), 122.28, 117.23 (d, JC–F = 22.4 Hz), 116.01 (d, JC–F = 21.9 Hz), 113.65, 111.76, 106.90 (d,JC–F = 3.5 Hz), 105.32 (d, JC–F = 18.5 Hz), 94.16, 73.57, 39.39, 37.24, 26.16;
HR-ESI-MS m/zcalcd for C32H25N4O5SF2+ [M + H]+ 615.1514, found 615.1500.
| HPLC Method | |
|---|---|
| column | Ascentis Express C18 2.7 μm (fused core), 100 mm × 4.6 mm |
| detection | UV at 210 nm |
| column temperature | 40 °C |
| flow rate | 1.8 mL/min |
| injection volume | 5.0 μL |
| gradient | 90% A to 5% A over 11 min, hold at 5% A for 2 min, 5% A back to 90% A over the next 0.1 min, and then hold at 90% A for 2.9 min |
| run time | 16 min |
| data collection | acquisition for the first 13 min |
| mobile phases | solvent A: water with 0.1% H3PO4 |
| solvent B: acetonitrile | |
| Retention Time Data | |
|---|---|
| identity | tR (min) |
| boronic acid 27 | 4.24 |
| desbromoarene 28 | 5.33 |
| MK-8876 (1) | 7.89 |
| chloropyridine starting material 2 | 8.03 |
| BHT | 10.22 |
SYNTHESIS



CONTD……………
MK 8876










MK 8876
Patent
Scheme 1

Scheme 2

Scheme 3

Q
Scheme 4

EXAMPLES
Example 1
Preparation of Compound 1
 THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876
THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876Step 1 – Synthesis of 2,6-dichloropyridin-3-ol

Η202 (1.60 g, 47.12 mmol) was added slowly to the solution of compound 2,6- dichloropyridin-3-ylboronic acid (3 g, 15.71 mmol) in CH2CI2 (30 mL) at 0 °C. After stirred at room temperature for about 15 hours, the mixture was quenched with sat. Na2S203 aqueous (50 mL) and adjusted to pH < 7 with IN HC1. The mixture was extracted with EtOAc (40 mL x 3). The organic layer was washed with brine (100 mL), dried over Na2S04, filtered and the solvent was evaporated to provide2,6-dichloropyridin-3-ol (2.34 g, yield: 91.4%). 1H-NMR (CDC13, 400 MHz) δ 7.30 (d, / = 8.4 Hz, 1H), 7.19 (d, / = 8.4 Hz, 1H), 5.70 (br, 1H).
– Synthesis of 2,6-dichloro- -methoxypyridine
To a solution of 2,6-dichloropyridin-3-ol (16.3 g, 0.1 mol) and K2C03 (41.4 g, 0.3 mol) in DMF (200 mL) were added Mel (21.3 g, 0.15 mol). The mixture was allowed to stir at 80 °C for 2 hours. The mixture was then diluted with water (200 mL) and extracted with EtOAc (200 mL x 3). The organic layer was washed with brine (200 mL x 3), dried over Na2S04, filtered and the solvent was evaporated to provide 2,6-dichloro-3-methoxypyridine (17.0 g, yield: 96.0%). 1H-NMR (CDC13, 400 MHz) δ 7.12-7.18 (m, 2H), 3.86 (s, 3H). Step 3 – Synthesis of2-(6-chloro-3-methoxypyridin-2-yl)-lH-indole

To a degassed solution of compound 2,6-dichloro-3-methoxypyridine (8.9 g, 0.05 mol), (l-(tert-butoxycarbonyl)-lH-indol-2-yl)boronic acid (13 g, 0.05 mol) and K3PO4 (31.8 g, 3.0 mol) in DMF (100 mL) was added Pd(dppf)Cl2 (3.65 g, 0.005 mol) under N2. The mixture was heated at 60 °C for about 15 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the resulting residue was purified using prep-HPLC to provide the desired product of 2-(6-chloro-3-methoxypyridin-2-yl)-lH-indole (9.0 g, yield:
69.8%). 1H-NMR (CDC13, 400 MHz) δ 9.52 (s, 1H), 7.65 (d, / = 7.6 Hz, 1H), 7.38-7.43 (m, 2H), 7.07-7.26 (m, 4H), 4.03 (s, 3H).
Step 4 – Synthesis of6-chlor -2-(lH-indol-2-yl)pyridin-3-ol

BBr3 (0.4 mL, 0.39 mmol) was added to the solution of 2-(6-chloro-3- methoxypyridin-2-yl)-lH-indole (50 mg, 0.194 mmol) in CH2C12 (0.5 mL) at -78 °C under N2. The mixture was allowed to stir at room temperature for 3 hours. The mixture was then quenched with CH3OH (10 mL) at -78 °C. After being concentrated in vacuo, the resulting residue was purified using prep-TLC (PE : EtOAc = 2.5 : 1) to afford the desired product of 6- chloro-2-(lH-indol-2-yl)pyridin-3-ol (40 mg, yield: 85.1%). 1H-NMR (CDC13, 400 MHz) δ 10.09 (s, 1H), 9.72 (s, 1H), 7.50 (d, / = 7.9 Hz, 1H), 7.17-7.32 (m, 3H), 7.08-7.14 (m, 1H), 6.87-6.96 (m, 2H).
Step 5 – Synthesis of 2-chlo -6H-pyrido[2′ ,3′ : 5 ,6] [ 1 ,3]oxazino[3 ,4-a]indole

To a solution of chloroiodomethane (3.51 g, 20.0 mmol) and K2CO3 (1.38 g, 10.0 mmol) in DMF (50 mL) was allowed to stir at 100 °C, 6-chloro-2-(lH-indol-2-yl)pyridin-3-ol (480 mg, 2.0 mmol) in DMF (50 mL) was added dropwise. After addition, the mixture was allowed to stir for another 0.5 hours. The mixture was then diluted with water (100 mL) and extracted with EtOAc (100 mL x 3). The organic layer was washed with brine (100 mL x 3), dried over Na2S04 and concentrated. The residue was purified using prep-TLC (PE : EtOAc = 3 1) to afford the desired product of 2-chloro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (260 mg, yield: 50.7%). 1H-NMR (CDC13, 400 MHz) δ 7.63 (d, / = 8.0 Hz, 1H), 7.22-7.27 (m, 3H), 7.19 (d, / = 2.4 Hz, 1H), 7.08-7.12 (m, 2H), 5.86 (s, 2H).
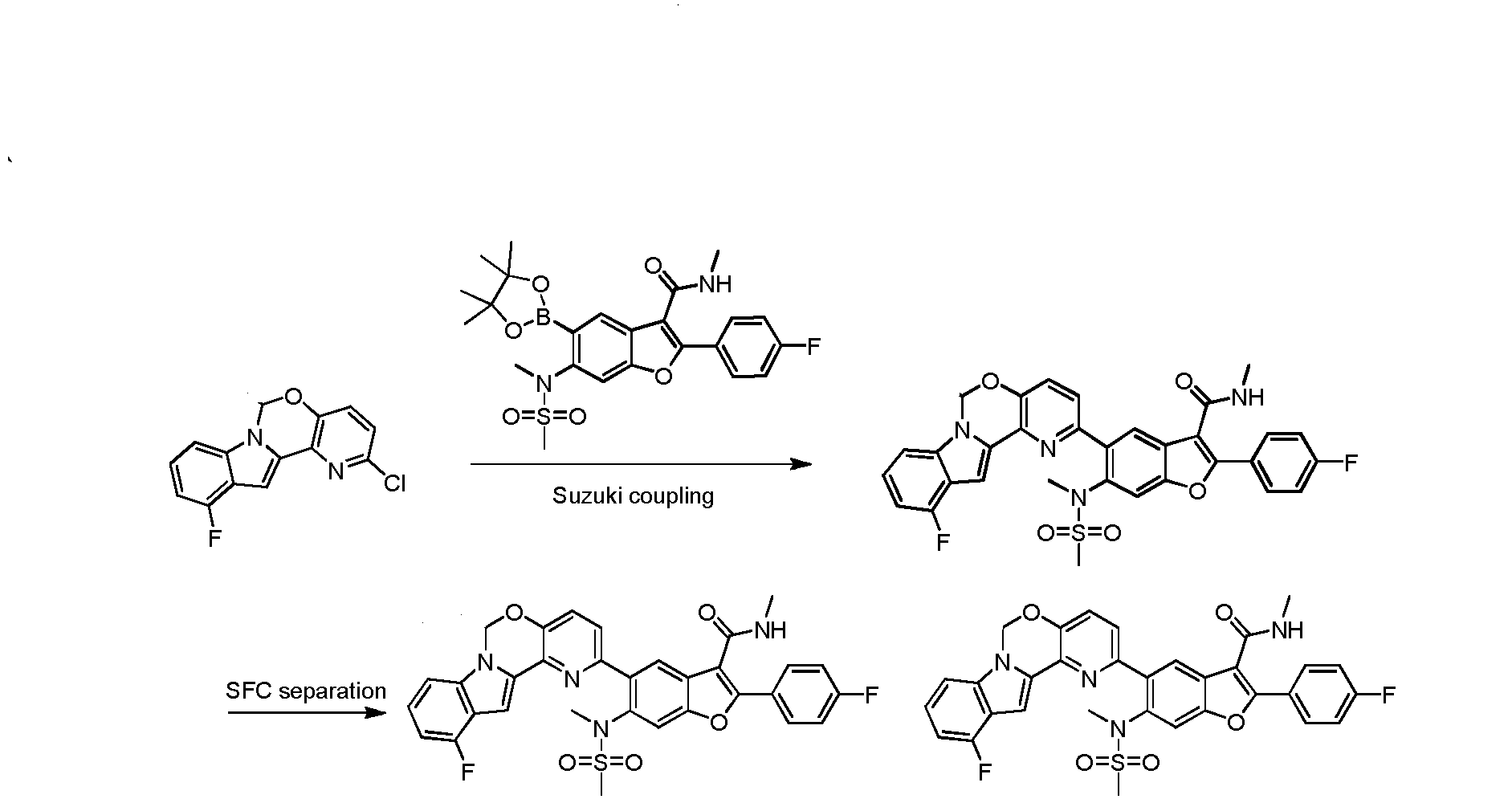
Step 6 – Synthesis of2-(4-fluowphenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(6H- pyridol 2 ‘,3’:5,6][ l, mpound 1 )
 THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876
THIS COMPD HAS ONE FLUORO MISSING, APPLY TO YOUR MK 8876To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzofuran-3- carboxamide (502 mg, 1.0 mmol), 2-chloro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (256 mg, 1.0 mmol) and K3PO4 (636 mg, 3.0 mmol) in dioxane : H20 (1.5 mL : 0.4 mL) was added Pd2(dba)3 (91 mg, 0.1 mmol) and X-phos (91 mg, 0.2 mmol) under N2. The mixture was heated to 110 °C for 3 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the resulting residue was purified using prep-HPLC to provide the desired product of Compound 1 (275 mg, yield: 46.1%). 1H-NMR (CDC13, 400 MHz) δ 7.88-7.94 (m, 3H), 7.61-7.63 (m, 2H), 7.40 (s, 2H), 7.09-7.28 (m, 6H), 5.94 (s, 2H), 5.86 (d, / = 4.4 Hz, 1H), 3.29 (s, 3H), 2.92 (d, / = 5.2 Hz, 3H), 2.65 (s, 3H). MS (M+H)+: 596.
Compounds 2-15, depicted in the table below, were prepared using the method described above.
COMPD 2 IS MK 8876

PATENT
Example 81
Preparation of Compound 2
Synthesis of ethyl 3- 4-fluorophenyl)-3-oxopropanoate
Diethyl carbonate (130 g, 1.1 mol) was dissolved in a suspension ofNaH (60% in oil, 50.2 g, 1.3 mol) in anhydrous tetrahydrofuran (1.5 L), and then l-(4-fluorophenyl)ethanone (150 g, 1.09 mol) was added dropwise at 70 °C. The resulting mixture was stirred at 70 °C for 3 hours. After the reaction mixture was cooled to room temperature and poured into HCl (1 N). The mixture was extracted with EtOAc, the organic phase was dried with anhydrous NaS04 and concentrated in vacuo. The resulting residue was purified using column chromatography (eluted with petroleum ether / EtOAc = 50 / 1) to provide ethyl 3-(4-fluorophenyl)-3-oxopropanoate (217 g, yield: 95%). 1H-NMR (CDC13, 400 MHz) δ 7.92-7.97 (m, 2H), 7.07-7.13 (m, 2H), 4.14-4.20 (m, 2H), 3.93 (s, 2H), 1.22 (d, J= 7.2 Hz, 3H). MS (M+H)+: 211. Step 2 – Synthesis of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A solution of ethyl 3-(4-fluorophenyl)-3-oxopropanoate (130 g, 0.6 mol), 4- bromophenol (311 g, 1.8 mol) and FeCl3-6H20 (19.5 g, 0.09 mol) in DCE (700 mL) was heated to reflux, and then 2-(tert-butylperoxy)-2-methylpropane (193 g, 1.32 mol) was added dropwise under nitrogen. After 6 hours of refluxing, the mixture was cooled to RT, quenched with saturated NaHS03 and extracted with dichloromethane. The organic phases were washed with water, brine and dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether / dichloromethane = 15 / 1) to provide the crude product, which was crystallized from cold MeOH to provde ethyl 5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (37 g, yield: 14.3%) as solid. 1H- MR (CDC13, 400 MHz) δ 8.12 (s, 1H), 7.97-8.01 (m, 2H), 7.37 (d, J= 4.0 Hz, 1H), 7.32 (d, J= 8.0 Hz, 1H), 7.11 (t, J= 8.0 Hz, 2H), 4.32-4.38 (m, 2H), 1.36 (t, J= 8.0 Hz, 3H). MS (M+H)+: 363 / 365.
Step 3 – Synthesis of eth l 5-bromo-2-(4-fluorophen -6-nitrobenzofuran-3-carboxylate
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate (50 g,
137.6 mmol) in CHC13 (500 mL), fuming HN03 (50 mL) was added dropwise at -15 °C and the mixture was stirred for 0.5 hour. The reaction mixture was poured into ice water and extracted with CH2C12. The organic layer was washed with a.q. sat. NaHC03 and brine, after removed the most of solvent, the resulting residue was crystallized with petroleum ether / dichloromethane = 20 / 1 to provide product of ethyl 5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3-carboxylate (35 g, yield: 66%). 1H- MR (CDC13, 400 MHz) δ 8.36 (s, 1H), 8.02-8.04 (m, 3H), 7.13-7.18 (m, 2H), 4.36-4.41 (m, 2H), 1.37 (t, J= 4.0 Hz, 3H). MS (M+H)+: 408 / 410.
Step 4 – Synthesis of ethyl 6-amino-5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A mixture of ethyl 5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3-carboxylate (52 g, 127 mmol), iron filings (21.3 g, 382.2 mmol) and H4C1 (41 g, 764.4 mmol) in MeOH / THF / H20 (2 / 2 / 1, 500 mL) was stirred at reflux for 3 hour. After filtered and concentrated, the resulting residue was purified using column chromatography (petroleum ether / EtOAc / dichloromethane = 20 : 1 : 20) to provide ethyl 6-amino-5-bromo-2-(4-fluorophenyl) benzofuran-3-carboxylate (40 g, yield: 82%). 1H- MR (CDC13, 400 MHz) δ 8.01 (s, 1H), 7.94-7.98 (m, 2H), 7.08 (t, J= 8.0 Hz, 2H), 6.83 (s, 1H), 4.32-4.36 (m, 2H), 4.18 (s, 2H), 1.35 (t, J= 8.0 Hz, 3H). MS (M+H)+: 378 / 380.
Step 5 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid eth l ester
MsCI (31.7 g, 277.5 mmol) was added to a solution of ethyl 6-amino-5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (35 g, 92.5 mmol) and pyridine (60 mL) in
dichloromethane (300 mL) at 0 °C. After stirred overnight at room temperature, the mixture was diluted with water and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo, the resulting residue was purified using crystallized with EtOAc to provde the pure product of ethyl 5-bromo-2-(4-fluorophenyl)-6- (methylsulfonamido)benzofuran-3-carboxylate (35 g, yield: 82%). 1H- MR (CDC13, 400 MHz) δ 8.27 (s, 1H), 8.01-8.05 (m, 2H), 7.87 (s, 1H), 7.15-7.19 (m, 2H), 6.87 (s, 1H), 4.38-4.43 (m, 2H), 3.00 (s, 3H), 1.40 (t, J= 40 Hz, 3H). MS (M+H)+: 456 / 458.
Step 6 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido) benzofuran-3-carboxylate (53 g, 0.23 mol) in dioxane / H20 (5 / 1, 600 mL) was added
LiOH-H20 (25 g, 1.17 mol), and the mixture was stirred at 100 °C for 3 hours. After
concentrated, the resulting residue was dissolved in H20, 1 N HCl was added until pH reached 3, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed to provide the product of 5-bromo-2-(4- fluorophenyl)-6-(methylsulfonamido)benzofuran-3-carboxylic acid (48 g, yield: 96%).1H- MR (DMSO- e, 400 MHz) δ 13.49 (s, 1H), 9.67 (s, 1H), 8.30 (s, 1H), 8.12-8.17 (m, 2H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H). MS (M+H)+: 428 / 430. Step 7 – Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino-benzofuran-3- carboxylic acid methylamide

A solution of 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido) benzofuran-3- carboxylic acid (33 g, 77 mmol), HOBT (15.6 g, 115.5 mmol) and EDCI (22.2 g, 115.5 mmol) in DMF (250 mL) was stirred at room temperature. After 2 hours, Et3N (50 mL) and CH3 H2 (HC1 salt, 17.7 g, 231 mmol) was added to the mixture, and the mixture was stirred overnight. After the solvent was removed, H20 was added and the mixture was extracted with ethyl acetate. The combined organic layer was washed with H20, brine and concentrated in vacuo. The resulting residue was washed with EtOAc to provide the product of 5-bromo-2-(4-fluorophenyl)-N- methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (32 g, yield: 94%). 1H- MR (DMSO- ck, 400 MHz) δ 9.55 (br s, 1H), 8.46-8.48 (m, 1H), 8.12-8.17 (m, 2H), 7.96 (s, 1H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H), 2.93 (d, J= 8.4 Hz, 3H). MS (M+H)+: 441 / 443.
Step 8 – Synthesis of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido benzofuran-3-carboxamide

CH3I (31.6 g, 223 mmol) was added to a mixture of 5-bromo-2-(4-fluorophenyl)- N-methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (32 g, 74 mmol), K2C03 (25.6 g, 186 mmol) and KI (246 mg, 1.5 mmol) in DMF (150 mL) under N2 protection. The mixture was stirred at 80-90 °C overnight. After concentrated in vacuo, the resulting residue was washed with water (200 mL) and EtOAc (200 mL) to provide the product of 5-bromo-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (31.5 g, 94%). 1H- MR (CDCI3, 400 MHz) δ 8.16 (s, 1H), 7.88-7.92 (m, 2H), 7.70 (s, 1H), 7.18-7.23 (m, 2H), 5.78 (br s, 1H), 3.34 (s, 3H), 3.09 (s, 3H), 3.00 (d, J= 4.8 Hz, 3H). MS (M+H)+: 455 / 457. Step 9 – Synthesis of 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(4, 4, 5, 5- tetramethyl-1 -dioxaborolan-2-yl)benzofuran-3-carboxamide
a degassed solution of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (1.0 g, 2.2 mmol) and pinacol diborane (2.79 g, 11.0 mmol) in 1,4-Dioxane (25 mL) was added KOAc (647 mg, 6.6 mmol) under N2 and stirred for 4 hours at room temperature. Then Pd(dppf)Cl2 (60 mg) was added, and the mixture was stirred for another 30 minutes. Then the mixture was put into a pre-heated oil-bath at 130 °C and stirred for another 1 hour under N2. The reaction mixture was cooled to room
temperatureand concentrated and extracted with EtOAc. The organic layers were washed with brine, dried over Na2S04. After concentrated, the crude product of the boronic ester was purified using column chromatography (petroleum ether / EtOAc = 5 / 1 to 2 / 1) to obtain 2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)benzofuran-3-carboxamide as white solid (700 mg, yield: 64%). 1H- MR (CDCI3, 400 ΜΗζ) δ 8.17 (s, 1H), 7.87-7.91 (m, 2H), 7.52 (s, 1H), 7.11 (t, 7= 7.6 Hz, 2H), 5.81 (d, 7= 2.8 Hz, 1H), 3.30 (s, 3H), 2.97 (d, 7= 5.2 Hz, 3H), 2.90 (s, 3H), 1.31 (s, 12H). MS (M+H)+: 503.
Step 10 – Synthesis of tert-butyl 4-fluoro-lH-indole-l -car boxy late
To a solution of 4-fluoro-lH-indole (5 g, 0.11 mol) and DMAP (150 mg, 3%Wt) in THF (50 mL) was added (Boc)20 (8.5 g, 0.04 mol) dropwise. The mixture was stirred at room temperature for 2 hours. The organic solvent was removed in vacuo, and the resulting residue was purified using column chromatography (pure petroleum ether) to provide tert-butyl 4-fluoro- lH-indole-l-carboxylate (8.3 g, yield: 96%). 1H- MR (CDC13, 400 MHz) δ 7.92 (d, J= 8.4 Hz, 1H), 7.55 (d, J= 3.6 Hz, 1H), 7.23 (m, 1H), 6.90 (m, 1H), 6.66 (d, J= 3.6 Hz, 1H), 1.67 (s, 9H). MS (M+H)+: 236.
Step 11 – Synthesis of (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid

To a solution of diisopropylamine (7.5 mL, 0.11 mol) in THF (35 mL) at 0 °C was added «-BuLi (21 mL, 0.055 mol) dropwise. The mixture was stirred at 0 °C for 40 minutes. Then the mixture was cooled to -78 °C. Tert-butyl 4-fluoro-lH-indole-l-carboxylate (5 g, 0.02 mol) in THF (13 mL) was added dropwise slowly. After addition, the mixture was stirred at -78 °C for 2 hours. Then triisopropyl borate (3.29 g, 0.03 mol) was added. The mixture was stirred at -78 °C for another 40 minutes. The reaction was monitored using TLC. When the reaction was completed, the mixture was adjusted to pH = 6 with 1 N HC1. After extracted with EtOAc (25 mL x 3), the combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered and concentrated in vacuo. The obtained solid was recrystallized with EtOAc and petroleum ether to provide (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid (4.5 g, yield: 76.7%, which might be unstable at high temp, work up, store in fridge). 1H- MR (CDC13, 400 MHz) δ 7.77 (d, J= 8.4 Hz, 1H), 7.57 (s, 1H), 7.44 (s, 2H), 7.24 (m, 1H), 6.90 (m, 1H), 1.66 (s, 9H). MS (M+H)+: 280.
Step 12 – Synthesis of 6-chloro-2-iodopyridin-3-ol
6-chloropyridin-3-ol (5.0 g, 38.6 mmol) was dissolved in water (50 mL) and placed under an N2 atmosphere. Na2C03 (8.2 g, 77.4 mmol) was added followed by iodine (9.8 g, 38.8 mmol). The reaction mixture was stirred at room temperature for 2 hours. The mixture was poured into 1M Na2S203 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04 and concentrated to provide the product of 6-chloro-2- iodopyridin-3-ol (7.0 g, yield: 70.9%). 1H- MR (CDC13, 400 MHz) δ 7.17 (d, J= 8.4 Hz, 1H), 7.06 (d, J= 8.4 Hz, 1H). MS (M+H)+: 256 / 258.
Step 13 – Synthesis of 6-chloro-2-(4-fluoro-lH-indol-2-yl)pyridin-3-ol
A mixture of (l-(tert-butoxycarbonyl)-4-fluoro-lH-indol-2-yl)boronic acid (5 g, 18.0 mmol), 6-chloro-2-iodopyridin-3-ol (3.82 g, 15.0 mol) and NaHC03 (3.78 g, 45.0 mol) in 1, 4-dioxane (76 mL) and water (7 mL) was stirred at room temperature for 15 minutes. Then Pd(PPh3)2Cl2 (527 mg, 0.75 mmol) was added under nitrogen atmosphere, and the mixture was heated at 100 °C under N2 for 16 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc (50 mL), filtered and concentrated in vacuo. The resulting residue was diluted with H20 (60 mL) and EtOAc (30 mL), and the layer was separated, the aqueous layer was extracted with EtOAc (3*30 mL). The combined organic layers were washed with brine (50 mL), dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether / EtOAc = 20 / 1 ~ 3 / 1) to provide 6-chloro-2- (4-fluoro-lH-indol-2-yl)pyridin-3-ol (3 g, yield: 76.5%). 1H- MR (MeOD, 400 MHz) δ 7.36 (s, 1H), 7.23-7.27 (m, 2H), 7.03-7.11 (m, 2H), 6.63-6.68 (m, 1H). MS (M+H)+: 263 / 265.
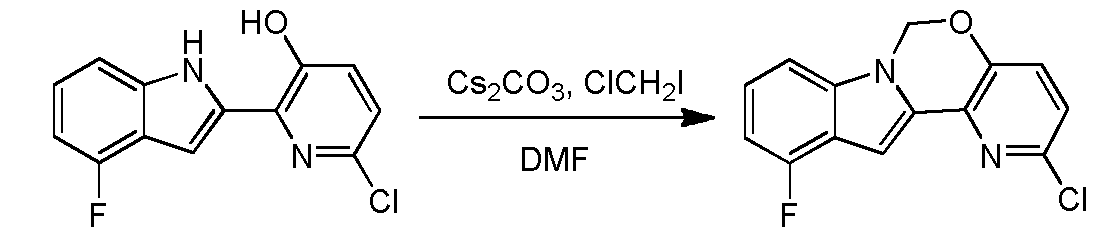
Ste 14 – Synthesis of 2-chloro-ll-fluoro-6H-pyrido[2′,3′:5, 6][l,3]oxazino[3,4-a]indole
A solution of 6-chloro-2-(4-fluoro-lH-indol-2-yl)pyridin-3-ol (2 g, 7.6 mmol) and Cs2C03 (7.46 g, 22.89 mmol) in DMF (100 mL) was stirred at 100 °C (internal temperature) for 15 min, and then chloroiodomethane (2.85 g, 15.3 mmol) in DMF (2 mL) was added dropwise. After the reaction was completed, the mixture was filtered and concentrated in vacuo. The resulting residue was diluted with water (50 mL) and extracted with ethyl acetate (30 mL x 3). The organic layer was washed with brine, dried over Na2S04 and concentrated in vacuo. The resulting residue was purified using column chromatography (petroleum ether:EA=10: l) to provde 2-chloro-l l-fluoro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4-a]indole (1.8 g, yield: 86.1%). 1H- MR (DMSO-i¾, 400 MHz) δ 7.64 (d, J= 8.8 Hz, 1H), 7.39-7.46 (m, 2H), 7.21-7.25 (m, 1H), 7.06 (s, 1H), 6.88-6.92 (m, 1H), 6.18 (s, 2H). MS (M+H)+: 275 / 277. Step 15 – Synthesis of5-(ll-fluoro-6H-pyrido[2 3′:5, 6][l,3]oxazino[3,4-a]indol-2-yl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxam
To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzofuran-3- carboxamide (100 mg, 0.199 mmol), 2-chloro-l l-fluoro-6H-pyrido[2′,3′:5,6][l,3]oxazino[3,4- a]indole (56 mg, 0.199 mmol) and Κ3Ρ04·3Η20 (159 mg, 0.597 mmol) in dioxane / H20 (0.8 mL / 0.2 mL) was added Pd2(dba)3 (9 mg, 0.01 mmol) and X-Phos (9 mg, 0.02 mmol) under N2. The mixture was heated at 80 °C for 1 hour. The mixture was then diluted with water (30 mL) and extracted with EtOAc (15 mL x 3). The organic layer was washed with brine (20 mL), dried over Na2S04 and concentrated in vacuo. The resulting residue was purified using prep-TLC (petroleum ether / EtOAc = 1 : 1.5) to provde the pure product of 5-(l l-fluoro-6H- pyrido [2′, 3 ‘ : 5 , 6] [ 1 , 3 ]oxazino [3 ,4-a]indol-2-yl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (60 mg, 48.8%). 1H- MR (CDC13, 400 MHz) δ: 7.99 (s, 1H), 7.93-7.96 (m, 2H), 7.65 (s, 1H), 7.45-7.50 (m, 2H), 7.17-7.21 (m, 4H), 7.10 (d, J= 8.0 Hz, 1H), 6.81-6.85 (m, 1H), 5.98 (s, 3H), 3.35 (s, 3H), 2.98 (d, J= 4.8 Hz, 3H), 2.72 (s, 3H). MS (M+H)+: 615.
Paper

We describe the route development and multikilogram-scale synthesis of an HCV NS5B site D inhibitor, MK-8876. The key topics covered are (1) process improvement of the two main fragments; (2) optimization of the initially troublesome penultimate step, a key bis(boronic acid) (BBA)-based borylation; (3) process development of the final Suzuki–Miyaura coupling; and (4) control of the drug substance form. These efforts culminated in a 28 kg delivery of the desired active pharmaceutical ingredient.
Process Development of the HCV NS5B Site D Inhibitor MK-8876
*E-mail: qinghao.chen@merck.com
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00405
PAPER

Using the Teasdale method, purge factor estimates for six impurities identified as mutagenic alerts in the synthesis of MK-8876 are compared to actual measured amounts of these impurities determined via appropriate analytical methods. The results from this comparison illustrate the conservative nature of purge factor estimates, meaning that overprediction of mutagenic impurity purging is unlikely when using this method. Industry and regulatory acceptance of the purge factor estimation method may help minimize analytical burden in pharmaceutical development projects.
Evaluation and Control of Mutagenic Impurities in a Development Compound: Purge Factor Estimates vs Measured Amounts
*E-mail: mark_mclaughlin@merck.com.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00263?journalCode=oprdfk
| WO2004041201A2 * | Oct 31, 2003 | May 21, 2004 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2011106992A1 * | Mar 2, 2011 | Sep 9, 2011 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| WO2004041201A2 * | Oct 31, 2003 | May 21, 2004 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2010030592A1 * | Sep 8, 2009 | Mar 18, 2010 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2011106992A1 * | Mar 2, 2011 | Sep 9, 2011 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus ns5b polymerase |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014123794A1 * | Feb 3, 2014 | Aug 14, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| WO2014123795A2 * | Feb 3, 2014 | Aug 14, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| WO2014123795A3 * | Feb 3, 2014 | Oct 30, 2014 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis c |
| US9242998 | Feb 3, 2014 | Jan 26, 2016 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds and methods of use thereof for the treatment of hepatitis C |
//////MK-8876, 1426960-33-9, Merck & Co, Antivirals, Phase I, Hepatitis C
Fc7cccc6c7cc2n6COc1ccc(nc12)c3cc4c(cc3N(C)S(C)(=O)=O)oc(c4C(=O)NC)c5ccc(F)cc5
Ravidasvir, PPI-668, BI 238630

Ravidasvir dihydrochloride
C42H50N8O6.2(HCl), 835.83
CAS 1303533-81-4
Phase II/IIIHepatitis C

Ravidasvir
PPI-668 free base; BI 238630;
CAS:1242087-93-9
C42H50N8O6, 762.38
Chemical Name:methyl N-[(1S)-1-({(2S)-2-[5-(6-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]- 3- methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}naphthalen-2-yl) -1H- benzimidazol- 2-yl]pyrrolidin-1-yl}carbonyl)-2-methylpropyl]carbamate
Mechanism of Action:NS5A Inhibitor
Indication: hepatitis C
Development Stage: Phase II
Developer:Presidio Pharmaceuticals, Inc
- OriginatorXTL Biopharmaceuticals
- Developer Pharco Corporation; Presidio Pharmaceuticals
- Class Antivirals; Benzimidazoles; Carbamates; Naphthalenes; Pyrrolidines; Small molecules
- Mechanism of Action Hepatitis C virus NS 5 protein inhibitors; Hepatitis C virus replication inhibitors
- 31 Aug 2015 Ascletis plans to initiate the phase II EVEREST trial for Hepatitis C (Combination therapy; Treatment-naive) in Taiwan
- 31 Aug 2015 Taiwan Food and Drug Administration approves Clinical Trial Application to initiate a phase II trial for interferon free regimen comprising danoprevir and ravidasvir in Hepatitis C
- 24 Jun 2015 Efficacy data from a phase IIa trial in Hepatitis C released by Ascletis
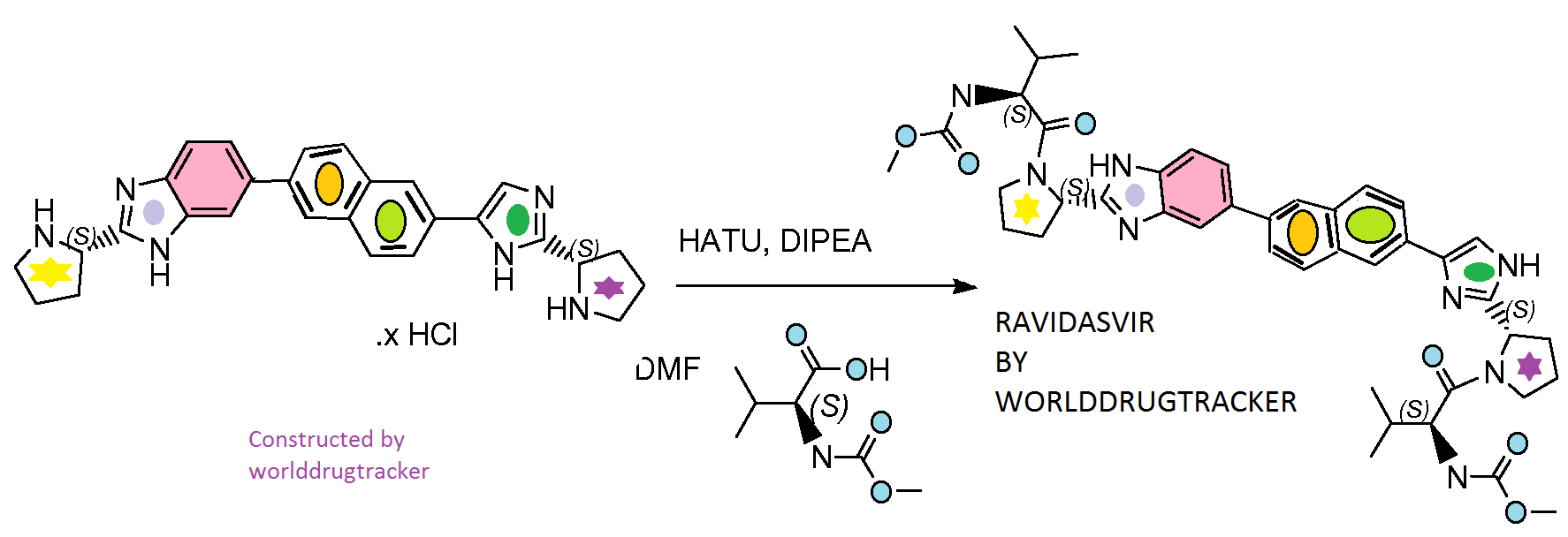
Ravidasvir [Methyl N-[(1S)-1-({(2S)-2-[5-(6-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]- 3- methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}naphthalen-2-yl) -1H- benzimidazol- 2-yl]pyrrolidin-1-yl}carbonyl)-2-methylpropyl]carbamate] is an Nonstructural protein 5A (NS5A) inhibitor. It is an antiviral agent that is being developed as a potential treatment for hepatitis C virus infection.
PPI-668, a non-structural 5A (NS5A) protein of hepatitis C virus (HCV) inhibitor, is in phase II clinical studies at Presidio Pharmaceuticals for the treatment of chronic genotype 1 hepatitis C virus infection.
Ravidasvir has 50% inhibitory concentrations (EC50s) values of 0.02-1.3 nM in replicon assays for HCV genotypes 1-7 (gt1-gt7).
Hepatitis C virus infection is a major health problem worldwide and no vaccine has yet been developed against this virus. The standard therapy of pegylated-interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferon are currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Nonstructural protein 5A (NS5A) is a zinc-binding and proline-rich hydrophilic phosphoprotein that plays a key role in Hepatitis C virus RNA replication.
A number of direct-acting antiviral agents (DAAs) are under development for the treatment of chronic HCV infection. These agents block viral production by directly inhibiting one of several steps of the HCV lifecycle. several viral proteins involved in the HCV lifecycle, such as the non-structural (NS)3/4A serine protease, the NS5B RNA-dependent RNA polymerase (RdRp), and the NS5A protein, have been targeted for drug development. Two NS3/4A protease inhibitors already approved for clinical use, numerous other protease inhibitors are being developed as well as inhibitors of viral replication, including nucleoside/nucleotide analogue inhibitors of HCV RdRp, non-nucleoside inhibitors of RdRp, cyclophilin inhibitors, and NS5A inhibitors.
Inhibition of NS5A at picomolar concentrations has been associated with significant reductions in HCV RNA levels in cell culture-based models, which makes these agents among the most potent antiviral molecules yet developed.
Activity:
This NS5A inhibitor has been shown to possess high efficacy against HCV genotype 1, with up to 3.7 log10 mean HCV RNA reductions, in a Phase Ib clinical trial. Activity was demonstrated against variants harbouring the L31M substitution. In an added genotype-2/3 cohort, the first 2 patients achieved mean 3.0 log10 RNA level reductions [1].
Results from the Phase IIa study involving a combination therapy with Faldaprevir and Deleobuvir plus Ravidasvir came with positive news where the said combination cured 92 percent of those with genotype 1a of hepatitis C virus (HCV) when given with ribavirin. The results presented at the 49th annual meeting of the European Association for the Study of the Liver (EASL) in London [2, 3].
The 36 study participants were randomly dived into three even cohorts of 12 each: The first received 600 mg of Deleobuvir twice a day as well as once-daily doses of Faldaprevir (120 mg), Ravidasvir and Ribavirin. The second group received the same regimen except the Faldaprevir dose was 400 mg. The third group took the regimen with the higher dose of Faldaprevir, but without Ribavirin. All participants were treated for 12 weeks with follow up for next 24 weeks.
Ninety-two percent of the first and second cohorts (11 out of 12 in both cases) achieved a sustained virologic response 12 weeks after completing therapy (SVR12, considered a cure). In the end, 14 participants were required for the third cohort, because one was incarcerated early on during treatment and another experienced viral rebound at week eight as a result of not adhering to the treatment regimen. Of the other 12 participants, eight, or two-thirds, have achieved an SVR12, while one more participant stopped taking the therapy at week eight but has since achieved an SVR8.
PATENT
WO 2011054834
http://www.google.co.in/patents/WO2011054834A1?cl=en
Scheme 1

GOING TO PRODUCT USING STRUCTURES FROM PATENT
 IIa
IIa
 IIIa one of side chain
IIIa one of side chain
DO NOT MISS OUT synthesis of XIIIa or XIII’a, this is needed in one of side chain
 L-boc-prolinol
L-boc-prolinol
 Z-boc-prolinal
Z-boc-prolinal
 XXIV
XXIV
 XIIIa
XIIIa
or

 XVIb
XVIb


MY CONSTRUCTION of 3
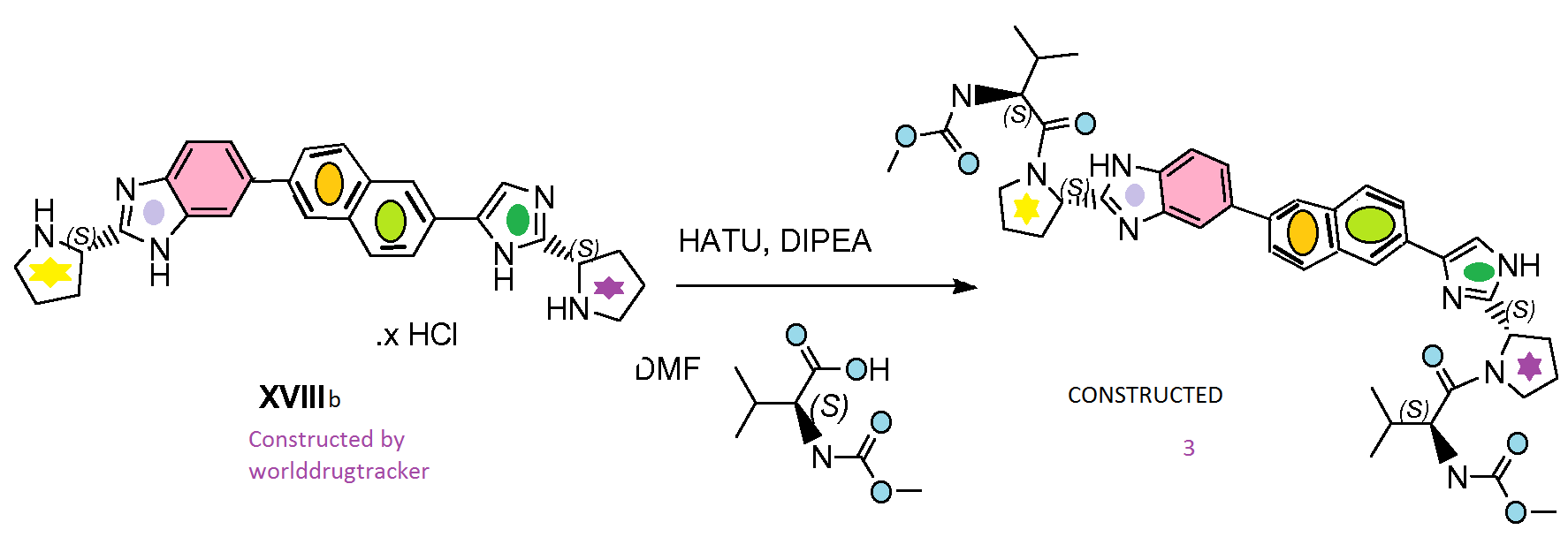

Compound 3 was prepared following the procedure reported for the synthesis of compound 1 using intermediate XVIIIb instead of intermediate XVIIIa. see my construction below

Compound 3. BASE
1H NMR (400 MHz, DMSO-d6) δ ppm 8.34 (2 H, s), 8.21 (1 H, s), 8.19
(1 H, d, J=8.69 Hz), 8.06 – 8.11 (2 H, m), 8.00 (1 H, dd, J=8.88, 1.61 Hz), 7.88 – 7.96
(2 H, m), 7.86 (1 H, d, J=8.48 Hz), 7.32 (1 H, d, J=8.48 Hz), 7.34 (1 H, d, J=8.53 Hz), 5.27 (1 H, dd, J=8.17, 5.33 Hz), 5.17 (1 H, t, J=7.00 Hz), 4.15 (2 H, t, J=7.95 Hz), 3.84
– 3.96 (4 H, m), 3.56 (6 H, s), 2.38 – 2.47 (2 H, m), 1.95 – 2.30 (8 H, m), 0.86 (3 H, d,
J=6.70 Hz), 0.85 (3 H, d, J=6.70 Hz), 0.81 (6 H, d, J=6.63 Hz).
[a] 2°= -148.98 0 (c 0.3336 w/v %, MeOH)
Alternative preparation of compound 3 and the corresponding HC1 salt
N-methoxycarbonyl-L- Valine (3.09 g, 17.7 mmol, 2.1 equiv) was dissolved in dichloro- methane (300 mL). Triethylamine (11.7 mL, 84.1 mmol, 10 equiv) and (l-cyano-2- ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluoro- phosphate were added (7.57 g, 17.7 mmol, 2.1 eq). The reaction mixture was stirred at room temperature for 5 minutes, after which XVIIIb was added (5 g, 8.41 mmol in case x.HCl equals 4 HC1). Stirring was continued for 30 minutes. HC1 in iPrOH (6N) was added to the mixture (until pH = 2), and the resulting mixture was stirred for 5 minutes. The solution was then washed with saturated aqueous sodium carbonate (2 x 200 mL) and once with brine (200 mL). The organic layer was separated, dried on magnesium sulphate and filtrated. After removal of the solvent in vacuum, the obtained residue was further dried in vacuum to afford an orange powder (6.84 g)
The powder was purified by silica gel column chromatography using gradient elution with 0 to 10 % MeOH (7N NH3) in dichloromethane, resulting in compound 3 (2.81 g) as a foam.
Compound 3 was dissolved in iPrOH (40 mL) and HC1 (6N in iPrOH, 10 mL) was added. The volatiles were removed in vacuum. Then, iPrOH (30 mL) was added and the mixture was heated at reflux. The solution was cooled to room temperature and stirred at room temperature for 4 days. tBuOMe (100 mL) was added to the solution, resulting in white precipitation, which was filtered, washed immediately with tBuOMe (3 x 10 mL) under nitrogen atmosphere and dried under vacuum at 40°C. The residue was mixed with acetonitrile and evaporated to dryness (2x). The residue was stirred in acetonitrile (150 mL) and the mixture was sonicated for 10 minutes. The precipitate was filtered under nitrogen atmosphere, washed twice with acetonitrile (50 mL) and dried in vacuum at 40°C, resulting in a slightly yellow powder (4 g).
HCL salt of compound 3:
[a] *° = -110.02 ° (589 nm, 20 °C, c 0.429 w/v%, MeOH)
1H NMR (600 MHz, DIMETHYLFORMAMIDE- y, 280K) δ ppm 0.86 (d, J=6.6 Hz, 6 H), 0.95 (d, J=7.0 Hz, 6 H), 2.03 – 2.20 (m, 2 H), 2.26 – 2.37 (m, 3 H), 2.39 – 2.61 (m, 5 H), 3.61 – 3.63 (m, 6 H), 3.93 – 4.01 (m, 2 H), 4.23 – 4.32 (m, 2 H), 4.32 – 4.39 (m, 2 H), 5.49 (t, J=7.5 Hz, 1 H), 5.52 (dd, J=8.3, 5.3 Hz, 1 H), 7.22 (d, J=8.8 Hz, 1 H), 7.27 (d, J=8.8 Hz, 1 H), 7.98 (d, J=8.6 Hz, 1 H), 8.01 (dd, J=8.6, 1.1 Hz, 1 H), 8.03 (dd, J=8.8, 1.8 Hz, 1 H), 8.09 (d, J=8.8 Hz, 1 H), 8.19 (d, J=8.8 Hz, 1 H), 8.22 (dd, J=8.4, 1.8 Hz, 1 H), 8.25 (s, 1 H), 8.32 (s, 1 H), 8.41 (s, 1 H), 8.88 (s, 1 H).
Anal. Calcd for C42H5oN806 . 2 HCl . 4 H20: C 55.56, H 6.66 , N 12.34. Found: C 55.00, H 6.60, N 12.30
Going reverse…………………..
Intermediate XVIIIb
2.8 preparation of intermediate XVIIIb (A=

To a solution of XVIIb (960 mg, 1.48 mmol) in CH2C12 (25mL) was added HCI (5-6 M in isopropanol, 5 mL). The mixture was stirred at room temperature overnight. The solvent was evaporated, the obtained solid was dried in vacuum and used as such in the next step. 2.8a Alternative preparation of intermediate XVIIIb (A=
XVIIb (19.52 g, 30.1 mmol, 1.00 equiv.) was dissolved in dichloromethane (200 mL) and HCI in isopropanol (5-6 N, 300 mL) was added. The reaction mixture was stirred for 1 hour at room temperature. tBuOMe (1000 mL) was added to the suspension and the slurry was stirred at roomtemperature for 30 minutes. The filtered solid was rinced with tBuOMe (2x 100 mL) and dried under vacuum overnight to afford XVIIIb as a powder (15.2 g). 1H NMR (400 MHz, MeOD-d4) δ ppm 2.15 – 2.37 (m, 2 H), 2.37 – 2.52 (m, 2 H), 2.52 – 2.69 (m, 2 H), 2.69 – 2.88 (m, 2 H), 3.56 – 3.71 (m, 4 H), 5.19 – 5.41 (m, 2 H), 7.90 – 8.02 (m, 3 H), 8.05 (dd, J= 8.6, 1.6 Hz, 1 H), 8.10 – 8.25 (m, 4 H), 8.30 (d, J=1.4 Hz, 1 H), 8.47 (d, J=1.2 Hz, 1 H)
INTERMEDIATE XVIIb
2.7 reparation of intermediate XVIIb (A= PG= Boc)
To boronic ester XVIb (1.22 g, 2.26 mmol), bromide Xllla (1072 mg, 3.39 mmol), sodium bicarbonate (380 mg, 4.52 mmol), Pd(dppf)Cl2 (166 mg, 0.226 mmol) in toluene (50 mL), was added water (1 mL). The resulting mixture was heated at reflux overnight. The reaction mixture was filtered, evaporated to dryness and purified by column chromatography by gradient elution with heptane to ethyl acetate. The collected fractions containing the product were pooled and the volatiles were removed under reduced pressure. The residue (960 mg, 65 %) was used as such in the next reaction.
2.7a Alternative preparation of intermediate XVIIb (A= . PG= Boc)

XVIb (10 g, 18.5 mmol), Xlll’a (8.76 g, 24 mmol), NaHC03 (9.32 g, 111 mmol) and Pd(dppf)Cl2 (lg) were stirred in dioxane/water (140 mL, 6/1) under argon. The mixture was heated to 85 °C for 15 hours. Brine (100 mL ) was added and the mixture was extracted with CH2CI2, after drying on MgSC^, filtration and evaporation of the solvent, the residue was purified by column chromotography by gradient elution with CH2CI2 to EtOAc to afford XVIIb (7 g, 58 %).

To a stirred, deoxygenated solution of Vlllb (20.0 g, 45.2 mmol, 1.00 equiv.), Ilia (20.6 g, 49.7 mmol, 1.1 equiv.) and sodium bicarbonate (11.4 g, 136 mmol, 3.0 equiv.) in 1 ,4-dioxane/water (500 mL, 5: 1) under nitrogen, was added l.,.r-Bis(diphenyi~ phosphmo)ferrocene-paiIadium(]I)dichloride dichJoromethane complex (2.50 g, 4.52 mmol, 0.1 equiv.). The mixture was heated at 80°C under argon for 15 hours and cooled to room temperature. The reaction mixture was diluted with dichloromethane (500 mL) and washed with brine (2 x 150 mL) dried on magnesium sulphate; filtered and evaporated to dryness to afford a dark brown foam (43 g). The foam was purified using silicagel column chromatography (gradient elution with 0-6% MeOH in CH2CI2) to afford XVIIb (19.52 g, 65%) as an off-white powder.
INTERMEDIATE XVIb

Bromide XVb (1890 mg, 3.83 mmol), 4,4,4\4\5,5,5\5*-octamethyl-2,2′-bis(l,3,2- dioxaborolane) (2437 mg, 9.59 mmol), KF (390 mg; 6.71 mmol) and (dppf)PdCl2 (281 mg, 0.384 mmol) were dissolved in toluene (50 mL) and heated 3 days at reflux.
The solids were removed by filtration over dicalite and the filtrate was evaporated to dryness on silica. The residue was purified by column chromatography using a heptane to ethylacetate gradient. The fractions containing the product were pooled and the solvent was removed under reduced pressure. The residue (1.22 g, 59 %) was used as such in the next reaction
Under nitrogen, Ilia (25 g, 60.5 mmol), 6-bromonaphthalen-2-yl trifluoromethane- sulfonate (20 g, 56.7 mmol), K3P04 (36.65 g, 173 mmol) and (PPh3)4Pd (717 mg, 0.62 mmol) were stirred in THF (60 mL) and water (15 mL) with the heating mantle at 85 °C (reflux) for 2 hours. CH2CI2 (50 mL) was added and the water layer was separated. The organic layer was dried on MgS04 and after filtration, the filtrate was concentrated resulting in a sticky solid. The residue was purified by column
chromatography (petroleum ether/Ethyl acetate 15/1 to 1/1) to afford XVb (20 g;
40.6 mmol). Compound XVb (1 g, 2.0 mmol), potassium acetate (0.5 g, 5.0 mmol), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bis(l,3,2-dioxaborolane) (1.29 g, 5.0 mmol), and Pd(dppf)Cl2 (0. lg) were stirred in DMF (15 mL) under argon. The mixture was heated at 60°C for 5 hours. After cooling, CH2CI2 (50 mL) was added and the mixture was washed with saturated NaHC03. The water layer was separated and extracted with CH2CI2. The organic layers were combined and dried on MgSC^. After filtration the solvent was removed and the product was purified by column chromatography (gradient elution with petroleum ether/ethyl acetate 10/1 to 1/1) to give of XVIb (0.7 g,1.3 mmol, 65 %) as light yellow solid.
INTERMEDIATE XVb

2,6-Dibromonaphthalene (6.92 g, 24.2 mmol), boronic ester Ilia (2 g, 4.84 mmol), NaHC03 (813 mg, 9.68 mmol), (dppf)PdCl2(710 mg, 0.968 mmol) were dissolved in toluene (75 mL). Water (1 mL) was added and the mixture was heated for 7 hours at reflux. The solids were removed by filtration over dicalite and the filtrate was evaporated to dryness on silica. The residue was purified by column chromatography by gradient elution with heptane to ethylacetate. The appropriate fractions were pooled and the solvent was removed under reduced pressure. The residue (1.89 g, 79 %) was used as such in the next step.
1.2 Preparation of intermediate IIIa (PG= Boc)
IIIaTo a mixture of Ila (200 g, 546 mmol), potassium acetate (160.8 g, 1.64 mol) and 4,4,4*,4*,5,5,5*,5*-octamethyl-2,2,-bis(l,3,2-dioxaborolane) (416 g, 1.64 mol) in DMF (3L) was added Pd(dppf)Cl2 (20 g) under nitrogen gas. The reaction mixture was stirred at 85°C for 15 hours. The mixture was diluted with ethyl acetate, washed with water and brine, dried over magnesium sulfate, the solids removed by filtration, and the solvents of the filtrate were removed under reduced pressure. The residue was purified by silica column chromatography (petroleum ether : ethyl acetate 10: 1 to 2: 1) to afford 125 g of Ilia as a white solid (contains 15% of boronic acid).
INT IIa
1.1 preparation of intermediate Ila (PG= Boc; X= Br)
Ma
To a solution of Boc-Z-Proline (2669 mg, 12.4 mmol) in pyridine/DMF (30 mL, 1/1) was added di(lH-imidazol-l-yl)ketone (2205 mg, 13.6 mmol). The mixture was stirred at 45°C for 2 hours. 4-bromobenzene-l,2-diamine (2319 mg, 12.4 mmol) was added and the mixture was stirred at ambient temperature overnight. The solvent was removed and the residue heated in acetic acid (15 mL) at 100°C for 30 minutes. After
concentration of the residue, the mixture was partitioned between ethyl acetate and a saturated sodium bicarbonate solution. The organic phase was separated and washed with water, after drying over Na2SC”4, the mixture was filtrated and the filtrate was concentrated in vacuum. The obtained residue was purified by flash chromatography using CH2Cl2/EtOAc 90/10 to 50/50, resulting in compound Ila (3.146 g, 69 %).
DO NOT MISS OUT synthesis of XIIIa or XIII’a, this is needed in one of side chain
2.1 preparation of L-boc-prolinol
Borane-methyl sulfide complex (180 mL, 1.80 mol) was added dropwise to a solution of N-Boc- L-Proline (300 g, 1.39 mol) in anhydrous THF (3.0 L) which was cooled to 0°C. When gas evolution ceased, the ice bath was removed and the solution was stirred at 10°C for 18 hours. Thin layer chromatography (TLC) showed that no starting material remained and that the desired product was formed. The solution was cooled to 0°C and methanol (2.4 L) was slowly added. The solvents were removed under reduced pressure. The residue was reconstituted in dichloromethane (1 L), washed with
NaHC03 (500 mL, saturated, aqueous) and brine (500 mL), dried over MgS04, the solids were removed via filtration, and the solvents of the filtrate were removed under reduced pressure to afford a white solid, 260 g (93%), used in the next step without further purification.
2.2 preparation of Z-boc-prolinal
To a solution of Z-boc-prolinol (100 g, 500 mmol) in CH2CI2 (1.5 L) at 0°C were added successively, under vigorous stirring, 2,2,6,6-tetramethylpiperidine-l-oxyl (TEMPO; 1.56 g, 10 mmol) and NaBr (5.14 g, 50 mmol). To the resulting mixture was added dropwise a solution of NaHC03 (6.3 g, 75 mmol) and 6% NaCIO in active chlorine (750 mL, 750 mmol) at 0°C over a period of 1 hour. TLC showed no starting material remained and that the desired product was formed. The mixture was rapidly extracted with dichloromethane (2 x 1.5 L). The organic layers were combined, washed with NaHS04 (10%, 1 L) and KI (4%, 200 mL), then with Na2S203 (10%, 1 L) and brine (1.5 L), dried over MgS04, the solids were removed via filtration, and the solvents evaporated to afford a yellow oil, Z-boc-prolinal, (89 g, 92%>), used in the next step without further purification.
2.3 preparation of intermediate XXIV
ammonia
XXIV
Aqueous ammonia (25~28%>, 200 mL) was added dropwise to a solution of L-boc- prolinal (89 g, 0.44 mol) and glyoxal (183 mL of 40% in water) in methanol (1 L). The reaction mixture was sealed and reacted at 10°C. After 16 hours, additional glyoxal (20 mL) and aqueous ammonia (20 mL) were added and reacted for an additional 6 hours. The solvents were removed under reduced pressure, and the crude was reconstituted in ethyl acetate (1.0 L), washed with water and brine, dried over MgSC^, the solids were removed via filtration and the solvents were removed under reduced pressure. The crude was purified by column chromatography (silica gel, dichloromethane to methanol/dichloromethane 1 :70) to obtain 73 g (70%) intermediate XXIV as a white solid.
1H NMR: (CD3OD 400 MHz) δ 6.95 (s, 2H), 4.82-4.94 (m, 1H), 3.60-3.70 (m, 1H), 3.41-3.50 (m, 1H), 2.20-2.39 (m, 1H), 1.91-2.03 (m, 3H), 1.47 (s, 3H), 1.25 (s, 6H)
2.4 preparation of intermediate XHIa (PG= Boc)
XXIV Xllla
N-Bromosuccinimide (47.2 g, 0.26 mol) was added portion wise over 1 hour to a cooled (ice-ethanol bath, -10 °C) solution of XXIV (63.0 g, 0.26 mol) in CH2C12 (1.5 L) and stirred at similar temperature for 2 hours. The reaction mixture was concentrated in vacuum and the residue was purification by preparatory HPLC to provide 25.3 g (30%) of Xllla as a pale yellow solid.
1H NMR: CD3OD 400Mhz
δ 6.99-7.03 (s,lH), 4.77-4.90 (m, 1H), 3.61-3.68 (m, 1H), 3.42-3.50 (m, 1H), 2.20-2.39 (m, 1H), 1.89-2.05 (m, 3H), 1.47 (s, 3H), 1.27 (s, 6H).
2.4a preparation of intermediate XHI’a (PG= Boc)
To a solution of iodine (43.3 g, 170.5 mmol, 2 eq) in chloroform (210 mL) in a round bottomed flask (1L) a suspension of XXIV (20 g, 84.3 mmol) in an aqueous NaOH solution (2M, 210 mL) was added. The mixture was stirred at room temperature for 15 hours. To the resulting reaction mixture was added a saturated aqueous Na2S2C”3 solution (100 mL) and the organic layer was separated. The aqueous layer was extracted with chloroform (4x 150 mL). The organic layers were combined, washed with water and dried on magnesium sulphate. The solids were filtered and the solution was evaporated to dryness to afford diiodide (38.61 g, 89 %).
The above obtained intermediate diiodide (2.24 g, 4.58 mmol) and sodium sulfite (4.82 g, 38 mmol) were placed in a round bottomed flask (100 mL) and suspended in 30% EtOH/water (80 mL). The resulting mixture was refluxed for 40 hours. The solvent was removed and after addition of H20 (20 mL), the mixture was stirred at room temperature overnight. The solids were filtered, washed with water and dried in a vacuum oven to afford compound XHI’a (1.024 g, 61 %).
1H NMR (400 MHz, DMSO-d6) δ ppm 1.16 and 1.38 (2x br. s., 9 H), 1.68 – 2.02 (m, 3 H), 2.02 – 2.27 (m, 1 H), 3.18 – 3.38 (m, 1 H), 3.38 – 3.59 (m, 1 H), 4.53 – 4.88 (m, 1 H), 6.81 (m, -0.1 H), 7.05 – 7.28 (m, -0.9 H), 11.90 – 12.20 (m, -0.9 H), 12.22 – 12.40 (m, -0.1 H)
PATENT
WO 2011149856
http://www.google.co.in/patents/WO2011149856A1?cl=en
1st scheme

IN ABOVE SCHEME CONVERSION OF f to g N-methoxycarbonyl-L-Val-OH is used,
USE R =H IN LAST STEP TO GET RAVIDASVIR
EXAMPLE 1 – Synthesis of compounds of Formula lie
Scheme 1-1 describes preparation of target molecules and their analogs with symmetrical and non-symmetrical functionalized ends.
[0341] Step a. To a solution of 2-bromonaphthane a (62.0 g, 300 mmol) in DCM (1 L) was added A1C13 (44.0 g, 330 mmol) and 2-chloroacetyl chloride (34.0 g, 330 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h and then H20 added (500 mL) and extracted. The organic layer was washed with H20, dried over anhydrous Na2S04, evaporated under reduced pressure to give 80 g crude product, which was purified by re-crystallization from 10% EtOAc- hexane (v/v) to yield b (28 g, 36% yield) as a white solid: JH NMR (500 MHz, CDC13) δ 8.44 (s, 1H), 8.07 (s, 1H), 8.04 (d, J= 11.0 Hz, 1H), 7.84 (d, J= 8.5 Hz, 2H), 7.66 (d, J= 8.5 Hz, 1H), 4.81 (s, 2H) ppm; LCMS (ESI) m/z 282.9 (M + H)+.
Step b. To a solution of b (28.0 g, 100 mmol) in DCM (500 mL) was added N-Boc- L-Pro-OH (24.7 g, 115 mmol) and Et3N (70.0 mL, 500 mmol) and the mixture was stirred at rt for 2 h. The mixture was concentrated under reduced pressure to afford crude c which was used for the next step without further purification. LC-MS (ESI) m/z 462.1 (M + H)+.
Step c. To a solution of c (46.0 g, 100 mmol) in toluene (500 mL) was added
NH4OAc (77 g, 1.0 mol) and the mixture was stirred at 110 °C overnight, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc l :l(v/v)) to afford d (30 g, 68% yield) as a yellow solid: LC-MS (ESI) m/z 442 A (M + H)+.
Step d. To a solution of d (10.0 g, 23.0 mmol) in anhydrous DME (200 mL) and equal molar of boronate e was added PPh3 (1.2 g, 4.6 mmol), Pd(PPh3)4 (1.6 g, 2.3 mmol), and 2.0 M Na2C03 solution. The mixture was refluxed under argon overnight. The organic solvent was removed under reduced pressure and the residue was treated with H20, extracted with EtOAc (2 x 200 mL). The combined organic phase was dried, filtered, and concentrated in vacuo to give a residue, which was purified by silica gel column chromatography (petroleum
ether/EtOAc 3: l(v/v)) to afford f (10 g, 96% yield) as a yellow solid. LC-MS (ESI): m/z 709.3 (M+H)+.
Step e. To a stirred solution of f (150 mg, 0.29 mmol) in dioxane (3 mL) was added 4.0 N HCl in dioxane (3 mL) dropwise. The mixture was stirred at rt for 4 h, and then
concentrated to yield a yellowish solid (134 mg), which was used directly for the next step. The residue (134 mg, 0.290 mmol) was suspended in THF (5 mL) and DIPEA (0.32 mL) was added and followed by addition of N-methoxycarbonyl-L-Val-OH (151 mg, 0.860 mmol). After stirring for 15 min, HATU (328 mg, 0.860 mmol) was added and the mixture was stirred at rt for another 2 h and then concentrated. The residue was purified by prep-HPLC to obtain g (40 mg, 19% yield).
2nd scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
Note 9 is not final product pl ignore it
Step a. Referring to Scheme 1-2, to a solution of compound 3 (2.0 g, 4.5 mmol) in dioxane (25 mL) was added 4.0 N HCl in dioxane (25 mL). After stirring at rt for 4 h, the reaction mixture was concentrated and the residue was dried in vacuo to give a yellowish solid (2.1 g), which was used directly for the next step without further purification.
[0347] Step b. To the residue of step a (4.5mmol) was added DMF (25 mL), followed by adding HATU (2.1 g, 5.4 mmol), DIPEA (3.7 mL, 22.5 mmol) and N-methyl carbamate-L-valine (945 mg, 5.4 mmol). After stirring at rt for 15 min, the reaction mixture was added slowly to H20 (400 mL). A white solid precipitated was filtered and dried to give compound 6 (2.2 g, 98% yield). LC-MS (ESI): m/z 499.1 (M+H)+.
[0348] Step c. To a mixture of compound 6 (800 mg, 1.6 mmol), compound 7 (718 mg, 1.6 mmol), and NaHC03 (480 mg, 5.7 mmol) in 1 ,2-dimethoxyethane (15mL) and H20 (5mL) was added Pd(dppf)Cl2 (59 mg, 0.08 mmol). After stirring at 80°C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20%
methanol/CHCl3 (100 mL) and H20 (100 mL). The organic phase was separated and the aqueous phase was extracted with 20% methanol/CHCl3 (100 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum
ether/EtOAc=15: l(v/v)) to give compound 8 (1.0 g, 85% yield) as a yellow solid. LC-MS (ESI): m/z 732.4 (M+H)+.
Step d. To a solution of compound 8 (200 mg, 0.27 mmol) in dioxane (3.0 mL) was added 4 N HCl in dioxane (3.0 mL). After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to give an HCl salt in quantitative yield, which was used directly for the next step without further purification…………..CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
CAUTION SIMILAR BUT NOT SAME……..Step e. To a solution of the salt (0.27 mmol) in DMF (5.0 mL) was added DIPEA (0.47mL, 2.7 mmol), followed by adding N,N-dimethyl-D-phenyl glycine (59 mg, 0.33 mmol) and HATU (125 mg, 0.33 mmol). After stirring at rt for lh, the reaction mixture was partitioned between H20 and DCM. The organic phase was washed successively with H20 and brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by prep-HPLC to give compound 9……..CAUTION SIMILAR BUT NOT SAME. LC-MS (ESI): m/z 793.4 (M+H)+.
3rd scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
15 NOT THE COMPD PL IGNORE IT IF YOU NEED RAVIDASVIR
Step a. To a mixture of compound 3 (3.2 g, 7.2 mmol), bis(pinacolato)diboron (3.86 g, 15.2 mmol), and KOAc (1.85g, 18.8mmol) in 1,4-dioxane (100 mL) was added Pd(dppf)Cl2 (440 mg, 0.6 mmol). After stirring at 80 °C for 3 h under an atmosphere of N2, the reaction mixture was concentrated. The residue was purified with silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 11 (2.8 g, 80% yield) as a white solid. LC- MS (ESI): m/z 490.3 (M+H)+.
[0352] Step b. To a mixture of compound 11 (626 mg, 1.27 mmol), compound 12 (570 mg, 1.27 mmol), and NaHC03 (420 mg, 4.99 mmol) in 1, 2-dimethoxyethane (30 mL) and H20 (10 mL) was added Pd(dppf)Cl2 (139 mg, 0.19 mmol). After stirring at 80°C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20% methanol/CHCl3 (100 mL) and H20 (100 mL). The aqueous phase was extracted with 20% methanol/CHCl3 (100 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 13 (635 mg, 68% yield) as a yellow solid. LC-MS (ESI): m/z 732.4 (M+H)+.
Step c. To a solution of compound 13 (200 mg, 0.27 mmol) in dioxane (3.0 mL) was added 4 N HC1 in dioxane (3.0 mL). After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to yield the HC1 salt of compound 14 in quantitative yield, which was used directly for the next step without further purification…..CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
CAUTION SIMILAR BUT NOT SAME………Step d. To a solution of the salt (0.27 mmol) in DMF (5.0 mL) was added DIPEA (0.47 mL, 2.7 mmol), followed by adding N,N-dimethyl-D-phenyl glycine (59 mg, 0.33 mmol) and HATU (125 mg, 0.33 mmol). After stirring at rt for lh, the reaction mixture was partitioned between H20 and DCM. The organic phase was consequently washed with H20 and brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by prep-HPLC to give compound 15..CAUTION SIMILAR BUT NOT SAME. LC-MS (ESI): m/z 793.4 (M+H)+.
4 th scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
5 NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
4 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
EXAMPLE 2 – Synthesis of compounds of Formula Hie
Step a. Referring to Scheme 2-1, to a mixture of compound 1 (5.05 g, 13.8 mmol), bis(pinacolato)diboron (7.1 g, 27.9 mmol), and KOAc (3.2 g, 32.5 mmol) in 1,4-dioxane (100 mL) was added Pd(dppf)Cl2 (400 mg, 0.5 mmol). After stirring at 80 °C for 3 h under an atmosphere of N2, the reaction mixture was concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 2 (3.0 g, 53% yield) as a gray solid. LC-MS (ESI): m/z 414.2 (M+H)+.
Step b. To a mixture of compound 2 (522 mg, 1.26 mmol), compound 3 (500 mg, 1.13 mmol), and NaHC03 (333 mg, 3.96 mmol) in 1, 2-dimethoxyethane (30 mL) and H20 (10 mL) was added Pd(dppf)Cl2 (74 mg, 0.1 mmol). After stirring at 80°C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20% methanol/CHCl3 (100 mL) and H20 (100 mL). The organic phase was separated and the aqueous phase was extracted with 20% methanol/CHCl3 (100 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (DCM/MeOH=50:l (v/v)) to give compound 4 (450 mg, 55% yield) as a yellow solid. LC-MS (ESI): m/z 649.3 (M+H)+.
Step c. To a stirred solution of compound 4 (160 mg, 0.25 mmol) in dioxane (2.0 mL) was added 4N HCl in dioxane (2.0 mL). After stirring at rt for 3h, the reaction mixture was concentrated and the residue was dried in vacuo to give an HCl salt in quantitative yield, which was used directly for the next step without further purification.4 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
SCHEME SIMILAR UPTO PENULTIMATE STEP
5 NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
scheme ……..CAUTION SIMILAR BUT NOT SAME
5 th scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
18NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
17 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
Step a. Referring to Scheme 2-2, to a mixture of compound 2 (1.16 g, 2.32 mmol), compound 6 (1.40 g, 3.39 mmol), and NaHC03 (823 mg, 9.8 mmol) in 1, 2-dimethoxyethane (30 mL) and H20 (10 mL) was added Pd(dppf)Cl2 (103 mg, 0.14 mmol). After stirring at 80 °C over night under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20% methanol/CHCl3 (150 mL) and H20 (150 mL). The aqueous phase was extracted with 20% methanol/CHCl3 (150 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/acetone=1.5/l (v/v)) to give compound 16 (1.32g, 80% yield) as a yellow solid. LC-MS (ESI): m/z 706.4 (M + H)+.
tep b. To a solution of compound 16 (200 mg, 0.28 mmol) in dioxane (3.0 mL) was added 4 N HC1 in dioxane (3.0 mL). After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to give the HC1 salt of compound 17 in quantitative yield, which was used directly for the next step…….17 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
6 th scheme
 scheme 2-3
scheme 2-3
SCHEME SIMILAR UPTO PENULTIMATE STEP
22NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
21 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
Scheme 2-3
Step a. Referring to Scheme 2-3, to a solution of compound 1 (4.0 g, 10.9 mmol) in dioxane (40 mL) was added 4 N HC1 in dioxane (40 mL). After stirring at rt overnight, the reaction mixture was concentrated. The residue was washed with DCM, filtered, and dried in vacuo to afford a hydrochloride salt in quantitative yield, which was used for the next step without further purification.
Step b. To a solution of the salt (10.9 mmol) in DMF (30 mL) was added DIPEA (5.8 mL, 33.0 mmol), followed by adding N-methoxycarbonyl-L-valine (2.1 g, 12.1 mmol) and HATU (4.6 g, 12.1 mmol). After stirring at rt for lh, the reaction mixture was partitioned between H20 and DCM. The organic phase was consequently washed with H20 and brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (DCM/Petroleum ether=4/l (v/v)) to give compound 19 (3.0 g, 65% yield). LC- MS (ESI): m/z 423.1 (M+H)+.
Step c. To a mixture of compound 11 (800 mg, 1.9 mmol), compound 19 (700 mg, 1.7 mmol), and NaHC03 (561 mg, 6.6 mmol) in 1, 2-dimethoxyethane (60 mL) and H20 (20 mL) was added Pd(dppf)Cl2 (183 mg, 0.25 mmol). After stirring at 80 °C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was then partitioned between 20% methanol/CHCl3 (100 mL) and H20 (100 mL). The aqueous phase was extracted with 20% methanol/CHCl3(100 mL) again. The combined organic phase was consequently washed with brine, dried with Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 20 (600 mg, 52% yield) as a yellow solid. LC-MS (ESI): m/z 706.4 (M+H)+.
Step d. To a solution of compound 20 (200 mg, 0.28 mmol) in dioxane (3.0 mL) was added 4N HC1 in dioxane (3.0 mL). After stirring at rt for 2h, the reaction mixture was concentrated and the residue was dried in vacuo to yield the HC1 salt of compound 21 in quantitative yield, which was used directly for the next step without further purification.
21 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
7 th scheme

Scheme 6-2
SCHEME SIMILAR UPTO n-2 STEP in above scheme
84, 85 NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
83 CAN BE USED AS early PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
Step a. Referring to Scheme 6-2, a solution of compound 78 (50.0 g, 0.30 mol) in THF (500 mL) and H20 (500 mL) was added K2C03 (83 g, 0.60 mol) and (Boc)20 (73. Og, 0.330 mol). After stirring at rt overnight, the reaction mixture was concentrated and the residue was extracted with EtOAc (250 mL x 3). The extracts were combined, washed with brine, and dried with anhydrous Na2S04. The solvent was removed and the residue was dried in vacuo to give crude compound 78 (62 g), which was used for the next step without further purification. LC-MS (ESI) m/z 230.1 (M + H)+.
[0453] Step b. To a solution of compound 78 (60.0 g, 260 mmol) in EtOH (1 L) was slowly added NaBH4 (50.0 g, 1.30 mol) at rt. After stirring at rt overnight, the reaction was quenched by adding acetone (10 mL). The resulting mixture was concentrated and the residue was diluted with EtOAc (500 mL). The mixture was washed with brined and dried in vacuo. The solvent was removed and the residue was purified by silica gel column chromatography (Petroleum ether/EtOAc = 1/1 (v/v)) to give compound 79 (42.0 g, 80% yield) as a white solid. LC-MS (ESI) m/z 202 A (M + H)+.
[0454] Step c. To a solution of compound 79 (30.0 g, 150 mmol) and DMSO (35.0 g, 450 mmol) in DCM (1 L) was added oxalyl chloride (28.0 g, 220 mmol) at -78 °C. After stirring at – 78 °C for 4 h, the reaction mixture was added Et3N (60.0 g, 600 mol) and the resulting mixture was stirred for another 1 h at -78 °C. Subsequently, the reaction was quenched by adding H20. The organic layer was separated and the aqueous layer was extracted with DCM (200mL x 2). The extracts were combined, washed with brine, and dried with Na2S04. The solvent was removed and the residue was dried in vacuo to give crude compound 80 (22.0 g) as a colorless oil, which was used immediately without further purification. LC-MS (ESI) m/z 200.1 (M + H)+.
[0455] Step d. A mixture of compound 80 (7.7 g, 38.5 mmol), 6-bromopyridine-2,3-diamine (8.0 g, 42.8 mmol) (PCT Intl. Appl. WO 2008021851) , and iodine (1.08 g, 4.28 mmol) in AcOH (30 mL) was stirred at rt overnight. The reaction mixture was neutralized by adding saturated aqueous NaHC03. The resulting mixture was extracted with EtOAc (200 mL x 3). The extracts were combined, washed with brine, and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel column chromatography (DCM/MeOH = 80/1 (v/v)) to give compound 81 (7.8 g, 55% yield). LC-MS (ESI) m/z 367.1 (M + H)+.
[0456] Step e. A mixture of compound 82 (10.0 g, 20.1 mmol), bis(pinacolato)diboron (7.65 g, 30.1 mmol), potassium acetate (6.89 g, 70.3 mmol), and Pd(dppf)Cl2-CH2Cl2 (886 mg, 1.0 mmol) in 1,4-dioxane (200 mL) was stirred at 80 °C for 3 h under an atmosphere of N2. The reaction mixture was filtered through CELITE™ 545 and the filtered cake was washed with EtOAc (200 mL x 3). The filtrate was washed with brine and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel column chromatography
(DCM/MeOH = 50/1 (v/v)) to give compound 83 (9.8 g, 89% yield) as a white solid: LC-MS (ESI) m/z 547.3 (M + H)+.83 CAN BE USED AS early PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
PATENT
CN 102796084
http://www.google.com/patents/CN102796084A?cl=en
Step One: Formula (2) compounds strokes trichloride catalyst (AlCl3), chloroacetyl chloride (2-chloroacetylchloride) at room temperature to obtain a compound of formula (3),

(3);
wherein the reaction temperature is room temperature, the solvent is methylene chloride. Material I (i.e., formula (2) compound) and chloroacetyl chloride (2-chloroacetyl chloride) was slowly added, higher yields can be obtained. (3) The compound was recrystallized from ether to obtain.
In the present embodiment, the 20.5 g of formula (2) compound (0. Imol) and 26.2 g AlCl3 (0.2mol) was added to 200ml of dichloromethane, cooled to room temperature, stirring speed slowly was added 13.4 g of chloroacetyl chloride (I. 2mol), within three hours after the addition and then mixed by stirring maintained at room temperature for 3 hours. Was slowly added 50 ml of ice water, the precipitate was collected by filtration. The filter cake was washed with 10 ml of water and 10 ml petroleum ether (twice). The filtrate and the organic layer together with 50 ml of dichloromethane and extracted twice with 50 ml brine and then paint extraction solution, the extract was dried over magnesium sulfate, the solution was removed, the solid with 100 ml of diethyl ether and recrystallized to afford 20g (71% yield compounds) of formula (3).
Step II: Formula (3) with a compound of formula (4) compound under acidic conditions and chloroform (CCl3H) heating the reaction, and the reaction system reached reflux to give a compound of formula (5),

(5);
[0042] wherein, the formula (3) with a compound of formula (4) compound in acetonitrile (chloroform (CCl3H), the reaction system must be reached reflux, and must be reacted under acidic conditions to give the compound of formula (5). [0043] In this embodiment, the compound (3) (0. Imol) 28. 2 克 formula and the compound (4) (0. Imol) 21. 5 克 style with 3 g of trifluoroacetic acid was added to 200 ml of chloroform, in was stirred at reflux under nitrogen for 17 hours. After cooling to room temperature, spin-dry, to give 46. I g of a yellow solid of formula (5) compound (99% yield).
Step three: (5) the compound obtained in toluene (toluene) and ammonium acetate (NH4OAc) reflux (6) of
Thereof,

Compound of formula (5) is ammonium acetate with toluene under reflux conditions for ring closure.
In the present embodiment, the compound (0. Imol) and 10 g of ammonium acetate (NH4OAc) was added 46. I g of formula (5) to IJ 200ml of toluene, heated under reflux for 3 hours with stirring. Was slowly added 50 ml of ice water, filtered, washed with 100 ml of toluene and extracted twice with 50 ml brine and then paint extraction solution, the extract was dried over magnesium sulfate, the solution was removed, the solid with 100 ml of diethyl ether and recrystallized to afford 40g (89% compound yield) of the formula (6).
Step Four: (6) compound in the catalyst and the associated button pinacolato ester (Bis (pinacolato) diboron) reacting a compound of formula (7),

wherein, Pd (dppf) 2Cl2 can be replaced by another of a palladium catalyst, a palladium catalyst with the other, the same effect.
In the present embodiment, 44 g of the compound of formula (6) (0. Imol) and 3 g Pd (dppf) 2C12,25. 4 克 United pinacolato ester (0. Imol) and 8.4 g of sodium bicarbonate (0. Imol) was added to a 200 ml I. 4- dioxane, stirred at reflux for 24 hours. Diatomaceous earth filtration, spin dry. Spin-dry 100 ml of ethyl acetate dissolved. Anhydrous magnesium sulfate and spin dry. Recrystallization from ether to yield 40 g (82% yield) of a yellow solid of formula (7) compound.
Step Five: formula (7) under palladium catalyst compound and the compound (8) obtained by reacting the compound of formula (9),

wherein, Pd (dppf) 2Cl2 can be replaced by another of a palladium catalyst, a palladium catalyst with the other, the same effect.
In the present embodiment, 48.9 g of the compound of formula (7) (0. Imol) and 3 g Pd compound (8) (0. Imol) (dppf) 2C12,41. 3 and 8 克 style. 4 g of sodium hydrogen carbonate (0. Imol) was added to a 200 ml I. 4- dioxane, stirred at reflux for 24 hours. Diatomaceous earth filtration, spin dry. Spin-dry 100 ml of ethyl acetate dissolved. Anhydrous magnesium sulfate and spin dry. Recrystallized from ether to give compound 55 g (85% yield) of a yellow solid of formula (9).
[0056] Step Six: formula (9) compound deprotected under acidic conditions to give a compound of formula (10),
[0057]

In the present embodiment, the 64.8 grams of formula (9) compound (0. Imol) was added to 100 ml I. 4_ dioxane was stirred, 100 ml of 5M / L of I under nitrogen 4- dioxane solution of hydrochloric acid. Spin-dry for 24 hours later, get 52. I g of pale yellow solid formula (10) compound (99% yield).
Step 7: Formula (10) with a compound (11) in a condensing agent is 2- (7-azo BTA) -N, N, N ‘, N’- tetramethyluronium hexafluorophosphate phosphate (HATU) under condensation reaction conditions to give the final product compound C0S-101, i.e. the compound of formula (I):

In the present embodiment, the compound of formula 52. I g of (10) (0. Imol) was added to a 200 ml N, N- dimethylformamide (DMF) cooled to 0 ° with stirring, in a nitrogen atmosphere was added 20.2 g of triethylamine (0. 2mol) 0 After 10 minutes of stirring, was added 19 g of formula (11) compound (0. Ilmol) was added followed by 26 g HATU (0. 2mol), stirred at room temperature for 32 hours . Was slowly added 50 ml of ice water, the precipitate was collected by filtration. The filter cake was washed with 10 ml of water and 50 ml dichloromethane twice. Together with the filtrate and the organic layer was extracted 2 times 50 ml of dichloromethane, and then washed with 50 ml brine solution, the extract was dried over magnesium sulfate, the solution was removed, solid was recrystallized from 100 ml of ethanol, to give 50g (66% yield) The pale yellow compound C0S-101.
In summary this compound on C0S-101 non-structural protein 5A inhibitor, or a pharmaceutically acceptable salt thereof, the treatment of hepatitis C active substance. A compound of formula (3) Friedel-Crafts reaction occurs directly from 2-bromo-naphthalene chloride and chlorine. A compound of formula (3) with a compound of formula (4) condensing a compound of formula (5). The compound of formula (5) self-condensation of a compound of formula (6). Of formula (6) is reacted with boronic acid pinacol ester linking reaction of the compound of formula (7). A compound of formula (7) with a compound of formula (8) coupling reaction of a compound of formula (9). Off compound under acidic conditions (9) protect the compound of formula (10) and formula (10) compound condensation of the final product C0S-101, method of operation of the invention is simple, mild conditions, process maturity, yield and high purity suitable for industrial production.
PATENT
WO 2013123092
http://www.google.com/patents/WO2013123092A1?cl=en

Scheme 3

3-3 2HCI salt
Step 1. Referring to Scheme 3, compounds l-5a (1.3 kg , 1.0 eq.), 2-2a (975.0 g, 1.0 eq.), NaHCOs (860.0 g, 3.80 eq.), Pd(dppf)Cl2 (121.7 g, 0.05 eq.), purified water (5.2 L, 4.0 volume) and 1 ,2-dimethoxy ethane (DME) (24.7 L, 19.0 volume) were charged into a 50.0 L 4-necked round bottom flask under argon atmosphere. After being degassed using argon for a period of 30 min, the reaction mass was slowly heated to ~ 80 °C and stirred at this temperature for 12 – 14 hrs. HPLC analysis indicated that > 97% of compound 2-2a was consumed. Next, the reaction mass was concentrated to completely remove DME under vacuum (600 mmHg) at 40 – 45 °C and the residue was diluted with 20% (v/v) MeOH in DCM (13.0 L , 10 volume) and purified water (13.0 L, 10.0 volume) with stirring. The organic layer was separated and the aqueous layer was extracted with 20% (v/v) MeOH in DCM (6.5 L x 2, 10.0 volume). The combined organic extracts were washed twice with water (6.5 L x 2, 10.0 volume) and once with saturated brine (6.5 L, 5.0 volume) and dried over anhydrous Na2S04. The solvent was removed under vacuum (600 mmHg) and the residue was purified by flash column chromatography using silica gel with hexanes/EtOAc as eluent to give compound 3-1 (1.0 kg, 63% yield) as off white solid with a purity of > 98.0%> determined by HPLC analysis. LC-MS (ESI): m/z 649.3 [M + H]+. 1H NMR (400 MHz, d6– DMSO): δ 12.26 – 12.36 (m, 1H), 11.88 – 11.95 (m, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 7.91 (m, 3H), 7.85 – 7.87 (m, 2H), 7.51 – 7.81 (m, 3H), 4.78 -4.99 (m, 2H), 3.55 – 3.59 (m, 2H), 3.35 – 3.44 (m, 2H), 2.30 – 2.47 (m, 2H), 1.85 – 2.01 (m, 6H), 1.39, 1.14, 1.04 (s, s, s, 18H) ppm. Alternatively, compound 3-1 can be obtained following the same procedure and using compounds l-4a and 2-3a instead of compounds l-5a and 2-2a as the Suzuki coupling components.
Step 2. Compound 3-1 (1.0 kg, 1.0 eq.) and IPA (7.0 L, 7.0 volume) were charged into a 20.0 L four-necked RB flask under nitrogen atm. The reaction mass was cooled to 18 – 20°C and 3.0 N HC1 in isopropyl alcohol (7.0 L, 7.0 volume) was added over a period of 90 – 120 min under nitrogen atmosphere. After stirring at 25 – 30 °C for 10 – 12 hrs under nitrogen atmosphere, HPLC analysis indicated that > 98%> compound 3-1 was consumed. Next, the reaction mass was concentrated to remove IPA under vacuum at 40 – 45 °C. The semi solid obtained was added to acetone (2.0 L, 2.0 volume) with stirring and the resulting suspension was filtered under nitrogen atmosphere. The solid was washed with acetone (2.0 L, 2.0 volume) and dried in a vacuum tray drier at 40 – 45 °C for 10 hrs to give compound 3- 2 (860 g, 94%o yield) as pale yellow solid with a purity of > 98.0%> determined by HPLC analysis. LC-MS (ESI): m/z 449.2 [M + H]+. 1H NMR (400 MHz, -DMSO): δ 10.49 – 10.59 (m, 2H), 10.10 and 9.75 (m, m, 2H), 8.60 (s, 1H), 8.31 (s, 2H), 8.15 (m, 1H), 8.13 – 8.15 (m, 2H), 7.96 – 8.09 (m, 2H), 7.82 (s, 2H), 5.08 (m, 2H), 3.39 – 3.53 (m, 4H), 2.47 – 2.54 (m, 3H), 2.37 (m, 1H), 2.14 – 2.21 (m, 2H), 2.08 (m, 2H) ppm.
Step 3. Compound 3-2 (2.2 kg, 1.0 eq.) was added to a four necked round bottom flask charged with DMF (4.4 L, 20.0 volume) under a nitrogen atmosphere. After stirring for 15 min, the mixture was added N-Moc-L-Valine (226.2 g, 3.52 eq.) in one lot at 25 – 30 °C. Next, the mixture was cooled to -20 to -15 °C, followed by adding HATU (372.9 g, 2.0 eq.) portion wise over 30 min. After stirring for 10 min, a solution of DIPEA (238.9 g, 5.0 eq.) in DMF (1.1 L, 5.0 volume) was added over 45 min. Subsequently, the reaction mass was warmed to 25 – 30 °C with stirring. After stirring for 1 hr, HPLC analysis indicated that > 99%) of compound 3-2 was consumed. The reaction mixture was poured into water (38.0 L) and the mixture was extracted with DCM (10.0 L x 3, 45.0 volume). The combined organic extracts were washed with water (10.0 L x 3, 45.0 volume) and saturated brine (10 L, 45.0 volume) and dried over anhydrous Na2S04. The solvent was removed at 40 – 45 °C under vacuum (600 mmHg) and the residue was purified by column chromatography on silica gel using DCM and MeOH as the eluent to give compound 3-3 (1.52 kg, 47% yield) as off white solid with a purity of > 97.0% determined by HPLC analysis. LC-MS (ESI): m/z 763.4 [M + H]+. 1H NMR (400 MHz, -DMSO): δ 8.60 (s, 1H), 8.29 (s, 1H), 8.20 (s, 1H), 8.09 – 8.14 (m, 2H), 7.99 – 8.05 (m, 2H), 7.86 – 7.95 (m, 3H), 7.20-7.21 (m, 2H), 5.24 – 5.33 (m, 2H), 4.06 – 4.18 (m, 4H), 3.83 (m, 2H), 3.53 (m, 6H), 2.26 – 2.55 (m, 10H), 0.85 (m, 6H), 0.78 (m, 6H) ppm. The transformation of 3-2 to 3-3 (Compound I) can be achieved via a range of conditions. One of these conditions is described below.
A reactor was charged with N-Moc-V aline (37.15 g, 0.211 mol), acetonitrile (750 mL) and DIPEA (22.5 g). The reaction mixture was agitated for 10 min and HOBT (35.3 g 0.361 mole) and EDCI (42.4 g, 0.221 mole) were added while keeping temperature < 2 °C. The reaction mixture was agitated for 30 min and DIPEA (22.5 g) and compound 3-2 (48.0 g, 0.092 mole) was added slowly to reactor over 30 min to keep temperature < 3 °C. The reaction mixture was agitated 4 hrs at 20 – 25 °C, and sample was submitted for reaction completion analysis by HPLC (IPC specification: < 1.0% area 3-2 remaining). At the completion of reaction as indicated by HPLC analysis, isopropyl acetate (750 mL) was added to the reactor and stirred for 10 min. The organic layer (product layer) was washed with brine (300 mL x 2) and 2% NaOH (200 mL). The organic solution was filtered through a silica gel pad to remove insoluble material. The silica gel pad was washed with isopropyl acetate and concentrated under vacuum (400 mm/Hg) to a minimum volume. The crude product was purified by column chromatography on silica gel using ethyl acetate and methanol as eluent to give compound 3-3 (38.0 g, 65%> yield) with purity of > 95 %>. LC-MS (ESI): m/z 763.4 [M + H]+.
Step 4. Compound 3-3 (132.0 g, 1.0 eq.) and ethanol (324.0 mL, 2.0 volume) were charged into a 10 L four-necked round bottom flask under nitrogen atmosphere. After stirring for 15 min, the suspension was cooled to 5 – 10 °C, to it was added 2.0 N HC1 in ethanol (190 mL, 1.5 volume) over 30 min. The resulting solution was allowed to warm to 25 – 30 °C. Acetone (3.96 L, 30.0 volume) was added over 90 min in to cause the slow precipitation. Next, the suspension was warmed to 60 °C and another batch of acetone (3.96 L, 30.0 volume) was added over 90 min. The temperature was maintained at 55 – 60 °C for 1 hr, and then allowed to cool to 25 – 30 °C. After stirring at 25 – 30 °C for 8 – 10 hrs, the mixture was filtered. The solid was washed with acetone (660.0 mL, 5.0 volume) and dried in a vacuum tray drier at 50 – 55 °C for 16 hrs to give the di-HCl salt of compound 3-3
(compound I) (101 g, 71% yield) as pale yellow solid with a purity of > 96.6% determined by HPLC analysis.
Preparation of N-Moc-L-Valine
N-Moc-L-Valine is available for purchase but can also be made. Moc-L-Valine was prepared by dissolving 1.0 eq of L-valine hydrochloride in 2-methyltetrahydrofuran (2- MeTHF) /water containing sodium hydroxide and sodium carbonate, and then treating with 1.0 eq of methyl chloroformate at 0 – 5°C for 6 hr. The reaction mixture was diluted with 2- MeTHF, acidified with HC1, and the organic layer was washed with water. The 2-MeTHF solution is concentrated and the compound is precipitated with n-heptane. The solid was rinsed with 2-MeTHF/ n-heptane and dried in vacuo to give N-Moc-L-Valine in 68% yield. Crystallization of Compound I to Yield Form A
Compound I Salt Formation and Crystallization, Example 1
Ethanol (3.19 L, 1.0 volume, 200 proof) was charged to the 230-L glass lined reactor under nitrogen atmosphere. Free base form of compound 3-3 (3.19 kg, 4.18 mol) was added to the flask with stirring, stir continued for an additional 20 to 30 min. To the thick solution of 3-3 in ethanol was added slowly 2.6 N HC1 in ethanol (3.19 L, 1.0 volume) to the above mass at 20 – 25 °C under nitrogen atmosphere. The entire mass was stirred for 20 min at rt, and then heated to 45 – 50 °C. Acetone (128.0 L, 40.0 volume) was added to the above reaction mass at 45 – 50 °C over a period of 3-4 hrs before it was cooled to ~25 °C and stirred for ~15 hrs. The precipitated solid was collected by filtration and washed with acetone (6.4 L x 2, 4.0 volume), suck dried for 1 hr and further dried in vacuum tray drier at 40 – 45 °C for 12 hrs. Yield: 2.5 kg (71.0% yield), purity by HPLC: 97.70%, XRPD: amorphous.
Isopropyl alcohol (7.5 L, 3.0 volume) was charged to a 50.0 L glass reactor protected under a nitrogen atmosphere. The amorphous di-HCl salt of 3-3 (2.5 kg) was added to the above reactor with stirring. The entire mass was heated to 60 – 65 °C to give a clear solution. Stir continued at 65 ± 2 °C for ~15 hrs, solid formation started during this time. The heating temperature was lowered to ~50 °C over a period of 3 hrs, methyl tertiary butyl ether (12.5 L, 5.0 volume) was added to the above mass slowly over a period of ~3 hrs with gentle agitation. The above reaction mass was further cooled to 25 – 30 °C over 2 – 3 hrs. The solid was collected by filtration, washed with 10.0% isopropyl alcohol in methyl tertiary butyl ether (6.25 L, 2.5 volume), suck dried for 1 hr and further dried in a tray drier at 45 – 50 °C under vacuum (600 mm/Hg) for 70 – 80 hrs. Yield: 2.13 kg (85.0% recovery, 61.0% yield based on the input of compound free base 3-3), purity by HPLC: 97.9%.
FIG. 1 : 1H NMR (500 MHz, -DMSO): δ 15.6 (bs, 2H), 14.7 (bs, 2H), 8.58 (s, 1H), 8.35 (s, 1H), 8.25 (s, 1H), 8.18 (d, J= 8.7 Hz, 1H), 8.13 (s, 1H), 8.06 (d, J= 8.6 Hz, 1H), 8.04 (s, 1H), 8.00 (s, 1H), 7.98 (d, J= 8.7 Hz, 1H), 7.91 (d, J= 8.6 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.33 (d, J= 8.6 Hz, 2H), 5.31 (m, 1H), 5.26 (m, 1H), 4.16 (d, J= 7.7 Hz, 1H), 4.04 (m, 2H), 3.87 (m, 2H), 3.55 (s, 6H), 2.42 (m, 2H), 2.22-2.26 (m, 4H), 2.07-2.14 (m, 4H), 0.86 (d, J= 2.6 Hz, 3H), 0.84 (d, J= 2.6 Hz, 3H), 0.78 (d, J= 2.2 Hz, 3H), 0.77 (d, J= 2.2 Hz, 3H), 3.06 (s, OMe of MTBE), 1.09 (s, t-Bu of MTBE), 1.03 (d, 2Me of IP A) ppm.
FIG. 2: 13C NMR (500 MHz, /-DMSO): δ 171.6, 171.5, 157.4, 156.1, 150.0, 138.2, 138.0, 133.5, 132.5, 131.3, 129.8, 129.4, 128.0, 127.0, 126.4, 125.6, 125.3, 124.4, 124.2, 115.8, 115.0, 112.5, 58.37, 58.26, 54.03, 53.34, 52.00 (2 carbons), 47.71 (2 carbons), 31.52, 31.47, 29.42 (2 carbons), 25.94, 25.44, 20.13, 20.07, 18.37, 18.36 ppm.
FIG. 3: FT-IR (KBr pellet): 3379.0, 2963.4, 2602.1, 1728.4, 1600.0, 1523.4, 1439.7, 1420.6, 1233.2, 1193.4, 1100.9, 1027.3 cm“1.
Elemental Analysis: Anal. Calcd for C42H52C12N806: C, 60.35; H, 6.27; N, 13.41; CI, 8.48. Found C, 58.63; H, 6.42; N, 12.65, CI, 8.2.
FIG. 1 is a representative 1H NMR spectrum of Compound I Form A.
FIG. 2 is a representative 13C NMR spectrum of Compound I Form A.
FIG. 3 is a representative FT-IR spectrum of Compound I Form A.
References:
1. Lalezari, J. P.; et. al. PPI-668, a potent new pan-genotypic HCV NS5A inhibitor: phase 1 efficacy and safety. Hepatology 2012, 56, 1065A-1066A.
- ClinicalTrials.govA Study of the Efficacy and Safety of PPI-668 (NS5A Inhibitor) Plus Sofosbuvir, With or Without Ribavirin, in Patients With Chronic Hepatitis C Genotype-4. NCT02371408(retrieved on 24-03-2015)
3. ClinicalTrials.gov Study of PPI-668, BI 207127 and Faldaprevir, With and Without Ribavirin, in the Treatment of Chronic Hepatitis C. NCT01859962 (retrieved on 15-09-2015)
4. Lalezari, J.; et. al. High rate of sustained virologic response in patients with hcv genotype-1a infection: a phase 2 trial of faldaprevir, deleobuvir and ppi-668, with and without ribavirin. EASL-The International Liver Congress 2014– 49th Annual Meeting of the European Association for the Study of the Liver London, United Kingdom April 9-13 (article here)
| US20070185175 * | 27 Jul 2006 | 9 Aug 2007 | Bristol-Myers Squibb Company | Benzothiazole and azabenzothiazole compounds useful as kinase inhibitors |
| US20080050336 * | 8 Aug 2007 | 28 Feb 2008 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| WO2012087976A2 * | 19 Dec 2011 | 28 Jun 2012 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| WO2013123092A1 * | 13 Feb 2013 | 22 Aug 2013 | Presidio Pharmaceuticals, Inc. | Solid forms comprising inhibitors of hcv ns5a, compositions thereof, and uses therewith |
| WO2013158776A1 * | 17 Apr 2013 | 24 Oct 2013 | Gilead Sciences, Inc. | Compounds and methods for antiviral treatment |
| US8765731 | 16 Nov 2012 | 1 Jul 2014 | Vertex Pharmaceuticals Incorporated | Benzimidazole analogues for the treatment or prevention of flavivirus infections |
| US8779156 | 24 Sep 2012 | 15 Jul 2014 | Vertex Pharmaceuticals Incorporated | Analogues for the treatment or prevention of flavivirus infections |
| US8809330 | 1 Nov 2013 | 19 Aug 2014 | Gilead Sciences, Inc. | Pyrazolo[1,5-A]pyrimidines for antiviral treatment |
| US8946238 | 20 Dec 2012 | 3 Feb 2015 | Gilead Sciences, Inc. | Pyrazolo[1,5-A]pyrimidines as antiviral agents |
| US8980878 | 17 Apr 2013 | 17 Mar 2015 | Gilead Sciences, Inc. | Compounds and methods for antiviral treatment |
| US20110274648 * | 4 Nov 2010 | 10 Nov 2011 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
////////////Phase III, Hepatitis C, RAVIDASVIR, PPI-668, BI 238630
BMS-791325, Beclabuvir In Phase 2 for Hepatitis C (HCV)
BMS-791325, Beclabuvir
IN PHASE 2 for Hepatitis C (HCV)
An NS5B inhibitor.

BMS-791325 preferably is


958002-33-0
958002-36-3 (as hydrochloride)
C36 H45 N5 O5 S, 659.838
Cycloprop(d)indolo(2,1-a)(2)benzazepine-9-carboxamide, 12-cyclohexyl-N-((dimethylamino)sulfonyl)-4b,5,5a,6-tetrahydro-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-, (4bS,5aR)-
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(1aR,12bS)-8-Cyclohexyl-N-(dimethylsulfamoyl)-11-methoxy-1a-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8-yl)carbonyl]-1,1a,2,12b-tetrahydrocyclopropa[d]indolo[2,1-a][2]benzazepine-5-carboxamide
Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-
Bristol-Myers Squibb (Originator)
RNA-Directed RNA Polymerase (NS5B) Inhibitors
UNII-MYW1X5CO9S
BMS-791325 is in phase II clinical studies at Bristol-Myers Squibb for the treatment of chronic hepatitis C. In 2013, the company received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and asunaprevir.
| Patent | WO 2007136982 |
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
http://www.google.com/patents/WO2007136982A1?cl=en
Scheme 1.

N-protected piperazines can also be coupled to the intermediate indolobenzazepine acids and the resultant piperazine carboxamides can be deprotected using methods known in the art and derivatized using a variety of synthetic protocols, some illustrative examples of which are shown below (See Scheme 2).
Scheme 2.

An intermediate useful for the synthesis of some compounds of the invention involves the preparation of the tert-butyl ester indolobenzazepine shown in Scheme 3. Scheme 3.

t-Butylation either:

This methodology involves base catalyzed hydrolysis of the indole methyl ester shown, followed by its reaction with either thionyl chloride and potassium tertiary butoxide, or alkylation with silver carbonate and tertiary butyl bromides. The resultant compound can be transformed using chemistry analogous to that outlined previously to provide the mixed ester indolobenzazepines shown above.
Scheme 4.

Some examples exist as stereoisomeric mixtures. The invention encompasses all stereoisomers of the compounds. Methods of fractionating stereoisomeric mixtures are well known in the art, and include but are not limited to; preparative chiral supercritical fluid chromatography (SFC) and chiral high performance liquid chromatography (HPLC). An example using this approach is shown in scheme 5. Scheme 5.

An additional method to achieve such separations involves the preparation of mixtures of diastereomers which can be separated using a variety of methods known in the art. One example of this approach is shown below (Scheme 6).
Scheme 6.

Diastereomers separated by reverse phase HPLC
Some diastereomeric amides can be separated using reverse phase HPLC. After hydroysis, the resultant optically active acids can be coupled with bridged piperazine derivatives (Scheme 6). For example, O-(lH-benzotriazol-l-yl)-N,N, N’,N’-tetramethyluronium tetrafluoroborate and diisopropyl ethyl amine in DMSO can be used to give the alkyl bridged piperazine carboxamides. Other standard acid amine coupling methods can also be used to give optically active carboxamides.

Schemes 7-9 illustrate other methods of making intermediates and compounds.

Scheme 8.

Scheme 9.


Biological Methods
The compounds demonstrated activity against HCV NS5B as determined in the following HCV RdRp assays.
DESCRIPTION OF SPECIFIC EMBODIMENTS
Unless otherwise specified, analytical LCMS data on the following intermediates and examples were acquired using the following columns and conditions. Stop time: Gradient time + 1 minute; Starting cone: 0% B unless otherwise noted; Eluent A: 5% CH3CN / 95% H2O with 10 mM NH4OAc (for columns A, D and E); 10 % MeOH / 90 % H2O with 0.1% TFA (for columns B and C); Eluent B: 95% CH3CN / 5% H2O with 10 mM NH4OAc (for columns A, D and E); 90 % MeOH / 10 % H2O with 0.1% TFA (for columns B and C); Column A:
Phenomenex lOμ 4.6 x 50 mm C18; Column B: Phenomenex C18 lOμ 3.0 x 50 mm; Column C: Phenomenex 4.6 x 50 mm C18 lOμ; Column D: Phenomenex Lina C18 5μ 3.0 x 50 mm; Column E: Phenomenex 5μ 4.6 x 50 mm Cl 8.
Intermediate 1

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester. Freshly recrystallized pyridinium tribromide (recrystallization from hot AcOH (5 mL per 1 g), rinsed with cold AcOH and dried under high vacuum over KOH) was added in portions (over 10 min.) to a stirring solution of methyl 3-cyclohexyl-lH-indole-6- carboxylate (60 g, 233 mmol) (prepared using procedures describe in WO2004/065367) in CHC1/THF (1: 1, 1.25 L) at 2o C. The reaction solution was stirred at 0-5 °C for 2.5h, and washed with sat. aq. NaHSO3 (1 L), 1 N HCl (1 L) and brine (1 L). The organic layer was dried (MgSO4) and concentrated. The resulting red oil was diluted with Et2θ and concentrated. The resulting pink solid was dissolved into Et2θ (200 mL) treated with hexanes (300 mL) and partially concentrated. The solids were collected by filtration and rinsed with hexanes. The mother liquor was concentrated to dryness and the procedure repeated. The solids were combined to yield lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester (64 g, 190 mmol, 82%) as a fluffy pink solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 8.47 (br s, IH), 8.03 (d, J = 1.4 Hz, IH), 7.74 (dd, J = 1.4, 8.8 Hz, IH), 7.69 (d, J = 8.8 Hz, IH), 3.92 (s, 3H), 2.82 (tt, J = 3.7, 11.7 Hz, IH), 1.98 – 1.72 (m, 7H), 1.50 – 1.27 (m, 3H). 13CNMR (75 MHz, CDC13) δ 168.2, 135.6, 130.2, 123.1, 120.8, 120.3, 118.7, 112.8, 110.7, 52.1, 37.0, 32.2(2), 27.0(2), 26.1. LCMS: m/e 334 (M-H)“, ret time 3.34 min, column A, 4 minute gradient.
Intermediate 2

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-. A solution of methyl 2- bromo-S-cyclohexyl-lH-indole-ό-carboxylate (20 g, 60 mmol) and LiOH (3.8 g, 160 mmol) in MeOΗ/TΗF/Η2O ( 1 : 1 : 1 , 300 mL) was heated at 90 °C for 2h. The reaction mixture was cooled in an ice/H2O bath, neutralized with IM HCl (-160 mL) diluted with H2O (250 mL) and stirred for Ih at rt. The precipitates were collected by filtration rinse with H2O and dried to yield lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (quant.) which was used without further purification.
An alternative procedure that can by used to provide lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl- is described below: A solution of methyl 2-bromo-3-cyclohexyl-lH-indole-6-carboxylate (117 g, 349 mmol) and LiOKH2O (26.4 g, 629 mmol) in MeOH/THF/H2O (1: 1: 1, 1.8 L) was heated at reflux for 3h. The reaction mixture was cooled in an ice/H2O bath to ~2 °C, neutralized with IM HCl (-650 mL) (added at such a rate that temperature did not exceed 5 °C), diluted with H2O (1 L) and stirred while warming to ambient temperature. The precipitates were collected by filtration rinsed with H2O and dried to yield the mono THF solvate of lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (135.5 g, 345 mmol, 99%) as a yellow solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 11.01 (br s, IH), 8.77 (s, IH), 8.07 (d, J = 1.5 Hz, IH), 7.82 (dd, J = 1.5, 8.8 Hz, IH), 7.72 (d, J = 8.8 Hz, IH), 3.84 – 3.74 (m, 4H), 2.89 (m, IH), 1.98 – 1.72 (m, HH), 1.50 – 1.24 (m, 3H). 13CNMR (75 MHz, CDC13) δ 172.7, 135.5, 130.7, 122.3, 120.9(2), 118.8, 113.3, 111.1, 67.9(2), 37.0, 32.2(2), 27.0(2), 26.1, 25.5(2). LCMS: m/e 320 (M-H)“, ret time 2.21 min, column A, 4 minute gradient.
Intermediate 3

lH-Indole-6-carboxamide, 2-bromo-3-cyclohexyl-N-
[(dimethylamino)sulfonyl]-. l,l’-Carbonyldiimidazole (1.17 g, 7.2 mmol) was added to a stirred solution of 2-bromo-3-cyclohexyl-lH-indole-6-carboxylic acid (2.03 g, 6.3 mmol) in THF (6 mL) at 22 °C. The evolution of CO2 was instantaneous and when it slowed the solution was heated at 50°C for 1 hr and then cooled to 220C. N,N-Dimethylsulfamide (0.94 g, 7.56 mmol) was added followed by the dropwise addition of a solution of DBU (1.34 g ,8.8 mmol) in THF (4 mL). Stirring was continued for 24 hr. The mixture was partitioned between ethyl acetate and dilute HCl. The ethyl acetate layer was washed with water followed by brine and dried over Na2SO4. The extract was concentrated to dryness to leave the title product as a pale yellow friable foam, (2.0 g, 74 %, >90 % purity , estimated from NMR). 1H NMR (300 MHz, DMSO-D6) δ ppm 1.28 – 1.49 (m, 3 H) 1.59 – 2.04 (m, 7 H) 2.74 – 2.82 (m, 1 H) 2.88 (s, 6 H) 7.57 (dd, J=8.42, 1.46 Hz, 1 H) 7.74 (d, J=8.78 Hz, 1 H) 7.91 (s, 1 H) 11.71 (s, 1 H) 12.08 (s, 1 H).
An alternative method for the preparation of lH-indole-6-carboxamide, 2- bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- is described below.
To a 1 L four necked round bottom flask equipped with a mechanical stirrer, a temperature controller, a N2 inlet , and a condenser, under N2, was added 2-bromo-3- cyclohexyl-lH-indole-6-carboxylic acid (102.0 g, 0.259 mol) and dry TΗF (300 mL). After stirring for 10 min, CDI (50.3 g, 0.31 mol) was added portion wise. The reaction mixture was then heated to 50 oC for 2 h. After cooling to 30 oC, N,N- dimethylaminosulfonamide (41.7 g, 0.336 mol) was added in one portion followed by addition of DBU (54.1 mL, 0.362 mol) drop wise over a period of 1 h. The reaction mixture was then stirred at rt for 20 h. The solvent was removed in vacuo and the residue was partitioned between EtOAc and 1 Ν HCl (1 : 1, 2 L). The organic layer was separated and the aqueous layer was extracted with EtOAc (500 mL). The combined organic layers were washed with brine (1.5 L) and dried over MgSO4. The solution was filtered and concentrated in vacuo to give the crude product (111.0 g). The crude product was suspended in EtOAc (400 mL) at 60 oC. To the suspension was added heptane (2 L) slowly. The resulting suspension was stirred and cooled to 0 oC. It was then filtered. The filter cake was rinsed with small amount of heptane and house vacuum air dried for 2 days. The product was collected as a white solid (92.0 g, 83%). 1H ΝMR (MeOD, 300 MHz) δ 7.89 (s, H), 7.77 (d, J= 8.4 Hz, IH), 7.55 (dd, J= 8.4 and 1.8 Hz, IH), 3.01 (s, 6H), 2.73-2.95 (m, IH), 1.81-2.05 (m, 8H), 1.39-1.50 (m, 2H); m/z 429 (M +H)+. Intermediate 4

lH-Indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. A mixture of the 2-Bromo-3-cyclohexyl- N- [(dimethylamino)sulfonyl]-lH-indole-6-carboxamide (4.28g, 0.01 mol), 4-methoxy- 2-formylphenyl boronic acid (2.1%, 0.015 mol), 2-dicyclohexylphosphino-2′,6′- dimethoxy-biphenyl (41 mg, 0.0001 mol), palladium acetate (11.2 mg), and finely ground potassium carbonate (4.24g, 0.02 mol) in toluene (30 mL) was stirred under reflux and under nitrogen for 30 min, at which time LC/MS analysis showed the reaction to be complete. The reaction mixture was then diluted with ethyl acetate and water, and then acidified with an excess of dilute HCl. The ethyl acetate layer was then collected and washed with dilute HCl, water and brine. The organic solution was then dried (magnesium sulfate), filtered and concentrated to give a gum. The gum was diluted with hexanes (250 ml) and ethyl acetate (25 mL), and the mixture was stirred for 20 hr at 22° C during which time the product was transformed into a bright yellow granular solid (4.8 g) which was used directly without further purification.
An alternative procedure for the preparation of lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- is provided below:
To a slurried solution of 2-bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- indole-6-carboxamide (54.0 g, 126 mmol), 4-methoxy-2-formylphenylboronic acid (29.5 g, 164 mmol) and LiCl (13.3 g, 315 mmol) in EtOH/toluene (1 : 1, 1 L) was added a solution of Na2CO3 (40.1 g, 379 mmol) in water (380 mL). The reaction mixture was stirred 10 min. and then Pd(PPh3)4 (11.3 g, 10.0 mmol) was added. The reaction solution was flushed with nitrogen and heated at 70 °C (internal monitoring) overnight and then cooled to rt. The reaction was diluted with EtOAc (1 L) and EtOH (100 mL), washed carefully with IN aqueous HCl (1 L) and brine (500 mL), dried (MgSO4), filtered and concentrated. The residual solids were stirred with Et20 (600 mL) for Ih and collected by filtration to yield lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- (52.8g, 109 mmol, 87%) as a yellow powder which was used without further purification. IHNMR (300 MHz, d6-DMSO) δ 11.66 (s, IH), 8.17 (s, IH), 7.75 (d, J = 8.4 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 7.59 (dd, J = 1.4, 8.4 Hz, IH), 7.23 – 7.16 (m, 2H), 7.08 (dd, J = 2.6, 8.4 Hz, IH), 6.54 (d, J = 8.8 Hz, IH), 3.86 (s, 3H), 3.22 – 3.08 (m, IH), 2.91 (s, 6H), 2.00 – 1.74 (m, 7H), 1.60 – 1.38 (m, 3H). 13CNMR (75 MHz, CDC13) δ 165.7, 158.8, 147.2, 139.1, 134.3, 132.0, 123.4, 122.0, 119.2, 118.2, 114.8, 112.3, 110.4, 109.8, 79.6, 45.9, 37.2(2), 34.7, 32.0(2), 25.9(2), 24.9. LCMS: m/e 482 (M- H)“, ret time 2.56 min, column A, 4 minute gradient.
Intermediate 5

6H-Isoindolo[2,l-a]indole-3-carboxamide, 11-cyclohexyl-N-
[(dimethylamino)sulfonyl]-6-ethoxy-8-methoxy-. To a 5 L four necked round bottom flask equipped with a temperature controller, a condenser, a N2 inlet and a mechanical stirrer, was charged toluene (900 mL), EtOH (900 mL), 2-bromo-3- cyclohexyl-N^NjN-dimethylsulfamoyiyiH-indole-ό-carboxamide (90 g, 0.21 mol), 2-formyl-4-methoxyphenylboronic acid (49.2 g, 0.273 mol) and LiCl (22.1 g, 0.525 mol). The resulting solution was bubbled with Ν2 for 15 mins. A solution of Na2CO3 (66.8 g, 0.63 mol) in Η2O (675 mL) was added and the reaction mixture was bubbled with N2 for another (10 mins). Pd(PPh3)4 (7.0 g, 6.3 mmol) was added and the reaction mixture was heated to 70 °C for 20 h. After cooling to 35 °C, a solution of 1 N HCl (1.5 L) was added slowly. The resulting mixture was transferred to a 6 L separatory funnel and extracted with EtOAc (2 X 1.5 L). The combined organic extracts were washed with brine (2 L), dried over MgSO4, filtered and concentrated in vacuo to give a yellow solid, which was triturated with 20% EtOAc in hexane (450 mL, 50 °C to 0 °C) to give 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4- methoxyphenyl)-lH-indole-6-carboxamide(65.9 g) as a yellow solid. HPLC purity, 98%.
The mother liquid from the trituration was concentrated in vacuo. The residue was refluxed with EtOH (50 mL) for 3 h. The solution was then cooled to 0 °C. The precipitates were filtered and washed with cooled TBME (5 °C) (20 mL). The filter cake was house vacuum air dried to give a further quantity of the title compound as a white solid (16.0 g). HPLC purity, 99%. 1H NMR (CDC13, 300 MHz) δ 8.75 (s, IH), 7.96 (s, IH), 7.73 (d, J= 8.4 Hz, IH), 7.67 (d, J= 8.4 Hz, IH), 7.45 (dd, J= 8.4 and 1.4 Hz, IH), 7.09 (d, J= 2.2 Hz, IH), 6.98 (dd, J= 8.4 and 2.2 Hz, IH), 6.50 (s, IH), 3.86 (s, 3H), 3.05 (s, 6H), 2.92-3.13 (m, 3H), 1.85-1.93 (m, 7 H), 1.40-1.42 (m, 3H), 1.05 (t, J= 7.1 Hz, 3H). m/z 512 (M + H)+.
Intermediate 6

lH-indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. 1 l-cyclohexyl-N-(N,N-dimethylsulfamoyl)-6-ethoxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide was dissolved in THF (75 mL). To the solution was added a solution of 2 N HCl (300 mL). The mixture was vigorously stirred under N2 at rt for 16 h. The resulting suspension was filtered and washed with cooled TBME (2 X 30 mL). the filer cake was vacuum air dried overnight to give the title compound as a yellow solid. HPLC purity, 99% 1H NMR (DMSO-d6, 300 MHz) δ 11.65 (s, IH), 8.16 (s, IH), 7.76 (d, J= 5.9 Hz, IH), 7.73 (d, J= 5.9 Hz, IH), 7.58 (dd, J= 8.5 and 1.5 Hz, IH), 7.17-7.20 (m, 2H), 7.08 (dd, J = 8.5 and 1.4 Hz, IH), 6.55 (d, J= 8.6 Hz, IH), 3.86 (s, 3H), 3.14-3.18 (m, IH), 2.91 (s, 6H), 1.75-1.99 (m, 7H), 1.48-1.60 (m, 3H); m/z 484 (M + H)+.
Intermediate 7

7H-Indolo[2, 1-a] ‘ [2] benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl] amino] carbonyl]-3-methoxy-, methyl ester. A mixture of the 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4-methoxyphenyl)-lH- indole-6-carboxamide (4.8g, 0.01 mol), methyl 2-(dimethoxyphosphoryl)acrylate (9.7 g, 0.02 mol) and cesium carbonate (7.1g, 0.02 mol) in DMF (28mL) was stirred for 20 hr at an oil bath temperature of 55 ° C. The mixture was poured into ice-water and acidified with dilute HCl to precipitate the crude product. The solid was collected, dried and flash chromatographed on Siθ2 (11Og) using an ethyl acetate and methylene chloride (1: 10) solution containing 2% acetic acid. Homogeneous fractions were combined and evaporated to afford the title compound as a pale yellow solid (3.9g, 71 % yield). MS: 552 (M=H+).
An alternate procedure for the preparation of 7H-indolo[2,l- a] [2]benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester is provided below. A solution of l l-cyclohexyl-N-[(dimethylamino)sulfonyl]-6-hydroxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide (cyclic hemiaminal) (63.0 g, 130 mmol), methyl 2-(dimethoxyphosphoryl)acrylate (60 g, 261 mmol), cesium carbonate (106 g, 326 mmol) in DMF (400 mL) was heated at 60 °C (bath temp) for 4.5h. Additional methyl 2-(dimethoxyphosphoryl)acrylate (15 g, 65 mmol) and cesium carbonate (21.2 g, 65 mmol) were added and the reaction was heated at 60 °C overnight then and cooled to rt. The stirring reaction mixture was diluted with H2O (1 L), slowly neutralized with IN aqueous HCl (800 mL), stirred 3h, and then the precipitates were collected by filtration. The solids were triturated with Et20 (800 mL) and dried to yield methyl 7H-indolo[2,l-a][2]benzazepine-6-carboxylic acid, 13- cyclohexyl-10-[[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester (70.2 g, 127 mmol, 98%) as a yellow solid which was used without further purification. IHNMR (300 MHz, CDC13) δ 8.67 (s, IH), 8.09 (s, IH), 7.86 (d, J = 8.4 Hz, IH), 7.80 (s, IH), 7.50 (d, J = 8.4 Hz, IH), 7.42 (d, J = 8.8 Hz, IH), 7.08 (dd, J = 2.6, 8.8 Hz, IH), 6.98 (d, J = 2.6 Hz, IH), 5.75 – 5.51 (m, IH), 4.29 – 4.01 (m, IH), 3.89 (s, 3H), 3.82 (s, 3H), 3.05 (s, 6H), 2.87 – 2.73 (m, IH), 2.11 – 1.12 (m, 10H). LCMS: m/e 550 (M-H)-, ret time 3.21 min, column A, 4 minute gradient.
Example 1

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (+/-)-. TBTU (43.7 mg, 0.136mmol) and DIPEA (0.095 mL, 0.544 mmol) were added to a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (50 mg, 0.0906 mmol) in DMSO (2.0 mL). The reaction mixture was stirred at rt for 15 min. 3-Methyl-3,8-diaza-bicyclo[3.2. l]octane dihydrochloride {J & W PharmLab, LLC Morrisville, PA 19067-3620}. (27.1 mg, 0. 136 mmol) was then added and the reaction mixture was stirred at rt for 3 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give the final product as a yellow solid, (32 mg, 46% yield). MS m/z 660(MH+), Retention time: 2.445 min IH NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H).
Example 4

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonylj ‘- 1 , Ia, 2, 12b-tetrahydro-ll-methoxy-la-[(8-methyl-3, 8- diazabicyclo[3.2.1]oct-3-yl)carbonyl]-, (+/-)-. To a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-5-carboxamide, 8-cyclohexyl-la-(3,8- diazabicyclo[3.2.1]oct-3-ylcarbonyl)-N-[(dimethylamino)sulfonyl]-l,la,2,12b- tetrahydro-11-methoxy- (54 mg, 0.071 mmol) in methanol (3 mL), paraformaldehyde (6.4 mg, 0.213 mmol), ZnCl2 (29 mg, 0.213 mmol) and
Na(CN)BH3 (13.4 mg, 0.213 mmol) were added. The resultant mixture was heated at 60°C for 2hr, and then cooled to rt. The solid present was removed by filtration, and the filtrate was concentrated under vacuum and the residue purified by preparative reverse phase HPLC to give the title compound as a light yellow colored solid, (37 mg, 67% yield). MS ml 660(MH+), Retention time: 2.495 min. IH NMR (500 MHz, MeOD) δ ppm 0.21 (m, 0.3 H) 1.13 (m, 0.3 H) 1.18 – 2.22 (m, 15.4 H) 2.58 (m, 0.3 H) 2.68 (m, 0.7 H) 2.76 – 3.11 (m, 11 H) 3.32 – 3.37 (m, 1 H) 3.63 (d, J=15.56 Hz, 0.7 H) 3.82 – 4.32 (m, 7.3 H) 4.88 – 4.92 (m, 0.3 H) 5.08 (d, J=15.56 Hz, 0.7 H) 7.00 – 7.08 (m, 1 H) 7.18 (d, J=2.14 Hz, 0.3 H) 7.21 (d, J=2.14 Hz, 0.7 H) 7.32 (d, J=8.55 Hz, 0.7 H) 7.35 (d, J=8.55 Hz, 0.3H) 7.57 (d, J=7.93 Hz, 0.7 H) 7.62 (dd, J=8.39, 1.37 Hz, 0.3 H) 7.91 (d, J=8.55 Hz, 0.7 H) 7.93 – 7.99 (m, 1 H) 8.09 (s, 0.3 H).
Example 6

Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-. To a solution of (-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (204 mg, 0.37 mmol) in DMSO (8.0 mL), TBTU (178 mg, 0.555 mmol) and DIPEA (0.39 mL, 2.22 mmol) were added. The reaction mixture was stirred at rt for 15 min. Then 3- methyl-3,8-diaza-bicyclo[3.2.1]octane dihydrochloride (111 mg, 0. 555 mmol) was added and the reaction mixture was stirred at rt for 2 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give a yellow solid as final TFA salt. (265 mg, 92% yield). Average Specific Rotation: -53.56° Solvent, MeOH.; Wavelength 589 nm; 50 cm cell. MS m/z 660(MH+), Retention time: 3.035 min. 1H NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 – 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 – 3.11 (m, 10 H) 3.28 – 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 – 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 – 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 – 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H). An alternate procedure for the synthesis of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)-is provided below. To a mixture of (-) cycloprop[<i]indolo[2,l-α][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (25.2 g, 45.68 mmol) and 3-methyl-3,8-diazabicyclo-[3.2.1]octane dihydrochloride (10.0 g, 50.22 mmol) in anhydrous MeCN (300 mL) was added DIPEA (23.62 g, 182.72 mmol) under N2. After 15 min, TBTU (16.12 g, 50.22 mmol) was added. The reaction solution was stirred for 30 min under N2. The ΗPLC indicated the disappearance of starting material. The solvent in the solution was evaporated to give a foam. This was dissolved in EtOAc (2.5 L), washed with H2O (1.5 L), H2O/brine (8:2) (1.5 L), brine (1.5 L), dried over Na2SO4 and evaporated to give 28.8 g of crude product. This solid was pooled with 45.4 g of material obtained from five separated reactions to afford a total of 74.2 g of crude product. This was passed through a pad of silica gel (E. Merck 230-400 mesh, 1 kg), eluting with MeOH/CH2Cl2 (2.5:97.5). After evaporation, it gave a foam, which was treated with EtOAc and hexane to turn into a solid. After drying at 50 °C under vacuum for 7 h, the GC analysis indicated it has 1.4% each of EtOAc and hexane. After further drying at 61-64 °C, the GC analysis indicated it still has 1.0% of hexane and 1.4% of EtOAc. The product was dissolved in Et2O and slowly evaporated in vacuum three times, dried at 60 °C under vacuum for 3 h to give 68.3 g. This was washed with H2O (900 mL) and redried at 68 °C under vacuum for 7 h to give 67.1 g (77% yield) of the compound of example 6. The GC analysis indicated it has 0.97% Of Et2O. HPLC conditions column: Cadenza CD-C18 3 x 250 mm; UV: 257 and 220 nm; 25 °C; flow rate: 0.5 mL/min; gradient time: 38 min, 0 – 80% B (0 – 35 min) and 80% B (35 – 38 min); solvent A: 25 nM CH3COONH4 at pH 4.7 in water, solvent B: MeCN. HPLC purity 99.7% (Rt 26.54 min); Chiral HPLC conditions column: Regis (S5S) Whelk-Ol 250 x 4.6 mm; UV 258nm; 35 °C; flow rate 2.0 mL/min; mobile phase C02/Me0H; gradient time 20 min, 30% MeOH (0 – 1 min), 30 – 48% MeOH (1 – 19 min), 48% MeOH (19 – 20 min). Chiral HPLC purity > 99.8% (Rt 16.60 min); LC/MS (ES+) 660.36 (M+H, 100); HRMS: calcd. 660.3220, found 660.3197; [α]D 25 C – 79.66 ° (c 1.06, MeOH); Anal. Calcd for C36H45N5O5S-O-O H2O»0.09 Et2O: C, 64.53; H, 7.00; N, 10.35; S, 4.74; H2O, 1.51; Et2O, 0.97. Found: C, 64.50; H, 7.12; N, 10.41; S, 5.14; H2O, 1.52; Et2O, 0.97. The absolute stereochemistry of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)- is as drawn above, and was determined from an x- ray crystal structure obtained on the (R)-camphorsulfonic acid salt.
Additionally, the following salts were prepared: hydrochloride, phosphate, acetate, sulfate, camsylate, sodium, calcium, and magnesium. The hydrochloride salt had the following characteristics. DSC: small, broad endotherm from 25°C to 75°C, and potential melt/degradation endotherm with peak at temperatures ranging between 253 °C and 258 °C; TGA: Early weight loss from 25°C to 75°C ranging between 0.003% and 1.5%, and degradation weight loss starting at approximately 200°C.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
| WO2006020082A1 * | Jul 15, 2005 | Feb 23, 2006 | Squibb Bristol Myers Co | Inhibitors of hcv replication |
| WO2006046030A2 * | Oct 25, 2005 | May 4, 2006 | Angeletti P Ist Richerche Bio | Tetracyclic indole derivatives as antiviral agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008111978A1 | Mar 13, 2007 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008112473A1 * | Mar 5, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112841A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112848A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112851A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv inhibitors |
| WO2008112863A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2009067108A1 * | Nov 20, 2007 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2009067392A1 * | Nov 17, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine derivatives for the treatment of hepatitis c |
| WO2009067481A1 * | Nov 19, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2013059265A1 * | Oct 17, 2012 | Apr 25, 2013 | Bristol-Myers Squibb Company | A compound for the treatment of hepatitis c |
| WO2014014885A1 * | Jul 16, 2013 | Jan 23, 2014 | Bristol-Myers Squibb Company | Novel methods and intermediates for the preparation of (4bs,5ar)-12-cyclohexyl-n-(n,n-dimethylsulfamoyl)-3-methoxy-5a-((1 r,5s) -3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo [3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide |
| CN101679442B | Mar 13, 2008 | Feb 20, 2013 | 百时美施贵宝公司 | Compounds for the treatment of hepatitis c |
| EP2518073A1 * | Nov 19, 2008 | Oct 31, 2012 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
The First Kilogram Synthesis of Beclabuvir, an HCV NS5B Polymerase Inhibitor
 , Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
, Kenneth J. Fraunhoffer, Zhinong Gao, Chao Hang, Yi Hsiao, Wenhao Hu, Kishta Katipally, Adam Littke, Aghogho Pedro, Yuping Qiu, Maria Sandoval, Richard Schild, Michelle Soltani, Anthony Tedesco, Dale Vanyo, Purushotham Vemishetti, and Robert E. Waltermire
The process development and kilogram-scale synthesis of beclabuvir (BMS-791325, 1) is described. The convergent synthesis features the use of asymmetric catalysis to generate a chiral cyclopropane fragment and coupling with an indole fragment via an alkylation. Subsequent palladium-catalyzed intramolecular direct arylation efficiently builds the central seven-membered ring. The target was prepared in 12 linear steps with five isolations in an overall yield of 8%.
Preparation of (4bS,5aR)-12-Cyclohexyl-N-(N,N-dimethylsulfamoyl)-3-methoxy-5a-((1R,5S)-3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo[3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide Hydrochloride (1·HCl)
BMS-791325·HCl (1·HCl) was isolated in 89.5% yield.
1H NMR (600 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 7.91 (br s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.55 (br d, J = 8.5 Hz, 1H), 7.29 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 2.5 Hz, 1H), 7.00 (dd, J = 8.5 Hz, 2.7 Hz, 1H), 5.03 (br d, J = 12.7 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 3.87 (s, 3H), 3.56 (d, J = 15.5 Hz, 1H), 3.40 (br s, 3H), 3.32–3.28 (m, 4H), 2.96 (s, 6H), 2.92 (tt, J= 12.2, 3.6 Hz, 1H), 2.59 (br t, J = 7.0 Hz, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.55 (br d, J= 12.2 Hz, 2H), 1.46–1.36 (m, 4H), 1.26 (t, J = 5.3 Hz, 2H), 1.23–1.15 (m, 2H);
minor rotamer: 8.05 (br s, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.58 (dd, J = 8.5, 1.4 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.15 (d, J = 2.6 Hz, 1H), 6.98 (d, 1H, overlap with major rotamer), 4.91 (d, J = 15.0 Hz, 1H), 4.58 (br d, J = 4.9 Hz, 2H), 4.11 (d, J = 15.0 Hz, 1H), 3.89 (s, 3H), 3.46 (br d, J = 12.5 Hz, 2H), 3.17 (br d, J = 12.5 Hz, 2H), 2.97 (s, 6H), 2.85 (br s, 3H), 2.76 (tt, J = 12.1, 3.5 Hz, 1H), 2.49 (br s, 1H), 2.05–1.90 (m, 2H), 1.79–1.71 (m, 4H), 1.46–1.36 (m, 6H), 1.23–1.15 (m, 2H), 1.10 (m, 1H), 0.03 (t, J = 6.1 Hz, 1H).
13C NMR (125 MHz, 10:1 v/v CD3CN/D2O): major rotamer: 170.1, 167.7, 161.0, 140.4, 139.3, 135.9, 133.6, 131.1, 124.9, 123.0, 121.7, 120.8, 119.0, 118.6, 114.3, 110.7, 59.2, 56.2, 53.1, 48.3, 44.5, 38.9, 37.6, 34.8, 33.77, 33.72, 27.92, 27.77, 26.82, 26.5, 23.6, 18.5;
minor rotamer: 168.3, 168.0, 161.3, 138.4, 137.5, 135.8, 134.2, 130.0, 125.4, 121.9, 120.0, 119.64, 119.58, 117.9, 113.3, 111.3, 59.6, 56.3, 53.1, 44.6, 42.2, 38.9, 38.3, 37.4, 33.8, 33.6, 28.3, 27.74, 26.79, 26.5, 24.84, 11.9.
HRMS (ESI) calcd for C36H45N5O5S (free base) [M + H]+660.3214, found m/z 660.3220.
////////BMS-791325, Beclabuvir, Phase 2, Hepatitis C, HCV,
Medivir AB :New Drug Application has been filed with FDA for Simeprevir (TMC435) for combination treatment of adult patients with genotype 1 chronic hepatitis C

Simeprevir
03/28/2013| Medivir AB announced that a new drug application (NDA) has been filed with the U.S. Food and Drug Administration (FDA) seeking approval for simeprevir. The filing is based on phase III data in treatment-naïve and treatment-experienced patients with compensated liver disease.
The filing of a regulatory application in the US triggers a milestone payment of ?10m to Medivir.
Simeprevir is jointly developed by Medivir and Janssen Pharmaceuticals, Inc. (Janssen), and is an investigational NS3/4A protease inhibitor, administered as a 150 mg capsule once daily with pegylated interferon and ribavirin for the treatment of genotype 1 chronic hepatitis C in adult patients.
“The filing in the U.S. is a very important milestone for simeprevir, the hepatitis C patients and for Medivir as a company. In addition it triggers a ? 10m milestone payment to us, which strengthens our solid financial situation even more.” comments Maris Hartmanis, CEO of Medivir.
The regulatory submission for simeprevir is supported in part by data from three pivotal phase III studies: QUEST-1 and QUEST-2 in treatment-naïve patients and PROMISE in patients who have relapsed after prior interferon-based treatment. In each study, participants were treated with one 150 mg simeprevir capsule once daily for 12 weeks plus pegylated interferon and ribavirin for 24 or 48 weeks. Primary efficacy data from the phase III studies will be presented at different upcoming medical meetings.
About Simeprevir
Simeprevir, an investigational next generation NS3/4A protease inhibitor jointly developed by Janssen R&D Ireland and Medivir AB, is currently in late phase III studies as a once-daily capsule (150 mg) taken in combination with pegylated interferon and ribavirin for the treatment of genotypes 1 and 4 HCV.
Global phase III studies of simeprevir include QUEST-1 and QUEST-2 in treatment-naïve patients, PROMISE in patients who have relapsed after prior interferon-based treatment and ATTAIN in null-responder patients. In parallel to these trials, phase III studies for simeprevir are ongoing in treatment-naïve and treatment-experienced HIV-HCV co-infected patients, HCV genotype 4 patients and Japanese HCV genotype 1 patients. Janssen recently announced the submission of a new drug application for simeprevir in Japan for the treatment of genotype 1 hepatitis C.
Simeprevir is being studied in phase II interferon-free trials with and without ribavirin in combination with:
- Janssen’s non-nucleoside inhibitor TMC647055 and ritonavir in treatment-naïve genotype 1a and 1b HCV patients;
- Gilead Sciences, Inc.’s nucleotide inhibitor sofosbuvir (GS-7977) in treatment-naïve and previous null-responder genotype 1 HCV patients; and
- Bristol-Myers Squibb’s NS5A replication complex inhibitor daclatasvir (BMS-790052) in treatment-naive and previous null-responder genotype 1 HCV patients.
In addition, Janssen has a non-exclusive collaboration with Vertex Pharmaceuticals to evaluate in a phase II study the safety and efficacy of an all-oral regimen of simeprevir and Vertex’s investigational nucleotide analogue polymerase inhibitor VX-135 for the treatment of HCV. As a first step, Janssen Pharmaceutical Inc. will conduct a drug-drug interaction (DDI) study with simeprevir and VX-135.
We also recently announced plans to initiate a phase II trial of an investigational interferon-free regimen with simeprevir, TMC647055 and Idenix’s IDX719, a once-daily, pan-genotypic NS5A inhibitor, with and without ribavirin.
About Hepatitis C
Hepatitis C, a blood-borne infectious disease of the liver and a leading cause of chronic liver disease and liver transplants, is a rapidly evolving treatment area with a clear need for innovative treatments. Approximately 150 million people are infected with hepatitis C worldwide, and 350,000 people per year die from the disease.
About Medivir AB
Medivir is an emerging research-based pharmaceutical company focused on infectious diseases. Medivir has world class expertise in polymerase and protease drug targets and drug development which has resulted in a strong infectious disease R&D portfolio. The Company’s key pipeline asset is simeprevir, a novel protease inhibitor in late phase III clinical development for hepatitis C that is being developed in collaboration with Janssen R&D Ireland.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed by Medivir and Johnson & Johnson’s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
- “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- [http://www.medivir.se/v4/en/ir_media/pressrelease.cfm Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
Phase 3-Gilead’s newly-acquired Sofosbuvir, GS-7977 shines in Hepatitis C trial

Sofosbuvir
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate

The Foster City, CA-based Gilead said that its experimental drug GS-7977, originally known as PSI-7977 before the acquisition, when combined with ribavirin, cured a group of genotype 1 hepatitis C patients after four weeks of treatment. The clinical study involved hepatitis C patients who either failed to respond to previous therapies or had not been treated before. The genotype 1 is the most common form of HCV in the United States. It affects 70 to 90 percent of the people in this country who have hepatitis C.
Norbert Bischofberger, chief scientific officer at Gilead said patients with genotype 1 hepatitis C had no detectable signs of the virus after treated with GS-7977 combination therapy for a course of close to a month. Previous study showed the drug candidate could also cure patients with genotype 2 and 3 HCV.
Gilead gained rights to GS-7977 through the $11 billion Pharmasset acquisition deal, which enable the company to be in an advanced position to compete with a few pharma companies seeking to develop an all-oral regimen for hepatitis C. The 100 percent cure rate data suggested that GS-7977 may be one of the most promising therapies for hepatitis C.
Last year, GS-7977, an oral uridine nucleotide analog polymerase inhibitor of HCV, received fast track designation from the U.S. FDA for the treatment of HCV infection.
The World Health Organization estimated that 3–4 million people are infected with HCV each year. Some 130–170 million people are chronically infected with HCV and at risk of developing liver cirrhosis and/or liver cancer, and more than 350,000 people die yearly from hepatitis C-related diseases.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]
- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S. et al. (2010). “Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus”. Journal of Medicinal Chemistry 53 (19): 7202–7218. doi:10.1021/jm100863x. PMID 20845908. edit
- “PSI-7977”. Gilead Sciences.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M. M.; Bao, D.; Chang, W.; Espiritu, C. et al. (2010). “Mechanism of Activation of PSI-7851 and Its Diastereoisomer PSI-7977”. Journal of Biological Chemistry 285 (45): 34337–34347. doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890. edit
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- Tom Murphy (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”. Bloomberg Businessweek.
- http://www.gilead.com/pr_1757156
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. Gane, E et al. New England Journal of Medicine 368:3444. January 3, 2013.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
