PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Monoclonal antibody Treatment and prophylaxis of SARS-CoV-2 infection
ANTIVIRAL
SARS-CoV-2 spike glycoprotein
REGN 10987
RG 6412
Fact Sheet – US Food and Drug Administration
https://www.fda.gov › media › download PDFBenefit of treatment with casirivimab and imdevimab has not been observed in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab.
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
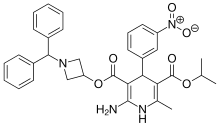
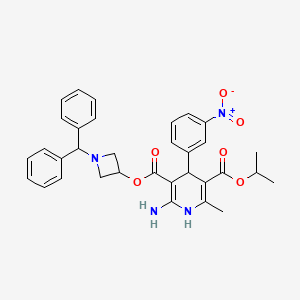
Azelnidipine is a dihydropyridine calcium channel blocker. It is marketed by Daiichi-Sankyo pharmaceuticals, Inc. in Japan. It has a gradual onset of action and produces a long-lasting decrease in blood pressure, with only a small increase in heart rate, unlike some other calcium channel blockers. It is currently being studied for post-ischemic stroke management.
Azelnidipine (INN; marketed under the brand name CalBlock — カルブロック) is a dihydropyridinecalcium channel blocker. Azelnidipine is L and T calcium channel blocker. It is sold in Japan by Daiichi-Sankyo pharmaceuticals, Inc. Unlike nicardipine, it has a gradual onset and has a long-lasting hypotensive effect, with little increase in heart rate. Drug Controller General Of India (DCGI) has approved the use of azelnipine in India. It is launched under the brand name Azusa (ajanta pharma ltd.)[1] In 2020.
Chemical Synthesis
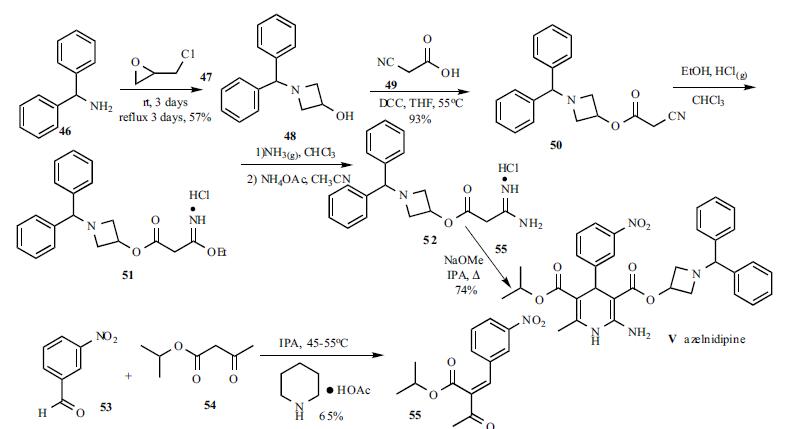
A solution of benzhydrylamine (46) and epichlorohydrin (47) was mixed without adding solvent to give azetidinol 48 in 57% yield. DCC coupling between cyanoacetic acid (49) and azetidinol 48 in hot THF gave ester 50 in 93% yield. Cyanoester 50 was treated with ethanol and HCl gas in chloroform to give imidate HCl salt 51, which was treated with ammonia gas in chloroform and ammonium acetate in acetonitrile to give the corresponding amidinoacetate 52. A modified Hantzsch reaction was employed to construct the 2-amino-1,4- dihydropyridine core structure. Compound 52 was condensed with 2-(3-nitrobenzylidene)acetic acid isopropyl ester (55) in the presence of NaOMe in refluxing isopropanol to give the cyclized product, azelnidipine (V) in 74% yield. Benzylideneacetoacetate 55 was obtained through the Knoevenagel reaction employing 3-nitrobenzaldehyde (53) and isopropyl acetoacetate (54) in isopropanol containing a catalytic amount of piperidinium acetate at 45-55oC in 65% yield.
PATENT
EP 266922
IN 201621044802
CN 106279109
CN 107188885
CN 105461691
CN 103509003
CN 103183663
CN 102382104
JP 2012020970 A
PAPER
Bioanalysis (2019), 11(4), 251-266.
PAPER
Asian Journal of Chemistry (2014), 26(15), 4675-4678.
Azelnidipine is designated chemically as 3-(1-benzhydrylazetidin-3-yl)-5-isopropyl-2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate. Its literature synthesis (Scheme-I) involves 3-nitrobenzaldehyde 5 with isopropyl acetoacetate 6. The product of (Z)-isopropyl 2-(3- nitrobenzylidene)-3-oxobutanoate (7a, b, c), on treatment with piperidine and acetic acid, coupling of (7) and 1-benzhydrylazetidin-3-yl 3-amino-3-iminopropanoate acetate (8) gave azelnidipine (1).
PAPER
International Research Journal of Pharmacy (2012), 3(8), 191-192.
Chemical & Pharmaceutical Bulletin (1995), 43(5), 797-817.
The invention belongs to the technical field of medicine and provides an important intermediate of dihydropyridine calcium antagonist adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidine The synthesis process of ester acetate. Background technique
Azelnidipine is a new type of dihydropyridine calcium channel blocker developed by Sankyo and Ube Industries of Japan. It was approved for sale in Japan in late May 2003 under the trade name Calblock. Adipine has a selective blockade of calcium channels in arterial smooth muscle cells, it can dilate blood vessels, reduce peripheral vascular resistance and arterial pressure, and is widely used clinically for mild or moderate essential hypertension, renal disorders with hypertension And treatment of severe hypertension. Compared with nicardipine and nifedipine dihydropyridine calcium channel blockers, adipine is superior in selectivity, long-lasting and long-lasting, and has little effect on the heart.
阿折地平的结构式
A flat floor structure
At present, references to the preparation of agdipine include: European patents EP0266922; Chinese patent CN201010516967.7; Chinese Journal of Medicinal Chemistry, 2010, 20 (3): 192-194; Chinese Journal of Pharmaceutical Industry, 2008, 39 (3): 163-165; Chemical Industry and Engineering, 2009, 26 ( 1 ): 15-18; Qilu Pharmacy, 2005, 24 (6): 365-366. The preparation method of adipine in these literatures is based on the reaction of epichlorohydrin and diphenylamine with N-alkylation, cyclization, esterification, Pinner synthesis, neutralization, and oxime reaction. The intermediate 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3-azetidinyl acetate is prepared first, followed by 2-(3-nitrobenzylidene)acetyl Acepinedipine was obtained by the Hantzsch condensation of isopropyl acetate.
The control of the solvent and reaction conditions in the esterification, Pinner synthesis and neutralization three-step reaction in this route is critical. Using the preparation methods provided by these documents, we found that the operation was cumbersome and the yield and purity were not satisfactory.
In the esterification reaction, according to the method specifically reported in the above literature, the highest yield of the obtained product is only 85%, and the purity is poor, it is difficult to purify, and it is difficult to obtain a solid product.
副产物 (7 )和(8 )结构式 发明内容 We have found that 3-amino-3-iminopropionic acid-1- (3) is prepared by a three-step reaction from cyanoacetate-1-diphenylhydrazin-3-azetidinyl ester (3) according to the method specifically reported in the above literature. Diphenylhydrazino)-3-azetidinyl acetate (6), the reaction operation is cumbersome, and it is easy to produce by-products of hydrolysis of ester bonds and hydrolysis of imid bonds (7) and (8), three-step reaction. The total yield is only 20~30%, and the purification of the product is difficult, which seriously affects the quality of the final product and greatly increases the production cost.
Byproducts (7) and (8) structural formula Summary of the invention
It is an object of the present invention to provide a process for the preparation of the key intermediate of adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate. The adipine intermediate of the present invention 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate acetate has the following structural formula:
The preparation method of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate of the present invention comprises the following steps: 1) Esterification: 1-diphenylhydrazin-3-azetidinol (2), cyanoacetic acid (1) and N,N-dicyclohexylcarbodiimide (DCC) in organic solvent at 0~ Reacting at 80 ° C, to obtain 7-diphenylindolyl-3-azetidinyl cyanoacetate (3);
2) Pinner reaction: Add intermediate (3), absolute ethanol to dichlorosilane, stir and cool
To -20~25 °C, dry hydrogen chloride gas is passed, and then the reaction solution is kept sealed at -20~25 °C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl) -3-azetidinyl ester hydrochloride (4);
3) Neutralization reaction: The intermediate (4) is dissolved in dichloromethane, and the base is added at -5 to 25 ° C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylhydrazine). Benzyl-3-azetidinyl ester (5);
4) Formation reaction: The intermediate (5) is dissolved in acetonitrile, ammonium acetate is added, and the temperature is raised to 40 to 60 ° C to obtain 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3. – azetidinium acetate compound (6). detailed description
Example
1. Preparation of cyanoacetic acid-1-diphenylhydrazine-3-azetidine (esterification)
Method 1: Add 1-diphenylhydrazin-3-azetidinol (2, 235 g, 0.983 mol) and cyanoacetic acid (1, 100 g, 1.18 mol) to 1.5 mL of dichloromethane, and stir until fully dissolved. Ν, Ν-dicyclohexylcarbodiimide (DCC, 243 g, 1.18 mol) was added at 0-10 ° C and allowed to react at room temperature for 3 h. After the completion of the reaction, the reaction mixture was cooled to 0 to 5 ° C, and filtered, filtered, washed with a small portion of dichloromethane. The organic solvent was evaporated to dryness under reduced pressure and dried to give 275 g of white solid.
Method 2: chloroform was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 5 hours, the HPLC purity was 98.7%, and the product yield was 95.3%.
Method 3: Ethyl acetate was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 2 h, the HPLC purity was 98.9%, and the product yield was 96.1%.
Method 4: Using hydrazine as the reaction solvent, the operation was the same as above, and the reaction was carried out at 55 ° C for 7 h, the HPLC purity was 98.5%, and the product yield was 94.7%. 2. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester hydrochloride (Pinner reaction)
Intermediate 3 (270 g, 0.882 mol), absolute ethanol (61.8 mL, 1.06 mol) was added to 1.5 L of dry dichloromethane, cooled to -5 to 0 ° C in a water salt bath, and dried. HC1 gas for 2.5 h, after the completion of the aeration, the reaction solution was kept under stirring at 0 ° C for 6 h.
Allow to stand overnight at 0-4 °C. After completion of the reaction, the solvent was evaporated under reduced pressure to give an oily viscous intermediate 4 .
3. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester
Method 1: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add dry diethylamine (182 mL, 1.76 mol) to the solution, adjust pH 7-8, continue to stir after the dropwise addition. 2h. The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo.
Method 2: Diamine is used for neutralization, and the operation is the same as above.
Method 3: Triethylamine is used for neutralization, and the operation is the same as above.
Method 4: Ethylenediamine is used for neutralization, and the operation is the same as above.
Method 5: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add potassium carbonate (242.88 g, 1.76 mol) to the solution in portions, adjust pH 7-8, continue stirring for 2 h. . The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo. Method 6: Neutralize with sodium carbonate, and operate as above.
Method 7: Neutralize with sodium hydroxide, and operate as above.
4. Preparation of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate (formed into 脒)
To the intermediate 5, 1.2 L of acetonitrile was added, and after dissolution, ammonium acetate (68.0 g, 0.882 mol) was added, and the mixture was heated to 55 ° C for 6 h. After the reaction, it was naturally cooled, crystallization, suction filtration, acetonitrile washing cake, and dried to give 236 g of a white solid. The total yield of the three-step reaction was 69.9 73.1%.
A protocol for the coupling of 3-iodoazetidines with Grignard reagents in the presence of an iron catalyst has been developed. A variety of aryl, heteroaryl, vinyl and alkyl Grignards were shown to participate in the coupling process to give the products in good to excellent yields. Furthermore, a short formal synthesis towards a pharmacologically active molecule was shown.
http://www.rsc.org/suppdata/cc/c4/c4cc09337b/c4cc09337b1.pdfPATENThttps://patents.google.com/patent/CN103509003A/zhAzelnidipine, whose chemical name is 3-(1-diphenylmethylazetidin-3-yl) 5-isopropyl 2-amino-1,4-dihydro-6-methyl 4-(3-nitrophenyl)-3,5-pyridinedicarboxylate, developed by Japan Sankyo Co., Ltd. and approved to be marketed in Japan in late May 2003. The existing synthesis method of azedipine is cumbersome, and the preparation of intermediate (VI) adopts column chromatography method, and the purification of product (I) also uses column chromatography method, which is not suitable for industrial production.
A method for preparing azeldipine, which is characterized in that it is prepared by the following steps.
[0006]
Description of the drawings:
Figure 1 is a flow chart of the synthesis process of azeldipine.
[0025] Example 12-Preparation of (3-nitrobenzylidene) isopropyl acetoacetate (III)
[0026] Add 2.1kg of 3-nitrobenzaldehyde and 5L of isopropanol to the reaction kettle, start stirring, add 3kg of isopropyl acetoacetate, and stir. Add 43ml of anhydrous piperidine and 12ml of glacial acetic acid, and continue to stir until the solid is completely dissolved. Heat the temperature to 45°C and keep the reaction for 6h, then lower the temperature, stir and crystallize for 16h. Filter and collect the resulting filter cake. Put the obtained filter cake and 16L ethanol (industrial) into the reaction kettle, start stirring, beating, filtering, and collecting the filter cake. Put the filter cake in the baking tray, put it in the oven, and dry at 70-80°C. Collect the product 2-(3-nitrobenzylidene) isopropyl acetoacetate (III), about 2.7 kg.
[0027] Example 21-Preparation of benzhydryl-3-hydroxyazetidine (Intermediate V)
[0028] 9.6L of methanol, 5.4kg of benzhydrylamine (IV) and 3.33kg of epichlorohydrin were added to the reaction kettle, stirred at room temperature for 48 hours, the reaction was completed, the temperature was raised to 68°C, and the reaction was refluxed for 72h. Cool to room temperature. Concentrate under reduced pressure to remove methanol, and collect the filter cake by filtration. The filter cake was put into the reaction kettle, 19.2L of ether and 13.75L of 3mol/L NaOH solution were added, stirred, and the water layer was released after standing still. The ether layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was collected. The ether was recovered under reduced pressure to dryness to obtain about 3.05 kg of 1-benzyl-3-hydroxyazetidine (Intermediate V).
[0029] Example 3 Preparation of cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI)
[0030] Put about 3.05g of intermediate (V), 27L of tetrahydrofuran and 1.7kg of cyanoacetic acid into the reactor, start stirring, turn on the chilled water of the reactor to cool down, and slowly add 3.1kgN, N’-dicyclohexyl to the reactor Diimine, control the temperature at IO0C -15°C, after the addition, close the chilled water in the reactor. Turn on the heating system, slowly increase the temperature to 55-60°C, and react for 10 hours. The material liquid was cooled to room temperature, filtered, and the filtrate was concentrated to dryness. Put 16.8L of ethyl acetate into the reaction kettle, stir to dissolve, then wash with water, dry with anhydrous sodium sulfate, filter, and collect the filtrate. Ethyl acetate was recovered under reduced pressure, petroleum ether was added to the solid residue, stirred, and filtered to obtain cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI), about 3.19 kg.
[0031] Example 4 Preparation of amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (VII)
[0032] Put 25L of dichloromethane, about 3.19kg of intermediate (VI), and 430g of ethanol into the reactor, start stirring, cool to below 0°C, and pass in hydrogen chloride gas until the temperature stabilizes below 0°C, at 0°C Let stand for 14 hours at °C. Concentrate under reduced pressure to remove most of the hydrogen chloride gas and recover the solvent dichloromethane. Add 25L of dichloromethane to the residue of the reaction kettle, stir, cool to below 0°C, and pass in ammonia until the temperature stabilizes below 0°C, and filter . The filtrate was poured into the reactor, concentrated under reduced pressure to recover the solvent to obtain a colorless liquid, added 22.8L of acetonitrile and 905g of amine acetate, heated to 55-60°C for 1.5 hours, stopped the reaction, filtered while hot, and recovered the filtrate under reduced pressure Solvent to dryness, add 3L of ether to the residue to crystallize, filter, and dry to obtain amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (Intermediate VII) about 3.2kg .
[0033] Example 5. Add about 3.2kg of Intermediate (VII), about 2.7kg of Intermediate (III), 21L of isopropanol and 585g of sodium methoxide to the reaction kettle, start stirring, heat to reflux and react for 4 hours, and cool to Below 10°C, filter, the filtrate is decompressed to recover the solvent to dryness, add 35L ethyl acetate to the residue to dissolve, wash with 6.5LX3 water, release the water layer, add anhydrous sodium sulfate to the ethyl acetate layer to dry, filter , Collect the filtrate, recover ethyl acetate under reduced pressure, add 4.2L of toluene to the residue,
3.4L of n-hexane was heated to dissolve, filtered, the filtrate was stirred to room temperature to crystallize, filtered and collected and dried, and the product was placed in an oven at 45-55°C to dry to obtain the crude azedipine (I), about 2.3kg.
[0034] Example 6, Refining
[0035] Put 8.8L ethyl acetate and 8.8L n-hexane into the reaction kettle, turn on the stirring, put about 2.3kg of the crude azeldipine into the reaction kettle, slowly heat up until the material is dissolved, add 180g of activated carbon and stir for 0.5h, while it is hot Filter, hydraulically filter the material to the crystallization dad, wash the filter cake with 5.5L ethyl acetate and 4.5L n-hexane solution, combine with the filtrate, cool to 0~5°C to crystallize, filter, collect the product, and place it in a hot air circulating oven After drying at 45-55°C, 2.2 g of azeldipine is obtained. The purity is 99.6% as measured by high performance liquid chromatography. The refined yield is 96.0%.
[0036] Example 7 Azedipin Refining
[0037] The mixed solvent was prepared according to the volume ratio of ethyl acetate and n-hexane of 2:1, 22L of the mixed solvent was put into the reactor, about 2.3kg of azedipine crude product was put into the reactor, and the temperature was slowly heated until the material was dissolved, Add 180g of activated carbon and stir for 0.5h, filter while hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid with the filtrate, cool to 0~5°C for crystallization, filter, collect the product, and circulate the hot air Dry in an oven at a temperature of 45-55°C to obtain 2.2 g of azeldipine fine product, with a purity of 99.7% measured by high performance liquid chromatography.
[0038] Example 8 prepared a mixed solvent at a volume ratio of ethyl acetate and n-hexane of 1.5:1, put 22L of the mixed solvent into the reactor, put about 2.3kg of crude azeldipine into the reactor, and slowly heated to Dissolve the material, add 180g of activated carbon and stir for 0.5h, filter while it is hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid and the filtrate, cool to 0~5°C to crystallize, filter, and collect the product. Dry in a hot air circulating oven at a temperature of 45-55°C to obtain
2.2g azeldipine is a fine product with a purity of 99.6% measured by high performance liquid chromatography.
Azelnidipine (Azelnidipine) is a new type of dihydropyridine calcium channel blocker jointly developed by Sankyo Co., Ltd. and Ube Industries Co., Ltd., which inhibits the entry of calcium ions into excitable tissues and causes peripheral blood vessels And coronary artery vasodilation plays a role in lowering blood pressure. Clinically, it is widely used in patients with mild or moderate symptoms of primary hypertension, hypertension with renal dysfunction, and severe hypertension. Compared with similar antihypertensive drugs, azeldipine has a slow and long-lasting antihypertensive effect.
[0004] The chemical structure of azeldipine is similar to that of nifedipine:
[0006] The Chinese patent CN87107150.9 reported the compound earlier and gave a detailed introduction to its synthesis; afterwards, most of the synthesis of azeldipine adopts this route:
[0008] The reaction takes o-nitrobenzaldehyde and isopropyl acetoacetate as raw materials to prepare intermediate compound 5; takes benzhydrylamine and epichlorohydrin as raw materials to prepare compound 2, compound 2 and cyanoacetic acid act in DCC Compound 3 is prepared by the next reaction. Compound 3 is added with ethanol under the action of hydrogen chloride gas, ammonia gas ammonolysis, and acetate anion exchange to obtain compound 4. Compound 4 and compound 5 are under the action of sodium methoxide to obtain compound 1, namely azeldipine.
[0009] Wherein: Compound 3 can be purchased as an industrial product, or can be prepared according to the traditional method reported in the literature; Compound 5 is prepared according to the traditional method reported in the literature.
[0010] In the process of preparing amidine 4 in the traditional reaction route, hydrogen chloride gas and ammonia gas need to be passed in successively. Therefore, the reaction requires anhydrous reagents. According to literature reports, the reaction yield is about 70%. From the perspective of industrial synthesis, The application of anhydrous reagents will undoubtedly increase the cost, while the use of gas will increase the difficulty of operation and require the use of high-pressure equipment. At the same time, post-reaction processing is difficult and industrial production is difficult. Therefore, this step of the reaction requires further improvement.
With acetonitrile as a solvent, the crude product of reaction 2) was stirred until dissolved, ammonium acetate was added, and acetate anion exchange was performed to obtain the amidine compound 4;
[0018] The second step: use toluene as a solvent, compound 4 and compound 5 in the use of sodium amide to obtain compound 1, namely azedipine
[0020] The preferred technical solution of the present invention is characterized in that the temperature of reaction 1) is controlled below _5°C
Example 1: Preparation of azeldipine
[0030] Add 50 g of compound 3, 1500 mL of dichloromethane, and 16.64 mL of absolute ethanol to a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5 °C to saturation, and after saturation, keep the reaction at -5 °C for 24 hours. Protect from light and nitrogen, slowly add the above reaction system to 1665ml of ammonia water with a concentration of 2.5-3.0% under the control of 0-5°C. After the addition, stir for 0.5h, stand for 0.5h, and separate the liquids. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C -60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 57.55 g of amidine 4, the yield was 91.2%, the HPLC purity was 99.63%, and the melting point was 130-132.3°C.
[0031] 50g amidine 4, 43.5g compound 5, 1000mL toluene, and 7.7g sodium amide were added into a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was purified once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 66.87g of α-crystal form of Azedipine, yield 88.2%, melting point: 121-123°C.
[0032] Example 2; Preparation of Azeldipine
[0033] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -6°C to -8°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to ammonia water with a concentration of 2.5-3.0%, adjust the pH to 7.8-8.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.0 lg of amidine 4 with a yield of 93.5%, an HPLC purity of 99.52%, and a melting point of 130.1-132.0°C.
[0034] 50g amidine 4, 43.5g compound 5, 1000mL toluene and 7.7g sodium amide were added to a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was refined once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 68.31 g of α-crystal azedipine, yield 90.01%, melting point: 121 -123 °C.
[0035] Example 3: Preparation of Amidine 4
[0036] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -7°C to -9°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to the ammonia water with a concentration of 2.5-3.0%, adjust the pH to 8.5-9.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.5 g of amidine 4, HPLC purity 99.78%, melting point: 130.7-132·2°C.
Step 2: Using toluene as a solvent, compound 4 and compound 5 under the action of sodium amide to obtain compound 1, namely azeldipine
[0007] In the synthesis workshop, benzhydrylamine is used as a raw material to be synthesized by addition, cyclization, esterification, acidification, ammoniation, condensation and other reactions. The crude azeodipine is refined, dried, mixed and packaged in a clean area. Fold the ground. The specific response is as follows:
[0008] 1. Addition and cyclization reaction
[0009] Methanol, benzhydrylamine, and epichlorohydrin were added to the reaction kettle, stirred at room temperature for 24hr, the reaction was completed, the reaction was heated to reflux for 24hr, cooled, filtered to collect the precipitated solid, and then the mother liquor was concentrated to recover the raw materials, and the heating was continued to reflux 18 After hours, collect the product, add dichloromethane and H2O to the obtained solid, adjust the pH to 10-11 with 40% NaOH while stirring in an ice bath, stand still, separate the organic layer, dry with anhydrous magnesium sulfate, and recover the dichloromethane under reduced pressure To dryness, a colorless solid compound III (1-benzyl-3-hydroxyazetidine) is obtained. After improvement, the raw materials are fully reacted, and the reaction yield of this step is improved. The mass yield is 75%. % Mentioned 85%.
[0010]
[0011] 2. Esterification reaction
[0012] Add THF, compound (III), and cyanoacetic acid to the reaction kettle, stir evenly, add DCC in batches under ice bath stirring, control the temperature at 10°C~15°C, after the addition, remove the ice water bath, and slowly heat up React at 55°C~60°C for 18h. After the reaction is complete, cool, filter to remove insoluble materials, concentrate the filtrate to dryness, add ethyl acetate to the residue to dissolve, wash with water, dry with anhydrous magnesium sulfate, and recover ethyl acetate under reduced pressure. The residue was added with petroleum ether and stirred for crystallization, and the solid was collected by filtration to obtain compound IV (1-diphenylmethyl-3-azetidinyl cyanoacetate).
[0013]
[0014] 3. Acidification and amination reaction
[0015] Dichloromethane, ethanol and intermediate (IV) were added to the reaction kettle respectively, mixed and stirred, cooled to about _5 ° C in an ice salt bath, and dried hydrogen chloride gas was introduced until saturation (about 1.5 hours) after . Let stand overnight at about -5°C, recover the solvent under reduced pressure at room temperature, add dichloromethane to the residue and stir, cool to about _5°C in an ice-salt bath, pass in the dried ammonia gas until saturation (about 3 hours) , Filtration to remove the insoluble matter, and the filtrate was decompressed to recover solvent at room temperature. Acetonitrile and ammonium acetate were added to the residue respectively, and the temperature was raised to 55~60°C for 2 hours with stirring. After the reaction was completed, it was cooled and filtered. 3-Azacyclobutanylamidinoacetate acetate), the reaction in this step is controlled at about _5°C, and the transesterification
The side reaction is reduced, and the reaction yield is improved.
[0016]
[0017] 4. Condensation reaction
[0018] Add isopropanol, intermediate (III’), sodium methoxide and compound V to the reaction kettle, mix and stir, heat to reflux and react for 5 hours. After the reaction is complete, cool and filter, and the filtrate is decompressed to recover the solvent to dryness, leaving residue Add ethyl acetate to dissolve, wash with water, dry with anhydrous magnesium sulfate, recover ethyl acetate under reduced pressure to 1/4 of the total volume, add n-hexane, and stir at 50°C for 30 min. After cooling and crystallization, the solid was collected by filtration, and air-dried at 45°C to obtain the crude azedipine (I). After the crude product was dissolved in ethyl acetate-n-hexane mixed solvent, activated carbon was added for decolorization and impurity removal to achieve the purpose of purification.
[0020] The refined product is dissolved in dioxane, refluxed with n-hexane, cooled and crystallized, and dried to obtain a solid that is boiled in cyclohexane, cooled and filtered, and dried to obtain α-crystalline form Azedipine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN102453023A *2010-10-212012-05-16大丰市天生药业有限公司Process for producing azelnidipineCN103130700A *2013-03-142013-06-05沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN103509003A *2012-06-272014-01-15威海威太医药技术开发有限公司Preparation method of azelnidipine JP3491506B2 *1997-10-142004-01-26宇部興産株式会社Method for producing dihydropyridine derivativeCN101475521B *2008-11-132010-11-10青岛黄海制药有限责任公司Method for synthesizing acetate of 1-benzhydryl-3-azetidine amidino acetic ester TitleLIU, JIAN-FENG ET AL.: “Improved Synthesis of Azelnidipine”, CHINESE JOURNAL OF MEDICINAL CHEMISTRY, vol. 20, no. 3, 30 June 2010 (2010-06-30), pages 192 – 194 *ZHANG, KAI ET AL.: “Synthesis of Azelnidipine”, CHINESE JOURNAL OF PHARMACEUTICALS, vol. 39, no. 3, 31 March 2008 (2008-03-31), pages 163 – 165, XP025959789, DOI: doi:10.1016/j.ejphar.2008.12.041 * CN103130700B *2013-03-142015-04-29沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN104860855B *2014-12-082017-06-16宁夏紫光天化蛋氨酸有限责任公司A kind of preparation method of the methylmercapto butyric acid ester of 2 hydroxyl of the D of high-purity, L 4CN105949102A *2016-06-202016-09-21许昌豪丰化学科技有限公司Production method of azelnidipine intermediatePublication numberPriority datePublication dateAssigneeTitleWO2014139410A1 *2013-03-142014-09-18Shenyang Zhonghai Pharmaceutical Co., Ltd.A kind of preparation method of azeldipine intermediateCN105461691A *2015-12-312016-04-06Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106279109A *2016-08-182017-01-04Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106543061A *2016-10-202017-03-29Weihai Disu Pharmaceutical Co., Ltd.Preparation method of N-diphenylmethylcyclobutane-3-alcohol
General information Recombinant von Willebrand Factor (rVWF) is co-expressed with recombinant Factor VIII (rFVIII) in Chinese hamster ovary (CHO) cells as part of the ADVATE (Centrally authorised product) manufacturing process. The rVWF protein is separated from the FVIII and further purified.
Vonicog alfa is expressed as a 2813 amino acid pro-VWF molecule. The pro-VWF is composed of A, B, C and D repeats, which contain various functional domains that have been identified. The mature VWF monomer is a 2050 amino acid protein. Every monomer contains a number of specific domains with a specific function. Elements of note are: • The D’/D3 domain, which binds to Factor VIII • The A1 domain, which binds to: Platelet gp1b-receptor, Heparin, Collagen • The A3 domain, which binds to collagen • The C1 domain, in which the RGD domain binds to platelet integrin αIIbβ3 when this is activated • The “cysteine knot” domain Monomers of pro-VWF are subsequently N-glycosylated, arranged into dimers via a C-terminal disulfide bond in the endoplasmic reticulum and into multimers by crosslinking of N-terminal cysteine residues via disulfide bonds.
Figure 1. Structure of Von Willebrand Factor Monomer/Dimer
After reduction of disulfide bonds in electrophoretic analysis, rVWF appears as a single predominant band having an apparent molecular weight of approximately 260 kDa. In low resolution agarose gel electrophoresis, rVWF shows a characteristic ladder of bands also known as multimers. In this analysis, rVWF contains as many distinct bands as VWF detectable in normal human plasma or VWF isolated from human plasma but in addition, has a zone with unresolved bands in the ultra-high molecular weight range. Highresolution electrophoresis shows a single band for all multimer levels without any satellite bands, as rVWF has never been exposed to ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) cleavage.
Vonicog alfa should not be used in the treatment of Hemophilia A.[4]
In the UK it is available only via a named patient access program.[7]
Vonicog alfa was approved for medical use in the United States in December 2015, in the European Union in August 2018, and in Australia in April 2020.[3][5][4][8] It was granted orphan drug designations in both the United States and the European Union.[4][1]
Medical uses
Vonicog alfa is indicated in adults with von Willebrand Disease (VWD), when desmopressin (DDAVP) treatment alone is ineffective or not indicated for the
The following side effects may occur during treatment with vonicog alfa: hypersensitivity (allergic) reactions, thromboembolic events (problems due to the formation of blood clots in the blood vessels), development of inhibitors (antibodies) against von Willebrand factor, causing the medicine to stop working and resulting in a loss of bleeding control.[4] The most common side effects with vonicog alfa (which may affect up to 1 in 10 patients) are dizziness, vertigo (a spinning sensation), dysgeusia (taste disturbances), tremor, rapid heartbeat, deep venous thrombosis (blood clot in a deep vein, usually in the leg), hypertension (high blood pressure), hot flush, vomiting, nausea (feeling sick), pruritus (itching), chest discomfort, sensations like numbness, tingling, pins and needles at the site of infusion, and an abnormal reading on the electrocardiogram (ECG).[4]
Singal M, Kouides PA: Recombinant von Willebrand factor: a first-of-its-kind product for von Willebrand disease. Drugs Today (Barc). 2016 Dec;52(12):653-664. doi: 10.1358/dot.2016.52.12.2570978. [PubMed:28276537]
Brown R: Recombinant von Willebrand factor for severe gastrointestinal bleeding unresponsive to other treatments in a patient with type 2A von Willebrand disease: a case report. Blood Coagul Fibrinolysis. 2017 Oct;28(7):570-575. doi: 10.1097/MBC.0000000000000632. [PubMed:28379876]
Gill JC, Castaman G, Windyga J, Kouides P, Ragni M, Leebeek FW, Obermann-Slupetzky O, Chapman M, Fritsch S, Pavlova BG, Presch I, Ewenstein B: Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015 Oct 22;126(17):2038-46. doi: 10.1182/blood-2015-02-629873. Epub 2015 Aug 3. [PubMed:26239086]
Lenting PJ, Christophe OD, Denis CV: von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015 Mar 26;125(13):2019-28. doi: 10.1182/blood-2014-06-528406. Epub 2015 Feb 23. [PubMed:25712991]
Chung MC, Popova TG, Jorgensen SC, Dong L, Chandhoke V, Bailey CL, Popov SG: Degradation of circulating von Willebrand factor and its regulator ADAMTS13 implicates secreted Bacillus anthracis metalloproteases in anthrax consumptive coagulopathy. J Biol Chem. 2008 Apr 11;283(15):9531-42. doi: 10.1074/jbc.M705871200. Epub 2008 Feb 8. [PubMed:18263586]
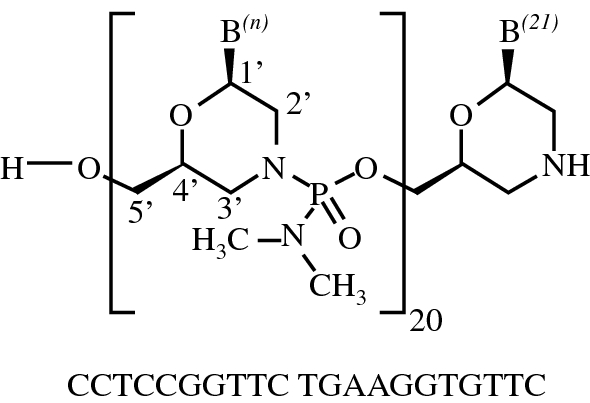
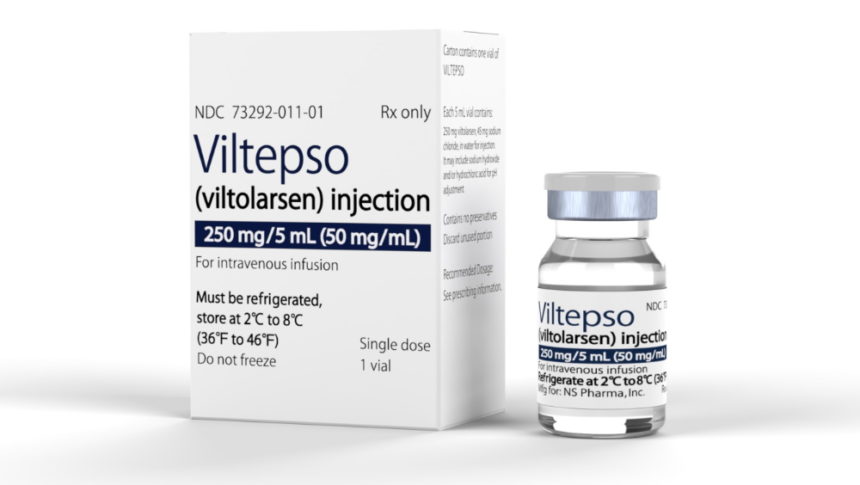
Viltolarsen was approved for medical use in the United States in August 2020.[3][4] After golodirsen was approved in December 2019, viltolarsen is the second approved targeted treatment for people with this type of mutation in the United States.[3][5] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[3]
Medical uses
Viltolarsen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[3][2]
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness.[3] It is the most common type of muscular dystrophy.[3] DMD is caused by mutations in the DMD gene that results in an absence of dystrophin, a protein that helps keep muscle cells intact.[3] The first symptoms are usually seen between three and five years of age and worsen over time.[3] DMD occurs in approximately one out of every 3,600 male infants worldwide; in rare cases, it can affect females.[3]
Adverse effects
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Although kidney toxicity was not observed in the clinical studies, the clinical experience is limited, and kidney toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides.[3]
History
Viltolarsen was evaluated in two clinical studies with a total of 32 participants, all of whom were male and had genetically confirmed DMD.[3] The increase in dystrophin production was established in one of those two studies, a study that included sixteen DMD participants, with eight participants receiving viltolarsen at the recommended dose.[3] In the study, dystrophin levels increased, on average, from 0.6% of normal at baseline to 5.9% of normal at week 25.[3] Trial 1 provided data for evaluation of the benefits of viltolarsen.[4] The combined populations from both trials provided data for evaluation of the side effects of viltolarsen.[4] Trial 1 was conducted at six sites in the United States and Canada and Trial 2 was conducted at five sites in Japan.[4] All participants in both trials were on a stable dose of corticosteroids for at least three months before entering the trials.[4]
The U.S. Food and Drug Administration (FDA) concluded that the applicant’s data demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in people with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping.[3] A clinical benefit of the drug has not been established.[3] In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of available therapies.[3]
The application for viltolarsen was granted priority review designation and the FDA granted the approval to NS Pharma, Inc.[3]
Hwang J, Yokota T (October 2019). “Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies”. Expert Rev Mol Med. 21: e5. doi:10.1017/erm.2019.5. PMID31576784.
Roshmi RR, Yokota T (October 2019). “Viltolarsen for the treatment of Duchenne muscular dystrophy”. Drugs Today. 55 (10): 627–639. doi:10.1358/dot.2019.55.10.3045038. PMID31720560.
Clinical trial number NCT02740972 for “Safety and Dose Finding Study of NS-065/NCNP-01 in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
Literature References: Polyol sweetener; relative sweetness compared to sucrose is 36%. Prepd by hydrogenation of lactose, q.v.: M. J. B. Senderens, Compt. Rend.170, 47 (1920); M. L. Wolfrom et al.,J. Am. Chem. Soc.60, 571 (1938). Pharmacology: D. H. Patil et al.,Br. J. Nutr.57, 195 (1987). Crystal structure: J. A. Kanters et al.,Acta Crystallogr.C46, 2408 (1990); J. Kivikoski et al.,Carbohydr. Res.223, 45 (1992). Toxicology: E. J. Sinkeldam et al.,J. Am. Coll. Toxicol.11, 165 (1992). Clinical trial in chronic hepatic encephalopathy: O. Riggio et al.,Hepatogastroenterology37, 524 (1990); as a laxative: L. Goovaerts, G. P. Ravelli, Acta Ther.19, 61 (1993). Review of properties and applications: J. A. van Velthuijsen, J. Agric. Food Chem.27, 680-686 (1979); of chemistry and use in foods: C. H. den Uyl, Dev. Sweeteners3, 65-81 (1987).
Properties: Crystals from absolute ethanol, mp 146°. [a]D23 +14° (c = 4 in water). Sol in water, dimethyl sulfoxide, N,N-dimethylformamide; slightly sol in ethanol, ether. Strongly hygroscopic.
Melting point: mp 146°
Optical Rotation: [a]D23 +14° (c = 4 in water)
Derivative Type: Monohydrate
CAS Registry Number: 81025-04-9
Trademarks: Importal (Novartis); Portolac (Zyma)
Properties: White, sweet, odorless, crystalline solid. Non-hygroscopic. mp 94-97° (van Velthuijsen), water of crystallization evaporates 145°-185°; also reported as mp 120° (den Uyl). [a]D22 +12.3°. Soly at 25° (g/100 g solvent): water 206; ethanol 0.75; ether 0.4; DMSO 233; DMF 39; at 50°: water 512; ethanol 0.88; at 75°: water 917.
Properties: White, sweet, odorless, crystalline powder. Data for food grade, mp 75°. [a]D25 +13.5-15.0°. pH of 10% solution 4.5 – 8.5. 140 g will dissolve in 100 ml water at 25°.
Melting point: mp 75°
Optical Rotation: [a]D25 +13.5-15.0°
Use: Sweetener in food.
Therap-Cat: Laxative. In treatment of hepatic encephalopathy.
Lactitol is listed as an excipient in some prescription drugs.[1][2]
Lactitol is a laxative and is used to prevent or treat constipation,[3] e.g., under the trade name Importal.[4][5]
In February 2020, Lactitol was approved for use in the United States as an osmotic laxative for the treatment of chronic idiopathic constipation (CIC) in adults.[6][7][8]
Lactitol in combination with Ispaghula husk is an approved combination for idiopathic constipation as a laxative and is used to prevent or treat constipation.[medical citation needed]
Like other sugar alcohols, lactitol causes cramping, flatulence, and diarrhea in some individuals who consume it. This is because humans lack a suitable beta-galactosidase in the upper gastrointestinal (GI) tract, and a majority of ingested lactitol reaches the large intestine,[9] where it then becomes fermentable to gut microbes (prebiotic) and can pull water into the gut by osmosis.{[medical citation needed] Those with health conditions should consult their GP or dietician prior to consumption.{[medical citation needed]
History
The U.S. Food and Drug Administration (FDA) approved Pizensy based on evidence from a clinical trial (Trial 1/ NCT02819297) of 594 patients with CIC conducted in the United States.[8] The FDA also considered other supportive evidence including data from Trial 2 (NCT02481947) which compared Pizensy to previously approved drug (lubiprostone) for CIC, and Trial 3 (NCT02819310) in which patients used Pizensy for one year as well as data from published literature.[8]
The benefit and side effects of Pizensy were evaluated in a clinical trial (Trial 1) of 594 patients with CIC.[8] In this trial, patients received treatment with either Pizensy or placebo once daily for 6 months.[8] Neither the patients nor the health care providers knew which treatment was being given until after the trials were completed.[8]
In the second trial (Trial 2) of three months duration, improvement in CSBMs was used to compare Pizensy to the drug lubiprostonewhich was previously approved for CIC.[8] The third trial (Trial 3) was used to collect the side effects in patients treated with Pizensy for one year.[8]
SYN
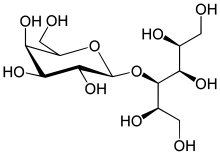
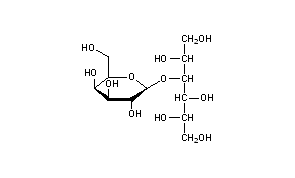
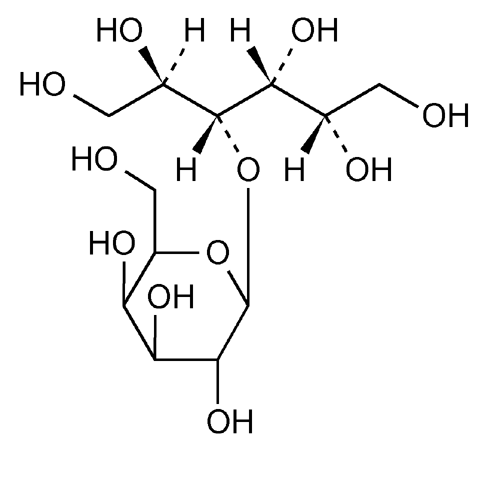
Lactitol (CAS NO.: 585-86-4), with its other name of 4-O-beta-D-Galactopyranosyl-D-glucitol, could be produced through many synthetic methods.
Following is one of the synthesis routes: Lactitol is obtained by catalytic hydrogenation of lactose (I) in the presence of either, nickel catalysts such as Raney nickel (1-9), or ruthenium catalysts (10). Alternatively, lactose (I) is reduced by employing NaBH4 (9).
Heavy metals 231— Dissolve 4 g of it in 25 mL of water: the limit is 5 µg per g.
Reducing sugars— Dissolve 500 mg of it in 2.0 mL of water in a 10-mL conical flask. Into a similar flask, pipet 2 mL of a dextrose solution containing 0.5 mg per mL. Concomitantly add 1 mL of alkaline cupric tartrate TS to each solution, heat to boiling, and cool: the lactitol solution shows no more turbidity than that produced in the dextrose solution, in which a reddish brown precipitate forms (0.2%, as dextrose).
Related compounds—
Standard solution— Dissolve an accurately weighed quantity of USP Lactitol RS in water to obtain a solution having a known concentration of about 0.3 mg per mL.
Chromatographic system— Proceed as directed in the Assay, except to chromatograph the Standard solution instead of the Standard preparation.
Test solution— Use the Assay preparation, prepared as directed in the Assay.
Procedure— Separately inject equal volumes (about 25 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the peak responses. The relative retention times are about 0.53 for lactose, 0.58 for glucose, 0.67 for galactose, 0.72 for lactulitol, 1.0 for lactitol, 1.55 for galactitol, and 1.68 for sorbitol. Calculate the percentages of galactitol, sorbitol, lactulitol, lactose, glucose, and galactose in the portion of Lactitol taken by the formula:
100(CV/W)(rU / rS)
in which C is the concentration, in mg per mL, of USP Lactitol RS in the Standard solution; V is the volume, in mL, of the Test solution; W is the weight, in mg, of Lactitol in the Test solution; rU is the peak response of the relevant related compound, if observed, obtained from the Test solution; and rS is the lactitol peak response obtained from the Standard solution. The total of the percentages of all related compounds is not more than 1.5%.
Assay—
Mobile phase— Use water.
Standard preparation— Dissolve an accurately weighed quantity of USP Lactitol RS in water to obtain a solution having a known concentration of about 10.0 mg per mL.
Assay preparation— Transfer about 1000 mg of Lactitol, accurately weighed, to a 100-mL volumetric flask, dissolve in and dilute with water to volume, and mix.
Chromatographic system (see Chromatography 621)—The liquid chromatograph is equipped with a refractive index detector and a 7.8-mm × 30-cm column that contains packing L34. The column is maintained at 85, and the flow rate is about 0.7 mL per minute. Chromatograph the Standard preparation, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 1.0% for lactitol.
Procedure— Separately inject equal volumes (about 25 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the peak responses. Calculate the quantity, in mg, of C12H24O11 in the portion of Lactitol taken by the formula:
100C(rU / rS)
in which C is the concentration, in mg per mL, of USP Lactitol RS in the Standard preparation, and rU and rS are the lactitol peak responses obtained from the Assay preparation and the Standard preparation, respectively.
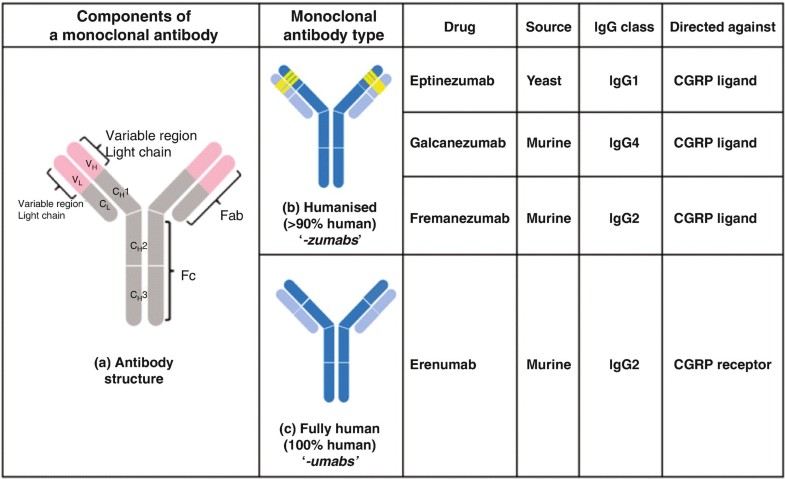
Immunoglobulin G1, anti-(calcitonin gene-related peptide) (human-oryctolagus cuniculus monoclonal ALD403 heavy chain), disulfide with human-oryctolagus cuniculus monoclonal ALD403 kappa-chain, dimer
Approved 2020 fda
ALD403, UNII-8202AY8I7H
Humanized anti-calcitonin gene-related peptide (CGRP) IgG1 antibody for the treatment of migraine.
Eptinezumab, sold under the brand name Vyepti, is a medication for the preventive treatment of migraine in adults.[2] It is a monoclonal antibody that targets calcitonin gene-related peptides (CGRP) alpha and beta.[3][4] It is administered by intravenous infusion every three months.[2]
Eeptinezumab-jjmr was approved for use in the United States in February 2020.[5]
^Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, et al. (November 2014). “Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial”. The Lancet. Neurology. 13 (11): 1100–1107. doi:10.1016/S1474-4422(14)70209-1. PMID25297013.
Alder BioPharmaceuticals has submitted a biologics license application (BLA) for eptinezumab, a humanized IgG1 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), for migraine prevention. If the US Food and Drug Administration grants approval, Alder will be on track to launch the drug in Q1 2020. The BLA included data from the PROMISE 1 and PROMISE 2 studies, which evaluated the effects of eptinezumab in episodic migraine patients (n=888) or chronic migraine patients (n=1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies.
Alder announced one-year results from the PROMISE 1 studyin June 2018, which indicated that, following the first quarterly infusion, episodic migraine patients treated with 300 mg eptinezumab experienced 4.3 fewer monthly migraine days (MMDs) from a baseline of 8 MMDs, compared to 3.2 fewer MMDs for placebo from baseline (p= 0.0001). At one year after the third and fourth quarterly infusions, patients treated with 300 mg eptinezumab experienced further gains in efficacy, with a reduction of 5.2 fewer MMDs compared to 4.0 fewer MMDs for placebo-treated patients. In addition, ~31% of episodic migraine patients achieved, on average per month, 100% reduction of migraine days from baseline compared to ~ 21% for placebo. New 6-month results from the PROMISE 2 study were also released in June 2018. These results indicated that, after the first quarterly infusion, chronic migraine patients dosed with 300 mg of eptinezumab experienced 8.2 fewer MMDs, from a baseline of 16 MMDs, compared to 5.6 fewer MMDs for placebo from baseline (p <.0001). A further reduction in MMDs was seen following a second infusion; 8.8 fewer MMDs for patients dosed with 300 mg compared to 6.2 fewer MMDs for those with placebo. In addition, ~ 21% of chronic migraine patients achieved, on average, 100% reduction of MMDs from baseline compared to 9% for placebo after two quarterly infusions of 300 mg of eptinezumab.
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....








































 Alder BioPharmaceuticals has submitted a biologics license application (BLA) for eptinezumab, a humanized IgG1 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), for migraine prevention. If the US Food and Drug Administration grants approval, Alder will be on track to launch the drug in Q1 2020. The BLA included data from the PROMISE 1 and PROMISE 2 studies, which evaluated the effects of eptinezumab in episodic migraine patients (n=888) or chronic migraine patients (n=1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies.
Alder BioPharmaceuticals has submitted a biologics license application (BLA) for eptinezumab, a humanized IgG1 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), for migraine prevention. If the US Food and Drug Administration grants approval, Alder will be on track to launch the drug in Q1 2020. The BLA included data from the PROMISE 1 and PROMISE 2 studies, which evaluated the effects of eptinezumab in episodic migraine patients (n=888) or chronic migraine patients (n=1,072), respectively. In PROMISE 1, the primary and key secondary endpoints were met, and the safety and tolerability were similar to placebo, while in PROMISE 2, the primary and all key secondary endpoints were met, and the safety and tolerability was consistent with earlier eptinezumab studies.

{kind=link}
{kind=link}
{kind=link}