



")
Anna Popova, Head of the Federal Service for Supervision of Consumer Protection and Welfare (Rospotrebnadzor)
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusABT-414 is in phase I/II clinical development at AbbVie for the treatment of squamous cell carcinoma. The product is also in early clinical development for the treatment of glioblastoma multiforme.
In 2014, orphan drug designation was received in the U.S. and E.U. by AbbVie for the treatment of glioblastoma multiforme.
EGFR antibody-drug conjugate (cancer), Abbott; ABT-414; EGFR antibody-drug conjugate (cancer), AbbVie; EGFR-ADC (cancer), AbbVie; ABT-806-MMAF conjugate; anti-EGFR antibody-MMAF conjugate, AbbVie; EGFR-ADC (cancer), Abbott
AbbVie’s glioblastoma multiforme therapy receives orphan drug designation
AbbVie has obtained orphan drug designation from the European Medicines Agency (EMA) and the US FDA for its anti-epidermal growth factor receptor monoclonal antibody drug conjugate, ABT-414, as a treatment for glioblastoma multiforme.
AbbVie has obtained orphan drug designation from the European Medicines Agency (EMA) and the US FDA for its anti-epidermal growth factor receptor monoclonal antibody drug conjugate, ABT-414, as a treatment for glioblastoma multiforme.
read at
AbbVie announced that the EMA and the FDA have granted orphan drug status to its investigational compound ABT-414, an anti-epidermal growth factor receptor antibody drug conjugate. It is currently being evaluated for safety and efficacy in patients with glioblastoma multiforme. Glioblastoma multiforme is the most common and most aggressive type of malignant primary brain tumor. Each year in the United States and Europe, two to three out of every 100,000 people are diagnosed with glioblastoma multiforme, which has a five-year survival rate of approximately 4 percent.
“The orphan drug designation is an important regulatory advancement as we further our development in recurrent glioblastoma multiforme, a disease that is uniformly fatal with limited treatment options,” said Gary Gordon, M.D., vice president, oncology clinical development, AbbVie. “We are pleased to continue developing ABT-414 in Phase II trials in patients with glioblastoma multiforme based on the results of our Phase I program.” Read the press release
AbbVie oncology clinical development vice-president Gary Gordon said: “The orphan drug designation is an important regulatory advancement as we further our development in recurrent glioblastoma multiforme, a disease that is uniformly fatal with limited treatment options.
“We are pleased to continue developing ABT-414 in Phase II trials in patients with glioblastoma multiforme based on the results of our Phase I programme.”
AbbVie is currently evaluating the safety and efficacy of ABT-414 in patients with glioblastoma multiforme, the most aggressive type of malignant primary brain tumour.
In May, the company presented results from the Phase I clinical trial evaluating ABT-414 in combination with temozolomide in patients with recurrent or unresectable glioblastoma multiforme.
The Phase I trial was designed to assess the toxicities, pharmacokinetics and recommended Phase II dose of ABT-414 when administered every other week in combination with temozolomide.
Other important assessments included adverse events, pharmacokinetic parameters, objective response and tumour tissue epidermal growth factor receptor biomarkers.
The study results showed four objective responses, including one complete response.
AbbVie has developed ABT-414 with components in-licenced from Life Science Pharmaceuticals and Seattle Genetics.
ABT-414 is also being evaluated in clinical trials for the treatment of patients with squamous cell tumours.
About ABT-414
ABT-414 is an anti-EGFR (epidermal growth factor receptor) monoclonal antibody drug conjugate (ADC). As an ADC, ABT-414 is designed to be stable in the bloodstream and only release the potent cytotoxic agent once inside targeted cancer cells. Developed by AbbVie researchers with components in-licensed from Life Science Pharmaceuticals, ABT-414 is currently being investigated for the treatment of glioblastoma multiforme, the most common and most aggressive malignant primary brain tumor. ABT-414 is also in clinical trials for the treatment of patients with squamous cell tumors. ABT-414 is an investigational compound and its efficacy and safety have not been established by the FDA.
About Glioblastoma Multiforme
Glioblastoma is the most common and most aggressive type of malignant primary brain tumor. Each year in the U.S. and Europe, two to three out of every 100,000 people are diagnosed with glioblastoma, which has a five year survival rate of less than 3 percent. Prior to diagnosis, most patients experience a serious symptom of glioblastoma, such as a seizure. Typically patients succumb to the disease approximately 15 months after diagnosis. Treatment for glioblastoma remains challenging and no long-term treatments are currently available. Standard treatment is surgical resection, radiotherapy and concomitant adjunctive chemotherapy. More than 8,700 patients are enrolled in industry-sponsored clinical studies.
ref………
A phase 1 study evaluating ABT-414 in combination with temozolomide (TMZ) for subjects with recurrent or unresectable glioblastoma (GBM)
50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2021
ABT-414: An anti-EGFR antibody drug conjugate for the treatment of glioblastoma patients
18th Annu Sci Meet Soc Neuro-Oncol (November 21-24, San Francisco) 2013, Abst ET-079
ABT-414: An anti-EGFR antibody-drug conjugate as a potential therapeutic for the treatment of patients with squamous cell tumors
25th EORTC-NCI-AACR Symp Mol Targets Cancer Ther (October 19-23, Boston) 2013, Abst A250
A Phase I/II Study Evaluating the Safety, Pharmacokinetics and Efficacy of ABT-414 in Subjects With Advanced Solid Tumors Likely to Over-Express the Epidermal Growth Factor Receptor (EGFR) (NCT01741727)
ClinicalTrials.gov Web Site 2012, December 07
Salk scientists find that a plant used for centuries by healers of São Tomé e Príncipe holds lessons for modern medicine
August 01, 2014
LA JOLLA—For hundreds of years, healers in São Tomé e Príncipe—an island off the western coast of Africa—have prescribed cata-manginga leaves and bark to their patients. These pickings from the Voacanga africana tree are said to decrease inflammation and ease the symptoms of mental disorders.
Now, scientists at the Salk Institute for Biological Studies have discovered that the power of the plant isn’t just folklore: a compound isolated from Voacanga africana protects cells from altered molecular pathways linked to Alzheimer’s disease, Parkinson’s disease and the neurodegeneration that often follows a stroke.
“What this provides us with is a source of potential new drug targets,” says senior author Pamela Maher, a senior staff scientist in Salk’s Cellular Neurobiology Laboratory. The results were published this week in the…
View original post 748 more words

ORITAVANCIN
August 6, 2014
The U.S. Food and Drug Administration today approved Orbactiv (oritavancin), a new antibacterial drug to treat adults with skin infections.
Orbactiv is approved to treat patients with acute bacterial skin and skin structure infections (ABSSSI) caused by certain susceptible bacteria, includingStaphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), various Streptococcus species and Enterococcus faecalis. Orbactiv is administered intravenously.
Orbactiv is the third new antibacterial drug approved by the FDA this year to treat ABSSSI. The agency approved Dalvance (dalbavancin) in May 2014 and Sivextro (tedizolid) in June 2014.
“The approval of several new antibacterial drugs this year demonstrates that we are making progress in increasing the availability of treatment options for patients and physicians,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “However, more work is needed in this area, and the FDA remains a committed partner to help promote the development of antibacterial drugs.”
Orbactiv is also the third new drug designated as a Qualified Infectious Disease Product (QIDP) to receive FDA approval. Under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act, Orbactiv was granted QIDP designation because it is an antibacterial or antifungal human drug intended to treat a serious or life-threatening infection.
As part of its QIDP designation, Orbactiv was given priority review, which provides an expedited review of the drug’s application. Orbactiv’s QIDP designation also qualifies it for an additional five years of marketing exclusivity to be added to certain exclusivity periods already provided by the Food, Drug, and Cosmetic Act.
Orbactiv’s safety and efficacy were evaluated in two clinical trials with a total of 1,987 adults with ABSSSI. Participants were randomly assigned to receive Orbactiv or vancomycin. Results showed Orbactiv was as effective as vancomycin for the treatment of ABSSSI.
The most common side effects identified in the clinical trials were headache, nausea, vomiting, the formation of skin and soft tissue abscesses on arms and legs and diarrhea. Orbactiv’s label also includes a warning regarding interference with coagulation tests and interaction with warfarin, a drug used to prevent blood clots.
Orbactiv is marketed by The Medicines Company, based in Parsippany, N.J.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Read more at: http://www.pharmatimes.com/Article/14-08-07/FDA_clears_third_antibacterial_for_skin_infections_this_year.aspx#ixzz39hKIT6nP
OLD ARTICLE CUT PASTE
Oritavancin
(4R)-22-O-(3-Amino-2,3,6-trideoxy-3-C-methyl-alpha-L-arabinohexopyranosyl)-N3-(p-(p-chlorophenyl)benzyl)vancomycin
(3S, 6R, 7R, 22R, 23S, 26S, 36R, 38aR) -22 – (3-Amino-2 ,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyloxy) -3 – (carbamoylmethyl ) -10,19-dichloro-44-[2-O-[3 – (4′-chlorobiphenyl-4-ylmethylamino) -2,3,6-trideoxy-3-C-methyl-alpha-L-mannopyranosyl] – beta-D-glucopyranosyloxy] –
| CAS No. | 171099-57-3 |
| CBNumber: | CB92451283 |
| Molecular Formula: | C86H97Cl3N10O26 |
| Formula Weight: | 1793.12 |
Also known as NDISACC-(4-(4-chlorophenyl)benzyl)A82846B and LY333328,N-(4-(4-chlorophenyl)benzyl)A82846B
Abbott (Supplier), Lilly (Originator), InterMune (Licensee)
The medicines company—

phx.corporate-ir.net/External.File?item…t=1
Jul 2, 2013 – Inhibits two key steps of cell wall synthesis: – Transglycosylation. – Transpeptidation. • Disrupts bacterial membrane integrity. Differentiated from …
FDA Accepts Filing of NDA for IV Antibiotic Oritavancin with Priority Review
PARSIPPANY, NJ — (Marketwired) — 02/19/14 — The Medicines Company (NASDAQ: MDCO) today announced that the U.S. Food and Drug Administration (FDA) has accepted the filing of a new drug application (NDA) for oritavancin, an investigational intravenous antibiotic, with priority review. The Medicines Company is seeking approval of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA), administered as a single dose.
In December 2013, the FDA designated oritavancin as a Qualified Infectious Disease Product (QIDP). The QIDP designation provides oritavancin priority review, and an additional five years of exclusivity upon approval of the product for the treatment of ABSSSI. Priority review means the FDA’s goal is to take action on the application within six months, compared to 10 months under standard review. The FDA action date (PDUFA date) for oritavancin is August 6, 2014.
Oritavancin (INN, also known as LY333328) is a novel semi-synthetic glycopeptide antibiotic being developed for the treatment of serious Gram-positive infections. Originally discovered and developed by Eli Lilly, oritavancin was acquired by InterMune in 2001 and then by Targanta Therapeuticsin late 2005.[1]
In Dec 2008 the FDA declined to approve it, and an EU application was withdrawn.
In 2009 the development rights were acquired by The Medicine Co. who are running clinical trials for a possible new FDA application in 2013.[2]
Its structure is similar to vancomycin[3] It is a lipoglycopeptide
Oritavancin is an investigational intravenous antibiotic for which The Medicines Company is seeking approval in the treatment of ABSSSI caused by susceptible gram-positive bacteria, including MRSA. In clinical trials, the most frequently reported adverse events associated with oritavancin were nausea, headache, vomiting and diarrhea. Hypersensitivity reactions have been reported with the use of antibacterial agents including oritavancin.

Oritavancin shares certain properties with other members of the glycopeptide class of antibiotics, which includes vancomycin, the current standard of care for serious Gram-positive infections in the United States and Europe.[4] Data presented at the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in September 2007 demonstrated that oritavancin possesses potent and rapid bactericidal activity in vitro against a broad spectrum of both resistant and susceptible Gram positive bacteria, including Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterococci, and Streptococci.[5] Two posters presented at the meeting also demonstrated that oritavancin was more active than either metronidazole or vancomycin against strains of Clostridium difficile tested.[6]
Anthrax : Research presented at the American Society for Microbiology (ASM) 107th Annual General Meeting in May 2007, suggested oritavancin’s potential utility as a therapy for exposure to Bacillus anthracis, the gram-positive bacterium that causes anthrax, having demonstrated efficacy in a mouse model both pre- and post-exposure to the bacterium[7]
 oritavancin
oritavancinThe 4′-chlorobiphenylmethyl group disrupts the cell membrane of gram positive bacteria.[8] It also acts by inhibition of transglycosylation and inhibition of transpeptidation.[9]
A Phase 2 clinical study was planned to run until May 2008 entitled “Single or Infrequent Doses for the Treatment of Complicated Skin and Skin Structure Infections (SIMPLIFI),” evaluating the efficacy and safety of either a single dose of oritavancin or an infrequent dose of oritavancin compared to the previously studied dosing regimen of 200 mg oritavancin given once daily for 3 to 7 days.[11] Results published May 2011.[12]
On February 11, 2008, Targanta submitted a New Drug Application (NDA) to the US FDA seeking approval of oritavancin;[13] in April 2008, the FDA accepted the NDA submission for standard review.[14] On 9 Dec 2008 the FDA said insufficient data for approval of oritavancin had been provided and they requested a further phase 3 clinical study to include more patients with MRSA.[15]
June 2008, Targanta’s Marketing Authorization Application (MAA) for oritavancin was submitted and accepted for review by the European Medicines Agency (EMEA),[16] but the company later withdrew the application in Aug 2009.[17]
The Medicines Company’s purpose is to save lives, alleviate suffering, and contribute to the economics of healthcare by focusing on 3,000 leading acute/intensive care hospitals worldwide. Its vision is to be a leading provider of solutions in three areas: acute cardiovascular care, surgery and perioperative care, and serious infectious disease care. The company operates in the Americas, Europe and the Middle East, and Asia Pacific regions with global centers today in Parsippany, NJ, USA and Zurich, Switzerland.
“We look forward to working with the FDA during the review process, and sharing the knowledge we have gained in our studies of oritavancin,” said Matthew Wikler, MD, Vice President and Medical Director, Infectious Disease Care for The Medicines Company. “We believe that upon approval, oritavancin, administered as a single dose for the treatment of ABSSSI, will offer new options for both physicians and their patients for the treatment of these infections.”
The oritavancin NDA is based on data from two Phase 3 clinical trials, SOLO I and SOLO II, which were conducted under a Special Protocol Assessment (SPA) agreement with the FDA. These Phase 3 trials evaluated the efficacy and safety of a single 1200mg dose of oritavancin compared to 7 to 10 days of twice-daily vancomycin in adults with ABSSSI, including infections caused by MRSA. The combined SOLO studies were conducted in 1,959 patients (modified intent-to -treat population, or mITT), with 405 of the patients suffering from an ABSSSI with a documented MRSA infection.
 oritavancin
oritavancin
Drug substance
Oritavancin diphosphate
CLINICAL TRIALS..http://clinicaltrials.gov/search/intervention=oritavancin
INTRODUCTION
Oritavancin
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.


vancomycin, desmethylvancomycin, eremomycin, teicoplanin (complex of five compounds), dalbavancin, oritavancin, telavancin, and A82846B (LY264826) having structures A, B, C, D, E, F, G and H:

R = B-2-Acetylamido-glucopyraπosyl- Attorney Docket No 33746-704 602


Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopeptides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglycopeptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, whileoritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci.
All three lipoglycopeptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clostridium spp. than dalbavancin, oritavancin or vancomycin. The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily administration. Dalbavancin and telavancin exhibit concentration-dependent activity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities.
Oritavancin’s activity is also considered concentration-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the pharmacodynamic parameter that best describes its activity. Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reactions, fever and diarrhoea were commonly observed withoritavancin therapy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Oritavancin diphosphate (oritavancin) is a semi-synthetic lipoglycopeptide derivative of a naturally occurring glycopeptide. Its structure confers potent antibacterial activity against gram-positive bacteria, including vancomycin-resistant enterococci (VRE), methicillin- and vancomycin-resistant staphylococci, and penicillin-resistant streptococci. The rapidity of its bactericidal activity against exponentially-growing S. aureus (≧3-log reduction within 15 minutes to 2 hours against MSSA, MRSA, and VRSA) is one of the features that distinguishes it from the prototypic glycopeptide vancomycin (McKay et al., J Antimicrob Chemother. 63(6):1191-9 (2009), Epub 2009 Apr. 15).
Oritavancin inhibits the synthesis of peptidoglycan, the major structural component of the bacterial cell wall by a mechanism that is shared with glycopeptides, such as vancomycin (Allen et al., Antimicrob Agents Chemother 41(1):66-71 (1997); Cegelski et al., J Mol Biol 357:1253-1262 (2006); Arhin et al., Poster C1-1471: Mechanisms of action of oritavancin in Staphylococcus aureus [poster]. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007; Chicago, Ill.). Oritavancin, like vancomycin, binds to the Acyl-D-Alanyl-D-Alanine terminus of the peptidoglycan precursor, lipid-bound N-acetyl-glucosamine-N-acetyl-muramic acid-pentapeptide (Reynolds, Eur J Clin Microbiol Infect Dis 8(11):943-950 (1989); Nicas and Allen, Resistance and mechanism of action.
In: Nagarajan R, editor. Glycopeptide antibiotics. New York: Marcel Dekker 195-215 (1994); Allen et al., Antimicrob Agents Chemother 40(10):2356-2362 (1996); Allen and Nicas, FEMS Microbiology Reviews 26:511-532 (2003); Kim et al., Biochemistry 45:5235-5250 (2006)). However, oritavancin inhibits cell wall biosynthesis even when the substrate is the altered peptidoglycan precursor that is present in VRE and vancomycin-resistant S. aureus (VRSA). Thus, the spectrum of oritavancin antibacterial activity extends beyond that of vancomycin to include glycopeptide-resistant enterococci and staphylococci (Ward et al., Expert Opin Investig Drugs 15:417-429 (2006); Scheinfeld, J Drugs Dermatol 6:97-103 (2007)). Oritavancin may inhibit resistant bacteria by interacting directly with bacterial proteins in the transglycosylation step of cell wall biosynthesis (Goldman and Gange, Curr Med Chem 7(8):801-820 (2000); Halliday et al., Biochem Pharmacol 71(7):957-967 (2006); Wang et al., Poster C1-1474: Probing the mechanism of inhibition of bacterial peptidoglycan glycotransferases by glycopeptide analogs. 47th Intersci Conf Antimicro Agents Chemo, Sep. 17-20, 2007). Oritavancin also collapses transmembrane potential in gram positive bacteria, leading to rapid killing (McKay et al., Poster C1-682: Oritavancin disrupts transmembrane potential and membrane integrity concomitantly with cell killing in Staphylococcus aureus and vancomycin-resistant Enterococci. 46th Intersci Conf Antimicro Agents Chemo, San Francisco, Calif., Sep. 27-30, 2006). These multiple effects contribute to the rapid bactericidal activity of oritavancin.
Vancomycin (U.S. Patent 3,067,099); A82846A, A82846B, and A82846C (U.S. Patent 5,312,738, European Patent Publication 256,071 A1); PA-42867 factors A, C, and D (U.S. Patent4,946,941 and European Patent Publication 231,111 A2); A83850 (U.S. Patent No. 5,187,082); avoparcm (U.S. Patent 3,338,786 and U.S. Patent 4,322,343); actmoidin, also known as K288 (J. Antibiotics Series A 14:141 (1961); helevecardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 86/157,397); galacardin (Chem. Abstracts 110:17188 (1989) and Japanese Patent Application 89/221,320); and M47767 (European Patent Publication 339,982).
Oritavancin is in clinical development against serious gram-positive infections, where administration of the drug is via intravenous infusion using several dosages administered over a series of days. The development of alternative dosing regimens for the drug could expand treatment options available to physicians. The present invention is directed to novel dosing regimens.
Means for the preparation of the glycopeptide antibiotics, including oritavancin and analogs thereof, may be found, for example, in U.S. Pat. No. 5,840,684,
SYNTHESIS
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
J Antibiot1996, 49, (6) :575-81
(3S,6R,7R,22R,23S,26S,36R,38aR)-3-(Carbamoylmethyl)-10,19-dichloro-7,28,30,32-tetrahydroxy-6-(N-methyl-D-leucylamido)-2,5,24,38,39-pentaoxo-22-(L-vancosaminyloxy)-44-[2-O-(L-vancosaminyl)-beta-D-glucopyranosyloxy]-2,3,4,5,6,7,23,24,25,26,36,37,38,38a-tetradecahydro-1H,22H-8,11:18,21-dietheno-23,36-(iminomethano)-13,16:31,36-dimetheno-[1,6,9]oxadiazacyclohexadecino[4,5-m][10,2,16]benzoxadiazacyclotetracosine-26-carboxylic acid; A82846B (I)
4′-chloro[1,1′-biphenyl]-4-carbaldehyde (II)
LY-333328 was synthesized by reductocondensation of the glycopeptide antibiotic A82846B (I) with 4′-chlorobiphenyl-4-carboxaldehyde (II) by means of sodium cyanoborohydride in refluxing methanol.
…………………..
EXAMPLE 4
Preparation of Compound 229
A three liter 3-necked flask was fitted with a
condenser, nitrogen inlet and overhead mechanical stirring apparatus. The flask was charged with pulverized A82846B acetate salt (20.0 g, 1.21 × 10-3 mol) and methanol (1000 mL) under a nitrogen atmosphere. 4′-chlorobiphenylcarboxaldehyde (2.88 g, 1.33 × 10-2 mol, 1.1 eq.) was added to this stirred mixture, followed by methanol (500 mL). Finally, sodium cyanoborohydride (0.84 g, 1.33 × 10-2 mol, 1.1 eq.) was added followed by methanol (500 mL). The resulting mixture was heated to reflux (about 65°C).
After 1 hour at reflux, the reaction mixture attained homogeneity. After 25 hours ac reflux, the heat source was removed and the clear reaction mixture was measured with a pH meter (6.97 at 58.0°C). 1 N NaOH (22.8 mL) was added
dropwise to adjust the pH to 9.0 (at 54.7°C). The flask was equipped with a distillation head and the mixture was concentrated under partial vacuum to a weight of 322.3 grams while maintaining the pot temperature between 40-45°C.
The distillation head was replaced with an addition funnel containing 500 mL of isopropanol (IPA). The IPA was added dropwise to the room temperature solution over 1 hour. After approximately 1/3 of the IPA was added, a granular precipitate formed. The remaining IPA was added at a faster rate after precipitation had commenced. The flask was weighed and found to hold 714.4 grams of the IPA/methanol slurry.
The flask was re-equipped with a still-head and
distilled under partial vacuum to remove the remaining methanol. The resulting slurry (377.8 g) was allowed to chill in the freezer overnight. The crude product was filtered through a polypropylene pad and rinsed twice with 25 mL of cold IPA. After pulling dry on the funnel for 5 minutes, the material was placed in the vacuum oven to dry at 40°C. A light pink solid (22.87 g (theory = 22.43 g) ) was recovered. HPLC analysis versus a standard indicated 68.0% weight percent of Compound 229 (4- [4-chlorophenyl] benzyl-A82846B] in the crude solid, which translated into a
corrected crude yield of 69.3%.
The products of the reaction were analyzed by reverse-phase HPLC utilizing a Zorbax SB-C18 column with ultraviolet light (UV; 230 nm) detection. A 20 minute gradient solvent system consisting of 95% aqueous buffer/5% CH3CN at time=0 minutes to 40% aqueous buffer/60% CH3CN at time=20 minutes was used, where the aqueous buffer was TEAP (5 ml CH3CN, 3 ml phosphoric acid in 1000 ml water).
………………….
Oritavancin (also termed N-(4-(4-chlorophenyl)benzyl)A82846B and LY333328) has the following Formula III:

Cooper, R.D.G.; Snyder, N.J.; Zweifel, M.J.; et al.; Reductive alkylation of glycopeptide antibiotics: Synthesis and antibacterial activity. J Antibiot 1996, 49, 6, 575-81.
Cooper, R.D.G.; Huff, B.E.; Nicas, T.I.; Quatroche, J.T.; Rodriguez, M.J.; Snyder, N.J.; Staszak, M.A.; Thompson, R.C.; Wilkie, S.C.; Zweifel, M.J. (Eli Lilly and Company); Glycopeptide antibiotic derivs. EP 0667353; EP 1016670; EP 1031576 .
| EP0435503A1 * | Dec 11, 1990 | Jul 3, 1991 | Eli Lilly And Company | Improvements in or relating to gylcopeptide derivatives |
| US4639433 * | Aug 14, 1985 | Jan 27, 1987 | Eli Lilly And Company | Glycopeptide derivatives |
| US4698327 * | Apr 18, 1986 | Oct 6, 1987 | Eli Lilly And Company | Novel glycopeptide derivatives |
| US20040106590 * | Aug 29, 2003 | Jun 3, 2004 | Barry Eisenstein | Methods and reagents for treating infections of clostridium difficile and diseases associated therewith |
| US20050197333 | Dec 22, 2004 | Sep 8, 2005 | Van Duzer John H. | Rifamycin analogs and uses thereof |
| US20070014849 | Sep 20, 2006 | Jan 18, 2007 | Daniela Jabes | Use of ramoplanin to treat diseases associated with the use of antibiotics |
| US20030176327 * | Oct 18, 2002 | Sep 18, 2003 | Cassell Gail Houston | Antibiotics for treating biohazardous bacterial agents |
| US20040147441 | Aug 25, 2003 | Jul 29, 2004 | Leach Timothy S. | Methods and reagents for preventing bacteremias |
| WO1999010006A1 | Aug 18, 1998 | Mar 4, 1999 | Lilly Co Eli | Therapy for staphylococcus aureus |
| WO2000066144A2 | Apr 19, 2000 | Nov 9, 2000 | Lilly Co Eli | Monthly doses of glycopeptide antibiotics for treatment of streptococcus pneumoniae infections |
| WO2008097364A2 | Sep 24, 2007 | Aug 14, 2008 | Targanta Therapeutics Corp | Use of oritavancin for prevention and treatment of anthrax |
| WO1998052592A1 * | May 5, 1998 | Nov 26, 1998 | Lilly Co Eli | Urea and thiourea derivatives of glycopeptides |
| WO2002036612A1 * | Nov 2, 2001 | May 10, 2002 | Univ Cambridge Tech | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane-associating elements |
| WO2007138999A1 | May 25, 2007 | Dec 6, 2007 | Shionogi & Co | Glycopeptide antibiotic derivative |
| WO2009081958A1 | Dec 25, 2008 | Jul 2, 2009 | Shionogi & Co | Glycosylated glycopeptide antibiotic derivative |
| EP2314599A1 | Nov 24, 2005 | Apr 27, 2011 | National University Corporation Nagoya University | Glycopeptide antibiotic monomer derivatives |
| US5919756 * | May 1, 1997 | Jul 6, 1999 | Eli Lilly And Company | Amides |
| US5919771 * | Apr 30, 1998 | Jul 6, 1999 | Eli Lilly And Company | Urea and thiourea derivatives of glycopeptides |
| US7078380 | Nov 2, 2001 | Jul 18, 2006 | Cambridge University Technical Services Limited | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane associating elements |
| US8481696 | Dec 25, 2008 | Jul 9, 2013 | Shionogi & Co., Ltd. | Glycosylated glycopeptide antibiotic derivatives |


How can one find a certain GMP requirement in the EU GMP Guide, in the FDA cGMP Guide and in the ISO 9001 without searching for a long time? The Good Practice Guide developed by the ECA has become a standard in many companies and is aimed at providing this information. A 26 pages matrix provides information about where to find a GMP requirement e.g. on Validation, QC Lab testing etc in the three major Guidelines. The comprehensive booklet with 500 pages contains a full text version of all three guidelines. You can find the GMP Matrix here.
http://www.gmp-compliance.org/eca_handbuecher.html
Publications

ECA Good Practice Guide – “GMP Matrix”
“FDA cGMP, EU GMP and ISO 9001 Matrix for a pharmaceutical Quality System – A GMP Roadmap”. (Version 15 of April 2014)
The revised ECA Good Practice Guide is a comprehensive juxtaposition containing the requirements laid down in FDA’s cGMP Guide, the EU GMP Guide and ISO 9001. The updated Matrix now has 26 pages as well as further 500 pages for the following three regulations
In addition, the Good Practice Guide contains a ISO 9001/ICH10 Matrix and the complete Part III to the EU GMP Guide.
Price*: € 149 Non ECA Members, € 99 ECA Members

Is it possible to use the results of collaborative trials for analytical methods to prove the laboratory- and product-specific validation of a method? From the perspective of this EMA reflection paper the concrete specifications are missing. These will be developed in the future. Find out more in this news.

GMP News: EMA publishes Document on the Validation of analytical Methods
On 26 June 2014, the European Medicines Agency (EMA) published the concept paper “Transferring quality control methods validated in collaborative trials to a product/laboratory specific context”.
To accept a method an authority always requires a scientific validation. The same applies when existing methods are to be replaced, reduced or to be optimized (3R = replacement, reduction, refinement). Many of these new methods principally represent an improvement compared to the old “standard” methods and therefore are acceptable from a regulatory perspective.
The scientific proof of validation also includes the evidence of the concept and the possibility to transfer a method between different laboratories as well as large scale collaborative studies indicating that a method is suitable for the intended purpose. After completing these steps successfully, it can ultimately result in a monograph of the European Pharmacopoeia (Ph. Eur.) or also in a guidance document for the WHO or the EMA.
This method’s validity has to be proven by the laboratory that proposes the new method. Moreover, this validation also needs to be proven specifically for the medicinal products it is supposed to be used for. Laboratories that participated in large scale collaborative studies before, usually already created plenty of data telling something about the function of this method.
This EMA concept paper now suggests that more guidance documents should be developed on this subject: how can these data from large scale collaborative studies be used to easier implement the laboratory- and product-specific validation of 3R methods (3R – see above)? The concrete specifications for this are currently still missing.
The issue is also to introduce an alternative method without necessarily having to show that the new method correlates with the existing Pharmacopoeia method.
To get additional details please see the complete Reflection Paper “Transferring quality control methods validated in collaborative trials to a product/laboratory specific context“.
The deadline for submission of comments is on 31 October 2014.


Momelotinib
414.47, C23H22N6O2,
1056634-68-4
FDA 2023, Ojjaara,
| To treat intermediate or high-risk myelofibrosis in adults with anemia Drug Trials Snapshot |
N-(Cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide
N-(Cyanomethyl)-4-[2-[4-(4-morpholinyl)phenylamino]pyrimidin-4-yl]benzamide
Jak2 tyrosine kinase inhibitor; Jak1 tyrosine kinase inhibitor
Inflammatory disease; Myelofibrosis; Myeloproliferative disorder; Pancreatic ductal adenocarcinoma; Polycythemia vera
CYT 387; CYT-387; momelotinib)
GS-0387
CYT387 sulfate saltCAS No: 1056636-06-6
 CYT387 Mesylate CAS No: 1056636-07-7
CYT387 Mesylate CAS No: 1056636-07-7
DI HCL SALT 1380317-28-1
Momelotinib, sold under the brand name Ojjaara among others, is an anticancer medication used for the treatment of myelofibrosis.[5] It is a Janus kinase inhibitor and it is taken by mouth.[5]
The most common adverse reactions include dizziness, fatigue, bacterial infection, hemorrhage, thrombocytopenia, diarrhea, and nausea.[8]
Momelotinib was approved for medical use in the United States in September 2023,[5][8][9] and in the European Union in January 2024.[6][10]
CYT387 is an ATP-competitive small molecule JAK1 / JAK2 inhibitor with IC50 of 11 and 18 nM for JAK1 and JAK2, respectively. CYT387 is useful for treatment of myeloproliferative disorders and anti-cancer.
CYT-387 is a potent, orally administered JAK1/JAK2/ Tyk2 inhibitor in phase III clinical studiest at Gilead for the treatment of post-polycythemia vera, for the treatment of primary myelofibrosis and for the treatment of post-essential thrombocythemia. Phase II studies are also ongoing, in combination with gemcitabine and nab-paclitaxel, in adults with untreated metastatic pancreatic ductal adenocarcinoma.
The compound possesses an excellent selectivity and safety profile. In 2010 and 2011, orphan drug designation was assigned by the FDA and the EMA, respectively, for the treatment of myelofibrosis. In 2011, orphan drug designation was assigned by the EMA for the treatment of post-essential thrombocythemia myelofibrosis and for the treatment of post-polycythemia vera myelofibrosis.
PAT
http://www.google.com.ar/patents/US8486941?cl=ja
N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide
| 3 |
 |
414.18 | 1H NMR (300 MHz, d6-DMSO): δ 9.47 (1 H, s), 9.32 (1 H, t, J = 5.5 Hz), 8.54 (1 H, d, J = 5.0 Hz), 8.27 (2 H, d, J = 8.7 Hz), 8.02 (2 H, d, J = 8.2 Hz), 7.67 (2 H, d, J = 9.1 Hz), 7.41 (1 H, d, J = 5.5 Hz), 6.93 (2 H, d, J = 9.1 Hz), 4.36 (2 H, d, J = 5.5 Hz), 3.75 (4 H, m), 3.05 (4 H, m). | m/z 415.3 [M + H]+ | N-(cyanomethyl)-4-(2-(4- morpholinophenylamino)pyrimidin- 4-yl)benzamide |
Example 1Synthesis of Compound 3
A mixture of 4-ethoxycarbonylphenyl boronic acid (23.11 g, 119 mmol), 2,4-dichloropyrimidine (16.90 g, 113 mmol), toluene (230 mL) and aqueous sodium carbonate (2 M, 56 mL) was stirred vigorously and nitrogen was bubbled through the suspension for 15 minutes. Tetrakis(triphenylphosphine)palladium[0] (2.61 g, 2.26 mmol) was added. Nitrogen was bubbled through for another 10 min., the mixture was heated to 100° C., then at 75° C. overnight. The mixture was cooled, diluted with ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. The aqueous layer was extracted with ethyl acetate (100 ml) and the two organic extracts were combined. The organics were washed with brine, filtered through sodium sulfate, concentrated, and the resultant solid was triturated with methanol (100 mL) and filtered. The solids were washed with methanol (2×30 mL) and air dried. This material was dissolved in acetonitrile (150 mL) and dichloromethane (200 mL), stirred with MP.TMT Pd-scavenging resin (Agronaut part number 800471) (7.5 g) over 2 days. The solution was filtered, the solids were washed with dichloromethane (2×100 mL), and the filtrate concentrated to give ethyl 4-(2-chloropyrimidin-4-yl)benzoate as an off-white solid (17.73 g, 60%)—additional washing with dichloromethane yielded a further 1.38 g and 0.5 g of product. 1H NMR (300 MHz, d6-DMSO) δ 8.89 (1H, d, J=5.0 Hz); 8.32 (2H, d, J=8.7 Hz); 8.22 (1H, d, J=5.5 Hz); 8.12 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=7.1 Hz); 1.34 (3H, t, J=7.1 Hz); LC-ESI-MS (method B): rt 7.3 min.; m/z 263.0/265.0 [M+H]+.
A mixture of ethyl 4-(2-chloropyrimidin-4-yl)benzoate (26.15 g, 99.7 mmol) and 4-morpholinoaniline (23.10 g, 129.6 mmol) was suspended in 1,4-dioxane (250 mL). p-Toluenesulfonic acid monohydrate (17.07 g, 89.73 mmol) was added. The mixture was heated at reflux for 40 h., cooled to ambient temperature, concentrated then the residue was partitioned between ethyl acetate and 1:1 saturated sodium bicarbonate/water (1 L total). The organic phase was washed with water (2×100 mL) and concentrated. The aqueous phase was extracted with dichloromethane (3×200 mL). The material which precipitated during this workup was collected by filtration and set aside. The liquid organics were combined, concentrated, triturated with methanol (200 mL) and filtered to yield additional yellow solid. The solids were combined, suspended in methanol (500 mL), allowed to stand overnight then sonicated and filtered. The solids were washed with methanol (2×50 mL) to give, after drying, ethyl 4-(2-(4-morphonlinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 88%). 1H NMR (300 MHz, d6-DMSO) δ 9.49 (1H, s); 8.54 (1H, d, J=5.0 Hz); 8.27 (2H, d, J=8.7 Hz); 8.10 (2H, d, J=8.7 Hz), 7.66 (2H, d, J=9.1 Hz); 7.38 (1H, d, J=5.0 Hz); 6.93 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=6.9 Hz), 3.73 (4H, m); 3.04 (4H, m); 1.34 (3H, t, J=6.9 Hz); LC-ESI-MS (method B): rt 7.5 min.; m/z 404.1 [M+H].
A solution of ethyl 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 87.6 mmol) in 3:1 methanol/tetrahydrofuran (350 mL) was treated with lithium hydroxide (4.41 g, 183.9 mmol) in water (90 mL). The mixture was heated at reflux for 2 h., cooled, concentrated and acidified with hydrochloric acid (2M, 92.5 mL, 185 mmol). The dark precipitate was filtered, washed with water, and dried under vacuum. The solid was ground to a powder with a mortar and pestle, triturated with methanol (500 mL) then filtered again to yield 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid as a muddy solid. This material was washed with ether, air dried overnight, and ground to a fine powder with mortar and pestle. On the basis of mass recovery (34.49 g) the yield was assumed to be quantitative. 1H NMR (300 MHz, d6-DMSO) δ 9.47 (1H, s); 8.53 (1H, d, J=5.2 Hz); 8.24 (2H, d, J=8.5 Hz); 8.08 (2H, d, J=8.8 Hz), 7.66 (2H, d, J=9.1 Hz); 7.37 (1H, d, J=5.2 Hz); 6.93 (2H, d, J=9.1 Hz); 3.73 (4H, m); 3.04 (4H, m). LC-ESI-MS (method C): rt 7.3 min.; m/z 377.1 [M+H]+.
To a suspension of 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid (theoretically 32.59 g, 86.6 mmol) in DMF (400 mL) was added triethylamine (72.4 mL, 519.6 mmol, 6 eq.) The mixture was sonicated to ensure dissolution. Aminoacetonitrile hydrochloride (16.02 g, 173.2 mmol) was added followed by N-hydroxybenzotriazole (anhydrous, 14.04 g, 103.8 mmol) and 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (19.92 g, 103.8 mmol). The suspension was stirred vigorously overnight. The solvent was evaporated under reduced pressure, the residue was diluted with 5% sodium bicarbonate (400 mL) and water (300 mL), giving a yellow solid, which was broken up and filtered. The solids were washed several times with 100 mL portions of water, triturated with hot methanol/dichloromethane (500 mL, 1:1), concentrated to a volume of approximately 300 mL), cooled and filtered. The solids were washed with cold methanol (3×100 mL), ether (200 mL) and hexane (200 mL) prior to drying to afford
Compound 3 (31.69 g, 88%). M.p. 238-243° C.
Microanalysis: Found C, 66.52; H, 5.41; N, 20.21. C23H26N6O10S2 requires C, 66.65; H, 5.35; N, 20.28%.
13C NMR (75.5 MHz, d6-DMSO) δ 166.04, 162.34, 160.26, 159.14, 146.14, 139.87, 134.44, 132.73, 127.80, 126.84, 120.29, 117.49, 115.50, 107.51, 66.06, 49.16, 27.68.
1H NMR GIVEN ABOVE
Example 6Salt Formation from Compound 3
Compound 3 (10.0 g) was suspended in methanol (1 L). Concentrated sulfuric acid (10.52 g, 90% w/w) was added dropwise to the stirring solution. A clear brown solution resulted and a solid lump formed. The solution was filtered quickly then allowed to continue stirring for 3 h (a second precipitate appeared within minutes). After this time the pale yellow precipitate was collected by filtration, washed with methanol (10 mL) then dried under vacuum overnight to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium hydrogensulfate, as a pale yellow solid (10.20 g, 69%). m.p. 205° C. Microanalysis: Found C, 45.18; H, 4.36; N, 13.84; S, 10.24. C23H26N6O10S2 requires C, 45.24; H, 4.29; N, 13.76; S 10.50%. 1H NMR (300 MHz, d6-DMSO) δ 9.85 (br. s, 1H), 9.34 (t, J=5.4 Hz, 1H), 8.59 (d, J=5.2 Hz, 1H), 8.27 (d, J=8.5 Hz, 2H), 8.03 (d, J=8.5 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 7.50 (d, J=5.2 Hz, 1H), 7.34 (br. s, 2H), 4.36 (d, J=5.4 Hz, 2H), 3.89 (br. s, 4H), 3.37 (br. s, 4H); 13C NMR (75.5 MHz, d6-DMSO) δ 166.07, 163.36, 159.20, 158.48, 140.19, 139.34, 136.45, 134.89, 128.00, 127.22, 121.13, 119.89, 117.59, 109.05, 64.02, 54.04, 27.82. LC-ESI-MS (method D): rt 10.0 min.; m/z 415.1 [M+H]+.
Compound 3 (0.25 g) was suspended in methanol (25 ml). Methane sulfonic acid (0.255 g) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 3 h, after which the volume was reduced to 9 ml. The resultant precipitate was collected and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium methanesulfonate as a pale yellow solid (0.22 g). m.p. 208° C. 1H NMR (300 MHz, d6-DMSO) δ 9.83 (br. s, 1H), 9.35 (t, J=5.3 Hz, 1H), 8.59 (d, J=5.1 Hz, 1H), 8.28 (d, J=8.5 Hz, 2H), 8.04 (d, J=8.5 Hz, 2H), 7.83 (d, J=9.0 Hz, 2H), 7.50 (d, J=5.5 Hz, 1H), 7.31 (d, J=9.0 Hz, 2H), 4.36 (d, J=5.5 Hz, 2H), 3.88 (m, 4H), 3.35 (br. s, 4H), 2.36 (s, 6H); LC-ESI-MS (method D): rt 10.2 min.; m/z 415.3 [M+H]+.
Compound 3 (0.50 g) was suspended in methanol (45 ml). A freshly prepared solution of hydrochloric acid in methanol (2.6 ml, HCl conc. 40 mg/ml) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 2 h, then the resultant precipitate was collected, washed with methanol (5 ml) and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium chloride a pale yellow solid (0.30 g). m.p. 210° C. 1H NMR (300 MHz, d6-DMSO) 1H NMR (300 MHz, DMSO) δ 9.92 (br. s, 1H), 9.42 (t, J=5.3, 1H), 8.62 (d, J=4.8, 1H), 8.29 (d, J=8.1, 2H), 8.06 (d, J=8.1, 2H), 7.89 (d, J=9.0, 2H), 7.53 (br. s, 3H), 4.36 (d, J=5.4, 2H), 3.82 (br. s, 4H), 3.43 (br. s, 4H)
LC-ESI-MS (method D): rt 10.3 min.; m/z 415.3 [M+H]+.
PAT
WO 2014114274
. [1] A Pardanani et al CYT387, a Selective JAK1 / JAK2 inhibitor: in vitroassessment of kinase selectivity and preclinical s using Cell lines and Primary cells from polycythemia vera Patients Leukemia (2009) 23, 1441-1445
Abstract
Somatic mutations in Janus kinase 2 (JAK2), including JAK2V617F, result in dysregulated JAK-signal transducer and activator transcription (STAT) signaling, which is implicated in myeloproliferative neoplasm (MPN) pathogenesis. CYT387 is an ATP-competitive small molecule that potently inhibits JAK1 / JAK2 kinases ( IC (50) = 11 and 18 nM, respectively), with significantly less activity against other kinases, including JAK3 (IC (50) = 155 nM). CYT387 inhibits growth of Ba / F3-JAK2V617F and human erythroleukemia (HEL) cells ( IC (50) approximately 1500 nM) or Ba / F3-MPLW515L cells (IC (50) = 200 nM), but has considerably less activity against BCR-ABL harboring K562 cells (IC = 58 000 nM). Cell lines harboring mutated JAK2 alleles (CHRF-288-11 or Ba / F3-TEL-JAK2) were inhibited more potently than the corresponding pair harboring mutated JAK3 alleles (CMK or Ba / F3-TEL-JAK3), and STAT-5 phosphorylation was inhibited in HEL cells with an IC (50) = 400 nM. …
[2]. Tyner Jeffrey W. et al CYT387, a novel JAK2 inhibitor, induces Hematologic Responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms Blood June 24, 2010vol. no 115. 255232-5240
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. IDENTIFIED We have an aminopyrimidine derivative ( CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting JAK2 inhibitors That May be Unable to Eliminate JAK2 (V617F) cells, Consistent with Preliminary results from Clinical Trials of JAK2 inhibitors in myelofibrosis. …
[3]. Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH.Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma.J Chromatogr B Analyt Technol Biomed Life Sci 2012 May 1; 895-896:. 174-7 Epub 2012 Mar 23..
abstract
A quantitative bioanalytical Liquid Chromatography-Tandem Mass spectrometric (LC-MS / MS) assay for the JAK2 inhibitor CYT387 WAS Developed and validated. Plasma samples Were Treated using pre-Protein precipitation with acetonitrile containing cediranib as Internal Standard. The extract WAS Directly Injected into the chromatographic system after dilution with water. This system consisted of a sub-2 μm particle, trifunctional bonded octadecyl silica column with a gradient using 0.005% (v / v) of formic acid in a mixture of water and methanol. The eluate was transferred into the electrospray interface with positive ionization and the analyte was detected in the selected reaction monitoring mode of a triple quadrupole mass spectrometer. The assay was validated in a 0.25-1000 ng / ml calibration range. Within day precisions were 3.0-13.5%, BETWEEN Day Precisions 5.7% and 14.5%. Accuracies Were BETWEEN 96% and 113% for the Whole Calibration range. The Drug WAS stable under All Relevant Analytical Conditions. Finally, the assay successfully WAS Used to ASSESS Drug Levels in mice.
[4] . Monaghan KA, Khong T, Burns CJ, Spencer A.The novel JAK inhibitor CYT387 suppresses Multiple Signalling pathways, and induces apoptosis in Prevents Proliferation phenotypically Diverse myeloma cells.Leukemia 2011 Dec; 25 (12):. 1891-9.
Abstract
Janus kinases (JAKs) are involved in various signalling pathways exploited by malignant cells. In multiple myeloma (MM), the interleukin-6 / JAK / signal transducers and activators of transcription (IL-6 / JAK / STAT) pathway has been the focus of research for a number of years and IL-6 has an established role in MM drug resistance. JAKs therefore make a rational drug target for anti-MM therapy. CYT387 is a novel, orally bioavailable JAK1 / 2 inhibitor, which has recently been described. This preclinical evaluation of CYT387 for treatment of MM demonstrated that CYT387 was able to prevent IL-6-induced phosphorylation of STAT3 and greatly decrease IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in human myeloma cell lines (HMCL). CYT387 inhibited MM proliferation in a time- and dose-dependent manner in 6/8 HMCL, and this was not abrogated by the addition of exogenous IL-6 (3/3 HMCL). Cell cycling was inhibited with a G (2) / M accumulation of cells, and apoptosis was induced by CYT387 in all HMCL tested (3/3). CYT387 synergised in killing HMCL when used in combination with the conventional anti-MM therapies melphalan and bortezomib. Importantly, WAS Also apoptosis induced in Primary Patient MM cells (N = 6) with CYT387 as a single agent, and synergy WAS Seen Again when Combined with Conventional therapies.
[5]. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW.CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms.Blood 2010 Jun 24; 115 (25):. 5232- 40. Epub 2010 Apr 12.
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. We have IDENTIFIED an aminopyrimidine derivative (CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting that JAK2 inhibitors may be unable to eliminate JAK2 (V617F) cells, consistent with preliminary results from clinical trials of JAK2 inhibitors in myelofibrosis. While the clinical benefit of JAK2 inhibitors may be substantial, not the least due to reduction of inflammatory cytokines and symptomatic improvement, our data add to increasing evidence that kinase inhibitor monotherapy of malignant disease is not curative, suggesting a need for drug combinations to optimally target the malignant cells.
JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to, amongst other things, cellular proliferation.
The central role played by the JAK family of protein tyrosine kinases in the cytokine dependent regulation of both proliferation and end function of several important cell types indicates that agents capable of inhibiting the JAK kinases are useful in the prevention and chemotherapeutic treatment of disease states dependent on these enzymes. Potent and specific inhibitors of each of the currently known four JAK family members will provide a means of inhibiting the action of the cytokines that drive immunological and inflammatory diseases.
Myeloproliferative disorders (MPD) include, among others, polycythemia vera (PV), primary myelofibrosis, thrombocythemia, essential thrombocythemia (ET), idiopathic myelofibrosis (IMF), chronic myelogenous leukemia (CML), systemic mastocystosis (SM), chronic neutrophilic leukemia (CNL), myelodisplastic syndrome (MDS) and systemic mast cell disease (SMCD). JAK2 is a member of the JAK family of kinases in which a specific mutation (JAK2V617F) has been found in 99% of polycythemia vera (PV) patients and 50% of essential thrombocytopenia (ET) and idiopathic myelofibrosis (MF). This mutation is thought to activate JAK2, giving weight to the proposition that a JAK2 inhibitor will be useful in treating these types of diseases.
Asthma is a complex disorder characterized by local and systemic allergic inflammation and reversible airway obstruction. Asthma symptoms, especially shortness of breath, are a consequence to airway obstruction, and death is almost invariably due to asphyxiation. Airway Hyper Responsiveness (AHR), and mucus hyper secretion by goblet cells are two of the principle causes of airway obstruction in asthma patients. Intriguingly recent work in animal experimental models of asthma has underscored the importance of IL-13 as a key player in the pathology of asthma. Using a specific IL-13 blocker, it has been demonstrated that IL-13 acts independently of IL-4 and may be capable of inducing the entire allergic asthma phenotype, without the induction of IgE (i.e. in a non-atopic fashion). This and other models have pointed to an important second tier mechanism for elicitating the pathophysiology of asthma, that is not dependent on the production of IgE by resident B-cells or the presence of eonisophils. A direct induction of AHR by IL-13, represents an important process that is likely to be an excellent target for intervention by new therapies. A contemplated effect of a JAK2 inhibitor to the lungs would result in the suppression of the local release of IL-13 mediated IgE production, and therefore reduction in histaminine release by mast cells and eosinophils. This and other consequences of the absence of IL-13 indicate that many of the effects of asthma may be alleviated through administration of a JAK2 inhibitor to the lungs.
Chronic Obstructive Pulmonary Disease (COPD) is a term which refers to a large group of lung diseases which can interfere with normal breathing. Current clinical guidelines define COPD as a disease state characterized by airflow limitation which is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles and gases, particularly cigarette smoke and pollution. Several studies have pointed to an association between increased production of IL-13 and COPD, lending support to the proposition that the potential alleviation of asthma symptoms by use of a JAK2 inhibitor, may also be achieved in COPD. COPD patients have a variety of symptoms including cough, shortness of breath, and excessive production of sputum. COPD includes several clinical respiratory syndromes including chronic bronchitis and emphysema.
Chronic bronchitis is a long standing inflammation of the bronchi which causes increased production of mucus and other changes. The patient’s symptoms are cough and expectoration of sputum. Chronic bronchitis can lead to more frequent and severe respiratory infections, narrowing and plugging of the bronchi, difficult breathing and disability.
Emphysema is a chronic lung disease which affects the alveoli and/or the ends of the smallest bronchi. The lung loses its elasticity and therefore these areas of the lungs become enlarged. These enlarged areas trap stale air and do not effectively exchange it with fresh air. This results in difficult breathing and may result in insufficient oxygen being delivered to the blood. The predominant symptom in patients with emphysema is shortness of breath.
Additionally, there is evidence of STAT activation in malignant tumors, among them lung, breast, colon, ovarian, prostate and liver cancer, as well as Hodgkins lymphoma, multiple myeloma and hepatocellular carcinoma. Chromosomal translocations involving JAK2 fusions to Tel, Bcr and PCM1 have been described in a number of hematopoietic malignancies including chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), chronic eosinophilic leukemia (CEL), myelodisplastic syndrome (MDS), myeloproliferative disease (MPD) and acute lymphocytic leukemia (ALL). This suggests treatment of hyperproliferative disorders such as cancers including multiple myeloma; prostate, breast and lung cancer; Hodgkin’s Lymphoma; CML; AML; CEL; MDS; ALL; B-cell Chronic Lymphocytic Leukemia; metastatic melanoma; glioma; and hepatoma, by JAK inhibitors is indicated.
Potent inhibitors of JAK2, in addition to the above, will also be useful in vascular disease such as hypertension, hypertrophy, cardiac ischemia, heart failure (including systolic heart failure and diastolic heart failure), migraine and related cerebrovascular disorders, stroke, Raynaud’s phenomenon, POEMS syndrome, Prinzmetal’s angina, vasculitides, such as Takayasu’s arteritis and Wegener’s granulomatosis, peripheral arterial disease, heart disease and pulmonary arterial hypertension.
Pulmonary arterial hypertension (PAH) is a pulmonary vascular disease affecting the pulmonary arterioles resulting in an elevation in pulmonary artery pressure and pulmonary vascular resistance but with normal or only mildly elevated left-sided filling pressures. PAH is caused by a constellation of diseases that affect the pulmonary vasculature. PAH can be caused by or associated with collagen vascular disorders such as systemic sclerosis (scleroderma), uncorrected congenital heart disease, liver disease, portal hypertension, HIV infection, Hepatitis C, certain toxins, splenectomy, hereditary hemorrhagic teleangiectasia, and primary genetic abnormalities. In particular, a mutation in the bone morphogenetic protein type 2 receptor (a TGF-b receptor) has been identified as a cause of familial primary pulmonary hypertension (PPH). It is estimated that 6% of cases of PPH are familial, and that the rest are “sporadic.” The incidence of PPH is estimated to be approximately 1 case per 1 million population. Secondary causes of PAH have a much higher incidence. The pathologic signature of PAH is the plexiform lesion of the lung which consists of obliterative endothelial cell proliferation and vascular smooth muscle cell hypertrophy in small precapillary pulmonary arterioles. PAH is a progressive disease associated with a high mortality. Patients with PAH may develop right ventricular (RV) failure. The extent of RV failure predicts outcome. The JAK/STAT pathway has recently been implicated in the pathophysiology of PAH. JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to proliferation of endothelial cells and smooth muscle cells, and cause hypertrophy of cardiac myocytes. There are three different isoforms of JAK: JAK1, JAK2, and JAK3. Another protein with high homology to JAKs is designated Tyk2. An emerging body of data has shown that the phosphorylation of STAT3, a substrate for JAK2, is increased in animal models of PAH. In the rat monocrotaline model, there was increased phosphorylation of the promitogenic transcription factor STAT3. In this same study pulmonary arterial endothelial cells (PAECs) treated with monocrotaline developed hyperactivation of STAT3. A promitogenic agent or protein is an agent or protein that induces or contributes to the induction of cellular proliferation. Therefore, one effect of JAK2 inhibition would be to decrease proliferation of endothelial cells or other cells, such as smooth muscle cells. A contemplated effect of a JAK2 inhibitor would be to decrease the proliferation of endothelial cells or other cells which obstruct the pulmonary arteriolar lumen. By decreasing the obstructive proliferation of cells, a JAK2 inhibitor could be an effective treatment of PAH.
Additionally the use of JAK kinase inhibitors for the treatment of viral diseases and metabolic diseases is indicated.
Although the other members of the JAK family are expressed by essentially all tissues, JAK3 expression appears to be limited to hematopoetic cells. This is consistent with its essential role in signalling through the receptors for IL-2, IL4, IL-7, IL-9 and IL-15 by non-covalent association of JAK3 with the gamma chain common to these multichain receptors. Males with X-linked severe combined immunodeficiency (XSCID) have defects in the common cytokine receptor gamma chain (gamma c) gene that encodes a shared, essential component of the receptors of interleukin-2 (IL-2), IL-4, IL-7, IL-9, and IL-15. An XSCID syndrome in which patients with either mutated or severely reduced levels of JAK3 protein has been identified, suggesting that immunosuppression should result from blocking signalling through the JAK3 pathway. Gene Knock out studies in mice have suggested that JAK3 not only plays a critical role in B and T lymphocyte maturation, but that JAK3 is constitutively required to maintain T cell function. Taken together with the biochemical evidence for the involvement of JAK3 in signalling events downstream of the IL-2 and IL-4 receptor, these human and mouse mutation studies suggest that modulation of immune activity through the inhibition of JAK3 could prove useful in the treatment of T-cell and B-cell proliferative disorders such as transplant rejection and autoimmune diseases. Conversely undesired inhibition of JAK3 could have a devastating effect on the immune status of an individual treated with drug.
Although the inhibition of various types of protein kinases, targeting a range of disease states, is clearly beneficial, it has been to date demonstrated that the identification of a compound which is selective for a protein kinase of interest, and has good “drug like” properties such as high oral bioavailability, is a challenging goal. In addition, it is well established that the predictability of inhibition, or selectivity, in the development of kinase inhibitors is quite low, regardless of the level sequence similarity between the enzymes being targeted.
The challenges in developing therapeutically appropriate JAK2 inhibitors for use in treatment kinase associated diseases such as immunological and inflammatory diseases including organ transplants; hyperproliferative diseases including cancer and myeloproliferative diseases; viral diseases; metabolic diseases; and vascular diseases include designing a compound with appropriate specificity which also has good drug-likeliness.
There is therefore a continuing need to design and/or identify compounds which specifically inhibit the JAK family of kinases, and particularly compounds which may preferentially inhibit one of the JAK kinases relative to the other JAK kinases, particularly JAK2. There is a need for such compounds for the treatment of a range of diseases.




join me on Linkedin
join me on Researchgate

join me on Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
N-(Cyanomethyl)-4-{2-[4-(morpholin-4-yl)anilino]pyrimidin-4-yl}benzamide
|
|
Other names
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank |
|
| KEGG | |
|
PubChem CID
|
|
| UNII |
|
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C23H22N6O2 | |
| Molar mass | 414.469 g·mol−1 |
| Pharmacology | |
| L01EJ04 (WHO) | |
| By mouth | |
| Legal status | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Clinical data | |
|---|---|
| Other names | Momelotinib hydrochloride hydrate (JAN JP), Momelotinib dihydrochloride (USAN US) |
| License data |
|
| Identifiers | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
//////////Momelotinib, APPROVALS 2023, FDA 2023, Ojjaara, high-risk myelofibrosis, anemia, APPROVALS 2024, EU 2024, EMA 2024
European Journal of Medicinal Chemistry 265 (2024) 116124
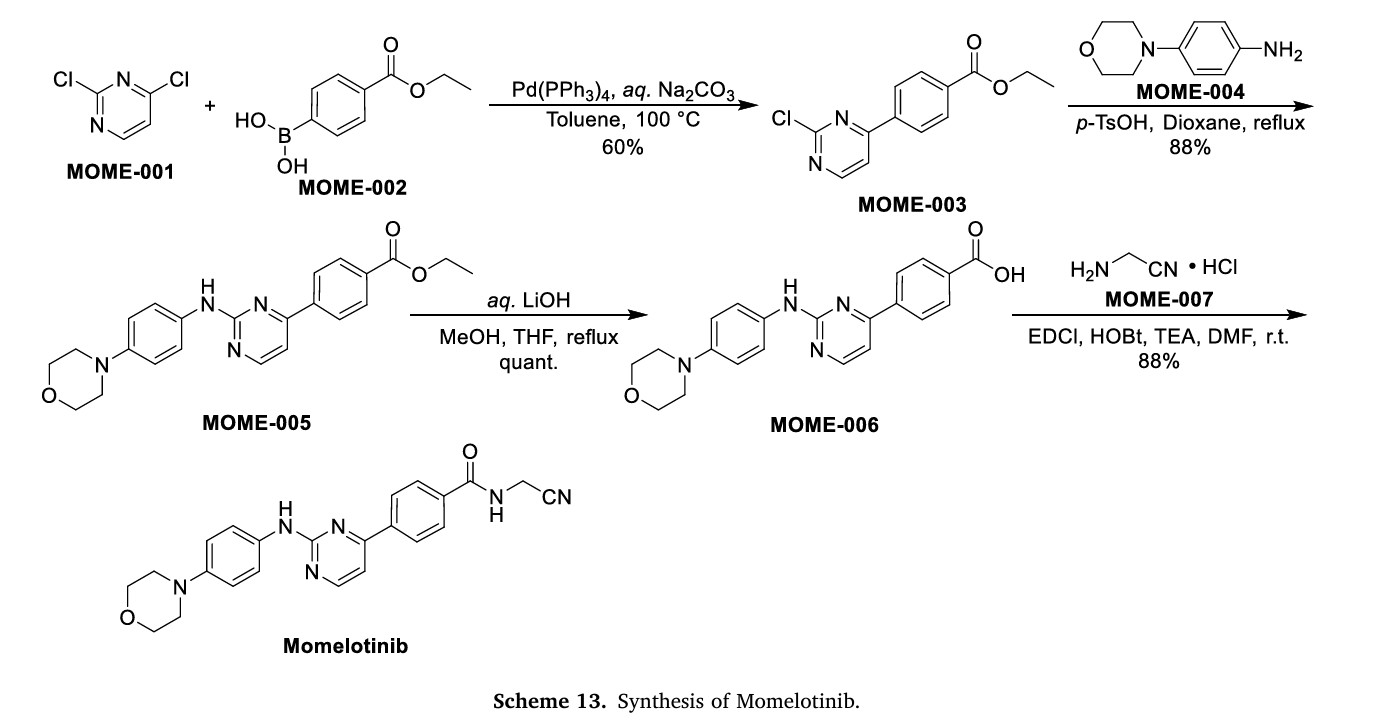
Scheme 13 illustrates the synthesis of Momelotinib Dihydrochloride [48]. The Pd(PPh3) 4-catalyzed Suzuki coupling reaction between 2,4-dichloropyrimidine (MOME-001) and boronic acid MOME-002
resulted in the formation of MOME-003. Subsequently, MOME-003 underwent a substitution reaction with aniline MOME-004 in the presence of p-toluenesulfonic acid (TsOH), yielding MOME-005.
MOME-005 was hydrolyzed by lithium hydroxide, leading to the formation of carboxylic acid MOME-006. MOME-006 underwent amidation with 2-aminoacetonitrile hydrochloride (MOME-007) to produce
Momelotinib.
[48] G.D. Smith, R. Fida, M.M. Kowalski, N-(cyanomethyl)-4-[2-[[4-(4-morpholinyl)
phenyl]amino]-4-pyrimidinyl]-benzamide [CYT387] or a Related Compound,
2012. WO2012071612A1.
.
Poziotinib
l-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazol in-6- yloxy)piperidin-l-yl)prop-2-en-l-one
: 1 – [4 – [[4 – [(3, 4 – dichloro – 2 – phenyl) amino] – 7 – methoxy – 6 – base] quinazoline oxygen radicals] – 1 – piperidine base] – 2 – acrylic – 1 – ketone
UNII-OEI6OOU6IK;
cas 1092364-38-9
NOV120101
Erbb2 tyrosine kinase receptor inhibitor; EGFR family tyrosine kinase receptor inhibitor
Non-small-cell lung cancer; Stomach tumor
for the treatment of Adenocarcinoma of Lung Stage IIIB or Adenocarcinoma of Lung Stage IV
http://www.centerwatch.com/clinical-trials/listings/external-studydetails.aspx?StudyID=NCT01819428
The purpose of this open-label, single-arm, multi-center phase II trial is to evaluate the efficacy and safety of novel pan-HER inhibitor, NOV120101 (Poziotinib), as a first-line monotherapeutic agent in patients with lung adenocarcinoma harboring EGFR mutation…….http://clinicaltrials.gov/show/NCT01819428

KR 1013319
………………………………………………………….
WO2013051883
http://www.google.com/patents/WO2013051883A2?cl=en
1 -(4-(4-(3 ,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6- yloxy)piperidin-l-yl)-prop-2-en-l-one hydrochloride of formula (I) below is an important drug having antiproliferative activities such as anti-tumor activity, which can be used for selectively and effectively treating drug resistance caused by tyrosine kinase mutation. Its free base form, i.e., l-(4-(4-(3,4-dichloro-2- fluoropheny lamino)-7-methoxyquinazolin-6-y loxy)piperidin- 1 -y l)-prop-2-en- 1 – one having formula (II) below is identified as CAS Registry Number 1092364-38-
9.


The compound of formula (II) may be prepared by, e.g., the method disclosed in Korean Patent No. 1013319, the reaction mechanism thereof being shown in Reaction Scheme 1 below. The compound of formula (II) prepared according to Reaction Scheme 1 may then be reacted with hydrochloric acid to produce the compound of formula (I).

wherein R is halogen.





formula (I):




In accordance with another aspect of the present invention, there are provided N-(3,4-dichloro-2-fluorophenyl)-7-methoxy-6-(piperidin-4- yloxy)quinazolin-4-amine dihydrochloride of formula (III), tert-butyl 4-(4-(3,4- dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-l- carboxylate of formula (IV) and 4-(3,4-dichloro-2-fluorophenylamino)-7- methoxyquinazolin-6-ol of formula (V), which can be used as intermediates for preparing the compound of formula (I).
Example 1: Preparation of 4-(3,4-dichloro-2-fluorophenyIamino)-7- methoxyquinazolin-6-yl acetate the compound of formula (VI))

7-methoxy-4-oxo-3,4-dihydroquinazolin-yl acetate (100 g) was added to toluene (850 ml) and NN-diisopropylethylamine (82.5 ml). Phosphorusoxy chloride (100 ml) was added thereto over 20 minutes at 75°C, followed by stirring for 3 hours. Toluene (450 ml) and 3,4-dichloro-2-fluoroaniline (84.6 g) were added to the resulting mixture, followed by stirring for 2 hours. Upon completion of the reaction, the resulting mixture was cooled to 25°C. The solid thus obtained was filtered under a reduced pressure and washed with toluene (400 ml). Isopropanol (1,000 ml) was added to the solid, which was then stirred for 2 hours. The resulting solid was filtered and washed with isopropanol (400 ml). The solid was dried at 40°C in an oven to produce the compound of formula (VI) (143 g, yield: 83%).
1H-NMR (DMSO-d6, 300 MHz, ppm) δ 8.92 (s, 1H), 8.76 (s, 1H), 7.69- 7.57 (m, 3H), 4.01 (s, 3H), 2.38 (s, 3H).
Example 2: Preparation of 4-(3,4-dichloro-2-fluorophenylamino)-7- methoxyquinazolin-6-ol (the com ound of formula (V))

4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yl acetate (100 g) was admixed with methanol (1,000 ml). The mixture was cooled to 10 to 15°C, added with an ammonia solution (460 g), and stirred for 3 hours at 25°C. The solid thus obtained was filtered and washed with a mixed solvent of methanol (200 ml) and water (200 ml). The resulting solid was dried at 40°C in an oven to produce the compound of formula (V) (74 g, yield: 83%).
1H-NMR (DMSO-d6, 300 MHz, ppm) 6 9.57 (br, 2H), 8.35 (s, 1H), 7.68 (s, 1H), 7.61-7.52 (m, 2H), 7.21 (s, 1H), 3.97 (s, 3H).
Example 3: Preparation of /er/-but l-4-(4-(3,4-dichloro-2- fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-l-carboxylate (the compound of formu

4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-ol (60 g) was admixed with N-dimethylformamide (360 ml) under stirring, followed by addition of tert-butyl 4-(tosyloxy)piperidin-l-carboxylate (120 g) and potassium carbonate (72 g) to the mixture. The reaction temperature was raised to 70°C, and the mixture was stirred for 14 hours. The temperature of the resulting solution was cooled to 25°C, and water (480 ml) was slowly added thereto. The solid thus obtained was filtered and dried. The solid was dissolved in a mixed solvent (600 ml) of dichloromethane and methanol. Active carbon (6 g) was then added thereto, followed by stirring for 30 minutes. The resulting mixture was filtered through a Celite pad, distilled under a reduced pressure, added with acetone (300 ml), and stirred for 2 hours. The resulting solid was filtered and washed with acetone (100 ml). The solid was dried at 40°C in an oven to produce the compound of formula (IV) (75 g, yield: 83%).
1H-NMR (DMSO-d6, 300 MHz, ppm) 6 8.69 (s, 1H), 8.47 (t, 1H), 7.34- 7.29 (m, 2H), 7.20 (s, 1H), 4.63-4.60 (m, 1H), 3.82 (s, 3H), 3.83-3.76 (m, 2H), 3.37-3.29 (m, 2H), 1.99-1.96 (m, 2H), 1.90-1.84 (m, 2H), 1.48 (s, 9H).
Example 4: Preparation of N-(3,4-dichIoro-2-fluorophenyi)-7- methoxy-6-(piperidin-4-yloxy)quinazoIin-4-amine dihydrochloride (the compound of formula (III))

Acetone (740 ml) was added to ter/-butyl 4-(4-(3,4-dichloro-2- fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-l-carboxylate (75 g), which was then stirred. The mixture was added with hydrochloric acid (145 ml) for 10 minutes and stirred for 5 hours. Upon completion of the reaction, the resulting mixture was filtered, and the solid thus obtained was washed with acetone (73 ml). The solid was dried at 30°C in an oven to produce the compound of formula (III) (71 g, yield: 99%).
1H-NMR (DMSO-d6, 300 MHz, ppm) 512.95 (bs, 1H), 9.42 (bs, 1H), 9.18 (bs, 1H), 9.01 (s, 1H), 8.86 (s, 1H), 7.69-7.56 (m, 2H), 7.45 (s, 1H), 5.11- 5.08 (m, 1H), 4.03 (s, 3H), 3.29-3.20 (m, 4H), 2.33-2.30 (m, 2H), 1.96-1.93 (m, 2H).
Example 5: Preparation of l-(4-(4-(3,4-dichloro-2- fluorophenylamino)-7-methoxyquinazoIin-6-yloxy)piperidin-l-yl)prop-2-en- 1-one (the compound of formula II))

N-(3,4-dichloro-2-fluorophenyl)-7-methoxy-6-(piperidin-4- yloxy)quinazolin-4-amine dihydrochloride (100 g) and sodium hydrogen carbonate (66 g) were added to a mixed solvent of tetrahydrofuran (630 ml) and water (1 L), and the temperature of the reaction mixture was cooled to 0°C with iced water. Acryloyol chloride (24 ml) diluted with tetrahydrofuran (370 ml) was slowly added to the reaction mixture over 30 minutes, followed by stirring at 0°C for 30 minutes. Upon completion of the reaction, aqueous acetone (2.0 L) was added to the resulting mixture, which was stirred for 12 hours and filtered to produce 1 -(4-(4-(3 ,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6- yloxy)piperidin-l-yl)prop-2-en-l-one (72 g, yield: 75%). The solid thus obtained was dissolved in a mixed solvent of dichloromethane (200 ml) and methanol (100 ml), added with ethyl acetate (1.2 L), and stirred for 12 hours. The resulting solid was filtered and washed with ethyl acetate (100 ml). The solid was dried at 40°C in an oven to produce the compound of formula (II) (55 g, yield: 76%, total yield = 57%).
Ή-NMR (CDC13, 300 MHz, ppm) 68.68(s, 1H), 8.39(t, 3H), 7.3 l(m, 3H), 6.61(m, 1H), 6.29(m, 1H), 5.72(m, 1H), 4.75(m, 1H), 4.02(s, 3H), 3.89(m, 2H), 3.60(m, 2H), 1.86(m, 4H). Example 6: Preparation of l-(4-(4-(3,4-dichloro-2- fluorophenylamino)-7-methoxyquinazolin-6-yIoxy)piperidin-l-yl)prop-2-en- 1-one hydrochloride (the com ound of formula (I))

1 -(4-(4-(3 ,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6- yloxy)piperidine-l-yl)prop-2-en-l-one (150 g) was added to methanol (700 ml). Hydrochloric acid (38.2 ml) diluted with methanol (300 ml) was added thereto, followed by stirring for 24 hours. The solid thus obtained was filtered and washed with acetone (100 ml). The resulting solid was dried at 40°C in an oven for 24 hours to produce the compound of formula (I) (131 g, yield: 81%).
1H-NMR (DMSO-d6, 300 MHz, ppm) 512.31 (bs, 1H), 8.83 (s, 1H), 8.67 (s, 1H), 7.64-7.55 (m, 2H), 7.39 (s, 1H), 6.87-6.78 (m, 1H), 6.12-6.06 (m, 1H), 5.68-5.64 (m, IH), 5.07-5.01 (m, IH), 4.06-3.88 (m, 5H), 3.51 (t, IH), 3.32 (t, IH), 2.10 (t, IH), 1.60 (t, IH).
…………………………………………………………..
WO-2014116070
http://www.sumobrain.com/patents/wipo/Method-preparing-1-4-34/WO2014116070.html
Process for preparing poziotinib – comprising the reaction of a 4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-ol compound with an N-acyl piperidine derivative.
A process for preparing poziotinib comprising the reaction of a 4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-ol compound with an N-acyl piperidine derivative in the presence of an inert polar protic solvent (eg N,N-dimethylformamide), and a base (eg sodium bicarbonate) is claimed. Also claimed are processes for preparing intermediates of poziotinib. Poziotinib is known to be an inhibitor of EGFR family, and Erbb2 tyrosine kinase receptors, useful for the treatment of stomach tumor and non-small-cell lung cancer. Novel method for preparing poziotinib. Follows on from WO2013051883 claiming method for preparing poziotinib and its intermediates. Hanmi, in collaboration with National Oncoventure, is developing poziotinib for the oral treatment of non small cell lung cancer and gastric cancer. As of August 2014, the drug is in phase 2 trials for both indications.
Compound of formula (II) is (I) and compound of formula (I) (poziotinib) is (II) (claim 1, page 13).The synthesis of (II) via intermediate (I) is described (example 1, pages 8-11).
Preparation Example 1: Preparation of 4-(3,4-dichloro-2-fluorophenylamino)- 7-methoxyquinazolin-6-ol, the compound of formula (II)
Step (i): Preparation of 4-(3,4-dichloro-2-fluorophenylamino)-7- methoxyquinazolin-6-yl acetate, the compound of formula (V)
7-methoxy-4-oxo-3,4-dihydroquinazolin-6-yl acetate (100 g) was added to toluene (850 mL) and NN-diisopropylethylamine (82.5 mL). Phosphorus oxychloride (100 mL) was added thereto over 20 minutes at 75°C, followed by stirring for 3 hours. Toluene (450 mL) and 3,4-dichloro-2-fluoroaniline (84.6 g) were added to the resulting mixture, followed by stirring for 2 hours. Upon completion of the reaction, the resulting mixture was cooled to 25°C, and the solid thus obtained was filtered under a reduced pressure and washed with toluene (400 mL). Isopropanol (1,000 mL) was added to the solid, and the resulting mixture was stirred for 2 hours. The solid thus obtained was filtered and washed with isopropanol (400 mL), and then was dried at 40°C in an oven to obtain the target compound (143 g, yield: 83%).
1H-NMR (DMSO-d 6 , 300 MHz, ppm) δ 8.92 (s, 1H), 8.76 (s, 1H), 7.69- 7.57 (m, 3H), 4.01 (s, 3H), 2.38 (s, 3H).
Step (ii): Preparation of 4-(3,4-dichloro-2-fluorophenylamino)-7- methoxyquinazolin-6-ol, the compound of formula (II)
4-(3,4-dichloro-2-fluorophenyIamino)-7-methoxyquinazolin-6-y l acetate (100 g) prepared in step (i) was admixed with methanol (1,000 mL). The mixture was cooled to 10 to 1 °C, added with an ammonia solution (460 g), and stirred for 3 hours at 25°C. The solid thus obtained was filtered and washed with a mixed solvent of methanol (200 mL) and water (200 mL). The resulting solid was dried at 40°C in an oven to obtain the target compound (74 g, yield: 83%). 1H-NMR (DMSO-d 6 , 300 MHz, ppm) 5 9.57 (br, 2H), 8.35 (s, 1H), 7.68 (s,
1H), 7.61-7.52 (m, 2H), 7.21 (s, 1H), 3.97 (s, 3H).
Example 1: Preparation of l-(4-(4-(3,4-dichIoro-2-fluorophenylamino)-7- methoxyquinazolin-6-yloxy)piperidin-l-yl)prop-2-en-l-one, the compound of formula (I) Step (1-1 : Preparation of l-acryloylpiperidin-4-yl 4- methylbenzenesulfonate. the compound of formula (HI)
Piperidin-4-yl 4-methylbenzenesulfonate hydrochloride (200 g, 685 mmol), tetrahydrofuran (THF, 1.6 L) and NaHCO 3 (172 g, 2047 mmol) were added to water (2 L), and the mixture was cooled to 0°C. A solution prepared by adding acryloyl chloride (56 mL, 519 mmol) to THF (0.4 L) was added thereto over 30 minutes, followed by stirring for 1 hour. Upon completion of the reaction, MeOH (0.4 L) was added thereto for quenching. The solution was extracted with ethyl ester (2 L), and washed with water (2 L). The organic layer was separated, distilled under a reduced pressure, and the residue thus obtained was recrystallized from dichloromethane-hexane to obtain the target compound (174 g, yield: 82%). 1H-NMR (300 MHz, DMSO-d 6 ) δ 7.82 (d, 2H), 7.48 (d, 2H), 6.80-6.71 (m,
1H), 6.10-6.03 (m, 1H), 5.67-5.62 (m, 1H), 4.76-4.71 (m, 1H), 3.70-3.68 (m, 2H), 3.43-3.31 (m, 2H), 2.42 (s, 3H), 1.73 (m, 2H), 1.52 (m, 2H).
Step (1-2): Preparation of l-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7- methoxyquinazolin-6-yloxy)piperidin-l-yl)prop-2-en-l-one, the compound of formula (I)
4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-o l (12 g, 34 mmol) prepared in Preparation Example 1, l-acryloylpiperidin-4-yl 4- methylbenzenesulfonate (16 g, 51 mmol) prepared in step (1-1), K 2 CO 3 (9.4 g, 68 mmol) and dimethylacetamide (DMAc, 300 mL) were admixed. The reaction temperature was raised to 70°C, and the mixture was stirred for 24 hours. Upon completion of the reaction, the mixture was cooled down to room temperature, extracted with ethyl ester (300 mL), and then washed with water (300 mL). The organic layer was separated, and distilled under a reduced pressure. The residue thus obtained was solidified by adding ethyl ester, filtered, and dried to obtain the target compound (12.8 g, yield: 77%). 1H-NMR (300 MHz, DMSO-d 6 ) δ 9.65 (bs, 1H), 8.40 (s, 1H), 7.88 (s, 1H),
7.64-7.56 (m, 2H), 7.24 (s, 1H), 6.89-6.80 (m, 1H), 6.15-6.08 (m, 1H), 5.70-5.66 (m, 1H), 4.78 (m, 1H), 3.94 (s, 3H), 3.87 (m, 2H), 3.48 (m, 2H), 2.03 (m, 2H), 1.70 (m, 1H). Example 2: Preparation of l-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7- methoxyquinazoIin-6-yloxy)piperidin-l-yl)prop-2-en-l-one, the compound of formula (I)
SEE
http://www.yuaigongwu.com/thread-8891-1-1.html
| WO2005030765A1 * | Sep 22, 2004 | Apr 7, 2005 | Astrazeneca Ab | Quinazoline derivatives as antiproliferative agents |
| WO2008150118A2 * | Jun 5, 2008 | Dec 11, 2008 | Hanmi Pharm Ind Co Ltd | Novel amide derivative for inhibiting the growth of cancer cells |
| WO2010122340A2 * | Apr 22, 2010 | Oct 28, 2010 | Astrazeneca Ab | Process 738 |
| US20070135463 * | Dec 6, 2006 | Jun 14, 2007 | Frank Himmelsbach | Bicyclic heterocycles, drugs containing said compounds, the use thereof and method for preparing same |



15 JUN 2012
ROCHESTER, Minn. — High doses of the herb American ginseng (Panax quinquefolius) over two months reduced cancer-related fatigue in patients more effectively than a placebo, a Mayo Clinic-led study found. Sixty percent of patients studied had breast cancer. The findings are being presented at the American Society of Clinical Oncology’s annual meeting.
Researchers studied 340 patients who had completed cancer treatment or were being treated for cancer at one of 40 community medical centers. Each day, participants received a placebo or 2,000 milligrams of ginseng administered in capsules containing pure, ground American ginseng root.
“Off-the-shelf ginseng is sometimes processed using ethanol, which can give it estrogen-like properties that may be harmful to breast cancer patients,” says researcher Debra Barton, Ph.D., of the Mayo Clinic Cancer Center.
At four weeks, the pure ginseng provided only a slight improvement in fatigue symptoms. However, at eight weeks, ginseng offered cancer…
View original post 249 more words
Anna Popova, Head of the Federal Service for Supervision of Consumer Protection and Welfare (Rospotrebnadzor)
MOSCOW, August 5 (RIA Novosti) – A Russian vaccine against Ebola hemorrhagic fever is now undergoing preclinical tests, Russian consumer rights watchdog, Rospotrebnadzor, head Anna Popova told journalists.
Russian Ebola Vaccine in Preclinical Trials
RIA Novosti
As of today, it is in a stage of preclinical drug trials. The works are intensified now,” Popova said. She added that there are currently no licensed drugs …
see link
http://en.ria.ru/russia/20140805/191739156/Russian-Ebola-Vaccine-in-Preclinical-Trials.html





