
Home » Uncategorized (Page 67)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New FDA Draft Guidance ‘Data Integrity and Compliance with cGMP’ published
DRUG REGULATORY AFFAIRS INTERNATIONAL

In the last years, the topic “data integrity” has become a priority for the FDA. Recently, the Agency has published the draft of a Guidance for Industry on the topic which presents the comprehensive opinion of the FDA on data integrity. Read more about the draft of the Guidance for Industry “Data Integrity and Compliance with cGMP”.
In recent years, the topic “data integrity” has become a priority for European and American inspectors. At the beginning of 2015, the British authority MHRA published a first paper on that topic. Also in 2015, the World Health Organisation WHO issued another significant draft document on data integrity. Recently, the US American FDA has released the draft of a Guidance for Industry entitled “Data Integrity and Compliance with cGMP”. Although the FDA describes the Guidance as a non-binding recommendation, one may assume that the document presents the current thinking of the…
View original post 543 more words
Five new General Chapters in the European Pharmacopoeia on Genotoxic Impurities in Pharmaceutical APIs
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg)
During the manufacture of APIs as sulfonate salts, esters of sulfonic acid may develop in undesired chemical side reactions. Recently, five new General Monographs have been included in the European Pharmacopoeia which describe how to cope with these impurities. Read more about these genotoxic impurities and the possibility to control them thanks to risk assessments.
Sulfonic acids are often used for the manufacture of pharmaceutical APIs. They serve as counterions in crystallisation processes, as protective groups or acid catalysts in API syntheses. Here, if short-chain alcohols such as methanol, ethanol or isopropanol are present, the formation of esters of these sulfonic acids can occur, which may have a genotoxic potential (alkylation of DNA).
The Mesilate Working Party which has been appointed in 2008 by the European Pharmacopoeia Commission has elaborated five General Chapters on different sulfonates which have been published in the European Pharmacopoeia Supplement 8.7 that came into force on 1 April 2016. The…
View original post 311 more words
FDA releases draft guidance on the use of comparability protocols for post approval changes
DRUG REGULATORY AFFAIRS INTERNATIONAL

The US FDA released a draft guidance for industry “Comparability Protocols for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information”. The guidance replaces the draft guidance published in February 2003. It provides recommendations on implementing postapproval changes through the use of comparability protocols (CPs). Read more about FDA´s draft guidance for industry “Comparability Protocols for Human Drugs and Biologics”.
On April 19, 2016, the US Food & Drug Administration (FDA) released a draft guidance for industry “Comparability Protocols for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information”. Comments and suggestions regarding the draft guideline should be submitted within 60 days of publication.
The guidance replaces the draft guidance published in February 2003. It provides recommendations on implementing postapproval changes through the use of comparability protocols (CPs). A CP is a comprehensive, prospectively written plan for assessing the effect of proposed CMC postapproval changes on the identity, strength…
View original post 418 more words
Cymipristone
Cymipristone
- Estra-4,9-dien-3-one, 11-[4-(cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propynyl)-, (11β,17β)- (9CI)
- (11β,17β)-11-[4-(Cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propyn-1-yl)estra-4,9-dien-3-one
- Saimisitong
NDA Filed china
Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
A progesterone receptor antagonist potentially for termination of intrauterine pregnancy.
![]()
CAS No.329971-40-6
- Molecular FormulaC34H43NO2
- Average mass497.711 Da
- Steroid Compounds, a Method for Preparation thereof, Pharmaceutical Compositions Containing the Same and Use thereof
-
This invention relates to steroid compounds and pharmaceutical acceptable salts thereof, a method for preparation thereof, pharmaceutical compositions containing the same as active component, and their use in the preparation of medicines for treating diseases associated with progestogen dependence and for fertility control, abortion or contraception and for anticancer use.
-
Mifepristone (11β-[4-(N,N-dimethylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in French Patent No. 2,497,807 to Rousell-Uclaf, published May 31, 1983. It is the first progesterone receptor antagonist put into clinical application and is a new type of anti-progestin. It binds to progesterone receptor and glucocorticoid receptor, having an affinity with progesterone receptor in rabbit endometrium five-fold higher than that of progesterone and thereby having strong anti-progesterone effect. It causes degeneration of pregnant villus tissue and decidual tissue, endogenous prostaglandin (PG) release, luteinizing hormone decrease, corpus luteum dissolution, and necrosis of embryo sac whose development depends on corpus luteum, leading to abortion. Therefore, it can be used as a non-surgical medicine for stopping early pregnancy. It can also be used, inter alia, in contraception and as an antineoplastic. (The Antiprogestin Steroid Ru486 and Human Fertility Control, 1985, New York: Plenum Press) .
-
Onapristone (11β-[4-(N,N-diemthylamino)phenyl]-17α-hydroxy-17β-(3-hydroxypropyl)-13α-4,9-estradiene-3-one), is a steroid compound which is disclosed in German Patent No. 3,321,826 to Schering AG, published Dec. 20, 1984. It has a strong antiprogestin activity and can be used in abortion (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), anticancer (Breast Cancer Research and Treatment, 1989, 14:275-288), etc. It was reported that onapristone had toxicity to human liver (European Journal of Cancer, 1999, 35(2):214-218).
-
Lilopristone (11β-[4-(N,N-dimethylamino) phenyl]-17α-[3-hydroxy-1(Z)-propenyl]-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in German Patent No. 3,347,126 to Schering AG, published July 11, 1985. It has a strong antiprogestin activity and can be used in abortion, contraception (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), etc. It was reported that the clinical effect of lilopristone in stopping early pregnancy was only equivalent to that of mifepristone (Human Reproduction, 1994, 9(1):57-63).
-
ZK112993 (11β-(4-acetylphenyl)-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is as steroid compound which is disclosed in German Patent No. 3,504,421 to Schering AG, published Aug. 7, 1986. It has a potent antiprogestin activity and can be used in, inter alia, anticancer (Anticancer Res., 1990, 10:683-688).
-
In European Patent No. 321,010 to Akzo NV, The Netherland published June 21, 1989 are disclosed “11-arylsteroid compounds” having a strong antiprogestin activity.
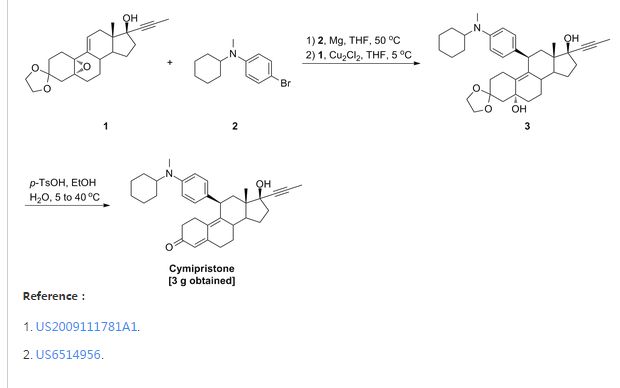
PATENT
WO 2001018026
http://www.google.com/patents/EP1219632A1?cl=en

The preparation method of the present invention includes the following single- or multi-step procedures:
1. Method for the preparation of 11β-[4-(N-methyl-N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (IV) which includes the following steps:
(1) Preparation of Grignard reagent (III)

4-bromo-N-methyl-N-cyclohexylaniline (II) is reacted with magnesium in tetrahydrofuran (THF) to obtain Grignard reagent of formula (III).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (III) prepared in step (1) are brought to an additive reaction to obtain compound of formula (V).
(3) Hydrolytic reaction

The compound of formula (V) prepared in step (2) is subjected to a hydrolytic reaction to obtain compound of form (VI).
2. Method for preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI) which includes the following steps:
(1) Preparation of Grignard reagent of formula (IX)

4-bromo-N-cyclohexylaniline (VII) is first protected by trimethylchlorosilane, then reacted with magnesium in THF to obtain Grignard reagent of formula (IX).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (IX) prepared in step (1) are brought to an additive reaction to obtain compound of formula (X).
(3) Hydrolytic reaction

The compound of formula (X) prepared in step (2) is subjects to a hydrolytic reaction to obtain compound of formula (XI).
Example 2:
-

-
9g 4-bromo-N-cyclohexylaniline (VII) (CA registration number [113388-04-8], see Synthetic Communications, 1986, 16(13): 1641-1645 for its preparation) was placed into a four-necked flask and 15 ml (1.5 mol/L) n-BuLi solution in n-hexane. The mixture was stirred for 30 min at room temperature. Then 8 g trimethylsilyl chloride (Me3SiCl) was added and the mixture was stirred for 1 hour. Solvent and excessive Me3SiCl was evaporated under reduced pressure to yield 4-bromo-(N-cyclohexyl-N-trimethylsilylaniline) (VIII) which was formulated into a solution with 7.5 ml anhydrous tetrahydrofuran for further use.
-
1.3 g magnesium was placed into a four-necked flask and a small amount of the above solution was added dropwise and slowly at 40°C. After completion of addition, the temperature was kept for 1 hour to yield a solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenylmagnesium bromide (IX) in tetrahydrofuran for further use.
- Preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI)(1) Preparation of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX)

(2) Preparation of 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cylohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).

- 5g 3,3-ethylenedioxy-5,10-epoxy-17α-(1-propinyl)-17β-hydroxy-9(11)-estrene (IV) was placed into a four-necked flask and 10 ml anhydrous tetrahydrofuran and a catalytic amount of cuprous chloride (Cu2Cl2) added. Then solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX) in tetrahydrofuran was added dropwise and slowly while controlling the temperature below 5°C. After completion of addition, the mixture was allowed to react for 2 hours at room temperature and to stand overnight. Saturated ammonium chloride aqueous solution was added and the tetrahydrofuran layer separated which was washed with saturated ammonium chloride solution. The solution in tetrahydrofuran was washed with saturated saline and dried over anhydrous sodium sulfate. Evaporation of tetrahydrofuran under reduced pressure yielded a residual which was chromatographed on silica gel column using cyclohexane: acetone (5:1) as developing agent to yield 3 g 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).
-
IR (KBr) cm-1: 3420 (C5, C17-OH), 1610, 1510 (benzene backbone), 840, 808 (ArH).
1H NMR (CDCl3) δ ppm: 0.52(3H, S, C13-CH3), 2.72(3H, S, N-CH3), 3.92(4H, m, -O-CH2CH2-O-), 4.24(1H, m, C11-H), 6.65-7.00 (4H, ArH).
(3) Preparation of 11β- [4- (N-cyclohexylamino)phenyl] -17α- (1-propinyl) -17β-hydroxy-4,9-estradiene-3-one (XI).

- 1.5g 3,3-ethylenedioxy-5,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene (X) and 0.75 g para-toluenesulfonic acid (PTS) were dissolved in 15 ml 90 % ethanol (v/v). The mixture was stirred for 2 hours while controlling the temperature at 40°C-50°C. After completion of the reaction, the reactant was poured into diluted sodium hydroxide aqueous solution, extracted with dichloroethane, washed with water to neutrality, and dried over anhydrous sodium sulfate. Evaporation of the solvent and chromatography on silica gel column using cyclohexane: ethyl acetate (5:1) as developing agent yielded 0.9 g 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI).
-
IR (KBr) cm-1: 3400 (C17-OH), 1658 (unsaturated ketone), 1613, 1514 (benzene backbone), 865, 810 (ArH).
1H NMR (CDCl3) δ ppm: 0.50 (3H, S, C13-CH3), 1.76 (3H, S, C≡C-CH3), 4.32(1H, S, C11-H), 5.75(1H, S, C4-H), 6.9-7.10 (4H, ArH).
PATENT
PATENT
Example 1
Race meters mifepristone synthetic routes:

Epoxy adduct match rice mifepristone
(N- hexylamino methylcyclohexyl) phenyl magnesium bromide (1) 4-
In the four-necked flask, 1.4 g of magnesium into pieces (Mg) and 10 ml of anhydrous tetrahydrofuran (THF), no iodine or add a little change, at about 50 ° C, a solution of 10.86 g of 4-bromo-methyl -N- cyclohexyl aniline (dissolved in 24 ml of anhydrous tetrahydrofuran) dropwise Bi, incubation was continued for 1 hour with stirring to give 4- (N- methyl-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran solution (to be used in the next step an addition reaction ).
(2) 3,3-ethylenedioxy -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propyl block-yl) -9 (10) – Preparation of estra-ene (adduct) of
In the four-necked flask, into 5 g of 3,3-ethylenedioxy-5,10-epoxy -17 α – (1- propynyl) – 17 (3 – hydroxy – 9 (11) – estra-ene (epoxy), 29.1 ml anhydrous tetrahydrofuran (THF) and 0.1 g cuprous chloride (of Cu 2 of Cl 2 ), a solution of 4- (N- methyl -N-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran
Nan solution, temperature control 5. C, the drop was completed, the incubation was continued for 5 hours, the reaction was completed, the reaction solution was poured into saturated aqueous ammonium chloride solution, points to the water layer, the organic layer was washed with saturated ammonium chloride solution, the aqueous layer extracted with ethyl acetate number times, the organic layers combined, washed with saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, and concentrated under reduced pressure, a silica gel column, eluent cyclohexane: acetone = (5: 1) to give 3,3-ethylene dioxo -5 α, 17 β – dihydroxy -11 β – [4- (- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – female steroidal women (adduct) solid 6 grams.
IR. ‘KBi cm- ^ SlS OI ^ ^ -OH lS jSlS benzene backbone), 819 (aromatic hydrogen). NMR Ή: (CDC1 3 ) ppm by [delta]: 0.47 (3H, the S, the C IR CH 3 ), 1.88 (3H, the S, the C ≡ the C-CH 3 ), 2.72 (3H, the S, the N-CH 3 ), 6.65- 7.03 (4H, ArH) O
(3) 11 β – [4- (N- methyl -N- cyclohexylamino) phenyl] -17 α – (1- propynyl) -17 β – hydroxy-estra-4,9-diene – Preparation of 3-one (match rice mifepristone) of
‘2.5 g of p-toluenesulfonic acid (PTS) and 5 grams of 3,3-ethylenedioxythiophene -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – estra-ene (adduct) was dissolved in 50 ml of ethanol 90% (V / V), and at 5 ° C – 40 ° C the reaction was stirred 3 hours, the reaction solution was poured into dilute aqueous sodium hydroxide solution, the precipitated solid was suction filtered, washed with water until neutral, the filter cake was dissolved in 50 ml of ethyl acetate, then with saturated aqueous sodium chloride solution to the water layer was evaporated part of the solvent, the precipitated solid was suction filtered, and dried to give a pale yellow solid 11 β – [4- (Ν- -N- methyl-cyclohexylamino)] -17 α – (1- propynyl) -17 β – hydroxy estra-4,9-dien-3-one (match rice mifepristone) 3 grams.
^ Cm & lt IRCKB 1 : 3447 (the C . 17 -OH), among 1655 (unsaturated ketone), 1607,1513 (benzene backbone), 865,819 (aromatic hydrogen).
NMR ¾: (CDC1 3 ) ppm by [delta]: 0.56 (3H, the S 5 the C 13 -CH 3 ), 1.89 (3H, the S 5 -C ≡ the C-the CH3), 2.74 (3H, the S, the N-the CH3), 4.34 ( lH, the S, the C N -H), 5.75 (lH, the S, the C 4 -H), 6.68-6.99 (4H, ArH).
PATENT
PATENT
PAPER
Volume 878, Issues 7–8, 1 March 2010, Pages 719–723
Determination of cymipristone in human plasma by liquid chromatography–electrospray ionization-tandem mass spectrometry
doi:10.1016/j.jchromb.2010.01.027
Abstract
A rapid, specific and sensitive liquid chromatography–electrospray ionization-tandem mass spectrometry method was developed and validated for determination of cymipristone in human plasma. Mifepristone was used as the internal standard (IS). Plasma samples were deproteinized using methanol. The compounds were separated on a ZORBAX SB C18 column (50 mm × 2.1 mm i.d., dp 1.8 μm) with gradient elution at a flow-rate of 0.3 ml/min. The mobile phase consisted of 10 mM ammonium acetate and acetonitrile. The detection was performed on a triple-quadruple tandem mass spectrometer by selective reaction monitoring (SRM) mode via electrospray ionization. Target ions were monitored at [M+H]+m/z 498 → 416 and 430 → 372 in positive electrospray ionization (ESI) mode for cymipristone and IS, respectively. Linearity was established for the range of concentrations 0.5–100 ng/ml with a coefficient correlation (r) of 0.9996. The lower limit of quantification (LLOQ) was identifiable and reproducible at 0.5 ng/ml. The validated method was successfully applied to study the pharmacokinetics of cymipristone in healthy Chinese female subjects.
CHEMICAL ABSTRACTS, vol. 115, no. 25, 23 December 1991 (1991-12-23) Columbus, Ohio, US; abstract no. 270851g, X. ZHAO ET AL.: “Synthesis and terminating early pregnancy effect of mifepristone derivatives” page 117; XP002219009 & ZHONGGUO YAOKE DAXUE XUEBAO, vol. 22, no. 3, 1991, pages 133-136,
//////////Cymipristone, Saimisitong, NDA Filed , china, Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
Idarucizumab

![]()
Idarucizumab
(Praxbind®) Approved
An antidote for rapid reversal of dabigatran-induced anticoagulation indicated for emergency surgery (urgent procedures) and life-threatening or uncontrolled bleeding in patients treated with dabigatran.
![]()
BI-655075
CAS No.1362509-93-0
Other Names
- BI 655075
- Idarucizumab
- Praxbind
Protein Sequence
Sequence Length: 444, 225, 219multichain; modified (modifications unspecified)
Idarucizumab, sold under the brand name Praxbind, is a monoclonal antibody designed for the reversal of anticoagulant effects ofdabigatran.[1][2]
This drug was developed by Boehringer Ingelheim Pharmaceuticals. A large study sponsored by the manufacturer found that idarucizumab effectively reversed anticoagulation by dabigatran within minutes.[3] It was FDA approved in October 2015.[4] In the United States the wholesale cost is $3500 US.[5]
References
- Statement On A Nonproprietary Name Adopted By The USAN Council – Idarucizumab, American Medical Association.
- World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 109” (PDF). WHO Drug Information 27 (2).
- Pollack, Charles V.; Reilly, Paul A.; Eikelboom, John; Glund, Stephan; Verhamme, Peter; Bernstein, Richard A.; Dubiel, Robert; Huisman, Menno V.; Hylek, Elaine M. (2015-08-06).“Idarucizumab for Dabigatran Reversal”. The New England Journal of Medicine 373 (6): 511–520. doi:10.1056/NEJMoa1502000. ISSN 1533-4406. PMID 26095746.
- “Press Announcements – FDA approves Praxbind, the first reversal agent for the anticoagulant Pradaxa”. http://www.fda.gov. Retrieved 2015-10-17.
- Elia, Joe. “Dabigatran-Reversal Agent Price Set”. Retrieved 20 October 2015.
| Monoclonal antibody | |
|---|---|
| Type | Fab fragment |
| Source | Humanized (from mouse) |
| Target | Dabigatran |
| Clinical data | |
| Trade names | Praxbind |
| Identifiers | |
| CAS Number | 1362509-93-0 |
| ATC code | V03AB37 (WHO) |
| IUPHAR/BPS | 8298 |
| ChemSpider | none |
| Chemical data | |
| Formula | C2131H3299N555O671S11 |
| Molar mass | 47.8 kg/mol |
/////Idarucizumab
Istradefylline

Istradefylline, KW-6002
(Nouriast®) Approved
A selective adenosine A2A receptor antagonist used to treat Parkinson’s disease.

KW-6002
CAS No. 155270-99-8
Istradefylline; 155270-99-8; KW-6002; KW 6002; 8-[(E)-2-(3,4-Dimethoxyphenyl)ethenyl]-1,3-diethyl-7-methyl-purine-2,6 -dione; (E)-8-(3,4-Dimethoxystyryl)-1,3-diethyl-7-methyl-1H-purine-2,6(3H,7H)-dione;
| Molecular Formula: | C20H24N4O4 |
|---|---|
| Molecular Weight: | 384.42896 g/mol |
Istradefylline (KW-6002) is a selective antagonist at the A2A receptor. It has been found to be useful in the treatment of Parkinson’s disease.[1] Istradefylline reduces dyskinesia resulting from long-term treatment with classical antiparkinson drugs such as levodopa. Istradefylline is an analog of caffeine.
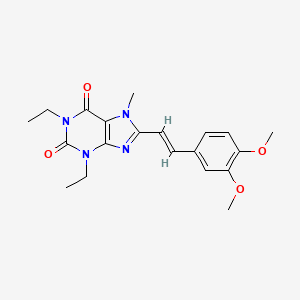
Kyowa Hakko Kirin is developing istradefylline, a selective adenosine A2A receptor antagonist, for the once-daily oral treatment of Parkinson’s disease (PD). Adenosine A2A receptors are considered to be present particularly in the basal ganglia of the brain; the degeneration or abnormality observed in PD is believed to occur in the basal ganglia, which is recognized to play a significant role in motor control.
Commercially available dopamine replacement therapies effectively treat the early motor symptoms of PD; however, these agents are associated with development of motor complications, limiting usefulness in late stages of the disease. Istradefylline is proposed to possess a clearly distinct action site from existing agents which act on dopamine metabolism or dopamine receptors. Kyowa Hakko Kirin has received approval for istradefylline in the adjunctive treatment of PD in Japan. A New Drug Application was filed in the USA, but the FDA issued a non-approvable letter in February 2008.
PATENT
US5484920A
http://www.google.co.in/patents/US5484920
PAPER
http://www.sciencedirect.com/science/article/pii/S0960894X13003983

Scheme 1.
Synthesis of KW 6002 (2). Reagents and conditions: (i) acetic anhydride, 80 °C, 2 h, 83%; (ii) sodium nitrite, 50% acetic acid, 60 °C, 15 min, 86%; (iii) sodium dithionite, NH4OH solution (12.5% (w/v)), 60 °C, 30 min, 98%; (iv) SOCl2, toluene, 75 °C, 2 h, 97%; (v) pyridine, DCM, rt, 16 h, 66%; (vi) HMDS, cat. (NH4)2SO4, CH3CN, 160 °C, microwave, 5 h, 100% followed by (vii) MeI, K2CO3, DMF, rt, 2 h, 75%.

Synthesis
(E)-8-(3,4-Dimethoxystyryl)-1,3-diethyl-7-methyl-1H-purine-2,6(3H,7H)-dione (2)3
- J. Hockemeyer; J. C. Burbiel; C. E. Müller, J. Org. Chem. 2004, 69, 3308.
(E)-8-(3,4-Dimethoxystyryl)-1,3-diethyl-1H-purine-2,6(3H,7H)-dione (1.11 g, 3.00 mmol) was taken up in dimethylformamide (15 mL) and potassium carbonate (828 mg, 6.00 mmol). To the milky white mixture was added iodomethane (468 µL, 7.50 mmol) and it was allowed to stir at room temperature for 2 h. The mixture was then filtered and washed with water (100 mL), leaving the title compound 2 as a pale yellow solid which was dried in the oven at 110 °C (863 mg, 75%), mp: 192 °C (lit.3 191 °C). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 15.7 Hz, 1H), 7.18 (dd, J = 8.4, 1.9 Hz, 1H), 7.09 (d, J = 1.9 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 15.7 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 4.12 – 4.04 (m, 5H), 3.95 (s, 3H), 3.93 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H), 1.26 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 155.0 (C), 150.8 (C), 150.4 (C), 150.3 (C), 149.2 (C), 148.2 (C), 138.1 (CH), 128.6 (C), 121.2 (CH), 111.2 (CH), 109.5 (CH), 109.3 (CH), 108.0 (C), 55.98 (CH3), 55.97 (CH3), 38.4 (CH2), 36.3 (CH2), 31.5 (CH3), 13.43 (CH3), 13.39 (CH3). LCMS: m/z (ESI 20 V) 385.2 (MH+, 100).
PATENT
http://www.google.com/patents/CN103254194A?cl=en
Specific synthetic route is as follows:

the above reaction is a synthetic Parkinson’s disease clinical drug KW-6002 against a yield of 83%.
Example 26 (a new synthetic method for anti-Parkinson’s disease in clinical drug KW-6002):
In addition to use in place of 3,4-dimethoxy-styryl boronic acid (0.4mmol, i.e., in formula IV, R5 is 3,4_-dimethoxy-styryl) benzene boronic acid in Example 23 and 1,3 – two-ethyl-8-phenylthio-9-methyl-xanthine (0.4mmol, i.e., Formula I, R1 is methyl, R2 and R3 are ethyl, R4 is a phenyl group) in place of Example 23 in 1 , 3,9-trimethyl xanthine -8- phenylthio, the remaining steps in Example 23 to give a white solid, yield 83%, mp = 101~103 ° C I1H NMR (⑶CI3, 600MHz): δ 7.71 (d, J = 15.6Hz, 1H), 7.17 (dd, J = 8.2,1.9Hz, 1H), 7.07 (d, J = L 9Hz, 1H), 6
• 88 (d, J = 8.2Hz, 1H), 6.74 (d, J = 15.8Hz, 1H), 4.19 (q, J = 7Hz, 2H), 4.07 (q, J = 7Hz, 2H), 4.03 (s , 3H), 3.93 (s, 3H), 3.90 (s, 3H), 1.36 (t, J = 7Hz, 3H), 1.23 (t, J = 7Hz, 3H); 13C NMR (150MHz, CDCl3): 155.1, 150.8,150.4,150.2,149.2,148.2,138.2,128.6,121.2, 111.2,109.5,109.3,108.0,56.0,55.9,38.4,36.3,31.5,13.4,13.4; HRMS: calcd for C20H25N4O4 (M + H) +385.187
6, Found385.1879. It indicates that the white solid was 8- (3,4-dimethoxy-styryl) structural formula shown KW-6002 (E) -1,3_ diethyl-7-methylxanthine.

In contrast, KW-6002 is a new drug to treat Parkinson’s disease developed by Kyowa Hakko in Japan, Japan and the United States is currently the second phase of clinical trials. Literature (. J.Hockemeyer, JCBurbiel andC.E.Muller, J.0rg.Chem, 2004,69,3308) through the following synthetic route:

The synthetic route requires five steps, with a total yield of 33%, and there is the use of environmentally unfriendly halogenated solvent methylene chloride, the reaction requires high pressure high temperature (170~180 ° C) and other shortcomings. By comparison, the present invention starting from 8- phenylthio xanthine coupling reaction catalyzed by palladium simple, a yield of 83% was synthesized KW6002, it is currently the most efficient synthesis route KW-6002’s. In particular, the multi-step synthesis route to avoid the complex operation of the reactor, but under relatively mild conditions (60 ° C) conduct, simple operation, suitable for scale synthesis.
PATENT
http://www.google.com/patents/CN104744464A?cl=en
itraconazole theophylline (Istradefylline, KW6002), the chemical name 8 – [(E) -2- (3, 4- dimethoxyphenyl) ethenyl] -1,3-diethyl -7 – methyl-purine-2,6-dione, CAS number: 155270-99-8, structural formula shown below.

itraconazole Theophylline is a selective adenosine A2a receptor antagonist, by changing the activity of neurons in Parkinson’s disease patients to improve motor function, for the treatment of Parkinson’s disease and Parkinson’s disease improve early dyskinesia.
The invention and JPH0940652A European Patent 0,590,919 discloses a method for preparing itraconazole and theophylline. WO 2004/099207 published good solubility stability of a particle size of less than 50 micrometers 8 – [(E) -2- (3, 4- dimethoxyphenyl) ethenyl] -1,3- diethyl-7-methyl-purine-2,6-dione crystallites.
Example 1 Preparation of theophylline itraconazole Example

ships equipped with a mechanical stirrer, a thermometer, a 2L 4-neck flask was added 30g8 – [(E) -2- (3, 4- dimethoxyphenyl) ethenyl] -1,3-diethyl- -7- hydrogen – purine-2,6-dione (Intermediate A), 400mL N, N- dimethylformamide and 15g of potassium carbonate, and 25g of methyl iodide and heated to 80 ° C after the reaction was stirred 8h, added 200mL water, cooled to room temperature, and stirring was continued crystallization 2h. The resulting suspension was suction filtered, washed with water after the cake was 800mL sash, 50 ° C under blast drying 24h, 32g give a pale yellow solid, for each polymorph of itraconazole theophylline preparation example the following examples.
References
- Peter A. LeWitt, MD, M. Guttman, James W. Tetrud, MD, Paul J. Tuite, MD, Akihisa Mori, PhD, Philip Chaikin, PharmD, MD, Neil M. Sussman, MD (2008). “Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces off time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005)”. Annals of Neurology 63 (3): 295–302. doi:10.1002/ana.21315. PMID 18306243.
Reference:1. EP0590919A1.
2. US5484920A.
3. US5543415A.
4. J. Org. Chem. 2004, 69, 3308-3318.
5. Bioorg. Med. Chem. Lett. 1997, 7, 2349-2352.
6. Bioorgan. Med. Chem. 2003, 11, 1299-1310.
7. Bioorg. Med. Chem. Lett. 2013, 23, 3427-3433.
8. Chinese Journal of Pharmaceuticals 2010, 41, 241-243.
9. JP0940652A.
10. Org. Biomo. Chem. 2010, 8, 4155-4157.
1. Chem. Commun. 2012, 48, 2864-2866.
2. CN103254194A.
| CN104744464A * | Nov 15, 2013 | Jul 1, 2015 | 南京华威医药科技开发有限公司 | Istradefylline crystal forms |
-
Istradefylline Systematic (IUPAC) name 8-[(E)-2-(3,4-dimethoxyphenyl)vinyl]-1,3-diethyl-7-methyl-3,7-dihydro-1H-purine-2,6-dioneIdentifiers CAS Number 155270-99-8 
ATC code none PubChem CID 5311037 IUPHAR/BPS 5608 ChemSpider 4470574 UNII 2GZ0LIK7T4 KEGG D04641 ChEMBL CHEMBL431770 Chemical data Formula C20H24N4O4 Molar mass 384.429 g/mol

//////Istradefylline, KW-6002, Nouriast®, Approved, A selective adenosine A2A receptor antagonist, Parkinson’s disease,
O=C2N(c1nc(n(c1C(=O)N2CC)C)\C=C\c3ccc(OC)c(OC)c3)CC
GMP Oversight of Medicines Manufacturers in the European Union
DRUG REGULATORY AFFAIRS INTERNATIONAL

A System of Equivalent Member States, a Coordinating Agency and a Centralized Institution
The regulatory system for supervision of pharmaceutical manufacturers and GMP inspection in the European Union is one of the most advanced in the world. Due to the globalization of pharmaceutical manufacture, it also affects industry, regulators and patients outside the European Union. This system, however, is often poorly understood beyond the EU borders.
What follows is an explanation of the EU system in order to increase awareness and facilitate cooperation on GMP between European Union regulators and those outside the European Union.

The European Union
The European Union includes 28 Member States located in Europe, which are: Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxemburg, Malta, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, Sweden, and United Kingdom. The EU total population is about 500 million people.
View original post 4,770 more words
GMP/GDP: When will I be inspected by the Authorities?
DRUG REGULATORY AFFAIRS INTERNATIONAL

Various competent authorities are performing inspections. But who is subject to such an inspection?
GMP Inspections are carried out at Manufacturer Licence Holders
A manufacturer of medicinal products must meet Good Manufacturing Practice (GMP) standards. These standards are defined in various laws and regulations. In the EU the compliance with these regulations is checked and assessed by the national competent authorities. The overall goal is to have medicinal products of consistent high quality that meet the requirements of the marketing authorisation (MA) or product specification.
If a company supplies product to the USA, the U.S. Food and Drug Administration (FDA) might inspect the site assuring that drugs, medical devices, certain active pharmaceutical ingredients (APIs) and biological products manufactured in foreign countries and intended for U.S. distribution are in compliance with the applicable U.S. law and regulations.
GDP Inspections are carried out at Wholesale Dealer Licence Holders
Good Distribution Practice…
View original post 312 more words
1R,2S-Methoxamine

1R,2S-methoxamine, also known as L-erythro-methoxamine
CAS 13699-29-1
- Molecular Weight, 211.26, C11 H17 N O3
HYDROCHLORIDE
(1R,2S)-isomer HCl salt of 1 -(2,5-dimethoxyphenyl)-2-amino-1 -propanol also called as (1R, 2S)methoxamine hydrochloride
CAS 16122-04-6
Used as a pressor agent, as a vasoconstrictor, as a nasal decongestant, in ophthalmology and also found very effective in the treatment of faecal incontinence.
treatment of relief of fecal incontinence and anal itch (pruritis ani) , particularly for patients who have had a major bowel resection and reanastomosis .
Anal or fecal incontinence is the inability to voluntarily control the passage of feces or gas through the anus. It may occur either as fecal soiling or as rare episodes of incontinence for gas or watery stools. It is a very distressing condition that can result in self-inflicted social isolation and despair.
Conventional treatments for fecal incontinence include drug therapy to improve stool consistency, such as morphine, loperamide and codeine phosphate to reduce gut motility, and laxatives to soften stools and relieve constipation. Biofeedback training is another treatment which involves muscle strengthening exercises to improve anal canal resting pressure, and squeeze pressure, and to teach symmetry of anal canal function. The most common form of treatment however, is surgical repair, such as the creation of a neo-sphincter which involves grafting on muscle from other parts of the anus, or a colostomy. (Gastroenterology in Practice, Summer 1995, pl8- 21; Dig Dis 1990; 8:179-188; and The New England Journal of Medicine, April 1992, pl002-1004) . In mild cases of anal leakage, the patient will often try and plug the anus with a ball of cotton wall.
In Gut, 1991, 32, p.345-346 it was reported that two thirds of patients with idiopathic faecal incontinence had a decreased anal resting pressure resulting from an abnormal internal sphincter function. In many incontinent patients, the internal anal sphincter was found to be abnormally thin, while others had an external anal sphincter defect. It has also been reported that in vi tro contractile response of the internal anal sphincter to noradrenaline is decreased in incontinence, (Br. J. Surg. 1992, vol 79, August, p829-832; Digestive Diseases and Sciences, vol 38, no. 11, Nov. 1993, pl961-1969) . A further discussion of the innervation and control of the internal anal sphincter and drugs which can increase or decrease the normal anal resting pressure, is discussed in the text book Coloproctology and the Pelvic Floor (Butterworths) , second edition, 1992, at chapter 3 p37-53; Automic Control of Internal Anal Sphincter; and Journal of Clinical Investigation 1990, 86: p424-429.
In Surgery 1990; 107: p311-315 sodium valproate was found to be useful in the treatment of minor incontinence after ileoanal anastomosis.
It has now surprisingly been found that fecal incontinence and anal itch can be resolved by treatment with α adrenergic agonists, nitric oxide synthase inhibitors, prostaglandins F2α, dopamine, morphine, β-blockers such as propranolol, and 5-Hydroxytryptamine (5-HT) .
This is surprising since it was always thought that once an anal sphincter began functioning abnormally, the patient would require major surgery.
In this way the anal leakage is reduced or eliminated without the patient having to undergo major surgery.
Accordingly in a first aspect of the invention there is provided use of a physiologically active agent selected from an α adrenergic agonist, nitric oxide synthase inhibitor, prostaglandin F2α, dopamine, morphine, β-blockers, and 5- Hydroxytryptamine in the preparation of a medicament for the treatment or prophylaxis of fecal incontinence or anal itch.
The agents of the invention appear to at least partially treat the incontinence by increasing the resting pressure of the internal anal sphincter. Preferred agents are λ adrenergic agonists, nitric oxide synthase inhibitors, and prostaglandins F2α.
Examples of suitable aλ adrenergic agonists are nor- adrenalin, methoxamine, but particularly preferred is phenylephrine .
Examples of suitable F2α prostaglandin are dinoprost and carboprost.
Examples of suitable NO synthase inhibitors are
NG-monnoommeetthhyyll–LL–aarrggiinn:ine (L-NMMA) , and NG-nitro-L-arginine methyl ester ( -NAME)
The medicament can contain a single active agent or a combination of any of the above active agents.
Nitric Oxide (NO) synthase inhibitors such as LNMMA have previously been suggested for the therapeutic treatment of septic shock.
The prostaglandins, along with thromboxanes and leukotrienes are all derived from 20 -carbon polyunsaturated fatty acids and are collectively termed eicosanoids. F2α prostaglandins are derived in vivo from the endoperoxide prostaglandin H2which is in turn derived from leukotrienes. Clinically, F2α prostaglandins such as dinoprost and carboprost are used as uterine stimulants in the termination of pregnancy, missed abortion or the induction of labour.
Phenylephrine (an αx adrenergic agonist) is used as a mydriatic in ophthalmology, and as a decongestant , for example, in cold and flu remedies.
However there has been no suggestion to the inventors knowledge of using any of these active agents to treat fecal incontinence or anal itch. As used herein “fecal incontinence” includes all types of anal leakage from minor leakage or ‘spotting’ through moderate leakage, to major instances of faecal incontinence, and includes neurogenic, active, urge and passive incontinence.
More particularly the class of incontinent patients who will benefit most from the present invention are those with idiopathic incontinence and those whose incontinence is at least partly due to a weakness of either the internal or external anal sphincter, especially those with a normal or low maximum anal pressure and a structurally intact internal anal sphincter muscle, such as with an abnormally thin sphincter. However patients with minor structural damage such as a fragmented sphincter would still benefit from the invention. Not only incontinent patients with a damaged or abnormal internal sphincter can be treated, but also patients with a damaged or abnormal external sphincter since the increase in the internal anal resting tone induced by the invention will compensate for a poorly functioning external sphincter.
Another class of patients who particularly benefit from the invention are post-surgical patients who have had major bowel resection and reanastomosis . For example patients with ileoanal pouch (restorative proctocolectomy) , coloanal (with or without colonic pouch) anostomosis, lower anterior resection, and colectomy with ileorectal anastomosis.
The damage to the sphincter could be caused by trauma, such as experienced in child birth, surgical operations, or road traffic accidents. Furthermore it is also believed that incontinence caused by primary internal anal degeneration can also be relieved by the invention.
Anal leakage also often leads to pruritis of the anus and therefore by reducing or eliminating the leakage, the pruritis or anal itch is also relieved or prevented. Furthermore, as a result of the increased anal resting pressure, the patient no longer has the discomfort of distended anal sphincter muscles.
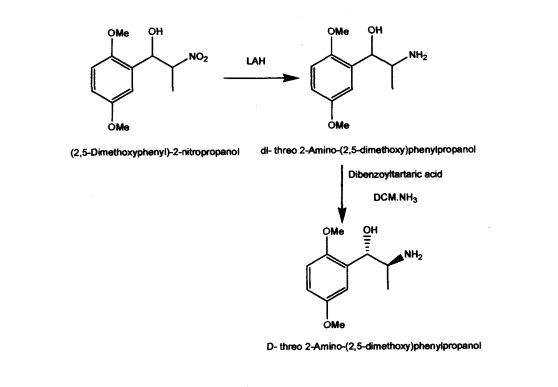
Methoxamine contains two chiral carbons and thus exists in four isomeric forms. Of all the isomeric forms, the studies revealed (1R,2S)- isomer to be therapeutically active.
US patent 2359707 describes the process for the synthesis of racemic β-(2,5-dimethoxy phenyl)-P-hydroxy-isopropyl amine in neutral, acid salt and its derivative from 2,5- dimethoxy propiophenone by treatment with methylnitrite in diethyl ether medium to obtain 2,5-dimethoxy-a-isonitrosopropiophenone hydrochloride. It is further reduced with palladium on carbon to yield β-(2,5-dimethoxyphenyl)-p-ketoisopropylamine hydrochloride and then with platinum black to get p-(2,5-dimethoxyphenyl)-β- hydroxyisopropyl amine hydrochloride. The described process for di-methoxamine HC1 is not cost-effective, due to the use of two expensive catalysts (platinum black and palladium carbon), solvent diethyl ether and involves more number of steps. The other drawback being it is racemic mixture and cannot be used directly as drug. The process described did not specify the quality of the product.
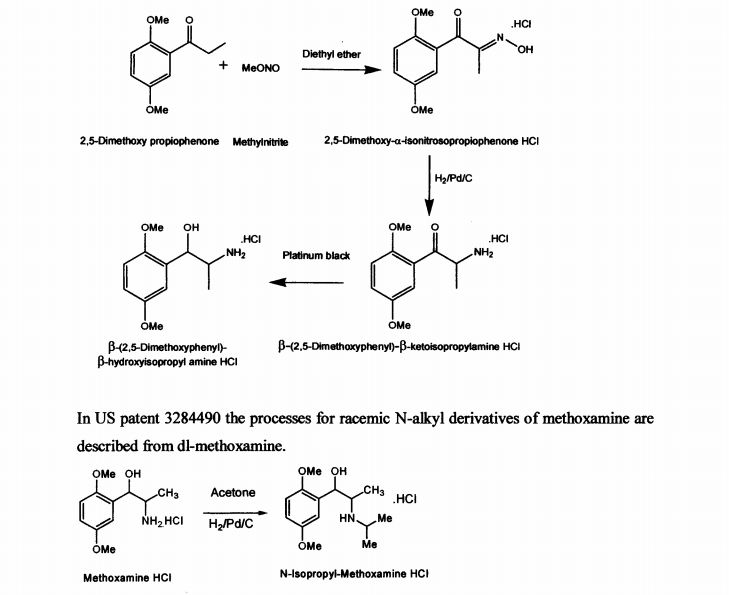
In US patent 3284490 the processes for racemic N-alkyl derivatives of methoxamine are described from dl-methoxamine.
JP 63165348 describes process for production of optically active l-(2,5- dimethoxyphenyl)-2-aminophenol by resolving racemic compound with the use of optically active L-N-acetylleucine as resolving agent. The disadvantages of the process are less yield, low quality and use of expensive naturally occurring amino acid, which prevents from employing this method on commercial scale.
WO 03/055474 A1 discloses mainly, the use of (1R, 2S)-methoxamine in the treatment of faecal incontinence at low doses without local or systemic side effects when used topically. The patent also described the synthesis of (1R, 2S)-methoxamine, from L- alanine, by protecting the amino group using methylchloroformate, converting carboxy
group of the N-protected alanine into an acid chloride insitu followed by reaction with an amine to produce an N-protected (S)-alanine amide and coupling that compound with a brominated 2,5-dimethoxybenzene in the presence of n-butyllithium or a magnesium based reagent to give (S)-amino-l-(2,5-dimethoxy-phenyl)-l-propanone, the amino group of which is protected .The reduction of the N-protected propanone was carried out using dimethylphenylsilane and the protecting group was removed by treatment with potassium hydroxide. Other method adopted in the patent to isolate (1R,2S)methoxamine is by separation of racemic methoxamine using chiral column.
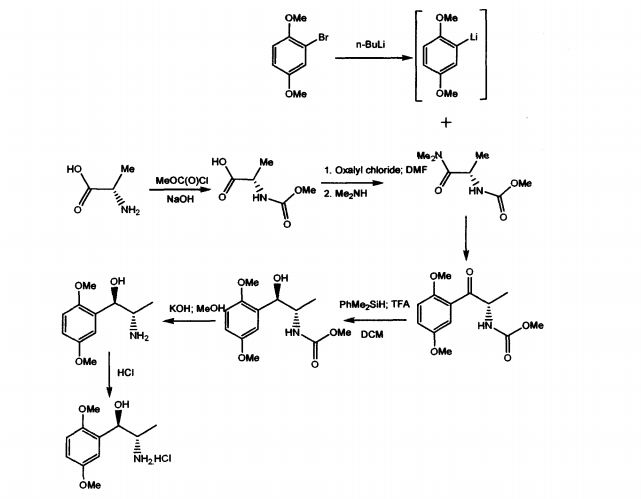
The prior art suffers with some of the disadvantages like using n-butyllithium, which is pyrophoric, expensive and causes hazards to commercial scale. Also, the separation of racemic Methoxamine using chiral column mentioned in the patent can be considered for
isolating small quantities of the required isomer for analytical purposes but cannot be adopted on commercial scale for production of the drug.
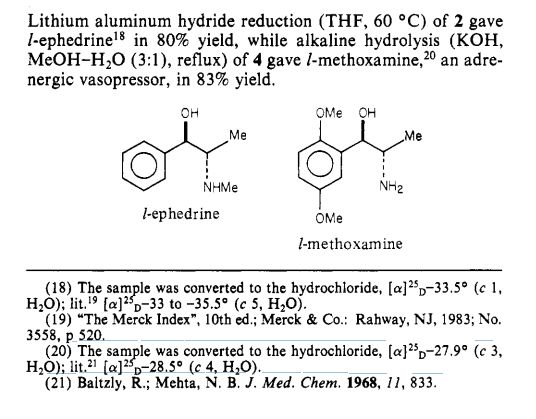
US Patent 5962737 described stereospecific synthesis of the racemic threo isomers of 2- nitro-1 -phenylpropanols by reacting benzaldehyde derivative with nitroalkane in the presence of a tertiary amine and reducing 2-nitro-l-phenylpropanols with lithium aluminium hydride to 2-amino-l-phenylpropanols. Also described is phase transfer resolution of racemic mixtures of 2-amino-l-phenylpropanol and its derivatives into their optically pure isomers by reacting with the mono alkali metal salt of tartaric acid ester in a two phase system of a hydrocarbon and water. The specification further describes optically pure isomer D-threo 2-amino-( 1 -dialkoxy or alkoxy)phenylpropanol by resolution of dl- threo 2-amino-( 1 -dialkoxy or alkoxy)phenylpropanol by using dibenzoyltartaric acid. The synthesis of the product (lS,2S)-threo 2-amino-(l-dialkoxy or alkoxy) phenyl propanol involves the use of expensive and hazardous chemicals like LAH making the process technically and commercially difficult for implementation.
Paper
Journal of the American Chemical Society (1984), 106(16), 4629-30
http://pubs.acs.org/doi/pdf/10.1021/ja00328a062
PATENT
http://www.google.com/patents/EP2275099A1?cl=en
EXAMPLE 3Synthesis of 1R,2S-Methoxamine(S)-N-Methoxycarbonyl alanine
To a stirred solution of L-alanine (300g, 3.37 mol sodium hydroxide (1N, 1800 cm3) at 0°C in an ice bath was added dropwise, over 2 hours, methyl chloroformate (274 cm3, 3.54 mol). The pH of the solution was maintained at 9 by the addition of sodium hydroxide (5N). The reaction mixture was stirred at 0°C for 3 hours whereupon it was acidified to pH 1 by the addition of phosphoric acid solution (15%) and extracted with diethyl ether (5 x 1000 cm3). The combined organic extracts were dried (MgSO4) and concentrated under reduced pressure to yield the product as a viscous green oil (386 g, 78%). 1H NMR (250 MHz; C2HCl3) 1.48 (3H, d, J7.25, CH3), 3.72 (3 H, s, COCH3), 4.40 (1 H, quintet, J7.25, CH), 5.31 (1 H, bs, NH).
(S)-N-Methoxycarbonyl alaninedimethylamide
To a stirred solution of MeOC-alanine (227 g, 1.54 mol) and dimethylformamide (DMF) (25 cm3) in dry dichlorourethane (DCM) (2000 cm3) at 0°C was added dropwise oxalyl chloride (146 cm3, 1.62 mol) over a period of 2 hours. The solution was stirred at 0°C until the evolution of gasses ceased whereupon a basic solution of dimethylamine (676 g, 7.70 mol) in NaOH (3 N, 2000 cm3) was added. The aqueous layer was extracted with diethyl ether (2 x 500 cm3) and the combined organic layers dried (MgSO4) and concentrated under reduced pressure to give the product as a white crystalline solid which required no further purification (230 g, 86%). 1H NMR (250 MHz; C2HCl3) 1.33 (3 H, d, J6.75, CH3), 2.99 3 H, s, OCH3) 3.08, (3 H, s, OCH3), 3.66 (3 H, s, COCH3), 4.66 (H, quintet, J7.00, CH), 5.75 (1 H, d, J5.75, NH).
(S)-2-[(Methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanone.
To a THF (1000 cm3) solution of bromo-2,5-dimethoxybenzene (55 g, 0.25 mol) at -20°C under nitrogen was addedn-butyl lithium (100 cm3, 2.5 M in hexanes, 0.25 mol). The mixture was stirred at -20°C for 0.75 hours, whereupon a THF (100 cm3) solution of amide (30 g, 0.17 mol) was added via cannula. The solution was stirred at -20°C for 2 hours and was then allowed to warm to room temperature over 1 hour and quenched by the addition of ammonium chloride solution (700 cm3). The solution was diluted with diethyl ether (1000 cm3) and the organic layer was dried (MgSO4) and concentrated under reduced pressure to give a yellow oil. The product was purified by dry flash chromatography on silica (eluant 4:1 hexane/ethyl acetate then 3:2 hexane/ethyl acetate) to give the product as a white crystalline solid (45 g, 98%). 1H NMR (250 MHz; C2HCl3) 1.36 (3 H, d, J7.0, CH3), 3.70 (3 H, s, COCH3), 3.82 (3 H, s, OCH3), 3.92 (3 H, s, OCH3), 5.43 (1 H, quintet, J 7.3, H-2), 5.80 (1 H, bs, NH), 6.94 (1 H, d, J 9.0, ArH), 7.10 (1 H, dd, J 9.0, 3.3, ArH), 7.32 (1 H, d, J 3.3, ArH).
(1R,2S)-2-[(Methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanol.
To a stirred solution of ketone i.e. (S)-2-[(methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanone (20 g, 74.9 mmol) and dimethylphenyl silane (10.7 g, 78.6 mmol) in dry DCM (500 cm3) at 0°C in an ice bath was added dropwise trithioroacetic acid (TFA) (50 cm3). The solution was stirred at 0°C for 1 h and then quenched by the addition of sodium hydroxide (500 cm3, 1 N). The organic layer was dried and concentrated under reduced pressure to give a yellow oil which solidified on standing. This solid was crystallized from ether/hexane to give the product as a white crystalline solid (15.6 g, 75%).1H NMR (250 MHz; C2HCl3) 1.03 (3 H, d, J7.0, CH3), 3.04 (1 H, d, J4.3, OH), 3.68 (3 H, s, COCH3), 3.78 (3 H, s, OCH3), 3.80 (3 H, s, OCH3), 3.94-3.99 (1 H, m, H-2), 5.05-5.15 (2 H, m, H-1 and NH), 6.72-6.85 (2 H, m, ArH) 6.97 (1 H, d, J 2.0, ArH).
(1,R,2S)-Methoxamine.
To a stirred solution of methoxycarbonyl (MeOC) protected alcohol i.e. (1R,2S)-2-[(methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanol (4.0 g, 14.9 mmol) in methanol (175 cm3) was added a solution of KOH (4.06 g, 72.8 mmol in water (60 cm3). The solution was cooled and acidified with phosphoric acid (15% v/v). The solution was extracted with DCM (2 x 50 cm3) and the aqueous layer basified by the addition of K2CO3. The aqueous layer was extracted with diethyl ether (5 x 50 cm3) and the combined ethereal extracts dried (MgSO4) and concentrated under reduced pressure to give the product as a clear yellow oil (1.9 g, 61%), 1H NMR (250 MHz; C2HCl3) 0.84 (3 H, d, J 7.0, CH3), 3.19-3.22 (1 H, m, H-2), 3.71 (6 H, s, 2 x OCH3), 4.67 (1 H, d, J 5.0, H-1), 6.66-6.72 (2 H, m, ArH), 6.92 (1 H, d, J 2.5, ArH).
(1R, 2S)-Methoxamine hydrochloride.
To an ice cooled solution of (1R,2S)-methoxamine (1.9 g, 9.00 mmol) in anhydrous diethyl ether (30 cm3) was passed a stream of dry HCl gas for 45 mins. The resultant precipitate was filtered by suction, washed with cold diethyl ether and dried under nitrogen to yield the title compound as a white solid. (1.5 g, 68%). 1H NMR (250 MHz; [C2H3]2SO) 0.89 (3 H, d, J 6.8, CH3), 3.37-3.42 (1 H,m,H-2), 3.71 (3 H, s, OCH3), 3.75 (3 H, s, OCH3), 5.12 (1 H, s, H-1), 5.92 (1 H, d, J 4.3, OH), 6.84 (1 H, dd, J 8.8, 3.0, ArH), 6.92-7.00 (2 H, m, ArH); HPLC.
Analytical Method for the Analysis of Methoxamine
The following method was used to analyse methoxamine samples.
Method
-
Column : Cyclobond I RSP 250 x 4.6 mm Column temperature : 23°C Mobile phase : 0.1% Tetraethylammonium pH 4.1* 95%v/v : Acetonitrile 5%v/v Flow rate : 0.6 ml/min Solution
Concentration :5 mg/l Injection volume : 2.5 µl to 20 µl Detection : UV 230 nm *Tetraethylammonium acetate pH 4.1 was prepared fresh daily.
Example 2 above allows the complete assignment of the methoxamine isomers as shown below:


PATENT
INDIAN 1020/CHE/2011
BY


The Managing Director of Malladi Drugs & Pharmaceuticals, Prashant Malladi (left), with the Chief Executive Officer, V. N. Gopalakrishnan

V.N Gopalakrishnan
CEO at Malladi Drugs & Pharmaceuticals Ltd

Prabhakaran Ranganathan
Vice President (Operations) at Malladi Drugs and Pharmaceuticals Limited

The present invention further provides an improved process for the preparation of (JS, 2S)-Methoxamine HC1 of formula (6) from (1R, 2S)-methoxamine by treating with acetic anhydride in toluene medium followed by acid hydrolysis and basification to obtain (IS, 2S)-Methoxamine base which is further acidified to form (1S,2S)- Methoxamine HC1 (6).
The present invention further provides an improved process for the preparation of (1R, 2R)-Methoxamine HC1 of formula (5) from its diastereomer (1S, 2R)-methoxamine HC1 of formula (2) by treating with acetic anhydride in toluene medium followed by acid hydrolysis and basification to obtain (1R, 2R)-Methoxamine base which is further acidified to form (1R, 2R)-Methoxamine HC1 (5).
The following examples illustrate the invention.
EXAMPLES
Example 1
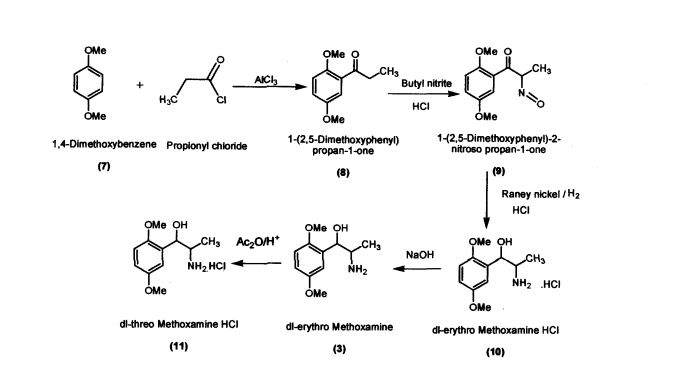
Preparation of l-(2,5-Dimethoxyphenyl)propan-l-one (8)
Aluminium chloride (127.4 g; 0.955 mol) was added to dichloromethane (420 mL) in a round bottomed flask under nitrogen atmosphere. The reaction mixture was cooled to -5 °C; 1,4-dimethoxybenzene (100 g; 0.724 mol) was added slowly within 15-30 minutes. Propionic chloride (87 g; 0.94 mol) dissolved in dichloromethane (245 mL) was added slowly within 2 hours. The reaction mass was allowed to stir for 2 hours and then was quenched in crushed ice (1 kilo) and HC1 (75 mL) at 0 – 5 °C. Separated the layers and the organic layer was washed with 5% sodium hydroxide solution, dried and concentrated (140 g; colorless liquid); Purity by HPLC : 99.04%
Spectroscopic interpretation
The structure of the product, l-(2,5-Dimethoxyphenyl)propan-l-one was confirmed with the help of the following spectroscopic data.
a) IR (cm-1) (KBr)
Aromatic C-H stretch at 3071, aliphatic C – H stretch at 2938, C = O stretch at 1674, benzenoid bands at 1609 and 1584, C – O stretch at 1223, C – H out of plane bending of tri-substituted benzene ring at 814,719.
b) 1H NMR(CDCb, 300 MHz) (δH)
1.16 (3H, t, -CH2-CH3), 3.0 (2H, q, -CH2-CH3), 3.78 (3H, s, -OCH3), 3.85 (3H, s, -OCH3), 6.83 – 7.72 (3H, m, aromatic protons)
c) 13C NMR (CDCb, 300 MHz) (δC)
8.44 (-CH2-CH3), 37.03 (-CH2-CH3), 55.74 (-OCH3), 56.01 (-OCH3), 113.09 – 153.41 (aromatic carbons), 202.96 (C=O)
d) Mass spectrum (ESI, methanol)
[M+Na]+ at m/z 217 (9), [M+H]+ at m/z 195 (100).
Example 2
Preparation of l-(2,5-Dimethoxyphenyl)-2-nitrosopropan-l-one (9) l-(2,5-Dimethoxyphenyl)propan-l-one (100 g; 0.515 mol) was added to dichloromethane (660 mL) in a round bottomed flask under nitrogen atmosphere. Butylnitrite (46.6 g; 0.52 mol) was slowly added in about 30 minutes at 30 – 35 °C. Diethyl ether (60.2 mL) was added to the reaction mixture and dry HC1 gas was purged for about 4 hours at 30 – 35 °C. The reaction mass was maintained for 12 hours and then concentrated under vacuum The residue obtained (60 g; Pale yellow crystalline powder); Purity by HPLC: 99.81%; mp: 104-107 °C
Spectroscopic interpretation
The structure of the product, l-(2,5-Dimethoxyphenyl)-2-nitrosopropan-l-one was confirmed with the help of the following spectroscopic data
a) IR (cm1) (KBr)
O-H stretch at 3250 (broad), aromatic C-H stretch at 3024, aliphatic C – H stretch at 2934, C = O stretch at 1688, C = N stretch at 1645, benzenoid bands at 1589 and 1504, C-O stretch at 1231, C-H out of plane bending of tri-substituted benzene ring at 745,702.
b) 1H NMR(CDCb, 300 MHz) (δh)
2.07 (3H, s, -C-CH3), 3.72 (3H, s, -OCH3), 3.76 (3H, s, -OCH3), 6.84-6.99 (3H, m, aromatic protons), 8.89 (1H, bs, OH)
c) 13C NMR (CDCb, 300 MHz) (δC)
9.16 (-C-CH3), 55.81 (-OCH3), 56.34 (-OCH3), 113.09 – 153.27 (aromatic carbons), 157.07 (C=N-OH); 193.32 (CO)
d) Mass spectrum (ESI, methanol) [M+H]+ at m/z 224 (100)
Example 3
Preparation of dl-erythro-methoxamine HC1 (10)
Raney nickel (50 g); iso-propyl alcohol (250 mL) were added to the autoclave. l-(2,5- Dimethoxyphenyl)-2-nitrosopropan-1 -one (100 g; 0.448 mol) was added slowly at 50 – 55 °C by simultaneously purging the flask with hydrogen at 2-3 Kilo pressure. When hydrogen consumption ceases, the catalyst was filtered and the filtrate was concentrated. iso-Propyl alcohol (200 mL) was added to the concentrated mass followed by acidification with HC1 to obtaindl-erythro-methoxamine HC1 (70 g; white crystalline solid)
Spectroscopic interpretation
The structure of the product, dl-erythro-methoxaxmne HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3409, aromatic C-H stretch at 3010, aliphatic C – H stretch at 2914, HN-H str. at 2574 and 2467, benzenoid bands at 1615 and 1569, C-N stretch at 1279, C-O stretch at 1216, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 812.
b) 1H NMR (DMSO-d6, 300 MHz) (δH)
1.0 (3H,d, -CH-CH3), 3.74 (3H, s, -OCH3), 3.77 (3H, s, -OCH3), 4.89 (1H, q, -CH-CH3),6.1 (1H, d, -CH-OH), 6.87-7.01 (3H, m, aromatic protons), 8.06 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δc)
14.75 (-CH-CH3), 52.12 (-OCH3), 55.70 (-OCH3), 55.70 (-CH-CH3), 67.25 (CH-OH), 111.89 – 153.16 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H)+ at m/z 212 (100), [M-H2O]+ at m/z 194 (56).
Example 4
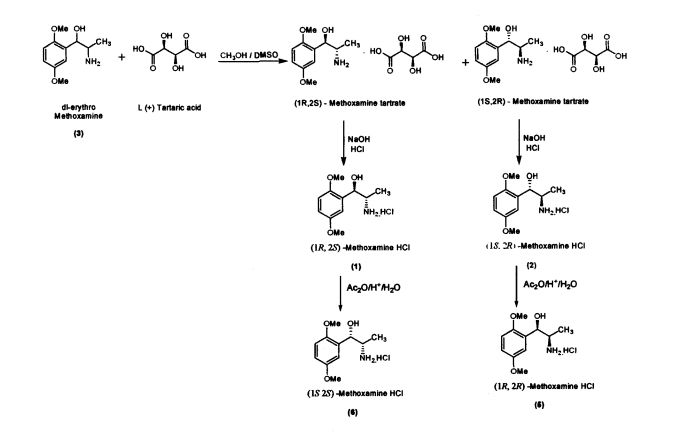
Preparation of(JR,2S)-Metboxamine HC1 (1) and (1S, 2R)-Methoxamine HC1 (2) dl-erythro-methoxamine HC1 (117g; 0.47 mol) was dissolved in water (350 mL) at 30-35 °C. The clear solution obtained was basified using 50% sodium hydroxide solution. dl-erythro-Methoxaumne (3) was extracted into dichloromethane (150 mL) and concentrated. Mixture of methanol/DMSO (4:1; 1650 mL) was added and the mass was heated to 50 °C. L-(+)-Tartaric acid (71.1g; 0.47mol) was added slowly and the temperature of the mass was further raised to 70 °C for complete dissolution. The mass was cooled to 35 °C and maintained for 48 hours. (IR,2.S)-Methoxamine tartrate complex (80 g) precipitated was filtered. From the filtrate on concentration was obtained (1S,2R)- methoxamine tartrate complex (82 g) (IR,25)-Methoxamine tartrate complex was added to water (250 mL) at 35 °C, basified to 12 – 13 pH with 50% sodium hydroxide solution. Dichloromethane (200 mL) was added and stirred for 30 min. Separated the org layer, dried over sodium sulphate and concentrated completely under vacuum at 45° C. Iso-Propyl alcohol (150 mL) was added, charcaolized and filtered. The clear filtrate was acidified with 20%IPA HC1 to yield (1R, 2S)-Methoxamine HC1 which was filtered and dried (48 g); White crystalline powder; Purity by HPLC : 100%; Chiral purity : 100 %; mp : 172-175 °C; [α]D: -47.94° (c = 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (1R,2S)-Methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3300, aromatic C-H stretch at 3065, aliphatic C-H stretch at 2938, HN-H str. at 2693 and 2580, benzenoid bands at 1609 and 1578, C-N stretch at 1277, C-O stretch at 1217, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 818.
b) 1H NMR (DMSO-d6 300 MHz) (δH)
0.91 (3H,d, -CH-CH3), 3.71 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 5.14 (1H, m, -CH- NH3+), 5.95 (1H, d, -CH-OH), 6.83-7.01 (3H, m, aromatic protons), 8.25 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δC)
II. 44 (-CH-CH3), 49.22 (-OCH3), 55.24 (-OCH3), 55.70 (-CH-CH3), 66.49 (CH-OH),
III. 41 – 153.03 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (15).
(IS, 2i?)-Methoxamine tartrate complex was added to water (275 mL) at 35 °C, basified
to 12 – 13 pH with 50% sodium hydroxide solution. Dichloromethane (250 mL) was added and stirred for 30 min. Separated the organic layer, dried over sodium sulphate and concentrated completely under vacuum at 45 °C. Iso-Propyl alcohol (175 mL) was added, charcaolized and filtered. The clear filtrate was acidified with 20%IPA HC1 to yield (1S, 2R)-Methoxamine HC1 which was filtered and dried (51 g) White crystalline powder; Purity by HPLC : 99.99%; Chiral purity . 100 %; mp . 172-175 °C;[α]D : + 47.9° (c = 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (1S, 2R)-Methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) m (cm1) (KBr)
O-H stretch at 3265, aromatic C-H stretch at 3059, aliphatic C-H stretch at 2997, HN-H str. at 2658 and 2567, benzenoid bands at 1611 and 1587,
C-N stretch at 1294, C-O stretch at 1217, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 818.
b) 1H NMR (DMSO-d6,300 MHz) (δH)
0.91 (3H,d, -CH-CH3), 3.71 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 5.14 (1H, m, -CH- NH3+), 5.97 (1H, d, -CH-OH), 6.83-7.01 (3H, m, aromatic protons), 8.19 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6,300 MHz) (δc)
II. 46 (-CH-CH3), 49.18 (-OCH3), 55.23 (-OCH3), 55.68 (-CH-CH3), 66.45 (CH-OH),
III. 42 – 153.02 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (15).
Example 5
Preparation of dl-threo-methoxamine HC1 (11)
dl-erythro-methoxamine HC1 (120g; 0.48 mol) was dissolved in DM water (500 mL) at 30 – 35 °C and cooled to 10 – 15 °C. The clear solution was basified using 50 % sodium hydroxide solution and extracted in dichloromethane (250 mL). The organic layer was separated and concentrated under vacuum. The residue thus obtained was dissolved in toluene (200 mL) and was added slowly to acetic anhydride (120 g; 1.17mol) at 65 – 70 °C. The reaction mass was maintained under stirring and further cooled to 10 – 20 °C. Conc.Sulphuric acid (57.6g; 0.58mol) was added to the reaction mass slowly by maintaining the reaction mass at 10 – 200 C. The reaction mass was heated to 35 – 400 C for 3 hours and concentrated under vacuum at below 80 °C.
The reaction mass was cooled to 10 – 15 °C and was dissolved in DM water (250 mL). The mass was maintained for 3 h at reflux temperature and again cooled to 10 – 15 °C.
The pH was adjusted to 12 – 13 using 50% sodium hydroxide solution and extracted the d/-threo-Methoxamine base in dichloromethane (250 mL). Separated the organic layer and concentrated under vacuum. The concentrated mass was triturated with iso-Propyl alcohol (150 mL); acidified using 20% HC1 in iso-propyl alcohol. Distilled the iso- propyl alcohol completely to the final traces and acetone (300 mL) was added. The material precipitated, crude dl-threo-methoxamine HC1 was filtered. (85 g) Off white powder; Purity by HPLC: 99.4%; mp: 221-223 °C Spectroscopic interpretation
The structure of the product, di-threo-methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm”1) (KBr)
O-H stretch at 3401, aromatic C-H stretch at 3005, aliphatic C-H stretch at 2924, HN-H str. at 2581 and 2490, benzenoid bands at 1609 and 1578, C-N stretch at 1277, C-0 stretch at 1215, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 802.
b) NMR (DMSO-d6,300 MHz) (δH)
1.2 (3H,d, -CH-CHs), 3.72 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 4.87 (1H, q, -CH-CH3),6.3 (1H, d, -CH-OH), 6.83-6.99 (3H, m, aromatic protons), 8.03 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δC)
14.76 (-CH-CH3), 52.15 (-OCH3), 55.89 (-OCH3), 67.34 (CH-OH), 111.96 – 153.21 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (52).
Example 6
Preparation of (1S,2S)- Methoxamine HC1 (6)
(IR, 2S)-Methoxamine HC1 (120 g; 0.48 mol) was dissolved in DM water (500 mL) at 30 -35 °C and cooled to 10 – 15 °C. The clear solution was basified using 50 % sodium hydroxide solution and extracted in dichloromethane (250 mL). The organic layer was separated and concentrated under vacuum. The residue thus obtained was dissolved in toluene (200 mL) and was added slowly to acetic anhydride (120 g; 1.17 mol) at 65 – 70 °C. The reaction mass was maintained under stirring and further cooled to 10 – 20 °C. Conc.sulphuric acid (57.6 g; 0.58 mol) was added to the reaction mass slowly by maintaining the reaction mass at 10 – 20 °C. The reaction mass was heated to 35 – 40 °C for 3 hours and concentrated under vacuum at below 80 °C.
The reaction mass was cooled to 10-15°C and was dissolved in DM water (250 mL). The mass was maintained for 3 h at reflux temperature and again cooled to 10 – 15 °C. The pH was adjusted to 12-13 using 50% sodium hydroxide solution and extracted the (1S, 2S)-Methoxamine base in dichloromethane (250 mL). Separated the organic layer and concentrated under vacuum The concentrated mass was triturated with iso-Propyl alcohol (150 mL); acidified using 20% HC1 in iso-propyl alcohol. Distilled the iso- propyl alcohol completely to the final traces and acetone (300 mL) was added. The material precipitated, crude (IS, 2S)-methoxamine HC1 was filtered. (86 g); White crystalline powder; Purity by HPLC . 99.8%; Chiral purity : 99.7%; mp : 172-175 °C; [α]D: + 30.739° (c = 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (IS, 2S)-methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3356, aromatic C-H stretch at 3080, aliphatic C-H stretch at 2999, HN-H str. at 2641 and 2583, benzenoid bands at 1611 and 1506, C-N stretch at 1302, C-O stretch at 1229, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 812.
b) 1H NMR (DMSO-d6 300 MHz) (δH)
1.04 (3H,d, -CH-CH3), 3.72 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 4.90 (1H, m, -CH- CH3),6.07 (1H, d, -CH-OH), 6.84-7.01 (3H, d, aromatic protons), 8.15 (3H, bs, HN-H)
The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6, 300 MHz) (δC)
14.75 (-CH-CH3), 52.18 (-OCH3), 55.21 (-OCH3), 55.69 (-CH-CH3), 67.32 (CH-OH), 111.38 -153.01 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (48).
Example 7
Preparation of (1R, 2R)-Methoxamine HC1 (5)
(IS, 2R)Methoxamine HC1 (120g; 0.48 mol) was dissolved in DM water (500 mL) at 30 – 35 °C and cooled to 10 – 15 °C. The clear solution was basified using 50 % sodium hydroxide solution and extracted in dichloromethane (250 mL). The organic layer was separated and concentrated under vacuum. The residue thus obtained was dissolved in toluene (200 mL) and was added slowly to acetic anhydride (120 g; 1.17mol) at 65 – 70 °C. The reaction mass was maintained under stirring and further cooled to 10 – 20 °C. Cone.Sulphuric acid (57.6g; 0.58mol) was added to the reaction mass slowly by maintaining the reaction mass at 10 – 20 °C. The reaction mass was heated to 35 – 40 °C for 3 hours and concentrated under vacuum at below 80 °C.
The reaction mass was cooled tol0-15°C and was dissolved in DM water (250 mL). The mass was maintained for 3 h at reflux temperature and again cooled to 10 – 15 °C. The pH was adjusted to 12-13 using 50% sodium hydroxide solution and extracted the (IR, 2i?)-Methoxamine base in dichloromethane (250 mL). Separated the organic layer and concentrated under vacuum. The concentrated mass was triturated with iso-Propyl alcohol (150 mL); acidified using 20% HC1 in iso-propyl alcohol Distilled the iso- propyl alcohol completely to the final traces and acetone (300 mL) was added. The material precipitated, crude (1R, 2R)-methoxamine HC1 was filtered. (90 g) White crystalline powder; Purity by HPLC: 99.1%, Chiral purity. 100%; mp: 172-175 °C;[α]D: -29.04° (c – 2% in MeOH)
Spectroscopic interpretation
The structure of the product, (1R, 2R)methoxamine HC1 was confirmed with the help of the following spectroscopic data.
a) IR (cm1) (KBr)
O-H stretch at 3356, aromatic C-H stretch at 3078, aliphatic C-H stretch at 2999, HN-H str. at 2619 and 2500, benzenoid bands at 1611 and 1508, C-N stretch at 1302, C-O stretch at 1229, C-H out of plane bending of 1,2,4-tri- substituted benzene ring at 812.
b) 1H NMR(DMSO-d6 300 MHz) (δH)
I. 04 (3H,d, -CH-CHa), 3.72 (3H, s, -OCH3), 3.75 (3H, s, -OCH3), 4.90 (1H, m, -CH- CH3),6.07 (1H, d, -CH-OH), 6.83-7.01 (3H, d, aromatic protons), 8.13 (3H, bs, HN-H) The -OH proton appears to have exchanged with the solvent.
c) 13C NMR (DMSO-d6 300 MHz) (δe)
II. 41 (-CH-CH3), 52.16 (-OCH3), 55.22 (-OCH3), 55.70 (-CH-CH3), 67.32 (CH-OH), III. 39-153.15 (aromatic carbons)
d) Mass spectrum (ESI, methanol)
[M+H]+ at m/z 212 (100), [M-H2O]+ at m/z 194 (44).
PATENT
http://www.google.com/patents/US8491931
(1,R,2S)-Methoxamine
To a stirred solution of methoxycarbonyl (MeOC) protected alcohol i.e. (1R,2S)-2-[(methoxycarbonyl)amino]-1-(2,5-dimethoxyphenyl)-1-propanol (4.0 g, 14.9 mmol) in methanol (175 cm3) was added a solution of KOH (4.06 g, 72.8 mmol in water (60 cm3). The solution was cooled and acidified with phosphoric acid (15% v/v). The solution was extracted with DCM (2×50 cm3) and the aqueous layer basified by the addition of K2CO3. The aqueous layer was extracted with diethyl ether (5×50 cm3) and the combined ethereal extracts dried (MgSO4) and concentrated under reduced pressure to give the product as a clear yellow oil (1.9 g, 61%), 1H NMR (250 MHz; C2HCl3) 0.84 (3H, d, J 7.0, CH3), 3.19-3.22 (1H, m, H-2), 3.71 (6H, s, 2×OCH3), 4.67 (1H, d, J 5.0, H-1), 6.66-6.72 (2H, m, ArH), 6.92 (1H, d, J 2.5, ArH).
(1R,2S)-Methoxamine hydrochloride
To an ice cooled solution of (1R,2S)-methoxamine (1.9 g, 9.00 mmol) in anhydrous diethyl ether (30 cm3) was passed a stream of dry HCl gas for 45 mins. The resultant precipitate was filtered by suction, washed with cold diethyl ether and dried under nitrogen to yield the title compound as a white solid. (1.5 g, 68%). 1H NMR (250 MHz; [C2H3]2SO) 0.89 (3H, d, J 6.8, CH3), 3.37-3.42 (1H,M,H-2), 3.71 (3H, s, OCH3), 3.75 (3H, s, OCH3), 5.12 (1H, s, H-1), 5.92 (1H, d, J 4.3, OH), 6.84 (1H, dd, J 8.8, 3.0, ArH), 6.92-7.00 (2H, m, ArH); HPLC.



//1R,2S-methoxamine
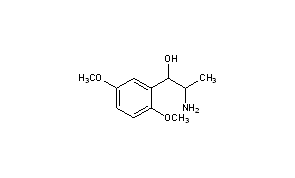
RACEMIC
 |
|
Title: Methoxamine
CAS Registry Number: 390-28-3
CAS Name: a-(1-Aminoethyl)-2,5-dimethoxybenzenemethanol
Additional Names: a-(1-aminoethyl)-2,5-dimethoxybenzyl alcohol; 2-amino-1-(2,5-dimethoxyphenyl)-1-propanol; b-hydroxy-b-(2,5-dimethoxyphenyl)isopropylamine; b-(2,5-dimethoxyphenyl)-b-hydroxyisopropylamine; 2,5-dimethoxynorephedrine
Molecular Formula: C11H17NO3
Molecular Weight: 211.26
Percent Composition: C 62.54%, H 8.11%, N 6.63%, O 22.72%
Literature References: a1-Adrenergic agonist. Prepn: Baltzly et al., US 2359707 (1944 to Burroughs Wellcome). Metabolism: A. Klutch, M. Bordun, J. Med. Chem. 10, 860 (1967). Clinical pharmacology: N. T. Smith, C. Whitcher, Anesthesiology 28, 735 (1967); P. D. Snashall et al., Clin. Sci. Mol. Med. 54, 283 (1978). HPLC determn in plasma: I. A. Al-Meshal et al., J. Liq. Chromatogr. 12, 1589 (1989). Therapeutic use: P. M. C. Wright et al., Anesth. Analg. 75, 56 (1992); L. Cabanes et al., N. Engl. J. Med. 326, 1661 (1992). Comprehensive description: A. M. Al-Obaid, M. M. El-Domiaty, Anal. Profiles Drug Subs. 20, 399-431 (1991).
Derivative Type: Hydrochloride
CAS Registry Number: 61-16-5
Trademarks: Vasoxine (Burroughs Wellcome); Vasoxyl (Burroughs Wellcome); Vasylox (Burroughs Wellcome)
Molecular Formula: C11H17NO3.HCl
Molecular Weight: 247.72
Percent Composition: C 53.33%, H 7.32%, N 5.65%, O 19.38%, Cl 14.31%
Properties: Crystals, mp 212-216°. pKa (25°C) 9.2. Very sol in water: One gram dissolves in 2.5 ml water, in 12 ml ethanol. Practically insol in ether, benzene, chloroform. pH of a 2% aq soln between 4.5 and 5.5.
Melting point: mp 212-216°
pKa: pKa (25°C) 9.2
Therap-Cat: Antihypotensive.
Keywords: a-Adrenergic Agonist; Antihypotensive.
|
Regulatory Approval Pathways: EU vs US
DRUG REGULATORY AFFAIRS INTERNATIONAL


Regulatory Approval Pathways: EU vs US
 Drug Authorization Procedures in the EU
Drug Authorization Procedures in the EU
Sponsors have several options when seeking market approval for a new drug in Europe: a national authorization procedure, a decentralized procedure, a mutual recognition procedure and a centralized procedure. Depending on a product’s eligibility, each of these authorization routes offers different advantages and disadvantages to the sponsor, and these should be considered when setting up the market strategy of a product.
National Procedure
This procedure is used whenever a company wants to commercialize a product in only one EU Member State.
The National procedure is specific to each country. That is, each country within the EU has its own procedures for authorizing a marketing application for a new drug. Sponsors can find information regarding the requirements and procedure of each country on the websites of the regulatory agencies.
ADVANTAGES of National Procedure
There are some advantages in submitting…
View original post 3,049 more words

{kind=link}