
Home » Uncategorized (Page 146)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DS-8587 (Daiichi Sankyo (Japan) a new broad-spectrum antibacterial agent, is in phase I clinical trials for the treatment of bacterial infection.

DS-8587
Daiichi Sankyo (Japan)
7-[3a(R)-Amino-6a(S)-fluoroperhydrocyclopenta[c]pyrrol-2-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropyl]-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride dihydrate
7-[(1S,6S)-1-amino-4-oxa-8-azabicyclo[4.3.0]nonan-8-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid
| C21 H22 F3 N3 O3 . Cl H . 2 H2 O | |
| Mw | 493.904 |
DS-8587, a new broad-spectrum antibacterial agent, is in phase I clinical trials at Daiichi Sankyo for the treatment of bacterial infection.
DS-8587, from Daiichi Sankyo, is a fluoroquinolone with improved activity against both Gram-negative and Gram-positive bacteria. The compound is especially effective against Acinetobacter baumannii but also has improved activity against streptococci, staphylococci, enterococci, E. coli, and anaerobes . The compound is currently under Phase I of clinical development .
DS-8587, a new generation of fluoroquinolone, against Acinetobacter baumannii. The MICs against clinical isolates and inhibitory activity against target enzymes of DS-8587 was superior to ciprofloxacin and levofloxacin. Furthermore, the antibacterial activity of DS-8587 was less affected by adeA/adeB/adeC or abeM efflux pumps and frequency of single-step mutations with DS-8587 was lower as compared to those with ciprofloxacin. DS-8587 might be an effective agent against A. baumanniiinfection.
WO 2008082009 or
http://www.google.com/patents/EP2540715A1?cl=en
- [Reference Example 71]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-


-
[(3S)-3-(3-Hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (46 g) and imidazole (11.9 g) were dissolved in dimethylformamide (600 mL). After addition of tert-butyldimethylsilyl chloride (23.2 g) under ice-cooling, the mixture was stirred at room temperature for 59.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 2:1) to give 29.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.11 Hz), 3.58 (2H, t, J=6.13 Hz), 3.34 (1H, d, J=10.05 Hz), 3.12 (1H, d, J=10.05 Hz), 2.94 (1H, d, J=16.91 Hz), 2.31 (1H, d, J=17.16 Hz), 1.86-1.74 (1H, m), 1.72-1.62 (1H, m), 1.51 (3H, d, J=7.11 Hz), 1.49-1.24 (2H, m), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).

[Reference Example 72]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-

-
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (30 g) was dissolved in tetrahydrofuran (280 mL), and the atmosphere was replaced with argon. Then, lithium hexamethyldisilazide (1.0 M solution in tetrahydrofuran) (78.0 mL) was added dropwise at -15°C, and the mixture was stirred at -5°C for 30 minutes. After cooling to -15°C again, a solution of N-fluorobenzenesulfonimide (26.6 g) in tetrahydrofuran (220 mL) was added dropwise, and the mixture was stirred at room temperature for 17 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2) to give 8.15 g of the title compound as a pale yellow solid. 1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.53-5.44 (1H, m), 5.18 (1H, d, J=51.72 Hz), 3.64-3.52 (2H, m), 3.32-3.19 (2H, m), 1.92-1.65 (2H, m), 1.55 (3H, d, J=4.66 Hz), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).
MS (FAB) m/z: 480 (M+H)+.
IR (ATR) ν: 3421, 2977, 2935, 2877, 1698, 1454, 1369, 1309, 1249, 1153, 1058, 1035, 1006, 842 cm-1.

[Reference Example 73]
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-

-
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (8.15 g) was dissolved in tetrahydrofuran (25.0 mL). Acetic acid (22.0 mL) and tetrabutylammonium fluoride (1.0 M solution in tetrahydrofuran) (25.0 mL) were added under ice-cooling, and the mixture was stirred at room temperature for 21.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 1:1) to give 5.77 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.03 Hz), 5.20 (1H, d, J=51.48 Hz), 3.69-3.59 (2H, m), 3.31-3.21 (2H, m), 1.95-1.72 (2H, m), 1.68-1.43 (2H, m), 1.56 (3H, d, J=7.11 Hz), 1.33 (9H, s).

[Reference Example 74]
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-

-
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (12.20 g) was dissolved in dichloromethane (400 mL). Benzenesulfonyl chloride (9.06 mL), triethylamine (10.7 mL), and 4-dimethylaminopyridine (2.04 g) were added under ice-cooling, and the mixture was stirred at room temperature for 12.5 hours. Saturated sodium bicarbonate water was added to the reaction solution, and the mixture was stirred for 30 minutes, followed by extraction with dichloromethane. The organic layer was sequentially washed with a 10% citric acid solution and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 8:2 -> 1:1) to give 11.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.94 – 7.87 (2H, m), 7.71-7.63 (1H, m), 7.60-7.53 (2H, m), 7.37-7.23 (5H, m), 5.46 (1H, q, J=7.11 Hz), 5.15 (1H, d, J=51.48 Hz), 4.10-3.98 (2H, m), 3.26-3.15 (2H, m), 1.88-1.50 (4H, m), 1.55 (3H, s), 1.30 (9H, s).

[Reference Example 75]
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester
-

-
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (10.9 g) was dissolved in tetrahydrofuran (350 mL), and the atmosphere was replaced with argon. Then, potassium hexamethyldisilazide (0.5 M solution in toluene) (86.5 mL) was added dropwise at – 15°C, and the mixture was stirred at 0°C for 1.5 hours. After cooling to -10°C, saturated aqueous ammonium chloride (100 mL) was added dropwise, and the mixture was stirred at room temperature for 30 minutes. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 7:1) to give 4.36 g of the title compound as a pale yellow solid.
1H-NMR (400 MHz, CDCl3) δ: 7.38-7.25 (5H, m), 5.58-5.49 (1H, m), 3.63 (1H, d, J=10.3 Hz), 2.91 (1H, dd, J=10.3, 3.2 Hz), 2.67-2.56 (1H, m), 2.50-2.38 (1H, m), 2.26-2.09 (1H, m), 2.06-1.94 (1H, m), 1.74-1.66 (1H, m), 1.54 (3H, d, J=7.1 Hz), 1.50-1.40 (1H, m), 1.34 (9H, s).

[Reference Example 76]
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane
-


-
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester (4.36 g, 12.5 mmol) was dissolved in dichloromethane (70 mL). Trifluoroacetic acid (70 mL) was added dropwise, and the mixture was stirred at room temperature for six hours. The solvent was evaporated under reduced pressure, and then the residue was azeotropically distilled with toluene to give carboxylic acid (3.70 g).
-
The resulting carboxylic acid was dissolved in toluene. Triethylamine (3.51 mL, 25.2 mmol) and diphenylphosphoryl azide (2.98 ml, 13.8 mmol) were added, and the mixture was heated to reflux for five hours. The solvent was evaporated under reduced pressure. Then, 1,4-dioxane (110 ml) and 6N hydrochloric acid (110 mL) were added to the residue, and the mixture was stirred at 60°C for 2.5 hours. After extraction with water and ethyl acetate, the aqueous layer was made alkaline with a saturated sodium hydroxide solution and extracted with chloroform twice. The organic layers were combined, dried over anhydrous sodium sulfate, and filtered, and then the solvent was evaporated under reduced pressure. Di-tert-butyl dicarbonate (11.05 g) was added to the residue, and the mixture was stirred at 75°C for six hours. The reaction solution was concentrated under reduced pressure, and then the residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 1:1) to give 3.69 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.50 (1H, q, J=7.1 Hz), 5.22 (1H, brs), 3.34 (2H, s), 2.49-2.37 (1H, m), 2.32-2.03 (3H, m), 2.02-1.90 (1H, m), 1.51 (3H, d, J=7.1 Hz), 1.55-1.48 (1H, m), 1.35 (9H, s).

[Reference Example 77]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane
-

-
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (3.69 g, 10.2 mmol) was dissolved in tetrahydrofuran (200 mL). A 1.20 M solution of a borane-tetrahydrofuran complex in tetrahydrofuran (42.4 mL, 50.9 mmol) was added dropwise under ice-cooling, and the mixture was stirred for two hours while gradually heating to room temperature. The solvent was evaporated under reduced pressure. Under ice-cooling, 90% aqueous ethanol (100 mL) and triethylamine (100 mL) were added to the residue, and the mixture was heated to reflux for two hours. The solvent was evaporated under reduced pressure, and then the residue was extracted with saturated sodium bicarbonate water and dichloromethane. Thereafter, the target substance was extracted from the aqueous layer with dichloromethane. The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure, and the resulting residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 95:5 -> 90:10) to give 3.33 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.32 – 7.18 (5H, m), 5.38 (1H, brs), 3.22 (1H, q, J=6.37 Hz), 2.92-2.57 (4H, m), 2.12-1.86 (4H, m), 1.80-1.67 (1H, m), 1.63-1.52 (3H, m), 1.42 (9H, s), 1.32 (3H, d, J=6.37 Hz)

- [Reference Example 78]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane
-

-
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (700 mg, 2.0 mmol) was dissolved in ethanol (30 mL). 10% palladium-carbon (50% wet) (1.01 g) was added, and the mixture was stirred in a hydrogen atmosphere at 50°C for 15 hours. The catalyst was removed by filtration, and then the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2 -> 95:5) to give 446 mg of the title compound as a pale yellow solid.
[α]D 23-15° (c=0.100, MeOH).
1H-NMR (400 MHz, CDCl3) δ: 5.29 (1H, brs), 3.47-3.18 (2H, m), 2.93-2.79 (2H, m), 2.15-1.71 (6H, m), 1.45 (9H, s).

- [Example 17]
7-[(1R,5S)-1-Amino-5-fluoro-3-azabicyclo[3.3.0]octan-3-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid
-

-
Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were added to a solution of (1R,5S)-1-(tert-butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane (250 mg, 1.02 mmol) in dimethyl sulfoxide (5.0 mL). The mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for ten days. Ethanol (6.0 mL), water (2.0 mL), and triethylamine (2.0 mL) were added to the reaction solution, and the mixture was stirred at 80°C for one hour. The solvent was evaporated under reduced pressure, and then the residue was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with water twice and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2), and the resulting fraction was concentrated under reduced pressure. Then, the residue was dissolved in concentrated hydrochloric acid (3.5 mL) under ice-cooling, and the solution was stirred at room temperature for one hour. The reaction solution was washed with chloroform five times, and then the aqueous layer was adjusted to pH 12 with a saturated sodium hydroxide solution. The basic solution was adjusted to pH 7.4 with hydrochloric acid, followed by extraction with chloroform. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the solvent was evaporated under reduced pressure. The resulting residue was purified by PTLC (developed with the lower layer of chloroform:methanol:water = 7:3:1). The resulting residue was solidified with isopropanol to give 7.1 mg of the title compound as a pale yellow solid.
-
1H-NMR (400 MHz, 0.1N NaOD) δ: 8.50 (1H, s), 7.77 (1H, d, J=13.73 Hz), 5.80-4.80 (1H, m), 4.22-4.10 (1H, m), 3.99-3.85 (1H, m), 3.68-3.47 (2H, m), 3.43-3.34 (1H, m), 2.68 (3H, s), 2.21-1.98 (3H, m), 1.97-1.56 (4H, m), 1.42-1.23 (1H, m).
MS (FAB); m/z: 422 (M+H)+.
Anal.Calcd C21H22F3N3O3·0.5H2O·0.25IPA: C, 58.65; H, 5.66; F, 12.80; N, 9.43. Found: C, 58.63; H, 5.35; F, 12.35; N, 9.22.
IR (ATR) ν: 2971, 2856, 1722, 1614, 1450, 1432, 1322, 1132, 1097, 987, 954, 929, 887, 856, 804 cm-1.

WO 2012018105
http://www.google.st/patents/WO2012018105A1?cl=en
The following structural formula

compound represented by, 7 – [(1R, 5S) -1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, . the 2S) -2 – fluoro-cyclopropane-1 – yl] -1,4 – dihydro-8 – methyl-4 – oxo-3-quinoline -) is referred to as carboxylic acid (hereinafter referred to as Compound A multi-agent containing quinolone resistance including resistant Gram-positive cocci resistant pneumococcus, etc., widely against gram-negative bacteria from Gram-positive bacteria, and, in addition to show strong antibacterial activity, convulsions, which is known in the art as a side effect of the antimicrobial agent of the present system potential cardiotoxicity light and toxicity-inducing activity (photosensitivity), has been reported recently in clinical further (QT prolongation), blood sugar abnormalities, and to express the side effects of delayed-type drug 疹等 is excellent safety low, Then, it is excellent oral absorbability and organ migration properties become apparent, is expected as an antimicrobial agent superior (Patent Document 1).
WO 2008/082009 pamphlet
The compound A, was synthesized according to the method described in Patent Document 1.
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A – [(1R, 5S) – 1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, 2S) -2 – fluoro-cyclo-1 – yl] -1 , 4 – dihydro-8 – methyl-4 – oxo-3-quinoline – was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue – was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 5.3,7.9,10.6,13.3,21.1,23.0,25.1,27.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A – was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
ref
- Higuchi, S.; Onodera, Y.; Chiba, M.; Hoshino, K.; Gotoh, N. Potent in vitro antibacterial activity of DS-8587, a new generation of broad spectrum quinolone, against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, doi:10.1128/AAC.02374-12.
- Daiichi Sankyo. Major R&D pipeline as of July, 2013. Available online: http://www.daiichisankyo.com/rd/pipeline/pdf/Pipeline_EN.pdf (accessed on 28 September 2013).
| EP0343524A1 | May 19, 1989 | Nov 29, 1989 | Shionogi Seiyaku Kabushiki Kaisha | Pyridonecarboxylic acids and antibacterial agents |
| JPH0395176A | Title not available | |||
| JPH02231475A | Title not available | |||
| JPH08225567A | Title not available | |||
| JPS6345261A | Title not available | |||
| JPS6456673A | Title not available | |||
| JPS61282382A | Title not available | |||
| US5017708 * | Sep 8, 1989 | May 21, 1991 | Shionogi & Co., Ltd. | Azabicycloalkanes |
| WO1994014794A1 | Dec 28, 1993 | Jul 7, 1994 | Hideki Ao | 8-methoxyquinolonecarboxylic acid derivative |
| WO1995021163A1 | Feb 2, 1995 | Aug 10, 1995 | Katsumi Chiba | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor |
| WO1996023782A1 | Feb 1, 1996 | Aug 8, 1996 | Daiichi Seiyaku Co | Heterocyclic compounds |

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.
Ozenoxacin in phase 3……topical formulation in the treatment of impetigo

1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid
1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
Ferrer Internacional (Spain), phase 3 Gram-positive
Ferrer Internacional has completed one Phase III clinical trial to evaluate the topical formulation of ozenoxacin in the treatment of impetigo [
|
poster……http://landing.quotientbioresearch.com/blog/bid/50380/Ozenoxacin-Activity-against-Atypical-Bacteria
Ozenoxacin is active against a great number of pathogens, such as Propionibacterium acnes, Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) including ciprofloxacin-resistant strains, methicillin-susceptible Staphylococcus epidermidis (MSSE), methicillin-resistant Staphylococcus epidermidis (MRSE), Streptococcus pyogenes, Group G Streptococci, penicillin-resistant Streptococcus pneumoniae, Beta-lactamase positive Haemophilus influenzae, non-typeable strains of Haemophilus influenzae, Beta-lactamase positive Moraxella catarrhalis, Neisseria meningitides, Legionella pneumophila, Mycoplasma pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Streptococcus agalactiae group B, Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma hominis, Ureaplasma urealyticum Helicobacter pylori, Bacteroides fragilis, Clostridium perfringens, Escherichia coli, quinolone-resistant Escherichia coli, Salmonella spp., Shigella spp., ciprofloxacin-susceptible Pseudomonas aeruginosa, Clostridium difficile, and Listeria monocytogenes.
Ozenoxacin is a novel non-fluorinated quinolone antibacterial agent. It is currently in late stage phase 3 trials for the topical treatment of impetigo. The bacterial action of ozenoxacin is through the dual inhibition of DNA gyrase and topoisomerase IV. Excellent in vitro and in vivo antibacterial activity has been demonstrated in pre-clinical and clinical studies against a broad range of bacterial organisms. This includes organisms with emerging resistance to quinolones. Phase I and II clinical trials have also shown that ozenoxacin is a safe and effective antibacterial agent. No evidence of adverse effects as linked to topically formulated halogenated quinolones has been shown.
Ozenoxacin (I) was firstly disclosed in US6335447, and equivalent patents. Its chemical name is 1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid. Its chemical formula is: H

Ozenoxacin (I)
Topical application of antimicrobial agents is a useful tool for therapy of skin and skin structures infections, sexually transmitted diseases and genital tract infections and some systemic infections susceptible to topical treatment. Topical antimicrobial therapy has several potential advantages compared with systemic therapy.
Firstly, it can avoid an unnecessary exposure of the gut flora which may exert selection for resistance. Secondly, it is expected that the high local drug concentration in topical application and the negligible systemic absorption should overwhelm many mutational resistances. Thirdly, topical applications are less likely than systemic therapy to cause side effects. Accordingly, some topical compositions comprising ozenoxacin have been reported in the art.
JP2002356426A discloses ointments and gels for skin. An ointment comprising ozenoxacin 1%, N-methyl-2-pyrrolidone 8%, propylene glycol 14.9%, oleic acid 0.9%, diisopropanolamine 2.3%, polyethylene glycol 400 20.2%, polyethylene glycol 4000 50.2%, and water 3.2% is reported in Example 2.
JP2003226643A discloses aqueous solutions comprising ozenoxacin, cyclodextrin, and a viscous agent.
EP1731138A1 discloses fine particle dispersion liquid comprising ozenoxacin to be used in the manufacture of pharmaceutical compositions.
WO2007015453A1 discloses lotions comprising ozenoxacin.
JP2007119456A discloses aqueous suspensions containing nanoparticles and solution granules of ozenoxacin to be used in the manufacture of pharmaceutical compositions. Ophthalmic solutions are mentioned preferably. A combined use of ozenoxacin, magnesium ions, and hydroxypropyl-β-cyclodextrin specially for ophthalmic use is disclosed in Yamakawa, T. et al., Journal of Controlled Release (2003), 86(1 ), 101-103.
Semisolid topical compositions are useful alternatives to liquid compositions, because of their better manipulation and consequent patient preferences. However, in spite of the great diversity of components present in the semisolid compositions disclosed in the art, no quantitative stability studies are available for them.
Thus, there is a need of proved stable semisolid topical compositions comprising ozenoxacin as active ingredient, wherein microbiological and therapeutic activities are warranted because of demonstrated durable and prolonged pharmaceutical stability.
Synthesis
US6335447
http://www.google.co.in/patents/US6335447
EXAMPLE 5
To a solution of 0.80 g of 7-[6-({[(benzyloxy)-carbonyl] (methyl)amino}methyl)-5-methyl-3-pyrdyl]-1-cyclo-propyl-8-methyl-4-oxo-1,4-dihydro-3-quinoline-carboxylic acid in 16 ml of acetic acid was added 0.20 g of 5% (w/w) palladium-carbon and the mixture was stirred at ambient temperature and atmospheric pressure for 2 hours under a hydrogen atmosphere. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in a mixed solvent consisting of 3.8 ml of ethanol and 3.8 ml of water. After adding 3.8 ml of an aqueous 1 mol/l sodium hydroxide solution thereto and adjusting the solution to pH 5.5with 1 mol/l hydrochloric acid, 10 ml of chloroform was added thereto. An organic layer was separated and dried over anhydrous magnesium sulfate and the solvent was evaporated under reduced pressure. Addition of diethyl ether to the obtained residue and filtration of crystals afforded 0.25 g of colorless crystals of 1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
IR (KBr) cm−1: 3322, 1721; NMR(d1-TFA) δ: 1.2-1.9 (4H, m), 2.94 (3H, s), 3.05 (3H, s), 3.29 (3H, s), 4.6-5.0 (1H, m), 5.12 (2H, s), 7.91 (1H, d, J=8.5 Hz), 8.6-9.0 (2H, m), 9.0-9.3 (1H, brs), 9.75 (1H, s). Melting point: 199° C.
- Ferrer Group. Key development projects. Available online: http://www.ferrergrupo.com/Innovation_Innovacion-Pipeline-de-proyectos-ENG (accessed on 15 April 2013).
- Yamakawa, T.; Mitsuyama, J.; Hayashi, K. In vitro and in vivo antibacterial activity of T-3912, a novel non-fluorinated topical quinolone. J. Antimicrob. Chemother. 2002, 49, 455–465, doi:10.1093/jac/49.3.455.
- Ferrer Internacional. Efficacy and safety of ozenoxacin 1% cream versus placebo in the treatment of patients with impetigo. Available online: http://clinicaltrials.gov/ct2/show/NCT01397461 (accessed on 13 April 2013).
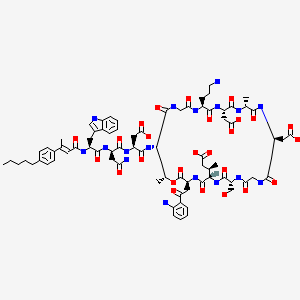
SUROTOMYCIN for Clostridium difficile-associated diarrhea

Surotomycin
Click to access surotomycin.pdf
N-[(2E)-3-(4-Pentylphenyl)-2-butenoyl]-D-tryptophyl-D-asparaginyl-N-[(3S,6S,9R,15S,18R,21S,24S,30S,31R)-3-[2-(2-aminophenyl)-2-oxoethyl]-24-(3-aminopropyl)-15,21-bis(carboxymethyl)-6-[(2R)-1-carboxy-2 -propanyl]-9-(hydroxymethyl)-18,31-dimethyl-2,5,8,11,14,17,20,23,26,29-decaoxo-1-oxa-4,7,10,13,16,19,22,25,28-nonaazacyclohentriacontan-30-yl]-L-α-asparagine
MOLECULAR FORMULA C77H101N17O26
MOLECULAR WEIGHT 1680.7
SPONSOR Cubist Pharmaceuticals, Inc.
CODE DESIGNATION CB-183,315
CB-315, CB-183315, CB-183,315
CAS REGISTRY NUMBER 1233389-51-9
U.S. – Fast Track (Treat Clostridium difficile-associated diarrhea (CDAD));
U.S. – Qualified Infectious Disease Program (Treat Clostridium difficile-associated diarrhea (CDAD))
| Company | Cubist Pharmaceuticals Inc. |
| Description | Oral antibacterial lipopeptide |
| Therapeutic Modality | Macrocycle |
| Latest Stage of Development | Phase III |
| Standard Indication | Diarrhea (infectious) |
| Indication Details | Treat Clostridium difficile-associated diarrhea (CDAD) |
EMEA……..
| Name | |||
|---|---|---|---|
| P/0096/2013: EMA decision of 29 April 2013 on the agreement of apaediatric investigation plan and on the granting of a deferral for surotomycin (EMEA-001226-PIP01-11) |
Surotomycin is an investigational oral antibiotic. This antibiotic is under investigation for the treatment of life-threatening Diarrhea, commonly caused by the bacteria Clostridium difficile.[1]
CB-183315 is an investigational antibacterial drug candidate in phase III clinical trials at Cubist for the treatment of Clostridium difficile-associated diarrhea. It is a potent, oral, cidal lipopeptide. In 2012, Qualified Infectious Disease Product Designation was assigned in the U.S. for the treatment of clostridium difficile-associated diarrhea (CDAD).
Surotomycin (CB-315)

 Surotomycin Overview
Surotomycin Overview  Surotomycin Fact Sheet
Surotomycin Fact SheetSurotomycin is an antibacterial lipopeptide discovered by Cubist scientists in our research laboratories in Lexington, Massachusetts. Surotomycin is both bactericidal against Clostridium difficile and more potent than vancomycin in vitro. Surotomycin stays at the site of infection in the bowel, with minimal systemic absorption and it does not interfere with normal bowel flora. Based on its features and its preclinical safety profile, Cubist filed an Investigational New Drug (IND) Application for surotomycin in December 2008.
Following safety and pharmacokinetic studies in healthy human volunteers, Cubist began a Phase 2 study in April 2010 to assess the safety and efficacy of surotomycin in patients with CDAD, in particular to assess its ability to reduce relapse rates. In this trial of 209 patients, two different doses of surotomycin were studied and compared with oral vancomycin. The higher dose demonstrated a high clinical cure rate as evidenced by resolution of diarrhea, comparable to oral vancomycin. The most interesting results in this study, however, relate to recurrence rates. The percent of patients who had an initial response to treatment but who subsequently had a recurrence or relapse was 36 percent in the oral vancomycin arm and was 17 percent in the surotomycin 250mg treatment group — about a 50% reduction in relapse rate, which was statistically significant. In this trial, 32% of patients were infected with the hypervirulent NAP-1 strain of C. difficile. The clinical response rate in the subset of patients infected with the NAP-1 strain was comparable across the surotomycin and oral vancomycin groups. Though not statistically significant, there was a modest reduction in the relapse rates in the subset of surotomycin patients infected with NAP-1 strains.
The ability to reduce relapses is important to both patients and health care providers. In the Phase 2 study we assessed the impact of surotomycin and oral vancomycin on normal bowel flora. Treatment with surotomycin had a very minimal impact on levels of Bacteroides, a key normal bowel bacterial species, compared to oral vancomycin which resulted in a marked depletion of stool levels of these bacteria during treatment. Why does this matter? The reason is — bowel flora like Bacteroides are critical in providing a competitive environment in the bowel that prevents C. difficile overgrowth. We believe that it is this difference in impact on normal bowel flora that helps explain the differences seen in recurrence rates following treatment with Surotomycin versus oral vancomycin.
Surotomycin’s Phase 3 program includes two identical global, randomized, double-blind, active-controlled, multi-center trials. The primary objective is to demonstrate non-inferiority of surotomycin versus the comparator, oral vancomycin, in clinical response at the end of treatment in adult subjects with CDAD, using a non-inferiority margin of 10%. We also have designed this trial to allow us to demonstrate that sustained clinical response to surotomycin at the end of the study is superior to oral vancomycin. Also, we will fully evaluate the safety of surotomycin in the study subjects.
In late 2012 Cubist received from the FDA a Qualified Infectious Disease Product (QIDP) designation for surotomycin. Additionally, in early 2013 Cubist was granted Fast track status for surotomycin. The QIDP designation and subsequent granting of Fast Track status was made possible by the GAIN Act, Title VIII (Sections 801 through 806) of the Food and Drug Administration Safety and Innovation Act. The GAIN Act provides pharmaceutical and biotechnology companies with incentives to develop new antibacterial and antifungal drugs for the treatment of life-threatening infectious diseases caused by drug resistant pathogens. Qualifying pathogens are defined by the GAIN Act to include multi-drug resistant Gram-negative bacteria, including Pseudomonas, Acinetobacter, Klebsiella, and Escherichia coli species; resistant Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus; multi-drug resistant tuberculosis; and Clostridium difficile.
About CDAD
CDAD is a disease caused by an overgrowth of, and subsequent toxin production by, C. difficile, a resident anaerobic spore-forming Gram-positive bacterium of the lower gastrointestinal tract. This overgrowth is caused by the use of antibiotics for the treatment of common community and hospital acquired infections (HAIs). Although they treat the underlying infection, many antibiotics disrupt the natural gut flora and allow C. difficile to proliferate. C. difficile produces enterotoxin and cytotoxin, which can lead to severe diarrhea, sepsis and even death. While most types of HAIs are declining, the infection caused by C. difficile remains at historically high levels. According to the latest data from the Centers for Disease Control, C. difficile continues to be the leading cause of death associated with gastroenteritis in the US. For CDAD alone, there was more than a five-fold increase in deaths between 1999 and 2007. C. difficile causes diarrhea linked to 14,000 American deaths each year. About 25% of C. difficile infections first show symptoms in hospital patients; 75% first show in nursing home patients or in people recently cared for in doctors’ offices and clinics. C. difficile infections cost at least $1 billion in extra health care costs annually.
SUROTOMYCIN
CB-183,315 is a cyclic lipopeptide antibiotic currently in Phase III clinical trials for the treatment of Clostridium difficile-associated disease (CDAD). As disclosed in International Patent Application WO 2010/075215, herein incorporated by reference in its entirety, CB-183,315 has antibacterial activity against a broad spectrum of bacteria, including drug-resistant bacteria and C. difficile. Further, the CB-183,315 exhibits bacteriacidal activity.
CB-183,315 (Figure 1) can be made by the deacylation of BOC-protected daptomycin, followed by acylation and deprotection as described in International Patent Application WO 2010/075215.
During the preparation and storage of CB-183,315, the CB-183,315 molecule can convert to structurally similar compounds as shown in Figures 2-4, leading to the formation of anhydro-CB-183,315 (Figure 3) and beta-isomer of CB-183,315 (“B- isomer CB183,315” in Figure 2). Accordingly, one measure of the chemical stability of CB- 183 ,315 is the amount of CB- 183 ,315 (Figure 1 ) present in the CB- 183 ,315 composition relative to the amount of structurally similar compounds including anhydro-CB-183,315 (Figure 3) and beta-isomer of CB-1 83,315 (Figure 2). The amount of CB-183,315 relative to the amount of these structurally similar compounds can be measured by high performance liquid chromatography (FIPLC) after reconstitution in an aqueous diluent (e.g., as described in Example 10). In particular, the purity of CB-183,315 and amounts of structurally similar compounds (e.g., Figures 2, 3 and 4) can be determined from peak areas obtained from HPLC (e.g., according to Example 10 herein), and measuring the rate of change in the amounts of CB-183,315 over time can provide a measure of CB-183,315 chemical stability in a solid form.
There is a need for solid CB-183,315 compositions with improved chemical stability in the solid form (i.e., higher total percent CB-183,315 purity over time), providing advantages of longer shelf life, increased tolerance for more varied storage conditions (e.g., higher temperature or humidity) and increased chemical stability.
……………..
WO2010075215A1
http://www.google.com/patents/WO2010075215A1?cl=en ………… copy paste link
Example 1
Preparation of N-{1 -[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl-D- asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D-alanyl-L-α- aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).


1003 1004

Step 1 : Preparation of (E)-ethyl 3-(4-pentylphenyl) but-2-enoate (1002).
A mixture of commercially available 1-(4-pentylphenyl)ethanone (5 g, 26.3 mmol) and (ethoxycarbonylmethylene)-triphenylphosphorane (18.3 g, 52.5 mmol) was stirred at 150 0C for 48 hours under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and diluted with ethyl acetate (50 ml_) and petroleum ether (200 ml_). The suspension was filtered through a fritted funnel. The concentrated filtrate was purified by flash column chromatography with silica gel (petroleum ether : ethyl acetate = 80:1 ) to give the title compound (1.6 g) having the following physical data: 1H NMR (300 MHz, δ, CDCI3) 0.90 (br, 3H), 1.36 (br, 7), 1.63 (br, 2H), 2.58 (s, 3H), 2.63 (br, 2H), 4.22 (q, 2H), 6.15 (s, 1 H), 7.20 (d, 2H), 7.41 (d, 2H).
Step 2: Preparation of (E)-3-(4-pentylphenyl) but-2-enoic acid (1003).
A solution of compound 1002 (1.5 g, 5.77 mmol) in ethanol (50 ml_) and 3N potassium hydroxide (25 ml_) was stirred at 45 0C for 3 hours. The reaction mixture was concentrated and the resulting residue was diluted with water (50 ml_). The aqueous solution was acidified to pH 2 with 1 N hydrochloric acid and extracted with EtOAc (2 * 30 ml_). The combined organic layers were dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (silica gel, petroleum ether : ethyl acetate = 10:1) to afford the title compound (0.95 g) having the following physical data: 1 H NMR (300 MHz, δ, CDCI3) 0.90 (br, 3H), 1.33 (br, 4H), 1.62 (br, 2H), 2.60 (br, 5H), 6.18 (s, 1 H), 7.18 (d, 2H), 7.42 (d, 2H).
Step 3: Preparation of (E)-3-(4-pentylphenyl)but-2-enoyl chloride (1004).
Oxalyl chloride (3.2 mL, 36.60 mmol) and DMF (50 μl_) were added drop wise to a solution of compound 1003 (5.0 g, 21.52 mmol) in dichloromethane (100 mL) at 0 0C. The reaction solution was warmed up to room temperature and stirred for 4 hours. The reaction mixture was concentrated in vacuum and the residue was dried under hi-vacuum for 3 hours. The crude product was used in the next step without further purification.
Step 4: Preparation of N-{1 -[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl-D- asparaginyl-L-α-aspartyl-L-threonylglycyl-L-[(N-tert-butoxycarbonyl)-ornithyl]-L-α- aspartyl-D-alanyl-L-α-aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2- diamino-γ-oxobenzenebutanoic acid (13-→4)-lactone (1005).
Deacylated BOC-protected daptomycin (3.5Og, 2.23 mmol) and sodium bicarbonate (1.13 g, 61.0 mmol) were dissolved in THF (130 mL) and water (50 mL). The deacylated BOC-protected daptomycin sodium bicarbonate solution was cooled to 0 0C. and a solution of compound 1004 (1.96 g, 7.82 mmol) in THF (20 mL) was then introduced. The reaction mixture was warmed to room temperature and stirred for 4 hours. The mixture was concentrated in vacuum to remove THF. The remaining aqueous solution was loaded on a C18 flash chromatography column (35mηnχ 300mm, Bondesil HF C18 resin purchased from Varian). The column was first washed with water to remove salt and then with methanol to wash out product. Crude compound 1005 (3.46 g) was afforded as a white solid after removal of methanol. MS m/z 1780.8 (M + H)+.
Steps 5-6: Preparation of N-{1-[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl- D-asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D-alanyl-L-α- aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).
TFA (10 ml_) was added to a solution of compound 1005 (3.46 g) in DCM (50 mL) at room temperature. The reaction mixture was stirred vigorously for 45 minutes and added slowly to vigorously stirring diethyl ether (100 mL). The resulting yellow precipitation was collected by filtration. The crude product was purified by Preparative HPLC to afford the TFA salt of compound 6 (0.75 g). MP carbonate resin (purchased from Biotage) was added to the solution of compound 6 TFA salt (0.70 g, 0.39 mmol) in anhydrous methanol (30.0 mL). The mixture was stirred at room temperature for 4 hours. The resins were removed by filtration and rinsed with methanol. The methanol solution was concentrated under vacuum to give product as off-white solid (408 mg). MS m/z 1680.7 (M + H)+.
Example 1 b
Alternative preparation of N-{1-[(E)-3-(4-pentylphenyl)but-2-enoyl]}- L-tryptophyl-D-asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D- alanyl-L-α-aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).
daptomycin,

1003

A solution of (E)-3-(4-pentylphenyl)but-2-enoic acid (1 100 g, 4.73 mol), Λ/-Ethyl-Λ/’-(3-dimethylaminopropyl)carbodiimide hydrochloride (907 g, 4.73 mol), HOBT (640 g, 4.73 mol) and 4-(dimethylamino)pyridine (22 g, 0.18 mol) in DMF (11 L) was stirred at room temperature for 4 hours at which point the activation of the (E)-3-(4-pentylphenyl)but-2-enoic acid was deemed complete by HPLC.
This reaction mixture was added to a suspension of Deacylated BOC- protected daptomycin (2600 g, 1.66 mol), sodium bicarbonate (804 g, 9.57 mol) in water (11.25 L) and 1 ,4-dioxane (33.75 L). The mixture was stirred at room temperature for 2.5 hours at which time HPLC indicated complete consumption of Deacylated BOC-protected daptomycin. The reaction mixture was diluted with water (22.5 L) and cooled with an ice bath. Concentrated hydrochloric acid (5.25 L) was added while maintaining the internal temperature below 30 0C. After the addition, the solution was stirred at room temperature for 5 days at which time HPLC indicated complete consumption of the Boc protected intermediate.
The reaction mixture was washed with methyl terf-butyl ether (90 L then approximately 60 L then approximately 45 L then approximately 45 L) to remove 1 ,4-dioxane. The remaining solution (approximately 44 L) was adjusted to pH 2.69 with 2N sodium hydroxide (11.3 L) and water (53.4 L). This material was processed by Tangential Flow Filtration (TTF) with a 1 K membrane until the total volume was reduced to 54 L.Water (120 L) was added in two portions and the solution was concentrated to 52 L by continued TTF. The aqueous solution (30 L of 52 L) was purified by chromatography using the following protocol: The aqueous solution was brought to three times of its volume (30 L→90l_) with 20% IPA in aqueous ammonium acetate solution (50 mM). The diluted solution was applied to a 38 L HP20SS resin column at 1.5 L/min. The column was eluted with IPA solution in aqueous 50 mM ammonium acetate (25%→30%→35%, 60 L each concentration).
Fractions (approximately 11 L) were collected and analyzed by HPLC. The fractions with HPLC purity less than 80% were combined and purified again using the same method. The key fractions from both chromatographic separations (with HPLC purity >80%) were combined and acidified with concentrated HCI to pH 2-3. The resulting solution was desalted on an ion exchange column (HP20SS resin, 16 L) which was eluted with WFI (until conductivity = 4.8 μS) followed by IPA in WFI (36 L 10%→ 40 L 60%). The yellow band which was eluted with 60% IPA (approximately 19L) was collected, adjusted to pH 2-3 with concentrated HCI and lyophilized to yield 636.5 g of Compound 49 (HPLC purity of 87.0%). MS m/z 1680.7 (M + H)+.
……………………………..
see formulation
| WO2012162567A1 | May 24, 2012 | Nov 29, 2012 | Cubist Pharmaceuticals, Inc. | Cb-183,315 compositions and related methods |
References
- http://www.cubist.com/downloads/Surotomycin-Fact-Sheet-13013.pdf
-
- Cubist Pharmaceuticals. Cubist products and pipeline. Available online: http://www.cubist.com/products/(accessed on 15 April 2013).
- Cubist Pharmaceuticals. Study of CB-183,315 in patients with Clostridium difficile associated diarrhea.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01597505 (accessed on 15 April 2013).
- Cubist Pharmaceuticals. A study of CB-183,315 in patients with Clostridium difficile associated diarrhea.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01598311 (accessed on 15 April 2013).
- Mascio, C.T.M.; Mortin, L.I.; Howland, K.T.; van, P.A.D.G.; Zhang, S.; Arya, A.; Chuong, C.L.; Kang, C.; Li, T.; Silverman, J.A. In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob. Agents Chemother. 2012, 56, 5023–5030, doi:10.1128/AAC.00057-12.
-
WO2012162567A1 May 24, 2012 Nov 29, 2012 Cubist Pharmaceuticals, Inc. Cb-183,315 compositions and related methods
-
WO2001097851A2 * Jun 18, 2001 Dec 27, 2001 Cubist Pharm Inc Compositions and methods to improve the oral absorption of antimicrobial agents WO2010075215A1 Dec 18, 2009 Jul 1, 2010 Cubist Pharmaceuticals, Inc. Novel antibacterial agents for the treatment of gram positive infections WO2011063419A2 * Nov 23, 2010 May 26, 2011 Cubist Pharmaceuticals Inc. Lipopeptide compositions and related methods
This Little Known Chinese Herb Kills 12,000 Cancer Cells For Every Healthy Cell

 artemisinin
artemisininWormwood
Other common name(s): absinthium, absinth wormwood
Scientific/medical name(s): Artemisia absinthium
Description
Wormwood is a shrubby perennial plant whose upper shoots, flowers, and leaves are used in herbal remedies and as a bitter flavoring for alcoholic drinks. It is native to Europe, northern Africa, and western Asia, and now also grows in North America.
Overview
Available scientific evidence does not support claims that wormwood is effective in treating cancer, the side effects of cancer treatment, or any other conditions. The plant contains a volatile oil with a high level of thujone (see Thuja). There are reports that taking large doses of wormwood internally can cause serious problems with the liver and kidneys. It can also cause nausea, vomiting, stomach pain, headache, dizziness, seizures, numbness of the legs and arms, delirium, and paralysis.
Wormwood, or Artemisia absinthium, should not be confused with sweet wormwood, or Artemisia annua. Although wormwood is related to sweet wormwood, they are used in different ways. Extracts of sweet wormwood have been used in traditional herbal medicine, and an active ingredient, artemisinin, is now used in conventional medical treatment of malaria.
How is it promoted for use?
Wormwood is promoted as a sedative and anti-inflammatory. There are also claims that it can treat loss of appetite, stomach disorders, and liver and gallbladder complaints. In folk medicine it is used for a wide range of stomach disorders, fever, and irregular menstruation. It is also used to fight intestinal worms. Externally, it is applied to poorly healing wounds, ulcers, skin blotches, and insect bites. It is used in Moxibustion treatments for cancer (seeMoxibustion). Available scientific evidence does not support these claims.
What does it involve?
Wormwood is taken in small doses for a short period of time, usually a maximum of 4 weeks. It is available as a capsule and as a liquid that can be added to water to make a tincture. The whole herb is sometimes brewed as a tea. Wormwood oil, washes, or poultices can also be used on the skin. Although pure wormwood is not available, “thujone-free” wormwood extract has been approved by the US Food and Drug Administration (FDA) for use in foods and as a flavoring in alcoholic drinks such as vermouth.
What is the history behind it?
Artemisia absinthium was used by Hippocrates, and the earliest references to wormwood in Western civilization can be found in the Bible. Extract of wormwood was also used in ancient Egypt. The herb is mentioned often in first-century Greek and Roman writings and reportedly was placed in the sandals of Roman soldiers to help soothe their sore feet. It was taken as a treatment for tapeworms as far back as the Middle Ages.
In 1797, Henri Pernod developed absinthe, an alcoholic drink containing distilled spirits of wormwood, fennel, anise and sometimes other herbs. Absinthe became very popular in Europe and the United States in the nineteenth century. It was eventually banned in several countries in the early twentieth century due to its purported ill effects and addictive qualities. More recent analysis has suggested that, when properly prepared and distilled, the thujone content in these drinks was very low. It appears more likely that the addictiveness and other ill effects of absinthe were due to its alcohol content, which is around 60% to 85%. Varying additives or impurities from different distillers may have also produced some of these effects. Even though absinthe is illegal in some countries, various types can be found in some European countries. However, their thujone content is strictly limited. Wormwood is also an ingredient in vermouth and other drinks.
What is the evidence?
Available scientific studies do not support the use of wormwood for the treatment of cancer or the side effects of conventional cancer treatment. There is not enough evidence available to support its use for other conditions. Wormwood oil has been tested in laboratory studies and appears to inhibit the growth of some fungi. However, human tests have not been completed.
Some derivatives of Artemisia annua, or sweet wormwood, a relative of wormwood, have been shown to be effective in the treatment of malaria. In fact, the World Health Organization approved artemisinin for use against malaria in Africa in 2004. These extracts also show some promise in laboratory studies as cancer treatment drugs. Further studies are required to find out whether the anti-cancer results apply to people. It is important to remember that extracted compounds are not the same as the whole herb, and study results are not likely to show the same effects.
Are there any possible problems or complications?
Wormwood should be avoided, especially by women who are pregnant or breast-feeding, by people who have had seizures, and by those with ulcers or stomach irritation. Thujone, a component of wormwood, is known to cause muscle spasms, seizures, and hallucinations if taken internally. In high doses it is known to damage the liver and the kidneys.
Because of its thujone content, large doses of wormwood taken internally can lead to vomiting, stomach and intestinal cramps, headaches, dizziness, nervous system problems, and seizures. Wormwood can also lead to liver failure. The New England Journal of Medicine reported that a man who ordered essential oil of wormwood over the Internet, thinking he had purchased absinthe, suffered liver failure shortly after drinking the oil. Wormwood may also make seizures more likely and may interfere with the anti-convulsant effects of medicines such as phenobarbital.
The plant is a relative of ragweed and daisies. Those with allergies to these types of plants may also be allergic to wormwood. Contact with wormwood can cause rash in some people.
Relying on this type of treatment alone and avoiding or delaying conventional medical care for cancer may have serious health consequences.
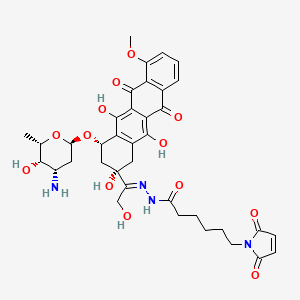
Aldoxorubicin…….Treatment of cancer …HIV-derived Kaposi’s Sarcoma, pancreatic cancer and for the treatment of soft tissue sarcoma.


Aldoxorubicin
Click to access aldoxorubicin.pdf
 in phase 3
in phase 3
(E)-N’-(1-((2S,4S)-4-(((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl)-2-hydroxyethylidene)-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanehydrazide hydrochloride
1H-Pyrrole-1-hexanoic acid, 2,5-dihydro-2,5-dioxo-, (2E)-2-[1-[(2S,4S)-4-[(3-amino-
2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-
7-methoxy-6,11-dioxo-2-naphthacenyl]-2-hydroxyethylidene]hydrazide
N’-[(1E)-1-{(2S,4S)-4-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-2,5,12-
trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl}-2-
hydroxyethylidene]-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanohydrazide
MOLECULAR FORMULA C37H42N4O13
MOLECULAR WEIGHT 750.7
SPONSOR CytRx Corp.
CODE DESIGNATION INNO-206
CAS REGISTRY NUMBER 1361644-26-9
CAS: 151038-96-9 (INNO-206); 480998-12-7 (INNO-206 HCl salt), 1361644-26-9

hydrochloride
CAS: 151038-96-9
Chemical Formula: C37H42N4O13
Exact Mass: 750.27484
Molecular Weight: 750.75
|
Certificate of Analysis: |
|
|
QC data: |
|
|
Safety Data Sheet (MSDS): |

|
In vitro protocol: |
Clin Cancer Res. 2012 Jul 15;18(14):3856-67 |
|
In vivo protocol: |
Clin Cancer Res. 2012 Jul 15;18(14):3856-67. Invest New Drugs. 2010 Feb;28(1):14-9. Invest New Drugs. 2012 Aug;30(4):1743-9. Int J Cancer. 2007 Feb 15;120(4):927-34. |
|
Clinical study: |
Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. |

Aldoxorubicin (INNO-206): Aldoxorubicin, also known as INNO-206, is the 6-maleimidocaproyl hydrazone derivative prodrug of the anthracycline antibiotic doxorubicin (DOXO-EMCH) with antineoplastic activity. Following intravenous administration, doxorubicin prodrug INNO-206 binds selectively to the cysteine-34 position of albumin via its maleimide moiety. Doxorubicin is released from the albumin carrier after cleavage of the acid-sensitive hydrazone linker within the acidic environment of tumors and, once located intracellularly, intercalates DNA, inhibits DNA synthesis, and induces apoptosis. Albumin tends to accumulate in solid tumors as a result of high metabolic turnover, rapid angiogenesis, hyervasculature, and impaired lymphatic drainage. Because of passive accumulation within tumors, this agent may improve the therapeutic effects of doxorubicin while minimizing systemic toxicity.
“Aldoxorubicin has demonstrated effectiveness against a range of tumors in both human and animal studies, thus we are optimistic in regard to a potential treatment for Kaposi’s sarcoma. The current standard-of-care for severe dermatological and systemic KS is liposomal doxorubicin (Doxil®). However, many patients exhibit minimal to no clinical response to this agent, and that drug has significant toxicity and manufacturing issues,” said CytRx President and CEO Steven A. Kriegsman. “In addition to obtaining valuable information related to Kaposi’s sarcoma, this trial represents another opportunity to validate the value and viability of our linker technology platform.” The company expects to announce Phase-2 study results in the second quarter of 2015.
Kaposi’s sarcoma is an orphan indication, meaning that only a small portion of the population has been diagnosed with the disease (fewer than 200,000 individuals in the country), and in turn, little research and drug development is being conducted to treat and cure it. The FDA’s Orphan Drug Act may grant orphan drug designation to a drug such as aldoxorubicin that treats a rare disease like Kaposi’s sarcoma, offering market exclusivity for seven years, fast-track status in some cases, tax credits, and grant monies to accelerate research
INNO-206 is an anthracycline in early clinical trials at CytRx Oncology for the treatment of breast cancer, HIV-related Kaposi’s sarcoma, glioblastoma multiforme, stomach cancer and pancreatic cancer. In 2014, a pivotal global phase 3 clinical trial was initiated as second-line treatment in patients with metastatic, locally advanced or unresectable soft tissue sarcomas. The drug candidate was originally developed at Bristol-Myers Squibb, and was subsequently licensed to KTB Tumorforschungs. In August 2006, Innovive Pharmaceuticals (acquired by CytRx in 2008) licensed the patent rights from KTB for the worldwide development and commercialization of the drug candidate. No recent development has been reported for research that had been ongoing for the treatment of small cell lung cancer (SCLC).
INNO-206 is a doxorubicin prodrug. Specifically, it is the 6-maleimidocaproyl hydrazone of doxorubicin. After administration, the drug candidate rapidly binds endogenous circulating albumin through the acid sensitive EMCH linker. Circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including the heart, bone marrow and the gastrointestinal tract. Once inside the acidic environment of the tumor cell, the EMCH linker is cleaved and free doxorubicin is released at the tumor site. Like other anthracyclines, doxorubicin inhibits DNA and RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly-growing cancer cells. It also creates iron-mediated free oxygen radicals that damage the DNA and cell membranes. In 2011, orphan drug designation was assigned in the U.S. for the treatment of pancreatic cancer and for the treatment of soft tissue sarcoma.
CytRx Corporation (NASDAQ:CYTR) has announced it has initiated a pivotal global Phase 3 clinical trial to evaluate the efficacy and safety of aldoxorubicin as a second-line treatment for patients with soft tissue sarcoma (STS) under a Special Protocol Assessment with the FDA. Aldoxorubicin combines the chemotherapeutic agent doxorubicin with a novel linker-molecule that binds specifically to albumin in the blood to allow for delivery of higher amounts of doxorubicin (3.5 to 4 times) without several of the major treatment-limiting toxicities seen with administration of doxorubicin alone.
According to a news from Medicalnewstoday.com; CytRx holds the exclusive worldwide rights to INNO-206. The Company has previously announced plans to initiate Phase 2 proof-of-concept clinical trials in patients with pancreatic cancer, gastric cancer and soft tissue sarcomas, upon the completion of optimizing the formulation of INNO-206. Based on the multiple myeloma interim results, the Company is exploring the possibility of rapidly including multiple myeloma in its INNO-206 clinical development plans.
According to CytRx’s website, In preclinical models, INNO-206 was superior to doxorubicin with regard to ability to increase dosing, antitumor efficacy and safety. A Phase I study of INNO-206 that demonstrated safety and objective clinical responses in a variety of tumor types was completed in the beginning of 2006 and presented at the March 2006 Krebskongress meeting in Berlin. In this study, doses were administered at up to 4 times the standard dosing of doxorubicin without an increase in observed side effects over historically seen levels. Objective clinical responses were seen in patients with sarcoma, breast, and lung cancers.
INNO-206 – Mechanism of action:
According to CytRx’s website, the proposed mechanism of action is as the follow steps: (1) after administration, INNO-206 rapidly binds endogenous circulating albumin through the EMCH linker. (2) circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including heart, bone marrow and gastrointestinal tract; (3) once albumin-bound INNO-206 reaches the tumor, the acidic environment of the tumor causes cleavage of the acid sensitive linker; (4) free doxorubicin is released at the site of the tumor.
INNO-206 – status of clinical trials:
CytRx has announced that, in December 2011, CytRx initiated its international Phase 2b clinical trial to evaluate the preliminary efficacy and safety of INNO-206 as a first-line therapy in patients with soft tissue sarcoma who are ineligible for surgery. The Phase 2b clinical trial will provide the first direct clinical trial comparison of INNO-206 with native doxorubicin, which is dose-limited due to toxicity, as a first-line therapy. (source:http://cytrx.com/inno_206, accessed date: 02/01/2012).
Results of Phase I study:
In a phase I study a starting dose of 20 mg/m2 doxorubicin equivalents was chosen and 41 patients with advanced cancer disease were treated at dose levels of 20–340 mg/m2 doxorubicin equivalents . Treatment with INNO-206 was well tolerated up to 200 mg/m2 without manifestation of drug-related side effects which is a ~3-fold increase over the standard dose for doxorubicin (60 mg/kg). Myelosuppression and mucositis were the predominant adverse effects at dose levels of 260 mg/m2 and became dose-limiting at 340 mg/m2. 30 of 41 patients were assessable for analysis of response. Partial responses were observed in 3 patients (10%, small cell lung cancer, liposacoma and breast carcinoma). 15 patients (50%) showed a stable disease at different dose levels and 12 patients (40%) had evidence of tumor progression. (source: Invest New Drugs (2010) 28:14–19)
|
References |
1: Kratz F, Azab S, Zeisig R, Fichtner I, Warnecke A. Evaluation of combination therapy schedules of doxorubicin and an acid-sensitive albumin-binding prodrug of doxorubicin in the MIA PaCa-2 pancreatic xenograft model. Int J Pharm. 2013 Jan 30;441(1-2):499-506. doi: 10.1016/j.ijpharm.2012.11.003. Epub 2012 Nov 10. PubMed PMID: 23149257.
2: Walker L, Perkins E, Kratz F, Raucher D. Cell penetrating peptides fused to a thermally targeted biopolymer drug carrier improve the delivery and antitumor efficacy of an acid-sensitive doxorubicin derivative. Int J Pharm. 2012 Oct 15;436(1-2):825-32. doi: 10.1016/j.ijpharm.2012.07.043. Epub 2012 Jul 28. PubMed PMID: 22850291; PubMed Central PMCID: PMC3465682.
3: Kratz F, Warnecke A. Finding the optimal balance: challenges of improving conventional cancer chemotherapy using suitable combinations with nano-sized drug delivery systems. J Control Release. 2012 Dec 10;164(2):221-35. doi: 10.1016/j.jconrel.2012.05.045. Epub 2012 Jun 13. PubMed PMID: 22705248.
4: Sanchez E, Li M, Wang C, Nichols CM, Li J, Chen H, Berenson JR. Anti-myeloma effects of the novel anthracycline derivative INNO-206. Clin Cancer Res. 2012 Jul 15;18(14):3856-67. doi: 10.1158/1078-0432.CCR-11-3130. Epub 2012 May 22. PubMed PMID: 22619306.
5: Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012 Jul 20;161(2):429-45. doi: 10.1016/j.jconrel.2011.11.028. Epub 2011 Dec 1. Review. PubMed PMID: 22155554.
6: Elsadek B, Kratz F. Impact of albumin on drug delivery–new applications on the horizon. J Control Release. 2012 Jan 10;157(1):4-28. doi: 10.1016/j.jconrel.2011.09.069. Epub 2011 Sep 16. Review. PubMed PMID: 21959118.
7: Kratz F, Fichtner I, Graeser R. Combination therapy with the albumin-binding prodrug of doxorubicin (INNO-206) and doxorubicin achieves complete remissions and improves tolerability in an ovarian A2780 xenograft model. Invest New Drugs. 2012 Aug;30(4):1743-9. doi: 10.1007/s10637-011-9686-5. Epub 2011 May 18. PubMed PMID: 21590366.
8: Boga C, Fiume L, Baglioni M, Bertucci C, Farina C, Kratz F, Manerba M, Naldi M, Di Stefano G. Characterisation of the conjugate of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin with lactosaminated human albumin by 13C NMR spectroscopy. Eur J Pharm Sci. 2009 Oct 8;38(3):262-9. doi: 10.1016/j.ejps.2009.08.001. Epub 2009 Aug 18. PubMed PMID: 19695327.
9: Graeser R, Esser N, Unger H, Fichtner I, Zhu A, Unger C, Kratz F. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest New Drugs. 2010 Feb;28(1):14-9. doi: 10.1007/s10637-008-9208-2. Epub 2009 Jan 8. PubMed PMID: 19148580.
10: Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008 Dec 18;132(3):171-83. doi: 10.1016/j.jconrel.2008.05.010. Epub 2008 May 17. Review. PubMed PMID: 18582981.
11: Unger C, Häring B, Medinger M, Drevs J, Steinbild S, Kratz F, Mross K. Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin. Clin Cancer Res. 2007 Aug 15;13(16):4858-66. PubMed PMID: 17699865.
12: Lebrecht D, Walker UA. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):108-13. Review. PubMed PMID: 17652814.
13: Kratz F. DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. Review. PubMed PMID: 17501697.
14: Kratz F, Ehling G, Kauffmann HM, Unger C. Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin. Hum Exp Toxicol. 2007 Jan;26(1):19-35. PubMed PMID: 17334177.
15: Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Kratz F, Walker UA. The 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damage. Int J Cancer. 2007 Feb 15;120(4):927-34. PubMed PMID: 17131338.
16: Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004 Dec;23(4-5):393-7. PubMed PMID: 15567293.
| EP0169111A1 * | Jun 18, 1985 | Jan 22, 1986 | Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0269188A2 * | Jun 18, 1985 | Jun 1, 1988 | Elf Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0306943A2 * | Sep 8, 1988 | Mar 15, 1989 | Neorx Corporation | Immunconjugates joined by thioether bonds having reduced toxicity and improved selectivity |
| EP0328147A2 * | Feb 10, 1989 | Aug 16, 1989 | Bristol-Myers Squibb Company | Anthracycline immunoconjugates having a novel linker and methods for their production |
| EP0398305A2 * | May 16, 1990 | Nov 22, 1990 | Bristol-Myers Squibb Company | Anthracycline conjugates having a novel linker and methods for their production |
| EP0457250A2 * | May 13, 1991 | Nov 21, 1991 | Bristol-Myers Squibb Company | Novel bifunctional linking compounds, conjugates and methods for their production |
Licorice मुलेठी, 甘草, شیرین بیان Inhibits 92% of Breast Cancer Cells & Slows Growth by 83% in Vivo:


 Isoliquiritigenin
IsoliquiritigeninLiquorice or licorice (/ˈlɪk(ə)rɪʃ/ lik-(ə-)rish or /ˈlɪk(ə)rɪs/ lik-(ə-)ris)[2] is the root of Glycyrrhiza glabra from which a somewhat sweet flavor can be extracted. The liquorice plant is a legume that is native to southern Europe and parts of Asia. It is not botanically related to anise, star anise, or fennel, which are sources of similar flavouring compounds. The word ‘liquorice’/’licorice’ is derived (via the Old French licoresse), from the Greek γλυκύρριζα (glukurrhiza), meaning “sweet root”,[3] from γλυκύς (glukus), “sweet”[4] + ῥίζα (rhiza), “root”,[5][6] the name provided by Dioscorides.[7]
Description
It is a herbaceous perennial, growing to 1 m in height, with pinnate leaves about 7–15 cm (3–6 in) long, with 9–17 leaflets. The flowers are 0.8–1.2 cm (⅓–½ in) long, purple to pale whitish blue, produced in a loose inflorescence. The fruit is an oblong pod, 2–3 cm (1 in) long, containing several seeds.[8]The roots are stoloniferous.[9]
Chemistry
The scent of liquorice root comes from a complex and variable combination of compounds, of which anethole is the most minor component (0-3% of total volatiles). Much of the sweetness in liquorice comes from glycyrrhizin, which has a sweet taste, 30–50 times the sweetness of sugar. The sweetness is very different from sugar, being less instant and lasting longer.
The isoflavene glabrene and the isoflavane glabridin, found in the roots of liquorice, are xenoestrogens.[10][11]

A, phase I metabolites of ILG formed during incubation with rat liver microsomes and NADPH. Based on accurate mass measurements, HPLC retention times, MS/MS analyses, and comparison with data reported by Guo et al. (18), the structures of metabolites M1, M2, M3, M4, M5, M6, and M7 were assigned as liquiritigenin, 7,8,4′-trihydroxychalcone, sulfuretin, 7,3′,4′-trihydroxychalcone, davidigenin, trans-6,4′-dihydroxyaurone, and cis-6,4′-dihydroxyaurone, respectively. B, structures of ILG glucuronide conjugates formed by rat liver microsomes in the presence of UDPGA.
Cultivation and uses
Liquorice grows best in deep valleys, well-drained soils, with full sun, and is harvested in the autumn, two to three years after planting.[8] Countries producing liquorice include Iran, Afghanistan, the People’s Republic of China, Pakistan, Iraq, Azerbaijan, Uzbekistan, Turkmenistan and Turkey.[12]
The world’s leading manufacturer of liquorice products is M&F Worldwide, which manufactures more than 70% of the worldwide liquorice flavors sold to end-users.[13]

Tobacco
Most liquorice is used as a flavoring agent for tobacco. For example, M&F Worldwide reported in 2011 that approximately 63% of its liquorice product sales are to the worldwide tobacco industry for use as tobacco flavor enhancing and moistening agents in the manufacture of American blend cigarettes, moist snuff, chewing tobacco and pipe tobacco.[12] American blend cigarettes made up a larger portion of worldwide tobacco consumption in earlier years,[14] and the percentage of liquorice products used by the tobacco industry was higher in the past. M&F Worldwide sold approximately 73% of its liquorice products to the tobacco industry in 2005,[15] and a consultant to M&F Worldwide’s predecessor company stated in 1975 that it was believed that well over 90% of the total production of liquorice extract and its derivatives found its way into tobacco products.[16]
Liquorice provides tobacco products with a natural sweetness and a distinctive flavor that blends readily with the natural and imitation flavoring components employed in the tobacco industry, represses harshness, and is not detectable as liquorice by the consumer.[16] Tobacco flavorings such as liquorice also make it easier to inhale the smoke by creating bronchodilators, which open up the lungs.[17] Chewing tobacco requires substantially higher levels of liquorice extract as emphasis on the sweet flavor appears highly desirable.[16]
Food and candy
Liquorice flavour is found in a wide variety of liquorice candies or sweets. In most of these candies the taste is reinforced by aniseed oil, and the actual content of liquorice is very low. Liquorice confections are primarily purchased by consumers in the European Union.[12]
In the Netherlands, where liquorice candy (“drop”) is one of the most popular forms of sweet, only a few of the many forms that are sold contain aniseed, although mixing it with mint, menthol or with laurel is quite popular. Mixing it with ammonium chloride (‘salmiak’) is also popular. The most popular liquorice, known in the Netherlands as zoute drop (salty liquorice) actually contains very little salt, i.e. sodium;[18] the salty taste is probably due to ammonium chloride, and the blood pressure raising effect is due to glycyrrhizin, see below. Strong, salty candies are popular in Scandinavia.
Pontefract in Yorkshire was the first place where liquorice mixed with sugar began to be used as a sweet in the same way it is in the modern day.[19] Pontefract cakes were originally made there. In County Durham, Yorkshire and Lancashire it is colloquially known as Spanish, supposedly because Spanish monks grew liquorice root at Rievaulx Abbey near Thirsk.[20]

Liquorice root

Various liquorice products.

Different flavoured liquorice sticks
Liquorice is popular in Italy (particularly in the South) and Spain in its natural form. The root of the plant is simply dug up, washed and chewed as a mouth freshener. Throughout Italy unsweetened liquorice is consumed in the form of small black pieces made only from 100% pure liquorice extract; the taste is bitter and intense. In Calabria a popular liqueur is made from pure liquorice extract. Liquorice is also very popular in Syria where it is sold as a drink. Dried liquorice root can be chewed as a sweet. Black liquorice contains approximately 100 calories per ounce (15 kJ/g).[21]
Medicine

Foliage

Glycyrrhiza glabra from Koehler’sMedicinal-Plants
The compound glycyrrhizin (or glycyrrhizic acid), found in liquorice, has been proposed as being useful for liver protection in tuberculosis therapy, however evidence does not support this use which may in fact be harmful.[22] Glycyrrhizin has also demonstrated antiviral, antimicrobial, anti-inflammatory, hepatoprotective and blood-pressure increasing effects in vitro and in vivo, as is supported by the finding that intravenous glycyrrhizin (as if it is given orally very little of the original drug makes it into circulation) slows the progression of viral and autoimmune hepatitis.[23][24][25][26] Liquorice has also demonstrated promising activity in one clinical trial, when applied topically, against atopic dermatitis.[27] Additionally liquorice has also proven itself effective in treating hyperlipidaemia (a high amount of fats in the blood).[28] Liquorice has also demonstrated efficacy in treating inflammation-induced skin hyperpigmentation.[29][30] Liquorice may also be useful in preventing neurodegenerative disorders and cavities.[31][32][33] Anti-ulcer, laxative, anti-diabetic, anti-inflammatory, immunomodulatory, antitumour and expectorant properties of liquorice have also been noted.[34][35][36]
In traditional Chinese medicine, liquorice (मुलेठी, 甘草, شیرین بیان) is commonly used in herbal formulae to “harmonize” the other ingredients in the formula and to carry the formula to the twelve “regular meridians”.[37]
Liquorice may be useful in conventional and naturopathic medicine for both mouth ulcers[38] and peptic ulcers.[39]
Its major dose-limiting toxicities are corticosteroid, in nature, due to the inhibitory effect its chief active constituents, glycyrrhizin and enoxolone have oncortisol degradation and include: oedema, hypokalaemia, weight gain or loss and hypertension.[40][41]
-

Sliver of liquorice root
-

Various liquorice root slivers
-

Liquorice root with bark
-

Inflorescence ofGlycyrrhiza glabra
.jpg)
.JPG)

References
- “Glycyrrhiza glabra information from NPGS/GRIN”. http://www.ars-grin.gov. Retrieved 6 March 2008.
- licorice. Merriam-Webster’s Medical Dictionary, © 2007 Merriam-Webster, Inc.
- γλυκύρριζα, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus
- γλυκύς, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus
- ῥίζα, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus<
- liquorice, on Oxford Dictionaries
- google books Maud Grieve, Manya Marshall – A modern herbal: the medicinal, culinary, cosmetic and economic properties, cultivation and folk-lore of herbs, grasses, fungi, shrubs, & trees with all their modern scientific uses, Volume 2 Dover Publications, 1982 & Pharmacist’s Guide to Medicinal Herbs Arthur M. Presser Smart Publications, 1 Apr 2001 2012-05-19
- Huxley, A., ed. (1992). New RHS Dictionary of Gardening. ISBN 0-333-47494-5
- Brown, D., ed. (1995). “The RHS encyclopedia of herbs and their uses”. ISBN 1-4053-0059-0
- Somjen, D.; Katzburg, S.; Vaya, J.; Kaye, A. M.; Hendel, D.; Posner, G. H.; Tamir, S. (2004). “Estrogenic activity of glabridin and glabrene from licorice roots on human osteoblasts and prepubertal rat skeletal tissues”. The Journal of Steroid Biochemistry and Molecular Biology 91(4–5): 241–246. doi:10.1016/j.jsbmb.2004.04.008. PMID 15336701.
- Tamir, S.; Eizenberg, M.; Somjen, D.; Izrael, S.; Vaya, J. (2001). “Estrogen-like activity of glabrene and other constituents isolated from licorice root”. The Journal of steroid biochemistry and molecular biology78 (3): 291–298. doi:10.1016/S0960-0760(01)00093-0.PMID 11595510.
- M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2010.
- M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2001.
- Erik Assadourian, Cigarette Production Drops, Vital Signs 2005, at 70.
- M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2005.
- Marvin K. Cook, The Use of Licorice and Other Flavoring Material in Tobacco (Apr. 10, 1975).
- Boeken v. Phillip Morris Inc., 127 Cal. App. 4th 1640, 1673, 26 Cal. Rptr. 3d 638, 664 (2005).
- [1] the online Dutch food composition database]
- “Right good food from the Ridings”. AboutFood.com. 25 October 2007.
- “Where Liquorice Roots Go Deep”. Northern Echo. Retrieved 9 December 2008.
- Licorice Calories
- Liu Q, Garner P, Wang Y, Huang B, Smith H (2008). “Drugs and herbs given to prevent hepatotoxicity of tuberculosis therapy: systematic review of ingredients and evaluation studies”. BMC Public Health (Systematic review) 8: 365. doi:10.1186/1471-2458-8-365. PMC 2576232.PMID 18939987.
- Sato, H; Goto, W; Yamamura, J; Kurokawa, M; Kageyama, S; Takahara, T; Watanabe, A; Shiraki, K (May 1996). “Therapeutic basis of glycyrrhizin on chronic hepatitis B.”. Antiviral Research 30 (2-3): 171–7.doi:10.1016/0166-3542(96)00942-4. PMID 8783808.
- van Rossum, TG; Vulto, AG; de Man, RA; Brouwer, JT; Schalm, SW (March 1998). “Review article: glycyrrhizin as a potential treatment for chronic hepatitis C.” (PDF). Alimentary Pharmacology & Therapeutics12 (3): 199–205. doi:10.1046/j.1365-2036.1998.00309.x.PMID 9570253.
- Chien, CF; Wu, YT; Tsai, TH (January 2011). “Biological analysis of herbal medicines used for the treatment of liver diseases.”. Biomedical Chromatography 25 (1-2): 21–38. doi:10.1002/bmc.1568.PMID 21204110.
- Yasui, S; Fujiwara, K; Tawada, A; Fukuda, Y; Nakano, M; Yokosuka, O (December 2011). “Efficacy of intravenous glycyrrhizin in the early stage of acute onset autoimmune hepatitis.”. Digestive Diseases and Sciences56 (12): 3638–47. doi:10.1007/s10620-011-1789-5.PMID 21681505.
- Reuter, J; Merfort, I; Schempp, CM (2010). “Botanicals in dermatology: an evidence-based review.”. American Journal of Clinical Dermatology11 (4): 247–67. doi:10.2165/11533220-000000000-00000.PMID 20509719.
- Hasani-Ranjbar, S; Nayebi, N; Moradi, L; Mehri, A; Larijani, B; Abdollahi, M (2010). “The efficacy and safety of herbal medicines used in the treatment of hyperlipidemia; a systematic review.”. Current pharmaceutical design 16 (26): 2935–47. PMID 20858178.
- Callender, VD; St Surin-Lord, S; Davis, EC; Maclin, M (April 2011). “Postinflammatory hyperpigmentation: etiologic and therapeutic considerations.”. American Journal of Clinical Dermatology 12 (2): 87–99. doi:10.2165/11536930-000000000-00000. PMID 21348540.
- Leyden, JJ; Shergill, B; Micali, G; Downie, J; Wallo, W (October 2011). “Natural options for the management of hyperpigmentation.”. Journal of the European Academy of Dermatology and Venereology 25 (10): 1140–5. doi:10.1111/j.1468-3083.2011.04130.x. PMID 21623927.
- Kannappan, R; Gupta, SC; Kim, JH; Reuter, S; Aggarwal, BB (October 2011). “Neuroprotection by spice-derived nutraceuticals: you are what you eat!” (PDF). Molecular Neurobiology 44 (2): 142–59.doi:10.1007/s12035-011-8168-2. PMC 3183139.PMID 21360003.
- Gazzani, G; Daglia, M; Papetti, A (April 2012). “Food components with anticaries activity.”. Current Opinion in Biotechnology 23 (2): 153–9.doi:10.1016/j.copbio.2011.09.003. PMID 22030309.
- Messier, C; Epifano, F; Genovese, S; Grenier, D (January 2012). “Licorice and its potential beneficial effects in common oro-dental diseases.”. Oral Diseases 18 (1): 32–9. doi:10.1111/j.1601-0825.2011.01842.x. PMID 21851508.
- Shibata, S (October 2000). “A drug over the millennia: pharmacognosy, chemistry, and pharmacology of licorice.”. Yakugaku Zasshi 120 (10): 849–62. PMID 11082698.
- Fiore, C; Eisenhut, M; Ragazzi, E; Zanchin, G; Armanini, D (July 2005). “A history of the therapeutic use of liquorice in Europe.”. Journal of Ethnopharmacology 99 (3): 317–24. doi:10.1016/j.jep.2005.04.015.PMID 15978760.
- Ming, LJ; Yin, AC (March 2013). “Therapeutic effects of glycyrrhizic acid.”. Natural Product Communications 8 (3): 415–8.PMID 23678825.
- Bensky, Dan; et al. (2004). Chinese Herbal Medicine: Materia Medica, Third Edition. Eastland Press. ISBN 0-939616-42-4.
- Das, S. K.; Das, V.; Gulati, A. K.; Singh, V. P. (1989). “Deglycyrrhizinated liquorice in aphthous ulcers”. The Journal of the Association of Physicians of India 37 (10): 647. PMID 2632514.
- Krausse, R.; Bielenberg, J.; Blaschek, W.; Ullmann, U. (2004). “In vitro anti-Helicobacter pylori activity of Extractum liquiritiae, glycyrrhizin and its metabolites”. Journal of Antimicrobial Chemotherapy 54 (1): 243–246.doi:10.1093/jac/dkh287. PMID 15190039.
- Olukoga, A; Donaldson, D (June 2000). “Liquorice and its health implications.”. The Journal of the Royal Society for the Promotion of Health 120 (2): 83–9. doi:10.1177/146642400012000203.PMID 10944880.
- Armanini, D; Fiore, C; Mattarello, MJ; Bielenberg, J; Palermo, M (September 2002). “History of the endocrine effects of licorice.”.Experimental and Clinical Endocrinology & diabetes 110 (6): 257–61.doi:10.1055/s-2002-34587. PMID 12373628.
National Institute of Health – Medline- PDRhealth.com – Profile of Deglycyrrhizinated Licorice (DGL)
- Chemical & Engineering News article on Licorice
- Non-profit site on the health aspects of licorice/liquorice
- Medical use of irritation on chest
Benefits of ginger in prostate cancer
Regular consumption of fruits, vegetables and Herbs & Spices are linked with some anti-cancer benefits.

Ginger (Zingiber officinale) is also coming under increasing scrutiny and is known to be an source of several bioactive phenolics (gingerols, paradols, shogaols and gingerones).
On these pages recently we have seen how Ginger has been studied and can display anti-inflammatory & antioxidant activities.
In this study, Karna et al investigated ginger extract and its growth-inhibitory and death-inductory effects in a spectrum of prostate cancer cells.
Comprehensive studies have confirmed that ginger extracts disturbed cell-cycle progression, impaired reproductive capacity, modulated cell-cycle and apoptosis regulatory molecules and induced apoptosis in human prostate cancer cells.
Tumour tissue from an animal model showed reduced proliferation index and widespread apoptosis compared with controls and did not exert any detectable toxicity in normal, rapidly dividing tissues such as gut and bone marrow.
The authors add that to the best of their knowledge…
View original post 23 more words
Researchers reverse bone loss in immune disorder

The absence of recruited neutrophils to the periodontal tissue in LAD patients leads to unrestrained local production of IL-23 and hence IL-17 and G-CSF. Increased IL-17 leads to inflammatory bone loss and dysbiosis, whereas increased G-CSF leads to excessive release of mature neutrophils from the bone marrow. In contrast, normal recruitment of neutrophils regulates the expression of the same cytokines maintaining homeostasis in terms of periodontal health and release of mature neutrophils from the bone marrow. Credit: Copyright Niki Moutsopoulos and George Hajishengallis
Patients with leukocyte adhesion deficiency, or LAD, suffer from frequent bacterial infections, including the severe gum disease known as periodontitis. These patients often lose their teeth early in life. New research by University of Pennsylvania School of Dental Medicine researchers, teaming with investigators from the National Institutes of Health, has demonstrated a method of reversing this bone loss and inflammation.
The work was led by Penn Dental Medicine’s…
View original post 671 more words
CHINESE MEDICINE..Scutellaria baicalensis fights cancer

Scutellaria baicalensis (or Baikal Skullcap, as opposed to Scutellaria lateriflora, a Skullcap native to North America) is a species of flowering plant in the Lamiaceae family.
Traditional Chinese medicine
It is one of the 50 fundamental herbs used in traditional Chinese medicine, where it has the name huáng qín (Chinese: 黄芩).[2] As a Chinese traditional medicine, Huang Qin usually refers to the dried root of Scutellaria baicalensis Georgi, S. viscidula Bge., S. amoena C.H. Wright, and S. ikoninkovii Ju.
Chemistry
Several chemical compounds have been isolated from the root; among them, baicalein, baicalin, wogonin, norwogonin, oroxylin A[3] and β-sitosterol are the major ones.
Etymology confusion
It is important to note the Latin name of the Skullcap being used as there are over 200 varieties, some used for various ailments, each with varying degrees of effectiveness. Sometimes Scutellaria lateriflora (North American Skullcap) is mistaken for Scutellaria baicalensis (Baikal Skullcap). This confusion can result in the intake of the lateriflora variety which is often processed and contaminated with other plants with high enough levels of toxicity to be of concern.
Baikal skullcap (scientific name Scutellaria baicalensis) is a plant. The root is used to make medicine. Common substitutions for Baikal skullcap in Chinese medicine include related plants whose scientific names are Scutellaria viscidula, Scutellaria amonea, and Scutellaria ikoninikovii.
Baikal skullcap is used to treat respiratory infections, hay fever, and fever. It is also used for gastrointestinal (GI) infections, as well as liver problems including viralhepatitis and jaundice.
Some people use Baikal skullcap for HIV/AIDS, kidney infections, pelvic inflammation, and sores or swelling. It is also used for scarlet fever, headache, irritability, red eyes, flushed face, seizures, epilepsy, hysteria, nervous tension, and to relieve a bitter taste in the mouth.

The active ingredient in Baikal skullcap, baicalin, is used in combination with shung hua (ephedra) to treat upper respiratory tract infections. In combination with other herbs, Baikal skullcap is used to treat attention deficit-hyperactivity disorder (ADHD),prostate cancer, a lung condition called bronchiolitis, arthritis, and hemorrhoids.
Baikal skullcap is also sometimes applied to the skin for psoriasis.
How does it work?
It is thought that the active chemicals in Baikal skullcap might be able to decrease inflammation, stop tumor growth, and prevent tumor cell reproduction.

Scutellaria baicalensis , also called Chinese skullcap, is a member of the mint family and has long been used in traditional Chinese herbal medicine . Chinese skullcap has been incorporated in herbal formulas designed to treat such widely varying conditions as cancer, liver disease, allergies, skin conditions, and epilepsy. The root is the part used medicinally.
Note: Chinese skullcap is substantially different from American skullcap ( Scutellaria lateriflora ).
Skullcap (Scutellaria baicalensis) has been widely used as a dietary ingredient and traditional herbal medicine owing to its anti-inflammatory and anticancer properties. In this study, we investigated the anti-allergic effects of skullcap and its active compounds, focusing on T cell-mediated responses ex vivoand in vivo. Splenocytes from mice sensitized with ovalbumin (OVA) were isolated for analyses of cytokine production and cell viability. Mice sensitized with OVA were orally administered skullcap or wogonin for 16 days, and then immunoglobulin (Ig) and cytokine levels were measured by enzyme-linked immunosorbent assays. Treatment with skullcap significantly inhibited interleukin (IL)-4 production without reduction of cell viability. Moreover, wogonin, but not baicalin and baicalein, suppressed IL-4 and interferon-gamma production. In vivo, skullcap and wogonin downregulated OVA-induced Th2 immune responses, especially IgE and IL-5 prediction. Wogonin as an active component of skullcap may be applied as a therapeutic agent for IgE- and IL-5-mediated allergic disorders…….http://www.mdpi.com/1420-3049/19/2/2536

References
- “Scutellaria baicalensis information from NPGS/GRIN”. USDA. Retrieved 2008-02-19.
- Zhang XW, Li WF, Li WW, Ren KH, Fan CM, Chen YY, Shen YL (2011). “Protective effects of the aqueous extract of Scutellaria baicalensis against acrolein-induced oxidative stress in cultured human umbilical vein endothelial cells”. Pharm Biol 49 (3): 256–261. doi:10.3109/13880209.2010.501803. PMID 20979538.
- Isolation and purification of baicalein, wogonin and oroxylin A from the medicinal plant Scutellaria baicalensis by high-speed counter-current chromatography. Hua-Bin Li and Feng Chen, Journal of Chromatography A, 13 May 2005, Volume 1074, Issues 1–2, pages 107–110, doi:10.1016/j.chroma.2005.03.088
- Scutellaria baicalensis List of Chemicals (Dr. Duke’s Databases)
- Scutellaria baicalensis (Plants for a Future)
- Sung Mun Jung et al., “Reduction of urate crystal-induced inflammation by root extracts from traditional oriental medicinal plants: elevation of prostaglandin D2 levels”, Arthritis Research & Therapy 2007, 9:R64 doi:10.1186/ar2222. Considers anti-inflammatory properties of dried roots from the species Angelica sinensis (Dong Quai), Acanthopanax senticosus (now known as Eleutherococcus senticosus, or Siberian Ginseng), andScutellaria baicalensis (Baikal Skullcap).
GREEN CHEMISTRY…Reduction of amides without hydride reagents

Clostridium sporogenes
Essentially all medicines and current drug candidates contain at least one basic nitrogen atom. A common approach to the synthesis of amines is to reduce the corresponding amide with a hydride reagent such as LiAlH4, DIBAL, RedAl, B2H6, Et3SiH, or polymethylhydroxysilane (PMHS).
The reaction survey reported that reduction of amides to amines was used in only 0.6% of chemical transformations; this number would surely be higher if safer methods for use on scale were available. The survey indicated that the number of amide reductions was equally split between diborane and hydride reagents.
Lithium aluminium hydride,

having a molecular weight of 38 and four hydrides per molecule, has the highest hydride density and is frequently used, even though it co-generates an inorganic by-product (lithium aluminum hydroxide) which is difficult to separate from the product. The workup procedure recommended by one bulk supplier (Chemetall) is to precipitate and filter the aluminum hydroxide salts. However, slow filtrations and product loss through occlusion or adsorption are typical problems that can be encountered.
Options for disposal of the cake include dissolving in water and sending to a waste water treatment plant or drying the cake and sending to a chemical waste dump that accepts solids.1 Both options have an environmental impact. Therefore, a generally applicable, safe, environmentally benign and economically viable method for the reduction of amides to amines would have an appreciable benefit to numerous processes.
Hydrogen gas is the ideal reductant because the only by-product is water. Thus, much research has been directed towards discovery of a transition metal catalyst selective for hydrogenation of amides. However, even with the best catalysts, both high temperature (![[similar]](https://i0.wp.com/www.rsc.org/images/entities/char_223c.gif) 150 °C) and pressure (>100 bar) are required. These conditions involve expensive high pressure hydrogenation equipment not typically available in a common pharmaceutical manufacturing plant.
150 °C) and pressure (>100 bar) are required. These conditions involve expensive high pressure hydrogenation equipment not typically available in a common pharmaceutical manufacturing plant.
The harsh conditions also preclude the use of these catalysts with substrates that contain other reducible or thermally labile functional groups. Recent research has led to the discovery of catalysts that are effective at lower temperature and pressure, giving encouragement that the goal of finding a selective, low pressure/temperature catalyst is realistic.2
Another approach would be to use a biotransformation to reduce the amide. It is notable that a number of bacteria and fungi reduce carboxylic acids to aldehydes or ketones.3 The usual fate of amides in biological pathways is hydrolysis. However, an anaerobic bacteria, Clostridium sporogenes, has been reported to reduce benzamide to benzylamine. 4

A key challenge in this technology area is gaining a detailed understanding of these complex enzyme-catalysed processes that require ATP/NADPH co-factor recycling, and getting the enzymes cloned and produced on a large scale in suitable expression systems.
The acylation/reduction strategy for N-alkylation avoids the need to handle alkylating agents and would be more widely used if a safer, more atom economical or preferably catalytic method for amide reduction were developed. The solution to this problem could be either chemical or biochemical.
- Chemetall brochures, Lithium Aluminum Hydride… strong, concentrated and economical, Oct. 2000, pp. 18–19 Search PubMed .
- A. A. Smith, P. Dani, P. D. Higginson and A. J. Pettman, World Pat., WO2005/066112 A1, 2005 Search PubMed .
- (a) A. Hage, H. E. Schoemaker and J. A. Field, Appl. Microbiol. Biotechnol., 1999, 52, 834–838 CrossRef CAS Search PubMed ; (b) A. He, T. Li, L. Daniels, I. Fotheringham and J. P. N. Rosazza, Appl. Environ. Microbiol., 2004, 70, 1874–1881 CrossRef CAS Search PubMed .
- O. Dipeolu, J. Gardiner and G. Stephens, Biotechnol. Lett., 2005, 27, 1803–1807 CrossRef CAS Search PubMed .
