PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Laszlo Czibula, Laszlo Dobay, Eva Werkne Papp, Judit Nagyne Bagdy, Ferenc Sebok, “High Purity Butoconazole Nitrate with Specified Particle Size and a Process for the Preparation Thereof.” U.S. Patent US20080221190, issued September 11, 2008.
Butoconazole nitrate (chemical name: l-[4-(4-chlorophenyl)-2-(2,6-dichloro- -phenylthio)-n-butyl]-imidazol nitrate) is a compound of the formula (I),
(I)
it belongs among the aryl-ethylimidazole compounds, has fungicidal activity and may be used for the treatment of vaginal infections caused primarily by Candida albicans. Azoles exert their antifungal effect via modifying the ergosterol synthesis of fungus cells; more particularly, imidazoles inhibit the 14α-demethylase enzyme, thereby bringing about an increased level of 14α-methyl sterols which, in turn, causes an alteration of cell membrane permeability leading to the destruction of the fungus cells (Tetrahedron: Asymmetry Vol 4, No. 7, pp. 1521-1526, 1993). The first process for the preparation of the butoconazole nitrate is a multistep synthesis disclosed in the US 4,078,071 patent specification. Here two reaction routes are given for the preparation of the key intermediate of the formula (TV) (l-[4-(4-chlorophenyl)-2-hydroxy-n- -butyl] -imidazole) .
(IN)
According to one of them first an epoxy compound is prepared from an aromatic aldehyde or from an olefinic compound having a terminal double bond; then the epoxy compound is reacted with imidazole to yield the key intermediate. The aromatic aldehyde (VIII)
(VIII)
is treated with expensive and hazardous reagents (trimethylsulfoxonium iodide and sodium hydride) in dry dimethyl sulfoxide and the epoxide formed in the reaction is isolated after a complicated work-up. The epoxide so obtained is converted to the imidazole derivate in a time consuming reaction in the presence of dimethylformamide, then the key intermediate of the formula (IN) (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) is isolated and purified in an additional step. From the compounds having terminal double bond (Nil)
(Nil) the epoxide is obtained via a highly explosive peracidic oxidation step and the epoxide is then converted into (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IV) in a manner described above. In the other reaction route a poisoning aromatic α-halo-keto compound is used as starting material which is reacted with imidazole to give the corresponding keto-imidazole which, in turn, is reduced with a complex metal hydride – a reagent with potential hazards – to yield the key intermediate (IN). The reaction mixture is worked up in an involved manner. The synthesis way described in J. Med. Chem., 1978, Vol. 21, No. 8, pp 840-843 is as follows: l-chloro-4-chlorophenyl-2-butanol (II)
(II) is treated with the imidazole (III)
(HI)
in the presence of sodium hydride reagent in dimethylformamide solvent. This substitution reaction takes a long time and gives the (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]- imidazole) (IN) with a poor yield (51.7 %). In the next step of the butoconazole nitrate synthesis
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is treated with thionyl chloride (which is at once a reagent and a solvent) at 65-70 °C to yield l-[4-(4-chlorophenyl)-2-chloro- -n-butyl] -imidazole of the formula (N).
(V)
The reaction mixture is then evaporated to dryness. The removal of the excess thionyl chloride, a highly corrosive substance, requires special equipment; the same applies to waste treatment, an operation which also involves an environmental risk. The residue is dissolved in dichloromethane, the solution is made alkaline by adding aqueous potassium carbonate solution. Phases are separated, the organic layer is washed with water, dried on magnesium sulphate and evaporated to give l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N), as a gum. Said gum is dissolved in acetone and reacted with 2,6-dichlorothiophenol in the presence of potassium carbonate with a long reaction time. After the reaction has been finished, the inorganic salts are removed by filtration, the solvent is evaporated, and the residue is partitioned between water and ether. Butoconazole nitrate is precipitated with nitric acid from the ethereal layer. The end-product crystals in white plates from a mixture of acetone and ethyl acetate (yield: 84 %). Our aim was to provide a process by which the active agent can be prepared in high purity via reaction steps producing good yields and besides that said steps require neither solvents that are highly flammable and explosive (ether), carcinogenic (dimethylformamide) or corrosive (thionylchloride), nor reagents (e. g. sodium hydride) that are highly flammable or explosive. We have surprisingly found that when the starting material l-chloro-4-chlorophenyl-2-
-butanol (II) is reacted with the imidazole (III) in a mixture of toluene and aqueous sodium hydroxide solution in the presence of a phase transfer catalyst, the
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) key intermediate is obtained with short reaction time and excellent yield (95 %). Next we studied alternative solvents to replace the thionyl chloride in solvent function in the reaction step converting (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) into (l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole) (N). In the inert solvents which could be taken into account such as dichloromethane, toluene, chlorobenzene and dimethylformamide, the chlorinating reaction yielded a sticky reaction mixture which couldn’t be processed. We have surprisingly found, however that when (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is dissolved in 1 ,2-dichloroethane and reacted with approximately equimolar amount of thionyl chloride reagent in the presence of catalytic amount of dimethylformamide at 30-35 °C temperature, a crystal suspension is obtained which is easy-to-stir during the whole reaction time, resulting in that chlorination proceeds completely giving l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N) in quantitative yield. Being the compound sufficiently pure, it is not isolated, but separated by extraction and reacted directly with 2,6-dichlorothiophenol in methyl isobutyl ketone to give 1 -[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]-imidazole (VI) (butoconazole).
(NI)
Example 1. Preparation of (1 4-(4-chlorophenyl)-2-hvdroxy-n-butyll-imidazole) (IV) To a solution of 56.7 g (0.26 mol) of l-chloro-4-chloroρhenyl-2-butanol (J. of Medicinal Chemistry, 1978. Nol. 21. No. 8. p. 842) in 200 ml of toluene 36.2 g (0.9 mol) of sodium hydroxide dissolved in 100 ml of water, 6.4 g (0.028 mol) of benzyltriethyammom‘um chloride and 35.2 g (0.51 mol) of imidazole (III) are added. The reaction mixture is heated at 93-95 °C for one hour then the temperature is returned to about 60 °C, the phases are separated and to the organic layer water (100 ml) is added. The mixture is first stirred at 22-25 °C for 1 hour then at 0-5 °C for two hours. The crystals are separated by filtration, washed with water (2 x 35 ml) of 0-5 °C to yield 74 g of wet (l-[4-(4-chloroρhenyl)-2-hydroxy-n-butyl]-imidazole) which is dried at maximum 50 °C in vacuo to give 61.6 g (95 %) of the product. Recrystallization from ethyl acetate gives 52.4 g (85 %) of dry product melting at 104-106 °C.
Example 2. Preparation of l-[4-(4-chlorophenvπ-2-(2,6-(McMorophenyl o)-n-butyl1-ϊmidazole nitrate (I) 25 g (0.1 mol) of l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole (IN) is suspended in 1,2-dichloroethane (125 ml), to this suspension dimethylformamide (1 ml) and thionyl chloride (13.6 g; 0.11 mol) are added at 30-32 °C and the reaction mixture is kept at 35-38 °C for 1.5 hour under stirring. After the chlorination has been finished the homogenous solution is cooled to 15-18 °C, the excess of thionyl choride is decomposed with water (10 ml) then again water (80 ml) is added to the solution. After stirring at 20-22 °C for 0.5 hour the phases are separated and the organic layer is extracted with water (30 ml). To the aqueous solution methyl isobutyl ketone (250 ml) is added and the pH of the mixture is adjusted to 8.5 – 9 with 15 g (0.14 mol) of sodium carbonate dissolved in water (70 ml). The mixture is stirred at 22-25 °C for 0.5 hour, phases are separated, from the organic layer an 50 ml portion is distilled off to remove water and to the remaining solution 26.8 g (0.15 mol) of 2,6-dichloro-thiophenol and 40 g (0.29 mol) of dry potassium carbonate are added. The suspension is stirred at 105 – 108 °C under nitrogen for 3-4 hours. After the reaction has been finished the inorganic salts are removed by filtration at 22-25 °C, the filtrate is washed and clarified with activated carbon and the pH of the clear solution is adjusted to 3 – 3.5 by adding about 8 – 9 ml of 65 % nitric acid. The solution is stirred at the same temperature for 1 hour then the temperature is lowered to 8 – 12 °C. The crystals obtained are filtered and washed to give 48 g of wet l-[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]- -imidazole nitrate corresponding to 42.6 g (90 %) of dry product.
HPLC
Details of the HPLC method: Type of the apparatus: Spectra System/TSP (manufacturer: Thermo Separation Products, USA) Column: LiChrospher RP-18, 250×4.0 mm ID., 5 μm (Merck, Germany, Cat. No. : 1.50983) Mobile phase: methanol : buffer = 8:2 Bujfer: 2.18 g KH2PO4 + 4.18 g K2HPO4-3H2O dissolved in 1000 ml of distilled water; MeOH (HPLC Gradient grade, Merck, Germany, Cat. No.: 1.06007.2500) KH2PO4 (p.a., Merck, Germany, Cat. No.: 1.04877.1000) K2HPO4-3H2O (p.a., Merck, Germany, Cat. No.: 1.05099.1000) Flow rate: 1.0 ml/min Temperature: 40 °C Detection: UN 229 nm Solvent for sampling: eluent Sample concentration: 1.0 mg/ml Injected volume: 10 μl Duration of analysis: 40 min Evaluation: area normalization method. Approximative retention time: 11.9 min B. Particle size: Particle size was determined by sieve analysis using an Alpine sieve operated by a jet of air.
……………………..
WALKER K A M ET AL: “1-[4-(4-Chlorophenyl)-2-(2,6-dichloro phenylthio)-n-butyl]-1H-imidazole nitrate, a new potent antifungal agent” JOURNAL OF MEDICINAL CHEMISTRY, vol. 21, no. 8, August 1978 (1978-08), pages 840-843,
I as colorless blades
(9.6 g, 84%): mp 162-163 “C (foaming). Anal. (C19H18C13N303S)
C, H, N. The free base prepared by neutralization of a suspension
of 1 in ether with aqueous potassium carbonate and recrystallization
from cyclohexane had mp 68-70.5 “C (foaming).
The chlorohydrin (II) is obtained by the reaction of p-chlorobenzylmagnesium chloride (I) with epichlorohydrin (A) in ether. This is then converted to the crystalline alcohol (III) by reaction with sodium imidazole (B) in DMF. On treatment with thionyl chloride is converted to the corresponding chloro compound (IV). When (IV) is reacted with 2,6-dichloro thiophenol (C) in the presence of anhydrous potassium carbonate in acetone, the free base of butoconazole is formed. Neutralization of the free base (V) with nitric acid gives butoconazole.
Imidazole derivative with antifungal properties. Prepn: K. A. M. Walker, US4078071 (1978 to Syntex).
Prepn, toxicity, activity vs Candida albicans in mice: K. A. M. Walker et al.,J. Med. Chem.21, 840 (1978).
In vitro comparison with other antifungal agents: F. C. Odds et al.,J. Antimicrob. Chemother.14, 105 (1984).
Clinical trials in treatment of vulvovaginal candidiasis: W. Droegemueller et al.,Obstet. Gynecol.64, 530 (1984); J. B. Jacobson et al.,Acta Obstet. Gynecol. Scand.64, 241 (1985).
Comparison with miconazole, q.v.: C. S. Bradbeer et al.,Genitourin. Med.61, 270 (1985).
FDA issues Guidance for a clear Identification of pharmaceutical Companies
In November the US FDA has issued a Guidance for a clear identification of pharmaceutical companies. The authority now definitely prefers the DUNS system. Get more information.
In our GMP News from September 2013 you learned about a draft of a FDA Guidance for Industry entitled “Specification of the Unique Facility Identifier (UFI) System for Drug Establishment Registration”. This document’s goal was to clearly identify pharmaceutical sites. The draft comprised (manageable) five pages – including the cover page. And in terms of volume this didn’t change. However, some of the alternatives still mentioned in the draft, are not stated any longer – as one can find out when contacting the authority in these cases. The method now wanted is a registration by a D-U-N-S- (Data Universal Numbering System) number. This number – which is a 9-digit code – is…
Lexipafant is a platelet-activating factor (PAF) antagonist that was in early clinical development at DevCo for the oral treatment of dementia and motor function disorders in HIV patients, intravenous treatment of acute pancreatitis, as well as for the prevention of certain serious renal and neurological complications experienced by patients undergoing cardiac surgery, including stroke. However, no recent developments of the drug candidate have been reported by the company.
Lexipafant was also being studied at British Biotech (now Vernalis) for the intravenous treatment of pancreatitis, but development for this indication was discontinued. In 2002, DevCo obtained from British Biotech exclusive rights to develop, manufacture and sell lexipafant for the treatment of human disease, excluding the fields of oncology and ophthalmology.
BMS-663068 is an HIV-1 attachment inhibitor in development for the treatment of HIV-1 infection. BMS-663068 is a prodrug for BMS-626529 which binds to the viral envelope glycoprotein gp120 and interferes with attachment of the virus to the cellular CD4 receptor. Administration of BMS-663068 for 8 days with or without ritonavir resulted in substantial declines in plasma HIV-1 RNA levels and was generally well tolerated. Longer-term clinical trials of BMS-663068 as part of combination antiretroviral therapy are warranted.
Fostemsavir (GSK3684934/BMS-663068) is an experimental HIVentry inhibitor and a prodrug of temsavir (BMS-626529). It is under development by [ViiV Healthcare / GlaxoSmithKline]] for use in the treatment of HIV infection. By blocking the gp120 receptor of the virus, it prevents initial viral attachment to the host CD4+ T cell and entry into the host immune cell; its method of action is a first for HIV drugs.[1] Because it targets a different step of the viral lifecycle, it offers promise for individuals with virus that has become highly resistant to other HIV drugs.[2] Since gp120 is a highly conserved area of the virus, the drug is unlikely to promote resistance to itself via generation of CD4-independent virus.[3]
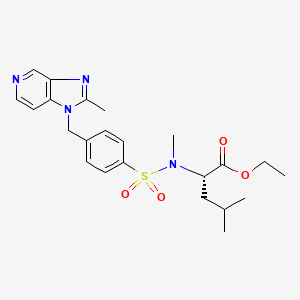
Example 6Preparation of Compound I from Compound D′ (Example 5)
N-Benzoylpiperazine HCl, Compound Db, (11.73 g, 51.74 mmol) was added to a mixture of Compound D′ (14.83 g, 47.03 mmol) (prepared in Example 5) in dry THF (265 mL) and dry DMF (29.5 mL). NaOt-Bu, 30% w/w (52.3 mL, 147 mmol) was added dropwise (30 min.) keeping the temperature at 17-21° C. The resulting yellow slurry was stirred at 17-20° for 1 h more, then cooled to about 5° C. The mixture was slowly poured into cold water (90 mL) and the flask rinsed with additional water (10 mL). The pH of the resulting yellow solution was adjusted to 6-7 with slow addition (˜20 min., 5-12° C.) of 1 N HCl (105 mL). The resulting slurry was warmed and stirred at room temperature for 1.5 h. The slurry was filtered and the cake washed with water (2×60 mL) then dried in vacuo at 65-70° C. for 5 h giving 18.4 g Compound I as a white solid (82.6%), HPLC AP 99.4. 1H NMR (400 MHz, d6-DMSO): δ 2.48 (s, 3H), 3.43 (b, 4H), 3.67 (b, 4H), 3.99 (s, 3H), 7.45 (s, 5H), 7.88 (s, 1H), 8.24 (s, 1H), 9.22 (s, 1H), 12.39 (s, 1H). 13C NMR (100 MHz, d6-DMSO): 13.85, 40.65, 45.22, 56.85, 114.19, 121.02, 122.78, 123.65, 127.06, 128.42, 129.61, 129.70, 135.51, 138.59, 142.18, 149.23, 161.38, 166.25, 169.30, 185.51.
If necessary, the product could be further purified by recrystallization from acetic acid-water-ethanol, ethanol-water, or acetone-water. For example: A mixture of Compound I (25.0 g), glacial acetic acid (260 mL) and DI water (13.8 mL) was heated to 80° C. and held with stirring (overhead) until a solution was obtained (40 min.). The batch was cooled to 70° C. and seeded (0.5 g). With slow agitation (100 rpm), EtOH (300 mL) was added slowly (1 h), keeping the temperature at 70° C. The resulting slurry was kept at 70° C. for 1 h more with very slow stirring. The slurry was cooled to 20° C. over 2 hours and held at 20° C. for over 4 hours. The slurry was filtered, the wet cake washed with EtOH (125 mL), and the solid dried in vacuo at 70° C. (≧16 h), giving 22.6 g Compound I as a white solid (88.4%).
The development of a short and efficient synthesis of a complex 6-azaindole, BMS-663068, is described. Construction of the 6-azaindole core is quickly accomplished starting from a simple pyrrole, via a regioselective Friedel–Crafts acylation, Pictet–Spengler cyclization, and a radical-mediated aromatization. The synthesis leverages an unusual heterocyclic N-oxide α-bromination to functionalize a critical C–H bond, enabling a highly regioselective copper-mediated Ullmann–Goldberg–Buchwald coupling to install a challenging triazole substituent. This strategy resulted in an efficient 11 step linear synthesis of this complex clinical candidate
Attachment inhibitor BMS-663068 is currently in clinical development for the treatment of HIV infection. Key steps in the synthesis depicted are (1) a radical-mediated redox-aromatization to generate the 6-azaindole (B → C) and (2) the regioselective bromination of an N-oxide using PyBroP (D → E).
High regioselectivity was observed in the copper(I)-mediated Ullmann–Goldberg–Buchwald coupling (H → K) using the diamine ligand J (N1/N2 = 22:1), whereas a thermal SNAr reaction gave N1/N2 = 1:1. Alternative conditions for the bromination of the N-oxide D led mainly to deoxygenation.
Procedure: To a solution of the acid 6-81 (3.01 g, 10 mmol) and benzoylpiperazine hydrochloride (3.39 g, 15 mmol) in DMF (50 mL) was added triethylamine (10.1 g, 100 mmol, 10 eq.), followed by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC; 5.75 g, 30 mmol) under N2 and the mixture stirred at room temperature for 22 h after sonication and at 40° C. for 2 h. The mixture was concentrated in vacuo to remove DMF and TEA, and to the residual solution was added water (200 mL) under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 2.8 g (5.9 mmol, Y. 59%) of the title compound IVc as off-white solid. The filtrate was extracted with CH2Cl2 (x2). The CH2Cl2 extracts were dried (Na2SO4), filtered and concentrated to gum which was triturated with Et2O to obtain a solid. This solid was suspended and triturated with MeOH to obtain 400 mg of the title compound IVc as off-white solid. Total yield: 3.2 g (6.8 mmol, Y. 68%): MS m/z 474 (MH); HRMS (ESI) m/z calcd for C24H24N7O4 (M+H) 474.1890, found 474.1884 (Δ-1.2 ppm); 1H NMR (DMSO-d6) δ ppm 2.50 (3H, s, overlapped with DMSO peaks), 3.43 (4H, br, CH2N), 3.68 (4H, br, CH2N), 3.99 (3H, s, CH3O), 7.46 (5H, br. s, Ar—Hs), 7.88 (1H, s, indole-H-5), 8.25 (1H, s, indole-H-2), 9.25 (1H, s, triazole-H-5), 12.40 (1H, s, NH); 13C-NMR (DMSO-d6) δ ppm 13.78 ,40.58, 45.11, 56.78, 114.11, 120.95, 122.71, 123.60, 126.98, 128.34, 129.6, 135.43, 138.52, 142.10, 149.15, 161.29, 166.17, 169.22, 185.42; UV (MeOH) λ max 233.6 nm (ε 3.43×104), 314.9 nm (ε 1.73×104); Anal: Calc for C24H24N7O4.1/5H2O; C, 60.42; H, 4.94; N, 20.55, Found; C 60.42, H 5.03, N 20.65; KF (H2O) 0.75%.
This reaction can also be performed by use of HATU and DMAP to provide more consistent yield of the title compound: To a suspension of the acid 6-81 (15.6 mmol) and HATU [O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophos phonate] (8.90 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) and CH2Cl2 (60 mL) was added a mixture of DMAP (5.72 g, 46.8 mmol, 3 eq.) and benzoylpiperazine hydrochloride (5.30 g, 23.4 mmol; 1.5 eq.) in DMF (60 mL) at room temperature and the mixture was stirred under nitrogen atmosphere for 4 hrs. The mixture was concentrated in vacuo to remove CH2Cl2 and most of DMF, and to the residual solution was added water under stirring and sonication. The precipitates formed were collected, washed with water and dried in vacuo to obtain 5.38 g (11.4 mmol, Y. 72.8%) of the title compound IVc as off-white solid: HPLC >95% (AP, uv at 254 nm)
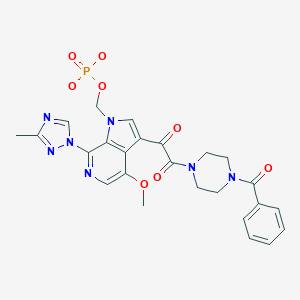
EXAMPLE 5Preparation of Ica, (Disodium Salt)
General Procedure: A suspension of IVc (0.24 g, 0.5 mmol) in anhydrous THF (4 mL) under nitrogen atmosphere was treated with sodium hydride (60% oil dispersion, 0.08 g, 2.0 mmol), and stirred until gas evolution ceased (approximately 5 minutes). The reaction mixture was treated with iodine (0.13 g, 0.5 mmol) and stirred for 2-3 minutes followed by addition of di-tert-butyl chloromethyl phosphate (1.6 g, 6.0 mmol, crude). A stream of nitrogen was allowed to pass over the reaction to facilitate the removal of much or all of the THF. The reaction mixture was stirred overnight. HPLC analysis of crude indicated starting IVc (ca. 56%) and desired adduct (ca. 32%).
Several crude reaction mixtures (a total of 6.7 mmol based on starting material IVc) were re-dissolved in dichloromethane, combined, concentrated in vacuo to remove any remaining THF. The residue was suspended in dichloromethane and TFA (1:1, approximately 40 mL total volume). The mixture was stirred for 1.5-2 hours and then solvent was removed in vacuo. The residue was suspended in dichloromethane and extracted into water (approximately 60 mL) made weakly basic with solid or aqueous sodium bicarbonate. The aqueous layer was reduced in volume by rotary evaporator if required and the solution was loaded onto a C-18 reverse phase column (approximately 80 g of C-18, YMC ODS-Aq, 50 micron) and eluted with water, followed by water containing 2.5% acetonitrile. Fractions containing pure product were pooled and organic solvent was removed by rotary evaporator. Purified product was recovered after lyophilization to give 1.00 g (1.30 mmol, 19% over 2 steps) of the title compound Ica (disodium salt) as an off-white powder: HPLC purity>99% AP at 254 nm (gradient 0-100% B/A; A 10% CH3CN-90% H2O-0.1% TFA, B 90% CH3CN-10% H2O-0.1 % TFA, gradient time 4 min, column YMC ODS-Aq 4.6×50 mm 3 micron); MS-ESI— m/z 482 (M−H minus 2Na)−; HRMS (ESI) m/z calcd for C25H27N7O8P (M+H minus 2Na)+584.1659, found 584.1651 (Δ-1.3 ppm); 1H NMR (D2O, 500 MHz) δ ppm 2.53, 2.54 (3H, 2s), 3.56 (2H, s, CH2N), 3.72 (2H, br.s, CH2N), 3.78, 3.83 (2H, 2br.s, CH2N), 3.94, 3.96 (2H, 2br.s, CH2N), 4.14 (3H, s, CH3O), 5.38, 5.40 (2H, 2d, J=11 Hz), 7.45-7.59 (5H, m, Ar—Hs), 8.07, 8.09 (1H, 2s, indole-H-5), 8.64, 8.67 (1H, 2s, indole-H-2), 8.87, 8.89 (1H, 2s, triazole-H-5); 13C NMR (125.7 MHz, D2O) δ ppm 15.43 (N-Me), 44.03, 44.47, 44.66, 45.05, 48.20, 48.82, 49.60, 50.23, 59.78 (OMe), 75.81 (NCH2O), 115.6, 126.0, 127.2, 129.6, 131.0, 131.7, 132.1, 133.5, 136.8, 147.6, 150.1, 154.2, 164.8, 170.4, 175.8, 189.2; UV (H2O) λmax 220 nm (ε 3.91×104), 249 nm (ε 2.00×104), 303 nm (ε 1.60×104); Anal: Calc for C25H24N7O8PNa2. 8H2O. 0.2NaHCO3; C, 38.39; H, 5.14; N, 12.44, P, 3.93, Na, 6.42 Found; C, 38.16; H, 4.81; N, 12.43, P, 3.72, Na, 6.05; KF (H2O) 17.3%. A less pure fractions were collected to obtain 0.22 g (0.29 mmol, Y. 4%) of the title compound Ica (disodium salt): HPLC purity>95% (AP at 254 nm).
EXAMPLE 7Preparation of Crystalline Ic (Free Acid Mono-Hydrate)
To a mixture of IVc (600 mg, 1.27 mmol) in anhydrous THF (10 ml) in an oven-dried round bottle flask under nitrogen at r.t. was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the white suspension stirred until no gas evolution was observed. The mixture was then added I2 (375 mg, 1.48 mmol), and stirred at r.t. for 3 h. To the reaction mixture was added NaH (153 mg, 6.38 mmol, dry powder, 95%), and the mixture stirred for about 5 to 10 min. The crude chloromethyl di-tert-butylphosphate (2.0 g, about 1.6 ml, 7.79 mmol) was added to the mixture, which was then stirred at r.t. for 15 h. LCMS analysis of the reaction showed a >97% conversion of the starting material. After evaporation of the volatiles, the residue was added CH2Cl2 (10 ml), cooled in an ice-water bath, slowly added TFA (10 ml) and stirred at r.t. for 3 h. The reaction mixture was then evaporated, and the residue partitioned between CH2Cl2 (50 ml) and H2O (50 ml). The CH2Cl2 layer was poured into the reaction flask that contained some undissolved brownish solid, and this mixture was extracted with a dilute aqueous NaHCO3 solution (50 ml). The aqueous mixture was purified by reverse phase preparative HPLC (solvent A: 10% MeOH-90% H2O-0.1% TFA; solvent B: 90% MeOH-10% H2O-0.1% TFA; start % B=0, final % B=100; gradient time=6 min; flow rate=45 ml/min; column: phenomenex-Luna 30×50 mm, S5; fraction collected: 3.65 to 4.05 min). The fractions collected were evaporated to dryness, and the residue dried under high vacuum to obtain the acid Ic as a pale yellow solid (356.6 mg); 1H NMR: (500 MHz, CD3OD) δ 9.05 (s, 1H), 8.46 (s, 1H), 8.04 (s, 1H), 7.47 (b s, 5H), 5.93 (d, J=12, 2H), 4.10 (s, 3H), 4.00-3.40 (b s, 8H), 2.53 (s, 3H); 19F NMR analysis showed that the material contained residual TFA, (the percentage was not quantified); Analytical HPLC method: Start % B=0, Final % B=100, Gradient time=2 min, Flow Rate=5 mL/min, Column: Xterra MS C18 7u 3.0×50 mm, LC/MS: (ES+) m/z (M+H)+=584, HPLC Rt=0.983.
172.2 mg of the purified acid Ic was dissolved in 1 ml of H2O and then about 0.3 ml of absolute EtOH (200 proof) was added. The mixture was left standing in a refrigerator (temperature about 3° C.) overnight, after which time, crystalline material was observed. The mixture was then warmed to ambient temperature, diluted with H2O to a volumn of 3 mL, and then 20 mL of MeCN was added slowly. Following the completion of addition, the mixture was stirred at r.t. for 2 h and then filtered. The solid collected (90 mg) was dried in vacuo, and then under high vacuum. This material was shown by powder x-ray studies to be crystalline; Elemental Analysis calculated for C25H26N7O8P.H2O: C 49.92; H 4.69; N 16.30; observed: C 49.66; H 4.62; N 15.99; mp=205° C. (measured by differential scanning calorimetry). The 1H NMR pattern for crystalline material was compared with that from the purified acid and both were consistent with the structure.
EXAMPLE 10Preparation of Icb (mono tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2, 3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1). The sequence of reactions is described in Scheme for Example 10.
Scheme for Example 10
Preparation of di-tert-butyl chloromethyl phosphate
A mixture of tetrabutylammonium di-tert-butyl phosphate (57 g, 0.126 mol, Digital Specialty Chemicals) and chloroiodomethane (221 g, 1.26 mol) was stirred at room temperature for four hours before the volatiles were removed under vacuum. 500 ml of ethyl ether was added to the residue and insoluble solid was filtered away. Concentration of the filtrate in vacuo and removal of remaining volatiles using a vacuum pump provided di-tert-butyl chloromethyl phosphate as a light brown or yellow oil, which was utilized in the next step without further purification.
Preparation of IIc: (3-(2-(4-benzoylpiperazin-1-yl)-2-oxoacetyl)-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl)methyl di-tert-butyl phosphate
NaH (2.6 g, 10.3 mmol, 95% in oil, Seq.) was added slowly into a suspension of IVc (10.0 g, 21.1 mmol) in dry THF (100 ml) and the mixture was allowed to stir for 0.5 hour at room temperature. A solution of iodine (5.27 g, 20.8 mmol) dissolved in dry THF (10 ml) was added slowly into the stirring solution at a rate which prevented foaming or a violent reaction. The resultant mixture was stirred for an additional 3 hours before a second 2.6 g portion of NaH was introduced. After 15 minutes at ambient temperature di-tert-butyl chloromethyl phosphate, the entire batch of di-tert-butyl chloromethyl phosphate, obtained from step one, was added. After stirring for 16 hours, the reaction mixture was poured into iced NH4OAc (30%) (120 ml), followed by extraction with EtOAc (3×300 ml). The combined organic extracts were washed with water (100 ml) and then brine (100 ml), dried over Na2SO4, and concentrated under vacuum to afford a residue, which was purified by silica gel chromatography (elution with EtOAc/Et3N (50/1) and then EtOAc/MeOH (100/1)) to give 8.0 g (˜75% AP, ˜41% yield) of diester IIc as a light yellow solid.
Preparation of Icb (mono L tromethamine salt): [3-[(4-benzoylpiperazin-1-yl)(oxo)acetyl]-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl]methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)propane-1,3-diol salt (1:1)
500 mg (˜p75 AP, 0.54 mmol) of diester IIc was dissolved in a mixture of water (2.5 ml) and acetone (2.5 ml). The resulting mixture was stirred at 40° C. for 16 hours to complete the solvolysis. To this reaction mixture was added 3.0M aqueous TRIS (mono tromethamine) solution to adjust pH to 3.32. Acetone (30 ml) was slowly added to the reaction mixture in 1 hour.* After complete addition of acetone, the solution was stirred overnight to complete the crystallization of Icb. The solid was collected by filtration and rinsed with 20:1 acetone-water (2×5 mL). The white crystalline solid was dried under house vacuum under nitrogen atomosphere at 50° C. for 24 h to afford 290 mg of Icb (>98.5 AP).
*After adding about 15 and 20 ml of acetone, the reaction mixture was seeded with crystalline Icb.
Obtained via other process (hydrolysis with TFA in methylene chloride), salt Icb is ˜1 molar mono tromethamine salt with 0.47% of water, 0.1% of acetone and 0.05% of methanol. 1H NMR (500 MHz, d6-DMSO, 30° C.) δ8.77 (s, 1H), 8.48 (s, 1H), 8.00 (s, 1H) 7.44 (b, 5H), 5.42 (d, 2H, J=15 Hz), 4.02 (s, 3H), 3.70-3.30 (m, 8H), 3.41 (s, 6H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3, 30° C.) δ184.8, 169.0, 165.8, 160.3, 150.4, 146.2, 143.2, 135.4, 129.4, 128.9, 128.2, 127.7, 126.9, 123.2, 122.2, 112.9, 72.3, 60.7, 59.0, 56.7, 13.4. MS m/z: (M-trisamine+H)+ calcd for C25H27N7O8P 584.2, found 584.0. Anal. Calcd. C, 49.11; H, 5.37; N, 15.76; P, 4.32; found: C, 48.88; H, 5.28; N, 15.71; P, 4.16. M.P. 201-205° C.
EXAMPLE 13Alternate preparation of Icb (Pro-drug of IVc)
To a 10 L reactor equipped with an overhead stirrer, thermocouple, distillation apparatus, and nitrogen inlet was charged IVc (200.00 g, 422.39 mmol), Cs2CO3 (344.06 g, 1.06 mol), KI (140.24 g, 844.81 mmol) and NMP (1.00 L, 10.38 mol). The reaction was stirred at room temperature resulting in a light brown heterogeneous suspension. Di-tert-butyl chloromethyl phosphate (273.16 g, 1.06 mol) was added via addition funnel and the reaction mixture was heated to 30° C. for 16-24 hours with stirring after which time the reaction was cooled to 5° C. To the reaction was added DCM (1.5 L) then the reaction was slowly quenched with water (3.5 L) maintaining the reaction temperature under 20° C. resulting in a biphasic mixture. The product rich bottom layer was separated, washed with water (3.5 L×3), then transferred back to the reactor. The solution was concentrated under vacuum to a volume of 1 L keeping the temperature below 25° C. IPA was added (2 L) then the reaction was concentrated under vacuum to a volume of 2 L keeping the temperature below 25° C. The reaction was then seeded with IIc (0.200 g), stirred overnight at room temperature resulting in a slurry. The slurry was filtered and the wet cake was washed with MTBE (1 L), dried in a vacuum oven at 50° C. overnight resulting in a yellow/white powder (207.1 g, 70%). 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 8.18 (s, 1H), 7.91 (s, 1H), 7.42 (s, 5H), 5.95 (d, J=14.2 Hz, 2H), 4.06 (s, 3H), 3.97-3.36 (m, 8H), 2.50 (s, 3H), 1.27 (s, 18H); 3C NMR (100 MHz, CDCl3) δ 184.64, 170.65, 165.91, 161.60, 150.82, 145.38, 141.89, 134.96, 130.20, 129.59, 128.68, 127.58, 127.10, 124.77, 122.64, 115.22, 83.90, 83.83, 73.69, 73.63, 56.95, 46.04, 41.66, 29.61, 29.56, 13.90; ES+ MS m/z (rel. intensity) 696 (MH+,10), 640 (MH+-isobutylene, 30), 584 (MH+-2 isobutylene, 100).
To a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (200.24 g, 287.82 mmol), acetone (800.00 ml, 10.88 mol) and water (800.00 ml, 44.41 mol). The reaction was heated to 40° C. and stirred for 18-24 hours. The reaction was cooled to 20° C. then tromethamine (33.62 g, 277.54 mmol) was added. The reaction was heated to 40° C. then stirred for an additional hour until all solids were dissolved. The reaction was cooled to 20° C. then filtered through a 10 micron cuno filter into a 10 L 4 neck reactor equipped with a thermocouple, overhead stirrer, and nitrogen inlet. Acetone (3 L) was added rapidly, followed by seeding with Icb (0.500 g), then additional acetone (3 L) was added. The reaction was stirred at room temperature overnight resulting in a slurry then filtered. The wet cake was washed with acetone (800 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (165.91 g, 82%).
Supplementary Information:
Isolation of the Free-Acid Intermediate IC:
In a 250 mL 3 neck reactor equipped with a thermocouple, overhead stirrer, condenser and nitrogen inlet was added IIc (10.0 g, 14.37 mmol), acetone (40.00 ml, 544.15 mmol) and water (40.00 ml, 2.22 mol). The reaction was heated to 40° C. and stirred for 14-24 hours. The reaction was cooled to 20° C. then stirred for three hours, resulting in a slurry. The slurry was filtered, then the wet cake washed with acetone (40.00 ml) then dried in a vacuum oven at 50° C. overnight resulting in a fluffy white powder (7.00 g, 83%). NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.47 (s, 1H), 8.06 (s, 1H), 7.45 (s, 5H), 5.81 (d, J=12.3 Hz, 2H), 4.03 (s, 3H), 3.91-3.19 (m, 8H), 2.39 (s, 3H); 13C NMR (500 MHz, DMSO-d6) δ 185.20, 169.32, 165.85, 160.75, 150.51, 146.30, 143.24, 135.53, 129.74, 129.22, 128.46, 127.34, 127.09, 123.67, 122.73, 113.94, 72.90 (d, 2JC-P=5 Hz), 57.01, 45.2 (bs), 40.8 (bs), 13.66. ES+ MS m/z (rel. intensity) 486 (MH+−H3PO4, 100).
Ripasudil hydrochloride hydrate (Glanatec® ophthalmic solution 0.4 %; hereafter referred to as ripasudil) is a small-molecule, Rho-associated kinase inhibitor developed by Kowa Company, Ltd. for the treatment of glaucoma and ocular hypertension. This compound, which was originally discovered by D. Western Therapeutics Institute, Inc., reduces intraocular pressure (IOP) by directly acting on the trabecular meshwork, thereby increasing conventional outflow through the Schlemm’s canal.
As a result of this mechanism of action, ripasudil may offer additive effects in the treatment of glaucoma and ocular hypertension when used in combination with agents such as prostaglandin analogues (which increase uveoscleral outflow) and β blockers (which reduce aqueous production).
The eye drop product has been approved in Japan for the twice-daily treatment of glaucoma and ocular hypertension, when other therapeutic agents are not effective or cannot be administered. Phase II study is underway for the treatment of diabetic retinopathy.
K-115 is a Rho-kinase inhibitor as ophthalmic solution originally developed by Kowa and D Western Therapeutics Institute (DWTI). The product candidate was approved and launched in Japan for the treatment of glaucoma and ocular hypertension in 2014.
In 2002, the compound was licensed to Kowa Pharmaceutical by D Western Therapeutics Institute (DWTI) in Japan for the treatment of glaucoma. The compound is currently in phase II clinical trials at the company for the treatment of age-related macular degeneration and diabetic retinopathy.
Use of (S)-(-)-1-(4- fluoro-5-isoquinoline-sulfonyl)-2-methyl-1,4-homopiperazine (ripasudil hydrochloride, first disclosed in WO9920620), in the form of eye drops, for the treatment of retinal diseases, particularly diabetic retinopathy or age-related macular degeneration.
Follows on from WO2012105674 by claiming a combination of the same compound. Kowa, under license from D Western Therapeutics Institute, has developed the Rho kinase inhibitor ripasudil hydrochloride hydrate (presumed to be Glanatek) as an eye drop formulation for the treatment of glaucoma and ocular hypertension which was approved in Japan in September 2014..
The company is also developing the agent for the treatment of diabetic retinopathy, for which it is in phase II trial as of October 2014.
…………………….
A Practical Synthesis of (S)-tert-butyl 3-methyl-1,4-diazepane-1-carboxylate, the key intermediate of Rho-kinase inhibitor K-115
Synthesis (Stuttgart) 2012, 44(20): 3171
practical synthesis of (S)-tert-butyl 3-methyl-1,4-diazepane-1-carboxylate has been established for supplying this key intermediate of Rho–kinase inhibitor K-115 in a multikilogram production. The chiral 1,4-diazepane was constructed by intramolecular Fukuyama–Mitsunobu cyclization of a N-nosyl diamino alcohol starting from the commercially available (S)- or (R)-2-aminopropan-1-ol. In the same manner, an enantiomeric pair of a structural isomer were prepared for demonstration of the synthetic utility.
The including prevention and treatment cerebral infarction, cerebral hemorrhage, subarachnoid hemorrhage, cerebrovascular disorders such as cerebral edema, the present invention relates to a salt thereof or isoquinoline derivatives useful as therapeutic agents, particularly glaucoma.
(S) – (-) -1 – (4 – fluoro-iso-5 – yl) sulfonyl – 2 – methyl -1,4 – diazepane the following formula (1):
It is a compound represented by the particular it is a crystalline water-soluble, not hygroscopic, because it is excellent in chemical stability, it is useful as a medicament has been known for its hydrochloride dihydrate ( refer to Patent Documents 1 and 2). -5 Isoquinoline of these – the sulfonamide compounds, that prophylactic and therapeutic agents for cerebral infarction, cerebral hemorrhage, subarachnoid hemorrhage, cerebrovascular disorders such as cerebral edema, is useful as a therapeutic agent for preventing and glaucoma in particular is known (1-5 see Patent Document 1).
Conventionally, for example, a method of manufacturing by the method described in Patent Document 1, as shown in the following production process has been reported preparation of said compound (Production Method 1-A).
That is, (S)-1-tert-butoxycarbonyl – 3 – by reacting the presence of triethylamine in methylene chloride-fluoro-isoquinoline (2) – methyl -1,4 – diazepane and 5 (3) – chloro-sulfonyl -4 by adding trifluoroacetic acid in methylene chloride compound (the first step), obtained following (4) to synthesize a compound (4) by deprotection to (second step) the desired compound (1) This is a method of manufacturing.
It is also an important intermediate for preparing the compound (1) (S)-1-tert-butoxycarbonyl – 3 – methyl-1 ,4 – diazepane to (3), for example, in the following manner (; see JP Production Process 1-B) that can be produced is known.
Further, on the other hand, the compound (1) (see Patent Document 1) to be manufactured manufacturing routes such as: Any (Process 2) are known.
WO 1999/20620 pamphlet WO 2006/057397 pamphlet WO 1997/028130 pamphlet JP Patent Publication No. 2006-348028 JP Patent Publication No. 2006-290827
However, it is possible to produce in the laboratory of a small amount scale, but you place the point of view for mass industrial production, environmentally harmful halogenated hydrocarbon solvent in the compound of the above-mentioned process for producing 1-A is ( problem because it is carried out coupling step (3) and 2), giving significant adverse environmental exists. Therefore, solvent of halogenated hydrocarbon other than those listed to the specification of the patent document 1, for example, I tried actually dioxane, tetrahydrofuran and the like, but the present coupling reaction will be some progress indeed, Problems reaction is not completed raw material remained even after prolonged reaction time, yield undesirably stays in at most 30% was found. Furthermore, it is hard to decompose in the environment, elimination is also difficult to dioxane is not preferred irritating to humans, and are known as compounds that potentially harmful brain, kidney and liver .
When we actually produced compound (3) by the above production method 1-B, can be obtained desired compound in good yield merged with reproducibility is difficult has further been found that. That is, in the production path, 1,4 – and is used sodium hydride with dimethyl sulfoxide in forming a diazepane ring, except that I actually doing this step, Tsu than the reproducibility of the desired compound It could not be obtained in high yield Te. Also, that this is due to the synthetic route through the unstable intermediate, that it would be converted into another compound easily found this way. limitations and potential problems of the present production process is exposed since this stability may affect the reproducibility of the reaction.
Meanwhile, an attempt to carry out mass production is actually in the Process 2, it encounters various problems. For example, it is stored as an impurity whenever I repeat step, by-products formed in each stage by tandem production process ranging from step 8 gave more complex impurity profile. Depending, it is necessary to repeat a complicated recrystallization purity obtained as a medicine until the purification, the yield in the laboratory be a good overall yield is significantly reduced in the mass production of actual example be away, it does not have industrial utility of true was found. It can be summarized as follows: Considering from the viewpoint of GMP process control required for pharmaceutical production these problems.
Requires control process and numerous complex ranging 1) to 8 step, 3 2) third step – amino-1 – in the step of reacting a propanol, a difficult to remove positional isomers are mixed, 3) The fourth step water is mixed by the minute liquid extraction operation at the time of return to the free base from oxalate require crystallization purification by oxalate in the removal of contaminants of positional isomers, in 4) fifth step, 5) sixth step The Mitsunobu by reproducibility poor require water control in the Mitsunobu reaction used in the ring closure compounds to (1) compounds in (6), 6) ring closure reaction, departing management of the reagent added or the like is generated, in 7) Seventh Step it takes a complicated purification in impurity removal after the reaction, resulting in a decrease in isolated yield. These are issues that must be solved in order to provide a stable supply of raw material for pharmaceuticals high chemical purity is required.
Thus, gentle salt thereof, or the environment isoquinoline derivative comprising a compound represented by the formula (1), the present invention provides a novel production method having good reproducibility and high purity easily and in high yield I intended.
As a result of intensive studies in view of such circumstances, the present inventors, in the manufacturing process of the final target compound shown by the following expression
(Wherein represents a fluorine, chlorine, bromine or iodine,may,R 3 and 1, R 2 R represents a C 1-4 alkyl group be the same or different from each other, and P, X 1 is a protecting group shows a, 0 to m represents an integer of 3, 0 to n is. represents an integer of 3)
Is a urea-based solvents nitrile solvents, amide solvents, sulfoxide or solvents, the solvent may be preferably used in the coupling step of the compound (III) and (II) are generally very short time With these solvents It has been found that can be converted to the desired product quantitatively. It is possible to carry out the coupling step Volume scale while maintaining a high yield by using these solvents, there is no need to use a halogenated hydrocarbon solvent to give significant adverse environment. In consideration of the process such as removal of the solvent after the reaction was further found that acetonitrile is the best among these solvents. Also, since by using hydrochloric acid with ethyl acetate solvent in step deprotection can be isolated as crystal of hydrochloride desired compound (I), without going through the manipulation of solvent evaporation complicated , it has been found that it is possible to obtain the object compound (I) is a simpler operating procedure. Since there is no need to use a halogenated hydrocarbon solvent in this deprotection step further, there is no possibility of harming the environment.
It has been found that it is possible in mass production of (II), leading to the target compound purity, in high yield with good reproducibility as compared with the conventional method compounds are important intermediates in the coupling step further. That is, was it possible to lead to the intermediate high purity and in high yield by eliminating the production of a harmful halogenated hydrocarbon solvent to the environment in this manner. 1,4 addition – in order to avoid the problems encountered in the reaction using sodium hydride in dimethyl sulfoxide in forming the diazepane ring, in order to allow the cyclization reaction at mild conditions more, as a protecting group By performing the Mitsunobu reaction using Noshiru group instead of the carbobenzyloxy group, in addition to one step shorten the manufacturing process of the whole, without deteriorating the optical purity was successfully obtained the desired compound desired.
Paul Murray Catalysis Consulting helps companies to save money and resources through more efficient chemical processes.
About
Dr Paul Murray is a world leading consultant scientist, providing expertise and training in the fields of Catalysis, Design of Experiments and Principal Component Analysis. Paul is an experienced scientist with an additional expertise in automation, multivariate data analysis, process development and problem solving. Paul has a proven track record of the timely delivery of innovative solutions to client projects resulting in significant reductions in costs and resources to customers.
Paul Murray Catalysis Consulting provides expertise in:
The development and optimisation of challenging catalytic reactions.
The use of Principal Component Analysis (PCA) to optimise ligand and solvent selection.
The use of advanced experimental design linking DoE with PCA for efficient…
OK it was an extended time away from posting — I totally blame the Turkey, Ham, Beer, Stuffing, Pie — at least I have tapered off over the years.
So what’s sitting on my desk — after several pontifications, I have gotten back to thinking about how chemists think about their chemistry and where it can go in flow processes — so, OK, retrosynthesis — but I often think in classes of fragments and what they can do (think of it as a review on enaminone transformations so to speak). In this case, Ian Baxendale got me thinking about ynones or alpha, beta-acteylenic ketones — used quite a bit right? furans, flavones, pyrazoles, pyrimidines and heck back at Bayer I used them in a number of dipolarcycloadditions and intramolecular cyclizations to isoxazoles and pyrroles……you get the point……if interested in a nice article on using a flow approach to ynones and…
In October 2014, the US FDA issued a Warning Letter to the company Hikma Pharmaceuticals justified by deficiencies in the visual inspection of vials. Read more here.
In October 2014, the US FDA issued a Warning Letter to the company Hikma Pharmaceuticals because of deficiencies in the visual inspection of vials and environmental monitoring.
Already in a Warning Letter issued in 2011, a deficiency in the visual inspection was noted as the detection and evaluation of particulate matter failed to be sufficient. Now, the current complaint in the area of visual control explicitly refers to the qualification of staff for the performance of the manual visual inspection. Here, the FDA inspectors noticed that visible markings were present on the qualifcation test sets which enabled operators for visual inspection to recognize – thanks to these markings – vials with particles. The qualification of staff…
I (Dr Anthony) seated with Dr Vijay Kirpalani CEO of Pi-Process Intensification Experts LLP
at CPhI Mumbai India 3rd Dec 2014
Pi-Process Intensification Experts LLP
provide
Process Intensification
Creating competitive advantage through Improved and consistent quality, high efficiencies and maximum flexibility.
Safer, Cleaner, Smaller, Cheaper and Smarter processes , The basic principle of Process Intensification is to fit the equipment to the process and not process to the equipment, as is the case now.
Process Intensification can achieve drastic improvement in the time cycle and yields as well as converting batch processes to continuous process using specialized set of equipment. The design philosophy in process intensification is to design a process which has Chemical Kinetics as its only limitation. See the illustration below
“Process Intensification by Kinetics alone controlling the reaction, using specialized equipments; modification / telescoping of process steps achieves drastic reduction in time cycles and converts batch processes to continuous ; Reduced energy consumption, Reduced by-product formation; sustainability , hazard-containment, compliance to QbD and PAT and importantly a much faster time-to-market”
Illustrative examples are as follows:
Watt’s aldol reaction: Time needed to reach 100 % conversion 20 minutes against 24 hours in batch process
Fisher Esterification: gives 83% yield against 15% in batch process
Grignard Reaction: gives 78% yield against 49% in batch process
Nitration Reaction: Product purity increase from 56% to 78% and yield of mononitrate increases 55% to 75%.
Other Reactions: Acetylation, Amine Protection, Carbonylation, Claisen Schmidt Reaction, Esterification, Hydrogenation, Hydrolysis, Methylation, Oxidation, Phosgenation, Sulphonation, Suzuki Coupling Ring Expansion
Benefits of Process Intensification (PI) Techniques
Sponsored Projects
Scale-up for Retrofitting in existing plant as well as greenfield projects based on flow chemistry data generated in our laboratory. A well-equipped Laboratory and Pilot Plant set-up is available at our “Pi-Lab” for carrying out “FLOW Chemistry” based Reactions and utilizing numerous Process Intensification techniquesfor Unit-Processes & Unit-Operations for the industry to reap the benefits of Process Intensification.
The laboratory and pilot plant data will be utilized to provide the plant scale design using specialized equipments like micro-reactors, micro-plate-reactors in SiC, monolithic loop reactors, spinning disk reactors-cum-heat exchangers, FUMI reactors, dynamic mixing reactors, oscillatory baffled reactors (OBR), Bio-catalytic impregnated membrane Reactors, and other modern state-of-the-art equipments enabling conversion of batch to continuous flow processes.
We handle hazardous chemistries with very high exotherms (upto 1300 J/gm) safely in the range of -70oC to + 250oC with pressures upto 200 bar, and with reaction times from 0.03 sec to 1 hour and reactor volumes from 0.2 ml to 100 ml (Lab) and 1 L (Pilot) — yielding from 20 gms to 8 Kgs/hour (Lab) and 500 gms to 200 Kgs/hour (Pilot).
Scale Up – Flexibility & Adaptability
…… will provide all the services for scale up to the sizes desired by clients by utilizing data from Laboratory trials.
Rental
A range of Flow Chemistry and Process Intensification equipments can be offered on rent. This enables the users to get the hands-on experience so as to select the apt equipments for their needs.
Vijay Kirpalani
CEO Pi-Process Intensification Experts LLP
Plot-W-33, M.I.D.C. Industrial Area
TALOJA – 410208, Navi Mumbai, INDIA
email : vk@pi-inc.co www.pi-inc.co
Tel: +91-9321342022 // +91-9821342022
some pics from hall 5 -H-47 at cphi mumbai india dec 3 2014
French drug maker Sanofi Tuesday said it has received approval from the U.S. Food and Drug Administration for its Priftin (rifapentine) tablets to treat latent tuberculosis infection, or LTBI.
Following a priority review, FDA has approved Priftin in combination with isoniazid, or INH, for a new indication for treatment of LTBI in patients two years of age and older at high risk of progression to tuberculosis or TB disease.
Rifapentine is an antibiotic drug used in the treatment of tuberculosis. It inhibits DNA-dependent RNA polymerase activity in susceptible cells. Specifically, it interacts with bacterial RNA polymerase but does not inhibit the mammalian enzyme.
Rifapentine was first synthesized in 1965 by the same company that produced rifampin. The drug was approved by the Food and Drug Administration (FDA) in June 1998.
Medical uses
A review of alternative regimens for prevention of active tuberculosis in HIV-negative individuals with latent TB found that a weekly, directly observed regimen of rifapentine with isoniazid for three months was as effective as a daily, self -administered regimen of isoniazid for nine months. But the rifapentine-isoniazid regimen had higher rates of treatment completion and lower rates of hepatotoxicity. However, the rate of treatment-limiting adverse events was higher in the rifapentine-isoniazid regimen. [1]
PRIFTIN (rifapentine) for oral administration contains 150 mg of the active ingredient rifapentine per tablet.
The 150 mg tablets also contain, as inactive ingredients: calcium stearate, disodium EDTA, FD&C Blue No. 2 aluminum lake, hydroxypropyl cellulose, hypromellose USP, microcrystalline cellulose, polyethylene glycol, pregelatinized starch, propylene glycol, sodium ascorbate, sodium lauryl sulfate, sodium starch glycolate, synthetic red iron oxide, and titanium dioxide.
Rifapentine is a rifamycin derivative antibiotic and has a similar profile of microbiological activity to rifampin (rifampicin). The molecular weight is 877.04.
The molecular formula is C47H64N4O12.
The chemical name for rifapentine is rifamycin, 3-[[(4-cyclopentyl-1-piperazinyl)imino]methyl]-or 3-[N-(4-Cyclopentyl – 1-piperazinyl)formimidoyl] rifamycin or 5,6,9,17,19,21-hexahydroxy-23-methoxy-2,4,12,16,18,20,22-heptamethyl-8-[N-(4-cyclopentyl-l-piperazinyl)-formimidoyl]-2,7-(epoxypentadeca[1,11,13]trienimino)naphtho[2,1-b]furan-1,11(2H)-dione 21-acetate. It has the following structure:
Use in special populations
Pregnancy
Rifapentine has been assigned a Pregnancy Category C by the FDA. Rifapentine in pregnant women has not been studied, but animal reproduction studies have resulted in fetal harm and were teratogenic. If rifapentine and rifampin are used together in pregnancy, coagulation should be monitored due to a possible increased risk of maternal postpartum hemorrhage and infant bleeding. [2]
Adverse effects
Common side effects are hyperuricemia, pyuria, hematuria, urinary tract infection, proteinuria, neutropenia, anemia, and hypoglycemia. [2]
Contraindications
Rifapentine should be avoided in patients with an allergy to the rifamycin class of drugs. [2] This drug class includes rifampin and rifabutin. [3]
Interactions
Rifapentine induces metabolism by CYP3A4, CYP2C8 and CYP2C9 enzymes. It may be necessary to adjust the dosage of drugs metabolized by these enzymes if they are taken with rifapentine. Examples of drugs that may be affected by rifapentine include warfarin, propranolol, digoxin, protease inhibitors and oral contraceptives.[2]
History
Rifapentine was first synthesized in 1965 by the same company that produced rifampin. The drug was approved by the Food and Drug Administration (FDA) in June 1998. It is synthesized in one step from rifampicine.
Rifapentine was first synthesized in 1965 by the same company that produced rifampin. The drug was approved by the Food and Drug Administration (FDA) in June 1998.
(7S,11S,12S,13S,14R,15S,16R,17R,18R,26E)-26-{[(4-Cyclopentyl-1-piperazinyl)amino]methylene}-2,15,17,29-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-6,23,27-trioxo-8,30-dioxa-24-azatetracyclo [23.3.1.14,7.05,28]triaconta-1(28),2,4,9,19,21,25(29)-heptaen-13-yl acetate. Rifapentine is an antibiotic drug used in the treatment of tuberculosis.
Preparation of Rifapentine: this chemical can be prepared by 3-aldehyde rifamycin SV with 1-Amino-4-cyclopentylpiperazine. This reaction needs reagent tetrahydrofuran. The yield is 55 %
References
Sharma SK et al . (2013). “Rifamycins (rifampicin, rifabutin and rifapentine) compared to isoniazid for preventing tuberculosis in HIV-negative people at risk of active TB.”. Cochrane Database of Systematic Reviews7: CD007545. doi:10.1002/14651858.CD007545.pub2. PMID23828580.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



























































![4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline NMR spectra analysis, Chemical CAS NO. 223645-67-8 NMR spectral analysis, 4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/466/2466109_1h.png)
![4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline NMR spectra analysis, Chemical CAS NO. 223645-67-8 NMR spectral analysis, 4-fluoro-5-[[(2S)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/466/2466109_13c.png)

























