
Home » Posts tagged 'GENERIC'
Tag Archives: GENERIC
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Dipivefrine, дипивефрин , ديبيفيفرين , 地匹福林 , ジピベフリン
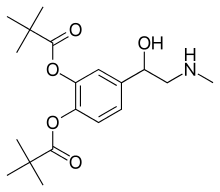
Dipivefrine
- Molecular FormulaC19H29NO5
- Average mass351.437 Da

Dipivefrine (INN) or dipivefrin (USAN), trade name Propine among others, is a prodrug of epinephrine, and is used to treat open-angle glaucoma.[1][2] It is available as a 0.1% ophthalmic solution. It is no longer available in the United States.[3]
Dipivefrin is a prodrug with little or no pharmacologically activity until it is hydrolyzed into epinephrine inside the human eye. The liberated epinephrine, an adrenergic agonist, appears to exert its action by stimulating α -and/or β2-adrenergic receptors, leading to a decrease in aqueous production and an enhancement of outflow facility. The dipivefrin prodrug delivery system is a more efficient way of delivering the therapeutic effects of epinephrine, with fewer side effects than are associated with conventional epinephrine therapy. Dipivefrin is used as initial therapy for the control of intraocular pressure in chronic open-angle glaucoma.

Contraindications
Use in narrow-angle glaucoma may be dangerous because it could make the eye susceptible to an attack of angle closure,[2] causing an increase in pressure and pain, and possibly loss of vision.
Side effects
The most common side effects of dipivefrine are burning, stinging and other irritations of the eye. Possible, but uncommon, side effects are those of epinephrine: tachycardia (fast heartbeat), hypertension (high blood pressure) and arrhythmias (irregular heartbeat).[2]
Pharmacology
Dipivefrine penetrates the cornea and is then hydrolysed to epinephrine by esterase enzymes. It increases outflow of the aqueous humour and also reduces its formation (mediated by its action on α1 and α2 receptors), thus reducing pressure inside the eye. It also increases the conductivity of trabecular filtering cells (a β2 receptor mediated action). It is preferred to epinephrine because it is longer acting, more consistent in its action and better tolerated.[1]
Patent
https://patents.google.com/patent/CN102153485A/en

Example 1 [0023] Embodiment
[0024] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 666g (6. 58mol) of triethylamine, and then was added dropwise 784g (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; to give 990g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96.2%. [0025] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to 0 ° C, was added dropwise 990g (2. 8mol) 4- (2- chloroacetyl) -I, DMF solution tank 2-phenyl pivalate ester. At room temperature was stirred for 4h.
[0026] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 923. Og. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.5%.
[0027] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. At room temperature was stirred for 4h. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 552. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.2%.
[0028] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 13g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0029] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0030] After the solution was washed with methanol hydrochloride salt to give an off-white solid 119. 9g, dipivefrin i.e., the content of 98.9%.
[0031] m.p. 161 ~162 ° C;
[0032] 1H NMR (CDCl3) δ: 1. 35 (s, 18Η), 2 68 (s, 3Η), 3 07-3 13 (m, 2Η), 5 36-5 39 (m….. , 1H),
[0033] 7. 06-7. 30 (m, 3H), 8. 61 (s, 1H), 9. 48 (s, 1H)
Dipivefrin prepared: Example 2 [0034] Embodiment
[0035] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 10 ° C, was added 666g (6. 58mol) of triethylamine, and then dropwise 78½ (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; 978. 2g to give yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96. 2% o
[0036] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to O0C, dropwise 978. 2g (2. 77mol) 4- (2- chloroacetyl) of DMF solution tank Laid-1,2-phenyl valerate. At room temperature was stirred for 4h.
[0037] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 910. 2g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.3%.
[0038] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 97g (l. SOmol) potassium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 532. 7g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0039] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 15g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0040] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0041] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 112. 8g, i.e., dipivefrin, content 98.6%.
3 [0042] Example 2: Preparation of dipivefrin
[0043] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 897g (6. 5mol) of potassium carbonate, and then drops was added 784g (6. 5mol) pivaloyl chloride addition was completed stirring was continued Syndrome. Filtered off with suction, the filtrate by rotary evaporation; to give 900g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 95.6%.
[0044] A 526g (4. 35mol) N_ methylbenzylamine, 414g (3. Omol) of potassium carbonate, 25g (0. 15mol) KI, 3L DMF force Λ IOL of four port flask. Cooled to O0C, was added dropwise 900g (2. 55mol) 4- (2- chloroacetyl) of DMF solution of 1,2-Shan Laid phenyl valerate. It was stirred at room temperature Mi.
[0045] The suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 820g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 95.6%.
[0046] Take 625g (1. 42mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 512. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0047] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 16g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0048] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0049] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 109. 8g, i.e., dipivefrin, content 98.5%.
SYN
SYN
2-chloro-3′,4′-dihydroxyacetophenone, 99-40-1
3′,4′-dihydroxy-2-methylaminoacetophenone, 99-45-6
2,2-dimethylpropanoic acid 4-[(methylamino)acetyl]-1,2-phenylene ester, 52245-00-8
Pivaloyl chloride, 3282-30-2
Trimethylacetyl chloride, 3282-30-2

1-(3,4-dipivaloyloxyphenyl)-2-(benzylmethylamino)ethan-1-one, 42146-03-2
SPECTROSCOPY

infrared spectral assignments for dipiveh hydrochloride
Wavelength (cm-1) Assignment
3255,2804,2475, 2397 RflHz+-NH stretch
2974-2875 sp3 C-H stretch
1273, 1258-1163 C-0-C stretch
3600-3400 0-H stretch
phenyl ester C=O stretch 1761
aromatic C-C stretch 1614, 1595, 1562, 1504
sp3 C-H bending and scissoring 1481, 1461, 1441, 1397
tert-butyl C-H bending1368, 1332
secondary alcohol C-0 stretch 1 124- 1028
out-of-plane bending for 1,substituted benzene ring 3,4 891,842

Ultraviolet absorption of dipivefrin hydrochloride
E (176, 1 cm)
Solvent 210 nm 264 Nn 270 nm
Acetonitrile 267.3 14.8 13.4
Ethanol 246.8 14.5 13.1
pH 3 Buffer 266.7 12.4 10.4
pH 7 Buffer 257.6 10.8 8.9
Water 278.0 18.0 16.2




References
- ^ Jump up to:a b KD Tripari. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
- ^ Jump up to:a b c Dipivefrin FDA Professional Drug Information.
- ^ Zhang L, Weizer JS, Musch DC (2017). “Perioperative medications for preventing temporarily increased intraocular pressure after laser trabeculoplasty”. Cochrane Database Syst Rev. 2: CD010746. doi:10.1002/14651858.CD010746.pub2. PMC 5477062. PMID 28231380.
-
- Hussain, A.; Truelove, J.E.: J. Pharm. Sci. (JPMSAE) 65, 1510 (1976).
- US 3 839 584.
- a DOS 2 343 657 (Interx Res. Corp.; appl. 30.8.1973; USA-prior. 31.8.1972).
- US 3 809 714 (Interx; 7.5.1974; prior. 31.8.1972) also racemate resolution.
- b DOS 2 152 058 (Klinge; appl. 19.10.1971).
 |
|
| Clinical data | |
|---|---|
| Trade names | Propine, Pivalephrine |
| Synonyms | Dipivefrin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686005 |
| Pregnancy category |
|
| Routes of administration |
Eye drops |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C19H29NO5 |
| Molar mass | 351.437 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////дипивефрин , ديبيفيفرين , 地匹福林 , Dipivefrine, antiglaucoma, GENERIC, ジピベフリン
SPIRONOLACTONE, спиронолактон , سبيرونولاكتون , 螺内酯 ,

Spironolactone
Spironolactone, Supra-puren, Suracton, спиронолактон, سبيرونولاكتون ,
螺内酯 , Abbolactone, Aldactide, SNL, Spiroctanie, Sprioderm, Verospirone, Opianin
7α-Acetylthio-17α-hydroxy-3-oxopregn-4-ene-21-carboxylic acid γ-lactone
(1’S,2R,2’R,9’R,10’R,11’S,15’S)-9′-(acetylsulfanyl)-2′,15′-dimethylspiro[oxolane-2,14′-tetracyclo[8.7.0.02,7.011,15]heptadecan]-6′-ene-5,5′-dione
| CAS 52-01-7 |
MF C24H32O4S, MW 416.573 Da
Spironolactone, marketed under the brand name Aldactone among others, is a medication primarily used to treatfluid build-up due to heart failure, liver scarring, or kidney disease.[1] Other uses include high blood pressure, low blood potassium that does not improve with supplementation, early puberty, excessive hair growth in women,[1] and as a component of hormone replacement therapy for transgender women.[6] It is taken by mouth.[1]
Common side effects include electrolyte abnormalities particularly high blood potassium, nausea, vomiting, headache, a rash, and a decreased desire for sex. In those with liver or kidney problems extra care should be taken.[1]Spironolactone has not been well studied in pregnancy and should not be used to treat high blood pressure of pregnancy.[7] It is a steroid that blocks mineralocorticoid receptors. It also blocks androgen, and blocks progesterone. It belongs to a class of medications known as potassium-sparing diuretics.[1]
Spironolactone was introduced in 1959.[8][9] It is on the World Health Organization’s List of Essential Medicines, the most important medications needed in a basic health system.[10] It is available as a generic medication.[1] The wholesale cost in the developing world as of 2014 is between 0.02 and 0.12 USD per day.[11] In the United States it costs about 0.50 USD per day.[1]
|
Title: Spironolactone
CAS Registry Number: 52-01-7
CAS Name: (7a,17a)-7-(Acetylthio)-17-hydroxy-3-oxopregn-4-ene-21-carboxylic acid g-lactone
Additional Names: 17-hydroxy-7a-mercapto-3-oxo-17a-pregn-4-ene-21-carboxylic acid g-lactone, acetate; 3-(3-oxo-7a-acetylthio-17b-hydroxy-4-androsten-17a-yl)propionic acid g-lactone
Manufacturers’ Codes: SC-9420
Trademarks: Aldactone (Pharmacia & Upjohn); Aquareduct (Azupharma); Practon (Pfizer); Osyrol (Aventis); Sincomen (Schering AG); Spirobeta (Betapharm); Spiroctan (Ferlux); Spirolone (APS); Spironone (Dexo); Verospiron (Richter Gedeon); Xenalon (Mepha)
Molecular Formula: C24H32O4S
Molecular Weight: 416.57
Percent Composition: C 69.20%, H 7.74%, O 15.36%, S 7.70%
Literature References: Aldosterone antagonist. Prepn: Cella, Tweit, J. Org. Chem. 24, 1109 (1959); US 3013012 (1961 to Searle); Tweit et al., J. Org. Chem. 27, 3325 (1962). Activity and metabolic studies: Gerhards, Engelhardt, Arzneim.-Forsch. 13, 972 (1963). Crystal and molecular structure: Dideberg, Dupont, Acta Crystallogr. B28, 3014 (1972). Comprehensive description: J. L. Sutter, E. P. K. Lau, Anal. Profiles Drug Subs. 4, 431-451 (1975). Review of carcinogenetic risk: IARC Monographs 24, 259-273 (1980). Review of antiandrogen effects and clinical use in hirsutism: R. R. Tremblay, Clin. Endocrinol. Metab. 15, 363-371 (1986); of clinical efficacy in hypertension: A. N. Brest, Clin. Ther. 8, 568-585 (1986). Review of pharmacology: H. A. Skluth, J. G. Gums,DICP Ann. Pharmacother. 24, 52-59 (1990). Clinical trial in congestive heart failure: B. Pitt et al., N. Engl. J. Med. 341, 709 (1999).
Properties: Crystals from methanol, mp 134-135° (resolidifies and dec 201-202°). [a]D20 -33.5° (chloroform). uv max: 238 nm (e20200). Practically insol in water. Sol in alcohol; freely sol in benzene, chloroform. LD50 in rats, mice, rabbits (mg/kg): 790, 360, 870 i.p. (IARC, 1980).
Melting point: mp 134-135° (resolidifies and dec 201-202°)
Optical Rotation: [a]D20 -33.5° (chloroform)
Absorption maximum: uv max: 238 nm (e 20200)
Toxicity data: LD50 in rats, mice, rabbits (mg/kg): 790, 360, 870 i.p. (IARC, 1980)
Therap-Cat: Diuretic.
Therap-Cat-Vet: Diuretic.
Keywords: Aldosterone Antagonist; Diuretic; Steroids
|
Medical uses
Spironolactone is used primarily to treat heart failure, edematous conditions such as nephrotic syndrome or ascites in people with liver disease, essential hypertension, hypokalemia, secondary hyperaldosteronism (such as occurs with hepatic cirrhosis), and Conn’s syndrome (primary hyperaldosteronism). On its own, spironolactone is only a weak diuretic because it primarily targets the distal nephron (collecting tubule), where only small amounts of sodium are reabsorbed, but it can be combined with other diuretics to increase efficacy.
Spironolactone is an antagonist of the androgen receptor (AR) as well as an inhibitor of androgen production. Due to the antiandrogenic effects that result from these actions, it is frequently used off-label to treat a variety of dermatological conditions in which androgens, such as testosterone and dihydrotestosterone (DHT), play a role. Some of these uses include androgenic alopecia in men (either at low doses or as a topical formulation) and women, and hirsutism, acne, and seborrhea in women.[12] Spironolactone is the most commonly used drug in the treatment of hirsutism in the United States.[13] Higher doses of spironolactone are not recommended in males due to the high risk of feminization and other side effects. Similarly, it is also commonly used to treat symptoms of hyperandrogenism in polycystic ovary syndrome.[14]
Spironolactone (SL) is known to be a potent aldosterone antagonist at mineralocorticoid steroid hormone receptors, and it is widely used in humans for the treatment of essential hypertension, congestive heat failure and refractory edema or hyperaldosteronism. However, the prolonged use of SL is associated with undesirable endocrine side effects such as gynecomastia and lose of libido in men and menstrual irregularities in women due to interaction of SL with gonadal steroid hormone biosynthesis and target cell gonadal steroid receptors.
The nature and prevalence of the undesirable side effects limit the usefulness of spironolactone as a therapeutic agent. Gynecomastia or tender breast enlargement has been found to occur in 10% of hypertensive patients using spironolactone for therapy as compared to 1% of men in the placebo group. Recent studies by Pitt, et al. with spironolactone have shown that in patients with congestive heart failure (CHF) taking digoxin and a loop diuretic—spironolactone therapy in conjunction with digitalis and ACE inhibitor—reduces mortality by 30%. See Pitt, B., et al., The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure, Randomized Aldactone Evaluation Study Investigors; N. Engl. J. Med., 1999, 341:709-717. These authors stated that the 30% reduction in the risk of death among patients in the group receiving spironolactone could be attributed to a lower risk of both death from progressive heart failure and sudden death from cardiac arrhythmic causes. In addition, they found that the frequency of hospitalization for worsening heart failure is 35% lower in the spironolacotone treated group than in the placebo group. These authors concluded that patients who received spironolactone had a significant improvement in the symptoms of severe heart failure caused by systolic left ventricular dysfunction. Overall, 8% of the patients in the spironolactone group discontinued treatment because of adverse events. The purpose of the present invention is to make available the individual chiral isomers of spironolactone that would be effective in treating CHF and in reducing hypertension, and at the same time would be devoid of undesirable side effects such as gynecomastia, lose of libido in men, and menstrual irregularities in women.
Spironolactone is the name commonly used for a specific spirolactone that has the full chemical name 17-hydroxy-7-alpha-mercapto-3-oxo-17-alpha-pregn-4-ene-21-carboxylic acid gamma-lactone acetate. The term “spirolactone” denotes that a lactone 10 ring (i.e., a cyclic ester) is attached to another ring structure in a spiro configuration (i.e., the lactone ring shares a single carbon atom with the other ring). Spirolactones that are coupled to steroids are the most important class of spirolactones from a pharmaceutical perspective, so they are widely referred to in the pharmaceutical arts simply as spirolactones. As used herein, “spironolactone” refers to a molecule comprising a lactone structure coupled via a spiro configuration to a steroid structure or steroid derivative.
Spironolactone, its activities, and modes of synthesis and purification are described in a number of U.S. patents, notably U.S. Pat. Nos. 3,013,012, 4,529,811 and 4,603,128.
Intracellular receptors (IRs) form a class of structurally-related genetic regulators that act as ligand-dependent transcription factors. See Evans, R. M., “The Steroid and Thyroid Hormone Receptor Superfamily”, Science, May 13, 1988; 240(4854):889-95. Steroid receptors are a recognized subset of the IRs, including the progesterone receptor (PR), androgen receptor (AR), estrogen receptor (ER), which can be referred to collectively as the gonadal steroid receptors, glucocorticoid receptor (GR), and mineralocorticoid receptor (MR). Regulation of a gene by such factors requires both the IR itself and a corresponding ligand that has the ability to selectively bind to the IR in a way that affects gene transcription.
Ligands for the IRs can include low molecular weight native molecules, such as the hormones aldosterone, progesterone, estrogen and testosterone, as well as synthetic derivative compounds such as medroxyprogesterone acetate, diethylstilbesterol and 19-nortestosterone. These ligands, when present the fluid surrounding a cell, pass through the outer cell membrane by passive diffusion and bind to specific IR proteins to create a ligand/receptor complex. This complex then translocates to the cell’s nucleus, where it binds to a specific gene or genes present in the cell’s DNA. Once bound to DNA, the complex modulates the production of the protein encoded by that gene. In this regard, a compound that binds to an IR and mimics the effect of the native ligand is referred to as an “agonist”, while a compound that binds to an IR and inhibits the effect of the native ligand is called an “antagonist”.
The therapeutic mechanism of action of spironolactone involves binding to intracellular mineralocorticoid receptors (MRs) in kidney epithelial cells, thereby inhibiting the binding of aldosterone. Spironolactone has been found to counteract the sodium reabsorption and potassium excretion effects of aldosterone and other mineralocorticoids. Spironolactone has also been shown to interfere with testosterone biosynthesis, has anti-androgen action and inhibits adrenal aldosterone biosynthesis. Large doses of spironolactone in children appear to decrease the testosterone production rate.
Spironolactone is found to exhibit intra-individual variability of pharmacokinetic parameters and it presumably belongs to the group of drugs with high inter-subject variability. Spironolactone has poor water solubility and dissolution rate.
In order to prolong the half-life and decrease the side effects associated with spironolactone, syntheses of spironolactone derivatives have been developed (e.g. synthesis of mexrenone, prorenone, spirorenone). Slight modifications of the spironolactone steroid skeleton, e.g. such as formation of 11β-allenic and epoxy compounds, have been shown to effect important variations in the affinity and specificity for the mineralocorticoid receptor. These results suggest that it is possible to develop spironolactone analogues that do not interact with the androgen receptor or cytochrome P-450 and are therefore free of spironolactone undesirable side-effects.
METABOLISM

SYNTHESIS
METHOD 1 REF 150

REF 130, 150
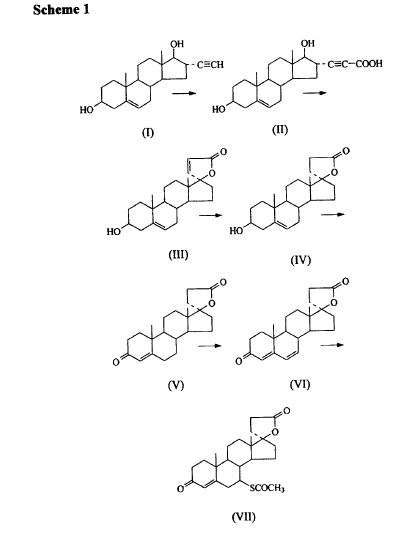

METHOD 2 REF 140
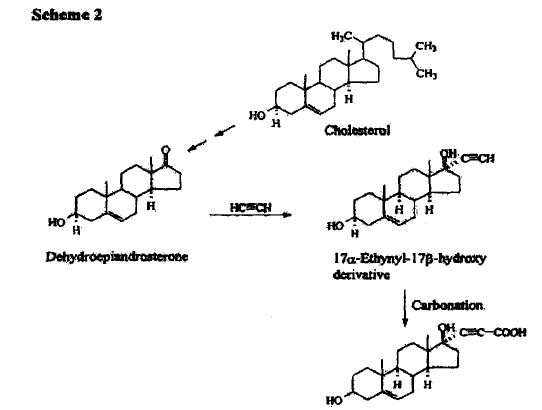
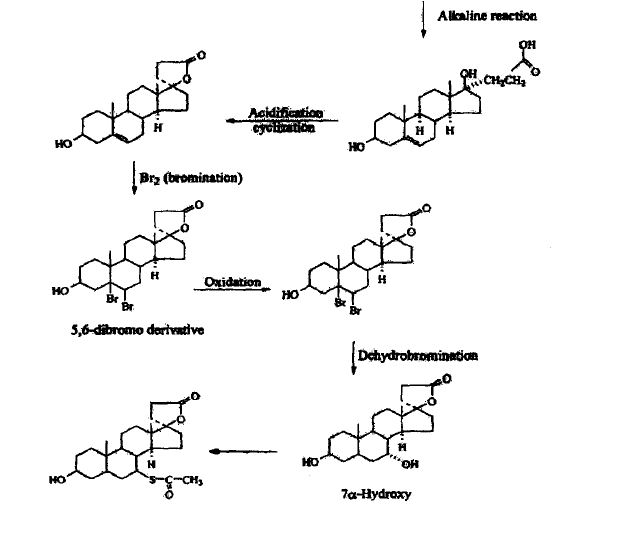

METHOD 3 REF 150
Synthesis
Cella, John A.; Tweit, Robert C. (1959). Journal of Organic Chemistry 24: 1109. doi:.
(See also part 1 and part 3)

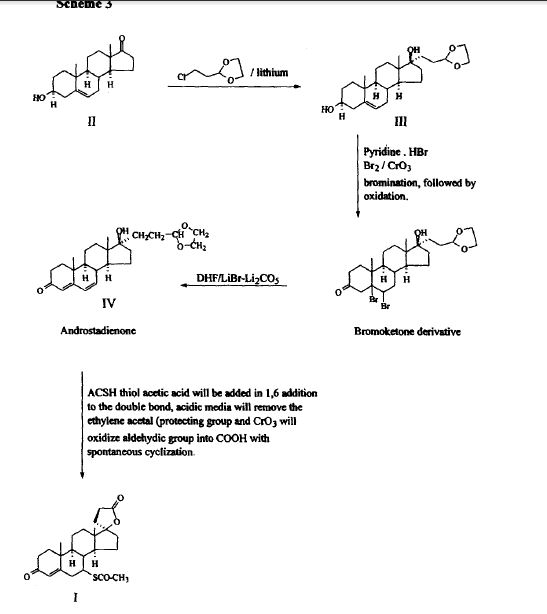
SPECTROSCOPY UV
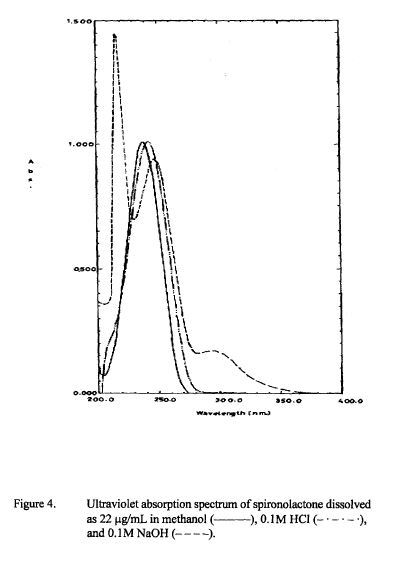
SPECTROSCOPY IR
KBR
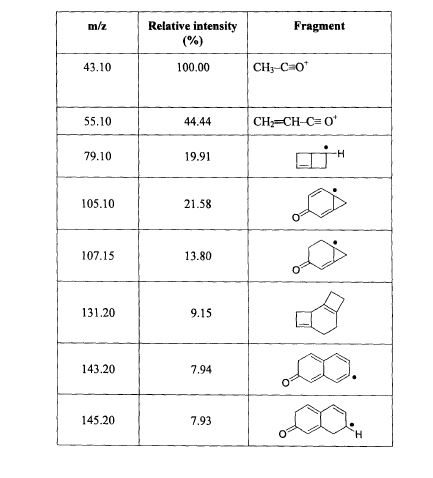
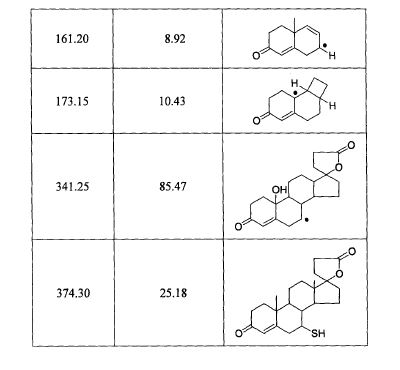
The principal absorption peaks of the spectrum shown in Figure 5 were noted at 1765,
1693, 1673, 1240, 1178, 1135, 1123 and 1193 cm -1.
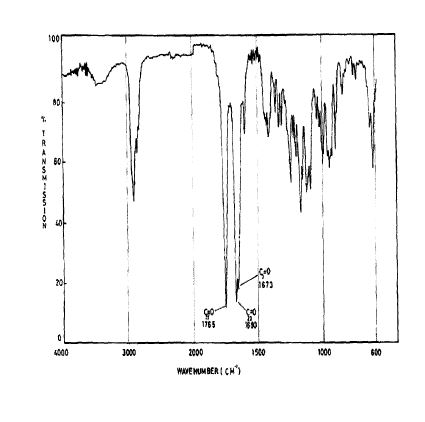
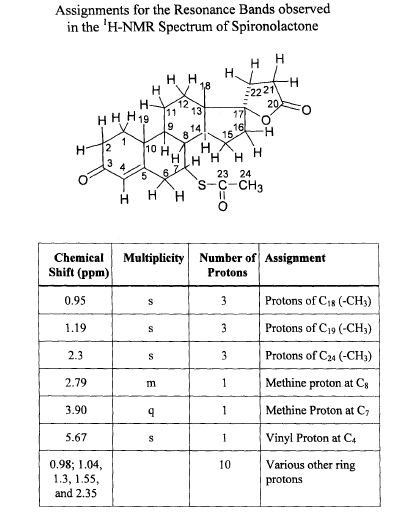
SPECTROSCOPY 1H NMR
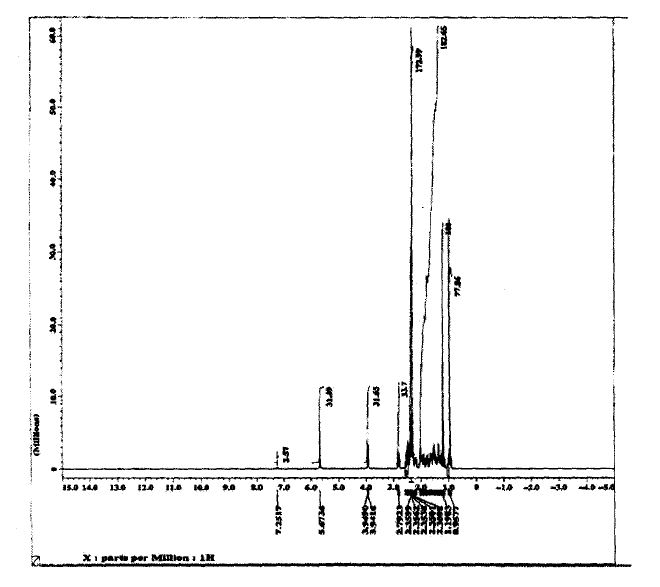
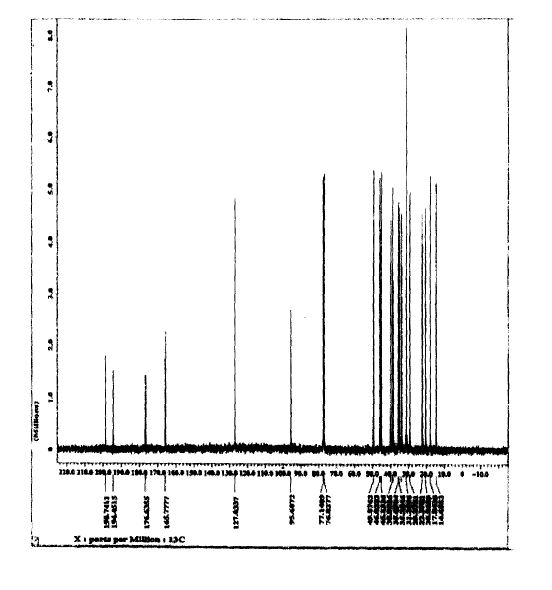
SPECTROSCOPY 13C NMR
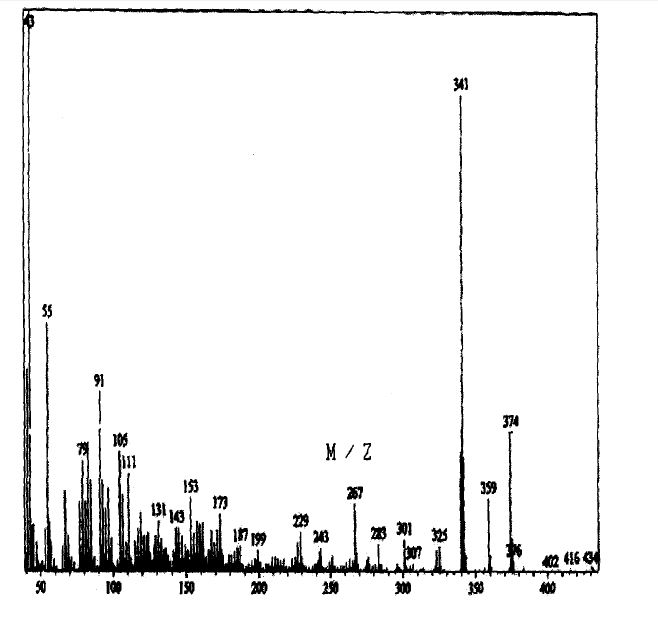
SPECTROSCOPY MASS SPECTRUM
130 J.A. Cola, E.A. Brown, and R.R. Burtner, 3. Org. Chem., 24, 1109(1959).
140 Remington’s: The Science and Practice of Pharmacy, 19 t~ edn.Volume II, K.G. Alfonso, ed.; Mack Publishing Co., Pennsylvania (1995) p.1048.
150. G. Anner and H. Wehrli (Ciba-Geigy, A.-G.), German Often 2,625,723 (cl.C07J21/00), Dec,1976; Swiss Appl. 75/7, 696, 13Jun. 1975; pp. 37.
ANALYTICAL
-
High-Performance Liquid Chromatographic Conditions Column LiChrosorb RP-8, 5 μm. 150 × 4.6 mm I.D. Eluent Acetonitrile-0.05 M phosphate buffer, pH 4 (45:55) Flow-rate 1 ml/min Temperature 25° C. Detector UV detector, wavelength 286 nm or 271 nm Recorder Chart speed 0.5 cm/min Sample loop 10 μl -
The concentration of canrenone is determined in plasma and urine samples by high-performance liquid chromatography (HPLC) with UV-detection. An aliquot of 300 ng of spironolactone derivative is added to the samples as internal standard, which are then extracted twice with 1 ml n-hexane-toluene (1:1, v/v). The organic phase is taken to dryness and re-dissolved in 250 μl HPLC eluent (methanol-water, 60:40, v/v). (25×4.6 mm; 5 μm). Detection is performed with the UV detector set at λ=285 nm.
Flurometric Method
- Five ml of water is a reagent blank and 5 ml of working standards containing 0.05 μg and 0.20 μg of SC-9376 are carried through the entire procedure. Lower sales are read vs. the 0.05 μg standard at full scale, and higher samples vs. the 0.20 μg standard. Fluorescence readings are proportional to the concentrations of the standards in this range.
- Pipette 0.2 ml of heparinized plasma into a 50-ml polyethylene-stoppered centrifuge tube, dilute to 5 ml with water and add 15 ml of methylene chloride (Du Pont refrigeration grade, redistilled). Shake for 30 seconds, centrifuge and discard the aqueous supernatant. Add 1 ml 0.1 N NaOH, shake 15 seconds, centrifuge and discard the supernatant. Transfer a 10-ml aliquot of the methylene chloride phase to another tube containing 2 ml of 65% aqueous sulfuric acid, shake 30 seconds, centrifuge and remove organic phase by aspiration. The material is allowed to stand at room temperature for about 1 hour and then about 1 ml of the sulfuric acid phase in transferred to a quartz cuvette. Fluorescence intensity is determined in an Aminco-Bowman spectrophotofluorometer (activation maximum, 465 nm).
- Gas Liquid Chromatography
- The GLC estimation is carried out on a Fractovap Model 251 series 2150 (Carlo Erba) instrument equipped with a Nickel-63 electron capture detector. A 6-foot, 0.4 mm internal diameter, U-shaped glass column, packed with OV-17 2% or XE-60 1% on gas chrom A, 100-120 mesh (Applied Science Lab) is conditioned for 3 days before use. Argon with 10% methane which passed through a molecular sieve before entering the column is used as the carrier gas. The conditions of analysis are: column 255° C., detector 275° C., carrier gas flow 30 ml/min. Samples are injected on the column with a 10 μl Hamilton syringe. The injector in not heated.
PATENT
https://www.google.com/patents/US20090325918
EXAMPLE 1Chiral Separation
The separation of 7 beta isomer of SL is schematically described below.
-
 Chromatographic Method for Isolation of SL IsomersThe basic method is described in Chan, Ky, et al., J. Chromatog, Nov. 15, 1991:571 (1-2) 291-297. The separation is performed using spectra-physics HPLC instrument and UV variable wavelength detector set at 254 nm. For chiral separation, the chromatographic column is either a pre-packed 25 mm×4.6 mm ID Cyclobond 1 (5 μm particle size), or a pre-packed 150 mm×4 mm ID Resolvosil BSA-7 column (5 μm) operated using the conditions described herein.Analysis of the isomers present in the peaks in the chromatograms and their chiral extract purity analysis can be determined in each case by high resolution NMR spectroscopy using a chiral shift reagent. Based on this information and the determination of molecular weight by mass spectrometry and/or optical activity, structural configuration is assigned to each isomer. Eluted samples of isomers may be re-chromatographed in order to obtain adequate quantities of isomers having desired optical purity for study. For future use, reference standards that are optically pure will be compared for confirmation of purity and identity to the isolated isomers that are obtained after their chromatographic separation.
Chromatographic Method for Isolation of SL IsomersThe basic method is described in Chan, Ky, et al., J. Chromatog, Nov. 15, 1991:571 (1-2) 291-297. The separation is performed using spectra-physics HPLC instrument and UV variable wavelength detector set at 254 nm. For chiral separation, the chromatographic column is either a pre-packed 25 mm×4.6 mm ID Cyclobond 1 (5 μm particle size), or a pre-packed 150 mm×4 mm ID Resolvosil BSA-7 column (5 μm) operated using the conditions described herein.Analysis of the isomers present in the peaks in the chromatograms and their chiral extract purity analysis can be determined in each case by high resolution NMR spectroscopy using a chiral shift reagent. Based on this information and the determination of molecular weight by mass spectrometry and/or optical activity, structural configuration is assigned to each isomer. Eluted samples of isomers may be re-chromatographed in order to obtain adequate quantities of isomers having desired optical purity for study. For future use, reference standards that are optically pure will be compared for confirmation of purity and identity to the isolated isomers that are obtained after their chromatographic separation.
EXAMPLE 2Chemical Synthesis of Optical Isomers
- As an example, the desire spironolactone 7-beta-isomer is synthesized following the scheme that is described below:
-
 Diene (i) is prepared from commercially available starting materials using methods well known in the art of chemical synthesis.Diene (i) is treated with acetic acid and the mixture is heated to reflux to yield 7-alpha-acetate ester (ii). The 7-alpha-ester (ii) is further subjected to nucleophilic substitution, followed by hydrolysis to obtain the 7-beta-isomer (iii). The 7-beta-isomer (iii) is then esterified with an acyl halide in the presence of a base to generate the desired spironolactone 7-beta-isomer (iv).
Diene (i) is prepared from commercially available starting materials using methods well known in the art of chemical synthesis.Diene (i) is treated with acetic acid and the mixture is heated to reflux to yield 7-alpha-acetate ester (ii). The 7-alpha-ester (ii) is further subjected to nucleophilic substitution, followed by hydrolysis to obtain the 7-beta-isomer (iii). The 7-beta-isomer (iii) is then esterified with an acyl halide in the presence of a base to generate the desired spironolactone 7-beta-isomer (iv).
EXAMPLE 3Preparation of Radiolabeled Probe Compounds of the Invention
- Using known methods, the compounds of the invention may be prepared as radiolabeled probes by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope. The radioisotope is preferably selected from at least one of carbon (preferably
14
- C), hydrogen (preferably
3
- H), sulfur (preferably
35
- S), or iodine (preferably I). Such radiolabeled probes are conveniently synthesized by a radioisotope supplier specializing in customer synthesis of radiolabeled probe compounds. Such suppliers include Amersham Corporation, Arlington Heights, Ill.; Cambridge Isotope Laboratories, Inc., Andover, Mass.; SRI International, Menlo Park, Calif.; Wizard Laboratories, West Sacramento, Calif.; ChemSyn Laboratories, Lexena, Kans.; American Radiolabeled Chemicals, Inc., St. Louis, Mo.; and Moravek Biochemicals Inc., Brea, Calif.
- Tritium labeled probe compounds are also conveniently prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas. Tritium labeled probe compounds can also be prepared, when appropriate, by sodium borotritide reduction. Such preparations are also conveniently carried out as a custom radiolabeling by any of the suppliers listed in the preceding paragraph using the compound of the invention as substrate.
- EXAMPLE 4Isolation and Purification Procedure
- The optical isomers of spironolactones may be isolated from fluid sample such as urine or blood as follows:
- Extraction from Urine
- The urine sample is extracted with dichloromethane and the extract washed with NaOH (0.1 N) and then with water to neutrality. The residue obtained after evaporation of the dichloromethane extract is purified on TLC in three different systems: benzene-acetone-water, (150:100:0.4); chloroform-ethanol, (90:10); ethyl acetate-cyclohexane-ethanol, (45:25:10), using aldosterone as reference standard.
- The extract is then purified by high performance liquid chromatography (HPLC) on a Waters 6000 A, 480 U.V. detector instrument with radial pressure. The extract is first run through a C
18
- 10μ column using methanol-water (70:30) as the eluent, followed by a silica 5μ column using dichloromethane-methanol (95:5). In both cases, the rate of the eluent is 1.5 ml/min. A small part of the extract is subjected to heptafluorobutyrylation for GLC investigation.
References
- “Spironolactone”. The American Society of Health-System Pharmacists. Retrieved Oct 24, 2015.
- “Spironolactone: MedlinePlus Drug Information”. Retrieved 2016-01-20.
- “Spironolactone”. Merriam-Webster Dictionary.
- “Spironolactone”. Dictionary.com Unabridged. Random House.
- Harry G. Brittain (26 November 2002). Analytical Profiles of Drug Substances and Excipients. Academic Press. p. 309. ISBN 978-0-12-260829-2. Retrieved 27 May 2012.
- Maizes, Victoria (2015). Integrative Women’s Health (2 ed.). p. 746.ISBN 9780190214807.
- “Spironolactone Pregnancy and Breastfeeding Warnings”. Retrieved 29 November2015.
- Camille Georges Wermuth (24 July 2008). The Practice of Medicinal Chemistry. Academic Press. p. 34. ISBN 978-0-12-374194-3. Retrieved 27 May 2012.
- Marshall Sittig (1988). Pharmaceutical Manufacturing Encyclopedia. William Andrew. p. 1385. ISBN 978-0-8155-1144-1. Retrieved 27 May 2012.
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- “Spironolactone”. International Drug Price Indicator Guide. Retrieved 29 November2015.
- Hughes BR, Cunliffe WJ (May 1988). “Tolerance of spironolactone”. The British Journal of Dermatology 118 (5): 687–91. doi:10.1111/j.1365-2133.1988.tb02571.x.PMID 2969259.
- Victor R. Preedy (1 January 2012). Handbook of Hair in Health and Disease. Springer Science & Business Media. pp. 132–. ISBN 978-90-8686-728-8.
- Loy R, Seibel MM (December 1988). “Evaluation and therapy of polycystic ovarian syndrome”. Endocrinology and Metabolism Clinics of North America 17 (4): 785–813.PMID 3143568.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
7α-Acetylthio-17α-hydroxy-3-oxopregn-4-ene-21-carboxylic acid γ-lactone
|
|
| Clinical data | |
| Pronunciation | /spɪˌroʊnəˈlæktoʊn, spaɪ–, spə–, –ˈrɒ–, –noʊ–/or /ˌspaɪrənoʊˈlæktoʊn/[2][3][4] |
| Trade names | Aldactone |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682627 |
| Pregnancy category |
|
| Routes of administration |
Oral[1] |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 90%+[5] |
| Metabolism | Hepatic CYP450 |
| Biological half-life | 1.3-2 hours |
| Excretion | Urine, bile |
| Identifiers | |
| CAS Number | 52-01-7 |
| ATC code | C03DA01 (WHO) |
| PubChem | CID 5833 |
| IUPHAR/BPS | 2875 |
| DrugBank | DB00421 |
| ChemSpider | 5628 |
| UNII | 27O7W4T232 |
| KEGG | D00443 |
| ChEBI | CHEBI:9241 |
| ChEMBL | CHEMBL1393 |
| Chemical data | |
| Formula | C24H32O4S |
| Molar mass | 416.574 g/mol |
///////Spironolactone, Supra-puren, Suracton, спиронолактон, سبيرونولاكتون ,
螺内酯 , Abbolactone, Aldactide, SNL, Spiroctanie, Sprioderm, Verospirone, Opianin
O=C5O[C@@]4([C@@]3([C@H]([C@@H]2[C@H](SC(=O)C)C/C1=C/C(=O)CC[C@]1(C)[C@H]2CC3)CC4)C)CC5
India’s Strides to buy Aspen’s Australian generic pharmaceutical business
![]()
India’s Strides to buy Aspen’s Australian generic pharmaceutical business
India-based Strides Arcolab has signed an agreement with subsidiaries of South African drugmaker Aspen Pharmacare Holdings to acquire its generic pharmaceutical business in Australia and certain branded pharmaceutical assets for around A$380m ($300m).
see
About Strides
-
 Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.
Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.

.
Chandos Street, St Leonards


///////////
FDA approves first generic Copaxone to treat multiple sclerosis

April 16, 2015
The U.S. Food and Drug Administration today approved the first generic version of Copaxone (glatiramer acetate injection), used to treat patients with relapsing forms of multiple sclerosis (MS).
Sandoz has received FDA approval to market generic glatiramer acetate in a 20 mg/1 ml daily injection.
“Health care professionals and patients can be assured that FDA-approved generic drugs have met the same rigorous standards of quality as the brand-name drug,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “Before approving this generic product, given its complexity, we reviewed additional information to make sure that the generic product is as safe and effective as the brand name product.”
The FDA applies the same rigorous and reliable standards to evaluate all generic drug products. As needed, the agency requires appropriate information to demonstrate sameness for complex active ingredients, such as glatiramer acetate. For this approval, FDA scientists established a thorough scientific approach for demonstrating active ingredient sameness that takes into consideration the complexity of glatiramer acetate.

MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function (relapses) are initially followed by recovery periods (remissions). Over time, recovery periods may be incomplete, leading to progressive decline in function and increased disability. MS patients often experience muscle weakness and difficulty with coordination and balance. Most people experience their first symptoms of MS between the ages of 20 and 40.
In the clinical trials for Copaxone, the most common adverse reactions reported by those taking Copaxone were skin problems at the injection site (redness, pain, swelling and itching), flushing (vasodilation), rash, shortness of breath and chest pain.
BLOG STATS OF NEW DRUG APPROVALS
MOSCOW



 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
NADIFLOXACIN, Jinofloxacin

-
(+-)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidino)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylic acid
- CCRIS 4066
- Jinofloxacin
- Nadifloxacin
- Nadifloxacine
- Nadifloxacine [INN-French]
- Nadifloxacino
- Nadifloxacino [INN-Spanish]
- Nadifloxacinum
- Nadifloxacinum [INN-Latin]
- Nadixa
- OPC-7251
- S-Nadifloxacin
- UNII-6CL9Y5YZEQ
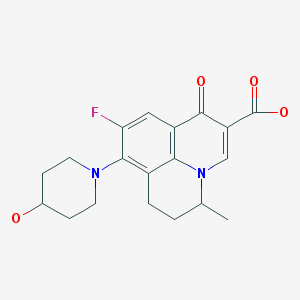
Nadifloxacin is chemically, 9-fluoro-6,7-dihydro-8-(4-hydroxy-l-pyperidinyl)-5-methyl- l-oxo-lH,5H-benzo(I,j)quinolizine-2-carboxylic acid of Formula I provided below.

FORMULA I Nadifloxacin is a synthetic quinolone with potent broad-spectrum anti-bacterial activity. Nadifloxacin inhibits the enzyme DNA gyrase that is involved in bacterial DNA synthesis and replication, thus inhibiting the bacterial multiplication. RS-nadifloxacin and S-nadifloxacin, in particular, exhibit strong antibacterial activity against Gram-positive, Gram-negative and anaerobic bacteria, resistant Gram-positive organisms such as methicillin-resistant Staphylococcus aureus (MRSA), quinolone-resistant Staphylococcus aureus, coagulase negative staphylococci, such as methicillin-resistant Staphylococcus epidermidis (MRSE), enterococci, betahemolytic streptococci and viridans group of streptococci, mycobacteria and newly emerging nosocomial pathogens such as Chryseobacterium meninges epticum, and Gram-negative pathogens such as E.coli, Klebsiella, Proteus, Serratia, Citrobacter and Pseudomonas. Recently, it has also been shown that S-(-)-nadifloxacin, in particular exhibits potent antibacterial activity against glycopeptide intermediate S. aureus (GISA), vancomycin intermediate S. aureus (VISA) and vancomycin-resistant S. aureus (VRSA). Nadifloxacin is also active against quinolone-resistant Staphylococci.
Nadifloxacin is marketed in the form of cream for topical application for the treatment of acne vulgaris, folliculitis and sycosis vulgaris. It is also indicated for the treatment of topical bacterial infections with susceptible bacteria.
The use of quinolone antibiotics to treat infections is known art in the field of ophthalmic pharmaceutical compositions and methods of treatment. Several quinolone antibacterial agents available in the market include gatifloxacin (available as Zymar®), Levofloxacin (available as Quixin® or Iquix®), Ciprofloxacin (available as Ciloxan®), Ofloxacin (available as Ocuflox®), Lomefloxacin (available as Lomeflox®), Moxifloxacin (available as Vigamox®) and Norfloxacin (available as Chibroxin®).
U.S. Patent No. 4,844,902 discloses a topically applicable formulation comprising by weight about 0.01 to 30% of an anti-bacterially active compound, 0.01 to 10% of a corticosteroid and a carrier. U.S. Patent No. 6,333,045 discloses liquid pharmaceutical compositions of gatifloxacin or salt thereof and disodium edetate.
U.S. Patent No. 6,716,830 discloses ophthalmic dosage forms of moxifioxacin or salts thereof in a concentration of 0.1% to 1% (w/w) and pharmaceutically acceptable vehicle.
U.S. Patent No. 6,359,016 relates to topical suspension formulations containing ciprofloxacin and dexamethasone.
U.S. Patent No 4,399,134 discloses processes for the preparation of nadifloxacin or salts thereof and antibacterially effective pharmaceutical compositions of nadifloxacin. Typical dosage forms include tablets, pills, powders, liquid preparations, suspensions, emulsions, granules, capsules, suppositories, and injectable preparations (solutions, suspensions, etc).
U.S. Patent No 6,884,768 discloses solid oral pharmaceutical compositions that includes nadifloxacin, an absorbefacient and taurine compounds.
U.S. Patent Application 20060183698 describes topical ophthalmic formulation that includes serum electrolytes; an antimicrobial compound and an anti-inflammatory or steroidal compound. Several antimicrobial agents have been disclosed including nadifloxacin.
U.S. Patent Application 20040176337 discloses topical . compositions of benzoquinolizine-2-carboxylic acid antimicrobial drug.
U.S. Patent Application 20040176321 discloses injectable pharmaceutical composition for intravenous delivery of an active agent that includes RS-(±)-nadifloxacin; S-(-)- nadifloxacin and hydrates thereof; or S~(-)-nadifloxacin arginine and salts thereof. PCT Publication WO 04/00360 describes pharmaceutical compositions of several active ingredients including nadifloxacin for topical use for treatment of dermatosis.
European Patent EP 275,515 and U.S. Patent No. 4,923,862 disclose aqueous pharmaceutical compositions of levofloxacin and ofloxacin or salts thereof.
PCT application WO 02/39993 discloses a stable pharmaceutical preparation of a combination drug, comprising an anti-infective agent, selected from the group consisting of quinolone derivatives, amino-glycoside derivatives and their pharmaceutically acceptable salts; an ant-inflammatory agent which is a corticosteroid; a complexation enhancing polymer; a solubilizer exhibiting an inclusion phenomena; pharmaceutically acceptable excipients within a suitable carrier system.
Journal of Ocular Pharmacology and Therapeutics, vol 23(3): 243-256, 2007 discloses (7- [(3R)-3 -aminohexahydro- 1 H-azepine- 1 -yl]-8-chloro- 1 -cyclopropyl-6-fluoro- 1 ,4-dihydro- 4-oxo-3-quinolinecarboxylivc acid as the topical agent for the treatment of ophthalmic infections.

5-Bromo-6-fluoro-2-methylquinoline (II)
5-Bromo-6-fluoro-2-methyl-1,2,3,4-tetrahydroquinoline (III)
diethyl 2-[(E)-ethoxymethylidene]succinate (IV)
8-Bromo-9-fluoro-5-methyl-1-oxo-6,7-dihydro-1H,5H-pyrido[3,2,1-ij]quinoline-2-carboxylic acid (V)
4-Piperidinol; 4-Hydroxypiperidine

______________________________________Elemental Analysis for C.sub.19 H.sub.21 N.sub.2 O.sub.4 F C H N______________________________________Calc'd (%): 63.32 5.87 7.78Found (%): 63.28 5.76 7.89______________________________________

Temozolomide 替莫唑胺

Temozolomide 替莫唑胺
Temozolomide is a DNA damage inducer.
4-methyl-5-oxo-2,3,4,6,8-pentazabicyclo[4.3.0]nona-2,7,9-triene-9-carboxamide
3,4-dihydro-3-methyl-4-oxoimidazo(5,1-d)-1,2,3,5-tetrazine-8-carboxamide
Methazolastone, Temodar, Temodal
CAS NO 85622-93-1
Molecular Weight: 194.15
MF C6H6N6O2
Cancer Research UK (Originator), Schering-Plough (Licensee), National Cancer Institute (Codevelopment)
NMR..http://file.selleckchem.com/downloads/nmr/S123702-Methazolastone-NMR-Selleck.pdf
HPLC.http://file.selleckchem.com/downloads/hplc/S123702-Methazolastone-HPLC-Selleck.pdf
Temozolomide is an antitumor agent indicated for treating patients with malignant glioma such as cancer, breast cancer, refractory anaplastic astrocytoma, i.e., patients at first relapse who have experienced disease progression in malignant glioma, glioblastoma multiform and anaplastic astrocytoma, on a drug regimen containing a nitrosourea and procarbazine.
Temozolomide preparations are sold on the US market as hard capsules containing 5 mg, 20 mg, 100 mg or 250 mg Temozolomide (marketed as Temodar® by Schering Corporation, Kenilworth, N.J., USA). In other markets it is sold as Temodal®.
Temozolomide (brand names Temodar and Temodal and Temcad) is an oral chemotherapy drug. It is an alkylating agent used for the treatment of Grade IV astrocytoma — an aggressive brain tumor, also known as glioblastoma multiforme — as well as for treating melanoma, a form of skin cancer.
Temozolomide is also indicated for relapsed Grade III anaplastic astrocytoma and not indicated for, but as of 2011 used to treatoligodendroglioma brain tumors in some countries, replacing the older (and less well tolerated) PCV (Procarbazine–Lomustine–Vincristine) regimen.
Temozolomide, 3-methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one, is a known antitumor drug; see for example Stevens et al., J. Med. Chem. 1984, 27, 196-201, and Wang et al., J. Chem. Soc., Chem. Commun.,1994,1687-1688. Temozolomide, the compound of formula 1:

is described in U.S. Pat. No. 5,260,291 (Lunt et al.).
The synthesis of 1 by the process described in J. Med. Chem. 1984, 27, 196-201 is depicted in the scheme I below.

In this process, 5-amino-1H-imidazole-4-carboxamide (A) is converted into 5-diazo-1H-imidazole-4-carboxamide (B), which is then cyclized with methylisocyanate in dichloromethane to provide a high yield of temozolomide. However, this process requires isolation of the unstable and potentially dangerous 5-diazo-1H-imidazole-4-carboxamide (B). Moreover, methylisocyanate is a difficult reagent to handle and ship, especially on the industrial scale, and indeed is better avoided in industrial manufacture. Furthermore, the cycloaddition of methylisocyanate requires a very long reaction time: Table I in J. Med Chem.1984, 27,196-201, suggests 20 days. Additionally, Stevens et al mention that the cycloaddition of the methylisocyanate to the compound of the formula (B) can proceed through two different intermediates:
The production of I by the two processes described in J. Chem. Soc., Chem. Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1H-imidazole-4-carboxamide (A): less than 20% (unoptimized—about 17% through 5-diazo-1H-imidazole-4-carboxamide (B) and about 15% through 5-amino-N1-(ethoxycarbonylmethyl)-1H-imidazole-1,4-dicarboxamide (C)); Scheme II below

The agent was developed by Malcolm Stevens[1] and his team at Aston University in Birmingham,[2][3] Temozolomide is a prodrug and animidazotetrazine derivative of the alkylating agent dacarbazine. It has been available in the US since August 1999, and in other countries since the early 2000s.
The therapeutic benefit of temozolomide depends on its ability to alkylate/methylate DNA, which most often occurs at the N-7 or O-6 positions ofguanine residues. This methylation damages the DNA and triggers the death of tumor cells. However, some tumor cells are able to repair this type of DNA damage, and therefore diminish the therapeutic efficacy of temozolomide, by expressing a protein O6-alkylguanine DNA alkyltransferase (AGT) encoded in humans by the O-6-methylguanine-DNA methyltransferase (MGMT) gene.[4] In some tumors, epigenetic silencing of the MGMT gene prevents the synthesis of this enzyme, and as a consequence such tumors are more sensitive to killing by temozolomide.[5] Conversely, the presence of AGT protein in brain tumors predicts poor response to temozolomide and these patients receive little benefit from chemotherapy with temozolomide.[6]
- Nitrosourea- and procarbazine-refractory anaplastic astrocytoma
- Newly diagnosed glioblastoma multiforme
- Malignant prolactinoma
Temozolomide (sometimes referred to as TMZ) is an imidazotetrazine derivative of the alkylating agent dacarbazine. It undergoes rapid chemical conversion in the systemic circulation at physiological pH to the active compound, 3-methyl-(triazen-1-yl)imidazole-4-carboxamide (MTIC). Temozolomide exhibits schedule-dependent antineoplastic activity by interfering with DNA replication. Temozolomide has demonstrated activity against recurrent glioma. In a recent randomized trial, concomitant and adjuvant temozolomide chemotherapy with radiation significantly improves, from 12.1 months to 14.6 months, progression free survival and overall survival in glioblastoma multiforme patients.
Formulations
Temozolomide is available in the United States in 5 mg, 20 mg, 100 mg, 140 mg, 180 mg & 250 mg capsules. Now also available in an IV form for people who can not swallow capsules or who have insurance that does not cover oral cancer agents.
A generic version is available in the UK.
Further improvement of anticancer potency
Laboratory studies and clinical trials are investigating whether it might be possible to further increase the anticancer potency of temozolomide by combining it with other pharmacologic agents. For example, clinical trials have indicated that the addition of chloroquine might be beneficial for the treatment of glioma patients.[8] In laboratory studies, it was found that temozolomide killed brain tumor cells more efficiently when epigallocatechin gallate (EGCG), a component of green tea, was added; however, the efficacy of this effect has not yet been confirmed in brain tumor patients.[9]More recently, use of the novel oxygen diffusion-enhancing compound trans sodium crocetinate (TSC) when combined with temozolomide and radiation therapy has been investigated in preclinical studies [10] and a clinical trial is currently underway.[11]
Because tumor cells that express the MGMT gene are more resistant to killing by temozolomide, it was investigated[according to whom?] whether the inclusion of [[O6-benzylguanine]] (O6-BG), an AGT inhibitor, would be able to overcome this resistance and improve the drug’s therapeutic effectiveness. In the laboratory, this combination indeed showed increased temozolomide activity in tumor cell culture in vitro and in animal models in vivo.[12] However, a recently completed phase-II clinical trial with brain tumor patients yielded mixed outcomes; while there was some improved therapeutic activity when O6-BG and temozolomide were given to patients with temozolomide-resistant anaplastic glioma, there seemed to be no significant restoration of temozolomide sensitivity in patients with temozolomide-resistant glioblastoma multiforme.[13]
There are also efforts to engineer hematopoietic stem cells expressing the MGMT gene prior to transplanting them into brain tumor patients. This would allow for the patients to receive stronger doses of temozolomide, since the patient’s hematopoietic cells would be resistant to the drug.[14]
High doses of temozolomide in high grade gliomas have low toxicity, but the results are comparable to the standard doses.[15]
A case report suggests that temozolomide may be of use in relapsed primary CNS lymphoma.[16] Confirmation of this possible use seems indicated.
Temozolomide, 3-methyl-8-aminocarbonyl-imidazo[5,1-d]- 1 ,2,3,5-tetrazin- 4(3H)-one, is a known antitumor drug; see for example Stevens et al., J. Med. Chem. 1984, 27, 196-201 , and Wang et al., J. Chem. Soc, Chem. Commυn., 1994, 1687-1688. Temozolomide, the compound of formula 1 :

1 is described in U.S. Patent No. 5,260,291 (Lunt et al.).
The synthesis of 1 by the process described in J. Med. Chem. 1984, 27, 196- 201 is depicted in the scheme I below. Scheme I:

In this process, 5-amino-1 H-imidazole-4-carboxamide (A) is converted into 5- diazo-1 H-imidazole-4-carboxamide (B), which is then cyclized with methylisocyanate in dichloromethane to provide a high yield of temozolomide.
However, this process requires isolation of the unstable and potentially dangerous 5-diazo-1 H-imidazole-4-carboxamide (B). Moreover, methylisocyanate is a difficult reagent to handle and ship, especially on the industrial scale, and indeed is better avoided in industrial manufacture.
Furthermore, the cycloaddition of methylisocyanate requires a very long reaction time: Table I in J. Med Chem. 1984, 27,196-201 , suggests 20 days. Additionally, Stevens et al mention that the cycloaddition of the methylisocyanate to the compound of the formula (B) can proceed through two different intermediates:
The production of I by the two processes described in J. Chem. Soc, Chem.
Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1 H- imidazole-4-carboxamide (A): less than 20% (unoptimized – about 17% through 5- diazo-1 H-imidazole-4-carboxamide (B) and about 15% through 5-amino-N1– (ethoxycarbonylmethyl)- 1 H-imidazole- 1 ,4-dicarboxamide (C)); Scheme II below
Scheme II:

Moreover, the unstable 5-diazo-1 H-imidazole-4-carboxamide (B) still has to be isolated in the branch of this process that uses it as an intermediate. Clearly, therefore, there is a need for synthetic methods that: a) are more convenient and higher yielding, especially on commercial scale; b) approach the synthesis of the temozolomide nucleus in novel ways; or c) improve the preparation or use of intermediates for the processes.
Temozolomide of formula I, is an antitumor drag and is chemically known as 3-methyl-8- aminocarbonyl-imidazole[5,l-d]-l,2,3,5-tetrazin-4(3H)-one.

Formula I
It is indicated for treating patients with malignant glioma such as cancer, breast cancer, refractory anaplastic, astrocytoma, i.e. patient at first relapse who have experienced disease progression in malignant glioma, glioblastoma multiform and anaplastic astrocytoma, on a drug containing a nitrosourea and procarbazine. It is sold in the US market as hard capsules containing 5 mg, 20 mg, 100 mg or 250 mg as Temodar® by Schering corporation.
Temozolomide and compounds having similar activity (higher alkyl analogues at the 3 -position) were first disclosed in US patent 5,260,291. According to said patent, temozolomide is prepared by the reaction of 5-diazoimidazole-4-carboxamide with methyl isocyanate in the presence of N- methylpyrrolid-2-one in dichloromethane at room temperature for three to four weeks. Melting point of temozolomide reported in above patent is 200 0C (recrystallized from acetonitrile); 21O0C with effervescence (recrystallized from acetone and water), and 2150C with effervescence and darkening (recrystallized from hot water). Major drawback of process is the longer reaction duration of three to four weeks for completion of reaction.
Further, the process described in the patent involves use of low boiling and extremely toxic, methyl isocyanate, which is very difficult to handle, especially on industrial scale, as its use should be avoided in the industrial synthesis. Further, cycloaddition reaction requires a very long period of 21 to 28 days, which makes the process unattractive for industrial scale.
US patent 5,003,099 discloses a process for preparation of aminocyanoacetamide, a key intermediate for the synthesis of temozolomide. According to the patent, aminocyanoacetamide is synthesized in two steps by the reaction of cyanoacetic acid alkyl ester using sodium nitrite in the presence of glacial acetic acid to form a hydroxyimino intermediate, which is then reduced in the presence of platinum on carbon to yield aminocyanoacetic acid alkyl ester, which is unstable.
The alkyl ester intermediate is then in situ reacted with aqueous ammonia to give the desired product. The main drawback of the above mentioned process is the use of aqueous ammonia, since aminocyanoacetamide, generated in reaction, is soluble in aqueous solution and hence difficult to extract from the reaction mass which results in lower yields. The patent is silent about the purity of intermediate and process needs extraction of the above mentioned intermediate from filtrate.
US patent 6,844,434 describes synthesis of temozolomide by cyclization of 5-amino-l-(N-rnethyl- hydrazinocarbonyl)-lH-imidazole-4-carboxylic acid in the presence of tetrabutyl nickel and periodic acid to form a reaction mixture which is concentrated under reduce pressure and resulting residue was treated with acetonitrile and filtered. The filtrate was concentrated and chromatographed on a column of silica gel to give temozolomide.
Use of time consuming and cumbersome technique i.e. column chromatography for isolation of product makes the process not suitable to employ at industrial level. US patent 7,087,751 discloses a process for the preparation of temozolomide from protected imidazole intermediate.
The process involves reaction of l-methyl-3-carbamoyliminomethyl-urea with JV- protected aminocyanoacetamide in the presence of acetic acid in a suitable solvent to form an JV- protected imidazole intermediate which is then cyclized in the presence of lithium chloride to minimize undesired cyclisation product. After cyclisation, the protected group has to be removed which makes the process more laborious with more number of steps.
As exemplified in example 1 of the above patent, yield of the JV-protected imidazole intermediate obtained is very low, almost half of the product goes in the filtrate which further needs extraction from the filtrate. After extraction of inteπnediate from the filtrate, the combined yield is only 67 %. The intermediate obtained is only 93 to 94% pure and requires additional purifications, crystallization using ethyl acetate and slurry wash with mixture of methyl tertiary butyl ether and isopropanol. These additional purification further takes away around 20 % yield of the inteπnediate thus yield of the pure intermediate, which is suitable for the further reaction, remains around 53 % which is very low from commercial point of view.
The patent also describes condensation of l-methyl-3-carbamoyliminomethyl-urea with unprotected aminocyanoacetamide in presence of acetic acid to give an imidazole intermediate. This patent fails to disclose the process of conversion of above imidazole intermediate to temozolomide, but only up to hydrolysis to prepare 5-amino-lH-imidazole-4-carboxamide hydrochloride is reported.
Another US patent no. 6,844,434 of same applicant (Schering) discloses a process for the conversion of 5-amino- lH-imidazole-4-carboxamide hydrochloride, which is prepared by the hydrolysis of above imidazole intermediate, to temozolomide. By combining the above two processes, this adds further four additional steps to the synthesis of temozolomide. The process of preparation of temozolomide is described by the following scheme:

It has been observed that for the preparation of unprotected imidazole intermediate as exemplified in US 7,087,751, use of excess amount of the acetic acid (around 21 times with respect to aminocyanoacetamide) is reported. Thereafter acetic acid is removed by distillation.
The inventors of the present invention have repeated example 2 as described in US 7,087,751 for the preparation of unprotected imidazole intermediate. As per the process, after the completion of the reaction, acetic acid has to be removed from the reaction mixture. It is noticed that removal of acetic acid is a very tedious move so as on commercial scale and leads to decomposition.
In a publication namely, Journal of Organic Chemistry, volume 62, no. 21, 7288-7294, a process is disclosed for the preparation of temozolomide by the hydrolysis of 8-cyano-3-methyl-[3H]-imidazole~ [5,l-d]-tetrazin-4-one in the presence of hydrochloric acid to give hydrochloride salt of temozolomide, which has to be neutralized to obtain temozolomide. In the same Journal, another process for the preparation of temozolomide is also described. Temozolomide is prepared by the nitrosative cyclization of imidazole intermediate using aqueous solution of sodium nitrite and tartaric acid to give temozolomide in 45 % yield in solution.
US patent publication 2007/0225496 exemplified a process for preparation of temozolomide by pyrolising N’-methyl-N,N-diphenyl urea to form vapor of methyl isocyanate which is then reacted with 5-diazo-5H-imidazole-4-carboxylic acid amide to form temozolomide.
The above described process involves use of methyl isocyanate, which is highly flammable and makes the process unsuitable for industrial synthesis, hi addition to this, isolation of temozolomide from the reaction mixture requires addition of large amount of ethyl acetate followed by addition of hexane and again ethyl acetate to isolate compound.
US patent publication 2009/0326028 describes a process for preparation of temozolomide by diazotization of imidazole intermediate in the presence of at least one metal halide, a source of nitrous acid and an acid to form acidic solution of temozolomide, wherein temozolomide forms a salt with acid. The desired product i.e. temozolomide is then isolated from the acidic solution by extraction with a solvent.
The process requires very strict reaction parameters including the addition of metal halide during diazotization as well as addition of pre-cooled reaction mixture to sodium nitrite solution to achieve desired level of selective cyclization. Patent application also describes two methods for the extraction of temozolomide.
US patent publication 2010/0036121 discloses a process for the preparation of temozolomide by reaction of 5-aminoimidazole-4-carboxamide with N-succinimidyl-N’-methylcarbamate to form carbamoyl 5~aminoimidazole-4-carboxamide which is then reacted with alkali or alkaline earth nitrile to give reaction mass containing temozolomide
-
Temozolomide, is a known antitumour drug, and is represented by formula I:

3-methyl-8-aminocarbonyl-imidazo [5,1-d]-1,2,3,5-tetrazin-4(3H)-one
-
It is described in US 5,260,291 together with compounds of broadly similar activity such as higher alkyl analogs at the 3-position.
-
J.Med.Chem. 1984, 27, 196-201 describes a process wherein 5-amino-1H-imidazole-4-carboxamide is converted into 5-diazo-1H-imidazole-4-carboxamide, which is then cyclised with methylisocyanate in dichloromethane to provide a high yield of temozolomide.
-
This process requires isolation of the unstable and potentially dangerous 5-diazo-1H-imidazole-4-carboxamide, methyl isocyanate is a difficult reagent to handle and ship, especially on the industrial scale. Furthermore, the cycloaddition of methylisocyanate requires a long reaction time (Table I in J.Med.Chem. 1984, 27, 196-201, suggests 20 days).
-
The product obtained by this process contains, high residual dichloromethane. It is essential to limit dichloromethane content in the final API below 600 ppm as per ICH guideline. Dichloromethane content can be reduced if one follows technique of US 5,260,291 .
-
US 5,260,291 discloses acetone-water recrystallisation of temozolomide, which results in low yield (60% recovery) due to decomposition of temozolomide to impurities like 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide, compound of formula V

and 5-amino-1H-imidazole-4-carboxamide.
-
The production of compound of formula I by the two processes described in J.Chem.Soc., Chem.Commun., 1994, 1687-1688 provides a low overall yield from 5-amino-1H-imidazole-4-carboxamide: less than 20% (about 17% through 5-diazo-1H-imidazole-4-carboxamide and about 15% through 5-amino-N1-(ethoxy carbonylmethyl)-1H-imidazole-1,4-dicarboxamide).
-
The unstable 5-diazo-1H-imidazole-4-carboxamide has to be isolated in the branch of this process that uses it as an intermediate.
-
US 2002/0133006 discloses a process for the preparation of compound of formula I using methyl hydrazine which is a toxic and flammable liquid, hence not feasible on industrial scale and the final isolation involves tedious workup including column chromatography.
-
J.Org.Chem. 1997, 62, 7288-7294 describes a process wherein the final step of diazotization provides equi-formation of aza-hypoxanthine and temozolomide, resulting in low yield. This literature does not provide the experimental procedure for work up.
-
US 2005/0131227 describes a process involving the use of a bulky protecting group on nitrogen of the primary amide for cyclisation in presence of LiCl to minimize the undesired cyclization product. After cyclization the protecting group has to be removed which makes the process more laborious with more number of steps (Scheme I).

U.S. Pat. No. 6,844,434 describes the preparation of Temozolomide, alkyl analogs and intermediates thereof. The process, which is depicted in Scheme 3 below, comprises reacting 5-amino-1H-imidazole-4-carboxamide hydrochloride (II) with 4-nitrophenyl chloroformate to afford compound (III), which is subsequently reacted with methyl hydrazine to obtain the corresponding compound (IV), which is cyclized to yield Temozolomide.

Another process for preparing Temozolomide is described in U.S. patent application having the Publication No. 2002/0095036 (see Scheme 4 below). In this process, the imine (V) is converted to 2-cyano-N-(1,1-dimethylethyl)-2-[(diphenyl-methylene)amino]-acetamide, which is converted to 2-amino-2-cyano-N-(1,1-dimethyl-ethyl)-acetamide hydrochloride.
The latter is reacted with compound (VI) to obtain 5-amino-N4-(1,1-dimethylethyl)-N1-methyl-1H-imidazole-1,4-dicarboxamide, which is converted to 3,4-dihydro-N-(1,1-dimethylethyl)-3-methyl-imidazo-[5,1-d]-1,2,3,5-tetrazine-8-carboxamide (tert-butyl-Temozolomide), which yields Temozolomide under acidic treatment with concentrated sulfuric acid.

Yet another synthesis of Temozolomide is described by Stevens et al. in J. Org. Chem., Vol. 62, No. 21, 7288-7294, 1997, wherein Temozolomide hydrochloride salt is obtained in 65% yield by the hydrolysis of 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one with hydrochloric acid, as shown in Scheme 5.

The main disadvantage of this process is the low yield in which Temozolomide hydrochloride is obtained (65%). It is assumed that the relatively elevated temperature of 60° C. used in the process increases the content of decomposition products.






…………………………
Synthesis
US Patent 8,232,392
Temozolomide (1) is a drug that was discovered more than 30 years ago. In the past 10 years, it has been used to treat aggressive brain tumors. S. Turchetta and co-inventors summarize several processes for preparing temozolomide, all of which use toxic reagents such as MeNCO or MeNHNH2or generate large amounts of chemical waste. They describe a safer route to 1.
The inventors’ method starts with the preparation of carbamoyl compound 4 from amide 2 by treating it with succinimidyl reagent 3 in the presence of a base. The product is isolated in 88% yield and 96.9% purity by HPLC. Reagent 3 is a nonexplosive, crystalline solid with comparatively low toxicity and is much safer than MeNCO for this reaction.

In the next stage, the amine group in 4 is converted to diazonium salt 5 via a diazotization reaction. The details of this reaction are not described, but reference is made to a method reported in 1997 (Wang, Y., et al. J. Org. Chem. 1997, 62, 7288–7294). Compound 5 is not isolated; when acid is added, it cyclizes by the reaction of the diazonium group with one of the two amide groups to give products 1 and 6 in approximately equal amounts. The desired product 1 is formed by the reaction of the secondary amide group; when the primary amide reacts, the product is its isomer, 6.
Products 1 and 6 are separated by passing the acidified reaction mixture from the diazotization reaction over a column of a polymeric adsorbent resin. The material used in the example is XAD 1600 from Rohm & Haas; other resins are covered in the claims. Compound 6 elutes from the column first; then 1 is eluted with acidified aq EtOH. After separation, 1 is recrystallized from acidified acetone and isolated in 30% yield with 99.9% purity.
The process provides an alternative, safer route to temozolomide, but half of intermediate 4 is lost as unwanted product 6. [Chemi S.p.A. [Cinisello Balsamo, Italy]. US Patent 8,232,392, July 31, 2012; )
………………..
SYNTHESIS
http://www.google.com/patents/WO2002057268A1?cl=en

EXAMPLE 1
Preparation of Temozolomide (1 ) Step A Preparation compound (3)

5-Amino-1 H-imidazole-4-carboxamide*HCI (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2CI2 (0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2CI2was added dropwise.
The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H20 (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol). 1H NMR (400MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1 H), 7.74 (d, 2H), 7.08 (bs, 1 H), 6.95 (bs, 1 H), 6.52 (s, 2H). Step B Preparation of compound (2)

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry
1 -liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0°C, and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5.000-1 ) was added dropwise.
The reaction mixture was stirred vigorously for 1 hour at 0°C and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol). 1H NMR (400MHz, DMSO-d6, δ): 7.62 (s, 1 H), 6.85 (bs, 1 H), 6.75 (bs,1 H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188°C (dec).
Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41.
Found: C, 36.46; H, 4.99; N, 42.12.
Step C Preparation of Temozolomide (1 )

2 1 (Temozolomide)
Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1 -liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen.
The reaction mixture was heated at 60°C for 20 mm and then cooled to room temperature. H5lθ6 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1 ) (280 mg). 1H NMR (400MHz, DMSO-d6, δ): 8.80 (s, 1 H), 7.80 (bs, 1 H), 7.66 (bs, 1 H), 3.43 (s,3H).
………………
SYNTHESIS

…………………
SYNTHESIS
http://www.google.com/patents/WO2010140168A1?cl=en
Accordingly, the present invention provides an improved process for the preparation of temozolomide of formula I,

Formula I which proves to be efficient and industrially advantageous.
The process comprises the step of: a), condensing compound of formula II,

Formula II with compound of formula III,
CH3 H CH3 Formula III in the presence of an acid in an alcoholic solvent to form a compound of formula IV;

Formula IV b). isolating the compound of formula IV from the reaction mixture by filtration; c). diazotizing and cyclizing the compound of formula IV in the presence of source of nitrous acid and a suitable acid; d). isolating temozolomide therefrom; and e). optionally purifying temozolomide of formula I.
Accordingly, the present invention provides an improved process for the preparation of temozolomide of formula I, process comprises the steps of: a), diazotizing and cyclizing the compound of formula IV in the presence of a source of nitrous acid and a suitable acid; b). optionally, cooling the reaction mixture; c). isolating precipitate of temozolomide from the reaction mixture; and d). purifying temozolomide of formula I with a suitable solvent
REFERENCE EXAMPLE:
Preparation* of S-Aøiino-N’-methyl-lH-imidazole-ljΦdicarboxamide (US 7,087,751) 2-Amino-2-cyanoacetamide (10 g), l-methyl-3-methylcarbamoyliminomethyl urea (19 g) and acetic acid (120 ml) were stirred together at ambient temperature under the positive pressure of nitrogen for 2 hours. Excess acetic acid was removed under reduced pressure and methyl tertiary butyl ether (25 ml) was added to the concentrated reaction mass, cooled to obtained crude solid.
The mixture was stirred for 30 minutes and the precipitate was collected by vacuum filtration. The solid was dried under vacuum at 20-250C for 18 hours to obtain 13 g of title compound as grayish solid. The crude product was stirred with water (66 ml) for 1 hour at 20-250C, filtered, suck dried and dried under vacuum at2O0C for 18 hours to obtain 11.2 g of title compound as greyish solid.
EXAMPLES
Example 1: Preparation of hydroxylirainocyano acetic acid ethyl ester
To a suspension of ethyl cyanoacetate (1.0 Kg, 8.84 mol) and sodium nitrite (0.735 kg, 10.65 mol) in water (0.80 L), acetic acid (0.70 kg, 11.66 mol) was added at 0-50C over a period of one hour.
Temperature was slowly raised to 23-270C and the reaction mixture was stirred for one hour at that temperature. After the complete consumption of ethyl cyanoacetate (monitored by TLC/GC), the reaction mixture was extracted with ethyl acetate (5 x 1.5 L). The combined organic layer was successively washed with 10% sodium bicarbonate (2 x 1.25 L) and brine solution (1.25 L), dried over sodium sulfate and filtered through hyflow bed. Solvent was removed under reduced pressure at 40-
450C. The resulting solid was stirred with cyclohexane (3.0 L) for 30 minutes at 25-300C, filtered and dried at 40-450C under vacuum to afford 1.14 kg (91.2 %) of title compound having purity 99.82% by
HPLC.
Example 2: Preparation of aminocyanoacetic acid ethyl ester
To a solution hydroxyliminocyano acetic acid ethyl ester (1.14 Kg, 8.02 mol) in methanol (11.4 L) was added 5% platinum on carbon (91.2 g, 50 % wet) and the mixture was hydrogenated at hydrogen gas pressure of 6.2-6.4 kg/cm2 over a period of 12 hours and the completion of reaction was checked by
TLC. The reaction mixture was filtered under nitrogen atmosphere to recover the catalyst. The filtrate was used as such for the next stage.
Example 3: Preparation of amimøcyanoacetamide
The solution of aminocyanoacetic acid ethyl ester (as prepared above) in methanol was cooled to 0-5
0C and ammonia gas was purged into it approximately for 1 hour. After the completion of the reaction
(monitored by TLC), the reaction mass was concentrated to 2.5-3.0 L under reduced pressure at 40-
45°C, cooled to 0-50C and stirred for 1 hour. The precipitated solid was filtered, washed with chilled methanol (200 ml) and dried at 35-400C under vacuum for 6 hours to obtain 572 g of title compound.
The resulting product was added to methanol (4.57 L) and heated to reflux till the solution become clear. Activated charcoal (25g) was added to the reaction mixture and refluxed for 15 minutes. The solution was filtered through hyflow bed, the bed was washed with methanol (500 ml) and the filtrate was concentrated to half of its original volume (approx 2.0 L). The mixture was cooled to 0-50C and stirred for 45 minutes. The resulting solid was filtered, washed with chilled methanol (250 ml) and dried at 40-450C under vacuum to obtain 425g (53.6%) of pure title compound having purity 99.46% by HPLC. Example 4: Preparation of l-methyl-3-methylcarbamoyliminomethyl urea
A suspension of monomethyl urea (1.5 kg, 20.27 mol) in triethyl orthoformate (4.5 L, 30.40 mol) was heated to reflux at 150-1600C for 12 hours. The reaction mixture was cooled to 5-100C, and stirred for 1 hour to ensure complete precipitation, of the product. The resulting solid was filtered, washed with ethyl acetate (350ml) and dried under vacuum at 45-5O0C to yield 1.08 kg (67.9%) of title compound having purity 93.82% by HPLC.
Exainple-5: Preparation of S-amino-N^methyl-lH-imidazole-l^-dicarboxamide Acetic acid (200 ml, 3.53 mol) was added to a suspension of aminocyanoacetamide (40Og, 4.04 mol) and l-methyl-3-methylcarbamoyliminomethyl urea (76Og, 4.8 mol) in methanol (2.0 L) at 20-250C and the mixture was stirred at 20-250C for 18 hours till completion of the reaction (monitored by HPLC). The reaction mixture was cooled to 0-50C, stirred for 1 hour and the resulting solid was filtered, washed with chilled methanol (450 ml), suck dried and finally dried under vacuum at 30-350C to afford 648 g (88.04%) of title compound as an off white colored solid having purity 99.21 % by HPLC. Example 6: Preparation of temozolomide
Acetic acid (450 ml, 7.95 mol) was added to a suspension of S-amino-N^methyl-lH-imidazole-l^- dicarboxamide (500g, 2.73mol) and sodium nitrite (25Og, 3.62mol) in water (5.0 L) at -5 to 00C at such a rate so that temperature does not rise above 5°C. The reaction mixture was stirred at 0 to 5°C for one hour and absence of starting material was checked by HPLC analysis. Ice bath was removed and powdered calcium chloride (1.25Kg) was added in small lots to the reaction mass and stirred at 25- 300C for 2 hours. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 50 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflow bed. Solvent was removed under reduced pressure below 4O0C and residual dimethylsulfoxide layer was degassed completely. The dimethylsulfoxide layer was cooled to 0 to – 100C and stirred for 1 hour. The resulting solid was filtered, washed with ethyl acetate (25OmL), and suck dried for 2 hours to afford 32Og of the title compound having purity 78.5% by HPLC. Example 7: Preparation of temozolomide
Acetic acid (9ml, 0.159mol) was added to a suspension of 5-ammo-N1 -methyl- lH-imidazole- 1,4- dicarboxamide (1Og, 0.054mol) and sodium nitrite (5g, 0.072mol) in water (100ml) at -5 to 00C at a rate so that temperature does not rise above 0-50C. The reaction mixture was stirred at 0-50C for one and half hour. Brine (30g) was added to the reaction mixture and stirred at room temperature for two hours to saturate the reaction mixture. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 1 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflow bed. Solvent was removed under reduced pressure and residual dimethylsulfoxide layer was degassed completely. The dimethylsulfoxide layer was cooled to 0 to -5°C and stirred for 1 hour. The resulting solid was filtered, washed with ethyl acetate (2x 5 ml), and suck dried for 2 hours to afford 5.0 g of the title compound having purity 81.6% by HPLC. Example 8: Preparation of temozolomide
Acetic acid (450ml) was added to a suspension of 5 -amino-N1 -methyl- lH-imidazole- 1,4- dicarboxamide (500g) and sodium nitrite (25Og) in water (5.0 L) at -5 to O0C at a rate so that temperature does not rise above 0-50C. The reaction mixture was stirred at 0-50C for one and half hour and the absence of starting material was checked by HPLC analysis. Ice bath was removed and powdered calcium chloride (1.25 kg) was added to the reaction mixture and stirred at room temperature for two hours. The reaction mass was extracted with a 2.5% solution of dimethylsulfoxide in dichloromethane (5 X 50 L). Combined organic layer was dried over sodium sulfate and filtered through a hyflo bed. Solvent was removed under reduced pressure at below 400C and residue at 35- 400C was filtered through a candle filter to remove suspended particles and the filtrate was then degassed completely. The residual dimethylsulfoxide layer was cooled to 0±2°C and stirred for one hours. The resulting solid was filtered and sucked dried. The solid was then washed with ethyl acetate (2x 250 ml), and suck dried for 1 hours to afford 240 g of the title compound.
………………………………….
SYNTHESIS
http://www.google.com/patents/US20020133006
Example 1
Preparation of Temozolomide (1)

5-Amino-1H-imidazole-4-carboxamide.HCl (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2Cl2(0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2Cl2 was added dropwise. The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H2O (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol).
1H NMR (400 MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1H), 7.74 (d, 2H), 7.08 (bs, 1H), 6.95 (bs, 1H), 6.52 (s, 2H).

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0° C., and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5,000-1) was added dropwise. The reaction mixture was stirred vigorously for 1 hour at 0° C. and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol).
1H NMR (400 MHz, DMSO-d6, δ): 7.62 (s, 1H), 6.85 (bs, 1H), 6.75 (bs,1H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188° C. (dec.).
Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41.
Found: C, 36.46; H, 4.99; N, 42.12.

Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60° C. for 20 mm and then cooled to room temperature. H5I06 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg).
1H NMR (400 MHz, DMSO-d6, δ): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).
…………………….
EXAMPLES
Example 1:
- Preparation of 3-Methyl-8-aminocarbonyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (Temozolomide).
-
Glacial acetic acid (25 ml), water (250 ml) and LiCl (225 g) were charged and the contents were stirred for 30 minutes and cooled to room temperature. 5-Amino-1-(N-methylcarbamoyl) imidazole-4-carboxamide (II) (25 g) was added and stirred the contents for further 30 minutes. The reaction mixture was cooled to 0°C and then added drop wise to NaNO2 solution (12.5 g in 50 ml water) at -10 to 5 °C. The reaction mass was stirred for 1 hr at 0-5 °C and then at room temperature for 5 hrs. To this reaction mixture, sodium thiosulphate solution (25 g in 250 ml of water) was added slowly and stirred for 20 minutes (solution A). This process yielded an acidic solution containing temozolomide.
……………………..
SYNTHESIS
EXAMPLES Example 1
A 250 ml reaction vessel equipped with a magnetic stirrer and a reflux condenser was charged with 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one (10 grams, 0.0568 mol) and hydrochloric acid (36.5-38%, 50 ml). The reaction mixture was heated to 32-35° C. and stirring was maintained at this temperature for about 3 hours. A sample was withdrawn and analyzed by HPLC to verify that the high conversion was received. (If the content of the starting material 8-cyano-3-methyl-[3H]-imidazo-[5,1-d]-tetrazin-4-one is more than 2.5% by area according to HPLC, the stirring may be continued for additional one hour).
The reaction mixture was then cooled to 20° C. and 50 ml of acetone were added drop-wise while maintaining the temperature at 20° C. Stirring was continued for 15-30 minutes. The precipitated white crystals were washed with cold acetone (20 ml) and dried at 40° C. in vacuum to obtain 11.7 grams (0.0507 mol) of Temozolomide hydrochloride (89.3% yield). Purity (by HPLC): 99.6%.
…………………………
SYNTHESIS
EXAMPLES
The following Examples illustrate but do not in any way limit the present invention. Chemicals obtained from Aldrich Chemical Company (Milwaukee, Wis.) are identified by their catalog number. It should be noted that nomenclature may differ slightly between this specification and the Aldrich catalog.
Example 1 Preparation of Temozolomide (1)
Step A Preparation Compound (3)

5-Amino-1H-imidazole-4-carboxamide.HCl (4) (25 g, 0.154 mol) (Aldrich 16,496-8), CH2Cl2(0.6 L) and Et3N (45 mL) (Aldrich, 13,206-3) were placed into a dry 2-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen at ambient temperature. The mixture was stirred, and a solution of 400 mL of 4-nitrophenyl chloroformate (34 g, 0.169 mol) (Aldrich, 16,021-0) in CH2Cl2was added dropwise. The reaction mixture was stirred vigorously for 4 hours and then left to stand for 18 hours at room temperature. The precipitate was collected by vacuum filtration and washed with H2O (1.5 L) to afford the product (3) as a pale yellow solid (42 g, 0.144 mol).
1H NMR (400 MHz, DMSO-d6, δ): 8.40 (d, 2H), 7.83 (s, 1H), 7.74 (d, 2H), 7.08 (bs, 1H), 6.95 (bs, 1H), 6.52 (s, 2H).
Step B Preparation of Compound (2)

Compound (3) (42 g, 0.144 mol) and DMF (0.27 L) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was cooled to 0° C., and methylhydrazine (10 mL, 0.188 mol) (Aldrich, M5,000-1) was added dropwise. The reaction mixture was stirred vigorously for 1 hour at 0° C. and was then poured into EtOAc (2.1 L). The precipitate was collected by vacuum filtration and was dried under vacuum (20 mm Hg, room temperature, 18 hours) to afford (2) as a tan solid (27.1 g, 0.137 mol).
1H NMR (400 MHz, DMSO-d6, δ): 7.62 (s, 1H), 6.85 (bs, 1H), 6.75 (bs,1H), 6.00 (s, 2H), 5.10 (s, 2H), 3.15, s, 3H).mp: 188° C. (dec.). Analysis: Calcd for C6H10N6O2: C, 36.36; H, 5.09; N, 42.41. Found: C, 36.46; H, 4.99; N, 42.12.
Step C Preparation of Temozolomide (1)

Compound (2) (500 mg, 2.5 mmol), Bu4NI (95 mg, 0.25 mmol), THF (250 mL) and CH3CN (250 mL) were placed into a dry 1-liter, three-necked flask equipped with dropping funnel, a gas inlet tube, a gas outlet tube, reflux condenser and mechanical stirrer, and maintained under a positive pressure of nitrogen. The reaction mixture was heated at 60° C. for 20 mm and then cooled to room temperature. H5IO6 (1.14 g, 5 mmol) was added and the reaction mixture was stirred vigorously at room temperature for 1 hour. The resulting solution was treated with saturated aqueous Na2S2O3 (5 mL) and was then concentrated under reduced pressure to dryness. The residue was treated with CH3CN (200 mL) and was filtered. The filtrate was concentrated and chromatographed on a column of silica gel (1.5% to 2% AcOH/EtOAc) to afford temozolomide (1) (280 mg).
1H NMR (400 MHz, DMSO-d6, δ): 8.80 (s, 1H), 7.80 (bs, 1H), 7.66 (bs, 1H), 3.43 (s, 3H).

TEMOZOLOMIDE
References
- Malcolm Stevens – interview, Cancer Research UK impact & achievements page
- Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C (January 1997). “Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials”. Cancer Treat. Rev. 23 (1): 35–61. doi:10.1016/S0305-7372(97)90019-0. PMID 9189180.
- Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, Baig G, Goddard C, Gibson NW, Slack JA et al. (November 1987). “Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine”. Cancer Res. 47 (22): 5846–52.PMID 3664486.
- Jacinto, FV; Esteller, M (August 2007). “MGMT hypermethylation: a prognostic foe, a predictive friend.”. DNA Repair 6 (8): 1155–60. doi:10.1016/j.dnarep.2007.03.013. PMID 17482895.
- Hegi ME, R, Hau, Mirimanoff et al. (March 2005). “MGMT gene silencing and benefit from temozolomide in glioblastoma”. N. Engl. J. Med. 352 (10): 997–1003. doi:10.1056/NEJMoa043331.PMID 15758010. More than one of
|last1=and|author=specified (help) - National Cancer Institute Of Canada Clinical Trials, Group; Hegi, ME; Mason, WP; Van Den Bent, MJ; Taphoorn, MJ; Janzer, RC; Ludwin, SK; Allgeier, A et al. (May 2009). “Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial”. Lancet Oncology 10 (5): 459–466. doi:10.1016/S1470-2045(09)70025-7. PMID 19269895.
- Sitbon Sitruk, L.; Sanson, M.; Prades, M.; Lefebvre, G.; Schubert, B.; Poirot, C. (2010). “Chimiothérapie à gonadotoxicité inconnue et préservation de la fertilité : Exemple du témozolomide☆”.Gynécologie Obstétrique & Fertilité 38 (11): 660–662. doi:10.1016/j.gyobfe.2010.09.002. PMID 21030284. edit
- Gilbert MR (March 2006). “New treatments for malignant gliomas: careful evaluation and cautious optimism required”. Ann. Intern. Med. 144 (5): 371–3. PMID 16520480.
- Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS (October 2007). “The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas”.Cancer Res. 67 (20): 9809–16. doi:10.1158/0008-5472.CAN-07-0625. PMID 17942911.
- Sheehan J, Cifarelli C, Dassoulas K, Olson C, Rainey J, Han S (2010). “Trans-sodium crocetinate enhancing survival and glioma response on magnetic resonance imaging to radiation and temozolomide”. Journal of Neurosurgery 113 (2): 234–239. doi:10.3171/2009.11.JNS091314. PMID 20001586.
- “Safety and Efficacy Study of Trans Sodium Crocetinate (TSC) With Concomitant Radiation Therapy and Temozolomide in Newly Diagnosed Glioblastoma (GBM)”. ClinicalTrials.gov. November 2011.
- Ueno T, Ko SH, Grubbs E et al. (March 2006). “Modulation of chemotherapy resistance in regional therapy: a novel therapeutic approach to advanced extremity melanoma using intra-arterial temozolomide in combination with systemic O6-benzylguanine”. Mol. Cancer Ther. 5 (3): 732–8. doi:10.1158/1535-7163.MCT-05-0098. PMID 16546988.
- Friedman, HS; Jiang, SX; Reardon, DA; Desjardins, A; Vredenburgh, JJ; Rich, JN; Gururangan, S; Friedman, AH et al. (March 2009). “Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma”. J. Clin. Oncol. 27 (8): 1262–7. doi:10.1200/JCO.2008.18.8417. PMC 2667825. PMID 19204199.
- http://labs.fhcrc.org/kiem/Hans-Peter_Kiem.html
- Dall’oglio S, D’Amico A, Pioli F, Gabbani M, Pasini F, Passarin MG, Talacchi A, Turazzi S, Maluta S (December 2008). “Dose-intensity temozolomide after concurrent chemoradiotherapy in operated high-grade gliomas”. J Neurooncol 90 (3): 315–9. doi:10.1007/s11060-008-9663-9. PMID 18688571.
- Osmani AH, Masood N; Masood (2012). “Temozolomide for relapsed primary CNS lymphoma”. J Coll Physicians Surg Pak 22 (9): 594–595. PMID 22980617.
- Chemotherapy Drug Shrinks Brain Tumors American Academy of Neurology, May 21, 2007
- Information for people undergoing treatment with temozolomide Cancer Research UK (CancerHelp UK)
Wang, et al., “Alternative Syntheses of the antitumor drug temozolomide avoiding the use of methyl isocyanates”, Journal of Chemical Society, Chemical Communication, Chemical Society, Letchworth, GB, p. 1687-1688 (1994).
Wang, et al., “Antitumor imidazotetrazines. Part 33. new syntheses of the antitumor drug temozolomide using ‘masked’ methyl isocyanates”, J. Chem. Soc., Perkin Trans. 1(21):2783-2787 (1995).
Wang, et al., “Synthetic studies of 8-carbamoylimidzo-‘5, 1-D!-1, 2, 3, 5-tetrazi n-4(3H)- one: a key derivative of antitumor drug temozolomide”, Bioorg. Med Chem. Lett., 6(2):185-188 (1996).
Yongfeng Wang, “A new route to the antitumor drug temozolomide, but not thiotemozolomide”, Chem. Commun., 4:363-364 (1997).
Wang, et al., “Antitumor Imidazotetrazines. 35. New Synthetic Routes to the Antitumor Drug Temozolomide”, J. org. Chem. 62(21):7228-7294 (1997).
Newlands, E.S., et al., “Temozolomide: a review of its discovery, chemical properties, pre-clinica development and clinical trials”, Cancer Treat. Rev. , 23(1):35-61 (1997).
Wang, et al., Antitumor Imidazotetrazines. Part 36. Conversion of 5-Amino-Imidazole-4-Carboxamide to . . . Journal of the Chemical Society, Perkin Transactions 1, Chemical Society, Letchworth, GB, 10:1669-1675 (1998).
1 Catapano CV, et al. Cancer Res. 1987, 47(18), 4884-4889.
[2] Sun S, et al. J Neurooncol. 2012.
[3] Bauer M, et al. PLoS One. 2012, 7(6):e39956.
[4] Wong ST, et al. Anticancer Res. 2012, 32(7), 2835-2841.
[5] Lin CJ, et al. PLoS One. 2012, 7(6), e38706.
[6] Gori JL, et al. Cancer Gene Ther. 2012.
| US5260291 | Oct 18, 1991 | Nov 9, 1993 | Cancer Research Campaign Technology Limited | Tetrazine derivatives |
| US20020133006 | Jan 16, 2002 | Sep 19, 2002 | Schering Corporation | Synthesis of temozolomide and analogs |
| US20050131227 | Jan 21, 2005 | Jun 16, 2005 | Schering Corporation | Synthesis of temozolomide and analogs |
| US20060183898 * | Feb 16, 2006 | Aug 17, 2006 | Olga Etlin | Process for preparing temozolomide |
| CN1487941A * | Jan 16, 2002 | Apr 7, 2004 | 先灵公司 | Synthesis of temozolomide and analogs |
| CN1706843A * | Apr 8, 2005 | Dec 14, 2005 | 江苏天士力帝益药业有限公司 | Temozolomide refining process |
| US20060183898 * | Feb 16, 2006 | Aug 17, 2006 | Olga Etlin | Process for preparing temozolomide |
| US20070225496 * | Mar 23, 2007 | Sep 27, 2007 | Palle Raghavendracharyulu Venk | rocess for preparing temozolomide |
| US8258294 * | Sep 28, 2007 | Sep 4, 2012 | Cipla Limited | Process for the preparation of temozolomide and analogs |
| EP2151442A2 | Jul 22, 2009 | Feb 10, 2010 | Chemi SPA | Process for preparing temozolomide |
| EP2374807A2 * | Sep 28, 2007 | Oct 12, 2011 | Cipla Limited | An improved process for the isolation of temozolomide |
| WO2008038031A1 | Sep 28, 2007 | Apr 3, 2008 | Cipla Ltd | An improved process for the preparation of temozolomide and analogs |
| WO2010140168A1 * | Jun 2, 2010 | Dec 9, 2010 | Ind-Swift Laboratories Limited | Improved process for preparing temozolomide |
| WO2011036676A2 | Sep 14, 2010 | Mar 31, 2011 | Ashwini Nangia | Stable cocrystals of temozolomide |
AVANAFIL …..A PDE5 inhibitor.

AVANAFIL
A phosphodiesterase (PDE5) inhibitor, used to treat erectile dysfunction.

Avanafil is a new phosphodiesterase-5 inhibitor that is faster acting and more selective than other drugs belonging to the same class. Chemically, it is a derivative of pyrimidine and is only available as the S-enantiomer. FDA approved on April 27, 2012.
CAS RN: 330784-47-9
4-{[(3-chloro-4-methoxyphenyl)methyl]amino}-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide
| (S)-2-(2-Hydroxymethyl-1-pyrrolidinyl)-4-(3-chloro-4-methoxybenzylamino)-5-[(2-pyrimidinylmethyl)carbamoyl]pyrimidine |
| 4-[[(3-Chloro-4-methoxyphenyl)methyl]amino]-2-[(2S)-2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide |
| TA 1790 |
Molecular Formular: C23H26ClN7O3
Molecular Mass: 483.95064
- Stendra
- TA 1790
- TA-1790
- UNII-DR5S136IVO
- NDA 202276
INNOVATOR — VIVUS
APPROVED FDA 27/4/2-12
| Patent No | Patent Expiry | patent use code |
|---|---|---|
| 6656935 | Sep 13, 2020 | U-155 |
| 7501409 | May 5, 2023 |
U 155… TREATMENT OF ERECTILE DYSFUNCTION
| Exclusivity Code | Exclusivity_Date |
|---|---|
| NCE | Apr 27, 2017 |
Stendra (avanafil) was given the green light by the US Food and Drug Administration 27/4/2012, but there has been no launch yet as Vivus has been seeking a partner. The latest data should be attractive to potential suitors and could help Stendra take on other phosphodiesterase type 5 (PDE5) inhibitors, notably Pfizer’s Viagra (sildenafil) but also Eli Lilly’s Cialis (tadalafil) and Bayer’s Levitra (vardenafil).
read all at
http://www.pharmatimes.com/Article/13-06-20/Vivus_ED_drug_gets_to_work_in_less_than_15_mins.aspx
STENDRA (avanafil) is a selective inhibitor of cGMP-specific PDE5.
。

Avanafil is designated chemically as (S)-4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2pyrimidinylmethyl)-5-pyrimidinecarboxamide and has the following structural formula:
 |
Avanafil occurs as white crystalline powder, molecular formula C23H26ClN7O3 and molecular weight of 483.95 and is slightly soluble in ethanol, practically insoluble in water, soluble in 0.1 mol/L hydrochloric acid. STENDRA, for oral administration, is supplied as oval, pale yellow tablets containing 50 mg, 100 mg, or 200 mg avanafil debossed with dosage strengths. In addition to the active ingredient, avanafil, each tablet contains the following inactive ingredients: mannitol, fumaric acid, hydroxypropylcellulose, low substituted hydroxypropylcellulose, calcium carbonate, magnesium stearate, and ferric oxide yellow.
AVANAFIL

Avanafil is a PDE5 inhibitor approved for erectile dysfunction by FDA on April 27, 2012 [1] and by EMA on June 21, 2013.[2] Avanafil is known by the trademark names Stendra and Spedra and was developed by Vivus Inc. In July 2013 Vivus announced partnership with Menarini Group, which will commercialise and promote Spedra in over 40 European countries plus Australia and New Zealand.[3] Avanafil acts by inhibiting a specificphosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors. It is absorbed quickly, reaching a maximum concentration in about 30–45 minutes.[4] About two-thirds of the participants were able to engage in sexual activity within 15 minutes.[4]
Avanafil is a highly selective PDE5 inhibitor that is a competitive antagonist of cyclic guanosine monophosphate. Specifically, avanafil has a high ratio of inhibiting PDE5 as compared with other PDE subtypes allowing for the drug to be used for ED while minimizing adverse effects. Absorption occurs quickly following oral administration with a median Tmax of 30 to 45 minutes and a terminal elimination half-life of 5 hours. Additionally, it is predominantly metabolized by cytochrome P450 3A4. As such, avanafil should not be co-administered with strong cytochrome P450 3A4 inhibitors. Dosage adjustments are not warranted based on renal function, hepatic function, age or gender. Five clinical trials suggest that avanafil 100 and 200 mg doses are effective in improving the Sexual Encounter Profile and the Erectile Function Domain scores among men as part of the International Index of Erectile Function. A network meta-analysis comparing the PDE5 inhibitors revealed avanafil was less effective on Global Assessment Questionnaire question 1 while safety data indicated no major differences among the different PDE5 inhibitors. The most common adverse effects reported from the clinical trials associated with avanafil were headache, flushing, nasal congestion, nasopharyngitis, sinusitis, and dyspepsia.
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
…………………………………
DESCRIPTION IN A PATENT
EXAMPLE 92-145
The corresponding starting compounds are treated in a similar manner to give the compounds as listed in the following Table 7.
| TABLE 7 |
 |
 |
 |
Amorphous MS(m/z): 484(MH+) |
ENTRY 98 IS AVANAFIL
…………………………………………………….
The invention discloses a preparation method of Avanafil (Avanafil, I), which comprises the following steps: carrying out a substitution reaction on 6-amino-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (XII) and 3-chloro-4-methoxy benzyl chloride (XIII) so as to obtain 6-(3-chloro-4-methoxy benzyl amino)-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (IXV); carrying out condensation on the compound (IXV) and S-hydroxymethyl pyrrolidine (II) so as to generate 4-[(3-chloro-4-methoxy benzyl) amino]-2-[2-(hydroxymethyl)-1-pyrrole alkyl] pyrimidine-5-carboxylic acid ethyl ester (XI); and carrying out hydrolysis on the compound (XI) and then carrying out an acylation reaction on the compound (XI) and the compound (XI) so as to obtain Avanafil (I). The preparation method is simple in process, economic and environmental-friendly, suitable for the requirements of industrialization amplification.
……………………………………………………
The invention discloses a method for preparing avanafil (Avanafil, I). The method comprises the steps of taking cytosine as an initial material; and orderly carrying out replacement, halogen addition and condensation reaction on a side chain 3-chlorine-4-methoxy benzyl halide (III), N-(2-methylpyrimidine) formamide (IV) and S-hydroxymethyl pyrrolidine (II), so as to obtain a target product avanafil (I). The preparation method is available in material, concise in technology, economic and environment-friendly, and suitable for the demands of industrial amplification.
…………………………………………………….
SYNTHESIS
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative REF 5:Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709

- ………………………………………………………
- SYNTHESIS
- A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 – and 4 – positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 – position, but does not guarantee the 2 – side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 – bit, and a good leaving group active chlorine occupy four – position, thus ensuring a high regioselectivity of the reaction. 4 – position after completion of the reaction, then the 2 – position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.
AVANAFIL
- …………………………….

-
- FDA approves Stendra for erectile dysfunction” (Press release). Food and Drug Administration (FDA). April 27, 2012.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002581/human_med_001661.jsp&mid=WC0b01ac058001d124
- http://ir.vivus.com/releasedetail.cfm?releaseid=775706
- Kyle, Jeffery; Brown, Dana (2013). “Avanafil for Erectile Dysfunction”. Annals of Pharmacotherapy (Sage Publishing). doi:10.1177/1060028013501989. Retrieved 28 September 2013.
- Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
-
- Peterson CA. Hemodynamic effect of avanafil and glyceryl trinitrate coadministration. , Drugs Context , Volume 2013 , 2013 Feb 26
- Gur S. The Effect of Intracavernosal Avanafil, a Newer Phosphodiesterase-5 Inhibitor, on Neonatal Type 2 Diabetic Rats With Erectile Dysfunction. , Urology , 2013 Dec 9
- Hill JK. Avanafil for erectile dysfunction. , Ann Pharmacother , Volume 47 , Issue 10 , 2013 Oct
- Sanford M. Avanafil: a review of its use in patients with erectile dysfunction. , Drugs Aging , Volume 30 , Issue 10 , 2013 Oct
- Hellstrom WJ. PDE5 inhibitors: considerations for preference and long-term adherence. , Int J Clin Pract , Volume 67 , Issue 8 , 2013 Aug
- Aversa A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. , Expert Opin Pharmacother , Volume 14 , Issue 10 , 2013 Jul
- Oelke M. Phosphodiesterase inhibitors in clinical urology. , Expert Rev Clin Pharmacol , Volume 6 , Issue 3 , 2013 May
- Kukreja RC. Sildenafil and cardioprotection. , Curr Pharm Des , Volume 19 , Issue 39 , 2013
- Day WW. An open-label, long-term evaluation of the safety, efficacy and tolerability of avanafil in male patients with mild to severe erectile dysfunction. , Int J Clin Pract, Volume 67 , Issue 4 , 2013 Apr
- Tang J. Comparative effectiveness and safety of oral phosphodiesterase type 5 inhibitors for erectile dysfunction: a systematic review and network meta-analysis. , Eur Urol , Volume 63 , Issue 5 , 2013 May
United States APPROVED 6656935 2012-04-27 EXPIRY 2020-09-13 United States 7501409 2012-04-27 2023-05-05 - Faster-Working Erectile Dysfunction Drug?. CBS News. November 24, 2009.
- Vivus says men taking avanafil were more likely to be ready for sex within 15 minutes. The Gaea Times. January 11, 2010.
- “Avanafil is the New Player in The Erectile Dysfunction Field”. June 28, 2011.
-
- • Hatzimouratidis, K., et al.: Drugs, 68, 231 (2008)
-
4-20-2011Tablets quickly disintegrated in oral cavity7-16-2010Combination treatment for diabetes mellitus8-28-2009Roflumilast for the Treatment of Pulmonary Hypertension1-32-2008Cyclic compounds
US5242391 Oct 30, 1991 Sep 7, 1993 ALZA Corporation Urethral insert for treatment of erectile dysfunction US5474535 Jul 19, 1993 Dec 12, 1995 Vivus, Inc. Dosage and inserter for treatment of erectile dysfunction US5773020 Oct 28, 1997 Jun 30, 1998 Vivus, Inc. Treatment of erectile dysfunction US6656935 Aug 10, 2001 Dec 2, 2003 Tanabe Seiyaku Co., Ltd. Aromatic nitrogen-containing 6-membered cyclic compounds Update nov 2015
NEW PATENT WO 2015177807

WO 2015177807
Suryakant Shivaji Pol; Nitin Sharadchandra Pradhan; Shashikant Balu Padwal; Vihar Raghunath Telange; Nitn Shankar Bondre
Wanbury ltd

The present invention relates to a novel compound of Formula (II), and its use in preparation of Avanafil, [Formula should be inserted here] wherein R is -OH, -CI or -OR1 and R1 is C1 to C3 alkyl group
It having been developed and launched by VIVUS and JW Pharmaceutical, under license from Mitsubishi Tanabe Pharma, and Auxilium Pharmaceuticals, for treating ED.
A process for preparation of Avanafil was first disclosed in US 6,797,709 (depicted in Scheme I), wherein 4-chloro-5-ethoxycarbonyl-2-methylthio-pyrimidine is coupled with 3-chloro-4-methoxybenzylamine in presence of triethylamine to provide compound of Formula (A), which on oxidization provides a sulfonyl compound of Formula (B). Said compound of Formula (B) is reacted with L-prolinol and exert compound of Formula (C). The resulting compound of Formula (C) undergoes column chromatographic purification and crystallization, while further subjected to hydrolysis to obtain compound of Formula (D). The compound of Formula (D) is coupled with 2-aminomethylpyrimidine to obtain Avanafil of Formula (I). The final product obtained is purified by column chromatography. The need to purify the intermediate compound of Formula (C) and final product, by column chromatography makes this process cumbersome, time consuming and unviable for large scale production thereby contributing to main disadvantages of the process.
Scheme I

Formula (A)m-CPBA/chloroform

Formula (C) Formula (B)
NaOH/DMSO

Formula (D) Formula (I)CN 103254179, discloses a process for preparation of Avanafi, wherein 3-chloro-4-methoxybenzylhalide is coupled with cytosine to result compound of Formula (E), later on condensation with L-prolinol yields 4-[(3-chloro-4-methoxy benzyl)amino-2-(2-hydroxymethyl)-l -pyrrolinyl]pyrimidine of Formula (F). The compound of Formula (F) is then condensed with N-(2-pyrimidylmethyl)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme II
Scheme II

Formula (F) Formula (I)
CN 103254180 describes an alternate process for preparation of Avanafil of Formula (I), wherein a substitution reaction on 6-amino-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester and 3-chloro-4-methoxybenzylchloride provides 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester of Formula (G) which on condensation with L-prolinoI generates 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid ethyl ester of Formula (H). The compound of Formula (H) is then hydrolysed and coupled with N-(2-pyrimidylmethyI)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme III
Scheme III

Formula (H) Formula (Γ)
In all the prior art discussed above, chiral compound L-prolinol is coupled in molecule in earlier steps of synthesis. This approach seems to be less feasible for large scale production; the insertion of L-prolinol in early stage may need to exert number of purifications for intermediates. Further the main shortcoming in such process is that the chirality of molecule is disturbed by inserting L-prolinol in early stages because there are number of operations in line in process to obtain the target compound.
CN 103483323, discloses a synthetic method for preparation of avanafil, wherein amidation of pyrimidine-5-carbonyl chlorides with 2-(aminomethyl)pyrimidine at temperature ranging from -10 to 5°C resulted an amide (intermediates A); which underwent condensation with 3-chloro-4-methoxybenzylamine at the temperature ranging from 0 -3°C to give 4-[(3-chloro-4-methoxybenzyl)amino]-5-
pyrimidinecarboxamides (intermediates B), which further on condensation with L-prolinol gave avanafil. The disadvantage of this process is the need to maintain the reaction temperature in range of – 10 to 5°C which adds up to cost of process and makes the process complicated. The process is depicted in Scheme IV.
Scheme IV

Intermediate (A)

wherein, R’ & R2 are independently, hydrogen, halogen, alkoxy, alkoxyalkyl, cyno group, amino group
Hence, to overcome shortcomings of prior art the inventors of present invention have skillfully designed a process with novel intermediate which concomitantly result Avanafil compound of Formula (I), substantially free from impurities. Further this invention encompass L-proline in last stage of molecule in order to avoid the number of purifications of intermediate which relent the economic significances by taking into account yield of each stage.
Object of the invention
1. The main object of the invention is to provide a novel compound of Formula
(ID-
2. Another object of present invention is to provide a process for preparation of a novel compound of Formula (II).
3. Yet another object of present invention is to provide a process for preparation of Avanafil of Formula (I), in high yield and purity using a novel compound of Formula (II).
4. Yet another object of the present invention to provide simple, economic and industrially scalable process for the preparation of Avanafil o Formula (I).
Summary of the invention
According to an aspect of present invention, there is provided a novel compound of Formula (II).

Formula (II)
wherein R is -OH, -CI or -OR and R is Q to C3 alkyl group
The invention will be specifically described below with reference to Examples but it should not be construed that the scope of the invention is limited thereto. Since the starting compound was produced by a modified method from that described in prior art, it will be described as Referential Example 1 to 3. Here synthesis routes of Referential Example 1 to 3 and Example 1 to 10 are illustrated below in Scheme (V).
Scheme (V)

Formula (I) Referential Examples
Referential Example 1 – Preparation of ethyl 4-[(3-chloro-4-methoxybenzyl)amino]-2-(methyl sulfanyl)pyrimidine-5-carboxylate
To 600ml of methylene dichloride was added l OOg of ethyl 4-chloro-2-(methylsulfanyl) pyrimidine-5-carboxylate and 91.2g of 3-chloro-4-methoxybenzylamine. The reaction mixture was stirred and 500ml of water, 48g of sodium carbonate and Ig of tetra-butylammonium bromide were added to it. The reaction mixture was then maintained overnight at 25-30°C. After completion of reaction, methylene dichloride layer was separated, washed with water and evaporated to obtain 145g of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate having 95% of HPLC purity.
Above reaction can also be carried out using ammonia or triethylamine in same reaction conditions and parameters, in place of sodium carbonate.
Referential Example 2 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid
To 600ml of methanol was added l OOg of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate (Referential Example 1) and an aqueous solution of sodium hydroxide (15g of NaOH in 140ml of water). The reaction mixture was heated to reflux temperature. After completion of reaction, the pH of mixture was adjusted to 1 -2 using concentrated hydrochloric acid followed by stirring the mixture for 1 hour at 10-15°C. The solid product obtained was filtered, washed sequentially with water and methanol, and dried overnight at 70-75°C to get 87g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid.
Referential Example 3 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III)
To a mixture of 400ml of toluene and 0.5ml of dimethyl formamide was added 50g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid (Referential Example 2) and 70g of thionyl chloride, and the reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain yellow solid mass. The solid mass thus obtained, was cooled to 15-20°C followed by addition of 1 75ml of methylene dichloride, 36. l g of 2-amino methyl pyrimidine mesylate and 35.55g of triaethylamine. The reaction mixture was stirred overnight at 25-30°C. After completion of reaction, methylene dichloride was distilled out to get residue. The residue was washed sequentially with 2.5% sodium carbonate solution and water. The residue was then treated with methanol to obtain 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) having HPLC purity of more than 95% (yield: 80%)
Referential Example 4 – Preparation of 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)-l -pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Avanafil)
Step i)
To 200ml of dichloromethane was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyI)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting dichloromethane layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the dichloromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of 125ml solution of L-prolinol in dichloromethane (2.46g of L-prolinol in 125ml of dichlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichloromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
Examples
Example 1 : Preparation of Compound of Formula (II) (wherein R is -OH)
Step i)
To 200ml of methylene dichloride was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting methylene dichloride layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the methylene dichloride layer comprising compound of Formula (IV) obtained in step i), was added 5g of triethylamine followed by slow addition of 125ml solution of L-proline in methylene dichloride (2.8g of L-proline in 125ml of methylene dichloride). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water and 5% sodium carbonate solution, followed by evaporation of methylene dichloride to obtain an oily mass. The oily mass obtained was stripped with 50ml acetone to yield 9g of compound of Formula (II) having HPLC purity 98%.
Example 2: Preparation of Compound of Formula (II) (wherein R is -OC2H5)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filtrate containing product. The ethanol in filtrate is completely distilled out to isolate 10.45g of esterified compound of Formula (II).
Example 3 : Preparation of Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of toluene and 0.5ml of dimethylformamide was added 50g of compound of Formula (II) obtained in example 1 , and 70g of thionyl chloride. The reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain 50.5g of oily carboxylic acid chloride compound of Formula (II).
Example 4: Preparation of Avanafil of Formula (I)
In an inert atmosphere, a solution of 30g of compound of Formula (II) obtained in example 1 or 2, in 150 ml of tetrahydrofuran was dropwise added to 180ml of suspension of 1.0M lithium aluminium hydride solution in tetrahydrofuran, The reaction mixture was refluxed for 5 hours. After completion of reaction, the mixture was cooled in ice-bath and saturated aqueous solution of sodium sulfate was added to decompose excess of lithium aluminium hydride. The mixture was then diluted with 200ml of methylene dichloride and thus formed organic layer was separated. The organic layer was washed with water (3 χ 100 ml), dried over MgS04 and concentrated to collect crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 22.8g of Avanafil of Formula (I) having HPLC purity of 99.20%.
Example 5 : Preparation of Avanafil of Formula (I)
To a mixture of 1.3g sodium borohydride, 1 ml methanesulfonic acid and 50ml ethanol was added l Og of compound of Formula (II) obtained in example 1 or 2, and the mixture was stirred at 25-30°C for 5 hours. After completion of reaction, 100ml water was added and the mixture was extracted with 1 00ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with ethyl acetate at 45- 50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 6 to Example 8
The procedure is carried out as in example 5 except for instead of methanesulfonic acid other reducing agents are used in combination with sodium borohydride. The results are given in Table I
Table I

Example 9: Preparation of Avanafil of Formula (I)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filterate containing product. To the fi Iterate was added 1.2g of sodium borohydride and 2.6g of lithium bromide, and the mixture was stirred for 5 hours. After complete conversion of ester to final product, l OOml water was added and the mixture was extracted with 100ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with 25ml ethyl acetate at 45-50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7.5g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 10: Preparation of Avanafil of Formula (I) from Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of tetrahydrofuran and 50g of carboxylic acid chloride compound of Formula (II) obtained in example 3, was added 12g sodium borohydride at 0-5°C. After completion of reaction, water was added to reaction mixture to decompose excess of sodium borohydride present. The reaction mixture was then concentrated and a solution of 30g of potassium hydroxide in 200 ml of water was added. The mixture was heated to 60-70°C and maintained for 15-18 hours. The mixture was then cooled to 25-30°C and 500 ml of methylene dichloride was added. The organic layer thus formed, was separated and evaporated to yield crude Avanafil
of Formula (I) which was then subjected to purification using methanol as solvent to obtain 40g of Avanafil of Formula (I) having HPLC purity of 99.01%.

Mr. K. Chandran Wholetime Director & Vice Chairman EXTRAS
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof. In one aspect, the PDE5 inhibitor is tadalafil.
“Tadalafil” or “TAD” is described in U.S. Pat. Nos. 5,859,006 and 6,821,975. It refers to the chemical compound, (6R-trans)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione and has the following chemical formula:

Tadalafil is currently marketed in pill form for treating erectile dysfunction (ED) under the trade name Cialis® and under the trade name Adcirca® for the treatment of PAH.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
“Lodenafil” refers to the chemical compound, bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate and has the following chemical formula:

More information about lodenafil is available at Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95. Lodenafil is manufactured by Cristália Produtos Químicose Farmacêuticos in Brazil and sold there under the brand-name Helleva®. It has undergone Phase III clinical trials, but is not yet approved for use in the United States by the U.S. FDA.
“Mirodenafil” refers to the chemical compound, 5-Ethyl-3,5-dihydro-2-[5-([4-(2-hydroxyethyl)-1-piperazinyl]sulfonyl)-2-propoxyphenyl]-7-propyl-4H-pyrrolo[3,2-d]pyrimidin-4-one and has the following chemical formula:

More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80. Mirodenafil is not currently approved for use in the United States but clinical trials are being conducted.
“Sildenafil citrate,” marketed under the name Viagra®, is described in U.S. Pat. No. 5,250,534. It refers to 1-[4-ethoxy-3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)phenylsulfonyl]-4-methylpiperazine and has the following chemical formula:

Sildenafil citrate, sold as Viagra®, Revatio® and under various other trade names, is indicated to treat erectile dysfunction and PAH.
“Vardenafil” refers to the chemical compound, 4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one and has the following chemical formula:

Vardenafil is described in U.S. Pat. Nos. 6,362,178 and 7,696,206. Vardenafil is marketed under the trade name Levitra® for treating erectile dysfunction.
“Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:

More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States.



THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family







Iroko Pharmaceuticals Receives FDA Approval for ZORVOLEX™
![]()
Philadelphia, Pennsylvania, October 18, 2013 – Iroko Pharmaceuticals, LLC, a global specialty pharmaceutical company dedicated to advancing the science of analgesia, today announced that the U.S. Food and Drug Administration (FDA) has approved ZORVOLEX™ (diclofenac) capsules, a nonsteroidal anti-inflammatory drug (NSAID), for the treatment of mild to moderate acute pain in adults[i]. ZORVOLEX was approved at dosage strengths that are 20 percent lower than currently available diclofenac products. FDA approval of ZORVOLEX was supported by data from a Phase 3 multi-center, randomized study in which patients treated with ZORVOLEX reported significant pain relief compared with patients receiving placebo
Glenmark receives final ANDA approval for Desoximetasone Ointment USP, 0.25%

Desoximetasone 382-67-2 cas

September 23, 2013: Glenmark Generics Inc., USA the subsidiary of Glenmark Generics Limited has been granted final abbreviated new drug approval (ANDA) from the United States Food
and Drug Administration (U.S. FDA) for Desoximetasone Ointment USP, 0.25% their generic version of Topicort® by Taro Pharmaceuticals USA Inc and shipping will commence immediately.
Desoximetasone Ointment is indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses. According to IMS Health sales data for the
12 month period ending June 2013, Desoximetasone Ointment garnered annual sales of approximately USD 40 million.
more info
Desoximetasone is a medication belonging to the family of medications known as topical corticosteroids. It is used for the relief of various skin conditions, including rashes. It helps to reduce redness, itching, and irritation. Desoximetasone is a synthetic corticosteroid, a class of primarily synthetic steroids used as anti-inflammatory and anti-pruritic agents.
There are two brand name products:
- Topicort Emollient Cream (0.25% desoximetasone)
- Topicort LP Emollient Cream (0.05% desoximetasone)
When using desoximetasone, some of the medication may be absorbed through the skin and into the bloodstream. Too much absorption can lead to unwanted side effects elsewhere in the body. To keep this problem to a minimum, avoid using large amounts of desoximetasone over large areas, do not use it for extended periods of time, and do not cover it with airtight dressings such as plastic wrap or adhesive bandages unless specifically told to by your doctor. Children may absorb more medication than adults do. Desoximetasone is for use only on the skin and should be kept out of the eyes.
Desoximetasone can also be used to treat some types of psoriasis.


Actavis submits ANDAs for two more generic version of Bayer’s Safyral and Fresenius Kabi’s Diprivan (propofol) injection

Actavis submits ANDAs for two more generic version of

Bayer’s Safyral and

Fresenius Kabi’s Diprivan (propofol) injection
Actavis has filed for US approval for generic versions of a contraceptive and a sedative/anaesthetic.http://www.gabionline.net/Generics/News/Actavis-submits-ANDAs-for-two-more-generics
