
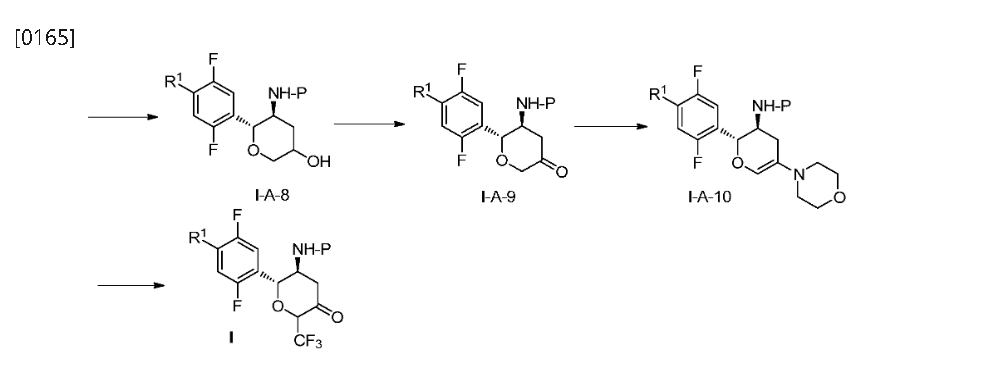
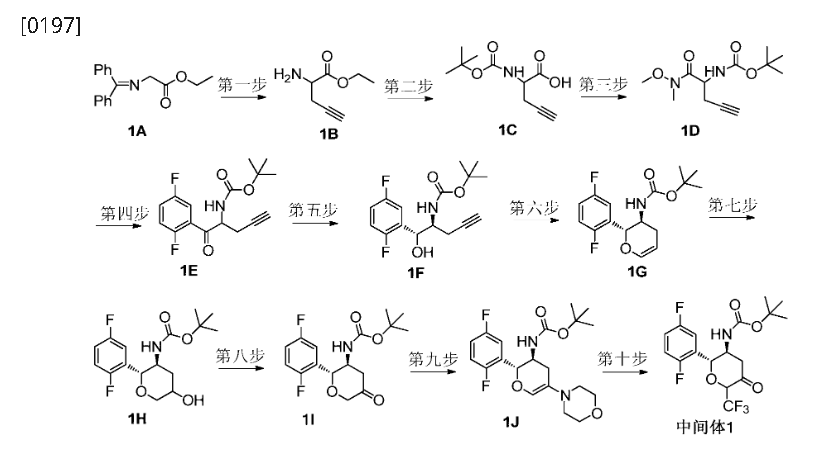
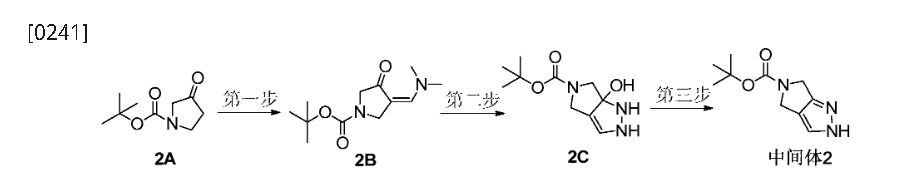
Home » Posts tagged 'DIABETES'
Tag Archives: DIABETES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Rongliflozin, Olorigliflozin
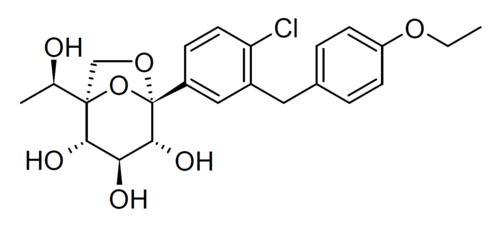
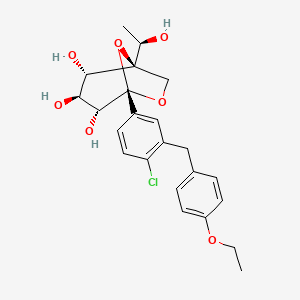
Rongliflozin
Olorigliflozin, 6FP3NST6ZQ, DJT1116PG
Cas 2035989-50-3
450.9 g/mol, C23H27ClO7
(1R,2S,3S,4R,5S)-5-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-[(1R)-1-hydroxyethyl]-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- (1R,2S,3S,4R,5S)-5-(4-Chloro-3-(4-ethoxybenzyl)phenyl)-1-((R)-1-hydroxyethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- 1,6-Anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-beta-L-idopyranose
- beta-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-
Rongliflozin 화학구조
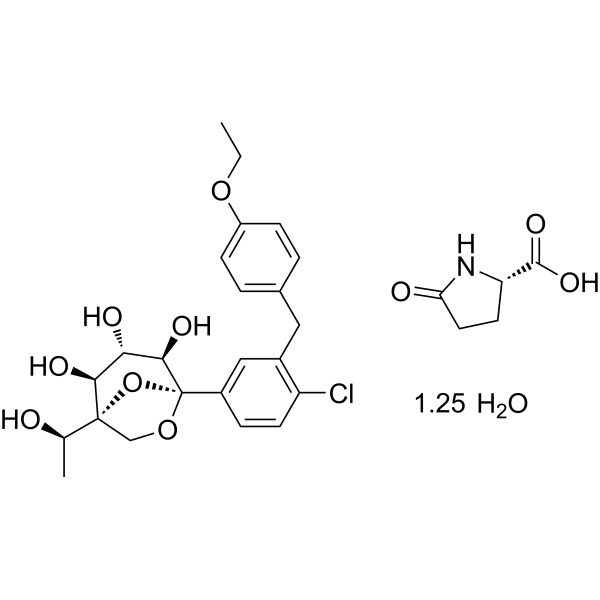
CAS No. : 2648020-91-9
| MW | 602.55 |
|---|---|
| MF | C23H27ClO7.C5H7NO3.5/4H2O |
- OriginatorHEC Pharm
- DeveloperSunshine Lake Pharma
- ClassAntihyperglycaemics; Small molecules
- Mechanism of ActionSodium-glucose transporter 2 inhibitors
- PreregistrationType 2 diabetes mellitus
- 04 Sep 2025Chemical structure information added.
- 31 Dec 2023Preregistration for Type 2 diabetes mellitus in China (PO), in December 2023
- 31 Dec 2023Efficacy and adverse events data from a phase IIIa trial in Type 2 diabetes mellitus released by Sunshine Lake Pharma, before December 2023
Rongliflozin is an SGLT2 inhibitor developed as a potential treatment for diabetes.[1][2]
Rongliflozin (DJT1116PG) is a selective and orally active inhibitor of sodium-glucose co-transporter-2 (SGLT-2). Rongliflozin can be used for the research of type 2 diabetes mellitus (T2DM).
PAT
- (1R,2S,3S,4R,5S)-5-(4-Chloro-3-(4-ethoxybenzyl)phenyl)-1-((R)-1-hydroxyethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- 1,6-Anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-beta-L-idopyranose
- beta-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-
- Complexes of glucopyranosyl derivatives and methods for their preparation and usePublication Number: JP-2018535237-APriority Date: 2015-11-27
- Complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: US-10555930-B2Priority Date: 2015-11-27Grant Date: 2020-02-11
- Complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: US-2018344689-A1Priority Date: 2015-11-27
- A complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: WO-2017088839-A1Priority Date: 2015-11-27
- Glucopyranosyl derivative complex and its preparation method and usePublication Number: JP-6916180-B2Priority Date: 2015-11-27Grant Date: 2021-08-11
- Preparation method and intermediate of glucopyranosyl derivativesPublication Number: CN-113195510-BPriority Date: 2019-01-08Grant Date: 2022-12-23
- Crystalline forms of glucopyranosyl derivativesPublication Number: CN-107778336-BPriority Date: 2016-08-24Grant Date: 2022-09-27
- Glucopyranosyl derivative compound, preparation method and applicationPublication Number: CN-106810582-APriority Date: 2015-11-27
- Glucopyranosyl derivative compound, preparation method and applicationPublication Number: CN-106810582-BPriority Date: 2015-11-27Grant Date: 2019-12-31
- A complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: EP-3371199-A1Priority Date: 2015-11-27
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: WO-2022007838-A1Priority Date: 2020-07-08
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: EP-4178970-A1Priority Date: 2020-07-08
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: US-2023250121-A1Priority Date: 2020-07-08
- Preparation methods of glucopyranosyl derivatives and intermediates thereofPublication Number: CN-113912567-BPriority Date: 2020-07-08Grant Date: 2024-01-16
- Preparation method for glucopyranosyl derivative and intermediate thereofPublication Number: WO-2020143653-A1Priority Date: 2019-01-08
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: WO-2022036506-A1Priority Date: 2020-08-17
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: EP-4197543-A1Priority Date: 2020-08-17
- Compositions of SGLT-2 inhibitors and angiotensin receptor antagonists and uses thereofPublication Number: KR-20230057388-APriority Date: 2020-08-17
- Composition and application of SGLT-2 inhibitor and angiotensin receptor blockerPublication Number: CN-116490178-APriority Date: 2020-08-17
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: US-2023346817-A1Priority Date: 2020-08-17
- Nintedanib targeted combinationPublication Number: CN-118021812-APriority Date: 2023-12-30
- Preparation method of L-pyroglutamic acid co-crystal of glucopyranosyl derivativesPublication Number: CN-115141235-APriority Date: 2021-03-30
- Preparation method of L-pyroglutamic acid cocrystal of pyranose glucopyranose derivativePublication Number: CN-115141235-BPriority Date: 2021-03-30Grant Date: 2024-08-09
- Fixed-dose combination of sglt-2 inhibitor and angiotensin converting enzyme inhibitor, and use thereofPublication Number: WO-2022104621-A1Priority Date: 2020-11-19
- Compositions and uses of fixed-dose SGLT-2 inhibitors and angiotensin-converting enzyme inhibitorsPublication Number: CN-116234545-APriority Date: 2020-11-19
SYN
https://pubs.rsc.org/en/content/articlelanding/2021/ce/d1ce01305j/unauth
Rongliflozin L-pyroglutamic acid, a highly active SGLT-2 inhibitor cocrystal discovered and developed by our group, is currently undergoing clinical trials for the treatment of diabetes. Here, we report and design a simple and robust process to obtain a single and pure crystalline form I (1) of the cocrystal, containing Rongliflozin (2) with L-pyroglutamic acid (L-PA), based on coformer-induced purification (CoIP). Extensive experiments showed that the addition of L-pyroglutamic acid in the eluent was key to suppression of the dissociation equilibrium of the cocrystal during lessivation, with high efficiency. Importantly, based in this profile, this process exhibited strong robustness and margin of safety at multigram and multikilogram scales
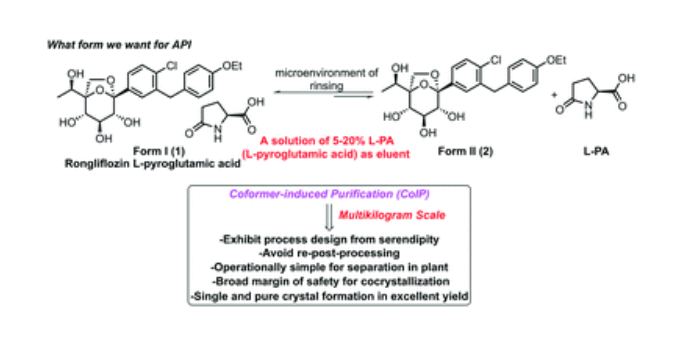
Kilogram scale Process of 1
A mixture of (1R,2S,3S,4R,5S)-5-(4-chloro-3-(4-ethoxybenzyl) phenyl)-1-((R)-1-
hydroxyethyl)-6,8-dioxabicyclo [3.2.1] octane-2,3,4-triol ethanolate form III (3) (23.45 kg, 47.3
mol), L-pyroglutamic acid (24.31 kg, 4.0 equiv.), EtOH (35.9 L) and H2O (70 L) was added into a
300 L reactor at room temperature. The slurry was heated to 65 °C and stirred until it is clear. The
clear solution was cooled to 35±5 °C typically. Seed crystal form I (1) (0.70 kg, 3% g/g) was added
when the solution was cooled to 34 °C and maintained for 1.5 h. Gradually, the slurry was cool to
30 °C and 25 °C in 3 hours, and finally stirred at 25 °C for 24 h. The slurry was collected on a
centrifuge filter. The filter cake was washed with a mixed solution of EtOH (31.3 L)/H2O (62.7 L)
with L-pyroglutamic acid (1.64 kg, 7% g/g) pre-cooled to -15°C. The wet cake was dried under
vacuum at 45 °C for 8 h. Pure cocrystal form I (1) was obtained as a white solid (24.91 kg, yield
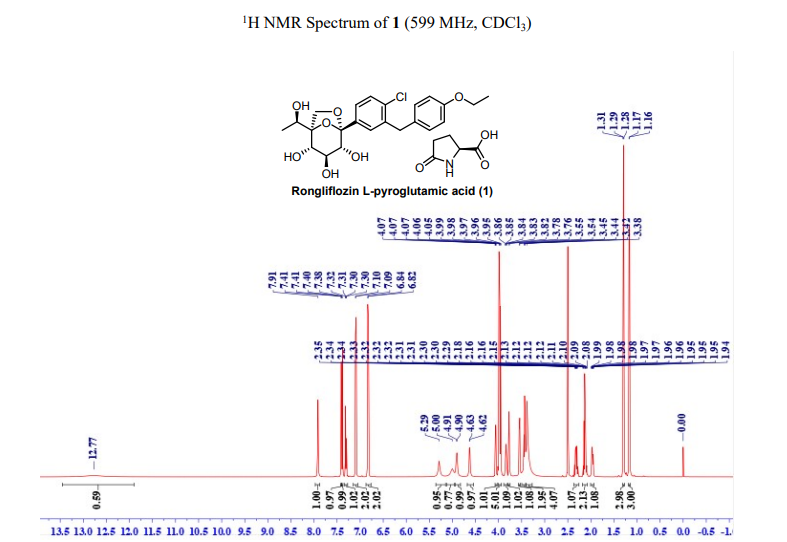
91%). MP (DSC onset) = 96.91 ℃. 1H NMR (599 MHz, DMSO-d6) δ 12.77 (br, 1H), 7.91 (s, 1H),
7.41 (d, J = 2.0 Hz, 1H), 7.39 (d, J = 12.0 Hz, 1H), 7.31 (dd, J = 12.0, 2.0 Hz, 1H), 7.10 (d, J = 2.0
Hz , 2H), 6.83 (d, J = 2.0 Hz, 2H), 5.29 (s, 1H), 5.00 (s, 1H), 4.91 (d, J = 6.7 Hz, 1H), 4.63 (d, J =
6.1 Hz, 1H), 4.06 (dd, J = 12.0, 6.0 Hz, 1H), 3.99– 3.95 (m, 5H), 3.84 (p, J = 6.0 Hz, 1H), 3.77 (d,
J = 12.0 Hz, 1H), 3.55 (d, J = 6.0 Hz, 1H), 3.44 (t, J = 12.0 Hz, 2H), 3.38 (s, 4H), 2.35-2.29 (m,
1H), 2.18-2.08 (m, 2), 1.99-1.94 (m, 1H), 1.29 (t, J = 12.0 Hz, 3H), 1.17 (d, J = 6.0 Hz, 3H). 13C
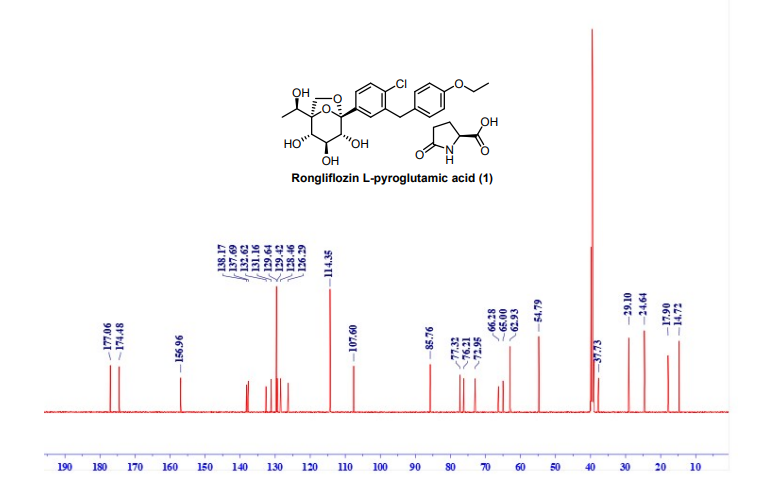
NMR (151 MHz, DMSO-d6) δ 177.06, 174.48, 156.96, 138.17, 137.69, 131.16, 129.64, 129.42,
128.46, 126.29, 114.35, 107.60, 85.76, 77.32, 76.21, 72.95, 66.28, 65.00, 62.93, 54.79, 37.73, 29.10,
24.64, 17.90, 14.72. HRMS: (ESI) Calcd for C23H27ClO7 [M+NH4]+: 468.1784, C5H7NO3 [M+H]+
:130.0499; Found: 468.1774, 130.0490 respectively. IR (KBr, cm-1): 3257, 2986, 2927, 1750, 1648,
1513, 1476, 1371, 1264, 1239, 1223, 1206, 1088, 1061, 821
13C NMR



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Zhang H, Liu J, Zhu X, Li X, Chen H, Wu M, et al. (May 2020). “A Phase I Study on the Pharmacokinetics and Pharmacodynamics of DJT1116PG, a Novel Selective Inhibitor of Sodium-glucose Cotransporter Type 2, in Healthy Individuals at Steady State”. Clinical Therapeutics. 42 (5): 892–905.e3. doi:10.1016/j.clinthera.2020.03.007. PMID 32265061.
- Zhang H, Zhu X, Li X, Chen H, Wu M, Li C, et al. (February 2020). “Pharmacokinetics and pharmacodynamics of rongliflozin, a novel selective inhibitor of sodium-glucose co-transporter-2, in people with type 2 diabetes mellitus”. Diabetes, Obesity & Metabolism. 22 (2): 191–202. doi:10.1111/dom.13887. PMID 31588657.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2035989-50-3 |
| PubChem CID | 122660464 |
| UNII | 6FP3NST6ZQ |
| ChEMBL | ChEMBL5314927 |
| Chemical and physical data | |
| Formula | C23H27ClO7 |
| Molar mass | 450.91 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Rongliflozin, diabetes, Olorigliflozin, 6FP3NST6ZQ, 2035989-50-3, DJT1116PG, DJT 1116PG,
Velagliflozin
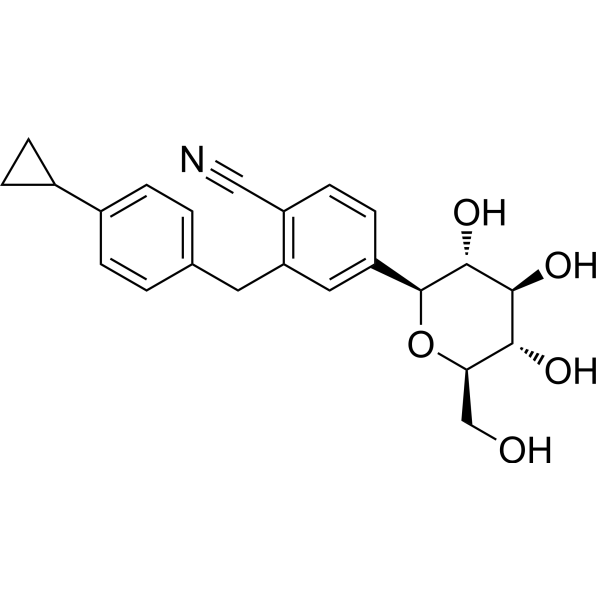
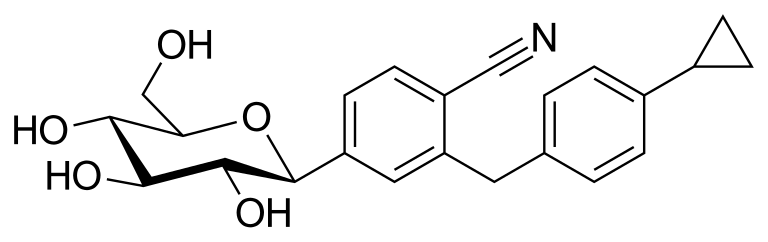
Velagliflozin
VETERINARY DRUG
- Cas 946525-65-1
- FV2YU8SL0P
- 2-((4-cyclopropylphenyl)methyl)-4-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl)benzonitrile
- 2-((4-Cyclopropylphenyl)methyl)-4-beta-D-glucopyranosylbenzonitrile
- 395.4 g/mol, C23H25NO5
2-[(4-cyclopropylphenyl)methyl]-4-[(2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]benzonitrile
- 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYLBENZONITRILE
- BENZONITRILE, 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYL-
Velagliflozin L-proline H2O
Velagliflozin, sold under the brand name Senvelgo, is an antidiabetic medication used for the treatment of cats.[2][4][5] Velagliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor.[6] It is taken by mouth.[2]
Velagliflozin is the active ingredient of the first oral liquid medication approved by the Food and Drug Administration for the treatment of diabetes in cats. This compound belongs to the known class of sodium-glucose cotransporter 2 inhibitors approved to treat diabetes in human.
- Application: NADA 141-568Drug: Senvelgo®Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Patent(s): 7776830 (Exp: 05/01/2027); 8557782 (Exp: 05/01/2027); 9145434 (Exp: 09/07/2033); 10617666 (Exp: 06/06/2035); 11896574 (Exp: 12/17/2034); 10220017 (Exp: 09/29/2036); 10709683 (Exp: 08/24/2036); 11225500 (Exp: 12/17/2038)
- [Indication for Use] To improve glycemic control in otherwise healthy cats with diabetes mellitus not previously treated with insulin.Application: NADA 141-568Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Freedom of Information: FOIA Summary 14320Approval Date: August 10, 2023
APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO
Velagliflozin (brand name Senvelgo) is a veterinary medication approved for treating diabetes in cats, not humans.
Approved countries and years for velagliflozin:
- United States (US): Approved by the FDA in August 2023.
- European Union (EU): Received marketing authorization in November 2023.
- Switzerland: Approved in 2023.
- Great Britain: Approved in 2023.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US310904480&_cid=P11-METCZG-99171-1
SYN
US7776830
https://patentscope.wipo.int/search/en/detail.jsf?docId=US41880220&_cid=P11-METD0X-00376-1
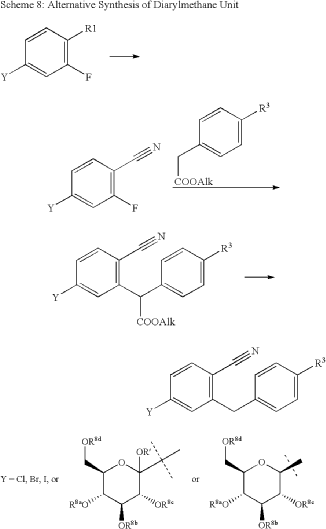
| The following compound is obtained analogously to Example XXIV: |
(1) 1-Cyano-2-(4-cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzene
EXAMPLE 17
2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile
| The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner. |
SYN
WO2007128749
https://patents.google.com/patent/WO2007128749A1/en
The following compound is obtained analogously to Example XXIV:
(1 ) 1 -Cvano-2-(4-cvclopropyl-benzyl)-4-(3-D-glucopyranos-1 -vD-benzene

Mass spectrum (ESI“): m/z = 413 [M+H] + Advantageously, the reduction of the anomeric carbon center of the appropriate intermediate obtained during the synthesis of this compound is conducted with the oxygen functionalities on the pyranose ring protected. Preferred protective groups are benzyl, p-methoxybenzyl, trimethylsilyl, triethylsilyl, terfbutyldimethylsilyl, triisopropylsilyl and allyl.
Example XXV

1-Cyano-2-(4-cyclopropyl-benzyl)-4-(tetra-O-acetyl-β-D-glucopyranos-1-yl)-benzene To a flask charged with a stir bar, 4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1-yl)-2-(4- trifluoromethylsulfonyloxy-benzyl)-benzonitrile (4.4 g), degassed toluene (12 ml.) and degassed water (8 ml.) and kept under argon atmosphere is added cyclopropylboronic acid (0.20 g), potassium phosphate (5.0 g), tricyclohexylphosphine (0.19 g) and at last palladium(ll)acetate (76 mg). The mixture is stirred at 1 10 °C for 6 h meanwhile cyclopropylboronic acid is added after each hour (5x 0.20 g). After cooling to room temperature, the mixture is diluted with aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is removed under reduced pressure. The residue is chromatographed on silica gel (cyclohexane/ethyl acetate 20:1 -> 1 :1 ). Yield: 3.2 g (87% of theory ) Mass spectrum (ESI+): m/z = 581 [M+NH4] +
Example XXVI

4-(1 -Hvdroxy-cvclopropyD-phenylboronic acid A 3.0 M solution of ethylmagnesium bromide in diethylether (7.6 ml.) is added to a stirred solution of titanium(IV) isopropoxide (2.2 ml.) in diethylether (70 ml.) chilled to -78 °C. The resultant solution is stirred at -78 °C for 1.5 h, before 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa borolan-2-yl)-benzoic acid methyl ester (2.0 g) is added. The reaction mixture is warmed to ambient temperature and stirred for an additional 12 h. Then, 1 M aqueous hydrochloric acid is added and the resulting mixture is extracted with ethyl acetate. The combined organic extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is dissolved in acetone (60 ml.) and 0.1 M aqueous NH4OAc solution (50 ml.) followed by NaIO4 (2.3 g) is added. The resulting reaction mixture is stirred at room temperature for 18 h. After removal of the acetone, the residue is extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is purified by chromatography on silicagel (cyclohexane/ethyl acetate). Yield: 0.45 g (33% of theory) Mass spectrum (ESI“): m/z = 223 [M+HCOO]“ Preparation of the end compounds:
Example 17: 2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile

Mass spectrum (ESI+): m/z = 413 [M+NH4]+
The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner.
Yield: 83% of theory
Alternatively this compound is obtained as described in Example XXIV(I ).
The compound of example 17 is also obtained by employing the following procedure:
A solution of 2-(4-cyclopropyl-benzyl)-4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1 -yl)- benzonitrile (0.80 g) in methanol (5 ml.) and THF (5 ml.) is treated with aqueous potassium hydroxide solution (4 mol/l, 5 ml_). The reaction solution is stirred at ambient temperature for 1 h and then neutralized with 1 M hydrochloric acid. The organic solvents are evaporated and the residue is diluted with brine and extracted with ethyl acetate. The organic extracts are dried (sodium sulphate) and the solvent is removed. The residue is chromatographed on silica gel (dichloromethane/methanol 1 :0 -> 9:1 ). Yield: 0.54 g (96% of theory)
SYN
Synthesis 2024, 56, 906–943
In 2007, Boehringer-Ingelheim Vetmedica GmbH pioneered the development of velagliflozin (15), subsequently submitting a patent application in the United States with the identification number US7776830B2.72a More recently, through clinical investigations, this compound has demonstrated its efficacy as an SGLT2 inhibitor, proving adept at curtailing glucose reabsorption, encouraging glucosuria,
and leading to reductions in both blood glucose and insulin levels.
The initial synthesis of velagliflozin (15) was also disclosed in the above patent,72a and in patent
WO2007128749A1.72b The synthesis, depicted in Scheme46, comprises of nine-steps starting with the readily available raw material 2-bromo-5-iodobenzoic acid (250), which undergoes reduction using LiBH4 to form the corresponding alcohol 251. Subsequently, chlorination is carried out using thionyl chloride, resulting in the formation of chloride 252. O-Alkylation of phenol with compound 252 is
then conducted in a basic medium, yielding intermediate 253.The C-glycosylation of 253 with 2,3,4,6-tetrakis-O(trimethylsilyl)-D-glucopyranone 22 in the presence of turbo Grignard reagent (isopropylmagnesium chloride and LiCl) and methanesulfonic acid in methanol gives compound
254 with an impressive 93% yield. The hydroxy group of in termediate 254 is protected using acetic anhydride, and themethoxy group is subsequently removed via Lewis acid (BF3·Et2O, Et3SiH) treatment, providing compound 255 in a yield of 60%. A metal-catalyzed cyano group installation is then performed on intermediate 255, leading to the formation of compound 256 in 84% yield. The subsequent steps involve benzylic bromination followed by coupling with cyclopropylphenyl boronic acid 260, resulting in the formation of intermediate 258. Finally, deacetylation of intermediate 258 using aqueous KOH produces the desired product
The overall yield obtained for velagliflozin (15) is calculated to be 11.3%, with this synthetic route providing a systematic and efficient approach. The highlight of the route is high-yielding chemical transformations. However, the drawback is the use of two palladium-mediated couplings
that increase the possibility of leaching of the toxic metal in scale-up batches. Additionally, the synthetic route requires a large number of chemical transformations and not best suited for commercial production.
The same authors reported an alternative method (Scheme 47) for the synthesis of velagliflozin (15) in the product patent.72 The aglycone intermediate 265 is accessed in two steps starting from ethyl 2-(4-bromophenyl)acetate (262). O-Glycosylation takes place with the aglycone
4-bromo-2-(4-cyclopropylbenzyl)benzonitrile (265) using 2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone 22 in the presence of tert-butyllithium in pentane (1.7 M), resulting in the formation of compound 266. Reduction of compound 266 using boron trifluoride–diethyl etherate yields
the final API velagliflozin (15). This truncated synthetic route is well suited for scale-up due to the significantly low er number of transformations compared to the previous route. Unfortunately, the specific yields were not clearly in dicated for this process. This method presents an alternative approach to the synthesis of velagliflozin (15), providing a potential pathway for its preparation in 5 steps with
an overall yield of 40%.
(72) (a) Eckhardt, M.; Himmelsbach, F.; Eickelmann, P.; Sauer, A.;
Thomas, L. US7776830B2, 2010. (b) Eckhardt, M.; Himmelsbach,
F.; Eickelmann, P.; Sauer, A.; Thomas, L. WO2007128749A1,
2007.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Velagliflozin is indicated to improve glycemic control in otherwise healthy cats with diabetes not previously treated with insulin.[2][4][6]
References
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- “Senvelgo- velagliflozin solution”. DailyMed. 8 November 2023. Retrieved 13 December 2023.
- “Senvelgo Product information”. Union Register of veterinary medicinal products. 22 November 2023. Retrieved 29 August 2024.
- “NADA 141-568 Senvelgo (velagliflozin oral solution) Cats”.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Cook AK, Behrend E (January 2025). “SGLT2 inhibitor use in the management of feline diabetes mellitus”. Journal of Veterinary Pharmacology and Therapeutics. 48 Suppl 1 (Suppl 1): 19–30. doi:10.1111/jvp.13466. PMC 11736986. PMID 38954371.
- “Dear Veterinarian Letter regarding important safety conditions associated with the use of Senvelgo (velagliflozin oral solution) for improving glycemic control in certain cats with diabetes mellitus”. U.S. Food and Drug Administration. 4 December 2023. Retrieved 13 December 2023. This article incorporates text from this source, which is in the public domain.
| Clinical data | |
|---|---|
| Trade names | Senvelgo |
| License data | US DailyMed: Velagliflozin |
| Routes of administration | By mouth |
| ATCvet code | QA10BK90 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 946525-65-1 |
| PubChem CID | 24862817 |
| ChemSpider | 58827717 |
| UNII | FV2YU8SL0PEQE2P2T77I |
| Chemical and physical data | |
| Formula | C23H25NO5 |
| Molar mass | 395.455 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous systemPublication Name: Translational NeurodegenerationPublication Date: 2024-08-09PMCID: PMC11312905PMID: 39123214DOI: 10.1186/s40035-024-00431-y
- Demographic, morphologic, hormonal and metabolic factors associated with the rate of improvement from equine hyperinsulinaemia-associated laminitisPublication Name: BMC Veterinary ResearchPublication Date: 2022-01-18PMCID: PMC8764787PMID: 35042535DOI: 10.1186/s12917-022-03149-z
- The efficacy and safety of velagliflozin over 16 weeks as a treatment for insulin dysregulation in poniesPublication Name: BMC Veterinary ResearchPublication Date: 2019-02-26PMCID: PMC6390376PMID: 30808423DOI: 10.1186/s12917-019-1811-2
- The sodium-glucose co-transporter 2 inhibitor velagliflozin reduces hyperinsulinemia and prevents laminitis in insulin-dysregulated poniesPublication Name: PLOS ONEPublication Date: 2018-09-13PMCID: PMC6136744PMID: 30212530DOI: 10.1371/journal.pone.0203655
- Effects of the sodium‐glucose cotransporter 2 (<scp>SGLT</scp>2) inhibitor velagliflozin, a new drug with therapeutic potential to treat diabetes in catsPublication Name: Journal of Veterinary Pharmacology and TherapeuticsPublication Date: 2017-11-15PMID: 29139146DOI: 10.1111/jvp.12467
/////////Velagliflozin, APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO, DIABETES, SENVELGO,
Cetagliptin
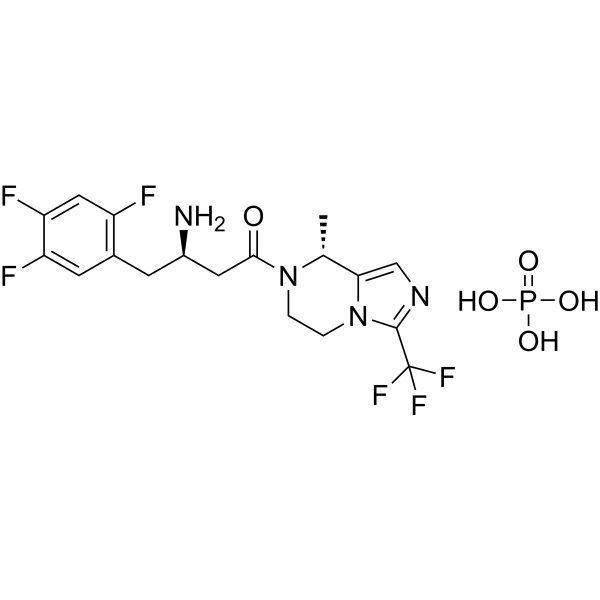
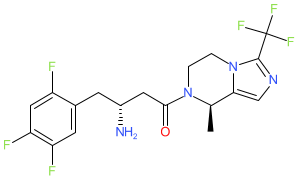
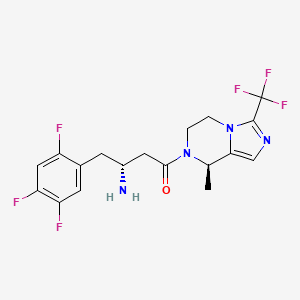
Cetagliptin
CAS No. FREE FORM : 2243737-33-7 C18H18F6N4O, 420.4 g/mol
[ Cetagliptin Phosphate 2243737-33-7 ]
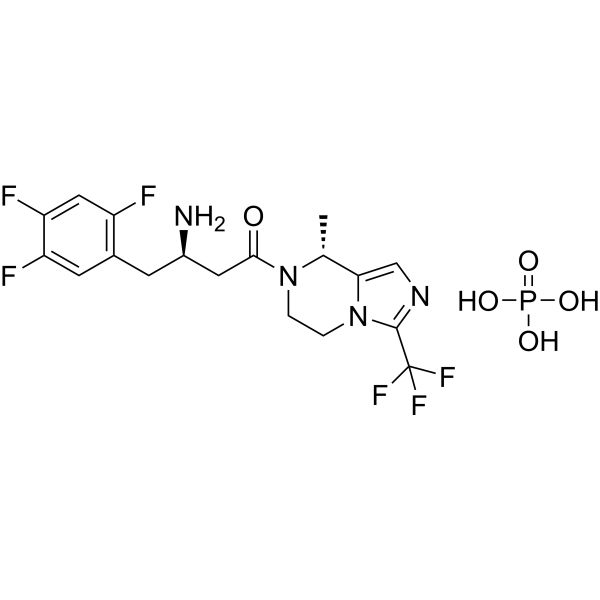
| 분자량 MW | 518.35 |
|---|---|
| 화학식 MF | C18H21F6N4O5P |
(3R)-3-amino-1-[(8R)-8-methyl-3-(trifluoromethyl)-6,8-dihydro-5H-imidazo[1,5-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one
(3R)-3-amino-1-[(8R)-8-methyl-3-(trifluoromethyl)-6,8-dihydro-5H-imidazo[1,5-a]pyrazin-7-yl]-4-(2,4,5-trifluorophenyl)butan-1-one
CHINA 2024, APPROVALS 2024, CGeneTec, DIABETES,
- GTPL13952
- CGT8012
- OriginatorCGeneTech
- Class2 ring heterocyclic compounds; Amines; Antihyperglycaemics; Fluorobenzenes; Imidazoles; Ketones; Pyrazines; Small molecules
- Mechanism of ActionDipeptidyl peptidase 4 inhibitors
RegisteredType 2 diabetes mellitus CHINA 2024
- 01 Dec 2024Registered for Type 2 diabetes mellitus in China (PO) – First global approval
- 20 Mar 2024Chemical structure information added
- 28 Jun 2023No recent reports of development identified for phase-I development in Type-2-diabetes-mellitus(In volunteers) in China (PO, Tablet)
- Cetagliptin is an orally active inhibitor for dipeptidyl peptidase 4 (DPP-4) and CYP2D6 (IC50 of 6 µM). Cetagliptin is a substrate for P-glycoprotein. Cetagliptin reduces the GLP-1 degradation, maintains the level of postprandial blood sugar, and can be used in type 2 diabetes mellitus research.
Cetagliptin (CGT-8012) is an orally bioavailable, dipeptidyl peptidase 4 enzyme (DPP-4) inhibitor (‘gliptin’) class drug. It was designed as an antihyperglycemic agent to treat type 2 diabetes mellitus (T2DM) via inhibition of DPP-4-mediated catbolism of incretin hormones including glucagon-like peptide-1 (GLP-1) [2].
- A DPP-4 inhibitor pharmaceutical composition and its preparation method and usePublication Number: CN-118557538-APriority Date: 2024-08-01
- A kind of preparation method of DPP-IV inhibitor and its key intermediatePublication Number: CN-114057751-APriority Date: 2022-01-17
- A kind of preparation method of DPP-IV inhibitor and its key intermediatePublication Number: CN-114057751-BPriority Date: 2022-01-17Grant Date: 2022-04-12
- A kind of preparation method of DPP-IV inhibitor and key intermediate thereofPublication Number: TW-202330535-APriority Date: 2022-01-17
- A preparation method of a DPP-IV inhibitor and its key intermediatePublication Number: TW-I842342-BPriority Date: 2022-01-17Grant Date: 2024-05-11
- Salt of cetagliptin, preparation method thereof, pharmaceutical composition, and use thereofPublication Number: US-2020123164-A1Priority Date: 2018-04-26
- Salt of cetagliptin, preparation method therefor, pharmaceutical composition, and use thereofPublication Number: EP-3785713-A1Priority Date: 2018-04-26
- Salt of cetagliptin, preparation method thereof, pharmaceutical composition, and use thereofPublication Number: US-11046701-B2Priority Date: 2018-04-26Grant Date: 2021-06-29
- Tetrahydro-imidaz0[1,5-a]pyrazine derivatives, preparation process and medicinal use thereofPublication Number: US-2010273786-A1Priority Date: 2007-12-26
- Tetrahydro-imidazo[1,5-α]pyrazine derivatives, preparation process and medicinal use thereofPublication Number: US-8207161-B2Priority Date: 2007-12-26Grant Date: 2012-06-26
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84092509&_cid=P20-MERZ31-36806-1
SYN
CN103351391
https://patents.google.com/patent/CN103351391A/en
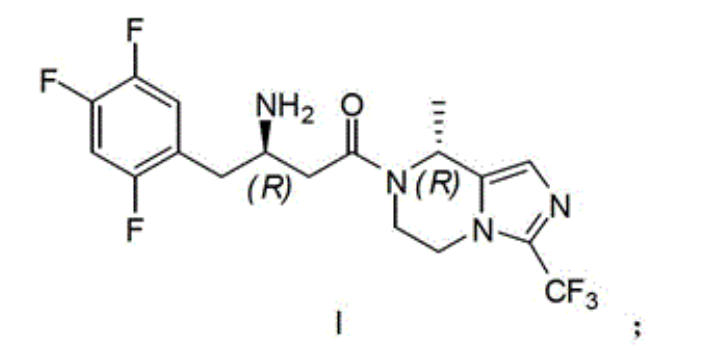
Synthetic route and the concrete steps of compound (I) are as follows:
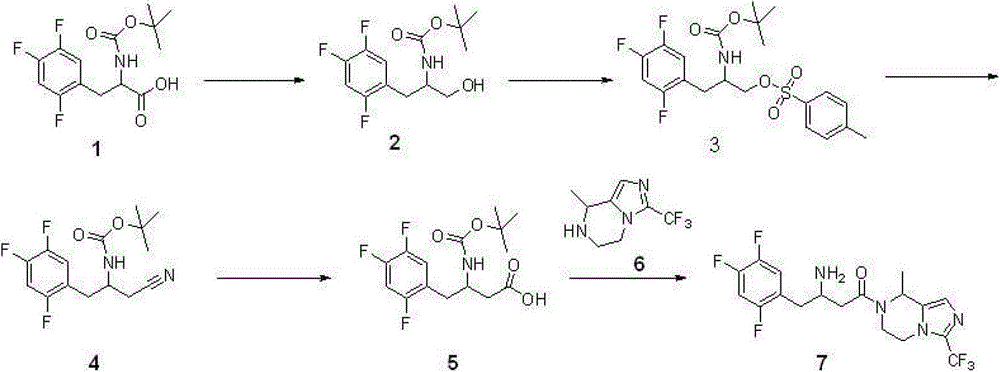
Step 1: synthetic compound 2
With 11.8 gram (0.037 mole) compound 1{DL N-[(1,1-dimethyl oxyethyl group) carbonyl]-2,4; 5-trifluorophenyl-L-Ala, DL N-[(1,1-dimethylethoxy) carbonyl]-2; 4; 5-trifluorophen yl-alanine, CAS:1367740-01-9, reference: synthetic chemistry; 2011; 19 (4), 557-560} is dissolved among 40 milliliters of THF, adds 5.8 milliliters of triethylamines (0.042 mole) again; reaction is cooled to 0 ℃; add 4.0 milliliters of Vinyl chloroformates (0.041 mole), 0 ℃ was reacted 1 hour under nitrogen protection, after the filtration filtered liquid was cooled to 0 ℃; slowly add sodium borohydride (1.4 grams; 0.057 the mole) mixed solution in 15 ml waters, stirring is spent the night, and adds 1N HCl acidifying; ethyl acetate extraction three times; merge organic phase, sodium hydrogen carbonate solution is washed, the saturated salt washing; anhydrous sodium sulfate drying; the concentrated 7.6 gram products that obtain, namely compound 2, yield 67%.Repeat this step, make more compound 2, use for subsequent step.
Step 2: synthetic compound 3
8.2 gram (0.027 mole) compounds 2 are dissolved in 40 milliliters of methylene dichloride; add again 4.2 milliliters of triethylamines (0.030 mole); the catalytic amount DMAP; reaction is cooled to 0 ℃; add Tosyl chloride (6.8 grams; 0.035 mole); 0 ℃ is arrived room temperature reaction 2 hours under nitrogen protection, adds 1N HCl acidifying, dichloromethane extraction three times; merge organic phase; sodium hydrogen carbonate solution is washed, saturated salt washing, anhydrous sodium sulfate drying; concentrate and obtain crude product, namely compound 3.Repeat this step, make more compound 3, use for subsequent step.
Step 3: synthetic compound 4
12.4 gram (0.027 mole) compounds 3 are dissolved in 40 milliliters of dimethyl formamides, slowly add the mixed solution of sodium cyanide (4.5 grams, 0.092 mole) in 30 milliliters of dimethyl formamides, room temperature reaction 48 hours, pour in 100 milliliters of frozen water, ethyl acetate extraction three times merges organic phase, the saturated salt washing, anhydrous sodium sulfate drying, concentrated rear column chromatography purification obtains 7.8 gram products, be compound 4, yield 92%.
Step 4: synthetic compound
5
3.1 gram (0.010 mole) compounds 4 are dissolved in 15 milliliters of 6N hydrochloric acid, and reflux is spent the night, and adds the neutralization of 2N sodium hydroxide solution, cooling drying.The gained solid is dissolved among 30 milliliters of THF, adds 20 milliliters of 0.5N sodium hydroxide solutions, adds tert-Butyl dicarbonate (2.4 grams again, 0.011 mole), room temperature reaction
16 hours, concentrated, add the neutralization of 10% sodium bisulfate, ethyl acetate extraction three times merges organic phase, the saturated salt washing, anhydrous sodium sulfate drying, the concentrated 3.3 gram products that obtain, namely, compound
5, yield 99%.
Step 5: synthetic compound 7
Compound 6{5; 6; 7; 8-tetrahydrochysene-8-methyl-3-(trifluoromethyl)-imidazo [1,5-a] pyrazine, 5; 6; 7,8-tetrahydro-8-methyl-3-(trifluoromethyl)-imidazo[1,5-a] pyrazine; synthesize and see CN103087067; 2.1 gram, 0.010 mole } be dissolved in 8 milliliters of methylene dichloride, add triethylamine 1.2 grams (0.012 mole); compound 5 (3.3 grams; 0.010 mole), EDCI2.3 restrains (0.012 mole), room temperature reaction is 24 hours under nitrogen protection; pour in 100 milliliters of frozen water; organic phase is washed saturated salt washing, anhydrous sodium sulfate drying; the concentrated crude product that obtains; be dissolved in 100 milliliters of the 2N HCl/ methanol solutions (anhydrous HCl gas is dissolved in the solution of methyl alcohol), room temperature reaction 4 hours is spin-dried for; cooling; pour in 100 milliliters of frozen water, transfer PH to 9, ethyl acetate extraction three times; merge organic phase; and wash saturated salt washing, anhydrous sodium sulfate drying; concentrated; column chromatography purification obtains 2.8 gram products, and namely compound 7, yield 66%.
Compound 7 comprises four optical isomers, and route and the concrete steps of their separation and purification are as follows:
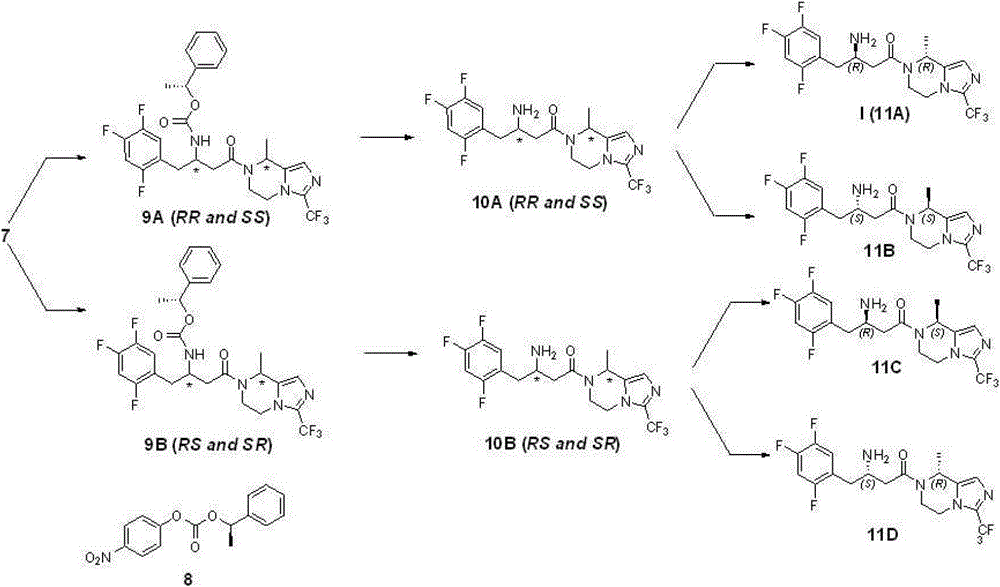
Step 6: preparation compound 9A and 9B
2.8 gram (6.67 mmole) compounds 7 are dissolved in 50 milliliters of acetonitriles; add triethylamine 1.2 grams (8.0 mmole); add again compound 8 (1.9 grams; 6.67 mmole; reference: J.Org.Chem.1995; 60 (3); 730), reflux is spent the night under nitrogen protection, and is concentrated; add ethyl acetate; the 1N sodium hydroxide solution is washed, and ethyl acetate milliliter extraction three times merges organic phase; the saturated salt washing; anhydrous sodium sulfate drying, the evaporating column chromatography purification obtains 1.6 gram 9A (43%) and 1.4 gram 9B (39%) products (de>98%); structural analysis determines that tentatively 9A is RR and SS mixture of enantiomers, and 9B is RS and SR mixture of enantiomers.Gained compound 9A and 9B give over to respectively next step and use.
Step 7: preparation compound 10A and 10B
1.5 gram (2.64 mmole) compound 9A are dissolved in 50 milliliters of methylene dichloride, reaction is cooled to 0 ℃, adds HBr solution (2M, 2.6 milliliters, 5.2 mmole), be dissolved in ethyl acetate after concentrated, sodium hydrogen carbonate solution is washed, the saturated salt washing, anhydrous sodium sulfate drying, the concentrated product that obtains, namely compound 10A (RR and SS mixture of enantiomers) gives over to next step and uses.
According to same reaction principle, condition and step, take compound 9B as starting raw material, obtain compound 10B (RS and SR mixture of enantiomers), give over to next step and use.
Step 8: preparation compound 11A, 11B and 11C, 11D
Resulting compound 10A in the step 7 (1.1 gram) is dissolved in 20 milliliters of ethanol, adds D-tartrate 0.4 gram (2.64 moles), reflux 0.5 hour, cooling, filter, obtain white solid, again with behind ten times of amount ethyl alcohol recrystallizations 2 times, obtain white solid, free with saturated sodium bicarbonate aqueous solution, obtain 0.29 and digest compound 11A, be i.e. compound (I), yield 26% is surveyed ee value>95%.
PAPER
https://www.tandfonline.com/doi/full/10.1080/00498254.2022.2091494
SYN
https://patents.google.com/patent/US11046701B2/en
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Cetagliptin phosphate, developed by CGeneTec, is a DPP-4 inhibitor designed for the treatment of T2DM. In 2024, the NMPA approved cetagliptin phosphate for managing T2DM. As a member of the DPP-4inhibitor, Cetagliptin exerts its effect on glycemic regulation by impeding the breakdown of incretin hormones. This action leads to a glucose-dependent increase in insulin secretion and a concurrent decrease in glucagon levels. Multiple clinical investigations have attested to the effectiveness and safety profile of sitagliptin. In a particular instance, a randomized, double-blind, placebo-controlled Phase 3 study was carried out to assess the use of sitagliptin as a single-agent treatment in patients diagnosed with type 2 diabetes [67]. The study found that cetagliptin significantly reduced HbA1c levels compared to placebo, with a greater proportion of patients achieving target glycemic control.
The treatment was generally well tolerated, with a safety profile comparable to placebo [68,69]. Regarding toxicity, cetagliptin was well tolerated in clinical studies, with no significant increase in adverse effects compared to placebo. No drug-related hypoglycemia was reported,
indicating a favorable safety profile [70].
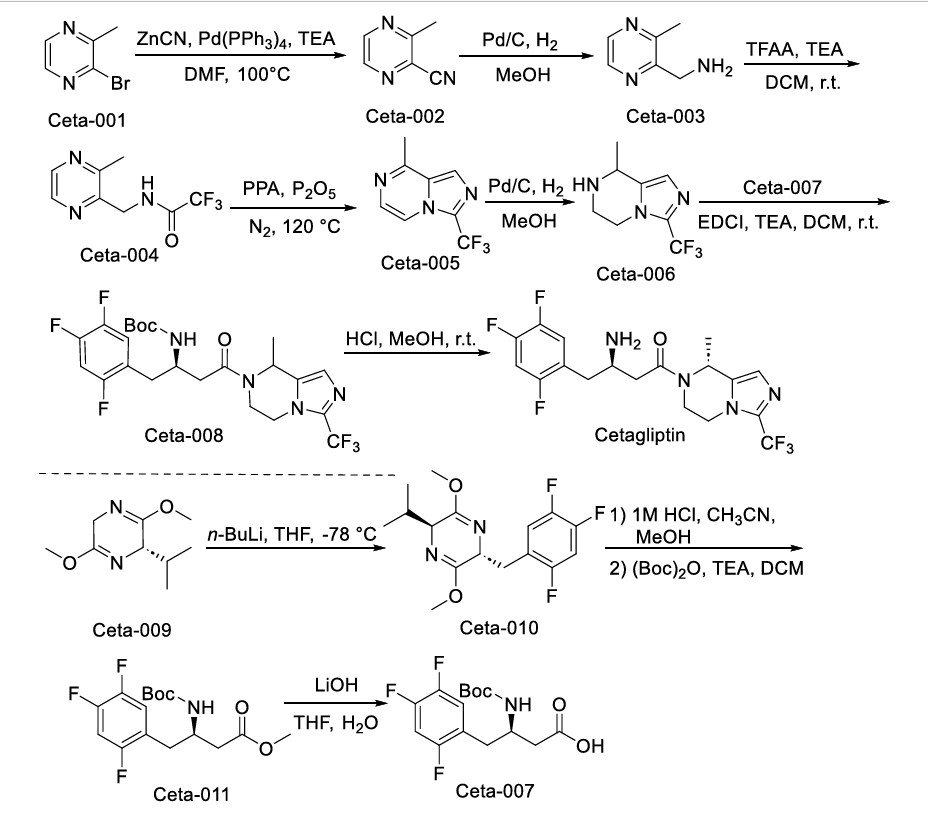
The synthesis of Cetagliptin, depicted in Scheme 16, initiates with Ceta-001 cyanidation affording Ceta-002, whose hydrogenative reduction yields Ceta-003 [71]. Subsequent amidation constructs Ceta-004,
followed by cyclization rearrangement producing Ceta-005. Hydrogenation delivers Ceta-006, which undergoes coupling with Ceta-007 assembling Ceta-008. Final TFA-mediated deprotection achieves
Cetagliptin. Concurrently, the side route involves Ceta-009 nucleophilic substitution forming Ceta-010. Sequential imine hydrolysis/protection converts Ceta-010 to Ceta-011, whose controlled hydrolysis ultimately delivers Ceta-007
67-70
[67] J. Lu, J. Zhao, D. Xie, J. Ding, Q. Yu, T. Wang, Use of a PK/PD model to select
Cetagliptin dosages for patients with type 2 diabetes in phase 3 trials, Clin.
Pharmacokinet. 63 (2024) 1463–1476.
[68] L. Guo, F. Tian, L. Liu, M. Chen, C. Jiang, S. Li, C. Liu, Y. Zhang, J. Qin, D. Yu,
Y. Zong, W. Dai, Retagliptin as add-on therapy to metformin in Chinese patients
with type 2 diabetes inadequately controlled with metformin: a multicentre,
randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab
26 (2024) 2830–2838.
[69] C. Hu, J. Zheng, J. Miao, F. Liu, T.T. Hu, J.K. Gu, S.Q. Shu, Y. Wang, X.H. Zhu, M.
Z. Liang, [Pharmacokinetics of Phosphate Retagliptin Tabletin in Patients with
Renal Dysfunction], Sichuan Da Xue Xue Bao Yi Xue Ban 49 (2018) 74–80.
[70] A. Cahn, S. Cernea, I. Raz, An update on DPP-4 inhibitors in the management of
type 2 diabetes, Expert Opin Emerg Drugs 21 (2016) 409–419.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
https://en.cgenetech.com.cn/news/55.html
Since the listing application of a class of innovative drug Cetagliptin independently developed by CGeneTech was accepted by the State Food and Drug Administration, it has received great attention in the industry. Recently, the well-known industry media “Shell News Agency” also took this opportunity to comprehensively sort out the hot track and broad market prospects of domestic DPP-4 inhibitors. This article is shared with you. In the face of the high expectations given by the industry, CGeneTech will continue to run the last “one kilometer” of product launch with a scientific and rigorous attitude.
Diabetes (DM), as a chronic disease, has attracted much attention. Diabetes drugs have become the second largest drug market after tumor drugs, and it is also a place for pharmaceutical enterprises to compete.
With the development of medicine, some new drugs with different mechanism of action from traditional oral hypoglycemic drugs have emerged in recent years. Dipeptidyl peptidase-4 (DPP-4) inhibitor is one of them. At present, there are dozens of DPP-4 inhibitors, which are collectively known as “gliptin drugs”. In the future, the market size of gliptin drugs in China will exceed 30 billion yuan.
Cetagliptin seven-year long run
On February 2, CGeneTech submitted to the National Drug Administration (NMPA) the marketing application (NDA) of Cetagliptin, a DPP-4 inhibitor, which was mainly used to treat type 2 diabetes. This means that the domestic DPP-4 inhibitor market will usher in new members, and the official website of CGeneTech will also publicize the progress of Cetagliptin research and development in the product pipeline for the first time, and the listing has been confirmed.
Cetagliptin is a Class 1 innovative drug independently developed by CGeneTech, and once was its own fist product in its pipeline. It has also experienced a seven-year long run since its launch of research and development, and is about to hit the line successfully.
In 2006, the targeted hypoglycemic drug Sigliptin was approved by the FDA of the United States, which is undoubtedly a major event in the industry. Ten years later, CGeneTech completed the pre-clinical study of head-to-head comparison of Cetagliptin and Xigliptin.
At the beginning of 2018, CGeneTech launched the phase I clinical trial of head-to-head comparison of Cetagliptin and Xigliptin. Among nearly 200 patients in the Phase I clinical trial completed by Cetagliptin, the data showed that when the intake of Cetagliptin reached 50 mg, it was able to achieve the DPP-4 inhibition capacity equivalent to the intake of 100 mg of Xigliptin. Cetagliptin is administered once a day. It can reach the peak within 1 to 2 hours after administration, and has a longer half-life than Sigliptin, which can maintain stable glucose reduction for a longer time.
Diabetes requires long-term medication, and safety is the first factor to be considered when doctors choose drugs when prescribing. In the safety study, the adverse effects of the intake of Cetagliptin on the body of patients were almost undetectable, lower than that of the blank group and Sigliptin group. In addition, although Cetagliptin has a long half-life, there is no accumulation of residual drugs in the body in the phase I clinical trial, which reflects the high selectivity and strong inhibition of Cetagliptin. The beautiful phase I clinical trial data have provided the foundation for the later clinical trial research of Cetagliptin.
In 2019, Cetagliptin was officially approved by the National Drug Evaluation Center to “exempt Phase II clinical trials from Phase III trials”, becoming the first DPP-4 inhibitor in the world to pass the quantitative pharmacological model, exempt Phase II clinical trials, and directly carry out Phase III confirmatory trials, which attracted the attention of experts in the field of diabetes at home and abroad.
In October 2022, the unblinding results of Cetagliptin phase III clinical trial showed that the reduction of glycosylated hemoglobin (HbA1c) in Cetagliptin tablet 50mg group reached the main clinical end point at the end of the 24th week, which was significantly superior to the control group. After 28 weeks, the Cetagliptin 100mg dose group also showed good drug safety, and the incidence of adverse reactions was similar to that of the placebo group. The clinical trial has shown the advantages of halving the dose but the same efficacy as similar products.
In February 2023, the marketing application (NDA) of Cetagliptin has been accepted by NMPA for the treatment of type 2 diabetes.
The approval of Cetagliptin has attracted much attention, which means that CGeneTech will officially participate in the domestic hot track of DPP-4 inhibitors, and the market of 10 billion statins will usher in new members.
DPP-4 inhibitor track is hot
DPP-4 inhibitors play a hypoglycemic role mainly by inhibiting the degradation of glucagon-like peptide-1 (GLP-1) by DPP-4 enzyme, promoting insulin and glucose dependent secretion, and inhibiting glucagon secretion, which can improve β Cell dysfunction does not increase the risk of hypoglycemia and body weight of patients. Moreover, DPP-4 inhibitor is a “mild and versatile”. It is mild, versatile and safe in reducing blood sugar. It is an oral drug that can be combined with various drugs in the whole process of management.
As the current mainstream hypoglycemic drug, DPP-4 inhibitor has become a hot spot in the eyes of major pharmaceutical enterprises. At present, there are five kinds of DPP-4 inhibitors that are taken daily on the market in China: Sigliptin, Viggliptin, Shagliptin, Aggliptin and Liggliptin, and these “five golden flowers” are included in the national health insurance list.
After entering medical insurance, the sales of several products have increased significantly. It is understood that from 2016 to 2022, the annual sales of DPP-4 inhibitors showed a continuous growth trend, with the highest year-on-year growth rate in 2018. In 2021 alone, the domestic sales of DPP-4 inhibitors reached nearly 7 billion yuan.
Sigliptin
Sigliptin is the first oral DPP-4 inhibitor on the market in the world, developed by MSD. It was approved by FDA for listing in October 2006; Sigliptin was approved for listing in China in September 2009; In July 2012, its compound preparation was approved for registration in China.
According to MSD’s annual report, the global market share of Sigliptin has been stable at more than US $3 billion in the past four years, ranking first in the global sales of DPP-4 inhibitors. At present, there are 14 pharmaceutical enterprises in China, including Zhengda Tianqing, Qilu Pharmaceutical, Kelun Pharmaceutical and Zhejiang Pharmaceutical, which have been copied and approved for production.
Viggliptin is the second DPP-4 inhibitor in the world developed by Novartis. In September 2007, Viggliptin was first approved for listing by the European Commission; In August 2011, it was officially approved for listing in China.
According to Novartis annual report, the global sales volume of Vigiletin has fluctuated steadily in recent years, basically maintaining at about 1.1 billion US dollars. The imitative production of Viggliptin in the domestic market is also hot. At present, 18 pharmaceutical enterprises such as Qilu Pharmaceutical, Yangzijiang Pharmaceutical, Jiangsu Haosen Pharmaceutical, Shandong Langnuo Pharmaceutical and Nanjing Shenghe Pharmaceutical have been approved for production. They are worthy of the title of the king of domestic imitative drugs for DPP-4 inhibitors.
Shagliptin was jointly developed by Bristol-Myers Squibb and AstraZeneca. It was approved by FDA for listing in July 2009; In May 2011, Shagliptin was approved for listing in China. Shagliptin’s overseas market share exceeded 20%. At present, there are five pharmaceutical enterprises in China, including Zhengda Tianqing, Qilu Pharmaceutical and Jiangsu Aosaikang Pharmaceutical, whose generic drugs have been approved for production.
Liggliptin was developed by BI. In May 2011, it was approved for listing by the FDA of the United States, and was jointly sold by Berger Ingelheim and Lilly. In March 2013, China approved the import registration of liggliptin. Liggliptin’s overseas market share exceeds 15%. At present, there are 6 pharmaceutical enterprises in China, including Guangdong East Sunshine Pharmaceutical, Yangzijiang Pharmaceutical and Kelun Pharmaceutical, which have been approved for production.
Agiletin
Agiletin was developed by Takeda Pharmaceutical of Japan. Approved for listing in Japan in April 2010; In January 2013, it was approved by the US FDA for listing; In July of the same year, Agiletin obtained the import registration certificate of China. According to the statistics of IQVIA, the sales amount of Agiletin in the Chinese market in 2022 was 52.36 million yuan. At present, 11 pharmaceutical enterprises such as Yabao Pharmaceutical, Ruiyang Pharmaceutical and Guorui Pharmaceutical of the National Pharmaceutical Group have been approved for production.
Throughout the domestic market of DPP-4 inhibitors, the original drugs and generic drugs of the “five golden flowers” are all in the Jianghu. In order to break the competition pattern, pharmaceutical enterprises have also invested in innovative self-research teams.
At present, the research and development of innovative DPP-4 inhibitors is also advancing rapidly. According to the data, in addition to the approval of CGeneTech’s Cetagliptin, many innovative DPP-4 inhibitors (excluding compound preparations) have entered the clinical research stage in China.
TQ-F3083 of Nanjing Shunxin, Shingliptin of Chenxin Pharmaceutical, and Boggliptin of Shandong Baiji Dichang Pharmaceutical are in clinical phase II; Fugliptin of Xinritai, DBPR108 of Shiyao Group, HSK7653 of Hisco and Unigliptin of Yuandong Biological are all in clinical phase III; Hengrui Pharmaceutical’s Retagliptin has submitted its listing application.
Although there are only a few “Ting” who have been approved to market independently developed DPP-4 inhibitors in China, the approval of Cetagliptin will take the lead in ushering in the harvest period of domestic innovative DPP-4 inhibitors, break the monopoly of non-self-developed DPP-4 inhibitors again, and give great confidence to pharmaceutical enterprises engaged in the research and development of DPP-4 inhibitors.
epilogue
The huge market potential of diabetes is like a magnet, attracting pharmaceutical enterprises to participate in the hot domestic track of DPP-4 inhibitors.
As the first oral DPP-4 inhibitor launched in the world and China, Sigliptin has been in the Chinese market for more than ten years, and still dominates the market. According to the Phase I clinical trial study, Cetagliptin has obtained significantly better data than Sigliptin in terms of efficacy, safety, half-life, toxicology and pathology, which will have considerable market persuasion and is expected to help it become a similar Best-in-class product, or change the curve overtaking into a competitive pattern.
Cetagliptin is only one step away from its listing. Not only is CGeneTech full of expectations for it, but also the industry has high expectations. Cetagliptin can be expected in the future, and we also expect more home-made original new “Ting” to come out.
reference material:
1. CGeneTech official website, official account
2. New weapon for treating diabetes (I) – DPP-4 inhibitor, Department of General Medicine, Shenzhen Hospital, University of Hong Kong, December 9, 2020
3. Unique Mechanism, Multi-dimensional Benefits – Mechanism and Clinical Application of DPP-4 Inhibitor, China Medical Forum Endocrinology Today, April 9, 2020
4. DPP-4 inhibitor market may add new force. Can CGeneTech break the “five giants” pattern
5. Market | DPP-4 inhibitor market pattern seen from the withdrawal of the first generic antidiabetic drug from the network of East Sunshine, CPHI Pharmaceutical Online, November 17, 2022
- [1]. Zhou C, et al., Safety, tolerability, pharmacokinetics and pharmacokinetic-pharmacodynamic modeling of cetagliptin in patients with type 2 diabetes mellitus. Front Endocrinol (Lausanne). 2024 Mar 11;15:1359407. [Content Brief][2]. Lu J, et al., In vitro study of the drug-drug interaction potential of cetagliptin and clinical study of pharmacokinetic interaction of cetagliptin and metformin in healthy volunteers. Xenobiotica. 2021 Oct;51(10):1122-1131. [Content Brief]
/////////Cetagliptin, CHINA 2024, APPROVALS 2024, CGeneTec, DIABETES, GTPL13952, CGT 8012,
Cofrogliptin
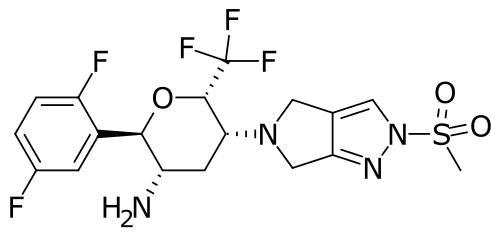
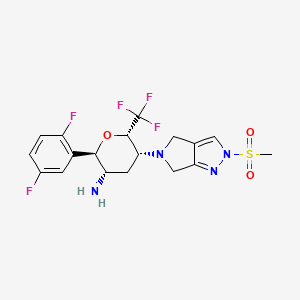
Cofrogliptin
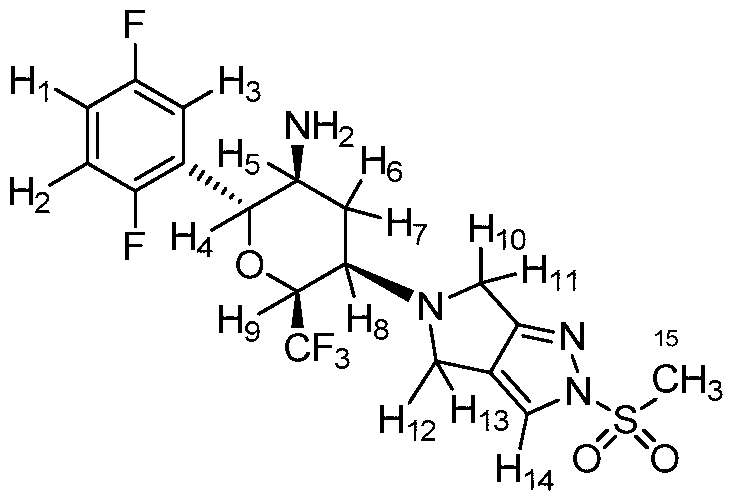
HSK 7653
- Haisco HSK 7653
- CAS 1844874-26-5
- 466.4 g/mol
- C18H19F5N4O3S
(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)-6-(trifluoromethyl)oxan-3-amine
- (2R,3S,5R,6S)-2-(2,5-Difluorophenyl)-5-(2- (methanesulfonyl)-2,6-dihydropyrrolo(3,4-C)pyrazol- 5(4H)-yl)-6-(trifluoromethyl)oxan-3-amine
- (2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-[2- (methanesulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol- 5(4H)-yl]-6-(trifluoromethyl)oxan-3-amine
- (6R)-5-Amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-(2,6-dihydro-2-(methylsulfonyl)pyrrolo(3,4-C)pyrazol-5(4H)-yl)-1,1,1-trifluoro-D-arabino-hexitol
- (6R)-5-Amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-[2,6-dihydro-2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(4H)-yl]-1,1,1-trifluoro-D-arabino-hexitol
- D-Arabino-hexitol, 5-amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-(2,6-dihydro-2-(methylsulfonyl)pyrrolo(3,4-C)pyrazol-5(4H)-yl)-1,1,1-trifluoro-, (6R)-
- (2r,3s,5r,6s)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4h)-yl]-6-(trifluoromethyl)-tetrahydro-2h-pyran-3-amine
APPROVALS 2024, CHINA 2024, Haisco Pharmaceutical Group Co, Beichangping, DIABETES
Cofrogliptin (developmental name HSK7653) is a long-acting DPP4 inhibitor dosed once every two weeks.[1][2][3][4]
Cofrogliptin (HSK7653) (compound 2), a tetrahydropyran derivative, is a potent oral dipeptidyl aminopeptidase 4 (DPP-4) inhibitor with Long-acting antidiabetic efficacy. Cofrogliptin (compound 2) has a great potential for type 2 diabetes mellitus (T2DM) .
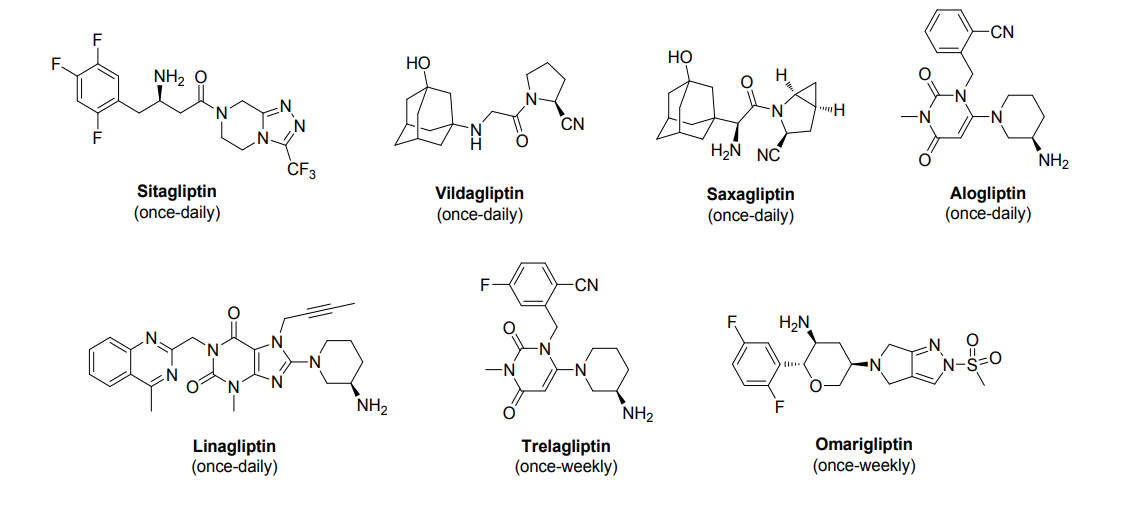
SYN
J Med Chem. 2020 Jul 9;63(13):7108-7126
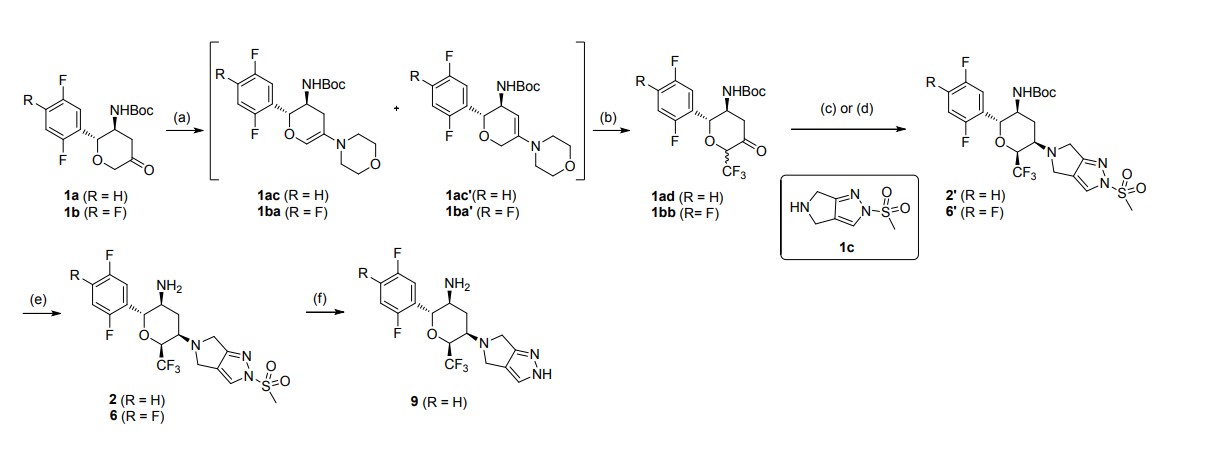
aReagents and conditions: (a) morpholine, toluene, reflux in Dean-Stark appartus; (b)
Umemoto’s reagent, DMAP, DMAc; (c) step 1: 1c, toluene, reflux; step 2: NaBH(OAc)3, CH3COOH, 1,2-DCE; (d) step 1: 1c, CHCl3, reflux in Dean-Stark apparatus; step 2:
NaBH(OAc)3, CH3COOH, 1,2-DCE; (e) TFA, DCM; (f) t-BuOK, THF
Step 2: To a stirred solution of tert-butyl N-[(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-
(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)-6-
(trifluoromethyl)tetrahydropyran-3-yl]carbamate (2′) (407.5 mg, 0.72 mmol) in DCM (6
mL) was added CF3COOH (2 mL) under nitrogen at 0 ℃. After the addition, the reaction
mixture was allowed to warm to room temperature and stirred for 2 h. The reaction mixture
was quenched with a saturated solution of Na2CO3 (15 mL), and extracted with DCM (15
mL × 2). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo.
The residue was purified by flash column chromatography (Eluent: DCM/MeOH = 80:1–
30:1) to afford the desired product 2 (301.9 mg, yield: 90%). White solid. Mp: 150.1–152.0
℃. [α]D20 = +17.6 (c = 2.000 in MeOH). Rf= 0.40 (1:15 MeOH/CH2Cl2, TLC).
1H NMR
(400 MHz, CDCl3) δ = 7.71 (s, 1H), 7.20 – 7.12 (m, 1H), 7.10 – 6.97 (m, 2H), 4.63 (d, J =
10.0 Hz, 1H), 4.49 – 4.38 (m, 1H), 4.07 – 3.97 (m, 2H), 3.93 – 3.81 (m, 2H), 3.53 – 3.42
(m, 1H), 3.29 (s, 3H), 3.01 – 2.91 (m, 1H), 2.45 – 2.35 (m, 1H), 2.07 – 1.93 (m, 1H), 1.19
(br. s, 2H). 13C NMR (100 MHz, CDCl3) δ = 163.6, 159.1 (dd, J = 2.3 Hz, 235.8 Hz), 156.6
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0223523424003441
SYN
WO2015192701
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015192701&_cid=P20-MEQV3M-18104-1
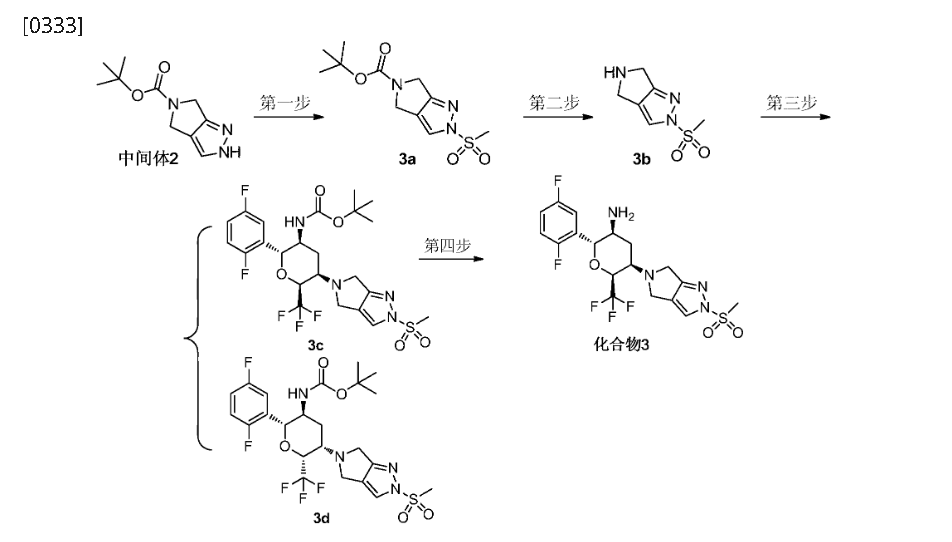
Step 4: (2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)-pyrrolo[3,4]pyrazol-5(2H,4H,6H)-yl)-6-(trifluoromethyl)tetrahydro-2H-pyran-3-amine (Compound 3)
[0345]
(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(2H,4H,6H)-yl)-6-(trifluoromethyl)tetrahydro-2H-pyran-3-amine
[0346]3c (410 mg, 0.72 mmol) was dissolved in 6 mL of dichloromethane and 2 mL of trifluoroacetic acid and stirred at room temperature for 1 hour. After completion, saturated aqueous sodium bicarbonate (30 mL) was added to quench the reaction. After separation, the aqueous phase was extracted with ethyl acetate (30 mL x 2). The combined organic phases were dried over anhydrous sodium sulfate, and concentrated. Purification by silica gel column chromatography (dichloromethane/methanol (v/v) = 30:1) afforded compound 3 (250 mg, 75% yield) as a white powder.
[0347]MS m/z(ESI): 467.1[M+1];
[0348]
1H NMR(400MHz,DMSO-d 6):δ7.96(m,1H),7.35–7.04(m,3H),4.86–4.63(qd,1H),4.50(d,1H),3.95(dd,2H),3.78(dd,2H),3.49(s,3H),3.45(m,1H),3.00(ddd,1H),2.33(m,1H),1.82(m,1H),1.48(br,2H)。
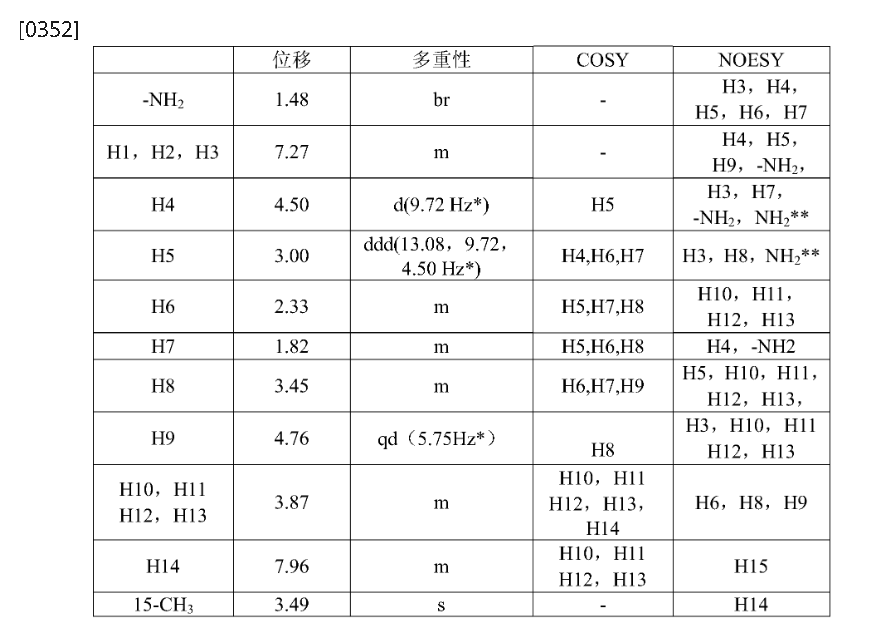
SYN
Cofrogliptin, developed by Haisco Pharmaceutical Group Co., Ltd., is a novel, ultra-long-acting dipeptidyl peptidase-4 (DPP-4) inhibitor designed for the treatment of T2DM. It is marketed under the brand name (Beichangping). In 2024, the NMPA approved Cofrogliptin for improving blood glucose control in adult patients with T2DM [59].Cofrogliptin acts pharmacologically by inhibiting DPP-4, an enzyme tasked with degrading incretin hormones like glucagon-like peptide-1(GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). By obstructing the degradation of these hormones, it amplifies their activity. This leads to a glucose-dependent rise in insulin secretion and a
corresponding decrease in glucagon release, which in turn improves glycemic control. The clinical efficacy of Cofrogliptin was demonstrated in Phase III, randomized, double-blind, non-inferiority trial
(NCT04556851), where its efficacy and safety were compared to those of daily linagliptin in patients with T2DM whose blood sugar was not well-controlled by metformin. The study reported that Cofrogliptin
administered once every two weeks achieved a reduction in HbA1c comparable to that of daily linagliptin, with a mean decrease of approximately 0.96 % over 24 weeks. Regarding toxicity, Cofrogliptin
was generally well-tolerated [60,61]. The incidence of hypoglycemia was low, and no severe hypoglycemic events directly attributed to the drug were reported.
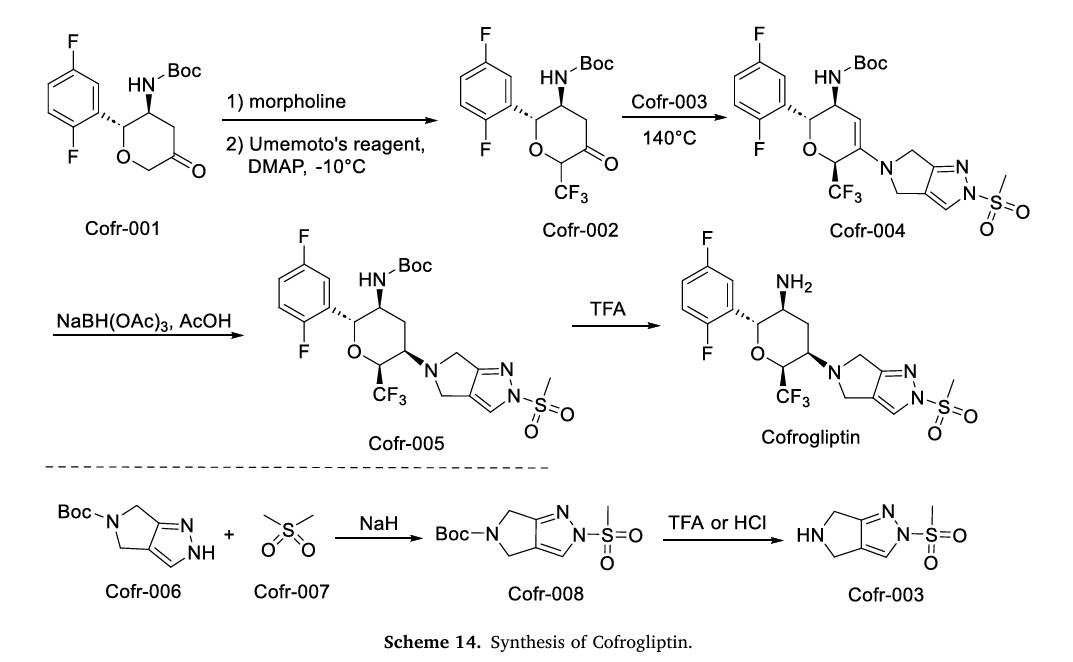
The synthesis of Cofrogliptin, illustrated in Scheme 14, initiates with trifluoromethylation of Cofr-001 via oxidation, affording Cofr-002 [62]. Nucleophilic addition of Cofr-003 to Cofr-002 yields Cofr-004, followed by NaBH(OAc)3 reduction to Cofr-005. TFA-mediated deprotection of Cofr-005 ultimately delivers Cofrogliptin. Concurrently, Cofr-006 undergoes nucleophilic substitution with Cofr-007 to form Cofr-008, whose deprotection regenerates Cofr-003
[59] L. Gao, F. Bian, T. Pan, H. Jiang, B. Feng, C. Jiang, J. Sun, J. Xiao, P. Yan, L. Ji,
Efficacy and safety of cofrogliptin once every 2 weeks in Chinese patients with type
2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes
Obes Metab 27 (2025) 280–290.
[60] C. Cui, F. Cao, I.I. Kong, Q. Wu, F. Li, H. Li, D. Liu, A model-informed approach to
accelerate the clinical development of cofrogliptin (HSK7653), a novel ultralong-
acting dipeptidyl peptidase-4 inhibitor, Diabetes Obes Metab 26 (2024) 592–601.
[61] Q. Ren, L. Li, X. Su, X. Hu, G. Qin, J. Han, Y. Liu, J. Wang, L. Ji, Cofrogliptin once
every 2 weeks as add-on therapy to metformin versus daily linagliptin in patients
with type 2 diabetes in China: a randomized, double-blind, non-inferiority trial,
Diabetes Obes Metab 26 (2024) 5013–5024.
[62] C. Zhang, J. Wang, C. Li, Y. Wei, Amino Pyranoid Ring Derivative as DPP-IV
Inhibitor and Its Preparation, 2015. WO2015192701A1.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji, Linong; Bian, Fang; Pan, Tianrong; Jiang, Hongwei; Jiang, Chengxia; Ren, Qian (20 June 2023). “55-OR: HSK7653, a Novel Ultralong-Acting DPP-4 Inhibitor, as Monotherapy in Patients With Type 2 Diabetes—A Randomized, Double-Blind, Placebo-Controlled Phase III Trial”. Diabetes. 72 (Supplement_1). doi:10.2337/db23-55-OR. S2CID 259433641.
- Zhang, Miao; Zhang, Shudong; Yu, Zhiheng; Yao, Xueting; Lei, Zihan; Yan, Pangke; Wu, Nan; Wang, Xu; Hu, Qin; Liu, Dongyang (October 2023). “Dose decision of HSK7653 oral immediate release tablets in specific populations clinical trials based on mechanistic physiologically-based pharmacokinetic model”. European Journal of Pharmaceutical Sciences. 189 106553. doi:10.1016/j.ejps.2023.106553. PMC 10485820. PMID 37532063.
- Liu, Yang; Yan, Shuai; Liu, Jie; Liu, Hongzhong; Song, Ling; Yao, Xueting; Jiang, Ji; Li, Fangqiong; Du, Ke; Liu, Dongyang; Hu, Pei (May 2023). “Development and validation of an HPLC coupled with tandem mass spectrometry method for the determination of HSK7653, a novel super long-acting dipeptidyl peptidase-4 inhibitor, in human plasma and urine and its application to a pharmacokinetic study”. Biomedical Chromatography. 37 (5): e5607. doi:10.1002/bmc.5607. PMID 36802077. S2CID 257048524.
- Bai, Nan; Wang, Jin; Liang, Wenxin; Gao, Leili; Cui, Wei; Wu, Qinghe; Li, Fangqiong; Ji, Linong; Cai, Yun (6 November 2023). “A Multicenter, Randomized, Double-Blind, Placebo-Controlled, and Dose-Increasing Study on the Safety, Tolerability and PK/PD of Multiple Doses of HSK7653 by Oral Administration in Patients with Type 2 Diabetes Mellitus in China”. Diabetes Therapy. 15 (1): 183–199. doi:10.1007/s13300-023-01496-0. PMC 10786778. PMID 37930584.
| Clinical data | |
|---|---|
| Other names | HSK7653 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1844874-26-5 |
| PubChem CID | 118613788 |
| ChemSpider | 115037226 |
| UNII | LH4G6K6NKP |
| ChEMBL | ChEMBL4646510 |
| Chemical and physical data | |
| Formula | C18H19F5N4O3S |
| Molar mass | 466.43 g·mol−1 |
- [1]. International Nonproprietary Names for Pharmaceutical Substances (INN)[2]. Chen Zhang, et al. Design, Synthesis, and Evaluation of a Series of Novel Super Long-Acting DPP-4 Inhibitors for the Treatment of Type 2 Diabetes. J Med Chem. 2020 Jul 9;63(13):7108-7126. [Content Brief]
///////Cofrogliptin, APPROVALS 2024, CHINA 2024, Haisco Pharmaceutical Group Co, Beichangping, DIABETES, HSK 7653, Haisco HSK 7653, 1844874-26-5
Dorzagliatin
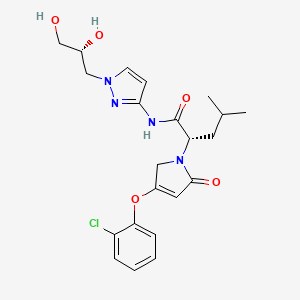
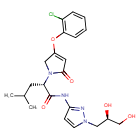
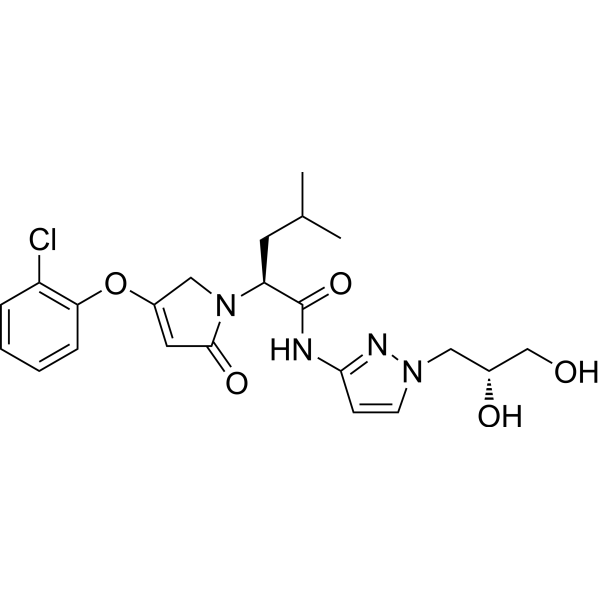
Dorzagliatin
- CAS 1191995-00-2
- HMS5552
- Sinogliatin
- HMS-5552
- MW 462.9 g/mol MF C22H27ClN4O5
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methylpentanamide
- RO5305552
- RO-5305552
- X59W6980E8
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methyl-pentanamide
- 1H-PYRROLE-1-ACETAMIDE, 4-(2-CHLOROPHENOXY)-N-(1-((2R)-2,3-DIHYDROXYPROPYL)-1H-PYRAZOL-3-YL)-2,5-DIHYDRO-.ALPHA.-(2-METHYLPROPYL)-2-OXO-, (.ALPHA.S)-
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM). CHINA 2022
Dorzagliatin is a glucokinase activator that is being developed to treat diabetes.[1] Unlike other diabetes drugs, it is intended to increase insulin sensitivity.[2]
Dorzagliatin is under investigation in clinical trial NCT03173391 (Long-term Efficacy and Safety of HMS5552 in T2DM).
PATENT
https://patents.google.com/patent/CN112062754A/en
(R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is a very important medical intermediate for synthesizing Dorzagliatin. Dorzagliatin is a novel medicine for treating type 2 diabetes mellitus, and (R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is an essential intermediate in the synthetic process of the medicine, and along with the steady promotion of new Dorzagliatin medicines to the market, the demand of the chiral intermediate in the market is required to be rapidly increased.
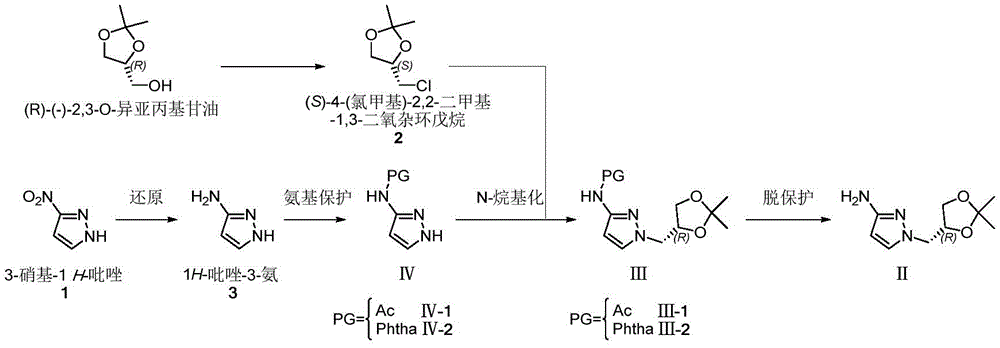
The main production method of the key chiral intermediate is shown as follows: reducing nitro in 3-nitro-1H-pyrazole substrate into amino, protecting free amino, carrying out N-alkylation reaction with (R) – (-) -2, 3-O-isopropylidene glycerol-OH derivative active intermediate, and deprotecting to obtain the final product. The synthetic route needs to be subjected to an N-protection process, so that route steps are added, and the cost is increased. The synthesis of N-protected substrate iv is reported: in the patent US2013203802, 1H-pyrazole-3-ammonia is protected by acetic anhydride, and in WO2017040757, N-acetyl-1H-pyrazole-3-ammonia is obtained by an N- (1-benzyl-1H-pyrazole-3-yl) acetamide debenzylation method; the protection of the N-benzoyl group of 1H-pyrazol-3-amine is reported in the patent US 6118008; in addition, WO2009106209, US2012095064, mention the phthalimide protection strategy of 1H-pyrazole-3-ammonia with phthalic anhydride.
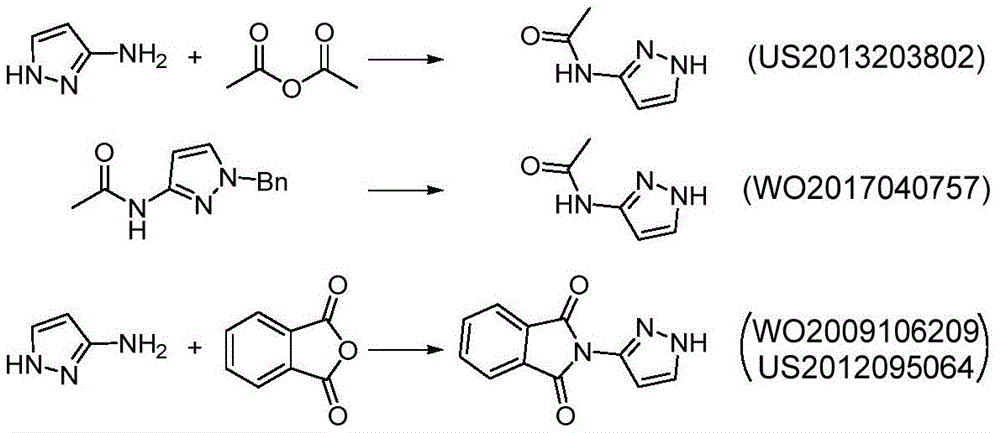
Example 1
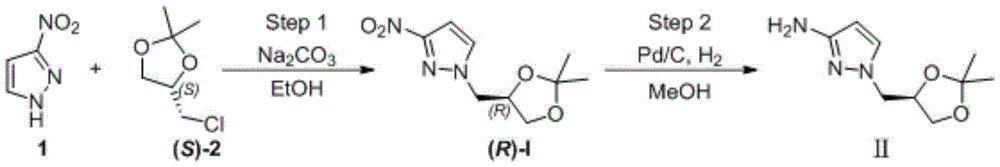
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine
The first step is as follows: intermediate (R) -I preparation:
under the protection of nitrogen, 3-nitro-1H-pyrazole (1) (100.00g,0.884mol), ethanol (1.0L) and sodium carbonate (133.90g, 1.26mol) are sequentially added into a 3L reaction bottle, and the system is stirred for 0.5H at room temperature; (S) – (-) -4-chloromethyl-2, 2-dimethyl-1, 3-dioxolane ((S) -2) (126.84g, 0.842mol) was dissolved and diluted with 634ml of ethanol and then added dropwise to the reaction flask. After the dropwise addition, the temperature is raised to 50 ℃ and the reaction is stirred for 5 hours. Ethanol was distilled off under reduced pressure, and the residue was diluted with (1.0L) of water and then extracted twice with dichloromethane (500ml × 2); the organic phase was washed with water and then with saturated sodium chloride brine. Concentrating under reduced pressure to remove dichloromethane to obtain crude oily substance; the crude product was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate mixed system) to give 166.5g of a pale yellow oily product, with a yield of 87% and an ee value of 98% or more.
The second step is that: reducing nitro to obtain target product
A2L autoclave was charged with (R) -I substrate (150g, 0.66mol), methanol (750mL), Pd/C (0.75g, 0.5% W/W), and the mixture was subjected to nitrogen substitution three times, then hydrogen substitution three times, under a hydrogen-charging pressure of 2.0MPa, at a temperature of 50 ℃ for reaction for 8 hours. Filtering, filtering to remove Pd/C catalyst, concentrating the filtrate to remove methanol to obtain 123.70g of light yellow oily matter, wherein the yield is 95%, and the ee value is more than or equal to 98%.
Example 2
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by Raney-Ni reduction system
The first step is the same as in example 1.
The second step is that: reduction of nitro groups by Rany-Ni
The intermediate (R) -I (150g, 0.66mol) obtained in the first step was charged into a 2L reactor, and ethanol (1.2L) was added thereto and stirred, followed by adding Rany-Ni (75g) and stirring at room temperature for reaction for 15 hours. Filtering, filtering to remove the solid catalyst, and concentrating the filtrate to dryness to obtain 106.77g of light yellow oily substance with yield of 82% and ee value of more than or equal to 97%.
Example 3
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by hydrazine hydrate system
The first step is the same as in example 1.
The second step is that: A2L reaction flask was charged with intermediate (R) -I (150g, 0.66mol), ferric trichloride (528mg, 3.3mmol), and ethanol (1.2L), stirred, charged with hydrazine hydrate (39.5g, 0.79mol), and heated to reflux for 6 h. Ethanol was removed by concentration under reduced pressure, the residue was diluted with 750ml of water and extracted twice with ethyl acetate (250 ml. times.2). The organic phase was washed with water and then with saturated brine. The ethyl acetate is removed by concentration to obtain 110.7g of crude light yellow oily substance, the yield is 85 percent, and the ee value is more than or equal to 97 percent.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374J.Med.Chem.2024,67,4376−4418
Dorzagliatin(HuaTangNing).
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM).133 This first-in-class, small
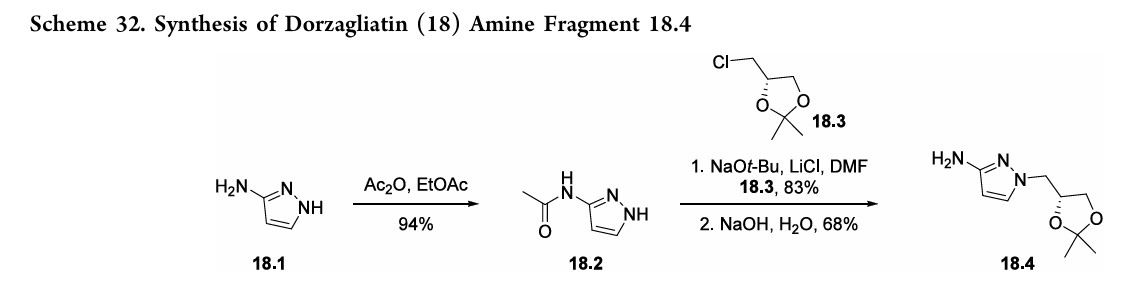
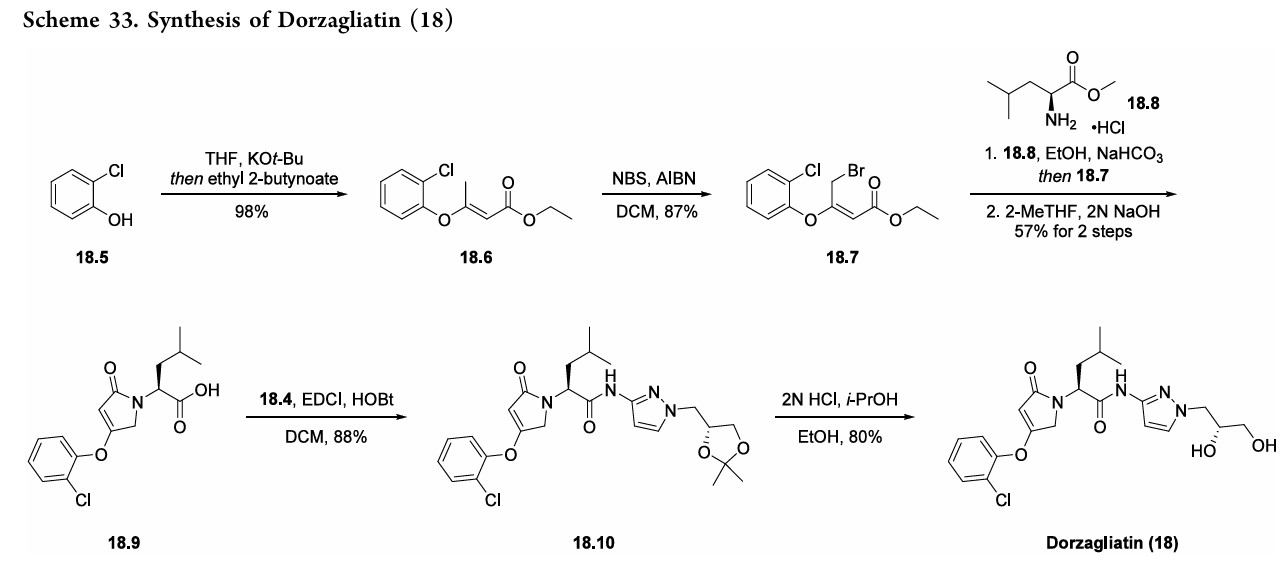
molecule,oral,glucokinaseactivator(GKA)wasfirst approved in ChinainSeptember2022foradultpatientswithT2DMasa monotherapy and in combination with metformin (an antidiabetic medication).134 Expression of glucokinase is reduced for individuals with T2DM, thus GKAs such as dorzagliatin serve as a novel class of antidiabetic treatment options.135,136 Theinitialpatent thatdisclosesthesynthesisofdorzagliatin (18)began fromreadily availablematerials 3-aminopyrazole
(18.1) and 2-chlorophenol (18.5). The synthetic strategy reliedonaconvergentamidecouplingofamine18.4(Scheme32) and carboxylic acid 18.9 (Scheme 33).137 A later disclosure provided an updated route toward amine 18.4 (Scheme 32), detailing the synthetic improvements with respect to yield and purity.138 This later disclosure also detailed the synthesis of dorzagliatinonmultikilogramscale fromtheamidationofacid18.9withamine18.4,yieldingover
10kgoftheactivepharmaceutical ingredient.Acetylationof3 aminopyrazole (18.1) with acetic anhydride provided the protectedpyrazole18.2(Scheme32). Subsequent alkylation with alkyl chloride 18.3 followed by base-mediated deprotectionyieldedamine18.4. The synthesis of acid 18.9 began with base-mediated
alkenylationof2-chlorophenol (18.5)withethyl 2-butynoate toprovideester18.6(Scheme33). Subsequentbromination withNBSandAIBNyieldsallylbromide18.7.Next,subjection
ofL-leucinemethylesterhydrochloride(18.8)tobaseresulted ina freeamine thatunderwent allylationwithbromide18.7. Acid 18.9was subsequently generated froma cyclization
condensation sequence and saponification reaction with NaOH. Final amidebondformationwas facilitatedbyEDCI andHOBt toprovideamide18.10, anddorzagliatin(18)was generatedonthemultikilogramscale followingacid-mediated acetonidedeprotectiontoreveal the1,2-diol.
(133) Syed, Y. Y. Dorzagliatin: First approval. Drugs 2022, 82,
1745−1750.
(134) Xu, H.; Sheng, L.; Chen, W.; Yuan, F.; Yang, M.; Li, H.; Li, X.;
Choi, J.; Zhao, G.; Hu, T.; et al. Safety, tolerability, pharmacokinetics,
and pharmacodynamics of novel glucokinase activator HMS5552:
results from a first-in-human single ascending dose study. Drug Des.
Devel. Ther. 2016, 10, 1619−26.
(135) Ren, Y.; Li, L.; Wan, L.; Huang, Y.; Cao, S. Glucokinase as an
emerging anti-diabetes target and recent progress in the development
of its agonists. J. Enzyme Inhib. Med. Chem. 2022, 37, 606−615.
(136) Toulis, K. A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.
H.; Tahrani, A. A. Glucokinase activators for type 2 diabetes:
Challenges and future developments. Drugs 2020, 80, 467−475.
(137) Berthel, S. J.; Brinkman, J. A.; Hayden, S.; Haynes, N.-E.;
Kester, R. F.; McDermott, L. A.; Qian, Y.; Sarabu, R.; Scott, N. R.;
Tilley, J. W. Pyrrolidinone as glucokinase activators and their
preparation, pharmaceutical compositions and use in the treatment
of metabolic disorders. WO 2009127546, 2009.
(138) Chen, J.; Ren, Y.; She, J.; Wang, L. Process for the preparation
of 1-([1,3]dioxolan-4-ylmethyl)-1h-pyrazol-3-ylamine. U.S. Patent US
20150315176, 2015.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Chow, Elaine; Wang, Ke; Lim, Cadmon K.P.; Tsoi, Sandra T.F.; Fan, Baoqi; Poon, Emily; Luk, Andrea O.Y.; Ma, Ronald C.W.; Ferrannini, Ele; Mari, Andrea; Chen, Li; Chan, Juliana C.N. (1 February 2023). “Dorzagliatin, a Dual-Acting Glucokinase Activator, Increases Insulin Secretion and Glucose Sensitivity in Glucokinase Maturity-Onset Diabetes of the Young and Recent-Onset Type 2 Diabetes”. Diabetes. 72 (2): 299–308. doi:10.2337/db22-0708. PMC 9871194.
- Zhu, Dalong; Li, Xiaoying; Ma, Jianhua; Zeng, Jiao’e; Gan, Shenglian; Dong, Xiaolin; Yang, Jing; Lin, Xiaohong; Cai, Hanqing; Song, Weihong; Li, Xuefeng; Zhang, Keqin; Zhang, Qiu; Lu, Yibing; Bu, Ruifang; Shao, Huige; Wang, Guixia; Yuan, Guoyue; Ran, Xingwu; Liao, Lin; Zhao, Wenjuan; Li, Ping; Sun, Li; Shi, Lixin; Jiang, Zhaoshun; Xue, Yaoming; Jiang, Hongwei; Li, Quanmin; Li, Zongbao; Fu, Maoxiong; Liang, Zerong; Guo, Lian; Liu, Ming; Xu, Chun; Li, Wenhui; Yu, Xuefeng; Qin, Guijun; Yang, Zhou; Su, Benli; Zeng, Longyi; Geng, Houfa; Shi, Yongquan; Zhao, Yu; Zhang, Yi; Yang, Wenying; Chen, Li (May 2022). “Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial”. Nature Medicine. 28 (5): 965–973.
- [1]. Zhu XX, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic β-cell function in patients with type 2 diabetes: A 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018 Sep;20(9):2113-2120. [Content Brief][2]. Wang P, et al. Effects of a Novel Glucokinase Activator, HMS5552, on Glucose Metabolism in a Rat Model of Type 2 Diabetes Mellitus. J Diabetes Res. 2017;2017:5812607. [Content Brief]
//////////Dorzagliatin, APPROVALS 22, CHINA 22, DIABETES, Hua Medicine, 1191995-00-2, HMS 5552, Sinogliatin, HMS-5552, RO 5305552, RO-5305552, X59W6980E8
Chiglitazar
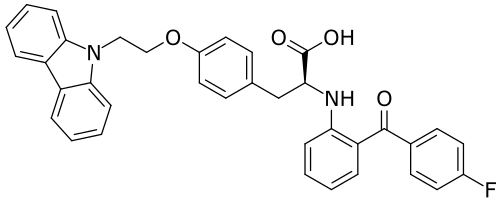
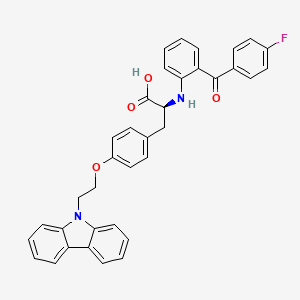
Chiglitazar
CAS 743438-45-1
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Chiglitazar sodium, (S)- | YN12H6OCV6 | 2390374-10-2 | RMVIEXHXRDCWBT-UCRKPPETSA-M |
- CS 038
- Carfloglitazar, (s)-
- E6EJV1J6Y0
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- C36H29FN2O4
- 572.6 g/mol
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- (2S)-3-(4-(2-CARBAZOL-9-YLETHOXY)PHENYL)-2-(2-(4-FLUOROBENZOYL)ANILINO)PROPANOIC ACID
- (2s)-3-(4-(2-carbazol-9-ylethoxy)phenyl)-2-(2-(4-fluorobenzoyl)anilino)propanoic acid
- Carfloglitazar, (s)-
- L-tyrosine, o-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-
- O-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-l-tyrosine
Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.
Chiglitazar (trade name Bilessglu) is a drug for the treatment of type 2 diabetes.[1] It is a peroxisome proliferator-activated receptor (PPAR) agonist.
In China, chiglitazar is approved for glycemic control in adult patients with type 2 diabetes when used in combination with diet and exercise.[2]
Chiglitazar is under investigation in clinical trial NCT06125587 (Chiglitazar/metformin in Non-obese Women With PCOS).
SYN
WO 2004048333
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004048333&_cid=P12-MDMUOB-48741-1
Example 15
Preparation of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-carbazolylethoxy)-phenyl]
-propionic acid (compound CS038)
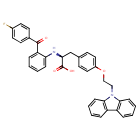
To a solution of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-bromoethoxy)-phenyl] -propionic acid methyl ester (0.25 g, 0.49 mmol) and carbazole (0.082 g, 0.49 mmol) in benzene (10 ml) is added tetrabutyl ammonium bromide (0.08 g) and 50% NaOH aqueous solution (0.084 g, 1.08 mmol), then the mixture is heated to reflux for 10 h. After cooled, benzene (30ml) is added, and the mixture is washed with water (3×30 ml). Then the solvent is evaporated under a vacuum. The crude product is purified by silica gel chromatography using CHCl3/MeOH (4:1) as eluent to give the title compound (0.10 g, 36%). HRMS calcd for C36H29FN204: 572.6357. Found: 572.6354. MA calcd for C36H29FN204: C, 75.51%; H, 5.11%; N, 4.89%. Found: C, 75.83%; H, 5.10%; N, 4.90%.
PATENT
US 10640465
https://patentscope.wipo.int/search/en/detail.jsf?docId=US249083802&_cid=P12-MDMUQY-52500-1
The pharmacological activity of the compound is described in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157. 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is able to selectively activate PPAR-α, PPAR-γ and PPAR-6, and can be used to treat the diseases associated with metabolic syndrome such as diabetes, hypertension, obesity, insulin resistance, hypertriglyceridemia, hyperglycemia, high cholesterol, arteries atherosclerosis, coronary heart disease, etc. A preparation method of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is disclosed in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157, and the synthetic route thereof is as follows:
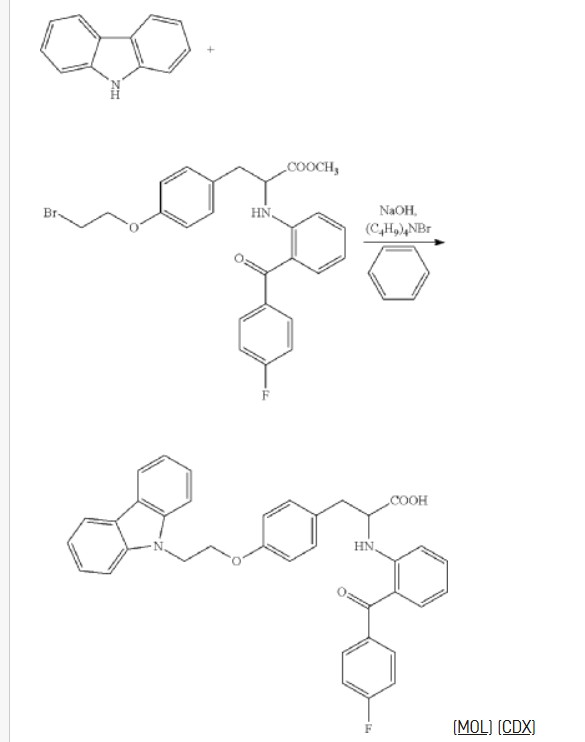
EXAMPLES
Example 1: Preparation of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic Acid
SYN
J. Med. Chem. 2024, 67, 4376−4418
Chiglitazar (Bilessglu). Chiglitazar (17), a novel nonthiazolidinedione pan-agonist of α, δ, and γ peroxisome proliferator-activated receptors (PPARs), has shown promise for the treatment of type 2 diabetes. 126 Type 2 diabetes impacts over 374 million patients worldwide and continues to
rise in incidence and prevalence globally. 127 Chiglitazar preferentially regulates expression of ANGPTL4 and PDK4 genes, which are involved in glucose and lipid metabolism. 128 Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.129 Thesynthesisof17beganwithimineformationbetweenL
tyrosine methyl ester (17.1) and 2-(4-fluorobenzoyl) cyclohexanone(17.2)with tandemaromatizationunderPd/C catalysis to generate aniline derivative 17.3 (Scheme31).130,131 Alkylation of the phenol moiety of 17.3 with mesylate17.4furnishedphenyl alkyl etherderivative17.5.132
Hydrolysisof themethylester in17.5withlithiumhydroxide followedbyacidificationwithhydrochloricacidandrecrystal lization fromacetonitrile afforded chiglitazar (17) in 42% overall yield from17.3.Thisprocessdeliveredchiglitazar in 99.4%purityat24gscale.
(126) Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.;
Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a
novel peroxisome proliferator-activated receptor pan-agonist, in
patients with type 2 diabetes: a randomized, double-blind, placebo
controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571−1580.
(127) Chatterjee, S.; Khunti, K.; Davies, M. J. Type 2 diabetes.
Lancet 2017, 389, 2239−2251.
(128) Pan, D.-S.; Wang, W.; Liu, N.-S.; Yang, Q.-J.; Zhang, K.; Zhu,
J.-Z.; Shan, S.; Li, Z.-B.; Ning, Z.-Q.; Huang, L.; Lu, X.-P. Chiglitazar
preferentially regulates gene expression via configuration-restricted
binding and phosphorylation inhibition of PPARγ. PPAR Research
2017 2017, 2017, 1−16.
(129) Deeks, E. D. Chiglitazar: First approval. Drugs 2022, 82, 87−
92.
(130) Li, Z.; Lu, X.-P.; Liao, C.; Shi, L.; Liu, Z.; Ma, B. Substituted
arylalcanoic acid derivatives as PPAR pan agonists with potent
antihyperglycemic and antihyperlipidemic activity. WO 2004048333
A1, 2004.
(131) Sutter, M.; Sotto, N.; Raoul, Y.; Métay, E.; Lemaire, M.
Straightforward heterogeneous palladium catalyzed synthesis of aryl
ethers and aryl amines via a solvent free aerobic and non-aerobic
dehydrogenative arylation. Green Chem. 2013, 15, 347−352.
(132) Lu, X.; Li, Z.; Wang, X. Method for preparing phenylalanine
compound. U.S. Patent US 10640465 B2, 2020.
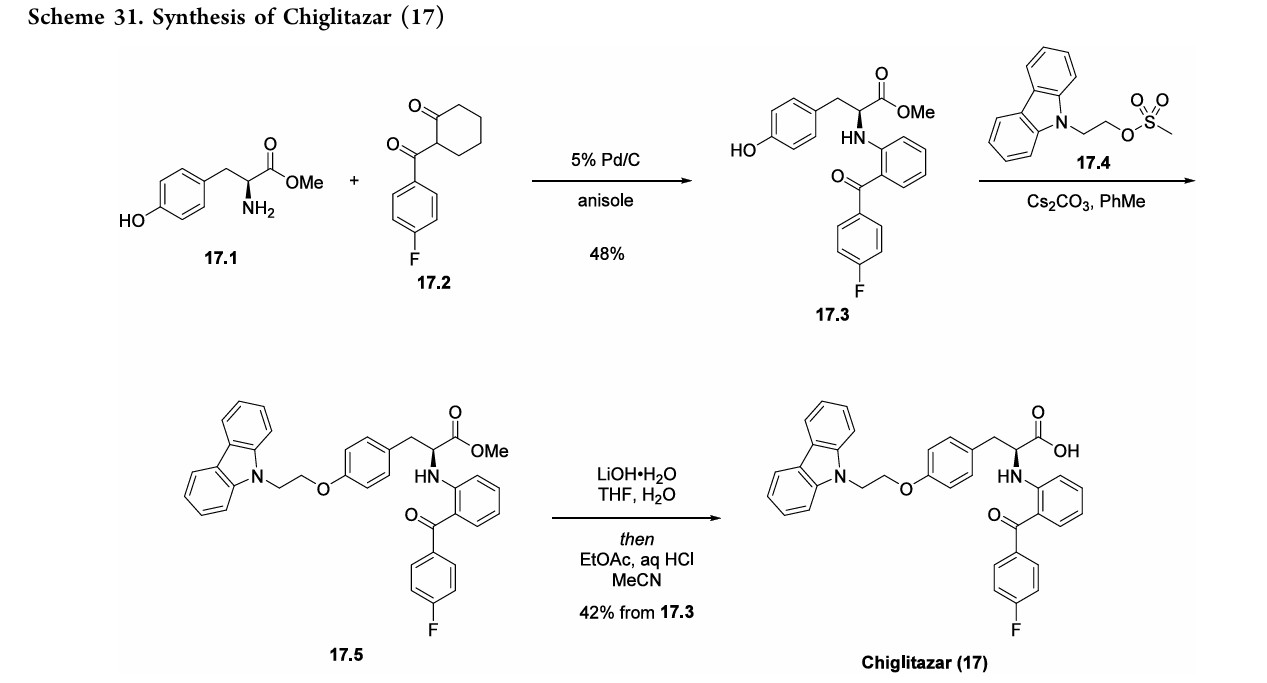
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji L, Song W, Fang H, Li W, Geng J, Wang Y, et al. (August 2021). “Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial (CMAP)”. Science Bulletin. 66 (15): 1571–1580. Bibcode:2021SciBu..66.1571J. doi:10.1016/j.scib.2021.03.019. PMID 36654286. S2CID 233650336.
- Deeks ED (January 2022). “Chiglitazar: First Approval”. Drugs. 82 (1): 87–92. doi:10.1007/s40265-021-01648-1. PMID 34846697. S2CID 244716275.
| Clinical data | |
|---|---|
| Trade names | Bilessglu |
| Other names | Carfloglitazar |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 743438-45-1 |
| PubChem CID | 71402018 |
| ChemSpider | 57523239 |
| UNII | E6EJV1J6Y0 |
| ChEMBL | ChEMBL4650349 |
| CompTox Dashboard (EPA) | DTXSID00225352 |
| Chemical and physical data | |
| Formula | C36H29FN2O4 |
| Molar mass | 572.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Chiglitazar, Chipscreen Biosciences, CHINA 2021, DIABETES, CS 038, Carfloglitazar, (s)-, E6EJV1J6Y0,
Meglimin hydrochloride

Meglimin hydrochloride
Imeglimin
hydrochloride
Twymeeg
| Formula | C6H13N5. HCl |
|---|---|
| CAS | 775351-61-6 (HCl). , C6H14ClN5 191.66CAS 775351-65-0, FREEFORM 155.20 |
| Mol weight | 191.6619 |
AntidiabeticAPPROVED PMDA JAPAN2021/6/23, イメグリミン塩酸塩
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
1,3,5-Triazine-2,4-diamine,1,6-dihydro-N,N,6-trimethyl-,(+)-(9CI)
(4R)-6-N,6-N,4-trimethyl-1,4-dihydro-1,3,5-triazine-2,6-diamine
JAPAN
Twymeeg Tablets 500 mg
(Sumitomo Dainippon Pharma Co., Ltd.)

Imeglimin is an experimental drug being developed as an oral anti-diabetic.[1][2] It is an oxidative phosphoryl
Imeglimin (brand name Twymeeg) is an oral anti-diabetic medication.[1][2] It was approved for use in Japan in June 2021.[3]
It is an oxidative phosphorylation blocker that acts to inhibit hepatic gluconeogenesis, increase muscle glucose uptake, and restore normal insulin secretion. It is the first approved drug of this class of anti-diabetic medication.


PATENT
https://patents.google.com/patent/WO2012072663A1/enEXAMPLESExample 1 : Synthesis and isolation of (+)-2-amino-3,6-dihydro-4-dimethylamino-6- methyl-l,3,5-triazine hydrochloride by the process according to the invention
Preliminary step: Synthesis of racemic 2-amino-3,6-dihydro-4-dimethylamino- 6-methyl-l,3,5-triazine hydrochloride:
Metformin hydrochloride is suspended in 4 volumes of isobutanol. Acetaldehyde diethylacetal (1.2 eq.) and para-toluenesulfonic acid (PTSA) (0.05 eq) are added and the resulting suspension is heated to reflux until a clear solution is obtained. Then 2 volumes of the solvent are removed via distillation and the resulting suspension is cooled to 20°C. The formed crystals are isolated on a filter dryer and washed with isobutanol (0.55 volumes). Drying is not necessary and the wet product can be directly used for the next step.Acetaldehyde diethylacetal can be replaced with 2,4,6-trimethyl-l,3,5-trioxane (paraldehyde).- Steps 1 and 2: formation of the diastereoisomeric salt and isolation of the desired diastereoisomer

Racemic 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine hydrochloride wet with isobutanol (obtained as crude product from preliminary step without drying) and L-(+)-Tartaric acid (1 eq.) are dissolved in 2.2 volumes of methanol at 20-40°C. The obtained clear solution is filtered and then 1 equivalent of triethylamine (TEA) is added while keeping the temperature below 30°C. The suspension is heated to reflux, stirred at that temperature for 10 minutes and then cooled down to 55°C. The temperature is maintained at 55°C for 2 hours and the suspension is then cooled to 5- 10°C. After additional stirring for 2 hours at 5-10°C the white crystals are isolated on a filter dryer, washed with methanol (2 x 0.5 Vol) and dried under vacuum at 50°C. The yield after drying is typically in the range of 40-45%
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt

γ ethanol HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt is suspended in 2 volumes of ethanol and 1.02 equivalents of HCl-gas are added under vacuum (-500 mbar). The suspension is heated to reflux under atmospheric pressure (N2) and 5% of the solvent is removed via distillation. Subsequent filtration of the clear colourless solution into a second reactor is followed by a cooling crystallization, the temperature is lowered to 2°C. The obtained suspension is stirred at 2°C for 3 hours and afterwards the white crystals are isolated with a horizontal centrifuge. The crystal cake is washed with ethanol and dried under vacuum at 40°C. The typical yield is 50-55% and the mother liquors can be used for the recovery of about 25-30%) of (+)-2-amino- 3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate.Example 2: Modification of the solvent of steps 3 and 4
– Steps 3 and 4: transformation of the isolated diastereoisomer of the tartrate salt into the hydrochloride salt and recovery of the salt
HN^NH acetone HN^NH(+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt synthesized according to steps 1 and 2 of example 1 is suspended in 1 volume (based on total amount of (+) 2-amino-3,6-dihydro-4-dimethylamino-6-methyl-l,3,5-triazine tartrate salt) of acetone at 20°C. To this suspension 1.01 equivalents of 37% Hydrochloric acid are added. The suspension is heated to reflux under atmospheric pressure (N2) and water is added until a clear solution is obtained. 1.5 vol of acetone are added at reflux temperature. The compound starts crystallising and the obtained suspension is kept at reflux for 2 hours followed by a cooling crystallization to 0°C. The obtained suspension is stirred at 0°C for 2 hours and the white crystals are isolated by centrifugation. The crystal cake is washed with isopropanol and dried under vacuum at 40°C in a continuous drying oven.
References
- ^ Vuylsteke V, Chastain LM, Maggu GA, Brown C (September 2015). “Imeglimin: A Potential New Multi-Target Drug for Type 2 Diabetes”. Drugs in R&D. 15 (3): 227–32. doi:10.1007/s40268-015-0099-3. PMC 4561051. PMID 26254210.
- ^ Dubourg J, Fouqueray P, Thang C, Grouin JM, Ueki K (April 2021). “Efficacy and Safety of Imeglimin Monotherapy Versus Placebo in Japanese Patients With Type 2 Diabetes (TIMES 1): A Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Multicenter Phase 3 Trial”. Diabetes Care. 44 (4): 952–959. doi:10.2337/dc20-0763. PMID 33574125.
- ^ Poxel SA (June 23, 2021). “Poxel and Sumitomo Dainippon Pharma Announce the Approval of TWYMEEG® (Imeglimin hydrochloride) for the Treatment of Type 2 Diabetes in Japan” (Press release).
| Clinical data | |
|---|---|
| Trade names | Twymeeg |
| Legal status | |
| Legal status | Rx-only in Japan |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 775351-65-0 |
| PubChem CID | 24812808 |
| ChemSpider | 26232690 |
| UNII | UU226QGU97 |
| CompTox Dashboard (EPA) | DTXSID50228237 |
| Chemical and physical data | |
| Formula | C6H13N5 |
| Molar mass | 155.205 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Imeglimin hydrochloride, Twymeeg, JAPAN 2021, APPROVALS 2021, Antidiabetic, イメグリミン塩酸塩, ATI DIABETES, DIABETES, Imeglimin
CC1N=C(NC(=N1)N(C)C)N.Cl
NEW DRUG APPROVALS
ONE TIME
$10.00
GLUCAGON
glucagon
EMA……Ogluo (glucagon), a hybrid medicine for the treatment of severe hypoglycaemia in diabetes mellitus. Hybrid applications rely in part on the results of pre-clinical tests and clinical trials of an already authorised reference product and in part on new data.
On 10 December 2020, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product Ogluo, intended for the treatment of severe hypoglycaemia in diabetes mellitus. The applicant for this medicinal product is Xeris Pharmaceuticals Ireland Limited.
Ogluo will be available as 0.5 and 1 mg solution for injection. The active substance of Ogluo is glucagon, a pancreatic hormone (ATC code: H04AA01); glucagon increases blood glucose concentration by stimulating glycogen breakdown and release of glucose from the liver.
The benefits with Ogluo are its ability to restore blood glucose levels in hypoglycaemic subjects. The most common side effects are nausea and vomiting.
Ogluo is a hybrid medicine1 of GlucaGen/GlucaGen Hypokit; GlucaGen has been authorised in the EU since October 1962. Ogluo contains the same active substance as GlucaGen but is available as a ready-to-use formulation intended for subcutaneous injection.
The full indication is:
Ogluo is indicated for the treatment of severe hypoglycaemia in adults, adolescents, and children aged 2 years and over with diabetes mellitus.
Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
1 Hybrid applications rely in part on the results of pre-clinical tests and clinical trials for a reference product and in part on new data.
Glucagon is a peptide hormone, produced by alpha cells of the pancreas. It works to raise the concentration of glucose and fatty acids in the bloodstream, and is considered to be the main catabolic hormone of the body.[3] It is also used as a medication to treat a number of health conditions. Its effect is opposite to that of insulin, which lowers extracellular glucose.[4] It is produced from proglucagon, encoded by the GCG gene.
The pancreas releases glucagon when the amount of glucose in the bloodstream is too low. Glucagon causes the liver to engage in glycogenolysis: converting stored glycogen into glucose, which is released into the bloodstream.[5] High blood-glucose levels, on the other hand, stimulate the release of insulin. Insulin allows glucose to be taken up and used by insulin-dependent tissues. Thus, glucagon and insulin are part of a feedback system that keeps blood glucose levels stable. Glucagon increases energy expenditure and is elevated under conditions of stress.[6] Glucagon belongs to the secretin family of hormones.
Function
Glucagon generally elevates the concentration of glucose in the blood by promoting gluconeogenesis and glycogenolysis.[7] Glucagon also decreases fatty acid synthesis in adipose tissue and the liver, as well as promoting lipolysis in these tissues, which causes them to release fatty acids into circulation where they can be catabolised to generate energy in tissues such as skeletal muscle when required.[8]
Glucose is stored in the liver in the form of the polysaccharide glycogen, which is a glucan (a polymer made up of glucose molecules). Liver cells (hepatocytes) have glucagon receptors. When glucagon binds to the glucagon receptors, the liver cells convert the glycogen into individual glucose molecules and release them into the bloodstream, in a process known as glycogenolysis. As these stores become depleted, glucagon then encourages the liver and kidney to synthesize additional glucose by gluconeogenesis. Glucagon turns off glycolysis in the liver, causing glycolytic intermediates to be shuttled to gluconeogenesis.
Glucagon also regulates the rate of glucose production through lipolysis. Glucagon induces lipolysis in humans under conditions of insulin suppression (such as diabetes mellitus type 1).[9]
Glucagon production appears to be dependent on the central nervous system through pathways yet to be defined. In invertebrate animals, eyestalk removal has been reported to affect glucagon production. Excising the eyestalk in young crayfish produces glucagon-induced hyperglycemia.[10]
Mechanism of action
Metabolic regulation of glycogen by glucagon.
Glucagon binds to the glucagon receptor, a G protein-coupled receptor, located in the plasma membrane of the cell. The conformation change in the receptor activates G proteins, a heterotrimeric protein with α, β, and γ subunits. When the G protein interacts with the receptor, it undergoes a conformational change that results in the replacement of the GDP molecule that was bound to the α subunit with a GTP molecule. This substitution results in the releasing of the α subunit from the β and γ subunits. The alpha subunit specifically activates the next enzyme in the cascade, adenylate cyclase.
Adenylate cyclase manufactures cyclic adenosine monophosphate (cyclic AMP or cAMP), which activates protein kinase A (cAMP-dependent protein kinase). This enzyme, in turn, activates phosphorylase kinase, which then phosphorylates glycogen phosphorylase b (PYG b), converting it into the active form called phosphorylase a (PYG a). Phosphorylase a is the enzyme responsible for the release of glucose 1-phosphate from glycogen polymers. An example of the pathway would be when glucagon binds to a transmembrane protein. The transmembrane proteins interacts with Gɑβ𝛾. Gɑ separates from Gβ𝛾 and interacts with the transmembrane protein adenylyl cyclase. Adenylyl cyclase catalyzes the conversion of ATP to cAMP. cAMP binds to protein kinase A, and the complex phosphorylates phosphorylase kinase.[11] Phosphorylated phosphorylase kinase phosphorylates phosphorylase. Phosphorylated phosphorylase clips glucose units from glycogen as glucose 1-phosphate. Additionally, the coordinated control of glycolysis and gluconeogenesis in the liver is adjusted by the phosphorylation state of the enzymes that catalyze the formation of a potent activator of glycolysis called fructose 2,6-bisphosphate.[12] The enzyme protein kinase A (PKA) that was stimulated by the cascade initiated by glucagon will also phosphorylate a single serine residue of the bifunctional polypeptide chain containing both the enzymes fructose 2,6-bisphosphatase and phosphofructokinase-2. This covalent phosphorylation initiated by glucagon activates the former and inhibits the latter. This regulates the reaction catalyzing fructose 2,6-bisphosphate (a potent activator of phosphofructokinase-1, the enzyme that is the primary regulatory step of glycolysis)[13] by slowing the rate of its formation, thereby inhibiting the flux of the glycolysis pathway and allowing gluconeogenesis to predominate. This process is reversible in the absence of glucagon (and thus, the presence of insulin).
Glucagon stimulation of PKA also inactivates the glycolytic enzyme pyruvate kinase in hepatocytes.[14]
Physiology
Production
A microscopic image stained for glucagon
The hormone is synthesized and secreted from alpha cells (α-cells) of the islets of Langerhans, which are located in the endocrine portion of the pancreas. Production, which is otherwise freerunning, is suppressed/regulated by amylin, a peptide hormone co-secreted with insulin from the pancreatic β cells.[15] As plasma glucose levels recede, the subsequent reduction in amylin secretion alleviates its suppression of the α cells, allowing for glucagon secretion.
In rodents, the alpha cells are located in the outer rim of the islet. Human islet structure is much less segregated, and alpha cells are distributed throughout the islet in close proximity to beta cells. Glucagon is also produced by alpha cells in the stomach.[16]
Recent research has demonstrated that glucagon production may also take place outside the pancreas, with the gut being the most likely site of extrapancreatic glucagon synthesis.[17]
Regulation
Secretion of glucagon is stimulated by:
- Hypoglycemia
- Epinephrine (via β2, α2,[18] and α1[19] adrenergic receptors)
- Arginine
- Alanine (often from muscle-derived pyruvate/glutamate transamination (see alanine transaminase reaction).
- Acetylcholine[20]
- Cholecystokinin
- Gastric inhibitory polypeptide
Secretion of glucagon is inhibited by:
- Somatostatin
- Amylin [21]
- Insulin (via GABA)[22]
- PPARγ/retinoid X receptor heterodimer.[23]
- Increased free fatty acids and keto acids into the blood.[24]
- Increased urea production
- Glucagon-like peptide-1
Structure
Glucagon is a 29-amino acid polypeptide. Its primary structure in humans is: NH2–His–Ser–Gln–Gly–Thr–Phe–Thr–Ser–Asp–Tyr–Ser–Lys–Tyr–Leu–Asp–Ser–Arg–Arg–Ala–Gln–Asp–Phe–Val–Gln–Trp–Leu–Met–Asn–Thr–COOH.
The polypeptide has a molecular mass of 3485 daltons.[25] Glucagon is a peptide (nonsteroid) hormone.
Glucagon is generated from the cleavage of proglucagon by proprotein convertase 2 in pancreatic islet α cells. In intestinal L cells, proglucagon is cleaved to the alternate products glicentin, GLP-1 (an incretin), IP-2, and GLP-2 (promotes intestinal growth).[26]
Pathology
Abnormally elevated levels of glucagon may be caused by pancreatic tumors, such as glucagonoma, symptoms of which include necrolytic migratory erythema,[27] reduced amino acids, and hyperglycemia. It may occur alone or in the context of multiple endocrine neoplasia type 1[28]
Elevated glucagon is the main contributor to hyperglycemic ketoacidosis in undiagnosed or poorly treated type 1 diabetes. As the beta cells cease to function, insulin and pancreatic GABA are no longer present to suppress the freerunning output of glucagon. As a result, glucagon is released from the alpha cells at a maximum, causing rapid breakdown of glycogen to glucose and fast ketogenesis.[29] It was found that a subset of adults with type 1 diabetes took 4 times longer on average to approach ketoacidosis when given somatostatin (inhibits glucagon production) with no insulin. Inhibiting glucagon has been a popular idea of diabetes treatment, however some have warned that doing so will give rise to brittle diabetes in patients with adequately stable blood glucose.[citation needed]
The absence of alpha cells (and hence glucagon) is thought to be one of the main influences in the extreme volatility of blood glucose in the setting of a total pancreatectomy.
History
In the 1920s, Kimball and Murlin studied pancreatic extracts, and found an additional substance with hyperglycemic properties. They described glucagon in 1923.[30] The amino acid sequence of glucagon was described in the late 1950s.[31] A more complete understanding of its role in physiology and disease was not established until the 1970s, when a specific radioimmunoassay was developed.[citation needed]
Etymology
Kimball and Murlin coined the term glucagon in 1923 when they initially named the substance the glucose agonist.[32]
References
- ^ Jump up to:a b c GRCh38: Ensembl release 89: ENSG00000115263 – Ensembl, May 2017
- ^ “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Voet D, Voet JG (2011). Biochemistry (4th ed.). New York: Wiley.
- ^ Reece J, Campbell N (2002). Biology. San Francisco: Benjamin Cummings. ISBN 978-0-8053-6624-2.
- ^ Orsay J (2014). Biology 1: Molecules. Examkrackers Inc. p. 77. ISBN 978-1-893858-70-1.
- ^ Jones BJ, Tan T, Bloom SR (March 2012). “Minireview: Glucagon in stress and energy homeostasis”. Endocrinology. 153 (3): 1049–54. doi:10.1210/en.2011-1979. PMC 3281544. PMID 22294753.
- ^ Voet D, Voet JG (2011). Biochemistry (4th ed.). New York: Wiley.
- ^ HABEGGER, K. M., HEPPNER, K. M., GEARY, N., BARTNESS, T. J., DIMARCHI, R. & TSCHÖP, M. H. (2010). “The metabolic actions of glucagon revisited”. Nature Reviews. Endocrinology. 6 (12): 689–697. doi:10.1038/nrendo.2010.187. PMC 3563428. PMID 20957001.
- ^ Liljenquist JE, Bomboy JD, Lewis SB, Sinclair-Smith BC, Felts PW, Lacy WW, Crofford OB, Liddle GW (January 1974). “Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men”. The Journal of Clinical Investigation. 53 (1): 190–7. doi:10.1172/JCI107537. PMC 301453. PMID 4808635.
- ^ Leinen RL, Giannini AJ (1983). “Effect of eyestalk removal on glucagon induced hyperglycemia in crayfish”. Society for Neuroscience Abstracts. 9: 604.
- ^ Yu Q, Shuai H, Ahooghalandari P, Gylfe E, Tengholm A (July 2019). “Glucose controls glucagon secretion by directly modulating cAMP in alpha cells”. Diabetologia. 62 (7): 1212–1224. doi:10.1007/s00125-019-4857-6. PMC 6560012. PMID 30953108.
- ^ Hue L, Rider MH (July 1987). “Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues”. The Biochemical Journal. 245 (2): 313–24. doi:10.1042/bj2450313. PMC 1148124. PMID 2822019.
- ^ Claus TH, El-Maghrabi MR, Regen DM, Stewart HB, McGrane M, Kountz PD, Nyfeler F, Pilkis J, Pilkis SJ (1984). “The role of fructose 2,6-bisphosphate in the regulation of carbohydrate metabolism”. Current Topics in Cellular Regulation. 23: 57–86. doi:10.1016/b978-0-12-152823-2.50006-4. ISBN 9780121528232. PMID 6327193.
- ^ Feliú JE, Hue L, Hers HG (August 1976). “Hormonal control of pyruvate kinase activity and of gluconeogenesis in isolated hepatocytes”. Proceedings of the National Academy of Sciences of the United States of America. 73 (8): 2762–6. Bibcode:1976PNAS…73.2762F. doi:10.1073/pnas.73.8.2762. PMC 430732. PMID 183209.
- ^ Zhang, Xiao-Xi (2016). “Neuroendocrine Hormone Amylin in Diabetes”. World J Diabetes. 7 (9): 189–197. doi:10.4239/wjd.v7.i9.189. PMC 4856891. PMID 27162583.
- ^ Unger RH, Cherrington AD (January 2012). “Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover”. The Journal of Clinical Investigation. 122(1): 4–12. doi:10.1172/JCI60016. PMC 3248306. PMID 22214853.
- ^ Holst JJ, Holland W, Gromada J, Lee Y, Unger RH, Yan H, Sloop KW, Kieffer TJ, Damond N, Herrera PL (April 2017). “Insulin and Glucagon: Partners for Life”. Endocrinology. 158(4): 696–701. doi:10.1210/en.2016-1748. PMC 6061217. PMID 28323959.
- ^ Layden BT, Durai V, Lowe WL (2010). “G-Protein-Coupled Receptors, Pancreatic Islets, and Diabetes”. Nature Education. 3 (9): 13.
- ^ Skoglund G, Lundquist I, Ahrén B (November 1987). “Alpha 1- and alpha 2-adrenoceptor activation increases plasma glucagon levels in the mouse”. European Journal of Pharmacology. 143 (1): 83–8. doi:10.1016/0014-2999(87)90737-0. PMID 2891547.
- ^ Honey RN, Weir GC (October 1980). “Acetylcholine stimulates insulin, glucagon, and somatostatin release in the perfused chicken pancreas”. Endocrinology. 107 (4): 1065–8. doi:10.1210/endo-107-4-1065. PMID 6105951.
- ^ Zhang, Xiao-Xi (2016). “Neuroendocrine Hormone Amylin in Diabetes”. World J Diabetes. 7 (9): 189–197. doi:10.4239/wjd.v7.i9.189. PMC 4856891. PMID 27162583.
- ^ Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, Liu S, Wendt A, Deng S, Ebina Y, Wheeler MB, Braun M, Wang Q (January 2006). “Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system”. Cell Metabolism. 3 (1): 47–58. doi:10.1016/j.cmet.2005.11.015. PMID 16399504.
- ^ Krätzner R, Fröhlich F, Lepler K, Schröder M, Röher K, Dickel C, Tzvetkov MV, Quentin T, Oetjen E, Knepel W (February 2008). “A peroxisome proliferator-activated receptor gamma-retinoid X receptor heterodimer physically interacts with the transcriptional activator PAX6 to inhibit glucagon gene transcription”. Molecular Pharmacology. 73 (2): 509–17. doi:10.1124/mol.107.035568. PMID 17962386. S2CID 10108970.
- ^ Johnson LR (2003). Essential Medical Physiology. Academic Press. pp. 643–. ISBN 978-0-12-387584-6.
- ^ Unger RH, Orci L (June 1981). “Glucagon and the A cell: physiology and pathophysiology (first two parts)”. The New England Journal of Medicine. 304 (25): 1518–24. doi:10.1056/NEJM198106183042504. PMID 7015132.
- ^ Orskov C, Holst JJ, Poulsen SS, Kirkegaard P (November 1987). “Pancreatic and intestinal processing of proglucagon in man”. Diabetologia. 30 (11): 874–81. doi:10.1007/BF00274797 (inactive 2020-10-11). PMID 3446554.
- ^ John AM, Schwartz RA (December 2016). “Glucagonoma syndrome: a review and update on treatment”. Journal of the European Academy of Dermatology and Venereology. 30 (12): 2016–2022. doi:10.1111/jdv.13752. PMID 27422767. S2CID 1228654.
- ^ Oberg K (December 2010). “Pancreatic endocrine tumors”. Seminars in Oncology. 37 (6): 594–618. doi:10.1053/j.seminoncol.2010.10.014. PMID 21167379.
- ^ Fasanmade OA, Odeniyi IA, Ogbera AO (June 2008). “Diabetic ketoacidosis: diagnosis and management”. African Journal of Medicine and Medical Sciences. 37 (2): 99–105. PMID 18939392.
- ^ Kimball C, Murlin J (1923). “Aqueous extracts of pancreas III. Some precipitation reactions of insulin”. J. Biol. Chem. 58 (1): 337–348.
- ^ Bromer W, Winn L, Behrens O (1957). “The amino acid sequence of glucagon V. Location of amide groups, acid degradation studies and summary of sequential evidence”. J. Am. Chem. Soc. 79 (11): 2807–2810. doi:10.1021/ja01568a038.
- ^ “History of glucagon – Metabolism, insulin and other hormones – Diapedia, The Living Textbook of Diabetes”. http://www.diapedia.org. Archived from the original on 2017-03-27. Retrieved 2017-03-26.
External links
- PDBe-KB provides an overview of all the structure information available in the PDB for Human Glucagon
| GCG | ||
|---|---|---|
| Available structuresPDBHuman UniProt search: PDBe RCSBshowList of PDB id codes | ||
| Identifiers | ||
| Aliases | GCG, GLP1, glucagon, GRPP, GLP-1, GLP2 | |
| External IDs | OMIM: 138030 HomoloGene: 136497 GeneCards: GCG | |
| hideGene location (Human)Chr.Chromosome 2 (human)[1]Band2q24.2Start162,142,882 bp[1]End162,152,404 bp[1] | ||
| hideRNA expression patternMore reference expression data | ||
| showGene ontology | ||
| Orthologs | ||
| Species | Human | Mouse |
| Entrez | 2641 | n/a |
| Ensembl | ENSG00000115263 | n/a |
| UniProt | P01275 | n/a |
| RefSeq (mRNA) | NM_002054 | n/a |
| RefSeq (protein) | NP_002045 | n/a |
| Location (UCSC) | Chr 2: 162.14 – 162.15 Mb | n/a |
| PubMed search | [2] | n/a |
| Wikidata | ||
| View/Edit Human |
///////////GLUCAGON, DIABETES, PEPTIDE, HORMONE
SY-008


![Acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91758795&t=l)
SY-008
CAS 1878218-66-6
FREE FORM 1480443-32-0
SGLT1 inhibitor (type 2 diabetes),
β-D-Glucopyranoside, 4-[[4-[(1E)-4-(2,9-diazaspiro[5.5]undec-2-yl)-1-buten-1-yl]-2-methylphenyl]methyl]-5-(1-methylethyl)-1H-pyrazol-3-yl, acetate (1:1)
acetic acid;(2S,3R,4S,5S,6R)-2-[[4-[[4-[(E)-4-(2,9-diazaspiro[5.5]undecan-2-yl)but-1-enyl]-2-methylphenyl]methyl]-5-propan-2-yl-1H-pyrazol-3-yl]oxy]-6-(hydroxymethyl)oxane-3,4,5-triol
MF H50 N4 O6 . C2 H4 O2
MW 58.8 g/mol,C35H54N4O8
Originator Eli Lilly
- Developer Eli Lilly; Yabao Pharmaceutical Group
- Class Antihyperglycaemics; Small molecules
- Mechanism of Action Sodium-glucose transporter 1 inhibitors
- Phase I Diabetes mellitus
- 28 Aug 2018 No recent reports of development identified for phase-I development in Diabetes-mellitus in Singapore (PO)
- 24 Jun 2018 Biomarkers information updated
- 12 Mar 2018 Phase-I clinical trials in Diabetes mellitus (In volunteers) in China (PO) (NCT03462589)
-
Eli Lilly is developing SY 008, a sodium glucose transporter 1 (SGLT1) inhibitor, for the treatment of diabetes mellitus. The approach of inhibiting SGLT1 could be promising because it acts independently of the beta cell and could be effective in both early and advanced stages of diabetes. Reducing both glucose and insulin may improve the metabolic state and potentially the health of beta cells, without causing weight gain or hypoglycaemia. Clinical development is underway in Singapore and China.
As at August 2018, no recent reports of development had been identified for phase-I development in Diabetes-mellitus in Singapore (PO).
Suzhou Yabao , under license from Eli Lilly , is developing SY-008 , an SGLT1 inhibitor, for the potential oral capsule treatment of type 2 diabetes in China. By April 2019, a phase Ia trial was completed
PATENT
WO 2013169546
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 201 1 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLTl is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLTl may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLTl inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 201 1/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides certain novel inhibitors of SGLTl which may be suitable for the treatment of diabetes.
Accordingly, the present invention provides a compound of Formula II:
Preparation 1
Synthesis of (4-bromo-2-methyl-phenyl)methanol.
Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). 1H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)methanol.
Borane-dimethyl sulfide complex (2M in THF; 1 16 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.1 13 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
Synthesis of 4-bromo- l-2-methyl-benzene.
Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4-bromo-2-methyl-phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and
-Cl-
dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. XH NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo- 1 -chloromethyl-2-methyl-benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). XH NMR (300.1 1 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
Synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol.
Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo- l-chloromethyl-2-methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction.
Extract with ethyl acetate (200 mL), wash extract with water (200 rnL) and brine (200 mL), dry over a2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l).
Alternative synthesis of 4-r(4-bromo-2-methyl-phenyl)methyl1-5-isopropyl- !H-pyrazol- 3-oL
A solution of 4-bromo- 1 -chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (1 1.94 g, 71.94 mmol) and methyl 4-methyl-3-oxo valerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2O5 at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/31 1 (M+l).
Preparation 4
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (ml 2): 889.2 (M+l), 887.2 (M-l).
Preparation 5
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside.
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
Synthesis of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2-dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+1).
Preparation 8
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D- glucopyranoside dihydrochloride.
Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3is)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
Example 1
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40 °C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 um C18XBridge ODB column, solvent A – 1¾0 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Preparation 9
Synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra- O-acetyl-beta-D-glucopyranoside.
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammomum chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate
(32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l).
Preparation 10
Synthesis of 4- {4-[( lis)-4-hydroxybut- 1 -en- 1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- 1H- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol). MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
Synthesis of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0- acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4- {4-[(lis)-4-oxybut- 1 -en-1 -yl]-2-methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2-dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
Synthesis oftert-butyl 2-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,8- diazaspiro[4.5]decane-8-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 852.8, 853.6 (M+l), 850.8, 851.6 (M-l).
Preparation 14
Synthesis oftert-butyl 9-{(3E)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-3,9- diazaspiro[5.5]undecane-3-carboxylate.
The title compound is prepared essentially by the method of Preparation 12. S (m/z): 866.8, 867.6 (M+l), 864.8, 865.6 (M-l).
Preparation 15
Synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2- methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D- glucopyranoside dihydrochloride.
Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Preparation 16
Synthesis of 4-{4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5- (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
dihydrochloride.
The title compound is prepared essentially by the method of Preparation 15. MS (m/z): 752.8, 753.8 (M+1), 750.8 (M-1).
First alternative synthesis of Example 1
First alternative synthesis of 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en- 2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4-{4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 urn C18XBridge ODB column, solvent A – H20 w NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+1), 596.8 (M-1). 1H MR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=1.3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.1 1 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Example 2
Synthesis of 4- {4-[(lE)-4-(2,8-diazaspiro[4.5]dec-2-yl)but-l-en-l-yl]-2-methylbi
(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
O H
The title compound is prepared essentially by the method of the first alternative synthesis of Example 1. MS (m/z): 584.7 (M+l), 582.8 (M-l).
Example 3
Synthesis of 4- {4-[( 1 E)-4-(3 ,9-diazaspiro[5.5]undec-3 -yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)- lH-pyrazol-3-yl beta-D-glucopyranoside.
The title compound is prepared essentially by first treating the compound of Prearation 14 with HC1 as discussed in Preparation 15 then treating the resulting hydrochloride salt with triethyl amine as discussed in the first alternative synthesis of Example 1. MS (m/z): 598.8, 599.8 (M+l), 596.8, 597.8 (M-l).
Example 1 Preparation 17
Synthesis of tert-butyl 4-but-3- nyl-4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgSC^, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). iH MR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H),
2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 18
Synthesis of tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]- 4,9-diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5, 5-tetramethyl-1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield), !H NMR (300.1 1 MHz, CDC13): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H),
3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 19
Synthesis of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]- lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate.
Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2-dicyclohexylphosphino-2′,4′,6′-tri-i-propyl-l, r-biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).
Second alternative Synthesis of Example 1
Second alternative synthesis of 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2- {(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200ml) and concentrated in vacuo. This is repeated several times give the title compound (12.22 g, yield 96%). MS (m/z): 599 (M+l). [a]D20 = -12 ° (C=0.2, MeOH).
PATENT
WO 2015069541
https://patents.google.com/patent/WO2015069541A1
4-{4-[(1 E)-4-(2,9-DIAZASPIRO[5.5]UNDEC-2-YL)BUT-1 -EN-1
-YL]-2-METHYLBENZYL}-5-(PROPAN-2-YL)-1 H-PYRAZOL-3-YL
BETA-D- GLUCOPYRANOSIDE ACETATE
The present invention relates to a novel SGLT1 inhibitor which is an acetate salt of a pyrazole compound, to pharmaceutical compositions comprising the compound, to methods of using the compound to treat physiological disorders, and to intermediates and processes useful in the synthesis of the compound.
The present invention is in the field of treatment of diabetes and other diseases and disorders associated with hyperglycemia. Diabetes is a group of diseases that is characterized by high levels of blood glucose. It affects approximately 25 million people in the United States and is also the 7th leading cause of death in U.S. according to the 2011 National Diabetes Fact Sheet (U.S. Department of Health and Human Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the absorption of carbohydrates, such as glucose. More specifically, SGLT1 is responsible for transport of glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 may result in reduced absorption of glucose in the small intestine, thus providing a useful approach to treating diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives with human SGLT1 inhibitory activity which are further disclosed as useful for the prevention or treatment of a disease associated with hyperglycemia, such as diabetes. In addition, WO 2011/039338 discloses certain pyrazole derivatives with SGLT1/SGLT2 inhibitor activity which are further disclosed as being useful for treatment of bone diseases, such as osteoporosis.
There is a need for alternative drugs and treatment for diabetes. The present invention provides an acetate salt of a pyrazole compound, which is an SGLT1 inhibitor, and as such, may be suitable for the treatment of certain disorders, such as diabetes. Accordingly, the present invention provides a compound of Formula I:

or hydrate thereof.
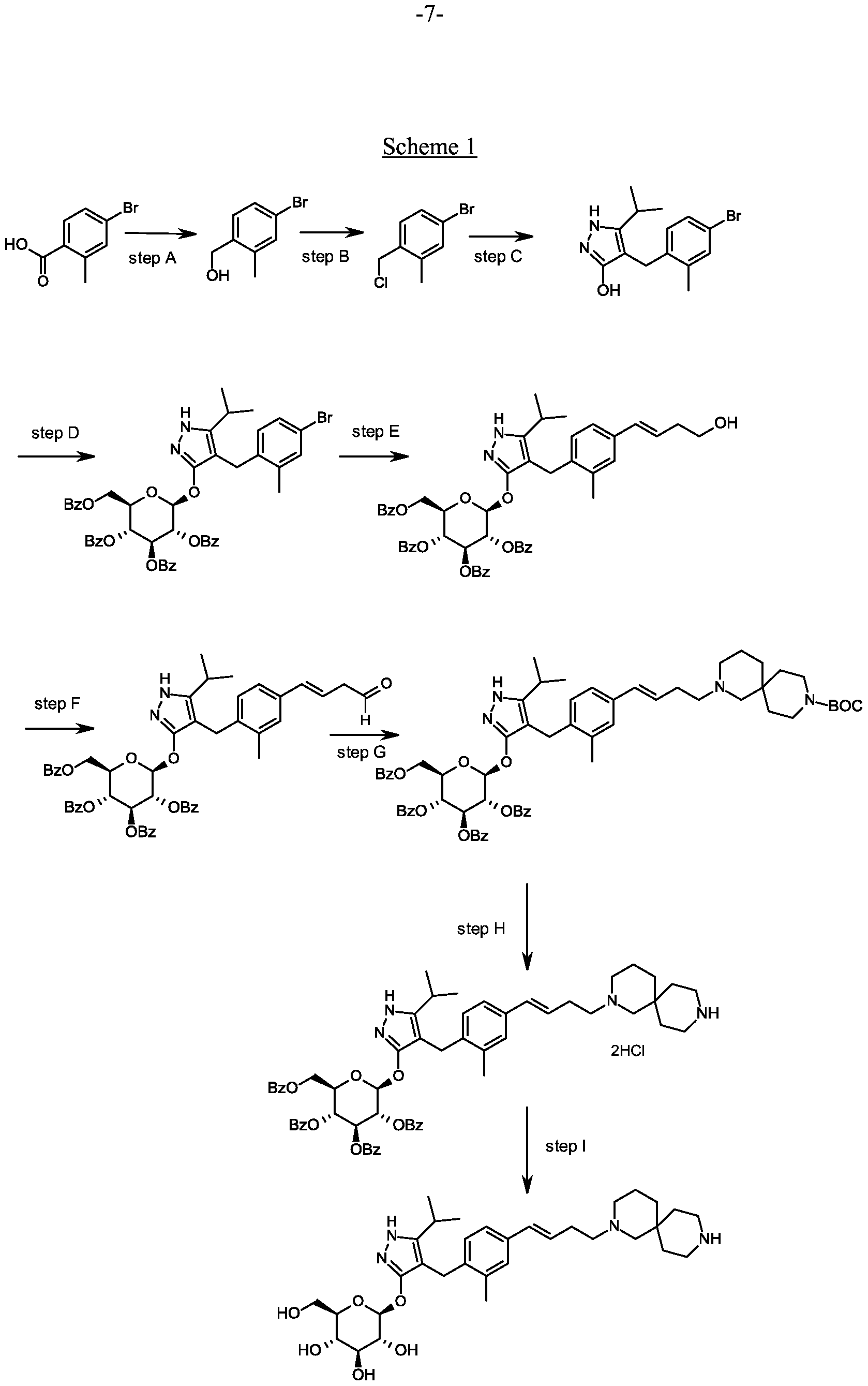
Preparation 1
(4-bromo-2-methyl-phenyl)methanol

Scheme 1, step A: Add borane-tetrahydrofuran complex (0.2 mol, 200 mL, 1.0 M solution) to a solution of 4-bromo-2-methylbenzoic acid (39 g, 0.18 mol) in
tetrahydrofuran (200 mL). After 18 hours at room temperature, remove the solvent under the reduced pressure to give a solid. Purify by flash chromatography to yield the title compound as a white solid (32.9 g, 0.16 mol). !H NMR (CDCI3): δ 1.55 (s, 1H), 2.28 (s, 3H), 4.61 (s, 2H), 7.18-7.29 (m, 3H).
Alternative synthesis of (4-bromo-2-methyl-phenyl)mefhanol.
Borane-dimethyl sulfide complex (2M in THF; 116 mL, 0.232 mol) is added slowly to a solution of 4-bromo-2-methylbenzoic acid (24.3 g, 0.113 mol) in anhydrous tetrahydrofuran (THF, 146 mL) at 3 °C. After stirring cold for 10 min the cooling bath is removed and the reaction is allowed to warm slowly to ambient temperature. After 1 hour, the solution is cooled to 5°C, and water (100 mL) is added slowly. Ethyl acetate (100 mL) is added and the phases are separated. The organic layer is washed with saturated aqueous NaHC03 solution (200 mL) and dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by filtration through a short pad of silica eluting with 15% ethyl acetate/iso-hexane to give the title compound (20.7 g, 91.2% yield). MS (m/z): 183/185 (M+l-18).
Preparation 2
4-bromo- 1 -chloromethyl -2 -methyl -benzene

Scheme 1, step B: Add thionyl chloride (14.31 mL, 0.2 mol,) to a solution of (4- bromo-2 -methyl -phenyl)methanol (32.9 g, 0.16 mol) in dichloromethane (200 mL) and dimethylformamide (0.025 mol, 2.0 mL) at 0°C. After 1 hour at room temperature pour the mixture into ice-water (100 g), extract with dichloromethane (300 mL), wash extract with 5% aq. sodium bicarbonate (30 mL) and brine (200 mL), dry over sodium sulfate, and concentrate under reduced pressure to give the crude title compound as a white solid (35.0 g, 0.16 mol). The material is used for the next step of reaction without further purification. !H NMR (CDC13): δ 2.38 (s, 3H), 4.52 (s, 2H), 7.13-7.35 (m, 3H).
Alternative synthesis of 4-bromo-l-chloromethyl-2-methyl -benzene. Methanesulfonyl chloride (6.83 mL, 88.3 mmol) is added slowly to a solution of (4-bromo-2-methyl-phenyl)methanol (16.14 g, 80.27 mmol) and triethylamine (16.78 mL; 120.4 mmol) in dichloromethane (80.7 mL) cooled in ice/water. The mixture is allowed to slowly warm to ambient temperature and is stirred for 16 hours. Further
methanesulfonyl chloride (1.24 mL; 16.1 mmol) is added and the mixture is stirred at ambient temperature for 2 hours. Water (80mL) is added and the phases are separated. The organic layer is washed with hydrochloric acid (IN; 80 mL) then saturated aqueous sodium hydrogen carbonate solution (80 mL), then water (80 mL), and is dried over Na2S04. Filtration and concentration under reduced pressure gives a residue which is purified by flash chromatography (eluting with hexane) to give the title compound (14.2 g; 80.5% yield). !H NMR (300.11 MHz, CDC13): δ 7.36-7.30 (m, 2H), 7.18 (d, J= 8.1 Hz, 1H), 4.55 (s, 2H), 2.41 (s, 3H).
Preparation 3
4- [(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-3 -ol

Scheme 1, step C: Add sodium hydride (8.29 g, 0.21 mol, 60% dispersion in oil) to a solution of methyl 4-methyl-3-oxovalerate (27.1 mL, 0.19 mol) in tetrahydrofuran at 0°C. After 30 min at room temperature, add a solution of 4-bromo-l-chloromethyl-2- methyl-benzene (35.0 g, 0.16 mol) in tetrahydrofuran (50 mL). Heat the resulting mixture at 70 °C overnight (18 hours). Add 1.0 M HC1 (20 mL) to quench the reaction. Extract with ethyl acetate (200 mL), wash extract with water (200 mL) and brine (200 mL), dry over Na2S04, filter and concentrate under reduced pressure. Dissolve the resulting residue in toluene (200 mL) and add hydrazine monohydrate (23.3 mL, 0.48 mol). Heat the mixture at 120 °C for 2 hours with a Dean-Stark apparatus to remove water. Cool and remove the solvent under the reduced pressure, dissolve the residue with dichloromethane (50 mL) and methanol (50 mL). Pour this solution slowly to a beaker with water (250 mL). Collect the resulting precipitated product by vacuum filtration. Dry in vacuo in an oven overnight at 40 °C to yield the title compound as a solid (48.0 g, 0.16 mol). MS (m/z): 311.0 (M+l), 309.0 (M-l). Alternative synthesis of 4-[(4-bromo-2-methyl-phenyl)methyl] -5 -isopropyl- lH-pyrazol-
3-ol.
A solution of 4-bromo-l-chloromethyl-2-methyl-benzene (13.16 g, 59.95 mmoles) in acetonitrile (65.8 mL) is prepared. Potassium carbonate (24.86 g, 179.9 mmol), potassium iodide (11.94 g, 71.94 mmol) and methyl 4-methyl-3-oxovalerate (8.96 mL; 62.95 mmol) are added. The resulting mixture is stirred at ambient temperature for 20 hours. Hydrochloric acid (2N) is added to give pH 3. The solution is extracted with ethyl acetate (100 ml), the organic phase is washed with brine (100 ml) and dried over Na2S04. The mixture is filtered and concentrated under reduced pressure. The residue is dissolved in toluene (65.8 mL) and hydrazine monohydrate (13.7 mL, 0.180 mol) is added. The resulting mixture is heated to reflux and water is removed using a Dean and Stark apparatus. After 3 hours the mixture is cooled to 90 °C and additional hydrazine monohydrate (13.7 mL; 0.180 mol) is added and the mixture is heated to reflux for 1 hour. The mixture is cooled and concentrated under reduced pressure. The resulting solid is triturated with water (200 mL), filtered and dried in a vacuum oven over P2Os at 60°C. The solid is triturated in iso-hexane (200 mL) and filtered to give the title compound (14.3 g; 77.1% yield). MS (m/z): 309/311 (M+l).
Preparation 4
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl- beta-D-glucopyranoside
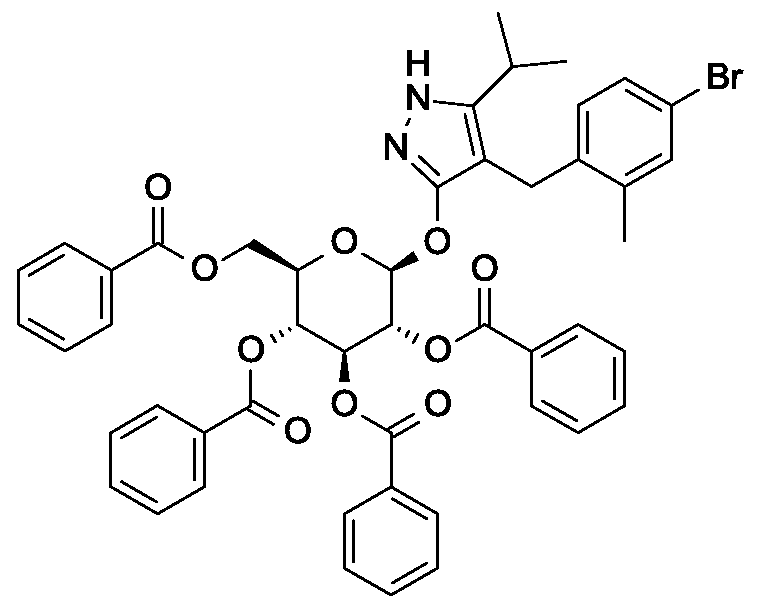
Scheme 1, step D: To a 1L flask, add 4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (20 g, 64.7 mmol), alpha-D-glucopyranosyl bromide tetrabenzoate (50 g, 76 mmol), benzyltributylammonium chloride (6 g, 19.4 mmol), dichloromethane (500 mL), potassium carbonate (44.7 g, 323 mmol) and water (100 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (500mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the residue by flash chromatography to yield the title compound (37 g, 64 mmol). MS (m/z): 889.2 (M+l), 887.2 (M-l).
Preparation 5
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
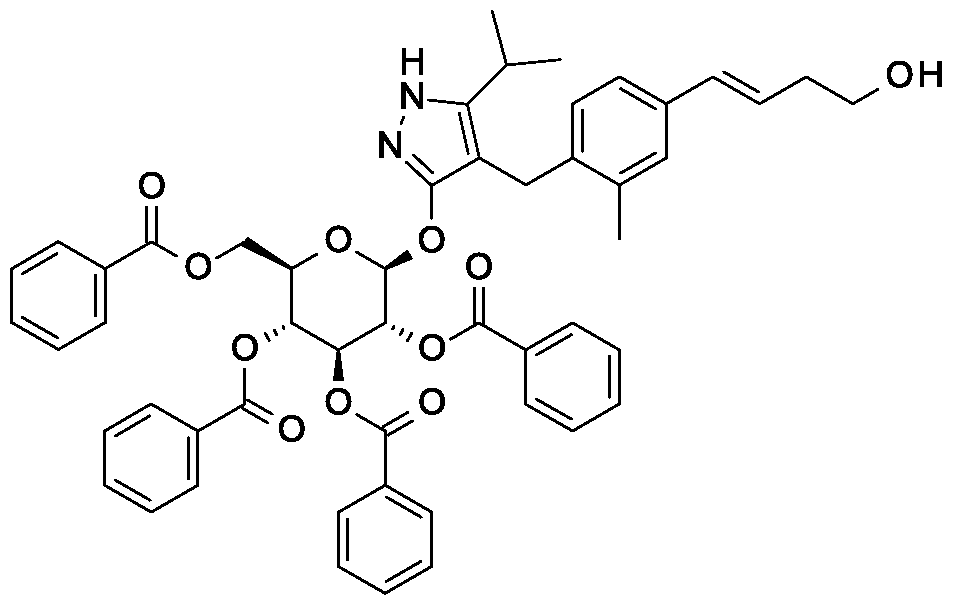
Scheme 1, step E: Add 3-buten-l-ol (0.58 mL, 6.8 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- lH-pyrazol-3 -yl 2,3 ,4,6-tetra-O-benzoyl-beta-D- glucopyranoside (3 g, 3.4 mmol) in acetonitrile (30 mL) and triethylamine (20 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (205 mg, 0.67 mmol) and palladium acetate (76 mg, 0.34 mmol). Reflux at 90 °C for 2 hours. Cool to room temperature and concentrate to remove the solvent under the reduced pressure. Purify the residue by flash chromatography to yield the title compound (2.1 g, 2.4 mmol). MS (m/z): 878.4 (M+l).
Preparation 6
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside
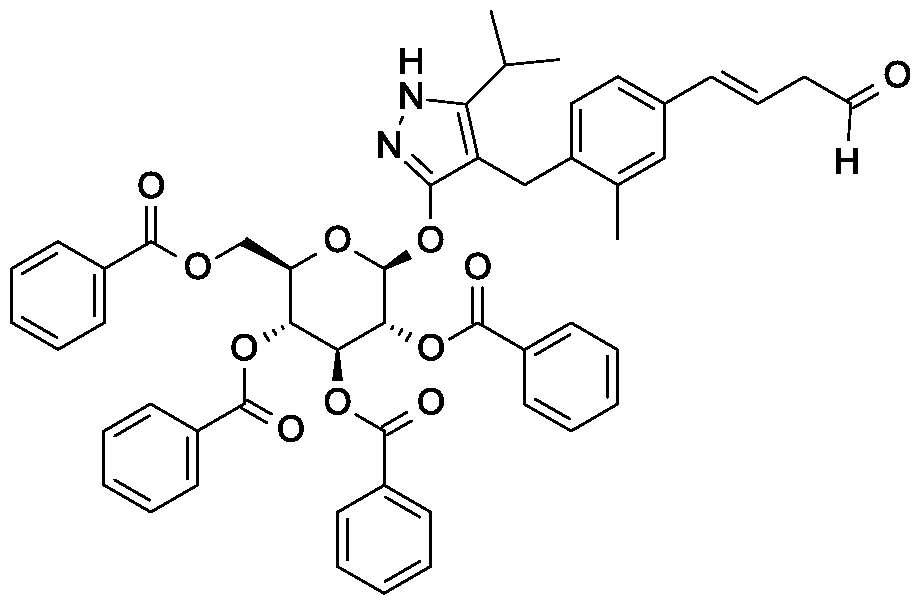
Scheme 1, step F: Add 3,3,3-triacetoxy-3-iodophthalide (134 mg, 0.96 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (280 mg, 0.32 mmol) and sodium bicarbonate (133.8 mg, 1.6 mmol) in dichloromethane (20 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (270 mg, 0.31 mmol). MS (m/z): 876.5 (M+l), 874.5 (M-l).
Preparation 7
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-benzoyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate

Scheme 1, step G: Add sodium triacetoxyborohydride (98 mg, 0.46 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside (270 mg, 0.31 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (179 mg, 0.62 mmol) in 1,2- dichloroethane (5 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (10 mL). Extract with dichloromethane (30 mL). Wash extract with water (30 mL) and brine (40 mL), dry organic phase over sodium sulfate, filter and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (275 mg, 0.25 mmol).
MS (m/z): 1115.6 (M+l).
Preparation 8
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride

Scheme 1, step H: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 0.6 mL, 2.4 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-benzoyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (275 mg, 0.25 mmol) in dichloromethane (5 mL). After overnight (18 hours) at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (258 mg, 0.24 mmol). MS (m/z): 1015.6 (M+l).
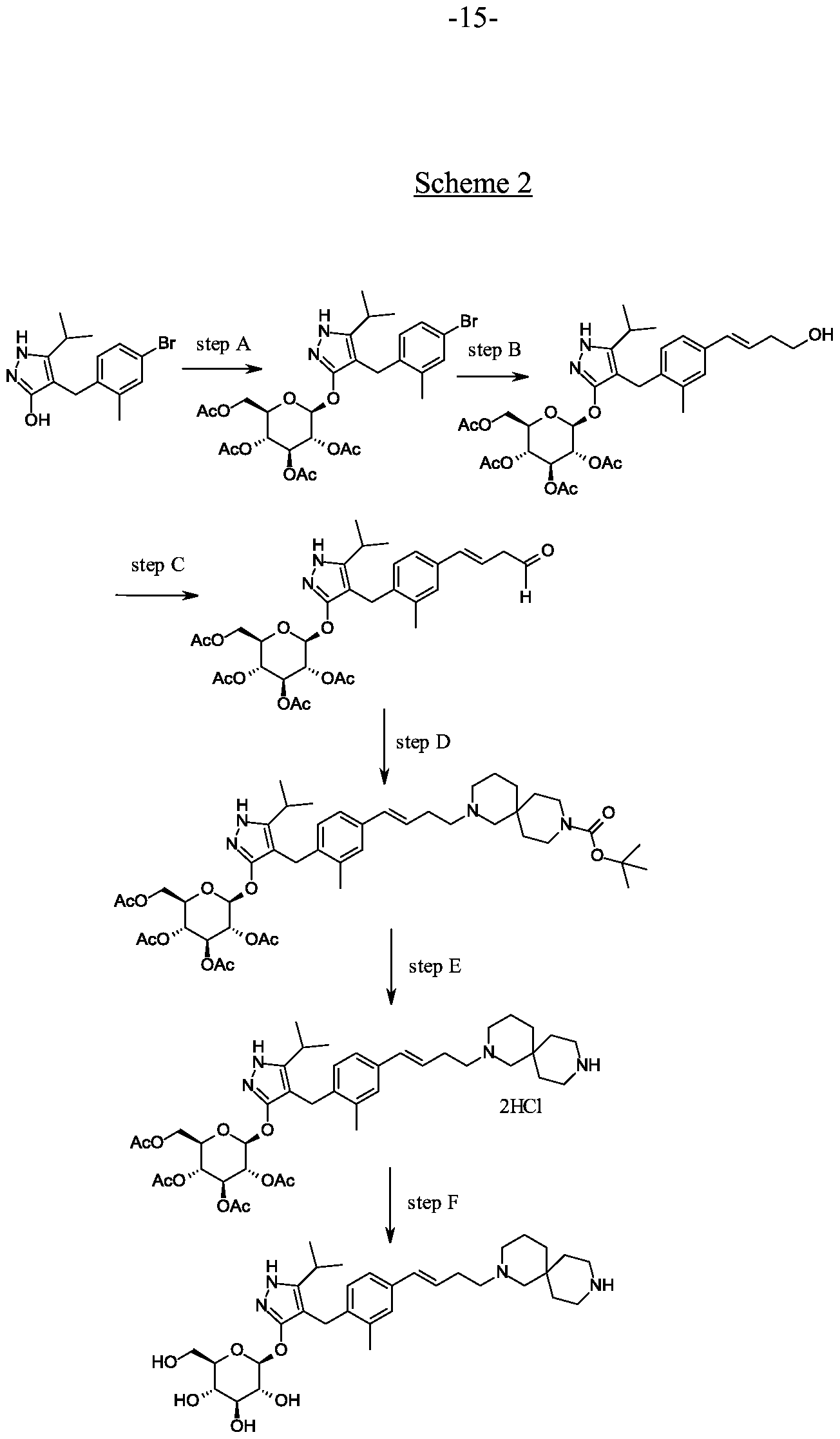
Preparation 9
4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl- beta-D-glucopyranoside.
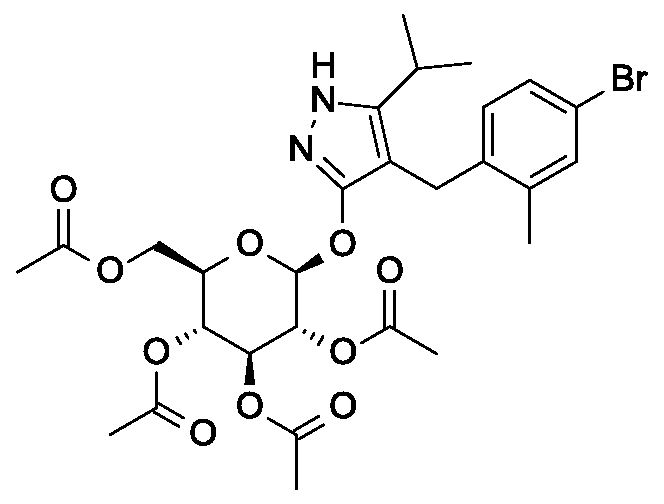
Scheme 2, step A: To a 1 L flask, add 4-[(4-bromo-2-methyl-phenyl)mefhyl]-5- isopropyl-lH-pyrazol-3-ol (24 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D- glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (5 g, 15.5 mmol), dichloromethane (250 mL), potassium carbonate (32 g, 323 mmol) and water (120 mL). Stir the reaction mixture overnight at room temperature. Extract with dichloromethane (450 mL). Wash extract with water (300 mL) and brine (500 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (36.5 g, 57 mmol). MS (m/z): 638.5 (M+l), 636.5 (M-l).
Alternative synthesis of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside.
Reagents 4-[(4-bromo-2-methyl-phenyl)methyl]-5-isopropyl-lH-pyrazol-3-ol (24.0 g, 77.6 mmol), 2,3,4,6-tetra-O-acetyl-alpha-D-glucopyranosyl bromide (50.4 g, 116 mmol), benzyltributylammonium chloride (4.94 g, 15.52 mmol), potassium carbonate (32.18 g, 232.9 mmol), dichloromethane (250 mL) and water (120 mL) are combined and the mixture is stirred at ambient temperature for 18 hours. The mixture is partitioned between dichloromethane (250 mL) and water (250 mL). The organic phase is washed with brine (250 mL), dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting residue is purified by flash chromatography (eluting with 10% ethyl acetate in dichloromethane to 70% ethyl acetate in dichloromethane) to give the title compound (36.5 g, 74% yield). MS (m/z): 639/641 (M+l). Preparation 10
4- {4- [(lis)-4-hydroxybut- 1 -en- 1 -yl] -2-methylbenzyl } -5 -(propan-2-yl)- lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside
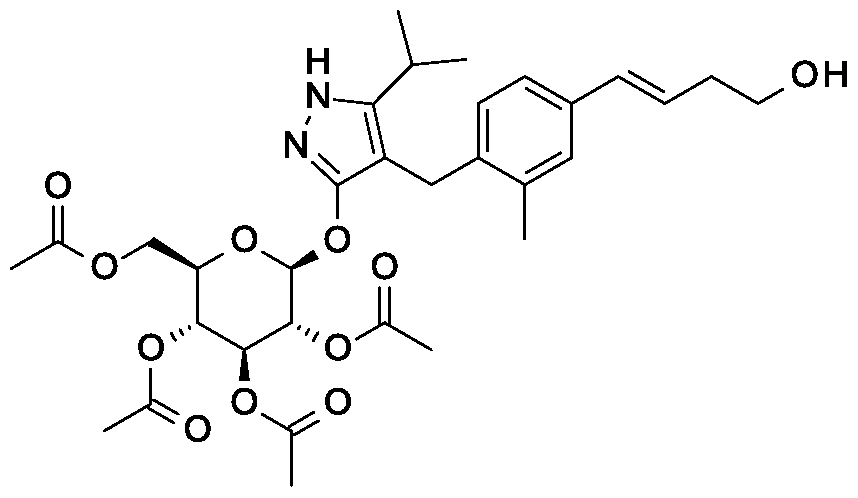
Scheme 2, step B: Add 3-buten-l-ol (6.1 mL, 70 mmol) to a solution of 4-(4- bromo-2-methylbenzyl)-5 -(propan-2-yl)- 1 H-pyrazol-3 -yl 2,3 ,4,6-tetra-O-acetyl-beta-D- glucopyranoside (15 g, 23.5 mmol) in acetonitrile (200 mL) and triethylamine (50 mL). Degas the solution with nitrogen over 10 minutes. Add tri-o-tolylphosphine (1.43 g, 4.7 mmol) and palladium acetate (526 mg, 2.35 mmol). After refluxing at 90 °C for 2 hours, cool, and concentrate to remove the solvent under the reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (7.5 g, 11.9 mmol) MS (m/z): 631.2 (M+l), 629.2 (M-l).
Preparation 11
4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol-3-yl
2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside

Scheme 2, step C: Add 3,3,3-triacetoxy-3-iodophthalide (2.1g, 4.76 mmol) to a solution of 4-{4-[(l£)-4-hydroxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH- pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside ( 1.5 g, 2.38 mmol) and sodium bicarbonate (2 g, 23.8 mmol) in dichloromethane (50 mL) at 0 °C. After 15 minutes at room temperature, quench the reaction with saturated aqueous sodium thiosulfate (10 mL). Extract with dichloromethane (30 mL), wash extract with water (30 mL) and brine (40 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (0.95 g, 1.51 mmol). MS (m/z): 628.8(M+1), 626.8 (M-l).
Preparation 12a
tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy] -lH-pyrazol-4-yl}methyl)phenyl]but-3-en- 1 -yl} -2,9- diazaspiro[5.5]undecane-9-carboxylate
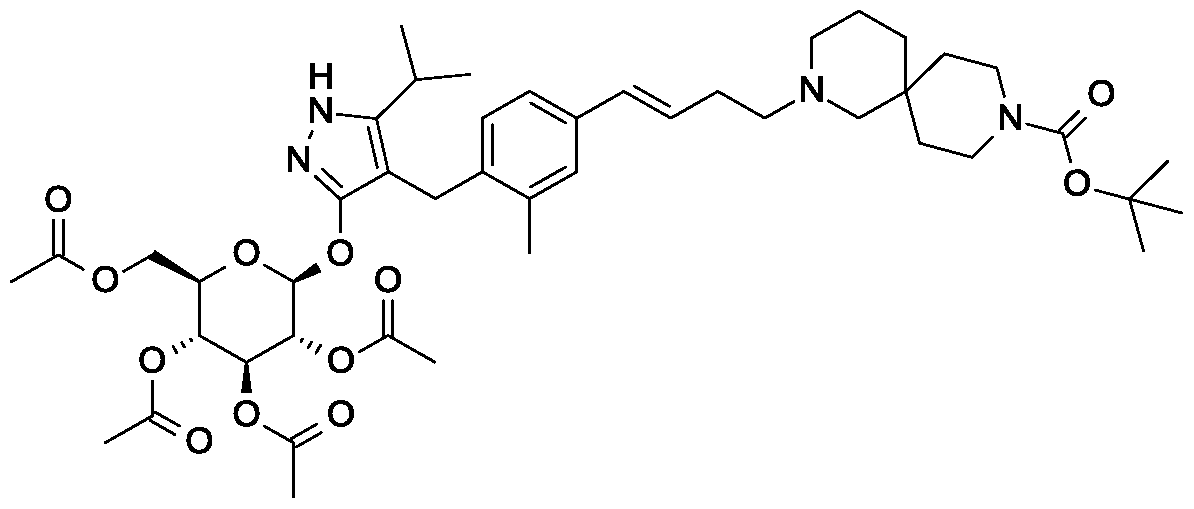
Scheme 2, Step D: Add sodium triacetoxyborohydride (303 mg, 1.4 mmol) to a solution of 4-{4-[(l£)-4-oxybut-l-en-l-yl]-2-methylbenzyl}-5-(propan-2-yl)-lH-pyrazol- 3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (600 mg, 0.95 mmol) and tert-butyl 2,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (333 mg, 1.2 mmol) in 1,2- dichloroethane (30 mL). After 30 minutes at room temperature, quench the reaction with saturated aqueous sodium bicarbonate (15 mL). Extract with dichloromethane (60 mL). Wash extract with water (30 mL) and brine (60 mL). Dry organic phase over sodium sulfate, filter, and concentrate under reduced pressure. Purify the resulting residue by flash chromatography to yield the title compound (500 mg, 0.58 mmol).
MS (m/z): 866.8, 867.8 (M+l), 864.8, 865.8 (M-l).
Preparation 13
4- {4- [( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan- 2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride

Scheme 2, step E: Add hydrogen chloride (4.0 M solution in 1,4-dioxane, 1.5 mL, 5.8 mmol) to a solution of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6- tetra-0-acetyl-beta-D-glucopyranosyl)oxy] – lH-pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 – yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (500 mg, 0.58 mmol) in dichloromethane (20 mL). After 2 hours at room temperature, concentrate to remove the solvent under reduced pressure to yield the title compound as a solid (480 mg, 0.57 mmol).
MS (m/z): 767.4 (M+l).
Scheme 3
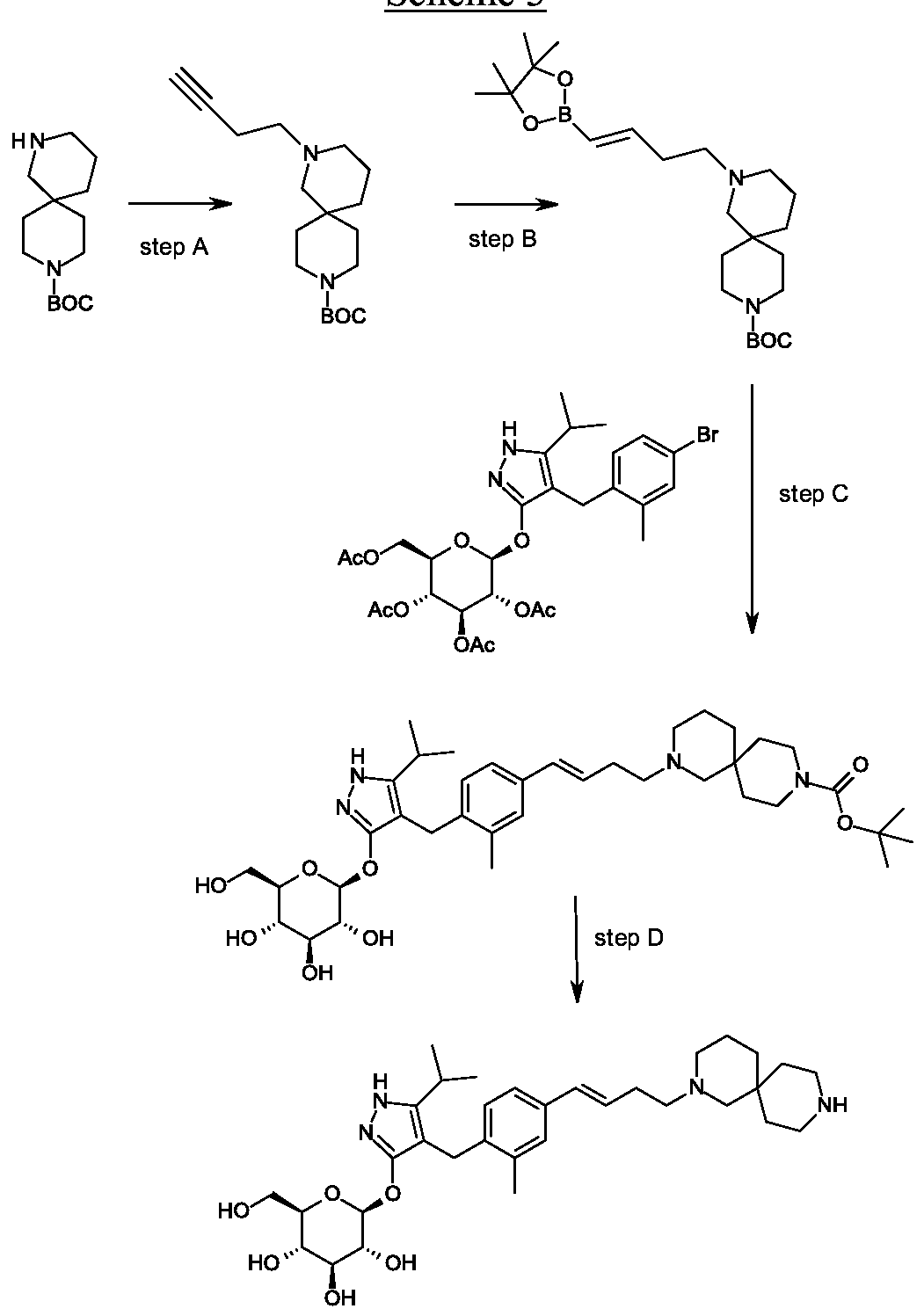
Preparation 14
tert-butyl 4-but-3-ynyl-4,9-diazas iro[5.5]undecane-9-carboxylate

Scheme 3, step A: Cesium carbonate (46.66 g, 143.21 mmol) is added to a suspension of tert-butyl 4,9-diazaspiro[5.5]undecane-9-carboxylate hydrochloride (16.66 g, 57.28 mmoles) in acetonitrile (167 mL). The mixture is stirred for 10 minutes at ambient temperature then 4-bromobutyne (6.45 mL, 68.74 mmol) is added. The reaction is heated to reflux and stirred for 18 hours. The mixture is cooled and concentrated under reduced pressure. The residue is partitioned between water (200 mL) and ethyl acetate (150 mL). The phases are separated and the aqueous layer is extracted with ethyl acetate (100 mL). The combined organic layers are washed with water (200 mL), then brine (150 mL), dried over MgS04, filtered, and concentrated under reduced pressure to give the title compound (17.2 g, 98% yield). lH NMR (300.11 MHz, CDC13): δ 3.43-3.31 (m, 4H), 2.53-2.48 (m, 2H), 2.37-2.29 (m, 4H), 2.20 (s, 2H), 1.94 (t, J= 2.6 Hz, 1H), 1.44 (s, 17H).
Preparation 15
tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)but-3-enyl]-4,9- diazaspiro[5.5]undecane-9-carboxylate
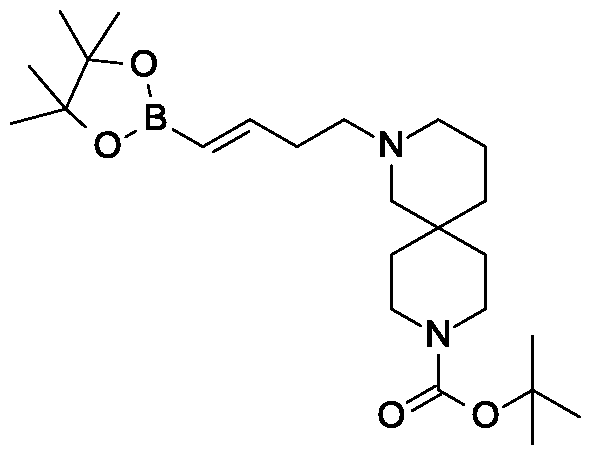
Scheme 3, step B: Triethylamine (5.62 mmoles; 0.783 mL), 4,4,5,5-tetramethyl- 1,3,2-dioxaborolane (8.56 mL, 59.0 mmol) and zirconocene chloride (1.45 g, 5.62 mmoles) are added to tert-butyl 4-but-3-ynyl-4,9-diazaspiro[5.5]undecane-9-carboxylate (17.21 g, 56.16 mmoles). The resulting mixture is heated to 65 °C for 3.5 hours. The mixture is cooled and dissolved in dichloromethane (150 mL). The resulting solution is passed through a ~4cm thick pad of silica gel, eluting with dichloromethane (2 x 200 mL). The filtrate is concentrated under reduced pressure to give the title compound (21.2 g, 87% yield). 1H NMR (300.11 MHz, CDCI3): δ 6.65-6.55 (m, 1H), 5.49-5.43 (m, 1H), 3.42-3.29 (m, 4H), 2.40-2.27 (m, 6H), 2.25-2.08 (m, 2H), 1.70 – 1.13 (m, 29H).
Preparation 16
tert-butyl 2-{(3£’)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D-glucopyranosyl)oxy]-lH- pyrazol-4-yl} methyl)phenyl]but-3 -en- 1 -yl} -2,9-diazaspiro [5.5]undecane-9-carboxylate

Scheme 3, step C: A solution of 4-(4-bromo-2-methylbenzyl)-5-(propan-2-yl)- lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside (20 g, 31.3 mmol), tert- butyl 4-[(£)-4-(4,4,5 ,5 -tetramethyl- 1 ,3,2-dioxaborolan-2-yl)but-3 -enyl] -4,9- diazaspiro[5.5]undecane-9-carboxylate (16.3 g, 37.5 mmol) and potassium carbonate (12.97 g, 93.82 mmol) in tetrahydrofuran (200 mL) and water (40 mL) is degassed for 15 min by bubbling nitrogen gas through it. Pd(OAc)2 (140 mg, 625 μιηοΐ) and 2- dicyclohexylphosphino-2′,4′,6′-tri-i -propyl- Ι, -biphenyl (0.596 g, 1.25 mmol) are added and the reaction is heated to reflux for 16 h. The solution is cooled to ambient temperature and methanol (200 mL) is added. After 30 minutes the solvent is removed under reduced pressure. The mixture is partitioned between ethyl acetate (500 mL) and brine (500 ml) adding aqueous MgS04 (1M; 500 ml) to aid the phase separation. The layers are separated and the organic layer is dried over MgS04 and filtered through a 10 cm pad of silica gel, eluting with ethyl acetate (-1.5 L). The filtrate is discarded and the silica pad is flushed with 5% MeOH in THF (2 L). The methanolic filtrate is concentrated under reduced pressure to give the title compound (20. lg, 92%).
MS (m/z): 699 (M+l).

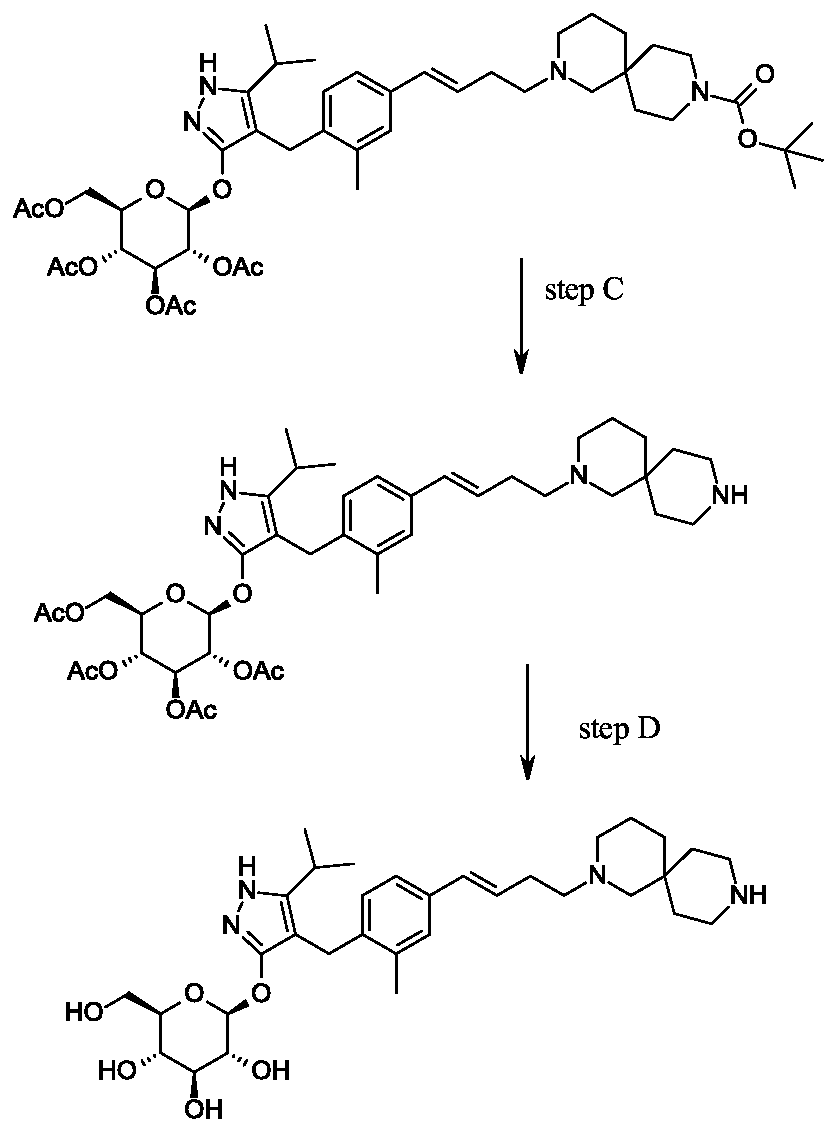
Preparation 17
tert-butyl 4- [(E)-4- [4- [(3 -hydroxy-5-isopropyl- 1 H-pyrazol-4-yl)methyl] -3 -methyl- phenyl]but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate

Scheme 4, step A: Add tert-butyl 4-[(£)-4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)but-3-enyl]-4,9-diazaspiro[5.5]undecane-9-carboxylate (35.8 kg, 82.4 mol) in methanol (130 L) to a solution of (4-[(4-bromo-2-methyl-phenyl)methyl]-5- isopropyl-lH-pyrazol-3-ol (23.9 kg, 77.3 mol) in methanol (440 L) at room temperature. Add water (590 L) and tripotassium phosphate (100 kg, 471.7 mol) and place the reaction under nitrogen atmosphere. To the stirring solution, add a suspension of
tris(dibenzylideneacetone) dipalladium (1.42 kg, 1.55 mol) and di-tert- butylmethylphosphonium tetrafluoroborate (775 g, 3.12 mol) in methanol (15 L). The resulting mixture is heated at 75 °C for 2 hours. Cool the mixture and filter over diatomaceous earth. Rinse the the filter cake with methanol (60 L), and concentrate the filtrate under reduced pressure. Add ethyl acetate (300 L), separate the layers, and wash the organic layer with 15% brine (3 x 120 L). Concentrate the organic layer under reduced pressure, add ethyl acetate (300 L), and stir the mixture for 18 to 20 hours. Add heptane (300 L), cool the mixture to 10 °C, and stir the mixture for an additional 18 to 20 hours. Collect the resulting solids by filtration, rinse the cake with ethyl acetate/heptane (2:3, 2 x 90 L), and dry under vacuum at 40°C to give the title compound (29.3 kg, 70.6% yield) as a white solid. lH NMR (400 MHz, CD3OD): δ 7.14 (s, 1H), 7.07 (d, J= 8.0 Hz, 1H), 6.92 (d, J= 7.6 Hz, 1H), 6.39 (d, J= 16.0 Hz, 1H), 6.25-6.12 (m, 1H), 3.63 (s, 2H), 3.45-3.38 (bs, 3H), 3.34 (s, 3 H), 3.33 (s, 3H), 2.85-2.75 (m, 1H), 2.49-2.40 (m, 5 H), 2.33 (s, 3H), 1.68-1.62 (m, 2H), 1.60-1.36 (m, 15H), 1.11 (s, 3H), 1.10 (s, 3H).
Preparation 12b
Alterternative preparation of tert-butyl 2-{(3£)-4-[3-methyl-4-({5-(propan-2-yl)-3- [(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but- 3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate.

Scheme 4, step B: Combine tert-butyl 4-[(E)-4-[4-[(3-hydroxy-5-isopropyl-lH- pyrazol-4-yl)methyl] -3-methyl-phenyl]but-3 -enyl] -4,9-diazaspiro [5.5]undecane-9- carboxylate (17.83 kg, 33.2 moles), acetonitrile (180 L), and benzyltributylammonium chloride (1.52 kg, 4.87 moles) at room temperature. Slowly add potassium carbonate (27.6 kg, 199.7 moles) and stir the mixture for 2 hours. Add 2,3,4,6-tetra-O-acetyl-alpha- D-glucopyranosyl bromide (24.9 kg, 60.55 mol), warm the reaction mixture to 30°C and stir for 18 hours. Concentrate the mixture under reduced pressure and add ethyl acetate (180 L), followed by water (90 L). Separate the layers, wash the organic phase with 15% brine (3 x 90 L), concentrate the mixture, and purify using column chromatography over silica gel (63 kg, ethyl acetate/heptanes as eluent (1 :2→1 :0)) to provide the title compound (19.8 kg, 94% purity, 68.8% yield) as a yellow foam, !H NMR (400 MHz, CDC13): δ 7.13 (s, 1H), 7.03 (d, J= 8.0 Hz, 1H), 6.78 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 16.0,
1H), 6.25-6.13 (m, 1H), 5.64 (d, J= 8.0 Hz, 1H), 5.45-5.25 (m, 2H), 5.13-4.95 (m, 2H), 4.84-4.76 (m, 1H), 4.25-4.13 (m, 2H), 4.10-4.00 (m, 2H), 3.90-3.86 (m, 1H), 3.58-3.50 (m, 2H), 3.40-3.22 (m, 4H), 2.89-2.79 (m, 1H), 2.10-1.90 (m, 18 H), 1.82 (s, 3H), 1.62- 0.82 (m, 22H).
Preparation 18
2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-[(2,3,4,6-tetra-0-acetyl-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl} -2,9- diazaspiro[5.5]undecane

Scheme 4, step C: Combine tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)- 3-[(2,3,4,6-tetra-0-acetyl-beta-D-glucopyranosyl)oxy]-lH-pyrazol-4- yl}methyl)phenyl]but-3-en-l-yl}-2,9-diazaspiro[5.5]undecane-9-carboxylate (19.6 kg, 22.6 moles) with dichloromethane (120 L) and cool to 0°C. Slowly add trifluoroacetic acid (34.6 L, 51.6 kg, 452 moles) and stir for 9 hours. Quench the reaction with ice water (80 L), and add ammonium hydroxide (85-90 L) to adjust the reaction mixture to pH (8- 9). Add dichloromethane (120 L), warm the reaction mixture to room temperature, and separate the layers. Wash the organic layer with water (75 L), brine, and concentrate under reduced pressure to provide the title compound (16.2 kg, 95.0% purity, 93% yield) as a yellow solid. lH NMR (400 MHz, CDC13): δ 7.08 (s, IH), 6.99 (d, J= 8.0 Hz, IH),
6.76 (d, J= 7.6 Hz, IH), 6.38 (d, J=15.6 Hz, IH), 6.00-5.83 (m, IH), 5.31 (d, J= 7.6 Hz, IH), 5.25-5.13 (m, 4H), 4.32 (dd, J= 12.8, 9.2 Hz, IH), 4.14 (d, J= 11.2 Hz, IH), 3.90 (d, J= 10.0 Hz, IH), 3.75-3.50 (m, 3H), 3.30-3.00 (m, 5 H), 2.85-2.75 (m, IH), 2.70-2.48 (m, 3H), 2.25 (s, IH), 2.13-1.63 (m, 19H), 1.32-1.21 (m, IH), 1.14 (s, 3H), 1.13 (s, 3H), 1.12 (s, 3H), 1.10 (s, 3H).
Example 1
Hydrated crystalline 4- {4-[(l£)-4-(2,9-diazaspiro[5.5]undec-2-yl)but- 1 -en- 1 -yl]-2- methylbenzyl} -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside acetate
First alternative preparation of 4-{4-[(l£’)-4-(2.9-diazaspiro[5.5]undec-2-yl)but-l-en-l- yl]-2-methylbenzyl| -5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (free base).
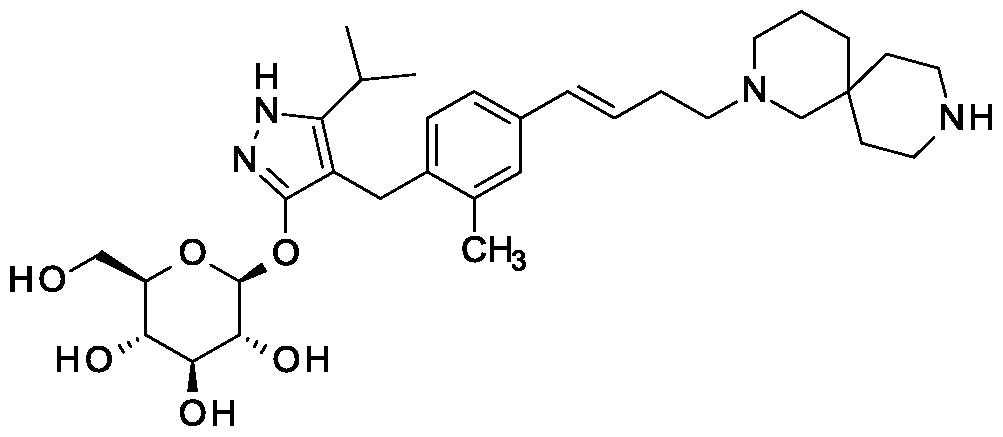
Scheme 1, step I: Add sodium hydroxide (0.5 mL, 0.5 mmol, 1.0 M solution) to a solution of 4- {4-[( l£)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} – 5-(propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-benzoyl-beta-D-glucopyranoside dihydrochloride (258 mg, 0.24 mmol) in methanol (2 mL). After 2 hours at 40°C, concentrate to remove the solvent under reduced pressure to give a residue, which is purified by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H.0 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (46 mg, 0.08 mmol). MS (m/z): 598.8 (M+l), 596.8 (M-l).
Second alternative preparation of 4-{4-r(l-£’)-4-(2.9-diazaspiror5.51undec-2-yl)but-l-en- 1 -yl] -2-methylbenzyl I -5 -(propan-2-yl)- lH-pyrazol-3 -yl beta-D-glucopyranoside (free base“).

Scheme 2, step F: Add methanol (5 mL), triethylamine (3 mL), and water (3 mL) to 4- {4-[( lJE)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl 2,3,4,6-tetra-O-acetyl-beta-D-glucopyranoside dihydrochloride (480 mg, 0.24 mmol). After 18 hours (overnight) at room temperature, concentrate to dryness under reduced pressure. Purify the resulting residue by preparative HPLC method: high pH, 25% B for 4 min, 25-40 B % for 4 min @ 85 mL/min using a 30 x 75 mm, 5 μιη C18XBridge ODB column, solvent A – H20 with NH4HCO3 @ pH 10, solvent B – MeCN to yield the title compound (free base) as a solid (50 mg, 0.08 mmol).
MS (m/z): 598.8 (M+l), 596.8 (M-l). 1H NMR (400.31 MHz, CD3OD): δ 7.11 (d, J=1.3
Hz, 1H), 7.04 (dd, J=l .3,8.0 Hz, 1H), 6.87 (d, J= 8.0 Hz, 1H), 6.36 (d, J= 15.8 Hz, 1H), 6.16 (dt, J= 15.8, 6.3 Hz, 1H), 5.02 (m, 1H), 3.81 (d, J= 11.7 Hz, 1H), 3.72 (d, J= 16.8 Hz, 1H), 3.68 (d, J= 16.8 Hz, 1H) , 3.64 (m, 1H), 3.37-3.29 (m, 4H), 2.79 (m, 1H), 2.72 (t, J= 5.8 Hz, 4H), 2.44-2.33 (m, 6H), 2.30 (s, 3H), 2.26 ( broad s, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.43 (m, 2H), 1.36 (m, 2H), 1.11 (d, J= 7.0 Hz, 3H), 1.10 (d, J= 7.0 Hz, 3H).
Third alternative preparation of 4-{4-[(l£,)-4-(2,9-diazaspiro[5.51undec-2-yl)but-l-en-l- yll-2-methylbenzyl|-5-(propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside.
Scheme 3, step D: Trifluoroacetic acid (32.2 mL; 0.426 mol) is added to a solution of tert-butyl 2-{(3JE)-4-[3-methyl-4-({5-(propan-2-yl)-3-beta-D- glucopyranosyl)oxy]-lH-pyrazol-4-yl}methyl)phenyl]but-3-en-l-yl}-2,9- diazaspiro[5.5]undecane-9-carboxylate (14.87 g; 21.28 mmol) in dichloromethane (149 mL) cooled in iced water. The solution is allowed to warm to room temperature. After 30 minutes, the mixture is slowly added to ammonia in MeOH (2M; 300 mL), applying cooling as necessary to maintain a constant temperature. The solution is stirred at room temperature for 15 min. The mixture is concentrated under reduced pressure and the residue is purified using SCX-2 resin. The basic filtrate is concentrated under reduced pressure and the residue is triturated/sonicated in ethyl acetate, filtered and dried. The resulting solid is dissolved in MeOH (200mL) and concentrated in vacuo. This is repeated several times to give the title compound (free base) (12.22 g, yield 96%). MS (m/z): 599 (M+l); [a]D 20 = -12 ° (C=0.2, MeOH).
Preparation of final title compound, hydrated crystalline 4-{4-|YlE)-4-(2.9- diazaspiro [5.5“|undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-vD- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.

4- {4- [(1 E)-4-(2,9-diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl } -5 – (propan-2-yl)-lH-pyrazol-3-yl beta-D-glucopyranoside (902 mg) is placed in a round bottom flask (100 mL) and treated with wet ethyl acetate (18 mL). [Note – wet ethyl acetate is prepared by mixing ethyl acetate (100 mL) and dionized water (100 mL). After mixing, the layers are allowed to separate, and the top wet ethyl acetate layer is removed for use. Acetic acid is a hydrolysis product of ethyl acetate and is present in wet ethyl acetate.] The compound dissolves, although not completely as wet ethyl acetate is added. After several minutes, a white precipitate forms. An additional amount of wet ethyl acetate (2 mL) is added to dissolve remaining compound. The solution is allowed to stir uncovered overnight at room temperature during which time the solvent partially evaporates. The remaining solvent from the product slurry is removed under vacuum, and the resulting solid is dried under a stream of nitrogen to provide the final title compound as a crystalline solid. A small amount of amorphous material is identified in the product by solid-state NMR. This crystalline final title compound may be used as seed crystals to prepare additional crystalline final title compound.
Alternative preparation of final title compound, hvdrated crystalline 4-{4-[(lE)-4-(2.,9- diazaspiro [5.5]undec-2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl I -5-(propan-2-yl)- 1 H-pyrazol-3 – yl beta-D-glucopyranoside acetate.
Under a nitrogen atmosphere combine of 4-{4-[(lE)-4-(2,9-diazaspiro[5.5]undec- 2-yl)but- 1 -en- 1 -yl] -2-methylbenzyl} -5-(propan-2-yl)- 1 H-pyrazol-3-yl 2,3,4,6-tetra-O- acetyl-beta-D-glucopyranoside (2.1 kg, 2.74 mol), methanol (4.4 L), tetrahydrofuran (4.2 L), and water (210 mL). Add potassium carbonate (460 g, 3.33 moles) and stir for four to six hours, then filter the reaction mixture to remove the solids. Concentrate the filtrate under reduced pressure, then add ethanol (9.0 L) followed by acetic acid (237 mL, 4.13 mol) and stir at room temperature for one hour. To the stirring solution add wet ethyl acetate (10 L, containing approx. 3 w/w% water) slowly over five hours, followed by water (500 mL). Stir the suspension for twelve hours and add wet ethyl acetate (4.95 L, containing approx. 3 w/w% water) over a period of eight hours. Stir the suspension for twelve hours and add additional wet ethyl acetate (11.5 L, containing approx. 3 w/w% water) slowly over sixteen hours. Stir the suspension for twelve hours, collect the solids by filtration and rinse the solids with wet ethyl acetate (3.3 L, containing approx. 3 w/w% water). Dry in an oven under reduced pressure below 30°C to give the title compound as an off-white crystalline solid (1.55 kg, 2.35 mol, 96.7% purity, 72.4 w/w% potency, 68.0% yield based on potency). HRMS (m/z): 599.3798 (M+l).
PATENT
CN105705509
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN175101669&tab=PCTDESCRIPTION
The present invention is in the field of treatment of diabetes and other diseases and conditions associated with hyperglycemia. Diabetes is a group of diseases characterized by high blood sugar levels. It affects approximately 25 million people in the United States, and according to the 2011 National Diabetes Bulletin, it is also the seventh leading cause of death in the United States (US Department of Health and Human Resources Services, Centers for Disease Control and Prevention). Sodium-coupled glucose cotransporters (SGLT’s) are one of the transporters known to be responsible for the uptake of carbohydrates such as glucose. More specifically, SGLT1 is responsible for transporting glucose across the brush border membrane of the small intestine. Inhibition of SGLT1 can result in a decrease in glucose absorption in the small intestine, thus providing a useful method of treating diabetes.
Alternative medicines and treatments for diabetes are needed. The present invention provides an acetate salt of a pyrazole compound which is an SGLT1 inhibitor, and thus it is suitable for treating certain conditions such as diabetes.
U.S. Patent No. 7,655,632 discloses certain pyrazole derivatives having human SGLT1 inhibitory activity, which are also disclosed for use in the prevention or treatment of diseases associated with hyperglycemia, such as diabetes. Moreover, WO 2011/039338 discloses certain pyrazole derivatives having SGLT1/SGLT2 inhibitor activity, which are also disclosed for use in the treatment of bone diseases such as osteoporosis.
PATENT
WO-2019141209
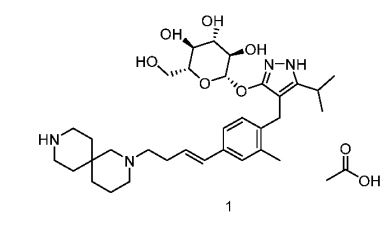
Process for preparing pyranoglucose-substituted pyrazole compound, used as a pharmaceutical intermediate in SGLT inhibitor for treating diabetes.

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9573970 | 4–5-(PROPAN-2-YL)-1H-PYRAZOL-3-YL BETA-D GLUCOPYRANOSIDE ACETATE | 2014-10-30 | 2016-07-28 |
/////////////SY-008 , SY 008 , SY008, ELI LILY, PHASE 1, GLT1 inhibitor, type 2 diabetes, Yabao Pharmaceutical, CHINA, DIABETES
CC(=O)O.Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4O
Cc5cc(\C=C\CCN2CCCC1(CCNCC1)C2)ccc5Cc3c(nnc3C(C)C)O[C@@H]4O[C@H](CO)[C@@H](O)[C@H](O)[C@H]4
O
FDA approves new treatment Victoza (liraglutide) for pediatric patients with type 2 diabetes
The U.S. Food and Drug Administration today approved Victoza (liraglutide) injection for treatment of pediatric patients 10 years or older with type 2 diabetes. Victoza is the first non-insulin drug approved to treat type 2 diabetes in pediatric patients since metformin was approved for pediatric use in 2000. Victoza has been approved to treat adult patients with type 2 diabetes since 2010.
“The FDA encourages drugs to be made available to the widest number of patients possible when there is evidence of safety and efficacy,” said Lisa Yanoff, M.D, acting director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research. “Victoza has now been shown to improve blood sugar control in pediatric patients with type 2 diabetes. The expanded indication provides an additional treatment option at a time when
- June 17, 2019
The U.S. Food and Drug Administration today approved Victoza (liraglutide) injection for treatment of pediatric patients 10 years or older with type 2 diabetes. Victoza is the first non-insulin drug approved to treat type 2 diabetes in pediatric patients since metformin was approved for pediatric use in 2000. Victoza has been approved to treat adult patients with type 2 diabetes since 2010.
“The FDA encourages drugs to be made available to the widest number of patients possible when there is evidence of safety and efficacy,” said Lisa Yanoff, M.D, acting director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research. “Victoza has now been shown to improve blood sugar control in pediatric patients with type 2 diabetes. The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with this disease.”
Type 2 diabetes is the most common form of diabetes, occurring when the pancreas cannot make enough insulin to keep blood sugar at normal levels. Although type 2 diabetes primarily occurs in patients over the age of 45, the prevalence rate among younger patients has been rising dramatically over the past couple of decades. The Diabetes Report Card published by the U.S. Centers for Disease Control and Prevention estimates that more than 5,000 new cases of type 2 diabetes are diagnosed each year among U.S. youth younger than age 20.
Victoza improves blood sugar levels by creating the same effects in the body as the glucagon-like peptide (GLP-1) receptor protein in the pancreas. GLP-1 is often found in insufficient levels in type 2 diabetes patients. Like GLP-1, Victoza slows digestion, prevents the liver from making too much glucose (a simple sugar), and helps the pancreas produce more insulin when needed. As noted on the label, Victoza is not a substitute for insulin and is not indicated for patients with type 1 diabetes or those with diabetic ketoacidosis, a condition associated with diabetes where the body breaks down fat too quickly because there is inadequate insulin or none at all. Victoza is also indicated to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease; however, its effect on major adverse cardiovascular events in pediatrics was not studied and it is not indicated for this use in children.
The efficacy and safety of Victoza for reducing blood sugar in patients with type 2 diabetes was studied in several placebo-controlled trials in adults and one placebo-controlled trial with 134 pediatric patients 10 years and older for more than 26 weeks. Approximately 64% of patients in the pediatric study had a reduction in their hemoglobin A1c (HbA1c) below 7% while on Victoza, compared to only 37% who achieved these results with the placebo. HbA1c is a blood test that is routinely performed to evaluate how well a patient’s diabetes is controlled, and a lower number indicates better control of the disease. These results occurred regardless of whether the patient also took insulin at the same time. Adult patients who took Victoza with insulin or other drugs that increase the amount of insulin the body makes (e.g., sulfonylurea) may have an increased risk of hypoglycemia (low blood sugar). Meanwhile, pediatric patients 10 years and older taking Victoza had a higher risk of hypoglycemia regardless of whether they took other therapies for diabetes.
The prescribing information for Victoza includes a Boxed Warning to advise health care professionals and patients about the increased risk of thyroid C-cell tumors. For this reason, patients who have had, or have family members who have ever had medullary thyroid carcinoma (MTC) should not use Victoza, nor should patients who have an endocrine system condition called multiple endocrine neoplasia syndrome type 2 (MEN 2). In addition, people who have a prior serious hypersensitivity reaction to Victoza or any of the product components should not use Victoza. Victoza also carries warnings about pancreatitis, Victoza pen sharing, hypoglycemia when used in conjunction with certain other drugs known to cause hypoglycemia including insulin and sulfonylurea, renal impairment or kidney failure, hypersensitivity and acute gallbladder disease. The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion and constipation.
The FDA granted this application Priority Review. The approval of Victoza was granted to Novo Nordisk.
//////Victoza, liraglutide, FDA 2019, Priority Review, Novo Nordisk, DIABETES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}