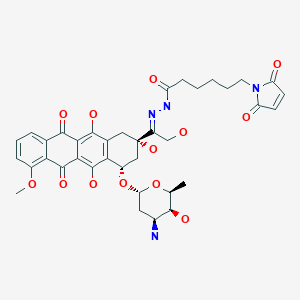
Idasanutlin(RG-7388)
cas 1229705-06-9
идасанутлин [Russian]
إيداسانوتلين [Arabic]
依达奴林 [Chinese]
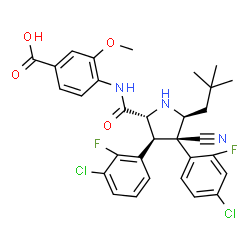
4-{ [(2R,3S,4R,5S)-4-(4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2- dimethyl-prop yl)-pyrrolidine-2-carbonyl] -amino }-3-methoxy-benzoic acid (C31H29Cl2F2N304)
4-{[(2R,3S,4R,5S)-4-(4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carbonyl]-amino}-3-methoxy-benzoic acid
MW 616.4973
F. Hoffmann-La Roche Ag, Hoffmann-La Roche Inc.ROCHE PHASE1
for the oral treatment of cancer, including solid tumors and hematological tumors, including acute myelogenous leukemia
Acute myelogenous leukemia; Cancer; Prostate tumor
Mdm2 p53-binding protein inhibitor


- INTRO
- RG7388 is a MDM2 inhibitor with superior potency and selectivity
- RG7388 is an oral, selective, small molecule MDM2 antagonist that inhibits binding of MDM2 to p53.
RG7388 is the second generation inhibitor of P53-MDM2 interaction. It is orally active, potently and selectively antagonizing the P53-MDM2 interaction with Ki at low nM. It is designed to selectively target MDM2, a key negative regulator of the p53 tumor suppressor protein. Blocking this essential interaction may lead to apoptosis via activation of p53 in tumor cells with functional p53 signaling. It is currently in clinical evaluation.
Description:
Value IC50: 30 nM (IC50 Average of three wt-p53 SJSA1 Cancer cell lines, RKO, HCT116)
. RG7388 is an Oral, Selective, small molecule antagonist that inhibits binding of MDM2 to p53 MDM2 Blocking the MDM2-p53 Interaction stabilizes p53 and activates p53-mediated cell death and inhibition of cell Growth.
RG7388 Showed all the Characteristics expected of an MDM2 inhibitor in terms of speci? c binding to the target, mechanistic outcomes Resulting from Activation of the p53 pathway, and in vivo ?. Although e cacy Mechanism of Action of the cellular is identical to that of RG7388 RG7112, it is much More potent and Selective.
Tumor suppressor p53 is a powerful growth suppressive and pro-apoptotic protein that plays a central role in protection from tumor development.A potent transcription factor, p53 is activated following cellular stress and regulates multiple downstream genes implicated in cell cycle control, apoptosis, DNA repair, and senescence.While p53 is inactivated in about 50% of human cancers by mutation or deletion, it remains wild-type in the remaining cases but its function is impaired by other mechanisms. One such mechanism is the overproduction of MDM2, the primary negative regulator of p53, which effectively disables p53 function.An E3 ligase, MDM2 binds p53 and regulates p53 protein levels through an autoregulatory feedback loop. Stabilization and activation of wild-type p53 by inhibition of MDM2 binding has been explored as a novel approach for cancer therapy.
PAPER
J. Med. Chem., 2013, 56 (14), pp 5979–5983
DOI: 10.1021/jm400487c
Restoration of p53 activity by inhibition of the p53–MDM2 interaction has been considered an attractive approach for cancer treatment. However, the hydrophobic protein–protein interaction surface represents a significant challenge for the development of small-molecule inhibitors with desirable pharmacological profiles. RG7112 was the first small-molecule p53–MDM2 inhibitor in clinical development. Here, we report the discovery and characterization of a second generation clinical MDM2 inhibitor, RG7388, with superior potency and selectivity.

compd 12
1H NMR (400 MHz, DMSO-d6)
δ12.86 (s, 1 H), 10.46 (s, 1 H), 8.35 (d, J = 8.86 Hz, 1 H), 7.71 (t, J = 6.95 Hz, 1 H), 7.48 – 7.61 (m,4 H), 7.29 – 7.42 (m, 3 H), 4.53 – 4.61 (m, 2 H), 4.38 (br. s., 1 H), 3.86 – 3.99 (m, 4 H), 1.62 (dd,J = 9.87, 14.00 Hz, 1 H), 1.24 (d, J = 14.00 Hz, 1 H), 0.95 (s, 9 H) ppm;
13C NMR (101 MHz, DMSO-d6) δ 171.2, 166.9, 160.8, 158.3, 156.8, 154.4, 147.5, 134.8, 134.7, 131.0, 130.8, 130.0,
128.6, 126.1, 125.9, 125.6, 125.3, 122.7, 119.6, 119.4, 119.2, 119.1, 117.7, 117.4, 117.3, 117.2, 111.0,
64.7, 63.4, 63.3, 63.3, 63.2, 55.8, 50.2, 43.9, 30.1, 29.5, 25.5 ppm;
HRMS (ES+) m/z CalcdC31H29Cl2F2N3O3+ H [M+H]+: 616.1576, found: 616.1574.
Anal. Calcd for C31H29Cl2F2N3O3: C, 60.4; H, 4.74; Cl, 11.5; F, 6.16; N, 6.82. Found: C, 60.3; H, 4.79; Cl, 11.3; F, 6.02; N, 6.82.
PATENT
see
WO-2014128094
http://www.google.com/patents/WO2014128094A1?cl=en
F Hoffmann-La Roche AG; Hoffmann-La Roche Inc
Asymmetric synthesis of a substituted pyrrolidine-2-carboxamide
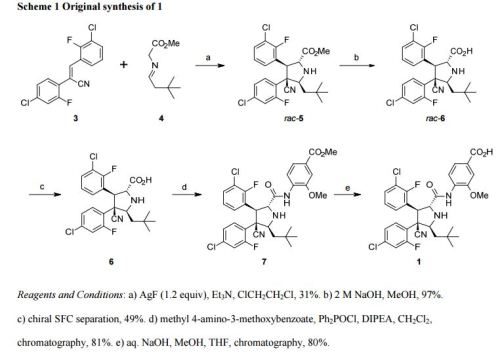
Process for the preparation of RG-7388 and their novel intermediates. Roche is developing idasanutlin (RG-7388), a small-molecule MDM2 antagonist that inhibits binding of MDM2 to p53, for the oral treatment of cancer, including solid tumors and hematological tumors, including acute myelogenous leukemia, as of September 2014, the drug is in Phase 1 trials. See WO2014114575 claims physically stable solid dispersion comprising a compound eg idasanutlin, with an aqueous solubility of less than 1 μg/ml and an ionic polymer eg copovidone, for treating cancer.
Compound I.
Scheme 2
process to produce a compound of the formula
which comprises
a) reacting a compound of the formula (IV)
with a compound of the formula (V)
in the presence of a silver catalyst; b) isomerising the product of (a) by reaction with a suitable base selected from a strong amine or with an insoluble base in the above solvents at a temperature range of from about 20 to 80 °C; and c) hydrolyzing the product of (b) in any suitable hydroxide in a solvent having water miscibility at a temperature between about 20 to about 80°C to obtain a compound of formula I; wherein
R1 is a non-tertiary alkyl or benzyl, or other ester protecting group.
Example 1 : (Z)-3-(3-Chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenvl)-acrvlonitrile
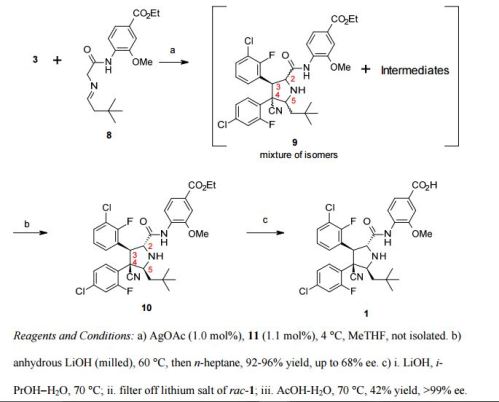
A 250-L glass-lined reactor was charged with 2-(4-chloro-2-fluorophenyl)acetonitrile (15.0 kg, 88.5 mol, Eq: 0.988), 3-chloro-2-fluorobenzaldehyde (14.2 kg, 89.6 mol, Eq: 1.00), MeOH (140 L). In one portion, a solution of sodium hydroxide [prepared from 50 wt% solution (0.23 L, 4.4 mmol, Eq: 0.05) diluted in methanol (10 L)] was added. The resulting mixture was heated to 50 °C for 4.5 h, and then the resulting thick slurry was cooled down to 20 °C. Consumption of 3- chloro-2-fluorobenzaldehyde was monitored by HPLC analysis. The solid product was isolated by filtration via a 0.3 m filter/dryer and the cake washed with methanol (58 L). The product was dried under vacuum with N2 purge at 60°C to provide the stilbene as a white powder, 24.2 kg (88.5% yield) with 99.87% purity by HPLC analysis.
1H NMR (300 MHz, CDC13) δ 8.10-8.15 (1H, m), 7.79 (1H, s), 7.48-7.59 (2H, m), 7.20-7.28 (3H, m).
Compound 5: 1H NMR (400 MHz, DMSO-d6) δ: 12.89 (br. s., 1H), 10.50 (s, 1H), 8.39 (d, J = 8.8 Hz, 1H), 7.75 (t, J = 6.8 Hz, 1H), 7.51 – 7.64 (m, 4H), 7.33 – 7.46 (m, 3H), 4.57 – 4.66 (m, 2H), 4.36 – 4.47 (m, 1H), 3.95 – 4.03 (m, 1H), 3.94 (s, 3H), 1.66 (dd, J = 14.2, 9.9 Hz, 1H), 1.28 (d, J = 13.8 Hz, 1H), 0.99 (s, 9H).

A 500-mL, round bottomed flask equipped with a magnetic stirrer and nitrogen inlet/bubbler was charged with copper(II) acetate (150 mg, 0.826 mmol), (R)-BINAP (560 mg, 0.899 mmol), and 2-methyltetrahydrofuran (120 mL). The suspension was stirred at room temperature under N2 for 3 h when a clear blue solution was obtained. Then 12.0 mL (68.7 mmol) of N,N- diisopropylethylamine was added, followed by 20.0 g (64.5 mmol) of Compound (1) and 24.0 g (71.8 mmol) of Compound (2). The suspension was stirred at room temperature under N2 for 18 h, and LCMS analysis indicated complete reaction. The reaction mixture was diluted with 100 mL of 5% ammonium acetate solution and stirred for 15 min, then poured into a 500-mL separatory funnel. The organic phase separated was washed with an additional 5% ammonium acetate solution (100 mL), then with 100 mL of 5% sodium chloride solution (100 mL), and „
– 27 – concentrated at 40 °C under reduced pressure to a thick syrup (ca. 60 g ). This syrup (containing 6 and 7) was dissolved in tetrahydrofuran (120 mL), methanol (60.0 mL), and water (6.00 mL). Then sodium hydroxide (50% solution, 6.00 mL, 114 mmol) was added dropwise. The mixture was stirred at room temperature for 18 h. LCMS and chiral HPLC indicated complete hydrolysis and isomerization. The reaction mixture was acidified with 20.0 mL (349 mmol) of acetic acid, and then concentrated at 40 °C under reduced pressure to remove ca. 80 mL of solvent. The residue was diluted with 2-propanol (200 mL), and further concentrated to remove ca. 60 mL of solvent, and then water (120 mL) was added. The slurry was stirred under reflux for 1 h, at room temperature overnight, then filtered and the flask was rinsed with of 2-propanol- water (2: 1) (20.0 mL). The filter cake was washed with 2-propanol- water (1: 1) (2 x 100 mL = 200 mL), and with water (2 x 200 mL = 400 mL), then vacuum oven dried at 60 °C to give 33.48 g (84.2% yield) of crude Compound 5 as a white solid ; 99.26% pure and 87.93% ee as judged by LCMS and chiral HPLC analysis. Compound 6 (exo cycloaddition product, 2,5-cis): 1H NMR (400 MHz, CDC13) δ 9.66 (brs, 1H),
8.42 (d, J = 8.3 Hz, 1H), 7.89 (m, 1H), 7.65 (dd, J = 8.6, 1.8 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.40 (m, 1H), 7.32 (td, J = 8.3, 1.5 Hz, 1H), 7.22-7.15 (m, 3H), 4.45 (m, 2H), 4.36 (q, J = 7.2 Hz, 2H), 4.25 (m, 1H), 3.91 (s, 3H), 1.39 (t, J = 7.2 Hz, 3H), 1.30 (dd, J = 14.2, 9.3 Hz, 1H), 0.92 (s, 9H), 0.84 (d, J = 14.2 Hz, 1H).
Compound 7 (endo cycloaddition product, 2,5-cis): 1H NMR (400 MHz, CDC13) δ 9.97 (brs, 1H), 8.30 (d, J = 8.4 Hz, 1H), 7.65 (dd, J = 8.3, 1.8 Hz, 1H), 7.56 (d, J = 1.7 Hz, 1H), 7.51 (m, 1H),
7.43 (t, J = 8.4 Hz, 1H), 7.23 (m, 1H), 7.17 (dd, J =12.6, 2.0 Hz, 1H), 7.11 (m, 1H), 6.89 (td, J = 8.1, 1.2 Hz, 1H), 5.05 (dd, J = 10.8, 2.1 Hz, 1H), 4.53 (d, J = 10.8 Hz, 1H), 4.37 (q, J = 7.2 Hz, 2H), 4.22 (d, J = 8.7 Hz, 1H), 3.95 (s, 3H), 1.85 (dd, J = 14.1, 8.7 Hz, 1H), 1.48 (d, J =14.1 Hz, 1H), 1.40 (t, J = 7.2 Hz, 1H), 0.97 (s, 9H).
…………………
WO2014114575A1
http://www.google.com/patents/WO2014114575A1?cl=en
The compound 4-{ [(2R,3S,4R,5S)-4- (4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)- pyrrolidine-2-carbonyl] -amino }-3-methoxy-benzoic acid (Compound A), as well as methods for making it, is disclosed in U.S. Patent No. 8,354,444 and WO2011/098398.

4-{ [(2R,3S,4R,5S)-4-(4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2- dimethyl-prop yl)-pyrrolidine-2-carbonyl] -amino }-3-methoxy-benzoic acid (C31H29Cl2F2N304) (Compound A) is a potent and selective inhibitor of the p53-MDM2 interaction that activates the p53 pathway and induces cell cycle arrest and/or apoptosis in a variety of tumor types expressing wild-type p53 in vitro and in vivo. Compound A belongs to a novel class of MDM2 inhibitors having potent anti-cancer therapeutic activity, in particular in leukemia such as AML and solid tumors such as for example non-small cell lung, breast and colorectal cancers.
The above-identified international patent application and US Patent describe Compound A in crystalline form and is herein incorporated by reference in its totality. The crystalline form of the compound has an on- set melting point of approximately 277 °C. The crystalline forms have relatively low aqueous solubility (<0.05 μg/mL in water) at physiological pHs (which range from pHl.5-8.0) and consequently less than optimal bioavailability (high variability)
…………………………..
WO2013139687A1
Compound A is an orally administered pyrrolidine that inhibits the binding of MDM2 to p53 and is thus useful in the treatment of cancer. It has the following chemical structure:
Molecular Weight =616.4973
Molecular Formula =C31 H29CI2F2N304
Compound A recently entered into phase I clinical trials for the treatment of solid tumors. See ClinicalTrials.gov, identifier NCT01462175. This compound is disclosed in US Pub 2010/0152190 A1 . To the extent necessary, this patent publication is herein incorporated by reference. The Compound A, as well as a method for making it, is also disclosed in WO201 1/098398.
Applicants have discovered that Compound A is especially effective, and best tolerated, in cancer therapy when administered in the specific doses and pursuant to the specific protocols herein described.
PATENT
http://www.google.com/patents/US20100152190
Example 52a Preparation of intermediate (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile
In a manner similar to the method described in Example 1b, 4-chloro-2-fluorophenylacetonitrile (5 g, 30 mmol) was reacted with 3-chloro-2-fluorobenzaldehyde (5 g, 32 mmol), methanolic solution (25 wt %) of sodium methoxide (21 mL, 92 mmol) in methanol (200 mL) at 45° C. for 5 h to give (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile as a white powder (9 g, 97%).
Example 52b Preparation of intermediate rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester
In a manner similar to the method described in Example 1c, [3-methyl-but-(E)-ylideneamino]-acetic acid tert-butyl ester prepared in Example 1a (2.3 g, 11 mmol) was reacted with (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile (2.5 g, 8 mmol) prepared in Example 52a, AgF (0.7 g, 5.5 mmol), and triethylamine (2.9 g, 29 mmol) in dichloromethane (200 mL) at room temperature for 18 h to give rac-(2R,3S,4R,5S)-3-(3-Chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester as a white foam (3 g, 64%).
Example 52c Preparation of intermediate rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid
In a manner similar to the method described in Example 25a, rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester prepared in Example 52b (0.4 g, 0.8 mmol) was reacted with trifluoroacetic acid in dichloromethane at room temperature to give rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid as a white solid (0.5 g, 100%).
HRMS (ES+) m/z Calcd for C23H22Cl2F2N2O2+H [(M+H)+]: 467.1099, found: 467.1098.
Example 137 Preparation of rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid amide
In a manner similar to the method described in Examples 1e, rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid prepared in Example 52c (0.5 g, 0.86 mmol) was reacted with a dioxane solution (0.5 M) of ammonia (2 mL, 1 mmol), HATU (0.38 g, 1 mmol) and iPr2NEt (0.6 g, 4.6 mmol) in CH2Cl2 at room temperature for 20 h to give rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid amide as a white solid (0.3 g, 75%).
HRMS (ES+) m/z Calcd for C23H23Cl2F2N3O+H [(M+H)+]: 466.1259, found: 466.1259.
Example 52a Preparation of intermediate (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile
In a manner similar to the method described in Example 1b, 4-chloro-2-fluorophenylacetonitrile (5 g, 30 mmol) was reacted with 3-chloro-2-fluorobenzaldehyde (5 g, 32 mmol), methanolic solution (25 wt %) of sodium methoxide (21 mL, 92 mmol) in methanol (200 mL) at 45° C. for 5 h to give (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile as a white powder (9 g, 97%).
Example 52b Preparation of intermediate rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester
In a manner similar to the method described in Example 1c, [3-methyl-but-(E)-ylideneamino]-acetic acid tert-butyl ester prepared in Example 1a (2.3 g, 11 mmol) was reacted with (Z)-3-(3-chloro-2-fluoro-phenyl)-2-(4-chloro-2-fluoro-phenyl)-acrylonitrile (2.5 g, 8 mmol) prepared in Example 52a, AgF (0.7 g, 5.5 mmol), and triethylamine (2.9 g, 29 mmol) in dichloromethane (200 mL) at room temperature for 18 h to give rac-(2R,3S,4R,5S)-3-(3-Chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester as a white foam (3 g, 64%).
Example 52c Preparation of intermediate rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid
In a manner similar to the method described in Example 25a, rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid tert-butyl ester prepared in Example 52b (0.4 g, 0.8 mmol) was reacted with trifluoroacetic acid in dichloromethane at room temperature to give rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid as a white solid (0.5 g, 100%).
HRMS (ES+) m/z Calcd for C23H22Cl2F2N2O2+H [(M+H)+]: 467.1099, found: 467.1098.
Example 137 Preparation of rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid amide
In a manner similar to the method described in Examples 1e, rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid trifluoroacetic acid prepared in Example 52c (0.5 g, 0.86 mmol) was reacted with a dioxane solution (0.5 M) of ammonia (2 mL, 1 mmol), HATU (0.38 g, 1 mmol) and iPr2NEt (0.6 g, 4.6 mmol) in CH2Cl2 at room temperature for 20 h to give rac-(2R,3S,4R,5S)-3-(3-chloro-2-fluoro-phenyl)-4-(4-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carboxylic acid amide as a white solid (0.3 g, 75%).
HRMS (ES+) m/z Calcd for C23H23Cl2F2N3O+H [(M+H)+]: 466.1259, found: 466.1259.

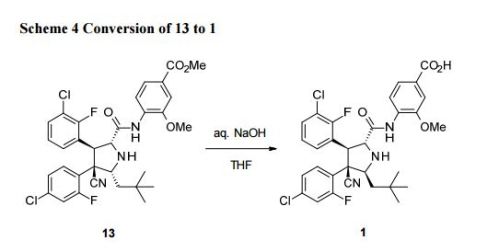
Example 447 Preparation of 4-{[(2R,3S,4R,5S)-4-(4-chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carbonyl]-amino}-3-methoxy-benzoic acid methyl ester
In a 25 mL round-bottomed flask, (2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxylic acid (250 mg, 535 μmol), was combined with CH2Cl2 (5 ml). DIPEA (277 mg, 374 μl, 2.14 mmol) and dipenylphospenic chloride (380 mg, 306 μl, 1.6 mmol) were added and the reaction was stirred at RT for 20 minutes. Methyl 4-amino-3-methoxybenzoate (100 mg, 552 μumol) was added and the reaction mixture was stirred at RT overnight.
The crude reaction mixture was concentrated in vacuum. The crude material was purified by flash chromatography (silica gel, 40 g, 5% to 25% EtOAc/Hexanes) to give the desired product as a white solid (275 mg, 81% yield).
Example 448 Preparation of 4-{[(2R,3S,4R,5S)-4-(4-Chloro-2-fluoro-phenyl)-3-(3-chloro-2-fluoro-phenyl)-4-cyano-5-(2,2-dimethyl-propyl)-pyrrolidine-2-carbonyl]-amino}-3-methoxy-benzoic acid
In a 25 mL round-bottomed flask, methyl 4-((2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxamido)-3-methoxybenzoate (150 mg, 238 μmol, Eq: 1.00) was combined with CH2Cl2 (2 ml) to give a colorless solution. Aluminum bromide (Aldrich, 254 mg, 952 μmol, Eq: 4) and dimethyl sulfide (1.69 g, 2 mL, 27.2 mmol, Eq: 114) were added. The reaction mixture was stirred for overnight.
The reaction mixture was diluted with CH3CN (6 ml), EtOAc (10 ml) and water (10 ml), stirred and layers separated. The aqueous layer was extracted with EtOAc (2×10 mL). The organic layers were combined, washed with saturated NaCl (1×15 mL), dried over MgSO4 and concentrated in vacuum.
The crude material was dissolved in DMSO (4 ml) and was purified by preparative HPLC (70-100% ACETONITRILE/water). The fractions were combined, concentrated and freeze dried to give a white powder as desired product (75 mg, 51% yield). (ES+) m/z Calcd: [(M+H)+]: 616, found: 616.
Alternatively, the title compound could be prepared by the following method.
In a 500 mL round-bottomed flask, methyl 4-((2R,3S,4R,5S)-3-(3-chloro-2-fluorophenyl)-4-(4-chloro-2-fluorophenyl)-4-cyano-5-neopentylpyrrolidine-2-carboxamido)-3-methoxybenzoate (3.74 g, 5.93 mmol, Eq: 1.00) was combined with THF (140 ml) and MeOH (160 ml) at 50° C. to give a colorless solution. 1 N NaOH (23.7 ml, 23.7 mmol, Eq: 4) was added. The reaction mixture was stirred at 40° C. for 18 hrs.
The reaction mixture was concentrated to remove about ½ of the solvent, filtered to removed the insoluble, acidified with 1N HCl to PH=4-5 and the resulting solid was collected by filtration and was washed with water, small amount of MeOH and diethyl ether. It was then dried in vacuum oven (60° C.) overnight. Obtained was a white solid as the desired product (2.96 g, 80.5% yield). H1NMR and LC/MASS data were the same as that in the above procedure.
Paper
see
Bioorganic & Medicinal Chemistry (2014), 22(15), 4001-4009
Update………
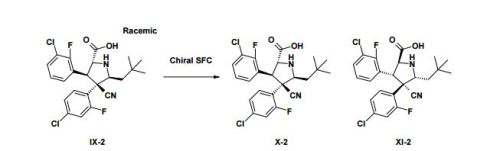
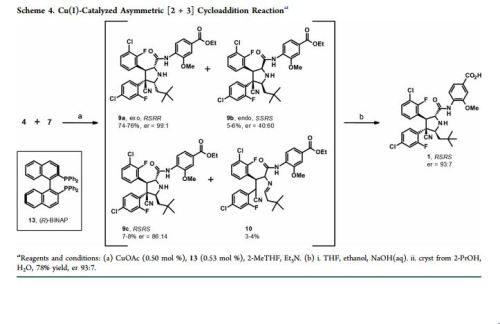
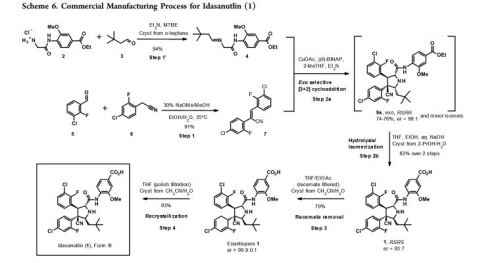
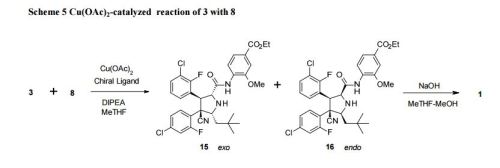
Practical Synthesis of MDM2 Antagonist RG7388. Part 2: Development of the Cu(I) Catalyzed [3 + 2] Asymmetric Cycloaddition Process for the Manufacture of Idasanutlin
Gösta Rimmler†, Andre Alker‡, Marcello Bosco†, Ralph Diodone†, Dan Fishlock†, Stefan Hildbrand†, Bernd Kuhn‡, Christian Moessner†, Carsten Peters†, Pankaj D. Rege*†, and Markus Schantz†
† Small Molecules Technical Development, F. Hoffmann-La Roche Ltd., 4070 Basel, Switzerland
‡ Therapeutic Modalities, Roche Pharma Research and Early Development, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., 4070 Basel, Switzerland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00319
Publication Date (Web): October 31, 2016
Copyright © 2016 American Chemical Society
*Address: F. Hoffmann-La Roche Ltd., Small Molecules Technical Development, Grenzacherstrasse 124, Bldg 86/Room 602, 4070 Basel, Switzerland. Phone:
+41 61 687 39 34. E-mail:
pankaj.rege@roche.com.
Abstract
A concise catalytic asymmetric synthesis of idasanutlin (1) was developed in which the key pyrrolidine core, containing four contiguous stereocenters, was constructed via a Ag/MeOBIPHEP promoted [3 + 2] cycloaddition reaction. Further development of the [3 + 2] cycloaddition reaction resulted in an improvement in diastereoselectivity and enantioselectivity by changing the catalyst system to Cu(I)/BINAP. While producing equivalent high quality API, the copper(I) catalyzed process not only increased the overall yield but also demonstrated benefit with respect to cycle times, waste streams, and processability. The optimized copper(I) catalyzed process has been used to prepare more than 1500 kg of idasanutlin (1).

PAPER
| WO1996038131A1 * |
May 30, 1996 |
Dec 5, 1996 |
James Matthew Butler |
Method of producing a solid dispersion of a poorly water soluble drug |
| WO2010114928A2 * |
Mar 31, 2010 |
Oct 7, 2010 |
F.Hoffmann-La Roche Ag |
Compositions and uses thereof |
| WO2013139687A1 * |
Mar 15, 2013 |
Sep 26, 2013 |
F. Hoffmann-La Roche Ag |
Method for administration of an anti tumor agent |
| WO2013149981A1 * |
Apr 2, 2013 |
Oct 10, 2013 |
F. Hoffmann-La Roche Ag |
Pharmaceutical composition with improved bioavailability, safety and tolerability |
| CN102871950A * |
Jul 15, 2011 |
Jan 16, 2013 |
上海睿智化学研究有限公司 |
一种熊果酸固体分散体及其制备方法 |
| US20100152190 * |
Feb 9, 2010 |
Jun 17, 2010 |
David Joseph Bartkovitz |
Substituted Pyrrolidine-2-Carboxamides |
| US8354444 |
Feb 9, 2010 |
Jan 15, 2013 |
Hoffmann-La Roche Inc. |
Substituted pyrrolidine-2-carboxamides |
| US8709419 |
Aug 10, 2011 |
Apr 29, 2014 |
Hoffmann-La Roche, Inc. |
Combination therapy |
| US20130245089 * |
Feb 5, 2013 |
Sep 19, 2013 |
Hoffmann-La Roche Inc. |
Method for administration |
| WO2011098398A1 * |
Feb 4, 2011 |
Aug 18, 2011 |
F. Hoffmann-La Roche Ag |
Substituted pyrrolidine-2-carboxamides |
| WO2012007409A1 * |
Jul 11, 2011 |
Jan 19, 2012 |
F. Hoffmann-La Roche Ag |
N-substituted pyrrolidines |
| WO2013135648A1 |
Mar 12, 2013 |
Sep 19, 2013 |
F. Hoffmann-La Roche Ag |
Substituted pyrrolidine-2-carboxamides |
| WO2013139687A1 * |
Mar 15, 2013 |
Sep 26, 2013 |
F. Hoffmann-La Roche Ag |
Method for administration of an anti tumor agent |
| WO2013139724A1 |
Mar 18, 2013 |
Sep 26, 2013 |
F. Hoffmann-La Roche Ag |
Combination therapy (vemrufenib and a mdm2 inhibitor) for the treatment proliferative disorders |
| WO2013178570A1 |
May 27, 2013 |
Dec 5, 2013 |
F. Hoffmann-La Roche Ag |
Substituted pyrrolidine-2-carboxamides |
| WO2014114575A1 * |
Jan 20, 2014 |
Jul 31, 2014 |
F. Hoffmann-La Roche Ag |
Pharmaceutical composition with improved bioavailability |
REFERENCES
1 Discovery of RG7388, a Potent and Selective p53-MDM2 Inhibitor in Clinical Development. By Ding, Qingjie; Zhang, Zhuming; Liu, Jin-Jun; Jiang, Nan; Zhang, Jing; Ross, Tina M.; Chu, Xin-Jie; Bartkovitz, David; Podlaski, Frank; Janson, Cheryl; et al From Journal of Medicinal Chemistry (2013), 56(14), 5979-5983.
2. Pyrrolo[1,2-c]imidazolone derivatives as inhibitors of MDM2-p53 interactions and their preparation and use for the treatment of cancer. By Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Zhuming From U.S. Pat. Appl. Publ. (2012), US 20120065210 A1 20120315.
3. Pyrrolidine-2-carboxamide derivatives and their preparation and use as anticancer agents. By Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Zhuming. From U.S. Pat. Appl. Publ. (2012), US 20120010235 A1 20120112.
4. Preparation of substituted pyrrolidine-2-carboxamides as anticancer agents. By Bartkovitz, David Joseph; Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Jing; Zhang, Zhuming
From PCT Int. Appl. (2011), WO 2011098398 A1 20110818.
5. Preparation of substituted pyrrolidine-2-carboxamides as anticancer agents. By Bartkovitz, David Joseph; Chu, Xin-Jie; Ding, Qingjie; Jiang, Nan; Liu, Jin-Jun; Ross, Tina Morgan; Zhang, Jing; Zhang, Zhuming
From U.S. Pat. Appl. Publ. (2010), US 20100152190 A1 20100617.
6 B. Higgins, et al, Antitumor Activity of the MDM2 Antagonist RG7388, Mol Cancer Ther 2013;12(11 Suppl):B55
Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development
J Med Chem 2013, 46(14): 5979
update
Org. Process Res. Dev., 2016, 20 (12), pp 2057–2066
Several polymorphs, pseudopolymorphs, hydrates, and an amorphous form of
1 are known, and the most relevant forms are summarized in
Table 1. The access to the different polymorphic forms is mainly determined by the solvate present in the crystallization process. The use of acetonitrile in the crystallization process of
1 (vide supra) results in the formation of a corresponding low soluble acetonitrile solvate. The acetonitrile in the solvate can easily be removed by drying, resulting in the solvate free Form III.
The final recrystallization process is the following: a water–acetonitrile mixture (1:2.5% w/w) is added to a polish-filtered THF solution of 1 (14% w/w). Crystallization of 1 starts after addition of approximately 1/3 of the aqueous mixture. The isolated acetonitrile solvate (Form IX) of 1 is dried at 80 °C under vacuum to obtain 1 in 93% yield. The acetonitrile in the solvate is easily removed by drying, resulting in the solvate free Form III. As listed in Table 1, Form III is a slightly hygroscopic polymorph of 1 and is reliably produced during clinical supply.
Pathways for Formation of Genotoxic Impurities 16 and 17

/////////

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO





























 STEREOCENTERS SHOWN
STEREOCENTERS SHOWN


 A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430
A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430


, click to zoom.")
















 in phase 3
in phase 3