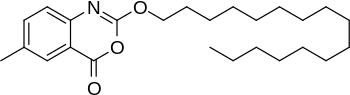
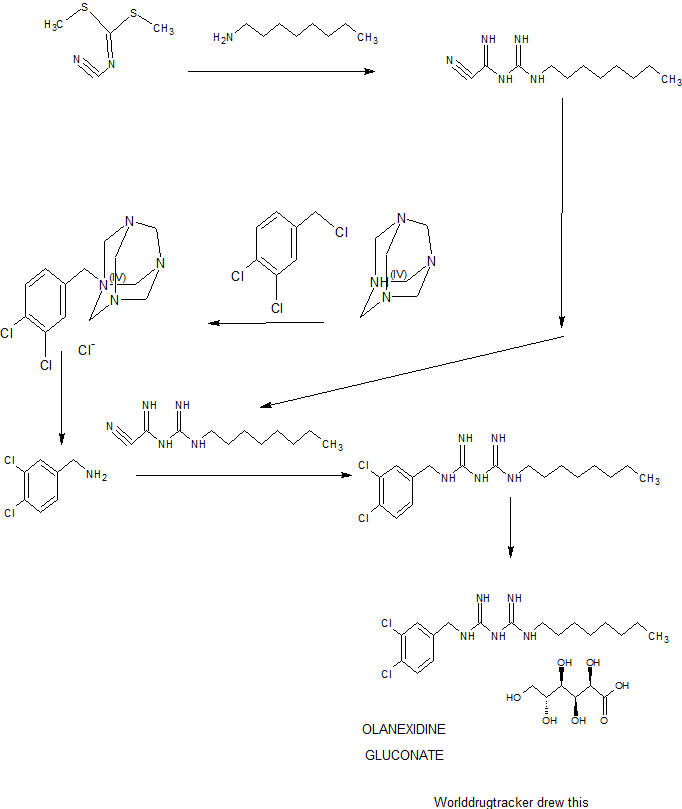
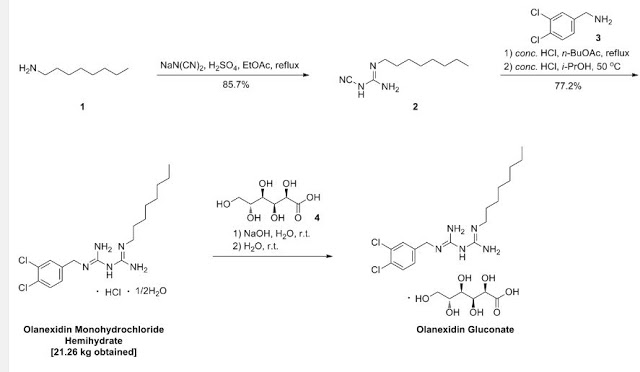
>Botulinum Toxin Type A Sequence
MPFVNKQFNYKDPVNGVDIAYIKIPNVGQMQPVKAFKIHNKIWVIPERDTFTNPEEGDLN
PPPEAKQVPVSYYDSTYLSTDNEKDNYLKGVTKLFERIYSTDLGRMLLTSIVRGIPFWGG
STIDTELKVIDTNCINVIQPDGSYRSEELNLVIIGPSADIIQFECKSFGHEVLNLTRNGY
GSTQYIRFSPDFTFGFEESLEVDTNPLLGAGKFATDPAVTLAHELIHAGHRLYGIAINPN
RVFKVNTNAYYEMSGLEVSFEELRTFGGHDAKFIDSLQENEFRLYYYNKFKDIASTLNKA
KSIVGTTASLQYMKNVFKEKYLLSEDTSGKFSVDKLKFDKLYKMLTEIYTEDNFVKFFKV
LNRKTYLNFDKAVFKINIVPKVNYTIYDGFNLRNTNLAANFNGQNTEINNMNFTKLKNFT
GLFEFYKLLCVRGIITSKTKSLDKGYNKALNDLCIKVNNWDLFFSPSEDNFTNDLNKGEE
ITSDTNIEAAEENISLDLIQQYYLTFNFDNEPENISIENLSSDIIGQLELMPNIERFPNG
KKYELDKYTMFHYLRAQEFEHGKSRIALTNSVNEALLNPSRVYTFFSSDYVKKVNKATEA
AMFLGWVEQLVYDFTDETSEVSTTDKIADITIIIPYIGPALNIGNMLYKDDFVGALIFSG
AVILLEFIPEIAIPVLGTFALVSYIANKVLTVQTIDNALSKRNEKWDEVYKYIVTNWLAK
VNTQIDLIRKKMKEALENQAEATKAIINYQYNQYTEEEKNNINFNIDDLSSKLNESINKA
MININKFLNQCSVSYLMNSMIPYGVKRLEDFDASLKDALLKYIYDNRGTLIGQVDRLKDK
VNNTLSTDIPFQLSKYVDNQRLLSTFTEYIKNIINTSILNLRYESNHLIDLSRYASKINI
GSKVNFDPIDKNQIQLFNLESSKIEVILKNAIVYNSMYENFSTSFWIRIPKYFNSISLNN
EYTIINCMENNSGWKVSLNYGEIIWTLQDTQEIKQRVVFKYSQMINISDYINRWIFVTIT
NNRLNNSKIYINGRLIDQKPISNLGNIHASNNIMFKLDGCRDTHRYIWIKYFNLFDKELN
EKEIKDLYDNQSNSGILKDFWGDYLQYDKPYYMLNLYDPNKYVDVNNVGIRGYMYLKGPR
GSVMTTNIYLNSSLYRGTKFIIKKYASGNKDNIVRNNDRVYINVVVKNKEYRLATNASQA
GVEKILSALEIPDVGNLSQVVVMKSKNDQGITNKCKMNLQDNNGNDIGFIGFHQFNNIAK
LVASNWYNRQIERSSRTLGCSWEFIPVDDGWGERPL
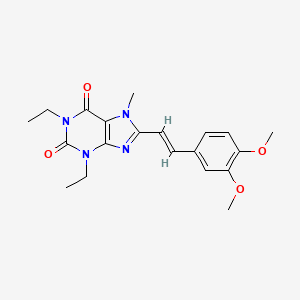
Prabotulinumtoxin A
プラボツリナムトキシンA;

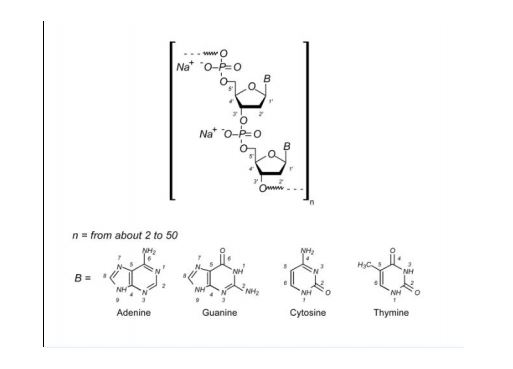
| Formula |
C6760H10447N1743O2010S32
|
| CAS |
93384-43-1
|
| Mol weight |
149320.8333
|
AGN 191622 / ANT-1207 / ANT-1401 / ANT-1403 / NT 201
-
-
-
APPROVED , FDA 2019, Jeuveau, 2019/2/1

- Purified botulinum toxin from Clostridium botulinum, purified from culture via dialysis and acid precipitation.
- Originator Daewoong Pharmaceutical
- Developer Daewoong Pharmaceutical; Evolus
- Class Analgesics; Antidepressants; Antimigraines; Antispasmodics; Bacterial proteins; Bacterial toxins; Botulinum toxins; Eye disorder therapies; Muscle relaxants; Skin disorder therapies; Urologics
- Mechanism of Action Acetylcholine inhibitors; Glutamate antagonists; Membrane transport protein modulators; Neuromuscular blocking agents
- Phase III Muscle spasticity
- Phase II/III Blepharospasm; Facial wrinkles
- 27 Feb 2019 Evolus plans to launch prabotulinumtoxin A for Glabellar lines in USA (IM)
- 01 Feb 2019 Registered for Glabellar lines in USA (IM)
- 26 Nov 2018 Daewoong Pharmaceutical expects to launch prabotulinumtoxin A for Glabellar lines in eight Middle Eastern countries, including UAE and Kuwait in 2018 (Parenteral)
- AbobotulinumtoxinA
- Botulinum A neurotoxin
- Botulinum toxin A
- Botulinum toxin type A
- BTX-A
- Evabotulinumtoxina
- IncobotulinumtoxinA
- OnabotulinumtoxinA
- Prabotulinumtoxin A
- Toxina botulínica A
- Toxine botulinique A
For the treatment of cervical dystonia in adults to decrease the severity of abnormal head position and neck pain associated with cervical dystonia. Also for the treatment of severe primary axillary hyperhidrosis that is inadequately managed with topical agents and for the treatment of strabismus and blepharospasm associated with dystonia, including benign essential blepharospasm or VII nerve disorders in patients 12 years of age and above. Also used cosmetically to temporarily improve the appearance of moderate-to-severe frown lines between the eyebrows (glabellar lines) as well as for the treatment of excessive underarm sweating.
Botulinum toxin (BTX) is a neurotoxic protein produced by the bacterium Clostridium botulinum and related species.[1] It prevents the release of the neurotransmitter acetylcholine from axon endings at the neuromuscular junction and thus causes flaccid paralysis.[2]Infection with the bacterium causes the disease botulism. The toxin is also used commercially in medicine, cosmetics and research.
Botulinum is the most acutely lethal toxin known, with an estimated human median lethal dose (LD50) of 1.3–2.1 ng/kg intravenously or intramuscularly and 10–13 ng/kg when inhaled.[3][clarification needed]
There are eight types of botulinum toxin, named type A–H. Types A and B are capable of causing disease in humans, and are also used commercially and medically.[4] Types C–G are less common; types E and F can cause disease in humans, while the other types cause disease in other animals.[5] Type H is considered the deadliest substance in the world – an injection of only 2 ng can cause death to an adult.[6] Botulinum toxin types A and B are used in medicine to treat various muscle spasms and diseases characterized by overactive muscle. Commercial forms are marketed under the brand names Botox and Dysport, among others.[7][8]
Medical uses
Botulinum toxin is used to treat a number of problems.
Muscle spasticity
Botulinum toxin is used to treat a number of disorders characterized by overactive muscle movement, including post-stroke spasticity, post-spinal cord injury spasticity, spasms of the head and neck,[9] eyelid,[10] vagina,[11] limbs, jaw, and vocal cords.[12] Similarly, botulinum toxin is used to relax clenching of muscles, including those of the oesophagus,[13] jaw,[14]lower urinary tract and bladder,[15] or clenching of the anus which can exacerbate anal fissure.[16] It may also be used for improper eye alignment.[17] Botulinum toxin appears to be effective for refractory overactive bladder.[18]
Other muscle disorders
Strabismus is caused by imbalances in the actions of muscles that rotate the eyes, and can sometimes be relieved by weakening a muscle that pulls too strongly, or pulls against one that has been weakened by disease or trauma. Muscles weakened by toxin injection recover from paralysis after several months, so it might seem that injection would then need to be repeated. However, muscles adapt to the lengths at which they are chronically held,[19] so that if a paralyzed muscle is stretched by its antagonist, it grows longer, while the antagonist shortens, yielding a permanent effect. If there is good binocular vision, the brain mechanism of motor fusion, which aligns the eyes on a target visible to both, can stabilize the corrected alignment.
In January 2014, botulinum toxin was approved by UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) for treatment of restricted ankle motion due to lower limb spasticity associated with stroke in adults.[20]
On July 29, 2016, Food and Drug Administration (FDA), of the United States of America approved abobotulinumtoxinA for injection for the treatment of lower limb spasticity in pediatric patients two years of age and older.[21] AbobotulinumtoxinA is the first and only FDA-approved botulinum toxin for the treatment of pediatric lower limb spasticity. In the United States of America, the FDA approves the text of the labels of prescription medicines. The FDA approves which medical conditions the drug manufacturer may sell the drug for. However, those approved by the FDA to prescribe these drugs may freely prescribe them for any condition they wish, called off-label use. Botulinum toxins have been used off-label for several pediatric conditions, including infantile esotropia.[22]
Excessive Sweating
Khalaf Bushara and David Park were the first to demonstrate a nonmuscular use of BTX-A while treating patients with hemifacial spasm in England in 1993, showing that botulinum toxin injections inhibit sweating, and so are useful in treating hyperhidrosis (excessive sweating).[23] BTX-A has since been approved for the treatment of severe primary axillary hyperhidrosis (excessive underarm sweating of unknown cause), which cannot be managed by topical agents.[12][24]
Migraine
In 2010, the FDA approved intramuscular botulinum toxin injections for prophylactic treatment of chronic migraine headache.[25]
Cosmetics
Botulinum toxin injected in human face
In cosmetic applications, botulinum toxin is considered safe and effective for reduction of facial wrinkles, especially in the uppermost third of the face.[26] Injection of botulinum toxin into the muscles under facial wrinkles causes relaxation of those muscles, resulting in the smoothing of the overlying skin.[26] Smoothing of wrinkles is usually visible three days after treatment and is maximally visible two weeks following injection.[26] The treated muscles gradually regain function, and generally return to their former appearance three to four months after treatment.[26] Muscles can be treated repeatedly to maintain the smoothed appearance.[26]
Other
Botulinum toxin is also used to treat disorders of hyperactive nerves including excessive sweating,[24] neuropathic pain,[27] and some allergysymptoms.[12] In addition to these uses, botulinum toxin is being evaluated for use in treating chronic pain.[28]
Side effects
While botulinum toxin is generally considered safe in a clinical setting, there can be serious side effects from its use. Most commonly, botulinum toxin can be injected into the wrong muscle group or spread from the injection site, causing paralysis of unintended muscles.
Side effects from cosmetic use generally result from unintended paralysis of facial muscles. These include partial facial paralysis, muscle weakness, and trouble swallowing. Side effects are not limited to direct paralysis however, and can also include headaches, flu-like symptoms, and allergic reactions.[29] Just as cosmetic treatments only last a number of months, paralysis side-effects can have the same durations.[citation needed] At least in some cases, these effects are reported to dissipate in the weeks after treatment.[citation needed] Bruising at the site of injection is not a side effect of the toxin but rather of the mode of administration, and is reported as preventable if the clinician applies pressure to the injection site; when it occurs, it is reported in specific cases to last 7–11 days.[citation needed] When injecting the masseter muscle of the jaw, loss of muscle function can result in a loss or reduction of power to chew solid foods.[29]
Side effects from therapeutic use can be much more varied depending on the location of injection and the dose of toxin injected. In general, side effects from therapeutic use can be more serious than those that arise during cosmetic use. These can arise from paralysis of critical muscle groups and can include arrhythmia, heart attack, and in some cases seizures, respiratory arrest, and death.[29] Additionally, side effects which are common in cosmetic use are also common in therapeutic use, including trouble swallowing, muscle weakness, allergic reactions, and flu-like syndromes.[29]
In response to the occurrence of these side effects, in 2008 the U.S. Food and Drug Administration notified the public of the potential dangers of the botulinum toxin as a therapeutic. Namely, they warned that the toxin can spread to areas distant from the site of injection and paralyze unintended muscle groups, especially when used for treating muscle spasticity in children treated for cerebral palsy.[30] In 2009, the FDA announced that boxed warnings would be added to available botulinum toxin products, warning of their ability to spread from the injection site.[31] Additionally, the FDA announced name changes to several botulinum toxin products, meant to emphasize that the products are not interchangeable and require different doses for proper use. Botox and Botox Cosmetic were renamed onabotulinumtoxinA, Myobloc was renamed rimabotulinumtoxinB, and Dysport name renamed abobotulinumtoxinA.[31] In conjunction with this, the FDA issued a communication to health care professionals reiterating the new drug names and the approved uses for each.[32] A similar warning was issued by Health Canada in 2009, warning that botulinum toxin products can spread to other parts of the body.[33]
Role in disease
Botulinum toxin produced by Clostridium botulinum is the cause of botulism.[10] Humans most commonly ingest the toxin from eating improperly-canned foods in which C. botulinumhas grown. However, the toxin can also be introduced through an infected wound. In infants, the bacteria can sometimes grow in the intestines and produce botulinum toxin within the intestine and can cause a condition known as floppy baby syndrome.[34] In all cases, the toxin can then spread, blocking nerves and muscle function. In severe cases, the toxin can block nerves controlling the respiratory system or heart, resulting in death.[1] Botulism can be difficult to diagnose, as it may appear similar to diseases such as Guillain–Barré syndrome, myasthenia gravis, and stroke. Other tests, such as brain scan and spinal fluid examination, may help to rule out other causes. If the symptoms of botulism are diagnosed early, various treatments can be administered. In an effort to remove contaminated food which remains in the gut, enemas or induced vomiting may be used.[35] For wound infections, infected material may be removed surgically.[35] Botulinum antitoxin is available and may be used to prevent the worsening of symptoms, though it will not reverse existing nerve damage. In severe cases, mechanical respiration may be used to support patients suffering from respiratory failure.[35] The nerve damage heals over time, generally over weeks to months.[5] With proper treatment, the case fatality rate for botulinum poisoning can be greatly reduced.[35]
Two preparations of botulinum antitoxins are available for treatment of botulism. Trivalent (A,B,E) botulinum antitoxin is derived from equine sources using whole antibodies. The second antitoxin is Heptavalent (A,B,C,D,E,F,G) botulinum antitoxin, which is derived from equine antibodies which have been altered to make them less immunogenic. This antitoxin is effective against all known strains of botulism.
Mechanism of action

Target molecules of botulinum neurotoxin (abbreviated BoNT) and tetanus neurotoxin (TeNT), toxins acting inside the axon terminal.[36]
Botulinum toxin exerts its effect by cleaving key proteins required for nerve activation. First, the toxin binds specifically to nerves which use the neurotransmitter acetylcholine. Once bound to the nerve terminal, the neuron takes up the toxin into a vesicle by receptor-mediated endocytosis.[37] As the vesicle moves farther into the cell, it acidifies, activating a portion of the toxin which triggers it to push across the vesicle membrane and into the cell cytoplasm.[1] Once inside the cytoplasm, the toxin cleaves SNARE proteins, meaning that the acetylcholine vesicles can’t bind to the intracellular cell membrane,[37] preventing the cell from releasing vesicles of neurotransmitter. This stops nerve signaling, leading to paralysis.[1]
The toxin itself is released from the bacterium as a single chain, then becomes activated when cleaved by its own proteases.[12] The active form consists of a two-chain protein composed of a 100-kDa heavy chain polypeptide joined via disulfide bond to a 50-kDa light chain polypeptide.[38] The heavy chain contains domains with several functions: it has the domain responsible for binding specifically to presynaptic nerve terminals, as well as the domain responsible for mediating translocation of the light chain into the cell cytoplasm as the vacuole acidifies.[1][38] The light chain is a zinc metalloprotease and is the active part of the toxin. It is translocated into the host cell cytoplasm where it cleaves the host protein SNAP-25, a member of the SNARE protein family which is responsible for fusion. The cleaved SNAP-25 is unable to mediate fusion of vesicles with the host cell membrane, thus preventing the release of the neurotransmitteracetylcholine from axon endings.[1] This blockage is slowly reversed as the toxin loses activity and the SNARE proteins are slowly regenerated by the affected cell.[1]
The seven toxin types (A-G) have different tertiary structures and sequence differences.[38][39] While the different toxin types all target members of the SNARE family, different toxin types target different SNARE family members.[36] The A, B, and E serotypes cause human botulism, with the activities of types A and B enduring longest in vivo (from several weeks to months).[38]
History
In 1820, Justinus Kerner, a small-town German medical officer and romantic poet, gave the first complete description of clinical botulism based on extensive clinical observations of so-called “sausage poisoning”.[40] Following experiments on animals and on himself, he concluded that the toxin acts by interrupting signal transmission in the somatic and autonomic motor systems, without affecting sensory signals or mental functions. He observed that the toxin develops under anaerobic conditions, and can be lethal in minute doses.[41] His prescience in suggesting that the toxin might be used therapeutically earned him recognition as the pioneer of modern botulinum toxin therapy.[42]
In 1895 (seventy-five years later), Émile van Ermengem, professor of bacteriology and a student of Robert Koch, correctly described Clostridium botulinum as the bacterial source of the toxin. Thirty-four attendees at a funeral were poisoned by eating partially salted ham, an extract of which was found to cause botulism-like paralysis in laboratory animals. Van Ermengem isolated and grew the bacterium, and described its toxin,[43] which was later purified by P Tessmer Snipe and Hermann Sommer.[44]
Food canning
Over the next three decades, 1895-1925, as food canning was approaching a billion-dollar-a-year industry, botulism was becoming a public health hazard. Karl Friedrich Meyer, a prodigiously productive Swiss-American veterinary scientist created a center at the Hooper Foundation in San Francisco, where he developed techniques for growing the organism and extracting the toxin, and conversely, for preventing organism growth and toxin production, and inactivating the toxin by heating. The California canning industry was thereby preserved.
World War II
With the outbreak of World War II, weaponization of botulinum toxin was investigated at Fort Detrick in Maryland. Carl Lamanna and James Duff[45] developed the concentration and crystallization techniques that Edward J. Schantz used to create the first clinical product. When the Army’s Chemical Corps was disbanded, Schantz moved to the Food Research Institute in Wisconsin, where he manufactured toxin for experimental use and generously provided it to the academic community.
The mechanism of botulinum toxin action – blocking the release from nerve endings of the neurotransmitter acetylcholine – was elucidated in the mid-1900s,[46] and remains an important research topic. Nearly all toxin treatments are based on this effect in various body tissues.
Strabismus
Ophthalmologists specializing in eye muscle disorders (strabismus) had developed the method of EMG-guided injection (using the electromyogram, the electrical signal from an activated muscle, to guide injection) of local anesthetics as a diagnostic technique for evaluating an individual muscle’s contribution to an eye movement.[47] Because strabismus surgery frequently needed repeating, a search was undertaken for non-surgical, injection treatments using various anesthetics, alcohols, enzymes, enzyme blockers, and snake neurotoxins. Finally, inspired by Daniel Drachman’s work with chicks at Johns Hopkins,[48] Alan B. Scott and colleagues injected botulinum toxin into monkey extraocular muscles.[49]The result was remarkable: a few picograms induced paralysis that was confined to the target muscle, long in duration, and without side-effects.
After working out techniques for freeze-drying, buffering with albumin, and assuring sterility, potency, and safety, Scott applied to the FDA for investigational drug use, and began manufacturing botulinum type A neurotoxin in his San Francisco lab. He injected the first strabismus patients in 1977, reported its clinical utility in 1980,[50] and had soon trained hundreds of ophthalmologists in EMG-guided injection of the drug he named Oculinum (“eye aligner”).
In 1986, Oculinum Inc, Scott’s micromanufacturer and distributor of botulinum toxin, was unable to obtain product liability insurance, and could no longer supply the drug. As supplies became exhausted, patients who had come to rely on periodic injections became desperate. For 4 months, as liability issues were resolved, American blepharospasm patients traveled to Canadian eye centers for their injections.[51]
Based on data from thousands of patients collected by 240 investigators, Allergan received FDA approval in 1989 to market Oculinum for clinical use in the United States to treat adult strabismus and blepharospasm, using the trademark Botox.[52] This was under the 1983 US Orphan Drug Act.[53]
Cosmetics
Richard Clark, a plastic surgeon from Sacramento (CA), was the first to document a cosmetic use for botulinum toxin.[54] He treated forehead asymmetry caused by left sided forehead nerve paralysis that occurred during a cosmetic facelift. Since the injured nerve could possibly regenerate by 24 months, a two-year waiting period was necessary before definitive surgical treatment could be done. Clark realized that botulinum toxin, which had been previously used only for cross eyed babies and facial tics, could also be injected to smooth the wrinkles of the right forehead to match her paralyzed left. He received FDA approval for this cosmetic application of the toxin and successfully treated the person and published the case study in 1989.[54]
Marrying ophthalmology to dermatology, Jean and Alistair Carruthers observed that blepharospasm patients who received injections around the eyes and upper face also enjoyed diminished facial glabellar lines (“frown lines” between the eyebrows), thereby initiating the highly-popular cosmetic use of the toxin.[55] Brin, and a group at Columbia University under Monte Keen made similar reports.[56] In 2002, following clinical trials, the FDA approved Botox Cosmetic, botulinum A toxin to temporarily improve the appearance of moderate-to-severe glabellar lines.[57] The FDA approved a fully in vitro assay for use in the stability and potency testing of Botox in response to increasing public concern that LD50testing was required for each batch sold in the market.[58][59]
Chronic pain
William J. Binder reported in 2000 that patients who had cosmetic injections around the face reported relief from chronic headache.[60] This was initially thought to be an indirect effect of reduced muscle tension, but it is now known that the toxin inhibits release of peripheral nociceptive neurotransmitters, suppressing the central pain processing systems responsible for migraine headache.[61][62]
Society and culture
Economics
As of 2013, botulinum toxin injections are the most common cosmetic operation, with 6.3 million procedures in the United States, according to the American Society of Plastic Surgeons. Qualifications for Botox injectors vary by county, state and country. Botox cosmetic providers include dermatologists, plastic surgeons, aesthetic spa physicians, dentists, nurse practitioners, nurses and physician assistants.
The global market for botulinum toxin products, driven by their cosmetic applications, is forecast to reach $2.9 billion by 2018. The facial aesthetics market, of which they are a component, is forecast to reach $4.7 billion ($2 billion in the U.S.) in the same timeframe.[63]
Bioterrorism
Botulinum toxin has been recognized as a potential agent for use in bioterrorism.[64] It can be absorbed through the eyes, mucous membranes, respiratory tract, or non-intact skin.[65]
The effects of botulinum toxin are different from those of nerve agents involved insofar in that botulism symptoms develop relatively slowly (over several days), while nerve agent effects are generally much more rapid and can be instantaneous.[citation needed] Evidence suggests that nerve exposure (simulated by injection of atropine and pralidoxime) will increase mortality by enhancing botulinum toxin’s mechanism of toxicity.[citation needed]
With regard to detection, current protocols using NBC detection equipment (such as M-8 paper or the ICAM) will not indicate a “positive” when samples containing botulinum toxin are tested.[citation needed] To confirm a diagnosis of botulinum toxin poisoning, therapeutically or to provide evidence in death investigations, botulinum toxin may be quantitated by immunoassay of human biological fluids; serum levels of 12–24 mouse LD50 units per milliliter have been detected in poisoned patients.[66]
The Japanese doomsday cult Aum Shinrikyo produced botulinum toxin and spread it as an aerosol in downtown Tokyo during the 1990s, but the attacks caused no fatalities.[67]
During the early 1980s, the German and French newspapers reported that the police had raided a Baader-Meinhof gang safe house in Paris and had found a makeshift laboratory that contained flasks full of Clostridium botulinum, which makes botulinum toxin. Their reports were later found to be incorrect; no such lab was ever found.[68]
Brand names
Botulinum toxin A is marketed under the brand names Botox and Xeomin. Botulinum toxin B is marketed under the brand name Myobloc.
In the United States, botulinum toxin products are manufactured by a variety of companies, for both therapeutic and cosmetic use. A U.S. supplier reported in its company materials in 2011 that it could “supply the world’s requirements for 25 indications approved by Government agencies around the world” with less than one gram of raw botulinum toxin.[69]Myobloc or Neurobloc, a botulinum toxin type B product, is produced by Solstice Neurosciences, a subsidiary of US WorldMeds. AbobotulinumtoxinA), a therapeutic formulation of the type A toxin manufactured by Galderma in the United Kingdom, is licensed for the treatment of focal dystonias and certain cosmetic uses in the U.S. and other countries.[32]
Besides the three primary U.S. manufacturers, there are numerous other botulinum toxin producers. Xeomin, manufactured in Germany by Merz, is also available for both therapeutic and cosmetic use in the U.S.[70] Lanzhou Institute of Biological Products in China manufactures a BTX-A product; as of 2014 it was the only BTX-A approved in China.[70] BTX-A is also sold as Lantox and Prosigne on the global market.[71] Neuronox, a BTX-A product, was introduced by Medy-Tox Inc. of South Korea in 2009;[72]
Toxin production
Botulism toxins are produced by bacteria of the genus Clostridium, namely Clostridium botulinum, C. butyricum, C. baratii and C. argentinense,[73] which are widely distributed, including in soil and dust. As well, the bacteria can be found inside homes on floors, carpet, and countertops even after cleaning.[citation needed] Some food products such as honey can contain amounts of the bacteria.[citation needed]
Food-borne botulism results, indirectly, from ingestion of food contaminated with Clostridium spores, where exposure to an anaerobic environment allows the spores to germinate, after which the bacteria can multiply and produce toxin.[citation needed] Critically, it is ingestion of toxin rather than spores or vegetative bacteria that causes botulism.[citation needed]Botulism is nevertheless known to be transmitted through canned foods not cooked correctly before canning or after can opening, and so is preventable.[citation needed] Infant botulism cases arise chiefly as a result of environmental exposure and are therefore more difficult to prevent.[citation needed] Infant botulism arising from consumption of honey can be prevented by eliminating honey from diets of children less than 12 months old.[74]
Organism and toxin susceptibilities
Proper refrigeration at temperatures below 3 °C (38 °F) retards the growth of Clostridium botulinum. The organism is also susceptible to high salt, high oxygen, and low pH levels.[5]The toxin itself is rapidly destroyed by heat, such as in thorough cooking.[75] The spores that produce the toxin are heat-tolerant and will survive boiling water for an extended period of time.[76]
The botulinum toxin is denatured and thus deactivated at temperatures greater than 80 °C (176 °F).[77] As a zinc metalloprotease (see below), the toxin’s activity is also susceptible, post-exposure, to inhibition by protease inhibitors, e.g., zinc-coordinating hydroxamates.[38][78]
Research
Blepharospasm and strabismus
University-based ophthalmologists in the USA and Canada further refined the use of botulinum toxin as a therapeutic agent. By 1985, a scientific protocol of injection sites and dosage had been empirically determined for treatment of blepharospasm and strabismus.[79] Side effects in treatment of this condition were deemed to be rare, mild and treatable.[80]The beneficial effects of the injection lasted only 4–6 months. Thus, blepharospasm patients required re-injection two or three times a year.
In 1986, Scott’s micromanufacturer and distributor of Botox was no longer able to supply the drug because of an inability to obtain product liability insurance. Patients became desperate, as supplies of Botox were gradually consumed, forcing him to abandon patients who would have been due for their next injection. For a period of four months, American blepharospasm patients had to arrange to have their injections performed by participating doctors at Canadian eye centers until the liability issues could be resolved.[51]
In December 1989, Botox was approved by the US Food and Drug Administration (FDA) for the treatment of strabismus, blepharospasm, and hemifacial spasm in patients over 12 years old.[52]
Botox has not been approved for any pediatric use.[32] It has, however, been used off-label by physicians for several conditions. including spastic conditions in pediatric patients with cerebral palsy, a therapeutic course that has resulted in patient deaths.[32] In the case of treatment of infantile esotropia in patients younger than 12 years of age, several studies have yielded differing results.[22][better source needed]
Cosmetic
The cosmetic effect of BTX-A on wrinkles was originally documented by a plastic surgeon from Sacramento, California, Richard Clark, and published in the journal Plastic and Reconstructive Surgery in 1989.[54] Canadian husband and wife ophthalmologist and dermatologist physicians, JD and JA Carruthers, were the first to publish a study on BTX-A for the treatment of glabellar frown lines in 1992.[55] Similar effects had reportedly been observed by a number of independent groups (Brin, and the Columbia University group under Monte Keen.[56]) After formal trials, on April 12, 2002, the FDA announced regulatory approval of botulinum toxin type A (Botox Cosmetic) to temporarily improve the appearance of moderate-to-severe frown lines between the eyebrows (glabellar lines).[57] Subsequently, cosmetic use of botulinum toxin type A has become widespread.[81] The results of Botox Cosmetic can last up to four months and may vary with each patient.[82] The US Food and Drug Administration approved an alternative product-safety testing method in response to increasing public concern that LD50 testing was required for each batch sold in the market.[58][59]
BTX-A has also been used in the treatment of gummy smiles,[83][84] the material is injected into the hyperactive muscles of upper lip, which causes a reduction in the upward movement of lip thus resulting in a smile with a less exposure of gingiva.[85] Botox is usually injected in the three lip elevator muscles that converge on the lateral side of the ala of the nose; the levator labii superioris (LLS), the levator labii superioris alaeque nasi muscle (LLSAN), and the zygomaticus minor (ZMi).[86][87]
Upper motor neuron syndrome
BTX-A is now a common treatment for muscles affected by the upper motor neuron syndrome (UMNS), such as cerebral palsy, for muscles with an impaired ability to effectively lengthen. Muscles affected by UMNS frequently are limited by weakness, loss of reciprocal inhibition, decreased movement control and hypertonicity (including spasticity). In January 2014, Botulinum toxin was approved by UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) for the treatment of ankle disability due to lower limb spasticity associated with stroke in adults.[20] Joint motion may be restricted by severe muscle imbalance related to the syndrome, when some muscles are markedly hypertonic, and lack effective active lengthening. Injecting an overactive muscle to decrease its level of contraction can allow improved reciprocal motion, so improved ability to move and exercise.
Sweating
Khalaf Bushara and David Park were the first to demonstrate a nonmuscular use of BTX-A while treating patients with hemifacial spasm in England in 1993, showing that botulinum toxin injections inhibit sweating, and so are useful in treating hyperhidrosis (excessive sweating).[23] BTX-A has since been approved for the treatment of severe primary axillary hyperhidrosis (excessive underarm sweating of unknown cause), which cannot be managed by topical agents.[12][24]
Cervical dystonia
BTX-A is commonly used to treat cervical dystonia, but it can become ineffective after a time. Botulinum toxin type B (BTX-B) received FDA approval for treatment of cervical dystonia on December 21, 2000. Trade names for BTX-B are Myobloc in the United States, and Neurobloc in the European Union.[70]
Chronic migraine
Onabotulinumtoxin A (trade name Botox) received FDA approval for treatment of chronic migraines on October 15, 2010. The toxin is injected into the head and neck to treat these chronic headaches. Approval followed evidence presented to the agency from two studies funded by Allergan showing a very slight improvement in incidence of chronic migraines for migraine sufferers undergoing the Botox treatment.[88][89]
Since then, several randomized control trials have shown botulinum toxin type A to improve headache symptoms and quality of life when used prophylactically for patients with chronic migraine[90] who exhibit headache characteristics consistent with: pressure perceived from outside source, shorter total duration of chronic migraines (<30 years), “detoxification” of patients with coexisting chronic daily headache due to medication overuse, and no current history of other preventive headache medications.[91]
Depression
A few small trials have found benefits in people with depression.[92][93]
Premature ejaculation
The drug is under development for the treatment of premature ejaculation.[93]
References
- ^ Jump up to:a b c d e f g Montecucco C, Molgó J (June 2005). “Botulinal neurotoxins: revival of an old killer”. Current Opinion in Pharmacology. 5 (3): 274–79. doi:10.1016/j.coph.2004.12.006. PMID 15907915.
- ^ Figgitt, David P.; Noble, Stuart (2002). “Botulinum Toxin B”. Drugs. 62 (4): 705–722. doi:10.2165/00003495-200262040-00011. PMID 11893235.
- ^ Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K (February 2001). “Botulinum toxin as a biological weapon: medical and public health management”. JAMA. 285 (8): 1059–70. doi:10.1001/jama.285.8.1059. PMID 11209178.
- ^ American Society of Health-System Pharmacists (October 27, 2011). “Botulinum Toxin Type A”. drugs.com. Retrieved 4 March 2015.
- ^ Jump up to:a b c “Fact Sheet: Botulism”. World Health Organization. Retrieved 4 October 2016.
- ^ Weller C (15 October 2013). “New Botulinum Toxin Deemed Deadliest Substance Ever: Sniffing 13-Billionths of a Gram Can Kill”. Medical Daily.
- ^ Egan M (17 November 2014). “Botox maker bought for $66 billion in biggest deal of 2014”. cnn.com.
- ^ “Actavis plc is now Allergan plc”. Allergan. 15 June 2015. Retrieved 16 June 2015.
- ^ “Cervical dystonia”. Mayo Clinic. 2014-01-28. Retrieved 2015-10-14.
- ^ Jump up to:a b Shukla HD, Sharma SK (2005). “Clostridium botulinum: a bug with beauty and weapon”. Critical Reviews in Microbiology. 31 (1): 11–18. doi:10.1080/10408410590912952. PMID 15839401.
- ^ Pacik PT (December 2009). “Botox treatment for vaginismus”. Plastic and Reconstructive Surgery. 124 (6): 455e–56e. doi:10.1097/PRS.0b013e3181bf7f11. PMID 19952618.
- ^ Jump up to:a b c d e Felber ES (October 2006). “Botulinum toxin in primary care medicine”. The Journal of the American Osteopathic Association. 106 (10): 609–14. PMID 17122031.
- ^ Stavropoulos SN, Friedel D, Modayil R, Iqbal S, Grendell JH (March 2013). “Endoscopic approaches to treatment of achalasia”. Therapeutic Advances in Gastroenterology. 6 (2): 115–35. doi:10.1177/1756283X12468039. PMC 3589133. PMID 23503707.
- ^ Long H, Liao Z, Wang Y, Liao L, Lai W (February 2012). “Efficacy of botulinum toxins on bruxism: an evidence-based review”. International Dental Journal. 62 (1): 1–5. doi:10.1111/j.1875-595X.2011.00085.x. PMID 22251031.
- ^ Mangera A, Andersson KE, Apostolidis A, Chapple C, Dasgupta P, Giannantoni A, Gravas S, Madersbacher S (October 2011). “Contemporary management of lower urinary tract disease with botulinum toxin A: a systematic review of botox (onabotulinumtoxinA) and dysport (abobotulinumtoxinA)”. European Urology. 60 (4): 784–95. doi:10.1016/j.eururo.2011.07.001. PMID 21782318.
- ^ Trzciński R, Dziki A, Tchórzewski M (2002). “Injections of botulinum A toxin for the treatment of anal fissures”. The European Journal of Surgery = Acta Chirurgica. 168 (12): 720–23. doi:10.1080/11024150201680030. PMID 15362583.
- ^ Kowal L, Wong E, Yahalom C (December 2007). “Botulinum toxin in the treatment of strabismus. A review of its use and effects”. Disability and Rehabilitation. 29 (23): 1823–31. doi:10.1080/09638280701568189. PMID 18033607.
- ^ Duthie JB, Vincent M, Herbison GP, Wilson DI, Wilson D (December 2011). “Botulinum toxin injections for adults with overactive bladder syndrome”. The Cochrane Database of Systematic Reviews (12): CD005493. doi:10.1002/14651858.CD005493.pub3. PMID 22161392.
- ^ Scott AB (1994). “Change of eye muscle sarcomeres according to eye position”. Journal of Pediatric Ophthalmology and Strabismus. 31 (2): 85–88. PMID 8014792.
- ^ Jump up to:a b UK Approves New Botox Use Archived 2014-02-22 at the Wayback Machine. dddmag.com. February 4, 2014
- ^ “FDA Approved Drug Products – Dysport”. http://www.accessdata.fda.gov. U.S. Food and Drug Administration. Retrieved 2016-11-07.
- ^ Jump up to:a b Ocampo VV, Foster CS (May 30, 2012). “Infantile Esotropia Treatment & Management”. Medscape. Retrieved April 6, 2014.
- ^ Jump up to:a b Bushara KO, Park DM (November 1994). “Botulinum toxin and sweating”. Journal of Neurology, Neurosurgery, and Psychiatry. 57 (11): 1437–38. doi:10.1136/jnnp.57.11.1437. PMC 1073208. PMID 7964832.
- ^ Jump up to:a b c Eisenach JH, Atkinson JL, Fealey RD (May 2005). “Hyperhidrosis: evolving therapies for a well-established phenomenon”. Mayo Clinic Proceedings. 80 (5): 657–66. doi:10.4065/80.5.657. PMID 15887434.
- ^ “FDA Approves Botox to Treat Chronic Migraines”. WebMD. Retrieved 12 May 2017.
- ^ Jump up to:a b c d e Small R (August 2014). “Botulinum toxin injection for facial wrinkles”. American Family Physician. 90 (3): 168–75. PMID 25077722.
- ^ Mittal SO, Safarpour D, Jabbari B (February 2016). “Botulinum Toxin Treatment of Neuropathic Pain”. Seminars in Neurology. 36 (1): 73–83. doi:10.1055/s-0036-1571953. PMID 26866499.
- ^ Charles PD (November 2004). “Botulinum neurotoxin serotype A: a clinical update on non-cosmetic uses”. American Journal of Health-System Pharmacy. 61 (22 Suppl 6): S11–23. doi:10.1093/ajhp/61.suppl_6.S11. PMID 15598005.
- ^ Jump up to:a b c d Coté TR, Mohan AK, Polder JA, Walton MK, Braun MM (September 2005). “Botulinum toxin type A injections: adverse events reported to the US Food and Drug Administration in therapeutic and cosmetic cases”. Journal of the American Academy of Dermatology. 53 (3): 407–15. doi:10.1016/j.jaad.2005.06.011. PMID 16112345.
- ^ “FDA Notifies Public of Adverse Reactions Linked to Botox Use”. Fda.gov. 8 February 2008. Retrieved May 6, 2012.
- ^ Jump up to:a b “FDA Gives Update on Botulinum Toxin Safety Warnings; Established Names of Drugs Changed”. FDA Press Announcement. August 3, 2009. Retrieved 21 September 2016.
- ^ Jump up to:a b c d “Information for Healthcare Professionals: OnabotulinumtoxinA (marketed as Botox/Botox Cosmetic), AbobotulinumtoxinA (marketed as Dysport) and RimabotulinumtoxinB (marketed as Myobloc)”. fda.gov. Food and Drug Administration. Retrieved 1 September 2015.
- ^ “Botox chemical may spread, Health Canada confirms”. CBC News. January 13, 2009. Archived from the original on 21 February 2009.
- ^ “Kinds of Botulism”. Centers for Disease Control and Prevention. Retrieved 4 October2016.
- ^ Jump up to:a b c d “Botulism – Diagnosis and Treatment”. Centers for Disease Control and Prevention. Retrieved 5 October 2016.
- ^ Jump up to:a b Barr JR, Moura H, Boyer AE, Woolfitt AR, Kalb SR, Pavlopoulos A, McWilliams LG, Schmidt JG, Martinez RA, Ashley DL (October 2005). “Botulinum neurotoxin detection and differentiation by mass spectrometry”. Emerging Infectious Diseases. 11 (10): 1578–83. doi:10.3201/eid1110.041279. PMC 3366733. PMID 16318699.
- ^ Jump up to:a b Dressler, D; Saberi, FA; Barbosa, ER (March 2005). “Botulinum toxin: mechanisms of action”. Arquivos de Neuro-Psiquiatria. 63 (1): 180–5. doi:10.1159/000083259. PMID 15830090.
- ^ Jump up to:a b c d e Li B, Peet NP, Butler MM, Burnett JC, Moir DT, Bowlin TL (December 2010). “Small molecule inhibitors as countermeasures for botulinum neurotoxin intoxication”. Molecules. 16 (1): 202–20. doi:10.3390/molecules16010202. PMC 6259422. PMID 21193845.
- ^ Hill KK, Smith TJ (2013). “Genetic Diversity Within Clostridium botulinum Serotypes, Botulinum Neurotoxin Gene Clusters and Toxin Subtypes”. In Rummel A, Binz T. Botulinum neurotoxins. Current Topics in Microbiology and Immunology. 364. Heidelberg: Springer. pp. 1–20. doi:10.1007/978-3-642-33570-9_1. ISBN 978-3-642-33569-3. PMID 23239346.
- ^ Kerner J (1820). Neue Beobachtungen über die in Württemberg so häufig vorfallenden tödlichen Vergiftungen durch den Genuss geräucherter Würste. Tübingen: Osiander.
- ^ Kerner J (1822). Das Fettgift oder die Fettsäure und ihre Wirkungen auf den thierischen Organismus, ein Beytrag zur Untersuchung des in verdorbenen Würsten giftig wirkenden Stoffes. Stuttgart, Tübingen: Cotta. pp. 681–87. OCLC 863571562.
- ^ Erbguth FJ, Naumann M (November 1999). “Historical aspects of botulinum toxin: Justinus Kerner (1786–1862) and the “sausage poison““. Neurology. 53 (8): 1850–53. doi:10.1212/wnl.53.8.1850. PMID 10563638.
- ^ van Ermengem E (1979). “Classics in infectious diseases. A new anaerobic bacillus and its relation to botulism. E. van Ermengem. Originally published as “Ueber einen neuen anaëroben Bacillus und seine Beziehungen zum Botulismus” in Zeitschrift für Hygiene und Infektionskrankheiten 26: 1–56, 1897″. Reviews of Infectious Diseases (in German). 1 (4): 701–19. PMID 399378. Original doi:10.1007/BF02220526
- ^ Snipe PT, Sommer H (August 1928). “Studies on Botulinus Toxin: 3. Acid Precipitation of Botulinus Toxin”. The Journal of Infectious Diseases. 43 (2): 152–60. doi:10.1093/infdis/43.2.152. JSTOR 30083772.
- ^ Lamanna C, McElroy OE, Eklund HW (May 1946). “The purification and crystallization of Clostridium botulinum type A toxin”. Science. 103 (2681): 613–14. Bibcode:1946Sci…103..613L. doi:10.1126/science.103.2681.613. PMID 21026141.
- ^ Burgen AS, Dickens F, Zatman LJ (August 1949). “The action of botulinum toxin on the neuro-muscular junction”. The Journal of Physiology. 109 (1–2): 10–24. doi:10.1113/jphysiol.1949.sp004364. PMC 1392572. PMID 15394302.
- ^ Magoon E, Cruciger M, Scott AB, Jampolsky A (May 1982). “Diagnostic injection of Xylocaine into extraocular muscles”. Ophthalmology. 89 (5): 489–91. doi:10.1016/s0161-6420(82)34764-8. PMID 7099568.
- ^ Drachman DB (August 1964). “Atrophy of skeletal muscle in chick embryos treated with botulinum toxin”. Science. 145 (3633): 719–21. Bibcode:1964Sci…145..719D. doi:10.1126/science.145.3633.719. PMID 14163805.
- ^ Scott AB, Rosenbaum A, Collins CC (December 1973). “Pharmacologic weakening of extraocular muscles”. Investigative Ophthalmology. 12 (12): 924–27. PMID 4203467.
- ^ Scott AB (October 1980). “Botulinum toxin injection into extraocular muscles as an alternative to strabismus surgery”. Ophthalmology. 87 (10): 1044–49. doi:10.1016/s0161-6420(80)35127-0. PMID 7243198.
- ^ Jump up to:a b Boffey, Philip M. (October 14, 1986). “Loss Of Drug Relegates Many To Blindness Again”. The New York Times. Retrieved July 14, 2010.
- ^ Jump up to:a b United States Department of Health and Human Services (April 30, 2009). “Re: Docket No. FDA-2008-P-0061” (PDF). Food and Drug Administration. Retrieved July 26, 2010.
- ^ Wellman-Labadie O, Zhou Y (May 2010). “The US Orphan Drug Act: rare disease research stimulator or commercial opportunity?”. Health Policy. 95 (2–3): 216–28. doi:10.1016/j.healthpol.2009.12.001. PMID 20036435.
- ^ Jump up to:a b c Clark RP, Berris CE (August 1989). “Botulinum toxin: a treatment for facial asymmetry caused by facial nerve paralysis”. Plastic and Reconstructive Surgery. 84 (2): 353–55. doi:10.1097/01.prs.0000205566.47797.8d. PMID 2748749.
- ^ Jump up to:a b Carruthers JD, Carruthers JA (January 1992). “Treatment of glabellar frown lines with C. botulinum-A exotoxin”. The Journal of Dermatologic Surgery and Oncology. 18 (1): 17–21. doi:10.1111/j.1524-4725.1992.tb03295.x. PMID 1740562.
- ^ Jump up to:a b Keen M, Kopelman JE, Aviv JE, Binder W, Brin M, Blitzer A (April 1994). “Botulinum toxin A: a novel method to remove periorbital wrinkles”. Facial Plastic Surgery. 10 (2): 141–46. doi:10.1055/s-2008-1064563. PMID 7995530.
- ^ Jump up to:a b “Botulinum Toxin Type A Product Approval Information – Licensing Action 4/12/02”. Food and Drug Administration. October 29, 2009. Retrieved July 26, 2010.
- ^ Jump up to:a b “Allergan Receives FDA Approval for First-of-Its-Kind, Fully in vitro, Cell-Based Assay for BOTOX and BOTOX Cosmetic (onabotulinumtoxinA)”. Allergan. June 24, 2011. Archived from the original on June 26, 2011. Retrieved June 26, 2011.
- ^ Jump up to:a b “In U.S., Few Alternatives To Testing On Animals”. Washington Post. April 12, 2008. Retrieved June 26, 2011.
- ^ Binder WJ, Brin MF, Blitzer A, Schoenrock LD, Pogoda JM (December 2000). “Botulinum toxin type A (BOTOX) for treatment of migraine headaches: an open-label study”. Otolaryngology–Head and Neck Surgery. 123 (6): 669–76. doi:10.1067/mhn.2000.110960. PMID 11112955.
- ^ Jackson JL, Kuriyama A, Hayashino Y (April 2012). “Botulinum toxin A for prophylactic treatment of migraine and tension headaches in adults: a meta-analysis”. JAMA. 307 (16): 1736–45. doi:10.1001/jama.2012.505. PMID 22535858.
- ^ Ramachandran R, Yaksh TL (September 2014). “Therapeutic use of botulinum toxin in migraine: mechanisms of action”. British Journal of Pharmacology. 171 (18): 4177–92. doi:10.1111/bph.12763. PMC 4241086. PMID 24819339.
- ^ Chapman, Paul (May 10, 2012). “The global botox market forecast to reach $2.9 billion by 2018”. Archived from the original on 2012-08-06. Retrieved October 5, 2012.
- ^ Koirala J, Basnet S (July 14, 2004). “Botulism, Botulinum Toxin, and Bioterrorism: Review and Update”. Medscape. Cliggott Publishing. Retrieved July 14, 2010.
- ^ Clostridium botulinum – Public Health Agency of Canada. Phac-aspc.gc.ca (April 19, 2011). Retrieved on May 6, 2012.
- ^ Baselt RC (2014). Disposition of toxic drugs and chemicals in man. Seal Beach, Ca.: Biomedical Publications. pp. 260–61. ISBN 978-0-9626523-9-4.
- ^ http://www.emsworld.com/article/10324792/botulinum-toxin-a-bioterrorism-weapon
- ^ McAdams D, Kornblet S (15 July 2011). “Baader-Meinhof Group (OR Baader-Meinhof Gang”. In Pilch RF, Zilinskas RA. Encyclopedia of Bioterrorism Defense. Wiley-Liss. doi:10.1002/0471686786.ebd0012.pub2. ISBN 978-0-471-68678-1.
- ^ “2011 Allergan Annual Report” (PDF). Allergan. Retrieved May 3, 2012. See PDF p. 7.
- ^ Jump up to:a b c Walker TJ, Dayan SH (February 2014). “Comparison and overview of currently available neurotoxins”. The Journal of Clinical and Aesthetic Dermatology. 7 (2): 31–39. PMC 3935649. PMID 24587850.
- ^ “Botulinum Toxin Type A”. Hugh Source (International) Limited. Retrieved July 14, 2010.
- ^ Petrou I (Spring 2009). “Medy-Tox Introduces Neuronox to the Botulinum Toxin Arena”(PDF). The European Aesthetic Guide. Archived from the original (PDF) on 2013-03-20. Retrieved 2009-12-09.
- ^ Schantz EJ, Johnson EA (March 1992). “Properties and use of botulinum toxin and other microbial neurotoxins in medicine”. Microbiological Reviews. 56 (1): 80–99. PMC 372855. PMID 1579114.
- ^ Botulism, General Information – NCZVED. Cdc.gov. Retrieved on May 6, 2012.
- ^ Licciardello JJ, Nickerson JT, Ribich CA, Goldblith SA (March 1967). “Thermal inactivation of type E botulinum toxin”. Applied Microbiology. 15 (2): 249–56. PMC 546888. PMID 5339838.
- ^ Setlow P (April 2007). “I will survive: DNA protection in bacterial spores”. Trends in Microbiology. 15 (4): 172–80. doi:10.1016/j.tim.2007.02.004. PMID 17336071.
- ^ Jay JM, Loessner MJ, Golden DA (2005). “Chapter 24: Food Poisoning Caused by Gram-Positive Sporeforming Bacteria”. Modern Food Microbiology: Seventh Edition. New York: Springer. p. 581. ISBN 978-0-387-23180-8.
- ^ Capková K, Salzameda NT, Janda KD (October 2009). “Investigations into small molecule non-peptidic inhibitors of the botulinum neurotoxins”. Toxicon. 54 (5): 575–82. doi:10.1016/j.toxicon.2009.03.016. PMC 2730986. PMID 19327377.
- ^ Flanders M, Tischler A, Wise J, Williams F, Beneish R, Auger N (June 1987). “Injection of type A botulinum toxin into extraocular muscles for correction of strabismus”. Canadian Journal of Ophthalmology. 22 (4): 212–17. PMID 3607594.
- ^ “Botulinum toxin therapy of eye muscle disorders. Safety and effectiveness. American Academy of Ophthalmology”. Ophthalmology. Suppl: 37–41. September 1989. doi:10.1016/s0161-6420(89)32989-7. PMID 2779991.
- ^ Giesler M (2012). “How Doppelgänger Brand Images Influence the Market Creation Process: Longitudinal Insights from the Rise of Botox Cosmetic”. Journal of Marketing. 76(6): 55–68. doi:10.1509/jm.10.0406.
- ^ “BOTOX Cosmetic (onabotulinumtoxinA) Product Information”. Allergan. January 22, 2014.
- ^ Nayyar P, Kumar P, Nayyar PV, Singh A (December 2014). “BOTOX: Broadening the Horizon of Dentistry”. Journal of Clinical and Diagnostic Research : JCDR. 8 (12): ZE25–9. doi:10.7860/JCDR/2014/11624.5341. PMC 4316364. PMID 25654058.
- ^ Al-Fouzan AF, Mokeem LS, Al-Saqat RT, Alfalah MA, Alharbi MA, Al-Samary AE (June 2017). “Botulinum Toxin for the Treatment of Gummv Smile”. The Journal of Contemporary Dental Practice. 18 (6): 474–478. doi:10.5005/jp-journals-10024-2068. PMID 28621277.
- ^ Hwang WS, Hur MS, Hu KS, Song WC, Koh KS, Baik HS, Kim ST, Kim HJ, Lee KJ (January 2009). “Surface anatomy of the lip elevator muscles for the treatment of gummy smile using botulinum toxin”. The Angle Orthodontist. 79 (1): 70–7. doi:10.2319/091407-437.1. PMID 19123705.
- ^ Gracco A, Tracey S (May 2010). “Botox and the gummy smile”. Progress in Orthodontics. 11 (1): 76–82. doi:10.1016/j.pio.2010.04.004. PMID 20529632.
- ^ Mazzuco R, Hexsel D (December 2010). “Gummy smile and botulinum toxin: a new approach based on the gingival exposure area”. Journal of the American Academy of Dermatology. 63 (6): 1042–51. doi:10.1016/j.jaad.2010.02.053. PMID 21093661.
- ^ Walsh S (October 15, 2010). “FDA approves Botox to treat chronic migraine”. FDA Press Releases. Retrieved October 15, 2010.
- ^ Watkins T (October 15, 2010). “FDA approves Botox as migraine preventative”. CNN (US).
- ^ Dodick DW, Turkel CC, DeGryse RE, Aurora SK, Silberstein SD, Lipton RB, Diener HC, Brin MF (June 2010). “OnabotulinumtoxinA for treatment of chronic migraine: pooled results from the double-blind, randomized, placebo-controlled phases of the PREEMPT clinical program”. Headache. 50 (6): 921–36. doi:10.1111/j.1526-4610.2010.01678.x. PMID 20487038.
- ^ Ashkenazi A (March 2010). “Botulinum toxin type a for chronic migraine”. Current Neurology and Neuroscience Reports. 10 (2): 140–46. doi:10.1007/s11910-010-0087-5. PMID 20425239.
- ^ Magid M, Keeling BH, Reichenberg JS (November 2015). “Neurotoxins: Expanding Uses of Neuromodulators in Medicine – Major Depressive Disorder”. Plastic and Reconstructive Surgery. 136 (5 Suppl): 111S–19S. doi:10.1097/PRS.0000000000001733. PMID 26441090.
- ^ Jump up to:a b http://adisinsight.springer.com/drugs/800008810
External links
////////////Prabotulinumtoxin A, プラボツリナムトキシンA ,APPROVED , FDA 2019, Jeuveau, AGN 191622, ANT-1207, ANT-1401, ANT-1403, NT 201

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











.jpg)