
Home » Posts tagged 'CLINICAL TRIAL'
Tag Archives: CLINICAL TRIAL
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
XL 114, AUR 104 and XL 102, AUR 102 (NO CONCLUSIONS, ONLY PREDICTIONS)

XL 114
FOR BOTH, JUST PREDICTION
PREDICTIONS
or


N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
![(2S)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117943691&t=l)

(2S)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
CAS 2305027-62-5
C12 H20 N6 O7, 360.32Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-, (2S,3ξ)-N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
ALSO SEE


![(2S,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](http://drugapprovalsint.com/wp-content/uploads/2021/10/str1-10.jpg)
1673534-76-3C12 H20 N6 O7, 360.32
L-Threonine, N-[[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]
(2S,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acidN-[[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]amino]carbonyl]-L-threonine
CAS 1673534-76-3
PD-1-IN-1 free base, EX-A1918, CS-6240, NSC-799645, CA-170 (AUPM-170)|PDL1 inhibitor, HY-101093, PD-1-IN-1
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)[C@@H](C)O)CC(N)=O
XL 114, AUR 104
A novel covalent inhibitor of FABP5 for cancer therapy
XL 102, AUR 102
A potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7)
NO CONCLUSIONS, ONLY PREDICTIONS
PREDICTIONS MORE
![(2R,3R)-2-[[(1S)-3-Amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155529077&t=l)

(2R,3R)-2-[[(1S)-3-amino-1-[3-[(1R)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
C12H20N6O7, 360.32
![(2S,3S)-2-[[(1S)-3-Amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=155339943&t=l)

(2S,3S)-2-[[(1S)-3-amino-1-[3-[(1S)-1-amino-2-hydroxyethyl]-1,2,4-oxadiazol-5-yl]-3-oxopropyl]carbamoylamino]-3-hydroxybutanoic acid
XL102, AUR 102
XL102 is a potent, selective and orally bioavailable covalent inhibitor of CDK7, which is an important regulator of the cellular transcriptional and cell cycle machinery. CDK7 helps regulate cell cycle progression, with overexpression observed in multiple cancers, such as breast, prostate and ovarian cancers. In preclinical studies, XL102 revealed potent anti-proliferative activity, induced cell death in a large panel of cancer cell lines and caused tumor growth inhibition and regression in xenograft models, demonstrating its potential as a targeted antitumor agent.
In late 2020, Exelixis exercised its option to in-license XL102 (formerly AUR102) from Aurigene per the companies’ July 2019 collaboration, option and license agreement. Exelixis has assumed responsibility for the future clinical development, manufacturing and commercialization of XL102. Aurigene retains limited development and commercial rights for India and Russia.
SYN

ABOUT Fatty acid-binding proteins (FABPs)
Fatty acid-binding proteins (FABPs) are involved in binding and storing hydrophobic ligands such as long-chain fatty acids, as well as transporting them to the appropriate compartments in the cell. Epidermal fatty acid-binding protein (FABP5) is an intracellular lipid-binding protein that is abundantly expressed in adipocytes and macrophages. Previous studies have revealed that the FABP5 expression level is closely related to malignancy in various types of cancer. However, its precise functions in the metabolisms of cancer cells remain unclear. Here, we revealed that FABP5 knockdown significantly induced downregulation of the genes expression, such as hormone-sensitive lipase (HSL), monoacylglycerol lipase (MAGL), elongation of long-chain fatty acid member 6 (Elovl6), and acyl-CoA synthetase long-chain family member 1 (ACSL1), which are involved in altered lipid metabolism, lipolysis, and de novo FA synthesis in highly aggressive prostate and breast cancer cells. Moreover, we demonstrated that FABP5 induced inflammation and cytokine production through the nuclear factor-kappa B signaling pathway activated by reactive oxygen species and protein kinase C in PC-3 and MDA-MB-231 cells. Thus, FABP5 might regulate lipid quality and/or quantity to promote aggressiveness such as cell growth, invasiveness, survival, and inflammation in prostate and breast cancer cells. In the present study, we have revealed for the first time that high expression of FABP5 plays a critical role in alterations of lipid metabolism, leading to cancer development and metastasis in highly aggressive prostate and breast cancer cells.
Fatty acid-binding protein, epidermal is a protein that in humans is encoded by the FABP5 gene
Function
This gene encodes the fatty acid binding protein found in epidermal cells, and was first identified as being upregulated in psoriasis tissue. Fatty acid binding proteins are a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. It is thought that FABPs roles include fatty acid uptake, transport, and metabolism.[6]
The phytocannabinoids (THC and CBD) inhibit endocannabinoid anandamide (AEA) uptake by targeting FABP5, and competition for FABPs may in part or wholly explain the increased circulating levels of endocannabinoids reported after consumption of cannabinoids.[7] Results show that cannabinoids inhibit keratinocyte proliferation, and therefore support a potential role for cannabinoids in the treatment of psoriasis.[8]
Interactions
FABP5 has been shown to interact with S100A7.[
ABOUT CD47/SIRPa axis
CD47/SIRPa axis is established as a critical regulator of myeloid cell activation and serves as an immune checkpoint for macrophage mediated phagocytosis. Because of its frequent upregulation in several cancers, CD47 contributes to immune evasion and cancer progression. CD47 regulates phagocytosis primarily through interactions with SIRPla expressed on macrophages. Blockade of SIRPla/CD47 has been shown to dramatically enhance tumor cell phagocytosis and dendritic cells maturation for better antigen presentation leading to substantially improved antitumor responses in preclinical models of cancer (M. P. Chao et al. Curr Opin Immunol. 2012 (2): 225-232). Disruption of CD47-SIRPa interaction is now being evaluated as a therapeutic strategy for cancer with the use of monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys.
CD47 is expressed on virtually all non-malignant cells, and blocking the CD47 or the loss of CD47 expression or changes in membrane distribution can serve as markers of aged or damaged cells, particularly on red blood cells (RBC). Alternatively, blocking SIRPa also allows engulfment of targets that are not normally phagocytosed, for those cells where pre-phagocytic signals are also present. CD47 is a broadly expressed transmembrane glycoprotein with a single Ig-like domain and five membrane- spanning regions, which functions as a cellular ligand for SIRPa with binding mediated through the NH2-terminal V-like domain of SIRPa. SIRPa is expressed primarily on myeloid cells, including macrophages, granulocytes, myeloid dendritic cells (DCs), mast cells, and their precursors, including hematopoietic stem cells.
CD47 is also constitutively upregulated on a number of cancers such as Non-Hodgkin Lymphoma (NHL), Acute myeloid leukemia (AML), breast, colon, glioblastoma, glioma, ovarian, bladder and prostate cancers, etc. Overexpression of CD47 by tumor cells, which efficiently helps them to escape immune surveillance and killing by innate immune cells. However, in most of the tumor types, blockade of the CD47-SIRPa interaction as a single agent may not be capable of inducing significant phagocytosis and antitumor immunity, necessitating the need to combine with other therapeutic agents. The concomitant engagement of activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors (collectively known as “eat-me” signals) may be necessary for exploiting the maximum potential of the CD-47-SIPRa pathway blockade.
The role of engagement of prophagocytic receptors is proved by inefficiency to trigger phagocytosis either by anti-CD47 F(ab) fragments, single chain variable fragments of CD-47 or non-Fc portion- containing SIRPa proteins in blocking of the CD47-SIRPa interaction. When activating prophagocytic receptors are engaged, as evident in the case of using Fc portion-containing blocking anti-CD47 antibodies, CD47- SIRPa blockade is able to trigger more efficient phagocytosis. Combining CD47-SIRPa blocking agents with therapeutic antibodies (Fc-containing) targeting tumor antigens stimulate activating Fc receptors (FcRs) leading to efficient phagocytosis. The Fc portion of therapeutic antibody targeting tumor antigen also induces antibody-dependent cellular cytotoxicity (ADCC), which also adds to the therapeutic efficacy. Hence antibodies selected from the group consisting of rituximab, herceptin, trastuzumab, alemtuzumab, bevacizumab, cetuximab and panitumumab, daratumumab due to its tumor targeting nature and ADCC, can trigger more efficient phagocytosis.
Earlier approaches to disrupt CD47- SIRPa interaction utilized monoclonal antibodies targeting CD47 or SIRPa and engineered receptor decoys fused to Fc fragment. However, a concern with this approach is that CD47 is highly expressed on both hematopoietic and non-hematopoietic normal cells. Hence along with tumor cells CD47-SIRPa blocking agents containing Fc-portion may also target many normal cells potentially leading to their elimination by macrophages. The interaction of blocking antibodies with normal cells is considered as a major safety issue resulting in anemia, thrombocytopenia, and leukopenia. These agents may also affect solid tissues rich in macrophages such as liver, lung, and brain. Hence it may be ideal to block the CD47- SIRPa interaction by agents devoid of Fc portion, such as small
molecules, peptides, Fab fragments etc. while activating prophagocytic receptors in tumor cells by appropriate combinations to induce efficient phagocytosis of tumor cells.
Apart from Fc Receptors, a number of other prophagocytic receptors are also reported to promote engulfment of tumor cells in response to CD47-SIRPa blockade by triggering the phagocytosis. These include receptors for SLAMF7, Mac-l, calreticulin and possibly yet to identified receptors. B cell tumor lines such as Raji and other diffuse large B cell lymphoma express SLAMF7 and are implicated in triggering prophagocytic signals during CD47-SIRPa blockade.
Therapeutic agents known to activate prophagocytic receptors are also therefore ideal partners for use in combination with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These agents include proteasome inhibitors (bortezomib, ixazomib and carfilzomib), Anthracyclines (Doxorubicin, Epirubicin, Daunorubicin, Idarubicin, Mitoxantrone) Oxaliplatin, Cyclophosphamide, Bleomycin, Vorinostat, Paclitaxel, 5-Fluorouracil, Cytarabine, BRAF inhibitory drugs (Dabrafenib, Vemurafenib), PI3K inhibitor, Docetaxel, Mitomycin C, Sorafenib, Tamoxifen and oncolytic viruses.
Apart from the specific agents known to have effect on‘eat me’ signals other agents including Abiraterone acetate, Afatinib, Aldesleukin, Aldesleukin, Alemtuzumab, Anastrozole, Axitinib, Belinostat, Bendamustine, Bicalutamide, Blinatumomab, Bosutinib, Brentuximab, Busulfan, Cabazitaxel, Capecitabine, Carboplatin, Carfilzomib, Carmustine, Ceritinib, Clofarabine, Crizotinib, Dacarbazine, Dactinomycin, Dasatinib, Degarelix, Denileukin, Denosumab, Enzalutamide, Eribulin, Erlotinib, Everolimus, Exemestane, Exemestane, Fludarabine, Fulvestrant, Gefitinib, Goserelin, Ibritumomab, Imatinib, Ipilimumab, Irinotecan, Ixabepilone, Lapatinib, Lenalidomide, Letrozole, Leucovorin, Leuprolide, Lomustine, Mechlorethamine, Megestrol, Nelarabine, Nilotinib, Nivolumab, Olaparib, Omacetaxine, Palbociclib, Pamidronate, Panitumumab, Panobinostat, Pazopanib, Pegaspargase, Pembrolizumab, Pemetrexed Disodium, Pertuzumab, Plerixafor, Pomalidomide, Ponatinib, Pralatrexate, Procarbazine, Radium 223, Ramucirumab, Regorafenib, rIFNa-2b, Romidepsin, Sunitinib, Temozolomide, Temsirolimus, Thiotepa, Tositumomab, Trametinib, Vinorelbine, Methotrexate, Ibrutinib, Aflibercept, Toremifene, Vinblastine, Vincristine, Idelalisib, Mercaptopurine and Thalidomide could potentially have effect on‘eat me’ signal pathway on combining with CD-47-SIRPa blocking agents.
In addition to the therapeutic agents mentioned above, other treatment modalities that are in use in cancer therapy also activate prophagocytic receptors, and thus can be combined with CD47-SIRPa blocking agents to achieve efficient phagocytosis. These include Hypericin-based photodynamic therapy (Hyp-PDT), radiotherapy, High-hydrostatic pressure, Photofrin-based PDT and Rose Bengal acetate -based PDT.
However, there is an unmet need for combining small molecule CD-47-SIRPa pathway inhibitors with agents capable of stimulating activating receptors such as Fc-receptors (FcRs) or other prophagocytic receptors, or combining with other treatment modalities that are in use in cancer therapy to activate prophagocytic receptors for exploiting the maximum potential of the CD-47- SIRPa pathway blockade.
CLIP
Exelixis In-Licenses Second Anti-Cancer Compound from Aurigene Following FDA Acceptance of Investigational New Drug Application for Phase 1 Clinical Trial in Non-Hodgkin’s Lymphoma
– Robust preclinical data support Exelixis’ clinical development of XL114, with phase 1 trial in Non-Hodgkin’s lymphoma expected to begin in the coming months –
– Exelixis will make an option exercise payment of $10 million to Aurigene –
https://www.businesswire.com/news/home/20211014005549/en/Exelixis-In-Licenses-Second-Anti-Cancer-Compound-from-Aurigene-Following-FDA-Acceptance-of-Investigational-New-Drug-Application-for-Phase-1-Clinical-Trial-in-Non-Hodgkin%E2%80%99s-LymphomaOctober 14, 2021 08:00 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today announced that Exelixis has exercised its exclusive option under the companies’ July 2019 agreement to in-license XL114 (formerly AUR104), a novel anti-cancer compound that inhibits the CARD11-BCL10-MALT1 (CBM) signaling pathway, which promotes lymphocyte survival and proliferation. Exelixis has now assumed responsibility for the future clinical development, commercialization and global manufacturing of XL114. Following the U.S. Food and Drug Administration’s (FDA) recent acceptance of its Investigational New Drug (IND) application, Exelixis will soon initiate a phase 1 clinical trial evaluating XL114 monotherapy in patients with Non-Hodgkin’s lymphoma (NHL). At the American Association of Cancer Research Annual Meeting in April of this year, Aurigene presented preclinical data (Abstract 1266) demonstrating that XL114 exhibited potent anti-proliferative activity in a large panel of cancer cell lines ranging from hematological cancers to solid tumors with excellent selectivity over normal cells. In addition, oral dosing of XL114 resulted in significant dose-dependent tumor growth inhibition in diffuse large B-cell lymphoma (DLBCL) and colon carcinoma models.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline”
XL114 is the second molecule that Exelixis in-licensed from Aurigene under the companies’ July 2019 collaboration, option and license agreement. Exelixis previously exercised its option to in-license XL102, a potent, selective and orally bioavailable inhibitor of cyclin-dependent kinase 7 (CDK7), from Aurigene in December 2020 and initiated a phase 1 trial of XL102 as a single agent and in combination with other anti-cancer agents in patients with advanced or metastatic solid tumors in January 2021.
“We are pleased that our agreement with Aurigene has generated a second promising compound that warrants advancement into clinical development and believe the collaboration will continue to play an important role in expanding our pipeline,” said Peter Lamb, Ph.D., Executive Vice President, Scientific Strategy and Chief Scientific Officer, Exelixis. “XL114 has shown potent anti-proliferative activity in lymphoma cell lines that have aberrant activation of the CBM signaling pathway and may have a differentiated profile and potential as a best-in-class molecule that could improve outcomes for patients with Non-Hodgkin’s lymphoma and other hematologic cancers.”
XL114 was identified to have anti-proliferative activity in cell lines with constitutive activation of CBM signaling, including activated B-cell-like DLBCL (ABC-DLBCL), mantle cell lymphoma and follicular lymphoma cell lines. Further characterization of XL114 in cell-based assays demonstrated a functional role in B-cell (BCR) signaling pathways. Additionally, XL114 showed dose-dependent tumor growth inhibition in an ABC-DLBCL mouse xenograft tumor model. In preclinical development, XL114 also demonstrated a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. While the precise molecular mechanism underlying XL114’s function in repressing BCR signaling and MALT1 activation has yet to be characterized, the fatty acid-binding protein 5 (FABP5) has been identified as a prominent XL114-binding target.
“XL114 is the second molecule that Exelixis has opted to in-license under our July 2019 agreement, underscoring the significant potential of our approach to the discovery and preclinical development of innovative cancer therapies that target novel mechanisms of action,” said Murali Ramachandra, Ph.D., Chief Executive Officer, Aurigene. “Exelixis has a track record of success in the clinical development and commercialization of anti-cancer therapies that provide patients with important new treatment options, and we are pleased that the continued advancement of XL114 will be supported by the company’s extensive clinical, regulatory and commercialization infrastructure.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to obtain an exclusive license from Aurigene to three preexisting programs, including the compounds now known as XL102 and XL114. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for an additional upfront payment of $2.5 million per program. The collaboration was expanded in 2021 to include three additional early discovery programs. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all nine programs. Exelixis may exercise its option for a program at any time up until the first IND for the program becomes effective. Having exercised options on two programs thus far (XL102 and XL114), if and when Exelixis exercises a future option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. To exercise its option for XL114, Exelixis will make an option exercise payment to Aurigene of $10 million. Once Exelixis exercises its option for a program, Aurigene will be eligible for clinical development, regulatory and sales milestones, as well as royalties on future potential sales of the compound. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene Discovery Technologies Limited is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY, NSEIFSC: DRREDDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the U.S. and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of the Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. In November 2020, the company was named to Fortune’s 100 Fastest-Growing Companies list for the first time, ranking 17th overall and the third-highest biopharmaceutical company. For more information about Exelixis, please visit www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.

Dinesh Chikkanna
Director, Medicinal Chemistry Aurigene Discovery Technologies

Murali Ramachandra
CEO at Aurigene Discovery Technologies

CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1266
Abstract 1266: Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapyDinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar and Murali Ramachandra
DOI: 10.1158/1538-7445.AM2021-1266 Published July 2021
Proceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Dysregulated fatty acid metabolism is thought to be a hallmark of cancer, wherein fatty acids function both as an energy source and as signals for enzymatic and transcriptional networks contributing to malignancy. Fatty acid-binding protein 5 (FABP5) is an intracellular protein that facilitates transport of fatty acids and plays a role in regulating the expression of genes associated with cancer progression such as cell growth, survival, and metastasis. Overexpression of FABP5 has been reported to contribute to an aggressive phenotype and a poor survival correlation in several cancers. Therefore, inhibition of FABP5 is considered as a therapeutic approach for cancers. Phenotypic screening of a library of covalent compounds for selective sensitivity of cancer cells followed by medicinal chemistry optimization resulted in the identification of AUR104 with desirable properties. Chemoproteomic-based target deconvolution revealed FABP5 as the cellular target of AUR104. Covalent adduct formation with Cys43 of FABP5 by AUR104 was confirmed by mass spectrometry. Target occupancy studies using a biotin-tagged AUR104 demonstrated potent covalent binding to FABP5 in both cell-free and cellular conditions. Ligand displacement assay with a fluorescent fatty acid probe confirmed the competitive binding mode of AUR104 with fatty acids. Binding at the fatty acid site and covalent bond formation with Cys43 were also demonstrated by crystallography. Furthermore, AUR104 showed a high degree of selectivity against a broad safety pharmacology panel of enzymes and receptors. AUR104 exhibited potent anti-proliferative activity in a large panel of cell lines derived from both hematological and solid cancers with a high degree of selectivity over normal cells. Anti-proliferative activity in lymphoma cell lines correlated with inhibition of MALT1 pathway activity, cleavage of RelB/Bcl10 and secretion of cytokines, IL-10 and IL-6. AUR104 displayed desirable drug-like properties and dose-dependent oral exposure in pharmacokinetic studies. Oral dosing with AUR104 resulted in dose-dependent anti-tumor activity in DLBCL (OCI-LY10) and NSCLC (NCI-H1975) xenograft models. In a repeated dose MTD studies in rodents and non-rodents, AUR104 showed good tolerability with an exposure multiple of >500 over cellular EC50 for up to 8 hours. In summary, we have identified a novel covalent FABP5 inhibitor with optimized properties that showed anti-tumor activity in in vitro and in vivo models with acceptable safety profile. The data presented here strongly support clinical development of AUR104.
Citation Format: Dinesh Chikkanna, Leena Khare Satyam, Sunil Kumar Pnaigrahi, Vinayak Khairnar, Manoj Pothuganti, Lakshmi Narayan Kaza, Narasimha Raju Kalidindi, Vijaya Shankar Nataraj, Aditya Kiran Gatta, Narasimha Rao Krishnamurthy, Sandeep Patil, DS Samiulla, Kiran Aithal, Vijay Kamal Ahuja, Nirbhay Kumar Tiwari, KB Charamannna, Pravin Pise, Thomas Anthony, Kavitha Nellore, Sanjeev Giri, Shekar Chelur, Susanta Samajdar, Murali Ramachandra. Discovery and preclinical evaluation of a novel covalent inhibitor of FABP5 for cancer therapy [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1266.
Patent
US20200147054 – COMBINATION OF SMALL MOLECULE CD-47 INHIBITORS WITH OTHER ANTI-CANCER AGENTS
Muralidhara Ramachandra
Pottayil Govindan Nair Sasikumar
Girish Chandrappa Daginakatte
Kiran Aithal Balkudru
PATENT
WO 2020095256
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020095256
Example- 1: The synthetic procedures for the preparation of compounds described in the present invention were described in co-pending Indian provisional patent application 201841001438 dated 12* Jan 2018, which is converted as PCT application
PCT/IB2019/050219, the contents of which are hereby incorporated by reference in their entirety.
PATENT
WO 2018178947https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018178947&tab=PCTDESCRIPTION
PATENT
WO 2019138367
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019138367
PATENT
WO 2019073399
https://patents.google.com/patent/WO2019073399A1/en
Priority to IN201741036169
Example 4 of WO 2015/033299
PATENT
https://patents.google.com/patent/BR112020014202A2/en

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
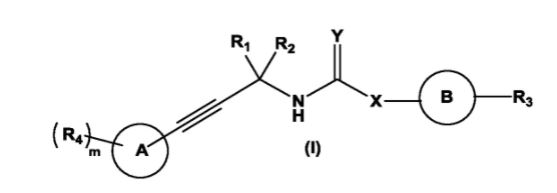
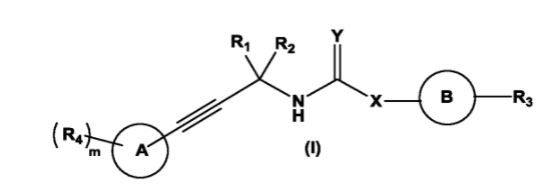
The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
Patent
Example 1
(((S)-4-amino-1-(3-((S)-1,5-diaminopentyl)-1,2,4-oxadiazol-5-yl)-4-oxobutyl)carbamoyl)-L-proline (Compound 1)
Synthesis of Compound 1 b
Synthesis of Compound 1C
Synthesis of Compound 1d
Synthesis of Compound 1f
Synthesis of Compound 1g
Synthesis of Compound 1h
Synthesis of Compound 1i
Synthesis of compound 1j
Synthesis of Compound 1
Example 2
(S)-4-(3-((S)-1-amino-4-guanidinobutyl)-1,2,4-oxadiazol-5-yl)-4-(3-((S)-1-carboxy-2-phenylethyl) ureido)butanoic acid (Compound 7)
Synthesis of Compound 2b
Synthesis of Compound 2c
Synthesis of Compound 2d
Synthesis of Compound 2f
Synthesis of Compound 2g
Synthesis of Compound 2h
Synthesis of Compound 2i
Synthesis of Compound 2j
Synthesis of Compound 7
PATENT
WO 2015/033299
https://patents.google.com/patent/WO2015033299A1/en?oq=WO+2015%2f033299
Pottayil Govindan Nair SasikumarMuralidhara RamachandraSeetharamaiah Setty Sudarshan Naremaddepalli
Example 1: Synthesis of Compound 1

Step la:

Ethylchloroformate (1.5 g, 13.78 mniol) and N-Methylmorpholine ( 1.4 g, 13.78 mmol) were added to a solution of compound la (3 g, 11.48 mmol) in THE (30 mL) arid stirred at -20 °C. After 20 min. Liquid ammonia (0.77 g, 45.92 mmol) was added to the active mixed anhydride formed in- situ and stirred at 0-5 °C for 20 min. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOs, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to get 2.9 g of compound lb (Yield: 96.3%). LCMS: 261.0 ( Vi+H ; .
Step lb:

1 b 1cTrifluroacetic anhydride (9.7 g, 46.0 mmol) was added to a solution of compound lb (8 g, 30.7 mmol) in pyridine (24.3 g, 307.0 mmol) and stirred at room temperature for 3 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCO?,, citric acid, brine solution, dried over Na2-S04 and evaporated under reduced pressure to afford 7 g of compound lc (Yield: 94.0%). LCMS: 187.2 (M-¾u )+.
Step lc:

1 c 1dHydroxylamine hydrochloride (3 g, 43.37 mmol) and potassium carbonate (6 g, 43.37 mmol) were added to a solution of compound lc (7 g, 28.91 mmol) in EtOH (70 m L) and stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with brine solution, dried over Na2S04 and evaporated under reduced pressure. The crude compound was purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to get 4.2 g of compound Id (Yield: 52.8%). LCMS: 276.4 (M+H)+.Step Id:

Deoxo-Fluor® (1.83 g, 8.3 mmol) was added to a solution of Fmoc-Asn(Trt)-OH (4.5 g, 7.5 mmol) in CH2Q2 (50 mL) and stirred at 0 °C for 3 h. Then CH2CI2 was evaporated and triturated with hexane, decanted and evaporated under vacuum to get the corresponding acid fluoride. NMM (1.17 g, 1 1.6 mmol) and compound Id (1.6 g, 5.8 mmol) in THF were added to the acid fluoride and stirred at room temperature for 12 h. Then THF was evaporated and sodium acetate (0.72 g, 8.7 mmol) was added followed by EtOH (50 mL). The reaction mixture was stirred at 90 °C for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure and partitioned between water and ethyl acetate. Organic layer was washed with NaHCOa, citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure, which was further purified by silica gel column chromatography (Eluent: 0-5% ethyl acetate in hexane) to afford 2.8 g of compound le (Yield: 44.4%). LCMS: 836.4 (M+Hf .Step le:
Ph3

To compound le (2.3 g, 2.7 mmol) in CH2CI2 (10 mL) diethyiarnine (10 mL) was added and the reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get gummy residue. The crude compound was purified by neutral alumina column chromatography (Eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to get 1.4 g of If (Yield: 90 %). LCMS: 636.5 (M+Na)+.

1f 1To a solution of compound If (0.45 g) in CH2CI2 (5 mL), trifluoroacetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred for 3 h at room temperature to remove the acid sensitive protecting groups. The resulting solution was concentrated in vacuum to afford 0.29 g of crude compound 1 which was purified using prep-HPLC method described under experimental conditions. \H NMR (DMSQ-d6, 400 MHz): δ 2.58 (m, 2H), 3.53 (m, 3H), 3.91 (t, 1H), 4.36 (t, 1H), 6.91 (s, 1H), 7.45 (s, 1H); 1 C NMR (DMSO-de, 400 MHz): δ 20.85, 45.71 , 50.23, 65.55, 171.03, 171 .41, 181.66. LCMS: 216.2 (Μ+ΗΓ; HPLC: tR = 13.1 min.Example 2: Synthesis of Co

Step 2a:

1f2a
The urea linkage was carried out by the coupling compound If (2.7 g, 4.39 mmoi) in THF (30 mL) at room temperature with compound 2b (1.67 g, 4.39 mmoi). The coupling was initiated by the addition of TEA (0.9 g, 8.78 mmoi) in THF (10 m L) and the resultant mixture was stirred at room temperature. After completion of 20 h, THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get compound 2a, which was further purified by silica gel column chromatography (Fluent: 0-50% ethyl acetate in hexane) to afford 3.46 g of compound 2a (Yield: 92.10%). LCMS 857.4 (M+H)+.

2aTo a solution of compound 2a (0.22 g, 0.25 mmol) in 0¾ί¾ (5 m L), trifluoroaeetic acid (5 mL) and catalytic amount of triisopropyisilane were added and stirred for 3h at room, temperature. The resulting solution was concentrated under reduced pressure to obtain 0.35 g of crude compound. The crude solid material was purified using preparative- HPLC method described under experimental conditions. LCMS: 347.1 (M+H)+; HPLC: tR = 12.9 min.
Synthesis of

2bTo the compound H-Ser(tBu)-OiBu (2 g, 9.2 mmol) in C I I■■(.‘{■ (20 mL), triethylamine (1.39 g, 13.8 mmol) was added and the solution was stirred at room temperature for 5-10 min. To this mixture, solution of 4-Nitrophenyl chioro formate (2.22 g, 11.04 mmol) in CH2CI2 was added and the resultant mixture was stirred at room temperature for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction, reaction mixture was diluted with CH2CI2 and washed with water and 5.0 M citric acid solution, dried over Na2SC>4 and evaporated under reduced pressure to get crude compound 2b, which was further purified by silica gel column chromatography (Eiuent: 0-20% ethyl acetate in hexane) to yield 2.1 g (58.9%) of 2b.Example 3: Synthesis of Compound 3

The compound was synthesised using similar procedure as depicted in Example 1 (compound 1) and D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH (compound la, Example 1) and Fmoc-D- Asn(trt)-OH in place of Fmoc-Asn(trt)-OH to yield 0.15 g crude material of the title compound 3. LCMS: 230.1 (M+H)+.Example 4: Synthesis of Co
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using

instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.Example 5: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)- OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.Example 6: Synthesis of Compound 6

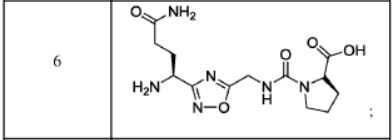
The compound was synthesised using similar procedure as depicted in Example 2 by using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.2 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 375.1 (M+H)+, HPLC: tR = 1.84 min.Example 7: Synthesis of Compound 7

Step 7a:

1f7aThe compound 7a was synthesised using similar procedure as for compound 2a (Example 2, step 2a) using H-Thr(‘Bu)-OMe instead of H-Ser(‘Bu)-OtBu to get crude material which was further purified by silica gel column chromatography (Eluent: 0-50% ethyl acetate in he ane) to get 2.0 g of compound 7a (Yield: 74 %). LCMS: 829.2 (M+H)+.Step 7b:

7a 7bTo a solution of compound 7a (0.35 g, 4.0 mmol) in THF (5 mL) was added lithium hydroxide (0.026 g, 0.63 mmol) at 0 °C and the mixture was stirred for 2 h at room temperature. The completion of the reaction was confirmed by TLC analysis. THF was evaporated from the reaction mass, and partitioned between water and ethyl acetate. Organic layer was washed with citric acid, brine solution, dried over Na2S04 and evaporated under reduced pressure to afford 7b, which was further purified by silica gel column chromatography (Eluent: 0-5% methanol in DCM) to get 0.3 g of product 7b (Yield: 86.7%). LCMS 815.2 (M+H)+.
Step 7c:

7b 7Compound 7b (0.295 g, 0.39 mmol) was anchored to Rink amide resin (0.7 g, 0.55 mmol/g) using HOBT (0.072 g, 0.54 mmol) and DIC (0.068 g, 0.54 mmol) method in DMF (10 mL). The resin was stirred for 12 h at room temperature. The resin was washed with DCM, DMF and DCM and dried. The target compound was cleaved from the rink amide resin using TFA (5 mL) and catalytic amount of TIPS. The resin was allowed to remain at room temperature for 2 h with occasional stirring. After 2 h, TFA and TIPS were evaporated under nitrogen atmosphere and the resulting residue was washed with diethyl ether to yield 0.1 g crude material of the title compound 7. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 360.0 (M+H)+, HPLC: tR = 13.88 min.Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324&tab=FULLTEXT
Patenthttps://patents.google.com/patent/WO2019067678A1/enPATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019061324
PATENThttps://patents.google.com/patent/WO2018073754A1/en
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
PAPERSScientific Reports (2019), 9(1), 1-19. https://www.nature.com/articles/s41598-019-48826-6
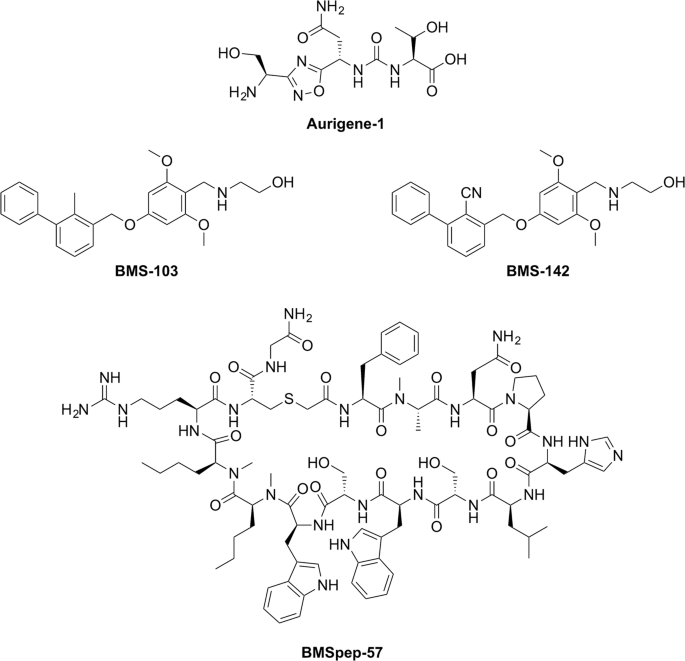
Chemical structures of PD-L1 inhibitors developed by Aurigene (Aurigene-1) and Bristol-Meyers Squibb (BMSpep-57, BMS-103, and BMS-142). Chemical structures were generated using ChemDraw Professional 15. PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019087087
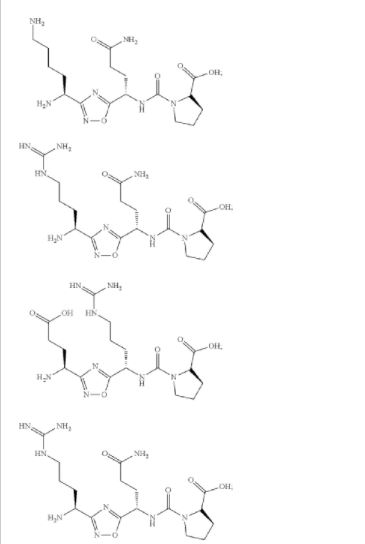
L-threonine’ mentioned in compound of formula (I) thereof can be represented by any one of the following formulae:
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020289477-A1 | Conjoint therapies for immunomodulation | 2017-11-06 | |
| WO-2019073399-A1 | CRYSTALLINE FORMS OF 1,2,4-OXADIAZOLE SUBSTITUTED IN POSITION 3 | 2017-10-11 | |
| AU-2018341583-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| WO-2019061324-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 | |
| WO-2019067678-A1 | CRYSTALLINE FORMS OF IMMUNOMODULATORS | 2017-09-29 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020247766-A1 | Crystal forms of immunomodulators | 2017-09-29 | |
| US-2020061030-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| WO-2018073754-A1 | Dual inhibitors of vista and pd-1 pathways | 2016-10-20 | |
| US-2020361880-A1 | 1,2,4-Oxadiazole and Thiadiazole Compounds as Immunomodulators | 2015-03-10 | |
| EP-3041827-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2018-04-18 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-3363790-B1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-02-19 |
| US-10173989-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2019-01-08 |
| US-10590093-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2020-03-17 |
| US-2015073024-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2017101386-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018072689-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2019144402-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-2020199086-A1 | 1,2,4-Oxadiazole Derivatives as Immunomodulators | 2013-09-06 | |
| US-9771338-B2 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 | 2017-09-26 |
| WO-2015033299-A1 | 1,2,4-oxadiazole derivatives as immunomodulators | 2013-09-06 |
////////////Investigational New Drug Application, Phase 1, Clinical Trial, Non-Hodgkin’s Lymphoma, XL 114, AUR 104, aurigene, Exelixis
N[C@@H](CO)c1nc(on1)[C@@H](NC(=O)N[C@H](C(=O)O)C(C)O)CC(N)=O
https://patentscope.wipo.int/search/en/result.jsf?inchikey=HFOBENSCBRZVSP-WHFCDURNSA-N

NEW DRUG APPROVALS
ONE TIME
$10.00
PATENT


The present invention relates to substituted alkynylene compounds represented by compound of formula (I) pharmaceutically acceptable salts and stereoisomers thereof. The present invention further provides the methods of preparation of compound of formula (I) and therapeutic uses thereof as anti-cancer agents.
XL 102
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
EXELIXIS AND AURIGENE ANNOUNCE THAT PROMISING PRECLINICAL DATA TO BE PRESENTED AT THE ENA SYMPOSIUM SUPPORT THE CLINICAL DEVELOPMENT OF A NOVEL CDK7 INHIBITOR
Exelixis and Aurigene Announce That Promising Preclinical Data to Be Presented at the ENA Symposium Support the Clinical Development of a Novel CDK7 Inhibitor
– Detailed characterization of an oral inhibitor of CDK7 demonstrates potent activity against multiple hematologic and solid tumor cell lines, as monotherapy and in combination with chemotherapies –
October 09, 2020 03:02 AM Eastern Daylight Time
ALAMEDA, Calif.–(BUSINESS WIRE)–Exelixis, Inc. (Nasdaq: EXEL) and Aurigene Discovery Technologies Limited (Aurigene) today disclosed new preclinical data showing that AUR102 has potent anti-tumor activity in a large panel of cancer cell lines. AUR102 is a potent, selective, and orally bioavailable covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which is an important regulator of the cellular transcriptional and cell cycle machinery. Exelixis has an exclusive option for AUR102 under its July 2019 exclusive collaboration, option and license agreement with Aurigene. The new data will be presented in a poster (Abstract 170) at the 32nd EORTC-NCI-AACR (ENA) Symposium, which is being held virtually on October 24-25, 2020.
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy”
“CDK7 plays a critical role in regulating cellular transcription and cell cycle machinery, making it an exciting target for cancer therapy,” said Murali Ramachandra, Ph.D., Chief Executive Officer of Aurigene. “The data to be presented at ENA 2020 demonstrate that AUR102 effectively engages CDK7 and inhibits a key mediator of the cell cycle and transcription. The ability to inhibit CDK7 activity with an orally available therapeutic such as AUR102 holds great potential to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma.”
The abstract provides a summary of results from a detailed characterization of AUR102 in cancer cell lines and animal tumor models. Additional data will be presented in the poster. Key findings included in the abstract are:
• AUR102 exhibited potent anti-proliferative activity in a large panel of cell lines with induction of cell death in cell lines derived from multiple cancer types.
• The observed anti-proliferative activity correlated with cellular CDK7 target engagement and decreased levels of P-Ser5 RNAPII, a key mediator of transcription.
• AUR102 studies showed synergy when used in combination with multiple chemotherapies.
• Oral dosing with AUR102 resulted in dose-dependent anti-tumor activity, including complete tumor regression in diffuse large B-cell lymphoma, acute myeloid leukemia, and triple-negative breast cancer xenograft models.
• Inhibition of tumor growth was accompanied by complete target engagement as demonstrated in a parallel PK-PD study.
• AUR102 significantly impacts several pathways and key cancer driver and immune-response genes.
The study authors conclude that the data support clinical evaluation of AUR102 as a single agent and in combination with chemotherapies for the treatment of cancer.
“The exciting AUR102 data to be presented at ENA 2020 provide further validation of our partnering strategy, which gives us multiple opportunities to build a pipeline of best-in-class cancer therapies,” said Peter Lamb, Ph.D., Executive Vice President of Scientific Strategy and Chief Scientific Officer of Exelixis. “AUR102 could be the subject of an Investigational New Drug filing later this year, which would be an important value driver for the program itself and for our collaboration with Aurigene. We commend the Aurigene team on their ongoing success in building a robust body of data supporting the broad clinical potential of AUR102.”
Under the terms of the July 2019 agreement, Exelixis made an upfront payment of $10 million for exclusive options to license three preexisting programs from Aurigene. In addition, Exelixis and Aurigene initiated three Aurigene-led drug discovery programs on mutually agreed upon targets, in exchange for additional upfront option payments of $2.5 million per program. Exelixis is also contributing research funding to Aurigene to facilitate discovery and preclinical development work on all six programs. As the programs mature, Exelixis will have the opportunity to exercise an exclusive option for each program up until the time of Investigational New Drug (IND) filing acceptance. If Exelixis decides to exercise an option, it will make an option exercise payment to Aurigene and assume responsibility for that program’s future clinical development and commercialization including global manufacturing. Aurigene will be eligible for clinical development, regulatory, and sales milestones, as well as royalties on sales. Under the terms of the agreement, Aurigene retains limited development and commercial rights for India and Russia.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision-oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene’s programs currently in clinical development include an oral ROR-gamma inhibitor AUR101 for moderate to severe psoriasis in phase 2 under a U.S. FDA IND and a PD-L1/ VISTA antagonist CA-170 for non-squamous non-small cell lung cancer in phase 2b/3 in India. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has also partnered with several large and mid-pharma companies in the United States and Europe and has multiple programs in clinical development. For more information, please visit Aurigene’s website at http://www.aurigene.com.
About Exelixis
Founded in 1994, Exelixis, Inc. (Nasdaq: EXEL) is a commercially successful, oncology-focused biotechnology company that strives to accelerate the discovery, development and commercialization of new medicines for difficult-to-treat cancers. Following early work in model system genetics, we established a broad drug discovery and development platform that has served as the foundation for our continued efforts to bring new cancer therapies to patients in need. Our discovery efforts have resulted in four commercially available products, CABOMETYX® (cabozantinib), COMETRIQ® (cabozantinib), COTELLIC® (cobimetinib) and MINNEBRO® (esaxerenone), and we have entered into partnerships with leading pharmaceutical companies to bring these important medicines to patients worldwide. Supported by revenues from our marketed products and collaborations, we are committed to prudently reinvesting in our business to maximize the potential of our pipeline. We are supplementing our existing therapeutic assets with targeted business development activities and internal drug discovery – all to deliver the next generation of Exelixis medicines and help patients recover stronger and live longer. Exelixis is a member of Standard & Poor’s (S&P) MidCap 400 index, which measures the performance of profitable mid-sized companies. For more information about Exelixis, please visit http://www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
Exelixis Forward-Looking Statements
This press release contains forward-looking statements, including, without limitation, statements related to: Exelixis’ and Aurigene’s plans to present preclinical data in support of the continued development of AUR102 in a poster as part of the 32nd ENA Symposium; the potential for AUR102 to improve care and outcomes for patients with diverse cancer indications, including breast cancer, prostate cancer, leukemia and lymphoma; the potential for AUR102 to be the subject of an Investigational New Drug filing later in 2020; Exelixis’ potential future financial and other obligations under the exclusive collaboration, option and license agreement with Aurigene; and Exelixis’ plans to reinvest in its business to maximize the potential of the company’s pipeline, including through targeted business development activities and internal drug discovery. Any statements that refer to expectations, projections or other characterizations of future events or circumstances are forward-looking statements and are based upon Exelixis’ current plans, assumptions, beliefs, expectations, estimates and projections. Forward-looking statements involve risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in the forward-looking statements as a result of these risks and uncertainties, which include, without limitation: the availability of data at the referenced times; the level of costs associated with Exelixis’ commercialization, research and development, in-licensing or acquisition of product candidates, and other activities; uncertainties inherent in the drug discovery and product development process; Exelixis’ dependence on its relationship with Aurigene, including Aurigene’s adherence to its obligations under the exclusive collaboration, option and license agreement and the level of Aurigene’s assistance to Exelixis in completing clinical trials, pursuing regulatory approvals or successfully commercializing partnered compounds in the territories where they may be approved; the continuing COVID-19 pandemic and its impact on Exelixis’ research and development operations; complexities and the unpredictability of the regulatory review and approval processes in the U.S. and elsewhere; Exelixis’ and Aurigene’s continuing compliance with applicable legal and regulatory requirements; Exelixis’ and Aurigene’s ability to protect their respective intellectual property rights; market competition; changes in economic and business conditions; and other factors affecting Exelixis and its product pipeline discussed under the caption “Risk Factors” in Exelixis’ Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission (SEC) on August 6, 2020, and in Exelixis’ future filings with the SEC. All forward-looking statements in this press release are based on information available to Exelixis as of the date of this press release, and Exelixis undertakes no obligation to update or revise any forward-looking statements contained herein, except as required by law.
Exelixis, the Exelixis logo, CABOMETYX, COMETRIQ and COTELLIC are registered U.S. trademarks. MINNEBRO is a registered Japanese trademark.
Sage Kills 51% of Melanoma Cells in Vitro and Reduces Risk in Humans

Sage Kills 51% of Melanoma Cells in Vitro and Reduces Risk in Humans: The herb sage is rich in the powerful anti-cancer compound thujone, which in this study was shown to kill 51% of human melanoma cells in vitro.
But does sage reduce melanoma risk in humans? Yes, according to a study out of the Italy, where this herb is often consumed as part of the traditional diet: people eating sage at least once weekly had 32% less risk of melanoma. Apart from thujone, this super-herb contains several other compounds with proven health benefits, including cineole, carnosol, caffeic acid and chlorogenic acid.
Sage has been used in traditional medicine for centuries to treat a variety of conditions, and it’s many health benefits are now being confirmed by some remarkable new clinical trials. One trial showed that 333 mg of sage extract daily significantly improved memory performance in older adults while in another clinical trial, 60 drops daily of liquid extract significantly enhanced cognitive performance in patients with Alzheimer’s.
Sage is also such a powerful natural antibacterial that one clinical trial (out of Switzerland) showed that a spray of sage + echinacea performed as well as chlorhexidine + lidocaine in treating sore throats! Sage can be consumed as supplements, prepared as a tea, or used generously as a herb in a variety of dishes.
http://www.ncbi.nlm.nih.gov/pubmed/21647317


Sage – Nature Wonder Herb
(Salvia officinalis) is cultivated as a spice and medicinal herb. This plant is known from parts of Europe, especially the Balkans, where is used for obtaining essential oils.
The Latin name of the whole genus Salvia comes from the Latin word “salvare“, which means “rescue saving curing …” because the Romans 2000 years ago appreciated and used sage for healing.

The effectiveness of the leaves is due primarily to the presence of etheric oil (1.5 – 2.5 percent), which has antimicrobial and anti-inflammatory action.
It has been proven that etheric oil destroys bacteria Eshericia colli, Schigela sonei, Salmonela, and has a slightly weaker activity against bacteria of a group of staphylococci and streptococci.
It is effective in destroying certain types of fungi as: cadida albicas, cadida krusei, cadidapseudotropicalis, torulopsis glabrata and cyptococcus eoformas.

Etheric sage oil is colorless or yellow-green liquid with an aromatic and bitter taste.
Because of this effect, sage is a useful herb for treating inflammation and infections of the mucous membranes in the mouth, gums and throat. For these diseases gargle from sage tea is recommended.
 -Put 2 tablespoons of crushed dried sage leaves in 2 cups of boiling water water. Cover it and let it stay like that for 20 or 30 minutes, then drain it.
-Put 2 tablespoons of crushed dried sage leaves in 2 cups of boiling water water. Cover it and let it stay like that for 20 or 30 minutes, then drain it.
For treatment to be successful, you must do gargle regularly , every 3 hours .
Sage softens the mucous secretions from the inflamed mucosa of the respiratory organs. Because of that you can drink sage tea against bronchitis.
-The tea is prepared with 1 tablespoon dried leaves and 2 cups of boiling water. Drink it three times a day.
You should avoid long term drinking tea because etheric oil from sage contains toxic substance thujone.
Dry leaves are used as a spice because it improves the taste and scent of food, and helps the digestion too.
Tea can be used for improving the functions of bile and liver, because bitter substances and etheric oil increase the secretion of digestive juices in the body.
Sage tea has a diuretic effect, which is poorly expressed and due to the presence of flavonoids . It can help with chronic disease of urinary tract .

From a long time ago is known that sage tea is very effective cure for sweating. This power of sage it’s explained by its effect on the nervous center which regulates the glands that secrete sweat.
Commonly is recommended against perspiration of neuromuscular origin or the heat waves that occur during menopause.
Applied on the skin , tea tightens skin and calms inflammation. Especially good for oily skin with open pores and irritated skin.

Sage is a remarkable tool for whitening teeth, strengthening gums and aid in periodontitis. A small spoon of sage leaves is mixed with 1 drop of peppermint etheric oil and abit of baking soda. With this mixture rub the teeth and gums twice a week.
BI launches COPD drug Striverdi, olodaterol in UK and Ireland

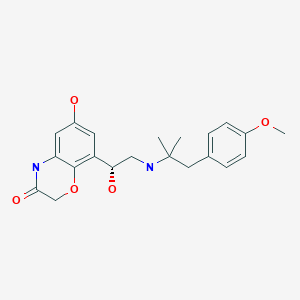
Olodaterol
BI-1744
BI-1744-CL (hydrochloride) marketed as drug
Boehringer Ingelheim Pharma innovator
synthesis…..http://wendang.baidu.com/view/d4f95541e518964bcf847c22.html
Olodaterol (trade name Striverdi) is a long acting beta-adrenoceptor agonist used as an inhalation for treating patients with chronic obstructive pulmonary disease (COPD), manufactured by Boehringer-Ingelheim.[1]
see……….https://www.thieme-connect.de/DOI/DOI?10.1055/s-0029-1219649 ……… synfacts
Olodaterol is a potent agonist of the human β2-adrenoceptor with a high β1/β2 selectivity. Its crystalline hydrochloride salt is suitable for inhalation and is currently undergoing clinical trials in man for the treatment of asthma. Olodaterol has a duration of action that exceeds 24 hours in two preclinical animal models of bronchoprotection and it has a better safety margin compared with formoterol.
Olodaterol hydrochloride [USAN]
Bi 1744 cl
Bi-1744-cl
Olodaterol hydrochloride
Olodaterol hydrochloride [usan]
UNII-65R445W3V9
868049-49-4 [RN] FREE FORM
CAS 869477-96-3 HCL SALT
R ENANTIOMER
2H-1,4-Benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
2H-1,4-benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
6-Hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)-1,1-dimethylethyl)amino)ethyl)- 2H-1,4-benzoxazin-3(4H)-one hydrochloride
clinical trialshttp://clinicaltrials.gov/search/intervention=Olodaterol+OR+BI+1744
Boehringer Ingelheim has launched a new chronic obstructive pulmonary disease drug, Striverdi in the UK and Ireland.
Striverdi (olodaterol) is the second molecule to be licenced for delivery via the company’s Respimat Soft Mist inhaler, following the COPD blockbuster Spiriva (tiotropium). The drug was approved in Europe in November based on results from a Phase III programme that included more than 3,000 patients with moderate to very severe disease.http://www.pharmatimes.com/Article/14-07-01/BI_launches_COPD_drug_Striverdi_in_UK_and_Ireland.aspx
Olodaterol hydrochloride is a drug candidate originated by Boehringer Ingelheim. The product, delivered once-daily by the Respimat Soft Mist Inhaler, was first launched in Denmark and the Netherlands in March 2014 for the use as maintenance treatment of chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. In 2013, approval was obtained in Russia and Canada for the same indication, and in the U.S, the product was recommended for approval. Phase III clinical trials for the treatment of COPD are ongoing in Japan.
| Systematic (IUPAC) name | |
|---|---|
| 6-hydroxy-8-{(1R)-1-hydroxy-2-{[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino}ethyl}-4H-1,4-benzoxazin-3-one | |
| Clinical data | |
| Trade names | Striverdi |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy cat. | No experience |
| Legal status | POM (UK) |
| Routes | Inhalation |
| Identifiers | |
| CAS number | 868049-49-4; 869477-96-3 (hydrochloride) |
| ATC code | R03AC19 |
| PubChem | CID 11504295 |
| ChemSpider | 9679097 |
| UNII | VD2YSN1AFD |
| ChEMBL | CHEMBL605846 |
| Synonyms | BI 1744 CL |
| Chemical data | |
| Formula | C21H26N2O5 free form C21 H26 N2 O5 . Cl H; of hcl salt |
| Mol. mass | 386.44 g/mol free form; 422.902 as hyd salt |
Medical uses
Olodaterol is a once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD including chronic bronchitis and/or emphysema, and is administered in an inhaler called Respimat Soft Mist Inhaler.[2][3][4][5][6][7]
As of December 2013, olodaterol is not approved for the treatment of asthma. Olodaterol monotherapy was previously evaluated in four Phase 2 studies in asthma patients. However, currently there are no Phase 3 studies planned for olodaterol monotherapy in patients with asthma.
In late January 2013, Olodaterol CAS# 868049-49-4 was the focus of an FDA committee reviewing data for the drug’s approval as a once-daily maintenance bronchodilator to treat chronic obstructive pulmonary disease (COPD), as well as chronic bronchitis and emphysema. The FDA Pulmonary-Allergy Drugs Advisory Committee recommended that the clinical data from the Boehringer Ingelheim Phase III studies be included in their NDA.
Also known as the trade name Striverdi Respimat, Olodaterol is efficacious as a long-acting beta-agonist, which patients self-administer via an easy to use metered dose inhaler. While early statistics from clinical trials of Olodaterol were encouraging, a new set of data was released earlier this week, which only further solidified the effectual and tolerable benefits of this COPD drug.
On September 10, 2013 results from two Phase 3 studies of Olodaterol revealed additional positive results from this formidable COPD treatment. The conclusion from these two 48 week studies, which included over 3,000 patients, showed sizable and significant improvements in the lung function of patients who were dosed with Olodaterol. Patients in the aforementioned studies were administered either a once a day dosage of Olodaterol via the appropriate metered-dose inhaler or “usual care”. The “usual care” included a variety of treatment options, such as inhaled corticosteroids (not Olodaterol), short and long acting anticholinergics, xanthines and beta agonists, which were short acting. The clinical trial participants who were dosed with Olodaterol displayed a rapid onset of action from this drug, oftentimes within the first five minutes after taking this medication. Additionally, patients dispensed the Olodaterol inhaler were successfully able to maintain optimum lung function for longer than a full 24 hour period. The participants who were given Olodaterol experienced such an obvious clinical improvement in their COPD symptoms, and it quickly became apparent that the “usual care” protocol was lacking in efficacy and reliability.
A staggering 24 million patients in the United States suffer from chronic obstructive pulmonary disease, and this patient population is in need of an effectual, safe and tolerable solution. Olodaterol is shaping up to be that much needed solution. Not only have the results from studies of Olodaterol been encouraging, the studies themselves have actually been forward thinking and wellness centered. Boehringer Ingelheim is the first company to included studies to evaluate exercise tolerance in patients with COPD, and compare the data to those patients who were dosed with Olodaterol. By including exercise tolerance as an important benchmark in pertinent data for Olodaterol, Boehringer Ingelheim has created a standard for COPD treatment expectations. The impaired lung function for patients with COPD contributes greatly to their inability to exercise and stay healthy. Patients who find treatments and management techniques to combat the lung hyperinflation that develops during exercise have a distinct advantage to attaining overall good health.
– See more at: http://www.lgmpharma.com/blog/olodaterol-offers-encouraging-results-patients-copd/#sthash.DOjcrGxc.dpuf
Data has demonstrated that Striverdi, a once-daily long-acting beta2 agonist, significantly improved lung function versus placebo and is comparable to improvements shown with the older LABA formoterol. The NHS price for the drug is £26.35 for a 30-day supply.
Boehringer cited Richard Russell at Wexham Park Hospital as saying that the licensing of Stirverdi will be welcomed by clinicians as it provides another option. He added that the trial results showing improvements in lung function “are particularly impressive considering the study design, which allowed participants to continue their usual treatment regimen. This reflects more closely the real-world patient population”.
Significantly, the company is also developing olodaterol in combination with Spiriva, a long-acting muscarinic antagonist. LAMA/LABA combinations provide the convenience of delivering the two major bronchodilator classes.
 Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.Adverse effects
Adverse effects generally were rare and mild in clinical studies. Most common, but still affecting no more than 1% of patients, were nasopharyngitis (running nose), dizziness and rash. To judge from the drug’s mechanism of action and from experiences with related drugs, hypertension (high blood pressure), tachycardia (fast heartbeat), hypokalaemia (low blood levels of potassium), shaking, etc., might occur in some patients, but these effects have rarely, if at all, been observed in studies.[1]
Interactions
Based on theoretical considerations, co-application of other beta-adrenoceptor agonists, potassium lowering drugs (e. g. corticoids, many diuretics, and theophylline), tricyclic antidepressants, and monoamine oxidase inhibitors could increase the likelihood of adverse effects to occur. Beta blockers, a group of drugs for the treatment of hypertension (high blood pressure) and various conditions of the heart, could reduce the efficacy of olodaterol.[1] Clinical data on the relevance of such interactions are very limited.
Pharmacology
Mechanism of action
Like all beta-adrenoceptor agonists, olodaterol mimics the effect of epinephrine at beta-2 receptors (β₂-receptors) in the lung, which causes the bronchi to relax and reduces their resistance to airflow.[3]
Olodaterol is a nearly full β₂-agonist, having 88% intrinsic activity compared to the gold standard isoprenaline. Its half maximal effective concentration (EC50) is 0.1 nM. It has a higher in vitro selectivity for β₂-receptors than the related drugs formoterol and salmeterol: 241-fold versus β₁- and 2299-fold versus β₃-receptors.[2] The high β₂/β₁ selectivity may account for the apparent lack of tachycardia in clinical trials, which is mediated by β₁-receptors on the heart.
Pharmacokinetics
Once bound to a β₂-receptor, an olodaterol molecule stays there for hours – its dissociation half-life is 17.8 hours –, which allows for once-a-day application of the drug[3] like with indacaterol. Other related compounds generally have a shorter duration of action and have to be applied twice daily (e.g. formoterol, salmeterol). Still others (e. g. salbutamol, fenoterol) have to be applied three or four times a day for continuous action, which can also be an advantage for patients who need to apply β₂-agonists only occasionally, for example in an asthma attack.[8]
History
On 29 January 2013 the U.S. Food and Drug Administration (FDA) Pulmonary-Allergy Drugs Advisory Committee (PADAC) recommended that the clinical data included in the new drug application (NDA) for olodaterol provide substantial evidence of safety and efficacy to support the approval of olodaterol as a once-daily maintenance bronchodilator treatment for airflow obstruction in patients with COPD.[9]
On 18 October 2013 approval of olodaterol in the first three European countries – the United Kingdom, Denmark and Iceland – was announced by the manufacturer.[10]

Figure Chemical structures of salmeterol, formoterol, inda- caterol, and emerging once-daily long-acting β2-agonists
CLIP
Synthetic approaches to the 2013 new drugs – ScienceDirect
Synthesis of olodaterol hydrochloride (XVI).

Olodaterol hydrochloride was approved for long-term, once-daily maintenance treatment of chronic
obstructive pulmonary disease (COPD) in 2013 in the following countries: Canada, Russia, United
Kingdom, Denmark, and Iceland.142, 143 The drug has been recommended by a federal advisory panel for
approval by the FDA.142, 143 Developed and marketed by Boehringer Ingelheim, olodaterol is a longacting
β2-adrenergic receptor agonist with high selectivity over the β1- and β3-receptors (219- and 1622-fold, respectively).144 Upon binding to and activating the β2-adrenergic receptor in the airway, olodaterol
stimulates adenyl cyclase to synthesize cAMP, leading to the relaxation of smooth muscle cells in the
airway. Administered by inhalation using the Respimat®
Soft Mist inhaler, it delivers significant
bronchodilator effects within five minutes of the first dose and provides sustained improvement in
forced expiratory volume (FEV1) for over 24 hours.143 While several routes have been reported in the
patent and published literature,144-146 the manufacturing route for olodaterol hydrochloride disclosed in
2011 is summarized in Scheme 19 below.147
Commercial 2’,5’-dihydroxyacetophenone (122) was treated with one equivalent of benzyl bromide
and potassium carbonate in methylisobutylketone (MIBK) to give the 5’-monobenzylated product in
76% yield. Subsequent nitration occurred at the 4’-position to provide nitrophenol 123 in 87% yield.
Reduction of the nitro group followed by subjection to chloroacetyl chloride resulted in the construction
of benzoxazine 124 in 82% yield. Next, monobromination through the use of tetrabutylammonium
tribromide occurred at the acetophenone carbon to provide bromoketone 125, and this was followed by
asymmetric reduction of the ketone employing (−)-DIP chloride to afford an intermediate bromohydrin,
which underwent conversion to the corresponding epoxide 126 in situ upon treatment with aqueous
NaOH. This epoxide was efficiently formed in 85% yield and 98.3% enantiomeric excess. Epoxide
126 underwent ring-opening upon subjection to amine 127 to provide amino-alcohol 128 in in 84-90%
yield and 89.5-99.5% enantiomeric purity following salt formation with HCl. Tertiary amine 127 was
itself prepared in three steps by reaction of ketone 129 with methylmagnesium chloride, Ritter reaction
of the tertiary alcohol with acetonitrile, and hydrolysis of the resultant acetamide with ethanolic
potassium hydroxide. Hydrogenative removal of the benzyl ether within 128 followed by
recrystallization with methanolic isopropanol furnished olodaterol hydrochloride (XVI) in 63-70%
yield. Overall, the synthesis of olodaterol hydrochloride required 10 total steps (7 linear) from
commercially available acetophenone 122.
142. Gibb, A.; Yang, L. P. H. Drugs 2013, 73, 1841.
143. http://www.boehringeringelheim.com/news/news_releases/press_releases/2013/18_october_2013_olodaterol.html.
144. Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine,
C.; Buettner, F. H.; Schnapp, A.; Konetzki, I. Bioorg. Med. Chem. Lett. 2010, 20, 1410.
145. Trunk, M. J. F.; Schiewe, J. US Patent 20050255050A1, 2005.
146. Lustenberger, P.; Konetzki, I.; Sieger, P. US Patent 20090137578A1, 2009.
147. Krueger, T.; Ries, U.; Schnaubelt, J.; Rall, W.; Leuter, Z. A.; Duran, A.; Soyka, R. US Patent
20110124859A1, 2011.
PATENT
WO 2004045618 or
http://www.google.com/patents/EP1562603B1?cl=en
Example

a)
To a solution of 3.6 g 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine in 100 mL of ethanol at 70 ° C. 7.5 g of (6-benzyloxy-4H-benzo [1,4] oxazin-3-one )-glyoxal added and allowed to stir for 15 minutes. Then within 30 minutes at 10 to 20 ° C. 1 g of sodium borohydride added. It is stirred for one hour, with 10 mL of acetone and stirred for another 30 minutes. The reaction mixture is diluted with 150 mL ethyl acetate, washed with water, dried with sodium sulfate and concentrated. The residue is dissolved in 50 mL of methanol and 100 mL ethyl acetate and acidified with conc. Hydrochloric acid. After addition of 100 mL of diethyl ether, the product precipitates. The crystals are filtered, washed and recrystallized from 50 mL of ethanol. Yield: 7 g (68%; hydrochloride), mp = 232-234 ° C.
b)
6.8 g of the above obtained benzyl compound in 125 mL of methanol with the addition of 1 g of palladium on carbon (5%) was hydrogenated at room temperature and normal pressure. The catalyst is filtered and the filtrate was freed from solvent. Recrystallization of the residue in 50 mL of acetone and a little water, a solid is obtained, which is filtered and washed.
Yield: 5.0 g (89%; hydrochloride), mp = 155-160 ° C.
The (R) – and (S)-enantiomers of Example 3 can be obtained from the racemate, for example, by chiral HPLC (for example, column: Chirobiotic T, 250 x 1.22 mm from the company Astec). As the mobile phase, methanol with 0.05% triethylamine and 0.05% acetic acid. Silica gel with a grain size of 5 microns, to which is covalently bound the glycoprotein teicoplanin can reach as column material used. Retention time (R enantiomer) = 40.1 min, retention time (S-enantiomer) = 45.9 min. The two enantiomers can be obtained by this method in the form of free bases. According to the invention of paramount importance is the R enantiomer of Example 3
PATENT
WO 2005111005
http://www.google.fm/patents/WO2005111005A1?cl=en
Scheme 1.




Scheme 1:
Example 1 6-Hydroxy-8-{(1-hydroxy-2-r2-(4-methoxy-phenyl) – 1, 1-dimethyl-ethylamino]-ethyl)-4H-benzor 41oxazin-3-one – Hvdrochlorid

a) l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone
To a solution of 81.5 g (0.34 mol) l-(5-benzyloxy-2-hydroxy-phenyl)-ethanone in 700 ml of acetic acid are added dropwise under cooling with ice bath, 18 mL of fuming nitric acid, the temperature does not exceed 20 ° C. increases. The reaction mixture is stirred for two hours at room temperature, poured onto ice water and filtered. The product is recrystallized from isopropanol, filtered off and washed with isopropanol and diisopropyl ether. Yield: 69.6 g (72%), mass spectroscopy [M + H] + = 288
b) l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone
69.5 g (242 mmol) of l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone are dissolved in 1.4 L of methanol and in the presence of 14 g of rhodium on carbon (10%) as catalyst at 3 bar room temperature and hydrogenated. Then the catalyst is filtered off and the filtrate concentrated. The residue is reacted further without additional purification. Yield: 60.0 g (96%), R f value = 0.45 (silica gel, dichloromethane).
c) 8-acetyl-6-benzyloxy-4H-benzoπ .4] oxazin-3-one
To 60.0 g (233 mmol) of l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone and 70.0 g (506 mmol) of potassium carbonate while cooling with ice bath, 21.0 ml (258 mmol) of chloroacetyl chloride added dropwise. Then stirred overnight at room temperature and then for 6 hours under reflux. The hot reaction mixture is filtered and then concentrated to about 400 mL and treated with ice water. The precipitate is filtered off, dried and purified by chromatography on a short silica gel column (dichloromethane: methanol = 99:1). The product-containing fractions are concentrated, suspended in isopropanol, diisopropyl ether, and extracted with
Diisopropyl ether. Yield: 34.6 g (50%), mass spectroscopy [M + H] + = 298
d) 6-Benzyloxy-8-(2-chloro-acetyl)-4H-benzoFl, 4] oxazin-3-one 13.8 g (46.0 mmol) of 8-benzyloxy-6-Acetyl-4H-benzo [l, 4] oxazin -3-one and 35.3 g (101.5 mmol) of benzyltrimethylammonium dichloriodat are stirred in 250 mL dichloroethane, 84 mL glacial acetic acid and 14 mL water for 5 hours at 65 ° C. After cooling to room temperature, treated with 5% aqueous sodium hydrogen sulfite solution and stirred for 30 minutes. The precipitated solid is filtered off, washed with water and diethyl ether and dried. Yield: 13.2 g (86%), mass spectroscopy [M + H] + = 330/32.
e) 6-Benzyloxy-8-((R-2-chloro-l-hydroxy-ethyl)-4H-benzori ,41-oxazin-3-one The procedure is analogous to a procedure described in the literature (Org. Lett ., 2002, 4, 4373-4376).
To 13:15 g (39.6 mmol) of 6-benzyloxy-8-(2-chloro-acetyl)-4H-benzo [l, 4] oxazin-3-one and 25.5 mg (0:04 mmol) Cρ * RhCl [(S, S) -TsDPEN] (Cp * = pentamethylcyclopentadienyl and TsDPEN = (lS, 2S)-Np-toluenesulfonyl-l ,2-diphenylethylenediamine) in 40 mL of dimethylformamide at -15 ° C and 8 mL of a mixture of formic acid and triethylamine (molar ratio = 5: 2) dropwise. It is allowed for 5 hours at this temperature, stirring, then 25 mg of catalyst and stirred overnight at -15 ° C. The reaction mixture is mixed with ice water and filtered. The filter residue is dissolved in dichloromethane, dried with sodium sulfate and the solvent evaporated. The residue is recrystallized gel (dichloromethane / methanol gradient) and the product in diethyl ether / diisopropyl ether. Yield: 10.08 g (76%), R f value = 00:28 (on silica gel, dichloromethane ethanol = 50:1).
f) 6-Benzyloxy-8-(R-oxiranyl-4H-benzo [“L4] oxazin-3-one 6.10 g (30.1 mmol) of 6-benzyloxy-8-((R)-2-chloro-l-hydroxy- ethyl)-4H-benzo [l, 4] oxazin-3-one are dissolved in 200 mL of dimethylformamide. added to the solution at 0 ° C with 40 mL of a 2 molar sodium hydroxide solution and stirred at this temperature for 4 hours. the reaction mixture is poured onto ice water, stirred for 15 minutes, and then filtered The solid is washed with water and dried to give 8.60 g (96%), mass spectroscopy [M + H] + = 298..
g) 6-Benyloxy-8-{(R-l-hydroxy-2-r2-(4-methoxy-phenyl)-dimethyl-ll-ethvIaminol-ethyl)-4H-benzo-3-Tl A1oxazin
5.25 g (17.7 mmol) of 6-benzyloxy-8-(R)-oxiranyl-4H-benzo [l, 4] oxazin-3-one and 6.30 g (35.1 mmol) of 2 – (4-methoxy-phenyl 1, 1 – dimethyl-ethyl to be with 21 mL
Of isopropanol and stirred at 135 ° C for 30 minutes under microwave irradiation in a sealed reaction vessel. The solvent is distilled off and the residue chromatographed (alumina, ethyl acetate / methanol gradient). The product thus obtained is purified by recrystallization from a mixture further Diethylether/Diisopropylether-. Yield: 5:33 g (63%), mass spectroscopy [M + H] + = 477 h) 6-Hydroxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino] – ethyl}-4H-benzo [1, 4, 1 oxazin-3-one hydrochloride
A suspension of 5:33 g (11.2 mmol) of 6-Benyloxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino]-ethyl}-4H -benzo [l, 4] oxazin-3-one in 120 mL of methanol with 0.8 g of palladium on carbon (10%), heated to 50 ° C and hydrogenated at 3 bar hydrogen pressure. Then the catalyst is filtered off and the filtrate concentrated. The residue is dissolved in 20 mL of isopropanol, and 2.5 mL of 5 molar hydrochloric acid in isopropanol. The product is precipitated with 200 mL of diethyl ether, filtered off and dried. Yield: 4.50 g (95%, hydrochloride), mass spectroscopy [M + H] + = 387
PATENT
WO 2007020227
http://www.google.com.ar/patents/WO2007020227A1?cl=en
PATENT
WO 2008090193
or
http://www.google.com/patents/EP2125759B1?cl=en
PAPER
Discovery of olodaterol, a novel inhaled beta(2)-adrenoceptor agonist with a 24h bronchodilatory efficacy
Bioorg Med Chem Lett 2010, 20(4): 1410
http://www.sciencedirect.com/science/article/pii/S0960894X09018101
The discovery of the β2-adrenoceptor agonist (R)-4p designated olodaterol is described. The preclinical profile of the compound suggests a bronchoprotective effect over 24 h in humans.

CLIP
Australia
http://www.tga.gov.au/pdf/auspar/auspar-olodaterol-140327-pi.pdf
CLIP
DUTCH
http://mri.medagencies.org/download/NL_H_2498_001_PAR.pdf
FDA
Click to access 203108Orig1s000ChemR.pdf
NDA 203108
Striverdi® Respimat® (olodaterol) Inhalation Spray
Boehringer Ingelheim Pharmaceuticals, Inc.
References
- Striverdi UK Drug Information
- Bouyssou, T.; Casarosa, P.; Naline, E.; Pestel, S.; Konetzki, I.; Devillier, P.; Schnapp, A. (2010). “Pharmacological Characterization of Olodaterol, a Novel Inhaled 2-Adrenoceptor Agonist Exerting a 24-Hour-Long Duration of Action in Preclinical Models”. Journal of Pharmacology and Experimental Therapeutics 334 (1): 53–62. doi:10.1124/jpet.110.167007. PMID 20371707.
- Casarosa, P.; Kollak, I.; Kiechle, T.; Ostermann, A.; Schnapp, A.; Kiesling, R.; Pieper, M.; Sieger, P.; Gantner, F. (2011). “Functional and Biochemical Rationales for the 24-Hour-Long Duration of Action of Olodaterol”. Journal of Pharmacology and Experimental Therapeutics 337 (3): 600–609. doi:10.1124/jpet.111.179259. PMID 21357659.
- Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine, C.; Büttner, F. H.; Schnapp, A.; Konetzki, I. (2010). “Discovery of olodaterol, a novel inhaled β2-adrenoceptor agonist with a 24h bronchodilatory efficacy”. Bioorganic & Medicinal Chemistry Letters 20 (4): 1410–1414. doi:10.1016/j.bmcl.2009.12.087. PMID 20096576.
- Joos G, Aumann JL, Coeck C, et al. ATS 2012 Abstract: Comparison of 24-Hour FEV1 Profile for Once-Daily versus Twice-Daily Treatment with Olodaterol, A Novel Long-Acting ß2-Agonist, in Patients with COPD[dead link]
- Van Noord, J. A.; Smeets, J. J.; Drenth, B. M.; Rascher, J.; Pivovarova, A.; Hamilton, A. L.; Cornelissen, P. J. G. (2011). “24-hour Bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD”. Pulmonary Pharmacology & Therapeutics 24 (6): 666–672. doi:10.1016/j.pupt.2011.07.006. PMID 21839850.
- van Noord JA, Korducki L, Hamilton AL and Koker P. Four Weeks Once Daily Treatment with BI 1744 CL, a Novel Long-Acting ß2-Agonist, is Effective in COPD Patients. Am. J. Respir. Crit. Care Med. 2009; 179: A6183[dead link]
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Hollis A (31 January 2013). “Panel Overwhelmingly Supports Boehringer COPD Drug Striverdi”. FDA News/Drug Industry Daily.
- “New once-daily Striverdi (olodaterol) Respimat gains approval in first EU countries”. Boehringer-Ingelheim. 18 October 2013.
External links
The active moiety olodaterol is a selective beta2-adrenergic bronchodilator. The drug substance, olodaterol hydrochloride, is chemically described as 2H-1,4- Benzoxazin-3H(4H)-one, 6-hydroxy-8-[(1R)-1-hydroxy-2-[[2-(4-methoxyphenyl)-1,1-dimethylethyl]-amino]ethyl]-, monohydrochloride. Olodaterol hydrochloride is a white to off-white powder that is sparingly-slightly soluble in water and slightly soluble in ethanol. The molecular weight is 422.9 g/mole (salt): 386.5 g/mole (base), and the molecular formula is C21H26N2O5 x HCl as a hydrochloride. The conversion factor from salt to free base is 1.094.
The structural formula is:
 |
The drug product, STRIVERDI RESPIMAT, is composed of a sterile, aqueous solution of olodaterol hydrochloride filled into a 4.5 mL plastic container crimped into an aluminum cylinder (STRIVERDI RESPIMAT cartridge) for use with the STRIVERDI RESPIMAT inhaler.
Excipients include water for injection, benzalkonium chloride, edetate disodium, and anhydrous citric acid. The STRIVERDI RESPIMAT cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler. The STRIVERDI RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow-moving aerosol cloud of medication from a metered volume of the drug solution. The STRIVERDI RESPIMAT inhaler has a yellow-colored cap.
When used with the STRIVERDI RESPIMAT inhaler, each cartridge containing a minimum of 4 grams of a sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (1 dose equals 2 actuations) from the STRIVERDI RESPIMAT inhaler delivers 5 mcg olodaterol in 22.1 mcL of solution from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
| WO2002030928A1 | 28 Sep 2001 | 11 Apr 2003 | Boehringer Ingelheim Pharma | Crystalline monohydrate, method for producing the same and the use thereof in the production of a medicament |
| WO2003000265A1 | 8 Jun 2002 | 3 Jan 2003 | Boehringer Ingelheim Pharma | Crystalline anticholinergic, method for its production, and use thereof in the production of a drug |
| WO2004045618A2 * | 11 Nov 2003 | 3 Jun 2004 | Boehringer Ingelheim Pharma | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| EP0073505A1 * | 28 Aug 1982 | 9 Mar 1983 | Boehringer Ingelheim Kg | Benzo-heterocycles |
| EP0321864A2 * | 15 Dec 1988 | 28 Jun 1989 | Boehringer Ingelheim Kg | Ammonium compounds, their preparation and use |
| US4460581 | 12 Oct 1982 | 17 Jul 1984 | Boehringer Ingelheim Kg | Antispasmodic agents, antiallergens |
| US4656168 * | 13 Oct 1983 | 7 Apr 1987 | Merck & Co., Inc. | Vision defects; adrenergic blocking and hypotensive agents |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com
PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF)

ATALUREN
PTC 124
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
| MF C15H9FN2O3 | ||
| Molecular Weight | 284.24 | |
| CAS Registry Number | 775304-57-9 |
PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF) – MarketWatch

Ataluren, formerly known as PTC124, is a small-molecular agent designed by PTC Therapeutics and sold under the trade nameTranslarna. It makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. There is ongoing debate over whether Ataluren is truly a functional drug (inducing codon read-through), or if it is nonfunctional, and the result was a false-positive hit from a biochemical screen based on luciferase.[1]
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[2][3] such as some people with cystic fibrosis and Duchenne muscular dystrophy. In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[4] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug. However, other phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[5] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[6]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[7]
Ataluren appears to be most effective for the stop codon ‘UGA’.[2]
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[8]

It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).


References
- Derek (2013-09-18). “The Arguing Over PTC124 and Duchenne Muscular Dystrophy. In the Pipeline:”. Pipeline.corante.com. Retrieved 2013-11-28.
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (May 2007). “PTC124 targets genetic disorders caused by nonsense mutations”. Nature 447 (7140): 87–91.doi:10.1038/nature05756. PMID 17450125.
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL (Apr 2007). “Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers”. Journal of clinical pharmacology 47 (4): 430–444. doi:10.1177/0091270006297140. PMID 17389552.
- “PTC THERAPEUTICS AND GENZYME CORPORATION ANNOUNCE PRELIMINARY RESULTS FROM THE PHASE 2B CLINICAL TRIAL OF ATALUREN FOR NONSENSE MUTATION DUCHENNE/BECKER MUSCULAR DYSTROPHY (NASDAQ:PTCT)”. Ptct.client.shareholder.com. Retrieved 2013-11-28.
- Wilschanski, M.; Miller, L. L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; Cohen-Cymberknoh, M.; Miller, N. L.; Reha, A.; Northcutt, V. J.; Hirawat, S.; Donnelly, K.; Elfring, G. L.; Ajayi, T.; Kerem, E. (2011). “Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis”. European Respiratory Journal 38 (1): 59–69. doi:10.1183/09031936.00120910. PMID 21233271. Sermet-Gaudelus, I.; Boeck, K. D.; Casimir, G. J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; Miller, N. L.; Constantine, S.; Reha, A.; Ajayi, T.; Elfring, G. L.; Miller, L. L. (November 2010). “Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis”. American Journal of Respiratory and Critical Care Medicine 182 (10): 1262–1272.doi:10.1164/rccm.201001-0137OC. PMID 20622033.
- “PTC Therapeutics Completes Enrollment of Phase 3 Trial of Ataluren in Patients with Cystic Fibrosis (NASDAQ:PTCT)”. Ptct.client.shareholder.com. 2010-12-21. Retrieved 2013-11-28.
- Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery medicine 15 (81): 127–133. PMID 23449115.
- http://www.marketwatch.com/story/ptc-therapeutics-receives-positive-opinion-from-chmp-for-translarna-ataluren-2014-05-23
External links
other sources
rINN: Ataluren
Other Names
PTC124®, 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
Pharmacological Information
Pharmacology Images

Ataluren Molecule

Web information on Ataluren
Relevant Clinical Literature
UK Guidance
Regulatory Literature
Other Literature
Orphan drug under investigation for treatment of genetic conditions where nonsense mutations result in premature termination of polypeptides. This drug, which is convenient to deliver orally, appears to allow ribosomal transcription ofRNA to continue past premature termination codon mutations with correct reading of the full normal transcript which then terminates at the proper stop codon. Problematically it has been postulated that assay artifact may have complicated evaluation of its efficacy which appears to be less than gentamicin.[1] Faults of this class in the transcription process are involved in several inherited diseases.
Some forms of cystic fibrosis and Duchenne muscular dystrophy are being targeted in the development stage of the drug.[2] Phase I and II trials are promising for cystic fibrosis.[3][4] In a mouse model of Duchenne muscular dystrophy, restoration of muscle function occurred.[5]
A potential issue is that there may be parts of the human genome whose optimal gene function through evolution has resulted from relatively recent in evolutionary terms insertion of a premature termination codon and so functional suboptimal transcripts of other proteins or functional RNAs might result.
References
- ↑ Roberts RG. A read-through drug put through its paces. PLoS biology. 2013; 11(6):e1001458.(Link to article – subscription may be required.)
- ↑ Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. Journal of clinical pharmacology. 2007 Apr; 47(4):430-44.(Link to article– subscription may be required.)
- ↑ Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, Elfring GL, Northcutt VJ, Miller LL, Kerem B, Wilschanski M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008 Aug 30; 372(9640):719-27.(Link to article – subscription may be required.)
- ↑ Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL. Ataluren (PTC124) Induces Cystic Fibrosis Transmembrane Conductance Regulator Protein Expression and Activity in Children with Nonsense Mutation Cystic Fibrosis. American journal of respiratory and critical care medicine. 2010 Nov 15; 182(10):1262-72.(Link to article – subscription may be required.)
- ↑ Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007 May 3; 447(7140):87-91.(Link to article – subscription may be required.)
old cut paste
A large-scale, multinational, phase 3 trial of the experimental drug ataluren has opened its first trial site, in Cincinnati, Ohio.
The trial is recruiting boys with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) caused by anonsense mutation — also known as a premature stop codon — in the dystrophin gene. This type of mutation causes cells to stop synthesizing a protein before the process is complete, resulting in a short, nonfunctional protein. Nonsense mutations are believed to cause DMD or BMD in approximately 10 to 15 percent of boys with these disorders.
Ataluren — sometimes referred to as a stop codon read-through drug — has the potential to overcome the effects of a nonsense mutation and allow functional dystrophin — the muscle protein that’s missing in Duchenne MD and deficient in Becker MD — to be produced.
The orally delivered drug is being developed by PTC Therapeutics, a South Plainfield, N.J., biotechnology company, to whichMDA gave a $1.5 million grant in 2005.
PTC124 has been developed by PTC Therapeutics.
India approved 26 drugs without clinical trials
New Delhi: Officials in the Indian health ministry have admitted that about 26 new drug molecules were given approval since 2010 without conducting any proper clinical trials on local population to test their safety and efficacy. Despite strict instructions by the parliamentary standing committee on health, so many new drugs have continued to make their way into the market.
19 August 2013 Officials in the Indian health ministry has accepted that about 26 new drugs were permitted for sale in the country without holding any clinical trials on Indian patients to test their safety and efficacy –
Read more at: http://www.biospectrumasia.com/biospectrum/news/193708/india-approved-26-drugs-clinical-trials#.UhHPwaI3CSo

DR A.M. CRASTO
Oramed Enrolls First Patient in its Phase 2a U.S. Oral Insulin Clinical Trial
![]()

Nadav Kidron
Marks initiation of Oramed’s first FDA trial for its flagship ORMD-0801 oral insulin product
JERUSALEM, July 8, 2013
Oramed Pharmaceuticals Inc. (NASDAQCM: ORMP) (http://www.oramed.com), a developer of oral drug delivery systems, announced today that the first patient has been enrolled in a Phase 2a trial of ORMD-0801, an orally ingestible insulin capsule, on patients with type 2 diabetes. The current trial is to be a randomized, double-blind study designed to assess the safety of ORMD-0801.
Read more at

In addition to ORMD-0801, Oramed is also developing an oral GLP-1 analog, known as exenatide, and a combination therapy of ORMD-0801 and exenatide.
GLP-1, or glucagon-like peptide-1, possesses a number of physiological properties that make it and its analogs the subject of intensive investigation as a potential treatment for diabetes. Among other things, it aids in the balance of blood sugar levels by decreasing glucose levels, especially after a meal; promotes weight loss; and does not cause hypoglycemia.
Sandoz launches Phase III clinical trial for biosimilar etanercept

Etanercept
is made from the combination of two naturally occurring soluble human 75-kilodalton TNF receptors linked to an Fc portion of an IgG1. The effect is an artificially engineered dimeric fusion protein.
Sandoz launches Phase III clinical trial for biosimilar etanercept
Trial expected to support registration in the U.S. and European Union
• Sandoz continues to advance biosimilar pipeline with seven Phase III trials across five molecules
• Global program underscores Sandoz’s leadership in biosimilarsHolzkirchen, Germany, June 24, 2013 – Sandoz, the global leader in biosimilars, announced it has initiated a major Phase III clinical trial with its biosimilar version of etanercept (Amgen’s Enbrel®).
Read more at
• Sandoz continues to advance biosimilar pipeline with seven Phase III trials across five molecules
• Global program underscores Sandoz’s leadership in biosimilarsHolzkirchen, Germany, June 24, 2013 – Sandoz, the global leader in biosimilars, announced it has initiated a major Phase III clinical trial with its biosimilar version of etanercept (Amgen’s Enbrel®).
Read more at
http://www.drugs.com/news/novartis-begins-enbrel-phase-iii-trial-45414.html
| Etanercept (trade name Enbrel) is a biopharmaceutical that treats autoimmune diseases by interfering with tumor necrosis factor (TNF; a soluble inflammatory cytokine) by acting as a TNF inhibitor. It has U.S. F.D.A. approval to treat rheumatoid, juvenile rheumatoid andpsoriatic arthritis, plaque psoriasis and ankylosing spondylitis. TNF-alpha is the “master regulator” of the inflammatory (immune) response in many organ systems. Autoimmune diseases are caused by an overactive immune response. Etanercept has the potential to treat these diseases by inhibiting TNF-alpha. Etanercept is a fusion protein produced by recombinant DNA. It fuses the TNF receptor to the constant end of the IgG1 antibody. First, the developers isolated the DNA sequence that codes the human gene for soluble TNF receptor 2, which is a receptor that binds to tumor necrosis factor-alpha. Second, they isolated the DNA sequence that codes the human gene for the Fc end of immunoglobulin G1 (IgG1). Third, they linked the DNA for TNF receptor 2 to the DNA for IgG1 Fc. Finally, they expressed the linked DNA to produce a protein that links the protein for TNF receptor 2 to the protein for IgG1 Fc. The prototypic fusion protein was first synthesized and shown to be highly active and unusually stable as a modality for blockade of TNF in vivo in the early 1990s by Bruce A. Beutler, an academic researcher then at the University of Texas Southwestern Medical Center at Dallas, and his colleagues.[2][3][4] These investigators also patented the protein, selling all rights to its use to Immunex, a biotechnology company that was acquired by Amgen in 2002. It is a large molecule, with a molecular weight of 150 kDa., that binds to TNFα and decreases its role in disorders involving excess inflammation in humans and other animals, including autoimmune diseases such as ankylosing spondylitis, juvenile rheumatoid arthritis, psoriasis, psoriatic arthritis, rheumatoid arthritis, and, potentially, in a variety of other disorders mediated by excess TNFα. In North America, etanercept is co-marketed by Amgen and Pfizer under the trade name Enbrel in two separate formulations, one in powder form, the other as a pre-mixed liquid. Wyeth is the sole marketer of Enbrel outside North America excluding Japan whereTakeda Pharmaceuticals markets the drug. Etanercept is an example of a protein-based drug created using the tools of biotechnologyand conceived through an understanding afforded by modern cell biology. |
Clinical trial starts in the UK to heal hearts with genetic virus

A group of UK heart failure patients have been enrolled on a new clinical trial to see if a genetically engineered virus can help heal their ailing hearts.
Researchers at Imperial College London will introduce a genetic virus into the heart muscles of the 200 participants, in a bid to reverse the heart muscle’s decline.
READ ALL AT
