
Home » Uncategorized (Page 23)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MONENSIN
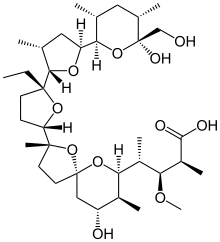
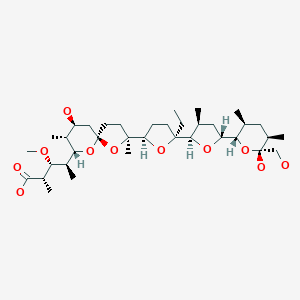
モネンシン;
MONENSIN
- Molecular FormulaC36H62O11
- Average mass670.871 Da
1,6-dioxaspiro[4.5]decane-7-butanoic acid, 2-[(2S,2’R,3’S,5R,5’R)-2-ethyloctahydro-3′-methyl-5′-[(2S,3S,5R,6R)-tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl][2,2′-bifuran]-5-yl]-9-hydroxy-β-methoxy-α,γ,2,8-tetramethyl-, (αS,βR,γS,2S,5R,7S,8R,9S)-
17090-79-8[RN]
241-154-0[EINECS]
(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-{(2S,2’R,3’S,5R,5’R)-2-Ethyl-5′-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyltetrahydro-2H-pyran-2-yl]-3′-methyloctahydro-2,2′-bifuran-5-yl}-9-hydroxy-2,8-di methyl-1,6-dioxaspiro[4.5]dec-7-yl]-3-methoxy-2-methylpentanoic acid
монензин[Russian]
مونانسين[Arabic]
莫能星[Chinese]
Antibiotic, Antifungal, Antiprotozoal

Synonym(s):
Monensin A sodium salt
Empirical Formula (Hill Notation):C36H61NaO11
CAS Number:22373-78-0
Molecular Weight:692.85
Beilstein:4122200
Title: Monensin
CAS Registry Number: 17090-79-8
CAS Name: 2-[5-Ethyltetrahydro-5-[tetrahydro-3-methyl-5-[tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl]-2-furyl]-2-furyl]-9-hydroxy-b-methoxy-a,g,2,8-tetramethyl-1,6-dioxaspiro[4.5]decane-7-butyric acid
Additional Names: monensic acid (obsolete)
Manufacturers’ Codes: A-3823A
Molecular Formula: C36H62O11, Molecular Weight: 670.87
Percent Composition: C 64.45%, H 9.32%, O 26.23%
Literature References: Polyether antibiotic. Major factor in antibiotic complex isolated from Streptomyces cinnamonensis. Discovery and isolation: Haney, Hoehn, Antimicrob. Agents Chemother.1967, 349. Production: Haney, Hoehn, US3501568 (1970 to Lilly). Structure: Agtarap et al.,J. Am. Chem. Soc.89, 5737 (1967). Crystal structure studies: Lutz et al.,Helv. Chim. Acta53, 1732 (1970); ibid.54, 1103 (1971). Fermentation studies: Stark et al.,Antimicrob. Agents Chemother.1967, 353. Chemistry: Agtarap, Chamberlin, ibid. 359. Stereocontrolled total synthesis: T. Fukuyama et al.,J. Am. Chem. Soc.101, 262 (1979); D. B. Collum et al.,ibid.102, 2117, 2118, 2120 (1980). 13C-NMR study: J. A. Robinson, D. L. Turner, Chem. Commun.1982, 148. Biosynthesis: Day et al.,Antimicrob. Agents Chemother.4, 410 (1973). Review: Stark, “Monensin, A New Biologically Active Compound Produced by a Fermentation Process”, in Fermentation Advances, Pap. Int. Ferment. Symp., 3rd, 1968, D. Perlman, Ed. (Academic Press, New York, 1969) pp 517-540.
Properties: Crystals, mp 103-105° (monohydrate). [a]D +47.7°. pKa 6.6 (in 66% DMF). Very stable under alkaline conditions. Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents. LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn).
Melting point: mp 103-105° (monohydrate)
pKa: pKa 6.6 (in 66% DMF)
Optical Rotation: [a]D +47.7°
Toxicity data: LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn)
Derivative Type: Sodium salt
Trademarks: Coban (Elanco); Romensin (Elanco); Rumensin (Elanco)
Molecular Formula: C36H61NaO11, Molecular Weight: 692.85
Percent Composition: C 62.41%, H 8.87%, Na 3.32%, O 25.40%
Properties: mp 267-269°. [a]D +57.3° (methanol). Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents.
Melting point: mp 267-269°
Optical Rotation: [a]D +57.3° (methanol)
Therap-Cat-Vet: Coccidiostat. Feed additive to improve feed efficiency in ruminants.
Monensin is a polyether antibiotic isolated from Streptomyces cinnamonensis.[1] It is widely used in ruminant animal feeds.[1][2]
The structure of monensin was first described by Agtarap et al. in 1967, and was the first polyether antibiotic to have its structure elucidated in this way. The first total synthesis of monensin was reported in 1979 by Kishi et al.[3]
SYN

SYN
Production / synthesis Monensin is produced in vivo by Streptomyces cinnamonensis as a natural defense against competing bacteria. Monensin presents a formidable challenge to synthetic chemists as it possesses 17 asymmetric centers on a backbone of only 26 carbon atoms. Although its total synthesis has been described (e.g., Kishi et al., 1979), the high complexity of monensin makes an extraction from the bacterium the most economical procedure for its production. The total synthesis has 56 steps and a yield of only 0.26%. The chemical precursors are 2-allyl-1,3-propanediol and 2- (furan-2-yl)acetonitrile. The method used for synthesizing monensin is based on the principle of “absolute asymmetric synthesis”. Molecules are constructed out of prefabricated building blocks in the correct conformation, aiming for higher yields of the desired enantiomer. New stereocenters are also introduced. Using this method, monensin is assembled in two parts, a larger right side and a smaller left one. The penultimate step is connecting the left and the right halves of monensin, which are independently generated, in an Aldol-condensation. The two halves’ keto end groups (C7/ C8) are linked by eliminating a water molecule. The C7 atom is favored over the C1 atom, because it is more reactive. For catalyzing this step, Yoshito Kishi’s group used iPr2NMgBr (Hauser base) and THF to coordinate it at a temperature of − 78°C. Thus, they were able to isolate the molecule in the right conformation at a ratio of 8:1. Due to the low temperature required for a high yield of the correct enantiomer, the reaction is very solw. One of the most difficult steps is the last one: the connection of the spiro center. This is due to a characteristic feature of spiro compounds; they open and close very easily. Therefore, the conditions for forming the right conformation must be optimal in the last step of synthesis. The biosynthesis in a cell culture of Streptomyces cinnamonensis involves a complex medium containing, among other components, glucose, soybean oil, and grit. Cultivation is carried out for a week at a temperature of 30°C and under constant aeration. Product isolation requires filtration, acidification to pH3, extraction with chloroform and purification with activated carbon. In this way, a few grams per liter of monensin are produced and isolated. For crystallization, azeotropic distillation is necessary. In vivo, polyether backbones are assembled by modular polyketide synthases and are modified by two key enzymes, epoxidase and epoxide hydrolase, to generate the product. Precursors of the polyketide pathway are acetate, butyrate and propionate.
SYN
The final-stage aldol addition in Yoshito Kishi‘s 1979 total synthesis of monensin. (1979). “Synthetic studies on polyether antibiotics. 6. Total synthesis of monensin. 3. Stereocontrolled total synthesis of monensin”. J. Am. Chem. Soc. 101 (1): 262–263. DOI:10.1021/ja00495a066.

SYN
A polyether antibiotic, Monensin was the first member of this class of molecules to be structurally characterized.1 The structural features of these polyethers comprise of a terminal carboxylic acid, multiple cyclic ether rings (ex. Tetrahydrofuran and tetrahydropyran), a large amount of stereocenters and (for many of these molecules) one or more spiroketal moieties.2 Monensin was introduced into the market in 1971 and is used to fight coccidial infections in poultry and as an additive in cattle feed.3 Of the 26 carbon atom’s in Monensin’s backbone, 17 are stereogenic and six of those are contiguous. Coupled with a spiroketal moiety, three hydrofuran rings and two hydropyran rings, the molecule was an attractive synthetic target.
1. Agtarap, A.; Chamberlain, J.W.; Pinkerton, M.; Stein-rauf, L. J. Am. Chem. Soc. 1967, 89, 5737 2. Polyether Antibiotics : Naturally Occurring Acid Ionophores. Westley J.W.; Marcel Dekker: New York (1982) Vol. 1-2. 3. Stark, W.M. In Fermentation Advances, Perlman, D., Ed., Academic Press: New York, 1969, 517
Retrosynthetic Analysis of Monensin
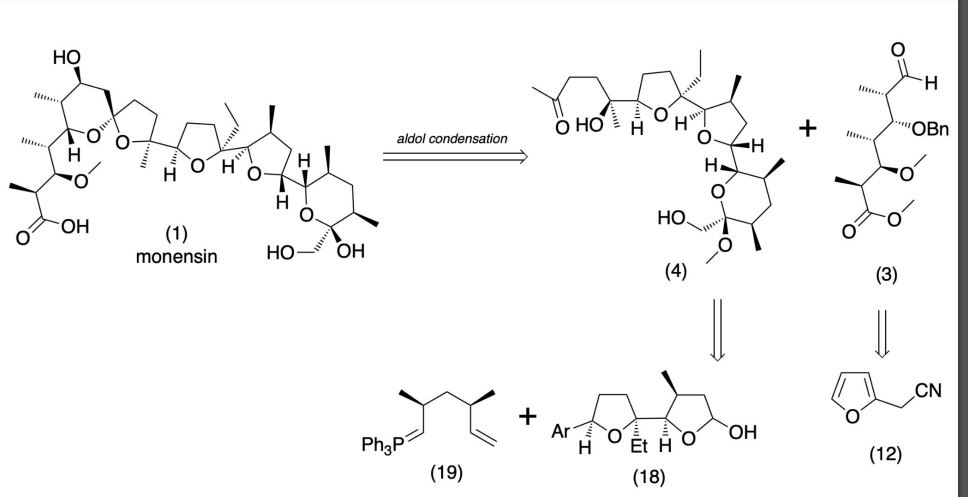
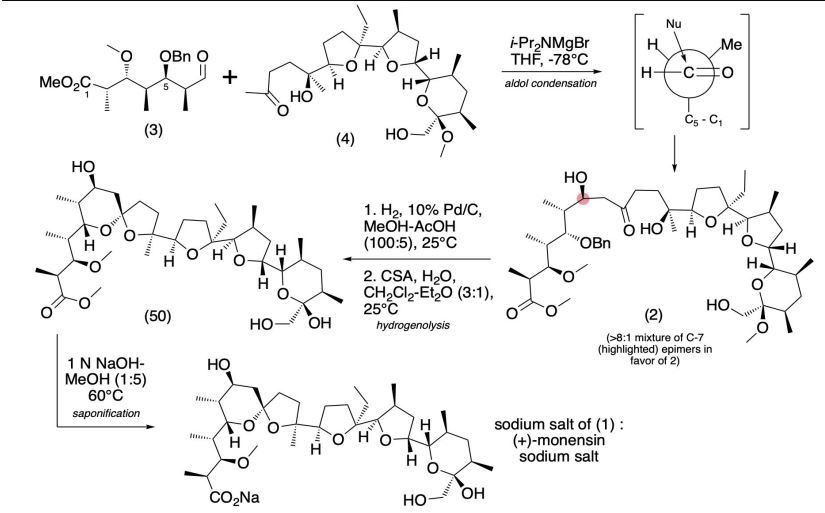
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Mechanism of action

The structure of the sodium (Na+) complex of monensin A.
Monensin A is an ionophore related to the crown ethers with a preference to form complexes with monovalent cations such as: Li+, Na+, K+, Rb+, Ag+, and Tl+.[4][5] Monensin A is able to transport these cations across lipid membranes of cells in an electroneutral (i.e. non-depolarizing) exchange, playing an important role as an Na+/H+ antiporter. Recent studies have shown that monensin may transport sodium ion through the membrane in both electrogenic and electroneutral manner.[6] This approach explains ionophoric ability and in consequence antibacterial properties of not only parental monensin, but also its derivatives that do not possess carboxylic groups. It blocks intracellular protein transport, and exhibits antibiotic, antimalarial, and other biological activities.[7] The antibacterial properties of monensin and its derivatives are a result of their ability to transport metal cations through cellular and subcellular membranes.[8]
Uses
Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, increase the production of propionic acid and prevent bloat.[9] Furthermore, monensin, but also its derivatives monensin methyl ester (MME), and particularly monensin decyl ester (MDE) are widely used in ion-selective electrodes.[10][11][12]
In laboratory research, monensin is used extensively to block Golgi transport.[13][14][15]
Toxicity
Monensin has some degree of activity on mammalian cells and thus toxicity is common. This is especially pronounced in horses, where monensin has a median lethal dose 1/100th that of ruminants. Accidental poisoning of equines with monensin is a well-documented occurrence which has resulted in deaths.[16]
References
- ^ Jump up to:a b Daniel Łowicki and Adam Huczyński (2013). “Structure and Antimicrobial Properties of Monensin A and Its Derivatives: Summary of the Achievements”. BioMed Research International. 2013: 1–14. doi:10.1155/2013/742149. PMC 3586448. PMID 23509771.
- ^ Butaye, P.; Devriese, L. A.; Haesebrouck, F. (2003). “Antimicrobial Growth Promoters Used in Animal Feed: Effects of Less Well Known Antibiotics on Gram-Positive Bacteria”. Clinical Microbiology Reviews. 16 (2): 175–188. doi:10.1128/CMR.16.2.175-188.2003. PMC 153145. PMID 12692092.
- ^ Nicolaou, K. C.; E. J. Sorensen (1996). Classics in Total Synthesis. Weinheim, Germany: VCH. pp. 185–187. ISBN 3-527-29284-5.
- ^ Huczyński, A.; Ratajczak-Sitarz, M.; Katrusiak, A.; Brzezinski, B. (2007). “Molecular structure of the 1:1 inclusion complex of Monensin A lithium salt with acetonitrile”. J. Mol. Struct. 871 (1–3): 92–97. Bibcode:2007JMoSt.871…92H. doi:10.1016/j.molstruc.2006.07.046.
- ^ Pinkerton, M.; Steinrauf, L. K. (1970). “Molecular structure of monovalent metal cation complexes of monensin”. J. Mol. Biol. 49 (3): 533–546. doi:10.1016/0022-2836(70)90279-2. PMID 5453344.
- ^ Huczyński, Adam; Jan Janczak; Daniel Łowicki; Bogumil Brzezinski (2012). “Monensin A acid complexes as a model of electrogenic transport of sodium cation”. Biochim. Biophys. Acta. 1818 (9): 2108–2119. doi:10.1016/j.bbamem.2012.04.017. PMID 22564680.
- ^ Mollenhauer, H. H.; Morre, D. J.; Rowe, L. D. (1990). “Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity”. Biochim. Biophys. Acta. 1031 (2): 225–246. doi:10.1016/0304-4157(90)90008-Z. PMC 7148783. PMID 2160275.
- ^ Huczyński, A.; Stefańska, J.; Przybylski, P.; Brzezinski, B.; Bartl, F. (2008). “Synthesis and antimicrobial properties of Monensin A esters”. Bioorg. Med. Chem. Lett. 18 (8): 2585–2589. doi:10.1016/j.bmcl.2008.03.038. PMID 18375122.
- ^ Matsuoka, T.; Novilla, M.N.; Thomson, T.D.; Donoho, A.L. (1996). “Review of monensin toxicosis in horses”. Journal of Equine Veterinary Science. 16: 8–15. doi:10.1016/S0737-0806(96)80059-1.
- ^ Tohda, Koji; Suzuki, Koji; Kosuge, Nobutaka; Nagashima, Hitoshi; Watanabe, Kazuhiko; Inoue, Hidenari; Shirai, Tsuneo (1990). “A sodium ion selective electrode based on a highly lipophilic monensin derivative and its application to the measurement of sodium ion concentrations in serum”. Analytical Sciences. 6 (2): 227–232. doi:10.2116/analsci.6.227.
- ^ Kim, N.; Park, K.; Park, I.; Cho, Y.; Bae, Y. (2005). “Application of a taste evaluation system to the monitoring of Kimchi fermentation”. Biosensors and Bioelectronics. 20 (11): 2283–2291. doi:10.1016/j.bios.2004.10.007. PMID 15797327.
- ^ Toko, K. (2000). “Taste Sensor”. Sensors and Actuators B: Chemical. 64 (1–3): 205–215. doi:10.1016/S0925-4005(99)00508-0.
- ^ Griffiths, G.; Quinn, P.; Warren, G. (March 1983). “Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki Forest virus”. The Journal of Cell Biology. 96 (3): 835–850. doi:10.1083/jcb.96.3.835. ISSN 0021-9525. PMC 2112386. PMID 6682112.
- ^ Kallen, K. J.; Quinn, P.; Allan, D. (1993-02-24). “Monensin inhibits synthesis of plasma membrane sphingomyelin by blocking transport of ceramide through the Golgi: evidence for two sites of sphingomyelin synthesis in BHK cells”. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 1166 (2–3): 305–308. doi:10.1016/0005-2760(93)90111-l. ISSN 0006-3002. PMID 8443249.
- ^ Zhang, G. F.; Driouich, A.; Staehelin, L. A. (December 1996). “Monensin-induced redistribution of enzymes and products from Golgi stacks to swollen vesicles in plant cells”. European Journal of Cell Biology. 71 (4): 332–340. ISSN 0171-9335. PMID 8980903.
- ^ “Tainted feed blamed for 4 horse deaths at Florida stable”. 2014-12-16.
| Names | |
|---|---|
| Preferred IUPAC name(2S,3R,4S)-4-[(2S,5R,7S,8R,9S)-2-{(2S,2′R,3′S,5R,5′R)-2-Ethyl-5′-[(2S,3S,5R,6R)-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyloxan-2-yl]-3′-methyl[2,2′-bioxolan]-5-yl}-9-hydroxy-2,8-dimethyl-1,6-dioxaspiro[4.5]decan-7-yl]-3-methoxy-2-methylpentanoic acid | |
| Other namesMonensic acid | |
| Identifiers | |
| CAS Number | 17090-79-8 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:27617 |
| ChEMBL | ChEMBL256105 |
| ChemSpider | 389937 |
| ECHA InfoCard | 100.037.398 |
| E number | E714 (antibiotics) |
| KEGG | D08228 |
| PubChemCID | 441145 |
| UNII | 906O0YJ6ZP |
| CompTox Dashboard (EPA) | DTXSID4048561 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C36H62O11 |
| Molar mass | 670.871 g/mol |
| Appearance | solid state, white crystals |
| Melting point | 104 °C (219 °F; 377 K) |
| Solubility in water | 3×10−6 g/dm3 (20 °C) |
| Solubility | ethanol, acetone, diethyl ether, benzene |
| Pharmacology | |
| ATCvet code | QA16QA06 (WHO) QP51AH03 (WHO) |
| Related compounds | |
| Related | antibiotics, ionophores |
| Related compounds | Monensin A methyl ester, |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
///////////MONENSIN, Elancoban, VETERINARY, Coccidiostat, A-3823A, A 3823A

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN THIS BLOG SUBSCRIPTION AND KEEP GOING
$10.00
Ferric derisomaltose

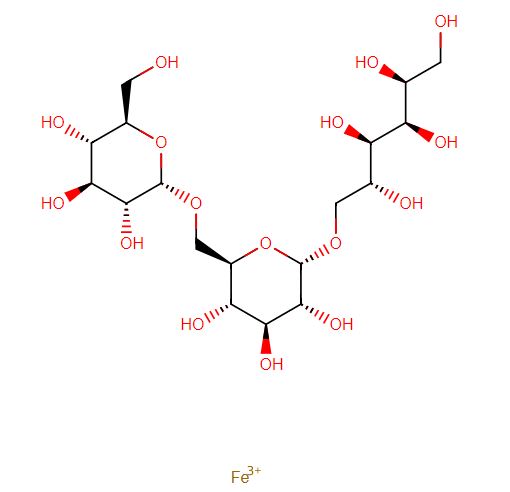
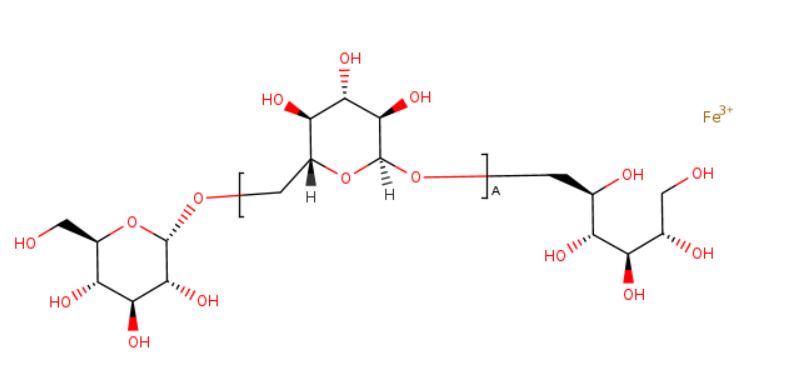
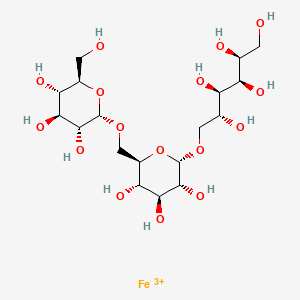
Ferric derisomaltose
WeightAverage: 562.297
Monoisotopic: 562.117975Chemical FormulaC18H34FeO16
Monover, JAPAN 2022, 2022/3/28
Monoferric (TN);
Monover (TN)
Anti-anemic, Hematinic, Supplement (iron)
CAS 1345510-43-1
デルイソマルトース第二鉄
- NS32
- WHO 9712
- UNII-AHU547PI9H
| Originator Company | Pharmacosmos |
| Active Companies | Nippon Shinyaku Co Ltd;Pharmacosmos A/S;Wasserburger Arzneimittelwerk Gmbh;Zealand University Hospital |
iron(3+) (2S,3R,4R,5R)-6-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-({[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}methyl)oxan-2-yl]oxy}hexane-1,2,3,4,5-pentol
- alpha-D-Glucan, (1-6)-, reduced, reaction products with iron hydroxide (Fe(OH)3)
Ferric derisomaltose is an iron injection used in the treatment of iron deficiency anemia.
Ferric derisomaltose, sold under the brand name Monoferric, is a medication for the treatment of iron deficiency anemia (IDA) in adults who have intolerance to oral iron or have had unsatisfactory response to oral iron or who have non-hemodialysis dependent chronic kidney disease (NDD-CKD).[1] It was approved for use in the United States in January 2020.[1][2][3] It is given intravenously.[1]
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life.3
Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020.8,9 Clinical trials show that it is non-inferior to iron sucrose, another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.1,4
This drug is indicated for the treatment of iron deficiency anemia in adult patients who have experienced intolerance to oral iron preparations or insufficient clinical response to orally administered iron. Ferric derisomaltase is also indicated for patients with non-hemodialysis dependent chronic kidney disease.8 In Australia and United Kingdom, ferric derisomaltase is indicated for cases in which rapid delivery of iron is required.10,11
Iron deficiency is an extremely common condition and is the most frequent cause of anemia worldwide. Iron deficiency results when iron intake, iron stores, and loss of iron from the body do not adequately support production of erythrocytes, also known as red blood cells. Though it is generally considered non life-threatening, iron deficiency may considerably affect quality of life. Ferric derisomaltose is a form of iron used in the treatment of iron deficiency. This drug is a complex of iron (III) hydroxide and derisomaltose. The latter is an iron carbohydrate oligosaccharide that works to release iron. Ferric derisomaltose was developed by Pharmacosmos Therapeutics ad was granted FDA approval in January 2020. Clinical trials show that it is non-inferior to [iron sucrose], another form of iron that is often administered in iron deficiency, and less likely to cause serious hypersensitivity that is associated with other forms of injectable iron.
Monoferric is an iron replacement product containing ferric derisomaltose for intravenous infusion. Ferric derisomaltose is an iron carbohydrate complex with a matrix structure composed of interchanging layers of ferric hydroxide and the carbohydrate derisomaltose. Derisomaltose consists of linear, hydrogenated isomaltooligosaccharides with an average molecular weight of 1000 Da and a narrow molecular weight distribution that is almost devoid of mono-and disaccharides.
Ferric derisomaltose has an average molecular weight of 155,000 Da and has the following empirical formula:
{FeO(1-3X) (OH)(1+3X) (C6H5O73-)X}, (H20)T, –
(C6H10O6)R(-C6H10O5-)Z(C6H13O5)R, (NaCl)Y
X= 0.0311; T = 0.25; R = 0.14; Z = 0.49; Y = 0.14
Iron atoms placed in the electronegative cavities of the 3-D structure between and within the derisomaltose molecules. A schematic representation is presented below
 |
Monoferric is a sterile, dark brown, non-transparent aqueous solution with pH 5.0-7.0, containing ferric derisomaltose dissolved in water for injections and filled into Type I glass vials.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
| Mkt. Status | Active Ingredient | Proprietary Name | Appl. No. | Dosage Form | Route | Strength | TE Code | RLD | RS | Applicant Holder |
|---|---|---|---|---|---|---|---|---|---|---|
| RX | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 1GM/10ML (100MG/ML) | RLD | RS | PHARMACOSMOS AS | |
| DISCN | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 100MG/ML (100MG/ML) | RLD | PHARMACOSMOS AS | ||
| DISCN | FERRIC DERISOMALTOSE | MONOFERRIC | N208171 | SOLUTION | INTRAVENOUS | 500MG/5ML (100MG/ML) | RLD | PHARMACOSMOS AS | ||
| Mkt. Status | Active Ingredient | Proprietary Name | Appl. No. | Dosage Form | Route | Strength | TE Code | >RLD | RS | Applicant Holder |
MONOFERRIC (FERRIC DERISOMALTOSE)
1GM/10ML (100MG/ML)
Marketing Status: Prescription
Active Ingredient: FERRIC DERISOMALTOSE
Proprietary Name: MONOFERRIC
Dosage Form; Route of Administration: SOLUTION; INTRAVENOUS
Strength: 1GM/10ML (100MG/ML)
Reference Listed Drug: Yes
Reference Standard: Yes
TE Code:
Application Number: N208171
Product Number: 003
Approval Date: Jan 16, 2020
Applicant Holder Full Name: PHARMACOSMOS AS
Marketing Status: Prescription
Patent and Exclusivity Information
Patent and Exclusivity for: N208171
Product 003
FERRIC DERISOMALTOSE (MONOFERRIC) SOLUTION 1GM/10ML (100MG/ML)
Patent Data
| Product No | Patent No | Patent Expiration | Drug Substance | Drug Product | Patent Use Code | Delist Requested | Submission Date |
|---|---|---|---|---|---|---|---|
| 003 | 8815301 | 08/14/2029 | DS | DP | U-2734 | 02/14/2020 | |
| 003 | 10414831 | 03/25/2029 | DS | DP | 02/14/2020 |
PATENT
AU2009342799B2
US10414831B2
US2012010166A1
US2014303364A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d “Monoferric- ferric derisomaltose solution”. DailyMed. 24 January 2020. Retrieved 16 February 2020.
- ^ “Monoferric approval letter” (PDF). U.S. Food and Drug Administration (FDA). 16 January 2020. Retrieved 16 February 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Monoferric Injection”. U.S. Food and Drug Administration (FDA). 7 May 2020. Retrieved 13 August 2020.
External links
- “Ferric derisomaltose”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Monoferric |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Ferric_derisomaltose |
| Routes of administration | Intravenous (IV) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1345510-43-1 |
| PubChem CID | 86278348 |
| DrugBank | DB15617 |
| UNII | AHU547PI9H |
| KEGG | D11808 |
| Chemical and physical data | |
| Formula | C18H34FeO16+3 |
| Molar mass | 562.299 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////////Ferric derisomaltose, デルイソマルトース第二鉄 , APPROVALS 2022, JAPAN 2022, NS32, WHO 9712
[Fe+3].OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO[C@H]1O[C@H](CO[C@H]2O[C@H](CO)[C@@H](O)[C@H](O)[C@H]2O)[C@@H](O)[C@H](O)[C@H]1O

NEW DRUG APPROVALS
one time
$10.00
Difamilast
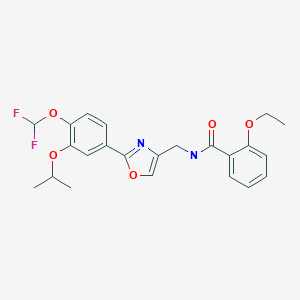
{kind=link}
Difamilast
FDA 2026, APPROVALS 2026, difamilast, Adquey, 2/12/2026, To treat mild to moderate atopic dermatitis
PMDA Moizerto, JAPAN APPROVED 2021/9/27
ジファミラスト
ディファミラスト;
地法米司特
N-({2-[4-(difluoromethoxy)-3-(propan-2-yloxy)phenyl]-1,3- oxazol-4-yl}methyl)-2-ethoxybenzamide
OPA-15406
| Formula |
C23H24F2N2O5
|
|---|---|
| CAS |
937782-05-3
|
| Mol weight |
446.4439
|
MM 36; MM-36-Medimetriks-Pharmaceuticals; Moizerto; OPA-15406
| Efficacy |
Anti-inflammatory, Phosphodiesterase IV inhibitor
|
|---|---|
| Comment |
Treatment of atopic dermatitis
|
OriginatorOtsuka Pharmaceutical Development & Commercialization- DeveloperMedimetriks Pharmaceuticals; Otsuka Pharmaceutical Development & Commercialization
- ClassBenzamides; Nonsteroidal anti-inflammatories; Oxazoles; Skin disorder therapies
- Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
- RegisteredAtopic dermatitis
- 27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
- 11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
- 28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)

Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211&_cid=P10-MLOK7B-25338-2
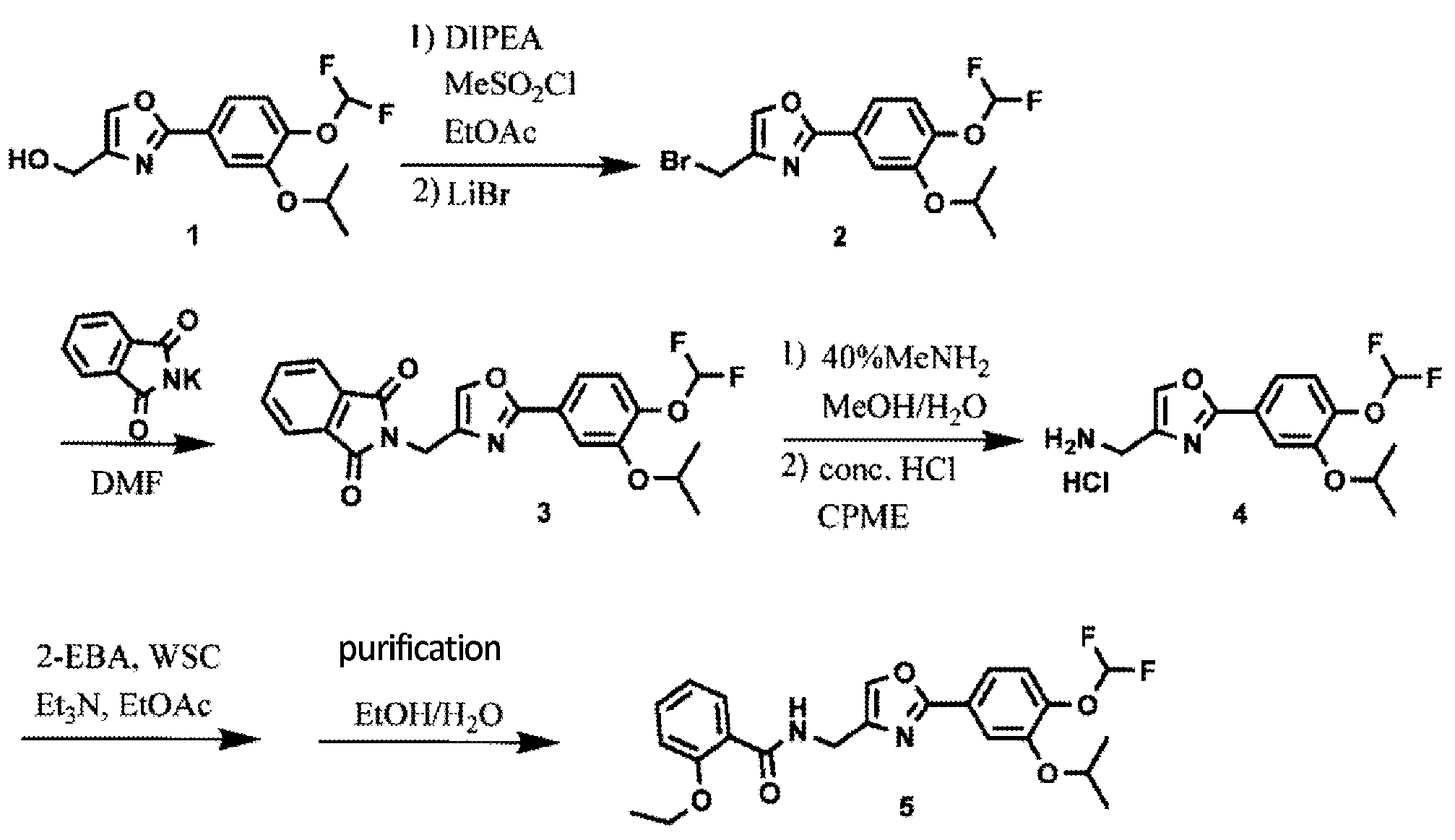
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
SYN
https://www.chemicalbook.com/article/how-is-difamilast-synthesised.htm
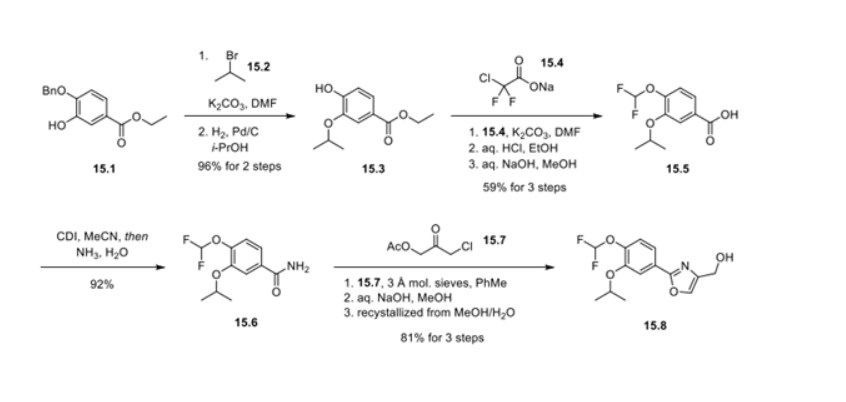
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
PATENT
JP 2021059538
https://patentscope.wipo.int/search/en/detail.jsf?docId=JP322244172&_cid=P20-L1WXG6-04592-1
patcit 2 : International Publication No. 2014/034958 (Japanese Publication No. 2015-528433 )
patcit 3 : International Publication No. 2017/115780
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
N−({2−[4−(difluoromethoxy)−3−isopropoxyphenyl]oxazol−4−yl}methyl)−2−ethoxybenzamide
: white powder.
1H NMR (400 MHz, CDCl3): δ = 8.56 (br s,
1H, NH), 8.23 (dd, J = 7.6 Hz, 1.6 Hz, 1H, ArH), 7.66 (s, 1H, ArH), 7.63 (d, J = 2.0 Hz, 1H, ArH), 7.58 (dd, J = 8.4 Hz, 2.0 Hz, 1H, ArH), 7.44−7.39 (m, 1H, ArH), 7.21 (d, J = 8.0 Hz, 1H, ArH), 7.08−7.04 (m, 1H, ArH), 6.94 (d, J = 8.0 Hz, 1H, ArH), 6.61 (t, J = 75.2 Hz, 1H, CHF 2), 4.68 (sept, J = 6.0 Hz, 1H, CH), 4.62
(d, J = 6.0 Hz, 2H, CH 2), 4.17 (q, J = 6.93, 2H, CH 2), 1.48 (t, J = 7.2 Hz, 3H,
CH 3), 1.39 (d, J = 5.6 Hz, 6H, 2CH 3).
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007058338&_cid=P10-MLOK7P-25609-1
Example 352
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
1H-NMR (CDCl3) δ: 8.57 (1H, br s), 8.24 (1H, dd, J = 7.5, 1.8 Hz), 7.67 (1H, s), 7.65-7.57 (2H, m), 7.46-7.40 (1H, m), 7.26-7.21 (1H, m), 7.08 (1H, t, J = 7.5 Hz), 6.95 (1H, d, J = 8.4 Hz), 6.63 (1H, t, J = 75 Hz), 4.74-4.62 (3H, m), 4.19 (2H, q, J = 6.9 Hz), 1.49 (3H, t, J = 6.9 Hz), 1.40 (6H, d, J = 6.3 Hz)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017115780
Compound (3) was produced in accordance with the following reaction scheme.
1H-NMR (CDCl 3) δ: 7.70 (2H,dd,J = 6.4 Hz,2.0 Hz),7.22 (1H,d,J = 9.2 Hz),6.66 (1H,t,J = 74.8 Hz),4.66(1H,sept,J = 6.0 Hz),1.39 (6H,d,J = 6.0 Hz).
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
Compound (7) was produced in accordance with the following reaction scheme.
Compound (11) was produced in accordance with the following reaction scheme.
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019194211
*DIPEA: Diisopropylethylamine, CPME: Cyclopentyl methyl ether,
DMF: N,N-dimethylformamide, 2-EBA: 2-Ethoxybenzoic acid,
WSC: 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Difamilast, JAPAN 2021, APPROVALS 2021, ジファミラスト , MM 36, MM-36-Medimetriks-Pharmaceuticals, Moizerto, OPA-15406, OPA 15406, 地法米司特
O=C(NCC1=COC(C2=CC=C(OC(F)F)C(OC(C)C)=C2)=N1)C3=CC=CC=C3OCC
INTrmediate No.CAS No.DIFAM-001177429-27-5DIFAM-00293652-48-3DIFAM-0031574285-26-9DIFAM-00470-23-5DIFAM-0051574285-28-1DIFAM-0061574285-30-5DIFAM-0071574285-32-7DIFAM-0081574285-36-1DIFAM-0091574285-38-3DIFAM-010DIFAM-0111574285-40-7DIFAM-0121574285-43-0DIFAM-013134-11-2Difamilast937782-05-3
TOLDIMFOS SODIUM
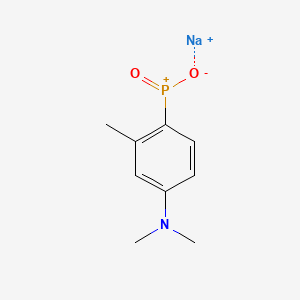

TOLDIMFOS SODIUM
C9H12NNaO2P+ , 220.16
Toldimfos sodium
575-75-7
Sodium (4-(dimethylamino)-2-methylphenyl)phosphinate
UNII-6139240O1E
sodium;[4-(dimethylamino)-2-methylphenyl]-oxido-oxophosphanium
Sodium (4-(dimethylamino)-2-methylphenyl)phosphinate
sodium;[4-(dimethylamino)-2-methylphenyl]-oxido-oxophosphanium
Phosphinic acid, [4-(dimethylamino)-2-methylphenyl]-, sodium salt
Phosphinic acid, (4-(dimethylamino)-2-methylphenyl)-, sodium salt
Phosphinic acid, P-(4-(dimethylamino)-2-methylphenyl)-, sodium salt (1:1)
Toldimfos is an aromatic phosphorus compound which falls between phosphorous itself and phosphoric acid in the stages of oxidation. Toldimfos sodium is the sodium salt of 2- methyl-4-(dimethylamino)phenylphosphinic acid. It is used to treat and prevent diseases associated with parturition and peri-partum period, developmental and nutritional disorders in young animals, and bone growth disorders and tetany or paresis caused by calcium, magnesium, and phosphorus metabolism disorders. Toldimfos has been used as a human medicine since 1920. While it is no longer indicated for human use, it is used in horses, cattle, sheep, pigs, and goats, and administered by intravenous, intramuscular, or subcutaneous injection. No specific data on the pharmacodynamic action of toldimfos was submitted. The precise mode of action of toldimfos is unknown and it is questionable whether the effect of toldimfos is simply a matter of the substitution of deficient phosphorus. It appears more likely that the effect of toldimfos arises due to multiple stimulation of metabolism with the body.
Toldimfos sodium trihydrate
5787-63-3
Toldimfos [INN:BAN]
57808-64-7
Toldimfos Sodium
CAS Registry Number: 575-75-7
CAS Name: (4-Dimethylamino-o-tolyl)phosphonous acid sodium salt
Additional Names: sodium (4-dimethylamino-o-tolyl)phosphonate; p-dimethylamino-o-toluenephosphonous acid sodium salt
Trademarks: Foston (Hoechst); Tonofosfan (Hoechst)
Molecular Formula: C9H13NNaO2P, Molecular Weight: 221.17
Percent Composition: C 48.87%, H 5.92%, N 6.33%, Na 10.39%, O 14.47%, P 14.00%
Literature References: Prepd from N,N-dimethyl-m-toluidine and phosphorus trichloride: Benda, Schmidt, DE397813 (1924 to Cassella), Frdl.14, 1409.
Derivative Type: Trihydrate
CAS Registry Number: 5787-63-3
Properties: Scales, needles, or prisms from alc. Freely sol in cold water, hot alcohol.
Therap-Cat-Vet: Phosphorus source.
SYN

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011026571
PATENT
https://patents.google.com/patent/CN103830261A/en
China’s animal husbandry fast development is the important motivity that promotes China’s agricultural and rural economy development, improves farmers’ income.The disease relevant with Nutrition and Metabolism of serious harm animal health is in rising trend in recent years, the direct economic loss that raising poultry nutritive metabolic disease causes over ten billion to China’s animal husbandry every year, and indirect economic loss is difficult to estimate.
Due to the life-time service of chemicals, will cause some poultrys, poultry product drug residue is serious, this is harm humans healthy not only, also affecting the export of farm produce earns foreign exchange, therefore, tackle this problem, research and development are efficient, the new Nutrition and Metabolism medicine of low toxicity, wide spectrum will have huge market.
Toldimfos (Toldimfos Sodium) belongs to the nutritional supplementation medicine of phosphorus supplement, can be used as benzenephosphonic acid (Phenylphosphinicacid, BPA) succedaneum uses, can be used for treating the disease relevant with childbirth and perinatal stage of the food animals such as horse, cattle, pig, sheep, the diseases such as the bone lengthening obstacle being caused by calcium, magnesium, phosphorus metabolism obstacle.
Toldimfos has higher water solublity, mainly excretes through urine rapidly with the former medicine form of not metabolism in animal body, and the half-life is short, can in tissue, not accumulate.
Toldimfos is developed by German Hoechst company, the symptom such as since nineteen twenty, once physical weakness, chronic stress, depression, mental muscle power postoperative for human treatment, that catch was overtired.Now be not used in the mankind, be mainly used in animal.Its commodity are called onofosfan.
In sum, toldimfos, as a kind of nutritional supplementation medicine of new and effective noresidue, has wide market prospect in China.The development of this product will be made outstanding contributions to the sound development of China’s animal husbandry, remarkable economic and social benefits with application.
PATENT
https://patents.google.com/patent/WO2004003198A1/ja
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
//////////TOLDIMFOS SODIUM, HOECHST, Foston, Tonofosfan,
O.O.O.[Na+].CN(C)c1ccc(c(C)c1)P(=O)[O-]
NEW DRUG APPROVALS
ONE TIME
$10.00
CRENOLANIB

Crenolanib
- Molecular FormulaC26H29N5O2
- Average mass443.541 Da
1-(2-{5-[(3-Methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl}-8-quinolinyl)-4-piperidinamine
1-(2-{5-[(3-methyloxetan-3-yl)methoxy]-1H-benzimidazol-1-yl}quinolin-8-yl)piperidin-4-amine
1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-4-piperidinamine
4-Piperidinamine, 1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-
670220-88-9[RN]
9459
UNII-LQF7I567TQ
креноланиб
كرينولانيب
克拉尼布
CP-868,596-26 or AR-868,596-26
Crenolanib besylate
CAS#: 670220-93-6 (besylate)
Chemical Formula: C32H35N5O5S
Molecular Weight: 601.72
Crenolanib besylate (CP-868,596-26 or AR-868,596-26, 4-piperidinamine, 1-[2-[5-[(3-Methyl-3-oxetanyl) methoxy]-1H-benzimidazol-1-yl]- 8-quinolinyl]-, monobenzenesulfonate) is an investigational inhibitor being developed by AROG Pharmaceuticals, LLC. The compound is currently being evaluated for safety and efficacy in clinical trials for various types of cancer, including acute myeloid leukemia (AML),[1][2] gastrointestinal stromal tumor (GIST),[3] and glioma.[4] Crenolanib is an orally bioavailable benzamidazole that selectively and potently inhibits signaling of wild-type and mutant isoforms of class III receptor tyrosine kinases (RTK) FLT3 (FMS-like Tyrosine Kinase 3), PDGFR α (Platelet-Derived Growth Factor Receptor), and PDGFR β. Unlike most RTK inhibitors, crenolanib is a type I mutant-specific inhibitor that preferentially binds to phosphorylated active kinases with the ‘DFG in’ conformation motif.[5]
CN 109678849

PATENT
WO/2022/060421CRENOLANIB FOR TREATING TRK KINASE ASSOCIATED PROLIFERATIVE DISORDERS
PATENT
WO/2022/060422CRENOLANIB FOR TREATING PAIN
PAPER
https://www.nature.com/articles/s41598-018-21839-3
PAPER
Chembiochem : a European journal of chemical biology (2019), 20(14), 1783-1788.
PATENT
CN 109678849
PATENT
WO 2018118598
https://patents.google.com/patent/WO2018118598A1/en
PAT
US 20170121321
PAT
CN 107382984
https://patents.google.com/patent/CN107382984A/en
Embodiment is as follows:
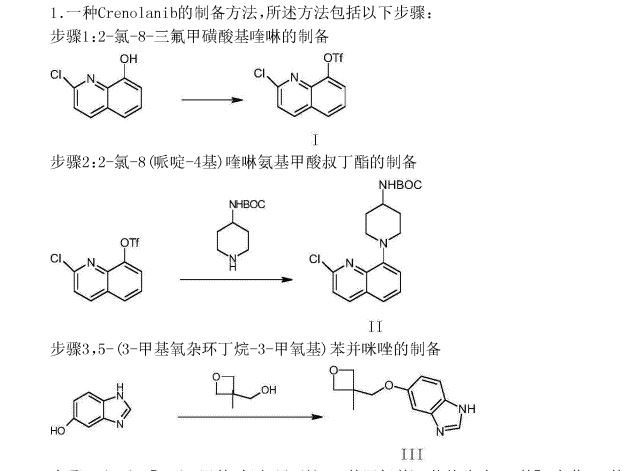
The synthesis of the chloro- 8- trifluoromethanesulfonic acids base quinoline (Ι) of 2-
Compound 2- chloro-8-hydroxyquinolines 50g, DMF150ml, trifluoromethanesulfchloride chloride 53g, triethylamine 25g are added to 250ml In three-necked bottle, stir.Temperature control reacts 20~30h at 25~30 DEG C.After reaction completely, the solid of precipitation is filtered, filter cake is used Wash washing, 40 DEG C of forced air dryings, the chloro- 8- trifluoromethanesulfonic acids base benzimidazoles of gained off-white powder 2- in n-hexane 20ml × 3 83.39g yield 95.78%.
The synthesis of (base of piperidines -4) the quinoline t-butyl carbamates of 2- chloro- 8 (II)
BINAP 0.2g, toluene 70ml are added into 250ml three-necked bottles, temperature control stirs 1h at 20~25 DEG C.Added again into bottle The chloro- 8- trifluoromethanesulfonic acids base quinoline 10g of 2-, piperidin-4-yl t-butyl carbamate 6.41g, potassium carbonate 7.8g, stir lower by instead Answer liquid to be heated to 80 DEG C~100 DEG C, keep 20~30h of this thermotonus.TLC is detected, and whether reaction is complete.Reaction is complete, stops Only heat.20~30 DEG C are cooled to, dichloroethanes 50ml is added, adds diatomite to filter out the solid in reaction solution, filter cake second Acetoacetic ester 150ml is washed., 20~25 DEG C of stirring 8h.The solid separated out in solution is filtered out, filtrate is molten with 5% disodium hydrogen phosphate Liquid 2x50ml is washed.Organic phase is concentrated to dryness again, adds acetonitrile 50ml, 20~25 DEG C of 10~20h of stirring and crystallizing.Filtering analysis The solid gone out, 40 DEG C of forced air dryings obtain the tertiary fourth of yellow solid 10.69g, 2- chloro- 8 (piperidin-4-yl) benzimidazole carbamic acid Ester, yield 92.3%.
The synthesis of 5- (3- methy oxetane -3- methoxyl groups) benzimidazole (III)
Compound 3- methyl -3- oxetane methanols 30.77g, THF140ml, metallic sodium 6.95g are added to the necks of 250ml tri- In bottle, 66 DEG C of backflow 4h are heated under stirring, 55 DEG C is cooled to, then adds 5- hydroxybenzimidazole 40.4g, stir lower heat Backflow, react 20~24h.
Ethyl acetate 100ml is added into reaction bulb, 0.5h dissolvings are stirred at 30~50 DEG C, are then reduced to -5 DEG C, are added dropwise just Hexane 30ml, stirs 1h, and suction filtration obtains light yellow solid, 40 DEG C of dryings to constant weight, obtains 56.41 grams, yield 85.8%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl }-piperazine The synthesis of pyridine -4- bases-t-butyl carbamate (IV)
II (50 grams), III (30.14 grams), potassium carbonate 80g, DIPHOS 4.3g, toluene 700ml, are added in 2L three-necked bottles, add Enter acid chloride 0.9g, stir.Stirring is lower to heat up, and temperature control reacts 24~30h at 80~100 DEG C.After the completion of reaction, it is cooled to 55 DEG C add dichloroethanes 700ml.10min is stirred, adds the solid in diatomite filtering reacting liquid, the filter cake chloroethenes of 500ml bis- Alkane rinses.Concentrate the filtrate to it is dry, add ethyl acetate 480ml, be heated to flowing back, be cooled to 20~25 DEG C of 10~20h of crystallization. The solid separated out is filtered, 50 DEG C of forced air dryings, obtains white solid, the amount of obtaining 70.51g, yield 93.90%.
(1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline-8-yl } -4- The synthesis of amino piperidine (V)
By compound (1- { 2- [5- (3- methvl-oxetan -3- ylmethoxies)-benzimidazole -1- bases]-quinoline -8- Base }-piperidin-4-yl-t-butyl carbamate 5g, caustic alcohol 2.8g, 2- methyltetrahydrofuran 30ml and water 0.08ml be added to In 100ml three-necked bottles, stir.The mixture is heated to flowing back, and stirs 3~4h under reflux.
TLC is detected, and reaction is complete.Stop heating, add purified water 60ml, extracting and demixing.Aqueous phase is extracted with 2 × 20ml of ethyl acetate Take, merge organic phase, washed with saturated nacl aqueous solution 20ml.Be concentrated under reduced pressure organic phase, and 30ml is added into condensate residue Ethyl acetate, in 20~25 DEG C of stirring and crystallizing 6h.The solid separated out is filtered, filtrate decompression is concentrated to dryness.Added into residue 24ml ethyl acetate, in 20~25 DEG C of 10~12h of stirring and crystallizing.Filter the solid separated out, dry white solid product, the amount of obtaining 3.68g, yield 90.3%.
1H NMR test (referring to accompanying drawing)
(d6-DMSO):δ 9.176 (s, 1H), δ 8.88-8.91 (d, 1H, J=8.7Hz), δ 8.51-8.53 (d, 1H, J= 9.0Hz), δ 8.13-8.15 (d, 1H, J=9.0Hz), δ 7.6 (d, 1H, J=7.5Hz), 7.49 (t, 1H, J=7.9Hz) 7.39 (d, 1H, J=2.4Hz), 7.29 (d, 1H, J=7.6Hz), 7.19 (dd, 1H, J=9.2hz, 2.5Hz) 4.56 (d, 2H, J= 5.6Hz), 4.34 (d, 2H, J=5.7Hz), 4.14 (s, 2H), 3.74 (d, 2H, J=10.1Hz), 2.77 (m, 3H), 1.91 (d, 2H, J=11.1Hz), 1.68 (m, 2H), 1.41 (s, 3H)


SET 2




///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Background
Type III Receptor tyrosine kinase, including FLT3, PDGFRα and PDGFRβ, have been directly implicated in the pathogenesis of epithelial, mesenchymal, and hematological malignancies.[6]
Mutations of FLT3 comprise one of the most frequently identified types of genetic alterations in Angiomyolipoma.[7][8] Approximately one-third of AML patients present with a mutation in this gene.[9] The majority of these mutations result in constitutive activation of downstream signaling pathways and aberrant cell growth.[7] Mutations in FLT3 have also been reported in acute lymphoblastic leukemia (ALL)[10] and myelodysplastic syndrome (MDS).[11]
Activating mutations in PDGFRA have been detected in 5-12% of Gastrointestinal stromal tumor.[12] Fusion of PDGFRA has been found to be responsible for hematological malignances like hypereosinophilic syndrome.[13] The amplification of chromosome 4q12, the site of the PDGFRA gene[citation needed], has been identified in 13-29% of adult gliomas[citation needed] and in 29% to 36% of diffuse intrinsic pontine gliomas (DIPG)[citation needed], a subset of high-grade gliomas (HGG) in pediatric patients. Activation of PDGFRB, a third member of the type III RTK family, has been implicated in the development of chronic myelomonocytic leukemia due to the fusion of PDGFRB with the TEL gene.[13] Furthermore, PDGFB translocation to the COL1A1 gene locus has been identified to be responsible for dermatofibrosarcoma protuberans (DFSP).[13] In cancer cells, PDGFR promotes tumor development and migration via proto-oncogenic downstream mediators like AKT and MEK[citation needed]. In stromal fibroblasts, PDGFRα activation leads to local tissue invasion, production and secretion of VEGF, and elevated intratumoral interstitial pressure[citation needed]. In stromal pericytes, PDGFRβ activation mediates vascular stability.[13] Thus, either FLT3 or PDGF/PDGFR pathway is the primary driver of oncogenesis in the above malignancies and can be targeted by crenolanib therapy[citation needed].
Mechanism
FLT3: wild-type and mutant
Crenolanib inhibits both wild type FLT3 and its constitutively active mutations. In vitro studies have shown that crenolanib has low Kd for the FLT3 enzyme with constitutively activating internal tandem duplication (ITD) mutations and tyrosine kinase domain (TKD) mutations, D835H and D835Y, as compared to wild type. Crenolanib tightly binds to FLT3-ITD, FLT3-D835H and FLT3-D835Y with Kd of 0.74 nM, 0.4 nM, and 0.18 nM, respectively.[14] Crenolanib inhibits the phosphorylation of the FLT3-ITD receptor in transfected TF-1 cells and the FLT3-D835Y TKD mutation in transfected Ba/F3 cells at nanomolar IC50 concentrations of 1.3 nM and 8.8 nM, respectively.[15] Immunoblot experiments performed in the Molm14 FLT3-ITD positive cell line show that crenolanib inhibits downstream signaling of FLT3 at a concentration of 10 nM.[15] MTT assay measurements of crenolanib cytotoxicity evaluated in the FLT3-ITD expressing cell lines Molm14 and MV411, showed that crenolanib is toxic at IC50 concentrations of 7 nM and 8 nM, respectively.[15]
PDGFRα: wild-type and mutant
Crenolanib has been shown to inhibit PDGFRα with an IC50 of 0.4 ng/mL in porcine aortic epithelial cell lines. In Chinese hamster ovary (CHO) cells expressing PDGFRα, crenolanib inhibited the phosphorylation of wild type PDGFRα at an IC50 of 10 nM.[16] Additionally, crenolanib completely blocked PDGFRα phosphorylation and downstream AKT signaling at a concentration between 0.1 and 1 uM in Ink4a/Arf-/- mouse astrocytes transfected to stably co-express both human PDGFRα and PDGF AA.[17] The lung cancer cell line H1703, which is reported to have amplification of both PDGFRA (4q12) and PDGFC (4q32) genes on chromosome 4, and also overexpress PDGFRα, was sensitive to crenolanib with an IC50 of ~80 nM.[18] In CHO cells expressing an activating exon 18 (D842V) PDGFRα mutation, crenolanib was effective at an IC50 of 6nM and IC90 of 25nM. In addition, crenolanib also inhibited phosphorylation of the double mutants PDGFRα (V561D + D842V and T674I + D842V).[16]
PDGFRβ: wild-type
Crenolanib has been shown to inhibit PDGFRβ with an IC50 of 0.8 ng/mL in porcine aortic epithelial cell lines. Crenolanib inhibits the ability of recombinant PDGFRβ to phosphorylate a synthetic tyrosine substrate (poly-glutamic acid-tyrosine), with an IC50 of 0.4 ng/mL. Evaluation of the antitumor activity of crenolanib in a genetically engineered BSG DIPG mouse model showed that it is highly selective for PDGFRβ with an IC50 of 10 nM when measured by BrdU assay and 1.25 uM by MTT assay.
C-Kit: wild-type and mutant
Crenolanib has been shown to have IC50 and Kd values of 67 nM and 78 nM, respectively, for wild type c-KIT in in vitro assays[citation needed]. Similar assays show that crenolanib inhibits c-KIT activating mutations D816H and D816V with IC50 concentrations of 5.4 and 2.5 nM, respectively.[14][citation needed] Human bone marrow progenitor cell growth assays showed that crenolanib has modest effects on GM-CSF and BFUE driven colony formation at the IC50 concentration of 20 nM.[15]
Clinical
Phase I single-agent[19] and Phase Ib combination[20] studies have investigated the clinical pharmacology of crenolanib in patients with cancer. Pharmacokinetic and safety studies of Crenolanib administered alone or in combination with docetaxel with or without axitinib have been completed. Results suggest that Crenolanib is well tolerated as a single agent, and can also be safely combined with docetaxel and axitinib due to their non-overlapping toxicity profiles.
Clinical trials
- Clinical trial number NCT01229644 for “A Phase II Study of Crenolanib (CP-868,596), a Selective and Potent Inhibitor of PDGFR, for the Treatment of Adult Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT01243346 for “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene” at ClinicalTrials.gov
- Clinical trial number NCT01393912 for “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma” at ClinicalTrials.gov
- Clinical trial number NCT01522469 for “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations” at ClinicalTrials.gov
- Clinical trial number NCT01657682 for “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations” at ClinicalTrials.gov
References
- ^ “A Phase II Study of Crenolanib in Relapsed/Refractory Acute Myeloid Leukemia Patients With FLT3 Activating Mutations – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “Phase II Study of Crenolanib in Subjects With Relapsed/Refractory AML With FLT3 Activating Mutations – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “Phase II Study of Crenolanib (CP-868,596), for the Treatment of Patients With Advanced Gastrointestinal Stromal Tumors With the D842-related Mutations and Deletions in the PDGFRA Gene – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ “PDGFR Inhibitor Crenolanib in Children/Young Adults With Diffuse Intrinsic Pontine Glioma or Recurrent High-Grade Glioma – Full Text View”. ClinicalTrials.gov. Retrieved 2014-04-08.
- ^ A. Ramachandran; H. Marshall; V. Jain. “CRENOLANIB, A NOVEL TYPE I, MUTANT -SPECIFIC INHIBITOR OF CLASS III RECEPTOR TYROSINE KINASES, PREFERENTIALLY BINDS TO PHOSPHORYLATED KINASES” (PDF). gistsupport.org. Retrieved 2014-04-08.
- ^ Lemmon, Mark A.; Schlessinger, Joseph (2010). “Cell Signaling by Receptor Tyrosine Kinases”. Cell. 141 (7): 1117–34. doi:10.1016/j.cell.2010.06.011. PMC 2914105. PMID 20602996.
- ^ Jump up to:a b Takahashi, S (2011-04-01). “Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications”. J Hematol Oncol. 4: 13. doi:10.1186/1756-8722-4-13. PMC 3076284. PMID 21453545.
- ^ Cancer Genome Atlas Research Network; Ley, T. J.; Miller, C.; Ding, L.; Raphael, B. J.; Mungall, A. J.; Robertson, A.; Hoadley, K.; Triche Jr, T. J.; Laird, P. W.; Baty, J. D.; Fulton, L. L.; Fulton, R.; Heath, S. E.; Kalicki-Veizer, J.; Kandoth, C.; Klco, J. M.; Koboldt, D. C.; Kanchi, K. L.; Kulkarni, S.; Lamprecht, T. L.; Larson, D. E.; Lin, L.; Lu, C.; McLellan, M. D.; McMichael, J. F.; Payton, J.; Schmidt, H.; Spencer, D. H.; et al. (2013). “Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia”. New England Journal of Medicine. 368 (22): 2059–2074. doi:10.1056/NEJMoa1301689. ISSN 0028-4793. PMC 3767041. PMID 23634996.
- ^ “The Impact of FLT3 Mutations on the Development of Acute Myeloid Leukemias”. Hindawi.com. Retrieved 2014-04-08.
- ^ Xu, F; Taki, T; Yang, HW; Hanada, R; Hongo, T; Ohnishi, H; Kobayashi, M; Bessho, F; Yanagisawa, M; Hayashi, Y (2014-01-24). “Tandem duplication of the FLT3 gene is found in acute lymphoblastic leukaemia as well as acute myeloid leukaemia but not in myelodysplastic syndrome or juvenile chronic myelogenous leukaemia in children”. Br. J. Haematol. 105 (1): 155–62. doi:10.1111/j.1365-2141.1999.01284.x. PMID 10233379. S2CID 40898615.
- ^ Yokota, S; Kiyoi, H; Nakao, M; Iwai, T; Misawa, S; Okuda, T; Sonoda, Y; Abe, T; Kahsima, K; Matsuo, Y; Naoe, T (2014-01-24). “Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines”. Leukemia. 11 (10): 1605–9. doi:10.1038/sj.leu.2400812. PMID 9324277.
- ^ Heinrich, M. C.; Corless, CL; Duensing, A; McGreevey, L; Chen, CJ; Joseph, N; Singer, S; Griffith, DJ; Haley, A; Town, A; Demetri, GD; Fletcher, CD; Fletcher, JA (2003). “PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors”. Science. 299 (5607): 708–10. doi:10.1126/science.1079666. PMID 12522257. S2CID 11725958.
- ^ Jump up to:a b c d Östman, Arne; Heldin, Carl‐Henrik (2007). PDGF Receptors as Targets in Tumor Treatment. Advances in Cancer Research. Vol. 97. pp. 247–274. doi:10.1016/S0065-230X(06)97011-0. ISBN 9780120066971. PMID 17419949.
- ^ Jump up to:a b Muralidhara, C.; Ramachandran, A.; Jain, V. K. (2012). “Abstract 3683: Crenolanib, a novel Type I, mutant-specific inhibitor of Class III receptor tyrosine kinases, preferentially binds to phosphorylated kinases”. Cancer Research. 72 (8 Supplement): 3683. doi:10.1158/1538-7445.AM2012-3683.
- ^ Jump up to:a b c d Galanis, A.; Rajkhowa, T.; Muralidhara, C.; Ramachandran, A.; Levis, M. (2012). “Abstract 3660: Crenolanib: A next generation FLT3 inhibitor”. Cancer Research. 72 (8 Supplement): 3660. doi:10.1158/1538-7445.am2012-3660.
- ^ Jump up to:a b Heinrich, M. C.; Griffith, D.; McKinley, A.; Patterson, J.; Presnell, A.; Ramachandran, A.; Debiec-Rychter, M. (2012). “Crenolanib Inhibits the Drug-Resistant PDGFRA D842V Mutation Associated with Imatinib-Resistant Gastrointestinal Stromal Tumors”. Clinical Cancer Research. 18 (16): 4375–84. doi:10.1158/1078-0432.CCR-12-0625. PMID 22745105.
- ^ Yang, X.-L.; Mashimo, T.; Su, Y.; Vemireddy, V.; Guntipalli, P.; Ramachandran, A.; Chaudhary, P.; Mickey, B.; Hatanpaa, K.; Maher, E.; Bachoo, R. M. (2011). “Abstract 1111: Preclinical evaluation of CP868,596, a novel PDGFR Inhibitor for treatment of glioblastoma”. Cancer Research. 71 (8 Supplement): 1111. doi:10.1158/1538-7445.am2011-1111.
- ^ Peyton, M.; Chaudhary, P.; Ramachandran, A.; Minna, J. (2011). “Abstract 3601: CP-868,596, a highly potent and selective PDGFR TKI inhibits growth of PDGFR -driven lung cancer cells”. Cancer Research. 71 (8 Supplement): 3601. doi:10.1158/1538-7445.am2011-3601.
- ^ Lewis, N. L.; Lewis, L. D.; Eder, J. P.; Reddy, N. J.; Guo, F.; Pierce, K. J.; Olszanski, A. J.; Cohen, R. B. (2009). “Phase I Study of the Safety, Tolerability, and Pharmacokinetics of Oral CP-868,596, a Highly Specific Platelet-Derived Growth Factor Receptor Tyrosine Kinase Inhibitor in Patients with Advanced Cancers”. Journal of Clinical Oncology. 27 (31): 5262–9. doi:10.1200/jco.2009.21.8487. PMC 2773478. PMID 19738123.
- ^ Michael, M; Vlahovic, G; Khamly, K; Pierce, K J; Guo, F; Olszanski, A J (2010). “Phase Ib study of CP-868,596, a PDGFR inhibitor, combined with docetaxel with or without axitinib, a VEGFR inhibitor”. British Journal of Cancer. 103 (10): 1554–61. doi:10.1038/sj.bjc.6605941. PMC 2990584. PMID 20959830.
External links
- “PDGFR Inhibitor CP-868596 (Code C64639)”, National Cancer Institute Thesaurus.
- “PDGFR and Human Cancer” , AROG Pharmaceuticals LLC.
| Names | |
|---|---|
| IUPAC name1-(2-{5-[(3-methyloxetan-3-yl)methoxy]-1H-benzimidazol-1-yl}quinolin-8-yl)piperidin-4-amine | |
| Other namesCP-868,596; AR-868,596-26 | |
| Identifiers | |
| CAS Number | 670220-88-9 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:145365 |
| ChEMBL | ChEMBL2105728 ChEMBL2146086 |
| ChemSpider | 8541584 |
| IUPHAR/BPS | 7882 |
| KEGG | D10102 |
| PubChemCID | 10366136 |
| UNII | LQF7I567TQ |
| CompTox Dashboard (EPA) | DTXSID50985873 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C26H29N5O2 |
| Molar mass | 443.551 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
{kind=link}
//////////Crenolanib, UNII-LQF7I567TQ, креноланиб , كرينولانيب , 克拉尼布, CP-868,596-26, AR-868,596-26
CC1(COc2ccc3c(c2)ncn3c4ccc5cccc(N6CCC(N)CC6)c5n4)COC1.OS(=O)(=O)c7ccccc7

NEW DRUG APPROVALS
ONE TIME
$10.00
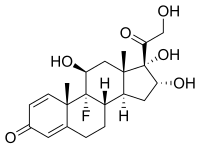
TRIAMCINOLONE

TRIAMCINOLONE
- Molecular FormulaC21H27FO6
- Average mass394.434 Da
(11β,16α)-9-Fluoro-11,16,17,21-tetrahydroxypregna-1,4-diene-3,20-dione
(8S,9R,10S,11S,13S,14S,16R,17S)-9-Fluor-11,16,17-trihydroxy-17-(hydroxyacetyl)-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-on
(8S,9R,10S,11S,13S,14S,16R,17S)-9-fluoro-11,16,17-trihydroxy-17-(hydroxyacetyl)-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one
(8S,9R,10S,11S,13S,14S,16R,17S)-9-fluoro-11,16,17-trihydroxy-17-(hydroxyacétyl)-10,13-diméthyl-6,7,8,9,10,11,12,13,14,15,16,17-dodécahydro-3H-cyclopenta[a]phénanthrén-3-one
(8S,9R,10S,11S,13S,14S,16R,17S)-9-Fluoro-17-glycoloyl-11,16,17-trihydroxy-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one
124-94-7[RN]
16a-Hydroxy-9a-fluoroprednisolone
1ZK20VI6TY
204-718-7[EINECS]
755
9a-Fluoro-16a-hydroxyprednisolone
TU3850000
トリアムシノロン[Japanese]
去炎松[Chinese]
Triamcinolone
CAS Registry Number: 124-94-7
CAS Name: (11b, 16a)-9-Fluoro-11,16,17,21-tetrahydroxypregna-1,4-diene-3,20-dione
Additional Names: D1-9a-fluoro-16a-hydroxyhydrocortisone; 9a-fluoro-16a-hydroxyprednisolone; D1-16a-hydroxy-9a-fluorohydrocortisone; 16a-hydroxy-9a-fluoroprednisolone
Manufacturers’ Codes: CL-19823
Trademarks: Aristocort (Lederle); Kenacort (BMS); Ledercort (tabl.) (Lederle); Omcilon (BMS); Tricortale (Bergamon); Volon (BMS)
Molecular Formula: C21H27FO6, Molecular Weight: 394.43
Percent Composition: C 63.95%, H 6.90%, F 4.82%, O 24.34%
Literature References: Prepn: Bernstein et al.,J. Am. Chem. Soc.78, 5693 (1956); 81, 1689 (1959); Thoma et al.,ibid.79, 4818 (1957); Bernstein et al., Allen et al.,US2789118; US3021347 (1957, 1962, both to Am. Cyanamid). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 367-396, 423-442 (1972); D. H. Sieh, ibid.11, 593-614, 651-661 (1982).
Properties: Crystals, mp 269-271°. mp also reported as 260-262.5°. [a]D25 +75° (acetone). uv max: 238 nm (e 15800).
Melting point: mp 269-271°; mp also reported as 260-262.5°
Optical Rotation: [a]D25 +75° (acetone)
Absorption maximum: uv max: 238 nm (e 15800)
………………………………
Derivative Type: 16,21-Diacetate
CAS Registry Number: 67-78-7
CAS Name: (11b,16a)-16,21-Bis(acetyloxy)-9-fluoro-11,17-dihydroxypregna-1,4-diene-3,20-dione
Additional Names: 16a,21-diacetoxy-9a-fluoro-11b,17a-dihydroxy-1,4-pregnadiene-3,20-dione
Trademarks: Cenocort (Central Pharm.); CINO-40 (Tutag); Tracilon (Savage)
Molecular Formula: C25H31FO8, Molecular Weight: 478.51
Percent Composition: C 62.75%, H 6.53%, F 3.97%, O 26.75%
Properties: Solvated crystals, mp 186-188° (with effervescence, mp 235° after drying). [a]D25 +22° (chloroform). uv max: 239 nm (e 15200).
Melting point: Solvated crystals, mp 186-188° (with effervescence, mp 235° after drying)
Optical Rotation: [a]D25 +22° (chloroform)
Absorption maximum: uv max: 239 nm (e 15200)
Therap-Cat: Glucocorticoid., Therap-Cat-Vet: Glucocorticoid., Keywords: Glucocorticoid.
///////////////////////
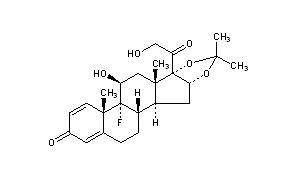
Triamcinolone Acetonide
CAS Registry Number: 76-25-5
CAS Name: (11b,16a)-9-Fluoro-11,21-dihydroxy-16,17-[1-methylethylidenebis(oxy)]pregna-1,4-diene-3,20-dione
Additional Names: 9a-fluoro-11b,16a,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone; 9a-fluoro-16a-hydroxyprednisolone acetonide; triamcinolone 16a,17-acetonide; 9a-fluoro-11b,21-dihydroxy-16a,17a-isopropylidenedioxy-1,4-pregnadiene-3,20-dione; 9a-fluoro-16a,17-isopropylidenedioxyprednisolone
Trademarks: Adcortyl (BMS); Azmacort (Aventis); Delphicort (Lederle); Extracort (Basotherm); Ftorocort (Gedeon Richter); Kenacort-A (BMS); Kenalog (Apothecon); Ledercort Cream (Lederle); Nasacort (Aventis); Respicort (Mundipharma); Rineton (Sanwa); Solodelf (Cyanamid); Tramacin (J & J); Triam (Lichtenstein); Tricinolon (Kaken); Vetalog (Solvay); Volon A (BMS); Volonimat (BMS)
Molecular Formula: C24H31FO6, Molecular Weight: 434.50
Percent Composition: C 66.34%, H 7.19%, F 4.37%, O 22.09%
Literature References: Prepd by stirring a suspension of triamcinolone in acetone in the presence of a trace of perchloric acid: Fried et al.,J. Am. Chem. Soc.80, 2338 (1958); Bernstein et al.,ibid.81, 1689 (1959); Bernstein, Allen, US2990401 (1961 to Am. Cyanamid). Alternate synthesis using 2,3-dibromo-5,6-dicyanoquinone: Hydorn, US3035050 (1962 to Olin Mathieson). Clinical trial in chronic asthma: I. L. Bernstein et al.,Chest81, 20 (1982). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.1, 397-421 (1972); D. H. Sieh, ibid.11, 615-649 (1982).
Properties: Crystals, mp 292-294°. [a]D23 +109° (c = 0.75 in chloroform). uv max (abs alc.): 238 nm (e 14600). Sparingly sol in methanol, acetone, ethyl acetate.
Melting point: mp 292-294°
Optical Rotation: [a]D23 +109° (c = 0.75 in chloroform)
Absorption maximum: uv max (abs alc.): 238 nm (e 14600)
………………..
Derivative Type: 21-Acetate
Properties: Crystals, mp 268-270°. [a]D23 +92° (c = 0.59 in chloroform).
Melting point: mp 268-270°
Optical Rotation: [a]D23 +92° (c = 0.59 in chloroform)
Derivative Type: 21-Disodium phosphate
CAS Registry Number: 1997-15-5
Trademarks: Aristosol (Lederle)
Molecular Formula: C24H30FNa2O9P, Molecular Weight: 558.44
Percent Composition: C 51.62%, H 5.41%, F 3.40%, Na 8.23%, O 25.79%, P 5.55%
………………….
Derivative Type: 21-Hemisuccinate
Trademarks: Solutedarol (Specia)
Molecular Formula: C28H35FO9, Molecular Weight: 534.57
Percent Composition: C 62.91%, H 6.60%, F 3.55%, O 26.94%
Therap-Cat: Glucocorticoid; antiasthmatic (inhalant); antiallergic (nasal).
Therap-Cat-Vet: Glucocorticoid.
Keywords: Antiallergic (Steroidal, Nasal); Antiasthmatic (Steroidal, Inhalant); Glucocorticoid.
//////////////////////////
Title: Triamcinolone Benetonide
CAS Registry Number: 31002-79-6
CAS Name: (11b,16a)-21-[3-(Benzoylamino)-2-methyl-1-oxopropoxy]-9-fluoro-11-hydroxy-16,17-[(1-methylethylidene)bis(oxy)]pregna-1,4-diene-3,20-dione
Additional Names: 9-fluoro-11b,16a,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone 21-ester with N-benzoyl-2-methyl-b-alanine; 9a-fluoro-16a-hydroxyprednisolone 16a,17a-acetonide 21-(b-benzoylamino)isobutyrate; triamcinolone acetonide b-benzoylaminoisobutyrate; TBI
Trademarks: Tibicorten (Stiefel)
Molecular Formula: C35H42FNO8, Molecular Weight: 623.71
Percent Composition: C 67.40%, H 6.79%, F 3.05%, N 2.25%, O 20.52%
Literature References: Prepn: C. Cavazza et al.,DE2047218; eidem,US3749712 (1971, 1973 both to Sigma-Tau). Pharmacology: E. T. Ordonez, Arzneim.-Forsch.21, 248 (1971). Percutaneous absorption by rats and rabbits: W. H. Down et al.,Toxicol. Lett.1, 95 (1977). Clinical study: D. J. Tazelaar, J. Int. Med. Res.5, 338 (1977). HPLC analysis: S. Muck et al.,Boll. Chim. Farm.120, 240 (1981). For structure see Triamcinolone Acetonide.
Properties: Crystalline powder, mp 203-207°. [a]D20 +96 ±3° (c = 1 in ethanol). Sol in methanol, acetone, ethanol, dioxane, pyridine, DMF, chloroform. Insol in water.
Melting point: mp 203-207°
Optical Rotation: [a]D20 +96 ±3° (c = 1 in ethanol)
Therap-Cat: Glucocorticoid; anti-inflammatory (topical).
Keywords: Glucocorticoid
////////////////////////
Triamcinolone Hexacetonide
CAS Registry Number: 5611-51-8
CAS Name: (11b,16a)-21-(3,3-dimethyl-1-oxobutoxy)-9-fluoro-11-hydroxy-16,17-[(1-methylethylidene)bis(oxy)]pregna-1,4-diene-3,20-dione
Additional Names: 9-fluoro-11b,16a,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone, 21-(3,3-dimethylbutyrate); 21-tert-butylacetate-9a-fluoro-11b-hydroxy-16a,17a-(isopropylidenedioxy)pregna-1,4-diene-3,20-dione; 21-(3,3-dimethylbutyryloxy)-9a-fluoro-11b-hydroxy-16a,17a-(isopropylidenedioxy)pregna-1,4-diene-3,20-dione; triamcinolone acetonide tert-butyl acetate; TATBA
Manufacturers’ Codes: CL-34433
Trademarks: Aristospan (Fujisawa); Hexatrione (Lederle); Lederlon (Lederle); Lederspan (Lederle)
Molecular Formula: C30H41FO7, Molecular Weight: 532.64
Percent Composition: C 67.65%, H 7.76%, F 3.57%, O 21.03%
Literature References: The hexacetonide ester of the potent glucocorticoid, triamcinolone, q.v. Prepn of syringeable suspension: Nash, Naeger, US3457348 (1969 to Am. Cyanamid). Anti-inflammatory activity in rabbits: I. M. Hunneyball, Agents Actions11, 490 (1981). Early clinical studies: Bilka, Minn. Med.50, 483 (1967); Layman, Peterson, ibid. 669. Clinical studies of intra-articular therapy in arthritis: R. C. Allen et al.,Arthritis Rheum.29, 997 (1986); M. Talke, Fortschr. Med.104, 742 (1986). Toxicity study: Tonelli, Steroids8, 857 (1966). Comprehensive description: V. Zbinovsky, G. P. Chrekian, Anal. Profiles Drug Subs.6, 579-595 (1977). For structure see Triamcinolone Acetonide.
Properties: Fine, white, needle-like crystals, mp 295-296° (dec), also reported as mp 271-272° (dec). uv max (ethanol): 238 nm (e 15500). [a]D25 +90±2° (c = 1.13% in chloroform). Soly in g/100 ml at 25°: chloroform and dimethylacetamide >5; ethyl acetate 0.77, methanol 0.59, diethyl carbonate 0.50, glycerin 0.42, propylene glycol 0.13; absolute alcohol 0.03; water 0.0004.
Melting point: mp 295-296° (dec); mp 271-272° (dec)
Optical Rotation: [a]D25 +90±2° (c = 1.13% in chloroform)
Absorption maximum: uv max (ethanol): 238 nm (e 15500)
Therap-Cat: Anti-inflammatory.
Keywords: Glucocorticoid.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Triamcinolone acetonide | F446C597KA | 76-25-5 | YNDXUCZADRHECN-JNQJZLCISA-N |
| Triamcinolone diacetate | A73MM2Q32P | 67-78-7 | XGMPVBXKDAHORN-RBWIMXSLSA-N |
| Triamcinolone hexacetonide | I7GT1U99Y9 | 5611-51-8 | TZIZWYVVGLXXFV-FLRHRWPCSA-N |
Triamcinolone is a glucocorticoid used to treat a wide variety of inflammatory conditions of organ systems and tissues.
Triamcinolone is a glucocorticoid used to treat certain skin diseases, allergies, and rheumatic disorders among others.[6] It is also used to prevent worsening of asthma and COPD.[6] It can be taken in various ways including by mouth, injection into a muscle, and inhalation.[6]
Common side effects with long-term use include osteoporosis, cataracts, thrush, and muscle weakness.[6] Serious side effects may include psychosis, increased risk of infections, adrenal suppression, and bronchospasm.[6] Use in pregnancy is generally safe.[7] It works by decreasing inflammation and immune system activity.[6]
Triamcinolone was patented in 1956 and came into medical use in 1958.[8] It is available as a generic medication.[9] In 2019, it was the 107th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[10][11]
PATENT
Skin is the layer of usually soft, flexible outer tissue covering the body of a vertebrate animal, with three main functions: protection, regulation, and sensation. Skin diseases are the medical condition that affects the skin, hair, nails and related muscle and glands.
Skin disorders vary greatly in symptoms and severity. They can be temporary or permanent, and may be painless or painful. Some have situational causes, while others may be genetic. Some skin conditions are minor, and others can be lifethreatening.
There are many different types of skin disorders which include rashes, dermatoses or skin eruptions. Such rashes, dermatoses or skin eruptions include acute, inflammatory reactions of the skin caused by an allergic or irritant reaction, other forms of eczema, lichen simplex chronicus. Chronic nature includes seborrheic dermatitis, psoriasis, and atopic dermatitis or caused by infection, irritation or aggravation of another condition such as occurs with acne, other rashes, dermatoses or skin eruptions, inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses, contact dermatitis, impetigo, urticarial and scabies.
Typical symptoms of the skin disorders include but not limited to raised bumps that are red or white, a rash, which might be painful or itchy, scaly or rough skin peeling skin, ulcers, open sores or lesions, dry, cracked skin, discolored patches of skin, fleshy bumps, warts, or other skin growths, changes in mole color or size a loss of skin pigment, excessive flushing or the like.
Atopic dermatitis (AD), also known as eczema or atopic eczema, is a type of inflammation of the skin (dermatitis). Atopic dermatitis (AD) is common worldwide. People of all ages from newborns to adults and older live with this condition. Symptoms range from excessively dry, itchy skin to painful, itchy rashes that cause sleepless nights and interfere with everyday life.
Topical corticosteroids have been the mainstay of treatment for atopic dermatitis over the past years, further the cure for atopic dermatitis involves Lifestyle modification, balanced diet intake, self-care measures, phototherapy, wet wrap therapy, use of medications like tacrolimus, pimecrolimus, crisaborole, dupilumab, ciclosporin, methotrexate, interferon gamma- lb, mycophenolate mofetil, and azathioprine or the like.
Triamcinolone Acetonide is a synthetic corticosteroid. Chemically it is [Pregna-1, 4-diene-3, 20-dione, 9-fluoro-l l, 21 -dihydroxy- 16, 17-[(1 methylethylidene) bis-(oxy)]-, (HP, 16a)-] with the empirical formula C24H31FO6 and molecular weight 434.50. Triamcinolone Acetonide is represented by compound of structural formula I
Triamcinolone Acetonide topical cream and ointment with strengths 0.025%, 0.1% and 0.5% (containing 0.25 mg/gm, 1 mg/gm & 5 mg/gm Triamcinolone Acetonide respectively) were approved in USA prior to Jan 1, 1982 under the trade name “Triamcinolone Acetonide” and were indicated for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses.
The commercially available products or product known in the prior art produces side effects such as burning, itching, irritation, or dryness of skin at site of application, folliculitis, hypertrichosis, acneiform eruptions, hypopigmentation, perioral dermatitis, allergic contact dermatitis, maceration of the skin, secondary infection, skin atrophy, striae and miliaria.
Pediatric patients may demonstrate greater susceptibility to topical triamcinolone -induced HPA axis suppression and Cushing’s syndrome than mature patients because of a larger skin surface area to body weight ratio. Hypothalamic -pituitary-adrenal (HPA) axis suppression, Cushing’s syndrome and intracranial hypertension have been reported in children receiving topical triamcinolone. Manifestations of adrenal suppression in children include linear growth retardation, delayed weight gain, low plasma cortisol levels, and absence of response to ACTH stimulation. Manifestations of intracranial hypertension include bulging fontanelles, headaches, and bilateral papilledema. Chronic corticosteroid therapy may interfere with the growth and development of children.
Making low dose compositions can present technical and economic challenges that are not present for higher dose formulations.
Examples
The following table 1 shows cream formulation containing lOO.OOmcg per gm, 50.00mcg per gm and 25.00mcg per gm of Triamcinolone Acetonide
Table – 1: cream
Drug Strength IQOmcg/gm 50mcg/gm 25mcg/gm
lOO.OOmcg per gm and for lOOgm, it is lO.OOmg*
50.00mcg per gm and for lOOgm, it is 5.00mg*
25.00mcg per gm and for lOOgm, it is 2.50mg**
Manufacturing process:
a) Dispensing following excipients – isopropyl myristate, glyceryl monostearate and white soft paraffin in vessel I;
b) Dispensing the following excipients – polysorbate 40 and purified water in vessel II;
c) Dispensing the following excipients methyl paraben, propylene glycol in vessel III; wherein methyl paraben is dissolved in propylene glycol to form a clear solution;
d) Dispensing the following active & excipients triamcinolone acetonide or salt thereof, propylene glycol in vessel IV; wherein triamcinolone acetonide or salt thereof is dissolved in propylene glycol to form clear solution;
e) Adding content of step (c) into content of step (b) and stirring to form uniform and homogeneous emulsion;
f) Heating content of step (b) and step (a) at about 75 °C and stirring to form a homogenous uniform emulsion;
g) Cooling the above emulsion gradually to temperature of about 25 °C – 30°C h) Adding the content of step (d) to the primary emulsion of (f) with constant stirring; and
i) Making up the volume of the emulsion with purified water to the required quantity.
SYN
DOI: 10.1021/ja01516a043

CLIP
Corticosteroids
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Triamcinolone
Triamcinolone, 9a-fluoro-11b,16a,17,21-tetrahydroxypregna-1, 4-dien-3,20-dione (27.1.61), differs from dexamethsone in terms of chemical structure in that the a methyl group at C16 is replaced with a hydroxyl group. It is synthesized from the 21-O-acetate of hydrocortisone 27.1.17. In the first stage, both carbonyl groups of this compound undergo ketalization by ethylene glycol. Next, the hydroxyl group in the resulting diketal 27.1.53 is replaced with chlorine using thionyl chloride, and the product undergoes dehydrochlorination using an alkaline, during which the 21-O-acetyl group also is hydrolyzed. Acetylating the hydroxyl group once again with acetic anhydride gives a triene 27.1.54. Reacting this with osmium tetroxide gives the vicinal diol 27.1.55. The secondary hydroxyl group at C16 of this product undergoes acetylation by acetic anhydride in pyridine, which forms the diacetate 27.1.56. Treating the product with N-bromoacetamide in chloric acid gives a bromohydrin (27.1.57), which upon reaction with potassium acetate is transformed to an epoxide (27.1.58). Opening of the epoxide ring, using hydrofluoric acid, gives the corresponding 9-fluoro-11-hydroxy derivative 27.1.59. Upon microbiological dehydrogenation, the C1–C2 bond is oxidized to a double bond, forming triamcinolone acetate (27.1.60), the acetyl group of which is hydrolyzed, forming the desired triamcinolone (27.1.61) [30–32].

Triamcinolone is similar to dexamethasone in terms of pharmacological action, and it is better tolerated in some cases. Synonyms of this drug are ledercort, cenocort, delsolon, and others.
SYN
Drugs for Treating Respiratory System Diseases
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Triamcinolone–Nasacort
The synthesis of triamcinolone (23.2.1) starts from ketalization of cortisol 21-acetate (23.2.8) using ethylene glycol. Dehydration of the obtained compound (23.2.9) for creation of a double bond in position 16,17 of the steroid skeleton through the series of sequential reactions of chlorination, dehydrochlorination, hydrolysis, and acetylation produces 21-acetoxy-4,9(11),16-pregnatriene-3,20-dione (23.2.10), treatment of which with osmium tetroxide in benzene and pyridine produced diol (23.2.11), the secondary hydroxyl group of which, in position 16, was acetylated with acetic anhydride in pyridine to produce the diacetate (23.2.12). The obtained compound in dioxane and water was treated with N-bromoacetamide and 10% perchloric acid to yield bromohydrine (23.2.13). Dehydrobromination of the bromohydrine (23.2.13) with anhydrous potassium acetate in refluxing ethanol produced the epoxy-derivative (23.2.14). Opening of the epoxide ring in (23.2.14) with anhydrous hydrogen fluoride in chloroform produced (23.2.15). Microbiological dehydrogenation of the obtained product with Corynebacterium simplex produced crude diacetate (23.2.16), saponification of which produced triamcinolone (23.2.1) [108-110] (Scheme 23.7.).

Scheme 23.7. Synthesis of triamcinolone.
Triamcinolone is commonly used in the treatment of respiratory inflammation and improves airway reactivity, decreasing respiratory problems. Strangely, there are only few reviews of the pharmacotherapy of triamcinolone [111-113].
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 426-39-1 | C25H33FO8 | 16α,21-diacetoxy-11β,17-dihydroxy-3,20-dioxo-9-fluoro-4-pregnene | Pregn-4-ene-3,20-dione, 16,21-bis(acetyloxy)-9-fluoro-11,17-dihydroxy-, (11β,16α)- |
| 96670-24-5 | C25H30O8 | 16α,21-diacetoxy-3,20-dioxo-17-hydroxy-9β,11β-epoxy-1,4-pregnadiene | 9β-Pregna-1,4-diene-3,20-dione, 9,11β-epoxy-16α,17,21-trihydroxy-, 16,21-diacetate |
SYN
https://patents.google.com/patent/WO2016120891A1/en
Glucocorticoids have a number of diverse effects in different body tissues. Glucocorticoids, in topical, oral and inhaled formulations, are useful for their anti-inflammatory and immunosuppressive properties. Several glucocorticoids such as budesonide and ciclesonide are used for treatment of several disorders.
The synthesis and purification of glucocorticoids have been disclosed at different instances. However, most of these synthetic procedures involve toxic solvents or long reaction times and are ineffective for large scale synthesis. For instance, US 3,92,9768 discloses a process for preparation of budesonide by reacting 16, 17-dihydroxy compound with aldehyde in solvents such as dioxane, methylene chloride or their combinations.
DE 4129535 discloses a process for the synthesis of Ciclesonide which involves the intermediate 16A, 17-[(7?,S)-cyclohexylmethylenedioxy]-l 13, 21-dihydroxy-pregna-l 4- dien-3,20-one which is obtained by an acid catalysed reaction of 11 , 16 , 17, 21-tetra hydroxypregna-l,4-dien-3,20-one with cyclohexane aldehyde.
WO 02/38584 discloses the synthesis of Ciclesonide by reacting corresponding 16, 17-ketals with a cyclohexane aldehyde in the presence of 70% perchloric acid, 1-nitropropane as solvent. However, perchloric acid is a dangerous solvent and can cause serious accidents with fatal consequences.
US Patent No. 6169178 relates to a process for the preparation of budesonide and of 16, 17- acetals of pregnane derivatives structurally co-related thereto comprising treating 16, 17-dios or of 16, 17-ketals or cyclic acetals with aldehydes in the presence of aqueous hydrobromic acid or hydroiodic acid used as reaction catalyst or solvents. However, hydroiodic and other hydrohalic solvents are corrosive, light sensitive and expensive. Further, these acids also post environmental problems. Notwithstanding the use of hydrohalo acids requires use of special equipment since they are extremely corrosive and consequently increase the cost of production.
US 5,55,6964 discloses a process for the preparation of budesonide by reacting 16 – Hydroxy Prednisolone in acetonitrile in the presence of /^-toluene sulfonic acid as a catalyst. There are certain other patents that use alkyl sulfonic acid instead of aryl sulfonic acid for the synthesis of budesonide or similar compounds. However, sulfonic acids are hazardous solvents and FDA has expressed significant concern over the presence or traces of sulfonic acid in pharmaceutical products. Hence, there is a need to have a process for the synthesis 16, 17- acetals of pregnane compounds that is industrially scalable and which does not involve the use of harmful solvents.


Example- 1: Process for preparation of 16-HPN from 3TR
Stage-I


Stage- 1 Stage-I I

Stage-IV

1 6-HPN acetate 1 6-HPN
Scheme 2: Synthesis of 16HPN from 3TR
Stage-I (oxidation)
Charge 750L of acetone (50 volume), 39L of purified water (2.60 volume) and 15 Kg of 3TR (40.93mol) in a SS Reactor at ambient temperature. Cool to -7°C to -5°C than added 6.0L of formic acid (159.03 mol) and 9.0 kg of potassium permanganate (56.95 mol). Maintain at – 5°Cto -3°C for 30 minutes. In-process check by TLC, 3TR should be less than 1.0%. Added 1.5kg sodium metabisulphite (7.89 mol solution in 12L of purified water at -5°C to -3°C then added 3.0 kg of hyflow super cell at 15°C (+2°C) and filter through 10.0 kg of hyflowbed at 27°C(+3°C) and wash with 150L of acetone Added 1.5 kg of activated charcoal, Stir and filter through hyflow bed and wash with 60L of acetone. Total filtrate was distilled under reduced pressure, while maintaining temperature below 45°C. Added 81L of purified water and cool to 5°C+5°C. Filter through centrifuge and wash with 156L of purified water. Wet material is dry at 60°+5°C till moisture less than 0.50%, Yield=15 kg, HPLC purity=98%.
Stage-II (Bromination)
Charge 75L of tetrahydrofuran, 16L of purified water and 15.0 kg of Stage-I (37.46 mol) in a glass reactor. Cool to -6°C (+2°C) and added 7.50 kg of dibromantin (26.23 mol) and 0.60L of perchloric acid (9.38 mol) and maintain at -6°C (+2°C) for one hour. In-process check by TLC, stage-I should be less than 0.50%. Reaction mass is quench in 390L of purified water at ~5°C. Raised the temperature to 25°C and maintained for 01 hour, filter through centrifuge and wash with 828L of purified water or till neutral pH. Wet material is dry at 40°C+5°C till moisture content should be less than 10%, Yield=21.0kg, HPLC purity=97%.
Stage-Ill (Debromination)
Charge 68.0L of N, N-dimethyl formamide(3.238volume) and 21.0kg of stage-II (42.22 mol) in glass reactor, start argon gas purging and cool to -5°C. Charge 13.0L of N,N- dimethylformamide (0.619volume) , 9.70L of dimethylsulfoxide(0.462volume), 1.62kg of chromium chloride hexahydrate (6.51 mol) and 1.94 kg of zinc dust (0.703 mol). Cool to – 10°Cand added 5.50L of thioglycolic acid (79.21 mol). Maintain for one hour while maintaining temperature around -10°C. In-process check by TLC, stage-II should be less than 1.0%. Added 310 L of purified water and cool to 0°C. Filter through centrifuge and wash with 1600L of purified water. Wet material is dry at 60°C+ (5°C) till moisture content less than 6.0%, Yield=15.0kg, HPLC Purity=90%.
Charge 150L of methylene chloride (10 volume), 150L of methanol (10 volume.) and 15.0kg (30.16 mol) of stage-Ill in a SS Reactor. Heat to clear solution then added 3.0 kg of activated charcoal (20%) and reflux for 04 hours, Filter through hyflow bed and wash with 75L of methylene chloride (5 volume), and 75L of methanol (5 volume) mixture. Total filtrate is distilled till last drop and added 75L (5 volume) of methylene dichloride, reflux for 04 hours than cool to 40°C+(5°C), Filter through centrifuge and wash with 15L (one volume) of methylene chloride. Wet material is dry at 60°C (+5°C) till moisture contents less than 1.0% (Yield =13.0kg, HPLC Purity=96%). Further charge 65.0L (5volume) of ethyl acetate and 13.0 kg (1.0 mol) of purified material. Heat to reflux and maintain for 04 hours under reflux, then cool to 40°C. Filter through centrifuge and wash with 13.0L (one volume) of ethyl acetate. Wet material is dry at 60°C (+5°C) till moisture contents less than 0.50%, Yield=12.0kg, HPLC Purity=98.6%.
Stage-IV (Deacetylation)
Charge 5.83L of methanol (10 volume) and 5.83L of methylene chloride (10 volume) in a glass flask and added 583 gm of 16-HPN acetate(1.397mol) at RT. Start argon gas purging and cool to 0°C to 5°C under argon purging. Prepare 11.66 gm of sodium hydroxide (0.2915mol) solution in 0.583L of methanol (one volume) under argon purging and cool to 0°Cto 5°C. Sodium hydroxide solution is charge in reaction mass at 0°C to 5°C. Maintained the reaction mass at 0°C to 5°C for one hour, In-process check by TLC against 16-HPN acetate it should be nil. Adjust pH to neutral by 21.40ml of acetic acid (0.3742 mol); distill under reduced pressure while maintaining temperature below 40°C, till dry. Cool to ambient temperature and added 1.166L of purified water (02 volume). Cool to 0°C and maintain for one hour. Filter and wash with 300ml of purified water. Dry at 60°C (+5°C) till moisture content less than 1.0%, Yield=490gm (93.50%), HPLC Purity=98.97%, Single impurity= 0.40%. Example 2: Process of synthesis of Budesonide from 16-HPN

16-HPN Budesonide
Charge 800 ml of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to -5°C and maintain for 15 min. then added 100 gm of stage-I (0.27 mol) at -5°C and stir for 15 min., added 30 ml of N-butyraldehyde (0.33 mol) while maintaining temperature -5°C to 0°C in around 30 minutes and maintain at 0°C to 5°C for 150 min. under stirring. In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 1200 ml of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 100 kg of sodium bicarbonate (1.19 mol) and 1 ml of purified water (10 volume) in reaction mass at 5°C to 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =110 gm (96.49%), HPLC purity=96.45%, single impurity=1.29%, Epimer-A=47.76%, Epimer-B=49.69%.
(Purification)
Charge 2.5 L of methanol (25 volume) in a Glass flask and added 100 gm of above mentioned crude product. Dissolved at 25°C+5°C till clear solution, added 10 gm of activated charcoal and stir for 30 min. than filter through hyflow bed and wash with 200 ml of methanol (2 volume). Combined filtrates charged in a Glass flask and cool to 10°C to 15°C and added 5.40 L of purified water (54 volume) at 5°Cto 10°C, stir for 15min., filter and wash with purified water. Wet material is dry at 50°C (+5°C) under vacuum till moisture content less than 0.50%, Output=90.0gm, HPLC purity=99.66%, single impurity=0.1%, Epimer-A=44.47%, Epimer-B=55.01%.
Example 2.1: Scale-up process of manufacturing of Budesonide from 16-HPN
Charge 40 L of aqueous hydrochloric acid (8 volume) in a glass flask, start nitrogen gas purging and Cool to – 10°C and maintain for 15 min. then added 5.0 kg of stage-I (13.315 mol) at – 10°C and stir for 45 min. added 1.5 L of N-butyraldehyde (16.68 mol) while maintaining temperature -7°C to – 11°C in around 30 minutes and maintain at -2°C to -6°C for 60 min. under stirring In-process check by TLC against stage-I, it should be nil. Reaction mass is quench in 60 L of purified water (12 volume) at 5°C to 10°C and stir for 15 min. Added solution of 5.0 kg of sodium bicarbonate (59.525 mol) and 50L of purified water (10 volume) in reaction mass at 5°Cto 10°C. Stir at 5°C to 10°C for 15 min. Filter and wash with purified water till neutral pH. Wet material is dry at 50°C (+5°C) till moisture contents less than 1.0 %, Yield =5.293 kg (94.46%), HPLC purity=95.45%, single impurity=1.45%, Epimer-A=53.51 %, Epimer-B=43.78% Effect of temperature and its variation on epimer ratio (A and B) with respect to batch size (From lab to commercial batch)
Example 3: Process for synthesis ofCiclesonide from 16HPN
Preparation of cyclohexane carboxaldehydemetabisulphite complex
200gm of Cyclohexane carboxaldehyde (1.786 mol) was dissolved in 3.0L of denatured sprit (15 volume) and a solution of 190gm of sodium metabisulphite (1.827 mol) in 300ml of purified water (1.5 volume) was added. The resulting precipitate was filtered and washed with 1.0L of denatured sprit(5.0 volume) and dried under vacuum at 50°C, till moisture content less than 6.00%, Yield=400gm (97 %)
Stage I: Preparation of stage-I from 16-HPN

Cyclohexane carboxaldehyde
sodium metabisulphite complex
170gm of 16-HPN (0.4528 mol) was suspended in 3.40L of dichloromethane (20 volume) and treated with 340ml of 70% perchloric acid. (5.65 mol) and 110.5gm of cyclohexane carboxaldehyde metabisulphite complex (0.512 mol) was added in lots while maintaining the temperature between 0°Cto 5°C. The reaction mass was stirred at 0°C to 5°C for 03 hours. In- process check by TLC 16-HPN should be nil and then neutralized with 10% aqueous sodium bicarbonate solution. The organic layer was separated and concentrated under vacuum to obtain a residue which was stripped with methanol (1.0 volume). The solvent was concentrated and the residue was dissolved by refluxing in methanol (5.0 volume). The clear solution was cooled to 0°C to 5.0°C and the resulting solid was filtered and dried at 50°C till moisture content less than 0.50%, Yield=170.0gm (80.0%), HPLC purity=91.68%.
Stage -II Preparation of Ciclesonide from Stage -I

Stage-I Ciclesonide
158gm of stage-I (0.34mol) was suspended in 1.58L of methylene chloride (10.0 volume) at 25°C to 30°C. The reaction mass was chilled to 0°C to 5°C and 81.0ml of triethylamine(0.581 mol) was added, followed by the addition of 79.0ml of isobutyryl chloride [0.75 mol; diluted with 79.0 ml of methylene chloride (0.50 volume)] slowly at 0° to 5°C and maintained at same temperature for 60min. In-process check by TLC, Stage-I should be nil. The reaction mass was diluted with 2.53L of purified water (16.0 volume) , the organic layer was separated and washed with purified water till neutral pH, than organic layer was separated and concentrated under vacuum to obtained a residue. The residue was dissolved by refluxing in 948ml of methanol (6.0 volume); the clear solution was cooled to 0°C to 5°C under stirring and filtered. The product was dried under vacuum at ~50°C till moisture contents comes less than 0.50%, Yield=158.0 gm (87.0%), HPLC purity=95.74%.
(Purification)
120gm of Ciclesonide crude was dissolved by refluxing in 600ml of methanol. The clear solution was chilled to 20°C under stirring and filtered. The product was dried under vacuum at 90°C till moisture content less than 0.50%. Yield=105 gm (87.50%), HPLC purity=99.7 %.
Example 4: Process for synthesis of Desonide from 16HPN acetate
Stage-I : Preparation of Desonide acetate from 16 HPN acetate

Desonide acetate
16HPN acetate 190.0 ml of acetone (7.0 volume) was charged in a glass flask under nitrogen blanketing than added 27 gm of 16HPN acetate (0.0645mol) at ambient temperature. Temperature raised to 28°C (+2°C) and stir for 20 minutes. 1.35 ml of perchloric acid 70% (0.02 lmol) was added at 28°C (+2°C) and stir for 30 minutes. Temperature further raised to 35°C and stir for 60 minutes. In-process check by TLC against 16HPN acetate, it should be nil. Reaction mass cooled to 10°C, filtered and washed with purified water till neutral pH (~7) and finally washed with acetone. Wet material dried at 50°C+5°C till moisture content less than 0.50% to get stage-I. Yield =23gm (77.76%), HPLC Purity=98.28%
Stage-II: Preparation of Desonide from Desonide acetate

Desonide
Desonide acetate
200 ml of methanol (10 volume) and 200ml of methylene dichloride (10 volume) was charged in a glass flask and start argon gas purging. 20 gm of stage- 1st (0.0436mol) was added at ambient temperature. Cool to 0°C+5°C. 0.40gm of sodium hydroxide (O.Olmol) solution in 20ml of methanol (l.Ovolume) was added at 0°C+5°C. Stir at 0°C+5°C for 120 minutes. In-process check by TLC against stage- 1st it should be nil. Adjust pH to neutral (~7) by 2.0ml of acetic acid at 0°C+5°C. Distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume get reduced to 3 to 4 volume of the input. Cool to 0°C and further added 60ml of purified water and stir for 30 minutes. Filtered, washed with purified water till neutral pH (~7). Wet material dried at 50°C+5°C till moisture content less than 0.50% to get crude Desonide. Yield =14.70gm (80.92%), HPLC Purity=88.15%.
(Purification)
140 ml of methanol (10 volume) and 140 ml of methylene chloride (10 volume) was charged in a glass flask and added 14.0 gm of crude material (0.034mol) than stir till clear solution. Added 1.5 gm of activated charcoal and stir for 30 minutes than filtered through hyflow supercel bed and washed with 30ml of methanol and 30ml of methylene chloride mixture. Combined filtrate and distilled the solvent from reaction mass under vacuum while maintaining temperature below 40°C till the volume reduced to 3 to 4 volume of the input. Cool to 0°C. Filtered the reaction mass and washed with 10ml of precooled methanol. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get Desonide. Yield=8.60gm, HPLC Purity= 99.43%

lOOgm of 3TR (0.27 mol.)was suspended in 1300ml (13 volume) acetone. Cooled it to -5°C to -10°C than added 4.0 ml (0.062 mol.) perchloric acid solution and 50gm of dibromantin. Maintained the reaction at same temperature for 02 hours. In-process check by TLC against 3TR it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol.) in 5 lots and reaction was maintained at 35°C+2°C. In-process check by TLC against step-I reaction mass, it should be nil. Cooled to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol.). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50%. Yield =87gm, (83.36%), HPLC Purity=97.883%.
Stage – II:

80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permagnate (0.30 mol.) at -5°C (+2°C). Reaction was maintained at – 5°C+2°Cfor one hour. In-process check by TLC against stage-I it should be nil. Added 8gm of sodium metabisulphite (0.042 mol.) In 80 ml purified water (01 volume) solution at -5°C (+2°C). Temperature raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4volume of stage-I than cool to 0°C to 5°C and added 480ml of purified water stir and filter and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0%. Yield =78.30gm, (89.88%), HPLC Purity=99.178%. Stage -III:

Stage-ll Stage-
300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction was maintained at -45°C to -50°C for 02 hours. In-process check by TLC against before acetone reaction mass. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at ~20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C (+2°C), stir and filter and washed with purified water till neutral pH. Wet material was dried at 45°C to 50°C, Yield =78.50gm, (91.48%), HPLC Purity=91.593%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol.) was dissolved in 760ml of methylene chloride (lOvolume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stir till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than stir for 30minutes, filter through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and stir for 02 hours. Filtered and washed with minimum precooled methanol, Wet material was dried 45°C to 50°C till moisture contents less than 0.50%, Yield=62gm, HPLC Purity=98.633%.
Stage – IV (Process for synthesis of Triamcinolone acetonide from Stage – III):

Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.13 mol) was dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 1.2gm of sodium hydroxide (0.03mol.) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C (+2°C) for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C (+2°C). Reaction mass was distilled at below 40°C under vacuum till 3 to 4 volume of input. Cool to 30°C and added 120ml of purified water, stir for one hour than filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield =52gm, (95.04%), HPLC Purity=99.21%
(Purification)
50gm of crude material (0.12 mol.) was dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stir for one hour at same temperature, Filter through hyflow bed and washed with 120ml of acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°Cand maintained for one hour at same temperature. Filter and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%, Yield=43gm, HPLC Purity=99.40%.
Example 6: Process for synthesis of Flunisolide from 16HPN acetate Stage -I (Preparation of Desonide acetate from 16HPN acetate):

1 6 H PN acetate eson e acetate
140ml of acetone (7 volume) was charged in glass flask and start argon blanketing than added 20 gm of 16-HPN acetate (0.048mol) at ambient temperature. Cooled to 28°C (+2°C). 1.0ml of perchloric acid 70% (0.016mol) was added at 28°C (+2°) C and stirred for 30 minutes. Temperature raised up to 35°Cand stirred for 60 minutes. In-process check by TLC against 16-HPN acetate, it should be nil. Reaction mass was cooled to 10°C (+2°C). Reaction mass was filtered and washed with purified water till neutral pH (~7) to get wet material. Wet material was dried at 50°C+5°C till moisture content less than 0.50% to get stage-lst. Yield=17.40gm, (79.40%), HPLC Purity=98.241%.
Stage -II (Preparation of Desonide from Desonide acetate):

170ml of methanol (lOvolume) and 170ml of methylene chloride (lOvolume) was charged in a glass flask and start inert atmosphere. 17gm of stage-lst (0.037mol) was added at ambient temperature. Cooled to -5°C. 0.4gm of sodium hydroxide (O.Olmol) solution in 17ml of methanol was added at 0°C (+5°C). Reaction mass was stirred for 02 hours at 0°C (+5°C). In- process check by TLC against stage- 1st it should be nil. Neutral pH (~7) was adjusted by acetic acid. Reaction mass was distilled under vacuum at below 40°C till ~ 100ml. Concentrated mass was cooled to 0°C (+5°C) and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get stage-2nd. Yield=14.0gm, (90.67%), HPLC Purity=99.426%, Single impurity=0.136%.
Stage -III (Preparation of Flunisolide acetate from Desonide):

Desonide Flunisolide acetate
50ml of isopropenyl acetate (5 volume) was charged in a glass flask and added lOgm of stage-2nd (0.024mol) at ambient temperature than heated to 65°C and added 1.5ml of methane sulphonic acid (0.023mol) and temperature raised up to 80°C and stir for one hour. In-process check by TLC against stage-2, it should be nil. Reaction mass cooled to 25°C and adjust pH neutral (~7) by triethylamine. Reaction mass was distilled under vacuum till last drop and degases with acetonitrile. 90ml of acetonitrile (09 volume) was added and cooled to -5°C and than further added 10ml of purified water. lOgm of selectfluor(0.028mol) was added in two lots at 0°C(+5°C) in 02 volume of acetonitrile. Reaction mass was stirred at 10°C to 15°C for 12 hours. In-process check by TLC against before selectfluor reaction mass it should be nil. Adjust pH neutral (~7) by liq. ammonia solution at 0°C+5°C. Reaction mass was quenched in 500ml of purified water (lOOvolume) at ambient temperature. Reaction mass was filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C+5°C till moisture content less than 0.50% to get stage-3rd. Yield=8.60gm, (75.17%), HPLC Purity= 94.12%.
Stage -IV (Preparation of Flunisolide from Flunisolide acetate):

Flunisolide acetate Flunisolide
80ml of methanol (lOvolume) and 80ml of methylene chloride (lOvolume) was charged in a glass flask under inert atmosphere at ambient temperature than added 8.0gm of stage-3r (0.017mol) at ambient temperature. Cooled to -5°C and added 0.16gm of sodium hydroxide (0.004mol) solution in 8ml of methanol at -5°C(+5°C) and stir for 02 hours at -5°C(+5°C). In-process check by TLC against stage-3 ‘ it should be nil. Adjust pH neutral(~7) by acetic acid and reaction mass was distilled under vacuum at below 40°C(+5°C) till ~40ml of volume. Cool to 0°C to 5°C and stir for one hour. Reaction mass was filtered and washed with precooled methanol to get wet material. Wet material was dried at 45°C (+5°C) till moisture content less than 0.50% to get Flunisolide crude. Yield=6.0gm, (82.30%), HPLC Purity=86.50%.
(Purification)
6.0gm of crude Flunisolide(0.014mol) was dissolved in 65ml of ethyl acetate (10.83volume) and 35ml of n-hexane (5.83volume) mixture and clear solution was passed through 60gm of silica gel column. Column was washed with 975ml of ethyl acetate (162.5volume) and 525ml of ft-hexane (87.5volume) mixture. Eluted fraction was distilled under vacuum till 3 to 4 volume of input than cooled it to 0°C and filter to get wet material. Wet material was dried at 50°C (+5°C) till moisture content less than 0.50% to get Flunisolide. Yield=4.28gm, HPLC Purity=95.60%.
Example 7: Process for synthesis of Triamcinolone from 3TR
S

lOOgm of 3TR (0.27mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to- 10°C than added 4.0 ml (0.062mol) perchloric acid solution and 50gm of dibromantin. Reaction maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C+5°Cand adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH (~7). Wet material was dried at 45°C (+2°C) till moisture content less than 0.50% to get stage-I. Yield=85.30gm, (81.74%), HPLC Purity=96.54%. Stage -II:

80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C (+2°C) than added 32ml of formic acid (0.85 mol.) and 48gm of potassium per magnate (0.30 mol) at -5°C (+2°C). Reaction was maintained at same temperature for one hour. In-process check by TLC against stage-I, it should be nil. Added sodiummetabisulphite solution (8 gm in 80 ml of water) at -5°C+2°C. Temperature was raised up to 27°C and filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than further cooled to 0°C to 5°C and added 480ml of purified water, stirred, filter and washed with purified water to get wet stage- II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-II. Yield=82gm, (94.13%), HPLC Purity=97.75%.
Stage -III:

Stage-II Triamcinolone acetate
160ml of hydrofluoric acid (70%) (6.72mol) was cooled at -25°C to -30°C than added 40gm of stage-II (0.096mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II, it should be nil. Added 280ml of purified water at 0°C and 650ml of liq. ammonia at 20°C than reaction mass was quenched in 200ml of liq. ammonia and 500ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH(~7). Wet material was dried at 45°C to 50°C to get stage-Ill Yield=40gm, (95.42%), HPLC Purity=88.71%
(Purification)
40gm of stage-Ill crude (0.0916 mol) was refluxed in 160ml of acetone. Cool to 0°C. Filtered and washed with minimum precooled acetone. Wet material was dried at 50°C+5°C till moisture content comes less than 0.50% to get stage-Ill. Yield=24.9gm HPLC Purity=95.17%.

24gm of stage-Ill (0.055mol) was dissolved in 240ml of methanol (lOvolume) and 240ml of methylene chloride (lOvolume) mixture under argon bubbling. Cool to -5°C+2°C and added 0.48gm of sodium hydroxide (0.012mol) solution in 24ml of methanol (Olvolume) at – 5°C+2°C. Reaction was maintaining at -5°C (+2°C) for 03hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral by adding 0.70ml of acetic acid at -5°C (+2°C). Reaction mass distilled at below 40°C under vacuum till 04-05 volume of input. Cooled to 0°C+5°Cand stir for one hour than filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50%. Yield=18.50gm, (85.29%), HPLC Purity=98.60%.
Example 8: Process for synthesis of Triamcinolone Hexacetonide from 3TR
S

lOOgm of 3TR (0.27288 mol) was suspended in 1300ml (13 volume) acetone. Cool to -5°C to -10°C than added 4.0 ml (0.0625 mol) perchloric acid solution and 50gm of dibromantin. Reaction was maintained at same temperature for 02 hours. In-process check by TLC against 3TR, it should be nil. Added lOOgm of potassium carbonate solution (0.723 mol) in 5 lots and reaction was maintained at 35°C (+2°C). In-process check by TLC against step-I reaction mass, it should be nil. Cool to 0°C (+5°C) and adjust pH neutral (~7) by 36ml of acetic acid (0.63 mol). Added 3.0L of purified water (30 volume). Filter and washed with purified water till neutral pH. Wet material was dried at45°C(+2°C) till moisture content less than 0.50% to get stage-lst. Yield =87gm, (83.36%), HPLC Purity=97.883%. Stage-II :

80gm of stage-I (0.21 mol) was dissolved in 4.0L of acetone (50 volume) and 208ml of purified water (2.6 volume). Cool to -5°C than added 32ml of formic acid (0.85 mol.) and 48gm of potassium permanganate (0.30 mol) at -5°C+2°C. Reaction maintained at -5°C (+2°C) for one hour. In-process check by TLC against stage-I, it should be nil. Added sodium metabisulphite solution (8 gm in 80 ml water) at -5°C (+2°C). Temperature raised up to 27°Cand filtered through hyflow bed and washed with acetone. Acetone was distilled under vacuum till 3 to 4 volume of stage-I than cooled to 0°C to 5°C and added 480ml of purified water, stirred, filtered and washed with purified water to get wet stage-II. Wet material was dried at 50°C (+5°C) till moisture content less than 3.0% to get stage-2nd. Yield=78.30gm, (89.88%), HPLC Purity=99.18%.
Stage – III:

300ml of hydrofluoric acid (12.60mol) was cooled at -25°C to -30°C than added 75gm of stage-II (0.180mol). Reaction was maintained at -25°C to -30°C for 04 hours. In-process check by TLC against stage-II. It should be nil. Reaction mass was cooled to -50°C than added 45ml of acetone (0.60volume) at -45°C to -50°C. Reaction maintained at -45°Cto – 50°C for 02 hours. In-process check by TLC against reaction input, it should be nil. Added 565ml of purified water at 0°C and 340ml of liq. ammonia at 20°C than reaction mass was quenched in 410ml of liq. ammonia and 735ml of purified water solution at 15°C(+2°C), stirred, filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°Cto get stage-3rd. Yield=78.50gm, (91.48%), HPLC Purity=91.59%.
(Purification)
76 gm of stage-Ill Crude (0.16 mol) was dissolved in 760ml of methylene chloride (01 volume) and 760ml of methanol (lOvolume) mixture at ambient temperature. Stirred till clear solution and added 7.6gm of activated charcoal (0. lOvolume) than further stir for 30 minutes and filtered through hyflow bed and washed with methanol (one volume) and methylene chloride (one volume) mixture. Total filtrate was distilled under vacuum till 3 to 4 volume of input. Cooled to 0°C to 5°Cand stir for 02 hours. Filtered and washed with minimum precooled methanol. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-3rd. Yield=62gm, HPLC Purity=98.633%
Stage -IV : (Preparation of Triamcinolone acetonide from Stage – III)

Stage- Ill Triamcinolone acetonide
60gm of stage-Ill (0.1259 mol) dissolved in 600ml of methanol (lOvolume) and 600ml of methylene chloride (lOvolume) mixture under inert atmosphere. Cool to -5°C and added 1.2gm of sodium hydroxide (0.03mol) solution in 60ml of methanol (Olvolume) at -5°C (+2°C). Reaction maintained at -5°C+2°C for 03 hours. In-process check by TLC against stage-Ill, it should be nil. Adjust pH neutral (~7) by adding 1.8ml of acetic acid at -5°C+2°C. Reaction mass was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to
30°C and added 120ml of purified water, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get stage-4111 (Triamcinolone acetonide). Yield=52gm, (95.04%), HPLC Purity=99.21%.
(Purification)
50gm of crude material (0.12 mol) dissolved in 1100ml of acetone (22volume) and 100ml of purified water (02volume) at 50°C than added 2.5gm of activated charcoal and stirred for one hour at same temperature. Filter through hyflow bed and washed with 120ml acetone (2.40volume). Filtrate was distilled below 40°C under vacuum till 3 to 4 volume of input. Cool to 0°C to 5°C and maintained for one hour at same temperature. Filtered and washed with water. Wet material was dried at 45°C to 50°C till moisture content less than 0.50% to get purified stage-4th. Yield =43gm, HPLC Purity=99.40%
-V: (Preparation of Triamcinolone Hexacetonide from Triamcinolone acetonide):

50ml of pyridine (lOvolume) charged in a glass flask and added lOgm of Triamcinolone acetonide (0.023mol) at ambient temperature. Heated to 80°C to 90°C than added 10ml of 3, 3-dimethyl butyryl chloride (O.l lmol) at 80°C to 90°C. Stirred at 80°C to 90°C for 02 hours. In-process check by TLC against Triamcinolone acetonide, it should be nil. Reaction mass cooled to ambient temperature and reaction mass was quenched in 1000ml of purified water (lOOvolume) at ambient temperature, stir for one hour than filtered and washed with purified water till neutral pH (~7). Wet material was dried at 50°C (+5°C) till moisture content less than 1.0% to get stage-5th (Triamcinolone Hexacetonide). Yield=12gm, (97.90%), HPLC Purity=98.63%.
(Purification)
120ml of methanol and 120ml of methylene chloride charged in a glass flask and added 12gm of crude material, stir till clear solution than added 1.2gm of activated charcoal and stir for 30 minutes. Filtered through hyflow bed and washed with 12ml of methanol and 12ml of methylene chloride mixture. Total filtrate was distilled under vacuum at below 40°C till 5 to 6 volume of crude. Cooled to 0°C+5°C and stir for one hour. Filtered and washed with 12ml of precooled methanol. Wet material was dried at 40°C+5°C till moisture content less than 0.50% to get TrimcinolneHexacetonide. Yield=8.8gm, HPLC Purity=99.625%//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses

Aristocort brand triamcinolone cream
Triamcinolone is used to treat a number of different medical conditions, such as eczema, alopecia areata, lichen sclerosus, psoriasis, arthritis, allergies, ulcerative colitis, lupus, sympathetic ophthalmia, temporal arteritis, uveitis, ocular inflammation, keloids, urushiol-induced contact dermatitis, aphthous ulcers (usually as triamcinolone acetonide), central retinal vein occlusion, visualization during vitrectomy and the prevention of asthma attacks.[12][13][14]
The derivative triamcinolone acetonide is the active ingredient in various topical skin preparations (cream, lotion, ointment, aerosol spray) designed to treat skin conditions such as rash, inflammation, redness, or intense itching due to eczema[15] and dermatitis.[16]
Contraindications
Contraindications for systemic triamcinolone are similar to those of other corticoids. They include systemic mycoses (fungal infections) and parasitic diseases, as well as eight weeks before and two weeks after application of live vaccines. For long-term treatment, the drug is also contraindicated in people with peptic ulcers, severe osteoporosis, severe myopathy, certain viral infections, glaucoma, and metastasizing tumours.[17]
There are no contraindications for use in emergency medicine.[4]
Side effects
Further information: Glucocorticoid § Side effects
Side effects of triamcinolone are similar to other corticoids. In short-term treatment up to ten days, it has very few adverse effects; however, sometimes gastrointestinal bleeding is seen, as well as acute infections (mainly viral) and impaired glucose tolerance.[4]
Side effects of triamcinolone long-term treatment may include coughing (up to bronchospasms), sinusitis, metabolic syndrome–like symptoms such as high blood sugar and cholesterol, weight gain due to water retention, and electrolyte imbalance, as well as cataract, thrush, osteoporosis, reduced muscle mass, and psychosis.[5][6][17] Triamcinolone injections can cause bruising and joint swelling.[5] Symptoms of an allergic reaction include rash, itch, swelling, severe dizziness, trouble breathing,[18] and anaphylaxis.[17]
Overdose
No acute overdosing of triamcinolone has been described.[17]
Interactions
Drug interactions are mainly pharmacodynamic, that is, they result from other drugs either adding to triamcinolone’s corticoid side effects or working against its desired effects. They include:[4][17]
- Atropin and other anticholinergics can substantially increase pressure in the eyes.
- Antidiabetic drugs can become less effective because triamcinolone causes diabetes-like symptoms.
- Aspirin and other NSAIDs, as well as anticoagulants such as warfarin, add to the risk of gastrointestinal bleeding.
- Diuretics that excrete potassium (such as loop diuretics and thiazides) can increase the risk of hypokalemia and thus lead to abnormal heart rhythm.
- Cardiac glycosides may have more adverse effects due to reduced potassium levels in the blood.
- The risk for blood count changes is increased when combining triamcinolone with ACE inhibitors.
Triamcinolone and other drugs can also influence each other’s concentrations in the body, amounting to pharmacokinetic interactions such as:[4][17]
- Rifampicin, phenytoin, carbamazepine and other inducers of the liver enzyme CYP3A4[19] speed up metabolization of triamcinolone and can therefore reduce its effectiveness.
- Conversely, CYP3A4 inhibitors such as ketoconazole and itraconazole can increase its concentrations in the body and the risk for adverse effects.
- Blood concentrations of ciclosporin can be increased.
Pharmacology
Mechanism of action
Further information: Glucocorticoid § Mechanism of action
Triamcinolone is a glucocorticoid that is about five times as potent as cortisol, but has very little mineralocorticoid effects.[4]
Pharmacokinetics
When taken by mouth, the drug’s bioavailability is over 90%. It reaches highest concentrations in the blood plasma after one to two hours and is bound to plasma proteins to about 80%. The biological half-life from the plasma is 200 to 300 minutes; due to stable complexes of triamcinolone and its receptor in the intracellular fluid, the total half-life is significantly longer at about 36 hours.[4][5]
A small fraction of the substance is metabolized to 6-hydroxy- and 20-dihydro-triamcinolone; most of it probably undergoes glucuronidation, and a smaller part sulfation. Three quarters are excreted via the urine, and the rest via the faeces.[4][17]
Due to corticoids’ mechanism of action, the effects are delayed as compared to plasma concentrations. Depending on the route of administration and the treated condition, the onset of action can be from two hours up to one or two days after application; and the drug can act much longer than its elimination half-life would suggest.[4][5]
Chemistry
Triamcinolone is a synthetic pregnane corticosteroid and derivative of cortisol (hydrocortisone) and is also known as 1-dehydro-9α-fluoro-16α-hydroxyhydrocortisone or 9α-fluoro-16α-hydroxyprednisolone as well as 9α-fluoro-11β,16α,17α,21-tetrahydroxypregna-1,4-diene-3,20-dione.[20][21]
The substance is a light-sensitive, white to off-white, crystalline powder, or has the form of colourless, matted crystals. It has no odour or is nearly odourless. Information on the melting point varies, partly due to the substance’s polymorphism: 260 to 263 °C (500 to 505 °F), 264 to 268 °C (507 to 514 °F), or 269 to 271 °C (516 to 520 °F) can be found in the literature.[4]
Solubility is 1:500 in water and 1:240 in ethanol; it is slightly soluble in methanol, very slightly soluble in chloroform and diethylether, and practically insoluble in dichloromethane. The specific rotation is {\displaystyle [\alpha ]_{D}^{20}}![{\displaystyle [\alpha ]_{D}^{20}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b67b300e0d318e964fcdab92c872ed89d1d4f43f)
Society and culture
In 2010, TEVA and Perrigo launched the first generic inhalable triamcinolone.[22]
According to Chang et al. (2014), “Triamcinolone acetonide (TA) is classified as an S9 glucocorticoid in the 2014 Prohibited List published by the World Anti-Doping Agency, which caused it to be prohibited in international athletic competition when administered orally, intravenously, intramuscularly or rectally”.[23]
See also
- Glucocorticoid (a chart comparing various glucocorticoids)
References
- ^ “Kenalog Intra-articular / Intramuscular Injection – Summary of Product Characteristics (SmPC)”. (emc). 10 June 2020. Retrieved 20 August 2020.
- ^ “Nasacort Allergy 55 micrograms/dose Nasal Spray suspension – Summary of Product Characteristics (SmPC)”. (emc). 30 August 2018. Retrieved 20 August 2020.
- ^ “Adcortyl Intra-Articular/Intradermal Injection 10mg/ml – Summary of Product Characteristics (SmPC)”. (emc). 11 December 2017. Retrieved 20 August 2020.
- ^ Jump up to:a b c d e f g h i j k l m n Dinnendahl V, Fricke U, eds. (2004). Arzneistoff-Profile (in German). Vol. 10 (19 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. Triamcinolon. ISBN 978-3-7741-9846-3.
- ^ Jump up to:a b c d e f Triamcinolone (systemic) Professional Drug Facts. Accessed 2020-08-19.
- ^ Jump up to:a b c d e f g “Triamcinolone Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ “Triamcinolone Use During Pregnancy”. Drugs.com. Retrieved 3 March 2019.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 486. ISBN 978-3-527-60749-5.
- ^ Vallerand, April Hazard (2018). Davis’s Drug Guide for Nurses. F.A. Davis. p. 365. ISBN 978-0-8036-7000-6.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Triamcinolone – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ Triamcinolone – Drugs.com
- ^ Triamcinolone Inhalation – Drugs.com
- ^ Alcon Receives FDA Approval of Triesence Injectable Triamcinolone Suspension for Use in Eye Surgery – Drugs.com
- ^ Chong M, Fonacier L (December 2016). “Treatment of Eczema: Corticosteroids and Beyond”. Clinical Reviews in Allergy & Immunology. 51 (3): 249–262. doi:10.1007/s12016-015-8486-7. PMID 25869743. S2CID 44337035.
- ^ Eichenfield LF, Tom WL, Berger TG, Krol A, Paller AS, Schwarzenberger K, et al. (July 2014). “Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies”. Journal of the American Academy of Dermatology. 71 (1): 116–32. doi:10.1016/j.jaad.2014.03.023. PMC 4326095. PMID 24813302.
Topical corticosteroids (TCS) are used in the management of AD in both adults and children and are the mainstay of anti-inflammatory therapy.
- ^ Jump up to:a b c d e f g Haberfeld H, ed. (2020). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Volon 4 mg-Tabletten.
- ^ “Drugs and Treatments – Nasacort AQ Nasl – Patient Handout”. WebMD. Retrieved 2008-03-24.
- ^ Moore CD, Roberts JK, Orton CR, et al. (2012). “Metabolic Pathways of Inhaled Glucocorticoids by the CYP3A Enzymes”. Drug Metab. Dispos. 41 (2): 379–389. doi:10.1124/dmd.112.046318. PMC 3558858. PMID 23143891.
- ^ Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1228–. ISBN 978-1-4757-2085-3.
- ^ Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 1054–. ISBN 978-3-88763-075-1.
- ^ Perrigo Announces Launch Of Generic Version Of Nasacort AQ – CBS Detroit
- ^ Chang CW, Huang TY, Tseng YC, Chang-Chien GP, Lin SF, Hsu MC (November 2014). “Positive doping results caused by the single-dose local injection of triamcinolone acetonide”. Forensic Science International. 244: 1–6. doi:10.1016/j.forsciint.2014.07.024. PMID 25126738.
External links
- “Triamcinolone”. Drug Information Portal. U.S. National Library of Medicine.
- “Triamcinolone Topical”. MedlinePlus.
- “Triamcinolone Nasal Spray”. MedlinePlus.
- “Triamcinolone Acetonide Cream”. HealthClubFinder.
{kind=link}
{kind=link}
///////////////TRIAMCINOLONE, TU3850000, トリアムシノロン , 去炎松 , Glucocorticoid
[H][C@@]12C[C@@H](O)[C@](O)(C(=O)CO)[C@@]1(C)C[C@H](O)[C@@]1(F)[C@@]2([H])CCC2=CC(=O)C=C[C@]12C

NEW DRUG APPROVALS
ONE TIME
$10.00
CDSCO INDIA APPROVED 20.01.2022
Triamcinolone Hexacetonide injectable suspension
20mg/ml
For intraarticular, intra-synovial or
periarticular use in adults and adolescents for
the symptomatic treatment of subacute and
chronic inflammatory joint diseases including
rheumatoid arthritis and Juvenile Idiopathic
Arthritis (JIA), Osteoarthritis and posttramautic arthritis, Synovitis, tendinitis,
bursitis and epicondylitis.
Triamcinolone hexacetonide (brand name Aristospan; also known as triamcinolone acetonide 21-tebutate) is a synthetic glucocorticoid corticosteroid.[1][2][3]
References
- ^ Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 1228–. ISBN 978-1-4757-2085-3.
- ^ Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. p. 1657. ISBN 978-3-88763-075-1.
- ^ Morton IK, Hall JM (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 280–. ISBN 978-94-011-4439-1.
UPDATE
| Clinical data | |
|---|---|
| Trade names | Aristospan |
| Other names | Triamcinolone acetonide 21-tebutate; Triamcinolone acetonide 21-(tert-butylacetate); 9α-Fluoro-11β,16α,17α,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone, 21-(3,3-dimethylbutyrate); 9α-Fluoro-11β-hydroxy-16α,17α-((1-methylethylidene)bis(oxy))pregna-1,4-diene-3,20-dione 21-(3,3-dimethylbutyrate) |
| Drug class | Corticosteroid; Glucocorticoid |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 5611-51-8 |
| PubChem CID | 21826 |
| ChemSpider | 20516 |
| UNII | I7GT1U99Y9 |
| ChEBI | CHEBI:9670 |
| ChEMBL | ChEMBL1200878 |
| CompTox Dashboard (EPA) | DTXSID0048634 |
| ECHA InfoCard | 100.024.575 |
| Chemical and physical data | |
| Formula | C30H41FO7 |
| Molar mass | 532.649 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
{kind=link}
ENSITRELVIR

Ensitrelvir
S-217622, S 217622, Xocova, SHIONOGI,
6-[(6-chloro-2-methylindazol-5-yl)amino]-3-[(1-methyl-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-1,3,5-triazine-2,4-dione
CAS 2647530-73-0
| C22H17ClF3N9O2531.9 | |
| Synonyms | BDBM513874bioRxiv20220126.477782, S-217622 |
|---|
Ensitrelvir fumarate
CAS No. : 2757470-18-9
C22 H17 Cl F3 N9 O2 . C4 H4 O4
1,3,5-Triazine-2,4(1H,3H)-dione, 6-[(6-chloro-2-methyl-2H-indazol-5-yl)imino]dihydro-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-, (6E)-, (2E)-2-butenedioate (1:1)
| Formula: | C26H21ClF3N9O6 |
|---|---|
| M. Wt. : | 647.95 |
A Phase 1 study of S-217622 in healthy adult participants (jRCT2031210202)
Japan Registry of Clinical Trials Web Site 2021, July 16
PMDA APPROVED 2022/11/22, Xocova
Ensitrelvir[1] (code name S-217622, brand name Xocova)[2] is an antiviral drug developed by Shionogi in partnership with Hokkaido University, which acts as an orally active 3C-like protease inhibitor for the treatment of COVID-19 infection.[3][4] It is taken by mouth, and has been successfully tested against the recently emerged Omicron variant.[5]
About S-217622
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
J.Med.Chem.2024,67,4376−4418
Ensitrelvir fumaric acid (3), also referred to as S-217622, is an oral noncovalent SARS-CoV-2 main protease (Mpro) inhibitordeveloped by Shionogi & Co. that was approved by the japan Pharmaceuticals and Medical Devices Agency (PMDA)for the treatment of disease caused by SARS-CoV-2 (COVID
19) infection. Dosed once daily for 5 days, ensitrelvirsuppresses the replication of SARS-CoV-2 in infected patients as a result of its inhibition of the viral mpro.25,26
Ensitrelvir retains potent inhibitory activity against many of the most common M mutants and exhibits antiviral activity against a wide variety of circulating SARS-CoV-2 variants. 27is the second Mpro
Ensitrelvir inhibitor approved for the treatment of 28 disease caused by COVID-19. Unlike the first approved treatment, Paxlovid, ensitrelvir does not require coadministration with a CYP3A4 inhibitor to attenuate metabolism in vivo.
Furthermore, crystal structures of ensitrelvir in complex with the main proteases of three other human-infecting coronaviruses (MERS-CoV, SARS-CoV, and HCoV-NL63)
A convergent, kilogram-scale synthesis of ensitrelvir suitable for manufacturing has been described in the literature by researchers at Shionogi. 30 The synthetic approach involved the union of two key building blocks indazole 3.7 and 1,3,5triazinone 3.14, each necessitating development of a scale worthy route. The preparation of triazinone 3.14 necessitated construction of a triazolyl methylene chloride subunit which
began with the reduction of triazole ester 3.1 with aluminum hydride 3.2 (a less pyrophoric alternative to LAH yet still required aqueous Rochelle salt quench to chelate excess aluminum) 31to provide alcohol 3.3, which was then convertedto the corresponding chloride and isolated as the triazole HCl
salt 3.4 (Scheme 6). Assembly of indazole intermediate 3.7began with regioselective nitration of benzaldehyde 3.5followed by treatment with hydrazine hydrate in aqueous EtOHtoprovide indazole3.6(Scheme7).Fascinatingly, the Shionogi team isolated a variety of byproducts during the
conversionof3.5to3.6whichsupportedtheirhypothesisforareaction mechanism that likely equilibrated through a dibenzylidenehydrazine intermediateenroute tothedesired
indazole3.6.Ascreenofelectrophilicmethyl sourcesrevealed thatMeerwein’s salt facilitatedthebest conversionof 3.6to the correspondingN2-monomethylated indazole; subsequenthydrogenative nitro reduction furnished the key indazole intermediate 3.7.Construction of the ensitrelvir core started with reaction of carboximidamide 3.8 with t-butyl isocyanate followed by N,N′carbonyldiimidazole (CDI) to secure 1,3,5-triazinone 3.10(Scheme 8). Subsequent N-alkylation with bromide 3.11provided benzyl triazinone 3.12. Substitution of the pyrazolewith m-cresol was accomplished under acidic conditions. The
authors report that m-cresol was identified as a leaving group that facilitated introduction of indazole 3.7 with a minimal number of byproducts in a later step of the synthesis. The TFA-mediated reaction concomitantly removed the N-tertbutyl group providing compound 3.13 in 91% yield. Nalkylation with chloride 3.4 in the presence of a base resulted in intermediate 3.14 which was then treated with building
block 3.7 in the presence of anhydrous acetic acid. Isolation of ensitrelvir fumaric acid was achieved by exposure to fumaric acid in aqueous acetone.
(25) Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.;
Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.;
Mukae, H. A phase 2/3 study of S-217622 in participants with SARS
CoV-2 infection (Phase 3 part). Medicine 2023, 102, No. e33024.
(26) Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura,
T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.;
Takeda, Y.; Tsuge, Y.; Uehara, T. A randomized phase 2/3 study of
ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in
Japanese patients with mild-to-moderate COVID-19 or asymptomatic
SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents
Chemother. 2022, 66, No. 00697.
(27) Kawashima, S.; Matsui, Y.; Adachi, T.; Morikawa, Y.; Inoue, K.;
Takebayashi, S.; Nobori, H.; Rokushima, M.; Tachibana, Y.; Kato, T.
Ensitrelvir is effective against SARS-CoV-2 3CL protease mutants
circulating globally. Biochem. Biophys. Res. Commun. 2023, 645, 132−
136.
(28) Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.;
Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.;
et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL
protease inhibitor clinical candidate for treating COVID-19. J. Med.
Chem. 2022, 65, 6499−6512
(29) Lin, C.; Jiang, H.; Li, W.; Zeng, P.; Zhou, X.; Zhang, J.; Li, J.
Structural basis for the inhibition of coronaviral main proteases by
ensitrelvir. Structure 2023, 31, 1016.
(30) Kawajiri, T.; Kijima, A.; Iimuro, A.; Ohashi, E.; Yamakawa, K.;
Agura, K.; Masuda, K.; Kouki, K.; Kasamatsu, K.; Yanagisawa, S.; et al.
Development of a manufacturing process toward the convergent
synthesis of the COVID-19 antiviral Ensitrelvir. ACS Cent. Sci. 2023,
9, 836−843.
(31) Gugelchuk, M.; Silva, III, L. F.; Vasconcelos, R. S.; Quintiliano,
S. A. P. Sodium bis(2-methoxyethoxy)aluminum hydride. In
Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J.,
Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd., 2007.


Syn
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
View ORCID ProfileYuto Unoh, View ORCID ProfileShota Uehara, View ORCID ProfileKenji Nakahara, View ORCID ProfileHaruaki Nobori, Yukiko Yamatsu, View ORCID ProfileShiho Yamamoto, View ORCID ProfileYuki Maruyama, View ORCID ProfileYoshiyuki Taoda, View ORCID ProfileKoji Kasamatsu, View ORCID ProfileTakahiro Suto, Kensuke Kouki, View ORCID ProfileAtsufumi Nakahashi, Sho Kawashima, View ORCID ProfileTakao Sanaki, Shinsuke Toba, Kentaro Uemura, Tohru Mizutare, View ORCID ProfileShigeru Ando, View ORCID ProfileMichihito Sasaki, View ORCID ProfileYasuko Orba, View ORCID ProfileHirofumi Sawa, View ORCID ProfileAkihiko Sato, View ORCID ProfileTakafumi Sato, View ORCID ProfileTeruhisa Kato, View ORCID ProfileYuki Tachibana
doi: https://doi.org/10.1101/2022.01.26.477782
https://www.biorxiv.org/content/10.1101/2022.01.26.477782v1.full
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry

The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012

Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.

Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.

Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
(6E)-6-[(6-Chloro-2-methyl-2H-indazol-5-yl)imino]-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluorobenzyl)-1,3,5-triazinane-2,4-dione (3, S-217622)
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
- Supporting information[supplements/477782_file02.pdf]
see spectrum at end of page
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]

NEW DRUG APPROVALS
ONE TIME
$10.00
References
- ^ World Health Organization (2021). “International Nonproprietary Names for Pharmaceutical Substances. Proposed INN: List 126” (PDF). WHO Drug Information. 35 (4): 1135.
- ^ Xocova: Powerful New Japanese Pill for Coronavirus Treatment. BioPharma Media, February 2022
- ^ Jump up to:a b Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. (January 2022). “Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19”. bioRxiv. doi:10.1101/2022.01.26.477782. S2CID 246367525.
- ^ “Shionogi presents positive Ph II/III results for COVID-19 antiviral S-217622”. thepharmaletter.com. 31 January 2022.
- ^ Shionogi’s new COVID pill appears to ease omicron symptoms. Nikkei Asia, 21 December 2021
- ^ Japan to consider early approval for Shionogi COVID-19 pill. Japan Times, 8 February 2022
- ^ https://www.reuters.com/business/healthcare-pharmaceuticals/japans-shionogi-seeks-approval-oral-covid-19-drug-2022-02-25/[bare URL]
- ^ “Japan’s Shionogi seeks approval for COVID-19 pill”. Reuters. Reuters. 25 February 2022.
| Clinical data | |
|---|---|
| Other names | S-217622 |
| Identifiers | |
| showIUPAC name | |
| PubChem CID | 162533924 |
| Chemical and physical data | |
| Formula | C22H17ClF3N9O2 |
| Molar mass | 531.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
{kind=link}
Journal reference:
- Sasaki, M., Tabata, K., Kishimoto, M., et al. (2022). Oral administration of S-217622, a SARS-CoV-2 main protease inhibitor, decreases the viral load and accelerates recovery from clinical aspects of COVID-19. bioRxiv. doi:10.1101/2022.02.14.480338. https://www.biorxiv.org/content/10.1101/2022.02.14.480338v1.full.
///////////Ensitrelvir, S-217622, S 217622, Xocova, SHIONOGI, CORONA VIRUS, covid 19


Cyclobenzaprine


Cyclobenzaprine
- Molecular FormulaC20H21N
- Average mass275.387 Da
- MK-130
- TNX-102
1-(3-Dimethylaminopropylidene)-2,3:6,7-dibenzo-4-suberene
1-Propanamine, 3-(5H-dibenzo[a,d]cyclohepten-5-ylidene)-N,N-dimethyl-[ACD/Index Name]
206-145-8[EINECS]
3-(5H-Dibenzo[a,d]cyclohepten-5-ylidene)-N,N-dimethyl-1-propanamine
303-53-7[RN]
5-(3-Dimethylaminopropylidene)dibenzo[a,e]cycloheptatriene
циклобензаприн[Russian][INN]
سيكلوبنزابرين[Arabic][INN]
环苯扎林[Chinese][INN]
Cyclobenzaprine, CAS Registry Number: 303-53-7
CAS Name: 3-(5H-Dibenzo[a,d]cyclohepten-5-ylidene)-N,N-dimethyl-1-propanamine
Additional Names:N,N-dimethyl-5H-dibenzo[a,d]cyclohepten-D5,g-propylamine; 5-(3-dimethylaminopropylidene)dibenzo[a,e]cycloheptatriene; 1-(3-dimethylaminopropylidene)-2,3:6,7-dibenzo-4-suberene; proheptatriene
Manufacturers’ Codes: MK-130; Ro-4-1577; RP-9715
Molecular Formula: C20H21N, Molecular Weight: 275.39
Percent Composition: C 87.23%, H 7.69%, N 5.09%
Literature References: Prepn: GB858187 (1961 to Hoffmann-La Roche); Villani et al.,J. Med. Pharm. Chem.5, 373 (1962); Winthrop et al.,J. Org. Chem.27, 230 (1962). Pharmacology: C. D. Barnes, W. L. Adams, Neuropharmacology17, 445 (1978); N. N. Share, ibid. 721; and toxicology: J. Metysova et al.,Arch. Int. Pharmacodyn. Ther.144, 481 (1963). Metabolism: G. Belvedere et al.,Biomed. Mass Spectrom.1, 329 (1974); H. B. Hucker et al.,Drug Metab. Dispos.6, 184 (1978). Bioavailability: eidem,J. Clin. Pharmacol.17, 719 (1977). Clinical studies: J. V. Basmajian, Arch. Phys. Med. Rehabil.5, 58 (1978); B. R. Brown, J. Womble, J. Am. Med. Assoc.240, 1151 (1978). Comprehensive description: M. L. Cotton, G. R. B. Down, Anal. Profiles Drug Subs.17, 41-72 (1988).
Properties: bp1 175-180°. uv max: 224, 289 nm (log e 4.57, 4.02), (Villani et al.)
Boiling point: bp1 175-180°
Absorption maximum: uv max: 224, 289 nm (log e 4.57, 4.02), (Villani et al.)
Derivative Type: Hydrochloride
CAS Registry Number: 6202-23-9
Trademarks: Flexeril (Merck & Co.); Flexiban (Merck & Co.)
Molecular Formula: C20H21N.HCl, Molecular Weight: 311.85
Percent Composition: C 77.03%, H 7.11%, N 4.49%, Cl 11.37%
Literature References: Use as muscle relaxant: N. N. Share, FR2100873 (1972 to Frosst), C.A.78, 47801n (1973).
Properties: Crystals from isopropanol, mp 216-218°. Soly in water: >20 g/100 ml. Freely sol in water, methanol, ethanol; sparingly sol in isopropanol; slightly sol in chloroform, methylene chloride. Practically insol in hydrocarbons. uv max: 226, 295 nm (e 52300, 12000). LD50 in mice (mg/kg): 35 i.v., 250 orally (Metysova).
Melting point: mp 216-218°
Absorption maximum: uv max: 226, 295 nm (e 52300, 12000)
Toxicity data: LD50 in mice (mg/kg): 35 i.v., 250 orally (Metysova)
Therap-Cat: Muscle relaxant (skeletal).
Keywords: Muscle Relaxant (Skeletal).
Cyclobenzaprine, a centrally-acting muscle relaxant, was first synthesized in 196111 and has been available for human use since 1977.10 It was initially studied for use as antidepressant given its structural similarity to tricyclic antidepressants – it differs from Amitriptyline by only a single double bond.11,10 Since its approval, it has remained relatively popular as an adjunctive, short-term treatment for acute skeletal muscle spasms secondary to musculoskeletal injury.
Cyclobenzaprine (sold under the brand name Flexeril, among others) is a medication used for muscle spasms from musculoskeletal conditions of sudden onset.[6] It is not useful in cerebral palsy.[6] It is taken by mouth.[6] Use is not recommended for more than a few weeks.[6]
Common side effects include headache, feeling tired, dizziness, and dry mouth.[6] Serious side effects may include an irregular heartbeat.[6] There is no evidence of harm in pregnancy, but it has not been well studied in this population.[6] It should not be used with an MAO inhibitor.[6] How it works is unclear.[6]
Cyclobenzaprine was approved for medical use in the United States in 1977.[6] It is available as a generic medication.[6] In 2019, it was the 45th most commonly prescribed medication in the United States, with more than 15 million prescriptions.[7][8] It was not available in the United Kingdom as of 2012.[9]
Synthesis Reference
Villani, F.J.; US. Patent 3,409,640; November 5,1968; assigned to Schering Corporation.
Paper
By: Gowda, Narendra B.; Rao, Gopal Krishna; Ramakrishna, Ramesha A.
Tetrahedron Letters (2010), 51, (43), 5690-5693.
https://www.sciencedirect.com/science/article/abs/pii/S0040403910014668
A simple and convenient protocol for deoxygenation of aliphatic and aromatic N-oxides to the corresponding amines in good to excellent yield using sodium borohydride–Raney nickel in water is reported. Other functional moieties such as alkenes, halides, ethers, and amides are unaffected under the present reaction condition.
Graphical abstract

Cyclobenzaprine N-oxide, CAS RN: 6682-26-4
Dissolve (1 mmol) of cyclobenzaprine N-oxide in 2.5 mL of water at 60 °C. 2. Add Raney nickel (0.10 g, W6 grade) to the solution. 3. Stir the reaction mixture for 10 minutes. 4. Add (2 mmol) of sodium borohydride slowly in portions over 15-20 minutes to the reaction mixture. 5. Stir the reaction mixture at the same temperature for 2.5 hours (the completion of the reaction as monitored by TLC). 6. Once the reaction is completed, add chloroform (50 mL) to the reaction mixture. 7. Filter the resulted mixture to remove Raney nickel. 8. Dry the chloroform layer over anhydrous magnesium sulfate. 9. Filter the reaction mixture. 10. Evaporate the solvent under vacuum. 11. Purify the obtained residue through short path flash chromatography with silica gel and chloroform.
1H NMR (400 MHz, CDCl3) δ: 1.12 (s, 6H, N-CH3), 1.23- 1.34 (m, 4H, CH2), 4.58 (t, J= 4.0 Hz, 1H, CH), 5.82(d, J= 4.0 Hz, 2H, CH), 6.21- 6.33 (m, 8H, ArH).
13C NMR (100 MHz, CDCl3) δ: 27.89, 45.93, 60.12, 127.40, 127.55, 128.30, 128.59, 128.92, 129.33, 129.45, 129.67, 131.74, 131.96, 132.40, 134.63, 135.39, 137.97, 142.95, 143.30.
SYN
PATENT
https://patents.google.com/patent/WO2012098563A2/en
Cyclobenzaprine hydrochloride, chemically known as 5-(3-dimethylaminopropylidene)- dibenzo (a,e) cycloheptatriene hydrochloride (Formula I),

Formula I is a commonly prescribed tricyclic amine having muscle relaxant pharmaceutical activity. After sustaining an injury, muscle spasms may occur to stabilize the affected body part and prevent further damage. Cyclobenzaprine hydrochloride is used to treat such muscle spasm associated with acute, painful musculoskeletal conditions.
Few multistep processes for the preparation of this tricyclic amine are already available in the literature which involves isolation and purification of intermediate compounds. The conventional route of synthesis as reported in US3454643, ES8201950 includes preparation of Grignard reagent (GR) of 3-dimethylaminopropyl chloride in a first step, reacting with 5-dibenzosuberenone (Formulall) in a second step. The reaction mass was extracted with benzene, solid obtained was recrystallized from alcohol to produce 5- hydroxy intermediate (Formula III) and further dehydrated in third step using acetyl chloride or acetic anhydride in presence of chloroform as a solvent medium followed by purging HC1 gas to produce hydrochloride salt (Formula I). CH,
CI-(CH2)3 NS
CH,
Dimeth laminopropyl chloide

Di methy lam i nopropy I 5-dibenzosubrenone – y roxy compoun magnesium chloide
(Formula II) (Formula III)

Cyclobenzaprine base Cyclobenzaprine hydrochloride
(Formula IV) (Formula I)
The multistep synthesis is cumbersome and use of hazardous solvents and reagents like chloroform, benzene and acetyl chloride etc are not recommended for the preparation of pharmaceutical substances.
J. Org. Chem. Vol. 27, 230-240 (1961) also portrayed similar procedure for the synthesis of cyclobenzaprine hydrochloride, wherein 5-hydroxy compound of formula III was isolated and recrystallized before dehydration reaction.
Synthetic Comm. 11 (3), 241-246 (1981) described a process which involves isolation and purification of the intermediate at magnesium -complex stage. Hydrolysis of the isolated complex afforded desired tricyclic amine. GB858186 and GB858187 jointly described a process which comprises preparation of 5- hydroxy compound (Formula III) and subsequent conversion of the same to cyclobenzaprine hydrochloride. However the overall yield reported is significantly low.
In a different approach, a high temperature dehydrogenation of amitriptyline base resulting in formation of cyclobenzaprine hydrochloride is reported in Indian patent application 387/CHE/2005.

. EXAMPLE:
In a reaction vessel, THF (1 10ml), magnesium turnings 20gm (0.823mole) were charged and the mixture was warmed to 45-55°C for 20 min. A solution of l OOgm (0.823mole) of 3-dimethylaminopropyl chloride prepared in 1 10ml THF was added dropwise to the reaction mixture by controlling the reflux generated due to reaction initiation and maintained for 2hrs. The formed Grignard reagent was then cooled to 0-5°C and a solution of lOOgm (0.485mole) 5-dibenzosuberenone prepared in 220ml THF was charged to the reaction mass at temperature below 10°C. The reaction mass was stirred for 45 min at temperature 10-15°C. The absence of 5-dibenzosuberenone was checked by TLC and 770ml of 20% aq. HC1 was charged to the reaction mass at a temperature below 10°C. The reaction mass was then heated to 70-80°C for 3 hrs. The acidic mass was neutralized by using aqueous Na2C03 solution and extracted with 900ml methylene dichloride. The solvent was removed completely under reduced pressure and oil thus formed was dissolved in 450ml IPA and acidified with 240 ml of 20% IPA .HC1 solution and stirred for 2 hrs at 0-5°C for complete precipitation. The precipitate is filtered, recrystallized from IPA (800 ml) and dried to obtain 1 18 gm (78%) white crystalline cyclobenzaprine hydrochloride with purity 99.93% by HPLC.

PATENT
PATENT
CN 111393305
CLIP
Muscle Relaxants
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Cyclobenzaprine
Cyclobenzaprine, N,N-dimethyl-3-(dibenzo[a,d]cyclohepten-5-ylidene) propylamine (15.3.9), is synthesized by reacting 5H-dibenzo[a,d]cyclohepten-5-one with 3-dimethylaminopropylmagnesium chloride and subsequent dehydration of the resulting carbinol (15.3.8) in acidic conditions into cyclobenzaprine (15.3.9) [30–32].

Cyclobenzaprine is structurally similar to tricyclic antidepressants. It acts at the brain stem level. It is used as an adjuvant agent for relieving muscle spasms associated with severe diseased conditions of the muscle. A synonym of this drug is flexeril.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical use
Cyclobenzaprine is used, in conjunction with physical therapy, to treat muscle spasms that occur because of acute musculoskeletal conditions.[10] After sustaining an injury, muscle spasms to stabilize the affected body part occur, which may increase pain to prevent further damage. Cyclobenzaprine is used to treat such muscle spasms associated with acute, painful musculoskeletal conditions.[11] It decreases pain in the first two weeks,[12][13] peaking in the first few days, but has no proven benefit after two weeks.[12][14] Since no benefit is proven beyond that, therapy should not be continued long-term.[11] It is the best-studied muscle relaxer.[12] It is not useful for spasticity due to neurologic conditions such as cerebral palsy.[11][15]
A 2004 review found benefit for fibromyalgia symptoms, with a reported number needed to treat of 4.8 (meaning that 1 person out of every 4.8 benefits from treatment) for pain reduction, but no change in fatigue or tender points.[16] A 2009 Cochrane review found insufficient evidence to justify its use in myofascial pain syndrome.[17] It may also be used along with other treatments for tetanus.[18]
Side effects
Cyclobenzaprine results in increased rates of drowsiness (38%), dry mouth (24%), and dizziness (10%).[14] Drowsiness and dry mouth appear to intensify with increasing dose.[19] The sedative effects of cyclobenzaprine are likely due to its antagonistic effect on histamine, serotonin, and muscarinic receptors.[medical citation needed]
Agitation is a common side effect observed, especially in the elderly. Some experts[who?] believe that cyclobenzaprine should be avoided in elderly patients because it can cause confusion, delirium, and cognitive impairment.[20][21] In general, the National Committee for Quality Assurance recommends avoiding the use of cyclobenzaprine in the elderly because of the potential for more severe side effects.[22]
Dysphagia, a life-threatening side-effect, may rarely occur.[23] Treatment protocols and support should follow the same as for any structurally related tricyclic, such as tricyclic antidepressants.[24]
Overdose
The most common effects of overdose are drowsiness and tachycardia.[11] Rare but potentially critical complications are cardiac arrest, abnormal heart rhythms, severe low blood pressure, seizures, and neuroleptic malignant syndrome.[11] Life-threatening overdose is rare,[11] however, as the median lethal dose is about 338 milligrams/kilogram in mice and 425 mg/kg in rats.[11] The potential harm is increased when central nervous system depressants and antidepressants are also used; deliberate overdose often includes alcohol among other drugs.[11]
Interactions
Cyclobenzaprine has major contraindications with monoamine oxidase inhibitors (MAOIs). At least one study also found increased risk of serotonin syndrome when cyclobenzaprine was taken with the serotonergic drugs duloxetine or phenelzine.[25]
These substances may interact with cyclobenzaprine:
- Central nervous system depressants (e.g. alcohol, opioids, benzodiazepines, nonbenzodiazepines, phenothiazines, carbamates, barbiturates, major tranquilizers)
- Monoamine oxidase inhibitors taken within two weeks of cyclobenzaprine may result in serious, life-threatening side effects.[11]
Cyclobenzaprine may affect the medications used in surgical sedation and some surgeons request that patients temporarily discontinue its use prior to surgery.[26]
Pharmacology
Cyclobenzaprine is a centrally acting muscle relaxant.[27] Cyclobenzaprine is a 5-HT2 receptor antagonist; it relieves muscle spasm through action on the central nervous system at the brain stem, rather than targeting the peripheral nervous system or muscles themselves.[28]
Pharmacodynamics
| Site | CBP | NCBP | Action | Ref |
|---|---|---|---|---|
| 5-HT1A | 5.3 | 3.2 | Agonist | [29] |
| 5-HT2A | 5.2 | 13 | Antagonist | [29] |
| 5-HT2B | 100 | ??? | Antagonist | [29] |
| 5-HT2C | 5.2 | 43 | Antagonist | [29] |
| α1A | 5.6 | 34 | ND | [29] |
| α2A | 4.3 | 6.4 | Antagonist | [29] |
| α2B | 21 | 150 | ND | [29] |
| α2C | 21 | 48 | ND | [29] |
| H1 | 1.3 | 5.6 | ND | [29] |
| M1 | 7.9 | 30 | ND | [29] |
| Values are Ki (nM), unless otherwise noted. The smaller the value, the more strongly the drug binds to the site. |
Pharmacokinetics
Cyclobenzaprine has an oral bioavailability of about 55% and approximately 93% is bound to proteins in plasma. The half-life of the drug is 18 hours and it has a plasma clearance of 0.7 litres per minute.[27][30][31]
Comparison to other medications
Cyclobenzaprine has been found to be not inferior to tizanidine, orphenadrine, and carisoprodol in the treatment of acute lower back pain, although none have been proven to be effective for long-term use (beyond two weeks of treatment). No differences in pain or spasm scores were noted among these agents, nor when compared to benzodiazepines.[32] However, nonbenzodiazepine (including cyclobenzaprine) treatment was found to have a lower risk of medication abuse and continuation of use against medical advice.[medical citation needed] Side effects such as sedation and ataxia are also less pronounced with nonbenzodiazepine antispasmodics.[medical citation needed]
In a study on the treatment of musculoskeletal pain treatment with cyclobenzaprine alone or in combination with ibuprofen, no significant differences in pain scores were noted among the three treatment groups. Peak benefit was found to occur on day seven of the treatment for all groups.[33]
Formulations

Cyclobenzaprine 10mg tablets
By mouth, cyclobenzaprine is marketed as Apo-Cyclobenzaprin, Fexmid, Flexeril and Novo-Cycloprine. It is available in generic form. A once-a-day, extended-release formulation, Amrix, is available.[34] Cyclobenzaprine is also used by compounding pharmacies in topical creams.[citation needed]
References
- ^ Micromedex® 2010 – DRUGDEX Evaluations (Cyclobenzaprine Hydrochloride)
- ^ “Cyclobenzaprine Hydrochloride Tablets USP Revised: April 2005 Rx only”. nih.gov. Retrieved 1 October 2016.
- ^ Teva Pharmaceuticals USA, Inc (May 2016). “AMR40470 (Amrix) Prescribing Information” (PDF).
- ^ U.S. Food and Drug Administration. “NDA 17-821/S-045 Flexeril (Cyclobenzaprine HCl) Tablets” (PDF).
- ^ Teva Pharmaceuticals USA, Inc (May 2016). “AMR40470 (Amrix) Prescribing Information” (PDF).
- ^ Jump up to:a b c d e f g h i j k “Cyclobenzaprine Monograph for Professionals”. Drugs.com. AHFS. Retrieved 22 December 2018.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Cyclobenzaprine – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ “Fibromyalgia, psychiatric comorbidity, and the somatosensory cortex”. British Journal of Medical Practitioners. 5 (2): a522. 2012.
- ^ Yang YW, Macdonald JB, Nelson SA, Sekulic A (December 2017). “Treatment of vismodegib-associated muscle cramps with cyclobenzaprine: A retrospective review”. Journal of the American Academy of Dermatology. 77 (6): 1170–1172. doi:10.1016/j.jaad.2016.12.017. PMID 29132849. S2CID 8265576.
- ^ Jump up to:a b c d e f g h i “Cyclobenzaprine- cyclobenzaprine hydrochloride tablet, film coated”. DailyMed. 30 December 2019. Retrieved 26 September 2020.
- ^ Jump up to:a b c Chou R, Peterson K, Helfand M (August 2004). “Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review”. Journal of Pain and Symptom Management. 28 (2): 140–75. doi:10.1016/j.jpainsymman.2004.05.002. PMID 15276195.
- ^ van Tulder MW, Touray T, Furlan AD, Solway S, Bouter LM (2003). Van Tulder MW (ed.). “Muscle relaxants for non-specific low back pain”. The Cochrane Database of Systematic Reviews. 2 (2): CD004252. doi:10.1002/14651858.CD004252. PMC 6464310. PMID 12804507.
- ^ Jump up to:a b Browning R, Jackson JL, O’Malley PG (July 2001). “Cyclobenzaprine and back pain: a meta-analysis”. Archives of Internal Medicine. 161 (13): 1613–20. doi:10.1001/archinte.161.13.1613. PMID 11434793.
- ^ Ashby P, Burke D, Rao S, Jones RF (October 1972). “Assessment of cyclobenzaprine in the treatment of spasticity”. Journal of Neurology, Neurosurgery, and Psychiatry. 35 (5): 599–605. doi:10.1136/jnnp.35.5.599. PMC 494138. PMID 4563483.
- ^ Tofferi JK, Jackson JL, O’Malley PG (February 2004). “Treatment of fibromyalgia with cyclobenzaprine: A meta-analysis”. Arthritis and Rheumatism. 51 (1): 9–13. doi:10.1002/art.20076. PMID 14872449.
- ^ Leite FM, Atallah AN, El Dib R, Grossmann E, Januzzi E, Andriolo RB, da Silva EM (July 2009). “Cyclobenzaprine for the treatment of myofascial pain in adults”. The Cochrane Database of Systematic Reviews (3): CD006830. doi:10.1002/14651858.CD006830.pub3. PMC 6481902. PMID 19588406.
- ^ Smith BT (2014). Pharmacology for Nurses. Jones & Bartlett Publishers. p. 122. ISBN 9781449689407.
- ^ “Flexeril: Side effects”. RxList.com. Archived from the original on 12 September 2008. Retrieved 22 February 2010.
- ^ “Long-term Use of Cyclobenzaprine for Pain: A Review of the Clinical Effectiveness”. CADTH Rapid Response Reports. Ottawa, Ontario: Canadian Agency for Drugs and Technologies in Health. 23 February 2015. PMID 25763449.
- ^ Potentially inappropriate medications for the elderly according to the revised Beers criteria. 2012. Duke Clinical Research Institute website. [1]
- ^ “High risk medications” (PDF). National Committee for Quality Assurance. Archived from the original (PDF) on 1 February 2010. Retrieved 22 February 2010.
- ^ “MEDICATIONS AND DYSPHAGIA/ SWALLOWING RISKS” (PDF).
- ^ Chabria SB (July 2006). “Rhabdomyolysis: a manifestation of cyclobenzaprine toxicity”. Journal of Occupational Medicine and Toxicology. 1 (1): 16. doi:10.1186/1745-6673-1-16. PMC 1540431. PMID 16846511.
- ^ Keegan MT, Brown DR, Rabinstein AA (December 2006). “Serotonin syndrome from the interaction of cyclobenzaprine with other serotoninergic drugs”. Anesthesia and Analgesia. 103 (6): 1466–8. doi:10.1213/01.ane.0000247699.81580.eb. PMID 17122225.
- ^ Medical Practice of William H. Gorman, M.D. (18 February 2014). “Medications to Avoid, Continue, or Stop – Before & After Surgery”.
- ^ Jump up to:a b “Cyclobenzaprine”. http://www.drugbank.ca.
- ^ Kobayashi H, Hasegawa Y, Ono H (September 1996). “Cyclobenzaprine, a centrally acting muscle relaxant, acts on descending serotonergic systems”. European Journal of Pharmacology. 311 (1): 29–35. doi:10.1016/0014-2999(96)00402-5. PMID 8884233.
- ^ Jump up to:a b c d e f g h i j k “Cyclobenzaprine (CBP) and Its Major Metabolite Norcyclobenzaprine (nCBP) Are Potent Antagonists of Human Serotonin Receptor 2a (5HT2a), Histamine Receptor H-1 and á-Adrenergic Receptors: Mechanistic and Safety Implications for Treating Fibromyalgia Syndrome by Improving Sleep Quality”. ACR Meeting Abstracts. Retrieved 27 January 2022.
- ^ “Cyclobenzaprine”. pubchem.ncbi.nlm.nih.gov.
- ^ Winchell GA, King JD, Chavez-Eng CM, Constanzer ML, Korn SH (January 2002). “Cyclobenzaprine pharmacokinetics, including the effects of age, gender, and hepatic insufficiency”. Journal of Clinical Pharmacology. 42 (1): 61–9. doi:10.1177/0091270002042001007. PMID 11808825. S2CID 7749001.
- ^ “Medscape: Medscape Access”. medscape.com. Retrieved 1 October 2016.
- ^ Childers MK, Petri M, Laudadio C, Harrison D, Silber S, Bowen D (2004). “Comparison of cyclobenzaprine alone versus cyclobenzaprine plus ibuprofen in patients with acute musculoskeletal spasm and pain”. Annals of Emergency Medicine. 44 (4): S87–S88. doi:10.1016/j.annemergmed.2004.07.286.
- ^ “Patient Web site for Amrix (Cyclobenzaprine Hydrochloride Extended‐Release Capsules)”. amrix.com. Retrieved 1 October 2016.
External links
- “Cyclobenzaprine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Flexeril, Amrix, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682514 |
| License data | US DailyMed: Cyclobenzaprine |
| Routes of administration | By mouth |
| ATC code | M03BX08 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 33–55%[1][2] |
| Protein binding | 93% |
| Metabolism | major: CYP3A4, CYP1A2; minor: CYP2D6, N-demethylation[5] |
| Metabolites | Norcyclobenzaprine |
| Elimination half-life | 32 hours (extended-release, range 8-37 hours),[3] 18 hours (immediate release, range 8–37 hours)[4] |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 303-53-7 |
| PubChem CID | 2895 |
| IUPHAR/BPS | 7152 |
| DrugBank | DB00924 |
| ChemSpider | 2792 |
| UNII | 69O5WQQ5TI |
| KEGG | D07758 |
| ChEBI | CHEBI:3996 |
| ChEMBL | ChEMBL669 |
| CompTox Dashboard (EPA) | DTXSID0046933 |
| ECHA InfoCard | 100.005.588 |
| Chemical and physical data | |
| Formula | C20H21N |
| Molar mass | 275.395 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
{kind=link}
{kind=link}
///////////////cyclobenzaprine, циклобензаприн , سيكلوبنزابرين , 环苯扎林 , MK-130, TNX-102, Muscle Relaxant
CN(C)CCC=C1C2=CC=CC=C2C=CC2=CC=CC=C12

NEW DRUG APPROVALS
ONE TIME
$10.00
Pyritinol
Pyritinol
- Molecular FormulaC16H20N2O4S2
- Average mass368.471 Da
1098-97-1[RN]
1308
214-150-1[EINECS]
233-178-5[EINECS]
3,3′-[Dithiobis(methylene)]bis[5-hydroxy-6-methyl-4-pyridinemethanol]
4-Pyridinemethanol, 3,3′-[dithiobis(methylene)]bis[5-hydroxy-6-methyl-
пиритинол[Russian][INN]
بيريتينول[Arabic][INN]
吡硫醇[Chinese][INN]
Pyritinol, CAS Registry Number: 1098-97-1
CAS Name: 3,3¢-[Dithiobis(methylene)]bis[5-hydroxy-6-methyl-4-pyridinemethanol]
Additional Names: bis(4-hydroxymethyl-5-hydroxy-6-methyl-3-pyridylmethyl) disulfide; bis[(3-hydroxy-4-hydroxymethyl-2-methyl-5-pyridyl)methyl] disulfide; dipyridoxolyldisulfide; pyridoxine-5-disulfide; pyrithioxin
Molecular Formula: C16H20N2O4S2, Molecular Weight: 368.47
Percent Composition: C 52.15%, H 5.47%, N 7.60%, O 17.37%, S 17.40%
Literature References: Prepn: Zima, Schorre, US3010966 (1961 to E. Merck); Iwanami et al.,Bitamin36, 122 (1967); J. Vitaminol.14, 321, 326 (1968). HPLC determn in urine: K. Kitao et al.,Chem. Pharm. Bull.25, 1335 (1977). Pharmacokinetics and metabolism: Darge et al.,Arzneim.-Forsch.19, 5, 9, (1969); Nowak, Schorre, ibid. 11. Clinical trial in dementia: S. Hoyer et al.,ibid.27, 671 (1977); A. J. Cooper, R. V. Magnus, Pharmacotherapeutica2, 317 (1980); in cerebrovascular disorders: Y. Tazaki et al.,J. Int. Med. Res.8, 118 (1980).
Properties: Crystals, mp 218-220°.
Melting point: mp 218-220°
Derivative Type: Dihydrochloride monohydrate
Trademarks: Biocefalin (Benvegna); Bonifen (Merck KGaA); Enbol (Chugai); Encephabol (Merck KGaA); Enerbol (Polfa); Epocan (Merck KGaA); Life (SIT)
Molecular Formula: C16H20N2O4S2.2HCl.H2O, Molecular Weight: 459.41
Percent Composition: C 41.83%, H 5.27%, N 6.10%, O 17.41%, S 13.96%, Cl 15.43%
Properties: mp 184°. Note: Has no vitamin B6 activity.
Melting point: mp 184°
Therap-Cat: Nootropic.
Keywords: Nootropic.
Derivatives
Dihydrochloride monohydrate
- Formula:C16H20N2O4S2 • 2HCl • H2O
- MW:459.42 g/mol
- CAS-RN:10049-83-9
- EINECS:233-178-5
- LD50:221 mg/kg (M, i.v.); 5786 mg/kg (M, p.o.);
300 mg/kg (R, i.v.); 6 g/kg (R, p.o.)
Pyritinol has been used in trials studying the treatment of Dementia, Depression, Schizophrenia, Anxiety Disorders, and Psychosomatic Disorders.
Pyritinol also called pyridoxine disulfide or pyrithioxine (European drug names Encephabol, Encefabol, Cerbon 6) is a semi-synthetic water-soluble analog of vitamin B6 (Pyridoxine HCl). It was produced in 1961 by Merck Laboratories by bonding 2 vitamin B6 compounds (pyridoxine) together with a disulfide bridge. Since the 1970s, it has been a prescription and OTC drug in several countries for cognitive disorders, rheumatoid arthritis,[1] and learning disorders in children. Since the early 1990s it has been sold as a nootropic dietary supplement in the United States.

SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 39984-49-1 | C8H10Br3NO | 3,4-bis(bromomethyl)-5-hydroxy-6-methylpyridine hydrobromide | 3-Pyridinol, 4,5-bis(bromomethyl)-2-methyl- |
| 92147-37-0 | C11H15NO3S2 | ethylxanthic acid [5-hydroxy-4-(hydroxymethyl)-6-methyl-3-pyridyl]methyl ester | Xanthic acid, ethyl-, [5-hydroxy-4-(hydroxymethyl)-6-methyl-3-pyridyl]methyl ester |
| 140-89-6 | C3H5KOS2 | potassium ethylxanthogenate | Carbonodithioic acid, O-ethyl ester, potassium salt |

PATENT
PATENT
https://patents.google.com/patent/CN103992268A/en
Pyritinol, it is the derivative of vitamin B6, for nootropic agents, can promote glucose and amino acid metabolism in brain, improve whole body assimilation, increase Flow of carotid artery, improve cerebral blood flow (CBF), be applicable to the dizzy distending pain, insomnia, hypomnesis of cerebral trauma sequela, encephalitis and meningitis sequela etc., the improvement of absent minded, emotional change; Also for cerebral arteriosclerosis, senile dementia mental symptom etc.
The pyritinol of applying clinically at present, it is pyritinol hydrochloride, be specially the monohydrate of hydrochloride, its chemical name is 3,3-(dithio methylene radical) two (5-hydroxyl-6-methyl-pyridine methane) dihydrochloride monohydrate, has recorded in < < Chinese Pharmacopoeia version > > in 2010.The preparation of this product listing has sheet, capsule and sterile powder injection, and its injection easily causes venous stimulation when clinical application, has greatly limited clinical application.The powder injection of pyritinol hydrochloride easy caking after standing storage, not soluble or dissolve and thoroughly cause liquid unclarity, particulate matter to exceed standard and easily cause the untoward reactions such as Microembolization during use.
CN101003509A discloses hydrobromate and the mesylate of pyritinol, record its stability having had, solvability and bland advantage, but in fact, Hydrogen bromide pyritinol, methylsulfonic acid pyritinol store easy moisture absorption under normal condition, in purification refine, be difficult to separate out with conventional crystallization method, need loaded down with trivial details aftertreatment technology, Hydrogen bromide and methylsulfonic acid have strong corrodibility in addition, comparatively difficult to its suitability for industrialized production.
CN101066266A discloses organic acid salt of pyritinol and preparation method thereof, wherein preferred pyritinol nicotinate.Yet, in nicotinic acid pyritinol water solvability a little less than, and nicotinic acid pyritinol preparation technology used dry-out benzene, toxicity is larger, and aftertreatment technology is complicated, is not suitable for suitability for industrialized production.
Yet, existing pyritinol or its salt, or pyritinol salt exists defect in the use, or the production technique that obtains this pyritinol salt is unsuitable for suitability for industrialized production.For this reason, need to provide a kind of safe, pyritinol salt and production method thereof of stablizing, meeting industrialization production requirements.
Embodiment 1: pyritinol maleate synthetic
Get 5.0g pyritinol powder, drop in reaction flask, add 100ml purified water, then under agitation add toxilic acid 3.8g, finish, be heated to 60-65 ℃ and stir 30min and all dissolve to solid, remove heating fluid, stirred crystallization under room temperature, separate out a large amount of white solids, use a small amount of cold water washing, 45 ℃ of vacuum-dryings, obtain white powder 5.97g, yield 72.9%.Purity: 99.5%; M.p.:134~137 ℃; Ultimate analysis (C16H20N2O4S22C4H4O4): C:47.9%, H:4.8%, N:4.6%, S:10.6%, O:32.1% (theory: C:48.0%, H:4.7%, N:4.7%, S:10.7%, O:32.0%); 1H-NMR (600MHz, DMSO) δ: 2.39 (6H, s), 3.93 (4H, s), 4.76 (4H, s), 6.18 (4H, s), 7.87 (2H, s).By the 1H-NMR (Fig. 2) of toxilic acid pyritinol and the 1H-NMR (Fig. 1) of pyritinol contrast, in a part toxilic acid pyritinol, contain 2 molecule toxilic acids.
Embodiment 2: pyritinol maleate synthetic
Get 5.0g pyritinol powder, drop in reaction flask, add 100ml ethanol, then under agitation add toxilic acid 3.0g, finish, be heated to return stirring 30min and all dissolve to solid, remove heating fluid, stirred crystallization under room temperature, separate out a large amount of white solids, use a small amount of cold water washing, 45 ℃ of vacuum-dryings, obtain white powder 5.50g, yield 67.5%.After measured, the toxilic acid pyritinol that structure makes with embodiment 1.
PATENT
https://patents.google.com/patent/CN105153021A/en
Embodiment 1
Toxilic acid 3.8g is dissolved in 100ml ethanol, be warming up to 60 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 1 hour, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid toxilic acid pyritinol crystal form A 4.9g.X-ray powder diffraction analysis, as Fig. 1, its 2 θ value is as following table.
Embodiment 2
Toxilic acid 3.8g is dissolved in 100ml acetone, be warming up to 45 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 1.5 hours, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid 5.2g.It is toxilic acid pyritinol crystal form A that dry product does X-ray powder diffraction.
Embodiment 3
Toxilic acid 3.8g is dissolved in and adds 100ml Virahol, be warming up to 60 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 2 hours, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid 5.1g.It is toxilic acid pyritinol crystal form A that dry product does X-ray powder diffraction.
PATENT
https://patents.google.com/patent/CN101066266A/en
Specific embodiment:
Embodiment 1: nicotinic acid pyritinol salt synthetic
Get nicotinic acid 24.6g, fully be dissolved in the 300ml anhydrous benzene, heated and stirred is to molten entirely, under complete molten state, add pyritinol 40.5g, reflux mixture 3 hours, TLC thin layer identification (developing solvent: ethyl acetate: ethanol: glacial acetic acid=5: 6: 0.6) fully, the cooling back adds the 200ml dehydrated alcohol slightly, mixture is put into refrigerator fully cool off, sucking filtration is separated out white crystals, with a small amount of cold absolute ether washing solid.65 ℃ of vacuum dryings get 62.1g nicotinic acid pyritinol salt, yield 89.7%.Determination of acid-basetitration nicotinic acid and pyritinol content are measured moisture with the karl Fischer method.The result is: nicotinic acid 37.2%, and pyritinol 62.0%, water 5.8%, approaching with theoretical value, contain 2 water of crystallization.Elementary analysis: theoretical value C52.8% H5.3% O25.2%N6.6% S10.1%; Measured value C52.4% H5.2% O25.1%N6.5% S10.0%.
Embodiment 2: fumaric acid pyritinol salt synthetic
Get fumaric acid 11.6g, fully be dissolved in the 300ml anhydrous benzene, heated and stirred is to molten entirely, under complete molten state, add pyritinol 40.5g, reflux mixture 3 hours, TLC thin layer identification (developing solvent: ethyl acetate: ethanol: glacial acetic acid=5: 4: 0.8) fully, the cooling back adds the 200ml dehydrated alcohol slightly, mixture is put into refrigerator fully cool off, sucking filtration is separated out white crystals, with a small amount of cold absolute ether washing solid.65 ℃ of vacuum dryings get 49.9g fumaric acid pyritinol salt, yield 88.9%.Determination of acid-basetitration fumaric acid and pyritinol content are measured moisture with the karl Fischer method.The result is: fumaric acid 20.8%, and pyritinol 72.7%, water 6.5%, approaching with theoretical value, contain 2 water of crystallization.Elementary analysis: theoretical value C49.6% H5.0%O26.4% N5.8% S13.2%; Measured value C49.4% H5.2% O26.5% N5.9%S13.1%.
PATENT
https://patents.google.com/patent/CN102516297A/en
Embodiment 1: the preparation of compd A
With Pyrithioxine hydrochloride 10g, be dissolved in the 20ml pyridine, slowly drip POCl3 solution 10ml under the room temperature; Drip and finish, stirring at room reaction 12 hours slowly adds the 100g frozen water and stirred hydrolysis reaction 2 hours; Toluene gradation extraction 30ml * 3, water layer evaporated under reduced pressure, Virahol dissolution residual substance; Filter, evaporate to dryness gets compd A 4.2g.
Embodiment 2: the preparation of compd B
With Pyrithioxine hydrochloride 10g, be dissolved in the 40ml THF, add 4gNaH, 30 ℃ were stirred 2 hours; Add the 20ml POCl3, stirring reaction 16 hours slowly adds the 100g frozen water and stirred hydrolysis reaction 2 hours; ETHYLE ACETATE gradation extraction 30ml * 3, the water layer evaporated under reduced pressure adds 80ml Virahol dissolution residual substance; Add 40ml water, freezing crystallization gets compd B 5.6g.
Embodiment 3: the preparation of Compound C
With Pyrithioxine hydrochloride 10g, be dissolved in the 40ml THF, add 4gNaH, 30 ℃ were stirred 2 hours; Add the 20ml chloroiodomethane, stirring reaction 16 hours, 60 ℃ of evaporated under reduced pressure add 20ml acetonitrile dissolution residual substance; As midbody, other gets triethylamine 9ml and is dissolved in the 10ml acetonitrile, drips 3.6ml phosphoric acid, after dropping finishes; Stir down and slowly splash into midbody, continued 60 ℃ of stirring reactions 12 hours, steaming desolventizes; Residue adds water 20ml dissolving, and water layer filters clarification, and freeze-drying promptly gets compd B 6.7g.
Embodiment 4: the preparation of Compound D
Serine 3 grams, ethylene bromohyrin 2.5g, N with the BOC protection; N-Dimethylamino pyridine 3g and NSC 57182 3g are dissolved in the THF; Stirring at room 10 hours, vacuum concentration is with the thick product of chromatography purification (with the ETHYLE ACETATE/normal hexane wash-out of normal hexane to 30%); Merging filtrate, evaporate to dryness gets intermediate A; Pyrithioxine hydrochloride 2g and intermediate A 2.5g are dissolved with THF 30ml, add triphenyl phosphorus 2g, slowly drip diethyl azodiformate solution 2ml, room temperature reaction 5 hours; Reaction is finished, and evaporated under reduced pressure adds ETHYLE ACETATE 50ml dissolving, filters insolubles; With the thick product of chromatography purification (with the ETHYLE ACETATE/normal hexane wash-out of normal hexane to 10%), merging filtrate, evaporate to dryness dissolves with methylene dichloride 20ml then; Feed hydrogen chloride gas to saturated, stirring reaction 5 hours filters; Get the hydrochloride of Compound D, transferring pH behind the use dissolved in distilled water is about 8, and the water layer lyophilize gets Compound C 0.27g.
Embodiment 5: the preparation of compd E
Get compd A 10g, be dissolved in the 30ml Virahol, add 25gBoc-Ser-OBZL in batches, 50 ℃ of stirring reactions; HPLC monitoring react to compd B less than 5%, add 0.1M hydrochloric acid soln 20ml, 60 ℃ of heating hydrolysis 5 hours are regulated pH to 7; Evaporated under reduced pressure adds anhydrous alcohol solution, removes by filter insolubles, evaporated under reduced pressure; Add the 5ml water dissolution, filtering, lyophilize get compd E 6.9g
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Availability
It is approved for “symptomatic treatment of chronically impaired brain function in dementia syndromes” and for “supportive treatment of sequelae of craniocerebral trauma” in various European countries, including Austria, Germany, France, Italy, Portugal, and Greece. In France it is also approved for rheumatoid arthritis as a disease modifying drug, on the basis of the results of clinical trials. In many countries it is available over the counter and is widely advertised on the internet as being for “memory disturbances.”
Effects
review refs needed
Adverse effects
Adverse effects include nausea, headache,[2] and rarely allergic reaction (mild skin reactions).[3] A 2004 survey of six case reports suggested a link between pyritinol and severe cholestatic hepatitis when on several drugs for certain diseases.[4]
Other rare side effects: acute pancreatitis[5] and photoallergic eruption.[6]
References
- ^ Lemmel EM (May 1993). “Comparison of pyritinol and auranofin in the treatment of rheumatoid arthritis. The European Multicentre Study Group”. British Journal of Rheumatology. 32 (5): 375–82. doi:10.1093/rheumatology/32.5.375. PMID 8495257.
- ^ Nachbar F, Korting HC, Vogl T (1993). “Erythema multiforme-like eruption in association with severe headache following pyritinol”. Dermatology. 187 (1): 42–6. doi:10.1159/000247196. PMID 8324277.
- ^ de Groot, Anton C.; Nater, Johan Pieter; Weyland, J. Willem. Unwanted Effects of Cosmetics and Drugs Used in Dermatology.[full citation needed][page needed]
- ^ Maria V, Albuquerque A, Loureiro A, Sousa A, Victorino R (March 2004). “Severe cholestatic hepatitis induced by pyritinol”. BMJ. 328 (7439): 572–4. doi:10.1136/bmj.328.7439.572. PMC 381054. PMID 15001508.
- ^ Straumann A, Bauer M, Pichler WJ, Pirovino M (August 1998). “Acute pancreatitis due to pyritinol: an immune-mediated phenomenon”. Gastroenterology. 115 (2): 452–4. doi:10.1016/S0016-5085(98)70212-4. PMID 9679051.
- ^ Tanaka M, Niizeki H, Shimizu S, Miyakawa S (October 1996). “Photoallergic drug eruption due to pyridoxine hydrochloride”. The Journal of Dermatology. 23 (10): 708–9. doi:10.1111/j.1346-8138.1996.tb02685.x. PMID 8973037. S2CID 28810619.
External links
- Media related to Pyritinol at Wikimedia Commons
{kind=link}
| Clinical data | |
|---|---|
| ATC code | N06BX02 (WHO) |
| Pharmacokinetic data | |
| Elimination half-life | 2.5 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1098-97-1 |
| PubChem CID | 14190 |
| ChemSpider | 13561 |
| UNII | AK5Q5FZH2R |
| KEGG | D02160 |
| ChEMBL | ChEMBL488093 |
| CompTox Dashboard (EPA) | DTXSID3048362 |
| ECHA InfoCard | 100.012.864 |
| Chemical and physical data | |
| Formula | C16H20N2O4S2 |
| Molar mass | 368.473 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
{kind=link}
//////////////Pyritinol, пиритинол , بيريتينول , 吡硫醇 , Nootropic,

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Fabomotizole

Fabomotizole
Afobazole
- Molecular FormulaC15H21N3O2S
- Average mass307.411 Da
0F8K1X115C
173352-21-1[RN], 173352-21-1 (free base) 173352-39-1 (HCl) 189638-30-0 (2HCl)
1H-Benzimidazole, 6-ethoxy-2-[[2-(4-morpholinyl)ethyl]thio]-
Obenoxazine, Afobazol, Afobazole, Aphobazole, Fabomotizole dihydrochloride, CM-346, CM346, CM 346,
фабомотизол[Russian][INN]
فابوموتيزول[Arabic][INN]
法莫替唑[Chinese][INN]
Fabomotizole dihydrochloride
CAS#: 189638-30-0 (2HCl)
Chemical Formula: C15H23Cl2N3O2S
Molecular Weight: 380.33
Fabomotizole (also known as Afobazole) is a selective non-benzodiazepine anxiolytic which was developed in Russia and launched in 2006. The drug is used for the treatment of wide range of diseases: generalized anxious disorders, neurasthenia, adaptation disorders, sleep disorders, for alleviation of withdrawal syndrome. According to the drug label (in Russian), its action is related to the interaction with sigma-1 receptors.
Fabomotizole (INN;[1] brand name Afobazole) is an anxiolytic drug launched in Russia in the early 2000s. It produces anxiolytic and neuroprotective effects without any sedative or muscle relaxant actions.[citation needed] Its mechanism of action remains poorly defined however, with GABAergic, NGF– and BDNF-release-promoting, MT1 receptor agonism, MT3 receptor antagonism, and sigma agonism suggested as potential mechanisms. Fabomotizole was shown to inhibit MAO-A reversibly and there might be also some involvement with serotonin receptors.[2][3][4][5][6] Clinical trials have shown fabomotizole to be well tolerated and reasonably effective for the treatment of anxiety.[7]
Experiments of mice have shown antimutagenic and antiteratogenic properties.[8]
Fabomotizole has found little clinical use outside Russia and has not been evaluated by the FDA.
PATENT
WO 9534304
https://patents.google.com/patent/WO1995034304A1/en

PAPER
European Journal of Medicinal Chemistry (2021), 211, 113110
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310825?
A ligand-based virtual screening study to search for giardicidal compounds on a 6551 ChEMBL drugs database was carried out using molecular similarity. Three fingerprints implemented in MayaChemTools with different design and validated by ROC curves, were used. Twelve compounds were retrieved from this screening, from which, four representative compounds were selected to carry out biological assays. Whereas two compounds were commercially available, the additional two compounds were synthesized during the development of this work. The biological assays revealed that the compounds possess in vitro activity against five strains of Giardia intestinalis, each with different susceptibility/resistance rates to metronidazole, albendazole and nitazoxanide. Particularly, tenatoprazole showed the best effect against the WB and IMSS strains. Furthermore, fabomotizole, tenatoprazole and ipriflavone showed a higher activity against resistant strains than the reference drugs: metronidazole, albendazole and nitazoxanide.
Graphical abstract


///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Afobazole |
| Other names | Fabomotizole |
| Routes of administration | Oral |
| ATC code | N05BX04 (WHO) |
| Legal status | |
| Legal status | US: Unscheduled Not FDA approved |
| Pharmacokinetic data | |
| Bioavailability | 43.64%, pronounced first-pass effect |
| Metabolism | extensive hepatic |
| Onset of action | 0.85±0.13 hours |
| Elimination half-life | 0.82±0,54 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 173352-39-1 |
| PubChem CID | 9862937 |
| ChemSpider | 8038633 |
| UNII | HDO6HX6NZU |
| CompTox Dashboard (EPA) | DTXSID00169606 |
| Chemical and physical data | |
| Formula | C15H21N3O2S |
| Molar mass | 307.41 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
{kind=link}
{kind=link}
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 26 (1): 63. 2012. Retrieved 21 March 2015.
- ^ Neznamov, GG; Siuniakov, SA; Chumakov, DV; Bochkarev, VK; Seredenin, SB (2001). “Clinical study of the selective anxiolytic agent afobazol”. Eksperimental’naia i Klinicheskaia Farmakologiia. 64 (2): 15–9. PMID 11548440.
- ^ Silkina, IV; Gan’shina, TC; Seredin, SB; Mirzoian, RS (2005). “Gabaergic mechanism of cerebrovascular and neuroprotective effects of afobazole and picamilon”. Eksperimental’naia i Klinicheskaia Farmakologiia. 68 (1): 20–4. PMID 15786959.
- ^ Seredin, SB; Melkumian, DS; Val’dman, EA; Iarkova, MA; Seredina, TC; Voronin, MV; Lapitskaia, AS (2006). “Effects of afobazole on the BDNF content in brain structures of inbred mice with different phenotypes of emotional stress reaction”. Eksperimental’naia i Klinicheskaia Farmakologiia. 69 (3): 3–6. PMID 16878488.
- ^ Antipova, TA; Sapozhnikova, DS; Bakhtina, LIu; Seredenin, SB (2009). “Selective anxiolytic afobazole increases the content of BDNF and NGF in cultured hippocampal HT-22 line neurons”. Eksperimental’naia i Klinicheskaia Farmakologiia. 72 (1): 12–4. PMID 19334503.
- ^ Seredenin, SB; Antipova, TA; Voronin, MV; Kurchashova, SY; Kuimov, AN (2009). “Interaction of afobazole with sigma1-receptors”. Bulletin of Experimental Biology and Medicine. 148 (1): 42–4. doi:10.1007/s10517-009-0624-x. PMID 19902093. S2CID 37411324.
- ^ Medvedev, VE; Trosnova, AP; Dobrovol’skiĭ, AV (2007). “Psychopharmacotherapy of anxiety disorders in patients with cardio-vascular diseases: the use of aphobazole”. Zh Nevrol Psikhiatr Im S S Korsakova. 107 (7): 25–9. PMID 18379478.
- ^ Durnev AD, Zhanataev AK, Shreder OV, Seredenin SB (Jan–Feb 2009). “Antimutagenic and antiteratogenic properties of afobazole”. Eksp Klin Farmakol. 72 (1): 46–51. PMID 19334511.
//////////////Fabomotizole, Afobazole, фабомотизол , فابوموتيزول , 法莫替唑 , Obenoxazine, Afobazol, Afobazole, Aphobazole, Fabomotizole dihydrochloride, CM-346, CM346, CM 346,
CCOc1ccc2c(c1)[nH]c(n2)SCCN3CCOCC3.Cl.Cl

NEW DRUG APPROVALS
ONE TIMETO MAINTAIN THIS BLOG
$10.00