












































Rivastigmine, (sold under the trade name Exelon) is a parasympathomimetic orcholinergic agent for the treatment of mild to moderate dementia of the Alzheimer’s typeand dementia due to Parkinson’s disease. The drug can be administered orally or via atransdermal patch; the latter form reduces the prevalence of side effects, which typically include nausea and vomiting.The drug is eliminated through the urine, and appears to have relatively few drug-drug interactions.
Rivastigmine was developed by Marta Weinstock-Rosin of the Department of Pharmacology, at the Hebrew University of Jerusalem and sold to Novartis by Yissum for commercial development.(It is a semi-synthetic derivative of physostigmine) It has been available in capsule and liquid formulations since 1997. In 2006, it became the first product approved globally for the treatment of mild to moderate dementia associated withParkinson’s disease; and in 2007 the rivastigmine transdermal patch became the first patch treatment for dementia
PATENT
US 4,948,807
Patent 5,602,176
Issued: February 11, 1997
Inventor(s): Enz; Albert
Assignee(s): Sandoz Ltd.Patent expiration dates:
- February 11, 2014


-
Rivastigmine hydrogen tartrate is chemically known as (S)-N-Ethyl-N-methyl-3- [1-(dimethylamino) ethyl]-phenyl carbamate hydrogen- (2R, 3R)-tartrate (hereinafter referred to as “rivastigmine tartrate”) and has structural Formula I.

-
Rivastigmine hydrogen tartrate is administered for the inhibition of reversible cholinesterase and is marketed under the brand name EXELON™ as capsules containing 0.5, 3, 4.5 and 6 mg rivastigmine base equivalent.
-
U.S. Patent No. 4,948,807 describes the compound N-ethyl, N-methyl-3-[1-(dimethylamino)ethyl]phenyl carbamate and its pharmacologically acceptable salts along with a pharmaceutical composition useful for treating anticholinesterase activity in humans.
-
U.S. Patent No. 5,602,176 describes (S)-N-ethyl-3-[(1-dimethylamino)ethyl]-N-methyl-phenyl carbamate in free base or acid addition salt form as useful for its anticholinesterase activity.
-
International Application Publication No. WO 2004/037771 A1 and European Patent 193926 describe a process for the preparation of (S)-3-[1-(dimethylamino)-ethyl]-phenyl-N-ethyl-N-methyl carbamate by the reaction of optically active m-hydroxyphenylethyl dimethylamine with a carbamoylhalide
-
International application No. WO 2005/058804A1 describes a process for the preparation of rivastigmine by streoselective reduction.

The synthesis of rivastigmine was reported in U.S. Pat. No. 5,602,176, GB2409453, and Yonwen, Jiang et. al. [Journal of East China Normal University (Natural Science), 2001, 1, 61-65], in which the method is disclosed as: preparing racemic rivastigmine by a series of reactions, then salifying the result with D-(+)-O, o′-bis-p-tolyl formacyl tartaric acid monohydrate (D-DTTA) to separate the racemic mixture, and recrystallizing at least three time to obtain (S)-rivastigmine with an optical purity of above 99%. The final yield is only 5.14%.
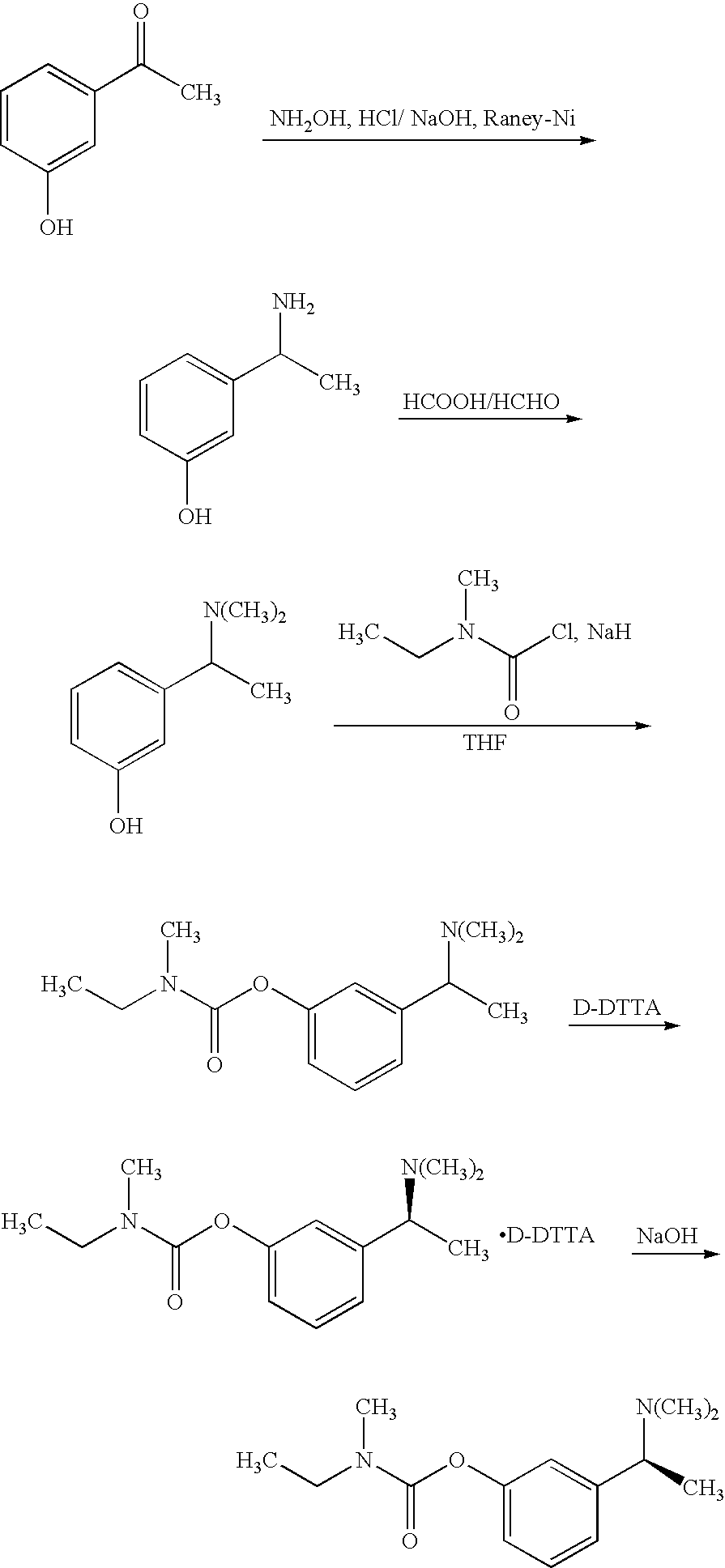
A method for resolution of a intermediate of rivastigmine is disclosed in WO200403771, in which S-(+)-camphor sulfonic acid is used to separate racemic intermediates of 3-(1-(S)—(N,N-dimethylamino) ethyl)phenol, and optically pure 3-(1-(S)—(N,N-dimethylamino) ethyl)phenol is obtained after three times recrystallization and then condensates with N-methyl-N-ethyl-amino formacyl chloride to obtain (S)-rivastigmine. The specific synthesis route is shown below:

A method for resolution of a intermediate of rivastigmine is also disclosed in WO2007014973, in which S-(+)-camphor sulfonic acid is used to separate racemic intermediates of 3-(1-(methylamino) ethyl)phenol, and the result condensates with N-methyl-N-ethyl-amino formacyl chloride to obtain N-methylethylcarbamino-3-[(S)-1-(methylamino)-ethyl]phenyl ester, and a methylation is then performed on the nitrogen atom followed by salifying with L-(+)-tartaric acid so that rivastigmine is obtained. The methylation needs a reduction system of sodium cyanoborohydride/formaldehyde, in which sodium cyanoborohydride is highly toxic, so that the method is not suitable for industrial production. The specific synthesis route is shown below:

The resolution methods mentioned above are time consuming with low yields, so that final yields are reduced and costs are increased, which are not beneficial for industrial production and the optical purity of rivastigmine cannot be guaranteed.
Pohang University of Science and Technology, Korea
Chemoenzymatic Synthesis of Rivastigmine via Dynamic Kinetic Resolution as a Key Step
J. Org. Chem. 2010, 75: 3105-3108

Rivastigmine (Exelon®) is an acetylcholinesterase inhibitor that is prescribed for the treatment of mild to moderate dementia in patients with Alzheimer’s disease and Parkinson’s disease. The key step in the synthesis depicted is a dynamic kinetic resolution of the benzylic secondary alcohol B involving a lipase (Novozyme 435) coupled with a polymer-bound racemization catalyst (C).

The polymer-bound racemization catalyst C was prepared by heating a polymer-bound benzoyl chloride with [Ph4(η4-C4CO]Ru(CO)3 in toluene for one day. The catalyst can be recycled several times. The enzymatic resolution was performed on a 1 mmol scale. For an alternative chemoenzymatic synthesis of rivastigmine, see: J. Mangas-Sánchez et al. J. Org. Chem. 2009, 74, 5304.
………………………….
http://www.google.com/patents/EP1980552A2

EXAMPLES
EXAMPLE 1:
-
To a solution of 200 g of 3-hydroxyacetophenone of Formula IX in 400 ml of acetone, 244 g of potassium carbonate were charged and stirred for about 10 minutes. To the above reaction mixture 204 g of dimethyl sulphate was added for about 60 minutes followed by heating to about 45 °C and stirred for about 1 hour. After completion of the reaction, the reaction mixture was quenched by charging of 800 ml of water. Organic and aqueous layers were separated and 370 g of ammonium formate was added to the organic layer. The contents were then heated to about 180 °C and stirred for about 2 hours. The reaction mixture was then cooled to about 30° C and 600 ml of water was charged. The mixture was extracted with ethyl acetate (1×400 ml, 2×150 ml). The organic layers were combined and charged 600 ml of hydrogen chloride in isopropanol (18% w/w) followed by heating to about 75 °C and stirred for about 3 hours. The mixtures was distilled completely at about 65 °C under vacuum and again charge 100 ml ethyl acetate and distilled completely at about 65°C to afford residue.
-
600 ml of ethyl acetate was charged to the residue and stirred for 30 minutes. Filtered the solid and was washed with 200 ml of ethyl acetate. The wet solid was then charged into 600 ml of water and pH was adjusted to about 11 by addition of 68.8 ml of 40% aqueous sodium hydroxide. The reaction mixture was extracted with ethyl acetate (1×200 ml, 2×100 ml). Organic and aqueous layers were separated and the organic layer was distilled off completely at about 65 °C under vacuum to afford 128 g of the title compound.
HPLC purity: 99.1%
- PREPARATION OF1-(3-METHOXY PHENYL) ETHYL AMINE (FORMULA VI).
EXAMPLE 2:
-
To a solution of 40 g of 1- (3-methoxyphenyl) ethyl amine of Formula VI in 1400 ml of isopropyl alcohol, 41.2 g of L-(+)-mandelic acid was added and stirred for about 15 minutes. The mixture was heated to about 75°C and stirred for about 45 minutes followed by cooling to about 37°C and stirred for about 10 minutes. The separated solid was filtered and the solid was washed 80 ml of isopropyl alcohol. The solid obtained was suck dried for 3 hours to obtain the wet compound of the diasteromeric salt of Formula V.
-
The obtained diasteromeric salt of Formula V was charged into a clean and dry round bottom flask containing 480 ml of isopropyl alcohol followed by heating to reflux. The resultant solution was stirred at reflux for about 45 minutes followed by cooling to about 37° C and stirred for about 10 minutes. Solid was separated by filtration and the solid was washed with 20 ml of isopropyl alcohol. The solid obtained was dried at about 55 °C for about 2 hours to yield 29 g of the title compound.
Purity by chiral HPLC: 99.9%.
- PREPARATION OF S-(-)-1-(3-METHOXY PHENYL) ETHYL AMINE MANDALATE (FORMULA V).
EXAMPLE 3:
-
To a solution of 200 g of S-(-)-1-(3-methoxyphenyl)ethyl amine L (+)-Mandalate (diasteromeric salt) of Formula V in 800 ml of water, charged 148 g of formaldehyde (40%), 182.1 g of formic acid (98%) and the contents were heated to about 100 °C. The resultant mixture was stirred at about 100 °C for about 5 hours. After the completion of the reaction, the mixture was cooled to about 30° C and washed with toluene (3x1000ml). Aqueous layer pH was adjusted to 10.5 using 160 ml of 40% aqueous sodium hydroxide solution and extracted with ethyl acetate (2×500 ml). The organic layers were combined and washed with water (2×400 ml). The organic layer was distilled completely at about 60 °C under vacuum to yield 108 g of the title compound.
Purity by HPLC: 98.15%.
- PREPARATION OF S-(-)-[1-(3-METHOXYPHENYL) ETHYL] DIMETHYL AMINE (FORMULA IV)
EXAMPLE 4:
-
50 g of S-(-)-[1-(3-methoxyphenyl) ethyl] dimethyl amine of Formula IV and 283 g of 48% aqueous HBr solution were charged into a clean and round bottom flask followed by heating to about 110° C and stirred for about 6 hours. After completion of the reaction, the mixture was cooled to about 30°C and charged 250 ml of water and pH was adjusted to about 10.5 using 162 ml caustic lye and the reaction mixture was extracted with ethyl acetate ((1×150 ml, 2×50 ml)). The organic layer thus obtained was washed with water (2×50 ml) and treated with activated charcoal. The organic layer is filtered through celite and washed with 100 ml of ethyl acetate. The filtrate was distilled completely at below 60° C under vacuum. To the residue charged 200 ml of n-heptane at about 50°C and stirred for about 90 minutes at about 25°C. The separated solid was filtered and washed the solid with n-heptane 50 ml and suck dried. The solid obtained was dried at about 50°C for about 5 hours to yield 41.5 g of the title compound.
Purity by HPLC: 99.07%.
- PREPARATION OF S-(-)-[1-(3-HYDROXYPHENYL) ETHYL] DIMETHYL AMINE (FORMULA III)
EXAMPLE 5:
-
6 kg of S-(-)-[1-(3-hydroxyphenyl) ethyl] dimethyl amine of Formula III and 12 L of Methyl Isobutyl Ketone(MIBK) were charged and stirred for about 10 minutes. To this reaction solution 3.44 kg of pyridine, 1.18 kg of tetrabutylammonium bromide were charged and stirred for about 15 minutes to form clear solution. 3.97 kg of N-ethyl, N-methyl carbomyl chloride was added to the reaction mixture for about 30 minutes. Heated the contents to about 30°C and stirred for about 15 hours. After completion of the reaction 48 lit of water was charged and pH was adjusted to about 1.5 using 3.72 lit of 36% aqueous hydrochloric acid. Stirred the contents for about 30 minutes at about 25°C and aqueous layer was separated. The aqueous layers were then washed with MIBK (2×12 lit) and separate the aqueous layer. Aqueous layer pH was adjusted to 12.5 using 6 lit of 40% aqueous sodium hydroxide solution and stirred for about 15 minutes. The aqueous layer was then extracted with MIBK (2×12 lit) and separated the organic layer. Washed the organic layer with water (2×12 lit) and separated the organic layer. The obtained organic layer was distilled off completely at about 60°C to afford residue.
-
To the obtained residue 48 lit of ethyl acetate was added and pH of the reaction solution was adjusted to about 2 by adding about 6 lit of f 18% hydrochloride in isopropyl alcohol at about 5°C and stirred for about 90 minutes for solid separation. The separated solid was filtered and washed with 6 lit of ethyl acetate. The obtained wet solid was again charged into a reaction containing 30 lit of water and adjusted the pH to about 12.5 using 1.8 lit of 40% aqueous sodium hydroxide solution(caustic lye). The reaction mass was extracted with MIBK (2×12 lit) and the combined organic layer was washed with water (2×12 lit). The organic layer was distilled completely at about 60°C to afford residue.
-
To the obtained residue 48 lit of ethyl acetate was added and pH of the reaction solution was adjusted to about 2 by adding about 6 lit of f 18% hydrochloride in isopropyl alcohol at about 5°C and stirred for about 90 minutes for solid separation. The separated solid was filtered and washed with 6 lit of ethyl acetate. The obtained wet solid was again charged into a reaction containing 30 lit of water and adjusted the pH to about 12.5 using 1.8 lit of 40% aqueous sodium hydroxide solution. The reaction mass was extracted with MIBK (2×12 lit) and the combined organic layer was washed with water (2x121it). The organic layer was distilled completely at about 60°C to afford the title compound
Purity by HPLC. 99.33%
- PREPARATION OF (S)-N-ETHYL-N-METHYL-3-[1-DIMETHYL-AMINO)-ETHYL]-PHENYL CARBAMATE (FORMULA II).
EXAMPLE 6
- : PREPARATION OF RIVASTIGMINE TARTRATE (FORMULA I)
-
3 kg of rivastigmine freebase of Formula II in 105 lit of acetone, 1.8 kg of L-(+)-Tartaric acid was charged and heated to about 60° C followed by stirring for about 30 minutes for complete dissolution. The resulting reaction solutions was passed through celite and wash the bed with 13.5 lit acetone to made particle free. The obtained clear solution was distilled off up to 50% of the initial volume and cooled to 30°C. 12 g of rivastigmine hydrogen tartrate was added and stirred for about 60 minutes. The reaction mixture was heated to reflux and stirred for about 60 minutes and cooled to about 30°C and stirred for about 60 minutes for solid separation. The separated solid was filtered and washed the solid with 3 lit of acetone. Solid obtained was dried at about 60° C for about 9 hours to afford 4.10 kg of the title compound.
Purity by HPLC: 97.37%.
………………………

2Department of Chemistry, Jawaharlal Nehru Technological University of Anantapur, Anantapur 515002, Andhra Pradesh, India,
3Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500 046, Andhra Pradesh, India
 Corresponding author email
Corresponding author email1H NMR (CDCl3, 300 MHz) δ 1.17-1.27 (m, 3H), 1.37 (d, 3H, J = 6.4 Hz, CH3), 2.21 (s, 6H), 3.04 (s, 3H, CH3), 3.25 (q, J1 = 7.2 Hz, J2 = 6.4 Hz), 3.43 (q, 1H, J1 = 7.2 Hz, J2 = 6.8 Hz), 3.48 (q, 1H, J1 = 6.8 Hz, J2 = 7.2 Hz), 7.01 (d, 1H, J = 8.0 Hz), 7 . 1 8 ( d , 1 H , J = 8 . 0 H z ) ,7.26 (s, 1H,), 7.33 (t, 1H, J = 8.0 Hz);
13C NMR (CDCl3, 100 MHz) δ 154.4 (1C, C=O), 151.4 (CH), 129.3
(CH), 124.7 (CH), 121.2 (CH), 120.8 (2C, CH), 77.1 (1C), 66.0 (1C, CH2), 43.9 (2C, N-Me),34.6 (1C, Mecarbamoyl), 20.3 (1C, CH3), 12.4 (1C, Mecarbamoyl); M/z 251.20 (M+ H) +;
IR (cm -1, KBr) 2975, 1723 (C=O); HRMS (ESI): calcd for C14H22N2O2 (M+ H)+251.1760, found 251.1767;
[α]20D = -33.90 (C=1, CHCl3).

……………………………
http://www.google.com/patents/US8324429
SPECTRAL DATA FOR TARTRATE

Optical rotation [α]20 D=+6.0°, C=5, ethanol; mp 122.3-124.1
1H NMR (CDCl3) δ ppm: 1.24, 1.16 (2×t, 3H), 1.67 (d, 3H), 2.65 (s, 6H), 2.96, 3.05 (2×s, 3H), 3.37, 3.45 (2×q, 2H), 4.34 (q, 1H), 4.47 (s, 2H), 7.14 (t, 1H), 7.20 (s, 1H), 7.28 (d, 1H), 7.39 (t, 1H); MS (ESI) m/z: 251.2.
…………………………….
FREE BASE
Optical rotation [α]20 D=−32.1°, C=5, ethanol.
1H NMR (CDCl3) δ ppm: 1.22 (m, 3H), 1.35 (q, 3H), 2.20 (s, 6H), 3.02 (d, 3H), 3.25 (m, 1H), 3.44 (s, 2H), 7.05 (m, 3H), 7.27 (m, 1H); MS (ESI) m/z: 251.2 (M++1).

 DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D















