
Home » Posts tagged 'Antineoplastic' (Page 3)
Tag Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bosmolisib
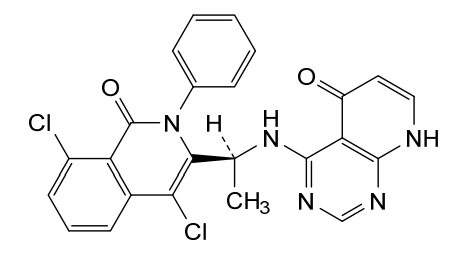
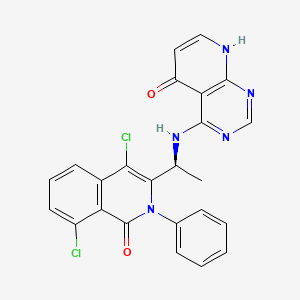
Bosmolisib
CAS 2055765-77-8
MF 2055765-77-8 MW478.3 g/mol
4-{[(1S)-1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl]amino}pyrido[2,3-d]pyrimidin-5(8H)-one
4-[[(1S)-1-(4,8-dichloro-1-oxo-2-phenylisoquinolin-3-yl)ethyl]amino]-8H-pyrido[2,3-d]pyrimidin-5-one
phosphatidylinositol 3-kinase (PI3K) inhibitor, antineoplastic, BR 101801, FJ5CTS1VNJ
- A Study of Bosmolisib (BR101801) in Participants With R/R PTCL.CTID: NCT07180771Phase: Phase 2Status: Not yet recruitingDate: 2025-09-18
- BR101801 in Adult Patients With Advanced Hematologic Malignancies(Phase I)CTID: NCT04018248Phase: Phase 1Status: CompletedDate: 2025-09-10
Bosmolisib is an orally bioavailable inhibitor of phosphoinositide 3-kinase delta (PI3-kinase subunit delta; PI3K-delta; PI3Kdelta) and DNA-dependent protein kinase (DNA-PK), with potential antineoplastic and immunomodulating activities. Upon oral administration, bosmolisib inhibits the activity of both PI3K-delta and DNA-PK. This prevents PI3K-mediated signaling pathways and may lead to the inhibition of cancer cell growth in PI3K-overexpressing tumor cells. Specifically, since PI3K regulates c-myc expression, inhibition of PI3K signaling may lead to a decrease in proliferation of c-myc-expressing tumor cells. Also, by inhibiting the activity of DNA-PK, bosmolisib interferes with the non-homologous end joining (NHEJ) process and prevents the repair of DNA double strand breaks (DSBs) caused by ionizing radiation or chemotherapeutic treatment. This increases chemo- and radiotherapy cytotoxicity by inhibiting the ability of tumor cells to repair damaged DNA. The PI3K pathway is upregulated in a variety of tumors and plays an important role in regulating cancer cell proliferation, growth, and survival. DNA-PK is activated upon DNA damage and plays a key role in repairing double-stranded DNA breaks. The enhanced ability of tumor cells to repair DSBs plays a major role in the resistance of tumor cells to chemo- and radiotherapy. In addition, bosmolisib is able to decrease Tregs and increase CD8 lymphocytes.
- OriginatorBoryung Pharmaceutical
- ClassAntineoplastics; Small molecules
- Mechanism of ActionDNA-activated protein kinase inhibitors; Phosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase IHaematological malignancies
- PreclinicalColorectal cancer
- 18 Sep 2025Boryung Pharmaceutical plans a phase II trial for Peripheral T Cell Lymphoma and Nodal T-follicular helper cell lymphoma (Second-line therapy or greater) in September 2025 (PO, Capsule) (NCT07180771)
- 06 Jan 2025Chemical structure information added.
- 09 Dec 2023Updated efficacy and adverse event data from a phase I trial in Hematological malignancies presented at the 65th American Society of Hematology Annual Meeting and Exposition (ASH-2023
SYN
WO 2016/204429.
SYN
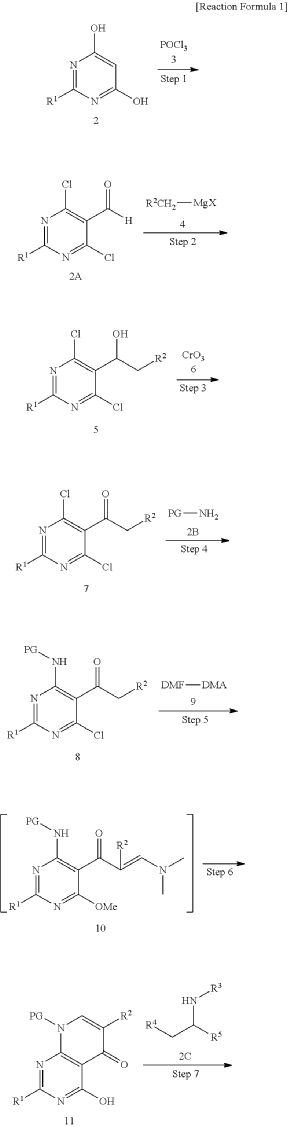
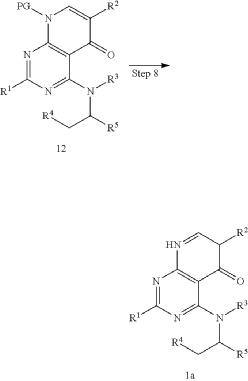
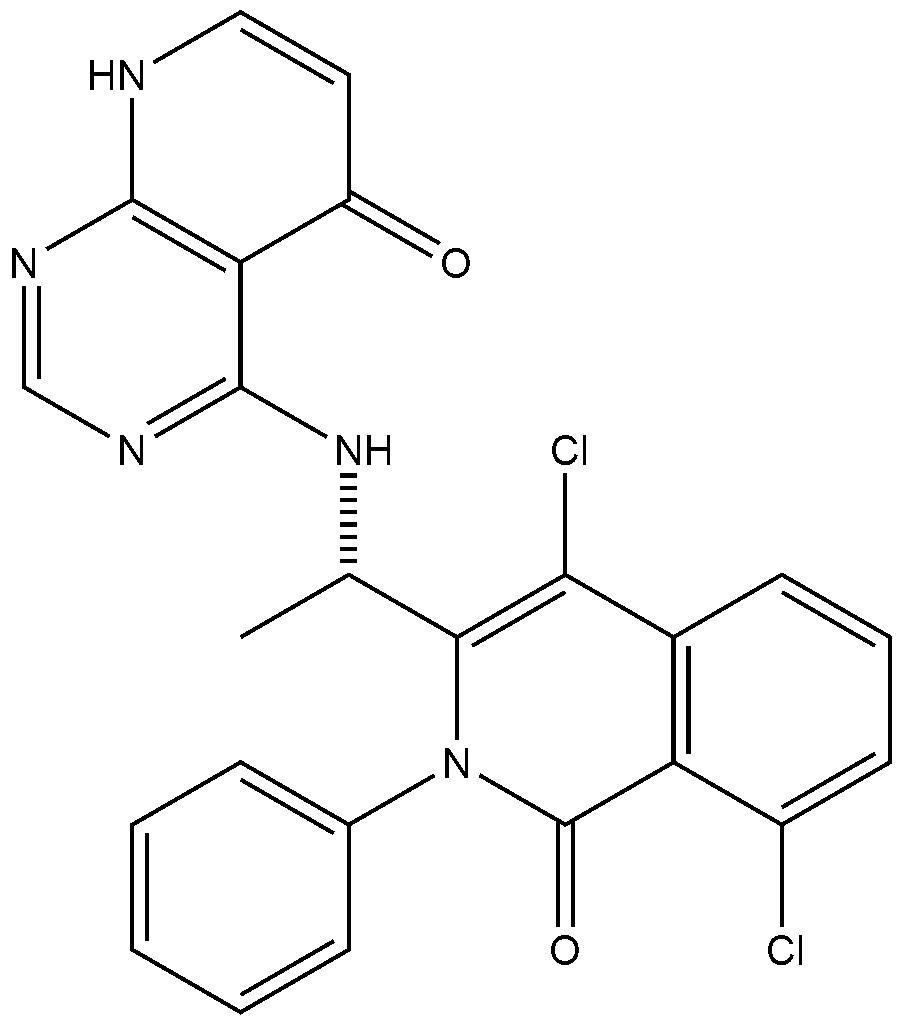
xample 1. Preparation of (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one
[116](S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one (4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin -3-yl)ethyl)amino)pyrido[2,3-d]pyrimidin-5(8H)-one) represented by the chemical formula 3 above was prepared by the same method as that described in Example 10 of International Patent Publication No.
WO 2016/204429.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016204429&_cid=P22-MK6A2W-95428-1
<Example 10> Preparation of (S)-4-((l-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido [2,3-d]pyrimidin-5(8H)-one
In Example 5, 50 mg (0.113 vol) of (S)-4— ((1-(8-chloro-1—oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido [2, 3-d]pyrimidin-5(8H)-one prepared was dissolved in 2 mL of acetic acid, and then 17 mg (0.124 vol) of N—chlorosuccinimide (NCS) was added. The mixture was stirred at 50 ° C for 15 hours, filtered under reduced pressure, neutralized using an aqueous sodium bicarbonate solution, and then the organic layer extracted by adding dichloromethane and water was dried (Na 2 SO 4 ), filtered, concentrated under reduced pressure, and separated by column chromatography (SiO 2 , eluent: dichloromethane/methanol, 30/1 -> dichloromethane/methanol, 10/1) to afford 25 mg (0.052 mmol, 46% yield) of compound (S)— 4-((1— (4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinolin-3-yl)ethyl)amino)pyrido[2, 3-d]pyramidin-5(8H)-one as a pale yellow solid.
LH NMR (300 MHz, CDC13) δ 10.99 (d, J = 4.8 Hz, 1Ή), 8.25 (s, 1H) , 7.95(dd, JJ = 1.9 Hz, J = 7.5 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H) , 7.46-7.62 (m, 6H), 7.20 (d, J = 6.7 Hz, 1H) , 6.3 (d, J = 7.5 Hz, 1H), 5.04 (t , J = 67.2 Hz, 1H) , 1.67 (d, J = 7.2 Hz, 3H) .
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US214732247&_cid=P22-MK69S5-86256-1
Example 10: Preparation of (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one
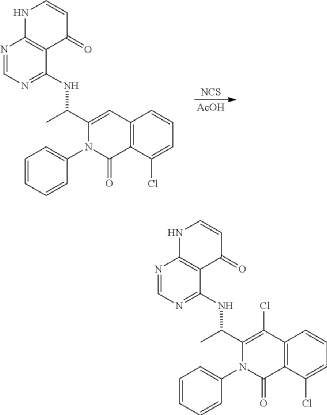
| 50 mg (0.113 mmol) of (S)-4-((1-(8-chloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one prepared in Example 5 was dissolved in 2 mL of acetic acid, to which 17 mg (0.124 mmol) of N-chlorosuccinimide (NCS) was added, followed by stirring at 50° C. for 15 hours. The reaction mixture was filtered under reduced pressure. Saturated sodiumbicarbonate aqueous solution was added thereto, followed by neutralization. Dichloromethane and water were added thereto, followed by extraction. The extracted organic layer was dried (Na 2SO 4), filtered, and concentrated under reduced pressure. The residue was separated by column chromatography (SiO 2, eluent: dichloromethane/methanol, 30/1→dichloromethane/methanol, 10/1) to give 25 mg of the target compound (S)-4-((1-(4,8-dichloro-1-oxo-2-phenyl-1,2-dihydroisoquinoline-3-yl)ethyl)amino)pyrido[2,3-d]pyrimidine-5(8H)-one as a pale yellow solid (0.052 mmol, yield: 46%). |
PAT
- A pharmaceutical composition for preventing or treating a heteroaryl derivative or a pharmaceutically acceptable salt thereof, a method for producing the same, and a PI3 kinase-related disease containing the heteroaryl derivative as an active ingredient.Publication Number: JP-6808905-B2Priority Date: 2015-06-18Grant Date: 2021-01-06
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, method of preparation thereof and pharmaceutical composition to prevent or treat diseases associated with PI3 kinases, which contains the same as active principlePublication Number: ES-2816050-T3Priority Date: 2015-06-18Grant Date: 2021-03-31
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical compostion for preventing or treating diseases associated with pi3 kinases, containing same as active ingredientPublication Number: US-2018105527-A1Priority Date: 2015-06-18
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical composition for preventing or treating diseases associated with pi3 kinases, containing same as active ingredientPublication Number: EP-3312175-B1Priority Date: 2015-06-18Grant Date: 2020-07-22
- Heteroaryl derivatives or pharmaceutically acceptable salts thereof, preparation method thereof and pharmaceutical composition for use in preventing or treating pi3 kinase related diseasesPublication Number: TW-I616446-BPriority Date: 2015-06-18Grant Date: 2018-03-01
- HETEROARYL DERIVATIVES OR PHARMACEUTICALLY ACCEPTABLE SALTS THEREOF, PROCESS FOR PRODUCING THE SAME, AND PHARMACEUTICAL COMPOSITIONS FOR PREVENTING OR TREATING PI3-KINASE RELATED DISEASES COMPRISING THE SAME AS THE ACTIVE INGREDIENTPublication Number: JP-2018522852-APriority Date: 2015-06-18
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method therefor, and pharmaceutical composition for preventing or treating diseases associated with PI3 kinases, containing same as active ingredientPublication Number: US-10526337-B2Priority Date: 2015-06-18Grant Date: 2020-01-07
- Heteroaryl derivative or a pharmaceutically acceptable salt thereof, a method for production thereof and a pharmaceutical composition for preventing or treating diseases associated with pi3 kinases, containing said active substancePublication Number: RU-2719367-C2Priority Date: 2015-06-18Grant Date: 2020-04-17
- Heteroaryl derivative or pharmaceutically acceptable salt thereof, preparation method thereof, and pharmaceutical composition comprising same as active ingredient for preventing or treating PI3 kinase-associated diseasesPublication Number: CN-107690433-APriority Date: 2015-06-18



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- PI3Kδ/γ inhibitor BR101801 extrinsically potentiates effector CD8+ T cell-dependent antitumor immunity and abscopal effect after local irradiationPublication Name: Journal for ImmunoTherapy of CancerPublication Date: 2022-03PMCID: PMC8921929PMID: 35288465DOI: 10.1136/jitc-2021-003762
- Synergistic radiosensitizing effect of BR101801, a specific DNA-dependent protein kinase inhibitor, in various human solid cancer cells and xenograftsPublication Name: American journal of cancer researchPublication Date: 2021PMCID: PMC8640799PMID: 34873471
/////////bosmolisib, phosphatidylinositol 3-kinase (PI3K) inhibitor, antineoplastic, BR 101801, FJ5CTS1VNJ
Beroterkib


Beroterkib
CAS 2095719-92-7
MF C29H31ClFN5O5 MW584.0 g/mol
(2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1,3-dihydro-2H-1-oxoisoindol-2-yl) -N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
(2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
(2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide
extracellular signal-regulated kinases (ERK) inhibitor, antineoplastic, ASTX029, ASTX 029, 14FDK6ISC9, Beroterkib anhydrous, AT 35029
Beroterkib Anhydrous is the anhydrous form of beroterkib, an orally bioavailable inhibitor of the extracellular signal-regulated kinases (ERK) 1 and 2, with potential antineoplastic activity. Upon administration, beroterkib specifically binds to and inhibits both ERK 1 and 2, thereby preventing the activation of mitogen-activated protein kinase (MAPK)/ERK-mediated signal transduction pathways. This results in the inhibition of ERK-dependent tumor cell proliferation and survival. The MAPK/ERK pathway is often upregulated in a variety of tumor cell types and plays a key role in the proliferation, differentiation and survival of tumor cells.
- Study of ASTX029 in Subjects With Advanced Solid TumorsCTID: NCT03520075Phase: Phase 1/Phase 2Status: CompletedDate: 2025-07-03
- Phase I/II Study of a Combination of Decitabine and Cedazuridine (ASTX727) and ASTX029, an ERK Inhibitor, for Patients With RAS Pathway Mutant Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative NeoplasmsCTID: NCT06284460Phase: Phase 1/Phase 2Status: WithdrawnDate: 2024-10-24
- A Phase 1 Study to Evaluate the Effect of Food on Pharmacokinetics of ASTX029CTID: NCT04466514Phase: Phase 1Status: CompletedDate: 2024-08-02
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017068412&_cid=P21-MK4TZX-17603-1
Example 685: (2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1-oxo-2,3-dihydro- 1H-isoindol-2-yl)-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide

A stirred solution of (R)-2-(6-(5-chloro-2-((oxan-4-yl)amino)pyrimidin-4-yl)-1-oxoisoindolin-2- yl)propanoic acid (70 mg, 0.168 mmol), (S)-2-amino-2-(3-fluoro-5-methoxyphenyl)ethanol, HCl (41 mg, 0.185 mmol) and triethylamine (0.094 ml, 0.672 mmol) in DMF (1 ml) was treated with TBTU (65 mg, 0.202 mmol) and stirred at room temperature overnight. The mixture was diluted with ethyl acetate (20 ml), was washed successively with 1M KHSO4 (10 ml), NaHCO3 (10 ml), brine (2x 10 ml) and then water (4x 10 ml), was dried (MgSO4) and evaporated. The residue was purified by chromatography (SiO2, 12 g column, 0- 5% EtOOH in EtOAc) to give a glass, which was triturated with ether (2 ml) to give a solid. The solid was collected by filtration, washed with ether (2x 1 ml) and dried under vacuum at 50°C overnight to give the titlecompound (64.3 mg, 64.3 %) as a cream solid. 1H NMR (DMSO, 400 MHz) δ 8.56 (1H, d), 8.44 (1H, s), 8.07 ‒ 8.00 (1H, m), 7.97 (1H, dd), 7.74 (1H, d), 7.61 (1H, s), 6.76 ‒ 6.64 (3H, m), 4.99 (1H, q), 4.91 (1H, t), 4.86 ‒ 4.70 (2H, m), 4.60 (1H, d), 4.00 ‒ 3.80 (3H, m), 3.76 (3H, s), 3.60 ‒ 3.47 (2H, m), 3.40 ‒ 3.33 (2H, m), 1.84 (2H, d), 1.59 ‒ 1.39 (5H, m). ). LCMS: [M+H]+ = 584.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US237389744&_cid=P21-MK4U5F-21416-1
Example 685: (2R)-2-(6-{5-chloro-2-[(oxan-4-yl)amino]pyrimidin-4-yl}-1-oxo-2,3-dihydro-1H-isoindol-2-yl)-N-[(1S)-1-(3-fluoro-5-methoxyphenyl)-2-hydroxyethyl]propanamide

SYN

PAT
- Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparationPublication Number: AU-2011276120-B2Priority Date: 2010-07-09Grant Date: 2013-12-19
- Conjugates comprising hydroxyalkyl starch and a cytotoxic agent and process for their preparationPublication Number: AU-2011276120-A1Priority Date: 2010-07-09
- Combustion modified flexible polyurethane foamPublication Number: GB-2124634-APriority Date: 1982-07-26
- Benzolactam compounds as protein kinase inhibitorsPublication Number: ES-2989326-T3Priority Date: 2015-10-21Grant Date: 2024-11-26
- Protein kinase inhibitor benzolactam compoundsPublication Number: CN-114948963-APriority Date: 2015-10-21
- Benzolactam compounds as protein kinase inhibitorsPublication Number: US-2024368136-A1Priority Date: 2015-10-21
- Protein Kinase Inhibitors Benzolactam CompoundsPublication Number: CN-108617166-BPriority Date: 2015-10-21Grant Date: 2022-05-17
- Benzolactam compounds as protein kinase inhibitorsPublication Number: CN-114948963-BPriority Date: 2015-10-21Grant Date: 2025-05-27
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
REF
- Discovery of ASTX029, a clinical candidate which modulates the phosphorylation and catalytic activity of ERK1/2Publication Name: Journal of Medicinal ChemistryPublication Date: 2021-10-06PMID: 34387469DOI: 10.1021/acs.jmedchem.1c00905
- ASTX029, a Novel Dual-mechanism ERK Inhibitor, Modulates Both the Phosphorylation and Catalytic Activity of ERKPublication Name: Molecular Cancer TherapeuticsPublication Date: 2021-07-30PMID: 34330842DOI: 10.1158/1535-7163.mct-20-0909
//////////////Beroterkib, extracellular signal-regulated kinases (ERK) inhibitor, antineoplastic, ASTX029, ASTX 029, 14FDK6ISC9, Beroterkib anhydrous, AT 35029
Atirmociclib



Atirmociclib
CAS 2380321-51-5
MF C22H27ClFN5O3,
463.9 g/mol
(3S,4R)-4-[[5-chloro-4-[7-fluoro-2-(2-hydroxypropan-2-yl)-3-propan-2-ylbenzimidazol-5-yl]pyrimidin-2-yl]amino]oxan-3-ol
(3S,4R)-4-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan2-yl)-1H-1,3-benzimidazol-6-yl]pyrimidin-2-yl}amino)oxan-3-ol
1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxpropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol
D-threo-Pentitol, 1,5-anhydro-3-[[5-chloro-4-[4-fluoro-2-(1-hydroxy-1-methylethyl)-1-(1-methylethyl)-1H-benzimidazol-6-yl]-2-pyrimidinyl]amino]-2,3-dideoxy-
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, PF 07220060, S743GOJ5LJ, CDK4/6-IN-6
Atirmociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 4 (CDK4), with potential antineoplastic activity. Upon administration, atirmociclib selectively inhibits CDK4, which inhibits the phosphorylation of retinoblastoma protein (Rb) early in the G1 phase, prevents CDK-mediated G1-S-phase transition and leads to cell cycle arrest. This suppresses DNA replication and inhibits tumor cell proliferation. CDK4, a serine/threonine kinase, is upregulated in many tumor cell types and plays a key role in the regulation of both cell cycle progression from the G1-phase into the S-phase and tumor cell proliferation.
Atirmociclib (development code PF-07220060) is an investigational orally bioavailable and CDK4-specific inhibitor being developed by Pfizer for the treatment of various solid tumors, particularly hormone receptor-positive, HER2-negative breast cancer.[1][2] The safety and efficacy of atirmociclib have not been established, as it remains in clinical development as of September 2025.[3][4][5]
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.5c02137


PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481329&_cid=P22-MK0K3I-13424-1

Example A94 (Scheme A-15): Preparation of 1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol

Step 8: Synthesis of 1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol (Example A94)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481329&_cid=P22-MK0KHW-23947-1

PAT
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: KR-102661053-B1Priority Date: 2018-04-26Grant Date: 2024-04-26
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: KR-20230152182-APriority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-11220494-B2Priority Date: 2018-04-26Grant Date: 2022-01-11
- CYCLINE-DEPENDENT KINASE INHIBITORSPublication Number: PE-20201202-A1Priority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-2022089580-A1Priority Date: 2018-04-26
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: HR-P20250254-T1Priority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-12378232-B2Priority Date: 2018-04-26Grant Date: 2025-08-05
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: EP-3784664-B1Priority Date: 2018-04-26Grant Date: 2025-02-19
- 2-Amino-pyridine or 2-amino-pyrimidine derivatives as cyclin-dependent kinase inhibitorsPublication Number: CN-112313219-BPriority Date: 2018-04-26Grant Date: 2024-04-26
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Mechanism of action
Atirmociclib is designed as a CDK4-specific inhibitor, distinguishing it from dual CDK4/6 inhibitors currently approved for cancer treatment.[6] The drug targets cyclin-dependent kinase 4, which plays a role in cell cycle regulation.[1][7][8]
Atirmociclib functions as a selective inhibitor of the CDK4/cyclin D complex, which plays a crucial role in cell cycle regulation.[4] The drug works by targeting the CDK4 kinase, rendering the retinoblastoma (Rb)/E2F transcription system inactive, which ultimately leads to cell cycle arrest in the G1 phase.[4] This mechanism is particularly effective in tumors that have lost Rb cell cycle-suppressive function, a common feature in various solid tumors.[5]
The selective nature of atirmociclib represents a significant advancement over existing dual CDK4/6 inhibitors.[6] By specifically targeting CDK4 while limiting CDK6 inhibition, atirmociclib is designed to maintain antitumor efficacy while potentially reducing dose-limiting hematologic toxicities, particularly neutropenia, which is believed to be primarily driven by CDK6 inhibition.[9]
Clinical development
Atirmociclib is currently being evaluated in clinical trials for the treatment of advanced solid tumors.[1] Clinical studies are ongoing with estimated completion dates extending to 2027–2028, reflecting the early stage of development for this investigational compound.[1]
Preclinical research published in Cancer Cell in March 2025 reported atirmociclib as a next-generation CDK4-selective inhibitor with enhanced anti-tumor activity and reduced predicted toxicity compared to FDA-approved dual CDK4/6 inhibitors, though these findings require validation in clinical studies.[6]
Preclinical studies
Preclinical research has demonstrated that atirmociclib exhibits enhanced anti-tumor activity compared to FDA-approved dual CDK4/6 inhibitors while showing reduced predicted toxicity.[6] Studies have shown that CDK4-selective inhibition can provide improved preclinical anti-tumor efficacy and safety profiles compared to dual CDK4/6 inhibition strategies.[10]
The preclinical development program has explored combination approaches with various therapeutic modalities, including endocrine therapy, CDK2 inhibition, HER2 antibodies, and immune checkpoint inhibitors.[6] These combination strategies are designed to counter resistance mechanisms to CDK4 inhibition and expand the potential therapeutic applications of cell cycle targeting therapy.[6]
Clinical trials
Atirmociclib has entered clinical development as part of Pfizer’s extensive oncology pipeline.[11] The clinical program is evaluating atirmociclib both as a single agent and in combination with other therapeutic approaches, particularly focusing on patients with hormone receptor-positive, HER2-negative breast cancer.[9][12][13][14][15][16][17]
Early clinical studies have included heavily pretreated patient populations, including those who have previously received CDK4/6 inhibitor therapy.[9] This approach allows for the evaluation of atirmociclib’s potential to overcome resistance to existing CDK4/6 inhibitors and provide therapeutic benefit in patients with limited treatment options.[9]
Safety profile and toxicity
One of the key differentiating features of atirmociclib is its potential for improved safety profile compared to existing dual CDK4/6 inhibitors.[6] The selective targeting of CDK4 while limiting CDK6 inhibition is specifically designed to reduce neutropenia, the most common dose-limiting toxicity associated with current CDK4/6 inhibitors.[18]
The rationale for this approach is based on preclinical evidence suggesting that neutropenia is primarily driven by CDK6 inhibition rather than CDK4 inhibition.[18] By selectively targeting CDK4, atirmociclib aims to maintain therapeutic efficacy while potentially allowing for higher or more sustained dosing without the dose-limiting hematologic toxicities that can compromise treatment outcomes with existing agents.[18]
Regulatory status
As of September 2025, atirmociclib remains an investigational drug that has not received approval from the FDA or other regulatory agencies.[5] The compound is part of Pfizer’s oncology development pipeline.[5]
References
- Pfizer (2 February 2025). A Phase 1/2A Study Evaluating the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Anti-Tumor Activity of Pf-07220060 as a Single Agent and as Part of Combination Therapy in Participants With Advanced Solid Tumors (Report). clinicaltrials.gov.
- Shapiro GI (March 2017). “The evolving role of cyclin-dependent kinase inhibitors in cancer management”. Clinical Advances in Hematology & Oncology. 15 (3): 174–177. PMID 28398270.
- “CDK4 inhibitor PF-07220060”. http://www.cancer.gov. 2 February 2011. Retrieved 3 September 2025.
- “Pfizer Pipeline”. Pfizer.
- “Atirmociclib PF-07220060”. Pfizer Oncology Development. Retrieved 3 September 2025.
- Chang J, Lu J, Liu Q, Xiang T, Zhang S, Yi Y, et al. (March 2025). “Single-cell multi-stage spatial evolutional map of esophageal carcinogenesis”. Cancer Cell. 43 (3): 380–397.e7. doi:10.1016/j.ccell.2025.02.009. PMID 40068596.
- Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, et al. (May 2019). “Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix”. Molecular Cell. 74 (4): 758–770.e4. doi:10.1016/j.molcel.2019.03.020. PMC 6800134. PMID 30982746.
- Helsten T, Kato S, Schwaederle M, Tomson BN, Buys TP, Elkin SK, et al. (July 2016). “Cell-Cycle Gene Alterations in 4,864 Tumors Analyzed by Next-Generation Sequencing: Implications for Targeted Therapeutics”. Molecular Cancer Therapeutics. 15 (7): 1682–1690. doi:10.1158/1535-7163.MCT-16-0071. PMID 27196769.
- “ESMO 2024 – combos could be the way forward for CDK2”. ApexOnco. 15 September 2024.
- Palmer CL, Boras B, Pascual B, Li N, Li D, Garza S, et al. (March 2025). “CDK4 selective inhibition improves preclinical anti-tumor efficacy and safety”. Cancer Cell. 43 (3): 464–481.e14. doi:10.1016/j.ccell.2025.02.006. PMID 40068598.
- “Pfizer Highlights Diverse Oncology Portfolio and Combination Approaches at ESMO 2024”. Pfizer. 2024.
- Pfizer (12 August 2025). A Phase 1/2a Dose Escalation and Expansion Study to Evaluate Safety, Tolerability, Pharmacokinetic, Pharmacodynamic, and Anti-Tumor Activity of Pf-07248144 in Participants With Advanced or Metastatic Solid Tumors (Report). clinicaltrials.gov.
- Pfizer (2 July 2025). An Interventional Safety and Efficacy Phase 1/2, Open-Label Study to Investigate Tolerability, Pk, and Antitumor Activity of Vepdegestrant (Arv-47/Pf-07850327), an Oral Proteolysis Targeting Chimera, in Combination With Pf-07220060 in Participants Aged 18 Years and Older With Er+/her2- Advanced or Metastatic Breast Cancer (Report). clinicaltrials.gov.
- Pfizer (14 November 2024). A Phase 1/2, Open-Label, Multicenter, Dose Escalation and Dose Expansion Study to Evaluate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Antitumor Activity of PF-07220060 in Combination With Pf-07104091 Plus Endocrine Therapy in Participants With Advanced Solid Tumors (Report). clinicaltrials.gov.
- Pfizer (17 June 2025). (FOURLIGHT-3) (Report). clinicaltrials.gov.
- Pfizer (13 March 2025). An Interventional, Open-Label, Randomized, Multicenter Phase 3 Study of PF-07220060 Plus Letrozole Compared to cdk4/6 Inhibitor Plus Letrozole in Participants Over 18 Years of Age With Hormone Receptor (Hr)-Positive, her2-Negative Advanced/Metastatic Breast Cancer Who Have Not Received Any Prior Systemic Anticancer Treatment for Advanced/Metastatic Disease (FOURLIGHT-1) (Report). clinicaltrials.gov.
- Pfizer (15 November 2024). An Interventional, Open-Label, Randomized, Multicenter, Phase 2 Study of Pf-07220060 Plus Letrozole Compared to Letrozole Alone in Postmenopausal Women 18 Years or Older With Hormone Receptor-Positive, her2-Negative Breast Cancer in the Neoadjuvant Setting (Report). clinicaltrials.gov.
- “Pfizer dials down its atirmociclib ambitions”. ApexOnco. 1 May 2025.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2380321-51-5 |
| PubChem CID | 146219790 |
| ChemSpider | 115009592 |
| UNII | S743GOJ5LJ |
| KEGG | D12834 |
| ChEMBL | ChEMBL5187755 |
| Chemical and physical data | |
| Formula | C22H27ClFN5O3 |
| Molar mass | 463.94 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Atirmociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, PF 07220060, S743GOJ5LJ, CDK4/6-IN-6
Asaretoclax


Asaretoclax
CAS 2363074-01-3
MF C47H57F2N7O7S, MW 902.1 g/mol
4-[4-[[2-[3-(difluoromethyl)-1-bicyclo[1.1.1]pentanyl]-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[4-[(4-hydroxy-4-methylcyclohexyl)methylamino]-3-nitrophenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)-N-((4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide

B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, GY6FD5FXA3, HY 159817, ABT 263
Asaretoclax is an orally bioavailable inhibitor of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2), with potential pro-apoptotic and antineoplastic activities. Upon oral administration, asaretoclax targets, binds to and inhibits the activity of Bcl-2. This restores apoptotic processes in tumor cells. Bcl-2 is overexpressed in many cancers and plays an important role in the negative regulation of apoptosis; its expression is associated with increased drug resistance and tumor cell survival.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US309776623&_cid=P21-MJZ42N-73938-1
Example 34
2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1l-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)-N-((4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrophenyl)sulfonyl)benzamide
Intermediate 18
Intermediate 18
4-((((1r,4r)-4-hydroxy-4-methylcyclohexyl)methyl)amino)-3-nitrobenzenesulfonamide

Intermediate 18 was prepared following a procedure described in WO2014/165044A1. LC/MS (ESI) m/z 344.1 [M+H] +.
Intermediate 30
Intermediate 30
2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)benzoic Acid
| Step 1: Methyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((2-(3-(difluoromethyl)bicyclo[1.1.1]pentan-1-yl)-4,4-dimethylcyclohex-1-en-1-yl)methyl)piperazin-1-yl)benzoate (Intermediate 30-1) was prepared following the procedure described in Step 1, Route C for Intermediate 28 using Intermediate 24 in place of Intermediate 22. LCMS (ESI) m/z 591.2 [M+H] +. |


Example 34 was prepared following General Procedure A using Intermediate 30 and Intermediate 18. 1H NMR (400 MHz, DMSO-d 6) δ 11.70 (s, 1H), 11.40 (br s, 1H), 8.59-8.49 (m, 2H), 8.04 (d, J=2.0 Hz, 1H), 7.78 (d, J=8.8 Hz, 1H), 7.53-7.48 (m, 3H), 7.06 (d, J=9.2 Hz, 1H), 6.72 (d, J=7.2 Hz, 1H), 6.38 (s, 1H), 6.25 (s, 1H), 5.99 (t, J=56.8 Hz, 1H), 4.25 (s, 1H), 3.33-3.25 (m, 2H), 3.18-3.05 (m, 4H), 2.97 (s, 2H), 2.40-2.28 (m, 4H), 2.05-1.95 (m, 2H), 1.94 (s, 6H), 1.71-1.59 (m, 5H), 1.58-1.49 (m, 2H), 1.39-1.28 (m, 2H), 1.27-1.20 (m, 2H), 1.18-1.09 (m, 2H), 1.10 (s, 3H), 0.83 (s, 6H); LC/MS (ESI) m/z 902.6 [M+H] +.
SYN
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US384526484&_cid=P21-MJZ3XL-69589-1
PAT
Publication Number: US-2021009543-A1
Priority Date: 2018-01-10
- Benzamide compoundsPublication Number: CN-118084904-APriority Date: 2018-01-10
- Benzamide compoundsPublication Number: EP-4556469-A1Priority Date: 2018-01-10
- Benzamide compounds as bci inhibitors for the treatment of hivPublication Number: EP-3740487-B1Priority Date: 2018-01-10Grant Date: 2025-01-08
- Benzamide compoundsPublication Number: US-11344546-B2Priority Date: 2018-01-10Grant Date: 2022-05-31
- Benzamide compoundsPublication Number: US-11318134-B2Priority Date: 2018-01-10Grant Date: 2022-05-03
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Asaretoclax, B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, GY6FD5FXA3, HY 159817, ABT 263
Zomiradomide


Zomiradomide

CAS 2655656-99-6
MF C45H48F3N7O6S MW871.97

- N-[2-[4-[[6-[2-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]amino]ethyl]-2-azaspiro[3.3]heptan-2-yl]methyl]cyclohexyl]-5-(2-hydroxypropan-2-yl)-1,3-benzothiazol-6-yl]-6-(trifluoromethyl)pyridine-2-carboxamide
- N-[2-[trans-4-[[6-[2-[[2-(2,6-Dioxo-3-piperidinyl)-2,3-dihydro-1,3-dioxo-1H-isoindol-4-yl]amino]ethyl]-2-azaspiro[3.3]hept-2-yl]methyl]cyclohexyl]-5-(1-hydroxy-1-methylethyl)-6-benzothiazolyl]-6-(trifluoromethyl)-2-pyridinecarboxamide
antineoplastic, IRAK degrader-1, AQ5UXV5646
Zomiradomide is an orally active PROTAC degrader for IRAK4 (DC50=6 nM), thereby inhibiting the NF-κB signaling pathway. Zomiradomide acts also as a molecular glue, recruiting Ikaros and Aiolos, and mediating their degradation (DC50 for Ikaros is 1 nM), thereby activating the type I IFN signaling pathway.
Zomiradomide is a small molecule protein degrader of interleukin-1 receptor-associated kinase 4 (IRAK4) and the immunomodulatory imide drug (IMiD) substrates Ikaros (IKZF1) and Aiolos (IKZF3), with potential immunomodulating and antineoplastic activities. Upon administration, zomiradomide modulates the E3 (ubiquitin) ligase and targets IRAK4, Ikaros and Aiolos for ubiquitination. This induces proteasome-mediated degradation of IRAK4, Ikaros and Aiolos. The degradation of IRAK4 inhibits IRAK4-mediated signaling and prevents the activation of IRAK4-mediated nuclear factor-kappa B (NF-kB) signaling and decreases the expression of inflammatory cytokines and certain pro-survival factors. This inhibits the proliferation of IRAK4-overactivated tumor cells, which are found in cells harboring MYD88 activating mutations or those with overactivated toll-like receptor (TLR) pathways. The degradation of the transcription factors Ikaros and Aiolos leads to a downregulation of other proteins, including interferon regulatory factor 4 (IRF4), which upregulates type I interferon signaling and further inhibits NF-kB activation. This leads to apoptosis and the inhibition of tumor cell proliferation. IRAK4, a serine/threonine-protein kinase that plays a key role in both the TLR and IL-1R signaling pathways, is activated though the adaptor protein MYD88 and links the TLR and IL-1R signaling pathway to the NF-kB pathway.

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022027058&_cid=P20-MJGJKA-81687-1
Example 1. Synthesis of N-[2-[4-[[6-[2-[[2-(2,6-dioxo-3-piperidyl)-1,3-dioxo-isoindolin-4-yl]amino]ethyl]-2- azaspiro[3.3]heptan-2-yl]methyl]cyclohexyl]-5-(1-hydroxy-1-methyl-ethyl)-1,3-benzothiazol-6-yl]-6-(trifluoromethyl)pyridine-2-carboxamide (Compound A)

[00349] To a solution of 4-[2-(2-azaspiro[3.3]heptan-6-yl)ethylamino]-2-(2,6-dioxo-3-piperidyl)isoindoline -1,3-dione (75.8 mg, 148 umol, TFA salt, Intermediate ATH) in THF (2 mL) was added TEA (15.0 mg, 148 umol), then the mixture stirred at 25 °C for 10 min. Next, HOAc (8.92 mg, 148 umol) and N-[2-(4-formylcyclohexyl)-5-(1-hydroxy-1-methyl-ethyl)-1,3-benzothiazol-6-yl]-6-(trifluoromethyl)pyridine-2-carboxamide (73.0 mg, 148 umol, Intermediate BAX) were added to the mixture and the mixture was stirred at 25 °C for 20 minutes, then NaBH(OAc)3 (62.9 mg, 297 umol) was added to the mixture at 0 °C. The reaction mixture was stirred at 0-25 °C for 2 hours. On completion, the reaction mixture was quenched with H2O (1 mL) and concentrated in vacuo. The residue was purified by prep-HPLC (column: Phenomenex Synergi C18150*25*10 um; mobile phase: [water(0.225%FA)-ACN]; B%: 31%-58%, 9 min) to give the title compound (59.1 mg, 41% yield) as a yellow solid.1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 11.09 (s, 1H), 9.06 (s, 1H), 8.49 – 8.44 (m, 1H), 8.38 (t, J = 8.0 Hz, 1H), 8.19 (d, J = 8.0 Hz, 1H), 7.88 (s, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.10 – 6.99 (m, 2H), 6.47 (t, J = 5.6 Hz, 1H), 6.07 (s, 1H), 5.05 (dd, J = 5.6, 12.8 Hz, 1H), 3.54 – 3.47 (m, 2H), 3.25 – 3.18 (m, 4H), 3.06 – 2.99 (m, 1H), 2.93 – 2.83 (m, 1H), 2.63 – 2.56 (m, 1H), 2.54 (s, 3H), 2.30 – 2.21 (m, 2H), 2.30 – 2.21 (m, 3H), 2.06 – 1.99 (m, 1H), 1.88 – 1.77 (m, 4H), 1.68 – 1.61 (m, 8H), 1.58 – 1.49 (m, 2H), 1.45 – 1.36 (m, 1H), 1.15 – 1.02 (m, 2H); LC-MS (ESI+) m/z 872.2 (M+H)+.
PAT
- Irak degraders and uses thereofPublication Number: US-2024131016-A1Priority Date: 2019-12-17
- Irak degraders and uses thereofPublication Number: EP-4076520-A1Priority Date: 2019-12-17
- IRAK degraders and uses thereofPublication Number: US-11779578-B2Priority Date: 2019-12-17Grant Date: 2023-10-10
- IRAK degraders and uses thereofPublication Number: US-11707457-B2Priority Date: 2019-12-17Grant Date: 2023-07-25
- Methods of treating mutant lymphomasPublication Number: US-2024316004-A1Priority Date: 2020-07-30
- Methods of treating mutant lymphomasPublication Number: US-2022054453-A1Priority Date: 2020-07-30
- Irak degraders and uses thereofPublication Number: US-2023144292-A1Priority Date: 2019-12-17
- Irak degraders and uses thereofPublication Number: WO-2021127190-A1Priority Date: 2019-12-17
- Irak degraders and uses thereofPublication Number: US-2021228562-A1Priority Date: 2019-12-17
- Irak4 degraders and uses thereofPublication Number: EP-4463166-A1Priority Date: 2022-01-14
- Methods of treating mutant lymphomasPublication Number: WO-2022027058-A1Priority Date: 2020-07-30
- Methods of treating mutated lymphomaPublication Number: CN-116133692-APriority Date: 2020-07-30
- Methods of treating mutant lymphomasPublication Number: US-11857535-B2Priority Date: 2020-07-30Grant Date: 2024-01-02
- Methods of treating mutant lymphomasPublication Number: EP-4188374-A1Priority Date: 2020-07-30
- Irak4 degraders and uses thereofPublication Number: WO-2024191788-A1Priority Date: 2023-03-10
- Formulations for treating cancerPublication Number: WO-2024163751-A1Priority Date: 2023-02-01
- Irak4 degraders and uses thereofPublication Number: WO-2024148049-A1Priority Date: 2023-01-04
- Irak4 degraders and uses thereofPublication Number: US-2023277519-A1Priority Date: 2022-01-14
- Irak4 degraders and uses thereofPublication Number: WO-2023137439-A1Priority Date: 2022-01-14
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////zomiradomide, antineoplastic, IRAK degrader-1, AQ5UXV5646
Zemirciclib



Zemirciclib
CAS 2057509-72-3
MF C22H28ClN5O2, 429.9 g/mol
(1S,3R)-3-acetamido-N-[5-chloro-4-(5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-
yl]cyclohexane-1-carboxamide
(1S,3R)-3-acetamido-N-[5-chloro-4-(5,5-dimethyl-4,6-dihydropyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-yl]cyclohexane-1-carboxamide
(1S,3R)-3-acetamido-N-(5-chloro-4-(5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-yl)cyclohexanecarboxamide
cyclin-dependent kinase inhibitor, antineoplastic, AZD 4573, UNII-E5XSP3X68B
Zemirciclib is a selective, short-acting inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon intravenous administration, zemirciclib binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins. This induces cell cycle arrest and apoptosis, and leads to a reduction in tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA polymerase II at serine 2 (p-Ser2-RNAPII). It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
AZD-4573 is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
- AZD4573 in Novel Combinations With Anti-cancer Agents in Patients With Advanced Blood CancerCTID: NCT04630756Phase: Phase 1/Phase 2Status: CompletedDate: 2025-04-09
- AZD4573 as Monotherapy or in Combinations With Anti-cancer Agents in Patients With r/r PTCL or r/r cHLCTID: NCT05140382Phase: Phase 2Status: CompletedDate: 2024-08-28
- Study to Assess Safety, Tolerability, Pharmacokinetics and Antitumor Activity of AZD4573 in Relapsed/Refractory Haematological MalignanciesCTID: NCT03263637Phase: Phase 1Status: CompletedDate: 2021-10-22
SYN
- Large-Scale Synthesis of AZD4573Publication Name: SynfactsPublication Date: 2022-05-17DOI: 10.1055/s-0041-1738312
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
- Discovery of AZD4573, a Potent and Selective Inhibitor of CDK9 That Enables Short Duration of Target Engagement for the Treatment of Hematological MalignanciesPublication Name: Journal of Medicinal ChemistryPublication Date: 2020-12-11PMID: 33306391DOI: 10.1021/acs.jmedchem.0c01754
- A comprehensive insight on the recent development of Cyclic Dependent Kinase inhibitors as anticancer agentsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2020-10-01PMID: 32707525DOI: 10.1016/j.ejmech.2020.112571
- Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An UpdatePublication Name: Journal of Medicinal ChemistryPublication Date: 2020-08-31PMID: 32866383DOI: 10.1021/acs.jmedchem.0c00744
- AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer CellsPublication Name: Clinical cancer research : an official journal of the American Association for Cancer ResearchPublication Date: 2020-02-14PMID: 31699827DOI: 10.1158/1078-0432.ccr-19-1853
- A New CDK9 Inhibitor on the Block to Treat Hematologic MalignanciesPublication Name: Clinical cancer research : an official journal of the American Association for Cancer ResearchPublication Date: 2020-02-14PMID: 31843752DOI: 10.1158/1078-0432.ccr-19-3670
- Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019)Publication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2019-10-15PMID: 31477350DOI: 10.1016/j.bmcl.2019.126637
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017001354&_cid=P22-MJC84G-87476-1


Example 14: (1S,3R)-3-acetamido-N-(5-chloro-4-(5,5-dimethyl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl)pyridin-2-yl)cyclohexanecarboxamide


PAT
- COMPOUNDS DERIVED FROM POLYCYCLIC AMIDE AS CDK9 INHIBITORS, COMPOSITION AND THEIR USESPublication Number: BR-122019013677-B1Priority Date: 2015-06-29
- Polycyclic amide derivatives as CDK9 inhibitorsPublication Number: KR-102663113-B1Priority Date: 2015-06-29Grant Date: 2024-05-02
- Methods of treating a ras protein-related disease or disorderPublication Number: US-2025049810-A1
- Chemical compoundsPublication Number: TW-I723028-BPriority Date: 2015-06-29Grant Date: 2021-04-01
- Chemical compoundsPublication Number: US-2021171541-A1Priority Date: 2015-06-29
- POLYCYCLIC AMIDA DERIVATIVES AS CDK9 INHIBITORSPublication Number: HR-P20211970-T1Priority Date: 2015-06-29
- Pyridine and pyrimidine derivativesPublication Number: US-11352369-B2Priority Date: 2015-06-29Grant Date: 2022-06-07
- Chemical compoundsPublication Number: US-2022340592-A1Priority Date: 2015-06-29
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Zemirciclib, cyclin-dependent kinase inhibitor, antineoplastic, AZD 4573, UNII-E5XSP3X68B
Zelenirstat



Zelenirstat
CAS 1215011-08-7
MF C24H30Cl2N6O2S, 537.5 g/mol
2,6-dichloro-N-[1,5-dimethyl-3-(2-methylpropyl)-1Hpyrazol-4-yl]-4-[2-(piperazin-1-yl)pyridin-4-yl]benzene-1-sulfonamide
N-myristoyltransferase inhibitor, antineoplastic, PCLX 001, DDD86481, CCI 002, DDD 86481
Zelenirstat (PCLX-001) is an investigational, oral small-molecule drug that inhibits N-myristoyltransferases (NMTs), enzymes crucial for adding fatty acids to proteins, a process vital for cell signaling and membrane attachment. Developed by Pacylex Pharmaceuticals, it’s being tested for various cancers, showing promise in hematologic cancers like AML and lymphomas, as well as solid tumors, by disrupting cancer cell survival and growth, with early trials indicating good safety and potential efficacy.
How it works:
- Targets NMT enzymes: Zelenirstat blocks NMT1 and NMT2, preventing myristoylation (adding a fatty acid) to proteins.
- Disrupts cancer cell processes: This inhibition interferes with essential cell signaling and stability, especially in cancer cells where NMT expression is altered, leading to cell death (apoptosis).
- Affects mitochondrial function: It also disrupts mitochondrial complex I and oxidative phosphorylation, vital for leukemia stem cell survival, notes Pacylex Pharmaceuticals.
Development & Status:
- Orphan Drug Status: Granted for Acute Myeloid Leukemia (AML).
- Clinical Trials: A Phase 1 trial demonstrated good safety and early signs of activity in patients with advanced solid tumors and lymphomas, leading to further development.
- New Drug Class: It represents a novel approach to cancer treatment, distinct from many existing therapies.
Potential Applications:
- Acute Myeloid Leukemia (AML)
- B-cell Lymphomas (like Diffuse Large B-Cell Lymphoma)
- Colorectal Carcinoma
- Other cancers, including breast, lung, bladder, and pancreatic cancers, show sensitivity in preclinical models.
Zelenirstat, also known as PCLX-001, is an investigational new drug that is being evaluated for the treatment of cancer and as an antiviral agent. It is a small molecule inhibitor targets both N-myristoyltransferase 1 (NMT1) and N-myristoyltransferase 2 (NMT2) proteins, which are responsible for myristoylation. Its dual mechanism of action disrupts both cell signaling and energy production in cancer cells.
Zelenirstat is a strong pan-N myristoyl transferase inhibitor, which prevents addition of myristic acid into penultimate glycine of protein with myristoylation signal, and initially has been introduced as anti-tumor drug.[1][2][3] It has completed phase I clinical trial and is going through escalation phase.[4] Its prototype DDD85646 as well as other NMT inhibitors such as IMP-1088 have strong antiviral activities against viruses that required myristoylated proteins to complete their life cycle, including hemorrhagic viruses, such as lassa and argentinian virus, and pox viruses, such as vaccinia and monkeypox.[5][6]
Zelenirstat is an orally bioavailable inhibitor of the enzyme N-myristoyl transferase (NMT), with potential antineoplastic activity. Upon oral administration, zelenirstat targets and binds to NMT, especially NMT type 2 (NMT2). This prevents NMT-mediated signaling and myristoylation. This inhibits proliferation of certain cancer cells in which NMT expression is lost. Zelenirstat also inhibits B-cell receptor (BCR) signaling and reduces the levels of Src-family tyrosine kinases (SFKs). NMTs mediate myristoylation, a key process by which the fatty acid myristate is added to proteins and allows proteins to interact with cell membranes and become part of the cell signaling system. NMT expression is lost in numerous cancers, such as blood cancer cells, thereby making these cells more sensitive to zelenirstat compared to normal cells. The loss of NMT expression may promote tumorigenesis.
Mechanism of action
Zelenirstat acts by inhibiting NMT I and II enzymes, which are required to complete the myristoylation of proteins. Without myristoylation, these proteins are targeted for proteasomal degradation.[7]
PCLX-001 is a first-in-kind N-Myristoyltransferase (NMT) inhibitor being developed by [Pacylex Pharmaceuticals](https://pacylex.com). Current studies have shown that PCLX-001 works differently than other known cancer drugs and has high activity and positive results in breast, lung, bladder and pancreas cancers.
- Study of PCLX-001 in R/R Advanced Solid Malignancies and B-cell LymphomaCTID: NCT04836195Phase: Phase 1Status: CompletedDate: 2025-04-17
- Study of Oral PCLX-001 in R/R Acute Myeloid LeukemiaCTID: NCT06613217Phase: Phase 1Status: RecruitingDate: 2025-03-10
REF
- Novel, First-in-Human, Oral PCLX-001 Treatment in a Patient with Relapsed Diffuse Large B-Cell LymphomaPublication Name: Current oncology (Toronto, Ont.)Publication Date: 2022-03-13PMCID: PMC8947478PMID: 35323358DOI: 10.3390/curroncol29030158
- N-myristoyltransferase proteins in breast cancer: prognostic relevance and validation as a new drug targetPublication Name: Breast Cancer Research and TreatmentPublication Date: 2021-01-04PMCID: PMC7940342PMID: 33398478DOI: 10.1007/s10549-020-06037-y
- Targeting N-myristoylation for therapy of B-cell lymphomasPublication Name: Nature CommunicationsPublication Date: 2020-10-22PMCID: PMC7582192PMID: 33093447DOI: 10.1038/s41467-020-18998-1
- Emerging New Targets for the Treatment of Resistant Fungal InfectionsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-01-02PMID: 29294275DOI: 10.1021/acs.jmedchem.7b01413
- Interrogating the Roles of Post-Translational Modifications of Non-Histone ProteinsPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-05-15PMID: 28505447DOI: 10.1021/acs.jmedchem.6b01817
SYN
DDD 86481
https://patentscope.wipo.int/search/en/detail.jsf?docId=US73438944&_cid=P12-MJAUPA-00022-1
INTERMEDIATE 23A
4-Bromo-2,6-dichloro-N-(3-isobutyl-1,5-dimethyl-1H-pyrazol-4-yl)-benzenesulfonamide
EXAMPLE DDD86481
2,6-Dichloro-N-(3-isobutyl-1,5-dimethyl-1H-pyrazol-4-yl)-4-(2-piperazin-1-yl-pyridin-4-yl)-benzenesulfonamide
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010026365&_cid=P12-MJAUOO-99381-1
PAT
N-myristoyl transferase inhibitors
Publication Number: WO-2010026365-A1
Priority Date: 2008-09-02
- N-myristoyl transferase inhibitorsPublication Number: ES-2546865-T3Priority Date: 2008-09-02Grant Date: 2015-09-29
- N-Myristoyl Transferase InhibitorsPublication Number: US-2011312921-A1Priority Date: 2008-09-02
- N-myristoyl transferase inhibitorsPublication Number: US-2016060224-A1Priority Date: 2008-09-02
- N-myristoyl transferase inhibitorsPublication Number: US-9156811-B2Priority Date: 2008-09-02Grant Date: 2015-10-13
- N-myristoyl transferase inhibitorsPublication Number: US-9828346-B2Priority Date: 2008-09-02Grant Date: 2017-11-28
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Gamma JM, Liu Q, Beauchamp E, Iyer A, Yap MC, Zak Z, et al. (January 2025). “Zelenirstat Inhibits N-Myristoyltransferases to Disrupt Src Family Kinase Signaling and Oxidative Phosphorylation, Killing Acute Myeloid Leukemia Cells”. Molecular Cancer Therapeutics. 24 (1): 69–80. doi:10.1158/1535-7163.MCT-24-0307. PMC 11694064. PMID 39382188.
- Sangha R, Jamal R, Spratlin J, Kuruvilla J, Sehn LH, Beauchamp E, et al. (August 2024). “A first-in-human phase I trial of daily oral zelenirstat, a N-myristoyltransferase inhibitor, in patients with advanced solid tumors and relapsed/refractory B-cell lymphomas”. Investigational New Drugs. 42 (4): 386–393. doi:10.1007/s10637-024-01448-w. PMC 11327210. PMID 38837078.
- Sangha RS, Jamal R, Spratlin J, Kuruvilla J, Sehn LH, Weickert M, et al. (June 2024). “Final results of a first-in-human phase I dose escalation trial of daily oral zelenirstat, a n-myristoyltransferase inhibitor, in patients with advanced solid tumors and relapsed/refractory B-cell lymphomas”. Journal of Clinical Oncology. 42 (16_suppl): 3082. doi:10.1200/JCO.2024.42.16_suppl.3082. ISSN 0732-183X.
- Spratlin JL, Sangha RS, Jamal R, Beauchamp E, Berthiaume LG, Mackey JR (20 January 2024). “A first-in-human, open-label, phase I trial of daily oral zelenirstat, an NMT inhibitor, in patients with relapsed/refractory advanced cancer including gastrointestinal cancers”. Journal of Clinical Oncology. 42 (3_suppl): 129–129. doi:10.1200/jco.2024.42.3_suppl.129. Retrieved 19 January 2025.
- Witwit H, Betancourt CA, Cubitt B, Khafaji R, Kowalski H, Jackson N, et al. (August 2024). “Cellular N-Myristoyl Transferases Are Required for Mammarenavirus Multiplication”. Viruses. 16 (9): 1362. doi:10.3390/v16091362. PMC 11436053. PMID 39339839.
- Witwit H, Cubitt B, Khafaji R, Castro EM, Goicoechea M, Lorenzo MM, et al. (January 2025). “Repurposing Drugs for Synergistic Combination Therapies to Counteract Monkeypox Virus Tecovirimat Resistance”. Viruses. 17 (1): 92. doi:10.3390/v17010092. ISSN 1999-4915. PMC 11769280.
- Witwit H, Betancourt CA, Cubitt B, Khafaji R, Kowalski H, Jackson N, et al. (August 2024). “Cellular N-Myristoyl Transferases Are Required for Mammarenavirus Multiplication”. Viruses. 16 (9): 1362. doi:10.3390/v16091362. PMC 11436053. PMID 39339839.
| Clinical data | |
|---|---|
| Other names | PCLX-001 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1215011-08-7 |
| PubChem CID | 58561243 |
| DrugBank | DB15567 |
| ChemSpider | 35034199 |
| UNII | 5HY8BYC3Q6 |
| ChEMBL | ChEMBL3357685 |
| Chemical and physical data | |
| Formula | C24H30Cl2N6O2S |
| Molar mass | 537.50 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////zelenirstat, N-myristoyltransferase inhibitor, antineoplastic, PCLX 001, DDD86481, CCI 002, DDD 86481
Zamzetoclax



Zamzetoclax
CAS 2388470-64-0
MF C38H46ClN5O6S MW736.32
N-[(3′R,4S,6′R,7′S,8′E,11′S)-7-chloro-7′-methoxy-11′-methyl-13′,15′-dioxospiro[2,3-dihydro-1H-naphthalene-4,22′-20-oxa-13λ6-thia-1,14-diazatetracyclo[14.7.2.03,6.019,24]pentacosa-8,13,16(25),17,19(24)-pentaene]-13′-yl]-3-methoxy-1-methylpyrazole-4-carboxamide
- 1H-Pyrazole-4-carboxamide, N-[(1’S,10S,12S,14E,16S,16aR,18aR)-6′-chloro-3′,4′,8,11,12,13,16,16a,17,18,18a,19-dodecahydro-16-methoxy-12-methyl-10-oxido-8-oxospiro[5,7-etheno-1H-10lambda4-cyclobut[i][1,4]oxazepino[3,4-f][1,2,7]thiadiazacyclohexadecine-2(3H),1′(2’H)-naphthalen]-10-yl]-3-methoxy-1-methyl-
- N-[(1’S,10S,12S,14E,16S,16aR,18aR)-6′-Chloro-3′,4′,8,11,12,13,16,16a,17,18,18a,19-dodecahydro-16-methoxy-12-methyl-10-oxido-8-oxospiro[5,7-etheno-1H-10lambda4-cyclobut[i][1,4]oxazepino[3,4-f][1,2,7]thiadiazacyclohexadecine-2(3H),1′(2’H)-naphthalen]-10-yl]-3-methoxy-1-methyl-1H-pyrazole-4-carboxamide

B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, RRS8GZU2UN
Zamzetoclax (compound 1) is a potential Mcl-1 inhibitor.
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2023-04-28
PMID: 37114951
DOI: 10.1021/acs.jmedchem.2c01953
SYN
WO 2019/222112
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019222112&_cid=P12-MJ7Z15-98773-1
Example 154

[0447] Example 154 was synthesized in the same manner as Example 18 using 3-methoxy-1-methyl-1H-pyrazole-4-carboxylic acid and Example 109. Example 109 (620 mg, 1.04 mmol) was dissolved in dichloromethane (12 mL). 3-Methoxy-1-methyl-1H-pyrazole-4-carboxylic acid (324 mg, 2.08 mmol, 2 equiv.) and N-(3-dimethylaminopropyl)-N¢-ethylcarbodiimide hydrochloride (400 mg, 2.08 mmol, 2 equiv.) were added. The reaction mixture was stirred for 5 minutes at room temperature before DMAP (253 mg, 2.08 mmol, 2 equiv.) was added in a single portion. The reaction mixture was stirred overnight at room temperature and the progress of the reaction was monitored by LCMS. Upon completion, the reaction mixture was concentrated under reduced pressure, and the residue was purified by Gilson reverse phase prep HPLC (60-100% ACN/H2O with 0.1% TFA) to give Example 154.1H NMR (400 MHz, methanol-d4) d 8.07 (s, 1H), 7.76 (d, J = 8.6 Hz, 1H), 7.34 (d, J = 8.2 Hz, 1H), 7.22– 7.10 (m, 3H), 6.92 (d, J = 8.2 Hz, 1H), 6.20– 6.05 (m, 1H), 5.63 (dd, J = 15.5, 8.0 Hz, 1H), 4.10 (d, J = 12.0 Hz, 1H), 4.06 (s, 4H), 3.91– 3.83 (m, 1H), 3.82 (s, 3H), 3.79 (s, 1H), 3.72 (d, J = 14.4 Hz, 1H), 3.38 (d, J = 14.5 Hz, 1H), 3.30 (s, 3H), 3.09 (dd, J = 15.1, 10.0 Hz, 1H), 2.89– 2.72 (m, 2H), 2.51 (d, J = 26.7 Hz, 2H), 2.24 (dd, J = 10.9, 6.0 Hz, 2H), 2.12 (d, J = 13.7 Hz, 1H), 2.02– 1.70 (m, 4H), 1.54– 1.40 (m, 1H), 1.14 (d, J = 6.1 Hz, 3H). LCMS-ESI+ (m/z): calcd for C38H46ClN5O6S: 735.28; found: 735.94.
SYN
WO 2019/222112 discloses novel 3′,4,4′,5-tetrahydro-2H,2′H-spiro[benzo[b][1,4]oxazepine-3,1′-naphthalene] derivatives that are active against MCL-1. For example, Compound 1 (below) has been shown to be an effective MCL-1 inhibitor

SYN
SYN
PAT
- Mcl-1 inhibitorsPublication Number: US-2019352271-A1Priority Date: 2018-05-14
- Mcl-1 inhibitorsPublication Number: US-2020331870-A1Priority Date: 2018-05-14
- MCL-1 inhibitorsPublication Number: US-10988451-B2Priority Date: 2018-05-14Grant Date: 2021-04-27
- MCL-1 inhibitorsPublication Number: US-11643400-B2Priority Date: 2018-05-14Grant Date: 2023-05-09
- Mcl-1 inhibitorsPublication Number: US-2023312490-A1Priority Date: 2018-05-14
- Processes and intermediates for preparing mcl1 inhibitorsPublication Number: US-2023013713-A1Priority Date: 2019-11-26
- Processes and intermediates for preparing MCL1 inhibitorsPublication Number: US-11760736-B2Priority Date: 2019-11-26Grant Date: 2023-09-19
- Mcl1 inhibitorsPublication Number: US-2021171543-A1Priority Date: 2019-11-12
- Mcl1 inhibitorsPublication Number: US-2023348494-A1Priority Date: 2019-11-12
- MCL-1 inhibitorsPublication Number: US-10703733-B2Priority Date: 2018-05-14Grant Date: 2020-07-07
- Salts and polymorphs of certain mcl-1 inhibitorsPublication Number: US-2023357274-A1Priority Date: 2022-05-04
- Combination mcl-1 inhibitors with anti-body drug conjugatesPublication Number: US-2022409736-A1Priority Date: 2021-06-11
- Processes and intermediates for preparing mcl1 inhibitorsPublication Number: US-2022177409-A1Priority Date: 2020-11-19
- Processes and intermediates for preparing mcl1 inhibitorsPublication Number: US-2021179570-A1Priority Date: 2019-11-26
- Processes and intermediates for preparing MCL1 inhibitorsPublication Number: US-11325891-B2Priority Date: 2019-11-26Grant Date: 2022-05-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////zamzetoclax, B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, RRS8GZU2UN
Vilzemetkib


Vilzemetkib
CAS 1363402-44-1
MF C36H36F2N4O5 MW 642.7 g/mol
1-N‘-[4-[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxyquinolin-4-yl]oxy-3-fluorophenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
- 1,1-Cyclopropanedicarboxamide, N-[4-[[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxy-4-quinolinyl]oxy]-3-fluorophenyl]-N’-(4-fluorophenyl)-
- 1-N’-[4-[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxyquinolin-4-yl]oxy-3-fluorophenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
- 1363402-44-1
- N-[4-[[7-[[1-(Cyclopentylamino)cyclopropyl]methoxy]-6-methoxy-4-quinolinyl]oxy]-3-fluorophenyl]-N’-(4-fluorophenyl)-1,1-cyclopropanedicarboxamide

hepatocyte growth factor receptor inhibitor, antineoplastic, AL 2846, FJ4Y6XP24Y
Vilzemetkib (also known as AL2846) is an investigational, orally active small-molecule drug that acts as a potent inhibitor of the c-Met receptor tyrosine kinase, a protein often overexpressed in cancers, aiming to block tumor growth, survival, and spread by disrupting key cellular signals. It’s being studied in clinical trials, often in combination with other agents like TQB2450 (a PD-L1 inhibitor), for advanced cancers such as esophageal and liver cancer, showing promise in immunotherapy-resistant patients.
How it Works:
- Targets c-Met: Vilzemetkib binds to the c-Met protein, preventing its phosphorylation (activation).
- Blocks Signaling: This action disrupts downstream pathways crucial for cancer cell proliferation, survival, invasion, metastasis, and new blood vessel formation (angiogenesis).
Development & Use:
- Developer: Developed by Advenchen Laboratories.
- Status: Investigational drug, currently in clinical trials.
- Research Focus: Studied for cancers like esophageal squamous cell carcinoma (ESCC) and hepatocellular carcinoma (HCC).
Key Information:
- Chemical Name: 1,1-Cyclopropanedicarboxamide, N-[4-[[7-[[1-(cyclopentylamino)cyclopropyl]methoxy]-6-methoxy-4-quinolinyl]oxy]-3-fluorophenyl]-N′-(4-fluorophenyl)-.
- Purpose: Potential anti-cancer (antineoplastic) activity.
- OriginatorAdvenchen Laboratories
- DeveloperAdvenchen Laboratories; Chia Tai Tianqing Pharmaceutical Group
- ClassAntineoplastics; Small molecules
- Mechanism of ActionReceptor protein-tyrosine kinase antagonists
- Phase IIINon-small cell lung cancer; Thyroid cancer
- Phase IILung cancer; Ovarian cancer
- Phase I/IIColorectal cancer; Neurofibromatosis 1; Pancreatic cancer
- No development reportedSolid tumours
- 28 Oct 2025No recent reports of development identified for phase-I development in Solid-tumours(Combination therapy, In the elderly, Late-stage disease, Second-line therapy or greater, In adults) in China (PO, Capsule)
- 10 Oct 2025700363489: CTP push: KDM and HE updated
- 26 Aug 2025Chemical structure information added.
Vilzemetkib is an orally bioavailable small molecule inhibitor of the oncoprotein c-Met (hepatocyte growth factor receptor; HGFR), with potential antineoplastic activity. Upon oral administration vilzemetkib targets and binds to the c-Met protein, prevents c-Met phosphorylation and disrupts c-Met-dependent signal transduction pathways. This may induce cell death in tumor cells overexpressing c-Met protein or expressing constitutively activated c-Met protein. c-Met protein is overexpressed or mutated in many tumor cell types and plays key roles in tumor cell proliferation, survival, invasion, metastasis, and tumor angiogenesis.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US73570351&_cid=P20-MJ6JF6-22611-1



EXAMPLE 6
N-(4-(7-((1-(cyclopentylamino)cyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-3-fluoro-phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
| The title compound was prepared by similar manner to Example 3, by using cyclopentanone instead of tetrahydro-4H-pyran-4-one. Mass: (M+1), 643 |
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022268158&_cid=P20-MJ6JJ7-25153-1
WO2012034055 discloses N-(4-((7-((1-(cyclopentylamino)cyclopropyl)methoxy)-6-methoxyquinolone-4-yl)oxy)-3-fluorophenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (hereinafter referred to as compound (I)) as a c-Met kinase inhibitor and its use in inhibiting tyrosine kinase activity. Compound (I) is a novel class of compounds with excellent pharmacological properties, capable of inhibiting the activity of various protein tyrosine kinases, such as c-Met, VEGFr, EGFr, c-kit, PDGF, FGF, SRC, Ron, Tie2, etc. This disclosure relates to the treatment of neurofibromatosis type I with compound (I).

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012034055&_cid=P20-MJ6JLU-26634-1
Example 6
N-(4-(7-((1-(cyclopentylamino)cyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-3-fluoro-phenyl)-N-(4-fluorophenyl)cyclopropane- 1,1-dicarboxamide
The title compound was prepared by similar manner to Example 3, by using cyclopentanone instead of tetrahydro-4H-pyran-4-one. Mass: (M + 1), 643
PAT
- Crystal of compound as c-met kinase inhibitor and preparation method therefor and use thereofPublication Number: EP-4279138-A2Priority Date: 2018-03-02
- Compounds As c-Met Kinase InhibitorsPublication Number: US-2012123126-A1Priority Date: 2010-09-12
- Compounds as c-Met kinase inhibitorsPublication Number: US-8664244-B2Priority Date: 2010-09-12Grant Date: 2014-03-04
- Compounds as c-met kinase inhibitorsPublication Number: WO-2012034055-A2Priority Date: 2010-09-12
- Use of compound as c-met kinase inhibitor for treatment of neurofibromatosis type iPublication Number: WO-2022268158-A1Priority Date: 2021-06-23
- Combined pharmaceutical composition of c-met kinase inhibitor and anti-pd-l1 antibodyPublication Number: US-2023263795-A1Priority Date: 2020-06-02
- Crystal of compound as c-met kinase inhibitor and preparation method therefor and use thereofPublication Number: EP-3766870-A1Priority Date: 2018-03-02
- Crystalline of compound as c-met kinase inhibitor and preparation method therefor and use thereofPublication Number: US-2021047272-A1Priority Date: 2018-03-02
- Crystalline of compound as c-Met kinase inhibitor and preparation method therefor and use thereofPublication Number: US-11279676-B2Priority Date: 2018-03-02Grant Date: 2022-03-22
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////Vilzemetkib, hepatocyte growth factor receptor inhibitor, antineoplastic, AL 2846, FJ4Y6XP24Y
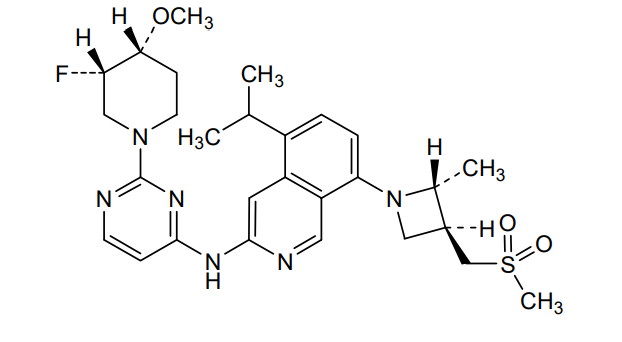
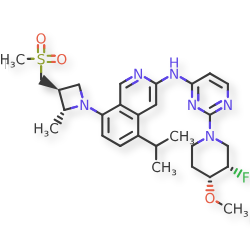
Tigozertinib
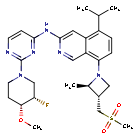
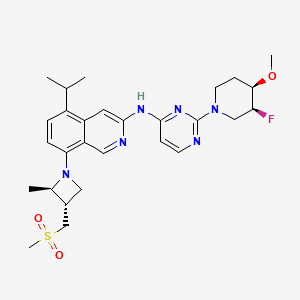
Tigozertinib
CAS 2660250-10-0
MF C28H37FN6O3S MW 556.7
3-Isoquinolinamine, N-[2-[(3S,4R)-3-fluoro-4-methoxy-1-piperidinyl]-4-pyrimidinyl]-5-(1-methylethyl)-8-[(2R,3S)-2-methyl-3- [(methylsulfonyl)methyl]-1-azetidinyl]-
N-{2-[(3S,4R)-3-fluoro-4-methoxypiperidin-1-yl]pyrimidin-4-yl}-8-{(2R,3S)-3-[(methanesulfonyl)methyl]-2-methylazetidin-1-yl}-5-(propan-2-yl)isoquinolin-3-amine
N-[2-[(3S,4R)-3-fluoro-4-methoxypiperidin-1-yl]pyrimidin-4-yl]-8-[(2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl]-5-propan-2-ylisoquinolin-3-amine
N-(2-((3S,4R)-3-fluoro-4-methoxypiperidin-l-yl)pyrimidin-4-yl)-5-isopropyl-8-(3-(methylsulfonylmethyl)azetidin-l-yl)isoquinolin-3-amine
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, antineoplastic, PA4PTH5HL9, BLU 945
Tigozertinib (BLU-945) is currently under investigation in clinical trial NCT04862780 (Phase 1/2 Study Targeting EGFR Resistance Mechanisms in NSCLC) for the treatment of NSCLC.
Tigozertinib is a fourth-generation, orally bioavailable, mutant-selective, epidermal growth factor receptor (EGFR) inhibitor, with potential antineoplastic activity. Upon oral administration, tigozertinib targets, binds to and inhibits the activity of EGFR with C797S triple mutations including ex19del/T790M/C797S and L858R/T790M/C797S, thereby preventing EGFR-mediated signaling. This may both induce cell death and inhibit tumor growth in EGFR-overexpressing tumor cells. EGFR, a receptor tyrosine kinase mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. BLU-945 inhibits mutated forms of EGFR with C797S mutation, which prevents covalent bond formation with third-generation EGFR inhibitors leading to drug resistance. BLU-945 may have enhanced anti-tumor effects in tumors with C797S-mediated resistance when compared to other EGFR tyrosine kinase inhibitors.Tigozertinib is a fourth-generation, orally bioavailable, mutant-selective, epidermal growth factor receptor (EGFR) inhibitor, with potential antineoplastic activity. Upon oral administration, tigozertinib targets, binds to and inhibits the activity of EGFR with C797S triple mutations including ex19del/T790M/C797S and L858R/T790M/C797S, thereby preventing EGFR-mediated signaling. This may both induce cell death and inhibit tumor growth in EGFR-overexpressing tumor cells. EGFR, a receptor tyrosine kinase mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. BLU-945 inhibits mutated forms of EGFR with C797S mutation, which prevents covalent bond formation with third-generation EGFR inhibitors leading to drug resistance. BLU-945 may have enhanced anti-tumor effects in tumors with C797S-mediated resistance when compared to other EGFR tyrosine kinase inhibitors.
- First-in-Human, Phase 1b/2a Trial of a Multipeptide Therapeutic Vaccine in Patients With Progressive GlioblastomaCTID: NCT04116658Phase: Phase 1/Phase 2Status: CompletedDate: 2025-11-28
- (SYMPHONY) Phase 1/2 Study Targeting EGFR Resistance Mechanisms in NSCLCCTID: NCT04862780Phase: Phase 1Status: TerminatedDate: 2025-02-10
- A Novel Therapeutic Vaccine (EO2401) in Metastatic Adrenocortical Carcinoma, or Malignant Pheochromocytoma/ParagangliomaCTID: NCT04187404Phase: Phase 1/Phase 2Status: TerminatedDate: 2024-11-12
REF
- Synthesis of BLU-945, an EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung CancerPublication Name: SynfactsPublication Date: 2022-09-20DOI: 10.1055/s-0041-1738705
- Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung CancerPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-07-15PMCID: PMC9340769PMID: 35838760DOI: 10.1021/acs.jmedchem.2c00704
- Effects of antithymocyte serum on lymph node cells participating in the graft-vs-host reactionPublication Name: Cellular ImmunologyPublication Date: 1976-06-01PMID: 7359DOI: 10.1016/0008-8749(76)90132-5
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021133809&_cid=P11-MJ29N8-15768-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021133809&_cid=P11-MJ292P-02302-1
Example 5, Compound 117: Synthesis of (3S,4R)-3-fluoro-l-(4-(5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-l-yl)isoquinolin-3-ylamino)pyrimidin-2-yl)-4-methylpiperidin-4-ol
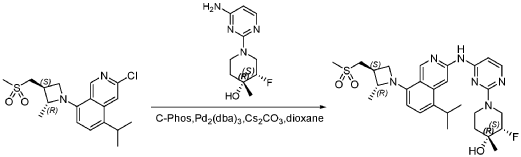
To a solution of 3-chloro-8-[(2R,3S)-3-(methanesulfonylmethyl)-2-methylazetidin-l-yl]-5-(propan-2-yl)isoquinoline(28 g,76.3mmol, from step 1 of Example 3), (3R,4S)-l-(4-aminopyrimidin-2-yl)-3-fluoro-4-methylpiperidin-4-ol(17.2g,76.3mmol, peak 1 from Example B12), CS2CO3 (49.8 g, 152 mmol),C-phos (4.27 g, 9.15mmol, 2-dicyclohexylphosphino-2’,6’-bis(N,N-dimethylamino)biphenyl) and Pd2(dba)3 (3.94 g, 3.81 mmol) in dioxane (400 mL) was heated to 100 °C for 16 h under N2 atmosphere. The mixture reaction was filtered and the filtrate was concentration under vacuum. The residue was applied onto a silica gel column with EA/PE (2: 1) to give product 28.8 g (67%) as a light-yellow solid.
OTHERS
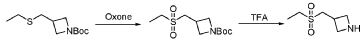

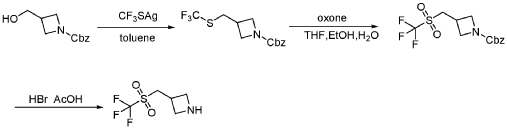
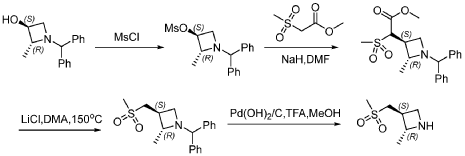
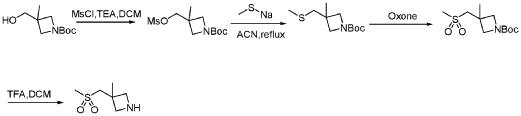
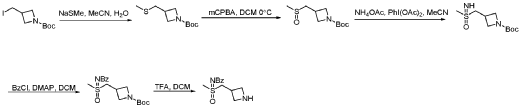
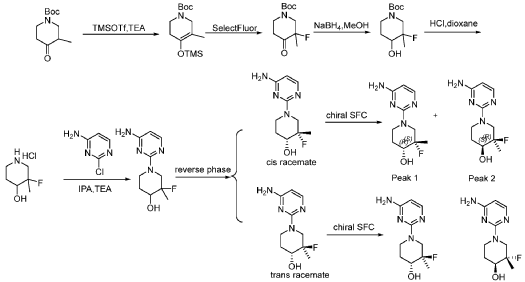
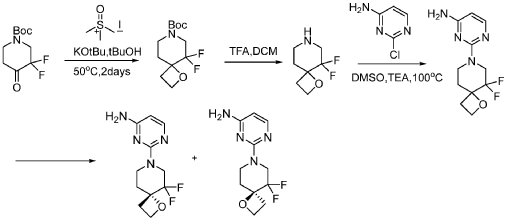
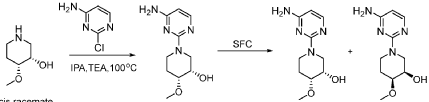
PAT
- Crystalline forms of cftr modulatorsPublication Number: US-2025051326-A1
- Small molecule inhibitors of dyrk/clk and uses thereofPublication Number: US-2025051325-A1
- RNAi agents for inhibiting expression of HIF-2 alpha (EPAS1), compositions thereof, and methods of usePublication Number: US-12221610-B2Grant Date: 2025-02-11
- Silicomagnesium-aluminate-hydrate gel and method of preparing the samePublication Number: US-3476692-APriority Date: 1966-02-04Grant Date: 1969-11-04
- InsecticidesPublication Number: DE-1542900-A1Priority Date: 1963-06-22
- Manufacture of conditioning compound for ground meat productsPublication Number: US-2634212-APriority Date: 1951-05-04Grant Date: 1953-04-07
- Kras modulators and uses thereofPublication Number: US-2025051365-A1
- Inhibitors of the myst family of lysine acetyl transferasesPublication Number: US-2025051343-A1
- Novel acid compoundsPublication Number: US-3945994-APriority Date: 1973-02-08Grant Date: 1976-03-23
- Production method of new phosphate ester compoundPublication Number: JP-S5930718-B2Priority Date: 1971-08-17
- Connection of a semiconductor device with a power sourcePublication Number: DE-2015923-A1Priority Date: 1969-04-23
- Ultrasonic doppler instrumentPublication Number: US-3732532-APriority Date: 1967-08-03Grant Date: 1973-05-08
- Electromechanical transducerPublication Number: US-3396285-APriority Date: 1966-08-10Grant Date: 1968-08-06
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Tigozertinib, antineoplastic, PA4PTH5HL9, BLU 945

{kind=link}
{kind=link}