
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64



FDA Approves Trulicity (dulaglutide) for Type 2 Diabetes
FDA Approves Trulicity (dulaglutide) for Type 2 Diabetes

DULAGLUTIDE
PRONUNCIATION doo” la gloo’ tide
THERAPEUTIC CLAIM Treatment of type II diabetes
CHEMICAL NAMES
1. 7-37-Glucagon-like peptide I [8-glycine,22-glutamic acid,36-glycine] (synthetic
human) fusion protein with peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
2. [Gly8,Glu22,Gly36]human glucagon-like peptide 1-(7-37)-peptidyltetraglycyl-Lseryltetraglycyl-L-seryltetraglycyl-L-seryl-L-alanyldes-Lys229-[Pro10,Ala16,Ala17]human immunoglobulin heavy constant γ4 chain H-CH2-CH3 fragment, (55-55′:58-58′)-bisdisulfide dimer
- Dulaglutide
- LY 2189265
- LY-2189265
- LY2189265
- UNII-WTT295HSY5
| GLP-1 immunoglobulin G (IgG4) Fc fusion protein with extended activity; a hypoglycemic agent. |
-
7-37-Glucagon-like peptide I (8-glycine,22-glutamic acid,36-glycine) (synthetic human) fusion proteinwith peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
sept 18 2014
The US Food and Drug Administration (FDA) has approved dulaglutide (Trulicity, Eli Lilly & Co), as a once-weekly injection for the treatment of type 2 diabetes.
A member of the glucagon-like peptide-1 receptor agonist class, dulaglutide joins liraglutide (Victoza, Novo Nordisk), exenatide (Byetta, AstraZeneca/Bristol-Myers Squibb), and albiglutide (Tanzeum, GlaxoSmithKline), on the US market.
Once-weekly dulaglutide was approved based on 6 clinical trials involving a total of 3342 patients who received the drug. It was studied as a stand-alone therapy and in combination withmetformin, sulfonylurea, thiazolidinedione, and prandial insulin.
In one trial the once-weekly dulaglutide was non-inferior to daily liraglutide and in another it topped the oral dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin (Januvia, Merck).
The most common side effects observed in patients treated with dulaglutide were nausea, diarrhea, vomiting, abdominal pain, and decreased appetite.
Dulaglutide should not be used to treat people with type 1 diabetes, diabetic ketoacidosis, or severe abdominal or intestinal problems, or as first-line therapy for patients who cannot be managed with diet and exercise.
As with others in its class, dulaglutide’s label will include a boxed warning that thyroid C-cell tumors have been observed in rodents but the risk in humans is unknown. The drug should not be used in patients with a personal or family history of medullary thyroid carcinoma (MTC) or multiple endocrine neoplasia type 2.
The FDA is requiring Lilly to conduct the following postmarketing studies for dulaglutide:
• A clinical trial to evaluate dosing, efficacy, and safety in children
• A study to assess potential effects on sexual maturation, reproduction, and central nervous system development and function in immature rats
• An MTC case registry of at least 15 years duration to identify any increase in MTC incidence with the drug
• A clinical trial comparing dulaglutide with insulin glargine on glycemic control in patients with type 2 diabetes and moderate or severe renal impairment
• A cardiovascular outcomes trial to evaluate the drug’s cardiovascular risk profile in patients with high baseline risk for cardiovascular disease.
The FDA approval also comes with a Risk Evaluation and Mitigation Strategy, including a communication plan to inform healthcare professionals about the serious risks associated with the drug.
STRUCTURAL FORMULA
Monomer
HGEGTFTSDV SSYLEEQAAK EFIAWLVKGG GGGGGSGGGG SGGGGSAESK 50
YGPPCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSQEDP 100
EVQFNWYVDG VEVHNAKTKP REEQFNSTYR VVSVLTVLHQ DWLNGKEYKC 150
KVSNKGLPSS IEKTISKAKG QPREPQVYTL PPSQEEMTKN QVSLTCLVKG 200
FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSRLT VDKSRWQEGN 250
VFSCSVMHEA LHNHYTQKSL SLSLG 275
Disulfide bridges location
55-55′ 58-58′ 90-150 90′-150′ 196-254 196′-254′
MOLECULAR FORMULA C2646H4044N704O836S18
MOLECULAR WEIGHT 59.67 kDa
MANUFACTURER Eli Lilly and Company
CODE DESIGNATION LY2189265
CAS REGISTRY NUMBER 923950-08-7
http://www.ama-assn.org/resources/doc/usan/dulaglutide.pdf
LY2189265 (dulaglutide), a glucagon-like peptide-1 analog, is a biologic entity being studied as a once-weekly treatment for type 2 diabetes.
Dulaglatuide works by stimulating cells to release insulin only when blood sugar levels are high.
Gwen Krivi, Ph.D., vice president, product development, Lilly Diabetes, said of the drug, “We believe dulaglutide, if approved, can bring significant benefits to people with type 2 diabetes.”
In fact, it might help to control both diabetics’ blood sugar and their high blood pressure.
Eli Lilly CEO John Lechleiter believes the drug has the potential to be a blockbuster. Lilly could be ready to seek approval by 2013.
For more information on dulaglutide clinical studies, click here.
PRESS RELEASES
Data Preseted at 49th EASD Annual Meeting Show Treatment with Lilly’s Investigational Dulaglutide Resulted in Improved Patient-Reported Health Outcomes – September 26, 2013
Lilly Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – April 16, 2013
Lilly Diabetes Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – October 22, 2012
Coconut water is an excellent sports drink — for light exercise
PHILADELPHIA, Aug. 20, 2012 — Coconut water really does deserve its popular reputation as Mother Nature’s own sports drink, a new scientific analysis of the much-hyped natural beverage concluded here today at the 244th National Meeting & Exposition of the American Chemical Society (ACS).
However, people who engage in strenuous exercise that involves a lot of sweat might want to take it all with a grain of salt ― literally ― or stick with a more traditional sports drink like Gatorade, said Chhandashri Bhattacharya, Ph.D. She presented a report on an analysis of coconut water to the ACS, the world’s largest scientific society, which is meeting here this week.
“Coconut water is a natural drink that has everything your average sports drink has and more,” said Bhattacharya. “It has five times more potassium than Gatorade or Powerade. Whenever you get cramps in your muscles, potassium will help you to get…
View original post 370 more words
Sun Pharma, Merck & Co Inc ink pact for Tildrakizumab

Sep 17, 2014,
Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of USD 80 million.
Pharma major Sun Pharmaceutical Industries today entered into a licensing agreement with Merck & Co Inc for investigational therapeutic antibody candidate, tildrakizumab to be used for treatment of plaque psoriasis. Under terms of the agreement, Sun Pharma will acquire worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of USD 80 million, the companies said in a joint statement. Tildrakizumab is being evaluated in Phase III registration trials for the treatment of chronic plaque psoriasis, a skin ailment. “Merck will continue all clinical development and regulatory activities, which will be funded by Sun Pharma. Upon product approval, Sun Pharma will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialisation of the approved product,” it added.


Sun Pharma managing director Dilip Shanghvi.


| Monoclonal antibody | |
|---|---|
| Source | Humanized (from mouse) |
| Target | IL23 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 1326244-10-3 |
| ATC code | None |
| Chemical data | |
| Formula | C6426H9918N1698O2000S46 |
| Mol. mass | 144.4 kDa |
Tildrakizumab is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system andautoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials will enroll a total of nearly 2000 patients, and preliminary results are expected in June, 2015. [2][3]
References
Talaglumetad hydrochloride
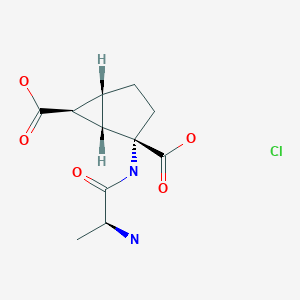
|
Talaglumetad hydrochloride
|
|
| Formula |
C11H16N2O5. HCl
|
|---|---|
| Exact mass |
292.0826
|
| Mol weight |
292.7161
|
CAS: 441765-97-5
441765-98-6 (free base)
IUPAC Name: (1R,4S,5S,6S)-4-[[(2S)-2-aminopropanoyl]amino]bicyclo[3.1.0]hexane-4,6-dicarboxylic acid hydrochloride
Synonyms: Talaglumetad HCl, Talaglumetad hydrochloride, LY 544344 hydrochloride,
UNII-X30300EU7I, D09008, 441765-97-5,
Bicyclo(310)hexane-2,6-dicarboxylic acid, 2-(((2S)-2-amino-1-oxopropyl)amino)-, monohydrochloride, (1S,2S,5R,6S)-
(1S,2S,5R,6S)-2-(L-Alanylamino)bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
(1S,2S,5R,6S)-2-[2(S)-Aminopropionamido]bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
(1S,2S,5R,6S)-2-[2(S)-Aminopropionamido]bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
Treatment of anxiety and stress disorders [metabotropic glutamate [mGlu] agonist]
Talaglumetad hydrochloride, a prodrug of the type II metabotropic glutamate receptor agonist eglumetad, reached phase III clinical studies for the treatment of anxiety at Lilly.

-
In recent years, with the repeated cloning of glutamate receptor genes, it has become clear that there are surprisingly many subtypes of glutamate receptors. At present, glutamate receptors are broadly classified into two types: the “ionotropic type”, in which the receptor has an ion channel structure, and the “metabotropic type”, in which the receptor is coupled to G-proteins (Science, 258, 597-603, 1992). Ionotropic receptors are classified pharmacologically into three types: N-methyl-D-asparaginic acid (NMDA), α-amino-3-hydroxy-5-methyl isoxazole-4-propionate AMPA), and kynate (Science, 258, 597-603, 1992). Metabotropic receptors are classified into eight types, type 1 through type 8 (J. Neurosci., 13, 1372-1378, 1993; Neuropharmacol., 34, 1-26, 1995).
-
The metabotropic glutamate receptors are classified pharmacologically into three groups. Of these, group 2 (mGluR2/mGluR3) bind with adenylcyclase, and inhibit the accumulation of the Forskolin stimulation of cyclic adenosine monophosphate (cAMP) (Trends Pharmacol. Sci., 14, 13 (1993)), which suggests that compounds that act on group 2 metabotropic glutamate receptors should be useful for the treatment or prevention of acute and chronic psychiatric and neurological disorders. As a substance that acts on group 2 metabotropic glutamate receptors, (+)-(1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid has been disclosed in Japanese Unexamined Patent Publication, No. Hei 8-188561 [1996].
-
Fluorine atoms tend to be strongly electron-attractive and to confer high fat solubility, and compounds into which fluorine atoms are introduced greatly change their physical properties. Thus introducing fluorine atoms might greatly affect the absorbability, metabolic stability, and pharmacological effects of a compound. But it is by no means easy to introduce fluorine atoms. In fact, Japanese Unexamined Patent Publication No. Hei 8-188561 [1996] does not even discuss the introduction of fluorine atoms into (+)-(1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid.
………………………………………………………….
Process development of (1S,2S,5R,6S)-spiro[bicyclo[3.1.0]hexane-2′,5′-dioxo-2,4′-imidazolidine]-6-carboxylic acid, (R)-alpha-methylbenzenemethanamine salt (LSN344309)
Org Process Res Dev 2006, 10(1): 28
http://pubs.acs.org/doi/abs/10.1021/op049829e
LY544344 hydrochloride 6 is Talaglumetad

Process development and a pilot-plant process for the synthesis of 4 and its resolution to obtain (1S,2S,5R,6S)-spiro[bicyclo[3.1.0]hexane-2‘,5‘-dioxo-2,4‘-imidazolidine]-6-carboxylic acid, (R)-α-methylbenzenemethanamine salt (5) are described. Starting from the inexpensive raw 2-cyclopenten-1-one and sulfur ylide 1 the racemic bicyclo keto ester 2 was synthesized. Reaction of 2 with potassium cyanide and ammonium carbonate under Bücherer−Berg’s reaction conditions affords racemic 3 in 80% yield. Hydrolysis of 3 followed by the resolution with (R)-(+)-α-methylbenzylamine gave 4 in excellent yield and purity under optimized conditions. The improvement of the original discovery process to accommodate safety and environmental requirements for scale-up in manufacturing facilities is also discussed.
LY544344 hydrochloride 6 is a new chemical entity under investigation by Eli Lilly & Company as a potential treatment of neurological or psychiatric disorders related to the mammalian central nervous system (CNS)

Scheme 1. Original process for the synthesis of LSN344309 an intermediate of Talaglumetad
…………………………………………………….
Journal of Medicinal Chemistry (2005), 48(16), 5305-5320
http://pubs.acs.org/doi/full/10.1021/jm050235r

…………………………………………………….
WO 2002055485
OR;
http://www.google.im/patents/US20040138304?cl=un

………………………………………………………….
http://www.google.com/patents/EP1052246A1?cl=en

………………………………………………
REFERENCES
New approaches in the development of orally bioavailable selective group 2 metabotropic glutamate receptor agonists
Drugs Fut 2002, 27(Suppl. A): Abst C39
Utility of influx transporters to enhance oral bioavailability
241st ACS Natl Meet (March 27-30, Anaheim) 2011, Abst MEDI 163
The intestinal absorption of a prodrug of the mGlu2/3 receptor agonist LY354740 is mediated by PEPT1: In situ rat intestinal perfusion studies
J Pharm Sci 2010, 99(3): 1574
Dipeptides as effective prodrugs of the unnatural amino acid (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740), a selective group II metabotropic glutamate receptor agonist
J Med Chem 2005, 48(16): 5305
An efficient synthesis of LY544344.HCl: A prodrug of mGluR2 agonist LY354740
Tetrahedron Lett 2005, 46(43): 7299
Pharmacodynamics of a novel anxiolytic (LY544344)
24th CINP Congr (June 20-24, Paris) 2004, Abst P02.269
| WO2000004010A1 * | Jul 14, 1999 | Jan 27, 2000 | Stephen Richard Baker | Bicyclohexane derivatives |
| EP0696577A1 * | Aug 11, 1995 | Feb 14, 1996 | Eli Lilly And Company | Synthetic excitatory amino acids |
| EP1052246A1 * | Jan 27, 1999 | Nov 15, 2000 | Taisho Pharmaceutical Co. Ltd | Fluorine-containing amino acid derivatives |
Complaints and Recalls: new EU-GMP Chapter 8 published
DRUG REGULATORY AFFAIRS INTERNATIONAL
GMP News: Complaints and Recalls: new EU-GMP Chapter 8 published
|
View original post 450 more words
If a Facility stores Medicinal Products for more than 36 Hours GDP will apply
DRUG REGULATORY AFFAIRS INTERNATIONAL
GMP News: If a Facility stores Medicinal Products for more than 36 Hours GDP will apply
Since the EU Good Distribution Practice (GDP) Guide has been revised, a number of questions regarding its interpretation have been raised. One of these questions relates to storage facilities and so called distribution hubs. In the past, many facilities which have been involved in the supply chain were not managed under GDP and didn’t posses a licence for their activities.
The British Medicines Authority MHRA published a press release on 18 August 2014 to explain what they consider to be a facility which must be licensed and which needs to implement the GDP requirements. According to the MHRA: “The GDP Inspectorate is raising awareness of the impact of the new regulations to those parties that are either directly or indirectly affected and any freight consolidator or freight forwarder either in the air, sea or road transport sector…
View original post 59 more words
FDA publishes ICH Q4B – Annex 6 on Uniformity of Dosage Units
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
GMP News: FDA publishes ICH Q4B – Annex 6 on Uniformity of Dosage Units
On 16 June 2014, the FDA published the ICH harmonised Guideline entitled “Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions on Uniformity of Dosage Units General Chapter (Q4B Annex 6)”. This ICH Guideline thus came into force in the USA, too.
The objective of the ICH Q4B Working Group is to reach mutual recognition by regulatory authorities in the ICH regions for all testing methods listed in the ICH Q6A Guideline on Specifications. Through this, comparable testing laid down in the different pharmacopeias shouldn’t be performed separately when it has been assessed by the authorities that those are similar and interchangeable.
The Annex 6 states that the following official texts :
- Ph.Eur. 2.9.40 (Uniformity of Dosage Units
- JP 6.02 Uniformity of Dosage Units
- USP General Chapter <905> Uniformity of Dosage Units
View original post 91 more words
Glenmark’s TRPA1 antagonist ‘GRC 17536’ shows positive data in a proof of concept study

MUMBAI, India, Sep 17, 2014
- Glenmark's first in class TRPA1 antagonist, GRC 17536, has shown positive data in a Phase 2a proof of concept study in patients with painful diabetic neuropathy
Glenmark Pharmaceuticals today announced that its first in class Transient Receptor Potential Ankyrin 1 (TRPA1) antagonist, GRC 17536 has shown positive data in a Phase 2a double blind, placebo controlled, multi-centre, proof of concept study conducted on 138 patients in Europe and India.
A statistically significant and clinically relevant response was seen in a prospectively-identified, substantial sub-group of patients with moderate to severe pain who had relatively intact sensory responses as detected by a standardized testing methodology. GRC 17536 was well-tolerated with no evidence of CNS or other drug related side effects.
Patrick Keohane, Chief Medical Officer, Glenmark stated “Diabetic neuropathy remains a difficult to manage chronic clinical condition with limited therapeutic options. These initial efficacy and safety data with GRC 17536, a peripherally acting novel therapeutic, are encouraging, and Glenmark intends to be ready to file for a Phase 2b dose range finding study in patients with neuropathic pain before the end of this financial year. This announcement also reaffirms our position globally in the development of novel pain therapies”.
Commenting on this result, Dr. Michael Buschle, Chief Scientific Officer & President – Biologics, Glenmark Pharmaceuticals mentioned, “This is very promising and GRC 17536 may be useful for several indications which we will pursue”.
The Glenmark TRPA1 program includes indications in pain as well as respiratory. Inhaled doses of GRC 17536 are also being tested in a Phase 2A proof of concept study in patients with Chronic Cough.
, Managing Director and CEO, along with Dr. Michael Buschle, President Biologics, Glenmark Pharmaceuticals at a press conference in Mumbai on Monday. Photo: Paul Noronha")
Glenn Saldanha (left), Managing Director and CEO, along with Dr. Michael Buschle, President Biologics, Photo: Paul Noronha
Note on TRPA1
TRPA1 is an ion channel expressed on peripheral and spinal sensory neurons and it mediates pain signal transmission. It functions as a cellular sensor for detecting painful mechanical, biochemical and thermal stimuli that cause sensory nerve hyperactivity during chronic pathologies including chronic pain, inflammation, itch and cough. TRPA1 receptor is shown to induce pain hypersensitivity in animal models of diabetic neuropathic pain and its blockade attenuates pain hypersensitivity as well as later loss of the nerve fibers and their function. GRC 17536 is a potent, selective and first in class antagonist of TRPA1 receptor. Preclinical studies have demonstrated its effectiveness in animal models of neuropathic and inflammatory pain including the peripheral diabetic neuropathic pain, osteoarthritic pain, postoperative pain and chemotherapy induced pain which supports potential utility of TRPA1 blockade in therapeutic pain management.
About Glenmark Pharmaceuticals Ltd
Glenmark Pharmaceuticals Ltd. (GPL) is a research-driven, global, integrated pharmaceutical company and ranked among the top 80 Pharma & Biotech companies of the world in terms of revenues as per SCRIP 100 Rankings. Glenmark is a leading player in the discovery of new molecules both NCEs and NBEs. Glenmark has several molecules in various stages of clinical development and primarily focused in the areas of Inflammation, Pain and Oncology. The company has significant presence in branded formulations across emerging economies including India. Its subsidiary, Glenmark Generics Limited services the requirements of the US and Western Europe markets.

CARMEGLIPTIN………….a DPP-4 inhibitor

(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
(2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one
813452-14-1 (di-HCl)
916069-91-5 (mono-HCl)
Roche…….innovator

CARMEGLIPTIN
813452-18-5
(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one
(S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| 分子式: | C20H28FN3O3 |
| 分子量: | 377 |
813452-18-5, Carmegliptin, R-1579;carmegliptin, Carmegliptin (USAN/INN), SureCN419289, UNII-9Z723VGH7J, CHEMBL591118, CHEBI:699093, Ro-4876904, D08631, R-1579, B1Q
Type 2 diabetes is a chronic, progressive metabolic disease, affecting about 4% of the world population. The main goal of the management of type 2 diabetes is to achieve glycemic control as close to the nondiabetic range as practicable, in order to reduce the risk of late-stage complications.However, the therapeutic effect provided by existing medications is often not sustainable, since the multi-organ defects responsible for the disease are only insufficiently addressed.
Dipeptidyl peptidase-IV (DPP-IV) inhibitors have emerged as a new therapeutic option to treat type 2 diabetes.
Their rapid rise in popularity is due to the favourable safety profile (no hypoglycemia, no weight gain, no gastrointestinal problems—typical side effects associated with established anti-diabetic agents). DPP-IV is a ubiquitous serine protease, the inhibition of which prevents the degradation of glucagon-like peptide 1 (GLP-1). The resulting higher levels of GLP-1 have a beneficial impact on major players involved in the pathogenesis of type 2 diabetes: β-cells, liver, α-cells, gut, and brain.
Long-term studies with DPP-IV inhibitors in patients are underway in order to confirm the safety and sustainability of these effects, and, in particular, their ability to prevent the progressive loss of β-cell function.
SYNTHESIS

aReagents and conditions: a) HCO2Me, Δ; b) POCl3, MeCN; c) HO2CCH2CO2Et, neat, 120 °C; d) ethyl acrylate, neat; e) t-BuOK, neat (5 steps); f) NH4OAc, MeOH; g) NaBH4, TFA, THF; h) Boc2O, CH2Cl2; i) KOH, aq THF; j) DPPA, Et3N, TMSCH2CH2OH, PhMe, 80 °C; k) Et4NF, MeCN; l) chiral HPLC; m) Et3N, CH2Cl2; n) NaH, DMF; o) HCl, dioxane; p) HCl, 2-PrOH.

Scheme 2.
Reagents and conditions: (a) NH4OAc, MeOH, rt, 95%; (b) NaBH4, TFA, THF, 0 °C; (c) Boc2O, CH2Cl2, 83% over 2 steps; (d) KOH, aq THF, rt; (e) DPPA, Et3N, 2-(trimethylsilyl)ethanol, toluene, 80 °C; (f) Et4NF, CH3CN, 50 °C, 56% over 3 steps; (g) Et3N, CH2Cl2, (h) NaH, cat. NaI, DMF; (i) HCl, 1,4-dioxane.
Carmegliptin (2.70) is an anti-diabetes drug which is currently in late stage clinical trials. It represents a further structural advancement from the other existing marketed drugs in this class, sitagliptin (2.71, Januvia) and vildagliptin (2.72, Zomelis, Figure 7). These compounds are all members of the dipeptidyl peptidase 4 class (DPP-4), a transmembrane protein that is responsible for the degradation of incretins; hormones which up-regulate the concentration of insulin excreted in a cell. As DPP-4 specifically cleaves at proline residues, it is unsurprising that the members of this drug class exhibit an embedded pyrrolidine ring (or mimic) and additional decoration (a nitrile or fluorinated alkyl substituent is present in order to reach into a local lipophilic pocket). One specific structural liability of the 2-cyano-N-acylpyrrolidinyl motif (2.73) is its inherent susceptibility towards diketopiperazine formation (2.74, Scheme 29) [80], however, one way to inhibit this transformation is to position a bulky substituent on the secondary amine nucleophile as is the case in vildagliptine (2.72).
![[1860-5397-9-265-7]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-7.png)
Figure 7: Structures of DPP-4 inhibitors of the gliptin-type.
![[1860-5397-9-265-i29]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i29.png)
Scheme 29: Formation of inactive diketopiperazines from cis-rotameric precursors.
A single crystal X-ray structure of carmegliptin bound in the human DPP-4 active site has been published indicating how the fluoromethylpyrrolidone moiety extends into an adjacent lipophilic pocket [81]. Additional binding is provided by π–π interaction between the aromatic substructure and an adjacent phenylalanine residue as well as through several H-bonds facilitated by the adjacent polar substituents (Figure 8).
![[1860-5397-9-265-8]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-8.png)
Figure 8: Co-crystal structure of carmegliptin bound in the human DPP-4 active site (PDB 3kwf).
The reported synthesis of carmegliptin enlists a Bischler-Napieralski reaction utilising the primary amine 2.76 and methyl formate to yield the initial dihydroquinoline 2.77 as its HCl salt (Scheme 30) [82]. This compound was next treated with 3-oxoglutaric acid mono ethyl ester (2.78) in the presence of sodium acetate. Decarboxylation then yields the resulting aminoester 2.79 which was progressed through an intramolecular Mannich-type transformation using aqueous formaldehyde to allow isolation of enaminoester 2.80 after treatment of the intermediate with ammonium acetate in methanol.
The next step involves a very efficient crystallisation-induced dynamic resolution of the racemic material using the non-natural (S,S)-dibenzoyl-D-tartaric acid ((+)-DBTA). It is described that the desired (S)-enantiomer of compound 2.81 can be isolated in greater than 99% ee and 93% overall yield. This approach is certainly superior to the original separation of the two enantiomers (at the stage of the final product) by preparative chiral HPLC that was used in the discovery route (albeit it should be noted that both enantiomers were required for physiological profiling at the discovery stage).
Next, a 1,2-syndiastereoselective reduction of enaminoester 2.81 occurs with high diastereocontrol imposed by the convexed presentation of the substrate for the formal conjugate addition and subsequent protonation steps. This is followed by Boc-protection and interconversion of the ethyl ester to its amide derivative 2.82 in 80% overall yield for this telescoped process. The primary amide in 2.82 was then oxidised via a modern variant of the classical Hoffmann rearrangement using phenyliodine diacetate (PIDA).
Following extensive investigation it was found that slowly adding this reagent in a mixture of acetonitrile/water to a suspension of amide 2.82 and KOH gave clean conversion to the amine product in high yield. This new procedure was also readily scalable offering a cleaner, safer and more reliable transformation when compared to other related rearrangement reactions. During a further telescoped procedure amine 2.83 was treated with lactone 2.84 to regenerate the corresponding lactam after mesylate formation. Finally, removal of the Boc-group with aqueous hydrochloric acid furnished carmegliptin as its HCl salt.
![[1860-5397-9-265-i30]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i30.png)
Scheme 30: Improved route to carmegliptin.
- Peters, J.-U. Curr. Top. Med. Chem. 2007, 7, 579–595……………..80
- Mattei, P.; Boehringer, M.; Di Gorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U.Bioorg. Med. Chem. Lett. 2010, 20, 1109–1113. doi:10.1016/j.bmcl.2009.12.024………..81
- Albrecht, S.; Adam, J.-M.; Bromberger, U.; Diodone, R.; Fettes, A.; Fischer, R.; Goeckel, V.; Hildbrand, S.; Moine, G.; Weber, M. Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207……….82
………………………………………………………………………………………………………………..
Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207
http://pubs.acs.org/doi/full/10.1021/op2000207

A short and high-yielding synthesis of carmegliptin (1) suitable for large-scale production is reported. The tricyclic core was assembled efficiently by a decarboxylative Mannich addition−Mannich cyclization sequence. Subsequent crystallization-induced dynamic resolution of enamine 7 using (S,S)-dibenzoyltartaric acid was followed by diastereoselective enamine reduction to give the fully functionalized tricyclic core with its three stereogenic centers. The C-3 nitrogen was introduced by Hofmann rearrangement of amide 28, and the resulting amine 10was coupled with (S)-fluoromethyl lactone 31. Following cyclization to lactam 13 and amine deprotection, 1 was obtained in 27−31% overall yield with six isolated intermediates.
Preparation of (2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride (1) CARMEGLIPTIN
A suspension of carbamate 13 (136 kg, 285 mol) in a mixture of H2O (112 kg) and acetone (122 kg) was treated at 50 °C within 60 min with 37% aq HCl (98.0 kg). After 90 min at 47−52 °C the solution was polish filtered through a 5 μm filter. The first reactor and the transfer lines were washed with a hot (47−52 °C) mixture of H2O (13.0 kg) and acetone (116 kg). The filtrate was cooled to 25 °C and treated at this temperature within 80 min with acetone (1600 kg) whereupon the product crystallized out. The resulting suspension was stirred for 1 h at 25 °C and subsequently centrifuged. The crystals were washed in two portions with acetone (391 kg) and dried at 50 °C and <30 mbar until constant weight to afford 122.4 kg (95%) of the title compound as colorless crystals with an assay (HPLC) of 98.8% (w/w).
1H NMR (400 MHz, D2O) δ 2.11−2.22 (m, 1H); 2.45 (dd, J = 17.6 Hz, 6.7 Hz; 1H); 2.76 (dd, J = 17.6 Hz, 9.55 Hz, 1H); 2.90−3.05 (m, 1H); 3.08−3.19 (m, 2H); 3.24−3.36 (m, 1H); 3.43 (dd, J = 9.8 Hz, 5.75 Hz, 1H); 3.49−3.58 (m, 1H); 3.70−3.84 (m, 4H); 3.87 (s, 3H); 3.88 (s, 3H); 4.12 (td, J = 11.6 Hz, 4.5 Hz, 1H); 4.45−4.55 (m, 1H); 4.56−4.68 (m, 3H); 6.91 (s, 1H), 6.95 (s, 1H).
IR (cm−1): 3237, 2925, 1682, 496, 478.
MS (ESI): m/z 378.3 ([M + H]+ (free amine)).
Anal. Calcd for C20H30Cl2FN3O3: C, 53.34; H, 6.71; N, 9.33; Cl, 15.74; F 4.22; O, 10.66. Found: C, 53.04; H, 6.43; N, 9.45; Cl, 15.66; F, 4.29; O, 11.09.
REF FOR ABOVE
Mattei, P.; Böhringer, M.; Di Giorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Kocer, B.; Kuhn, B.; Löffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U. Bioorg. Med. Chem. Lett. 2010, 20, 1109
Böhringer, M.; Kuhn, B.; Lübbers, T.; Mattei, P.; Narquizian, R.; Wessel,H. P. (F. Hoffmann-La Roche AG). U.S. Pat. Appl. 2004/0259902, 2004.
…………………………………………………..
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
Bioorg Med Chem Lett 2010, 20(3): 1109
Bioorg Med Chem Lett 2010, 20(3): 1109
http://www.sciencedirect.com/science/article/pii/S0960894X09017296
-
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
- Pages 1109-1113
- Patrizio Mattei, Markus Boehringer, Patrick Di Giorgio, Holger Fischer, Michael Hennig, Joerg Huwyler, Buelent Koçer, Bernd Kuhn, Bernd M. Loeffler, Alexander MacDonald, Robert Narquizian, Etienne Rauber, Elena Sebokova, Urs Sprecher
-

 Scheme 3.
Scheme 3.Reagents and conditions: (a) preparative HPLC (Chiralpak® AD column), heptane/2-propanol 85:15, 37% (b) BH3.Me2S, THF, 0 °C; (c) (MeOCH2CH2)2NSF3, CH2Cl2, 67% (2 steps); (d), SOCl2, ZnCl2, 80 °C, 72 h, 61%; (e) Et3N, CH2Cl2; (f) NaH, DMF, 56% (2 steps); (g) HCl, 1,4-dioxane, 91%; (h) HCl, 2-propanol, 86%.
The synthesis of 8p is outlined ABOVE and required the enantiopure building blocks (S,S,S)-5 and 12. (S,S,S)-5 was obtained from the racemate by preparative chiral HPLC. Acid chloride 12 was prepared starting from (S)-paraconic acid (9). Reduction of 9 with borane–dimethyl sulfide complex afforded hydroxymethyl lactone 10. Since 10 is known to racemise rather readily, it was immediately treated with bis(2-methoxyethyl)aminosulfur trifluoride, thereby affording fluoromethyl lactone 11. This was converted to 12 by reaction with thionyl chloride in the presence of zinc chloride. The (S)-4-fluoromethyl-pyrrolidinone 8p was isolated as the dihydrochloride salt, a highly water soluble white crystalline solid, mp >275 °C.
…………………………………………………….
US 2013109859
The most preferred product is (2S,3S,11bS)-2-tert.-Butoxycarbonylamino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H pyrido[2,1-a]isoquinoline-3-carboxylic acid amide having the following structure:

It has been found that during the amidation of the ester epimerization takes place at position 3 and thus the 3R-epimer of the formula IVb is transformed to a larger extent in the 3S-epimer of formula V.

e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
A 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
…………………………………………………….
US 2008071087







(2S,3S,11bS)-(3-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester (8)
Example 8
Transformation of (2S,3S,11bS)-(3-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl) ]-carbamic acid tert-butyl ester into (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl) -4-fluoromethyl-pyrrolidin-2-one.a)
Preparation of 4-fluoromethyl-5H-furan-2-oneA 6 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 500 g (4.38 mmol) 4-hydroxymethyl-5H-furan-2-one and 2.0 L dichloromethane. The solution was cooled to −10° C. and 1.12 kg (4.82 mol) bis-(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) was added during 50 min, maintaining the temperature at −5 to −10° C. with a cooling bath. During the addition a yellowish emulsion formed, which dissolved to an orange-red solution after completed addition. This solution was stirred for 1.5 h at 15-20° C., then cooled to −10° C. A solution of 250 ml water in 1.00 L ethanol was added during 30 min, maintaining the temperature between −5 and −10° C., before the mixture was allowed to reach 15-20° C. It was then concentrated in a rotatory evaporator to a volume of ca. 1.6 L at 40° C./600-120 mbar. The residue was dissolved in 2.0 L dichloromethane and washed three times with 4.0 L 1N hydrochloric acid. The combined aqueous layers were extracted three times with 1.4 L dichloromethane. The combined organic layers were evaporated in a rotatory evaporator to give 681 g crude product as a dark brown liquid. This material was distilled over a Vigreux column at 0.1 mbar, the product fractions being collected between 71 and 75° C. (312 g). This material was re-distilled under the same conditions, the fractions being collected between 65 and 73° C., to give 299 g 4-fluoromethyl-5H-furan-2-one (58% yield; assay: 99%).MS: m/e 118 M+, 74,59,41.b) Preparation of (S)-4-fluoromethyl-dihydro-furan-2-oneA 2 L autoclave equipped with a mechanical stirrer was charged with a solution of 96.0 g 4-fluoromethyl-5H-furan-2-one (8.27×10−1 mol) in 284 mL methanol. The autoclave was sealed and pressurized several times with argon (7 bar) in order to remove any traces of oxygen. At ˜1 bar argon, a solution of 82.74 mg Ru(OAc)2((R)-3,5-tBu-MeOBlPHEP) (6.62×10−5 mol) (S/C 12500) in 100 mL methanol was added under stirring from a catalyst addition device previously charged in a glove box (O2 content <2 ppm) and pressurized with argon (7 bar). The argon atmosphere in the autoclave was replaced by hydrogen (5 bar). At this pressure, the reaction mixture was stirred (˜800 rpm) for 20 h at 30° C. and then removed from the autoclave and concentrated in vacuo. The residue was distilled to afford 91.8 g (94%) (S)-4-fluoromethyl-dihydro-furan-2-one. The chemical purity of the product was 99.7% by GC-area.c) Preparation of (2S,3S,11bS)-3-(3-Fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 50 g (128 mmol) (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester, 500 mL toluene and 2.51 g (25.6 mmol) 2-hydroxypyridine. To this slightly brownish suspension, 22.7 g (192 mmol) of (S)-4-fluoromethyl-dihydro-furan-2-one was added dropwise at RT. No exothermy was observed during the addition. The dropping funnel was rinsed portionwise with totally 100 mL toluene. The suspension was heated to reflux, whereas it turned into a dear solution starting from 60° C., after 40 min under reflux a suspension formed again. After totally 23 h under reflux, the thick suspension was cooled to RT, diluted with 100 mL dichloromethane and stirred for 30 min at RT. After filtration, the filter cake was washed portionwise with totally 200 mL toluene, then portionwise with totally 100 mL dichloromethane. The filter cake was dried at 50° C./10 mbar for 20 h, to give 60.0 g product (94% yield; assay: 100%).


 UNDESIRED
UNDESIRED

 DESIRED
DESIRED
 UNDESIRED
UNDESIRED



MS: m/e 496 (M+H)+, 437.
d) Preparation of (2S,3S,11bS)-3-((4S)-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel, a cooling bath and a nitrogen inlet was charged with 28 g (56.5 mmol) of (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino) -9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and 750 mL THF. The mixture was cooled to 0° C. and a solution of 6.17 mL (79 mmol) methanesulfonic acid in 42 mL THF was added during 10 min, maintaining the temperature at 0-5° C. At 0° C. a solution of 12.6 mL (90.2 mmol) triethylamine in 42 mL THF was added during 15 min. The resulting suspension was stirred for 80 min at 0-5° C., whereas it became gradually thicker. Then 141 mL (141 mmol) 1 M lithium-bis(trimethylsilyl)amide were added to the mixture during 15 min, whereas the suspension dissolved. The solution was allowed to reach RT during 60 min under stirring. 500 mL water was added without cooling, the mixture was extracted and the aqueous phase was subsequently extracted with 500 mL and 250 mL dichloromethane. The organic layers were each washed with 300 mL half saturated brine, combined and evaporated on a rotatory evaporator. The resulting foam was dissolved in 155 mL dichloromethane, filtered and again evaporated to give 30.5 g crude product as a slightly brownish foam. This material was dissolved in 122 mL methanol, resulting in a thick suspension, which dissolved on heating to reflux. After 20 min of reflux the solution was allowed to gradually cool to RT during 2 h, whereas crystallization started after 10 min. After 2 h the suspension was cooled to 0° C. for 1 h, followed by −25° C. for 1 h. The crystals were filtered off via a pre-cooled glass sinter funnel, washed portionwise with 78 mL TBME and dried for 18 h at 45° C./20 mbar, to give 21.0 g of the title product as white crystals (77% yield; assay: 99.5%).
MS: m/e 478 (M+H)+, 437, 422.
e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one dihydrochlorideA 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
These compounds are useful intermediates for the preparation of DPP-IV inhibitors as disclosed in PCT International Patent Appl. WO 2005/000848. More preferably, the invention relates to a process for the preparation of (2S,3S,11bS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester.
XXXXXXX
According to still another embodiment (Scheme 2, below) the (S)-4-fluoromethyl-dihydro-furan-2-one (VII) is directly coupled with the amino-pyrido[2,1-a]isoquinoline derivative (VI) to form the hydroxymethyl derivative of the pyrido[2,1-a]isoquinoline (VIII), which is then subsequently cyclized to the fluoromethyl-pyrrolidin-2-one derivative (IX). The latter can be deprotected to yield the desired pyrido[2,1-a]isoquinoline derivative (I).

In a further preferable embodiment, the process for the preparation of (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one or of a pharmaceutically acceptable salt thereof comprises the subsequent steps:
- e) coupling of the (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (amine of formula VI, wherein R2 and R3 are methoxy, R4 is hydrogen and Prot is Boc) with the (S)-4-fluoromethyl-dihydro-furan-2-one of formula

- f) cyclization of the obtained (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester in the presence of a base, and
- g) deprotecting the obtained (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester.

………………………………………………………….
PATENT
http://www.google.com.ar/patents/US7122555?cl=pt-PT
Example 23
RACEMIC
1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

a) 4-Fluoromethyl-dihydro-furan-2-one
A solution of 4-hydroxymethyl-dihydro-furan-2-one (Tetrahedron 1994, 50, 6839; 1.02 g, 8.78 mmol) and bis(2-methoxyethyl)aminosulfur trifluoride (3.88 g, 17.6 mmol) in chloroform (4.4 mL) was stirred at 40° C. for 1 h, then poured onto ice and partitioned between sat. aq. sodium hydrogencarbonate solution and dichloromethane. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, heptane-ethyl acetate gradient) afforded the title compound (576 mg, 56%). Colourless liquid, MS (EI) 118.9 (M+H)+.
b) 3-Chloromethyl-4-fluoro-butyryl chloride
A mixture of 4-fluoromethyl-dihydro-furan-2-one (871 mg, 7.37 mmol), thionyl chloride (4.39 g, 36.9 mmol), and zinc chloride (60 mg, 0.44 mmol) was stirred 72 h at 80° C., then excess thionyl chloride was removed by distillation. Kugelrohr distillation of the residue (85° C., 0.2 mbar) afforded the title compound (450 mg, 35%). Colourless liquid, 1H-NMR (300 MHz, CDCl3): 4.65–4.55 (m, 1H), 4.50–4.40 (m, 1H), 3.70–3.60 (m, 2H), 3.25–3.05 (m, 2H), 2.80–2.60 (m, 1H).
c) (RS,RS,RS)-[3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (RS,RS,RS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 5b) and 3-chloromethyl-4-fluoro-butyryl chloride. White solid, MS (ISP) 514.5 (M+H)+.
d) (RS,RS,RS)-[3-(4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5d from (RS,RS,RS)-[3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Off-white foam, MS (ISP) 478.5 (M+H)+.
e) 1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
The title compound was produced in accordance with the general method of Example 1e from (RS,RS,RS)-[3-(4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Light yellow oil, MS (ISP) 378.5 (M+H)+.
Examples 28 and 29
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
 UNDESIRED
UNDESIREDand
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

The title compounds were produced from 1-((RS,RS,RS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one (Example 23) by chromatographic separation (SiO2, CH2Cl2/MeOH/NH4OH 80:1:0.2, then 95:5:0.25).
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Yellow oil, Rf=0.45 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Light yellow solid, Rf=0.40 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
Example 30
(S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one Dihydrochloride
 DESIRED
DESIREDa) [(S,S,S)-3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (S,S,S)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 16b) and 3-chloromethyl-4-fluoro-butyryl chloride (Example 23b). Off-white solid.
b) [(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
Sodium hydride (55–65% dispersion in oil, 1.14 g, 28.5 mmol) was added to a suspension of [(S,S,S)-3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (6.72 g, 13.1 mmol) in N,N-dimethylformamide (95 mL) at r.t., then after 1 h the reaction mixture was poured onto ice and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, cyclohexane/2-propanol 4:1) afforded [(S,S,S)-3-((S)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 38%) and the epimer, [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.73 g, 44%).
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.6 (SiO2, cyclohexane/2-propanol 1:1).
[(S,S,S)-3-((R)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.4 (SiO2, cyclohexane/2-propanol 1:1).
-
- c) (S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 5.02 mmol) was converted to (S)-1-((S,S,S)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one in accordance with the general method of Example 1e. The product was dissolved in 2-propanol (10 mL) and treated with hydrogen chloride (5–6 M in 2-propanol, 37 mL). The suspension formed was stirred for 64 h at r.t., then the precipitate was collected by filtration and dried, to afford the title compound (2.04 g, 91%). White solid, m.p. >300° C.
Example 31(R)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
 UNDESIRED
UNDESIREDThe title compound was produced in accordance with the general method of Example 30c from [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (Example 30b). White solid, m.p. >300° C.

DR ANTHONY MELVIN CRASTO
MY BLOGS ON MED CHEM
New Drug Approvals ALL ABOUT DRUGS WORLD DRUG TRACKER MEDICINAL CHEMISTRY INTERNATIONALDRUG SYN INTERNATIONALSCALEUP OF DRUGS ALL FOR DRUGS ON WEBEUREKAMOMENTS
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS, BRAVESITES, GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO, YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN, FRIENDFEED, NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING
FDA approves AstraZeneca’s constipation drug Movantik
September 16, 2014
The US Food and Drug Administration has approved AstraZeneca’s Movantik for opioid-induced constipation in adults with chronic non-cancer pain.
Movantik (naloxegol), an oral once-a-day treatment licensed from Nektar Therapeutics, belongs to a class of drugs called peripherally-acting mu-opioid receptor antagonists, which are used to decrease the constipating effects of opioids. The drug’s safety and effectiveness were established in two trials of 1,352 participants who had taken opioids for at least four weeks for non-cancer related pain and had opioid-induced constipation.
Read more at: http://www.pharmatimes.com/Article/14-09-16/FDA_approves_AstraZeneca_s_constipation_drug_Movantik.aspx#ixzz3DdGiFse8
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL
