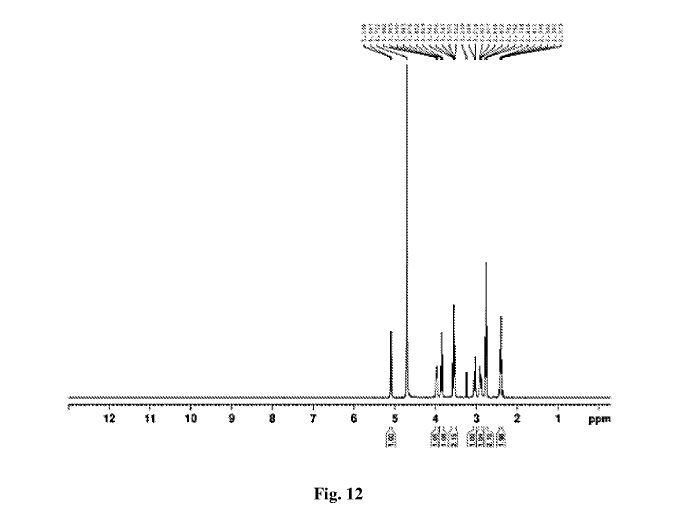
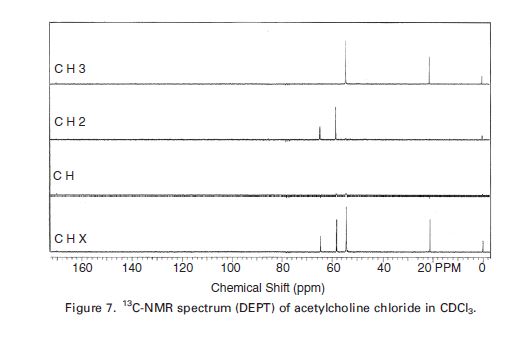
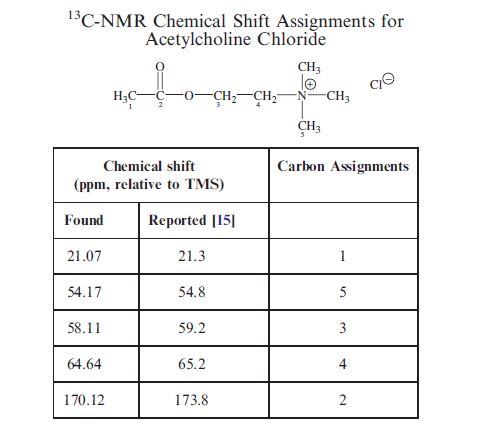
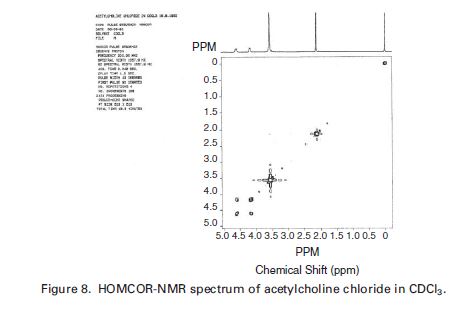
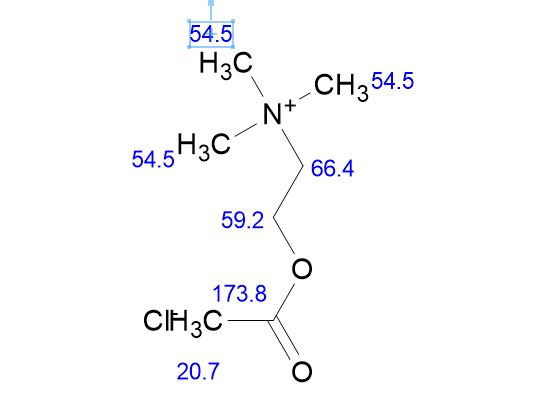
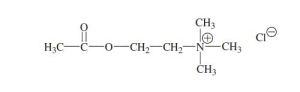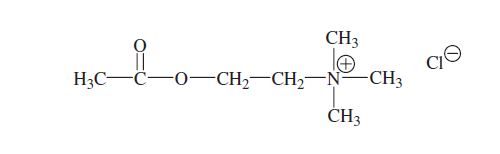SUGAMMADEX
| CAS number |
343306-71-8 |
| Weight |
Average: 2002.12
Monoisotopic: 2000.408874758 |
| Chemical Formula |
C72H112O48S8 |
WATCH OUT POST WILL BE UPDATED………

SUGAMMADEX

Bridion; UNII-ERJ6X2MXV7; Suγdex Sodium; Suγdex; 343306-79-6; Org 25969;Org25969; Org-25969; Org 25969; 361LPM2T56
Sugammadex (Org 25969, Bridion) is chemically known as Cyclooctakis-(l-→4)-[6-S-(2-carboxyethyl)-6-thio-a-D-glucopyranosyl].
Sugammadex
Sugammadex sodium, 343306-79-6
CAS No.:343306-71-8 free
Molecular Weight:2178.01
Molecular Formula:C72H104Na8O48S8
CAS 343306-79-6 sodium salt
Octanatrium-3,3′,3”,3”’,3””,3””’,3”””,3”””’-{[(1S,3S,5S,6S,8S,10S,11S,13S,15S,16S,18S,20S,21S,23S,25S,26S,28S,30S,31S,33S,35S,36S,38S,40S,41R,42R,43R,44R,45R,46R,47R,48R,49R,50R,51R,52R,53 ;R,54R,55R,56R)-41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56-hexadecahydroxy-2,4,7,9,12,14,17,19,22,24,27,29,32,34,37,39-hexadecaoxanonacyclo[36.2.2.23,6.28,11.213,16.218,21.223,26.228,31 .233,36]hexapentacontan-5,10,15,20,25,30,
Octasodium 3,3′,3”,3”’,3””,3””’,3”””,3”””’-{[(1S,3S,5S,6S,8S,10S,11S,13S,15S,16S,18S,20S,21S,23S,25S,26S,28S,30S,31S,33S,35S,36S,38S,40S,41R,42R,43R,44R,45R,46R,47R,48R,49R,50R,51R,52R,53R ;,54R,55R,56R)-41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56-hexadecahydroxy-2,4,7,9,12,14,17,19,22,24,27,29,32,34,37,39-hexadecaoxanonacyclo[36.2.2.23,6.28,11.213,16.218,21.223,26.228,31 .233,36]hexapentacontane-5,10,15,20,25,30,
Sugammadex Sodium
D05940,
SUYDEX SODIUM
UNII-ERJ6X2MXV7
Sugammadex (brand name Bridion) is marketed by Merck Sharp and Dohme, and was approved by the United States FDA on December 15, 2015.
Sugammadex sodium was first approved by European Medicine Agency (EMA) on July 25, 2008, then approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 20, 2010, and approved by the US. food and drug administration (FDA) on Dec 15, 2015. It was developed and marketed as Bridion® by Merck Sharp & Dohme.
Sugammadex is reversal of neuromuscular blockade without relying on inhibition of acetylcholinesterase. It is the first selective relaxant binding agent (SRBA). It is indicated in neuromuscular blockade induced by rocuronium or vecuronium in adults.
Bridion® is available as injection solution for intravenous use, containing 100 mg /mL of free Sugammadex. The recommended dose is 4 mg/kg for adults if recovery has reached at least 1-2 post-tetanic counts (PTC) following rocuronium or vecuronium induced blockade.
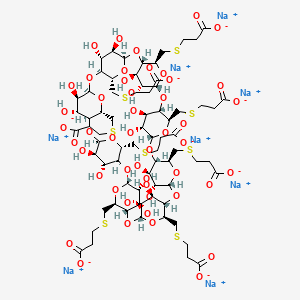
Sugammadex Sodium is the sodium salt form of the biologically inert, selective relaxant binding agent (SRBA) sugammadex, a modified, anionic gamma cyclodextrinderivative containing a hydrophilic exterior and a hydrophobic core, with neuromuscular blocking drug (NMBD) reversal activity. Upon administration, the negatively charged carboxyl-thio-ether groups of sugammadex selectively and reversibly bind to the positively charged quaternary nitrogen of a steroidal NMBD, which was administered at an earlier time for anesthetic purposes. The encapsulation of the NMBD by sugammadex blocks its ability to bind to nicotinic receptors in the neuromuscular junction and thereby reverses the NMBD-induced neuromuscular blockade. Sugammadex binds rocuronium, vecuronium, and to a lesser extent pancuronium.
Sugammadex is a selective relaxant binding agent indicated for reversal of neuromuscular blockade induced by
rocuronium bromide and
vecuronium bromide during surgery in adults.
Rocuronium bromide and
vecuronium bromide are neuromuscular blocking medications that cause temporary paralysis and are especially useful for general anesthesia, ventilation, or tracheal intubation that patients may require for surgery. Sugammadex provides a new treatment option to reverse the effects of those medications and possibly help patients recover sooner post-surgery. Sugammadex (brand name Bridion) is marketed by Merck Sharp and Dohme, and was approved by the United States FDA on December 15, 2015.
Sugammadex is used to reverse neuromuscular blockade after administration of non-depolarizing neuromuscular-blocking agentssuch as vecuronium or rocuronium.

Pharmacology
Pharmacodynamics
Sugammadex is a modified γ-cyclodextrin, with a lipophilic core and a hydrophilic periphery. This gamma cyclodextrin has been modified from its natural state by placing eight carboxyl thio ether groups at the sixth carbon positions. These extensions extend the cavity size allowing greater encapsulation of the rocuronium molecule. These negatively charged extensions electrostatically bind to the quaternary nitrogen of the target as well as contribute to the aqueous nature of the cyclodextrin. Sugammadex’s binding encapsulation of rocuronium is one of the strongest among cyclodextrins and their guest molecules. The rocuronium molecule (a modified steroid) bound within sugammadex’s lipophilic core, is rendered unavailable to bind to the acetylcholine receptor at the neuromuscular junction.
Left: Schematic of a sugammadex molecule encapsulating a rocuronium molecule.
Right:
Space-filling model of a sugammadex sodium molecule in the same orientation.
Sugammadex, unlike neostigmine, does not inhibit acetylcholinesterase so cholinergic effects are not produced and co-administration of an antimuscarinicagent (glycopyrronium bromide or atropine) is not needed. Sugammadex might therefore be expected to have fewer adverse effects than the traditional reversal agents.
When muscle relaxant with rapid onset and short duration of action is required, there has been little choice apart from suxamethonium but this drug has important contraindications; for example, it can trigger malignant hyperthermia in susceptible individuals, it has a prolonged duration of action in patients with pseudocholinesterase deficiency and it causes an increase in plasma potassium concentration which is dangerous in some circumstances. Rocuronium has a comparably quick onset in high dose (0.6 mg kg−1 to 1 mg kg−1) and can be rapidly reversed with sugammadex (16 mg kg−1), so this drug combination offers an alternative to suxamethonium.
‘Recurarisation’, a phenomenon of recurrence of neuromuscular block, may occur where the reversal agents wear off before a neuromuscular blocking drug is completely cleared. This is very unusual with all but the longest acting neuromuscular blocking drugs (such as gallamine, pancuronium or tubocurarine). It has been demonstrated to occur only rarely with sugammadex, and only when insufficient doses were administered.[1] The underlying mechanism is thought to be related to redistribution of relaxant after reversal. It may occur for a limited range of sugammadex doses which are sufficient for complex formation with relaxant in the central compartment, but insufficient for additional relaxant returning to central from peripheral compartments.[2]
Sugammadex has been shown to have affinity for two other aminosteroid neuromuscular blocking agents, vecuronium and pancuronium. Although sugammadex has a lower affinity for vecuronium than for rocuronium, reversal of vecuronium is still effective because fewer vecuronium molecules are present in vivo for equivalent blockade: vecuronium is approximately seven times more potent than rocuronium. Sugammadex encapsulates with a 1:1 ratio and therefore will adequately reverse vecuronium as there are fewer molecules to bind compared to rocuronium.[3] Shallow pancuronium blockade has been successfully reversed by sugammadex in phase III clinical trials.[4]
Efficacy
A study was carried out in Europe looking at its suitability in rapid sequence induction. It found that sugammadex provides a rapid and dose-dependent reversal of neuromuscular blockade induced by high-dose rocuronium.[5]
A Cochrane review on sugammadex concluded that “sugammadex was shown to be more effective than placebo (no medication) or neostigmine in reversing muscle relaxation caused by neuromuscular blockade during surgery and is relatively safe. Serious complications occurred in less than 1% of the patients who received sugammadex. The results of this review article (especially the safety results) need to be confirmed by future trials on larger patient populations”.[6]
Tolerability
Sugammadex was generally well tolerated in clinical trials in surgical patients or healthy volunteers. In pooled analyses, the tolerability profile of sugammadex was generally similar to that of placebo or neostigmine plus glycopyrrolate.[7]
History
Sugammadex was discovered by the pharmaceutical company Organon at the Newhouse Research Site in Scotland.[8] Organon was acquired by Schering-Plough in 2007; Schering-Plough merged with Merck in 2009. Sugammadex is now owned and sold by Merck.
The US Food and Drug Administration (FDA) initially rejected Schering-Plough’s New Drug Application for sugammadex in 2008,[9] but finally approved the medication for use in the United States on December 15, 2015.[10] Sugammadex was approved for use in the European Union on July 29, 2008.[11]
SYNTHESIS
Sugammadex (Trade name: Bridion) is first disclosed in US6670340 assigned to Akzo Nobel. Sugammadex sodium was approved in EMEA as an agent for reversal of neuromuscular blockade by the agent rocuronium in general anaesthesia in 2008 and is the first selective relaxant binding agent (SRBA).
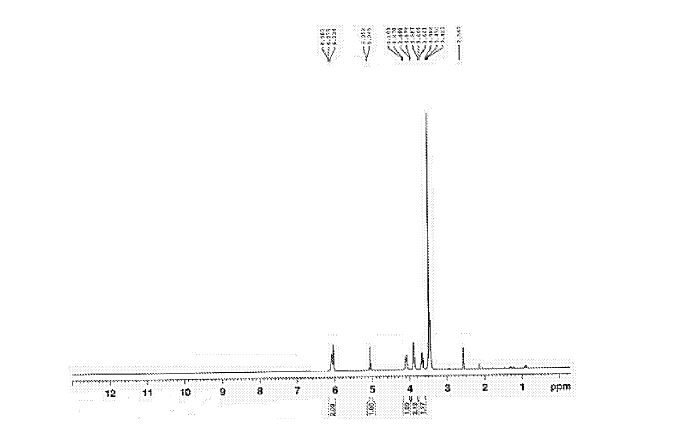
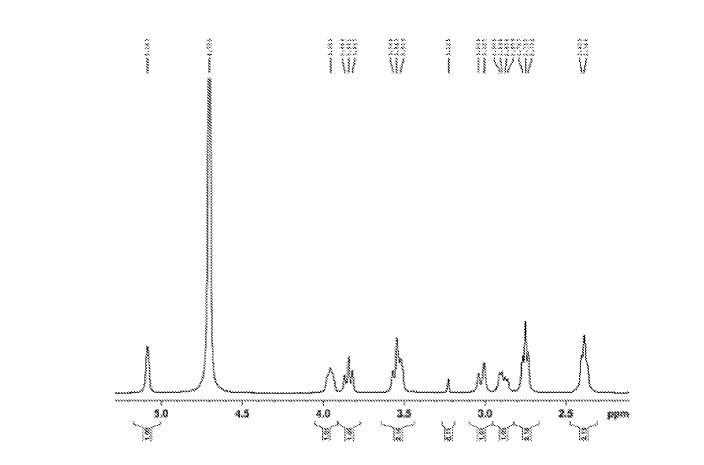
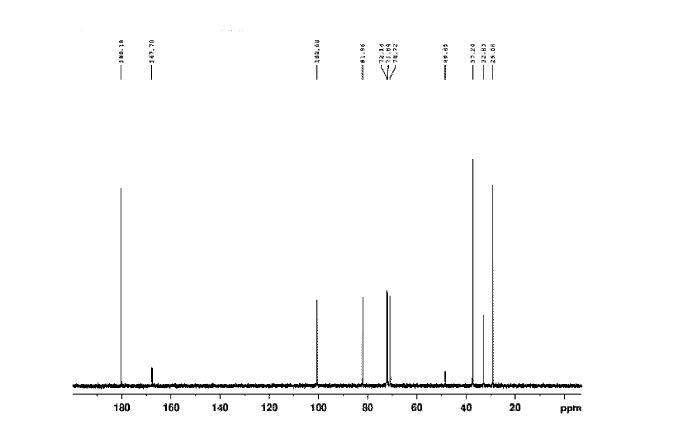
Sugammadex sodium contains 8 recurring glucose units each with 5 asymmetric carbon atoms, in total 40 asymmetric carbon atoms for the whole molecule. Sugammadex is a modified γ-cyclodextrin, with a lipophilic core and a hydrophilic periphery. The gamma cyclodextrin has been modified from its natural state by placing eight carboxyl thio ether groups at the sixth carbon positions.
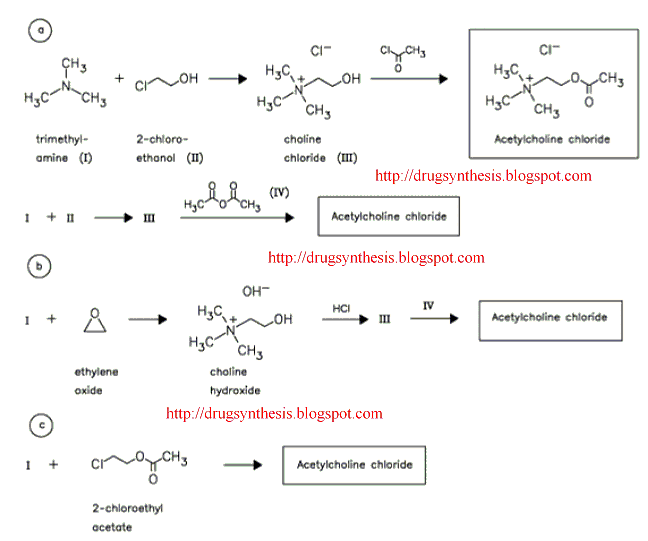
The US Patent 6670340 assigned to Akzo Nobel discloses a process for preparing Sugammadex sodium as depicted in Scheme-I:
Scheme-I
Sugammadex Sodium The first step in the process in the scheme-I involves the preparation of Vilsmeier Hack reagent by the reaction , of DMF, triphenylphosphine and Iodine. The triphenylphosphine oxide is formed as a byproduct of the first step. Removal of triphenylphosphine oxide from the product is very difficult from the reaction mass as it requires repeated washing with DMF under argon atmosphere, which leads to inconsistency in yield of final product Sugammadex. The second step involves the reaction of 6-perdeoxy-6-per-Iodo-Gamma cyclodextrin with 3-mercapto propionic acid in presence of alkali metal hydrides in an organic solvent to give 6-per-deoxy-6-per-(2-carboxyethyl)thio-y- cyclodextrin sodium salt.
The PCT publication WO2012/025937 discloses preparation of Sugammadex involving the reaction of gamma cyclodextrin with phosphorous halide in presence of organic solvent, thereby overcomes the formation of triphenyl phosphine oxide. The publication also discloses the use of 6-per deoxy-6-per- chloro-y-cyclodextrin in the preparation of the Sugammadex.
The purification techniques in the prior arts employ column chromatographic / membrane dialysis techniques which are costly and not convenient in large scale operations.


Sugammadex is a modified γ-cyclodextrin, with a lipophilic core and a hydrophilic periphery.
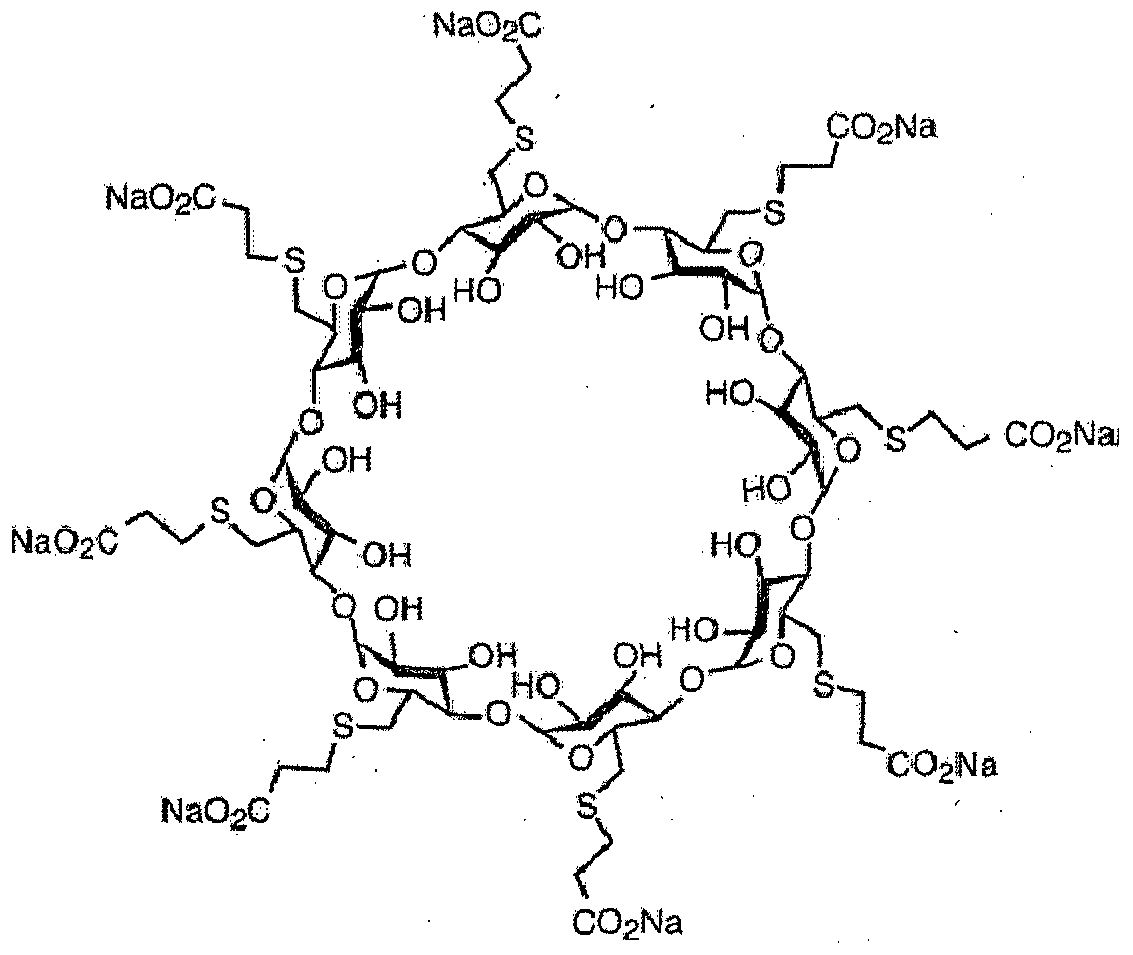
Sugammadex (designation Org 25969, trade name Bridion) is an agent for reversal of neuromuscular blockade by the agent rocuronium in general anaesthesia. It is the first selective relaxant binding agent (SRBA). This gamma cyclodextrin has been modified from its natural state by placing eight carboxyl thio ether groups at the sixth earbon positions. These extensions extend the cavity size allowing greater encapsulation of the rocuronium molecule. These negatively charged extensions electrostatically bind to the positively charged ammonium group as well as contribute to the aqueous nature of the cyclodextrin. Sugammadex’s binding encapsulation of rocuronium is one of the strongest among cyclodextrins and their guest molecules. The rocuronium molecule (a modified steroid) bound within Sugammadex’s lipophilic core, is rendered unavailable to bind to the acetylcholine receptor at the neuromuscular junction. Sugammadex sodium contains 8 recurring glucose units each with 5 asymmetric carbon atoms, in total 40 asymmetric carbon atoms for the whole molecule.
The Sugammadex was disclosed in US6670340 by Akzo Nobel. The process for preparing Sugammadex is there outlined as follows: (Scheme-I)
Scheme – 1 Suggamadex
above process step-1 involves the preparation of Vilsmeier Hack reagent by the reaction of DMF, triphenylphosphine and Iodine. Drawback associated with this step is formation of triphenylphosphine oxide as a byproduct. Removal of triphenylphosphine oxide is very difficult from the reaction; it requires repeated washing with DMF under argon atmosphere and leads to inconsistency in yield of final product. Due to this, process is lengthy and not feasible on commercial scale.
PATENT
https://www.google.com/patents/WO2012025937A1?cl=en

Suggamadex
The process of the invention is depicted in following
Formula – 1 Formula – lla
Scheme – III
scheme-IV
Suggamadex
Scheme – IV
[0018] The alkali metal hydrides are selected from the group consisting of sodium hydride, lithium hydride, potassium hydride preferably sodium hydride. [0019] The advantage of the present process is there that there is no formation of by product such as triphenylphosphine oxide, as present in prior art process. So, purification is not required which leads to better purity and yields for the intermediate as well as for final product.
[0020] Another advantage of the present invention is the significant difference between molecular weight of 6-per deoxy-6-per-chloro-y-cyclodextrin (Mol. wt. 1444) and the final product (Mol. wt. 2178). The use of 6-per deoxy-6-per- chforo-y-cyclodextrin instead of 6-per deoxy-6-per-bromo-y-cyclodextrin (Mol. wt. 1800) in the final stage‘ of the process would extend the scope of selection of appropriate dialysis membranes with precise molecular weight cut off and there by facilitate efficient purification of Sugammadex. The invention is further illustrated with following non-limiting examples:
Example: 1 Preparation of 6-perdeoxy-6-per-bromo Gamma Cyclodextrin
[0021] A portion of phosphorous pentachloride (256.5 g) was added in DMF (300 ml) at 0-5°C. Mixture was stirred at 20-25°C for lhr. A solution of gamma- cyclodextrin (50 g) in DMF (400ml) was added to above solution at 5-10°C under nitrogen. Mixture was stirred at 65 -70°C 14 hrs. The reaction mixture was cooled to 20 – 25°C and DMF was removed under vacuum. The viscous residue was diluted with water. 5M NaOH solution was added dropwise to the above solution at 5-10°C until PH=8, the resulting slurry was stirred for one hour at 20-25°C. The slurry was filtered under vacuum and washed with water and dried. The crude product was diluted with water and resulting slurry was stirred at 20-25uC for one hour. The slurry was filtered under vacuum and the solid dried at 55- 60°C under vacuum for 12hrs. (Yield – 94 – 98%, purity-98.5% by HPLC) Example: 2 Preparation of Sugammadax
[0022] To a mixture of sodium hydride (24.4 g) in DMF (150 ml) at 0-5°C, a solution of 3-mercapto propionic acid (23.7 ml, 10 eq) in DMF (50 ml) was added slowly under argon maintaining the temperature below 10 C. The resulting mixture was stirred at 20 -25°C for 30 mins. Then 6-deoxy-6-chloro gamma cyclodextrin (40 g) in DMF (400 ml) was added slowly at 5-10°C under argon and the resulting mixture was heated to 70-75°C for 12 hrs. Reaction mixture was cooled to 20 -25°C and DMF removed partially under vacuum and the reaction mixture is diluted with ethanol (600 ml). The resulting precipitate was stirred at 20 – 25°C for 1 hr and filtered under vacuum and the solid dried to afford the crude Sugammadex (wet) (100 g). The crude product was purified over silica gel and sephadex G-25 column using water as eluent. (Yield 60%)
CLIP
Sugammadex Sodium
- Synonyms:ORG-25969
- ATC:V03AB35
Use:Lorem
Chemical name:Lorem ipsum dolor sit amet, consetetur sadipscing elitr, sed diam
Formula:Lorem ipsum dolor sit amet,
- MW:2178.02 g/mol
- CAS-RN:343306-79-6
Derivatives
free acid
- Formula:C72H112O48S8
- MW:2002.17 g/mol
- CAS-RN:343306-71-8
Synthesis Path


Substances Referenced in Synthesis Path
| CAS-RN |
Formula |
Chemical Name |
CAS Index Name |
| 17465-86-0 |
C48H80O40 |
γ-cyclodextrin |
|
| 107-96-0 |
C3H6O2S |
3-mercaptopropionic acid |
Propanoic acid, 3-mercapto- |
| 53784-84-2 |
C48H72Br8O32 |
6A,6B,6C,6D,6E,6F,6G,6H-octabromo-A,6B,6C,6D,6E,6F,6G,6H-octadeoxy-γ-cyclodextrin |
|
| 168296-33-1 |
C48H72I8O32 |
6A,6B,6C,6D,6E,6F,6G,6H-octadeoxy-A,6B,6C,6D,6E,6F,6G,6H-octaiodo-γ-cyclodextrin |
|
Trade Names
| Country |
Trade Name |
Vendor |
Annotation |
| D |
Lorem |
Lorem ,2008 |
|
| GB |
Lorem |
Lorem ,2008 |
|
| USA |
Lorem |
Lorem |
|
References
-
- Adam, J. M. et al.: J. Med. Chem. (JMCMAR) 45, 1806-1818 (2002).
- Bom, A. et al.: Angew. Chem. Int. Ed. (ACIEF5) 41, 266-270 (2002)
- US 9 999 999 (Akzo Nobel; 30.12.2003; appl. 19.8.2002; EP-prior. 29.11.1999)
PATENT
http://www.google.co.in/patents/WO2001040316A1?cl=en


PATENT
https://www.google.com/patents/WO2014125501A1?cl=enhttps://www.google.com/patents/WO2014125501A1?cl=en
EXAMPLES Example: 1 Preparation of 6-perdeoxy-6-per-chloro Gamma Cyclodextrin
256.8 g (0.62 Moles) of Phosphorous pentachloride was added to 400 ml of Dimethyformamide (DMF) at 25-30 °C and mixture was maintained for 1 hour at the same temperature. 100 g (0.04 Moles) of Gamma-cyclodextrin was gradually added to the reaction mixture at 25-30 °C under nitrogen. The temperature of the reaction mixture was raised to 65 -70 °C and maintained at the same temperature for 14 to 16 hrs. The reaction mixture was then slowly added to chilled water at 0- 15 °C. The pH of the reaction mass was adjusted to 7-8 with 30% solution of sodium hydroxide in water. The contents were stirred at 25-30 °C at 2 hours. The resultant solid was filtered and washed with water (200 ml). The wet solid was repeatedly washed with purified water at 25-30 °C and dried at 65-70 °C till the moisture level was reduced to less than 4.0%. The yield of the obtained product was 90% Example: 2 Preparation of Sugammadex sodium
To a mixture of 1 10.2 g, (15 equ.) 3-mercapto propionic acid and 800 ml Dimethyl formamide (DMF) , a 30% solution of sodium methoxide (373.9 g, 30 equ) in methanol was added at 20-25°C and stirred for 1 hour at the same temperature. The compound from example- 1 (100 g) was added to the reaction mixture at 25-30°Cand heated to 75-80°C and maintained at the 75-80°C for 12 to 14 hours. After completion of the reaction, the reaction mass was cooled to 20- 25°C, then methanol (1000 ml) was added to the reaction mass and stirred for 2 hours at the same temperature. The resultant solid was filtered, washed with methanol (200 ml) and dried for 60-65°C for 8 hrs.
The crude product was dissolved in water (294 ml) and methanol (294 ml), treated with activated carbon (39.2 g, 20 % w/w) and was filtered through celite, washed the carbon cake with purified water (98 ml). The filtrate was heated to 50-55°C and slowly methanol (2646 ml) was added at the same temperature. The contents were cooled to 20 to 25°C and stirred for 2 hours at the same temperature. The resulted solid was washed with methanol (200 ml) and dried under vacuum at 60-65°C for 14 hours. The obtained product had yield of 70.34% and HPLC purity of 99.43 %.
Example-3
The Sugammadex prepared from example-2 was, dissolved in water ( 150 ml) and methanol (1 50 ml), treated with activated carbon (20 g) and filtered the carbon cake through celite bed and the carbon cake was washed with purified water (50 mL). The filtrate was heated to 50-55°C and added methanol (1350 ml) at the same temperature. The contents were cooled to 20 to 25°C and stirred for 2 hours at the same temperature. The resultant solid was washed with methanol (200 ml) and dried in vacuum at 70 – 75°C for 24 hrs. The obtained yield was 63%.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2012025937A1 |
Aug 23, 2011 |
Mar 1, 2012 |
Ramamohan Rao Davuluri |
Improved process for preparation of sugammadex |
| US5569756 * |
Mar 21, 1995 |
Oct 29, 1996 |
American Maize-Products Company |
Purification of chemically modified cyclodextrins |
| US6670340 |
Nov 23, 2000 |
Dec 30, 2003 |
Akzo Nobel |
6-Mercapto-cyclodextrin derivatives:reversal agents for drug-induced neuromuscular block |
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2001040316A1 * |
Nov 23, 2000 |
Jun 7, 2001 |
Akzo Nobel N.V. |
6-mercapto-cyclodextrin derivatives: reversal agents for drug-induced neuromuscular block |
| US6670340 |
Nov 23, 2000 |
Dec 30, 2003 |
Akzo Nobel |
6-Mercapto-cyclodextrin derivatives:reversal agents for drug-induced neuromuscular block |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2014125501A1 |
Apr 8, 2013 |
Aug 21, 2014 |
Neuland Laboratories Limited |
An improved process for preparation of sugammadex sodium |
| WO2015181224A1 |
May 27, 2015 |
Dec 3, 2015 |
Universitaet Des Saarlandes |
Novel water soluble 6-thioalkyl-cyclodextrins and uses thereof |
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 |
| Patent |
6949527 |
| Expiration |
Jan 27, 2021 |
| Applicant |
ORGANON SUB MERCK |
| Drug Application |
N022225 (Prescription Drug: BRIDION. Ingredients: SUGAMMADEX SODIUM) |
| FDA Orange Book Patents: 2 of 3 |
| Patent |
7265099 |
| Expiration |
Aug 7, 2020 |
| Applicant |
ORGANON SUB MERCK |
| Drug Application |
N022225 (Prescription Drug: BRIDION. Ingredients: SUGAMMADEX SODIUM) |
| FDA Orange Book Patents: 3 of 3 |
| Patent |
RE44733 |
| Expiration |
Jan 27, 2021 |
| Applicant |
ORGANON SUB MERCK |
| Drug Application |
N022225 (Prescription Drug: BRIDION. Ingredients: SUGAMMADEX SODIUM) |
References
- Jump up^ Miller R (2007). “Sugammadex: an opportunity to change the practice of anesthesiology?”. Anesth Analg. 104 (3): 477–8. doi:10.1213/01.ane.0000255645.64583.e8. PMID 17312188.
- Jump up^ Eleveld DJ; Kuizenga, K; Proost, JH; Wierda, JM (2008). “A Temporary Decrease in Twitch Response During Reversal of Rocuronium-Induced Muscle Relaxation with a Small Dose of Sugammadex”. Anesth Analg. 104 (3): 582–4. doi:10.1213/01.ane.0000250617.79166.7f. PMID 17312212.
- Jump up^ Welliver M (2006). “New drug sugammadex; A selective relaxant binding agent”. AANA J 74(5): 357–363. PMID 17048555
- Jump up^ Decoopman M (2007). “Reversal of pancuronium-induced block by the selective relaxant binding agent sugammadex”. Eur J Anaesthesiol. 24(Suppl 39):110-111.
- Jump up^ Pühringer FK, Rex C, Sielenkämper AW, et al. (August 2008). “Reversal of profound, high-dose rocuronium-induced neuromuscular blockade by sugammadex at two different time points: an international, multicenter, randomized, dose-finding, safety assessor-blinded, phase II trial”. Anesthesiology. 109 (2): 188–97. doi:10.1097/ALN.0b013e31817f5bc7. PMID 18648227.
- Jump up^ Abrishami A, Ho J, Wong J, Yin L, Chung F. (October 2009). Abrishami, Amir, ed. “Sugammadex, a selective reversal medication for preventing postoperative residual neuromuscular blockade”. Cochrane Database of Systematic Reviews (4): CD007362. doi:10.1002/14651858.CD007362.pub2. PMID 19821409.
- Jump up^ Yang LPH, Keam SJ.[1].Drugs 2009;69(7):919-942. doi:10.2165/00003495-200969070-00008.
- Jump up^ Naguib M (2007). “Sugammadex: another milestone in clinical neuromuscular pharmacology.”. Anesth Analg 104(3): 575–81. PMID 17312211
- Jump up^ “U.S. FDA Issues Action Letter for Sugammadex” (Press release). Schering-Plough. 2008-08-01. Retrieved 2008-08-02.
- Jump up^ “FDA approves Bridion to reverse effects of neuromuscular blocking drugs used during surgery” (Press release). Food and Drug Administration. 2015-12-15. Retrieved 2015-12-15.
- Jump up^ “BRIDION(R) (sugammadex) Injection – First and Only Selective Relaxant Binding Agent – Approved in European Union” (Press release). Schering-Plough. 2008-07-29. Retrieved 2008-08-02.
- http://www.aana.com/newsandjournal/documents/p357-363_sugammadex.pdf
REFERENCES
1: Takazawa T, Mitsuhata H, Mertes PM. Sugammadex and rocuronium-induced anaphylaxis. J Anesth. 2016 Apr;30(2):290-7. doi: 10.1007/s00540-015-2105-x. Epub 2015 Dec 8. Review. PubMed PMID: 26646837; PubMed Central PMCID: PMC4819478.
2: Abad-Gurumeta A, Ripollés-Melchor J, Casans-Francés R, Espinosa A, Martínez-Hurtado E, Fernández-Pérez C, Ramírez JM, López-Timoneda F, Calvo-Vecino JM; Evidence Anaesthesia Review Group. A systematic review of sugammadex vs neostigmine for reversal of neuromuscular blockade. Anaesthesia. 2015 Dec;70(12):1441-52. doi: 10.1111/anae.13277. Review. PubMed PMID: 26558858.
3: Ledowski T. Sugammadex: what do we know and what do we still need to know? A review of the recent (2013 to 2014) literature. Anaesth Intensive Care. 2015 Jan;43(1):14-22. Review. PubMed PMID: 25579285.
4: Partownavid P, Romito BT, Ching W, Berry AA, Barkulis CT, Nguyen KP, Jahr JS. Sugammadex: A Comprehensive Review of the Published Human Science, Including Renal Studies. Am J Ther. 2015 Jul-Aug;22(4):298-317. doi: 10.1097/MJT.0000000000000103. Review. PubMed PMID: 25299638.
5: Jahr JS, Miller JE, Hiruma J, Emaus K, You M, Meistelman C. Sugammadex: A Scientific Review Including Safety and Efficacy, Update on Regulatory Issues, and Clinical Use in Europe. Am J Ther. 2015 Jul-Aug;22(4):288-97. doi: 10.1097/MJT.0000000000000092. Review. PubMed PMID: 25299637.
6: de Boer HD, Shields MO, Booij LH. Reversal of neuromuscular blockade with sugammadex in patients with myasthenia gravis: a case series of 21 patients and review of the literature. Eur J Anaesthesiol. 2014 Dec;31(12):715-21. doi: 10.1097/EJA.0000000000000153. Review. PubMed PMID: 25192270.
7: Tsur A, Kalansky A. Hypersensitivity associated with sugammadex administration: a systematic review. Anaesthesia. 2014 Nov;69(11):1251-7. doi: 10.1111/anae.12736. Epub 2014 May 22. Review. PubMed PMID: 24848211.
8: Luxen J, Trentzsch H, Urban B. [Rocuronium and sugammadex in emergency medicine: requirements of a muscle relaxant for rapid sequence induction]. Anaesthesist. 2014 Apr;63(4):331-7. doi: 10.1007/s00101-014-2303-1. Review. German. PubMed PMID: 24595442.
9: Fuchs-Buder T, Meistelman C, Raft J. Sugammadex: clinical development and practical use. Korean J Anesthesiol. 2013 Dec;65(6):495-500. doi: 10.4097/kjae.2013.65.6.495. Epub 2013 Dec 26. Review. PubMed PMID: 24427454; PubMed Central PMCID: PMC3888841.
10: Dubois PE, Mulier JP. A review of the interest of sugammadex for deep neuromuscular blockade management in Belgium. Acta Anaesthesiol Belg. 2013;64(2):49-60. Review. PubMed PMID: 24191526.
11: Van Gestel L, Cammu G. Is the effect of sugammadex always rapid in onset? Acta Anaesthesiol Belg. 2013;64(2):41-7. Review. PubMed PMID: 24191525.
12: Schaller SJ, Fink H. Sugammadex as a reversal agent for neuromuscular block: an evidence-based review. Core Evid. 2013;8:57-67. doi: 10.2147/CE.S35675. Epub 2013 Sep 25. Review. PubMed PMID: 24098155; PubMed Central PMCID: PMC3789633.
13: Nag K, Singh DR, Shetti AN, Kumar H, Sivashanmugam T, Parthasarathy S. Sugammadex: A revolutionary drug in neuromuscular pharmacology. Anesth Essays Res. 2013 Sep-Dec;7(3):302-6. doi: 10.4103/0259-1162.123211. Review. PubMed PMID: 25885973; PubMed Central PMCID: PMC4173552.
14: Karalapillai D, Kaufman M, Weinberg L. Sugammadex. Crit Care Resusc. 2013 Mar;15(1):57-62. Review. PubMed PMID: 23432503.
15: Øberg E, Claudius C. [Possible clinical potential in reverting muscular block with sugammadex in anaesthesia and surgery]. Ugeskr Laeger. 2013 Feb 11;175(7):428-32. Review. Danish. PubMed PMID: 23402253.
16: Della Rocca G, Di Marco P, Beretta L, De Gaudio AR, Ori C, Mastronardi P. Do we need to use sugammadex at the end of a general anesthesia to reverse the action of neuromuscular bloking agents? Position Paper on Sugammadex use. Minerva Anestesiol. 2013 Jun;79(6):661-6. Epub 2012 Nov 29. Review. PubMed PMID: 23192221.
17: Stair C, Fernandez-Bustamante A. Sugammadex, the first selective relaxant binding agent for neuromuscular block reversal. Drugs Today (Barc). 2012 Jun;48(6):405-13. doi: 10.1358/dot.2012.48.6.1813474. Review. PubMed PMID: 22745926.
18: Baldo BA, McDonnell NJ, Pham NH. The cyclodextrin sugammadex and anaphylaxis to rocuronium: is rocuronium still potentially allergenic in the inclusion complex form? Mini Rev Med Chem. 2012 Jul;12(8):701-12. Review. PubMed PMID: 22512555.
19: Fuchs-Buder T, Meistelman C, Schreiber JU. Is sugammadex economically viable for routine use. Curr Opin Anaesthesiol. 2012 Apr;25(2):217-20. doi: 10.1097/ACO.0b013e32834f012d. Review. PubMed PMID: 22157200.
20: Baldo BA, McDonnell NJ, Pham NH. Drug-specific cyclodextrins with emphasis on sugammadex, the neuromuscular blocker rocuronium and perioperative anaphylaxis: implications for drug allergy. Clin Exp Allergy. 2011 Dec;41(12):1663-78. doi: 10.1111/j.1365-2222.2011.03805.x. Epub 2011 Jul 7. Review. PubMed PMID: 21732999.
BRIDION (sugammadex) injection, for intravenous use, contains sugammadex sodium, a modified gamma cyclodextrin chemically designated as 6A,6B,6C,6D,6E,6F,6G,6H-Octakis-S-(2-carboxyethyl)6A,6B,6C,6D,6E,6F,6G,6H-octathio-γ-cyclodextrin sodium salt (1:8) with a molecular weight of 2178.01. The structural formula is:
BRIDION is supplied as a sterile, non-pyrogenic aqueous solution that is clear, colorless to slightly yellow-brown for intravenous injection only. Each mL contains 100 mg sugammadex, which is equivalent to 108.8 mg sugammadex sodium. The aqueous solution is adjusted to a pH of between 7 and 8 with hydrochloric acid and/or sodium hydroxide. The osmolality of the product is between 300 and 500 mOsmol/kg.
BRIDION may contain up to 7 mg/mL of the mono OH-derivative of sugammadex [see CLINICAL PHARMACOLOGY]. This derivative is chemically designated as 6A,6B,6C,6D,6E,6F,6G-Heptakis-S-(2carboxyethyl)-6A,6B,6C,6D,6E,6F,6G-heptathio-γ-cyclodextrin sodium salt (1:7) with a molecular weight of 2067.90. The structural formula is:
/////////SUGAMMADEX, Sugammadex Sodium, D05940, SUYDEX SODIUM, UNII-ERJ6X2MXV7, fda 2015, bridion, Org25969, Org-25969, Org 25969, 361LPM2T56
O[C@@H]1[C@@H](O)[C@@H]2O[C@H]3O[C@H](CSCCC(O)=O)[C@@H](O[C@H]4O[C@H](CSCCC(O)=O)[C@@H](O[C@H]5O[C@H](CSCCC(O)=O)[C@@H](O[C@H]6O[C@H](CSCCC(O)=O)[C@@H](O[C@H]7O[C@H](CSCCC(O)=O)[C@@H](O[C@H]8O[C@H](CSCCC(O)=O)[C@@H](O[C@H]9O[C@H](CSCCC(O)=O)[C@@H](O[C@H]1O[C@@H]2CSCCC(O)=O)[C@H](O)[C@H]9O)[C@H](O)[C@H]8O)[C@H](O)[C@H]7O)[C@H](O)[C@H]6O)[C@H](O)[C@H]5O)[C@H](O)[C@H]4O)[C@H](O)[C@H]3O
O=C(O[Na])CCSC[C@@H]1OC(O[C@@H]2[C@H]([C@H](O)C(O[C@H]3[C@H](CSCCC(O[Na])=O)OC(O[C@H]4[C@H](CSCCC(O[Na])=O)OC5[C@@H](O)[C@@H]4O)[C@@H](O)[C@@H]3O)O[C@H]2CSCCC(O[Na])=O)O)[C@@H](O)[C@H](O)[C@H]1OC(O[C@@H](CSCCC(O[Na])=O)[C@H](OC6[C@@H](O)[C@H](O)[C@@H](OC7[C@@H](O)[C@H](O)[C@@H](OC8[C@@H](O)[C@H](O)[C@@H](O5)[C@H](CSCCC(O[Na])=O)O8)[C@H](CSCCC(O[Na])=O)O7)[C@H](CSCCC(O[Na])=O)O6)[C@H]9O)[C@H]9O
C(CSCC1C2C(C(C(O1)OC3C(OC(C(C3O)O)OC4C(OC(C(C4O)O)OC5C(OC(C(C5O)O)OC6C(OC(C(C6O)O)OC7C(OC(C(C7O)O)OC8C(OC(C(C8O)O)OC9C(OC(O2)C(C9O)O)CSCCC(=O)[O-])CSCCC(=O)[O-])CSCCC(=O)[O-])CSCCC(=O)[O-])CSCCC(=O)[O-])CSCCC(=O)[O-])CSCCC(=O)[O-])O)O)C(=O)[O-].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+]
A PRESENTATION
 The presentation will load below
The presentation will load below

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO