
Home » Posts tagged 'fda' (Page 7)
Tag Archives: fda
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bayer, Onyx win early FDA OK for Nexavar (sorafenib) in thyroid cancer
The U.S. Food and Drug Administration said on Friday it has expanded the approved use of the cancer drug Nexavar to include late-stage differentiated thyroid cancer.
Differentiated thyroid cancer is the most common type of thyroid cancer, the FDA said. The National Cancer Institute estimates that 60,220 people in the United States will be diagnosed with it and 1,850 will die from the disease in 2013.
The drug, made by Germany’s Bayer AG and Onyx Pharmaceuticals, is already approved to treat advanced kidney cancer and liver cancer that cannot be surgically removed. Onyx was acquired by Amgen Inc earlier this year.
READ ABOUT SORAFENIB IN MY EARLIER BLOGPOST
https://newdrugapprovals.wordpress.com/2013/07/16/nexavar-sorafenib/
SCRIP Awards 2013 -Best Company in an Emerging Market – Dr Reddy’s Laboratories – India, Novartis’s Bexsero, Best New Drug

The SCRIP Awards 2013 celebrated achievements in the global biopharma industry last night at the Lancaster, London.
Hosted by Justin Webb, the evening was a fantastic mix of dining, entertainment and awards.
Among the winners were:
- Novartis’s Bexsero, Best New Drug
- Genmab, Biotech Company of the Year
- Regeneron Pharmaceuticals and Sanofi’s Phase IIa study dupilumab in asthma, Clinical Advance of the Year
You can view the full roll of honour by clicking on the button below.
It was a great night and we would like to thank all those who entered and attended this year’s awards.
Finally congratulations to our winners and a huge thanks to our sponsors for helping us make it such a fantastic success.
Don’t forget to check our website in the next couple of days for all the pictures from the night.
2013 Winners
Best Company in an Emerging Market – Sponsored by Clinigen Group
- Dr Reddy’s Laboratories – India
Best Technological Development in Clinical Trials
- Quintiles’s Infosario Safety
Best Partnership Alliance
- AstraZeneca with Bristol-Myers Squibb and Amylin in diabetes
Financing Deal of the Year
- Mesoblast’s equity financing of Aus$170m
Best Advance in an Emerging Market
- Novartis’s Jian Kang Kuai Che Healthcare Project in China
Clinical Advance of the Year – Sponsored by Quintiles
- Regeneron Pharmaceuticals and Sanofi’s Phase IIa study dupilumab in in asthma
Licensing Deal of the Year – Sponsored by Hume Brophy
- AstraZeneca and Horizon Discovery for the development and commercialization of the HD-001 kinase target program for multiple cancer types
Executive of the Year
- Roch Doliveux, chairman and chief executive officer of UCB
Biotech Company of the Year
- Genmab
Best Contract Research Organization
- Quintiles
Management Team of the Year
- Regeneron Pharmaceuticals’ CEO Leonard S Schleifer and CSO George D Yancopoulos
Best New Drug – Sponsored by INC Research
- Novartis’ Bexsero (meningococcal group B vaccine)
Pharma Company of the Year – Sponsored by ICON
- Astellas
Lifetime Achievement Award
- Prof Dr Désiré Collen
…….read about bexero at

DR ANTHONY MELVIN CRASTO Ph.D

European Commission Approves Gilead’s VitektaTM, an Integrase Inhibitor for the Treatment of HIV-1 Infection

Elvitegravir
697761-98-1 CAS
FOSTER CITY, Calif.–(BUSINESS WIRE)–Nov. 18, 2013– Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission has granted marketing authorization for VitektaTM (elvitegravir 85 mg and 150 mg) tablets, an integrase inhibitor for the treatment of HIV-1 infection in adults without known mutations associated with resistance to elvitegravir. Vitekta is indicated for use as part of HIV treatment regimens that include a ritonavir-boosted protease inhibitor.http://www.pharmalive.com/eu-oks-gileads-vitekta Vitekta interferes with HIV replication by blocking the virus from integrating into the genetic material of human cells. In clinical trials, Vitekta was effective in suppressing HIV among patients with drug-resistant strains of HIV.http://www.pharmalive.com/eu-oks-gileads-vitekta

Elvitegravir (EVG, formerly GS-9137) is a drug used for the treatment of HIV infection. It acts as an integrase inhibitor. It was developed[1] by the pharmaceutical company Gilead Sciences, which licensed EVG from Japan Tobacco in March 2008.[2][3][4] The drug gained approval by U.S. Food and Drug Administration on August 27, 2012 for use in adult patients starting HIV treatment for the first time as part of the fixed dose combination known as Stribild.[5]
According to the results of the phase II clinical trial, patients taking once-daily elvitegravir boosted by ritonavir had greater reductions in viral load after 24 weeks compared to individuals randomized to receive a ritonavir-boosted protease inhibitor.[6]
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency disease syndrome (AIDS). After over 26 years of efforts, there is still not a therapeutic cure or an effective vaccine against HIV/AIDS. The clinical management of HIV-1 infected people largely relies on antiretroviral therapy (ART). Although highly active antiretroviral therapy (HAART) has provided an effective way to treat AIDS patients, the huge burden of ART in developing countries, together with the increasing incidence of drug resistant viruses among treated people, calls for continuous efforts for the development of anti-HIV-1 drugs. Currently, four classes of over 30 licensed antiretrovirals (ARVs) and combination regimens of these ARVs are in use clinically including: reverse transcriptase inhibitors (RTIs) (e.g. nucleoside reverse transcriptase inhibitors, NRTIs; and non-nucleoside reverse transcriptase inhibitors, NNRTIs), protease inhibitors (PIs), integrase inhibitors and entry inhibitors (e.g. fusion inhibitors and CCR5 antagonists).

- Gilead Press Release Phase III Clinical Trial of Elvitegravir July 22, 2008
- Gilead Press Release Gilead and Japan Tobacco Sign Licensing Agreement for Novel HIV Integrase Inhibitor March 22, 2008
- Shimura K, Kodama E, Sakagami Y, et al. (2007). “Broad Anti-Retroviral Activity and Resistance Profile of a Novel Human Immunodeficiency Virus Integrase Inhibitor, Elvitegravir (JTK-303/GS-9137)”. J Virol 82 (2): 764. doi:10.1128/JVI.01534-07. PMC 2224569. PMID 17977962.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Sax, P. E.; Dejesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J. E.; Liu, H. C.; Zhong, L.; Yale, K.; White, K.; Kearney, B. P.; Szwarcberg, J.; Quirk, E.; Cheng, A. K.; Gs-Us-236-0102 Study, T. (2012). “Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks”.The Lancet 379 (9835): 2439–2448. doi:10.1016/S0140-6736(12)60917-9. PMID 22748591. edit
- Thaczuk, Derek and Carter, Michael. ICAAC: Best response to elvitegravir seen when used with T-20 and other active agents Aidsmap.com. 19 Sept. 2007.

The life cycle of HIV-1. 1. HIV-1 gp120 binds to CD4 and co-receptor CCR5/CXCR4 on target cell; 2. HIV-1 gp41 mediates fusion with target cell; 3. Nucleocapsid containing viral genome and enzymes enters cells; 4. Viral genome and enzymes are released; 5. Viral reverse transcriptase catalyzes reverse transcription of ssRNA, forming RNA-DNA hybrids; 6. RNA template is degraded by ribonuclease H followed by the synthesis of HIV dsDNA; 7. Viral dsDNA is transported into the nucleus and integrated into the host chromosomal DNA by the viral integrase enzyme; 8. Transcription of proviral DNA into genomic ssRNA and mRNAs formation after processing; 9. Viral RNA is exported to cytoplasm; 10. Synthesis of viral precursor proteins under the catalysis of host-cell ribosomes; 11. Viral protease cleaves the precursors into viral proteins; 12. HIV ssRNA and proteins assemble under host cell membrane, into which gp120 and gp41 are inserted; 13. Membrane of host-cell buds out, forming the viral envelope; 14. Matured viral particle is released
Elvitegravir, also known as GS 9137 or JTK 303, is an investigational new drug and a novel oral integrase inhibitor that is being evaluated for the treatment of HIV-1 infection. After HIVs genetic material is deposited inside a cell, its RNA must be converted (reverse transcribed) into DNA. A viral enzyme called integrase then helps to hide HIVs DNA inside the cell’s DNA. Once this happens, the cell can begin producing genetic material for new viruses. Integrase inhibitors, such as elvitegravir, are designed to block the activity of the integrase enzyme and to prevent HIV DNA from entering healthy cell DNA. Elvitegravir has the chemical name: 6-(3-chloro-2-fluorobenzyl)-1-[(S)-1 -hydroxy -methyl-2- methylpropyl]-7-methoxy-4-oxo-1, 4-dihydroquinoline-3-carboxylic acid and has the following structural formula:

WO 2000040561 , WO 2000040563 and WO 2001098275 disclose 4-oxo-1 , 4-dihydro-3- quinoline which is useful as antiviral agents. WO2004046115 provides certain 4- oxoquinoline compounds that are useful as HIV Integrase inhibitors.
US 7176220 patent discloses elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and their method of treatment. The chemistry involved in the above said patent is depicted below in the Scheme A. Scheme-A
Toluene, DIPEA
SOCl2 ,COCl (S)-(+)-Valinol
Toluene
,4-Difluoro-5-iodo- benzoic acid


THF
dichlorobis(triphenylphosphine)
palladium argon stream,

Elvitegravir Form ] Elvitegravir (residue) US 7635704 patent discloses certain specific crystalline forms of elvitegravir. The specific crystalline forms are reported to have superior physical and chemical stability compared to other physical forms of the compound. Further, process for the preparation of elvitegravir also disclosed and is depicted below in the Scheme B. The given processes involve the isolation of the intermediates at almost all the stages.
Scheme B
2,
–

Zn THF,
CK Br THF CU “ZnBr dιchlorobis(trιphenylphos
phine)palladium

Elvitegravir WO 2007102499 discloses a compound which is useful as an intermediate for the synthesis of an anti-HIV agent having an integrase-inhibiting activity; a process for production of the compound; and a process for production of an anti-HIV agent using the intermediate.
WO 2009036161 also discloses synthetic processes and synthetic intermediates that can be used to prepare 4-oxoquinolone compounds having useful integrase inhibiting properties.
The said processes are tedious in making and the purity of the final compound is affected because of the number of steps, their isolation, purification etc., thus, there is a need for new synthetic methods for producing elvitegravir which process is cost effective, easy to practice, increase the yield and purity of the final compound, or that eliminate the use of toxic or costly reagents.
US Patent No 7176220 discloses Elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and ■ their method of treatment. US Patent No 7635704 discloses Elvitegravir Form II, Form III and processes for their preparation. The process for the preparation of Form Il disclosed in the said patent is mainly by three methods – a) dissolution of Elvitegravir followed by seeding with Form II, b) recrystallisation of Elvitegravir, and c) anti-solvent method.
The process for the preparation of Form III in the said patent is mainly by three methods – a) dissolution of Form Il in isobutyl acetate by heating followed by cooling the reaction mass, b) dissolution of Form Il in isobutyl acetate by heating followed by seeding with Form III, and c) dissolving Form Il in 2-propanol followed by seeding with Form III.
Amorphous materials are becoming more prevalent in the pharmaceutical industry. In order to overcome the solubility and potential bioavailability issues, amorphous solid forms are becoming front-runners. Of special importance is the distinction between amorphous and crystalline forms, as they have differing implications on drug substance stability, as well as drug product stability and efficacy.
An estimated 50% of all drug molecules used in medicinal therapy are administered as salts. A drug substance often has certain suboptimal physicochemical or biopharmaceutical properties that can be overcome by pairing a basic or acidic drug molecule with a counter- ion to create a salt version of the drug. The process is a simple way to modify the properties of a drug with ionizable functional groups to overcome undesirable features of the parent drug. Salt forms of drugs have a large effect on the drugs’ quality, safety, and performance. The properties of salt-forming species significantly affect the pharmaceutical properties of a drug and can greatly benefit chemists and formulators in various facets of drug discovery and development.



chemical synthesis from a carboxylic acid 1 starts after conversion to the acid chloride iodide NIS 2 , and with three condensation 4 . 4 and the amino alcohol 5 addition-elimination reaction occurs 6 , 6 off under alkaline conditions with TBS protected hydroxy get the ring 7 , 7 and zinc reagent 8 Negishi coupling occurs to get 9 , the last 9 hydrolysis and methoxylated
Elvitegravir dimer impurity, WO2011004389A2
Isolation of 1-[(2S)-1-({3-carboxy-6-(3-chloro-2-fluorobenzyl)-1 -[(2S)-I- hydroxy-3-methylbutan-2-yl]-4-oxo-1 , 4-dihydroquinolin-7-yl}oxy)-3- methylbutan-2-yl 6-(3-chloro-2-fluorobenzyl)-7-methoxy-4-oxo-1 , 4-dihydroquinoline-3-carboxylic acid (elvitegravir dimer impurity, 13)
After isolation of the elvitegravir from the mixture of ethyl acetate-hexane, solvent from the filtrate was removed under reduced pressure. The resultant residue purified by column chromatography using a mixture of ethyl acetate-hexane (gradient, 20-80% EtOAc in hexane) as an eluent. Upon concentration of the required fractions, a thick solid was obtained which was further purified on slurry washing with ethyl acetate to get pure elvitegravir dimer impurity (13). The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-Cf6, 300 MHz, ppm) – δ 0.79 (m, d=6.3 Hz, 6H, 20 & 2O’)\ 1.18 & 1.20 (d, J=6.3 Hz & J=6.2 Hz, 6H, 21 & 21′)1, 2.42-2.49 (m, 2H, 19 & 19′), 3.81-3.89 (m, 3H, T & 17’Ha), 3.94-4.01 (m, 1 H, 17’Hb), 4.01 (s, 3H, 23), 4.11 (s, 2H, 7), 4.83-4.85 (m, 3H, 17 & 18′), 5.22 (t, J=4.7 Hz, 1H, OH), 5.41-5.44 (m, 1 H, 18), 6.73-6.78 (t, J=7.1 Hz, 1 H, 11)1‘ 2, 6.92-6.98 (t, J=8.0 Hz, 1H, 3′) 1‘2, 7.12-7.22 (m, 2H, 1 & 3), 7.34-7.39 (m, 1H, 2′),
7.45-7.48 (m, 1 H, 2), 7.49, 7.56 (s, 2H, 15 & 15′), 7.99, 8.02 (s, 2H, 9 & 9′), 8.89, 9.01 (s, 2H, 13 & 13′), 15.30, 15.33 (s, 2H, COOH’ & COOH”).
13C-NMR (DMSO-Cf6, 75 MHz, ppm)- δ 18.87, 19.03 (2OC, 20’C), 19.11 , 19.24 (21 C, 21 ‘C), 27.94 (7’C), 28.40 (7C), 28.91 , 30.08 (19C, 19’C), 56.80(23C), 60.11 (171C), 63.59 (18C), 66.52 (18’C), 68.53 (17C), 97.86, 98.97 (15, 15′), 107.43, 108.16 (12C, 12’C),
118.77, 119.38 (1OC, 10’C), 119.57 (d, J=17.6 Hz, 41C), 119.61 (d, J=17.9 Hz, 4C),
124.88 (d, J=4.3 Hz, 31C), 125.18 (d, J=4.2 Hz, 3C), 126.59, 126.96 (9C1 9’C), 127.14 (8’C), 127.62 (d, J=15.9 Hz, 61C), 127.73 (8C), 127.99 (d, J=15.2 Hz, 6C), 128.66 (2’C),
128.84 (11C), 128.84 (2C), 130.03 (d, J=3.4 Hz, 1C), 142.14, 142.44 (14C, 14’C), 144.37, 145.56 (13C, 131C), 155.24 (d, J=245.1 Hz, 5’C)1 155.61 (d, J=245.1 Hz, 5C),
160.17 (16’C), 162.04 (16C), 166.00, 166.14 (22C, 22’C), 176.17, 176.22 (11C, 111C).
DIP MS: m/z (%)- 863 [M+H]+, 885 [M+Na]+.
Europace Publishes Data Supporting Use Of BRINAVESS™ (Vernakalant) As A First Line Agent For Pharmacological Cardioversion Of Atrial Fibrillation

Vernakalant, MK-6621, RSD 1235
(3R)-1-{(1R,2R)-2-[2-(3,4-dimethoxyphenyl)
ethoxy]cyclohexyl}pyrrolidin-3-ol
C20H31NO4 , 349.47, Brinavess , Kynapid
cas no 794466-70-9
748810-28-8 (HCl)
EMA:Link click here
PATENT WO 2004099137
VANCOUVER, Nov. 21, 2013 /PRNewswire/ – Cardiome Pharma Corp. (NASDAQ: CRME / TSX: COM) today announced that a publication titled, Pharmacological Cardioversion of Atrial Fibrillation with Vernakalant: Evidence in Support of the ESC Guidelines, was published in Europace, the official Journal of the European Heart Rhythm Association, and was made available in the advanced online article access section. The authors conclude that BRINAVESS is an efficacious and rapid acting pharmacological cardioversion agent, for recent-onset atrial fibrillation (AF,) that can be used first line in patients with little or no underlying cardiovascular disease and in patients with moderate disease, such as stable coronary and hypertensive heart disease.
Vernakalant (INN; codenamed RSD1235, proposed tradenames Kynapid and Brinavess) is an investigational drug under regulatory review for the acute conversion of atrial fibrillation. It was initially developed by Cardiome Pharma, and the intravenous formulation has been bought for further development by Merck in April 2009.[1] In September 2012, Merck terminated its agreements with Cardiom and has consequently returned all rights of the drug back to Cardiom.
On 11 December 2007, the Cardiovascular and Renal Drugs Advisory Committee of the USFood and Drug Administration (FDA) voted to recommend the approval of vernakalant,[2]but in August 2008 the FDA judged that additional information was necessary for approval.[1] The drug was approved in Europe on 1 September 2010.[3]
An oral formulation underwent Phase II clinical trials between 2005 and 2008.[4][5]
Like other class III antiarrhythmics, vernakalant blocks atrial potassium channels, thereby prolonging repolarization. It differs from typical class III agents by blocking a certain type of potassium channel, the cardiac transient outward potassium current, with increased potency as the heart rate increases. This means that it is more effective at high heart rates, while other class III agents tend to lose effectiveness under these circumstances. It also slightly blocks the hERG potassium channel, leading to a prolonged QT interval. This may theoretically increase the risk of ventricular tachycardia, though this does not seem to be clinically relevant.[6]
The drug also blocks atrial sodium channels.[6]
- “Merck and Cardiome Pharma Sign License Agreement for Vernakalant, an Investigational Drug for Treatment of Atrial Fibrillation”. FierceBiotech. 9 April 2009. Retrieved 12 October 2010.
- “FDA Advisory Committee Recommends Approval of Kynapid for Acute Atrial Fibrillation”. Drugs.com. Retrieved 2008-03-15.
- “BRINAVESS (vernakalant) for Infusion Approved in the European Union for Rapid Conversion of Recent Onset Atrial Fibrillation” (Press release). Merck & Co., Inc. 1 September 2010. Retrieved 28 September 2010.
- ClinicalTrials.gov NCT00267930 Study of RSD1235-SR for the Prevention of Atrial Fibrillation/Atrial Flutter Recurrence
- ClinicalTrials.gov NCT00526136 Vernakalant (Oral) Prevention of Atrial Fibrillation Recurrence Post-Conversion Study
- Miki Finnin, Vernakalant: A Novel Agent for the Termination of Atrial Fibrillation: Pharmacology, Medscape Today, retrieved 12 October 2010
- Arzneimittel-Fachinformation (EMA)
- Cheng J.W. Vernakalant in the management of atrial fibrillation. Ann Pharmacother, 2008, 42(4), 533-42Pubmed

- Dobrev D., Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet, 2010, 375(9721), 1212-23 Pubmed
- Finnin M. Vernakalant: A novel agent for the termination of atrial fibrillation. Am J Health Syst Pharm, 2010, 67(14), 1157-64 Pubmed
- Mason P.K., DiMarco J.P. New pharmacological agents for arrhythmias. Circ Arrhythm Electrophysiol, 2009, 2(5), 588-97 Pubmed
- Naccarelli G.V., Wolbrette D.L., Samii S., Banchs J.E., Penny-Peterson E., Stevenson R., Gonzalez M.D. Vernakalant – a promising therapy for conversion of recent-onset atrial fibrillation. Expert Opin Investig Drugs, 2008, 17(5), 805-10 Pubmed
- European Patent No. 1,560,812
- WO 2006138673
 , WO 200653037
, WO 200653037 - WO 200597203, WO 200688525
- Vernakalant HydrochlorideDrugs Fut 2007, 32(3): 234
//////////////////////////////////////////////////////

![]()
![]()

NMR
1H NMR (300 MHz, CDCI3) 5 6.75 (m, 3H), 4.22 (m, 1H), 3.87 (s, 3H), 3.85 (m, 3H), 3.74 (m, 1H), 3.57 (m, 1H), 3.32 (td, J =
7.7, 3.5, 1H), 2.96-2.75 (m, 5H), 2.64 (dd, J= 10.0, 5.0, 1H), 2.49-2.37 (m, 2H), 2.05-1.98 (m, 2H), 1.84 (m, 1H), 1.69-1.62 (m, 3H), 1.35-1.19 (m, 4H).
IN
WO 201240846
Arrhythmias are abnormal rhythms of the heart. The term “arrhythmia” refers to a deviation from the normal sequence of initiation and conduction of electrical impulses that cause the heart to beat. Arrhythmias may occur in the atria or the ventricles. Atrial arrhythmias are widespread and relatively benign, although they place the subject at a higher risk of stroke and heart failure. Ventricular arrhythmias are typically less common, but very often fatal.
Arrhythmia is a variation from the normal rhythm of the heart beat and generally represents the end product of abnormal ion-channel structure, number or function. Both atrial arrhythmias and ventricular arrhythmias are known. The major cause of fatalities due to cardiac arrhythmias is the subtype of ventricular arrhythmias known as ventricular fibrillation (VF). Conservative estimates indicate that, in the U.S. alone, each year over one million Americans will have a new or recurrent coronary attack (defined as myocardial infarction or fatal coronary heart disease). About 650,000 of these will be first heart attacks and 450,000 will be recurrent attacks. About one-third of the people experiencing these attacks will die of them. At least 250,000 people a year die of coronary heart disease within 1 hour of the onset of symptoms and before they reach a hospital. These are sudden deaths caused by cardiac arrest, usually resulting from ventricular fibrillation.
Atrial fibrillation (AF) is the most common arrhythmia seen in clinical practice and is a cause of morbidity in many individuals (Pritchett E.L., N. Engl. J. Med. 327(14):1031 Oct. 1, 1992, discussion 1031-2; Kannel and Wolf, Am. Heart J. 123(l):264-7 Jan. 1992). Its prevalence is likely to increase as the population ages and it is estimated that 3-5% of patients over the age of 60 years have AF (Kannel W.B., Abbot R.D., Savage D.D., McNamara P.M., N. Engl. J. Med. 306(17): 1018-22, 1982; Wolf P.A., Abbot R.D., Kannel W.B. Stroke. 22(8):983-8, 1991). While AF is rarely fatal, it can impair cardiac function and is a major cause of stroke (Hinton R.C., Kistler J.P., Fallon J.T., Friedlich A.L., Fisher CM., American Journal of Cardiology 40(4):509-13, 1977; Wolf P.A., Abbot R.D., Kannel W.B., Archives of Internal Medicine 147(9): 1561 -4, 1987; Wolf P. A., Abbot R.D., Kannel W.B. Stroke. 22(8):983-8, 1991; Cabin H.S., Clubb K.S., Hall C, Perlmutter R.A., Feinstein A.R., American Journal of Cardiology 65(16): 1112-6, 1990).
WO95/08544 discloses a class of aminocyclohexylester compounds as useful in the treatment of arrhythmias.
WO93/ 19056 discloses a class of aminocyclohexylamides as useful in the treatment of arrhythmia and in the inducement of local anaesthesia.
WO99/50225 discloses a class of aminocyclohexylether compounds as useful in the treatment of arrhythmias.
Antiarrhythmic agents have been developed to prevent or alleviate cardiac arrhythmia. For example, Class I antiarrhythmic compounds have been used to treat supraventricular arrhythmias and ventricular arrhythmias. Treatment of ventricular arrhythmia is very important since such an arrhythmia can be fatal. Serious ventricular arrhythmias (ventricular tachycardia and ventricular fibrillation) occur most often in the presence of myocardial ischemia and/or infarction. Ventricular fibrillation often occurs in the setting of acute myocardial ischemia, before infarction fully develops. At present, there is no satisfactory pharmacotherapy for the treatment and/or prevention of ventricular fibrillation during acute ischemia. In fact, many Class I antiarrhythmic compounds may actually increase mortality in patients who have had a myocardial infarction.
Class la, Ic and HI antiarrhythmic drugs have been used to convert recent onset AF to sinus rhythm and prevent recurrence of the arrhythmia (Fuch and Podrid, 1992; Nattel S., Hadjis T., Talajic M., Drugs 48(3):345-7l, 1994). However, drug therapy is often limited by adverse effects, including the possibility of increased mortality, and inadequate efficacy (Feld G.K., Circulation. <°3(<5):2248-50, 1990; Coplen S.E., Antman E.M., Berlin J.A., Hewitt P., Chalmers T.C., Circulation 1991; S3(2):714 and Circulation 82(4):1106-16, 1990; Flaker G.C., Blackshear J.L., McBride R., Kronmal R.A., Halperin J.L., Hart R.G., Journal of the American College of Cardiology 20(3):527-32, 1992; CAST, N. Engl. J. Med. 321:406, 1989; Nattel S., Cardiovascular Research. 37(3):567 -77, 1998). Conversion rates for Class I antiarrhythmics range between 50-90% (Nattel S., Hadjis T., Talajic M., Drugs 48(3)345-71, 1994; Steinbeck G., Remp T., Hoffmann E., Journal of Cardiovascular Electrophysiology. 9(8 Suppl):S 104-8, 1998). Class ILT antiarrhythmics appear to be more effective for terminating atrial flutter than for AF and are generally regarded as less effective than Class I drugs for terminating of AF (Nattel S., Hadjis T., Talajic M., Drugs. 48(3):345-71, 1994; Capucci A., Aschieri D., Villani G.Q., Drugs & Aging 13(l):5l- 70, 1998). Examples of such drugs include ibutilide, dofetilide and sotalol. Conversion rates for these drugs range between 30-50% for recent onset AF (Capucci A., Aschieri D., Nillani G.Q., Drugs & Aging J3(l):5l-70, 1998), and they are also associated with a risk of the induction of Torsades de Pointes ventricular tachyarrhythmias. For ibutilide, the risk of ventricular proarrhythmia is estimated at ~4.4%, with ~1.7% of patients requiring cardioversion for refractory ventricular arrhythmias (Kowey P.R., NanderLugt J.T., Luderer J.R., American Journal of Cardiology 78(8A):46-52, 1996). Such events are particularly tragic in the case of AF as this arrhythmia is rarely a fatal in and of itself.
Atrial fibrillation is the most common arrhythmia encountered in clinical practice. It has been estimated that 2.2 million individuals in the United States have paroxysmal or persistent atrial fibrillation. The prevalence of atrial fibrillation is estimated at 0.4% of the general population, and increases with age. Atrial fibrillation is usually associated with age and general physical condition, rather than with a specific cardiac event, as is often the case with ventricular arrhythmia. While not directly life threatening, atrial arrhythmias can cause discomfort and can lead to stroke or congestive heart failure, and increase overall morbidity.
There are two general therapeutic strategies used in treating subjects with atrial fibrillation. One strategy is to allow the atrial fibrillation to continue and to control the ventricular response rate by slowing the conduction through the atrioventricular (AV) node with digoxin, calcium channel blockers or beta-blockers; this is referred to as rate control. The other strategy, known as rhythm control, seeks to convert the atrial fibrillation and then maintain normal sinus rhythm, thus attempting to avoid the morbidity associated with chronic atrial fibrillation. The main disadvantage of the rhythm control strategy is related to the toxicities and proarrhythmic potential of the anti-arrhythmic drugs used in this strategy. Most drugs currently used to prevent atrial or ventricular arrhythmias have effects on the entire heart muscle, including both healthy and damaged tissue. These drugs, which globally block ion channels in the heart, have long been associated with life-threatening ventricular arrhythmia, leading to increased, rather than decreased, mortality in broad subject populations. There is therefore a long recognized need for antiarrhythmic drugs that are more selective for the tissue responsible for the arrhythmia, leaving the rest of the heart to function normally, less likely to cause ventricular arrhythmias.
One specific class of ion channel modulating compounds selective for the tissue responsible for arrhythmia has been described in U.S. Pat. No. 7,057,053, including the ion channel modulating compound known as vernakalant hydrochloride. Vernakalant hydrochloride is the non-proprietary name adopted by the United States Adopted Name (USAN) council for the ion channel modulating compound (1R,2R)-2-[(3R)-hydroxypyrrolidinyl]-1-(3,4-dimethoxyphenethoxy)-cyclohexane monohydrochloride, which compound has the following formula:

Vernakalant hydrochloride may also be referred to as “vernakalant” herein.
Vernakalant hydrochloride modifies atrial electrical activity through a combination of concentration-, voltage- and frequency-dependent blockade of sodium channels and blockade of potassium channels, including, e.g., the ultra-rapidly activating (lKur) and transient outward (lto) channels. These combined effects prolong atrial refractoriness and rate-dependently slow atrial conduction. This unique profile provides an effective anti-fibrillatory approach suitable for conversion of atrial fibrillation and the prevention of atrial fibrillation.

C20H32ClNO4, Mr = 385.9 g/mol
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review

Posaconazole, SCH 56592, Noxafil (Schering-Plough)
Posaconazole is a triazole antifungal drug that is used to treat invasive infections by Candida species and Aspergillus species in severely immunocompromised patients.
For prophylaxis of invasive Aspergillus and Candida infections in patients, 13 years of age and older, who are at high risk of developing these infections due to being severely immunocompromised as a result of procedures such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD), or due to hematologic malignancies with prolonged neutropenia from chemotherapy. Also for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole. Posaconazole is used as an alternative treatment for invasive aspergillosis, Fusarium infections, and zygomycosis in patients who are intolerant of, or whose disease is refractory to, other antifungals
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro-5-(1H-1,2,4-triazol-1 -ylmethyl)-3-furanyl]methoxy]phenyl]-1 -piperazinyl]phenyl]-2-[ (1S,2S)-1 -ethyl-2- hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8.
Posaconazole is used, for example, to prevent and/or treat invasive fungal infections caused by Candida species, Mucor species, Aspergillus species,Fusarium species, or Coccidioides species in immunocompromised patients and/or in patients where the disease is refractory to other antifungal agents such as amphothericin B, fluconazole, or itraconazole, and/or in patients who do not tolerate these antifungal agents.
CAS No. 171228-49-2
Posaconazole compounds have been described inU.S. Pat. Appl. No. 2003/0055067 for “Antifungal Composition with Enhanced Bioavailability,” U.S. Pat. Appl. No. 2004/0058974 for “Treating Fungal Infections,” and European Patent Publication1372394 (A1 ) for “Liquid Suspensions of Posaconazole (SCH 56592) with Enhanced Bioavailability for Treating Fungal Infections.”
| Synonyms: | Pcz;Pos;Noxafil;Sch 56592;Aids058495;Aids-058495;Posconazole;Posaconazole;Posaconazole for research;HYDROXYPROPYL]-2,4-DIHYDRO-3H-1,2,4-TRIAZOL-3-ONE |
| Molecular Formula: | C37H42F2N8O4 |
| Formula Weight: | 700.78 |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review
Marketing Authorization Application also Filed with the European Medicines Agency
WHITEHOUSE STATION, N.J., Nov. 18, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that its New Drug Application for an investigational intravenous (IV) solution formulation of the company’s antifungal agent, NOXAFIL® (posaconazole), has been accepted for priority review by the U.S. Food and Drug Administration (FDA).http://www.pharmalive.com/mercks-noxafil-nda-gets-fda-priority-review
Posaconazole (CAS Registry Number 171228-49-2; CAS Name: 2,5-anhydro-1 ,3,4-trideoxy-2- C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1 H-1 ,2,4-triazol-1-yl)-D-threo-pentitol) which is represented by the following general formula (I)

(I)
is known as an antifungal agent. It is available as an oral suspension (40 mg/ml) under the trademark NOXAFIL® from Schering Corporation, Kenilworth, NJ. WO95/17407 and WO 96/38443 disclose the compound having the general formula (I) and its use in treating fungal infections. Various pharmaceutical compositions comprising posaconazole and being adapted for oral, topical or parenteral use are described e.g. in WO 02/80678, U.S. Patent No. 5,972,381 , U.S. Patent No. 5,834,472, U.S. Patent No. 4,957,730 and WO 2005/117831. As was mentioned above, WO 95/17407 and WO 96/38443 disclose the compound having the general formula (I). However, during prosecution of the subsequently filed European patent application no. 98951994.7, now European patent EP 1 021 439 B1 , the applicant declared that the methods disclosed in these publications only lead to the compound of formula (I) as an amorphous solid.
Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and distinct and varying physical properties like melting point, XRPD pattern, IR-spectrum and solubility profile. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however, they may have distinct advantageous physical properties which can have a direct effect on the ability to process and/or manufacture the drug product, like flowability, as well as physical properties such as solubility, stability and dissolution properties which can have a direct effect on drug product stability, solubility, dissolution, and bioavailability.
Three polymorphic forms of posaconazole designated as forms I, Il and III are described and characterized in WO 99/18097 (US-B-6,713,481 , US-B-6,958,337). Crystalline forms Il and III were found to be unstable under the conditions investigated, so that crystalline form I was considered to be useful in the development of a pharmaceutical product.
A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering);
eidem, Tetrahedron Lett. 37, 5657 (1996).
SCH-56592, a novel orally active broad spectrum antifungal agent35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61
seeSaksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.-T.; Ganguly, A.K.; Bennett, F. (Schering Corp.) EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Process for the preparation of triazolonesWO 9633178
Mono N-arylation of piperazine(III): Metal-catalyzed N-arylation and its application to the novel preparations of the antifungal posaconazole and its advanced intermediateTetrahedron Lett 2002,43(18),3359
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000).
Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
synthesis 1

……………..

Synthesis of intermediate (XX): The reaction of 2-chloro-2′,4′-difluoroacetophenone (I) with sodium acetate and NaI in DMF gives 2-acetoxy-2′,4′-difluoroacetophenone (II), which by methylenation with methyltriphenylphosphonium bromide and sodium bis(trimethylsilyl)amide in THF yields 2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis of (III) with KOH in dioxane/water affords the corresponding alcohol (IV), which is regioselectively epoxidized with titanium tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to (S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of (V) with 1,2,4-triazole (VI) in DMF affords (R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII), which is selectively mesylated with methanesulfonyl chloride and triethylamine to the monomesylate (VIII). The cyclization of (VIII) with NaH in DMF gives the oxirane (IX), which is condensed with diethyl malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and (5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of (XI) with NaBH4 and LiCl in ethanol affords (R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl) pentane-1,4-diol (XII), which is selectively tosylated with tosyl chloride and triethylamine in THF to the bistosylate (XIII). The cyclization of (XIII) by means of NaH in refluxing toluene gives (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV) with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain compound (XVI), and the following reaction sequence (XVI) to (XVII) to (XVIII) to (XIX) to (5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one (XX) has been performed according to J Med Chem 1984, 27: 894-900.
………………….pat approved expiry
| United States | 5661151 | 1999-07-19 | 2019-07-19 |
| Canada | 2305803 | 2009-12-22 | 2018-10-05 |
| Canada | 2179396 | 2001-04-17 | 2014-12-20 |
| United States | 5703079 | 1994-08-26 | 2014-08-26 |
MORE INFO
| US Patent No | Patent expiry | |
|---|---|---|
| 5661151 | Jul 19, 2019 | |
| 5703079 | Aug 26, 2014 | |
| 6958337 | Oct 5, 2018 | |
| 8263600 | Apr 1, 2022 |
- Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D: Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007 Jan 25;356(4):348-59. Pubmed
- Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S: Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007 Jan 25;356(4):335-47. Pubmed
- Bhattacharya M, Rajeshwari K, Dhingra B: Posaconazole. J Postgrad Med. 2010 Apr-Jun;56(2):163-7. Pubmed
- Frampton JE, Scott LJ: Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs. 2008;68(7):993-1016.Pubmed
- Schiller DS, Fung HB: Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007 Sep;29(9):1862-86. Pubmed
- Kwon DS, Mylonakis E: Posaconazole: a new broad-spectrum antifungal agent. Expert Opin Pharmacother. 2007 Jun;8(8):1167-78.Pubmed
- Groll AH, Walsh TJ: Posaconazole: clinical pharmacology and potential for management of fungal infections. Expert Rev Anti Infect Ther. 2005 Aug;3(4):467-87. Pubmed
- Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH: Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008 Oct;42(10):1429-38. Epub 2008 Aug 19. Pubmed
- Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H: Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin Pharmacokinet. 2010 Jun;49(6):379-96. doi: 10.2165/11319340-000000000-00000. Pubmed
FDA panel backs Vanda body clock drug Tasimelteon for blind

Tasimelteon
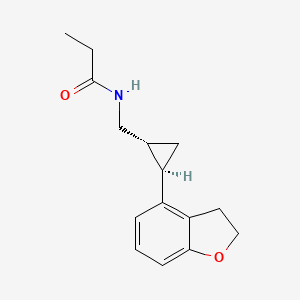
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide, 609799-22-6 cas
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
Tasimelteon (BMS-214,778) is a drug which is under development for the treatment of insomnia and other sleep disorders.[1] It is a selective agonistfor the melatonin receptors MT1 and MT2 in the suprachiasmatic nucleus of the brain, similar to older drugs such as ramelteon.[2] It has been through Phase III trials successfully and was shown to improve both onset and maintenance of sleep, with few side effects.[3]
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
A drug being developed to treat transient insomnia in circadian rhythm sleep disorders (eg jet-lag. The drug appears to be effective in the dose range of 20 to 100mg with an advance in the melatonin rhythm of 2-3 hours with the higher dose
- ‘Time-bending drug’ for jet lag. BBC News. 2 December 2008
- Vachharajani, Nimish N., Yeleswaram, Krishnaswamy, Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Shantha MW Rajaratnam, Mihael H Polymeropoulos, Dennis M Fisher, Thomas Roth, Christin Scott, Gunther Birznieks, Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Audio interview with Joseph Hull of Harvard, spring 2011
- Vanda Pharmaceuticals seeks FDA approval
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder
(N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.







WASHINGTON, June 5, 2013 /PRNewswire/ — Vanda Pharmaceuticals Inc. (Vanda) presented additional entrainment and patient-level clinical data at SLEEP 2013, the 27th Annual Meeting of Associated Professional Sleep Societies in Baltimore, from its SET (Safety and Efficacy of Tasimelteon) and RESET (Randomized-withdrawal study of the Efficacy and Safety of Tasimelteon to treat Non-24-Hour Disorder) Phase III studies of tasimelteon, a circadian regulator for the treatment of Non-24-Hour Disorder (Non-24) in totally blind individuals. Non-24 is a serious, rare and chronic circadian rhythm disorder that affects a majority of totally blind individuals who lack light perception and cannot entrain (synchronize) their master body clock to the 24-hour day. Currently there is no approved FDA treatment for Non-24.
In the SET study, tasimelteon achieved the primary endpoints of entrainment (synchronizing) of the melatonin (aMT6s) rhythm as compared to placebo and clinical response as measured by entrainment plus a score of greater than or equal to 3 on the Non-24 Clinical Response Scale (N24CRS). Tasimelteon also demonstrated significant improvement versus placebo across a number of sleep and wake parameters including measures of total sleep time, nap duration, and timing of sleep, as well as in the Clinical Global Impression of Change (CGI-C), an overall global functioning scale. In treated patients, daytime naps decreased by 46 minutes per day in the worst 25% of days in a cycle and nighttime sleep increased by 57 minutes per day during the worst 25% of nights in a cycle.
The RESET study demonstrated that continued treatment with 20mg of tasimelteon was required to maintain entrainment of melatonin and cortisol circadian rhythms in individuals with Non-24. Patients treated with tasimelteon maintained their clinical benefits while patients who received placebo showed significant deterioration in measures of nighttime sleep, daytime naps and timing of sleep. Furthermore, discontinuation of tasimelteon resulted in a rapid relapse of circadian entrainment and a return to misaligned circadian rhythms, reinforcing the importance of chronic therapy.
Study investigator, Steven W. Lockley, Ph.D., Associate Professor of Medicine, Division of Sleep Medicine, Brigham and Women’s Hospital, Harvard Medical School, commented, “the results clearly demonstrate that tasimelteon can entrain the circadian clock, and that continued treatment is necessary to maintain entrainment.”
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
UPDATED ON JAN 2014
TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
VEC-162, BMS-214778, 609799-22-6, Hetlioz, Tasimelteon (USAN/INN), Tasimelteon [USAN:INN], UNII-SHS4PU80D9,
Tasimelteon
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
References
- ‘Time-bending drug’ for jet lag. BBC News. 2 December 2008
- Vachharajani, Nimish N., Yeleswaram, Krishnaswamy, Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Shantha MW Rajaratnam, Mihael H Polymeropoulos, Dennis M Fisher, Thomas Roth, Christin Scott, Gunther Birznieks, Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet 373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Audio interview with Joseph Hull of Harvard, spring 2011
- Vanda Pharmaceuticals seeks FDA approval
- Recent progress in the development of agonists and antagonists for melatonin receptors.Zlotos DP.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
 TASIMELTION
TASIMELTION
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc. 1938, 60, 2794.
- Oppolzer, W.; Chapuis, C.; Bernardinelli, G. Helv. Chim. Acta 1984, 67, 1397.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, G.; Carroll,, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Oppolzer, W.; Mills, R. J.; Pachinger, W.; Stevenson, T. Helv. Chim. Acta 1986, 69, 1542; Oppolzer, W.; Schneider, P. Helv. Chim. Acta 1986, 69, 1817; Oppolzer, W.; Mills, R. J.; Réglier, M. Tetrahedron Lett. 1986, 27, 183; Oppolzer, W.; Poli. G.Tetrahedron Lett. 1986, 27, 4717; Oppolzer, W.; Poli, G.; Starkemann, C.; Bernardinelli, G. Tetrahedron Lett. 1988, 29, 3559.
- Oppolzer, W.; Barras, J-P. Helv. Chim. Acta 1987, 70, 1666.
- Curran, D. P.; Kim, B. H.; Daugherty, J.; Heffner, T. A. Tetrahedron Lett. 1988, 29, 3555.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
 |
- Org. Syn. Coll. Vol. 8, 110
- Org. Syn. Coll. Vol. 9, 212
-
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Reychler, M. A. Bull. Soc. Chim. III 1889, 19, 120.
- Armstrong, H. E.; Lowry, T. M. J. Chem. Soc., Trans. 1902, 81, 1441.
- Dauphin, G.; Kergomard, A.; Scarset, A. Bull. Soc. Chim. Fr. 1976, 862.
- Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc. 1982, 104, 5412.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I 1988, 1753.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
- For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
- Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
- Davis, F. A.; Sheppard, A. C. Tetrahedron 1989, 45, 5703.
- Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc. 1987, 109, 3370.
- Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron 1985, 41, 4747.
- Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett. 1986, 27, 5079.
- Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc. 1983, 105, 3123.
- Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett. 1987, 28, 5115.
- Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett. 1988, 29, 4269.
- Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem. 1986, 51, 2402.
- Davis, F. A.; Haque, M. S. J. Org. Chem. 1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem. 1989, 54, 2021.
- Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem. 1987, 52, 5288.
- Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett. 1989, 30, 779.
- Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc. 1990, 112, 6679.

back to home for more updates

DR ANTHONY MELVIN CRASTO Ph.D
FDA Approves Implanted Brain Stimulator for Epilepsy
epilepsy
THURSDAY Nov. 14, 2013 — The U.S. Food and Drug Administration on Thursday gave its approval to a new implanted device that lowers the rate of seizures among people with epilepsy
http://www.drugs.com/news/fda-approves-implanted-brain-stimulator-epilepsy-48978.html
Advaxis’s cancer vaccine gets FDA orphan status for treatment of HPV-associated head and neck cancer
US-based clinical-stage biotechnology firm Advaxis has received orphan drug designation from the US Food and Drug Administration (FDA) for its lead drug candidate ADXS-HPV to treat human papillomavirus (HPV) associated head and neck cancer patients.
Advaxis’s cancer vaccine gets FDA orphan status for treatment of HPV-associated head and neck cancer
PRINCETON, N.J., Nov 05, 2013 (BUSINESS WIRE) — Advaxis, Inc., /quotes/zigman/23528806/delayed/quotes/nls/adxs ADXS +2.61% , a leader in developing the next generation of cancer immunotherapies, announced that it has been granted Orphan Drug Designation from the U.S. Food and Drug Administration (FDA) Office of Orphan Products Development (OOPD) for ADXS-HPV, its lead drug candidate, for the treatment of human papillomavirus (HPV)-associated head and neck cancer.
Orphan Drug Designation is granted to drug therapies intended to treat diseases or conditions that affect fewer than 200,000 people in the United States. Orphan Drug Designation entitles the sponsor to clinical protocol assistance with the FDA, as well as federal grants, tax credits, and potentially a seven year market exclusivity period.
“We are very pleased to have been granted an orphan drug designation for ADXS-HPV in this unmet medical need,” commented Dr. Robert Petit, Chief Scientific Officer of Advaxis. “Patients with head and neck cancer have limited treatment options and we hope to improve their survival by developing ADXS-HPV for this indication. We plan to initiate an additional Phase 1/2 study in early stage head and neck cancer for ADXS-HPV with a nationally recognized center of excellence, and we will continue the ongoing Phase 1 study being sponsored by the University of Liverpool and Aintree University Hospitals NHS Foundation Trust that is evaluating the safety and efficacy of ADXS-HPV when combined with standard chemotherapy and radiation treatment in patients with head and neck cancer.”
“Receiving orphan drug designation for ADXS-HPV in head and neck cancer is excellent news for a technology that may offer the potential to treat an indication with few therapy options, and, importantly, it helps define a clear path forward to registration,” commented Daniel J. O’Connor, President and Chief Executive Officer of Advaxis.
About Orphan Drug Designation
Under the Orphan Drug Act (ODA), the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. The benefits of orphan drug designation can be substantial and include federal grants, tax credits, and potentially a seven year market exclusivity period once the product is approved, provided that the product is first to market.
In order for a sponsor to obtain orphan designation for a drug or biological product, an application must be submitted to OOPD, and the designation approved. The approval of an application for orphan designation is based upon the information submitted by the sponsor. A drug that has obtained orphan designation is said to have “orphan status.” Each designation request must stand on its own merit. Sponsors requesting designation of the same drug for the same indication as a previously designated product must submit their own data in support of their designation request. The approval of an orphan designation request does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and efficacy of a compound must be established through adequate and well-controlled studies.
About ADXS-HPV
ADXS-HPV is an immunotherapy that is designed to target cells expressing the HPV gene E7. Expression of the E7 gene from high-risk HPV variants is responsible for the transformation of infected cells into dysplastic and malignant tissues. Eliminating these cells can eliminate the dysplasia or malignancy. ADXS-HPV is designed to infect antigen-presenting cells and direct them to generate a powerful, cellular immune response to HPV E7. The resulting cytotoxic Tcells infiltrate and attack the tumors while specifically inhibiting tumor Tregs and MDSCs in the tumors that are protecting it.
About Head and Neck Cancer
Cancer of the head and neck includes cancers arising from mucosa lining the oral cavity, oropharynx, hypopharynx, larynx, sinonasal tract, and nasopharynx. The most common histologic type observed is squamous cell carcinoma; therefore, the term “head and neck squamous cell carcinoma” (HNSCC) is frequently used to imply squamous cell carcinomas involving these anatomical sites. Excessive tobacco and alcohol are important risk factors for HNSCCs overall, but human papillomavirus (HPV) is now recognized as the causative agent in a subset of HNSCCs.
While the incidence of head and neck cancers that are linked to alcohol and tobacco use as the primary risk factor has fallen in the past three decades, a trend attributed to decreasing tobacco use in the United States, the incidence of HPV-associated head and neck cancer has been increasing. The increase was observed particularly among young individuals (<60 years of age), men, and Caucasians. Studies have shown that oral HPV infection is likely to be sexually acquired, as the increase in the incidence of HPV-associated head and neck cancers may be attributed to changing sexual practices. According to the World Health Organization’s Human Papillomavirus and Related Cancers in the World Summary Report 2010, HPV is associated with 20-50% of oral squamous cell carcinomas. HPV-associated head and neck cancer is growing at an epidemic rate in western countries; and occurs more frequently (3:1) in men than women. In the United States, the number of HPV-positive head and neck cancer cases has already equaled the number of cervical cancer cases.
About Advaxis, Inc.
Advaxis is a clinical-stage biotechnology company developing the next generation of immunotherapies for cancer and infectious diseases. Advaxis immunotherapies are based on a novel platform technology using live, attenuated bacteria that are bio-engineered to secrete an antigen/adjuvant fusion protein(s) that is designed to redirect the powerful immune response all human beings have to the bacterium to the cancer itself.
ADXS-HPV is currently being evaluated in four clinical trials for human papillomavirus (HPV)-associated cancers: recurrent/refractory cervical cancer (India), locally advanced cervical cancer (GOG/NCI U.S. study, Clinical Trials.gov Identifier NCT01266460), head & neck cancer (CRUK study, Clinical Trials.gov Identifier NCT01598792), and anal cancer (BrUOG study, Clinical Trials.gov Identifier NCT01671488). Advaxis has over 15 distinct immunotherapies in various stages of development, developed directly by Advaxis and through strategic collaborations with recognized centers of excellence such as: the University of Pennsylvania, the Georgia Regents University Cancer Center, Brown University Oncology Group, and others.
ADXS-HPV is currently in Phase 1/2 clinical development for recurrent/refractory and advanced cervical cancer, HPV caused head and neck cancers, and anal cancer.
Links to ADXS-HPV trials:
ADXS-HPV is an immunotherapy that is designed to target cells expressing the HPV gene E7. Expression of the E7 gene from high-risk HPV variants is responsible for the transformation of infected cells into dysplastic and malignant tissues. Eliminating these cells can eliminate the dysplasia or malignancy. ADXS-HPV is designed to infect antigen-presenting cells and direct them to generate a powerful, cellular immune response to HPV E7. The resulting cytotoxic Tcells infiltrate and attack the tumors while specifically inhibiting tumor Tregs and MDSCs in the tumors that are protecting it.
The American Cancer Society estimates that there will be about 12,340 newly diagnosed cervical cancer cases and 7,060 newly diagnosed cases of anal cancer in the U.S. in 2013.
In 2009, the CDC reported that about 45% of women aged 20 to 24 had HPV. HPV causes a number of different types of cancer. The same types of genital HPV that cause cervical cancer (HPV-16, HPV-18) cause about 8 out of 10 squamous cell anal cancers. In addition, nearly half of cancers of the vulva and about 7 out of 10 vaginal cancers are HPV-related. Some other genital cancers (cancers of the penis and urethra) and some head and neck cancers (mostly the throat, tongue, and tonsils) are also related to high-risk types of HPV. For additional information about HPV, please visit: http://www.cancer.org/.
back to home for more updates
DR ANTHONY MELVIN CRASTO Ph.D
Phase III data show Boehringer Ingelheim’s faldaprevir was highly effective in a broad range of patients with genotype-1 hepatitis C

faldaprevir , 801283-95-4 cas no, BI-201335
(1R,2S)-1-{[(2S,4R)-4-[{8-bromo-7-methoxy-2-[2-(2-methylpropanamido)-1,3-thiazol-4-yl]quinolin-4-yl}oxy]-1-[(2S)-2-{[(cyclopentyloxy)carbonyl]amino}-3,3-dimethylbutanoyl]pyrrolidine-2-carboxamido]-2-ethenylcyclopropane-1-carboxylic acid
Molecular Formula: C40H49BrN6O9S
Molecular Weight: 869.82 g.mol-1
2 nd nov 2013
Boehringer Ingelheim today announced new data from its Phase III clinical trial programme, STARTVerso™, which evaluates faldaprevir* in combination with pegylated interferon and ribavirin (PegIFN/RBV). Patients with genotype-1 (GT-1) hepatitis C (HCV) who have not received previous treatment (treatment-naïve: STARTVerso™1&2),1 treatment-experienced patients (STARTVerso™3),2 and HIV co-infected patients (STARTVerso™4)3 participated in this study programme. The results from these and additional studies will be presented at the 64th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD), also known as The Liver Meeting®, taking place 1-5 November in Washington, D.C.
Faldaprevir (formerly BI 201335) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byBoehringer-Ingelheim and is currently in Phase III trials.[1]
Faldaprevir is a hepatitis C virus protease inhibitor.
Faldaprevir is being tested in combination regimens with pegylated interferon and ribavirin, and in interferon-free regimens with other direct-acting antiviral agents including BI 207127.
Data from the SOUND-C2 study, presented at the 2012 AASLD Liver Meeting, showed that a triple combination of faldaprevir, BI 207127, and ribavirin performed well in HCV genotype 1b patients.[2] Efficacy fell below 50%, however, for dual regimens without ribavirin and for genotype 1a patients.
- Efficacy and Safety of BI 201335 (Faldaprevir) in Combination With Pegylated Interferon-alpha and Ribavirin in Treatment-naïve Genotype 1 Hepatitis C Infected Patients (STARTverso 1). Cliicaltrials.gov. March 6, 2013.
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
- Bioorganic & Medicinal Chemistry Letters, Volume 23, Issue 14, 15 July 2013, Pages 4267–4271
Synthesis and optimization of a novel series of HCV NS3 protease inhibitors: 4-Arylproline analogs
The following Compound 1):

(1)
wherein B is

; L° is MeO-; L1 is Br; and R2 is and having the chemical name: l-{ [4-[8-Bromo-2-(2-isopropylcarbamoyl-thiazol-4-yl)-7- methoxy-quinolin-4-yloxy]-l-(R)-(2-cyclopentyloxycarbonyl amino-3,3-(S)-dimethyl- butyryl)-pyrrolidine-(S)-2-carbonyl]-amino}-2-(S)-vinyl-cyclopropane-(R)-carboxylic acid, is known as a selective and potent inhibitor of the HCV NS3 serine protease and useful in the treatment of HCV infection. Compound (1) falls within the scope of the acyclic peptide series of HCV inhibitors disclosed in U.S. Patents RE 40,525, 7,514,557 and 7,585,845. Compound (1) is disclosed specifically as Compound # 1055 in U.S. Patent 7,585,845, and as Compound # 1008 in U.S. Patent 7,514,557. Compound (1), and pharmaceutical formulations thereof, can be prepared according to the general procedures found in the above-cited references, all of which are herein incorporated by reference in their entirety. Preferred forms of Compound (1) include the crystalline forms, in particular the crystalline sodium salt form, which can be prepared as described in U.S. Patent Application Publication No. 2010/0093792, also incorporated herein by reference. Data demonstrating the activity of Compound (1) as an inhibitor of the HCV NS3 serine protease and its corresponding demonstrated utility in the treatment of HCV infection in mono-infected patients, can be found in U.S. Patent 7,585,845, as well as in numerous publications presenting the preclinical characterization or clinical trial results with Compound (1). See, e.g., Sulkowski MS, et al, Hepatol (2009), Vol. 50, pg. 2A, Abtract LB3; Sulkowski MS, et al., J Hepatol (2010) Vol. 52, Supp. 1, pgs. S462-S463, Abstract 1190; Berg et al., Hepatol (2010), Vol. 52, Supp. SI, Abstract 804; and White PW, et al., Antimicrob Agents Chemother (2010) 54(11):4611-4618.
Combination therapy regimens directed to administering Compound (1) with an interferon- alpha and ribavirin for the treatment of HCV infection are described in U.S. Patent Application Publication Nos. 2010/0068182 and 2011/0268700.
HIV/HCV coinfected persons tend to have higher HCV viral loads and are less likely to clear the HCV spontaneously. The urgency for treatment of persons who are coinfected is greater than it is for those with HCV infection alone. The course of liver disease is more rapid in HIV/HCV-coinfected persons, including an approximately 2-fold increased risk of cirrhosis, more rapid progression to decompensated liver disease and increased risk for hepatocellular carcinoma (Graham CS, et al., Clin Infect Dis (2001 );33:562-569) .
Treatment of HCV might improve the tolerability of highly active antiretroviral therapy (HAART) because HCV infection increases the risk of mitochondrial toxicity and hepatotoxicity from HAART (Sulkowski MS, et al., JAMA (2000);283:74-80; Lafeuil!ade A, et al., Lancet (2001);357:280-281 ). Although there is much less published information on treatment outcomes in those who are HIV/HCV-coinfected than in HCV mono-infected patients, all accumulated data demonstrate that sustained virological response (SVR) and cure from HCV infection with pegylated interferon alpha and ribavirin is achieved in a substantially lower proportion of HIV/HCV coinfected patients when compared to HCV mono-infected patients. Factors associated with a poor treatment response (e.g., a high baseline HCV viral load, cirrhosis, and African American race) are present in a higher proportion of HIV/HCV coinfected populations, when compared to HCV monoinfected populations. It is not clear to what extent HIV infection itself diminishes the SVR rate, and to what extent advanced immunosuppression (e.g., CD4+ T lymphocyte count <200/mm3) further reduces response to HCV treatment (Toriani FJ, et al., N Engl J Med (2004);351(5): 438 -50; Nunez M, et al., ARHR (2007); 23(8):972-982).
Thus, there is a continuing high unmet need in the art for therapies that are effective against HCV in patients that are co-infected with HIV.
Solanezumab, Eli Lilly’s anti-beta-amyloid monoclonal antibody for Alzheimer’s disease

- immunoglobulin G1-kappa, anti-[Homo sapiens amyloid-beta (Abeta)
peptide soluble monomer], humanized monoclonal antibody;
gamma1 heavy chain [humanized VH (Homo sapiens IGHV3-23*04
(87.60%) -(IGHD)-IGHJ4*01) [8.8.5] (1-112) -Homo sapiens
IGHG1*01, CH3 K130>del (113-441)], (215-219′)-disulfide with
kappa light chain (1’-219’) [humanized V-KAPPA (Homo sapiens
IGKV2-30*01 (90.00%) -IGKJ1*01) [11.3.9] (1′-112′) -Homo sapiens
IGKC*01 (113′-219′)]; (221-221″:224-224″)-bisdisulfide dimer
neuroprotective agent
C6396H9922N1712O1996S42 955085-14-0
Heavy chain / Chaîne lourde / Cadena pesada
EVQLVESGGG LVQPGGSLRL SCAASGFTFS RYSMSWVRQA PGKGLELVAQ 50
INSVGNSTYY PDTVKGRFTI SRDNAKNTLY LQMNSLRAED TAVYYCASGD 100
YWGQGTLVTV SSASTKGPSV FPLAPSSKST SGGTAALGCL VKDYFPEPVT 150
VSWNSGALTS GVHTFPAVLQ SSGLYSLSSV VTVPSSSLGT QTYICNVNHK 200
PSNTKVDKKV EPKSCDKTHT CPPCPAPELL GGPSVFLFPP KPKDTLMISR 250
TPEVTCVVVD VSHEDPEVKF NWYVDGVEVH NAKTKPREEQ YNSTYRVVSV 300
LTVLHQDWLN GKEYKCKVSN KALPAPIEKT ISKAKGQPRE PQVYTLPPSR 350
DELTKNQVSL TCLVKGFYPS DIAVEWESNG QPENNYKTTP PVLDSDGSFF 400
LYSKLTVDKS RWQQGNVFSC SVMHEALHNH YTQKSLSLSP G 441
Light chain / Chaîne légère / Cadena ligera
DVVMTQSPLS LPVTLGQPAS ISCRSSQSLI YSDGNAYLHW FLQKPGQSPR 50
LLIYKVSNRF SGVPDRFSGS GSGTDFTLKI SRVEAEDVGV YYCSQSTHVP 100
WTFGQGTKVE IKRTVAAPSV FIFPPSDEQL KSGTASVVCL LNNFYPREAK 150
VQWKVDNALQ SGNSQESVTE QDSKDSTYSL SSTLTLSKAD YEKHKVYACE 200
VTHQGLSSPV TKSFNRGEC 219
Disulfide bridges location / Position des ponts disulfure / Posiciones de los puentes disulfuro
Intra-H 22-96 139-195 256-316 362-420
22”-96” 139”-195” 256”-316” 362”-420”
Intra-L 23′-93′ 139′-199′
23”’-93”’ 139”’-199”’
Inter-H-L 215-219′ 215”-219”’
Inter-H-H 221-221” 224-224”
N-glycosylation sites / Sites de N-glycosylation / Posiciones de N-glicosilación
292, 292
Solanezumab, Eli Lilly’s anti-beta-amyloid monoclonal antibody for Alzheimer’s disease
The market for Alzheimer’s disease therapies is set to nearly triple between 2012 and 2022, despite increasing genericisation and the fact that few new product launches are expected during this time, according to new forecasts.
The key driver of growth in the AD market will be Eli Lilly’s anti-beta-amyloid monoclonal antibody solanezumab, the first potentially disease-modifying therapy (DMT) to launch for AD, according to the study, from Decision Resources. It reports that solanezumab is expected to launch in the seven major pharmaceutical markets – the US, France, Germany, Italy, Spain, the UK and Japan – starting in 2018 and that, by 2022, the drug is forecast to attain sales in excess of $5 billion in these markets.
More than 85% of solanezumab’s projected total use in 2022 will be in the mild AD market – the population in which the drug is currently being tested – followed by the pre-AD 1-2 years market segment, says the firm, which defines this latter population as those patients who will go on to develop overt AD within the next one to two years.
Solanezumab (proposed INN) is a monoclonal antibody being investigated by Eli Lilly as a neuroprotector[1] for patients withAlzheimer’s disease.[2][3]
It binds to the amyloid-β peptides that make up the protein plaques seen in the brains of people with the disease.
2012 results of the EXPEDITION 1 & 2 phase 3 clinical trials were only mildly encouraging.[4][5][6] but were said to be the “first evidence that targeting the amyloid cascade can slow the progression of disease.”[7]
- International Nonproprietary Names for Pharmaceutical Substances (INN, prepublication copy), World Health Organization.
- ClinicalTrials.gov NCT00749216 Solanezumab Safety Study in Japanese Patients With Alzheimer’s Disease
- ClinicalTrials.gov NCT00905372 Effect of LY2062430 on the Progression of Alzheimer’s Disease (EXPEDITION)
- “Lilly’s Solanezumab Slows Down Alzheimer’s Progression”. 9 Oct 2012.
- Solanezumab Did it actually work
- “Eli Lilly’s solanezumab faces grim prospects of attaining conditional FDA approval in mild Alzheimer’s”. 4 Sep 2012.
- “ALZHEIMER’S DRUG SLOWS MEMORY LOSS BY ONE THIRD”. 10 Oct 2012.

yellow coloured SOLANEZUMAB blocks beta amyloid from aa 16 to aa 25

Amyloid precursor protein (APP)
