
Home » Uncategorized (Page 27)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DOCUSATE
DOCUSATE
1,4-Bis(2-ethylhexyl) sulfosuccinate
- Molecular FormulaC20H38O7S
- Average mass422.577 Da
1,4-Bis[(2-ethylhexyl)oxy]-1,4-dioxobutane-2-sulfonic acid
10041-19-7[RN]
233-124-0[EINECS]
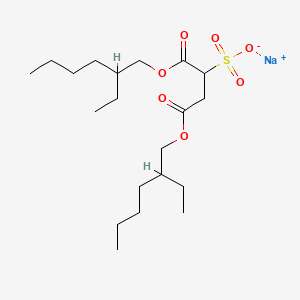
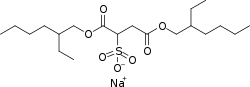
Docusate Sodium
Dioctyl sodium sulfosuccinate
sodium;1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate
CAS Registry Number: 577-11-7
CAS Name: Sulfobutanedioic acid 1,4-bis(2-ethylhexyl) ester sodium salt
Additional Names: sulfosuccinic acid 1,4-bis(2-ethylhexyl) ester S-sodium salt; bis(2-ethylhexyl)sodium sulfosuccinate; dioctyl sodium sulfosuccinate; sodium dioctyl sulfosuccinate; DSS
Trademarks: Aerosol OT (Cyanamid); Colace (Roberts); Comfolax (Searle); Coprola (Dunster); Dioctylal (Continental Pharma); Dioctyl (Medo); Diotilan (Chinoin); Disonate (Lannett); Doxinate (Hoechst); Doxol (Blair); Dulcivac (Harvey); Jamylène (Thaplix); Molatoc; Molcer (Wallace); Nevax; Regutol (Schering-Plough); Soliwax (Concept Pharm.); Velmol (Berlex); Waxsol (Norgine); Yal (Ritter)
Molecular Formula: C20H37NaO7S
Molecular Weight: 444.56
Percent Composition: C 54.03%, H 8.39%, Na 5.17%, O 25.19%, S 7.21%
Literature References: Prepn: Jaeger, US2028091; US2176423 (1936, 1939, both to Am. Cyanamid). Structure and wetting power: Caryl, Ind. Eng. Chem.33, 731 (1941). Comprehensive description: S. Ahuja, J. Cohen, Anal. Profiles Drug Subs.2, 199-219 (1973); 12, 713-720 (1983). For structure see Docusate calcium.
Properties: Available as wax-like solid, usually in rolls of tissue-thin material; also as 50-75% solns in various solvents. Soly in water (g/l): 15 (25°), 23 (40°), 30 (50°), 55 (70°). Sol in CCl4, petr ether, naphtha, xylene, dibutyl phthalate, liq petrolatum, acetone, alcohol, vegetable oils. Very sol in water + alcohol, water + water-miscible organic solvents. Stable in acid and neutral solns; hydrolyzes in alkaline solns.
Derivative Type: Docusate potassium
CAS Registry Number: 7491-09-0
Trademarks: Rectalad (Carter-Wallace)
Molecular Formula: C20H37KO7S
Molecular Weight: 460.67
Percent Composition: C 52.14%, H 8.10%, K 8.49%, O 24.31%, S 6.96%
NOTE: Ingredient of the laxative Peri-Colace (Roberts) which also contains casanthranol.Use: Sodium salt as pharmaceutic aid (surfactant); as wetting agent in industrial, pharmaceutical, cosmetic and food applications; dispersing and solubilizing agent in foods; adjuvant in tablet formation.
Therap-Cat: Stool softener.
Therap-Cat-Vet: Stool softener.
Keywords: Laxative/Cathartic.
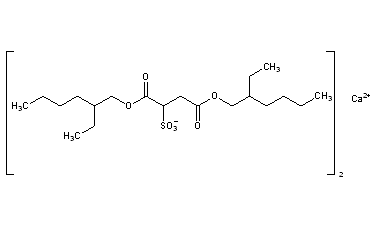
Docusate Calcium
CAS Registry Number: 128-49-4
CAS Name: Sulfobutanedioic acid 1,4-bis(2-ethylhexyl)ester calcium salt
Additional Names: bis[2-ethylhexyl]calcium sulfosuccinate; calcium dioctyl sulfosuccinate; dioctyl calcium sulfosuccinate
Trademarks: Surfak (HMR)
Molecular Formula: C40H74CaO14S2
Molecular Weight: 883.22
Percent Composition: C 54.40%, H 8.44%, Ca 4.54%, O 25.36%, S 7.26%
Literature References: Prepd from dioctyl sodium sulfosuccinate dissolved in isopropanol and from calcium chloride dissolved in methanol: Klotz, US3035973 (1962 to Lloyd Brothers).
Properties: White precipitate. Sol in mineral and vegetable oils, liq polyethylene glycol. Practically insol in glycerol. Claimed to have greater surface-active wetting properties than the sodium salt.
NOTE: Ingredient of Doxidan (HMR) which also contains phenolphthalein.
Therap-Cat: Stool softener.
Keywords: Laxative/Cathartic.
Derivatives
free acid
- Formula:C20H38O7S
- MW:422.58 g/mol
- CAS-RN:10041-19-7
- EINECS:233-124-0
calcium salt
- Formula:C40H74CaO14S2
- MW:883.23 g/mol
- CAS-RN:128-49-4
- EINECS:204-889-8
potassium salt
- Formula:C20H37KO7S
- MW:460.67 g/mol
- CAS-RN:7491-09-0
- EINECS:231-308-5
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 141-02-6 | C20H36O4 | bis(2-ethylhexyl) fumarate | 2-Butenedioic acid (E)-, bis(2-ethylhexyl) ester |
| C4H4O4 | (E)-2-butenedioic acid | ||
| 104-76-7 | C8H18O | 2-ethyl-1-hexanol | 1-Hexanol, 2-ethyl- |
SYN
https://scialert.net/fulltext/?doi=jas.2011.1396.1400
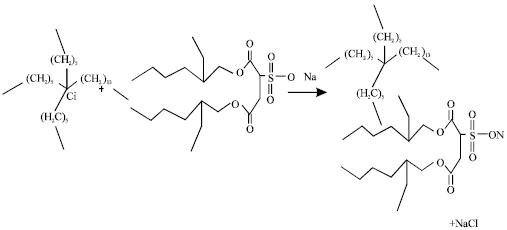 | |
| Fig. 1: | Synthesis of Trihexyltetradecylphosphonium octylsulfosuccinate [P6, 6, 6, 14][docusate] |
SYN

Docusate is the common chemical and pharmaceutical name of the anionbis(2-ethylhexyl) sulfosuccinate, also commonly called dioctyl sulfosuccinate (DOSS).[2][3][4]
Salts of this anion, especially docusate sodium, are widely used in medicine as laxatives and as stool softeners, by mouth or rectally.[1] It is on the World Health Organization’s List of Essential Medicines.[5][6] Some studies claim that docusate is not more effective than a placebo for improving constipation.[7][8][9][10] Other docusate salts with medical use include those of calcium and potassium.[11][1][2]
Docusate salts are also used as food additives, emulsifiers, dispersants, and wetting agents, among other uses.[12]
History
Sodium docusate was patented in 1937 by Coleman R. Caryl and Alphons O. Jaeger for American Cyanamid,[3] which commercialized it for many years as a detergent under the brand name Aerosol OT.
Its use for the treatment of constipation was first proposed in 1955 by James L. Wilson and David G. Dickinson,[4] and quicky popularized under the name Doxinate.[13]
Medical use
Constipation
The main medical use of docusate sodium is to treat constipation, acting as a laxative and stool softener. In painful anorectal conditions such as hemorrhoid and anal fissures, it can help avoid pain caused by straining during bowel movements.
When administered by mouth, a bowel movement often occurs in 1 to 3 days,[1] while rectal use may be effective within 20 minutes.[14]
Sodium docusate is recommended as a stool softener for children.[1]
However, its effectiveness for constipation is poorly supported by evidence.[7][8] Multiple studies have found docusate to be no more effective than a placebo for improving constipation.[7][8][9][10] Others have found it to be less useful for the treatment of chronic constipation than psyllium.[10][15][16]
The medication may be given to people who are receiving opioid medication, although prolonged use may cause irritation of the gastrointestinal tract.[10][16]
Other medical uses
Docusate sodium, when used with ear syringing, may help with earwax removal, particularly in the case of impaction.[17]
Sodium docusate is also used as a lubricant in the production of tablets and as an emulsifier in topical preparations and other suspensions.[18]
Precautions and contraindications
Docusate sodium is approved and recommended as safe during pregnancy and breastfeeding.[19][20]
Docusate is not recommended in people with appendicitis, acute abdomen, or ileus.[16]
When taken by mouth it should be ingested with plenty of water.
Side effects
Side effects are uncommon and typically mild,[1] and may include stomach pain, abdominal cramps or diarrhea,[1] Efficacy decreases with long-term use, and may cause poor bowel function.[11]
Serious allergic reactions may occur with the drug. The most severe side effect of docusate, although very rare, is rectal bleeding.[21]
Interactions
Docusate might increase resorption of other drugs, for example, dantron (1,8-dihydroxyanthraquinone).[16]
Mechanism of action
Docusate sodium works by allowing more water to be absorbed by the stool.[11][22]
Docusate does not stay in the gastrointestinal tract, but is absorbed into the bloodstream and excreted via the gallbladder[16] after undergoing extensive metabolism.
The effect of docusate may not necessarily be all due to its surfactant properties. Perfusion studies suggest that docusate inhibits fluid absorption or stimulates secretion in the portion of the small intestine known as the jejunum.
Pharmaceutical brand names
In the U.S., docusate sodium for pharmaceutical use is available under multiple brand names: Aqualax, Calube, Colace, Colace Micro-Enema, Correctol Softgel Extra Gentle, DC-240, Dialose, Diocto, Dioctocal, Dioctosoftez, Dioctyn, Dionex, Doc-Q-Lace, Docu Soft, Docucal, Doculax, Docusoft S, DOK, DOS, Doss-Relief, DSS, Dulcolax – Stool Softener (not to be confused with another drug marketed under the Dulcolax brand, bisacodyl, which is a stimulant laxative), Ex-Lax Stool Softener, Fleet Sof-Lax, Genasoft, Kasof, Laxa-basic, Modane Soft, Octycine-100, Pedia-Lax, Preferred Plus Pharmacy Stool Softener, Regulax SS, Sulfalax Calcium, Sur-Q-Lax, Surfak Stool Softener, and Therevac-SB. Generic preparations are also available.
In the UK, dioctyl sodium sulfosuccinate is sold under the brand name Docusol (Typharm Ltd) and DulcoEase (Boehringer Ingelheim).
In Australia, dioctyl sodium sulfosuccinate is sold as Coloxyl and Coloxyl with senna.
In India, preparations include Laxatin by Alembic, Doslax by Raptakos Laboratories, Cellubril by AstraZeneca, and Laxicon by Stadmed.
Other uses
Dioctyl sodium sulfosuccinate is used as a surfactant in a wide range of applications, often under the name Aerosol-OT.[4][23] It is unusual in that it is able to form microemulsions without the use of co-surfactants, and it has a rich variety of aqueous-phase behavior including multiple liquid crystalline phases.[24]
Food additive
Dioctyl sodium sulfosuccinate has been approved by the US FDA as a “generally recognized as safe” (GRAS) additive.[25] It is used in a variety of food products, as a surface active agent, stabilizer, thickener, wetting agent, processing aid, solubilizing agent, emulsifier, and dispersant. The highest amount found in food products is 0.5% by weight, which include pasteurized cheese spreads, cream cheeses and salad dressings.[26] The FDA also approved its use as a wetting agent or solubilizer for flavoring agents in carbonated and non-carbonated drinks at levels up to 10 parts per million.[25]
Microencapsulation
Sodium docusate is the most widely used surfactant in reverse micelleencapsulation studies.[27]
Non-medical brand names
As a surfactant, docusate sodium is or has been commercialized under many brand names, including DSSj Aerosol OT, Alphasol OT, Colace, Complemix, Coprol, Dioctylal, Dioctyl-Medo Forte, Diotilan, Diovac, Disonate, Doxinate, Doxol, Dulsivac, Molatoc, Molofac, Nevax, Norval, Regutol, Softili, Solusol, Sulfimel DOS, Vatsol OT, Velmol, and Waxsol[28]
Chemistry
Structure and properties
The structural formula of the docusate anion is R−O−C(=O)−CH(SO−
3)−CH
2−C(=O)−O−R, where R is the 2-ethylhexyl groupH
3C−(CH
2)
3−C(−CH
2−CH
3)H−CH
2−. The conjugate acid can be described as the twofold carboxylate ester of sulfosuccinic acid with 2-ethylhexanol.
The compound is a white, wax-like, plastic solid, with an odor suggestive of octyl alcohol. It starts to decompose at about 220 °C.[28]
Solubility of dioctyl sodium sulfosuccinate in water is 14 g/L at 25 °C, increasing to 55 g/L at 70 °C.[28] Solubility is better in less polar solvents: 1:30 in ethanol, 1:1 in chloroform and diethylether, and practically unlimited in petroleum ether (25 °C). It also is highly soluble in glycerol, although this is a rather polar solvent. It is also highly soluble in xylene, oleic acid, acetone, diacetone alcohol, methanol, isopropanol, 2-butanol, methyl acetate, ethyl acetate, furfurol, and vegetable oils.[28]
The ester groups are easily cleaved under basic conditions, but are stable against acids.[16]
Synthesis
Sodium dioctyl sulfosuccinate can be obtained by treating dioctyl maleate with sodium bisulfite. The bisulfite anion adds to the double bond:−CH=CH− + HSO−
3 → −CH(−SO−
3)−CH
2−
Toxicity
Ingestion may cause the side effects described above, such as diarrhea, intestinal bloating, and occasionally cramping pains. Dioctyl sodium sulfosuccinate is not known to be carcinogenic, mutagenic, or teratogenic.[29]
Marine species
Dioctyl sodium sulfosuccinate is of low toxicity for crustaceans such as the hermit crabClibanarius erythropus and the shrimp Crangon crangon. Toxicity for molluscs varies widely, with 48-hour LD50 found between 5 mg/l for the common limpet and 100 mg/l for the common periwinkle. Various species of phytoplankton have an LD50 around 8 mg/l.
In a 2010 study, dioctyl sodium sulfosuccinate exhibited higher toxicity against bacteria (Vibrio fischeri, Anabaena sp.) and algae (Pseudokirchneriella subcapitata) than did a number of fluorinated surfactants (PFOS, PFOA, or PFBS). Measuring bioluminescence inhibition of the bacteria and growth inhibition of the algae, the LD50 were in the range of 43–75 mg/l. Combinations of the fluorinated compounds with dioctyl sodium sulfosuccinate showed mid to highly synergistic effects in most settings, meaning that such combinations are significantly more toxic than the individual substances.[30]
Freshwater species
The substance is highly toxic for rainbow trout with a median lethal concentration (LC50) of 0.56 mg/l after 48 hours for the pure substance. It is only slightly to moderately toxic for rainbow trout fingerlings, and slightly toxic for harlequin rasboras (LC50 27 mg/l of a 60% formulation after 48 hours).

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e f g h “Docusate Salts”. The American Society of Health-System Pharmacists. Archived from the original on 23 September 2015. Retrieved 11 August 2015.
- ^ Jump up to:a b American Society of Health-System Pharmacists (15 August 2011). “Stool Softeners”. Archived from the original on 5 September 2015.
- ^ Jump up to:a b US 2181087, Caryl CR, Jaeger AO, “Detergent composition”, issued 21 November 1939, assigned to American Cyanamid
- ^ Jump up to:a b c Wilson JL, Dickinson DG (May 1955). “Use of dioctyl sodium sulfosuccinate (aerosol O.T.) for severe constipation”. Journal of the American Medical Association. 158 (4): 261–3. doi:10.1001/jama.1955.02960040019006a. PMID 14367076.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ “Docusate – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b c Fakheri RJ, Volpicelli FM (February 2019). “Things We Do for No Reason: Prescribing Docusate for Constipation in Hospitalized Adults”. Journal of Hospital Medicine. 14 (2): 110–113. doi:10.12788/jhm.3124. PMID 30785419.
- ^ Jump up to:a b c “Dioctyl Sulfosuccinate or Docusate (Calcium or Sodium) for the Prevention or Management of Constipation: A Review of the Clinical Effectiveness”. CADTH Rapid Response Reports. 26 June 2014. PMID 25520993.
- ^ Jump up to:a b Candy B, Jones L, Larkin PJ, Vickerstaff V, Tookman A, Stone P (May 2015). “Laxatives for the management of constipation in people receiving palliative care” (PDF). The Cochrane Database of Systematic Reviews. 13 (5): CD003448. doi:10.1002/14651858.CD003448.pub4. PMC 6956627. PMID 25967924.
- ^ Jump up to:a b c d Ramkumar D, Rao SS (April 2005). “Efficacy and safety of traditional medical therapies for chronic constipation: systematic review”. The American Journal of Gastroenterology. 100 (4): 936–71. PMID 15784043.
- ^ Jump up to:a b c 2013 Nurse’s Drug Handbook. Burlington, MA: Jones & Bartlett Learning. 2013. p. 366. ISBN 9781449642846.
- ^ Ash M, Ash I (2004). Handbook of preservatives. Endicott, N.Y.: Synapse information resources. p. 375. ISBN 9781890595661.
- ^ Friedman M (October 1956). “Dioctyl sodium sulfosuccinate (doxinate) in chronic functional constipation”. American Practitioner and Digest of Treatment. 7 (10): 1588–91. PMID 13362832.
- ^ “Docusate sodium”. 18 December 2004. Archived from the original on 21 July 2011. Retrieved 6 March 2019.
- ^ Portalatin M, Winstead N (March 2012). “Medical management of constipation”. Clinics in Colon and Rectal Surgery. 25 (1): 12–9. doi:10.1055/s-0032-1301754. PMC 3348737. PMID 23449608.
- ^ Jump up to:a b c d e f Dinnendahl V, Fricke U, eds. (2010). Arzneistoff-Profile(in German). 2 (23 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ^ “How effective is docusate as a cerumenolytic agent?”. GlobalRPH.com. Archived from the original on 23 November 2010.
- ^ Jasek W, ed. (2008). Austria-Codex Stoffliste (in German) (41 ed.). Vienna: Österreichischer Apothekerverlag. p. 316. ISBN 978-3-85200-190-6.
- ^ Yaffe SJ (2011). Drugs in pregnancy and lactation : a reference guide to fetal and neonatal risk (9 ed.). Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. p. 1651. ISBN 9781608317080.
- ^ Mahadevan U, Kane S (July 2006). “American gastroenterological association institute medical position statement on the use of gastrointestinal medications in pregnancy”. Gastroenterology. 131(1): 278–82. doi:10.1053/j.gastro.2006.04.048. PMID 16831610.
- ^ drugs.com: Docusate Archived 16 July 2010 at the Wayback Machine
- ^ Hamilton RJ (2013). Tarascon pocket pharmacopoeia : 2013 classic shirt-pocket edition (27 ed.). Burlington, Ma.: Jones & Bartlett Learning. p. 112. ISBN 9781449665869.
- ^ Whiffen AJ (1946). “Aerosol OT in the preparation of microscopic mounts of fungi”. Mycologia. 38: 346. doi:10.1080/00275514.1946.12024063. PMID 20983186.
- ^ Nave S, Eastoe J, Penfold J (November 2000). “What Is So Special about Aerosol-OT? 1. Aqueous Systems”. Langmuir. 16(23): 8733–8740. doi:10.1021/la000341q.
- ^ Jump up to:a b “GRAS Notice Inventory Agency Response Letter GRAS Notice No. GRN 000006”. Center for Food Safety and Applied Nutrition. 20 July 1998. Archived from the original on 31 October 2017. Retrieved 24 January 2020.
- ^ “CFR – Code of Federal Regulations Title 21”. http://www.accessdata.fda.gov. Retrieved 29 January 2020.
- ^ Flynn PF (2004). “Multidimensional multinuclear solution NMR studies of encapsulated macromolecules”. Prog. Nucl. Magn. Reson. Spectrosc. 45 (1–2): 31–51. doi:10.1016/j.pnmrs.2004.04.003.
- ^ Jump up to:a b c d Ahuja S, Cohen J (January 1973). “Dioctyl Sodium Sulfosuccinate”. InAnalytical Profiles of Drug Substances. Analytical Profiles of Drug Substances. 2. Academic Press. pp. 199–219. doi:10.1016/S0099-5428(08)60040-4. ISBN 9780122608025.
- ^ ScienceLab.com: Docusate sodium Material Safety Data SheetArchived 2006-10-17 at the Wayback Machine
- ^ Rosal R, Rodea-Palomares I, Boltes K, Fernández-Piñas F, Leganés F, Petre A (September 2010). “Ecotoxicological assessment of surfactants in the aquatic environment: combined toxicity of docusate sodium with chlorinated pollutants”. Chemosphere. 81 (2): 288–93. Bibcode:2010Chmsp..81..288R. doi:10.1016/j.chemosphere.2010.05.050. PMID 20579683.
External links
- “Docusate”. Drug Information Portal. U.S. National Library of Medicine.
- “Docusate sodium”. Drug Information Portal. U.S. National Library of Medicine.
- Stool Softeners at the N.I.H.PubMed Health resource.
{kind=link}
//////////DOCUSATE, Stool softener, Laxative, Cathartic,
CCCC(CC)COC(=O)CC(C(=O)OCC(CC)CCCC)S(=O)(=O)[O-].[Na+]

NEW DRUG APPROVALS
one time
$10.00
LIDOCAINE
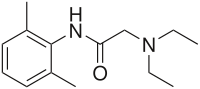
LIDOCAINE
CAS Registry Number: 137-58-6
CAS Name: 2-(Diethylamino)-N-(2,6-dimethylphenyl)acetamide
Additional Names: 2-diethylamino-2¢,6¢-acetoxylidide; w-diethylamino-2,6-dimethylacetanilide; lignocaine
Trademarks: Cuivasil (IDC); Lidoderm (Hind); LidoPosterine (Kade); Vagisil (Combe)
Molecular Formula: C14H22N2O
Molecular Weight: 234.34
Percent Composition: C 71.75%, H 9.46%, N 11.95%, O 6.83%
Literature References: Long-acting, membrane stabilizing agent against ventricular arrhythmia. Originally developed as a local anesthetic. Prepn: N. M. Löfgren, B. J. Lundqvist, US2441498 (1948 to Astra); A. D. H. Self, A. P. T. Easson, GB706409 (1954 to May & Baker); I. P. S. Hardie, E. S. Stern, GB758224 (1956 to J. F. Macfarlane & Co.); Zhuravlev, Nikolaev, Zh. Obshch. Khim.30, 1155 (1960). Toxicity studies: E. R. Smith, B. R. Duce, J. Pharmacol. Exp. Ther.179, 580 (1971); G. H. Kronberg et al.,J. Med. Chem.16, 739 (1973). Review of pharmacokinetics: N. L. Benowitz, W. Meister, Clin. Pharmacokinet.3, 177 (1978). Review of action as local anesthetic: Löfgren, Studies on Local Anesthetics: Xylocaine, A New Synthetic Drug (Hoeggstroms, Stockholm, 1948); Cooper, Pharm. J.171, 68 (1953). Reviews of anti-arrhythmic agents: J. L. Anderson et al.,Drugs15, 271 (1978); L. H. Opie, Lancet1, 861 (1980); E. Carmeliet, Ann. N.Y. Acad. Sci.427, 1 (1984). Comprehensive description: K. Groningsson et al.,Anal. Profiles Drug Subs.14, 207-243 (1985); M. F. Powell, ibid.15, 761-779 (1986). Review of use in treatment of postherpetic neuralgia: P. S. Davies, B. S. Galer, Drugs64, 937-947 (2004).Properties: Needles from benzene or alcohol, mp 68-69°. bp4 180-182°; bp2 159-160°. Insol in water. Sol in alcohol, ether, benzene, chloroform, oils. Partition coefficient (octanol/water, pH 7.4): 43.
Melting point: mp 68-69°
Boiling point: bp4 180-182°; bp2 159-160°
Log P: Partition coefficient (octanol/water, pH 7.4): 43
Derivative Type: Hydrochloride
CAS Registry Number: 73-78-9; 6108-05-0 (monohydrate)
Trademarks: Basicaina (Galenica); Batixim (So.Se.); Dynexan (Kreussler); Heweneural (Hevert); Licain (DeltaSelect); Lidesthesin (Ritsert); Lidofast (Angelini); Lidoject (Hexal); Lidrian (Baxter); Odontalg (Giovanardi); Sedagul (Wild); Xylocaine (AstraZeneca); Xylocard (AstraZeneca); Xylocitin (Jenapharm); Xyloneural (Strathmann)
Molecular Formula: C14H22N2O.HCl
Molecular Weight: 270.80
Percent Composition: C 62.09%, H 8.56%, N 10.34%, O 5.91%, Cl 13.09%
Properties: Crystals, mp 127-129°; monohydrate, mp 77-78°. Very sol in water, alcohol; sol in chloroform. Insol in ether. pH of 0.5% aq soln: 4.0-5.5. LD50 in mice (mg/kg): 292 orally (Smith, Duce); 105 i.p.; 19.5 i.v. (Kronberg).
Melting point: mp 127-129°; mp 77-78°
Toxicity data: LD50 in mice (mg/kg): 292 orally (Smith, Duce); 105 i.p.; 19.5 i.v. (Kronberg)
Therap-Cat: Anesthetic (local); antiarrhythmic (class IB).
Therap-Cat-Vet: Anesthetic (local).
Keywords: Anesthetic (Local); Antiarrhythmic.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CLIP
https://pubs.acs.org/doi/10.1021/ed076p1557
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=19_7_12
PATENT
https://patents.google.com/patent/CN102070483B/en#:~:text=The%20method%20comprises%20the%20following,as%20solvent%20and%20carbonate%20isPreparation method of the present invention, it can be two-step approach, comprises the steps:1) 2,6-xylidine is dissolved in the acetone, adds carbonate then, the back that stirs drips chloroacetyl chloride, and 20~35 ℃ (room temperature) be stirring reaction 3h down; After-filtration is finished in reaction, and after filter cake was washed with water to filtrate and is neutrality, drying made intermediate chloracetyl-2, the 6-xylidine, and yield is about about 94%;
2) intermediate that step 1) is made is dissolved in the acetone, adds carbonate then, and the back that stirs drips diethylamine, back flow reaction 8h; After-filtration is finished in reaction, and filtrate is recrystallization, drying after removing solvent under reduced pressure, makes lignocaine.Wherein, in the step 1) 2, the mol ratio of 6-xylidine, chloroacetyl chloride and carbonate is 1: 1.2~1.7: 1.3~2.0, is preferably 1: 1.5: 1.6.
Step 2) intermediate chloracetyl-2 in, the mol ratio of 6-xylidine, diethylamine and carbonate is 1: 1.5~2.5: 1.2~2.0, is preferably 1: 2: 1.5.In addition, preparation method of the present invention owing to all be that solvent, carbonate are catalyzer with acetone in the two-step reaction, therefore can further optimize reaction process on the basis of two-step approach, namely the intermediate of Sheng Chenging needn’t pass through aftertreatment, prepares lignocaine by one kettle way.Described one kettle way comprises the steps: 2,6-xylidine is dissolved in the acetone, adds carbonate then, after stirring, drips chloroacetyl chloride, and 20~35 ℃ (room temperature) be reaction 3h down; After reaction is finished, without processing, directly drip diethylamine, back flow reaction 8h, after-filtration is finished in reaction, and filtrate is recrystallization, drying after removing solvent under reduced pressure, makes lignocaine.Wherein, described 2, the mol ratio of 6-xylidine, chloroacetyl chloride, diethylamine and carbonate is 1: 1.2~1.7: 1.5~2.5: 2.5~3.5, is preferably 1: 1.5: 2: 2.5.
In addition, preparation method of the present invention adopts TLC monitoring reaction progress, and the developping agent of TLC is sherwood oil: ethyl acetate (V/V)=3: 1.The invention has the advantages that, the method synthesis technique for preparing lignocaine of the present invention is simple, do not need in the intermediate aftertreatment first pickling numerous and diverse step of alkali cleaning again, avoided unnecessary loss, therefore the yield of the intermediate that makes of the inventive method and lignocaine is all higher, and the lignocaine purity that makes is good, reaches more than 99%, has favorable industrial application prospect; In addition, the inventive method uses acetone to make solvent, and this solvent is nontoxic substantially non-stimulated, and can recycle, and is environmentally friendly.
EmbodimentBelow further specify the present invention by specific embodiment, but be not used for limiting the scope of the invention.
Embodiment 1 two-step approach prepares lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 200g salt of wormwood again, and mechanical stirring evenly back drips 100mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 156g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 94%, fusing point is 145.0~147.0 ℃.2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 112g salt of wormwood, drips the 60g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 90g white powder, is lignocaine, yield is 95%, and fusing point is 67.0~68.0 ℃, and content is 99.05%.
Embodiment 2 two-step approachs prepare lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 163g salt of wormwood again, and mechanical stirring evenly back drips 80mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 136g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 82%, fusing point is 145~146 ℃.
2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 90g salt of wormwood, drips the 45g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 84g white powder, is lignocaine, yield is 89%, and fusing point is 66~67 ℃, and content is 99.15%.
Embodiment 3 two-step approachs prepare lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 250g salt of wormwood again, and mechanical stirring evenly back drips 113mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 150g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 90%, fusing point is 147~148 ℃.
2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 150g salt of wormwood, drips the 75g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 88g white powder, is lignocaine, yield is 93%, and fusing point is 68~69 ℃, and content is 98.75%.
Embodiment 4 one kettle ways prepare lignocaineIn the 5000mL there-necked flask, add 305g 2, the 6-xylidine, make solvent with 2000mL acetone, add 700g salt of wormwood again, mechanical stirring evenly back slowly drips 230mL chloroacetyl chloride (1.5h drips off), room temperature (35 ℃) is stirring reaction 3h down, and TLC point plate (use sherwood oil: ethyl acetate (V/V)=3: 1 is made developping agent) demonstration reacts completely; Dropwise 5 50g diethylamine then, the back back flow reaction 8h that stirs, TLC monitoring (developping agent is the same) shows and reacts completely; The reaction solution suction filtration, filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, gets light yellow solid, uses sherwood oil recrystallization secondary then, makes 482g white lignocaine crystal, and total recovery is 82%, and fusing point is 68.0~69.0 ℃, and content is 99.75%.
Comparative example 1 existing method prepares lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineIn the 1000mL there-necked flask, with 102g 2, the 6-xylidine is dissolved in the 400mL glacial acetic acid, stirs slowly to add the 100mL chloroacetyl chloride down, is heated to 45 ℃, adds 200g solid sodium acetate (containing crystal water) then, reaction 2h; After reaction finished, ice bath was cooled to below 10 ℃, suction filtration, filter cake is washed with water to filtrate and is neutral, and drying is 1 hour under 100 ℃ of temperature, makes the 111g white powder, be intermediate chloracetyl-2,6-xylidine, yield are that 67% fusing point is 145.0~148.0 ℃.
2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL toluene, and the dissolving back drips 60g diethylamine, back flow reaction 3.5h fast; After reaction finished, ice bath was cooled to 5 ℃, suction filtration, filtrate is used the 3mol/L hcl as extraction agent, and the acid solution that extraction is obtained is cooled to 10 ℃ then, stirs slowly to add 6mol/L KOH solution down, be alkalescence (pH8~9) to solution, with pentane extraction, organic layer was after washing, Anhydrous potassium carbonate drying after ice bath was cooled to 20 ℃, vapor bath is steamed and is desolventized, make the 74g white powder, be lignocaine, yield is 78%, fusing point is 66.0~67.0 ℃, and content is 97.15%.Embodiment 1 compares with comparative example 1, its intermediate chloracetyl-2, and the yield of 6-xylidine obviously improves, and reaches 94%, and the total recovery of final product lignocaine also is significantly improved, and the content of lignocaine is brought up to about 99%.Embodiment 4 compares with comparative example 1, and the total recovery of lignocaine is significantly improved, and reach 82%, and content is brought up to more than 99%.
Though above with a general description of the specific embodiments, the present invention is described in detail, on basis of the present invention, can make some modifications or improvements it, this will be apparent to those skilled in the art.Therefore, these modifications or improvements all belong to the scope of protection of present invention without departing from theon the basis of the spirit of the present invention.
CLIPhttps://www.cerritos.edu/chemistry/chem_212/Documents/Lab/10_lidocaine.pdf
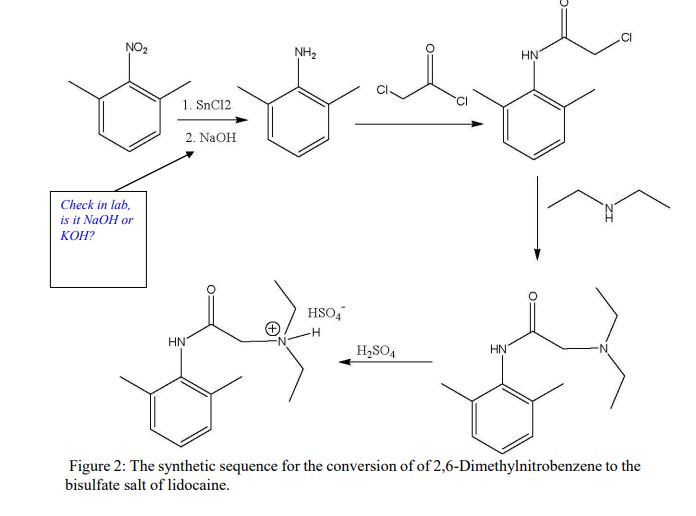
Procedure: (1st week)A: Synthesis of 2,6-Dimethylaniline via Reduction of 2,6-Dimethylnitrobenzene 1. Dissolve1.0 g of 2,6-dimethylnitrobenzene in 10 mL of glacial acetic acid in a 50 mL Erlenmeyer flask. 2. In a 25 mL flask, dissolve 4.6 grams of SnCl2 · 2H2O in 8 mL of concentrated HCl, inside the fume hood. 3. Add the SnCl2 solution in one portion to the nitroxylene solution, magnetically swirl and mix, and let the mixture stand for 15 minutes. 4. Cool the mixture and collect the crystalline salt (dimethylaniline in the salt form: C6H5NH3 +Cl- ) in a Buchner funnel. 5. Transfer the moist crystals to an Erlenmeyer flask, add 5-10 mL of water, and make the solution strongly basic (to remove the acid and change C6H5NH3 +Clback intoC6H5NH2) by adding 30% KOH solution (12 to 17 mL required). 6. After cooling extract with three 10 mL portions of ether, rinse the ether extracts twice with 10 mL of water, and dry over K2CO3. 7. Evaporate the dried and filtered solution to an oil, transfer and rinse into a 50 mL Erlenmeyer flask, complete evaporation, weigh, and calculate the %yield of 2,6-dimethylaniline.
B: Synthesis of α-Chloro-2,6-dimethylacetanilide (prepare for a steam bath ahead of time) 1. For every 7 grams (from this step on, you need to calculate proportionally how much you need to add according to the actual weight that you got) of dimethylaniline from the previous step, add 50 mL of glacial acetic acid, and 7.2 g (or 5.2 mL) of chloroacetyl chloride, in that order. 2. Warm the solution on a steam bath to (40–50)ºC, remove, and add a solution of 1 gram of sodium acetate in 100 mL of water. 3. Cool the mixture and collect the product in a Buchner funnel. 4. Transfer the product to a disk of medium–sized filter paper, finely divide it with a spatula, and let air dry until the next laboratory period. 5. Upon drying, measure the mass and the melting point. Also, calculate the % yield.
B: Synthesis of α-Chloro-2,6-dimethylacetanilide (prepare for a steam bath ahead of time) 1. For every 7 grams (from this step on, you need to calculate proportionally how much you need to add according to the actual weight that you got) of dimethylaniline from the previous step, add 50 mL of glacial acetic acid, and 7.2 g (or 5.2 mL) of chloroacetyl chloride, in that order. 2. Warm the solution on a steam bath to (40–50)ºC, remove, and add a solution of 1 gram of sodium acetate in 100 mL of water. 3. Cool the mixture and collect the product in a Buchner funnel. 4. Transfer the product to a disk of medium–sized filter paper, finely divide it with a spatula, and let air dry until the next laboratory period. 5. Upon drying, measure the mass and the melting point. Also, calculate the % yield.
D. Synthesis of the bisulfate salt of lidocaine 1. Dissolve the lidocaine in ether (10 mL per gram of lidocaine) and add 2 mL of 2.2 M sulfuric acid in ethanol per gram of lidocaine. 2. Stir and scratch with a glass rod to mix and induce crystallization. 3. Dilute the mixture with an equal volume of acetone to aid filtration and collect the salt in a small Buchner funnel. 4. Rinse the solid on the funnel with a few milliliters of acetone and air dry and weigh the product. 5. Calculate the % yield of this step. *** Overall % Yield The overall % YCLIPhttp://home.sandiego.edu/~khuong/chem302L/Handouts/Lidocaine_handout_Su07.pdfSynthetic Strategy Lidocaine will be prepared via a three-step linear synthesis starting from 2,6-dimethylnitrobenzene. The reduction of 2,6-dimethylnitrobenzene 1 with three equivalents of stannous chloride (SnCl2) yields the ammonium salt 2. It is very important that the reaction mixture is strongly acidic during this reaction because the reduction of nitrobenzene using different reducing reagents and conditions can afford a variety of functional groups: nitroso, hydroxylamine (zinc dust, pH 4), azoxy (sodium arsenite), azo (zinc, weakly basic), or hydrazo (zinc, strongly basic). In industrial settings, often iron or tin with hydrochloric acid is used instead of stannous chloride because iron and tin are cheaper, but the reduction takes much longer. In the workup portion of the reaction, the ammonium salt 2 is reacted with an aqueous potassium hydroxide solution, liberating the free 2,6-dimethylaniline 3 in an acid-base reaction.
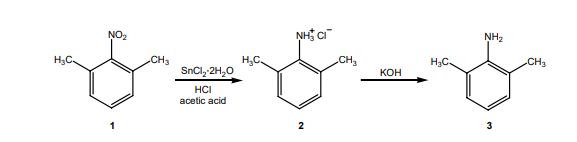
The reaction of 3 with the bifunctional α-chloroacetyl chloride leads to α-chloro-2,6-dimethylacetanilide 4. A slight excess of the acid chloride is used to ensure the complete conversion of the amine to the amide. The formation of the amide is a result of the significantly higher reactivity (~106 times) of the acyl chloride over the alkyl chloride. The addition of sodium acetate solution avoids the formation of HCl which would protonate unreacted 3 causing it to co-precipitate with the desired product 4.
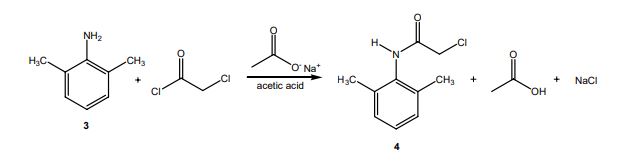
In the last step, diethylamine performs a nucleophilic substitution (SN2) on the remaining alkyl chloride. Diethylamine serves both as a nucleophile to form lidocaine 5, and as acid scavenger, leading to formation of NH2Et2 + Cl- in this reaction. Since diethylamine is not a very strong nucleophile, it is used in excess here to improve the yield and speed up the reaction. The unreacted amine is later removed by extraction with water. The aqueous extraction of lidocaine with acid separates the unreacted chloroanilide 4 and the lidocaine. After addition of a strong base like aqueous potassium hydroxide, crude lidocaine is obtained.
Procedure Synthesis of 2,6-dimethylaniline (3) Dissolve 15 g of SnCl2•2H2O in 27 mL of concentrated hydrochloric acid. If necessary, heat the mixture gently. Add this solution in one portion to a solution of 3 mL of 2,6-dimethylnitrobenzene in 34 mL of glacial acetic acid. Swirl the resulting mixture and then allow it to stand for 15 minutes before placing the mixture in an ice bath. Collect the formed precipitate by vacuum filtration. Place the wet precipitate obtained above in a beaker and add 20 mL of water. Neutralize the acidic mixture by carefully adding an 8 M aqueous potassium hydroxide with continuous stirring until basic to litmus. Place the mixture in an ice bath. Upon cooling to room temperature, extract the mixture three times with diethyl ether. Combine the organic layers and wash them twice with water and once with brine. Dry the organic layer over anhydrous potassium carbonate. Decant away from the drying agent and evaporate the diethyl ether from a dry, preweighed flask using a rotary evaporator. The oily residue will be your crude product 3. Obtain and record the following information: 1. crude product description (co2. crude weight/percent yieldSynthesis of α-chloro-2,6-dimethylacetanilide (4) Dissolve 3 in 17 mL of glacial acetic acid. Add 1.1 equivalents (based on the moles of 3) of α-chloroacetyl chloride to this solution. Heat the solution to 40-50 o C for ten minutes to complete the reaction. Upon cooling, add a solution of ~3.3 g sodium acetate trihydrate in 67 mL water and then place the resulting mixture in an ice bath. Collect the precipitate by vacuum filtration. Rinse the filter cake with copious amounts of water in order to remove the acetic acid. It is important that the product be completely free of acetic acid after this step (why?). The pH of the individual water rinses can be checked with litmus paper to determine if the product is acid free. Allow for the product to air-dry on a watch glass until the next meeting. There is a reasonable chance that you will not obtain a precipitate as described above. If this is the case, you can try “seeding” using a small sample of authentic product from a classmate. If this does not work, check the TLC to be sure that you have formed product and devise an extractive workup that will separate the unreacted aniline 3 from the desired product 4. (Make sure you understand how to do this even if you obtain a precipitate in the first place). After the aqueous workup and following removal of solvent, you should obtain a solid. If not, check the TLC, using a sample of authentic product from a classmate as a standard. If the product appears relatively pure, you can continue even though the material is not a solid. Obtain and record the following information: 1. crude product description (color, physical state, etc.) 2. crude weight/percent yield 3. mp (if a solid)
4. TLC analysis 5. IR (check for presence of amide functional group) Synthesis of lidocaine; α-(N,N-diethylamino)-2,6-dimethylacetanilide (5). In a round bottom flask, dissolve α-chloro-2,6-dimethylacetanilide 4 in 17 mL of toluene. Before continuing, spot several (4 to 5) TLC plates in advance with this solution of 4. Provide three lanes and spot the 4 on the “SM” and “CO-SPOT” lanes. You will use these plates to monitor the progress of this reaction. Add three equivalents of diethylamine to the round bottom flask, and reflux the mixture vigorously until the reaction is complete. The amount of time required for complete reaction depends on many factors but it will likely take anywhere from more than a few minutes up to several hours. If the reaction is not complete when your lab period ends, you can stopper the reaction and reflux it for additional time at the next period. Usually a white precipitate forms during the reflux. Upon cooling, transfer the reaction mixture to a separatory funnel and extract the mixture three times with water. Next, extract the organic layer with two portions of 3 M hydrochloric acid. Cool the combined acidic aqueous extracts in an ice bath and then add 8 M aqueous potassium hydroxide slowly until the mixture is strongly basic again. The formation of a thin, dark yellow oily layer on top or a white solid is observed at this point. Place the mixture in an ice bath. Once the mixture is chilled, try to initiate the crystallization of the final product if no solid has formed at this point. Collect the obtained precipitate by filtration using a Büchner funnel. Wash it with twice with water and then press it as dry as possible. Obtain and record the following information: 1. crude product description (color, physical state, etc.) 2. crude weight/percent yield 3. TLC analysis Recrystallize the crude product from hexanes. Regardless of the final physical state of your product (solid or oil), obtain and record the following: 1. pure product description (color, physical state, etc.) 2. pure product weight/percent yield 3. overall (three-step) percent yield (from starting material 1) 4. TLC analysis 5. melting point (if a solid) 6. IR 7. 1 H and 13C NMR spectra of lidocaine will be given to you. Turn in a sample of your final product.
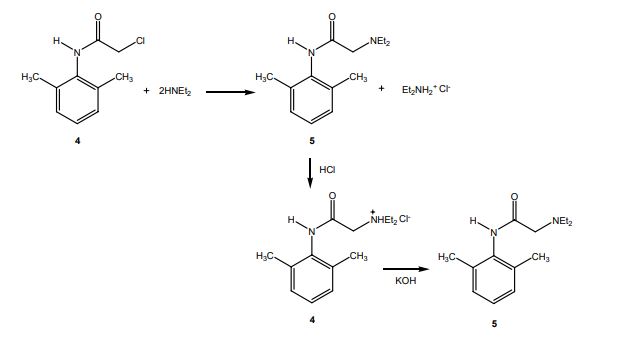
1H NMR
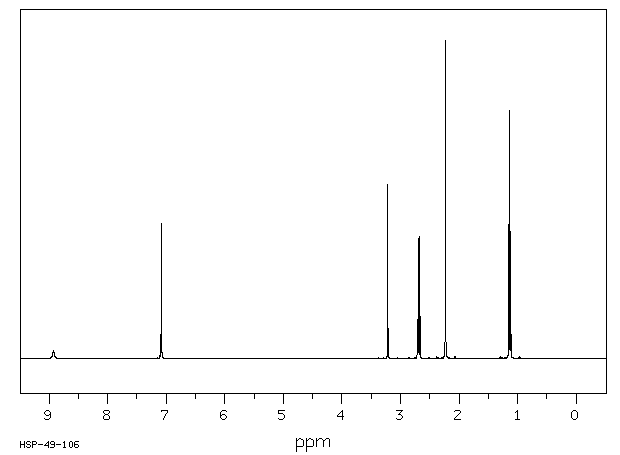
13C NMR
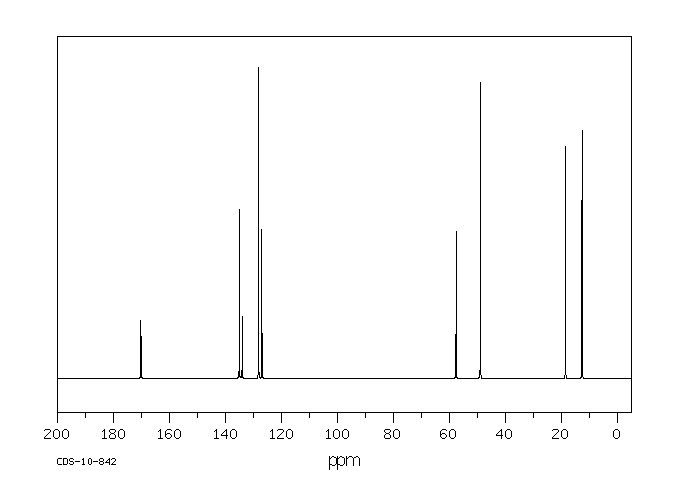
MS
IR KBR
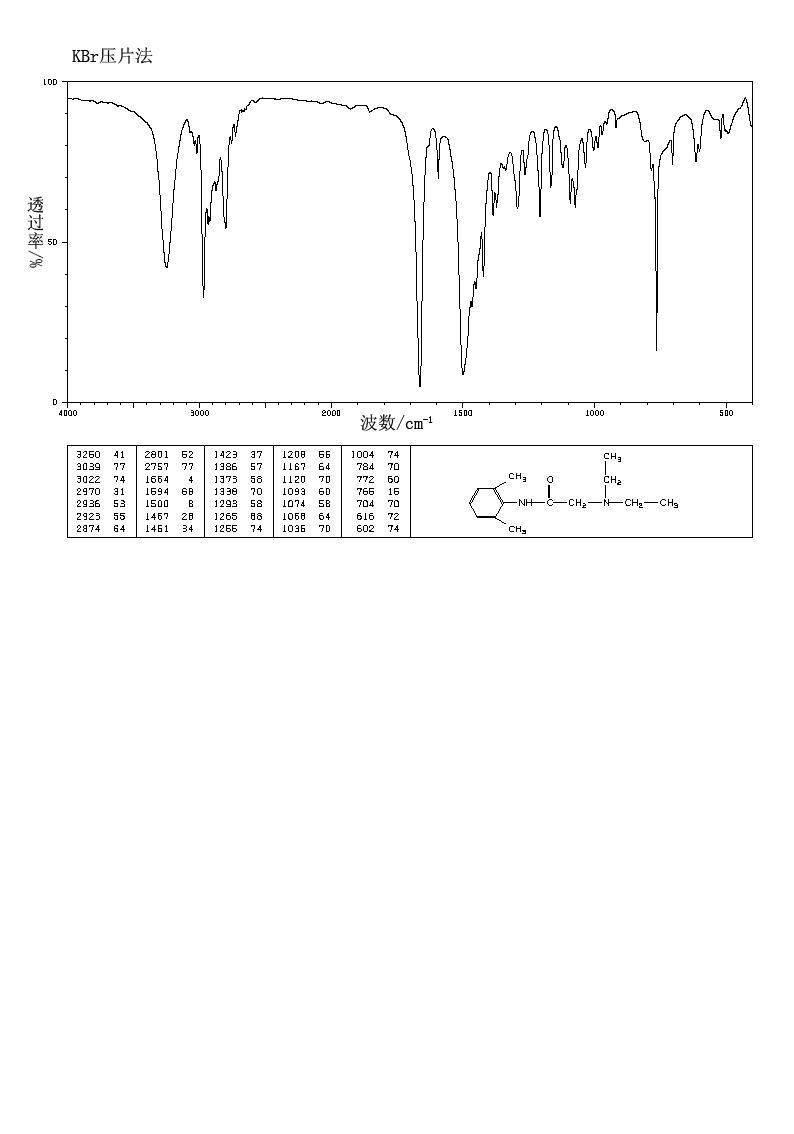
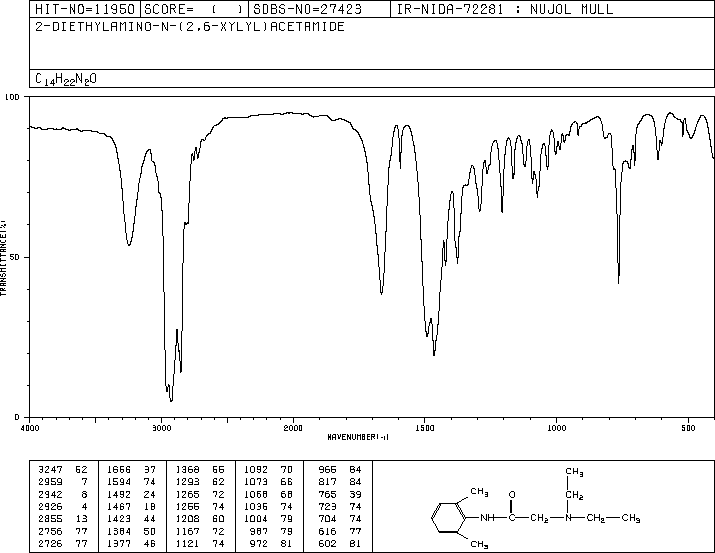
Lidocaine is an antiarrhythmic medicine and also serves as a local anaesthetic drug. It is utilized in topical application to relieve pain, burning and itching sensation caused from skin inflammations. This drug is mainly used for minor surgeries. Figure 1 shows the 1H NMR spectrum of 200 mM lidocaine in CDCl3.
.jpg)
Figure 1. Proton NMR spectrum of 200 mM lidocaine in CDCl3.
1H NMR Relaxation
Figures 2, 3 and 4 show the relaxation time measurements. It can be seen that the relaxation times are shortest for the CH2 protons and longest for the CH protons. The first data point amplitude increases with the number of protons for the related peak.
.jpg)
Figure 2. Proton T1 relaxation time measurement of 200 mM lidocaine in CDCl3.
.jpg)
Figure 3. Proton T2 relaxation time measurement of 200 mM lidocaine in CDCl3.
.jpg)
Figure 4. COSY spectrum of 200 mM lidocaine in CDCl3. The cross-peaks and corresponding exchanging protons are labeled by colour-coded arrows and ellipses.
2D COSY
Figure 4 shows the 2D COSY spectrum where two spin systems (6,7,8) to (10,11) can be clearly seen. For instance, the methyl groups at 10 and 11 positions bond to aromatic protons at 6 and 8 positions, while the methyl groups at 16 and 17 positions bond to the ethylene groups at 14 and 15 positions. No coupling occurs at positions (6,7,8) to (16,17) or (14,15).
2D Homonuclear J-Resolved Spectroscopy
The chemical shift in the 2D homonuclear j-resolved spectrum appears along the direct (f2) direction and the effects of coupling between protons appear along the indirect (f1) dimension. This enables the assignment of chemical shifts of multiplets and may help in measuring unresolved couplings. Also, a decoupled 1D proton spectrum is produced by the projection along the f1 dimension. The 2D homonuclear j-resolved spectrum of lidocaine, plus the 1D proton spectrum (blue line) are shown in Figure 5.
.jpg)
Figure 5. Homonuclear j-resolved spectrum of 200 mM lidocaine in CDCl3. The multiplet splitting frequencies for different couplings are colour- coded.
The projection which is vertical reveals how the multiplets disintegrate into a single peak, which makes the 1D spectrum more simplified. Peak multiplicities are produced by vertical traces from peaks in the 2D spectrum and help in determining the frequencies of proton-proton coupling. When coupling frequencies are compared between different peaks, information can be obtained regarding which peaks are bonded to each other. Also, Information regarding the coupling strength can be obtained from the size of the coupling frequencies. These couplings substantiate the results of the COSY experiment.
However, in this experiment, the effects of second order coupling appear in the f1 direction as additional peaks which are equidistant from the coupling partners detached from the zero frequency in the f1 dimension. These peaks provide proof of second order coupling partners, but are generally considered as artifacts. Figure 6 shows these coupling partners and additional peaks marked by colour-coded arrows and ellipses.
.jpg)
Figure 6. Homonuclear j-resolved spectrum of 200 mM lidocaine in CDCl3 showing the extra peaks due to strong couplings.
1D 13C Spectra
Figure 7 shows the 13C NMR spectra of 1 M lidocaine in CDCl3. Since the 1D Carbon experiment is highly susceptible to the 13C nuclei in the specimen, it easily and clearly resolves 9 resonances. In this experiment, only carbons coupled to protons are seen.
.jpg)
Figure 7. Carbon spectra of 1 M lidocaine in CDCl3.
Given the fact that the DEPT spectra do not display the peaks at 170 and 135ppm, they must be part of quaternary carbons. The DEPT-135 and the DEPT-45 experiments provide signals of CH3, CH2 and CH groups, while the DEPT-90 experiment provides only the signal of CH groups. However, in DEPT-135 the CH2 groups occur as negative peaks. It can thus be summed up that the peaks between 45 and 60ppm belong to ethylene groups; the peaks between 10 and 20ppm are part of the methyl groups; and the peaks between 125 and 130ppm belong to methyne groups. A similar study can be carried out on the C and CH peaks.
Heteronuclear Correlation
The Heteronuclear Correlation (HETCOR) experiment identifies the proton signal that appears along the indirect dimension and the carbon signal along the direct dimension. Figure 8 shows the HETCOR spectrum of 1 M lidocaine in CDCl3. in the 2D spectrum, the peaks reveal which proton is attached to which carbon. This experiment helps in resolving assignment uncertainty from the ID carbon spectra.
.jpg)
Figure 8. HETCOR spectrum of 1 M lidocaine in CDCl3.
Heteronuclear Multiple Quantum Coherence
Heteronuclear Multiple Quantum Coherence (HMQC) is similar to the HETCOR experiment and is utilized to associate proton resonances to the carbons that are coupled directly to those protons. But in the HMQC experiment, the proton signal appears along the direct dimension and the carbon signal along the indirect dimension. Figure 9 shows the HMQC spectrum of 1 M lidocaine in CDCl3. In the 2D spectrum, the peaks show which proton is attached to which carbon. For conclusive peak assignment, a similar study with the HETCOR spectrum can be carried out.
.jpg)
Figure 9. HMQC spectrum of 1 M lidocaine in CDCl3.
Heteronuclear Multiple Bond Correlation
The Heteronuclear Multiple Bond Correlation (HMBC) experiment can be employed to achieve long-range correlations of proton and carbon via two or three bond couplings. Similar to the HMQC experiment, the proton signal appears along the direct dimension and the carbon signal along the indirect dimension. Figure 10 shows the HMBC spectrum of 1 M lidocaine in CDCl3.
.jpg)
Figure 10. HMBC spectrum of 1 M lidocaine in CDCl3, with some of the long-range couplings marked.
The couplings amid the molecular positions appear analogous to the couplings seen in the COSY spectrum; however, the HMBC also displays couplings to quaternary carbons, which are not seen either in HMQC or COSY experiments. In addition, there is a correlation between protons and carbons. This is attributed to three-bond bonding from 14 and 15 and vice versa, as shown in light green in Figure 1.
SYN
Synthesis of lidocaine T. J. Reilly (1999). “The Preparation of Lidocaine”. J. Chem. Ed. 76 (11): 1557.

CLIP
The Present Synthesis Of Lidocaine Begins With 2,6-Dimethylnitrobenzene (1). This Compound Can Be Made From 1,3-Dimethylbenzene, Also Known As M-Xylene, Which Is More Difficult To Make. Luckily,
This problem has been solved!
See the answer
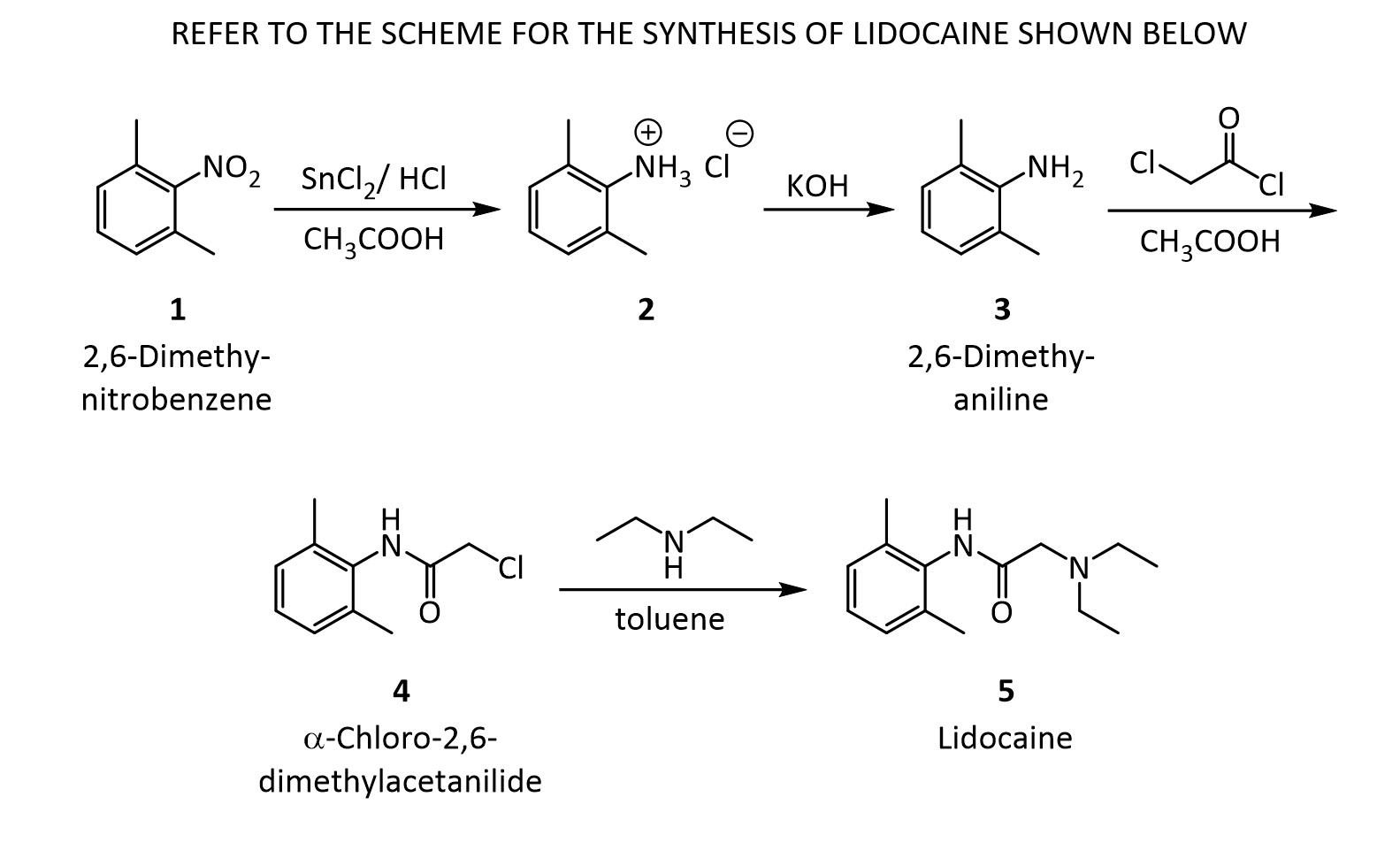
- The present synthesis of lidocaine begins with 2,6-dimethylnitrobenzene (1). This compound can be made from 1,3-dimethylbenzene, also known as m-xylene, which is more difficult to make. Luckily, m-xylene is commercially available, so a synthesis of 1 from m-xylene is a practical alternative if one wants to begin the synthesis of lidocaine with m-xylene. Suppose you want to prepare 1 from m-xylene. Show with chemical equations the reagents that you would use, and the possible isomers that would result.
2. The practical transformation of 1 into 3 is carried out by the following scheme:
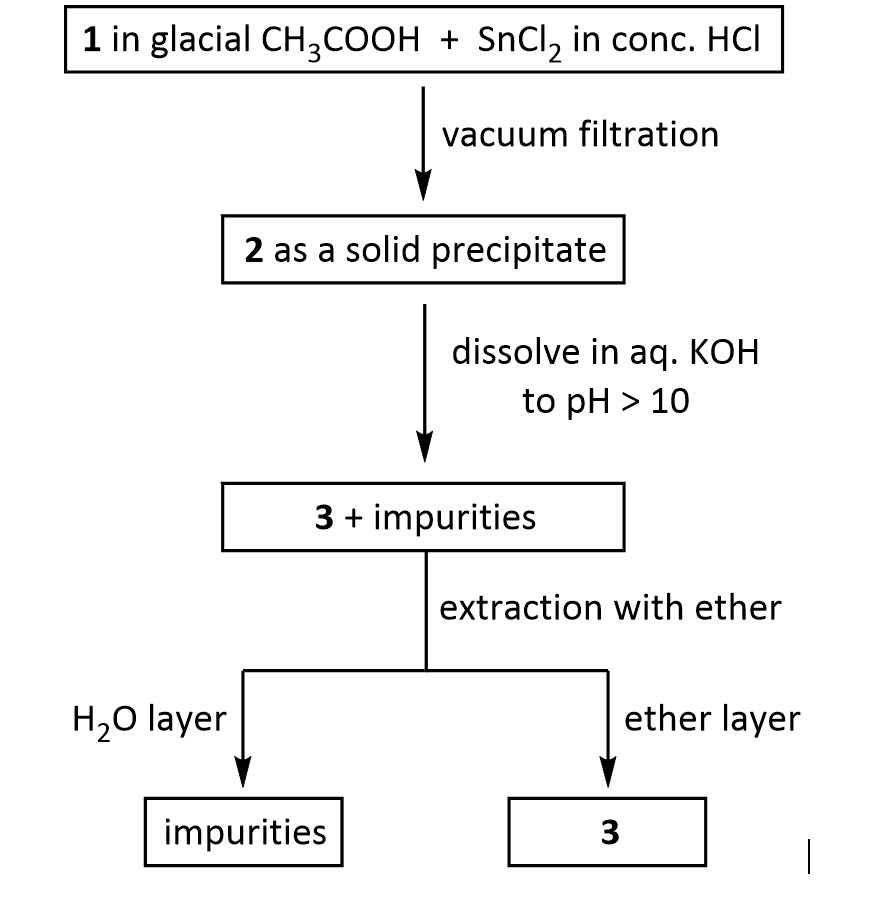
Suppose you dissolve the solid precipitate of 2 in water, but forget to include the KOH in the second step above. What would happen after the extraction with ether? Give your answer in terms of what would be found in the ether layer, and in the aqueous layer.
3. Suppose you’re out of acetic acid (CH3COOH) and decide to use ethanol (CH3 CH2OH) as the solvent in the transformation of 3 into 4. Would this be a wise choice, and why?
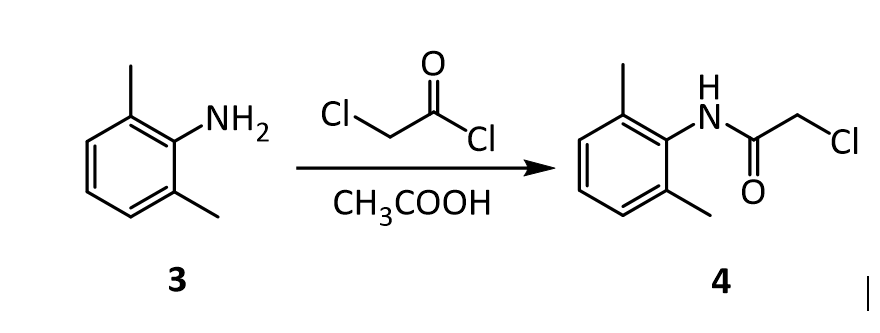
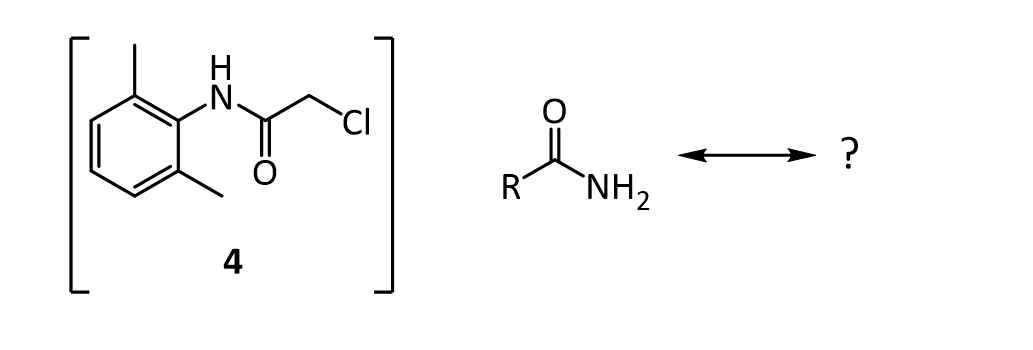
4.The amide 4 has a nitrogen attached to the benzene ring, and a chlorine attached to a primary carbon. Yet, it doesn’t react with itself in a nucleophilic displacement. Why is the nitrogen in the amide not nucleophilic? Give your answer in terms of the resonance forms of amides in general:
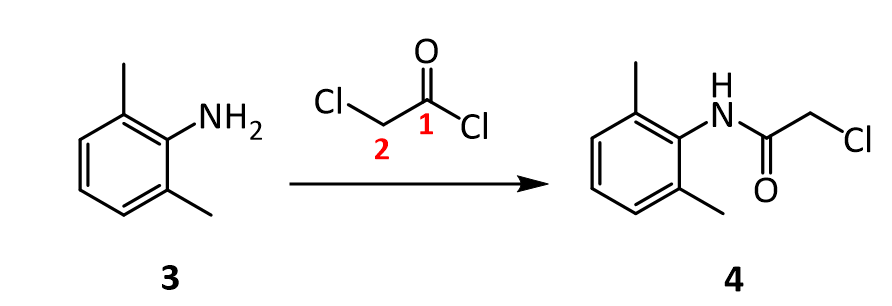
5. In the reaction below, what factors come into play to favor attack of the aniline 3 on the carbonyl carbon of the acid chloride (carbon 1 in red), rather than at the a-carbon (carbon 2 in red)?
6. Before carrying out the transformation below, compound 4 and the glassware used must be oven-dried. What would happen if the reaction was attempted using wet 4?
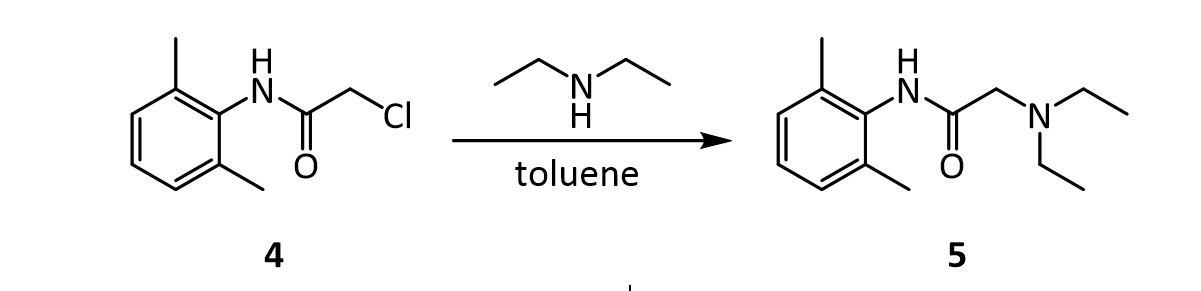
7.In the reaction below, what factors come into play to favor attack of diethylamine on the a-carbon (carbon 1 in red), rather than on the amide C=O carbon (carbon 2 in red)?
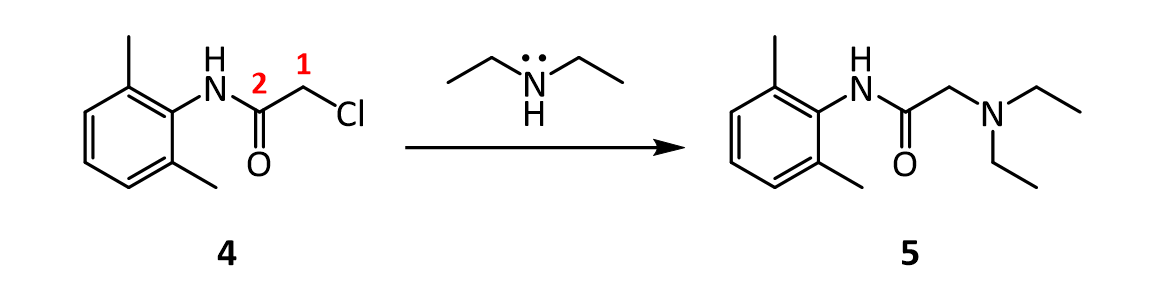
8. In the reaction below, why does the amine nitrogen (#1 in red) undergo protonation with H2SO4 preferentially over the amide nitrogen (#2 in red)? In other words, why is nitrogen 1 basic, but nitrogen 2 is not?
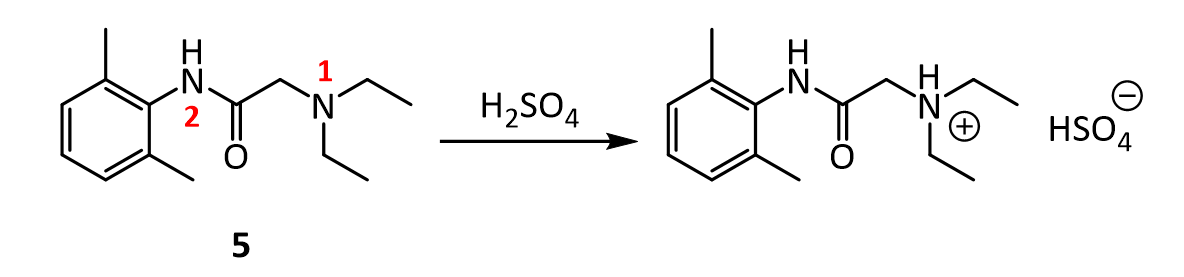
9.Lidocaine and other drugs containing amino groups are usually marketed as their hydrochloride or hydrogen sulfate salts, rather than as “free amines.” Provide two reasons why this practice makes sense.
10.Although lidocaine is marketed as its hydrochloride salt, it doesn’t exhibit the same level of physiological activity as the free amine. The free amine is more lipophilic and diffuses across a neuron cell membrane more rapidly than the ionic salt, resulting in a more rapid onset of anesthesia. Therefore, sodium bicarbonate (NaHCO3) is added to a solution of lidocaine prior to injection. How does the addition of sodium bicarbonate promote a faster anesthetic effect?
CLIP
CLIP
Lidocaine, also known as lignocaine and sold under the brand name Xylocaine among others, is a local anesthetic of the amino amide type. It is also used to treat ventricular tachycardia.[7][8] When used for local anaesthesia or in nerve blocks, lidocaine typically begins working within several minutes and lasts for half an hour to three hours.[8][9] Lidocaine mixtures may also be applied directly to the skin or mucous membranes to numb the area.[8] It is often used mixed with a small amount of adrenaline (epinephrine) to prolong its local effects and to decrease bleeding.[8]
If injected intravenously, it may cause cerebral effects such as confusion, changes in vision, numbness, tingling, and vomiting.[7] It can cause low blood pressure and an irregular heart rate.[7] There are concerns that injecting it into a joint can cause problems with the cartilage.[8] It appears to be generally safe for use in pregnancy.[7] A lower dose may be required in those with liver problems.[7] It is generally safe to use in those allergic to tetracaine or benzocaine.[8] Lidocaine is an antiarrhythmic medication of the class Ib type.[7] This means it works by blocking sodium channels and thus decreasing the rate of contractions of the heart.[7] When injected near nerves, the nerves cannot conduct signals to or from the brain.[8]
Lidocaine was discovered in 1946 and went on sale in 1948.[10] It is on the World Health Organization’s List of Essential Medicines.[11] It is available as a generic medication.[8][12] In 2018, it was the 233rd most commonly prescribed medication in the United States, with more than 2 million prescriptions.[13][14]
Medical uses
Local numbing agent
The efficacy profile of lidocaine as a local anaesthetic is characterized by a rapid onset of action and intermediate duration of efficacy. Therefore, lidocaine is suitable for infiltration, block, and surface anaesthesia. Longer-acting substances such as bupivacaine are sometimes given preference for spinal and epidural anaesthesias; lidocaine, though, has the advantage of a rapid onset of action. Adrenaline vasoconstricts arteries, reducing bleeding and also delaying the resorption of lidocaine, almost doubling the duration of anaesthesia.
Lidocaine is one of the most commonly used local anaesthetics in dentistry. It can be administered in multiple ways, most often as a nerve block or infiltration, depending on the type of treatment carried out and the area of the mouth worked on.[15]
For surface anaesthesia, several formulations can be used for endoscopies, before intubations, etc. Buffering the pH of lidocaine makes local numbing less painful.[16] Lidocaine drops can be used on the eyes for short ophthalmic procedures. There is tentative evidence for topical lidocaine for neuropathic pain and skin graft donor site pain.[17][18] As a local numbing agent, it is used for the treatment of premature ejaculation.[19]
An adhesive transdermal patch containing a 5% concentration of lidocaine in a hydrogel bandage, is approved by the US FDA for reducing nerve pain caused by shingles.[20] The transdermal patch is also used for pain from other causes, such as compressed nerves and persistent nerve pain after some surgeries.
Heart arrhythmia
Lidocaine is also the most important class-1b antiarrhythmic drug; it is used intravenously for the treatment of ventricular arrhythmias (for acute myocardial infarction, digoxin poisoning, cardioversion, or cardiac catheterization) if amiodarone is not available or contraindicated. Lidocaine should be given for this indication after defibrillation, CPR, and vasopressors have been initiated. A routine preventive dose is no longer recommended after a myocardial infarction as the overall benefit is not convincing.[21]
Epilepsy
A 2013 review on treatment for neonatal seizures recommended intravenous lidocaine as a second-line treatment, if phenobarbital fails to stop seizures.[22]
Other
Intravenous lidocaine infusions are also used to treat chronic pain and acute surgical pain as an opiate sparing technique. The quality of evidence for this use is poor so it is difficult to compare it to placebo or an epidural.[23]
Inhaled lidocaine can be used as a cough suppressor acting peripherally to reduce the cough reflex. This application can be implemented as a safety and comfort measure for patients who have to be intubated, as it reduces the incidence of coughing and any tracheal damage it might cause when emerging from anaesthesia.[24]
Lidocaine, along with ethanol, ammonia, and acetic acid, may also help in treating jellyfish stings, both numbing the affected area and preventing further nematocyst discharge.[25][26]
For gastritis, drinking a viscous lidocaine formulation may help with the pain.[27]
Adverse effects
Adverse drug reactions (ADRs) are rare when lidocaine is used as a local anesthetic and is administered correctly. Most ADRs associated with lidocaine for anesthesia relate to administration technique (resulting in systemic exposure) or pharmacological effects of anesthesia, and allergic reactions only rarely occur.[28] Systemic exposure to excessive quantities of lidocaine mainly result in central nervous system (CNS) and cardiovascular effects – CNS effects usually occur at lower blood plasma concentrations and additional cardiovascular effects present at higher concentrations, though cardiovascular collapse may also occur with low concentrations. ADRs by system are:
- CNS excitation: nervousness, agitation, anxiety, apprehension, tingling around the mouth (circumoral paraesthesia), headache, hyperesthesia, tremor, dizziness, pupillary changes, psychosis, euphoria, hallucinations, and seizures
- CNS depression with increasingly heavier exposure: drowsiness, lethargy, slurred speech, hypoesthesia, confusion, disorientation, loss of consciousness, respiratory depression and apnoea.
- Cardiovascular: hypotension, bradycardia, arrhythmias, flushing, venous insufficiency, increased defibrillator threshold, edema, and/or cardiac arrest – some of which may be due to hypoxemia secondary to respiratory depression.[29]
- Respiratory: bronchospasm, dyspnea, respiratory depression or arrest
- Gastrointestinal: metallic taste, nausea, vomiting
- Ears: tinnitus
- Eyes: local burning, conjunctival hyperemia, corneal epithelial changes/ulceration, diplopia, visual changes (opacification)
- Skin: itching, depigmentation, rash, urticaria, edema, angioedema, bruising, inflammation of the vein at the injection site, irritation of the skin when applied topically
- Blood: methemoglobinemia
- Allergy
ADRs associated with the use of intravenous lidocaine are similar to toxic effects from systemic exposure above. These are dose-related and more frequent at high infusion rates (≥3 mg/min). Common ADRs include: headache, dizziness, drowsiness, confusion, visual disturbances, tinnitus, tremor, and/or paraesthesia. Infrequent ADRs associated with the use of lidocaine include: hypotension, bradycardia, arrhythmias, cardiac arrest, muscle twitching, seizures, coma, and/or respiratory depression.[29]
It is generally safe to use lidocaine with vasoconstrictor such as adrenaline, including in regions such as the nose, ears, fingers, and toes.[30] While concerns of tissue death if used in these areas have been raised, evidence does not support these concerns.[30]
Interactions
Any drugs that are also ligands of CYP3A4 and CYP1A2 can potentially increase serum levels and potential for toxicity or decrease serum levels and the efficacy, depending on whether they induce or inhibit the enzymes, respectively. Drugs that may increase the chance of methemoglobinemia should also be considered carefully. Dronedarone and liposomal morphine are both absolutely a contraindication, as they may increase the serum levels, but hundreds of other drugs require monitoring for interaction.[31]
Contraindications
Absolute contraindications for the use of lidocaine include:
- Heart block, second or third degree (without pacemaker)
- Severe sinoatrial block (without pacemaker)
- Serious adverse drug reaction to lidocaine or amide local anesthetics
- Hypersensitivity to corn and corn-related products (corn-derived dextrose is used in the mixed injections)
- Concurrent treatment with quinidine, flecainide, disopyramide, procainamide (class I antiarrhythmic agents)
- Prior use of amiodarone hydrochloride
- Adams-Stokes syndrome[32]
- Wolff-Parkinson-White syndrome[32]
- Lidocaine viscous is not recommended by the FDA to treat teething pain in children and infants.[33]
Exercise caution in patients with any of these:
- Hypotension not due to arrhythmia
- Bradycardia
- Accelerated idioventricular rhythm
- Elderly patients
- Ehlers-Danlos Syndrome
- Pseudocholinesterase deficiency
- Intra-articular infusion (this is not an approved indication and can cause chondrolysis)
- Porphyria, especially acute intermittent porphyria; lidocaine has been classified as porphyrogenic because of the hepatic enzymes it induces,[34] although clinical evidence suggests it is not.[35] Bupivacaine is a safe alternative in this case.
- Impaired liver function – people with lowered hepatic function may have an adverse reaction with repeated administration of lidocaine because the drug is metabolized by the liver. Adverse reactions may include neurological symptoms (e.g. dizziness, nausea, muscle twitches, vomiting, or seizures).[36]
Overdosage
Overdoses of lidocaine may result from excessive administration by topical or parenteral routes, accidental oral ingestion of topical preparations by children (who are more susceptible to overdose), accidental intravenous (rather than subcutaneous, intrathecal, or paracervical) injection, or from prolonged use of subcutaneous infiltration anesthesia during cosmetic surgery.
Such overdoses have often led to severe toxicity or death in both children and adults. Lidocaine and its two major metabolites may be quantified in blood, plasma, or serum to confirm the diagnosis in potential poisoning victims or to assist forensic investigation in a case of fatal overdose.
Lidocaine is often given intravenously as an antiarrhythmic agent in critical cardiac-care situations.[37] Treatment with intravenous lipid emulsions (used for parenteral feeding) to reverse the effects of local anaesthetic toxicity is becoming more common.[38]
Postarthroscopic glenohumeral chondrolysis
Lidocaine in large amounts may be toxic to cartilage and intra-articular infusions can lead to postarthroscopic glenohumeral chondrolysis.[39]
Pharmacology
Mechanism of action
Lidocaine alters signal conduction in neurons by prolonging the inactivation of the fast voltage-gated Na+ channels in the neuronal cell membrane responsible for action potential propagation.[40] With sufficient blockage, the voltage-gated sodium channels will not open and an action potential will not be generated. Careful titration allows for a high degree of selectivity in the blockage of sensory neurons, whereas higher concentrations also affect other types of neurons.
The same principle applies for this drug’s actions in the heart. Blocking sodium channels in the conduction system, as well as the muscle cells of the heart, raises the depolarization threshold, making the heart less likely to initiate or conduct early action potentials that may cause an arrhythmia.[41]
Pharmacokinetics
When used as an injectable it typically begins working within four minutes and lasts for half an hour to three hours.[8][9] Lidocaine is about 95% metabolized (dealkylated) in the liver mainly by CYP3A4 to the pharmacologically active metabolites monoethylglycinexylidide (MEGX) and then subsequently to the inactive glycine xylidide. MEGX has a longer half-life than lidocaine, but also is a less potent sodium channel blocker.[42] The volume of distribution is 1.1 L/kg to 2.1 L/kg, but congestive heart failure can decrease it. About 60% to 80% circulates bound to the protein alpha1 acid glycoprotein. The oral bioavailability is 35% and the topical bioavailability is 3%.
The elimination half-life of lidocaine is biphasic and around 90 min to 120 min in most patients. This may be prolonged in patients with hepatic impairment (average 343 min) or congestive heart failure (average 136 min).[43] Lidocaine is excreted in the urine (90% as metabolites and 10% as unchanged drug).[44]
History
Lidocaine, the first amino amide–type local anesthetic, was first synthesized under the name ‘xylocaine’ by Swedish chemist Nils Löfgren in 1943.[45][46][47] His colleague Bengt Lundqvist performed the first injection anesthesia experiments on himself.[45] It was first marketed in 1949.
Society and culture
Dosage forms
Lidocaine, usually in the form of its hydrochloride salt, is available in various forms including many topical formulations and solutions for injection or infusion.[48] It is also available as a transdermal patch, which is applied directly to the skin.
- Lidocaine hydrochloride 2% epinephrine 1:80,000 solution for injection in a cartridge
- Lidocaine hydrochloride 1% solution for injection
- Topical lidocaine spray
- 2% viscous lidocaine
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Names
Lidocaine is the International Nonproprietary Name (INN), British Approved Name (BAN), and Australian Approved Name (AAN),[49] while lignocaine is the former BAN[citation needed] and AAN. Both the old and new names will be displayed on the product label in Australia until at least 2023.[50]
Xylocaine is a brand name.
Recreational use
As of 2021, lidocaine is not listed by the World Anti-Doping Agency as a substance whose use is banned in sport.[51] It is used as an adjuvant, adulterant, and diluent to street drugs such as cocaine and heroin.[52] It is one of the three common ingredients in site enhancement oil used by bodybuilders.[53]
Adulterant in cocaine
Lidocaine is often added to cocaine as a diluent.[54][55] Cocaine and lidocaine both numb the gums when applied. This gives the user the impression of high-quality cocaine, when in actuality the user is receiving a diluted product.[56]
Compendial status
Veterinary use
It is a component of the veterinary drug Tributame along with embutramide and chloroquine used to carry out euthanasia on horses and dogs.[58][59]
References
- ^ “Lidocaine”. Merriam-Webster Dictionary.
- ^ “Lidocaine”. Dictionary.com Unabridged. Random House.
- ^ “Poisons Standard February 2021”. Federal Register of Legislation. 1 January 2021. Retrieved 11 April 2021.
- ^ “Lidocaine Hydrochloride Injection BP 1% w/v – Summary of Product Characteristics (SmPC)”. (emc). 29 June 2020. Retrieved 11 April 2021.
- ^ “Xylocaine MPF- lidocaine hydrochloride injection, solution Xylocaine- lidocaine hydrochloride injection, solution Xylocaine- lidocaine hydrochloride,epinephrine bitartrate injection, solution”. DailyMed. Retrieved 11 April 2021.
- ^ “Ztlido- lidocaine patch”. DailyMed. Retrieved 11 April 2021.
- ^ Jump up to:a b c d e f g h i j k “Lidocaine Hydrochloride (Antiarrhythmic)”. The American Society of Health-System Pharmacists. Archivedfrom the original on 2015-08-10. Retrieved Aug 26, 2015.
- ^ Jump up to:a b c d e f g h i j “Lidocaine Hydrochloride (Local)”. The American Society of Health-System Pharmacists. Archived from the original on 2015-09-06. Retrieved Aug 26, 2015.
- ^ Jump up to:a b c J. P. Nolan; P. J. F. Baskett (1997). “Analgesia and anaesthesia”. In David Skinner; Andrew Swain; Rodney Peyton; Colin Robertson (eds.). Cambridge Textbook of Accident and Emergency Medicine. Project co-ordinator, Fiona Whinster. Cambridge, UK: Cambridge University Press. p. 194. ISBN 9780521433792. Archived from the original on 2017-09-08.
- ^ Scriabine, Alexander (1999). “Discovery and development of major drugs currently in use”. In Ralph Landau; Basil Achilladelis; Alexander Scriabine (eds.). Pharmaceutical Innovation: Revolutionizing Human Health. Philadelphia: Chemical Heritage Press. p. 211. ISBN 9780941901215. Archived from the original on 2017-09-08.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 22. ISBN 9781284057560.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Lidocaine – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “Local anaesthetic drugs”.
- ^ Cepeda MS, Tzortzopoulou A, Thackrey M, Hudcova J, Arora Gandhi P, Schumann R (December 2010). Tzortzopoulou A (ed.). “Adjusting the pH of lidocaine for reducing pain on injection”. The Cochrane Database of Systematic Reviews (12): CD006581. doi:10.1002/14651858.CD006581.pub2. PMID 21154371.(Retracted, see doi:10.1002/14651858.cd006581.pub3. If this is an intentional citation to a retracted paper, please replace
{{Retracted}}with{{Retracted|intentional=yes}}.) - ^ Derry S, Wiffen PJ, Moore RA, Quinlan J (July 2014). Derry S (ed.). “Topical lidocaine for neuropathic pain in adults”. The Cochrane Database of Systematic Reviews. 7 (7): CD010958. doi:10.1002/14651858.CD010958.pub2. PMC 6540846. PMID 25058164.
- ^ Sinha S, Schreiner AJ, Biernaskie J, Nickerson D, Gabriel VA (November 2017). “Treating pain on skin graft donor sites: Review and clinical recommendations”. The Journal of Trauma and Acute Care Surgery. 83 (5): 954–964. doi:10.1097/TA.0000000000001615. PMID 28598907. S2CID 44520644.
- ^ “Lidocaine/prilocaine spray for premature ejaculation”. Drug and Therapeutics Bulletin. 55 (4): 45–48. April 2017. doi:10.1136/dtb.2017.4.0469. PMID 28408390. S2CID 19110955.
- ^ Kumar M, Chawla R, Goyal M (2015). “Topical anesthesia”. Journal of Anaesthesiology Clinical Pharmacology. 31 (4): 450–6. doi:10.4103/0970-9185.169049. PMC 4676230. PMID 26702198.
- ^ Martí-Carvajal AJ, Simancas-Racines D, Anand V, Bangdiwala S (August 2015). “Prophylactic lidocaine for myocardial infarction”. The Cochrane Database of Systematic Reviews. 8 (8): CD008553. doi:10.1002/14651858.CD008553.pub2. PMC 8454263. PMID 26295202.
- ^ Slaughter LA, Patel AD, Slaughter JL (March 2013). “Pharmacological treatment of neonatal seizures: a systematic review”. Journal of Child Neurology. 28 (3): 351–64. doi:10.1177/0883073812470734. PMC 3805825. PMID 23318696.
- ^ Weibel S, Jelting Y, Pace NL, Helf A, Eberhart LH, Hahnenkamp K, et al. (June 2018). “Continuous intravenous perioperative lidocaine infusion for postoperative pain and recovery in adults”. The Cochrane Database of Systematic Reviews. 2018 (6): CD009642. doi:10.1002/14651858.cd009642.pub3. PMC 6513586. PMID 29864216.
- ^ Biller JA (2007). “Airway obstruction, bronchospasm, and cough”. In Berger AM, Shuster JL, Von Roenn JH (eds.). Principles and practice of palliative care and supportive oncology. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 297–307. ISBN 978-0-7817-9595-1.
Inhaled lidocaine is used to suppress cough during bronchoscopy. Animal studies and a few human studies suggest that lidocaine has an antitussive effect…
- ^ Birsa LM, Verity PG, Lee RF (May 2010). “Evaluation of the effects of various chemicals on discharge of and pain caused by jellyfish nematocysts”. Comp. Biochem. Physiol. C. 151 (4): 426–30. doi:10.1016/j.cbpc.2010.01.007. PMID 20116454.
- ^ Morabito R, Marino A, Dossena S, La Spada G (Jun 2014). “Nematocyst discharge in Pelagia noctiluca (Cnidaria, Scyphozoa) oral arms can be affected by lidocaine, ethanol, ammonia and acetic acid”. Toxicon. 83: 52–8. doi:10.1016/j.toxicon.2014.03.002. PMID 24637105.
- ^ James G. Adams (2012). “32”. Emergency Medicine: Clinical Essentials. Elsevier Health Sciences. ISBN 9781455733941. Archived from the original on 2017-09-08.
- ^ Jackson D, Chen AH, Bennett CR (October 1994). “Identifying true lidocaine allergy”. J Am Dent Assoc. 125 (10): 1362–6. doi:10.14219/jada.archive.1994.0180. PMID 7844301.
- ^ Jump up to:a b Australian Medicines Handbook. Adelaide, S. Aust: Australian Medicines Handbook Pty Ltd. 2006. ISBN 978-0-9757919-2-9.[page needed]
- ^ Jump up to:a b Nielsen LJ, Lumholt P, Hölmich LR (October 2014). “[Local anaesthesia with vasoconstrictor is safe to use in areas with end-arteries in fingers, toes, noses and ears]”. Ugeskrift for Laeger. 176(44). PMID 25354008.
- ^ “Lidocaine”. Epocrates. Archived from the original on 2014-04-22.
- ^ Jump up to:a b “Lidocaine Hydrochloride and 5% Dextrose Injection”. Safety Labeling Changes. FDA Center for Drug Evaluation and Research (CDER). January 2014. Archived from the original on 2013-04-03.
- ^ “Lidocaine Viscous: Drug Safety Communication – Boxed Warning Required – Should Not Be Used to Treat Teething Pain”. FDA Center for Drug Evaluation and Research (CDER). June 2014. Archived from the original on 2014-07-14.
- ^ “Table 96–4. Drugs and Porphyria” (PDF). Merck Manual. Merck & Company, Inc. 2011. Archived from the original on 2014-04-20.
- ^ “Lidocaine – N01BB02”. Drug porphyrinogenicity monograph. The Norwegian Porphyria Centre and the Swedish Porphyria Centre. Archived from the original on 2014-04-20.
strong clinical evidence points to lidocaine as probably not porphyrinogenic
- ^ Khan, M. Gabriel (2007). Cardiac Drug Therapy (7th ed.). Totowa, NJ: Humana Press. ISBN 9781597452380.
- ^ Baselt R (2008). Disposition of Toxic Drugs and Chemicals in Man(8th ed.). Foster City, CA: Biomedical Publications. pp. 840–4. ISBN 978-0-9626523-7-0.
- ^ Picard J, Ward SC, Zumpe R, Meek T, Barlow J, Harrop-Griffiths W (February 2009). “Guidelines and the adoption of ‘lipid rescue’ therapy for local anaesthetic toxicity”. Anaesthesia. 64 (2): 122–5. doi:10.1111/j.1365-2044.2008.05816.x. PMID 19143686. S2CID 25581037.
- ^ Gulihar A, Robati S, Twaij H, Salih A, Taylor GJ (December 2015). “Articular cartilage and local anaesthetic: A systematic review of the current literature”. Journal of Orthopaedics. 12 (Suppl 2): S200-10. doi:10.1016/j.jor.2015.10.005. PMC 4796530. PMID 27047224.
- ^ Carterall, William A. (2001). “Molecular mechanisms of gating and drug block of sodium channels”. Sodium Channels and Neuronal Hyperexcitability. Novartis Foundation Symposia. 241. pp. 206–225. doi:10.1002/0470846682.ch14. ISBN 9780470846681.
- ^ Sheu SS, Lederer WJ (Oct 1985). “Lidocaine’s negative inotropic and antiarrhythmic actions. Dependence on shortening of action potential duration and reduction of intracellular sodium activity”. Circulation Research. 57 (4): 578–90. doi:10.1161/01.res.57.4.578. PMID 2412723.
- ^ Lewin NA, Nelson LH (2006). “Chapter 61: Antidysrhythmics”. In Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA, Nelson LH (eds.). Goldfrank’s Toxicologic Emergencies(8th ed.). New York: McGraw-Hill. pp. 963–4. ISBN 978-0-07-143763-9.
- ^ Thomson PD, Melmon KL, Richardson JA, Cohn K, Steinbrunn W, Cudihee R, Rowland M (April 1973). “Lidocaine pharmacokinetics in advanced heart failure, liver disease, and renal failure in humans”. Ann. Intern. Med. 78 (4): 499–508. doi:10.7326/0003-4819-78-4-499. PMID 4694036.
- ^ Collinsworth KA, Kalman SM, Harrison DC (1974). “The clinical pharmacology of lidocaine as an antiarrhythymic drug”. Circulation. 50 (6): 1217–30. doi:10.1161/01.CIR.50.6.1217. PMID 4609637.
- ^ Jump up to:a b Löfgren N (1948). Studies on local anesthetics: Xylocaine: a new synthetic drug (Inaugural dissertation). Stockholm, Sweden: Ivar Heggstroms. OCLC 646046738.[page needed]
- ^ Löfgren N, Lundqvist B (1946). “Studies on local anaesthetics II”. Svensk Kemisk Tidskrift. 58: 206–17.
- ^ Wildsmith JAW (2011). “Lidocaine: A more complex story than ‘simple’ chemistry suggests” (PDF). The Proceedings of the History of Anaesthesia Society. 43: 9–16. Archived (PDF) from the original on 2014-04-22.
- ^ “Lidocaine international forms and names”. Drugs.com. Retrieved 29 October 2017.
- ^ “Lidocaine Ingredient Summary”. Therapeutic Goods Administration. Retrieved 20 September 2018.
- ^ “Updating medicine ingredient names – list of affected ingredients”. Therapeutic Goods Administration. 24 June 2019. Retrieved 16 February 2020.
- ^ “The 2021 Prohibited List International Standard” (PDF). The World Anti-Doping Code. World Anti-Doping Agency (WADA). 1 January 2021. Archived from the original (PDF) on 13 May 2021. Retrieved 18 May 2021.
- ^ “New York Drug Threat Assessment”. National Drug Intelligence Center. November 2002. Archived from the original on 2012-08-12.
- ^ Pupka A, Sikora J, Mauricz J, Cios D, Płonek T (2009). “[The usage of synthol in the body building]”. Polimery W Medycynie. 39(1): 63–5. PMID 19580174.
- ^ Bernardo NP; Siqueira MEPB; De Paiva MJN; Maia PP (2003). “Caffeine and other adulterants in seizures of street cocaine in Brazil”. International Journal of Drug Policy. 14 (4): 331–4. doi:10.1016/S0955-3959(03)00083-5.
- ^ “UNITED STATES of America, Plaintiff-Appellee, v. Luis A. CUELLO, Alvaro Bastides-Benitez, John Doe, a/k/a Hugo Hurtado, and Alvaro Carvajal, Defendants-Appellants”. Docket No. 78-5314. United States Court of Appeals, Fifth Circuit. 1979-07-25. Archived from the original on 2012-05-24.
- ^ Winterman, Denise (2010-09-07). “How cutting drugs became big business”. BBC News Online. BBC News Magazine. Archivedfrom the original on 2 February 2017. Retrieved 20 January 2017.
- ^ “Revision Bulletin: Lidocaine and Prilocaine Cream–Revision to Related Compounds Test”. The United States Pharmacopeial Convention. November 30, 2007. Archived from the original on May 1, 2013.
- ^ Peterson, Michael E.; Talcott, Patricia A. (2013-08-07). Small Animal Toxicology. Elsevier Health Sciences. ISBN 978-0323241984. Archived from the original on 2017-09-08.
- ^ “FDA Freedom of Information Summary – Tributame” (PDF). Archived from the original (PDF) on 2015-05-18.
External links
- “Lidocaine”. Drug Information Portal. U.S. National Library of Medicine.
- “Lidocaine Transdermal Patch”. MedlinePlus.
- US patent 2441498, Nils Magnus Loefgren & Bengt Josef Lundqvist, “Alkyl glycinanilides”, published 1948-05-11, issued 1948-05-11, assigned to ASTRA APOTEKARNES KEM FAB
| IUPAC name2-(diethylamino)- N-(2,6-dimethylphenyl)acetamide | |
| CAS Number | 137-58-6 as HCl: 73-78-9 |
|---|---|
| PubChem CID | 3676as HCl: 6314 |
| IUPHAR/BPS | 2623 |
| DrugBank | DB00281 as HCl: DBSALT001508 |
| ChemSpider | 3548 as HCl: 6075 |
| UNII | 98PI200987as HCl: EC2CNF7XFP |
| KEGG | D00358 as HCl: D02086 |
| ChEBI | CHEBI:6456 as HCl: CHEBI:50512 |
| ChEMBL | ChEMBL79 as HCl: ChEMBL541521 |
| PDB ligand | LQZ (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID1045166 |
| ECHA InfoCard | 100.004.821 |
| Chemical and physical data | |
| Formula | C14H22N2O |
| Molar mass | 234.343 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 68 °C (154 °F) |
| showSMILES | |
| showInChI |
////////LIDOCAINE, lignocaine

NEW DRUG APPROVALS
ONE TIME
$10.00
Divalproex sodium
Divalproex
- 44089
WeightAverage: 144.2114
Chemical FormulaC8H16O2
UNII614OI1Z5WI, CAS number99-66-1, 76584-70-8
2-propylpentanoic acid, DIVALPROEX SODIUM, 76584-70-8, Valproate semisodium, Epival, Depakote, Sodium divalproate, Semisodium Valproate, Abbott 50711, ValdisovalValproic Acid
CAS Registry Number: 99-66-1
CAS Name: 2-Propylpentanoic acid
Additional Names: 2-propylvaleric acid; di-n-propylacetic acid
Trademarks: Convulex (Pharmacia); Depakene (Abbott)
Molecular Formula: C8H16O2
Molecular Weight: 144.21
Percent Composition: C 66.63%, H 11.18%, O 22.19%
Literature References: Antiepileptic; increases levels of g-aminobutyric acid (GABA) in the brain. Prepn: B. S. Burton, Am. Chem. J.3, 385 (1882); E. Oberreit, Ber.29, 1998 (1896); M. Tiffeneau, Y. Deux, Compte Rend.212, 105 (1941). Anticonvulsant activity: H. Meunier et al.,Therapie18, 435 (1963). Toxicity data: Jenner et al.,Food Cosmet. Toxicol.2, 327 (1964). Comprehensive description: Z. L. Chang, Anal. Profiles Drug Subs.8, 529-556 (1979). Review of teratogenicity studies: H. Nau et al.,Pharmacol. Toxicol.69, 310-321 (1991); R. Alsdorf, D. F. Wyszynski, Expert Opin. Drug Safety4, 345-353 (2005). Review of pharmacology and clinical experience in epilepsy: E. M. Rimmer, A. Richens, Pharmacotherapy5, 171-184 (1985); in psychiatric disease: D. R. P. Guay, ibid.15, 631-647 (1995); in migraine prophylaxis: C. E. Shelton, J. F. Connelly, Ann. Pharmacother.30, 865-866 (1996). Review of pharmacodynamics and mechanisms of action: W. Löscher, Prog. Neurobiol.58, 31-59 (1999).
Properties: Colorless liquid with characteristic odor. bp 219.5°. nD24.5 1.425. d40 0.9215. pKa 4.6. Very sol in organic solvents. Soly in water: 1.3 mg/ml. LD50 orally in rats: 670 mg/kg (Jenner).
Boiling point: bp 219.5°
pKa: pKa 4.6
Index of refraction:nD24.5 1.425
Density: d40 0.9215
Toxicity data: LD50 orally in rats: 670 mg/kg (Jenner)
Derivative Type: Sodium salt (1:1)
CAS Registry Number: 1069-66-5
Additional Names: Sodium valproate
Trademarks: Depacon (Abbott); Depakin (Sanofi-Synthelabo); Dépakine (Sanofi-Aventis); Epilim (Sanofi-Aventis); Ergenyl (Sanofi-Synthelabo); Leptilan (Dolorgiet); Orfiril (Desitin)
Molecular Formula: C8H15NaO2
Molecular Weight: 166.19
Percent Composition: C 57.82%, H 9.10%, Na 13.83%, O 19.25%
Properties: White, odorless, crystalline, deliquescent powder. pKa 4.8. Hygroscopic. One gram is sol in 0.4 ml water; 1.5 ml ethanol; 5 ml methanol. Practically insol in common organic solvents. LD50 orally in mice: 1700 mg/kg (Meunier).
pKa: pKa 4.8
Toxicity data: LD50 orally in mice: 1700 mg/kg (Meunier)
Derivative Type: Sodium salt (2:1)
CAS Registry Number: 76584-70-8
Additional Names: Sodium hydrogen bis(2-propylpentanoate); divalproex sodium; valproate semisodium
Manufacturers’ Codes: Abbott 50711
Trademarks: Depakote (Abbott); Valcote (Abbott)
Molecular Formula: C16H31NaO4
Molecular Weight: 310.40
Percent Composition: C 61.91%, H 10.07%, Na 7.41%, O 20.62%
Derivative Type: Magnesium salt
Trademarks: Depamag (Sigma-Tau)
Molecular Formula: C16H30MgO4
Molecular Weight: 310.71
Percent Composition: C 61.85%, H 9.73%, Mg 7.82%, O 20.60%
Therap-Cat: Anticonvulsant; antimanic; antimigraine.Keywords: Anticonvulsant; Antimigraine; Antimanic.
Synthesis Reference
Daniel Aubert, Francis Blanc, Henri Desmolin, Michel Morre, Lucette Sindely, “Valproic acid preparations.” U.S. Patent US5017613, issued January, 1965.
Patent
https://patents.google.com/patent/WO2007004238A2/enDivalproex sodium is one of the most widely used epileptic agents presently available in the market. Both the constituents, valproic acid and sodium valproate themselves have also been used for the treatment of epileptic seizures and convulsions. But their utility has remained restricted since valproic acid is a liquid and is difficult to formulate for an oral dosage form whereas sodium valproate is a hygroscopic solid with poor stability characteristics. Divalproex sodium is an oligomer having 1:1 molar ratio of valproic acid and sodium valproate containing 4 to 6 units. The relevant prior art includes US 4,988,731 (’73I) relates to a non-hygroscopic stable sodium hydrogen divalproate oligomer. Its probable structure is shown in Fig 1

Fig 1 – Divalproex sodiumWhere M is a about 2.As can be seen from the displayed structure, one mole each of the valproic acid forms coordinate bonds with the sodium of the sodium valproate molecule, and the valproate ion is ionically bonded to the sodium atom. The structure is thus consistent with the unique characteristic of the compound. However the preferred mode of representing Divalproex sodium is by reference to single compound of the formula{(CH3CH2CH2)2CHCO2} {(CH3CH2CH2)2CHCO2}Na, HThe said patent also describes two alternative processes for the preparation of the oligomer. According to one aspect, the oligomer is produced by dissolving sodium valproate and valproic acid in equimolar amount in acetone and crystallizing from chilled acetone at around O0C. Alternatively Divalproex sodium can be isolated from a two component liquid medium, which includes acetone where in half equivalent of NaOH to the valproic acid present, preferable as a solution in an acetone miscible solvent eg. water. The new compound can be recovered from the liquid phase by evaporating the solvent(s) and, if desired, the new compound can be recrystallized, for instance from acetonitrile or others or the material may be spay-dried, lyophilized or purified by chromatography.US ‘731 claims yield of 90% of theory.Drawbacks of the above mentioned reported methods for the preparation of Divalproex sodium described in US 4988731 are difficult to reproduce on a large scale and provides inconsistent yields and the material obtained is not always free flowing in nature. The process involves the crystallization of a 1:1 mixture of valproic acid and sodium valproate from a chilled solution of acetone, followed by washing with chilled acetone. Divalproex sodium is as such fairly soluble in acetone at temperatures above 1O0C and extreme care has to be. taken while performing washing with chilled acetone as any rise in temperature would lead to the loss of yield. This problem actually comes to the fore while scaling up the process during commercialization since during centrifugation of the large volume the temperature of the mixture rises and acetone has to be cooled below O0C, which require large amount of liquid nitrogen or dry ice. Moreover it was also observed that due to the cooled nature of the solvent, the isolated Divalproex sodium absorbs considerable amount of moisture and therefore requires longer time to dry eventually leading to longer time cycle for the otherwise simple single step process. Also the high moisture content in the recovered acetone makes it unsuitable for reuse. Alternatively, to avoid absorption of water, the centrifugation had to be carried out under a blanket of dry nitrogen gas. These additional infrastructural loads add to input costs eventually making the otherwise single step low cost process becoming uncompetitive and economically unviable.Similarly the other process involves the addition of half molar equivalent of sodium hydroxide dissolved in water to valproic acid and the solvent has to be evaporated to obtain crude product, which has to be recrystallized to get Divalprox of the desired specification. The process is operationally tedious and requires the reduction in the level of water in the reaction mass via evaporation of the solvent followed by re- crystallization from acetonitrile making the process lengthy and economically unviable. There is therefore a need for operationally making this single step process more efficient and high yieldingExample I:To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH ( 13g; half molar) is added & the resulting reaction mixture is stirred at 40-500C for 1 hr. Then acetonitrile(600ml) is added to obtain clear solution at 40-500C and the solution is charcoalized at 40-500C followed by filtration at 40-500C through hyflo-bed. The resultant reaction mixture was stirred at 10-200C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. (102.25g, 95%)Example II;To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH (13g; half molar) is added & the resulting reaction mixture is stirred at 30-400C for 1 hr. Then acetone (600ml) is added to obtain clear solution at 30-400C and the material is charcoalized at 30-400C followed by filtration through hyflo-bed. The resultant reaction solution was stirred at -5°C to -1O0C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. ( 55g, 51.11%) Example III:To a solution of Valproic acid (10Og) in dichloromethane (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 30- 400C for 1 hr to get clear solution. Then acetonitrile (600ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (10Og; 93%)Example IV:To a solution of Valproic acid (10Og) in diisopropyl ether(200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (104g; 96.65%)Example V:To a solution of Valproic acid (10Og) in methyl tertiary butyl ether(200ml) at 20- 300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (102g;94.79%)Example VI:To a solution of Valproic acid (10Og) in toluene (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (101g; 93.87%)Example VII: A mixture of sodium valproate (6Og) and valproic acid (52.04g) was taken in acetonitrile (800ml) and heated at reflux to obtain a clear solution, which was filtered through hyflo-bed to remove suspended particles. Then the solution was stirred at 10- 200C for 2-3 hr. The solid, thus obtained, was filtered and washed with acetonitrile (100ml). The product was dried at 40-450C for 10-12 hr. (105g ; 93.75%)Example VIII;To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C5 caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring. The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-45°C for 10-12 hr.(102g; ~ 95%) Example IX:To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -1O0C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(54g; ~ 50.11%)Example X:To a solution of valproic acid (10Og) in ethanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring.The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-450C for 10-12 hr.(101g; ~ 93.87%)Example XI: To a solution of valproic acid (10Og) in ethanol (200ml) at 20-30°C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -100C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(55g; ~ 51%)ADVANTAGES:> The process is high yielding. > The process produces Divalproex sodium with improved flowability.> The process produces Divalproex sodium that is non-hygroscopic and more stable.> The process is industrially feasible, precise, reproducible and does not require sophisticated infrastructure.
Divalproex Sodium is a stable coordination compound comprised of sodium valproate and valproic acid with anticonvulsant and antiepileptic activities. Divalproex dissociates to the valproate ion in the gastrointestinal tract. This agent binds to and inhibits gamma-aminobutyric acid (GABA) transaminase and its anticonvulsant activity may be exerted by increasing brain concentration of GABA and by inhibiting enzymes that catabolize GABA or block the reuptake of GABA into glia and nerve endings. Divalproex may also work by suppressing repetitive neuronal firing through inhibition of voltage-sensitive sodium channels.
Valproate semisodium is a mixture of valproic acid and its sodium salt in a 1:1 molar ratio. It is used for the management and treatment of seizure disorders, mania, and prophylactic treatment of migraine headache. It has a role as an antimanic drug, an anticonvulsant and a GABA agent. It contains a valproic acid and a sodium valproate.
Divalproex sodium, valproate sodium, and valproic acid, are all similar medications that are used by the body as valproic acid. Therefore, the term valproic acid will be used to represent all of these medications in this discussion.
Valproate (VPA) and its valproic acid, sodium valproate, and valproate semisodium forms are medications primarily used to treat epilepsy and bipolar disorder and prevent migraine headaches.[2] They are useful for the prevention of seizures in those with absence seizures, partial seizures, and generalized seizures.[2] They can be given intravenously or by mouth, and the tablet forms exist in both long- and short-acting formulations.[2]
Common side effects of valproate include nausea, vomiting, sleepiness, and dry mouth.[2] Serious side effects can include liver failure, and regular monitoring of liver function tests is therefore recommended.[2] Other serious risks include pancreatitis and an increased suicide risk.[2] Valproate is known to cause serious abnormalities in babies if taken during pregnancy,[2][3] and as such it is not typically recommended for women of childbearing age who have migraines.[2]
Valproate’s precise mechanism of action is unclear.[2][4] Proposed mechanisms include affecting GABA levels, blocking voltage-gated sodium channels, and inhibiting histone deacetylases.[5][6] Valproic acid is a branched short-chain fatty acid (SCFA) made from valeric acid.[5]
Valproate was first made in 1881 and came into medical use in 1962.[7] It is on the World Health Organization’s List of Essential Medicines[8] and is available as a generic medication.[2] It is marketed under the brand names Depakote, among others.[2] In 2018, it was the 131st most commonly prescribed medication in the United States, with more than 5 million prescriptions.[9][10]
Terminology
Valproic acid (VPA) is an organic weak acid. The conjugate base is valproate. The sodium salt of the acid is sodium valproate and a coordination complex of the two is known as valproate semisodium.[11]
Medical uses
It is used primarily to treat epilepsy and bipolar disorder. It is also used to prevent migraine headaches.[12]
Epilepsy
Valproate has a broad spectrum of anticonvulsant activity, although it is primarily used as a first-line treatment for tonic–clonic seizures, absence seizures and myoclonic seizures and as a second-line treatment for partial seizures and infantile spasms.[12][13] It has also been successfully given intravenously to treat status epilepticus.[14][15]
Mental illness
Bipolar disorder
Valproate products are also used to treat manic or mixed episodes of bipolar disorder.[16][17]
Schizophrenia
A 2016 systematic review compared the efficacy of valproate as an add-on for people with schizophrenia:[18]
| There is limited evidence that adding valproate to antipsychotics may be effective for overall response and also for specific symptoms, especially in terms of excitement and aggression. Valproate was associated with a number of adverse events among which sedation and dizziness appeared more frequently than in the control groups.[18] |
| showOutcomeFindings in wordsFindings in numbersQuality of evidence |
Dopamine dysregulation syndrome
Based upon five case reports, valproic acid may have efficacy in controlling the symptoms of the dopamine dysregulation syndrome that arise from the treatment of Parkinson’s disease with levodopa.[19][20][21]
Migraines
Valproate is also used to prevent migraine headaches. Because this medication can be potentially harmful to the fetus, valproate should be considered for those able to become pregnant only after the risks have been discussed.[22]
Other
The medication has been tested in the treatment of AIDS and cancer, owing to its histone-deacetylase-inhibiting effects.[23]
Contraindications
Contraindications include:
- Pre-existing acute or chronic liver dysfunction or family history of severe liver inflammation (hepatitis), particularly medicine related.[24]
- Known hypersensitivity to valproate or any of the ingredients used in the preparation[24]
- Urea cycle disorders[24]
- Hepatic porphyria[24]
- Hepatotoxicity[24]
- Mitochondrial disease[24]
- Pancreatitis[24]
- Porphyria[25]
Adverse effects
See also: List of adverse effects of valproic acid and List of adverse effects of valproate semisodium
Most common adverse effects include:[22]
- Nausea (22%)
- Drowsiness (19%)
- Dizziness (12%)
- Vomiting (12%)
- Weakness (10%)
Serious adverse effects include:[22]
- Bleeding
- Low blood platelets
- Encephalopathy
- Suicidal behavior and thoughts
- Low body temperature
Valproic acid has a black box warning for hepatotoxicity, pancreatitis, and fetal abnormalities.[22]
There is evidence that valproic acid may cause premature growth plate ossification in children and adolescents, resulting in decreased height.[26][27][28][29] Valproic acid can also cause mydriasis, a dilation of the pupils.[30] There is evidence that shows valproic acid may increase the chance of polycystic ovary syndrome (PCOS) in women with epilepsy or bipolar disorder. Studies have shown this risk of PCOS is higher in women with epilepsy compared to those with bipolar disorder.[31] Weight gain is also possible.[32]
Pregnancy
Valproate causes birth defects;[33] exposure during pregnancy is associated with about three times as many major abnormalities as usual, mainly spina bifida with the risks being related to the strength of medication used and use of more than one drug.[34][35] More rarely, with several other defects, including a “valproate syndrome”.[36] Characteristics of this valproate syndrome include facial features that tend to evolve with age, including a triangle-shaped forehead, tall forehead with bifrontal narrowing, epicanthic folds, medial deficiency of eyebrows, flat nasal bridge, broad nasal root, anteverted nares, shallow philtrum, long upper lip and thin vermillion borders, thick lower lip and small downturned mouth.[37] While developmental delay is usually associated with altered physical characteristics (dysmorphic features), this is not always the case.[38]
Children of mothers taking valproate during pregnancy are at risk for lower IQs.[39][40][41] Maternal valproate use during pregnancy increased the probability of autism in the offspring compared to mothers not taking valproate from 1.5% to 4.4%.[42] A 2005 study found rates of autism among children exposed to sodium valproate before birth in the cohort studied were 8.9%.[43] The normal incidence for autism in the general population is estimated at less than one percent.[44] A 2009 study found that the 3-year-old children of pregnant women taking valproate had an IQ nine points lower than that of a well-matched control group. However, further research in older children and adults is needed.[45][46][47]
Sodium valproate has been associated with paroxysmal tonic upgaze of childhood, also known as Ouvrier–Billson syndrome, from childhood or fetal exposure. This condition resolved after discontinuing valproate therapy.[48][49]
Women who intend to become pregnant should switch to a different medication if possible or decrease their dose of valproate.[50] Women who become pregnant while taking valproate should be warned that it causes birth defects and cognitive impairment in the newborn, especially at high doses (although valproate is sometimes the only drug that can control seizures, and seizures in pregnancy could have worse outcomes for the fetus than exposure to valproate). Studies have shown that taking folic acid supplements can reduce the risk of congenital neural tube defects.[22] The use of valproate for migraine or bipolar disorder during pregnancy is contraindicated in the European Union, and the medicines are not recommended for epilepsy during pregnancy unless there is no other effective treatment available.[51]
Elderly
Valproate in elderly people with dementia caused increased sleepiness. More people stopped the medication for this reason. Additional side effects of weight loss and decreased food intake were also associated with one-half of people who become sleepy.[22]
Overdose and toxicity
| Form | Lower limit | Upper limit | Unit |
| Total (including protein bound) | 50[52] | 125[52] | µg/mL or mg/l |
| 350[53] | 700[53] | μmol/L | |
| Free | 6[52] | 22[52] | µg/mL or mg/l |
| 35[53] | 70[53] | μmol/L |
Excessive amounts of valproic acid can result in sleepiness, tremor, stupor, respiratory depression, coma, metabolic acidosis, and death.[54] In general, serum or plasma valproic acid concentrations are in a range of 20–100 mg/l during controlled therapy, but may reach 150–1500 mg/l following acute poisoning. Monitoring of the serum level is often accomplished using commercial immunoassay techniques, although some laboratories employ gas or liquid chromatography.[55] In contrast to other antiepileptic drugs, at present there is little favorable evidence for salivary therapeutic drug monitoring. Salivary levels of valproic acid correlate poorly with serum levels, partly due to valproate’s weak acid property (pKa of 4.9).[56]
In severe intoxication, hemoperfusion or hemofiltration can be an effective means of hastening elimination of the drug from the body.[57][58] Supportive therapy should be given to all patients experiencing an overdose and urine output should be monitored.[22] Supplemental L-carnitine is indicated in patients having an acute overdose[59][60] and also prophylactically[59] in high risk patients. Acetyl-L-carnitine lowers hyperammonemia less markedly[61] than L-carnitine.
Interactions
Valproate inhibits CYP2C9, glucuronyl transferase, and epoxide hydrolase and is highly protein bound and hence may interact with drugs that are substrates for any of these enzymes or are highly protein bound themselves.[24] It may also potentiate the CNS depressant effects of alcohol.[24] It should not be given in conjunction with other antiepileptics due to the potential for reduced clearance of other antiepileptics (including carbamazepine, lamotrigine, phenytoin and phenobarbitone) and itself.[24] It may also interact with:[22][24][62]
- Aspirin: may increase valproate concentrations. May also interfere with valproate’s metabolism.
- Benzodiazepines: may cause CNS depression and there are possible pharmacokinetic interactions.
- Carbapenem antibiotics: reduce valproate levels, potentially leading to seizures.
- Cimetidine: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
- Erythromycin: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
- Ethosuximide: valproate may increase ethosuximide concentrations and lead to toxicity.
- Felbamate: may increase plasma concentrations of valproate.
- Mefloquine: may increase valproate metabolism combined with the direct epileptogenic effects of mefloquine.
- Oral contraceptives: may reduce plasma concentrations of valproate.
- Primidone: may accelerate metabolism of valproate, leading to a decline of serum levels and potential breakthrough seizure.
- Rifampicin: increases the clearance of valproate, leading to decreased valproate concentrations
- Warfarin: valproate may increase free warfarin concentration and prolong bleeding time.
- Zidovudine: valproate may increase zidovudine serum concentration and lead to toxicity.
Pharmacology
Pharmacodynamics
Although the mechanism of action of valproate is not fully understood,[24] traditionally, its anticonvulsant effect has been attributed to the blockade of voltage-gated sodium channels and increased brain levels of gamma-aminobutyric acid (GABA).[24] The GABAergic effect is also believed to contribute towards the anti-manic properties of valproate.[24] In animals, sodium valproate raises cerebral and cerebellar levels of the inhibitory synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative enzymes, such as GABA transaminase, succinate-semialdehyde dehydrogenase and by inhibiting the re-uptake of GABA by neuronal cells.[24]
Prevention of neurotransmitter-induced hyperexcitability of nerve cells, via Kv7.2 channel and AKAP5, may also contribute to its mechanism.[63] Also, it has been shown to protect against a seizure-induced reduction in phosphatidylinositol (3,4,5)-trisphosphate (PIP3) as a potential therapeutic mechanism.[64]
It also has histone-deacetylase-inhibiting effects. The inhibition of histone deacetylase, by promoting more transcriptionally active chromatin structures, likely presents the epigenetic mechanism for regulation of many of the neuroprotective effects attributed to valproic acid. Intermediate molecules mediating these effects include VEGF, BDNF, and GDNF.[65][66]
Endocrine actions
Valproic acid has been found to be an antagonist of the androgen and progesterone receptors, and hence as a nonsteroidal antiandrogen and antiprogestogen, at concentrations much lower than therapeutic serum levels.[67] In addition, the drug has been identified as a potent aromatase inhibitor, and suppresses estrogen concentrations.[68] These actions are likely to be involved in the reproductive endocrine disturbances seen with valproic acid treatment.[67][68]
Valproic acid has been found to directly stimulate androgen biosynthesis in the gonads via inhibition of histone deacetylases and has been associated with hyperandrogenism in women and increased 4-androstenedione levels in men.[69][70] High rates of polycystic ovary syndrome and menstrual disorders have also been observed in women treated with valproic acid.[70]
Pharmacokinetics

Some metabolites of valproic acid. Glucuronidation and β-oxidation are the main metabolic pathways; ω-oxidation metabolites are considered hepatotoxic.[71][72] Details see text.
Taken by mouth, valproate is rapidly and virtually completely absorbed from the gut.[71] When in the bloodstream, 80–90% of the substance are bound to plasma proteins, mainly albumin. Protein binding is saturable: it decreases with increasing valproate concentration, low albumin concentrations, the patient’s age, additional use of other drugs such as aspirin, as well as liver and kidney impairment.[73][74] Concentrations in the cerebrospinal fluid and in breast milk are 1 to 10% of blood plasma concentrations.[71]
The vast majority of valproate metabolism occurs in the liver.[75] Valproate is known to be metabolized by the cytochrome P450 enzymes CYP2A6, CYP2B6, CYP2C9, and CYP3A5.[75] It is also known to be metabolized by the UDP-glucuronosyltransferase enzymes UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15.[75] Some of the known metabolites of valproate by these enzymes and uncharacterized enzymes include (see image):[75]
- via glucuronidation (30–50%): valproic acid β-O-glucuronide
- via beta oxidation (>40%): 2E-ene-valproic acid, 2Z-ene-valproic acid, 3-hydroxyvalproic acid, 3-oxovalproic acid
- via omega oxidation: 5-hydroxyvalproic acid, 2-propyl-glutaric acid
- some others: 3E-ene-valproic acid, 3Z-ene-valproic acid, 4-ene-valproic acid, 4-hydroxyvalproic acid
All in all, over 20 metabolites are known.[71]
In adult patients taking valproate alone, 30–50% of an administered dose is excreted in urine as the glucuronide conjugate.[75] The other major pathway in the metabolism of valproate is mitochondrial beta oxidation, which typically accounts for over 40% of an administered dose.[75] Typically, less than 20% of an administered dose is eliminated by other oxidative mechanisms.[75] Less than 3% of an administered dose of valproate is excreted unchanged (i.e., as valproate) in urine.[75] Only a small amount is excreted via the faeces.[71] Elimination half-life is 16±3 hours and can decrease to 4–9 hours when combined with enzyme inducers.[71][74]
Chemistry
Valproic acid is a branched short-chain fatty acid and the 2-n–propyl derivative of valeric acid.[5]
History
Valproic acid was first synthesized in 1882 by Beverly S. Burton as an analogue of valeric acid, found naturally in valerian.[76] Valproic acid is a carboxylic acid, a clear liquid at room temperature. For many decades, its only use was in laboratories as a “metabolically inert” solvent for organic compounds. In 1962, the French researcher Pierre Eymard serendipitously discovered the anticonvulsant properties of valproic acid while using it as a vehicle for a number of other compounds that were being screened for antiseizure activity. He found it prevented pentylenetetrazol-induced convulsions in laboratory rats.[77] It was approved as an antiepileptic drug in 1967 in France and has become the most widely prescribed antiepileptic drug worldwide.[78] Valproic acid has also been used for migraine prophylaxis and bipolar disorder.[79]
Society and culture
Valproate is available as a generic medication.[2]
Approval status
| Indications | FDA-labelled indication?[1] | TGA-labelled indication?[12] | MHRA-labelled indication?[80] | Literature support |
|---|---|---|---|---|
| Epilepsy | Yes | Yes | Yes | Limited (depends on the seizure type; it can help with certain kinds of seizures: drug-resistant epilepsy, partial and absence seizures, can be used against glioblastoma and other tumors both to improve survival and treat seizures, and against tonic–clonic seizures and status epilepticus).[81][82][83][84] |
| Bipolar mania | Yes | Yes | Yes | Limited.[85] |
| Bipolar depression | No | No | No | Moderate.[86] |
| Bipolar maintenance | No | No | No | Limited.[87] |
| Migraine prophylaxis | Yes | Yes (accepted) | No | Limited. |
| Acute migraine management | No | No | No | Only negative results.[88] |
| Schizophrenia | No | No | No | Weak evidence.[89] |
| Agitation in dementia | No | No | No | Weak evidence. Not recommended for agitation in people with dementia.[90] Increased rate of adverse effects, including a risk of serious adverse effects.[90] |
| Fragile X syndrome | Yes (orphan) | No | No | Limited.[66] |
| Familial adenomatous polyposis | Yes (orphan) | No | No | Limited. |
| Chronic pain & fibromyalgia | No | No | No | Limited.[91] |
| Alcohol hallucinosis | No | No | No | One randomised double-blind placebo-controlled trial.[92] |
| Intractable hiccups | No | No | No | Limited, five case reports support its efficacy, however.[93] |
| Non-epileptic myoclonus | No | No | No | Limited, three case reports support its efficacy, however.[94] |
| Cluster headaches | No | No | No | Limited, two case reports support its efficacy.[95] |
| West syndrome | No | No | No | A prospective clinical trial supported its efficacy in treating infantile spasms.[96] |
| HIV infection eradication | No | No | No | Double-blind placebo-controlled trials have been negative.[97][98][99] |
| Myelodysplastic syndrome | No | No | No | Several clinical trials have confirmed its efficacy as a monotherapy,[100] as an adjunct to tretinoin[100] and as an adjunct to hydralazine.[101] |
| Acute myeloid leukaemia | No | No | No | Two clinical trials have confirmed its efficacy in this indication as both a monotherapy and as an adjunct to tretinoin.[102][103][104] |
| Cervical cancer | No | No | No | One clinical trial supports its use here.[105] |
| Malignant melanoma | No | No | No | One phase II study has seemed to discount its efficacy.[106] |
| Breast cancer | No | No | No | A phase II study has supported its efficacy.[107] |
| Impulse control disorder | No | No | No | Limited.[108][109] |
Off-label uses
In 2012, pharmaceutical company Abbott paid $1.6 billion in fines to US federal and state governments for illegal promotion of off-label uses for Depakote, including the sedation of elderly nursing home residents.[110][111]
Some studies have suggested that valproate may reopen the critical period for learning absolute pitch and possibly other skills such as language.[112][113]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Formulations
| Clinical data | |
|---|---|
| Other names | valproate sodium (USAN US) |
| License data | US DailyMed: Valproate_sodium |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1069-66-5 |
| PubChem CID | 16760703 |
| DrugBank | DBSALT001257 |
| ChemSpider | 13428 |
| UNII | 5VOM6GYJ0D |
| KEGG | D00710 |
| ChEBI | CHEBI:9925 |
| ChEMBL | ChEMBL433 |
| CompTox Dashboard (EPA) | DTXSID6023733 |
| ECHA InfoCard | 100.002.525 |
| Chemical and physical data | |
| Formula | C8H15NaO2 |
| Molar mass | 166.196 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
{kind=link}
{kind=link}
| Clinical data | |
|---|---|
| Trade names | Depakote, others |
| Other names | semisodium valproate, divalproex sodium (USAN US) |
| License data | US DailyMed: Divalproex_sodium |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 76584-70-8 |
| PubChem CID | 23663956 |
| DrugBank | DBSALT000185 |
| ChemSpider | 48337 |
| UNII | 644VL95AO6 |
| KEGG | D00304 |
| ChEBI | CHEBI:4667 |
| ChEMBL | ChEMBL2105613 |
| CompTox Dashboard (EPA) | DTXSID6023733 |
| ECHA InfoCard | 100.002.525 |
| Chemical and physical data | |
| Formula | C16H31NaO4 |
| Molar mass | 310.410 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Valproate exists in two main molecular variants: sodium valproate and valproic acid without sodium (often implied by simply valproate). A mixture between these two is termed semisodium valproate. It is unclear whether there is any difference in efficacy between these variants, except from the fact that about 10% more mass of sodium valproate is needed than valproic acid without sodium to compensate for the sodium itself.[114]
Brand names of valproic acid
Branded products include:
- Absenor (Orion Corporation Finland)
- Convulex (G.L. Pharma GmbH Austria)
- Depakene (Abbott Laboratories in US and Canada)[115]
- Depakine (Sanofi Aventis France)
- Depakine (Sanofi Synthelabo Romania)
- Depalept (Sanofi Aventis Israel)
- Deprakine (Sanofi Aventis Finland)
- Encorate (Sun Pharmaceuticals India)
- Epival (Abbott Laboratories US and Canada)
- Epilim (Sanofi Synthelabo Australia and South Africa)
- Stavzor (Noven Pharmaceuticals Inc.)
- Valcote (Abbott Laboratories Argentina)
- Valpakine (Sanofi Aventis Brazil)
- Orfiril (Desitin Arzneimittel GmbH Norway)
Brand names of sodium valproate
Portugal
- Tablets – Diplexil-R by Bial.
United States
- Intravenous injection – Depacon by Abbott Laboratories.
- Syrup – Depakene by Abbott Laboratories. (Note Depakene capsules are valproic acid).
- Depakote tablets are a mixture of sodium valproate and valproic acid.
- Tablets – Eliaxim by Bial.
Australia
- Epilim Crushable Tablets Sanofi[116]
- Epilim Sugar Free Liquid Sanofi[116]
- Epilim Syrup Sanofi[116]
- Epilim Tablets Sanofi[116]
- Sodium Valproate Sandoz Tablets Sanofi
- Valpro Tablets Alphapharm
- Valproate Winthrop Tablets Sanofi
- Valprease tablets Sigma
New Zealand
- Epilim by Sanofi-Aventis
All the above formulations are Pharmac-subsidised.[117]
UK
- Depakote Tablets (as in USA)
- Tablets – Orlept by Wockhardt and Epilim by Sanofi
- Oral solution – Orlept Sugar Free by Wockhardt and Epilim by Sanofi
- Syrup – Epilim by Sanofi-Aventis
- Intravenous injection – Epilim Intravenous by Sanofi
- Extended release tablets – Epilim Chrono by Sanofi is a combination of sodium valproate and valproic acid in a 2.3:1 ratio.
- Enteric-coated tablets – Epilim EC200 by Sanofi is a 200-mg sodium valproate enteric-coated tablet.
UK only
- Capsules – Episenta prolonged release by Beacon
- Sachets – Episenta prolonged release by Beacon
- Intravenous solution for injection – Episenta solution for injection by Beacon
Germany, Switzerland, Norway, Finland, Sweden
- Tablets – Orfiril by Desitin Pharmaceuticals
- Intravenous injection – Orfiril IV by Desitin Pharmaceuticals
South Africa
- Syrup – Convulex by Byk Madaus[118]
- Tablets – Epilim by Sanofi-synthelabo
Malaysia
- Tablets – Epilim by Sanofi-Aventis
Romania
- Companies are SANOFI-AVENTIS FRANCE, GEROT PHARMAZEUTIKA GMBH and DESITIN ARZNEIMITTEL GMBH
- Types are Syrup, Extended release mini tablets, Gastric resistant coated tablets, Gastric resistant soft capsules, Extended release capsules, Extended release tablets and Extended release coated tablets
Canada
- Intravenous injection – Epival or Epiject by Abbott Laboratories.
- Syrup – Depakene by Abbott Laboratories its generic formulations include Apo-Valproic and ratio-Valproic.
Japan
- Tablets – Depakene by Kyowa Hakko Kirin
- Extended release tablets – Depakene-R by Kyowa Hakko Kogyo and Selenica-R by Kowa
- Syrup – Depakene by Kyowa Hakko Kogyo
Europe
In much of Europe, Dépakine and Depakine Chrono (tablets) are equivalent to Epilim and Epilim Chrono above.
Taiwan
- Tablets (white round tablet) – Depakine (Chinese: 帝拔癲; pinyin: di-ba-dian) by Sanofi Winthrop Industrie (France)
Iran
- Tablets – Epival 200 (enteric coated tablet) and Epival 500 (extended release tablet) by Iran Najo
- Slow release tablets – Depakine Chrono by Sanofi Winthrop Industrie (France)
Israel
Depalept and Depalept Chrono (extended release tablets) are equivalent to Epilim and Epilim Chrono above. Manufactured and distributed by Sanofi-Aventis.
India, Russia and CIS countries
- Valparin Chrono by Torrent Pharmaceuticals India
- Valprol CR by Intas Pharmaceutical (India)
- Encorate Chrono by Sun Pharmaceutical (India)
- Serven Chrono by Leeven APL Biotech (India)
Brand names of valproate semisodium
- Brazil – Depakote by Abbott Laboratories and Torval CR by Torrent do Brasil
- Canada – Epival by Abbott Laboratories
- Mexico – Epival and Epival ER (extended release) by Abbott Laboratories
- United Kingdom – Depakote (for psychiatric conditions) and Epilim (for epilepsy) by Sanofi-Aventis and generics
- United States – Depakote and Depakote ER (extended release) by Abbott Laboratories and generics[22]
- India – Valance and Valance OD by Abbott Healthcare Pvt Ltd, Divalid ER by Linux laboratories Pvt Ltd, Valex ER by Sigmund Promedica, Dicorate by Sun Pharma
- Germany – Ergenyl Chrono by Sanofi-Aventis and generics
- Chile – Valcote and Valcote ER by Abbott Laboratories
- France and other European countries — Depakote
- Peru – Divalprax by AC Farma Laboratories
- China – Diprate OD
References
- ^ Jump up to:a b c d “Depakene, Stavzor (valproic acid) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Archived from the original on 21 February 2014. Retrieved 13 February 2014.
- ^ Jump up to:a b c d e f g h i j k l “Valproic Acid”. The American Society of Health-System Pharmacists. Archived from the original on 2017-07-31. Retrieved Oct 23, 2015.
- ^ “Valproate banned without the pregnancy prevention programme”. GOV.UK. Retrieved 26 April 2018.
- ^ Owens MJ, Nemeroff CB (2003). “Pharmacology of valproate”. Psychopharmacol Bull. 37 Suppl 2: 17–24. PMID 14624230.
- ^ Jump up to:a b c Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, Altman RB, Klein TE (April 2013). “Valproic acid pathway: pharmacokinetics and pharmacodynamics”. Pharmacogenet. Genomics. 23 (4): 236–241. doi:10.1097/FPC.0b013e32835ea0b2. PMC 3696515. PMID 23407051.
- ^ “Valproic acid”. DrugBank. University of Alberta. 29 July 2017. Archived from the original on 31 July 2017. Retrieved 30 July2017.
- ^ Scott, D.F. (1993). The history of epileptic therapy : an account of how medication was developed (1. publ. ed.). Carnforth u.a.: Parthenon Publ. Group. p. 131. ISBN 9781850703914.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Divalproex Sodium – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Brayfield, Alison (ed.). Martindale: The Complete Drug Reference. London: Pharmaceutical Press. Retrieved March 3,2018.
- ^ Jump up to:a b c Rossi, S, ed. (2013). Australian Medicines Handbook(2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Löscher W (2002). “Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy”. CNS Drugs. 16 (10): 669–694. doi:10.2165/00023210-200216100-00003. PMID 12269861. S2CID 67999301.
- ^ Olsen KB, Taubøll E, Gjerstad L (2007). “Valproate is an effective, well-tolerated drug for treatment of status epilepticus/serial attacks in adults”. Acta Neurol. Scand. Suppl. 187: 51–4. doi:10.1111/j.1600-0404.2007.00847.x. PMID 17419829. S2CID 11159645.
- ^ Kwan SY (2010). “The role of intravenous valproate in convulsive status epilepticus in the future” (PDF). Acta Neurol Taiwan. 19(2): 78–81. PMID 20830628.[permanent dead link]
- ^ “Valproate Information”. Fda.gov. Archived from the original on 2015-05-03. Retrieved 2015-04-24.
- ^ Jochim, Janina; Rifkin-Zybutz, Raphael; Geddes, John; Cipriani, Andrea (7 October 2019). “Valproate for acute mania”. Cochrane Database of Systematic Reviews. 10: CD004052. doi:10.1002/14651858.CD004052.pub2. PMC 6797024. PMID 31621892.
- ^ Jump up to:a b Wang, Y; Xia, J; Helfer, B (2016). “Valproate for schizophrenia”. Cochrane Database of Systematic Reviews. 11: CD004028.pub4. doi:10.1002/14651858.CD004028.pub4. PMC 6734130. PMID 27884042. Archived from the originalon 2017-07-29. Retrieved 2017-07-27.
- ^ Pirritano D, Plastino M, Bosco D, Gallelli L, Siniscalchi A, De Sarro G (2014). “Gambling disorder during dopamine replacement treatment in Parkinson’s disease: a comprehensive review”. Biomed Res Int. 2014: 1–9. doi:10.1155/2014/728038. PMC 4119624. PMID 25114917.
- ^ Connolly B, Fox SH (2014). “Treatment of cognitive, psychiatric, and affective disorders associated with Parkinson’s disease”. Neurotherapeutics. 11 (1): 78–91. doi:10.1007/s13311-013-0238-x. PMC 3899484. PMID 24288035.
- ^ Averbeck BB, O’Sullivan SS, Djamshidian A (2014). “Impulsive and compulsive behaviors in Parkinson’s disease”. Annu Rev Clin Psychol. 10: 553–80. doi:10.1146/annurev-clinpsy-032813-153705. PMC 4197852. PMID 24313567.
- ^ Jump up to:a b c d e f g h i “Depakote- divalproex sodium tablet, delayed release”. Archived from the original on 5 March 2016. Retrieved 10 November 2015.
- ^ Činčárová L, Zdráhal Z, Fajkus J (2013). “New perspectives of valproic acid in clinical practice”. Expert Opin Investig Drugs. 22(12): 1535–1547. doi:10.1517/13543784.2013.853037. PMID 24160174. S2CID 11855893.
- ^ Jump up to:a b c d e f g h i j k l m n o “Valpro sodium valproate” (PDF). TGA eBusiness Services. Alphapharm Pty Limited. 16 December 2013. Retrieved 14 February 2014.
- ^ “Depakote 250mg Tablets – Summary of Product Characteristics”. electronic Medicines Compendium. Sanofi. 28 November 2013. Archived from the original on 1 February 2014. Retrieved 18 January 2014.
- ^ Wu S, Legido A, De Luca F (2004). “Effects of valproic acid on longitudinal bone growth”. J Child Neurol. 19 (1): 26–30. doi:10.1177/088307380401900105011. PMID 15032379. S2CID 19827846.
- ^ Robinson PB, Harvey W, Belal MS (1988). “Inhibition of cartilage growth by the anticonvulsant drugs diphenylhydantoin and sodium valproate”. Br J Exp Pathol. 69 (1): 17–22. PMC 2013195. PMID 3126792.
- ^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID 11580761. S2CID 25499280.
- ^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID 11580761. S2CID 25499280.
- ^ “Could Depakote cause Mydriasis”. eHealthMe.com. 2014-11-18. Archived from the original on 2014-12-05. Retrieved 2015-04-24.
- ^ Bilo, Leonilda; Meo, Roberta (October 2008). “Polycystic ovary syndrome in women using valproate: a review”. Gynecological Endocrinology. 24 (10): 562–70. doi:10.1080/09513590802288259. PMID 19012099. S2CID 36426338.
- ^ Chukwu, J; Delanty, N; Webb, D; Cavalleri, GL (January 2014). “Weight change, genetics and antiepileptic drugs”. Expert Review of Clinical Pharmacology. 7 (1): 43–51. doi:10.1586/17512433.2014.857599. PMID 24308788. S2CID 33444886.
- ^ New evidence in France of harm from epilepsy drug valproateArchived 2017-04-21 at the Wayback Machine BBC, 2017
- ^ Koch S, Göpfert-Geyer I, Jäger-Roman E, et al. (February 1983). “[Anti-epileptic agents during pregnancy. A prospective study on the course of pregnancy, malformations and child development]”. Dtsch. Med. Wochenschr. (in German). 108 (7): 250–7. doi:10.1055/s-2008-1069536. PMID 6402356.
- ^ Moore SJ, Turnpenny P, Quinn A, et al. (July 2000). “A clinical study of 57 children with fetal anticonvulsant syndromes”. J. Med. Genet. 37 (7): 489–97. doi:10.1136/jmg.37.7.489. PMC 1734633. PMID 10882750.
- ^ Ornoy A (2009). “Valproic acid in pregnancy: how much are we endangering the embryo and fetus?”. Reprod. Toxicol. 28 (1): 1–10. doi:10.1016/j.reprotox.2009.02.014. PMID 19490988.
- ^ Kulkarni ML, Zaheeruddin M, Shenoy N, Vani HN (2006). “Fetal valproate syndrome”. Indian J Pediatr. 73 (10): 937–939. doi:10.1007/bf02859291. PMID 17090909. S2CID 38371596.
- ^ Adab N, Kini U, Vinten J, et al. (November 2004). “The longer term outcome of children born to mothers with epilepsy”. J. Neurol. Neurosurg. Psychiatry. 75 (11): 1575–83. doi:10.1136/jnnp.2003.029132. PMC 1738809. PMID 15491979.
This argues that the fetal valproate syndrome constitutes a real clinical entity that includes developmental delay and cognitive impairments, but that some children might exhibit some developmental delay without marked dysmorphism.
- ^ Umur AS, Selcuki M, Bursali A, Umur N, Kara B, Vatansever HS, Duransoy YK (2012). “Simultaneous folate intake may prevent adverse effect of valproic acid on neurulating nervous system”. Childs Nerv Syst. 28 (5): 729–737. doi:10.1007/s00381-011-1673-9. PMID 22246336. S2CID 20344828.
- ^ Cassels, Caroline (December 8, 2006). “NEAD: In Utero Exposure To Valproate Linked to Poor Cognitive Outcomes in Kids”. Medscape. Archived from the original on July 31, 2011. Retrieved 2007-05-23.
- ^ Meador KJ, Baker GA, Finnell RH, Kalayjian LA, Liporace JD, Loring DW, Mawer G, Pennell PB, Smith JC, Wolff MC (2006). “In utero antiepileptic drug exposure: fetal death and malformations”. Neurology. 67 (3): 407–412. doi:10.1212/01.wnl.0000227919.81208.b2. PMC 1986655. PMID 16894099.
- ^ Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, Vestergaard M (2013). “Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism”. JAMA. 309 (16): 1696–1703. doi:10.1001/jama.2013.2270. PMC 4511955. PMID 23613074.
- ^ Rasalam AD, Hailey H, Williams JH, et al. (August 2005). “Characteristics of fetal anticonvulsant syndrome associated autistic disorder”. Dev Med Child Neurol. 47 (8): 551–5. doi:10.1017/S0012162205001076. PMID 16108456.
- ^ Autism Society of America: About Autism Archived 2011-01-10 at the Wayback Machine
- ^ I.Q. Harmed by Epilepsy Drug in Utero Archived 2015-12-29 at the Wayback Machine By RONI CARYN RABIN, New York Times, April 15, 2009
- ^ Meador KJ, Baker GA, Browning N, Clayton-Smith J, Combs-Cantrell DT, Cohen M, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, Loring DW (2009). “Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs”. N. Engl. J. Med. 360 (16): 1597–1605. doi:10.1056/NEJMoa0803531. PMC 2737185. PMID 19369666.
- ^ Valproate Products: Drug Safety Communication – Risk of Impaired Cognitive Development in Children Exposed In Utero (During Pregnancy) Archived 2011-09-02 at the Wayback Machine. FDA. June 2011
- ^ Luat, AF (20 September 2007). “Paroxysmal tonic upgaze of childhood with co-existent absence epilepsy”. Epileptic Disorders. 9(3): 332–6. doi:10.1684/epd.2007.0119 (inactive 31 May 2021). PMID 17884759.
- ^ Ouvrier, RA (July 1988). “Benign paroxysmal tonic upgaze of childhood”. Journal of Child Neurology. 3 (3): 177–80. doi:10.1177/088307388800300305. PMID 3209843. S2CID 38127378.
- ^ Valproate Not To Be Used for Migraine During Pregnancy, FDA Warns Archived 2013-07-09 at the Wayback Machine
- ^ “New measures to avoid valproate exposure in pregnancy endorsed”. European Medicines Agency. 31 May 2018.
- ^ Jump up to:a b c d Bentley, Suzanne (Dec 11, 2013). “Valproic Acid Level”. Medscape. Archived from the original on 2015-05-04. Retrieved 2015-06-05.
- ^ Jump up to:a b c d “Free Valproic Acid Assay (Reference – 2013.03.006) Notice of Assessment” (PDF). Canadian Agency for Drugs and Technologies in Health (CADTH) with INESSS’s permission. April 2014. Archived from the original (PDF) on 2016-03-03. Retrieved 2015-06-05.
- ^ Rissardo, Jamir Pitton; Caprara, Ana Letícia Fornari; Durante, Ícaro (2021). “Valproate-associated Movement Disorder: A Literature Review”. Prague Medical Report. 122 (3): 140–180. doi:10.14712/23362936.2021.14. ISSN 1214-6994.
- ^ Sztajnkrycer MD (2002). “Valproic acid toxicity: overview and management”. J. Toxicol. Clin. Toxicol. 40 (6): 789–801. doi:10.1081/CLT-120014645. PMID 12475192. S2CID 23031095.
- ^ Patsalos PN, Berry DJ (2013). “Therapeutic drug monitoring of antiepileptic drugs by use of saliva”. Ther Drug Monit. 35 (1): 4–29. doi:10.1097/FTD.0b013e31827c11e7. PMID 23288091. S2CID 15338188.
- ^ Thanacoody RH (2009). “Extracorporeal elimination in acute valproic acid poisoning”. Clin Toxicol. 47 (7): 609–616. doi:10.1080/15563650903167772. PMID 19656009. S2CID 13592043.
- ^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 1622–1626.
- ^ Jump up to:a b Lheureux PE, Penaloza A, Zahir S, Gris M (2005). “Science review: carnitine in the treatment of valproic acid-induced toxicity – what is the evidence?”. Crit Care. 9 (5): 431–440. doi:10.1186/cc3742. PMC 1297603. PMID 16277730.
- ^ Mock CM, Schwetschenau KH (2012). “Levocarnitine for valproic-acid-induced hyperammonemic encephalopathy”. Am J Health Syst Pharm. 69 (1): 35–39. doi:10.2146/ajhp110049. PMID 22180549.
- ^ Matsuoka M, Igisu H (1993). “Comparison of the effects of L-carnitine, D-carnitine and acetyl-L-carnitine on the neurotoxicity of ammonia”. Biochem. Pharmacol. 46 (1): 159–164. doi:10.1016/0006-2952(93)90360-9. PMID 8347126.
- ^ Herzog, Andrew; Farina, Erin (June 9, 2005). “Serum Valproate Levels with Oral Contraceptive Use”. Epilepsia. 46 (6): 970–971. doi:10.1111/j.1528-1167.2005.00605.x. PMID 15946343. S2CID 7696039.
- ^ Kay, Hee Yeon; Greene, Derek L.; Kang, Seungwoo; Kosenko, Anastasia; Hoshi, Naoto (2015-10-01). “M-current preservation contributes to anticonvulsant effects of valproic acid”. The Journal of Clinical Investigation. 125 (10): 3904–3914. doi:10.1172/JCI79727. ISSN 0021-9738. PMC 4607138. PMID 26348896.
- ^ Chang P, Walker MC, Williams RS (2014). “Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid”. Neurobiol. Dis. 62: 296–306. doi:10.1016/j.nbd.2013.10.017. PMC 3898270. PMID 24148856.
- ^ Kostrouchová M, Kostrouch Z, Kostrouchová M (2007). “Valproic acid, a molecular lead to multiple regulatory pathways” (PDF). Folia Biol. (Praha). 53 (2): 37–49. PMID 17448293. Archived from the original (PDF) on 2014-02-21. Retrieved 2014-02-13.
- ^ Jump up to:a b Chiu CT, Wang Z, Hunsberger JG, Chuang DM (2013). “Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder”. Pharmacol. Rev. 65 (1): 105–142. doi:10.1124/pr.111.005512. PMC 3565922. PMID 23300133.
- ^ Jump up to:a b Death AK, McGrath KC, Handelsman DJ (2005). “Valproate is an anti-androgen and anti-progestin” (PDF). Steroids. 70 (14): 946–53. doi:10.1016/j.steroids.2005.07.003. hdl:10453/16875. PMID 16165177. S2CID 25958985.
- ^ Jump up to:a b Wyllie E, Cascino GD, Gidal BE, Goodkin HP (17 February 2012). Wyllie’s Treatment of Epilepsy: Principles and Practice. Lippincott Williams & Wilkins. pp. 288–. ISBN 978-1-4511-5348-4. Archived from the original on 6 June 2014.
- ^ Uchida, Hiroshi; Maruyama, Tetsuo; Arase, Toru; Ono, Masanori; Nagashima, Takashi; Masuda, Hirotaka; Asada, Hironori; Yoshimura, Yasunori (2005). “Histone acetylation in reproductive organs: Significance of histone deacetylase inhibitors in gene transcription”. Reproductive Medicine and Biology. 4 (2): 115–122. doi:10.1111/j.1447-0578.2005.00101.x. ISSN 1445-5781. PMC 5891791. PMID 29662388.
- ^ Jump up to:a b Isojärvi, Jouko I T; Taubøll, Erik; Herzog, Andrew G (2005). “Effect of Antiepileptic Drugs on Reproductive Endocrine Function in Individuals with Epilepsy”. CNS Drugs. 19 (3): 207–223. doi:10.2165/00023210-200519030-00003. ISSN 1172-7047. PMID 15740176. S2CID 9893959.
- ^ Jump up to:a b c d e f Haberfeld H, ed. (2021). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Depakine chrono retard 300 mg Filmtabletten.
- ^ Kumar, S.; Wong, H.; Yeung, S. A.; Riggs, K. W.; Abbott, F. S.; Rurak, D. W. (2000). “Disposition of valproic acid in maternal, fetal, and newborn sheep. II: Metabolism and renal elimination”. Drug Metabolism and Disposition: The Biological Fate of Chemicals. 28(7): 857–864. PMID 10859160.
- ^ Schneemann; Young, Lloyd; Koda-Kimble, Mary Anne, eds. (2001). Angewandte Arzneimitteltherapie (in German). Springer. pp. 28–29. ISBN 3540413561.
- ^ Jump up to:a b Valproate FDA Professional Drug Information. Accessed 2021-08-06.
- ^ Jump up to:a b c d e f g h “Pharmacology”. Valproic Acid. DrugBank. University of Alberta. 31 August 2017.
- ^ Burton BS (1882). “On the propyl derivatives and decomposition products of ethylacetoacetate”. Am. Chem. J. 3: 385–395.
- ^ Meunier H, Carraz G, Neunier Y, Eymard P, Aimard M (1963). “[Pharmacodynamic properties of N-dipropylacetic acid]” [Pharmacodynamic properties of N-dipropylacetic acid]. Thérapie (in French). 18: 435–438. PMID 13935231.
- ^ Perucca E (2002). “Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience”. CNS Drugs. 16 (10): 695–714. doi:10.2165/00023210-200216100-00004. PMID 12269862. S2CID 803106.
- ^ Henry TR (2003). “The history of valproate in clinical neuroscience”. Psychopharmacol Bull. 37 Suppl 2: 5–16. PMID 14624229.
- ^ Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ Rimmer EM, Richens A (May–June 1985). “An update on sodium valproate”. Pharmacotherapy. 5 (3): 171–84. doi:10.1002/j.1875-9114.1985.tb03413.x. PMID 3927267. S2CID 7700266.
- ^ Glauser, Tracy A; Cnaan, Avital; Shinnar, Shlomo; Hirtz, Deborah G; Dlugos, Dennis; Masur, David; Clark, Peggy O; Capparelli, Edmund V; Adamson, Peter C (2010). “Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy”. New England Journal of Medicine. 362 (9): 790–9. doi:10.1056/NEJMoa0902014. PMC 2924476. PMID 20200383.
- ^ Jiang, Mei (6 April 2015). “Co-Administration of Valproic Acid and Lamotrigine in the Treatment of Refractory Epilepsy (P1.238)”. Neurology. 84 (14 Supplement): P1.238 – via http://www.neurology.org.
- ^ Berendsen, S.; Kroonen, J.; Seute, T.; Snijders, T.; Broekman, M. L. D.; Spliet, W. G. M.; Willems, M.; Artesi, M.; Bours, V.; Robe, P. A. (1 September 2014). “O9.06 * Prognostic Relevance and Oncogenic Correlates of Epilepsy in Glioblastoma Patients”. Neuro-Oncology. 16 (suppl_2): ii21. doi:10.1093/neuonc/nou174.77. PMC 4185847.
- ^ Vasudev K, Mead A, Macritchie K, Young AH (2012). “Valproate in acute mania: is our practice evidence based?”. Int J Health Care Qual Assur. 25 (1): 41–52. doi:10.1108/09526861211192395. PMID 22455007.
- ^ Bond DJ, Lam RW, Yatham LN (2010). “Divalproex sodium versus placebo in the treatment of acute bipolar depression: a systematic review and meta-analysis”. J Affect Disord. 124 (3): 228–334. doi:10.1016/j.jad.2009.11.008. PMID 20044142.
- ^ Haddad PM, Das A, Ashfaq M, Wieck A (2009). “A review of valproate in psychiatric practice”. Expert Opin Drug Metab Toxicol. 5(5): 539–51. doi:10.1517/17425250902911455. PMID 19409030. S2CID 74028228.
- ^ Frazee LA, Foraker KC (2008). “Use of intravenous valproic acid for acute migraine”. Ann Pharmacother. 42 (3): 403–7. doi:10.1345/aph.1K531. PMID 18303140. S2CID 207263036.
- ^ Wang, Yijun; Xia, Jun; Helfer, Bartosz; Li, Chunbo; Leucht, Stefan (2016). “Valproate for schizophrenia”. The Cochrane Database of Systematic Reviews. 11: CD004028. doi:10.1002/14651858.CD004028.pub4. ISSN 1469-493X. PMC 6734130. PMID 27884042.
- ^ Jump up to:a b Baillon, Sarah F.; Narayana, Usha; Luxenberg, Jay S.; Clifton, Andrew V. (2018-10-05). “Valproate preparations for agitation in dementia”. The Cochrane Database of Systematic Reviews. 2018(10): CD003945. doi:10.1002/14651858.CD003945.pub4. ISSN 1469-493X. PMC 6516950. PMID 30293233.
- ^ Gill D, Derry S, Wiffen PJ, Moore RA (2011). “Valproic acid and sodium valproate for neuropathic pain and fibromyalgia in adults”. Cochrane Database Syst Rev (10): CD009183. doi:10.1002/14651858.CD009183.pub2. PMC 6540387. PMID 21975791.
- ^ Aliyev ZN, Aliyev NA (July–August 2008). “Valproate treatment of acute alcohol hallucinosis: a double-blind, placebo-controlled study”. Alcohol Alcohol. 43 (4): 456–459. doi:10.1093/alcalc/agn043. PMID 18495806.
- ^ Jacobson PL, Messenheimer JA, Farmer TW (1981). “Treatment of intractable hiccups with valproic acid”. Neurology. 31 (11): 1458–60. doi:10.1212/WNL.31.11.1458. PMID 6796902. S2CID 1578958.
- ^ Sotaniemi K (1982). “Valproic acid in the treatment of nonepileptic myoclonus”. Arch. Neurol. 39 (7): 448–9. doi:10.1001/archneur.1982.00510190066025. PMID 6808975.
- ^ Wheeler SD (July–August 1998). “Significance of migrainous features in cluster headache: divalproex responsiveness”. Headache. 38 (7): 547–51. doi:10.1046/j.1526-4610.1998.3807547.x. PMID 15613172. S2CID 27948702.
- ^ Siemes H, Spohr HL, Michael T, Nau H (September–October 1988). “Therapy of infantile spasms with valproate: results of a prospective study”. Epilepsia. 29 (5): 553–60. doi:10.1111/j.1528-1157.1988.tb03760.x. PMID 2842127. S2CID 23789333.
- ^ Smith SM (2005). “Valproic acid and HIV-1 latency: beyond the sound bite” (PDF). Retrovirology. 2 (1): 56. doi:10.1186/1742-4690-2-56. PMC 1242254. PMID 16168066. Archived from the original (PDF) on 2015-09-24. Retrieved 2014-02-13.
- ^ Routy JP, Tremblay CL, Angel JB, Trottier B, Rouleau D, Baril JG, Harris M, Trottier S, Singer J, Chomont N, Sékaly RP, Boulassel MR (2012). “Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study”. HIV Med. 13 (5): 291–6. doi:10.1111/j.1468-1293.2011.00975.x. PMID 22276680. S2CID 27571864.
- ^ Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, Eron J, Cohen M, Margolis DM (2010). “Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection”. PLOS ONE. 5 (2): e9390. Bibcode:2010PLoSO…5.9390A. doi:10.1371/journal.pone.0009390. PMC 2826423. PMID 20186346.
- ^ Jump up to:a b Hardy JR, Rees EA, Gwilliam B, Ling J, Broadley K, A’Hern R (2001). “A phase II study to establish the efficacy and toxicity of sodium valproate in patients with cancer-related neuropathic pain” (PDF). J Pain Symptom Manage. 21 (3): 204–9. doi:10.1016/S0885-3924(00)00266-9. PMID 11239739.[permanent dead link]
- ^ Candelaria M, Herrera A, Labardini J, González-Fierro A, Trejo-Becerril C, Taja-Chayeb L, Pérez-Cárdenas E, de la Cruz-Hernández E, Arias-Bofill D, Vidal S, Cervera E, Dueñas-Gonzalez A (2011). “Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial”. Ann. Hematol. 90 (4): 379–387. doi:10.1007/s00277-010-1090-2. PMID 20922525. S2CID 13437134.
- ^ Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG (2005). “Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia”. Cancer. 104 (12): 2717–2725. doi:10.1002/cncr.21589. PMID 16294345. S2CID 1802132.
- ^ Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N (2006). “The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia”. Cancer. 106 (1): 112–119. doi:10.1002/cncr.21552. PMID 16323176. S2CID 43747497.
- ^ Fredly H, Gjertsen BT, Bruserud O (2013). “Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents” (PDF). Clin Epigenetics. 5 (1): 12. doi:10.1186/1868-7083-5-12. PMC 3733883. PMID 23898968. Archived from the original (PDF) on 2014-02-21. Retrieved 2014-02-13.
- ^ Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, Perez-Cardenas E, Taja-Chayeb L, Arias-Bofill D, Candelaria M, Vidal S, Dueñas-González A (2011). “A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results”. Med. Oncol. 28 Suppl 1: S540–6. doi:10.1007/s12032-010-9700-3. PMID 20931299. S2CID 207372333.
- ^ Rocca A, Minucci S, Tosti G, Croci D, Contegno F, Ballarini M, Nolè F, Munzone E, Salmaggi A, Goldhirsch A, Pelicci PG, Testori A (2009). “A phase I-II study of the histone deacetylase inhibitor valproic acid plus chemoimmunotherapy in patients with advanced melanoma”. Br. J. Cancer. 100 (1): 28–36. doi:10.1038/sj.bjc.6604817. PMC 2634690. PMID 19127265.
- ^ Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, Daud A, Neuger A, Minton S, Sullivan D (2009). “Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC” (PDF). Clin. Cancer Res. 15 (7): 2488–96. doi:10.1158/1078-0432.CCR-08-1930. PMID 19318486. S2CID 3230087.
- ^ Hicks CW, Pandya MM, Itin I, Fernandez HH (2011). “Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease”. Parkinsonism Relat. Disord. 17 (5): 379–81. doi:10.1016/j.parkreldis.2011.03.003. PMID 21459656.
- ^ Sriram A, Ward HE, Hassan A, Iyer S, Foote KD, Rodriguez RL, McFarland NR, Okun MS (2013). “Valproate as a treatment for dopamine dysregulation syndrome (DDS) in Parkinson’s disease”. J. Neurol. 260 (2): 521–7. doi:10.1007/s00415-012-6669-1. PMID 23007193. S2CID 21544457.
- ^ Aizenman, N. C. (7 May 2012). “Abbott Laboratories to pay $1.6 billion over illegal marketing of Depakote”. Washington Post. Retrieved 27 June 2018.
- ^ Schmidt, Michael; Thomas, Katie (8 May 2012). “Abbott settles marketing lawsuit”. New York Times. Retrieved 27 June 2018.
- ^ Gervain J, Vines BW, Chen LM, Seo RJ, Hensch TK, Werker JF, Young AH (2013). “Valproate reopens critical-period learning of absolute pitch”. Frontiers in Systems Neuroscience. 7: 102. doi:10.3389/fnsys.2013.00102. PMC 3848041. PMID 24348349.
- ^ Thomson, Helen. “Learning drugs reawaken grown-up brain’s inner child”. New Scientist. New Scientist Ltd. Retrieved 8 May2021.
- ^ David Taylor; Carol Paton; Shitij Kapur (2009). The Maudsley Prescribing Guidelines, Tenth Edition (10, revised ed.). CRC Press. p. 124. ISBN 9780203092835.
- ^ “Depakene- valproic acid capsule, liquid filled”. DailyMed. 19 September 2019. Retrieved 14 April 2020.
- ^ Jump up to:a b c d “Australian product information epilim (sodium valproate) crushable tablets, enteric-coated tablets, syrup, liquid” (PDF). TGA eBS. 15 April 2020. Retrieved 15 April 2020.
- ^ “Sodium valproate — Pharmaceutical Schedule”. Pharmaceutical Management Agency. Archived from the originalon 4 March 2016. Retrieved 22 June 2014.
- ^ South African Electronic Package Inserts: Convulex
External links
- “Valproic acid”. Drug Information Portal. U.S. National Library of Medicine.
- “Valproate sodium”. Drug Information Portal. U.S. National Library of Medicine.
- “Divalproex sodium”. Drug Information Portal. U.S. National Library of Medicine.
{kind=link}
{kind=link}
Patent
Publication numberPriority datePublication dateAssigneeTitleCA1144558A *1979-10-221983-04-12Francis E. FischerProcess for making sodium hydrogen divalproateUS4988731A *1979-08-201991-01-29Abbott LaboratoriesSodium hydrogen divalproate oligomerUS5212326A *1979-08-201993-05-18Abbott LaboratoriesSodium hydrogen divalproate oligomerWO2001032595A1 *1999-11-022001-05-10Cilag AgMethod for producing compounds of the valproinic acidUS20030018215A1 *2001-06-292003-01-23Procos S.P.A.Process for the preparation of sodium divalproatePublication numberPriority datePublication dateAssigneeTitleUS20110040122A1 *2009-08-112011-02-17Sci Pharmtech, Inc.Method for preparing metal salt of valproic acidCN102942467A *2012-10-172013-02-27山东方明药业集团股份有限公司Preparation method of divalproex sodiumCN103183600A *2011-12-302013-07-03北大方正集团有限公司Method for preparing divalproex sodium
////// divalproex, Anticonvulsant, Antimigraine, Antimanic, valproic acid, sodium valproate
CCCC(CCC)C(O)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
FAROPENEM
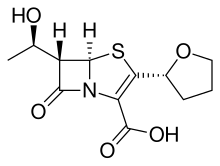
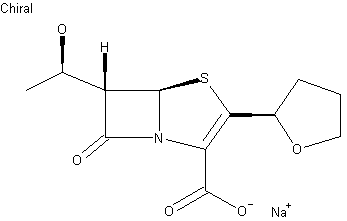
- Molecular FormulaC12H15NO5S
- Average mass285.316 Da
Faropenem
7086
(+)-(5R,6S)-6-((1R)-1-Hydroxyethyl)-7-oxo-3-((2R)-tetrahydro-2-furyl)-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic Acid
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydrofuran-2-yl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
106560-14-9[RN]
4-Thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, 6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-, (5R,6S)-
6α-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylic acid
4-Oxofenretinide
4-Oxo-N-(4-hydroxyphenyl)retinamide
6α-[(1R)-1-hydroxyethyl]-2-[(2R)-tetrahydrofuran-2-yl]-2,3-didehydropenam-3-carboxylic acid
7305146 [Beilstein]
FaropenemCAS Registry Number: 106560-14-9
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
Additional Names: fropenem; (5R,6S,8R,2¢R)-2-(2¢-tetrahydrofuryl)-6-hydroxyethylpenem-3-carboxylate
Molecular Formula: C12H15NO5S
Molecular Weight: 285.32
Percent Composition: C 50.51%, H 5.30%, N 4.91%, O 28.04%, S 11.24%
Literature References: Orally active, b-lactamase stable, penem antibiotic.Prepn: M. Ishiguro et al.,EP199446; eidem,US4997829 (1986, 1991 both to Suntory); eidem,J. Antibiot.41, 1685 (1988).Pharmacokinetics: A. Tsuji et al.,Drug Metab. Dispos.18, 245 (1990). In vitro antimicrobial spectrum: J. M. Woodcock et al.,J. Antimicrob. Chemother.39, 35 (1997). b-Lactamase stability: A. Dalhoff et al., Chemotherapy (Basel)49, 229 (2003).HPLC determn in plasma: R. V. S. Nirogi et al., Arzneim.-Forsch.55, 762 (2005). Clinical trial in urinary tract infections: S. Arakawa et al.,Nishinihon J. Urol.56, 300 (1994); in bacterial sinusitis: R. Siegert et al., Eur. Arch. Otorhinolaryngol.260, 186 (2003).
Derivative Type: Sodium salt
CAS Registry Number: 122547-49-3
Additional Names: Furopenem
Manufacturers’ Codes: ALP-201; SUN-5555; SY-5555; WY-49605
Trademarks: Farom (Daiichi)
Molecular Formula: C12H15NNaO5S
Molecular Weight: 308.31
Percent Composition: C 46.75%, H 4.90%, N 4.54%, Na 7.46%, O 25.95%, S 10.40%
Properties: [a]D22 +60° (c = 0.10).
Optical Rotation: [a]D22 +60° (c = 0.10)
Derivative Type: Daloxate
CAS Registry Number: 141702-36-5
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester
Additional Names: faropenem medoxomil
Manufacturers’ Codes: Bay-56-6854; SUN-208
Trademarks: Orapem (Replidyne)
Molecular Formula: C17H19NO8S
Molecular Weight: 397.40
Percent Composition: C 51.38%, H 4.82%, N 3.52%, O 32.21%, S 8.07%
Literature References: Prepn: H. Iwata et al., WO9203442; eidem, US5830889 (1992, 1998 both to Suntory).
Properties: Pale yellow crystals.
Therap-Cat: Antibacterial (antibiotics).
Keywords: Antibacterial (Antibiotics); ?Lactams; Penems.
Faropenem is an orally active beta-lactam antibiotic belonging to the penem group.[1] It is resistant to some forms of extended-spectrum beta-lactamase.[2] It is available for oral use.[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Forms
Faropenem was developed by Daiichi Asubio Pharma, which markets it in two forms.
- The sodium salt faropenem sodium, available under the trade name Farom, has been marketed in Japan since 1997. (CID 636379 from PubChem)
- The prodrug form faropenem medoxomil[4] (also known as faropenem daloxate) has been licensed from Daiichi Asubio Pharma by Replidyne, which plans to market it in conjunction with Forest Pharmaceuticals. The trade name proposed for the product was Orapem, but company officials recently announced this name was rejected by the FDA.[5]
Clinical use
As of 8 September 2015, Faropenem has yet to receive marketing approval in the United States, and was submitted for consideration by the United States Food and Drug Administration (FDA) on 20 December 2005. The new drug application dossier submitted included these proposed indications:
- acute bacterial sinusitis
- community-acquired pneumonia
- acute exacerbations of chronic bronchitis
- uncomplicated skin and skin structure infections
- urinary tract infections
History
The FDA refused to approve faropenem, an antibiotic manufactured by Louisville-based Replidyne. The FDA said the drug was “nonapprovable”, but did not refer to specific safety concerns about the product. The company will have to conduct new studies and clinical trials, lasting an estimated two more years, to prove the drug treats community-acquired pneumonia, bacterial sinusitis, chronic bronchitis, and skin infections.[citation needed]
In India it is available as Farobact 200/300ER CIPLA.
PATENT
https://patents.google.com/patent/WO2008035153A2/enFaropenem is an orally active β-lactam antibiotic belonging to the penem group. Faropenem is chemically known as 6-(l-hydroxyethyl)-7-oxo-3-(oxolan-2-yl)-4-thia-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid. The known forms of Faropenem are Faropenem sodium and the prodrug form, FaropenemMedoxomil (also known as Faropenem Daloxate). In view of the importance of the compound of the formula (I), several synthetic procedures to prepare the compound have been reported.US 4,997,829 provides process for the preparation of faropenem according to the following scheme. The process is exemplified with the allyl protected carboxyl group. One of the process involves the reaction of A- acetoxyazetidinone with tetrahydrothiofuroic acid, condensation with allyl glyoxalate in refluxing benzene, chlorination with thionyl chloride, reaction of triphenylphosphine with lutidine in hot THF, cyclization in refluxing toluene, deprotection of silyl protecting group with tetrabutylammonium fluoride, treating with triphenylphosphine and, treating with sodium 2-ethylhexanoate and (PP^)4Pd to result faropenem sodium. The process exemplified utilizes benzene as solvent, which is not environmentally acceptable. Tetrabutylammonium fluoride was used as desilylating agent that is expensive. Even though the description teaches that optically active compounds can be employed, the examples utilized the dl-compound of tetrahydrothiofuroic acid further requiring resolution.

Methods are provided for the synthesis of series of penem compounds in J Antibiotics 1988, 41(11), 1685-1693. The provided methods utilize sulfonylazetidinone as the starting materials. As one of the procedures gives lesser yield, another procedure was adopted which uses silver salts.Japanese patent, JP2949363 describes a process for deallylation and salt formation with an alkali metal salt of carboxylic acid in the presence of a catalytic amount of palladium complex for the preparation of faropenem.EP410727 describes a process for removing allyl group from a penem compound using cyclic 1,3-diketone such as dimedone.The yield and quality of the final product is always less in the above prior art methods. With the continued research, the present inventors have undertaken extensive studies for developing a process for the preparation of compound of formula (I), which is commercially viable, involves simple techniques such as crystallizations, with improved yields and quality of the product, and with lesser reaction time. None of the prior art suggests or teaches the techniques provided herein.The process is shown in Scheme-I as given below:

One-pot process for the preparation of Faropenem sodium:Sodium salt of R(+)-tetrahydrofuran-2-thiocarboxylic acid (67 g) in aqueous acetone was added slowly to a solution of AOSA (100 g) in acetone (200 mL) and stirred for 3 h at pH 8.0 to 8.5 using sodium bicarbonate solution.After completion of the reaction, the product was extracted with toluene. The combined toluene layer was washed with saturated sodium bicarbonate solution and brine solution. Toluene was removed under vacuum completely and the mass obtained, 3-(l’-tert-butyldimethylsilyloxyethyl)-4-(2′- tetrahydrofuranoylthio)-2-azetidinone was directly taken for next step.3-(r-tert-Butyldimethylsilyloxyethyl)-4-(2′-tetrahydrofuranoylthio)-2- azetidinone obtained was dissolved in toluene (1000 mL) and cooled to -10 to -5 °C under nitrogen. Triethylamine (124 mL) was added to it followed by allyl oxalyl chloride (82 g) at -10 to- 5 0C for 2 h. After completion of the reaction, cold water was added to the mass and washed with dilute hydrochloric acid and sodium bicarbonate solution. Toluene layer was separated and washed with purified water. The toluene layer containing compound of formula (VI) was concentrated under vacuum at 50 to 60 °C and taken for next step as such.Compound of formula (VI) (150 g) was dissolved in triethyl phosphite (150 mL), heated to 60 0C and stirred under nitrogen atmosphere. Toluene (3000 mL) was added, heated to 100 to 110 °C and stirred for 20- 24 h. Toluene was distilled under vacuum completely. Product obtained, allyl (1 ‘R,2″R,5R,6S)-6-(l 5-tert-butyldimethylsilyloxyethyl)-2-(2″-tetrahydrofuranyl) penem-3-carboxylate (VII) was directly taken for next step.Compound (VII) obtained was dissolved in DMF (700 mL) at 30 °C.Ammonium hydrogen difluoride (80 g) and NMP (210 mL) were added and stirred at room temperature for 25 to 35 h. The reaction mass was quenched into a mixture of water-ethyl acetate and stirred at room temperature. The ethyl acetate layer was separated and the aqueous layer extracted with ethyl acetate. ■ The combined ethyl acetate layer was washed with water followed by saturated sodium bicarbonate solution. The ethyl acetate layer was charcoal treated. The ethyl acetate layer containing allyl (l’R,2″R,5R,6S)-6-(l’-hydroxyethyl)-2-(2″- tetrahydrofuranyl)penem-3-carboxylate (XII) was partially distilled and taken for the next step.The ethyl acetate layer containing compound of formula (XII), Pd/C, sodium bicarbonate and purified water (1000 mL) were taken in an autoclave and maintained 5 to 10 kg pressure of hydrogen gas for 2-5 h. After completion of the reaction the Pd/C was filtered off and ethyl acetate layer separated. The pH of the mass was adjusted to 1.5 and extracted with ethyl acetate. The aqueous layer was extracted again with ethyl acetate twice. The combined ethyl acetate layer was carbon treated. Sodium-2-ethylhexanoate in ethyl acetate was added slowly and stirred. The precipitated title compound was filtered under vacuum, washed with acetone and dried. Dry weight of the product: 65-75 g.Example 9Purification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 0C. The solution was charcoalised. Acetone (1500 mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0 —2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).Example 9aPurification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 °C. Acetone (150O mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0-2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).
PATENT
https://patents.google.com/patent/CN103880864B/enFaropenem sodium is developed by Japanese Suntory companies, and first penemss antibiosis in listing in 1997 Element, it are similar to the several carbapenem antibiotics for listing, strong with has a broad antifungal spectrum, antibacterial activity, to beta-lactamase Stably, the features such as also having good action to extended spectrumβ-lactamase producing strains, citrobacter, enterococcus and anaerobe etc.. It is first orally active, penems antibiotics stable to beta-lactamase in the world so far.Its structural formula As follows:
Report about Faropenem sodium preparation method is a lot, mainly has several as follows:1st, J. Antibiotics 1988, the method that reports in 41,1685, see below row reaction equation:
Acyl group substitution reaction is carried out in the basic conditions with 4-AA and three beneze methane thiols and obtains thio trityl as protecting group Aza cyclo-butanone, then when 2-TETRAHYDROFUROYL chlorine is connected with lactams, using silver nitrate as condensing agent, but nitric acid Silver is expensive, and cost is too high, while the silver chloride for generating is difficult to filter, is not suitable for large-scale production.2nd, the classical preparation method of United States Patent (USP) US4997829 report:There is acyl with (R) tetrahydrofuran -2- thiocarboxylic acids Base substitution reaction generates thioesters, then through condensation, chlorine replacement, intramolecular Witting cyclization, slough hydroxyl protecting group and carboxylic Base protection group obtains product, and this synthetic route yield is very low, while side chain is thio-compoundss, abnormal smells from the patient is extremely smelly, and prepares complexity, There is-fixed harm to human body and environment.It is also required in chloro building-up process using pungent thionyl chloride, these factors are all It is unfavorable for industrialized production
3rd, the method that reports in Chinese patent CN1314691 is as follows:
Said method route is shorter, is produced using one kettle way, more convenient.But said method is related to some other salt such as acetate using heavy metal palladium in last operation The deprotecting regent of compound and triphenyl phosphorus together as pi-allyl, metal palladium reagent is expensive, while triphenyl phosphorus are most More difficult removing in step afterwards, increases operation difficulty, affects product quality.Allyloxy is used easily to produce as protection group simultaneously A kind of double bond olefinic polymerization species impurity of life, affects product quality, reduces yield.Embodiment one(R) tetrahydrofuran -2- thiocarboxylic acids (198g, 1.5 mol) are put in 3L reaction bulbs, plus 1 mol/L hydrogen-oxygens Change sodium body lotion (I.5 L) to be adjusted at 5 DEG C of pH 9- 10,0-, Deca 4AA(287g, 1. 0mo l) acetone (1 L) Solution, drop are finished, and are adjusted to pH 8 or so, 2 h of room temperature reaction with 1 mol/L sodium hydroxide. and add water (500 ml) dilution, second Acetoacetic ester (600 ml x3) is extracted, and merges organic layer, successively with 5 % sodium bicarbonate solutions (300 ml x 2) and water (300 m1 x 2) is washed, and anhydrous sodium sulphate is dried, and is filtered, and filtrate concentrates, and obtains pale yellow oil (about 360 g), directly Input the next step.Embodiment twoThe mixing of concentrated solution as obtained above, triethylamine (l70g, 1.7 mol) and dichloromethane (1.5 L), 0-5 DEG C Deca chlorine oxalic acid is finished to p-Nitrobenzyl (414.1 g, 1 .7 mo l), drop, and equality of temperature reacts 2 h, and add water (1 L) dilution, Extracted with dichloromethane (500 ml x 4), merge organic layer, molten with water (300m1 x 2) and 5 % sodium bicarbonate successively Liquid (300 m1 x 2) is washed, anhydrous sodium sulfate drying, is filtered, and concentration obtains pale yellow oil (about 530g), direct plunges into The next step..Embodiment threeAbove-mentioned gained grease, dimethylbenzene (4L) and NSC 5284 (500ml) are mixed, heating reflux reaction 5h , reduce pressure and boil off dimethylbenzene and NSC 5284, residue ethyl acetate-hexane (1:5,1 L) recrystallization, obtain yellowish Color solid (334.3g, 61%, in terms of 4AA).Example IVAbove-mentioned solid (0.60 mol of 330g.) is dissolved in methanol (2 L), adds 1.0M hydrochloric acid (0.4 L), adds palladium carbon (15.0 g), hydrogen is passed through, 40 DEG C of stirrings, response time are 16 h, and the pressure of system is 4atm, after reaction terminates, crosses and filters Catalyst is removed, is concentrated.Embodiment fiveThe product obtained after above-mentioned concentration is dissolved in tetrahydrofuran 600ml, the 2 ethyl hexanoic acid sodium of 100.0g is added Tetrahydrofuran(200ml)And water(200 ml)Mixed solution, 2 h are stirred at room temperature, have faint yellow solid generate, filter, be method Faropenem crude product 147.0g.Embodiment sixBy above-mentioned solid deionized water(2200ml)Acetone is slowly added under dissolved solution, stirring to start to become to solution Muddiness, when about adding acetone 750ml, solution starts to become cloudy, and stops adding, and continues stirring and allows its crystallize overnight, sucking filtration, acetone Washing, dries, and obtains the Faropenem sodium fine work 125.0g of white.
Syn
AU 8654460; EP 0199446; JP 1994128267; US 4997829
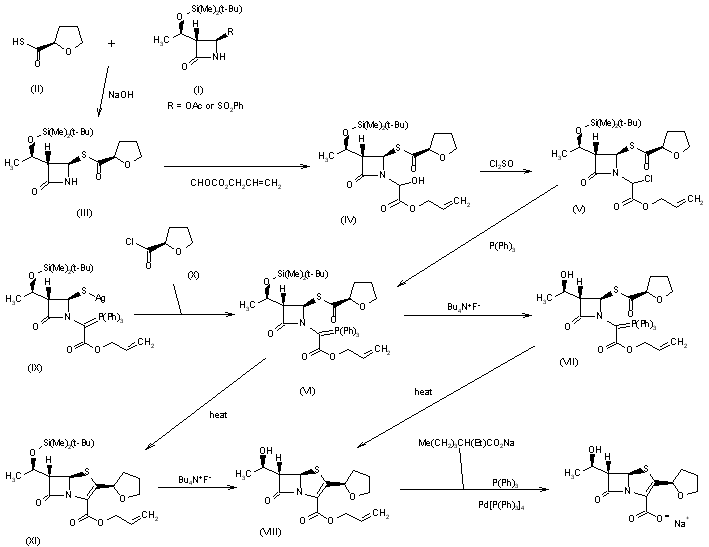
This compound is prepared by several related ways: 1) The reaction of silylated azetidinone (I) with tetrahydrofuran-2-thiocarboxylic acid (II) by means of NaOH in THF – water gives the azetidinone thioester (III), which is condensed with allyl glyoxylate in refluxing benzene yielding the hydroxyester (IV). The reaction of (IV) with SOCl2 affords the chloroester (V), which by reaction with triphenylphosphine by means of lutidine in hot THF is converted into the phosphoranylidene derivative (VI). The elimination of the silyl protecting group of (VI) with tetrabutylammonium fluoride gives the azetidinone (VII), which is cyclized in refluxing toluene yielding the (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxyli c acid allyl ester (VIII). Finally, this compound is hydrolyzed with triphenylphosphine, sodium 2-ethylhexanoate and Pd-tetrakis(triphenylphosphine). 2) The condensation of the silver salt of protected azetidinone (IX) with tetrahydrofuran-2(R)-carbonyl chloride (X) also yields the phosphoranylidene salt (VI). 3) Phosphoranylidene ester (VI) can also be cyclized first in refluxing benzene yielding the silylated penem ester (XI), which is deprotected with tetrabutylammonium fluoride to (VIII). 4) The hydrolysis of allyl ester (VIII) to the final product can also be performed with paladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethylacetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether. 5) The preceding hydrolysis can also be performed with triphenylphosphine and paladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone.
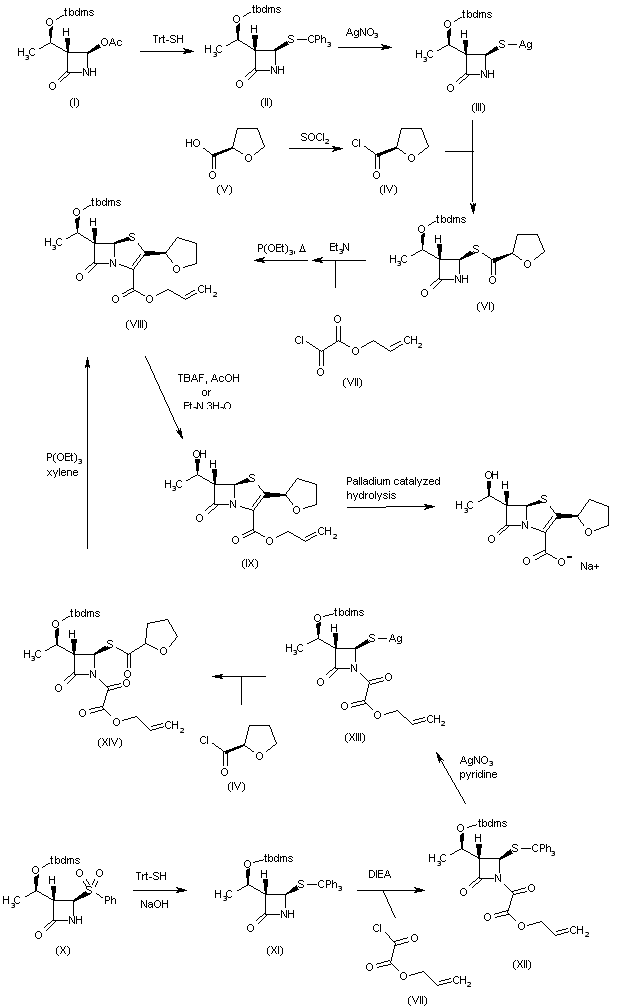
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanyl-azetidinone (II), which is converted into the silver salt (III) by reaction with AgNO3. Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) — obtained by treatment of carboxylic acid (V) with thionyl chloride — providing the azetidinone thioester (VI). Coupling of azetidinone (VI) with allyl oxalyl chloride (VII) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene, affords penem (VIII). Alternatively, compound (VIII) can also be obtained as follows: Substitution of phenyl sulfonyl group of azetidinone (X) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (XI), which is condensed with allyl oxalyl chloride (VII) by means of DIEA in CH2Cl2 to give the oxalyl amide (XII). Compound (XII) is then treated with AgNO3 and pyridine in acetonitrile, providing the silver mercaptide (XIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIV). Penem (VIII) is obtained by intramolecular Wittig cyclization of (XIV) with P(OEt)3 in refluxing xylene. Finally, faropenem sodium can be obtained by removal of the tbdms protecting group of (VIII) by means of either Et3N tris(hydrogen fluoride) in ethyl acetate or tetrabutylammonium fluoride (TBAF) and HOAc in THF to give compound (IX). This is followed by allyl ester group removal of (IX), which can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; or iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF.
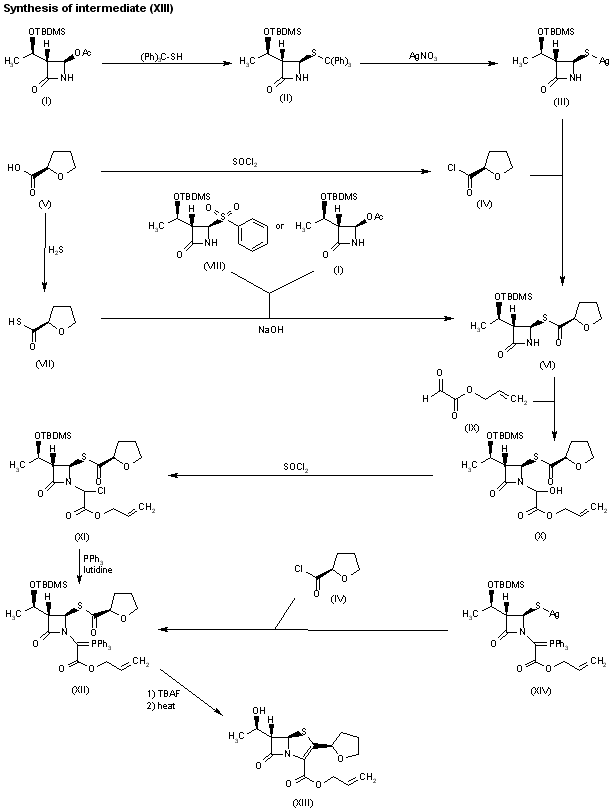
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanylazetidinone (II), which by reaction with AgNO3 is converted into the silver salt (III). Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) ?obtained by treatment of carboxylic acid (V) with thionyl chloride ?to provide the azetidinone thioester (VI). Alternatively, compound (VI) can be obtained by condensation of tetrahydrofuran-2(R)-thiocarboxylic S-acid (VII) ?obtained by treatment of carboxylic acid (V) with hydrogen sulfide ?with silylated azetidinones (I) or (VIII) by means of NaOH in THF/water. Condensation of azetidinone thioester (VI) with allyl glyoxylate (IX) in refluxing benzene gives the hydroxy ester (X), which is treated with SOCl2 to yield the chloro ester (XI). Reaction of compound (XI) with triphenylphosphine and lutidine in hot THF provides the phosphoranylidene derivative (XII), which is converted into (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxylic acid allyl ester, faropenem allyl ester (XIII) by removal of the silyl protecting group with tetrabutylammonium fluoride, followed by cyclization in refluxing toluene. Compound (XII) can also be obtained by condensation of the silver salt of protected azetidinone (XIV) with tetrahydrofuran-2(R)-carbonyl chloride (V).
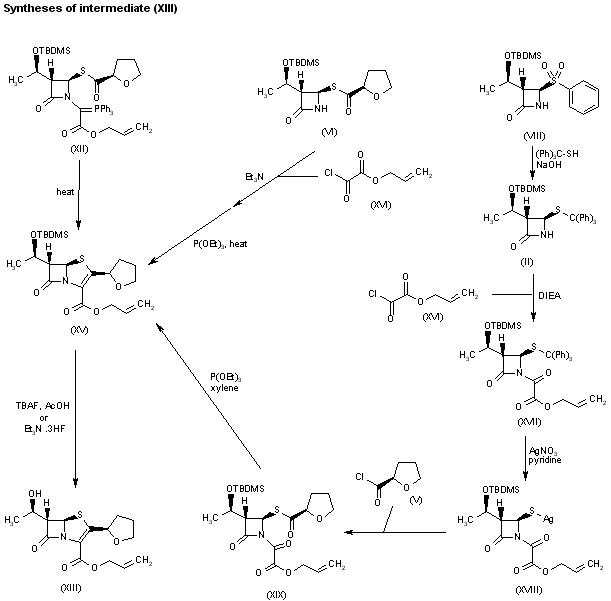
Alternatively, faropenem allyl ester (XIII) can also be prepared by cyclization of compound (XII) in refluxing benzene to yield silylated penem allyl ester (XV), which is then deprotected with either tetrabutylammonium fluoride in AcOH or triethylamine tris(hydrogen fluoride) in methyl isobutyl ketone or toluene. Penem (XV) can also be synthesized by several related ways: a) By coupling of azetidinone (VI) with allyl oxalyl chloride (XVI) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene. b) Substitution of phenyl sulfonyl group of azetidinone (VIII) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (II), which is condensed with allyl oxalyl chloride (XVI) by means of DIEA in CH2Cl2 to give the oxalyl amide (XVII). Compound (XVII) is then treated with AgNO3 and pyridine in acetonitrile to provide the silver mercaptide (XVIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIX). Finally, compound (XV) is obtained by intramolecular Wittig cyclization of (XX) with P(OEt)3 in refluxing xylene.
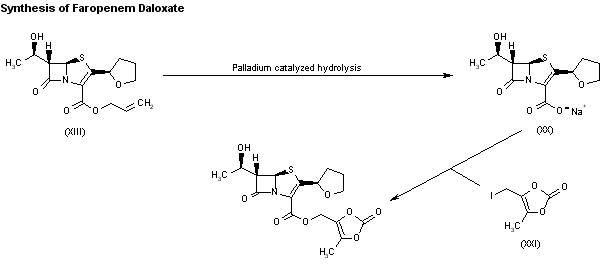
Hydrolysis of faropenem allyl ester (XIII) to faropenem sodium (XX) can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)- 5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; and iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF. Finally, faropenem daloxate can be directly obtained from faropenem sodium (XX) by esterification with 4-(iodomethyl)-5-methyl-1,3-dioxol-2-one (XXI) in DMF.
PATENT
https://patents.google.com/patent/CN103059046A/enFaropenem (Faropenem), chemistry (5R, 6S)-6-[(1R)-hydroxyethyl by name]-2-[(2R)-and tetrahydrofuran (THF)] penem-3-carboxylic acid list sodium salt, by the first exploitation listing in 1997 years of Japanese Suntory company.This medicine is a kind of atypical beta-lactam penems antibiotics, has very strong anti-microbial activity, especially to the anti-microbial activities of the anerobes such as the gram positive organisms such as golden Portugal bacterium, penicillin-fast streptococcus pneumoniae, streptococcus faecium and bacteroides fragilis apparently higher than existing cynnematin, anti-gram-negative bacteria is active similar to oral cephalosporin, and is stable to various β-lactamases.Various clinical studyes show that this medical instrument has clinical effectiveness good, safe, the advantage that renal toxicity and neurotoxicity are little.Its structural formula is as follows:
For synthesizing of Faropenem, existing many reports in the prior art, for example CN101125857A has reported following synthetic route:
Take (3R, 4R)-3-[(R)-1-tert-butyl dimethyl silica ethyl]-4-[(R)-and acetoxyl group] nitrogen heterocyclic din-2-ketone is as starting raw material, and warp gets intermediate compound I with R-(+)-sulfo-tetrahydrofuran (THF)-2-formic acid condensation; Intermediate compound I is carried out acylation reaction with monoene propoxy-oxalyl chloride under the catalysis of alkali, get intermediate II; Intermediate II cyclization under the effect of triethyl-phosphite gets intermediate III; Intermediate III is sloughed hydroxyl protecting group through the effect of tetrabutylammonium, gets intermediate compound IV; Intermediate compound IV decarboxylize protecting group under [four (triphenylphosphine)] palladium and triphenylphosphine effect gets Faropenem.Find that after deliberation the method for the present synthetic Faropenem of reporting is all similar with the disclosed method of above-mentioned CN101125857A, all need remove in two steps the protecting group of hydroxyl and carboxyl, reaction scheme is longer.When removing above-mentioned protecting group, need to use a large amount of tetrabutylammonium and [four (triphenylphosphine)] palladium and triphenylphosphine; these reagent costs are high, toxicity is large; be unfavorable for large industrial production; and can introduce the heavy metal palladium; so that the heavy metal remnants in the Faropenem exceed standard, be not suitable for the production of bulk drug.And when adopting aforesaid method deprotection base, the yield in per step only can reach 60%-75%, has further increased production cost.Embodiment 6The preparation of FaropenemWith intermediate 3(364.5g, 0.8mol) use the 700mL acetic acid ethyl dissolution, to open and stir, 0 ℃ of lower dropping with the 36g trifluoroacetic acid after the dilution of 100mL ethyl acetate dripped off in 1 hour, 0 ℃ of lower reaction 2h that continues.Stopped reaction stirs the sodium bicarbonate aqueous solution of lower dropping 5%, until reaction solution pH is neutral.Emit water layer from the reactor lower end, discard.In reactor, add gradually the ethanolic soln of sodium bicarbonate, until till no longer including solid and separating out.Suction filtration, filter cake gets white solid powder 230g(productive rate 93.7% with acetone-water (10:3, v/v) recrystallization), M.P. 163-164 ℃, detect through HPLC, purity is 99.8%Reference examples 1(5R, 6S)-6-[(R)-1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] preparation of penem-3-carboxylic acid propyleneWith (5R, 6S)-6-[(R)-the 1-tert-butyl dimethyl silica ethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (150g, 0.342mol) and ammonium bifluoride (59.5g, 1.025mmol) add successively among the 400mL DMF, 55~60 ℃ were reacted 5 hours, stopped reaction, suction filtration, filtrate adds water 800ml, uses ethyl acetate extraction, and organic phase is washed with 5% sodium hydrogen carbonate solution, anhydrous sodium sulfate drying, concentrated, gained incarnadine oily matter gets yellow solid 73g through the petrol ether/ethyl acetate recrystallization, yield 66%.Reference examples 2The preparation of Faropenem(the 5R that reference examples 1 is prepared, 6S)-6-[(R)-the 1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (73g, 0.224mol), 6.5g triphenylphosphine, 6.5g [four (triphenylphosphine)] palladium adds among the 500mL methylene dichloride l successively, the ethyl acetate solution that adds the 2 ethyl hexanoic acid sodium preparation of 500mL 0.5M, stirring at room 1 hour, stopped reaction adds 15mL water in reaction solution, stir 30min, suction filtration, this solid is dissolved in the 100mL water again, adds decolorizing with activated carbon 30min, filter, filtrate adds in the 500mL acetone, place crystallization, get Faropenem 66g, yield 96%.Find that by contrast the total recovery that two steps of reference examples remove hydroxyl and carboxyl-protecting group only has about 63.4%, and single stage method of the present invention removes the yield of hydroxyl and carboxyl-protecting group and can reach more than 90%.Preparation method of the present invention can the one-step removal hydroxyl and carboxyl on protecting group, shortened the production cycle, the deprotecting regent cost is low, toxicity is little, can not cause heavy metal remaining, and have higher reaction yield, is fit to very much the industrial production of raw material medicine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibioticCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemFamily To Family CitationsCN100522975C *2007-08-232009-08-05东北制药集团公司沈阳第一制药厂Method for preparing faropenemPublication numberPriority datePublication dateAssigneeTitleCN1884284A *2005-06-212006-12-27浙江金华康恩贝生物制药有限公司Process for the preparation of sodium faropenemCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumCN101125857A *2007-08-232008-02-20东北制药集团公司沈阳第一制药厂Method for preparing faropenemWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibiotic
Publication numberPriority datePublication dateAssigneeTitle
EP0410727A1 *1989-07-261991-01-30Suntory LimitedProcesses for removing allyl groupsUS4997829A *1985-03-091991-03-05Suntory LimitedPenem compounds, and use thereofEP0574940A1 *1992-06-181993-12-22Tanabe Seiyaku Co., Ltd.Method for removing the protecting group for carboxyl groupWO2007039885A1 *2005-10-052007-04-12Ranbaxy Laboratories LimitedA process for the preparation of faropenemFamily To Family Citations
Publication numberPriority datePublication dateAssigneeTitleCN102964357A *2012-11-112013-03-13苏州二叶制药有限公司Faropenem sodium and tablet thereofCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemCN103880864A *2014-03-252014-06-25江苏正大清江制药有限公司Method for synthesizing faropenem sodiumCN104086516A *2014-07-182014-10-08成都樵枫科技发展有限公司Synthetic method of R-(+)-sulfotetrahydrofuran-2-formic acidCN101941981B *2009-07-032015-01-21湖南华纳大药厂有限公司Catalyst composition and method for preparing faropenem sodiumCN106860405A *2015-12-142017-06-20山东新时代药业有限公司A kind of faropenem sodium granules and preparation method thereofCN108840877A *2018-06-122018-11-20赤峰迪生药业有限责任公司A kind of preparation method of oxygen cephalosporin intermediate
References
- ^ Critchley IA, Brown SD, Traczewski MM, Tillotson GS, Janjic N (December 2007). “National and regional assessment of antimicrobial resistance among community-acquired respiratory tract pathogens identified in a 2005-2006 U.S. Faropenem surveillance study”. Antimicrob. Agents Chemother. 51 (12): 4382–9. doi:10.1128/AAC.00971-07. PMC 2168020. PMID 17908940.
- ^ Mushtaq S, Hope R, Warner M, Livermore DM (May 2007). “Activity of faropenem against cephalosporin-resistant Enterobacteriaceae”. J. Antimicrob. Chemother. 59 (5): 1025–30. doi:10.1093/jac/dkm063. PMID 17353220.
- ^ Milazzo I, Blandino G, Caccamo F, Musumeci R, Nicoletti G, Speciale A (March 2003). “Faropenem, a new oral penem: antibacterial activity against selected anaerobic and fastidious periodontal isolates”. J. Antimicrob. Chemother. 51 (3): 721–5. doi:10.1093/jac/dkg120. PMID 12615878.
- ^ Gettig JP, Crank CW, Philbrick AH (January 2008). “Faropenem medoxomil”. Ann Pharmacother. 42 (1): 80–90. doi:10.1345/aph.1G232. PMID 18094341. Archived from the original on 2013-02-03.
- ^ (Q1 06 Investor Conf Call)(CID 6918218 from PubChem)
External links
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | J01DI03 (WHO) |
| Identifiers | |
| CAS Number | 106560-14-9 |
| PubChem CID | 65894 |
| ChemSpider | 59303 |
| UNII | F52Y83BGH3 |
| ChEBI | CHEBI:51257 |
| ChEMBL | ChEMBL556262 |
| CompTox Dashboard (EPA) | DTXSID0046430 |
| Chemical and physical data | |
| Formula | C12H15NO5S |
| Molar mass | 285.31 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
{kind=link}
///////////Faropenem, ALP-201, SUN-5555, SY-5555, WY-49605, ANTIBACTERIALS, DIICHI, Daiichi Asubio Pharma

NEW DRUG APPROVALS
ONE TIME
$10.00
AUR 101
AUR 101
AUR101-201
ANTIINNFLAMATORY
AUR-101, a ROR gamma inverse agonist for autoimmune disorders like psoriasis
AUR-101 is an ROR-gammaT inverse agonist in phase II clinical development at Aurigene for the treatment of patients with moderate-to-severe chronic plaque-type psoriasis.
- DrugsAUR 101 (Primary)
- IndicationsPlaque psoriasis
- FocusAdverse reactions; First in man
- AcronymsINDUS
- SponsorsAurigene Discovery Technologies
- OriginatorAurigene Discovery Technologies
- ClassAntipsoriatics; Small molecules
- Mechanism of ActionNuclear receptor subfamily 1 group F member 3 inverse agonists
- Phase IIPsoriasis
- 28 Aug 2021No recent reports of development identified for phase-I development in Psoriasis(In volunteers) in Australia (PO, Tablet)
- 23 Apr 2021Aurigene Discovery Technologies plans a phase II INDUS-3 trial for Psoriasis in USA (PO) in May 2021 (NCT04855721)
- 15 Apr 2021Aurigene Discovery Technologies completes a phase II trial in Psoriasis in India (PO) (NCT04207801)
- CDSCO
- https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/Aurigene20.pdf
- NCT04207801
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
AURIGENE ANNOUNCES FIRST PATIENT DOSED WITH AUR101 IN PHASE II STUDY IN PATIENTS WITH MODERATE TO SEVERE PSORIASIS
PRESS RELEASE
Aurigene Announces First Patient Dosed with AUR101 in Phase II Study in Patients with Moderate to Severe Psoriasis
Bangalore, February 17, 2020 — Aurigene, a development stage biotechnology company, today announced dose administration for the first patient in INDUS-2, a Phase II double blind placebo-controlled three-arm study of AUR101 in patients with moderate to severe psoriasis. AUR101 is an oral small molecule inverse agonist of RORγ and has shown desirable pharmacodynamic modulation of IL-17 and acceptable safety in a completed Phase I human study conducted in Australia.
“The initiation of this Phase II study under a US FDA IND represents a significant milestone for Aurigene, as it marks the first program which Aurigene has led from the bench side to the clinic all by itself,” said Murali Ramachandra, PhD, Chief Executive Officer of Aurigene. “We look forward to producing important clinical data by the end of 2020 to guide our future development plans and demonstrating Aurigene’s unique expertise in conducting Proof-of-Concept studies in a quality and fast-paced manner.”
About AUR101-201 and the Phase II Study of AUR101 in Patients with Moderate to Severe Psoriasis
The purpose of the Phase II multi-center, blinded, placebo-controlled, three-arm study is to evaluate the clinical activity of AUR101 in patients with moderate to severe psoriasis. In two of the arms, AUR101 will be administered twice daily, at 400 mg PO BID and 600 mg PO BID, for 12 weeks. Patients in the third arm will receive matched blinded placebo in a double dummy fashion. The trial is listed at clinicaltrials.gov with identifier NCT04207801.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY,NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene has also submitted an IND to DCGI, India for a Phase IIb/III trial of CA-170, a dual inhibitor of PD-L1 and VISTA, in non-squamous NSCLC. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/.
CLIP
Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways
https://onlinelibrary.wiley.com/doi/10.1111/bjd.20090
M.A.X. Tollenaere,J. Hebsgaard,D.A. Ewald,P. Lovato,S. Garcet,X. Li,S.D. Pilger,M.L. Tiirikainen,M. Bertelsen,J.G. Krueger,H. Norsgaard,First published: 01 April 2021 https://doi.org/10.1111/bjd.20090Citations: 2Funding sources LEO Pharma A/S funded this study.Conflicts of interest M.A.X.T., J.H., D.A.E., P.L., S.D.P., M.L.T., M.B. and H.N. are employees of LEO Pharma. J.G.K. received grants paid to his institution from Novartis, Pfizer, Amgen, Lilly, Boehringer, Innovaderm, BMS, Janssen, AbbVie, Paraxel, LEO Pharma, Vitae, Akros, Regeneron, Allergan, Novan, Biogen MA, Sienna, UCB, Celgene, Botanix, Incyte, Avillion and Exicure; and personal fees from Novartis, Pfizer, Amgen, Lilly, Boehringer, Biogen Idec, AbbVie, LEO Pharma, Escalier, Valeant, Aurigene, Allergan, Asana, UCB, Sienna, Celgene, Nimbus, Menlo, Aristea, Sanofi, Sun Pharma, Almirall, Arena and BMS.Data Availability Statement The gene array dataset described in this publication has been deposited in NCBI’s Gene Expression Omnibus and is accessible through GEO Series accession number GSE158448 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158448).
CLOP
https://www.drugdiscoverychemistry.com/Anti-Inflammatories/16
10:35 Small Molecule Inhibitors of RORgamma and IRAK4 for the Treatment of Autoimmune Disorders
 Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Although biologics such as anti-TNFα antibody are fairly successful in the treatment of autoimmune disorders, there is significant unmet need due to heterogeneity in diseases and lack of response to established therapies in some patients. While biologics typically target one cytokine signaling pathway, small molecule therapeutics directed towards intracellular target(s) can interfere in the signaling from multiple cytokines potentially leading to improved response. Development of small molecule oral inhibitors of IRAK4 and RORgamma to target TLR/IL-R and Th17 pathway respectively will be discussed.
PATENT
2448/CHE/2015 15.05.2015 IN
PATENT
PATENT
This application claims the benefit of Indian provisional application number 5641/CHE/2013 filed on 06th December 2013 which hereby incorporated by reference.
PATENT
- KOTRABASAIAH UJJINAMATADA, Ravi
- PANDIT, Chetan
2049005-13-0
2-Quinolinecarboxamide, 6-(2,6-dimethyl-4-pyrimidinyl)-N-[[4-(ethylsulfonyl)phenyl]methyl]-5,6,7,8-tetrahydro-6-methyl-5-oxo-, (6S)-
Molecular Weight492.59, C26 H28 N4 O4 S

EXAMPLE
PATENT
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0223523419301011
2013239366 CA 170

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////////////////AUR 101, AURIGENE, ROR, IL-17, PHASE 2, CDSCO, Ravi Ujjinamatada, KOTRABASAIAH UJJINAMATADA Ravi, PANDIT Chetan, AUR101-201, plaque-type psoriasis


Ravi Ujjinamatada
PIROXICAM

PIROXICAM
- Molecular FormulaC15H13N3O4S
- Average mass331.346 Da
1,1-Dioxyde de 4-hydroxy-2-méthyl-N-(2-pyridinyl)-2H-1,2-benzothiazine-3-carboxamide
13T4O6VMAM
252-974-3[EINECS]
2H-1,2-Benzothiazine-3-carboxamide, 4-hydroxy-2-methyl-N-2-pyridinyl-, 1,1-dioxide
36322-90-4[RN]37134
-Hydroxy-2-methyl-3-(pyrid-2-yl-carbamoyl)-2H-1,2-benzothiazine 1,1-dioxide
Piroxicam
CAS Registry Number: 36322-90-4
CAS Name: 4-Hydroxy-2-methyl-N-2-pyridinyl-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide
Additional Names: 3,4-dihydro-2-methyl-4-oxo-N-2-pyridyl-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide
Manufacturers’ Codes: CP-16171
Trademarks: Artroxicam (Coli); Baxo (Toyama); Bruxicam (Bruschettini); Caliment (Apotex); Erazon (Krka); Feldene (Pfizer); Flogobene (Farge); Geldene (Pfizer); Improntal (Kabi); Larapam (Lagap); Pirkam (DAK); Piroflex (Lagap); Reudene (ABC); Riacen (Chiesi); Roxicam (Gramon); Roxiden (Pulitzer); Sasulen (Andreu); Solocalm (Microsules); Zunden (Luitpold)Molecular Formula: C15H13N3O4S
Molecular Weight: 331.35
Percent Composition: C 54.37%, H 3.95%, N 12.68%, O 19.31%, S 9.68%
Literature References: Non-steroidal anti-inflammatory with long half-life. Prepn (keto form): J. Lombardino, DE1943265; idem,US3591584 (1970, 1971 to Pfizer).Synthesis and biological properties: J. Lombardino, E. Wiseman, J. Med. Chem.15, 848 (1972); J. Lombardino et al.,ibid.16, 493 (1973). Pharmacology: E. Wiseman et al.,Arzneim.-Forsch.26, 1300 (1976). Evaluation of ulcerogenic effects: G. Palacios et al.,Methods Find. Exp. Clin. Pharmacol.9, 353 (1987). Clinical pharmacology: L. Martinez et al.,ibid.10, 729 (1988). Review:eidem, in Pharmacological and Biochemical Properties of Drug Substancesvol. 3, M. E. Goldberg, Ed. (Am. Pharm. Assoc., Washington, DC, 1981) pp 324-346. Review of pharmacology and therapeutic efficacy: R. N. Brogden et al.,Drugs22, 165-187 (1981); eidem,ibid.28, 292-323 (1984). Symposium on clinical efficacy and safety: Am. J. Med.81, Suppl. 5B, 1-55 (1986). Comprehensive description: M. Mihalic et al.,Anal. Profiles Drug Subs.15, 509-531 (1986).
Properties: Crystals from methanol, mp 198-200°. pKa 6.3 (2:1 dioxane-water). LD50 orally in mice: 360 mg/kg (Wiseman).
Melting point: mp 198-200°
pKa: pKa 6.3 (2:1 dioxane-water)
Toxicity data: LD50 orally in mice: 360 mg/kg (Wiseman)
Derivative Type: Cinnamic acid ester
CAS Registry Number: 87234-24-0
Additional Names: Piroxicam cinnamate; cinnoxicam
Manufacturers’ Codes: SPA-S-510
Trademarks: Sinartrol (SPA); Zelis (Proter); Zen (Prophin)
Molecular Formula: C24H19N3O5S
Molecular Weight: 461.49
Percent Composition: C 62.46%, H 4.15%, N 9.11%, O 17.33%, S 6.95%
Derivative Type: Compd with b-cyclodextrinCAS Registry Number: 121696-62-6
Trademarks: Brexin (Chiesi); Cicladol (Master); Cycladol (Promedica)
Molecular Formula: C57H83N3O39S
Molecular Weight: 1466.33
Percent Composition: C 46.69%, H 5.71%, N 2.87%, O 42.55%, S 2.19%
Therap-Cat: Anti-inflammatory.
Keywords: Anti-inflammatory (Nonsteroidal); Thiazinecarboxamides.
- EINECS:252-974-3
- LD50:250 mg/kg (M, p.o.);
216 mg/kg (R, p.o.);
108 mg/kg (dog, p.o.)
Piroxicam is a nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class used to relieve the symptoms of painful inflammatory conditions like arthritis.[3][4] Piroxicam works by preventing the production of endogenous prostaglandins] which are involved in the mediation of pain, stiffness, tenderness and swelling.[3] The medicine is available as capsules, tablets and (not in all countries) as a prescription-free gel 0.5%.[5] It is also available in a betadex formulation, which allows a more rapid absorption of piroxicam from the digestive tract.[3] Piroxicam is one of the few NSAIDs that can be given parenteral routes.
It was patented in 1968 by Pfizer and approved for medical use in 1979.[6] It became generic in 1992,[7] and is marketed worldwide under many brandnames.[1]
Medical uses
It is used in the treatment of certain inflammatory conditions like rheumatoid and osteoarthritis, primary dysmenorrhoea, postoperative pain; and act as an analgesic, especially where there is an inflammatory component.[3] The European Medicines Agency issued a review of its use in 2007 and recommended that its use be limited to the treatment of chronic inflammatory conditions, as it is only in these circumstances that its risk-benefit ratio proves to be favourable.[5][8]
Adverse effects
See also: Nonsteroidal anti-inflammatory drug
As with other NSAIDs the principal side effects include: digestive complaints like nausea, discomfort, diarrhoea and bleeds or ulceration of the stomach, as well as headache, dizziness, nervousness, depression, drowsiness, insomnia, vertigo, hearing disturbances (such as tinnitus), high blood pressure, oedema, light sensitivity, skin reactions (including, albeit rarely, Stevens–Johnson syndrome and toxic epidermal necrolysis) and rarely, kidney failure, pancreatitis, liver damage, visual disturbances, pulmonary eosinophilia and alveolitis.[5] Compared to other NSAIDs it is more prone to causing gastrointestinal disturbances and serious skin reactions.[5]
In October 2020, the U.S. Food and Drug Administration (FDA) required the drug label to be updated for all nonsteroidal anti-inflammatory medications to describe the risk of kidney problems in unborn babies that result in low amniotic fluid.[9][10] They recommend avoiding NSAIDs in pregnant women at 20 weeks or later in pregnancy.[9][10]
Mechanism of action
See also: Nonsteroidal anti-inflammatory drug
Piroxicam is an NSAID and, as such, is a non-selective COX inhibitor possessing both analgesic and antipyretic properties.[5]
Chemical properties
Piroxicam exists as alkenol tautomer in organic solvents and as zwitterionic form in water.[11]
History
The project that produced piroxicam began in 1962 at Pfizer; the first clinical trial results were reported in 1977, and the product launched in 1980 under the brand name “Feldene”.[7][12] Major patents expired in 1992[7] and the drug is marketed worldwide under many brandnames.[1]
NMR
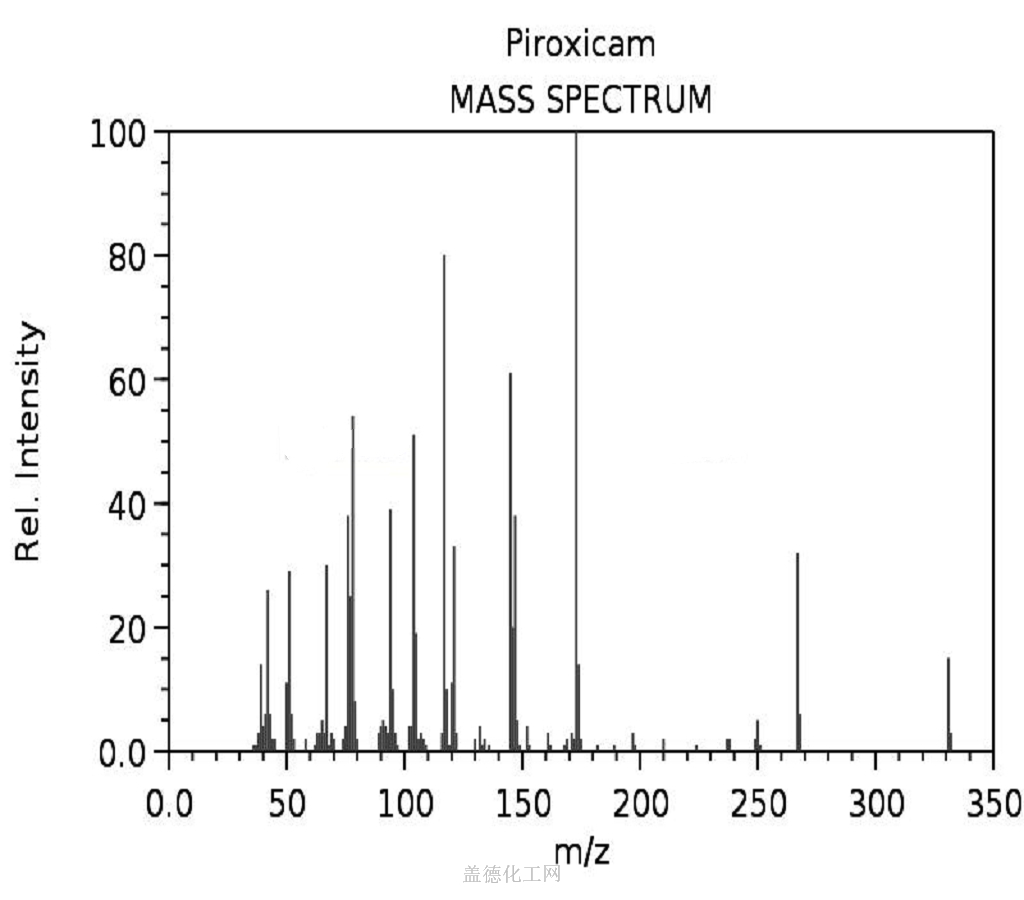
SYN
https://pubs.acs.org/doi/10.1021/jp1084444
Influence of Structure on the Spectroscopic Properties of the Polymorphs of Piroxicam
SYN
https://www.sciencedirect.com/science/article/abs/pii/S092420310400058X?via%3
PATENT

CN 101210013
https://patents.google.com/patent/CN101210013A/enIn the glassed steel reaction vessels of 2000L, add first ethyl ester thing 140Kg, dimethylbenzene 1500L, silica gel 10Kg.Be warming up to 100 ℃ of amino pyrrole 52Kg of adding 2-, continue to be warming up to the solvent refluxing temperature, keep refluxing slowly, steam the ethanol of reaction generation and the mixture of dimethylbenzene simultaneously, TLC follows the tracks of reaction, and reaction in 4.5-5 hour finishes.Underpressure distillation, the control temperature in the kettle is no more than 70 ℃, when the system volume be about cumulative volume 1/3 the time stop distillation, be cooled to normal temperature, stir 6-8h and filter, be i.e. crude product.Crude product adds methyl alcohol 1500L and adds the 15Kg gac, refluxes 30 minutes, filters, and is cooled to normal temperature, stirs 6-8h, methyl alcohol drip washing, 60-70 ℃ is dried by the fire 3-5h, measure product 140.5Kg, yield 85%.Press Cp2005 version standard detection, outward appearance; Off-white color, content 〉=99%.Methanol mother liquor reclaims methyl alcohol to overall 1/3 o’clock, and cooling stirring at normal temperature 6-8h filters and collects product, oven dry measure product 10Kg, yield 5.7%, this product meet the Cp2005 version and require to add up to yield.Add up to yield 90.7%.PAPER Bulletin of the Korean Chemical Society, 26(11), 1771-1775; 2005

SYN

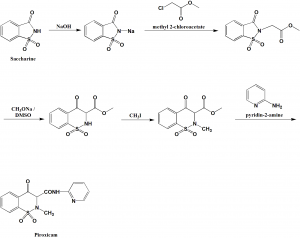


| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 504-29-0 | C5H6N2 | 2-aminopyridine | 2-Pyridinamine |
| 79-04-9 | C2H2Cl2O | chloroacetyl chloride | Acetyl chloride, chloro- |
| 29209-30-1 | C11H11NO5S | 3,4-dihydro-2-methyl-4-oxo-2H-1,2-benzothiazine-3-carboxylic acid methyl ester 1,1-dioxide | 2H-1,2-Benzothiazine-3-carboxylic acid, 3,4-dihydro-2-methyl-4-oxo-, methyl ester, 1,1-dioxide |
| 29209-29-8 | C10H9NO5S | 3-methoxycarbonyl-4-oxo-3,4-dihydro-2H-1,2-benzothiazine 1,1-dioxide | 2H-1,2-Benzothiazine-3-carboxylic acid, 3,4-dihydro-4-oxo-, methyl ester, 1,1-dioxide |
- Drebushchak, V. A.; Journal of Thermal Analysis and Calorimetry 2006, V84(3), P643-649
- Gehad, G. Mohamed; Vibrational Spectroscopy 2004, V36(1), P97-104
- Pajula, Katja; Molecular Pharmaceutics 2010, V7(3), P795-804
- Wassvik, Carola M.; European Journal of Pharmaceutical Sciences 2006, V29(3-4), P294-305
- Wassvik, Carola M.; Journal of Medicinal Chemistry 2008, V51(10), P3035-3039
- Zayed, M. A.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2004, V60A(12), P2843-2852
- Zia-ur-Rehman, Muhammad; Bulletin of the Korean Chemical Society 2005, V26(11), P1771-1775
- “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)
- Stulzer, H. K.; Pharmaceutical Chemistry Journal 2008, V42(4), P215-219 CAPLUS
- Drebushchak, V. A.; Journal of Thermal Analysis and Calorimetry 2006, V86(2), P303-309
- Hughes, Laura D.; Journal of Chemical Information and Modeling 2008, V48(1), P220-232
- Laban, Gunter; DD 260398 A3 1988
- Svoboda, Jiri; Collection of Czechoslovak Chemical Communications 1986, V51(5), P1133-9
- (26) Perillo, Isabel A.; Journal of Heterocyclic Chemistry 1983, V20(1), P155-60
- Zak, Bohumil; CS 276217 B6 1992 CAPLUS
- Dalla Croce, Piero; Journal of Chemical Research, Synopses 1986, (4), P150-1
- Vemavarapu, Chandra; Powder Technology 2009, V189(3), P444-453
- Sanghavi, N. M.; Indian Journal of Technology 1989, V27(2), P93-5
- “PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
- Mohamed, Gehad G.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2004, V60A(13), P3141-3154
- Zayed, M. A.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2006, V64A(1), P216-232
- Habibi-Yangjeh, Aziz; Bulletin of the Korean Chemical Society 2008, V29(4), P833-841
- Mahlin, Denny; Molecular Pharmaceutics 2011, V8(2), P498-506
- Kozjek, Franc; Acta Pharmaceutica Jugoslavica 1985, V35(4), P275-81
- Laban, Gunter; DD 258532 A3 1988
- Caira, Mino R.; Journal of Pharmaceutical Sciences 1998, V87(12), P1608-1614
- Mohamed, Gehad G.; Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 2005, V62A(4-5), P1165-1171
- Lin, Yannan; Journal of Pharmaceutical and Biomedical Analysis 2010, V51(4), P979-984
References
- ^ Jump up to:a b c Drugs.com Drugs.com international listings for piroxicamPage accessed July 3, 2015
- ^ https://www.ema.europa.eu/documents/psusa/piroxicam-list-nationally-authorised-medicinal-products-psusa/00002438/202004_en.pdf
- ^ Jump up to:a b c d e f g Brayfield, A, ed. (14 January 2014). “Piroxicam”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 June 2014.
- ^ “TGA Approved Terminology for Medicines, Section 1 – Chemical Substances” (PDF). Therapeutic Goods Administration, Department of Health and Ageing, Australian Government. July 1999: 97.
- ^ Jump up to:a b c d e Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. pp. 665, 673–674. ISBN 978-0-85711-084-8.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 519. ISBN 9783527607495.
- ^ Jump up to:a b c Lombardino, JG; Lowe, JA 3rd (2004). “The role of the medicinal chemist in drug discovery–then and now”. Nat Rev Drug Discov. 3 (10): 853–62. doi:10.1038/nrd1523. PMID 15459676. S2CID 11225541.. See: [1] Box 1: Discovery of piroxicam (1962–1980)
- ^ “COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) OPINION FOLLOWING AN ARTICLE 31(2) REFERRAL PIROXICAM CONTAINING MEDICINAL PRODUCTS” (PDF). European Medicines Agency. London, UK: European Medicines Agency. 20 September 2007. Retrieved 24 June 2014.
- ^ Jump up to:a b “FDA Warns that Using a Type of Pain and Fever Medication in Second Half of Pregnancy Could Lead to Complications”. U.S. Food and Drug Administration (FDA) (Press release). 15 October 2020. Retrieved 15 October 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “NSAIDs may cause rare kidney problems in unborn babies”. U.S. Food and Drug Administration. 21 July 2017. Retrieved 15 October 2020. This article incorporates text from this source, which is in the public domain.
- ^ Ivanova D, Deneva V, Nedeltcheva D, Kamounah FS, Gergov G, Hansen PE, Kawauchi S, Antonov L (2015). “Tautomeric transformations of piroxicam in solution: a combined experimental and theoretical study”. RSC Advances. 5 (40): 31852–31860. doi:10.1039/c5ra03653d.
- ^ Weintraub M, Jacox RF, Angevine CD, Atwater EC (1977). “Piroxicam (CP 16171) in rheumatoid arthritis: a controlled clinical trial with novel assessment techniques”. Journal of Rheumatology. 4 (4): 393–404. PMID 342691.
Further reading
- Dean L (2019). “Piroxicam Therapy and CYP2C9 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 30742401. Bookshelf ID: NBK537367.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Pronunciation | /paɪˈrɒksɪˌkæm/ |
| Trade names | Feldene, others[1] |
| Other names | Piroksikam, piroxikam |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a684045 |
| Pregnancy category | AU: C |
| Routes of administration | By mouth |
| ATC code | M01AC01 (WHO) M02AA07 (WHO), S01BC06 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only)US: ℞-onlyEU: Rx-only [2] |
| Pharmacokinetic data | |
| Protein binding | 99%[3] |
| Metabolism | Liver-mediated hydroxylation and glucuronidation[3] |
| Elimination half-life | 50 hours[3] |
| Excretion | Urine, faeces |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 36322-90-4 |
| PubChem CID | 54676228 |
| IUPHAR/BPS | 7273 |
| DrugBank | DB00554 |
| ChemSpider | 10442653 |
| UNII | 13T4O6VMAM |
| KEGG | D00127 |
| ChEBI | CHEBI:8249 |
| ChEMBL | ChEMBL527 |
| CompTox Dashboard (EPA) | DTXSID5021170 |
| ECHA InfoCard | 100.048.144 |
| Chemical and physical data | |
| Formula | C15H13N3O4S |
| Molar mass | 331.35 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
{kind=link}
{kind=link}
///////////PIROXICAM

NEW DRUG APPROVALS
ONE TIME
$10.00
PIRACETAM

Piracetam
- ATC:N06BX03
- MW:142.16 g/mol
- CAS-RN:7491-74-9
- InChI Key:GMZVRMREEHBGGF-UHFFFAOYSA-N
- InChI:InChI=1S/C6H10N2O2/c7-5(9)4-8-3-1-2-6(8)10/h1-4H2,(H2,7,9)
- EINECS:231-312-7
- LD50:9200 mg/kg (M, i.v.); 2 g/kg (M, p.o.)
CAS Registry Number: 7491-74-9
CAS Name: 2-Oxo-1-pyrrolidineacetamide
Additional Names: 2-pyrrolidoneacetamide; 2-pyrrolidinoneacetamide; 2-ketopyrrolidine-1-ylacetamide; 1-acetamido-2-pyrrolidinone
Manufacturers’ Codes: UCB-6215
Trademarks: Avigilen (Riemser); Axonyl (Pfizer); Cerebroforte (Azupharma); Encetrop (Alpharma); Gabacet (Sanofi-Synthelabo); Geram (UCB); Nootrop (UCB); Nootropil (UCB); Nootropyl (UCB); Norzetam (UCB); Normabraïn (UCB); Piracebral (Hexal); Piracetrop (Holsten); Sinapsan (Rodleben)Molecular Formula: C6H10N2O2
Molecular Weight: 142.16
Percent Composition: C 50.69%, H 7.09%, N 19.71%, O 22.51%
Literature References: Prepn: H. Morren, NL6509994; eidem,US3459738 (1966, 1969 both to U.C.B.). Pharmacology: Giurgea et al.,Arch. Int. Pharmacodyn. Ther.166, 238 (1967); Giurgea, Moyersoons, ibid.188, 401 (1970); Giurgea et al.,Psychopharmacologia20, 160 (1971). Metabolism and biochemical studies: Gobert, J. Pharm. Belg.27, 281 (1972). Clinical studies: W. J. Oosterveld, Arzneim.-Forsch.30, 1947 (1980); G. Chouinard et al.,Psychopharmacol. Bull.17, 129 (1981); in dyslexia: M. Di Ianni et al.,J. Clin. Psychopharmacol.5, 272 (1985).Properties: Crystals from isopropanol, mp 151.5-152.5°.
Melting point: mp 151.5-152.5°
Therap-Cat: Nootropic.
Keywords: Nootropic.
Piracetam is in the racetams group, with chemical name 2-oxo-1-pyrrolidine acetamide. It is a derivative of the neurotransmitter GABA[5] and shares the same 2-oxo-pyrrolidone base structure with pyroglutamic acid. Piracetam is a cyclic derivative of GABA (gamma-aminobutyric acid). Related drugs include the anticonvulsants levetiracetam and brivaracetam, and the putative nootropics aniracetam and phenylpiracetam.Piracetam is a drug marketed as a treatment for myoclonus[3] and a cognitive enhancer.[4] Evidence to support its use is unclear, with some studies showing modest benefits in specific populations and others showing minimal or no benefit.[5][6] Piracetam is sold as a medication in many European countries. Sale of piracetam is not illegal in the United States, although it is not regulated nor approved by the FDA so it must be marketed as a dietary supplement.[4]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Efficacy
Dementia
A 2001 Cochrane review concluded that there was not enough evidence to support piracetam for dementia or cognitive problems.[6] A 2005 review found some evidence of benefit in older subjects with cognitive impairment.[5] In 2008, a working group of the British Academy of Medical Sciences noted that many of the trials of piracetam for dementia were flawed.[7]
There is no good evidence that piracetam is of benefit in treating vascular dementia.[8]
Depression and anxiety
Some sources suggest that piracetam’s overall effect on lowering depression and anxiety is higher than on improving memory.[9] However, depression is reported to be an occasional adverse effect of piracetam.[10]
Other
Piracetam may facilitate the deformability of erythrocytes in capillary which is useful for cardiovascular disease.[5][3]
Peripheral vascular effects of piracetam have suggested its use potential for vertigo, dyslexia, Raynaud’s phenomenon and sickle cell anemia.[5][3] There is no evidence to support piracetam’s use in sickle cell crisis prevention[11] or for fetal distress during childbirth.[12] There is no evidence for benefit of piracetam with acute ischemic stroke,[13] though there is debate as to its utility during stroke rehabilitation.[14][15]
Anti-vasospasm
Piracetam has been found to diminish erythrocyte adhesion to vascular wall endothelium, making any vasospasm in the capillary less severe. This contributes to its efficacy in promoting microcirculation, including to the brain and kidneys.[5][3]
Side effects
Symptoms of general excitability, including anxiety, insomnia, irritability, headache, agitation, nervousness, tremor, and hyperkinesia, are occasionally reported.[10][16][17] Other reported side effects include somnolence, weight gain, clinical depression, weakness, increased libido, and hypersexuality.[10]
According to a 2005 review, piracetam has been observed to have the following side effects: hyperkinesia, weight gain, nervousness, somnolence, depression and asthenia.[5]
Piracetam reduces platelet aggregation as well as fibrinogen concentration, and thus is contraindicated to patients suffering from cerebral hemorrhage.[5][3]
Toxicity
Piracetam does not appear to be acutely toxic at the doses used in human studies.[6][18][19]
The LD50 for oral consumption in humans has not been determined.[20] The LD50 is 5.6 g/kg for rats and 20 g/kg for mice, indicating extremely low acute toxicity.[21] For comparison, in rats the LD50 of vitamin C is 12 g/kg and the LD50 of table salt is 3 g/kg.
Mechanisms of action
Piracetam’s mechanism of action, as with racetams in general, is not fully understood. The drug influences neuronal and vascular functions and influences cognitive function without acting as a sedative or stimulant.[5] Piracetam is a positive allosteric modulator of the AMPA receptor, although this action is very weak and its clinical effects may not necessarily be mediated by this action.[22] It is hypothesized to act on ion channels or ion carriers, thus leading to increased neuron excitability.[20] GABA brain metabolism and GABA receptors are not affected by piracetam[23]
Piracetam improves the function of the neurotransmitter acetylcholine via muscarinic cholinergic (ACh) receptors[citation needed], which are implicated in memory processes.[24] Furthermore, piracetam may have an effect on NMDA glutamate receptors, which are involved with learning and memory processes. Piracetam is thought to increase cell membrane permeability.[24][25] Piracetam may exert its global effect on brain neurotransmission via modulation of ion channels (i.e., Na+, K+).[20] It has been found to increase oxygen consumption in the brain, apparently in connection to ATP metabolism, and increases the activity of adenylate kinase in rat brains.[26][27] Piracetam, while in the brain, appears to increase the synthesis of cytochrome b5,[28] which is a part of the electron transport mechanism in mitochondria. But in the brain, it also increases the permeability of some intermediates of the Krebs cycle through the mitochondrial outer membrane.[26]
Piracetam inhibits N-type calcium channels. The concentration of piracetam achieved in central nervous system after a typical dose of 1200 mg (about 100 μM)[29] is much higher than the concentration necessary to inhibit N-type calcium channels (IC50 of piracetam in rat neurons was 3 μM).[30]
History
Piracetam was first made some time between the 1950s and 1964 by Corneliu E. Giurgea.[31] There are reports of it being used for epilepsy in the 1950s.[32]
Society and culture
In 2009 piracetam was reportedly popular as a cognitive enhancement drug among students.[33]
Legal status
Piracetam is an uncontrolled substance in the United States meaning it is legal to possess without a license or prescription.[34]
Regulatory status
In the United States, piracetam is not approved by the Food and Drug Administration.[1] Piracetam is not permitted in compounded drugs or dietary supplements in the United States.[35] Nevertheless, it is available in a number of dietary supplements.[4]
In the United Kingdom, piracetam is approved as a prescription drug Prescription Only Medicine (POM) number is PL 20636/2524[36] for adult with myoclonus of cortical origin, irrespective of cause, and should be used in combination with other anti-myoclonic therapies.[37]
In Japan piracetam is approved as a prescription drug.[38]
Piracetam has no DIN in Canada, and thus cannot be sold but can be imported for personal use in Canada.[39]
In Hungary, piracetam was a prescription-only medication, but as of 2020, no prescription is required and piracetam is available as an over-the-counter drug under the name Memoril Mite, and is available in 600 mg pills.

According to the literature reports, the synthetic route of piracetam can be divided into four synthetic methods: α-pyrrolidone method, glycine method, succinic anhydride method and one-step synthesis method:[0009] I. α-pyrrolidone method, 2-pyrrolidone is a lactam, which can react with a strong base (sodium hydride or potassium hydride, sodium methoxide) to generate pyrrolidone metal salt, which can be further combined with halogenated ester or halogen Substitute amide reaction to generate N-alkylated product.[0010] In 1966, a method for preparing piracetam by reacting pyrrolidone and chloroacetamide in 1,4-dioxane with sodium hydrogen as a strong base was reported. The specific synthetic route is shown in Scheme 1:[0011]

[0012] In this process, due to the high price of dioxane, industrial production is still difficult. On the basis of the above process, Xu Yungen used dimethyl sulfoxide as the solvent and sodium methoxide as the acid binding agent to synthesize piracetam in the presence of the phase transfer catalyst benzyltriethylammonium chloride. Due to the difficulty of solvent recovery, the cost of this route is relatively high.[0013] In 1981, Zhou Renxing et al. used sodium methoxide as a strong base to extract methanol in toluene by fractional distillation to convert pyrrolidone into the corresponding sodium salt, and then react with ethyl chloroacetate. The resulting ethyl pyrrolidone ethyl acetate was subjected to ammonolysis. Piracetam can be produced. The specific synthetic route is shown in Scheme 2.[00141

[0015] Because the ammonolysis is carried out in a methanol solution of ammonia, the calculated amount of ethanol generated during the ammonolysis contaminates the methanol solution of ammonia used, which affects the recycling of the methanol solution of ammonia, and is therefore not conducive to process production.[0016] 2. Glycine method, glycine and its derivatives can be used as starting materials for the synthesis of pyroacetamide. Glycine can be prepared by γ-chlorination butylation, amination and cyclization.[0017] According to a British patent report in 1979, glycine trimethylsilyl ester was first condensed with γ-chlorobutyryl chloride, and the corresponding acid chloride was subjected to ammonolysis, and finally cyclized to produce piracetam. The specific synthesis method is as Scheme 3 Shown[0018]

[0019] In this type of synthesis route, some raw materials are not easily available, which restricts industrial production.[0020] 3. Succinic acid method, succinic acid is heated and dehydrated to generate succinic anhydride, succinic anhydride then reacts with glycine to generate an aminolysis product, and the aminolysis product is reduced by sodium tetrafluoroborate, and piracetam can be synthesized by aminolysis , The specific synthetic route is shown in SCheme4. [0021]

[0022] Because sodium tetrafluoroborate is used as a reducing agent, it is expensive, and it is difficult to expand the scale of industrial production. Succinimide generates sodium salt under the action of metal sodium, and its sodium salt reacts with chloroacetamide to generate N-alkylated product. The alkylated product can be electrolytically reduced to obtain piracetam. Since electrolytic reduction is still in the research stage in our country, the production cost of this method is relatively high.[0023] 4. One-step synthesis method, using ethyl 4-chloro-n-butyrate in the presence of sodium bicarbonate, using anhydrous ethanol as a solvent, and glycinamide hydrochloride under heating and refluxing to obtain piracetam in one step, The specific synthetic route is shown in S Cheme5.[0024]

[0025] In this route, glycinamide hydrochloride is very easy to absorb moisture and agglomerate to affect the reaction rate, and the reaction is not easy to control, so it is difficult to achieve industrial production.
SYN

SYN
http://www.cjph.com.cn/EN/abstract/abstract373.shtml
With absolute ethanol as the solvent, ethyl 4-chloro-n-butanoate and glycinamide hydrochloride were refluxed for 20 h in the presence of sodium bicarbonate to obtain central stimulant piracetam. After recrystallization from isopropanol, the yield was about 58% with a purity of 99.6%.
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 79-07-2 | C2H4ClNO | 2-chloroacetamide | Acetamide, 2-chloro- |
| 105-39-5 | C4H7ClO2 | ethyl chloroacetate | Acetic acid, chloro-, ethyl ester |
| 61516-73-2 | C8H13NO3 | ethyl 2-oxo-1-pyrrolidineacetate | 1-Pyrrolidineacetic acid, 2-oxo-, ethyl ester |
| 616-45-5 | C4H7NO | 2-pyrrolidone | 2-Pyrrolidinone |
PATENT
https://patents.google.com/patent/CN104478779A/zh

Example 1[0036] A method for synthesizing piracetam, which includes the following steps:[0037] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 70°C, a methanol solution of sodium methoxide (28.4% (w/w); 114.0 g; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0038] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. When the temperature of the reaction system drops to 60°C, a toluene solution of 58 mL (0.66 mol) of methyl chloroacetate is slowly added dropwise, and the reaction temperature is controlled to 80-100°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain α-pyrrolidone methyl acetate, and measure its content by HPLC (area normalization method). [C18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1 . OmL/min; detection wavelength is 205nm; injection volume is 20yL][0039] Preparation of Piracetam: Put about 130 mL of methanol in a 500 mL three-necked flask, and vent ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 10 h, allowed to cool, filtered with suction, and the filter cake was dried.[0040] The purification of piracetam: 25.50g crude piracetam and 100mL isopropanol were sequentially added in a 500mL three-necked flask, heated to reflux for 40min, activated carbon was added, reflux stirring, hot filtration, and the resulting properties were all white As a powdery solid, the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.85 g of a white solid with a yield of 81.76% (calculated as α-pyrrolidone, the same below).Example 2[0042] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 100°C, a methanol solution of sodium methoxide (28.4% (w/w)); 114. Og; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the addition is complete, add toluene, increase the temperature, and distill at normal pressure until the distillate is completely distilled out, and the reaction is complete.[0043] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. When the temperature of the reaction system drops to 60°C, a mixed solution of 63 mL (0.72 mol) of methyl chloroacetate and 30 mL of toluene is slowly added dropwise, and the reaction temperature is controlled to 80-100°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1 .OmL/ min; detection wavelength is 205nm; injection volume is 20 μL][0044] Preparation of Piracetam: Put about 130 mL of methanol in a 250 mL three-necked flask, and ventilate ammonia to saturation. The obtained ammonia/methanol solution was mixed with 50.0 g of α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 12 hours, allowed to cool, filtered with suction, and the filter cake was dried.[0045] Purification of piracetam: 25.50g crude piracetam and 75mL methanol were sequentially added to a 500mL three-necked flask, heated to reflux for 40min, added activated carbon 0.5g, refluxed for 1h, hot filtered, magnetically stirred Under the conditions, the activated carbon was filtered out, and the properties were all white powdery solids, and the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 21.02g of white solids with a yield of 82.42%.Embodiment 3[0047] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionating column is connected with a thermometer, a condenser and a 1000 mL receiving bottle. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 70°C, a methanol solution of sodium methoxide (28.4% (w/w)); 114. Og; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0048] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. A mixed solution of 79 mL (0.90 mol) of methyl chloroacetate and 50 mL of toluene was slowly added dropwise, and the reaction temperature was controlled to 70-90°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C 18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1.0mL/min; The detection wavelength is 205nm; The injection volume is 20 μL)[0049] Preparation of Piracetam: Put about 130 mL of methanol in a 250 mL three-necked flask, and vent ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 14h, allowed to cool, filtered with suction, and the filter cake was dried.[0050] Purification of piracetam: 25.50g crude piracetam and 125mL ethanol were sequentially added in a 500mL three-necked flask, heated to reflux for 40min, added activated carbon 0.5g, refluxed for 1h, hot filtered, magnetically stirred Activated carbon was filtered off under conditions to obtain white powdery solids in all properties, and the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.24 g of white solids with a yield of 79.37%.Example 4[0052] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 60°C, a methanol solution of sodium methoxide (28.4% (w/w); 114.0 g; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0053] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. A mixed solution of 105 mL (1.20 mol) of methyl chloroacetate and 70 mL of toluene was slowly added dropwise, and the reaction temperature was controlled to be 60~70°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C 18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1.0mL/min; The detection wavelength is 205nm; The injection volume is 20 μL)[0054] Preparation of Piracetam: Put about 130 mL of methanol in a 500 mL three-necked flask, and ventilate ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 16h, allowed to cool, filtered with suction, and the filter cake was dried.[0055] The purification of piracetam: 25.50g crude piracetam and 100mL methanol were sequentially added into a 500mL three-necked flask, heated to reflux for 40min, added activated carbon, refluxed for dissolution, hot filtered, and the properties were all white powders The solid, the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.69 g of a white solid, with a yield of 81. 13%.[0056] Chemical analysis of the white crystals synthesized in each of the foregoing examples, and the obtained physical property values are as follows, thereby confirming that the synthesized product is piracetam.[0057] Melting point: 151.6-152. (TC[0058] ESI-MS m / z: 165. 06 [M + Na] +[0059] 1H-NMR (400MHz, DMS〇-d6, ppm) δ : 7. 38 (s, 1H), 7. 09 (s, 1H), 3. 74 (s, 2H), 3. 36 (t, J =7. 08Hz, 2H), 2. 23 (t, J = 7. 84Hz, 2H), I. 93 (m, 2H).[0060] 13C-NMR(100MHz, DMS0-d6, ppm) δ : 17. 80, 30. 42, 45. 28, 47. 74, 170. 21,174. 90.
PATENTCN110903230A *2019-12-042020-03-24Beijing Yuekang Kechuang Pharmaceutical Technology Co., Ltd.An industrialized preparation method of Pramiracetam sulfate
PATENTCN104478779A2015-04-01New synthetic method of nootropic drug Piracetam
References
- ^ Jump up to:a b “Piracetam”. DrugBank database.
- ^ Leaflet of Piracetam.
- ^ Jump up to:a b c d e “Nootropil Tablets 1200 mg”. (emc). 15 February 2017. Retrieved 14 April 2019.
- ^ Jump up to:a b c Cohen, Pieter A.; Zakharevich, Igor; Gerona, Roy (25 November 2019). “Presence of Piracetam in Cognitive Enhancement Dietary Supplements”. JAMA Internal Medicine. 180 (3): 458–459. doi:10.1001/jamainternmed.2019.5507. PMC 6902196. PMID 31764936.
- ^ Jump up to:a b c d e f g h i Winblad B (2005). “Piracetam: a review of pharmacological properties and clinical uses”. CNS Drug Reviews. 11 (2): 169–82. doi:10.1111/j.1527-3458.2005.tb00268.x. PMC 6741724. PMID 16007238.
- ^ Jump up to:a b c Flicker, L; Grimley Evans, G (2001). “Piracetam for dementia or cognitive impairment”. The Cochrane Database of Systematic Reviews (2): CD001011. doi:10.1002/14651858.CD001011. PMID 11405971.
- ^ Horne G, et al. (May 2008). Brain science, addiction and drugs(PDF) (Report). Academy of Medical Sciences. p. 145. ISBN 978-1-903401-18-7.
- ^ Farooq MU, Min J, Goshgarian C, Gorelick PB (September 2017). “Pharmacotherapy for Vascular Cognitive Impairment”. CNS Drugs(Review). 31 (9): 759–776. doi:10.1007/s40263-017-0459-3. PMID 28786085. S2CID 23271739.
Other medications have been considered or tried for the treatment of VCI or VaD. These include […] piracetam. There is no convincing evidence about the efficacy of these medications in the treatment of VCI.
- ^ Malykh AG, Sadaie MR (February 2010). “Piracetam and piracetam-like drugs: from basic science to novel clinical applications to CNS disorders”. Drugs. 70 (3): 287–312. doi:10.2165/11319230-000000000-00000. PMID 20166767. S2CID 12176745.
- ^ Jump up to:a b c Nootropil®. Arzneimittel-Kompendium der Schweiz. 2013-09-12. Retrieved 2013-10-27.
- ^ Al Hajeri A, Fedorowicz Z (February 2016). “Piracetam for reducing the incidence of painful sickle cell disease crises”. The Cochrane Database of Systematic Reviews. 2: CD006111. doi:10.1002/14651858.CD006111.pub3. PMC 7390168. PMID 26869149.
- ^ Hofmeyr, GJ; Kulier, R (13 June 2012). “Piracetam for fetal distress in labour”. The Cochrane Database of Systematic Reviews (6): CD001064. doi:10.1002/14651858.CD001064.pub2. PMC 7048034. PMID 22696322.
- ^ Ricci S, Celani MG, Cantisani TA, Righetti E (September 2012). “Piracetam for acute ischaemic stroke”. The Cochrane Database of Systematic Reviews (9): CD000419. doi:10.1002/14651858.CD000419.pub3. PMC 7034527. PMID 22972044.
- ^ Zhang J, Wei R, Chen Z, Luo B (July 2016). “Piracetam for Aphasia in Post-stroke Patients: A Systematic Review and Meta-analysis of Randomized Controlled Trials”. CNS Drugs. 30 (7): 575–87. doi:10.1007/s40263-016-0348-1. PMID 27236454. S2CID 22955205.
- ^ Yeo SH, Lim ZI, Mao J, Yau WP (October 2017). “Effects of Central Nervous System Drugs on Recovery After Stroke: A Systematic Review and Meta-Analysis of Randomized Controlled Trials”. Clinical Drug Investigation. 37 (10): 901–928. doi:10.1007/s40261-017-0558-4. PMID 28756557. S2CID 6520934.
- ^ Chouinard G, Annable L, Ross-Chouinard A, Olivier M, Fontaine F (1983). “Piracetam in elderly psychiatric patients with mild diffuse cerebral impairment”. Psychopharmacology. 81 (2): 100–106. doi:10.1007/BF00429000. PMID 6415738. S2CID 32702769.
- ^ Hakkarainen H, Hakamies L (1978). “Piracetam in the treatment of post-concussional syndrome. A double-blind study”. European Neurology. 17 (1): 50–55. doi:10.1159/000114922. PMID 342247.
- ^ Koskiniemi M, Van Vleymen B, Hakamies L, Lamusuo S, Taalas J (March 1998). “Piracetam relieves symptoms in progressive myoclonus epilepsy: a multicentre, randomised, double blind, crossover study comparing the efficacy and safety of three dosages of oral piracetam with placebo”. Journal of Neurology, Neurosurgery, and Psychiatry. 64 (3): 344–348. doi:10.1136/jnnp.64.3.344. PMC 2169975. PMID 9527146.
- ^ Fedi M, Reutens D, Dubeau F, Andermann E, D’Agostino D, Andermann F (May 2001). “Long-term efficacy and safety of piracetam in the treatment of progressive myoclonus epilepsy”. Archives of Neurology. 58 (5): 781–786. doi:10.1001/archneur.58.5.781. PMID 11346373.
- ^ Jump up to:a b c Gouliaev AH, Senning A (May 1994). “Piracetam and other structurally related nootropics”. Brain Research. Brain Research Reviews. 19 (2): 180–222. doi:10.1016/0165-0173(94)90011-6. PMID 8061686. S2CID 18122566.
- ^ “Piracetam Material Safety Sheet” (PDF). Spectrum.
- ^ Ahmed AH, Oswald RE (March 2010). “Piracetam defines a new binding site for allosteric modulators of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors”. Journal of Medicinal Chemistry. 53 (5): 2197–203. doi:10.1021/jm901905j. PMC 2872987. PMID 20163115.
- ^ Giurgea CE (January 1982). “The nootropic concept and its prospective implications”. Drug Development Research. 2 (5): 441–446. doi:10.1002/ddr.430020505. ISSN 1098-2299. S2CID 145059666.
- ^ Jump up to:a b Winnicka K, Tomasiak M, Bielawska A (2005). “Piracetam–an old drug with novel properties?”. Acta Poloniae Pharmaceutica. 62(5): 405–9. PMID 16459490.
- ^ Müller WE, Eckert GP, Eckert A (March 1999). “Piracetam: novelty in a unique mode of action”. Pharmacopsychiatry. 32 (Suppl 1): 2–9. doi:10.1055/s-2007-979230. PMID 10338102.
- ^ Jump up to:a b Grau M, Montero JL, Balasch J (1987). “Effect of Piracetam on electrocorticogram and local cerebral glucose utilization in the rat”. General Pharmacology. 18 (2): 205–11. doi:10.1016/0306-3623(87)90252-7. PMID 3569848.
- ^ Nickolson VJ, Wolthuis OL (October 1976). “Effect of the acquisition-enhancing drug piracetam on rat cerebral energy metabolism. Comparison with naftidrofuryl and methamphetamine”. Biochemical Pharmacology. 25 (20): 2241–4. doi:10.1016/0006-2952(76)90004-6. PMID 985556.
- ^ Tacconi MT, Wurtman RJ (1986). “Piracetam: physiological disposition and mechanism of action”. Advances in Neurology. 43: 675–85. PMID 3946121.
- ^ Yeh HH, Yang YH, Ko JY, Chen SH (July 2006). “Rapid determination of piracetam in human plasma and cerebrospinal fluid by micellar electrokinetic chromatography with sample direct injection”. J Chromatogr A. 1120 (1–2): 27–34. doi:10.1016/j.chroma.2005.11.071. PMID 16343512.
- ^ Bravo-Martínez J, Arenas I, Vivas O, Rebolledo-Antúnez S, Vázquez-García M, Larrazolo A, García DE (October 2012). “A novel CaV2.2 channel inhibition by piracetam in peripheral and central neurons”. Exp Biol Med (Maywood). 237 (10): 1209–18. doi:10.1258/ebm.2012.012128. PMID 23045722.
- ^ Li JJ, Corey EJ (2013). Drug Discovery: Practices, Processes, and Perspectives. John Wiley & Sons. p. 276. ISBN 9781118354469.
- ^ Schmidt D, Shorvon S (2016). The End of Epilepsy?: A History of the Modern Era of Epilepsy Research 1860-2010. Oxford University Press. p. 69. ISBN 9780198725909.
- ^ Medew J (1 October 2009). “Call for testing on ‘smart drugs'”. Fairfax Media. Retrieved 29 May 2014.
- ^ “Erowid Piracetam Vault: Legal Status”.
- ^ Jann Bellamy (26 September 2019). “FDA proposes ban on curcumin and other naturopathic favorites in compounded drugs”. Science-Based Medicine.
- ^http://www.mhra.gov.uk/home/groups/spcpil/documents/spcpil/con1547788739542.pdf
- ^ “Nootropil Tablets 800 mg”. (emc).
- ^ “UCB’s piracetam approved in Japan”. The Pharma Letter. 25 November 1999.
- ^ “Guidance Document on the Import Requirements for Health Products under the Food and Drugs Act and its Regulations (GUI-0084)”. Health Canada / Health Products and Food Branch Inspectorate. 1 June 2010. Retrieved 15 December 2019.
- UCB Pharma Limited (2005). “Nootropil 800 mg & 1200 mg Tablets and Solution”. electronic Medicines Compendium. Datapharm Communications. Archived from the original on 7 December 2006. Retrieved 8 December 2005.
External links
Gouliaev AH, Senning A (May 1994). “Piracetam and other structurally related nootropics”. Brain Research. Brain Research Reviews. 19 (2): 180–222. doi:10.1016/0165-0173(94)90011-6. PMID 8061686. S2CID 18122566.
| Clinical data | |
|---|---|
| Trade names | Breinox, Dinagen, Lucetam, Nootropil, Nootropyl, Oikamid, Piracetam and many others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth, parenteral, or vaporized |
| ATC code | N06BX03 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: UnscheduledUK: POM (Prescription only)US: Unscheduled (Not permitted as drug or supplement[1]) |
| Pharmacokinetic data | |
| Bioavailability | ~100% |
| Onset of action | Swiftly following administration. Food delays time to peak concentration by 1.5 h approximately to 2–3 h since dosing.[2] |
| Elimination half-life | 4–5 h |
| Excretion | Urinary |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 7491-74-9 |
| PubChem CID | 4843 |
| IUPHAR/BPS | 4288 |
| DrugBank | DB09210 |
| ChemSpider | 4677 |
| UNII | ZH516LNZ10 |
| KEGG | D01914 |
| ChEMBL | ChEMBL36715 |
| CompTox Dashboard (EPA) | DTXSID5044491 |
| ECHA InfoCard | 100.028.466 |
| Chemical and physical data | |
| Formula | C6H10N2O2 |
| Molar mass | 142.158 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 152 °C (306 °F) |
| showSMILES | |
| showInChI | |
| (verify) |
{kind=link}
{kind=link}
///////////UCB 6215, Nootropic, PIRACETAM

NEW DRUG APPROVALS
ONE TIME
$10.00
REACH, Registration, Evaluation, Authorisation and Restriction of Chemicals
DRUG REGULATORY AFFAIRS INTERNATIONAL

REACH
https://ec.europa.eu/environment/chemicals/reach/reach_en.htm#:~:text=REACH%20(EC%201907%2F2006),authorisation%20and%20restriction%20of%20chemicals.
REACH (EC 1907/2006)aims to improve the protection of human health and the environment through the better and earlier identification of the intrinsic properties of chemical substances. This is done by the four processes of REACH, namely the registration, evaluation, authorisation and restriction of chemicals. REACH also aims to enhance innovationand competitiveness of the EU chemicals industry.
“No data no market”: the REACH Regulation places responsibility on industry to manage the risks from chemicals and to provide safety information on the substances. Manufacturers and importers are required to gather information on the properties of their chemical substances, which will allow their safe handling, and to register the information in a central database in theEuropean Chemicals Agency (ECHA)in Helsinki. The Agency is the central point in the REACH system: it manages the databases necessary to operate the system, co-ordinates the in-depth evaluation of suspicious chemicals and is…
View original post 4,663 more words
VX 759
VX 759
CAS#: 478025-29-5
Chemical Formula: C22H27NO3S
Molecular Weight: 385.522
478025-29-5
Drug Name:VCH-759Research Code:VX-759; BCH-27759; VCH-759
VX-759; BCH-27759; VCH-759; VX759; BCH27759; VCH759; VX 759; BCH 27759; VCH 759; NNI-1
3-(N-isopropyl-4-methylcyclohexane-1-carboxamido)-5-phenylthiophene-2-carboxylic acid
- 3-[[(trans-4-Methylcyclohexyl)carbonyl](1-methylethyl)amino]-5-phenyl-2-thiophenecarboxylic acid
- 3-[Isopropyl(trans-4-methylcyclohexylcarbonyl)amino]-5-phenylthiophene-2-carboxylic acid
- NNI 1
MOA:NS5B inhibitorIndication:HCV infectionStatus:Phase Ⅱ (Discontinued)Company:Vertex (Originator)
| Molecular Formula | C26H32NNaO3S |
|---|---|
| Synonyms | VX-759 sodium saltBCP17193 |
| Molecular Weight | 461.6 |
VCH-759 had been in phase II clinical trials by ViroChem Pharma (acquired by Vertex in 2009) for the treatment of HCV infection. However, this research has been discontinued.
Infection with HCV is a major cause of human liver disease throughout the world. In the US, an estimated 4.5 million Americans are chronically infected with HCV. Although only 30% of acute infections are symptomatic, greater than 85% of infected individuals develop chronic, persistent infection. Treatment costs for HCV infection have been estimated at $5.46 billion for the US in 1997. Worldwide over 200 million people are estimated to be infected chronically. HCV infection is responsible for 40-60% of all chronic liver disease and 30% of all liver transplants. Chronic HCV infection accounts for 30% of all cirrhosis, end-stage liver disease, and liver cancer in the U.S. The CDC estimates that the number of deaths due to HCV will minimally increase to 38,000/year by the year 2010.
Due to the high degree of variability in the viral surface antigens, existence of multiple viral genotypes, and demonstrated specificity of immunity, the development of a successful vaccine in the near future is unlikely. Alpha-interferon (alone or in combination with ribavirin) has been widely used since its approval for treatment of chronic HCV infection. However, adverse side effects are commonly associated with this treatment: flu-like symptoms, leukopenia, thrombocytopenia, depression from interferon, as well as anemia induced by ribavirin (Lindsay, K. L. (1997) Hepatology 26 (suppl 1 ): 71 S-77S). This therapy remains less effective against infections caused by HCV genotype 1 (which constitutes -75% of all HCV infections in the developed markets) compared to infections caused by the other 5 major HCV genotypes. Unfortunately, only -50-80% of the patients respond to this treatment (measured by a reduction in serum HCV RNA levels and normalization of liver enzymes) and, of responders, 50-70% relapse within 6 months of cessation of treatment. Recently, with the introduction of pegylated interferon (Peg-IFN), both initial and sustained response rates have improved substantially, and combination treatment of Peg-IFN with ribavirin constitutes the gold standard for therapy. However, the side effects associated with combination therapy and the impaired response in patients with genotype 1 present opportunities for improvement in the management of this disease.
First identified by molecular cloning in 1989 (Choo, Q-L et al (1989) Science 244:359-362), HCV is now widely accepted as the most common causative agent of post-transfusion non A, non-B hepatitis (NANBH) (Kuo, G et al (1989) Science 244:362-364). Due to its genome structure and sequence homology, this virus was assigned as a new genus in the Flaviviridae family. Like the other members of the Flaviviridae, such as flaviviruses (e.g. yellow fever virus and Dengue virus types 1-4) and pestiviruses (e.g. bovine viral diarrhea virus, border disease virus, and classic swine fever virus) (Choo, Q-L et al (1989) Science 244:359-362; Miller, R.H. and R.H. Purcell (1990) Proc. Natl. Acad. Sci. USA 87:2057-2061 ), HCV is an enveloped virus containing a single strand RNA molecule of positive polarity. The HCV genome is approximately 9.6 kilobases (kb) with a long, highly conserved, noncapped 5′ nontranslated region (NTR) of approximately 340 bases which functions as an internal ribosome entry site (IRES) (Wang CY et al ‘An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region’ RNA- A Publication of the RNA Society. 1 (5): 526-537, 1995 JuL). This element is followed by a region which encodes a single long open reading frame (ORF) encoding a polypeptide of -3000 amino acids comprising both the structural and nonstructural viral proteins.
Upon entry into the cytoplasm of the cell, this RNA is directly translated into a polypeptide of -3000 amino acids comprising both the structural and nonstructural viral proteins. This large polypeptide is subsequently processed into the individual structural and nonstructural proteins by a combination of host and virally-encoded proteinases (Rice, CM. (1996) in B.N. Fields, D.M.Knipe and P.M. Howley (eds) Virology 2nd Edition, p931-960; Raven Press, N.Y.). Following the termination codon at the end of the long ORF, there is a 3′ NTR which roughly consists of three regions: an – 40 base region which is poorly conserved among various genotypes, a variable length poly(U)/polypyrimidine tract, and a highly conserved 98 base element also called the “3′ X-tail” (Kolykhalov, A. et al (1996) J. Virology 70:3363-3371 ; Tanaka, T. et al (1995) Biochem Biophys. Res. Commun. 215:744-749; Tanaka, T. et al (1996) J. Virology 70:3307-3312; Yamada, N. et al (1996) Virology 223:255-261 ). The 3′ NTR is predicted to form a stable secondary structure which is essential for HCV growth in chimps and is believed to function in the initiation and regulation of viral RNA replication.
The NS5B protein (591 amino acids, 65 kDa) of HCV (Behrens, S. E. et al (1996) EMBO J. 15:12-22), encodes an RNA-dependent RNA polymerase (RdRp) activity and contains canonical motifs present in other RNA viral polymerases. The NS5B protein is fairly well conserved both intra-typically (-95-98% amino acid (aa) identity across 1 b isolates) and inter-typically (-85% aa identity between genotype 1 a and 1 b isolates). The essentiality of the HCV NS5B RdRp activity for the generation of infectious progeny virions has been formally proven in chimpanzees (A. A. Kolykhalov et al.. (2000) Journal of Virology, 74(4): 2046-2051 ). Thus, inhibition of NS5B RdRp activity (inhibition of RNA replication) is predicted to be useful to treat HCV infection.
Although the predominant HCV genotype worldwide is genotype 1, this itself has two main subtypes, denoted 1a and 1 b. As seen from entries into the Los Alamos HCV database
(www.hcv.lanl.gov) (Table 1 ) there are regional differences in the distribution of these subtypes: while genotype 1 a is most abundant in the United States, the majority of sequences in Europe and Japan are from genotype 1 b.
Table 1
Based on the foregoing, there exists a significant need to identify synthetic or biological compounds for their ability to inhibit replication of both genotype 1 a and genotype 1 b of HCV.
PATENT
WO 2002100851
WO 2007071434
PATENT
WO 2009000818
Compound A
5-Phenyl-3-[[(frans-4-methylcyclohexyl)carbonyl](1-methylethyl)amino]-2-thiophenecarboxylic acid
To a mixture of methyl S-^trans^-methylcyclohexyOcarbonylKI-methylethy^aminol-S-phenyl-2-thiophenecarboxylate (Intermediate 31 ) (390 mg) in THF/MeOH/water (3:2:1, vol/vol, 40 ml. total) was added lithium hydroxide monohydrate (246 mg). The mixture was stirred at room temperature for 20 hours, the solvents removed in vacuo, and the residue partitioned between water (40 ml.) and ethyl acetate (40 ml_). The organic layer was dried
(Na2SC>4), evaporated and triturated with ether to give the title compound.
MS calcd for (C22H27NO3S+ H)+: 356
MS found (electrospray): (M+H)+ =356
Compounds A, B, C and D may be made according to the processes described in WO2002/100851 or as described hereinabove.
Structures of Compounds A, B, C and D are shown below for the avoidance of doubt.
The compounds of Formula (I) which have been tested demonstrate a surprisingly superior potency as HCV polymerase inhibitors, as shown by the IC5O values in the cell-based assays across both of the 1 a and 1 b genotypes of HCV, compared to Compounds A, B, C and D. Accordingly, the compounds of Formula (I) are of great potential therapeutic benefit in the treatment and prophylaxis of HCV.
PAPER
Bioorganic & Medicinal Chemistry Letters (2016), 26(18), 4536-4541.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X16300427
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
| Application Id | Application Number | Application Date | Country | Title |
| US333830037 | 16610899 | 04.05.2018 | US | IDENTIFICATION AND TARGETED MODULATION OF GENE SIGNALING NETWORKS |
| US77274496 | 13661508 | 26.10.2012 | US | COMPOUNDS AND METHODS FOR THE TREATMENT OR PREVENTION OF FLAVIVIRUS INFECTIONS |
| US73506396 | 13172477 | 29.06.2011 | US | Thiophene analogues for the treatment or prevention of flavivirus infections |
| EP29858255 | 10185737 | 11.06.2002 | EP | Thiophene derivatives as antiviral agents for flavivirus infection |
| US73326585 | 13081780 | 07.04.2011 | US | Compounds and methods for the treatment or prevention of <i>Flavivirus </i>infections |
| JP272602128 | 2010103706 | 28.04.2010 | JP | COMPOUND AND METHOD FOR TREATMENT OR PREVENTION OF FLAVIVIRUS INFECTION |
| EP11167270 | 09167203 | 11.06.2002 | EP | Thiophene derivatives as antiviral agents for flavivirus infection |
| CN83819886 | 200910139375.5 | 11.06.2002 | CN | Thiophene derivatives as antiviral agents for flavivirus infection |
| US42846304 | 12097840 | 20.12.2006 | US | Antiviral 2-Carboxy-Thiophene Compounds |
| JP272180287 | 2008546276 | 20.12.2006 | JP | 抗ウイルス性2-カルボキシ-チオフェン化合物 |
| WO2007071434 | PCT/EP2006/012442 | 20.12.2006 | WO | ANTIVIRAL 2-CARBOXY-THIOPHENE COMPOUNDS |
| US41429134 | 11042442 | 26.01.2005 | US | Compounds and methods for the treatment or prevention of <i>Flavivirus </i>infections |
| CN82792692 | 02815768.0 | 11.06.2002 | CN | Thiophene derivatives used as antiviral agent against flavivirus infections |
| JP270324690 | 2003503618 | 11.06.2002 | JP | FLAVIVIRUS感染の治療または予防のための化合物および方法 |
| EA95398282 | 200400022 | 11.06.2002 | EA | COMPOUNDS AND METHODS FOR THE TREATMENT OR PREVENTION OF FLAVIVIRUS INFECTIONS |
| US40367952 | 10166031 | 11.06.2002 | US | Compounds and methods for the treatment or prevention of Flavivirus infections |
| KR588271 | 1020037016240 | 11.12.2003 | KR | THIOPHENE DERIVATIVES AS ANTIVIRAL AGENTS FOR FLAVIVIRUS INFECTION |
| EP14092312 | 02742563 | 11.06.2002 | EP | THIOPHENE DERIVATIVES AS ANTIVIRAL AGENTS FOR FLAVIVIRUS INFECTION |
| WO2002100851 | PCT/CA2002/000876 | 11.06.2002 | WO | THIOPHENE DERIVATIVES AS ANTIVIRAL AGENTS FOR FLAVIVIRUS INFECTION |
////////////////VX-759, BCH-27759, VCH-759, VX759, BCH27759, VCH759, VX 759, BCH 27759, VCH 759, NNI-1
O=C(C1=C(N(C(C2CCC(C)CC2)=O)C(C)C)C=C(C3=CC=CC=C3)S1)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Afoxolaner
Afoxolaner
- Molecular FormulaC26H17ClF9N3O3
- Average mass625.870 Da
- A1443
- AH252723
1093861-60-9[RN]1-Naphthalenecarboxamide, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,2-trifluoroethyl)amino]ethyl]-4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide
Afoxolaner Merial
On 9 September 2021, the Committee for Medicinal Products for Veterinary Use (CVMP) adopted a positive opinion1, recommending the granting of a variation to the terms of the marketing authorisation for the veterinary medicinal product Frontpro. The marketing authorisation holder for this veterinary medicinal product is Boehringer Ingelheim Vetmedica GmbH. ,,,, https://www.ema.europa.eu/en/medicines/veterinary/summaries-opinion/frontpro-previously-known-afoxolaner-merial
Frontpro is currently authorised as chewable tablets for use in dogs. The variation concerns the change of legal status from prescription-only to non-prescription veterinary medicine. Additionally, the applicant is adding the list of local representatives to the package leaflet.
Detailed conditions for the use of this product are described in the summary of product characteristics (SPC), for which an updated version reflecting the changes will be published in the revised European public assessment report (EPAR) and will be available in all official European Union languages after the variation to the marketing authorisation has been granted by the European Commission.
| Name | Frontpro (previously known as Afoxolaner Merial) |
| Agency product number | EMEA/V/C/005126 |
| International non-proprietary name (INN) or common name | afoxolaner |
| Species | Dogs |
| Active substance | afoxolaner |
| Date opinion adopted | 09/09/2021 |
| Company name | Boehringer Ingelheim Vetmedica GmbH |
| Status | Positive |
| Application type | Post-authorisation |
| Medicine | Frontpro (previously known as Afoxolaner Merial) |
|---|---|
| Active Substance | afoxolaner |
| INN/Common name | afoxolaner |
| Pharmacotherapeutic Classes | Ectoparasiticides for systemic use |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2019-05-20 |
European Medicines Agency (EMA)
| Medicine | Nexgard Spectra |
|---|---|
| Active Substance | afoxolaner, milbemycin oxime |
| INN/Common name | afoxolaner, milbemycin oxime |
| Pharmacotherapeutic Classes | Endectocides, Antiparasitic products, insecticides and repellents, milbemycin oxime, combinations |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2015-01-15 |
| Medicine | NexGard |
|---|---|
| Active Substance | afoxolaner |
| INN/Common name | afoxolaner |
| Pharmacotherapeutic Classes | Isoxazolines, Ectoparasiticides for systemic use |
| Status | This medicine is authorized for use in the European Union |
| Company | Boehringer Ingelheim Vetmedica GmbH |
| Market Date | 2014-02-11 |
European Medicines Agency (EMA)
SYN WO2009126668,

SYN
IP .COM

PATENT
PATENT
https://patents.google.com/patent/WO2009126668A2/en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017176948
A particularly active isoxazoline compound, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,24rifluoroethyl)amino]ethyl]-l-naphthalenecarboxamide, is known by the nonproprietary name afoxolaner. Afoxolaner has the following chemical structure:
Afoxolaner
Other isoxazoline compounds that have been found to be highly active against parasitic insects and arachnids are known by the nonproprietary names fluralaner (see US 7,662,972, which is incorporated herein by reference), sarolaner (see US 8,466, 15, incorporated herein by reference) and lotilaner (see, for example US 8,383,659, incorporated herein by reference). The structures of these compounds are shown below:
In addition, published patent application nos. US 2010/0254960 Al, WO 2007/070606
A2, WO 2007/123855 A2, WO 2010/003923 Al, US7951828 & US7662972, US 2010/0137372 Al, US 2010/0179194 A2, US 2011/0086886 A2, US 2011/0059988 Al, US 2010/0179195 Al and WO 2007/075459 A2 and U.S. Patent No. 7,951,828 (all incorporated herein by reference) describe various other parasiticidal isoxazoline compounds.
It is known in the field that isoxazoline compounds having a chiral quaternary carbon atom such as the carbon atom adjacent to the oxygen on the isoxazoline ring of the compounds described above have at least two optical isomer (enantiomers) that are mirror images of each other. Furthermore, it is sometimes the case with biologically active compounds that one of the enantiomers is more active than the other enantiomer. In addition, it is sometimes the case that one enantiomer of a biologically active compound is less toxic than the other enantiomer.
Therefore, with optically active compounds it is desirable to utilize the enantiomer that is most active and less toxic (eutomer). However, isolating the most active enantiomer from a mixture can be costly and result in waste of up to half of the racemic mixture prepared.
Processes to prepare certain isoxazoline compounds enriched in an enantiomer using some cinchona alkaloid-derived phase transfer catalysts have been described. For example, US 2014/0206633 Al, US 2014/0350261 Al, WO 2013/116236 Al and WO 2014/081800 Al (incorporated herein by reference) describe the synthesis of certain isoxazoline active agents enriched in an enantiomer using cinchona alkaloid-based chiral phase transfer catalysts. Further, Matoba et al., Angew. Chem. 2010, 122, 5898-5902 describes the chiral synthesis of certain pesticidal isoxazoline active agents. However, these documents do not describe the processes and certain catalysts described herein.
Scheme 3
Example 7: Preparation of (S)-afoxolaner using chiral phase transfer catalyst (Ilia- 13-1):
(ΠΑ-1) (^-afoxolaner
1) Starting material (IIA-1) (200g, 1.Oeq, 94.0%) and DCM (6 L, 30 volumes) were placed into a 10 L reactor, the solid was dissolved completely.
2) The mixture was cooled to 0°C, and some starting material precipitated out.
3) The catalyst (Ilia- 13-1) (7.56g, 3% mol, 95.0%) was added to the mixture and the resulting mixture cooled further to -10° C.
4) Hydroxylamine (64.9 g, 3.0 eq, 50% solution in water) was added to a solution of NaOH (52.5g, 4. Oeq, in 5v water) in a separate reactor and stirred for 30 minutes.
5) The resulting hydroxylamine/NaOH solution was then added dropwise to the 10 L reactor containing (IIA-1) over about 4 hours.
6) The resulting mixture was stirred for 12 hours at -10°C and monitored for the extent of reaction until the amount of starting material was < 1.0% by HPLC.
7) The mixture was then warmed to 10°C, 1 liter of water was added and the mixture was stirred for 10 minutes.
8) The mixture was allowed to settle to separate the two phases, and the organic layer was collected.
9) The organic layer was then washed with 2 liters of water, the layers were allowed to separate again and the organic layer was collected.
10) The organic layer was washed with 1 liter of brine, the layers allowed to separate and the organic layer was collected and dried over Na2S04 (200 g).
11) The dried organic layer was concentrated under vacuum to about 2 volumes.
12) Toluene (2 L, 10 volumes) was charged to the concentrated mixture and concentration under vacuum was continued to about 5 volumes. Solvent exchange was repeated twice again.
13) The resulting solution was placed into a 2.0 L reactor and heated to 55-60°C.
14) Cyclohexane (300 ml, 1.5 volumes) was added at 55-60°C.
15) The mixture was then cooled to 40 °C over 1.5 hours and then stirred at 40°C for 3 hours.
16) The mixture was then cooled to 25 °C over 2 hours and stirred at 25°C for a further 3 hours.
17) The resulting mixture was cooled to 0-5 °C over 1 hour and stirred at 5 °C for 12 hours, at which time the mixture was filtered to isolate the product.
18) The filter cake was washed with cold toluene/ Cyclohexane (3 : 1, 1000 ml, 5 volumes).
19) The product was obtained as a white solid. (171.5g, chiral purity > 99.0% by area using the chiral HPLC method described in Example 3, chemical purity > 99.0% by area (HPLC), yield: 83.6%, assay purity: 92%). The 1H NMR and LCMS spectra are consistent with the structure of (^-afoxolaner as the toluene solvate. Figure 3 shows the 1H NMR spectra of (S)-afoxolaner in DMSO-d6 and Figure 4 shows the 1H NMR spectra of afoxolaner (racemic) for comparison. The chiral purity of the product was determined using the chiral HPLC method described in Example 3. Figure 5 shows the chiral HPLC chromatogram of afoxolaner (racemic) and Figure 6 shows the chiral HPLC chromatogram of the product (^-afoxolaner showing one enantiomer.
Example 8: Alternate Process to prepare (^-afoxolaner
An alternate process for the preparation of (S)-afoxolaner was conducted. Some of the key variations in the alternate process are noted below.
1. 1 kilogram of compound (IIA-1) (1 eq.) and 9 liters of DCM are charged to a reactor and stirred to dissolve the compound.
2. The mixture is cooled to about 0° C and 50 grams (5 mole %) of the chiral phase transfer catalyst (Ilia- 13-1) and 1 liter of DCM are charged and the resulting mixture is cooled to about -13° C.
3. A solution of 19% (w/w) hydroxylamine sulfate (294 g, 1.1 eq.) (made with 294 grams of ( H2OH)H2S04 and 141 grams of NaCl in 1112 mL of water) and 4.4 equivalents of NaOH as a 17.6% (w/w) solution (286 grams NaOH and 158 grams of NaCl in 1180 mL water) are charged to the reaction mixture simultaneously.
4. The resulting reaction mixture was aged about 20 hours at about -13° C and then checked for reaction conversion by HPLC (target < 0.5% by area);
5. After completion of the reaction, water (3 vol.) was added at about 0° C. Then, a solution of 709 g of KH2P04 in 4.2 liters of water are added to the mixture to adjust the pH (target 7-8) and the resulting mixture is stirred at about 20° C for 30 minutes.
6. The layers are allowed to settle, the aqueous layer is removed and the organic layer is washed with 3 liters of water twice.
Crystallization of Toluene Solvate
1. After the extraction/washing step, the dichloromethane is removed by distillation under vacuum to about 1-2 volumes and toluene (about 5-10 volumes) is added.
2. The volume is adjusted by further distillation under vacuum and/or addition of more toluene to about 5-6 volumes. The mixture is distilled further while maintaining the volume to completely remove the dichloromethane reaction solvent.
3. The mixture is then cooled to about 10° C and seeded with afoxolaner (racemic compound) and stirred at the same temperature for at least 2 hours;
4. The mixture is heated to about 55-65° C, aged for at least 17 hours and then the solid is filtered off. The filtered solid is washed with toluene;
5. The combined filtrate and wash is adjusted to a volume of about 5-6 volumes by
distillation under vacuum and/or toluene addition;
6. The resulting mixture is cooled to about 10° C and aged for at least 5 hours then filtered.
The cake is washed with toluene.
7. The cake is dried at 50° C under vacuum to obtain a toluene solvate of (S)-afoxolaner containing between about 6% and 8% toluene.
Re-crystallization from cyclohexane/ethanol
The toluene solvate of (S)-afoxolaner was subsequently re-crystallized from a mixture of cyclohexane and ethanol to remove the associated toluene and to further purify the product.
1. 591 grams of the (S)-afoxolaner toluene solvate were charged to a vessel along with 709 mL of ethanol (1.2 vol.) and 1773 mL of cyclohexane (3 vol.) and the mixture heated to about 60° C.
2. To the resulting mixture was added an additional 6383 mL of cyclohexane with stirring.
3. The resulting mixture was cooled to about 30° C and then heated again to 60° C. This process was repeated once.
4. The mixture was slowly cooled to 10° C and stirred for at least 5 hours.
5. The resulting slurry was filtered and the cake washed with cyclohexane.
6. The cake was dried at 50° C under vacuum to provide 453.7 grams of (S)-afoxolaner
Example 9: Comparative selectivity of benzyloxy-substituted chiral phase transfer catalyst (Illa-13) with other cinchona alkaloid-based chiral phase transfer catalysts.
The selectivity of the formation of (S)-afoxolaner from compound IIA-1 as shown above was studied with sixteen chiral phase transfer catalysts (PTC) of different structures. The reaction was conducted using conditions similar to those of example 7. The ratio of (^-afoxolaner and (R)-afoxolaner in the reaction mixture was determined by chiral HPLC using the method described in Example 3. The results of the study are provided in Table 2 below.
Table 2
No. Chiral PTC Ratio of (S)- to (R)-afoxolaner
16 50% : 50%
As shown in the table, the catalyst in which the group R in the structure of formula (Ilia) is 3,4,5-tribenzyloxy phenyl results in a surprising improved selectivity for the (S)-enantiomer compared with other quinine-based phase transfer catalysts in which the group corresponding to R in formula (Ilia) is another group.
Example 10: Improvement of Chiral Purity of (<S)-afoxolaner by Crystallization from Toluene
A sample of reaction mixture containing a ratio (HPLC area) of 92.1 :7.9, (^-afoxolaner to (R)-afoxolaner, was concentrated to dryness and the residue was crystallized from toluene and from ethanol/cyclohexane using a process similar to that described in Example 8. The isolated crystalline solid was analyzed by chiral HPLC to determine the relative amounts of (S)-afoxolaner and (R)-afoxolaner (HPLC method: column – Chiralpak AD-3 150 mm x 4.6 mm x 3.0 μηι, injection volume – 10 μΐ., temperature – 35° C, flow – 0.8 mL/minute, mobile phase -89% hexane/10% isopropanol/1% methanol, detection – 312 nm). The ratio of (^-afoxolaner to (R)-afoxolaner in the solid isolated from the toluene crystallization was found to be 99.0 : 1.0 while the ratio of (S)-afoxolaner to (R)-afoxolaner in the solid crystallized from ethanol/cyclohexane was found to be 95.0 : 5.0.
The example shows that the crystallization (^-afoxolaner from an aromatic solvent such as toluene results in a significant improvement of chiral purity of the product. This is very unexpected and surprising.
Example 1 1 : Comparative selectivity of benzyl oxy vs. alkoxy-substituted chiral phase transfer catalyst of Formula (Ilia- 13)
Three chiral phase transfer catalysts of Formula (IIIa-13), wherein the phenyl ring is substituted with three alkoxy groups and three benzyloxy groups (R = methyl, ethyl and benzyl); R’=OMe, W=vinyl and X=chloro were evaluated in the process to prepare of (,S)-IA from compound IIA-1
as shown below.
The amount of solvents and reagents and the reaction and isolation conditions were as described in Example 7 above. The same procedure was used for each catalyst tested. It was found that the selectivity of the tri-benzyloxy catalyst was surprisingly significantly better than the two alkoxy-substituted catalysts, as shown by the chiral purity of the product. Furthermore, it was found that using the tri-benzyloxy substituted phase transfer catalyst the resulting chemical purity was also much better. The superior selectivity of the benzyloxy-substituted catalyst is significant and surprising and cannot be predicted. Chiral phase transfer catalysts containing a phenyl substituted with benzyloxy and alkoxy groups were found to be superior to catalysts substituted with other groups such as electron-withdrawing groups and alkyl groups. The chiral purity and chemical purity of the product produced from the respective phase-transfer catalysts is shown in the Table 3 below:
Table 3
PATENT
WO 2009002809
WO 2009025983
WO 2009126668
WO 2017176948
WO 2018117034
CN 109879826
JP 2020023442
WO 2020158889
WO 2020171129
WO 2021013825
CN 112457267
CN 112679338
PAPER
IP.com Journal (2009), 9(9B), 35.
Afoxolaner (INN)[2] is an insecticide and acaricide that belongs to the isoxazoline chemical compound group.
It acts as an antagonist at ligand-gated chloride channels, in particular those gated by the neurotransmitter gamma-aminobutyric acid (GABA-receptors). Isoxazolines, among the chloride channel modulators, bind to a distinct and unique target site within the insect GABA-gated chloride channels, thereby blocking pre-and post-synaptic transfer of chloride ions across cell membranes. Prolonged afoxolaner-induced hyperexcitation results in uncontrolled activity of the central nervous system and death of insects and acarines.[3]
Marketing
Afoxolaner is the active principle of the veterinary medicinal products NexGard (alone) and Nexgard Spectra (in combination with milbemycin oxime).[4][5][6] They are indicated for the treatment and prevention of flea infestations, and the treatment and control of tick infestations in dogs and puppies (8 weeks of age and older, weighing 4 pounds (~1.8 kilograms) of body weight or greater) for one month.[7] These products are administered orally and poisons fleas once they start feeding.
The marketing authorization was granted by the European Medicines Agency in February 2014, for NexGard and January 2015, for Nexgard Spectra, after only 14[8] and 12[9] months of quality, safety and efficacy assessment performed by the Committee for Medicinal Products for Veterinary Use (CVMP).[10] Therefore, long-term effects are not known.
List of excipients
In NexGard[11] and NexGard Spectra:[3]
- Maize starch
- Soy protein fines
- Beef braised flavouring
- Povidone (E1201)
- Macrogol 400 (reputed laxatives)
- Macrogol 4000 (reputed laxatives)
- Macrogol 15 hydroxystearate (reputed laxatives)
- Glycerol (E422)
- Triglycerides, medium-chain
Additionally in NexGard Spectra:
Safety
Dosage
Afoxolaner is recommended to be administered at a dose of 2.7–7 mg/kg dog’s body weight.[11]
Toxicity for mammals
According to clinical studies performed prior marketing:
- The oral toxicity profile of afoxolaner consists of a diuretic effect (rats only), effects secondary to a reduction in food consumption (rats and rabbits only) and occasional vomiting and/or diarrhoea (dogs, 120 and 200 mg/kg bodyweight (bw)) following high oral doses. No treatment-related effects on vomiting or diarrhoea were noted following oral doses of up to 31.5 mg/kg bw in the pivotal target animal safety study, nor in the EU field trial.[9]
- mild gastrointestinal effects (vomiting, diarrhoea), pruritus, lethargy, anorexia, and neurological signs (convulsions, ataxia and muscle tremors) have been reported in less than 0.1% of 10,000 animals treated, including isolated reports, most reported adverse reactions being self-limiting and of short duration,[11]
- (in combination with milbemycin oxime): vomiting, diarrhoea, lethargy, anorexia, and pruritus were observed in 0.2 to 1% of 10,000 animals treated and were generally self-limiting and of short duration,[3]
- In vitro studies reported that afoxolaner can bind to dopamine and norepinephrine cellular transport receptor systems and the CB1 receptor; inhibition of these catecholaminergic systems and certain types of competitive binding at CB1 receptors may mediate pharmacodynamic effects of diuresis, decreased food consumption, and decreased body weight in animals.[9]
According to post-marketing safety experience:
- (in combination with milbemycin oxime): erythema and neurological signs (convulsions, ataxia and muscle tremors) have been reported in less than 0.1% of 10,000 animals treated, including isolated reports,[3]
- The US FDA reports[12] that some drugs in this class (isoxazolines), including afoxalaner, can have adverse neurologic effects on some dogs, such as muscle tremors, ataxia, and seizures.
- Extralabel use of afoxolaner in a pet pig has been described without any adverse effects.[13] Experimental use in commercial pigs also did not result in any adverse effects.[14]
Selectivity in insects over mammalians
In vivo studies (repeat-dose toxicology in laboratory animals, target animal safety, field studies) provided by MERIAL, the company that produces afoxolaner-derivative medicines, did not show evidence of neurological or behavioural effects suggestive of GABA-mediated perturbations in mammals. The Committee for Medicinal Products for Veterinary Use (CVMP) therefore concluded that binding to dog, rat or human GABA receptors is expected to be low for afoxolaner.[9]
Selectivity for insect over mammalian GABA-receptors has been demonstrated for other isoxazolines.[15] The selectivity might be explained by the number of pharmacological differences that exist between GABA-gated chloride channels of insects and vertebrates.[16]
GEN REF
- Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, Lahm GP, Long JK, Xu M, Wagerle T, Jones GS, Dietrich RF, Cordova D, Schroeder ME, Rhoades DF, Benner EA, Confalone PN: Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs. Vet Parasitol. 2014 Apr 2;201(3-4):179-89. doi: 10.1016/j.vetpar.2014.02.020. Epub 2014 Mar 14. [Article]
References
- ^ Jump up to:a b c “Frontline NexGard (afoxolaner) for the Treatment and Prophylaxis of Ectoparasitic Diseases in Dogs. Full Prescribing Information” (PDF) (in Russian). Sanofi Russia. Retrieved 14 November 2016.
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 70” (PDF). World Health Organization. pp. 276–7. Retrieved 14 November 2016.
- ^ Jump up to:a b c d “NexGard Spectra product information – Annex I “Summary of product characteristics”” (PDF). European Medicines Agency. Retrieved 13 November 2019.
- ^ Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, et al. (April 2014). “Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs”. Veterinary Parasitology. 201 (3–4): 179–89. doi:10.1016/j.vetpar.2014.02.020. PMID 24631502.
- ^ Beugnet F, deVos C, Liebenberg J, Halos L, Fourie J (25 August 2014). “Afoxolaner against fleas: immediate efficacy and resultant mortality after short exposure on dogs”. Parasite. 21: 42. doi:10.1051/parasite/2014045. PMC 4141545. PMID 25148564.
- ^ Beugnet F, Crafford D, de Vos C, Kok D, Larsen D, Fourie J (August 2016). “Evaluation of the efficacy of monthly oral administration of afoxolaner plus milbemycin oxime (NexGard Spectra, Merial) in the prevention of adult Spirocerca lupi establishment in experimentally infected dogs”. Veterinary Parasitology. 226: 150–61. doi:10.1016/j.vetpar.2016.07.002. PMID 27514901.
- ^ “Boehringer-Ingelheim companion-animals-product NexGard (afoxolaner)”. Boehringer Ingelheim International GmbH. Retrieved 13 November 2019.
- ^ “CVMP Assessment Report for NEXGARD SPECTRA(EMEA/V/C/003842/0000)” (PDF). European Medicines Agency. Retrieved 14 November 2019.
- ^ Jump up to:a b c d “CVMP assessment report for NexGard (EMEA/V/C/002729/0000)” (PDF). European Medicines Agency. Retrieved 14 November 2019.
- ^ “Committee for Medicinal Products for Veterinary Use (CVMP) – Section “Role of the CVMP””. European Medicines Agency. Retrieved 14 November 2019.
- ^ Jump up to:a b c “NexGard product information – Annex I “Summary of product characteristics”” (PDF). European Medicines Angency. Retrieved 14 November 2019.
- ^ Medicine, Center for Veterinary. “CVM Updates – Animal Drug Safety Communication: FDA Alerts Pet Owners and Veterinarians About Potential for Neurologic Adverse Events Associated with Certain Flea and Tick Products”. http://www.fda.gov. Retrieved 2018-09-22.
- ^ Smith, Joe S.; Berger, Darren J.; Hoff, Sarah E.; Jesudoss Chelladurai, Jeba R. J.; Martin, Katy A.; Brewer, Matthew T. (2020). “Afoxolaner as a Treatment for a Novel Sarcoptes scabiei Infestation in a Juvenile Potbelly Pig”. Frontiers in Veterinary Science. 7: 473. doi:10.3389/fvets.2020.00473. PMC 7505946. PMID 33102538.
- ^ Bernigaud, C.; Fang, F.; Fischer, K.; Lespine, A.; Aho, L. S.; Mullins, A. J.; Tecle, B.; Kelly, A.; Sutra, J. F.; Moreau, F.; Lilin, T.; Beugnet, F.; Botterel, F.; Chosidow, O.; Guillot, J. (2018). “Efficacy and Pharmacokinetics Evaluation of a Single Oral Dose of Afoxolaner against Sarcoptes scabiei in the Porcine Scabies Model for Human Infestation”. Antimicrobial Agents and Chemotherapy. 62 (9). doi:10.1128/AAC.02334-17. PMC 6125498. PMID 29914951.
- ^ Casida JE (April 2015). “Golden age of RyR and GABA-R diamide and isoxazoline insecticides: common genesis, serendipity, surprises, selectivity, and safety”. Chemical Research in Toxicology. 28 (4): 560–6. doi:10.1021/tx500520w. PMID 25688713.
- ^ Hosie AM, Aronstein K, Sattelle DB, ffrench-Constant RH (December 1997). “Molecular biology of insect neuronal GABA receptors”. Trends in Neurosciences. 20 (12): 578–83. doi:10.1016/S0166-2236(97)01127-2. PMID 9416671. S2CID 5028039.
| Clinical data | |
|---|---|
| Pronunciation | /eɪˌfɒksoʊˈlænər/ ay-FOK-soh-LAN-ər |
| Trade names | NexGard, Frontpro |
| Other names | 4-[(5RS)-5-(5-Chloro-α,α,α-trifluoro-m-tolyl)-4,5-dihydro-5-(trifluoromethyl)-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide |
| License data | US DailyMed: Afoxolaner |
| Routes of administration | By mouth (chewables) |
| ATCvet code | QP53BE01 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyOTC (RU)[1] |
| Pharmacokinetic data | |
| Bioavailability | 74% (Tmax = 2–4 hours)[1] |
| Elimination half-life | 14 hours[1] |
| Excretion | Bile duct (major route) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1093861-60-9 |
| PubChem CID | 25154249 |
| DrugBank | DB11369 |
| ChemSpider | 28651525 |
| UNII | 02L07H6D0U |
| KEGG | D10361 |
| ChEMBL | ChEMBL2219412 |
| CompTox Dashboard (EPA) | DTXSID50148921 |
| Chemical and physical data | |
| Formula | C26H17ClF9N3O3 |
| Molar mass | 625.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI |
{kind=link}
///////////// afoxolaner, A1443, AH252723
FC(F)(F)CNC(=O)CNC(=O)C1=C2C=CC=CC2=C(C=C1)C1=NOC(C1)(C1=CC(=CC(Cl)=C1)C(F)(F)F)C(F)(F)F


NEW DRUG APPROVALS
ONE TIME
$10.00