Home » Uncategorized (Page 45)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cp2TiCl: An Ideal Reagent for Green Chemistry?

The development of Green Chemistry inevitably involves the development of green reagents. In this review, we highlight that Cp2TiCl is a reagent widely used in radical and organometallic chemistry, which shows, if not all, at least some of the 12 principles summarized for Green Chemistry, such as waste minimization, catalysis, safer solvents, toxicity, energy efficiency, and atom economy. Also, this complex has proved to be an ideal reagent for green C–C and C–O bond forming reactions, green reduction, isomerization, and deoxygenation reactions of several functional organic groups as we demonstrate throughout the review.
Cp2TiCl: An Ideal Reagent for Green Chemistry?

View original post 2,879 more words
Lisdexamphetamine

Lisdexamfetamine
- Molecular FormulaC15H25N3O
- Average mass263.379 Da

CAS 608137-33-3
(2S)-2,6-Diamino-N-[(1S)-1-methyl-2-phenylethyl]hexanamide dimethanesulfonate
https://www.google.com/patents/WO2005032474A2
| Applicants: | NEW RIVER PHARMACEUTICALS INC. [US/US]; The Governor Tyler, 1881 Grove Avenue, Radford, VA 24141 (US) (For All Designated States Except US). MICKLE, Travis [US/US]; (US) (For US Only). KRISHNAN, Suma [US/US]; (US) (For US Only). MONCRIEF, James, Scott [US/US]; (US) (For US Only). LAUDERBACK, Christopher [US/US]; (US) (For US Only). MILLER, Christal [US/US]; (US) (For US Only) |
| Inventors: | MICKLE, Travis; (US). KRISHNAN, Suma; (US). MONCRIEF, James, Scott; (US). LAUDERBACK, Christopher; (US). MILLER, Christal; (US) |

MICKLE, Travis
Dr. Travis Mickle founded KemPharm, Inc. in late 2006. Prior to KemPharm, from January 2003 to October 2005, Dr. Mickle was Director of Drug Discovery and Chemical Development at New River Pharmaceuticals where he also served in a variety of other senior research roles since joining the firm in 2001. During his tenure at New River, Dr. Mickle was responsible for creating a strong preclinical and clinical pipeline of drugs in the areas of ADHD, pain and thyroid dysfunction. His contributions included, as principal inventor, the discovery and development of lisdexamfetamine dimesylate, the highly successful therapy for the treatment of ADHD known as Vyvanse®. In addition, Dr. Mickle was an active participant in FDA and DEA meetings representing the company’s discovery/chemistry group and was also called in as a critical scientific resource during New River’s financings and strategic partnering discussions. Before his departure, Dr. Mickle played an integral part in New River’s development into a successful publicly-traded company which was subsequently acquired for $2.6 billion by its marketing partner, Shire PLC. Dr. Mickle is also the author of more than 150 US and international patents and patent applications, as well as several research papers. Dr. Mickle holds a Ph.D in Bio-Organic Chemistry from the University of Iowa.
ABOUT SUMA KRISHNAN
Mrs. Krishnan has served as our Senior Vice President — Regulatory Affairs since 2012. From 2009 to 2011, Mrs. Krishnan served as Senior Vice President of Product Development at Pinnacle Pharmaceuticals, Inc. From 2007 to 2009, she served as Chief Financial Officer of Light Matters Foundation. Previously, Mrs. Krishnan was Vice President, Product Development at New River Pharmaceuticals Inc. from September 2002 until its acquisition by Shire plc in April 2007.
Mrs. Krishnan has 22 years’ experience in drug development. Prior to serving at New River Pharmaceuticals Inc., Mrs. Krishnan served in the following capacities: Director, Regulatory Affairs at Shire Pharmaceuticals, Inc., a specialty pharmaceutical company; Senior Project Manager at Pfizer, Inc., a multi-national pharmaceutical company; and a consultant at the Weinberg Group, a pharmaceutical and environmental consulting firm.
Mrs. Krishnan began her career as a discovery scientist for Janssen Pharmaceuticals, Inc., a subsidiary of Johnson & Johnson, a multi-national pharmaceutical company, in May 1991. Mrs. Krishnan received an M.S. in Organic Chemistry from Villanova University, an M.B.A. from Institute of Management and Research (India) and an undergraduate degree in Organic Chemistry from Ferguson University (India).
Lisdexamfetamine (contracted from L–lysine–dextroamphetamine) is a prodrug of the central nervous system (CNS) stimulantdextroamphetamine, a phenethylamine of the amphetamine class that is used in the treatment of attention deficit hyperactivity disorder (ADHD) and binge eating disorder.[4][5] Its chemical structure consists of dextroamphetamine coupled with the essential amino acid L-lysine. Lisdexamfetamine itself is inactive prior to its absorption and the subsequent rate-limited enzymaticcleavage of the molecule’s L-lysine portion, which produces the active metabolite (dextroamphetamine).
Lisdexamfetamine can be prescribed for the treatment of attention deficit hyperactivity disorder (ADHD) in adults and children six and older, as well as for moderate to severe binge eating disorder in adults.[4] The safety and the efficacy of lisdexamfetamine dimesylate in children with ADHD three to five years old have not been established.[6]
Lisdexamfetamine is a Class B/Schedule II substance in the United Kingdom and a Schedule II controlled substance in the United States (DEA number 1205)[7] and the aggregate production quota for 2016 in the United States is 29,750 kilograms of anhydrous acid or base.[8] Lisdexamfetamine is currently in Phase III trials in Japan for ADHD.[9]
Uses
Medical
Lisdexamfetamine is used primarily as a treatment for attention deficit hyperactivity disorder (ADHD) and binge eating disorder;[4] it has similar off-label uses as those of other pharmaceutical amphetamines.[4][5] Long-term amphetamine exposure at sufficiently high doses in some animal species is known to produce abnormal dopamine system development or nerve damage,[10][11] but, in humans with ADHD, pharmaceutical amphetamines appear to improve brain development and nerve growth.[12][13][14] Reviews of magnetic resonance imaging (MRI) studies suggest that long-term treatment with amphetamine decreases abnormalities in brain structure and function found in subjects with ADHD, and improves function in several parts of the brain, such as the right caudate nucleus of the basal ganglia.[12][13][14]
Reviews of clinical stimulant research have established the safety and effectiveness of long-term continuous amphetamine use for the treatment of ADHD.[15][16][17] Randomized controlled trials of continuous stimulant therapy for the treatment of ADHD spanning two years have demonstrated treatment effectiveness and safety.[15][17] Two reviews have indicated that long-term continuous stimulant therapy for ADHD is effective for reducing the core symptoms of ADHD (i.e., hyperactivity, inattention, and impulsivity), enhancing quality of life and academic achievement, and producing improvements in a large number of functional outcomes[note 1] across nine outcome categories related to academics, antisocial behavior, driving, non-medicinal drug use, obesity, occupation, self-esteem, service use (i.e., academic, occupational, health, financial, and legal services), and social function.[16][17] One review highlighted a nine-month randomized controlled trial in children with ADHD that found an average increase of 4.5 IQ points, continued increases in attention, and continued decreases in disruptive behaviors and hyperactivity.[15] Another review indicated that, based upon the longest follow-up studies conducted to date, lifetime stimulant therapy that begins during childhood is continuously effective for controlling ADHD symptoms and reduces the risk of developing a substance use disorder as an adult.[17]
Current models of ADHD suggest that it is associated with functional impairments in some of the brain’s neurotransmitter systems;[18] these functional impairments involve impaired dopamine neurotransmission in the mesocorticolimbic projection and norepinephrine neurotransmission in the noradrenergic projections from the locus coeruleus to the prefrontal cortex.[18]Psychostimulants like methylphenidate and amphetamine are effective in treating ADHD because they increase neurotransmitter activity in these systems.[19][18][20] Approximately 80% of those who use these stimulants see improvements in ADHD symptoms.[21] Children with ADHD who use stimulant medications generally have better relationships with peers and family members, perform better in school, are less distractible and impulsive, and have longer attention spans.[22][23] The Cochrane Collaboration‘s reviews[note 2] on the treatment of ADHD in children, adolescents, and adults with pharmaceutical amphetamines stated that while these drugs improve short-term symptoms, they have higher discontinuation rates than non-stimulant medications due to their adverse side effects.[25][26] A Cochrane Collaboration review on the treatment of ADHD in children with tic disorders such as Tourette syndrome indicated that stimulants in general do not make tics worse, but high doses of dextroamphetamine could exacerbate tics in some individuals.[27]
Individuals over the age of 65 were not commonly tested in clinical trials of lisdexamfetamine for ADHD.[4] Lisdexamfetamine is being investigated for possible treatment of cognitive impairment associated with schizophrenia and excessive daytime sleepiness.[28]
Cognitive
In 2015, a systematic review and a meta-analysis of high quality clinical trials found that, when used at low (therapeutic) doses, amphetamine produces modest yet unambiguous improvements in cognition, including working memory, long-term episodic memory, inhibitory control, and some aspects of attention, in normal healthy adults;[29][30] these cognition-enhancing effects of amphetamine are known to be partially mediated through the indirect activation of both dopamine receptor D1 and adrenoceptor α2 in the prefrontal cortex.[19][29] A systematic review from 2014 found that low doses of amphetamine also improve memory consolidation, in turn leading to improved recall of information.[31] Therapeutic doses of amphetamine also enhance cortical network efficiency, an effect which mediates improvements in working memory in all individuals.[19][32]Amphetamine and other ADHD stimulants also improve task saliency (motivation to perform a task) and increase arousal (wakefulness), in turn promoting goal-directed behavior.[19][33][34] Stimulants such as amphetamine can improve performance on difficult and boring tasks and are used by some students as a study and test-taking aid.[19][34][35]Based upon studies of self-reported illicit stimulant use, 5–35% of college students use diverted ADHD stimulants, which are primarily used for performance enhancement rather than as recreational drugs.[36][37][38] However, high amphetamine doses that are above the therapeutic range can interfere with working memory and other aspects of cognitive control.[19][34]
Physical
Amphetamine is used by some athletes for its psychological and athletic performance-enhancing effects, such as increased endurance and alertness;[39][40] however, non-medical amphetamine use is prohibited at sporting events that are regulated by collegiate, national, and international anti-doping agencies.[41][42] In healthy people at oral therapeutic doses, amphetamine has been shown to increase muscle strength, acceleration, athletic performance in anaerobic conditions, and endurance (i.e., it delays the onset of fatigue), while improving reaction time.[39][43][44] Amphetamine improves endurance and reaction time primarily through reuptake inhibition and effluxion of dopamine in the central nervous system.[43][44][45] Amphetamine and other dopaminergic drugs also increase power output at fixed levels of perceived exertion by overriding a “safety switch” that allows the core temperature limit to increase in order to access a reserve capacity that is normally off-limits.[44][46][47] At therapeutic doses, the adverse effects of amphetamine do not impede athletic performance;[39][43] however, at much higher doses, amphetamine can induce effects that severely impair performance, such as rapid muscle breakdown and elevated body temperature.[48][49][43]
Available forms
Vyvanse capsules are available in doses of 10 mg, 20 mg, 30 mg, 40 mg, 50 mg, 60 mg, and 70 mg of the active ingredient, lisdexamfetamine dimesylate.[50] Vyvanse capsules contain several inactive ingredients, including microcrystalline cellulose, croscarmellose sodium, and magnesium stearate.[50] The capsule shells contain gelatin and titanium dioxide, and may contain FD&C Red 3, FD&C Yellow 6, FD&C Blue 1, black iron oxide, and yellow iron oxide.[50]













DESCRIPTION/SYNTHESIS
Lisdexamfetamine dimesylate is approved and marketed in the United States for the treatment of attention-deficit hyperactivity disorder in pediatric patients. The active compound lisdexamfetamine contains D-amphetamine covalently linked to the essential amino acid L-lysine. Controlled release of D-amphetamine, a psychostimulant, occurs following administration of lisdexamfetamine to a patient. The controlled release has been reported to occur through hydrolysis of the amide bond linking D-amphetamine and L-lysine.
A procedure for making lisdexamfetamine hydrochloride is described in U.S. Pat. No. 7,223,735 to Mickle et al. (hereinafter Mickle). The procedure involves reacting D-amphetamine with (S)-2,5-dioxopyrrolidin-1-yl 2,6-bis(tert-butoxycarbonylamino)hexanoate to form a lysine-amphetamine intermediate bearing tert-butylcarbamate protecting groups. This intermediate is treated with hydrochloric acid to remove the tert-butylcarbamate protecting groups and provide lisdexamfetamine as its hydrochloride salt. However, this procedure suffers several drawbacks that are problematic when carrying out large scale reactions, such as manufacturing scale, to prepare lisdexamfetamine.



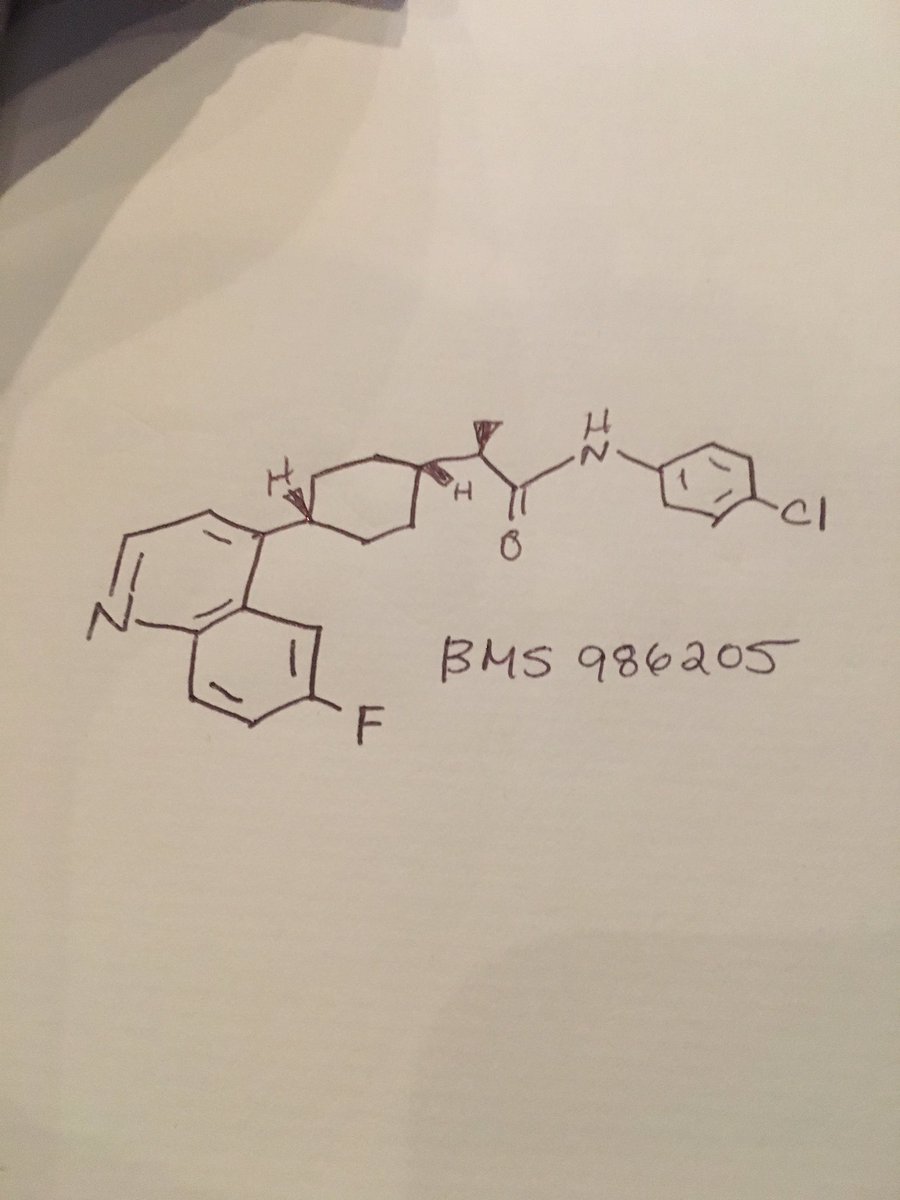
Lisdexamphetamine of formula I, is a conjugate of D-amphetamine and L-lysine and is chemically named as (2S)-2,6-diamino-N-[(lS)-methyl-2-phenylethyl]hexan amide.
Formula- I

Amphetamines stimulate central nervous system (CNS). Amphetamine is prescribed for treatment of various disorders, including attention deficit hyperactivity disorder (ADHD), obesity, nacrolepsy. It is approved as lisdextamphetamine dimesylate of formula IA and
Formula- IA

marketed under trade name Vyvanse for treatment of attention-deficit hyperactivity disorder in pediatric patients.
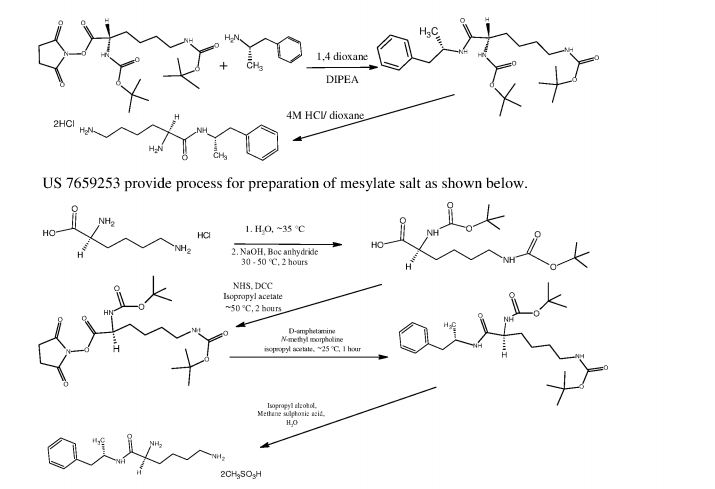
L-Lysine-D-amphetamine and its pharmaceutically acceptable salts were first disclosed in US patent 7,662,787 wherein it is exemplified as hydrochloride salt. Process for preparation of L- lysine-D-amphetamine includes reaction of BOC-Lys-(BOC)-hydroxysuccinimido ester with D-amphetamine in dioxane using diisopropyl ethyl amine (DIPEA) as a base to obtain BOC- protected lisdexamphetamine which is then purified using flash chromatography and further reacted with a mixture of 4M hydrochloric acid /dioxane to yield L-lysine-D-amphetamine hydrochloride. The process is as shown in following scheme:

4M HCI/dioxane

In equivalent patent US 7,659,253, process for preparation of mesylate salt is disclosed as shown below.

NHS;DCC;
isopropyl acetate
-50°C, 2 hr

Process includes preparation of BOC-Lys-(BOC)-hydroxysuccinimido ester wherein use of reagents like N-hydroxy-succinimide (NHS) and Ν,Ν-dicyclohexyl- carbodimiide (DCC) is carried out, The above processes involve use of flash column chromatography to purify crude BOC- protected L-lysine-D-amphetamine intermediate. Use of column chromatography is very cumbersome, tedoius and time consuming, therefore not advisable at commercial scale. Further Ν,Ν-dicyclohexylcarbodimiide is known to be highly toxic and moisture sensitive compound, and its use leads to formation of a large amount of Ν,Ν-dicyclohexyl urea (DCU) as bye product which has to be removed from reaction mixture.. Therefore use of DCC is not advisable at industrial scale.
International patent publication WO 2010/042120 discloses a process for preparing L-lysine- D-amphetamine or its salts by reacting D-amphetamine with protected lysine or its salt by using an alkylphosphonic acid anhydride as coupling agent in presence of a base and solvent. The process is as shown in following scheme:

The application discloses use of alkylphosphonic acid anhydrides, which are expensive, and needs additional testing to show absence of phosphic impurities in intermediate or final compound to meet regulatory requirements. So it is not appealing to use alkylphosphonic anhydrides for scale up operations.
International patent publication WO2010/148305 discloses a process for preparation of lisdexamphetamine by removal of chlorine from Ν,Ν’-bistrifluoroacetyl-chloro- lisdexamphetamine by using hydrogenation catalyst like Pd/C, under hydrogen gas to form Ν,Ν’-bistrifluoroacetyl-lisdexamphetamine which on further deprotection by using deprotecting agent to form lisdexamphetamine. Alternatively first deprotection by using deprotecting agent and then chlorine is removed by using hydrogenation catalyst like Pd/C under hydrogen gas. The process involves additional steps of inserting chloro group and thereafter removing chloro group; further Pd/C is an expensive reagent, hence not attractive option from cost point of view.
It is therefore, necessary to overcome problems associated with prior art and to provide an efficient process for preparation of lisdexamphetamine and its pharmaceutically acceptable salts using easily available, less expensive, easy to handle raw materials and avoid use of column chromatography.
PATENT
https://www.google.com/patents/US20120157706





-
-
- Example IPreparation of N,N′-Biscarbobenzyloxy-Lisdexamfetamine (LDX-(Cbz)2)
-

General Experimental Procedure: In an appropriately sized, inert jacketed reactor charge 378.3 g of Cbz-Lys(Cbz)-OSu and 2309 g of isopropyl acetate. Heat the stirred slurry to ˜50° C. In a second reactor mix 100.0 g of D-amphetamine into 165 g of isopropyl acetate. Add the D-amphetamine solution to the batch over 1.75-2.25 hours. After the addition is complete stir the heterogeneous mixture at 50-55° C. until the reaction is complete by HPLC analysis. Charge 1197 g of methanol and heat the batch at a vigorous reflux 1 hour. Cool the batch to 45-55° C. over 2 hours then hold at temperature for 14-16 hours. Cool the batch to 15-25° C. at a rate of 5-10° C. per hour. Filter the slurry. Rinse the wet cake with 449 g of methanol and dry on the filter under nitrogen. To a clean, dry reactor charge the crude solids and 1642 g of methanol. Stir the slurry and heat to a vigorous reflux for 2 hours. Cool the batch to 45-55° C. over 1-2 hours then hold at temperature 12-16 hours. Continue cooling to 15-25° C. at a rate of 5-10° C. per hour. Filter the slurry and wash the wet cake with 547 g of methanol. Vacuum dry the wet cake at ˜55° C. to give product as a white to off-white solid (332 g, 88 mol %).
The crude LDX-(Cbz)product isolated from the reaction mixture by crystallization had a purity of 99.96% according to HPLC analysis.1H NMR analysis of crude LDX-(Cbz)2product revealed the presence of 0.57% by weight N-hydroxysuccinimide. The purified LDX-(Cbz)2product obtained by re-crystallization from methanol had a purity of 99.99% according to HPLC analysis.1H NMR analysis of the purified LDX-(Cbz)2product obtained by re-crystallization from methanol revealed that the amount of N-hydroxysuccinimide in the purified LDX-(Cbz)2product was reduced to 0.05% by weight.
Example 2Preparation of Lisdexamfetamine Dimesylate

General Experimental Procedure: In an appropriately sized, inert autoclave charge 100.0 g of LDX-(Cbz)2, 1 g of 10% (50% wet) palladium on carbon and 607.5 g of n-butanol. Stir the mixture under 100-150 psi of hydrogen at 80-85° C. until the reaction is complete by HPLC analysis. Heat the batch to 95-97° C. and hot filter. Transfer the product rich filtrate to an appropriately sized glass reactor. Charge 7.6 g of methanesulfonic acid maintaining a batch temperature of 32-38° C. To the resulting solution add 1.3 g of LDX-2MSA seed crystal. Stir the batch 4-16 hours at 32-38° C. Charge 30.4 g of methanesulfonic acid to the slurry over not less than 2 hours maintaining a batch temperature of 32-38° C. After the addition stir 1-2 hours at 32-38° C. Charge 436 g of isopropyl acetate over not less than 2 hours, then stir 1-2 hours at 32-38° C. Cool the batch to 15-25° C. and hold 1 hour. Filter the slurry. Wash the wet cake with a premixed combination of n-butanol (91.5 g) and isopropyl acetate (32.7 g) followed by a wash of isopropyl acetate (87.2 g). Vacuum dry the wet cake at ˜50° C. to give product as a white to off-white solid (78.8 g, 92 mol %).
PATENT
Sun Pharmaceutical Industries Ltd
Process for preparation of lisdexamphetamine and its salts via a novel aziridine intermediate is claimed. Lisdexamphetamine attention deficit hyperactivity disorder (ADHD). Represents the first patenting to be seen from Sun pharmaceutical that focuses on lisdexamphetamine. At the time of publication Pawar and Patel are affiliated with Chattem chemicals .


PATENT
IN 2011DE02040
WO 2013011526
IN 2009CH01986
WO 2010042120
https://www.google.com/patents/WO2013011526A1?cl=en

Formula II A
Formula IV
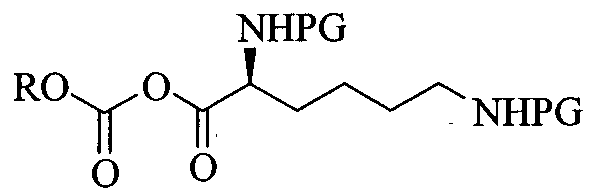
Formula IVA
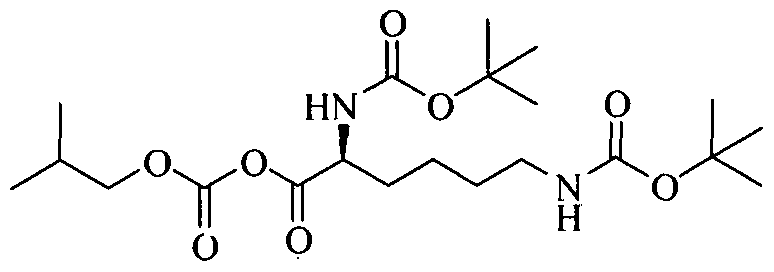
EXAMPLES
Examplel Preparation of 2,6-bis-tertiarvbutoxycarbonylamino hexanoic acid
To a solution of L-lysine monohydrochloride (25g, 0.14mol) and sodium hydroxide (15g) in water (250 ml), ditertiary butyl dicarbonate (70.0 g, 0.32 mol) was added at 15-25°C. The temperature was slowly raised to 55-60 °C and the reaction mixture was stirred for 12 hours.. After completion of reaction, ( monitored by TLC), the reaction mixture was cooled to 10-
15°C and pH was adjusted to 2.5-3.5 with 2N hydrochloric acid. The reaction mass’ was then extracted with dichloromethane (2 x 125 ml) and combined organic layer was successively washed with water (150 ml) and brine (150 ml). Dichloromethane layer was distilled under vacuum at 30-40 °C to obtain 29.7g of title compound as a viscous oily mass having purity 96.5% by HPLC.
Example 2: Preparation of (5-tert-butoxycarbonylamino-5-(l-methyl-2-phenyl- ethylcarba moyl)-pentyll-carbamic acid tert-butyl ester;
To a solution of 2,6-bis-tertbutoxy carbonylamino hexanoic acid (7.5g) in dichloromethane (150 ml) , triethyl amine (8.0 ml) was added at 25-30°C and the reaction mixture was stirred for 15 minutes. The solution was cooled to -15 to -10°C and isobutylchloroformate (4.35 g) was slowly added under nitrogen atmosphere and stirred for 30 minutes at -15°C to – 10°C. A solution of D-amphetamine (3.85 g) in dichloromethane (10 ml) was slowly added and. the reaction mixture was stirred at 15 to -10°C for 60 minutes. The reaction completion was checked by TLC. After completion of the reaction temperature was raised to 25-30°C and reaction mixture was successively washed with 0.5 N hydrochloric acid solution (2 x 75 ml), sodium bicarbonate solution (5%w/w 75ml), water (50 ml) and brine solution (50 ml). The combined dichloromethane layer was dried over sodium sulfate (10.0 g) and distilled at 30- 40°C to obtain a semisolid compound which was stirred with a mixture of n-heptane (85 ml) and ethyl acetate (5ml) at 25-30°C for 30 minutes. The solid, thus obtained, was filtered and dried to get 10.21 g of title compound having purity 89.77% by HPLC. The crude compound was dissolved in ethanol (45ml) at 50-55°C and water (50ml) was added. The reaction mixture was slowly cooled to 35-40°C, stirred for 30 minutes. The solid, thus obtained, was filtered and dried to get 7.35g of pure title compound as a white crystalline solid having purity 99.5 % by HPLC.
Example 3: Preparation of lisdexamphetamine dimesylate
(5-Tert-butoxycarbonylamino-5-( 1 -methyl-2-phenyl-ethylcarbamoyl)-pentyl]-carbamic acid tert-butyl ester (2.5g,) was dissolved in a mixture of isopropyl alcohol (10ml) and ethyl acetate (10ml) at 40-45°C and the reaction mass was cooled to 15-20°C. To this cold solution, methane sulphonic acid (2.5g) was added slowly and stirred for 12 hours at 15- 20°C. The reaction completion was checked by HPLC. The resulting solid was filtered, washed with a mixture of chilled isopropyl alcohol (5ml) and ethyl acetate (5ml) and dried under vacuum to obtain 1.68 g of title compound as a white crystalline solid having purity 99.72 % by HPLC.
Example 4: Preparation of lisdexamphetamine dimesylate:
(5-Tert-butoxycarbonylamino-5-( 1 -methyl-2-phenyl-ethylcarbamoyl)-pentyl]-carbamic acid tert-butyl ester (2.5 g,) was dissolved in ethanol (20 ml) at 25-30°C. To the reaction mixture, methane sulphonic acid (2.5 g) was slowly added and the reaction mixture was heated to 55- 60°C and stirred for 3 hours at 55-60°C. The reaction mixture was cooled to 25-30°C, stirred for 2 hours, filtered, washed with ethanol (10ml) and dried under vacuum to obtain 1.55g of lisdexamphetamine dimesylate having purity 99.61 % by HPLC.
Example 5: Purification of lisdexamphetamine dimesylate
Lisdexamphetamine dimesylate (1.40g,) was dissolved in ethanol (10 ml) at 50-55 °C and ethyl acetate (10 ml) was slowly added at 50-55 °C. The reaction mixture was cooled to 20- 25°C and stirred for 30 minutes. The resulting solid was filtered, washed with a mixture of ethanol and ethyl acetate (3 ml, 1: 1) and dried under vacuum at 55-60°C to obtain 1.28g of pure lisdexamphetamine dimesylate as a white crystalline solid having purity 99.90 % by HPLC.
Example 6: Preparation of lisdexamphetamine dimesylate
To a stirred solution of L-lysine monohydrochloride (50g) and sodium hydroxide (30 g) in water (500ml) at 15-25°C, ditertbutyl dicarbonate(140g) was added. The temperature was slowly raised to 55-60°C and reaction mixture was stirred for 12 hours. After completion of reaction, the reaction mixture was cooled to 10-15°C and pH was adjusted to 2.5-3.5 with 2N hydrochloric acid. The reaction mixture was then extracted with dichloromethane (2 x 250 ml) and combined dichloromethane layer was successively washed with water (300 ml) and brine (300 ml). To the organic layer triethyl amine (58g) in dichloromethane (500 ml) was added. The solution was cooled to -15 to -20°C and isobutylchloroformate (42.5 g) was slowly added at -15 to -20°C and stirred for 1 hour. A solution of D-amphetamine (41.85 g) in dichloromethane (100 ml) was slowly added to reaction mixture at -15 to -20 °C and stirred. After completion of the reaction, the reaction mixture was successively washed with 0.5 N hydrochloric solution (2 x 450 ml), sodium bicarbonate solution (5%w/w, 450 ml), water (450 ml) and brine solution (450 ml). The organic layer was dried over sodium sulfate and distilled under vacuum at 30-40°C to afford a residue. To this residue, ethanol (480 ml) was added followed by slow addition of methane sulphonic acid (55 g) under nitrogen atmosphere. The reaction temperature was raised 55-60°C and after completion of reaction, the reaction mixture was cooled to 20-25°C and stirred for 2 hours. The resulting solid was filtered, washed with ethanol (50 ml) and suck dried for 30 minutes, further washed with ethanol and dried to obtain lisdexamphetamine dimesylate.
Mechanism of action

Amphetamine enters the presynaptic neuron across the neuronal membrane or through DAT. Once inside, it binds to TAAR1 or enters synaptic vesicles through VMAT2. When amphetamine enters synaptic vesicles through VMAT2, it collapses the vesicular pH gradient, which in turn causes dopamine to be released into the cytosol (light tan-colored area) through VMAT2. When amphetamine binds to TAAR1, it reduces the firing rate of the dopamine neuron via potassium channels and activates protein kinase A (PKA) and protein kinase C (PKC), which subsequently phosphorylate DAT. PKA-phosphorylation causes DAT to withdraw into the presynaptic neuron (internalize) and cease transport. PKC-phosphorylated DAT may either operate in reverse or, like PKA-phosphorylated DAT, internalize and cease transport. Amphetamine is also known to increase intracellular calcium, an effect which is associated with DAT phosphorylation through a CAMKIIα-dependent pathway, in turn producing dopamine efflux.
Lisdexamfetamine is an inactive prodrug that is converted in the body to dextroamphetamine, a pharmacologically active compound which is responsible for the drug’s activity.[119] After oral ingestion, lisdexamfetamine is broken down by enzymes in red blood cells to form L-lysine, a naturally occurring essential amino acid, and dextroamphetamine.[4] The conversion of lisdexamfetamine to dextroamphetamine is not affected by gastrointestinal pH and is unlikely to be affected by alterations in normal gastrointestinal transit times.[4][120]
The optical isomers of amphetamine, i.e., dextroamphetamine and levoamphetamine, are TAAR1 agonists and vesicular monoamine transporter 2 inhibitors that can enter monoamine neurons;[121][122] this allows them to release monoamine neurotransmitters (dopamine, norepinephrine, and serotonin, among others) from their storage sites in the presynaptic neuron, as well as prevent the reuptake of these neurotransmitters from the synaptic cleft.[121][122]
Lisdexamfetamine was developed with the goal of providing a long duration of effect that is consistent throughout the day, with reduced potential for abuse. The attachment of the amino acid lysine slows down the relative amount of dextroamphetamine available to the blood stream. Because no free dextroamphetamine is present in lisdexamfetamine capsules, dextroamphetamine does not become available through mechanical manipulation, such as crushing or simple extraction. A relatively sophisticated biochemical process is needed to produce dextroamphetamine from lisdexamfetamine.[120] As opposed to Adderall, which contains roughly equal parts of racemic amphetamine and dextroamphetamine salts, lisdexamfetamine is a single-enantiomer dextroamphetamine formula.[119][123] Studies conducted show that lisdexamfetamine dimesylate may have less abuse potential than dextroamphetamine and an abuse profile similar to diethylpropion at dosages that are FDA-approved for treatment of ADHD, but still has a high abuse potential when this dosage is exceeded by over 100%.[120]
Pharmacokinetics
The oral bioavailability of amphetamine varies with gastrointestinal pH;[118] it is well absorbed from the gut, and bioavailability is typically over 75% for dextroamphetamine.[124] Amphetamine is a weak base with a pKa of 9.9;[125]consequently, when the pH is basic, more of the drug is in its lipid soluble free base form, and more is absorbed through the lipid-rich cell membranes of the gut epithelium.[125][118] Conversely, an acidic pH means the drug is predominantly in a water-soluble cationic (salt) form, and less is absorbed.[125] Approximately 15–40% of amphetamine circulating in the bloodstream is bound to plasma proteins.[126]
The half-life of amphetamine enantiomers differ and vary with urine pH.[125] At normal urine pH, the half-lives of dextroamphetamine and levoamphetamine are 9–11 hours and 11–14 hours, respectively.[125] An acidic diet will reduce the enantiomer half-lives to 8–11 hours; an alkaline diet will increase the range to 16–31 hours.[127][128] The biological half-life is longer and distribution volumes are larger in amphetamine dependent individuals.[128] The immediate-release and extended release variants of salts of both isomers reach peak plasma concentrations at 3 hours and 7 hours post-dose respectively.[125] Amphetamine is eliminated via the kidneys, with 30–40% of the drug being excreted unchanged at normal urinary pH.[125] When the urinary pH is basic, amphetamine is in its free base form, so less is excreted.[125] When urine pH is abnormal, the urinary recovery of amphetamine may range from a low of 1% to a high of 75%, depending mostly upon whether urine is too basic or acidic, respectively.[125] Amphetamine is usually eliminated within two days of the last oral dose.[127]
The prodrug lisdexamfetamine is not as sensitive to pH as amphetamine when being absorbed in the gastrointestinal tract;[129] following absorption into the blood stream, it is converted by red blood cell-associated enzymes to dextroamphetamine via hydrolysis.[129] The elimination half-life of lisdexamfetamine is generally less than one hour.[129]
CYP2D6, dopamine β-hydroxylase (DBH), flavin-containing monooxygenase 3 (FMO3), butyrate-CoA ligase (XM-ligase), and glycine N-acyltransferase (GLYAT) are the enzymes known to metabolize amphetamine or its metabolites in humans.[sources 9] Amphetamine has a variety of excreted metabolic products, including 4-hydroxyamphetamine, 4-hydroxynorephedrine, 4-hydroxyphenylacetone, benzoic acid, hippuric acid, norephedrine, and phenylacetone.[125][127][134] Among these metabolites, the active sympathomimetics are 4‑hydroxyamphetamine,[138] 4‑hydroxynorephedrine,[139] and norephedrine.[140] The main metabolic pathways involve aromatic para-hydroxylation, aliphatic alpha- and beta-hydroxylation, N-oxidation, N-dealkylation, and deamination.[125][127] The known metabolic pathways, detectable metabolites, and metabolizing enzymes in humans include the following:

Hydroxylation
Hydroxylation
Hydroxylation
Hydroxylation
Hydroxylation
Deamination
Conjugation
The primary active metabolites of amphetamine are 4-hydroxyamphetamine and norephedrine;[134] at normal urine pH, about 30–40% of amphetamine is excreted unchanged and roughly 50% is excreted as the inactive metabolites (bottom row).[125] The remaining 10–20% is excreted as the active metabolites.[125] Benzoic acid is metabolized by XM-ligase into an intermediate product, benzoyl-CoA,[136] which is then metabolized by GLYAT into hippuric acid.[137]
Chemistry
Comparison to other formulationsLisdexamfetamine dimesylate is a water-soluble (792 mg/mL) powder with a white to off-white color.[50]
Lisdexamfetamine dimesylate is one marketed formulation delivering dextroamphetamine. The following table compares the drug to other amphetamine pharmaceuticals.
| drug | formula | molecular mass [note 8] |
amphetamine base [note 9] |
amphetamine base in equal doses |
doses with equal base content [note 10] |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| (g/mol) | (percent) | (30 mg dose) | ||||||||
| total | base | total | dextro- | levo- | dextro- | levo- | ||||
| dextroamphetamine sulfate[142][143] | (C9H13N)2•H2SO4 | 368.49 | 270.41 | 73.38% | 73.38% | — | 22.0 mg | — | 30.0 mg | |
| amphetamine sulfate[144] | (C9H13N)2•H2SO4 | 368.49 | 270.41 | 73.38% | 36.69% | 36.69% | 11.0 mg | 11.0 mg | 30.0 mg | |
| Adderall | 62.57% | 47.49% | 15.08% | 14.2 mg | 4.5 mg | 35.2 mg | ||||
| 25% | dextroamphetamine sulfate[142][143] | (C9H13N)2•H2SO4 | 368.49 | 270.41 | 73.38% | 73.38% | — | |||
| 25% | amphetamine sulfate[144] | (C9H13N)2•H2SO4 | 368.49 | 270.41 | 73.38% | 36.69% | 36.69% | |||
| 25% | dextroamphetamine saccharate[145] | (C9H13N)2•C6H10O8 | 480.55 | 270.41 | 56.27% | 56.27% | — | |||
| 25% | amphetamine aspartate monohydrate[146] | (C9H13N)•C4H7NO4•H2O | 286.32 | 135.21 | 47.22% | 23.61% | 23.61% | |||
| lisdexamfetamine dimesylate[147] | C15H25N3O•(CH4O3S)2 | 455.49 | 135.21 | 29.68% | 29.68% | — | 8.9 mg | — | 74.2 mg | |
| amphetamine base suspension[note 11][56] | C9H13N | 135.21 | 135.21 | 100% | 76.19% | 23.81% | 22.9 mg | 7.1 mg | 22.0 mg | |
History, society, and culture
Lisdexamfetamine was developed by New River Pharmaceuticals, who were bought by Shire Pharmaceuticals shortly before lisdexamfetamine began being marketed. It was developed for the intention of creating a longer-lasting and less-easily abused version of dextroamphetamine, as the requirement of conversion into dextroamphetamine via enzymes in the red blood cells increases its duration of action, regardless of the route of ingestion.[148] The drug lisdexamfetamine dimesylate is the first prodrug of its kind.
On 23 April 2008, Vyvanse received FDA approval for the adult population.[149] On 19 February 2009, Health Canada approved 30 mg and 50 mg capsules of lisdexamfetamine for treatment of ADHD.[150] On 8 February 2012, Vyvanse received FDA approval for maintenance treatment of adult ADHD.[151] In February 2014, Shire announced that two late-stage clinical trials had shown that Vyvanse was not an effective treatment for depression.[152] Lisdexamfetamine was granted approval in a number of European countries for the treatment of ADHD in children and adolescents over the age of 6 years, as well as adults who are continuing treatment from childhood, after a positive outcome of the regulatory procedure.[153] Shire also recently announced receipt of a positive result from a European decentralised procedure for lisdexamfetamine for adult patients with ADHD in the United Kingdom, Sweden and Denmark, expanding the indication of lisdexamfetamine to include newly diagnosed adult patients.[154]
In January 2015, lisdexamfetamine was approved by the U.S. Food and Drug Administration for treatment of binge eating disorder in adults.[28][155][156]
In January 2017, a new dosage form of lisdexamfetamine in the form of a chewable tablet (as opposed to a capsule) was approved by the FDA.[157]
Brand names
Lisdexamfetamine is sold as Tyvense (IE), Elvanse (UK), Venvanse (BR), Vyvanse (CA, US).[158]
Clinical research
Some clinical trials that used lisdexamfetamine as an add-on therapy with a selective serotonin reuptake inhibitor (SSRI) or serotonin-norepinephrine reuptake inhibitor (SNRI) for treatment-resistant depression indicated that this is no more effective than the use of an SSRI or SNRI alone.[159] Other studies indicated that psychostimulants potentiated antidepressants, and were under-prescribed for treatment resistant depression. In those studies patients showed significant improvement in energy, mood, and psychomotor activity.[160]
Notes
- Jump up^ The ADHD-related outcome domains with the greatest proportion of significantly improved outcomes from long-term continuous stimulant therapy include academics (~55% of academic outcomes improved), driving (100% of driving outcomes improved), non-medical drug use (47% of addiction-related outcomes improved), obesity (~65% of obesity-related outcomes improved), self esteem (50% of self-esteem outcomes improved), and social function (67% of social function outcomes improved).[16]The largest effect sizes for outcome improvements from long-term stimulant therapy occur in the domains involving academics (e.g., grade point average, achievement test scores, length of education, and education level), self-esteem (e.g., self-esteem questionnaire assessments, number of suicide attempts, and suicide rates), and social function (e.g., peer nomination scores, social skills, and quality of peer, family, and romantic relationships).[16]Long-term combination therapy for ADHD (i.e., treatment with both a stimulant and behavioral therapy) produces even larger effect sizes for outcome improvements and improves a larger proportion of outcomes across each domain compared to long-term stimulant therapy alone.[16]
- Jump up^ Cochrane Collaboration reviews are high quality meta-analytic systematic reviews of randomized controlled trials.[24]
- Jump up^ The 95% confidence interval indicates that there is a 95% probability that the true number of deaths lies between 3,425 and 4,145.
- Jump up^ Transcription factors are proteins that increase or decrease the expression of specific genes.[93]
- Jump up^ In simpler terms, this necessary and sufficient relationship means that ΔFosB overexpression in the nucleus accumbens and addiction-related behavioral and neural adaptations always occur together and never occur alone.
- Jump up^ NMDA receptors are voltage-dependent ligand-gated ion channels that requires simultaneous binding of glutamate and a co-agonist (d-serine or glycine) to open the ion channel.[105]
- Jump up^ The review indicated that magnesium L-aspartate and magnesium chloride produce significant changes in addictive behavior;[71] other forms of magnesium were not mentioned.
- Jump up^ For uniformity, molecular masses were calculated using the Lenntech Molecular Weight Calculator[141] and were within 0.01g/mol of published pharmaceutical values.
- Jump up^ Amphetamine base percentage = molecular massbase / molecular masstotal. Amphetamine base percentage for Adderall = sum of component percentages / 4.
- Jump up^ dose = (1 / amphetamine base percentage) × scaling factor = (molecular masstotal / molecular massbase) × scaling factor. The values in this column were scaled to a 30 mg dose of dextroamphetamine sulfate. Due to pharmacological differences between these medications (e.g., differences in the release, absorption, conversion, concentration, differing effects of enantiomers, half-life, etc.), the listed values should not be considered equipotent doses.
- Jump up^ This product (Dyanavel XR) is an oral suspension (i.e., a drug that is suspended in a liquid and taken by mouth) that contains 2.5 mg/mL of amphetamine base.[56] The amphetamine base contains dextro- to levo-amphetamine in a ratio of 3.2:1,[56] which is approximately the ratio in Adderall. The product uses an ion exchange resin to achieve extended release of the amphetamine base.[56]
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20050038121 * | Jun 1, 2004 | Feb 17, 2005 | New River Pharmaceuticals Inc. | Abuse resistant lysine amphetamine compounds |
| US20110196173 * | Oct 9, 2008 | Aug 11, 2011 | Andreas Meudt | Process for the Synthesis of Amphetamine Derivatives |
| DE1493824A1 * | Nov 23, 1964 | May 22, 1969 | Hoffmann La Roche | Verfahren zur Herstellung von Aminocarbonsaeureamiden |
| Reference | ||
|---|---|---|
| 1 | * | “Benzyl Chloroformate” in Handbook of Reagents for Organic Synthesis – Activating Agents and Protecting Groups ; Pearson et al., eds., 1999 John Wiley & Sons, pp. 46-50 |
| 2 | * | DATABASE CAPLUS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; Database Accession No. 1966:19805, Abstract NL 6414901, 28 July 1965 |
| 3 | * | Smith and March. Advanced Organic Chemistry 6th ed. (501-502) |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8614346 | Jun 18, 2010 | Dec 24, 2013 | Cambrex Charles City, Inc. | Methods and compositions for preparation of amphetamine conjugates and salts thereof |
| WO2017003721A1 | Jun 17, 2016 | Jan 5, 2017 | Noramco, Inc. | Process for the preparation of lisdexamfetamine and related derivatives |
References
- Jump up^ “Public Assessment Report Decentralised Procedure” (PDF). Shire Pharmaceuticals Contracts Limited. p. 14. Retrieved 23 August 2014.
- ^ Jump up to:a b Millichap JG (2010). “Chapter 9: Medications for ADHD”. In Millichap JG. Attention Deficit Hyperactivity Disorder Handbook: A Physician’s Guide to ADHD (2nd ed.). New York, USA: Springer. p. 112. ISBN 9781441913968.
Table 9.2 Dextroamphetamine formulations of stimulant medication
Dexedrine [Peak:2–3 h] [Duration:5–6 h] …
Adderall [Peak:2–3 h] [Duration:5–7 h]
Dexedrine spansules [Peak:7–8 h] [Duration:12 h] …
Adderall XR [Peak:7–8 h] [Duration:12 h]
Vyvanse [Peak:3–4 h] [Duration:12 h] - ^ Jump up to:a b Brams M, Mao AR, Doyle RL (September 2008). “Onset of efficacy of long-acting psychostimulants in pediatric attention-deficit/hyperactivity disorder”. Postgrad. Med. 120 (3): 69–88. PMID 18824827. doi:10.3810/pgm.2008.09.1909.
Onset of efficacy was earliest for d-MPH-ER at 0.5 hours, followed by d, l-MPH-LA at 1 to 2 hours, MCD at 1.5 hours, d, l-MPH-OR at 1 to 2 hours, MAS-XR at 1.5 to 2 hours, MTS at 2 hours, and LDX at approximately 2 hours. … MAS-XR, and LDX have a long duration of action at 12 hours postdose
- ^ Jump up to:a b c d e f g h i j k l m “Vyvanse Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. January 2015. Retrieved 24 February 2015.
- ^ Jump up to:a b Heal DJ, Smith SL, Gosden J, Nutt DJ (June 2013). “Amphetamine, past and present – a pharmacological and clinical perspective”. J. Psychopharmacol. 27 (6): 479–496. PMC 3666194
 . PMID 23539642. doi:10.1177/0269881113482532.
. PMID 23539642. doi:10.1177/0269881113482532. - Jump up^ “Lisdexamfetamine dimesylate (generic).” Brown University Psychopharmacology Update 19.7 (2008): 1–2. Academic Search Premier. EBSCO. Web. 12 September 2010.
- Jump up^ “DEA – Department of Justice” (PDF). DEA – Department of Justice. Retrieved 1 July 2014.
- Jump up^ “DEA Office of Diversion Control” (PDF). DEA. Retrieved 1 July 2014.
- Jump up^ “Phase-III clinical trials in Attention-deficit hyperactivity disorder (In children, In adolescents) in Japan (PO)”. Retrieved 20 March 2016.
- ^ Jump up to:a b Carvalho M, Carmo H, Costa VM, Capela JP, Pontes H, Remião F, Carvalho F, Bastos Mde L (August 2012). “Toxicity of amphetamines: an update”. Arch. Toxicol. 86 (8): 1167–1231. PMID 22392347. doi:10.1007/s00204-012-0815-5.
- Jump up^ Berman S, O’Neill J, Fears S, Bartzokis G, London ED (October 2008). “Abuse of amphetamines and structural abnormalities in the brain”. Ann. N. Y. Acad. Sci. 1141: 195–220. PMC 2769923 . PMID 18991959. doi:10.1196/annals.1441.031.
- ^ Jump up to:a b Hart H, Radua J, Nakao T, Mataix-Cols D, Rubia K (February 2013). “Meta-analysis of functional magnetic resonance imaging studies of inhibition and attention in attention-deficit/hyperactivity disorder: exploring task-specific, stimulant medication, and age effects”. JAMA Psychiatry. 70 (2): 185–198. PMID 23247506. doi:10.1001/jamapsychiatry.2013.277.
- ^ Jump up to:a b Spencer TJ, Brown A, Seidman LJ, Valera EM, Makris N, Lomedico A, Faraone SV, Biederman J (September 2013). “Effect of psychostimulants on brain structure and function in ADHD: a qualitative literature review of magnetic resonance imaging-based neuroimaging studies”. J. Clin. Psychiatry. 74 (9): 902–917. PMC 3801446 . PMID 24107764. doi:10.4088/JCP.12r08287.
- ^ Jump up to:a b Frodl T, Skokauskas N (February 2012). “Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects.”. Acta psychiatrica Scand. 125 (2): 114–126. PMID 22118249. doi:10.1111/j.1600-0447.2011.01786.x.
Basal ganglia regions like the right globus pallidus, the right putamen, and the nucleus caudatus are structurally affected in children with ADHD. These changes and alterations in limbic regions like ACC and amygdala are more pronounced in non-treated populations and seem to diminish over time from child to adulthood. Treatment seems to have positive effects on brain structure.
- ^ Jump up to:a b c Millichap JG (2010). “Chapter 9: Medications for ADHD”. In Millichap JG. Attention Deficit Hyperactivity Disorder Handbook: A Physician’s Guide to ADHD (2nd ed.). New York, USA: Springer. pp. 121–123, 125–127. ISBN 9781441913968.
Ongoing research has provided answers to many of the parents’ concerns, and has confirmed the effectiveness and safety of the long-term use of medication.
- ^ Jump up to:a b c d e Arnold LE, Hodgkins P, Caci H, Kahle J, Young S (February 2015). “Effect of treatment modality on long-term outcomes in attention-deficit/hyperactivity disorder: a systematic review”. PLoS ONE. 10 (2): e0116407. PMC 4340791 . PMID 25714373. doi:10.1371/journal.pone.0116407.
The highest proportion of improved outcomes was reported with combination treatment (83% of outcomes). Among significantly improved outcomes, the largest effect sizes were found for combination treatment. The greatest improvements were associated with academic, self-esteem, or social function outcomes.
Figure 3: Treatment benefit by treatment type and outcome group - ^ Jump up to:a b c d Huang YS, Tsai MH (July 2011). “Long-term outcomes with medications for attention-deficit hyperactivity disorder: current status of knowledge”. CNS Drugs. 25 (7): 539–554. PMID 21699268. doi:10.2165/11589380-000000000-00000.
Recent studies have demonstrated that stimulants, along with the non-stimulants atomoxetine and extended-release guanfacine, are continuously effective for more than 2-year treatment periods with few and tolerable adverse effects. The effectiveness of long-term therapy includes not only the core symptoms of ADHD, but also improved quality of life and academic achievements. The most concerning short-term adverse effects of stimulants, such as elevated blood pressure and heart rate, waned in long-term follow-up studies. … In the longest follow-up study (of more than 10 years), lifetime stimulant treatment for ADHD was effective and protective against the development of adverse psychiatric disorders.
- ^ Jump up to:a b c Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 6: Widely Projecting Systems: Monoamines, Acetylcholine, and Orexin”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. pp. 154–157. ISBN 9780071481274.
- ^ Jump up to:a b c d e f Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 13: Higher Cognitive Function and Behavioral Control”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. pp. 318, 321. ISBN 9780071481274.
Therapeutic (relatively low) doses of psychostimulants, such as methylphenidate and amphetamine, improve performance on working memory tasks both in normal subjects and those with ADHD. … stimulants act not only on working memory function, but also on general levels of arousal and, within the nucleus accumbens, improve the saliency of tasks. Thus, stimulants improve performance on effortful but tedious tasks … through indirect stimulation of dopamine and norepinephrine receptors. …
Beyond these general permissive effects, dopamine (acting via D1 receptors) and norepinephrine (acting at several receptors) can, at optimal levels, enhance working memory and aspects of attention. - Jump up^ Bidwell LC, McClernon FJ, Kollins SH (August 2011). “Cognitive enhancers for the treatment of ADHD”. Pharmacol. Biochem. Behav. 99 (2): 262–274. PMC 3353150 . PMID 21596055. doi:10.1016/j.pbb.2011.05.002.
- Jump up^ Parker J, Wales G, Chalhoub N, Harpin V (September 2013). “The long-term outcomes of interventions for the management of attention-deficit hyperactivity disorder in children and adolescents: a systematic review of randomized controlled trials”. Psychol. Res. Behav. Manag. 6: 87–99. PMC 3785407 . PMID 24082796. doi:10.2147/PRBM.S49114.
Only one paper53 examining outcomes beyond 36 months met the review criteria. … There is high level evidence suggesting that pharmacological treatment can have a major beneficial effect on the core symptoms of ADHD (hyperactivity, inattention, and impulsivity) in approximately 80% of cases compared with placebo controls, in the short term.
- Jump up^ Millichap JG (2010). “Chapter 9: Medications for ADHD”. In Millichap JG. Attention Deficit Hyperactivity Disorder Handbook: A Physician’s Guide to ADHD (2nd ed.). New York, USA: Springer. pp. 111–113. ISBN 9781441913968.
- Jump up^ “Stimulants for Attention Deficit Hyperactivity Disorder”. WebMD. Healthwise. 12 April 2010. Retrieved 12 November 2013.
- Jump up^ Scholten RJ, Clarke M, Hetherington J (August 2005). “The Cochrane Collaboration”. Eur. J. Clin. Nutr. 59 Suppl 1: S147–S149; discussion S195–S196. PMID 16052183. doi:10.1038/sj.ejcn.1602188.
- ^ Jump up to:a b Castells X, Ramos-Quiroga JA, Bosch R, Nogueira M, Casas M (June 2011). Castells X, ed. “Amphetamines for Attention Deficit Hyperactivity Disorder (ADHD) in adults”. Cochrane Database Syst. Rev. (6): CD007813. PMID 21678370. doi:10.1002/14651858.CD007813.pub2.
- Jump up^ Punja S, Shamseer L, Hartling L, Urichuk L, Vandermeer B, Nikles J, Vohra S (February 2016). “Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents”. Cochrane Database Syst. Rev. 2: CD009996. PMID 26844979. doi:10.1002/14651858.CD009996.pub2.
- Jump up^ Pringsheim T, Steeves T (April 2011). Pringsheim T, ed. “Pharmacological treatment for Attention Deficit Hyperactivity Disorder (ADHD) in children with comorbid tic disorders”. Cochrane Database Syst. Rev. (4): CD007990. PMID 21491404. doi:10.1002/14651858.CD007990.pub2.
- ^ Jump up to:a b http://www.shire.com/shireplc/en/rd/pipeline
- ^ Jump up to:a b Spencer RC, Devilbiss DM, Berridge CW (June 2015). “The Cognition-Enhancing Effects of Psychostimulants Involve Direct Action in the Prefrontal Cortex”. Biol. Psychiatry. 77 (11): 940–950. PMC 4377121 . PMID 25499957. doi:10.1016/j.biopsych.2014.09.013.
The procognitive actions of psychostimulants are only associated with low doses. Surprisingly, despite nearly 80 years of clinical use, the neurobiology of the procognitive actions of psychostimulants has only recently been systematically investigated. Findings from this research unambiguously demonstrate that the cognition-enhancing effects of psychostimulants involve the preferential elevation of catecholamines in the PFC and the subsequent activation of norepinephrine α2 and dopamine D1 receptors. … This differential modulation of PFC-dependent processes across dose appears to be associated with the differential involvement of noradrenergic α2 versus α1 receptors. Collectively, this evidence indicates that at low, clinically relevant doses, psychostimulants are devoid of the behavioral and neurochemical actions that define this class of drugs and instead act largely as cognitive enhancers (improving PFC-dependent function). … In particular, in both animals and humans, lower doses maximally improve performance in tests of working memory and response inhibition, whereas maximal suppression of overt behavior and facilitation of attentional processes occurs at higher doses.
- Jump up^ Ilieva IP, Hook CJ, Farah MJ (January 2015). “Prescription Stimulants’ Effects on Healthy Inhibitory Control, Working Memory, and Episodic Memory: A Meta-analysis”. J. Cogn. Neurosci. 27: 1–21. PMID 25591060. doi:10.1162/jocn_a_00776.
Specifically, in a set of experiments limited to high-quality designs, we found significant enhancement of several cognitive abilities. … The results of this meta-analysis … do confirm the reality of cognitive enhancing effects for normal healthy adults in general, while also indicating that these effects are modest in size.
- Jump up^ Bagot KS, Kaminer Y (April 2014). “Efficacy of stimulants for cognitive enhancement in non-attention deficit hyperactivity disorder youth: a systematic review”. Addiction. 109 (4): 547–557. PMC 4471173 . PMID 24749160. doi:10.1111/add.12460.
Amphetamine has been shown to improve consolidation of information (0.02 ≥ P ≤ 0.05), leading to improved recall.
- Jump up^ Devous MD, Trivedi MH, Rush AJ (April 2001). “Regional cerebral blood flow response to oral amphetamine challenge in healthy volunteers”. J. Nucl. Med. 42 (4): 535–542. PMID 11337538.
- Jump up^ Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 10: Neural and Neuroendocrine Control of the Internal Milieu”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 266. ISBN 9780071481274.
Dopamine acts in the nucleus accumbens to attach motivational significance to stimuli associated with reward.
- ^ Jump up to:a b c Wood S, Sage JR, Shuman T, Anagnostaras SG (January 2014). “Psychostimulants and cognition: a continuum of behavioral and cognitive activation”. Pharmacol. Rev. 66 (1): 193–221. PMID 24344115. doi:10.1124/pr.112.007054.
- Jump up^ Twohey M (26 March 2006). “Pills become an addictive study aid”. JS Online. Archived from the original on 15 August 2007. Retrieved 2 December 2007.
- Jump up^ Teter CJ, McCabe SE, LaGrange K, Cranford JA, Boyd CJ (October 2006). “Illicit use of specific prescription stimulants among college students: prevalence, motives, and routes of administration”. Pharmacotherapy. 26 (10): 1501–1510. PMC 1794223 . PMID 16999660. doi:10.1592/phco.26.10.1501.
- Jump up^ Weyandt LL, Oster DR, Marraccini ME, Gudmundsdottir BG, Munro BA, Zavras BM, Kuhar B (September 2014). “Pharmacological interventions for adolescents and adults with ADHD: stimulant and nonstimulant medications and misuse of prescription stimulants”. Psychol. Res. Behav. Manag. 7: 223–249. PMC 4164338 . PMID 25228824. doi:10.2147/PRBM.S47013.
misuse of prescription stimulants has become a serious problem on college campuses across the US and has been recently documented in other countries as well. … Indeed, large numbers of students claim to have engaged in the nonmedical use of prescription stimulants, which is reflected in lifetime prevalence rates of prescription stimulant misuse ranging from 5% to nearly 34% of students.
- Jump up^ Clemow DB, Walker DJ (September 2014). “The potential for misuse and abuse of medications in ADHD: a review”. Postgrad. Med. 126 (5): 64–81. PMID 25295651. doi:10.3810/pgm.2014.09.2801.
Overall, the data suggest that ADHD medication misuse and diversion are common health care problems for stimulant medications, with the prevalence believed to be approximately 5% to 10% of high school students and 5% to 35% of college students, depending on the study.
- ^ Jump up to:a b c Liddle DG, Connor DJ (June 2013). “Nutritional supplements and ergogenic AIDS”. Prim. Care. 40 (2): 487–505. PMID 23668655. doi:10.1016/j.pop.2013.02.009.
Amphetamines and caffeine are stimulants that increase alertness, improve focus, decrease reaction time, and delay fatigue, allowing for an increased intensity and duration of training …
Physiologic and performance effects
• Amphetamines increase dopamine/norepinephrine release and inhibit their reuptake, leading to central nervous system (CNS) stimulation
• Amphetamines seem to enhance athletic performance in anaerobic conditions 39 40
• Improved reaction time
• Increased muscle strength and delayed muscle fatigue
• Increased acceleration
• Increased alertness and attention to task - ^ Jump up to:a b c d e f g h i j k l m n o p q r s Westfall DP, Westfall TC (2010). “Miscellaneous Sympathomimetic Agonists”. In Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s Pharmacological Basis of Therapeutics (12th ed.). New York, USA: McGraw-Hill. ISBN 9780071624428.
- Jump up^ Bracken NM (January 2012). “National Study of Substance Use Trends Among NCAA College Student-Athletes” (PDF). NCAA Publications. National Collegiate Athletic Association. Retrieved 8 October 2013.
- Jump up^ Docherty JR (June 2008). “Pharmacology of stimulants prohibited by the World Anti-Doping Agency (WADA)”. Br. J. Pharmacol. 154 (3): 606–622. PMC 2439527 . PMID 18500382. doi:10.1038/bjp.2008.124.
- ^ Jump up to:a b c d Parr JW (July 2011). “Attention-deficit hyperactivity disorder and the athlete: new advances and understanding”. Clin. Sports Med. 30 (3): 591–610. PMID 21658550. doi:10.1016/j.csm.2011.03.007.
In 1980, Chandler and Blair47 showed significant increases in knee extension strength, acceleration, anaerobic capacity, time to exhaustion during exercise, pre-exercise and maximum heart rates, and time to exhaustion during maximal oxygen consumption (VO2 max) testing after administration of 15 mg of dextroamphetamine versus placebo. Most of the information to answer this question has been obtained in the past decade through studies of fatigue rather than an attempt to systematically investigate the effect of ADHD drugs on exercise.
- ^ Jump up to:a b c Roelands B, de Koning J, Foster C, Hettinga F, Meeusen R (May 2013). “Neurophysiological determinants of theoretical concepts and mechanisms involved in pacing”. Sports Med. 43 (5): 301–311. PMID 23456493. doi:10.1007/s40279-013-0030-4.
In high-ambient temperatures, dopaminergic manipulations clearly improve performance. The distribution of the power output reveals that after dopamine reuptake inhibition, subjects are able to maintain a higher power output compared with placebo. … Dopaminergic drugs appear to override a safety switch and allow athletes to use a reserve capacity that is ‘off-limits’ in a normal (placebo) situation.
- Jump up^ Parker KL, Lamichhane D, Caetano MS, Narayanan NS (October 2013). “Executive dysfunction in Parkinson’s disease and timing deficits”. Front. Integr. Neurosci. 7: 75. PMC 3813949 . PMID 24198770. doi:10.3389/fnint.2013.00075.
Manipulations of dopaminergic signaling profoundly influence interval timing, leading to the hypothesis that dopamine influences internal pacemaker, or “clock,” activity. For instance, amphetamine, which increases concentrations of dopamine at the synaptic cleft advances the start of responding during interval timing, whereas antagonists of D2 type dopamine receptors typically slow timing;… Depletion of dopamine in healthy volunteers impairs timing, while amphetamine releases synaptic dopamine and speeds up timing.
- Jump up^ Rattray B, Argus C, Martin K, Northey J, Driller M (March 2015). “Is it time to turn our attention toward central mechanisms for post-exertional recovery strategies and performance?”. Front. Physiol. 6: 79. PMC 4362407 . PMID 25852568. doi:10.3389/fphys.2015.00079.
Aside from accounting for the reduced performance of mentally fatigued participants, this model rationalizes the reduced RPE and hence improved cycling time trial performance of athletes using a glucose mouthwash (Chambers et al., 2009) and the greater power output during a RPE matched cycling time trial following amphetamine ingestion (Swart, 2009). … Dopamine stimulating drugs are known to enhance aspects of exercise performance (Roelands et al., 2008)
- Jump up^ Roelands B, De Pauw K, Meeusen R (June 2015). “Neurophysiological effects of exercise in the heat”. Scand. J. Med. Sci. Sports. 25 Suppl 1: 65–78. PMID 25943657. doi:10.1111/sms.12350.
This indicates that subjects did not feel they were producing more power and consequently more heat. The authors concluded that the “safety switch” or the mechanisms existing in the body to prevent harmful effects are overridden by the drug administration (Roelands et al., 2008b). Taken together, these data indicate strong ergogenic effects of an increased DA concentration in the brain, without any change in the perception of effort.
- ^ Jump up to:a b c d e f g “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. p. 11. Retrieved 30 December 2013.
- ^ Jump up to:a b c d e f g h i j k l “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 4–8. Retrieved 30 December 2013.
- ^ Jump up to:a b c d “Vyvanse Prescribing Information” (PDF). Shire Inc. Retrieved 1 July 2014.
- ^ Jump up to:a b c d e f Heedes G; Ailakis J. “Amphetamine (PIM 934)”. INCHEM. International Programme on Chemical Safety. Retrieved 24 June 2014.
- ^ Jump up to:a b c d e f g “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 4–6. Retrieved 30 December 2013.
- Jump up^ “FDA Pregnancy Categories” (PDF). United States Food and Drug Administration. 21 October 2004. Retrieved 31 October 2013.
- Jump up^ “Dexamphetamine tablets”. Therapeutic Goods Administration. Retrieved 12 April 2014.
- ^ Jump up to:a b Vitiello B (April 2008). “Understanding the risk of using medications for attention deficit hyperactivity disorder with respect to physical growth and cardiovascular function”. Child Adolesc. Psychiatr. Clin. N. Am. 17 (2): 459–474. PMC 2408826 . PMID 18295156. doi:10.1016/j.chc.2007.11.010.
- Jump up^ Ramey JT, Bailen E, Lockey RF (2006). “Rhinitis medicamentosa” (PDF). J. Investig. Allergol. Clin. Immunol. 16 (3): 148–155. PMID 16784007. Retrieved 29 April 2015.
Table 2. Decongestants Causing Rhinitis Medicamentosa
– Nasal decongestants:
– Sympathomimetic:
• Amphetamine - ^ Jump up to:a b “FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in children and young adults”. United States Food and Drug Administration. 20 December 2011. Retrieved 4 November 2013.
- Jump up^ Cooper WO, Habel LA, Sox CM, Chan KA, Arbogast PG, Cheetham TC, Murray KT, Quinn VP, Stein CM, Callahan ST, Fireman BH, Fish FA, Kirshner HS, O’Duffy A, Connell FA, Ray WA (November 2011). “ADHD drugs and serious cardiovascular events in children and young adults”. N. Engl. J. Med. 365 (20): 1896–1904. PMC 4943074 . PMID 22043968. doi:10.1056/NEJMoa1110212.
- ^ Jump up to:a b “FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in adults”. United States Food and Drug Administration. 15 December 2011. Retrieved 4 November 2013.
- Jump up^ Habel LA, Cooper WO, Sox CM, Chan KA, Fireman BH, Arbogast PG, Cheetham TC, Quinn VP, Dublin S, Boudreau DM, Andrade SE, Pawloski PA, Raebel MA, Smith DH, Achacoso N, Uratsu C, Go AS, Sidney S, Nguyen-Huynh MN, Ray WA, Selby JV (December 2011). “ADHD medications and risk of serious cardiovascular events in young and middle-aged adults”. JAMA. 306 (24): 2673–2683. PMC 3350308 . PMID 22161946. doi:10.1001/jama.2011.1830.
- Jump up^ Montgomery KA (June 2008). “Sexual desire disorders”. Psychiatry (Edgmont). 5 (6): 50–55. PMC 2695750 . PMID 19727285.
- Jump up^ O’Connor PG (February 2012). “Amphetamines”. Merck Manual for Health Care Professionals. Merck. Retrieved 8 May 2012.
- ^ Jump up to:a b c d Shoptaw SJ, Kao U, Ling W (January 2009). Shoptaw SJ, Ali R, ed. “Treatment for amphetamine psychosis”. Cochrane Database Syst. Rev. (1): CD003026. PMID 19160215. doi:10.1002/14651858.CD003026.pub3.
A minority of individuals who use amphetamines develop full-blown psychosis requiring care at emergency departments or psychiatric hospitals. In such cases, symptoms of amphetamine psychosis commonly include paranoid and persecutory delusions as well as auditory and visual hallucinations in the presence of extreme agitation. More common (about 18%) is for frequent amphetamine users to report psychotic symptoms that are sub-clinical and that do not require high-intensity intervention …
About 5–15% of the users who develop an amphetamine psychosis fail to recover completely (Hofmann 1983) …
Findings from one trial indicate use of antipsychotic medications effectively resolves symptoms of acute amphetamine psychosis. - ^ Jump up to:a b Greydanus D. “Stimulant Misuse: Strategies to Manage a Growing Problem” (PDF). American College Health Association (Review Article). ACHA Professional Development Program. p. 20. Archived from the original (PDF) on 3 November 2013. Retrieved 2 November 2013.
- ^ Jump up to:a b Childs E, de Wit H (May 2009). “Amphetamine-induced place preference in humans”. Biol. Psychiatry. 65 (10): 900–904. PMC 2693956 . PMID 19111278. doi:10.1016/j.biopsych.2008.11.016.
This study demonstrates that humans, like nonhumans, prefer a place associated with amphetamine administration. These findings support the idea that subjective responses to a drug contribute to its ability to establish place conditioning.
- ^ Jump up to:a b Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 15: Reinforcement and Addictive Disorders”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. pp. 364–375. ISBN 9780071481274.
- ^ Jump up to:a b Spiller HA, Hays HL, Aleguas A (June 2013). “Overdose of drugs for attention-deficit hyperactivity disorder: clinical presentation, mechanisms of toxicity, and management”. CNS Drugs. 27 (7): 531–543. PMID 23757186. doi:10.1007/s40263-013-0084-8.
Amphetamine, dextroamphetamine, and methylphenidate act as substrates for the cellular monoamine transporter, especially the dopamine transporter (DAT) and less so the norepinephrine (NET) and serotonin transporter. The mechanism of toxicity is primarily related to excessive extracellular dopamine, norepinephrine, and serotonin.
- Jump up^ Collaborators (2015). “Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013” (PDF). Lancet. 385 (9963): 117–171. PMC 4340604 . PMID 25530442. doi:10.1016/S0140-6736(14)61682-2. Retrieved 3 March 2015.
Amphetamine use disorders … 3,788 (3,425–4,145)
- Jump up^ Kanehisa Laboratories (10 October 2014). “Amphetamine – Homo sapiens (human)”. KEGG Pathway. Retrieved 31 October 2014.
- ^ Jump up to:a b c d e f Nechifor M (March 2008). “Magnesium in drug dependences”. Magnes. Res. 21 (1): 5–15. PMID 18557129.
- ^ Jump up to:a b c d e Ruffle JK (November 2014). “Molecular neurobiology of addiction: what’s all the (Δ)FosB about?”. Am. J. Drug Alcohol Abuse. 40 (6): 428–437. PMID 25083822. doi:10.3109/00952990.2014.933840.
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure.
- ^ Jump up to:a b c d e Nestler EJ (December 2013). “Cellular basis of memory for addiction”. Dialogues Clin. Neurosci. 15 (4): 431–443. PMC 3898681 . PMID 24459410.
Despite the importance of numerous psychosocial factors, at its core, drug addiction involves a biological process: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. … A large body of literature has demonstrated that such ΔFosB induction in D1-type [nucleus accumbens] neurons increases an animal’s sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement … Another ΔFosB target is cFos: as ΔFosB accumulates with repeated drug exposure it represses c-Fos and contributes to the molecular switch whereby ΔFosB is selectively induced in the chronic drug-treated state.41. … Moreover, there is increasing evidence that, despite a range of genetic risks for addiction across the population, exposure to sufficiently high doses of a drug for long periods of time can transform someone who has relatively lower genetic loading into an addict.
- Jump up^ Robison AJ, Nestler EJ (November 2011). “Transcriptional and epigenetic mechanisms of addiction”. Nat. Rev. Neurosci. 12 (11): 623–637. PMC 3272277 . PMID 21989194. doi:10.1038/nrn3111.
ΔFosB serves as one of the master control proteins governing this structural plasticity.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v Olsen CM (December 2011). “Natural rewards, neuroplasticity, and non-drug addictions”. Neuropharmacology. 61 (7): 1109–1122. PMC 3139704 . PMID 21459101. doi:10.1016/j.neuropharm.2011.03.010.
Similar to environmental enrichment, studies have found that exercise reduces self-administration and relapse to drugs of abuse (Cosgrove et al., 2002; Zlebnik et al., 2010). There is also some evidence that these preclinical findings translate to human populations, as exercise reduces withdrawal symptoms and relapse in abstinent smokers (Daniel et al., 2006; Prochaska et al., 2008), and one drug recovery program has seen success in participants that train for and compete in a marathon as part of the program (Butler, 2005). … In humans, the role of dopamine signaling in incentive-sensitization processes has recently been highlighted by the observation of a dopamine dysregulation syndrome in some patients taking dopaminergic drugs. This syndrome is characterized by a medication-induced increase in (or compulsive) engagement in non-drug rewards such as gambling, shopping, or sex (Evans et al., 2006; Aiken, 2007; Lader, 2008).
- ^ Jump up to:a b c d Lynch WJ, Peterson AB, Sanchez V, Abel J, Smith MA (September 2013). “Exercise as a novel treatment for drug addiction: a neurobiological and stage-dependent hypothesis”. Neurosci. Biobehav. Rev. 37 (8): 1622–1644. PMC 3788047 . PMID 23806439. doi:10.1016/j.neubiorev.2013.06.011.
These findings suggest that exercise may “magnitude”-dependently prevent the development of an addicted phenotype possibly by blocking/reversing behavioral and neuroadaptive changes that develop during and following extended access to the drug. … Exercise has been proposed as a treatment for drug addiction that may reduce drug craving and risk of relapse. Although few clinical studies have investigated the efficacy of exercise for preventing relapse, the few studies that have been conducted generally report a reduction in drug craving and better treatment outcomes … Taken together, these data suggest that the potential benefits of exercise during relapse, particularly for relapse to psychostimulants, may be mediated via chromatin remodeling and possibly lead to greater treatment outcomes.
- ^ Jump up to:a b c Zhou Y, Zhao M, Zhou C, Li R (July 2015). “Sex differences in drug addiction and response to exercise intervention: From human to animal studies”. Front. Neuroendocrinol. 40: 24–41. PMID 26182835. doi:10.1016/j.yfrne.2015.07.001.
Collectively, these findings demonstrate that exercise may serve as a substitute or competition for drug abuse by changing ΔFosB or cFos immunoreactivity in the reward system to protect against later or previous drug use. … The postulate that exercise serves as an ideal intervention for drug addiction has been widely recognized and used in human and animal rehabilitation.
- ^ Jump up to:a b c Linke SE, Ussher M (January 2015). “Exercise-based treatments for substance use disorders: evidence, theory, and practicality”. Am. J. Drug Alcohol Abuse. 41 (1): 7–15. PMC 4831948 . PMID 25397661. doi:10.3109/00952990.2014.976708.
The limited research conducted suggests that exercise may be an effective adjunctive treatment for SUDs. In contrast to the scarce intervention trials to date, a relative abundance of literature on the theoretical and practical reasons supporting the investigation of this topic has been published. … numerous theoretical and practical reasons support exercise-based treatments for SUDs, including psychological, behavioral, neurobiological, nearly universal safety profile, and overall positive health effects.
- ^ Jump up to:a b Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 15: Reinforcement and Addictive Disorders”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 386. ISBN 9780071481274.
Currently, cognitive–behavioral therapies are the most successful treatment available for preventing the relapse of psychostimulant use.
- Jump up^ Greene SL, Kerr F, Braitberg G (October 2008). “Review article: amphetamines and related drugs of abuse”. Emerg. Med. Australas. 20 (5): 391–402. PMID 18973636. doi:10.1111/j.1742-6723.2008.01114.x.
- Jump up^ Albertson TE (2011). “Amphetamines”. In Olson KR, Anderson IB, Benowitz NL, Blanc PD, Kearney TE, Kim-Katz SY, Wu AH. Poisoning & Drug Overdose (6th ed.). New York: McGraw-Hill Medical. pp. 77–79. ISBN 9780071668330.
- Jump up^ “Glossary of Terms”. Mount Sinai School of Medicine. Department of Neuroscience. Retrieved 9 February 2015.
- Jump up^ Volkow ND, Koob GF, McLellan AT (January 2016). “Neurobiologic Advances from the Brain Disease Model of Addiction”. N. Engl. J. Med. 374 (4): 363–371. PMID 26816013. doi:10.1056/NEJMra1511480.
Substance-use disorder: A diagnostic term in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) referring to recurrent use of alcohol or other drugs that causes clinically and functionally significant impairment, such as health problems, disability, and failure to meet major responsibilities at work, school, or home. Depending on the level of severity, this disorder is classified as mild, moderate, or severe.
Addiction: A term used to indicate the most severe, chronic stage of substance-use disorder, in which there is a substantial loss of self-control, as indicated by compulsive drug taking despite the desire to stop taking the drug. In the DSM-5, the term addiction is synonymous with the classification of severe substance-use disorder. - Jump up^ Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 15: Reinforcement and Addictive Disorders”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. p. 368. ISBN 9780071481274.
Such agents also have important therapeutic uses; cocaine, for example, is used as a local anesthetic (Chapter 2), and amphetamines and methylphenidate are used in low doses to treat attention deficit hyperactivity disorder and in higher doses to treat narcolepsy (Chapter 12). Despite their clinical uses, these drugs are strongly reinforcing, and their long-term use at high doses is linked with potential addiction, especially when they are rapidly administered or when high-potency forms are given.
- Jump up^ Kollins SH (May 2008). “A qualitative review of issues arising in the use of psycho-stimulant medications in patients with ADHD and co-morbid substance use disorders”. Curr. Med. Res. Opin. 24 (5): 1345–1357. PMID 18384709. doi:10.1185/030079908X280707.
When oral formulations of psychostimulants are used at recommended doses and frequencies, they are unlikely to yield effects consistent with abuse potential in patients with ADHD.
- Jump up^ Stolerman IP (2010). Stolerman IP, ed. Encyclopedia of Psychopharmacology. Berlin, Germany; London, England: Springer. p. 78. ISBN 9783540686989.
- Jump up^ Coghill DR, Caballero B, Sorooshian S, Civil R (June 2014). “A systematic review of the safety of lisdexamfetamine dimesylate”. CNS Drugs. 28 (6): 497–511. PMC 4057639 . PMID 24788672. doi:10.1007/s40263-014-0166-2.
The prodrug formulation of LDX may also lead to reduced abuse potential of LDX compared with immediate-release d-AMP.
- Jump up^ “Amphetamines: Drug Use and Abuse”. Merck Manual Home Edition. Merck. February 2003. Archived from the original on 17 February 2007. Retrieved 28 February 2007.
- Jump up^ Perez-Mana C, Castells X, Torrens M, Capella D, Farre M (September 2013). Pérez-Mañá C, ed. “Efficacy of psychostimulant drugs for amphetamine abuse or dependence”. Cochrane Database Syst. Rev. 9: CD009695. PMID 23996457. doi:10.1002/14651858.CD009695.pub2.
- Jump up^ Hyman SE, Malenka RC, Nestler EJ (July 2006). “Neural mechanisms of addiction: the role of reward-related learning and memory”. Annu. Rev. Neurosci. 29: 565–598. PMID 16776597. doi:10.1146/annurev.neuro.29.051605.113009.
- ^ Jump up to:a b c d e f g h Robison AJ, Nestler EJ (November 2011). “Transcriptional and epigenetic mechanisms of addiction”. Nat. Rev. Neurosci. 12 (11): 623–637. PMC 3272277 . PLID 21989194. doi:10.1038/nrn3111.
- ^ Jump up to:a b <` href=”https://en.wikipedia.org/wiki/Lisdexamfetamine#cite_ref-@ddiction_genetics_101-2″>c d <` href=”httpr://en.wikipedia.org/wiki/Lisdexamfetamine#cite_ref-Addiction_genetics_101-4″>e Rteiner H, Van Waes V (January 2013). “Addiction-related gene regulation: risks of exposure to cognitive enhancers vs. other psychostimulants”. Prog. Neurobiol. 100: 60–80. PMC 3525776 . PMID 23085425. doi:10.1016/j.pneurobio.2012.10.001.
- Jump up^ Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 4: Signal Transduction in the Brain”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 94. ISBN 9780071481274.
- Jump up^ Kanehisa Laboratories (29 October 2014). “Alcoholism – Homo sapiens (human)”. KEGG Pathway. Retrieved 31 October 2014.
- Jump up^ Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P (February 2009). “Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens”. Proc. Natl. Acad. Sci. U.S.A. 106 (8): 2915–2920. PMC 2650365 . PMID 19202072. doi:10.1073/pnas.0813179106.
- Jump up^ Nestler EJ (January 2014). “Epigenetic mechanisms of drug addiction”. Neuropharmacology. 76 Pt B: 259–268. PMC 3766384 . PMID 23643695. doi:10.1016/j.neuropharm.2013.04.004.
- ^ Jump up to:a b Blum K, Werner T, Carnes S, Carnes P, Bowirrat A, Giordano J, Oscar-Berman M, Gold M (March 2012). “Sex, drugs, and rock ‘n’ roll: hypothesizing common mesolimbic activation as a function of reward gene polymorphisms”. J. Psychoactive Drugs. 44 (1): 38–55. PMC 4040958 . PMID 22641964. doi:10.1080/02791072.2012.662112.
- Jump up^ Pitchers KK, Vialou V, Nestler EJ, Laviolette SR, Lehman MN, Coolen LM (February 2013). “Natural and drug rewards act on common neural plasticity mechanisms with ΔFosB as a key mediator”. J. Neurosci. 33 (8): 3434–3442. PMC 3865508 . PMID 23426671. doi:10.1523/JNEUROSCI.4881-12.2013.
- Jump up^ Beloate LN, Weems PW, Casey GR, Webb IC, Coolen LM (February 2016). “Nucleus accumbens NMDA receptor activation regulates amphetamine cross-sensitization and deltaFosB expression following sexual experience in male rats”. Neuropharmacology. 101: 154–164. PMID 26391065. doi:10.1016/j.neuropharm.2015.09.023.
- Jump up^ Stoops WW, Rush CR (May 2014). “Combination pharmacotherapies for stimulant use disorder: a review of clinical findings and recommendations for future research”. Expert Rev Clin Pharmacol. 7 (3): 363–374. PMC 4017926 . PMID 24716825. doi:10.1586/17512433.2014.909283.
Despite concerted efforts to identify a pharmacotherapy for managing stimulant use disorders, no widely effective medications have been approved.
- Jump up^ Perez-Mana C, Castells X, Torrens M, Capella D, Farre M (September 2013). “Efficacy of psychostimulant drugs for amphetamine abuse or dependence”. Cochrane Database Syst. Rev. 9: CD009695. PMID 23996457. doi:10.1002/14651858.CD009695.pub2.
To date, no pharmacological treatment has been approved for [addiction], and psychotherapy remains the mainstay of treatment. … Results of this review do not support the use of psychostimulant medications at the tested doses as a replacement therapy
- Jump up^ Forray A, Sofuoglu M (February 2014). “Future pharmacological treatments for substance use disorders”. Br. J. Clin. Pharmacol. 77 (2): 382–400. PMC 4014020 . PMID 23039267. doi:10.1111/j.1365-2125.2012.04474.x.
- ^ Jump up to:a b Grandy DK, Miller GM, Li JX (February 2016). “”TAARgeting Addiction”-The Alamo Bears Witness to Another Revolution: An Overview of the Plenary Symposium of the 2015 Behavior, Biology and Chemistry Conference”. Drug Alcohol Depend. 159: 9–16. PMID 26644139. doi:10.1016/j.drugalcdep.2015.11.014.
When considered together with the rapidly growing literature in the field a compelling case emerges in support of developing TAAR1-selective agonists as medications for preventing relapse to psychostimulant abuse.
- ^ Jump up to:a b Jing L, Li JX (August 2015). “Trace amine-associated receptor 1: A promising target for the treatment of psychostimulant addiction”. Eur. J. Pharmacol. 761: 345–352. PMC 4532615 . PMID 26092759. doi:10.1016/j.ejphar.2015.06.019.
Existing data provided robust preclinical evidence supporting the development of TAAR1 agonists as potential treatment for psychostimulant abuse and addiction.
- ^ Jump up to:a b Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 5: Excitatory and Inhibitory Amino Acids”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. pp. 124–125. ISBN 9780071481274.
- ^ Jump up to:a b c Carroll ME, Smethells JR (February 2016). “Sex Differences in Behavioral Dyscontrol: Role in Drug Addiction and Novel Treatments”. Front. Psychiatry. 6: 175. PMC 4745113 . PMID 26903885. doi:10.3389/fpsyt.2015.00175.
Physical Exercise
There is accelerating evidence that physical exercise is a useful treatment for preventing and reducing drug addiction … In some individuals, exercise has its own rewarding effects, and a behavioral economic interaction may occur, such that physical and social rewards of exercise can substitute for the rewarding effects of drug abuse. … The value of this form of treatment for drug addiction in laboratory animals and humans is that exercise, if it can substitute for the rewarding effects of drugs, could be self-maintained over an extended period of time. Work to date in [laboratory animals and humans] regarding exercise as a treatment for drug addiction supports this hypothesis. … Animal and human research on physical exercise as a treatment for stimulant addiction indicates that this is one of the most promising treatments on the horizon. - ^ Jump up to:a b c d Shoptaw SJ, Kao U, Heinzerling K, Ling W (April 2009). Shoptaw SJ, ed. “Treatment for amphetamine withdrawal”. Cochrane Database Syst. Rev. (2): CD003021. PMID 19370579. doi:10.1002/14651858.CD003021.pub2.
- Jump up^ “Dexedrine Prescribing Information” (PDF). United States Food and Drug Administration. Amedra Pharmaceuticals LLC. October 2013. Retrieved 4 November 2013.
- Jump up^ “Adderall IR Prescribing Information” (PDF). United States Food and Drug Administration. Teva Pharmaceuticals USA, Inc. October 2015. Retrieved 18 May 2016.
- Jump up^ “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. Retrieved 30 December 2013.
- Jump up^ Advokat C (July 2007). “Update on amphetamine neurotoxicity and its relevance to the treatment of ADHD”. J. Atten. Disord. 11 (1): 8–16. PMID 17606768. doi:10.1177/1087054706295605.
- ^ Jump up to:a b c d Bowyer JF, Hanig JP (November 2014). “Amphetamine- and methamphetamine-induced hyperthermia: Implications of the effects produced in brain vasculature and peripheral organs to forebrain neurotoxicity”. Temperature (Austin). 1 (3): 172–182. PMC 5008711 . PMID 27626044. doi:10.4161/23328940.2014.982049.
Hyperthermia alone does not produce amphetamine-like neurotoxicity but AMPH and METH exposures that do not produce hyperthermia (≥40°C) are minimally neurotoxic. Hyperthermia likely enhances AMPH and METH neurotoxicity directly through disruption of protein function, ion channels and enhanced ROS production. … The hyperthermia and the hypertension produced by high doses amphetamines are a primary cause of transient breakdowns in the blood-brain barrier (BBB) resulting in concomitant regional neurodegeneration and neuroinflammation in laboratory animals. … In animal models that evaluate the neurotoxicity of AMPH and METH, it is quite clear that hyperthermia is one of the essential components necessary for the production of histological signs of dopamine terminal damage and neurodegeneration in cortex, striatum, thalamus and hippocampus.
- Jump up^ “Amphetamine”. Hazardous Substances Data Bank. United States National Library of Medicine – Toxicology Data Network. Retrieved 26 February 2014.
Direct toxic damage to vessels seems unlikely because of the dilution that occurs before the drug reaches the cerebral circulation.
- Jump up^ Malenka RC, Nestler EJ, Hyman SE (2009). “Chapter 15: Reinforcement and addictive disorders”. In Sydor A, Brown RY. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 370. ISBN 9780071481274.
Unlike cocaine and amphetamine, methamphetamine is directly toxic to midbrain dopamine neurons.
- Jump up^ Sulzer D, Zecca L (February 2000). “Intraneuronal dopamine-quinone synthesis: a review”. Neurotox. Res. 1 (3): 181–195. PMID 12835101. doi:10.1007/BF03033289.
- Jump up^ Miyazaki I, Asanuma M (June 2008). “Dopaminergic neuron-specific oxidative stress caused by dopamine itself” (PDF). Acta Med. Okayama. 62 (3): 141–150. PMID 18596830.
- Jump up^ Hofmann FG (1983). A Handbook on Drug and Alcohol Abuse: The Biomedical Aspects (2nd ed.). New York, USA: Oxford University Press. p. 329. ISBN 9780195030570.
- ^ Jump up to:a b c d “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 8–10. Retrieved 30 December 2013.
- ^ Jump up to:a b “Identification”. Lisdexamfetamine. DrugBank. University of Alberta. 16 September 2013. Retrieved 13 June 2014.
- ^ Jump up to:a b c Jasinski DR, Krishnan S (June 2009). “Abuse liability and safety of oral lisdexamfetamine dimesylate in individuals with a history of stimulant abuse”. J. Psychopharmacol. (Oxford). 23 (4): 419–427. PMID 19329547. doi:10.1177/0269881109103113.
- ^ Jump up to:a b Miller GM (January 2011). “The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity”. J. Neurochem. 116 (2): 164–176. PMC 3005101 . PMID 21073468. doi:10.1111/j.1471-4159.2010.07109.x.
- ^ Jump up to:a b Eiden LE, Weihe E (January 2011). “VMAT2: a dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse”. Ann. N. Y. Acad. Sci. 1216: 86–98. Bibcode:2011NYASA1216…86E. PMC 4183197 . PMID 21272013. doi:10.1111/j.1749-6632.2010.05906.x.
VMAT2 is the CNS vesicular transporter for not only the biogenic amines DA, NE, EPI, 5-HT, and HIS, but likely also for the trace amines TYR, PEA, and thyronamine (THYR) … [Trace aminergic] neurons in mammalian CNS would be identifiable as neurons expressing VMAT2 for storage, and the biosynthetic enzyme aromatic amino acid decarboxylase (AADC).
- Jump up^ “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. pp. 1–18. Retrieved 7 October 2013.
- Jump up^ “Pharmacology”. Dextroamphetamine. DrugBank. University of Alberta. 8 February 2013. Retrieved 5 November 2013.
- ^ Jump up to:a b c d e f g h i j k l m n “Adderall XR Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 12–13. Retrieved 30 December 2013.
- Jump up^ “Pharmacology”. Amphetamine. DrugBank. University of Alberta. 8 February 2013. Retrieved 5 November 2013.
- ^ Jump up to:a b c d “Pharmacology and Biochemistry”. Amphetamine. Pubchem Compound. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 12 October 2013.
- ^ Jump up to:a b “Metabolism/Pharmacokinetics”. AMPHETAMINE. United States National Library of Medicine – Toxicology Data Network. Hazardous Substances Data Bank. Retrieved 5 January 2014.
Plasma protein binding, rate of absorption, & volumes of distribution of amphetamine isomers are similar. … The biological half-life of amphetamine is greater in drug dependent individuals than in control subjects, & distribution volumes are increased, indicating that greater affinity of tissues for the drug may contribute to development of amphetamine tolerance. … Concentrations of (14)C-amphetamine declined less rapidly in the plasma of human subjects maintained on an alkaline diet (urinary pH > 7.5) than those on an acid diet (urinary pH < 6). Plasma half-lives of amphetamine ranged between 16-31 hr & 8-11 hr, respectively, & the excretion of (14)C in 24 hr urine was 45 & 70%.
- ^ Jump up to:a b c “Vyvanse Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. January 2017. pp. 18–21. Retrieved 16 February 2017.
- Jump up^ Glennon RA (2013). “Phenylisopropylamine stimulants: amphetamine-related agents”. In Lemke TL, Williams DA, Roche VF, Zito W. Foye’s principles of medicinal chemistry (7th ed.). Philadelphia, USA: Wolters Kluwer Health/Lippincott Williams & Wilkins. pp. 646–648. ISBN 9781609133450.
The simplest unsubstituted phenylisopropylamine, 1-phenyl-2-aminopropane, or amphetamine, serves as a common structural template for hallucinogens and psychostimulants. Amphetamine produces central stimulant, anorectic, and sympathomimetic actions, and it is the prototype member of this class (39). … The phase 1 metabolism of amphetamine analogs is catalyzed by two systems: cytochrome P450 and flavin monooxygenase. … Amphetamine can also undergo aromatic hydroxylation to p-hydroxyamphetamine. … Subsequent oxidation at the benzylic position by DA β-hydroxylase affords p-hydroxynorephedrine. Alternatively, direct oxidation of amphetamine by DA β-hydroxylase can afford norephedrine.
- Jump up^ Taylor KB (January 1974). “Dopamine-beta-hydroxylase. Stereochemical course of the reaction” (PDF). J. Biol. Chem. 249 (2): 454–458. PMID 4809526. Retrieved 6 November 2014.
Dopamine-β-hydroxylase catalyzed the removal of the pro-R hydrogen atom and the production of 1-norephedrine, (2S,1R)-2-amino-1-hydroxyl-1-phenylpropane, from d-amphetamine.
- Jump up^ Krueger SK, Williams DE (June 2005). “Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism”. Pharmacol. Ther. 106 (3): 357–387. PMC 1828602 . PMID 15922018. doi:10.1016/j.pharmthera.2005.01.001.
Table 5: N-containing drugs and xenobiotics oxygenated by FMO - Jump up^ Cashman JR, Xiong YN, Xu L, Janowsky A (March 1999). “N-oxygenation of amphetamine and methamphetamine by the human flavin-containing monooxygenase (form 3): role in bioactivation and detoxication”. J. Pharmacol. Exp. Ther. 288 (3): 1251–1260. PMID 10027866.
- ^ Jump up to:a b c Santagati NA, Ferrara G, Marrazzo A, Ronsisvalle G (September 2002). “Simultaneous determination of amphetamine and one of its metabolites by HPLC with electrochemical detection”. J. Pharm. Biomed. Anal. 30 (2): 247–255. PMID 12191709. doi:10.1016/S0731-7085(02)00330-8.
- Jump up^ Horwitz D, Alexander RW, Lovenberg W, Keiser HR (May 1973). “Human serum dopamine-β-hydroxylase. Relationship to hypertension and sympathetic activity”. Circ. Res. 32 (5): 594–599. PMID 4713201. doi:10.1161/01.RES.32.5.594.
Subjects with exceptionally low levels of serum dopamine-β-hydroxylase activity showed normal cardiovascular function and normal β-hydroxylation of an administered synthetic substrate, hydroxyamphetamine.
- ^ Jump up to:a b “Substrate/Product”. butyrate-CoA ligase. BRENDA. Technische Universität Braunschweig. Retrieved 7 May 2014.
- ^ Jump up to:a b “Substrate/Product”. glycine N-acyltransferase. BRENDA. Technische Universität Braunschweig. Retrieved 7 May 2014.
- Jump up^ “Compound Summary”. p-Hydroxyamphetamine. PubChem Compound. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 15 October 2013.
- Jump up^ “Compound Summary”. p-Hydroxynorephedrine. PubChem Compound. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 15 October 2013.
- Jump up^ “Compound Summary”. Phenylpropanolamine. PubChem Compound. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 15 October 2013.
- Jump up^ “Molecular Weight Calculator”. Lenntech. Retrieved 19 August 2015.
- ^ Jump up to:a b “Dextroamphetamine Sulfate USP”. Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- ^ Jump up to:a b “D-amphetamine sulfate”. Tocris. 2015. Retrieved 19 August 2015.
- ^ Jump up to:a b “Amphetamine Sulfate USP”. Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- Jump up^ “Dextroamphetamine Saccharate”. Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- Jump up^ “Amphetamine Aspartate”. Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- Jump up^ “Vyvanse Prescribing Information” (PDF). United States Food and Drug Administration. Shire US Inc. January 2015. pp. 12–16. Retrieved 24 February 2015.
- Jump up^ Lisdexamfetamine Dimesylate: A Prodrug Stimulant for the Treatment of ADHD in Children and Adults
- Jump up^ FDA Adult Approval of Vyvanse – FDA Label and Approval History
- Jump up^ Health Canada Notice of Compliance – Vyvanse. 19 February 2009, retrieved on 9 March 2009.
- Jump up^ [1]. 8 February 2012, retrieved on 9 February 2012.
- Jump up^ Hirschler, Ben (7 February 2014). “UPDATE 2-Shire scraps Vyvanse for depression after failed trials”. Reuters. Retrieved 13 February 2014.
- Jump up^ http://www.shire.com/shireplc/en/investors/irshirenews?id=684
- Jump up^ http://www.shire.com/shireplc/en/investors/investorsnews/irshirenews?id=1055
- Jump up^ http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm432543.htm
- Jump up^ Cassels, Caroline. “FDA Okays Vyvanse for Binge Eating Disorder”. medscape.com. Retrieved 30 January 2015.
- Jump up^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 14 February 2017.
- Jump up^ http://www.shire.com/shireplc/en/investors/investorsnews/irshirenews?id=684
- Jump up^ Dale E, Bang-Andersen B, Sánchez C (May 2015). “Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs”. Biochem. Pharmacol. 95 (2): 81–97. PMID 25813654. doi:10.1016/j.bcp.2015.03.011.
- Jump up^ “Psychostimulants in the therapy of treatment-resistant depression Review of the literature and findings from a retrospective study in 65 depressed patients”. Dialogues Clin Neurosci. 1: 165–74. 1999. PMC 3181580 . PMID 22034135.


////////
CC(CC1=CC=CC=C1)NC(=O)C(CCCCN)N
FDA approves first subcutaneous C1 Esterase Inhibitor to treat rare genetic disease
The U.S. Food and Drug Administration today approved Haegarda, the first C1 Esterase Inhibitor (Human) for subcutaneous (under the skin) administration to prevent Hereditary Angioedema (HAE) attacks in adolescent and adult patients. The subcutaneous route of administration allows for easier at-home self-injection by the patient or caregiver, once proper training is received.
HAE, which is caused by having insufficient amounts of a plasma protein called C1-esterase inhibitor (or C1-INH), affects approximately 6,000 to 10,000 people in the U.S. People with HAE can develop rapid swelling of the hands, feet, limbs, face, intestinal tract or airway. These attacks of swelling can occur spontaneously, or can be triggered by stress, surgery or infection.
“The approval of Haegarda provides a new treatment option for adolescents and adults with Hereditary Angioedema,” said Peter Marks, M.D., Ph.D., director of FDA’s Center for Biologics Evaluation and Research. “The subcutaneous formulation allows patients to administer the product at home to help prevent attacks.”
Haegarda is a human plasma-derived, purified, pasteurized, lyophilized (freeze-dried) concentrate prepared from large pools of human plasma from U.S. donors. Haegarda is indicated for routine prophylaxis to prevent HAE attacks, but is not indicated for treatment of acute HAE attacks.
The efficacy of Haegarda was demonstrated in a multicenter controlled clinical trial. The study included 90 subjects ranging in age from 12 to 72 years old with symptomatic HAE. Subjects were randomized to receive twice per week subcutaneous doses of either 40 IU/kg or 60 IU/kg, and the treatment effect was compared to a placebo treatment period. During the 16 week treatment period, patients in both treatment groups experienced a significantly reduced number of HAE attacks compared to their placebo treatment period.
The most common side effects included injection site reactions, hypersensitivity (allergic) reactions, nasopharyngitis (swelling of the nasal passages and throat) and dizziness. Haegarda should not be used in individuals who have experienced life-threatening hypersensitivity reactions, including anaphylaxis, to a C1-INH preparation or its inactive ingredients.
Haegarda received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs to treat rare diseases or conditions.
The FDA granted approval of Haegarda to CSL Behring LLC.
///////////Haegarda, C1 Esterase inhibitor, CSL Behring LLC, fda 2017, orphan drug
HNIW, CL 20, 六硝基六氮杂异伍兹烷

HNIW, CL-20
- Molecular FormulaC6H6N12O12
- Average mass438.185 Da
-
1,3,4,7,8,10-hexanitrooctahydro-1H-5,2,6-(epiminomethanetriylimino)imidazo[4,5-b]pyrazineCAS 135285-90-4
- Hexanitrohexaazaisowurtzitane
- 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane
- Octahydro-1,3,4,7,8,10-hexanitro-5,2,6-(iminomethenimino)-1H-imidazo[4,5-b]pyrazine
- HNIW
- 六硝基六氮杂异伍兹烷
ABOUT AUTHOR
Thermochemistry, Physical Chemistry, Materials Chemistry

Staff Paweł Maksimowski Wincenty Skupiński Wojciech Pawłowski Waldemar Tomaszewski Tomasz Gołofit Katarzyna Cieślak

Faculty of Chemistry, Division of High Energetic Materials, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
Hexanitrohexaazaisowurtzitane /ˈhɛksɑːˈnaɪtroʊˈhɛksɑːˌæzɑːˌaɪsoʊˈvʊərtsɪteɪn/, also called HNIW and CL-20, is a nitroamine explosive with the formula C6H6N12O12. The structure of CL-20 was first proposed in 1979 by Dalian Institute of Chemical Physics.[1]In 1980s, CL-20 was developed by the China Lake facility, primarily to be used in propellants. It has a better oxidizer-to-fuel ratio than conventional HMX or RDX. It releases 20% more energy than traditional HMX-based propellants, and is widely superior to conventional high-energy propellants and explosives.
Industrial production of CL-20 was achieved in China in 2011, and it was soon fielded in propellant of solid rockets.[2] While most development of CL-20 has been fielded by the Thiokol Corporation, the US Navy (through ONR) has also been interested in CL-20 for use in rocket propellants, such as for missiles, as it has lower observability characteristics such as less visible smoke.[3]
CL-20 has not yet been fielded in any production weapons system, but is undergoing testing for stability, production capabilities, and other weapons characteristics.
Synthesis


THEN CONVERTED TO CL20, HNIW



Synthesis of CL20, HNIW

| 505 | Synthesis of CL-20: By oxidative debenzylation with cerium(IV) ammonium nitrate (CAN)
IPC: Int.Cl.8 C07D
|
A simple debenzylation approach has been discussed for the synthesis of hexanitrohexaazaisowurtzitane (HNIW or CL-20) one of the most powerful high explosives of today with cerium ammonium (IV) nitrate. | ||
 |
||||
| G M Gore, R Sivabalan*, U R Nair, A Saikia,
S Venugopalan & B R Gandhe |
||||
First, benzylamine (1) is condensed with glyoxal (2) under acidic and dehydrating conditions to yield the first intermediate compound.(3). Four benzyl groups selectively undergo hydrogenolysis using palladium on carbon and hydrogen. The amino groups are then acetylated during the same step using acetic anhydride as the solvent. (4). Finally, compound 4 is reacted with nitronium tetrafluoroborate and nitrosonium tetrafluoroborate, resulting in HNIW.[4]
(3R,9R)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane
- Molecular FormulaC6H6N12O12
- Average mass438.185 Da
-
(3R,9R)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecan(3R,9R)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane(3R,9R)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatétracyclo[5.5.0.03,11.05,9]dodécane5,2,6-(Iminomethanetriylimino)-1H-imidazo[4,5-b]pyrazine, octahydro-1,3,4,7,8,10-hexanitro-, (5R,7aR)-
(3R,5S,9R,11S)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane
- Molecular FormulaC6H6N12O12
- Average mass438.185 Da
-
(3R,5S,9R,11S)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecan(3R,5S,9R,11S)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane(3R,5S,9R,11S)-2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatétracyclo[5.5.0.03,11.05,9]dodécane5,2,6-(Iminomethanetriylimino)-1H-imidazo[4,5-b]pyrazine, octahydro-1,3,4,7,8,10-hexanitro-, (3aR,5S,6R,7aS)
Cocrystal product with HMX
In August 2012, Onas Bolton et al. published results showing that a cocrystal of 2 parts CL-20 and 1 part HMX had similar safety properties to HMX, but with a greater firing power closer to CL-20. [5][6]
Cocrystal product with TNT
In August 2011, Adam Matzger and Onas Bolton published results showing that a cocrystal of CL-20 and TNT had
The synthesis of 2,4,6,8,10,12-hexabenzyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.05,9.03,11]-dodecane (HBIW) is the first stage in the production of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.05,9.03,11] dodecane (CL-20), which is the most potent explosive known today. Because of the high performance characteristics of CL-20, a number of research projects are being conducted worldwide on CL-20 synthesis, properties and applications
Scale-Up Synthesis of Hexabenzylhexaazaisowurtzitane, an Intermediate in CL-20 Synthesis


After successful synthesis of hexabenzylhexaazaisowurtzitane (HBIW) on a laboratory scale (0.25 L reactor), it was performed on a multilaboratory scale (10 L reactor) and subsequently in an experimental installation in which a 300 L reactor was built. Seven syntheses were carried out in the unit on a pilot scale to produce 250 kg of HBIW. The pilot-scale syntheses ran with a yield comparable to those observed for the processes conducted on a large-laboratory scale. Some modifications were suggested that allowed for reduction of the HBIW weight unit by approximately 50%
HBIW was 89%. FTIR υ (cm–1): 3022, 2835, 1954, 1669, 1602, 1492, 1451, 1396, 1351, 1302, 1264, 1208, 1169, 1140, 1122, 1072, 1057, 1028, 1017, 986, 926, 896, 828, 792, 781, 749, 732, 698. 1H NMR (CDCl3, 400 MHz): δ 7.39–7.42 (m, 30 H, phenyl CH), 4.33 (s, 4 H, CH2), 4.26–4.27 (d, 8 H, CH2), 4.21 (s, 4 H, CH), 3.75 (s, 2, H, CH).
References
- Jump up^ 王征, 和霄雯 (2016-04-19). “北理工的爆轰速度 中国力量的可靠基石”. 北京理工大学新闻网.
- Jump up^ 黎轩平 (2016-04-23). ““我们要在宇宙空间占一个位置!””. 北京理工大学新闻网.
- Jump up^ Yirka, Bob (9 September 2011). “University chemists devise means to stabilize explosive CL-20”. Physorg.com. Retrieved 8 July 2012.
- Jump up^ Nair, U. R.; Sivabalan, R.; Gore, G. M.; Geetha, M.; Asthana, S. N.; Singh, H. (2005). “Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-based formulations (review)”. Combust. Explos. Shock Waves. 41 (2): 121–132. doi:10.1007/s10573-005-0014-2.
- Jump up^ High Power Explosive with Good Sensitivity: A 2:1 Cocrystal of CL-20:HMX, Crystal Growth & Design (American Chemical Society), 2012, 12 (9), pp 4311–4314, DOI: 10.1021/cg3010882, Publication Date (Web): August 7, 2012, accessed 7 September 2012
- Jump up^ Powerful new explosive could replace today’s state-of-the-art military explosive, SpaceWar.com, 6 September 2012, accessed 7 September 2012
- Jump up^ Angewandte Chemie International Edition
- Jump up^ Things I Won’t Work With: Hexanitrohexaazaisowurtzitane
Further reading
- Bolton, Onas; Adam J. Matzger (September 12, 2011). “Improved Stability and Smart-Material Functionality Realized in an Energetic Cocrystal”. Angewandte Chemie. 123 (38): 9122–9125. doi:10.1002/ange.201104164. Retrieved 8 July 2012.
- Lowe, Derek (11 November 2011) “Things I won’t work with: Hexanitrohexaazaisowurtzitane”
////////////////CL 20, 135285-90-4, HNIW
|
|||
| Names | |||
|---|---|---|---|
| IUPAC name
2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane
|
|||
Other names
|
|||
| Identifiers | |||
|
3D model (JSmol)
|
|||
| Abbreviations | CL-20, HNIW | ||
| ChEBI | |||
| ChemSpider | |||
| ECHA InfoCard | 100.114.169 | ||
|
PubChem CID
|
|||
| Properties | |||
| C 6N 12H 6O 12 |
|||
| Molar mass | 438.1850 g mol−1 | ||
| Density | 2.044 g cm−3 | ||
| Explosive data | |||
| Detonation velocity | 9.38 km s−1 | ||
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|||


- 1.Bayat, Y.; Malmir, S.; Hajighasemali, F.; Dehghani, H. Cent. Eur. J. Energy Mater. 2015, 12, 439– 458
- 2.Bayat, Y.; Zarandi, M.; Khadiv-Parsi, P.; Salimi, A. Cent. Eur. J. Energy Mater. 2015, 12, 459– 472
- 3.Gołofit, T.; Zyśk, K. J. J. Therm. Anal. Calorim. 2015, 119, 1931– 1939, DOI: 10.1007/s10973-015-4418-2
- 4.Maksimowski, P.; Adamiak, J. Propellants, Explos., Pyrotech. 2010, 35, 353– 358, DOI: 10.1002/prep.200900057
TILOGLIPTIN
PRESENTING 2 MOLECULES………..I AM NOT SURE WHICH IS TITLE MOLECULE
EMAIL ME amcrasto@gmail.com
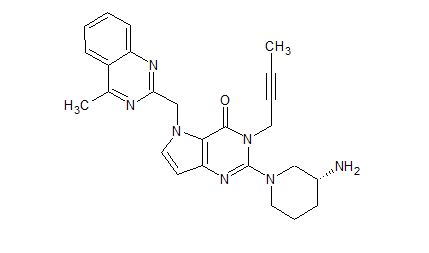
| Molecular Formula: | C25H27N7O |
|---|---|
| Molecular Weight: | 441.539 g/mol |
CAS 1428445-40-2
REF Bioorganic & Medicinal Chemistry (2013), 21(7), 1749-1755.
NEXT ONE………………

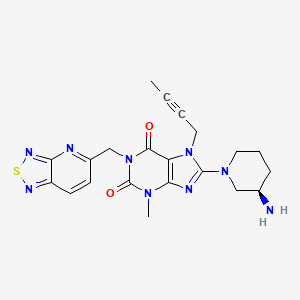
CAS 1415912-31-0
| Molecular Formula: | C21H23N9O2S |
|---|---|
| Molecular Weight: | 465.536 g/mol |
REF CN 102807568, CN 105315301, WO 2016019868
Salt………..
CAS 1874255-95-4
TILOGLIPTIN
HWH-ZGC-2-143
Guangzhou Institutes of Biomedicine and Health
![]()
Chia Tai Tianqing Pharmaceutical Group Co Ltd;

Non-insulin dependent diabetes
Dipeptidyl peptidase IV inhibitor (oral, type 2 diabetes),
DPP-IV inhibitors (oral, type 2 diabetes), Guangzhou Institutes of Biomedicine and Health/Jiangsu Chia Tai Tianqing Pharmaceutical ; HWH-ZGC-2-143 ;
Novel polymorphic forms of thiadiazole derivatives, preferably aglucin, sitagliptin, saxagliptin, vildagliptin, levaratine, useful for treating type II diabetes. Guangzhou Institutes of Biomedicine and Health , in collaboration with Jiangsu Chia Tai Tianqing Pharmaceutical , is investigating tilogliptin , an oral dipeptidyl peptidase IV inhibitor and a pyrrolopyrimidine analog, for treating type 2 diabetes.
As of June 2017, Centaurus BioPharma is developing diabetes therapy, CT-1006 and CT-1005 (in preclinical development) for treating diabetes mellitus.
See WO2016019868, claiming novel citric acid salt of 8-((R)-3-amino-piperidin-1-yl)-1-([1,2,5]-thiadiazolo [3,4-b] pyridine-5methyl)-7-(2-butyn-1-yl)-3-methyl-xanthine, coassigned to Lianyungang Runzhong Pharmaceutical .



PATENT
WO2016019868
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016019868
PATENT
Centaurus BioPharma Co Ltd; Chia Tai Tianqing Pharmaceutical Group Co Ltd
Discovery of potent dipeptidyl peptidase IV inhibitors through pharmacophore hybridization and hit-to-lead optimization
- a Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Science, 190 Kaiyuan Avenue, Guangzhou Science Park, Guangzhou 510530, China
- b Jiangsu Chia-Tai Tianqing Pharmaceutical Co. Ltd, No. 8 Julong North Rd., Xinpu Lianyungang, Jiangsu 222006, China
- c State Key Laboratory of Respiratory Disease, Guangzhou 510120, China
- https://doi.org/10.1016/j.bmc.2013.01.062

- A novel dipeptidyl peptidase IV inhibitor hit (5, IC50 = 0.86 μM) was structurally derived from our recently disclosed preclinical candidate 4 by replacing the cyanobenzyl with a butynyl based on pharmacophore hybridization. A hit-to-lead optimization effort was then initiated to improve its potency. Most N-substituted analogs exhibited good in vitro activity, and compound 18o (IC50 = 1.55 nM) was identified to be a potent dipeptidyl peptidase IV inhibitor with a significantly improved pharmacokinetic properties (bioavailablity: 41% vs 82.9%; T1/2: 2 h vs 4.9 h).
N[C@@H]1CCCN(C1)C5=Nc4ccn(Cc3nc2ccccc2c(C)n3)c4C(=O)N5CC#CC
N[C@@H]1CCCN(C1)c5nc4c(C(=O)N(Cc2ccc3nsnc3n2)C(=O)N4C)n5CC#CC
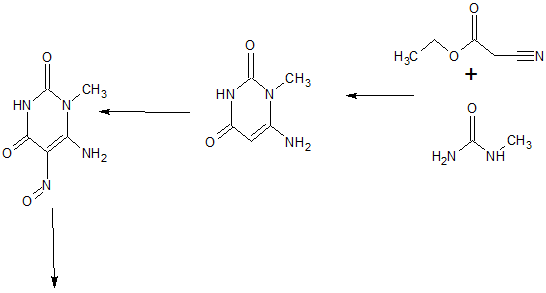
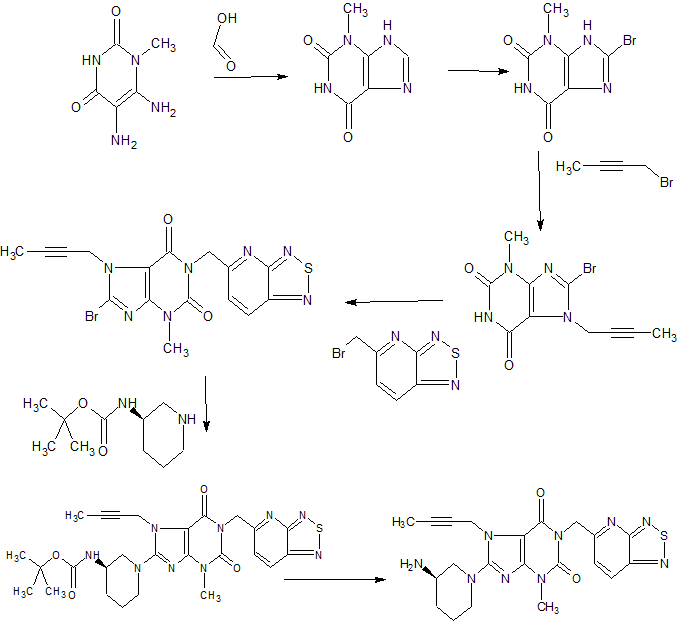
TEGAFUR
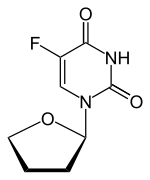
Tegafur
CAS 17902-23-7
- Molecular Weight,200.17, MF C8 H9 F N2 O3
-
172-173 °C
Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, 29(11), PG 3181-90
- Synonyms:Ftorafur
- ATC:L01BC03
- EINECS:241-846-2
- LD50:800 mg/kg (M, i.v.); 775 mg/kg (M, p.o.);
685 mg/kg (R, i.v.); 930 mg/kg (R, p.o.);
34 mg/kg (dog, p.o.)
Derivatives, monosodium salt
- Formula:C8H8FN2NaO3
- MW:222.15 g/mol
- CAS-RN:28721-46-2
Tegafur (INN, BAN, USAN) is a chemotherapeutic prodrug of 5-flourouracil (5-FU) used in the treatment of cancers. It is a component of the combination drug tegafur/uracil. When metabolised, it becomes 5-FU.[1]
Medical uses
As a prodrug to 5-FU it is used in the treatment of the following cancers:[2]
- Stomach (when combined with gimeracil and oteracil)
- Breast (with uracil)
- Gallbladder
- Lung (specifically adenocarcinoma, typically with uracil)
- Colorectal (usually when combined with gimeracil and oteracil)
- Head and neck
- Liver (with uracil)[3]
- Pancreatic
It is often given in combination with drugs that alter its bioavailability and toxicity such as gimeracil, oteracil or uracil.[2] These agents achieve this by inhibiting the enzyme dihydropyrimidine dehydrogenase (uracil/gimeracil) or orotate phosphoribosyltransferase (oteracil).[2]

Adverse effects
The major side effects of tegafur are similar to fluorouracil and include myelosuppression, central neurotoxicity and gastrointestinal toxicity (especially diarrhoea).[2] Gastrointestinal toxicity is the dose-limiting side effect of tegafur.[2] Central neurotoxicity is more common with tegafur than with fluorouracil.[2]

Pharmacogenetics
The dihydropyrimidine dehydrogenase (DPD) enzyme is responsible for the detoxifying metabolism of fluoropyrimidines, a class of drugs that includes 5-fluorouracil, capecitabine, and tegafur.[4] Genetic variations within the DPD gene (DPYD) can lead to reduced or absent DPD activity, and individuals who are heterozygous or homozygous for these variations may have partial or complete DPD deficiency; an estimated 0.2% of individuals have complete DPD deficiency.[4][5] Those with partial or complete DPD deficiency have a significantly increased risk of severe or even fatal drug toxicities when treated with fluoropyrimidines; examples of toxicities include myelosuppression, neurotoxicity and hand-foot syndrome.[4][5]
Mechanism of action
It is a prodrug to 5-FU, which is a thymidylate synthase inhibitor.[2]
Pharmacokinetics
It is metabolised to 5-FU by CYP2A6.[6][7]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]

The interactive pathway map can be edited at WikiPathways: “FluoropyrimidineActivity_WP1601”.



MASS SPECTRUM
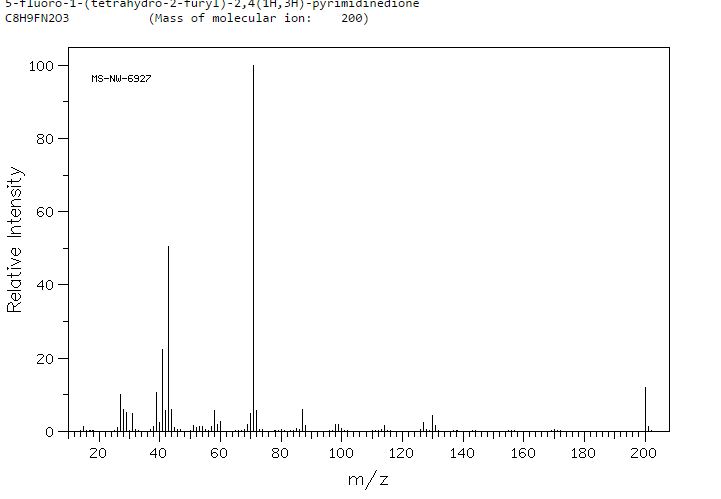
1H NMR
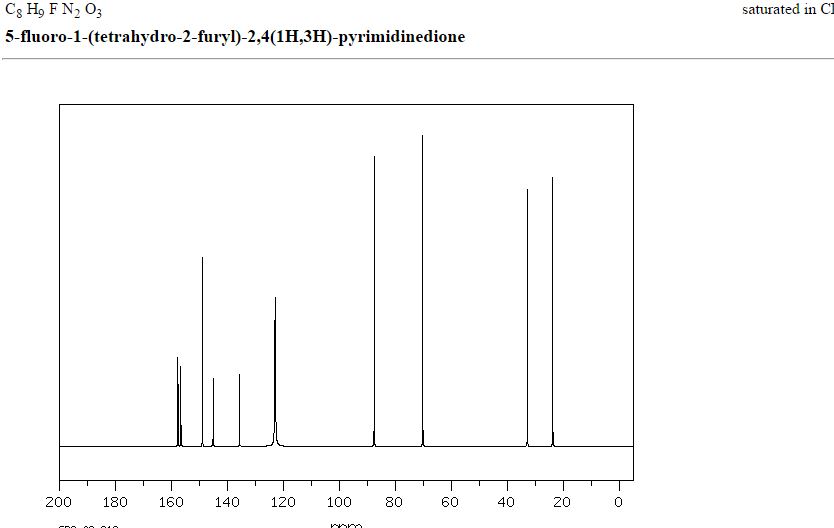
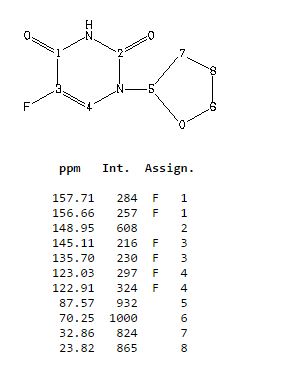
IR
13C NMR
RAMAN
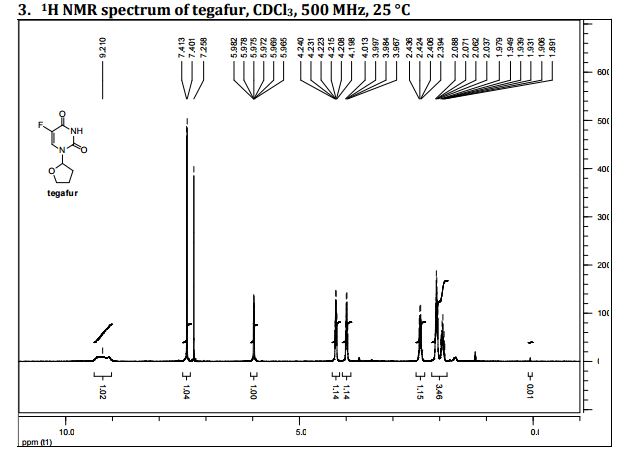
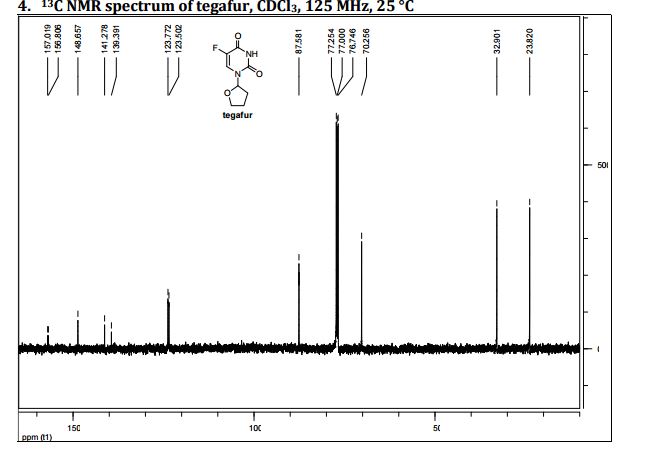
Synthesis
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 58138-78-6 | C10H19FN2O2Si2 | 1,3-bis(trimethylsilyl)fluorouracil | 2,4(1H,3H)-Pyrimidinedione, 5-fluoro-1,3-bis(trimethylsilyl)- |
| 13369-70-5 | C4H7ClO | 2-chlorotetrahydrofuran | Furan, 2-chlorotetrahydro- |
| 1191-99-7 | C4H6O | 2,3-dihydrofuran | Furan, 2,3-dihydro- |
| 51-21-8 | C4H3FN2O2 | 5-fluorouracil | 2,4(1H,3H)-Pyrimidinedione, 5-fluoro- |


SYN1
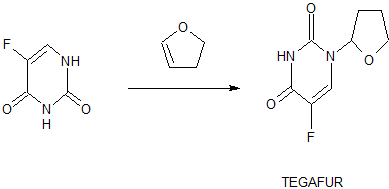
CN 106397416
SYN 2

Advanced Synthesis & Catalysis, 356(16), 3325-3330; 2014
PATENTS
CN 106397416
CN 104513230
CN 103159746
PATENT
tegafur is a derivative of 5-fluorouracil, and in 1967, Hiller of the former Soviet Union synthesized tegafur (SA Hiller, RA Zhuk, M. Yu. Lidak, et al. Substituted Uracil [ P, British Patent, 1168391 (1969)). In 1974, it was listed in Japan. China was successfully developed by Shandong Jinan Pharmaceutical Factory in 1979. Its present origin is Shanghai and Shandong provinces and cities. The anti-cancer effect of tegafur is similar to that of 5-fluorouracil and is activated in vivo by 5-fluorouracil through liver activation. Unlike 5-fluorouracil, tegafur is fat-soluble, has good oral absorption, maintains high concentrations in the blood for a long time and easily passes through the blood-brain barrier. Clinical and animal experiments show that tegafur on gastrointestinal cancer, breast cancer is better, the role of rectal cancer than 5-fluorouracil good, less toxic than 5-fluorouracil. Teflon has a chemotherapy index of 2-fold for 5-fluorouracil and only 1 / 4-1 / 7 of toxicity. So the addition of fluoride is widely used in cancer patients with chemotherapy.
[0003] The first synthesis of tegafur is Hiller ([SA Hiller, RA Zhuk, Μ. Yu. Lidak, et al. Substituted Uracil [P], British Patent, 1168391 (1969)]. 5-fluorouracil or 2,4-bis (trimethylsilyl) -5-fluorouracil (Me3Si-Fu, 1) and 2-chlorotetrahydrofuran (Thf-Cl), and it is reported that this synthesis must be carried out at low temperature (- 20 to -40 ° C), because Thf-Cl is unstable, and excess Thf-Cl results in a decomposition reaction, thereby reducing the yield of Thf-Fu.
[0004] Earl and Townsend also prepared 1_ (tetrahydro-2-furyl) uracil using Thf-Cl and 2,4-bis (trimethylsilyl) uracil, and then using trifluoromethyl fluorite to product Fluorination. Mitsugi Yasurnoto reacts with the Friedel-Crafts catalyst in the presence of 2,4-bis (trimethylsilyl) -5-fluorouracil (Me3Si-U, 1) 2-acetoxytetrahydrofuran (Thf-OAc, 2) (Kazu Kigasawa et al., 2-tert-Butoxy), & lt; RTI ID = 0.0 & gt;, & lt; / RTI & gt; (K. Kigasawa, M. Hiiragi, K. ffakisaka, et al. J. Heterocyclic Chem. 1977, 14: 473-475) was reacted with 5-Fu at 155-160 ° C. Reported in the literature for the fluoride production route there are the following questions: 1, high energy consumption. In the traditional synthesis method, in order to obtain the product, the second step of the reaction needs to continue heating at 160 ° C for 5-6 hours, high energy consumption; 2, difficult to produce, low yield: 5-fluorouracil as a solid powder The reaction needs to be carried out at a high temperature (160 ° C), which requires the use of a high boiling solvent N, N-dimethylformamide (DMF). But it is difficult to completely remove the fluoride from the addition of fluoride, because DMF can form hydrogen bonds with the fluoride molecules, difficult to separate from each other; 3, in order to unreacted 5-fluorouracil and tegafur separation and recycling , The use of carcinogenic solvent chloroform as a extractant in the conventional method to separate 5-fluorouracil and tegafur. However, the main role of chloroform on the central nervous system, with anesthesia, the heart, liver, kidney damage; the environment is also harmful to the water can cause pollution. Therefore, the use of volatile solvent chloroform, even if the necessary measures to reduce its volatilization, will still cause harm to human health and the environment; 4, low yield. Since both NI and N-3 in the 5-fluorouracil molecule react with 2-tert-butoxytetrahydrofuran, the addition of tegafur is also the addition of 1,3-bis (tetrahydro-2-furyl) -5 – Fluorouracil. Therefore, the improvement of the traditional production process of tegafur is a significant and imminent task.
Example 1 (for example, the best reaction conditions):
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 10. 0 mg CuCl2, microwave irradiation 250W at 25 ° C reaction 0. 6h. Cool to room temperature, add 1.95 g (15 mmol) of 5-fluorouracil (5-Fu), and microwave irradiation at 400 ° C for 100 ° C. After distilling off the low boiling solvent, the oil was obtained. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give 1.34349 g of product. Melting point: 160-165 ° C. The yield was 75%.
[0011] Example 2
Weigh 3,5 g (50 mmol) of 2,3-dihydrofuran and 3.8 g (100 mmol) of ethanol were added to a single-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 5mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cool to room temperature, add 1.95 g (15 mmol) of 5-fluorouracil (5-Fu), microwave irradiation 400W, reaction temperature 60 ° C under the reaction pool. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give the product 0. 46 g. Melting point: 160-165 ° C. The yield was 15%.
[0012] Example 3
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 15 ml of tetrahydrofuran (THF). And then weighed 20mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 2001, reaction temperature 1301: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give the product 1.81 g. Melting point: 160-165 ° C. The yield was 61%.
[0013] Example 4
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran and 19 g (500 mmol) of ethanol were added to a single-necked flask. To this was added 20 ml of tetrahydrofuran (THF). And then weighed IOmg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 2001, reaction temperature 1101: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give product U6g. Melting point: 160-165 ° C. The yield was 43%.
[0014] Example 5
Weigh 3,5 g (50 mmol) of 2,3-dihydrofuran and 9.5 g (250 mmol) of ethanol were added to a single-necked flask. To this was added 30 ml of tetrahydrofuran (THF). And then weighed IOmg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 6001, reaction temperature 1001: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from absolute ethanol to give 1.15 g of product. Melting point: 160-165 ° C. The yield was 38%.
[0015] Example 6
Weigh 3.5 g (50 mmol) of 2,3-dihydrofuran, 1.9 g (50 mmol) of ethanol was added to a one-necked flask. To this was added 25 ml of tetrahydrofuran (THF). And then weighed 15mg CuCl2, microwave irradiation 250W at 25 ° C for 0.6h. Cooled to room temperature, add 1.95 g (15 to 01) 5-fluorouracil (5 call 11), microwave irradiation 5001, reaction temperature 1101: reaction lh. The low boiling solvent was distilled off to give an oil. Rinsed with ether to give a white solid which was recrystallized from anhydrous ethanol to give product 2.10 g. Melting point: 160-165 ° C. The yield was 70%.
Paper
A novel protocol for preparation of tegafur (a prodrug of 5-fluorouracil) is reported. The process involves the 1,8-diazabicycloundec-7-ene-mediated alkylation of 5-fluorouracil with 2-acetoxytetrahydrofuran at 90 °C, followed by treatment of the prepurified mixture of the alkylation products with aqueous ethanol at 70 °C. The yield of the two-step process is 72%.
Synthesis of Tegafur by the Alkylation of 5-Fluorouracil under the Lewis Acid and Metal Salt-Free Conditions
Aleksandra Zasada, Ewa Mironiuk-Puchalska, and Mariola Koszytkowska-Stawińska*
Faculty of Chemistry, Warsaw University of Technology, ul. Noakowskiego 3, 00-664 Warszawa, Poland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00103
*E-mail: mkoszyt@ch.pw.edu.pl.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.7b00103
Click to access op7b00103_si_001.pdf
Tegafur, a prodrug of 5-fluorouracil (5-FUra), was discovered in 1967. The compound features high lipophilicity and water solubility compared to 5-FUra. Tegafur is used as a racemate since no significant difference in antitumor activity of enantiomers was observed.
The prodrug is gradually converted to 5-FUra by metabolism in the liver. Hence, a rapid breakdown of the released 5-FUra in the gastrointestinal tract is avoided.(6) In injectable form, tegafur provoked serious side effects, such as nausea, vomiting, or central nervous system disturbances.
The first generation of oral formulation of tegafur , UFT) is a combination of tegafur and uracil in a fixed molar ratio of 1:4, respectively. The uracil slows the metabolism of 5-FUra and reduces production of 2-fluoro-α-alanine as the toxic metabolite. UFT was approved in 50 countries worldwide excluding the USA.
S-1 is the next generation of oral formulation of tegafur.(7) It is a combination of tegafur, gimeracil, and oteracil in a fixed molar ratio of 1:0.4:1, respectively.
Gimeracil inhibits the enzyme responsible for the degradation of 5-FUra. Oteracil prevents the activation of 5-FUra in the gastrointestinal tract, thus minimizing the gastrointestinal toxicity of 5-FUra. S-1 is well-tolerated, but its safety can be influenced by schedule and dose, similar to any other cytotoxic agent. Since common side effects of S-1 can be managed with antidiarrheal and antiemetic medications, the drug can be administered in outpatient settings. S-1 was approved in Japan, China, Taiwan, Korea, and Singapore for the treatment of patients with gastric cancer.
In 2010, the Committee for Medicinal Products for Human Use (CHMP), a division of the European Medicines Agency (EMA), recommended the use of S-1 for the treatment of adults with advanced gastric cancer when given in a combination with cisplatin. Currently, S-1 has not been approved by the FDA in the United States.
There is a great interest in further examination of S-1 as an anticancer chemotherapeutic. Currently, 23 clinical trials with S-1 has been registered in National Institutes of Health (NIH). Combinations of S-1 and other anticancer agents have been employed in a majority of these trials.
5-Fluoro-1-(tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (Tegafur)
δH 1.89–2.10 (m, 3H), 2.38–2.45 (m, 1H), 3.97–4.01 (q-like m, 1H), 4.20–4.24 (dq-like m), 5.97–5.98 (m, 1H), 7.41 (d, 3JHF 6.1), 9.21 (bs, 1H, NH).
δC 23.82, 32.90, 70.26, 87.58, 123.63 (d, 2JCF 33.89), 140.33 (d, 1JCF 237.20) 148.66, 156.9 (d, 2JCF 26.81).
HRMS m/z calcd for C8H10N2O3F [M – H]+ 201.0670, found 201.0669.
Elemental analysis. Found C%, 46.42; H%, 4.45; N%, 13.35. Calcd for 3(C8H9N2O3F)·H2O: C%, 46.61; H%, 4.73; N%, 13.59.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN85108855A * | Nov 6, 1985 | Sep 24, 1986 | Central Chemical Research Institute | Preparation of 1- (2-tetrahydrofuryl) -5-fluorouracil |
| GB1168391A * | Title not available | |||
| JPS5452085A * | Title not available | |||
| JPS5455581A * | Title not available | |||
| JPS5459288A * | Title not available | |||
| JPS52118479A * | Title not available | |||
| JPS54103880A * | Title not available | |||
| US4256885 * | Dec 10, 1976 | Mar 17, 1981 | Mitsui Toatsu Kagaku Kabushiki Kaisha | Process for the preparation of 1- (2-tetrahydrofuryl) -5-fluorouracil |
| US5075446 * | Oct 12, 1990 | Dec 24, 1991 | Korea Advanced Institute Of Science & Technology | Synthesis of tetrahydro-2-furylated pyrimidine derivatives |
| Reference | ||
|---|---|---|
| 1 | * | KAZUO KIGASAWA, et al .: ” Studies on the Synthesis of Chemotherapeutics. Synthetic of 1- (2-Tetrahydrofuryl) -5-fluorouracil [Ftorafur] (Studies on the Syntheses of Heterocyclic Compound. Part 703) “, “J. HETEROCCLIC CHEM ., Vol. 14, 31 May 1977 (1977-05-31), pages 473 – 475 |
References
1
- El Sayed, YM; Sadée, W (1983). “Metabolic activation of R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil (ftorafur) to 5-fluorouracil by soluble enzymes”. Cancer Research. 43 (9): 4039–44. PMID 6409396.
- 2
- Sweetman, S, ed. (14 November 2011). “Martindale: The Complete Drug Reference”. Pharmaceutical Press. Retrieved 12 February 2014.
- 3
- Ishikawa, T (14 May 2008). “Chemotherapy with enteric-coated tegafur/uracil for advanced hepatocellular carcinoma.” (PDF). World Journal of Gastroenterology : WJG. 14 (18): 2797–2801. doi:10.3748/wjg.14.2797. PMC 2710718 . PMID 18473401.
- 4
- Caudle, KE; Thorn, CF; Klein, TE; Swen, JJ; McLeod, HL; Diasio, RB; Schwab, M (December 2013). “Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing.”. Clinical pharmacology and therapeutics. 94 (6): 640–5. doi:10.1038/clpt.2013.172. PMC 3831181 . PMID 23988873.
- 5
- Amstutz, U; Froehlich, TK; Largiadèr, CR (September 2011). “Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity.”. Pharmacogenomics. 12 (9): 1321–36. doi:10.2217/pgs.11.72. PMID 21919607.
- 6
- Nakayama, T; Noguchi, S (January 2010). “Therapeutic usefulness of postoperative adjuvant chemotherapy with Tegafur-Uracil (UFT) in patients with breast cancer: focus on the results of clinical studies in Japan.” (PDF). The Oncologist. 15 (1): 26–36. doi:10.1634/theoncologist.2009-0255. PMC 3227888 . PMID 20080863.
- 7
Matt P, van Zwieten-Boot B, Calvo Rojas G, Ter Hofstede H, Garcia-Carbonero R, Camarero J, Abadie E, Pignatti F (October 2011). “The European Medicines Agency review of Tegafur/Gimeracil/Oteracil (Teysuno™) for the treatment of advanced gastric cancer when given in combination with cisplatin: summary of the Scientific Assessment of the Committee for medicinal products for human use (CHMP).” (PDF). The Oncologist. 16 (10): 1451–1457. doi:10.1634/theoncologist.2011-0224. PMC 3228070 ![]() . PMID 21963999.
. PMID 21963999.
- (1) Hirose, Takashi; Oncology Reports 2010, V24(2), P529-536
- (2) Fujita, Ken-ichi; Cancer Science 2008, V99(5), P1049-1054
- (3) Tahara, Makoto; Cancer Science 2011, V102(2), P419-424
- (4) Chu, Quincy Siu-Chung; Clinical Cancer Research 2004, V10(15), P4913-4921
- (5) Tominaga, Kazunari; Oncology 2004, V66(5), P358-364
- (6) Peters, Godefridus J.; Clinical Cancer Research 2004, V10(12, Pt. 1), P4072-4076
- (7) Kim, Woo Young; Cancer Science 2007, V98(10), P1604-1608
- Hillers, Solomon; Puti Sinteza i Izyskaniya Protivoopukholevykh Preparatov 1970, VNo. 3, P109-12
- Grishko, V. A.; Trudy Kazakhskogo Nauchno-Issledovatel’skogo Instituta Onkologii i Radiologii 1977, V12, P110-14
- Ootsu, Koichiro; Takeda Kenkyushoho 1978, V37(3-4), P267-77
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Yabuuchi, Youichi; Oyo Yakuri 1971, V5(4), P569-84
- Germane, S.; Eksperimental’naya i Klinicheskaya Farmakoterapiya 1970, (1), P85-92
- JP 56046814 A 1981
MORE
- AIST: Integrated Spectral Database System of Organic Compounds. (Data were obtained from the National Institute of Advanced Industrial Science and Technology (Japan))
- ACD-A: Sigma-Aldrich (Spectral data were obtained from Advanced Chemistry Development, Inc.)
- Nomura, Hiroaki; Chemical & Pharmaceutical Bulletin 1979, V27(4), P899-906
- Sakurai, Kuniyoshi; Chemical & Pharmaceutical Bulletin 1978, V26(11), P3565-6
- Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, V29(11), P3181-90
- Lukevics, E.; Zhurnal Obshchei Khimii 1981, V51(4), P827-34
- Needham, F.; Powder Diffraction 2006, V21(3), P245-247
-
- Nomura, Hiroaki; Chemical & Pharmaceutical Bulletin 1979, V27(4), P899-906
- Sakurai, Kuniyoshi; Chemical & Pharmaceutical Bulletin 1978, V26(11), P3565-6
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Miyashita, Osamu; Chemical & Pharmaceutical Bulletin 1981, V29(11), P3181-90
- “PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
- Lukevics, E.; Zhurnal Obshchei Khimii 1981, V51(4), P827-34
- Lukevics, E.; Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija 1982, (3), P317-20
- Kruse, C. G.; Recueil des Travaux Chimiques des Pays-Bas 1979, V98(6), P371-80
- Lukevics, E.; Latvijas PSR Zinatnu Akademijas Vestis, Kimijas Serija 1981, (4), P492-3
- Kametani, Tetsuji; Heterocycles 1977, V6(5), P529-33
- Kametani, Tetsuji; Journal of Heterocyclic Chemistry 1977, V14(3), P473-5
- Hillers, S.; GB 1168391 1969
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Biological half-life | 3.9-11 hours |
| Identifiers | |
| Synonyms | 5-fluoro-1-(oxolan-2-yl)pyrimidine-2,4-dione |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.038.027 |
| Chemical and physical data | |
| Formula | C8H9FN2O3 |
| Molar mass | 200.16 g/mol |
| 3D model (Jmol) | |
///////////TEGAFUR
FC1=CN(C2CCCO2)C(=O)NC1=O
FDA approves first cancer treatment for any solid tumor with a specific genetic feature
May 23, 2017
Release
The U.S. Food and Drug Administration today granted accelerated approval to a treatment for patients whose cancers have a specific genetic feature (biomarker). This is the first time the agency has approved a cancer treatment based on a common biomarker rather than the location in the body where the tumor originated.
Keytruda (pembrolizumab) is indicated for the treatment of adult and pediatric patients with unresectable or metastatic solid tumors that have been identified as having a biomarker referred to as microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR). This indication covers patients with solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options and patients with colorectal cancer that has progressed following treatment with certain chemotherapy drugs.
“This is an important first for the cancer community,” said Richard Pazdur, M.D., acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research and director of the FDA’s Oncology Center of Excellence. “Until now, the FDA has approved cancer treatments based on where in the body the cancer started—for example, lung or breast cancers. We have now approved a drug based on a tumor’s biomarker without regard to the tumor’s original location.”
MSI-H and dMMR tumors contain abnormalities that affect the proper repair of DNA inside the cell. Tumors with these biomarkers are most commonly found in colorectal, endometrial and gastrointestinal cancers, but also less commonly appear in cancers arising in the breast, prostate, bladder, thyroid gland and other places. Approximately 5 percent of patients with metastatic colorectal cancer have MSI-H or dMMR tumors.
Keytruda works by targeting the cellular pathway known as PD-1/PD-L1 (proteins found on the body’s immune cells and some cancer cells). By blocking this pathway, Keytruda may help the body’s immune system fight the cancer cells. The FDA previously approved Keytruda for the treatment of certain patients with metastatic melanoma, metastatic non-small cell lung cancer, recurrent or metastatic head and neck cancer, refractory classical Hodgkin lymphoma, and urothelial carcinoma.
Keytruda was approved for this new indication using the Accelerated Approvalpathway, under which the FDA may approve drugs for serious conditions where there is unmet medical need and a drug is shown to have certain effects that are reasonably likely to predict a clinical benefit to patients. Further study is required to verify and describe anticipated clinical benefits of Keytruda, and the sponsor is currently conducting these studies in additional patients with MSI-H or dMMR tumors.
The safety and efficacy of Keytruda for this indication were studied in patients with MSI-H or dMMR solid tumors enrolled in one of five uncontrolled, single-arm clinical trials. In some trials, patients were required to have MSI-H or dMMR cancers, while in other trials, a subgroup of patients were identified as having MSI-H or dMMR cancers by testing tumor samples after treatment began. A total of 15 cancer types were identified among 149 patients enrolled across these five clinical trials. The most common cancers were colorectal, endometrial and other gastrointestinal cancers. The review of Keytruda for this indication was based on the percentage of patients who experienced complete or partial shrinkage of their tumors (overall response rate) and for how long (durability of response). Of the 149 patients who received Keytruda in the trials, 39.6 percent had a complete or partial response. For 78 percent of those patients, the response lasted for six months or more.
Common side effects of Keytruda include fatigue, itchy skin (pruritus), diarrhea, decreased appetite, rash, fever (pyrexia), cough, difficulty breathing (dyspnea), musculoskeletal pain, constipation and nausea. Keytruda can cause serious conditions known as immune-mediated side effects, including inflammation of healthy organs such as the lungs (pneumonitis), colon (colitis), liver (hepatitis), endocrine glands (endocrinopathies) and kidneys (nephritis). Complications or death related to allogeneic hematopoietic stem cell transplantation after using Keytruda has occurred.
Patients who experience severe or life-threatening infusion-related reactions should stop taking Keytruda. Women who are pregnant or breastfeeding should not take Keytruda because it may cause harm to a developing fetus or newborn baby. The safety and effectiveness of Keytruda in pediatric patients with MSI-H central nervous system cancers have not been established.
The FDA granted this application Priority Review designation, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted accelerated approval of Keytruda to Merck & Co.
///////////Keytruda, pembrolizumab, BIO MARKER, MERCK, FDA 2017
FDA approves first drug Actemra (tocilizumab) to specifically treat giant cell arteritis
May 22, 2017
Release
The U.S. Food and Drug Administration today expanded the approved use of subcutaneous Actemra (tocilizumab) to treat adults with giant cell arteritis. This new indication provides the first FDA-approved therapy, specific to this type of vasculitis.
“We expedited the development and review of this application because this drug fulfills a critical need for patients with this serious disease who had limited treatment options,” said Badrul Chowdhury, M.D., Ph.D., director of the Division of Pulmonary, Allergy, and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research.
Giant cell arteritis is a form of vasculitis, a group of disorders that results in inflammation of blood vessels. This inflammation causes the arteries to narrow or become irregular, impeding adequate blood flow. In giant cell arteritis, the vessels most involved are those of the head, especially the temporal arteries (located on each side of the head). For this reason, the disorder is sometimes called temporal arteritis. However, other blood vessels, including large ones like the aorta, can become inflamed in giant cell arteritis. Standard treatment involves high doses of corticosteroids that are tapered over time.
The efficacy and safety of subcutaneous (injected under the skin) Actemra for giant cell arteritis were established in a double-blind, placebo-controlled study with 251 patients with giant cell arteritis. The primary efficacy endpoint was the proportion of patients achieving sustained remission from Week 12 through Week 52. Sustained remission was defined as the absence of symptoms of giant cell arteritis, normalization of inflammatory laboratory tests, and tapering the use of prednisone (a steroid drug). A greater proportion of patients receiving subcutaneous Actemra with standardized prednisone regimens achieved sustained remission from Week 12 through Week 52 as compared to patients receiving placebo with standardized prednisone regimens. The cumulative prednisone dose was lower in treated patients with Actemra relative to placebo.
The overall safety profile observed in the Actemra treatment groups was generally consistent with the known safety profile of Actemra. Actemra carries a Boxed Warning for serious infections. Patients treated with Actemra who develop a serious infection should stop that treatment until the infection is controlled. Live vaccines should be avoided during treatment with Actemra. Actemra should be used with caution in patients at increased risk of gastrointestinal perforation. Hypersensitivity reactions, including anaphylaxis and death, have occurred. Laboratory monitoring is recommended due to potential consequences of treatment-related changes in neutrophils (type of white blood cell), platelets, lipids and liver function tests.
Subcutaneous Actemra was previously approved for the treatment of moderate to severely active rheumatoid arthritis. Intravenous Actemra was also previously approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis and polyarticular juvenile idiopathic arthritis. Intravenous administration is not approved for giant cell arteritis.
The FDA granted this application a Breakthrough Therapy designation and a Priority Review.
The FDA granted the supplemental approval of Actemra to Hoffman La Roche, Inc.
FDA expands approved use of Kalydeco IVACAFTOR to treat additional mutations of cystic fibrosis
For Immediate Release
May 17, 2017
Release
The U.S. Food and Drug Administration today expanded the approved use of Kalydeco (ivacaftor) for treating cystic fibrosis. The approval triples the number of rare gene mutations that the drug can now treat, expanding the indication from the treatment of 10 mutations, to 33. The agency based its decision, in part, on the results of laboratory testing, which it used in conjunction with evidence from earlier human clinical trials. The approach provides a pathway for adding additional, rare mutations of the disease, based on laboratory data.
“Many rare cystic fibrosis mutations have such small patient populations that clinical trial studies are not feasible,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “This challenge led us to using an alternative approach based on precision medicine, which made it possible to identify certain gene mutations that are likely to respond to Kalydeco.
Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery due to the movement of sufficient ions (chloride) and water in and out of the cells. People with the progressive disease have a defective cystic fibrosis transmembrane conductance regulator (CFTR) gene that can’t regulate the movement of ions and water, causing the secretions to become sticky and thick. The secretions build up in the lungs, digestive tract and other parts of the body leading to severe respiratory and digestive problems, as well as other complications such as infections and diabetes.
Results from an in vitro cell-based model system have been shown to reasonably predict clinical response to Kalydeco. When additional mutations responded to Kalydeco in the laboratory test, researchers were thus able to extrapolate clinical benefit demonstrated in earlier clinical trials of other mutations. This resulted in the addition of gene mutations for which the drug is now indicated.
Kalydeco, available as tablets or oral granules taken two times a day with fat-containing food, helps the protein made by the CFTR gene function better and as a result, improves lung function and other aspects of cystic fibrosis, including weight gain. If the patient’s genotype is unknown, an FDA-cleared cystic fibrosis mutation test should be used to detect the presence of a CFTR mutation followed by verification with bi-directional sequencing when recommended by the mutation test instructions for use.
Cystic fibrosis is a rare disease that affects about 30,000 people in the United States.Kalydeco is indicated for patients aged 2 and older who have one mutation in the CFTR gene that is responsive to drug treatment based on clinical and/or in vitro (laboratory) data. The expanded indication will affect another 3 percent of the cystic fibrosis population, impacting approximately 900 patients. Kalydeco serves as an example of how successful patient-focused drug development can provide greater understanding about a disease. For example, the Cystic Fibrosis Foundation maintains a 28,000-patient registry, including genetic data, which it makes available for research.
Common side effects of Kalydeco include headache; upper respiratory tract infection (common cold) including sore throat, nasal or sinus congestion, or runny nose; stomach (abdominal) pain; diarrhea; rash; nausea; and dizziness. Kalydeco is associated with risks including elevated transaminases (various enzymes produced by the liver) and pediatric cataracts. Co-administration with strong CYP3A inducers (e.g., rifampin, St. John’s wort) substantially decreases exposure of Kalydeco, which may diminish effectiveness, and is therefore not recommended.
Kalydeco is manufactured for Boston-based Vertex Pharmaceuticals Inc.
BMS 986205


BMS 986205
CAS: 1923833-60-6
Phase 1 cancer
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
- Originator Bristol-Myers Squibb
- Class Antineoplastics
- 01 Feb 2016 Phase-I/II clinical trials in Cancer (Combination therapy, Late-stage disease, Second-line therapy or greater) in Canada (PO) (NCT02658890)
- 31 Jan 2016 Preclinical trials in Cancer in USA (PO) before January 2016
- 01 Jan 2016 Bristol-Myers Squibb plans a phase I/IIa trial for Cancer (Late-stage disease, Combination therapy, Second-line therapy or greater) in USA, Australia and Canada (PO) (NCT02658890)

Hilary Beck
 EX Principal Investigator, Company NameFLX Bio, Inc.,
EX Principal Investigator, Company NameFLX Bio, Inc.,
CURRENTLY Director, Medicinal Chemistry at IDEAYA Biosciences, IDEAYA Biosciences, The University of Texas at Austin
![]()

Brian Wong
Chief Executive Officer at FLX Bio, Inc.
Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt

Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V

Juan Jaen

Jordan Fridman
Chief Scientific Officer at FLX Bio, Inc.

Rekha Hemrajani
Chief Operating Officer at FLX Bio, Inc

Max Osipov
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran