
Home » Preclinical drugs (Page 5)
Category Archives: Preclinical drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PF-04745637

PF-04745637
cas 1917294-46-2
MW 509.00, MF C27 H32 Cl F3 N2 O2
Cyclopentanecarboxamide, 1-(4-chlorophenyl)-N-[2-[4-hydroxy-4-(trifluoromethyl)-1-piperidinyl]-3-phenylpropyl]-
rac-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyDcyclopentanecarboxamide
|
PRODUCT PATENT WO-2016067143-A1
|
| Applicants: | PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US) |
| Inventors: | SWAIN, Nigel Alan; (GB). PRYDE, David Cameron; (GB). RAWSON, David James; (GB). RYCKMANS, Thomas; (GB). SKERRATT, Sarah Elizabeth; (GB). AMATO, George Salvatore; (US). MARRON, Brian Edward; (US). REISTER, Steven Michael; (US). |

TrpA1 is a member of the Transient Receptor Potential (Trp) family of ion channels. It was first described as being activated in response to noxious cold. It is activated by a number of exogenous chemical compounds and some endogenous inflammatory mediators. It has also been reported to be activated in response to mechanical stress.
There is substantial evidence for the involvement of TrpA1 in the physiology of pain, including neuropathic and inflammatory pain, and in pruritus (itch). For example, see:
Bautista, D.M. et al., “TRPA 1: A Gatekeeper for Inflammation” , Annu. Rev. Physiol.2013, 75, 181-200;
Bishnoi, M. & Premkumar, L.S., “Changes in TRP Channels Expression in Painful
Conditions”, Open Pain Journal 2013, 6(Suppl. 1), 10-22;Brederson, J.-D. et al., “Targeting TRP channels for pain relief, Eur. J. Pharmacol.2013, 716, 61-76;
Radresa, O. et al., “Roles of TRPAI in Pain Pathophysiology and Implications for the Development of a New Class of Analgesic Drugs”, Open Pain Journal 2013, 6(Suppl. 1), 137-153; and Toth, B.I. & Biro, T., “TRP Channels and Pruritus” , Open Pain Journal 2013, 6(Suppl.1), 62-80.
There is a continuing interest in finding new compounds that interact with TrpA1.
 Nigel Swain
Nigel Swain
PATENT
E8 that is 1-(4-chlorophenyl)-/V-[2-(4-hydroxy-4-(trifluoromethyl)piperidin-1-yl)-3-phenylpropyl]-cyclopentanecarboxamide, or a pharmaceutically acceptable salt thereof. This compound is represented by formula (lE).

Example 1
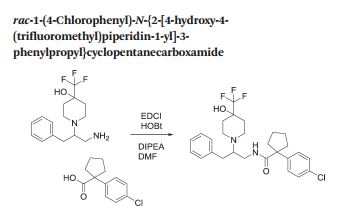
rac-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyDcyclopentanecarboxamide

Method 1
To a solution of rac-1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 2, 50 mg, 0.214 mmol) in DMF (1 mL) was added 1-(4-chlorophenyl)cyclopentanecarboxylic acid (37 mg, 0.165 mmol), DIPEA (0.035 mL, 0.198 mmol) and EDCI (38 mg, 0.198 mmol), followed by HOBt (30 mg, 0.198 mmol) and the reaction was stirred at room temperature for 18 hours. Water was added and the reaction stirred for a further 2 hours. DCM was added with further stirring for 1 hour followed by elution through a phase separation cartridge. The organic filtrate was concentrated in vacuo. The residue was dissolved in MeOH and treated with ethereal HCI with standing for 18 hours. The resulting suspension was filtered and triturated with EtOAc, heptanes and TBME to afford the title compound as the hydrochloride salt (69 mg, 82%).
1H NMR (400MHz, DMSO-d6): δ ppm 1.50-1.60 (m, 4H), 1.70-1.90 (m, 4H), 2.15-2.25 (m, 2H), 2.40-2.48 (m, 2H), 2.70-2.80 (m, 1 H), 3.05-3.25 (m, 6H), 3.47-3.62 (m, 2H), 6.38 (br s, 1 H), 7.20-7.40 (m, 9H), 7.80 (br m, 1 H).
MS m/z 509 [M+H]+
Example 1 may also be prepared according to the following method:
A mixture of 1-(4-chlorophenyl)cyclopentanecarboxylic acid (25.7 g, 114 mmol), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid-hexafluoro phosphate (49.4 g, 130 mmol) and N,N-diisopropylethylamine (40 mL, 229 mmol) in DMF (475 mL) was stirred at room temperature for 15 minutes. To this mixture was added a solution of 1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 2, 31.4 g, 104 mmol) in DMF (200 mL). The reaction was stirred at room temperature for 18 hours before partitioning between EtOAc (600 mL) and saturated aqueous sodium hydrogen carbonatesolution (600 mL). The aqueous layer was washed with EtOAc (2 x 600 mL). The combined organic layers were washed with water (600 mL), brine (600 mL), dried over sodium sulphate and concentrated in vacuo. The residue was purified using silica gel column chromatography eluting with 0: 1 to 1 : 1 EtOAc: heptanes to afford the title compound (44 g, 76%).
1H NMR (400MHz, CDCI3): δ ppm 1.35 (br s, 1 H), 1.49-1.85 (m, 6H), 1.90-1.99 (m, 2H), 2.25-2.55 (m, 7H), 2.56-2.70 (m, 1 H), 2.75-3.00 (m, 4H), 3.23-3.31 (m, 1 H), 5.87 (br s, 1 H), 7.07 (d, 2H), 7.16-7.30 (m, 7H).
MS m/z 509 [M+H]+
Examples 2 and 3
IS) and (R)-1-(4-Chlorophenyl)-N-f2-r4-hvdroxy-4-(trifluoromethyl)piperidin-1-vn-3-phenylpropyl)cyclopentanecarboxamide

Example 2
To a suspension of (S)-1-(1-amino-3-phenylpropan-2-yl)-4-(trifluoromethyl)piperidin-4-ol (Preparation 3, 70 mg, 0.232 mmol) and 1-(4-chlorophenyl)cyclopentanecarboxylic acid (57.3 mg, 0.255 mmol) in acetonitrile (0.8 mL) was added triethylamine (0.133 mL, 0.928 mmol) followed bypropylphosphonic anhydride (50% wt solution in EtOAc, 0.21 mL, 0.35 mmol). The reaction was stirred at room temperature for 1.5 hours after which the solution was purified directly by silica gel column chromatography eluting with 0-30% EtOAc in heptanes to afford the title compound (75 mg, 64%).
[a]D20 = +9.6 in DCM [20 mg/mL]
ee determination:
Column: ChiralTech AD-H, 250×4.6 mm, 5 micron.
Mobile phase A: CO2; Mobile phase B: MeOH with 0.2% ammonium hydroxide Gradient: 5% B at 0.00 mins, 60% B at 9.00 mins; hold to 9.5 mins and return to 5% B at 10 mins. Flow rate 3 mL/min.
Rt = 5.047 minutes, ee = 95%
Example 2 may also be prepared from rac-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4- (trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide(Example 1).
The racemate was separated into two enantiomers using preparative chiral chromatography as described below:
Chiralpak IA, 4.6x250mm, 5 micron.
Mobile phase: Hexane:DCM:EtOH:DEA 90:8:2:0.1
Flow rate: 1 mL/min
Rt = 8.351 minutes and Rt = 10.068 minutes
The first eluting isomer is Example 2: (S)-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide. ee = 100% The second eluting isomer is Example 3: (R)-1-(4-chlorophenyl)-N-{2-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]-3-phenylpropyl}cyclopentanecarboxamide. ee = 99.62% The compound of Example 2 prepared from the chiral separation method is identical by a-rotation and retention time to the compound of Example 2 prepared as the single enantiomer described above.
MS m/z 509 [M+H]+
1H NMR (400MHz, DMSO-d6): δ 1.30-1.80 (m, 10H), 2.20-2.30 (m, 1 H), 2.35-2.60 (m, 6H), 2.65-2.85 (m, 4H), 3.00-3.15 (m, 1 H), 5.50 (br s, 1 H), 6.95-7.00 (m, 1 H), 7.05-7.15 (m, 2H), 7.20-7.35 (m, 6H) ppm
PAPER
The discovery of a potent series of carboxamide TRPA1 antagonists
DOI: 10.1039/C6MD00387G, http://pubs.rsc.org/en/Content/ArticleLanding/2016/MD/C6MD00387G?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+rss%2FMD+%28RSC+-+Med.+Chem.+Commun.+latest+articles%29#!divAbstract
. Please note PF-6667294 is Compound 4 and PF-4746537 is Compound 8.
A series of potent and selective carboxamide TRPA1 antagonists were identified by a high throughput screen. Structure–activity relationship studies around this series are described, resulting in a highly potent example of the series. Pharmacokinetic and skin flux data are presented for this compound. Efficacy was observed in a topical cinnamaldehyde flare study, providing a topical proof of pharmacology for this mechanism. These data suggest TRPA1 antagonism could be a viable mechanism to treat topical conditions such as atopic dermatitis.

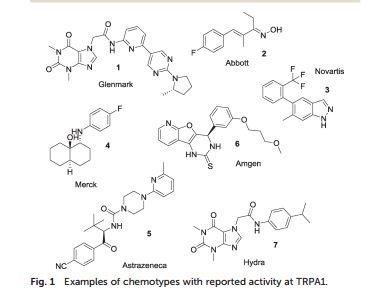


Discovery and development of TRPV1 antagonists
https://en.wikipedia.org/wiki/Discovery_and_development_of_TRPV1_antagonists
/////////////PF-04745637, PF 04745637, PF04745637, PFIZER, PRECLINICAL, TRPV1 antagonists, atopic dermatitis, 1917294-46-2
c1(ccccc1)CC(CNC(=O)C3(c2ccc(cc2)Cl)CCCC3)N4CCC(CC4)(O)C(F)(F)F
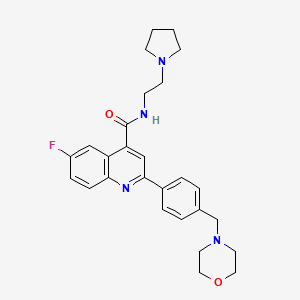
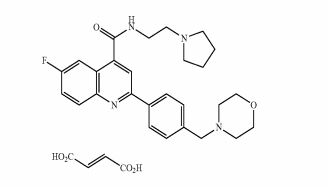
DDD 107498
DDD 107498, DDD 498
PATENT WO 2013153357, US2015045354
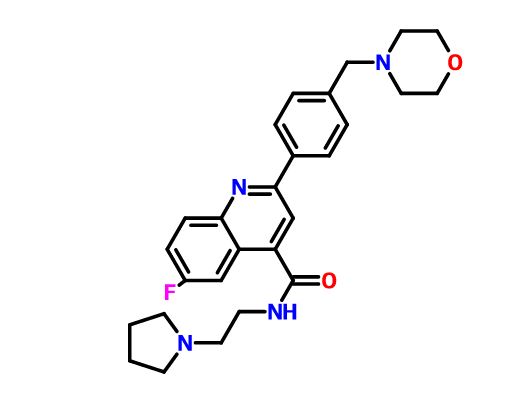
6-Fluoro-2-[4-(morpholinomethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide
6-Fluoro-2-[4-(4-morpholinylmethyl)phenyl]-N-[2-(1-pyrrolidinyl)ethyl]-4-quinolinecarboxamide
4-Quinolinecarboxamide, 6-fluoro-2-[4-(4-morpholinylmethyl)phenyl]-N-[2-(1-pyrrolidinyl)ethyl]-
CAS 1469439-69-7
CAS 1469439-71-1 SUCCINATE
| MF C27H31FN4O2 | |
| MW | 462.559043 g/mol |
|---|
- 6-fluoro-2-[4-(morpholin-4-ylmethyl)phenyl]-N-(2-pyrrolidin-1-ylethyl)quinoline-4-carboxamide
- Originator Medicines for Malaria Venture; University of Dundee
- Class Small molecules
- Mechanism of Action Protein synthesis inhibitors
Highest Development Phases
- No development reported Malaria
Most Recent Events
- 16 Jul 2016 No recent reports of development identified for preclinical development in Malaria in United Kingdom
- 01 Apr 2015 DDD 498 licensed to Merck Serono worldwide for the treatment of Malaria
| Inventors | Ian Hugh Gilbert, Neil Norcross, Beatriz Baragana Ruibal, Achim Porzelle |
| Original Assignee | University Of Dundee |

Prof Ian Gilbert:
Head of Biological Chemistry and Drug Discovery
BCDD, College of Life Sciences, University of Dundee, DD1 5EH, UK
Tel: +44 (0) 1382-386240


Merck Serono and MMV sign agreement to develop potential antimalarial therapy
Agreement further diversifies MMV’s partner base, strengthening our antimalarial research and development portfolio
 Photo © Merck Serono
Photo © Merck Serono
Merck Serono, the biopharmaceutical business of Merck, and MMV announced today that an agreement has been signed for Merck Serono to obtain the rights to the investigational antimalarial compound DDD107498 from MMV. This agreement underscores the commitment of Merck Serono to provide antimalarials for the most vulnerable populations in need.
“This agreement strengthens our Global Health research program and our ongoing collaboration with Medicines for Malaria Venture,” said Luciano Rossetti, Executive Vice President, Global Head of Research & Development at Merck Serono. “MMV is known worldwide for its major contribution to delivering innovative antimalarial treatments to the most vulnerable populations suffering from this disease, and at Merck Serono we share this goal.”
DDD107498 originated from a collaboration between MMV and the University of Dundee Drug Discovery Unit, led by Prof. Ian Gilbert and Dr. Kevin Read. The objective of the clinical program is to demonstrate whether the investigational compound exerts activity on a number of malaria parasite lifecycle stages, and remains active in the body long enough to offer potential as a single-dose treatment against the most severe strains of malaria.
While development and commercialization of the compound is under Merck Serono’s responsibility, MMV will provide expertise in the field of malaria drug development, including its clinical and delivery expertise, and provide access to its public and private sector networks in malaria-endemic countries.
Merck Serono has a dedicated Global Health R&D group working to address key unmet medical needs related to neglected diseases, such as schistosomiasis and malaria, with a focus on pediatric populations in developing countries. Its approach is based on public-private partnerships and collaborations with leading global health institutions and organizations in both developed and developing countries.
“Working with partners like Merck Serono is critical to the progress of potential antimalarial compounds, like DDD107498, through the malaria drug pipeline,” said Dr. Timothy Wells, Chief Scientific Officer at MMV. “Their Global Health Program is gaining momentum and we need more compounds to tackle malaria, a disease that places a huge burden on the world’s most vulnerable populations. DDD107498 holds great promise and we look forward to working with the Merck Serono team through the development phase.”
According to the World Health Organization, there were an estimated 198 million cases of malaria worldwide in 2013, and an estimated 584,000 deaths, primarily in young children from the developing world. The launch of the not-for-profit research foundation, MMV, in 1999 and a number of collaborations and partnerships, including those with Merck Serono, has contributed to reducing the major gap in malaria R&D investment and subsequent dearth of new medicines.
“It’s hugely encouraging to see the German pharmaceutical industry increasing their engagement in the development of novel antimalarials,” said global malaria expert Prof. Dr. Peter Kremsner, Director of the Institute for Tropical Medicine at the University of Tübingen, Germany. “The Merck Serono and MMV collaboration to develop DDD107498 is a great step. It’s a compound that offers lots of promise so I’m excited to see how it progresses.


Scots scientists in ‘single dose’ malaria treatment breakthrough
An antimalarial drug that could treat patients was discovered by Dundee university scientists
Scientists have discovered an antimalarial compound that could treat malaria patients in a single dose and help prevent the spread of the disease from infected people.
The compound DDD107498 also has the potential to treat patients with malaria parasites resistant to current medications, researchers say.
Scientists hope it could lead to treatments and protection against the disease, which claimed almost 600,000 lives amid 200 million reported cases in 2013.
The compound was identified through a collaboration between the University of Dundee’s drug discovery unit (DDU) and the Medicines for Malaria Venture (MMV), a separate organisation.
The compound is now undergoing further safety testing with a view to entering human clinical trials within the next year.
Details of the discovery have been published in the journal Nature.
Professor Ian Gilbert, head of chemistry at the DDU, who led the team that discovered the compound, said: “The publication describes the discovery and profiling of this exciting new compound.
“It reveals that DDD107498 has the potential to treat malaria with a single dose, prevent the spread of malaria from infected people and protect a person from developing the disease in the first place.
“There is still some way to go before the compound can be given to patients. However, we are very excited by the progress that we have made.”
The World Health Organisation reports that there were 200 million clinical cases of malaria in 2013, with 584,000 people dying from the disease. Most of these deaths were children under the age of five and pregnant women.
MMV chief executive officer Dr David Reddy said: “Malaria continues to threaten almost half of the world’s population – the half that can least afford it.
“DDD107498 is an exciting compound since it holds the promise to not only treat but also protect these vulnerable populations.
“The collaboration to identify and progress the compound, led by the drug discovery unit at the University of Dundee, drew on MMV’s network of scientists from Melbourne to San Diego.”The publication of the research is an important step and a clear testament to the power of partnership.”
MMV selected DDD107498 to enter preclinical development in October 2013 following the recommendation of its expert scientific advisory committee.
Since then, with MMV’s leadership, large quantities of the compound have been produced and it is undergoing further safety testing with a view to entering human clinical trials within the next year.
Merck Serono has recently obtained the right to develop and, if successful, commercialise the compound, with the input of MMV’s expertise in the field of malaria drug development and access and delivery in malaria-endemic countries.
Dr Michael Chew from the Wellcome Trust, which provides funding for the DDU and MMV, said: “The need for new antimalarial drugs is more urgent than ever before, with emerging strains of the parasite now showing resistance against the best available drugs.
“These strains are already present at the Myanmar-Indian border and it’s a race against time to stop resistance spreading to the most vulnerable populations in Africa.
“The discovery of this new antimalarial agent, which has shown remarkable potency against multiple stages of the malaria lifecycle, is an exciting prospect in the hunt for viable new treatments.”
PAPER


Discovery of a Quinoline-4-carboxamide Derivative with a Novel Mechanism of Action, Multistage Antimalarial Activity, and Potent in Vivo Efficacy

// https://tpc.googlesyndication.com/pagead/js/r20160906/r20110914/abg.js//
A single-dose treatment against malaria worked in mice to cure them of the disease. The drug also worked to block infection in healthy mice and to stop transmission, according to a study published in Nature today. The fact that the drug can act against so many stages of malaria is pretty new, but what’s even more exciting is the compound’s mode of action: it kills malaria in a completely new way, researchers say. The feature would make it a welcome addition to our roster of antimalarials — a roster that’s threatened by drug resistance.
RESEARCHERS SIFTED THROUGH A LIBRARY OF ABOUT 4,700 COMPOUNDS TO FIND THIS ONE
Malaria is an infectious disease that’s transmitted through mosquito bites; it’s also a leading cause of death in a number of developing countries. Approximately 3.4 billion people live in areas where malaria poses a real threat. As a result, there were 207 million cases of malaria in 2012 — and 627,000 deaths. There are drugs that can be used to prevent malaria, and even treat it, but drug resistance is halting the use of certain treatments in some areas.
A long search
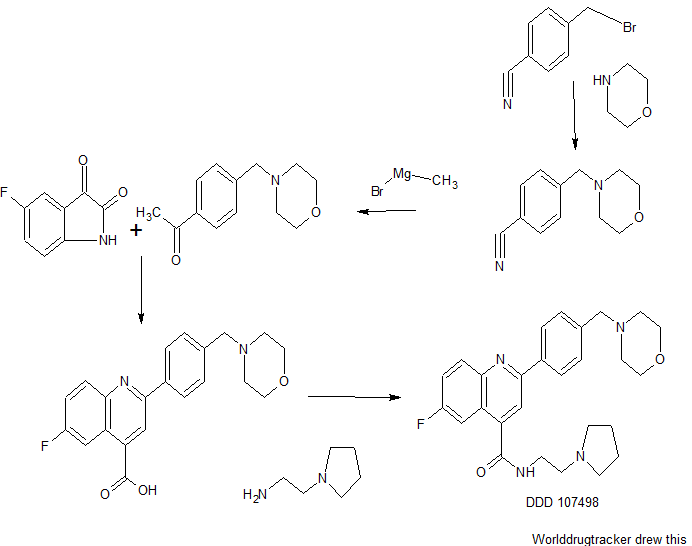
Searching for a new drug is all about trial and error. To find this particular compound, researchers sifted through a library of about 4,700 compounds, testing them to see if they were capable of killing the malaria parasite in a lab setting. When they found something that worked, they tweaked the drug candidate to see if it could perform more effectively. “We went through a lot of these cycles of testing and designing new compounds,” says Ian Gilbert, a medicinal chemist at the University of Dundee in the UK, and a co-author of the study. “Eventually we optimized to the compound which is the subject of the paper.” For now, that compound’s unwieldy name is DDD107498.
To make sure DDD107498 really had potential, the researchers tested it on mice that had already been infected with malaria. A single dose was enough to provoke a 90 percent reduction in the number of parasites in their blood. The scientists also gave the compound to healthy mice that were subsequently exposed to malaria. DDD107498 helped the mice evade infection with a single dose, but it’s unclear how long that effect would last in humans. Finally, the researchers looked at whether the compound could prevent the transmission from an infected mouse to a mosquito. A day after receiving the treatment, mice were put in contact with mosquitoes. The scientists noted a 91 percent reduction in infected mosquitoes.
“IT HAS THE ABILITY TO BE A ONE-DOSE [DRUG], IN COMBINATION WITH ANOTHER MOLECULE.”
“What’s exciting about this molecule is obviously the fact that it has the ability to be a one-dose [drug], in combination with another molecule to cure blood stage malaria,” says Kevin Read, a drug researcher also at the University of Dundee and a co-author of the study. The fact that the compound has the ability to block transmission and protect against infection is equally thrilling. But the way in which DDD107498 kills malaria might be its most interesting feature. It halts the production of proteins — which are necessary for the parasite’s survival. No other malaria drug does that right now, Read says. “So, in principle, there’s no resistance out there already to this mechanism.”
The drug hasn’t been tested in humans yet, so it may not be nearly as good in the field. But Read says DDD107498 looks promising. “From all the pre-clinical or non-clinical data we’ve generated, it is comparable or better than any of the current marketed anti-malarials in those studies.” And at $1 per treatment, the price of the drug should fall “within the range of what’s acceptable,” he says.
“It looks like an excellent study, and the results look very important,” says Philip Rosenthal, a malaria drug researcher at The University of California-San Francisco who didn’t participate in the study. This is a big shift for Rosenthal’s field. Five years ago, “we had very little going on in anti-malarial drug discovery,” he says. Now, there’s quite a bit going on for malaria researchers, and a number of promising compounds are moving along. DDD107498 “is another player, and it’s got a number of positive features,” he says.
OTHER TREATMENTS HAVE TO BE TAKEN FOR A FEW DAYS
One of the features is the drug’s potency. It’s very active against cultured malaria parasites, Rosenthal says. But what’s perhaps most intriguing about DDD107498 is that the drug works against the mechanism that enables protein synthesis the malaria parasite’s cells. No other malaria drug does that right now, Read says. “Considering challenges of treating malaria, which is often in rural areas and developing countries, a single dose would be a big plus,” he says. “In addition, because of it’s long half life, it may also work to prevent malaria with once a week dosing, which is also a benefit.”
Still, no drug is perfect. The data suggests that DDD107498 doesn’t kill malaria as quickly as some other drugs, Rosenthal says. And when the researchers tested it to see how long it might take for resistance to develop, the results weren’t as promising as he would like. The parasites figured out a way to become resistant to the compound “relatively easily,” he says. That shouldn’t be “deal-killer,” however. “Its slow onset of action probably means it should be combined with a faster-acting drug,” he says.
BUT IT’S SLOW-ACTING
The compound is going through safety testing now. If everything goes well, it should hit human trials within the next year, Read says. Chances are, it will have to be used in combination with other malaria drugs, Gilbert says. “All anti-malarials are given in combination because it slows down resistance.”
“When you’re treating infectious diseases, you know that drug resistance is always a potential problem, so having a number of choices to treat malaria is a good thing,” Rosenthal says. In this case, the drug’s new mode of action may hold lead to an entirely new weapon against malaria. “Obviously it’s got a long way to go,” Read says. But the compound is “very exciting,” nonetheless.
// https://tpc.googlesyndication.com/pagead/js/r20160906/r20110914/abg.js//
//
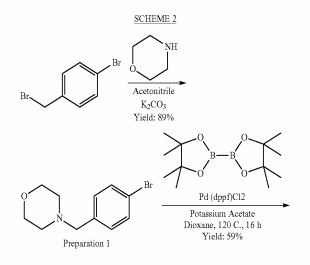
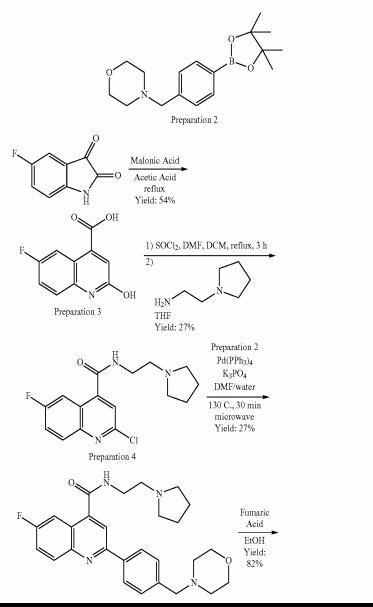

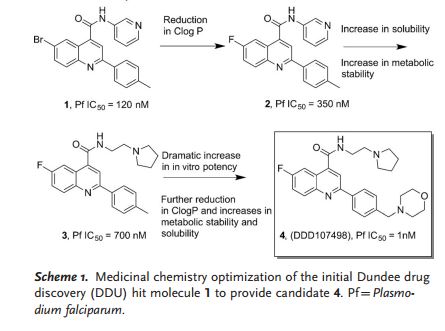
SCHEME 1

SCHEME 2

Preparation 4Yield: 54% Preparation 3
Yield: 27%

SCHEME 4 B

Yield: 72% Yield: 70% Preparation 6

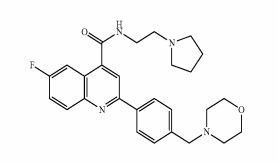
Example 1 : 6-Fluoro-2-r4-(morpholinomethyl)phenyll-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide, Example compound 1 in Scheme 2

In a sealed microwave tube, a suspension of 2-chloro-6-fluoro-N-(2-pyrrolidin-1- ylethyl)quinoline-4-carboxamide (preparation 4) (2.00 g, 6 mmol), [4- (morpholinomethyl)phenyl]boronic acid, hydrochloride, available from UORSY, (3.20 g, 12 mmol), potassium phosphate (2.63 g, 12 mmol) and tetrakis(triphenylphosphine)palladium (0) (0.21 g, 0.19 mmol) in DMF/Water 3/1 (40 ml) was heated at 130°C under microwave irradiation for 30 min. The reaction was filtered through Celite™ and solvents were removed under reduced pressure. The resulting residue was taken up in DCM (150 ml) and washed twice with NaHC03 saturated aqueous solution (2 x 100 ml). The organic layer was separated, dried over MgS04and concentrate to dryness under reduced pressure. The reaction crude was purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 1 min hold 100% A, followed by a 30 min ramp to 10 % B, and then 15 min hold at 10% B. The fractions containing product were pooled together and concentrated to dryness under vacuum to obtain the desired product as an off-white solid (1 g). The product was dissolved in methanol (100 ml) and 3-mercaptopropyl ethyl sulfide Silica (Phosphonics, SPM-32, 60- 200 uM) was added. The suspension was stirred at room temperature over for 2 days and then at 50°C for 1 h. After cooling to room temperature, the scavenger was filtered off and washed with methanol (30 ml). The solvent was removed under reduced pressure and the product was further purified by preparative HPLC. The fractions containing product were pooled together and freeze dried to obtain the desired product as a white solid (0.6 g, 1.3 mmol, Yield 20%).
1 H NMR (500 MHz; CDCI3) δ 1.81-1.84 (m, 4H), 2.50-2.52 (m, 4H), 2.63 (brs, 4H), 2.82 (t, 2H, J = 5.9 Hz), 3.61 (s, 2H), 3.71 (dd, 2H, J = 5.4 Hz, J = 1 1.4 Hz), 3.74-3.76 (m, 4H), 6.84 (brs, 1 H), 7.52-7.57 (m, 3H), 7.97-8.00 (m, 2H), 8.13 (d, 2H, J = 8.2 Hz), 8.21 (dd, 1 H, J = 5.5 Hz, J = 9.2 Hz) ppm . 19 F NMR (407.5 MHz; CDCI3) δ -11 1.47 ppm. Purity by LCMS (UV Chromatogram, 190-450nm) 99 %, rt = 5.7 min, m/z 463 (M+H)+ HRMS (ES+) found 463.2501 [M+H]+, C27H32F1 N402 requires 463.2504.
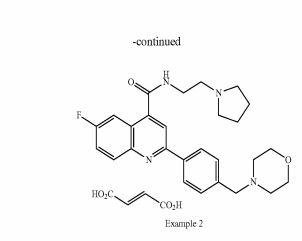
Example 2: 6-Fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2-pyrrolidin-1-ylethyl)quinoline- 4-carboxamide; fumaric acid salt, compound (IB) in Scheme 2

The starting free base (example 1) (0.58 g, 1 mmol) was dissolved in dry ethanol (10 ml) and added dropwise to a stirred solution of fumaric acid (0.15 g, 1 mmol) in dry ethanol (9 ml). The mixture was stirred at room temperature for 1 h. The white precipitate was filtered, washed with ethanol (20 ml) and then dissolved in 10 ml of water and freeze dried to obtain the desired salt as a white solid (0.601 g, 1 mmol, Yield 82%).
1 H NMR (500 MHz; d6-DMSO) δ 1.83-1.86 (m, 4H), 2.41 (brs, 4H), 2.94 (brs, 4H), 3.03 (t, 2H, J = 6.2 Hz), 3.57 (s, 2H), 3.60-3.65 (m, 6H), 6.47 (s, 2H), 7.51 (d, 2H, J = 8.25), 7.74-7.78 (m, 1 H), 8.06 (dd, 1 H, J = 2.9 Hz, J = 10.4 Hz), 8.17 (dd, 1 H, J = 5.7 Hz, J = 9.3 Hz), 8.24-8.26 (m, 3H), 9.24 (t, 1 H, J = 5.5 Hz) ppm. 19 F NMR (407.5 MHz; d6- DMSO) δ -112.30 ppm.
Purity by LCMS (UV Chromatogram, 190-450nm) 99 %, rt = 5.3 min, m/z 463 (M+H)+
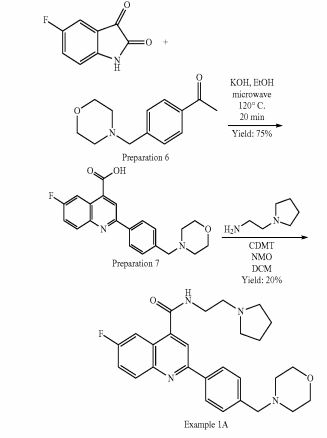
Example 1A: Alternative synthesis of 6-fluoro-2-[4-(morpholinomethyl)phenyl1-N-(2- pyrrolidin-1-ylethyl)quinoline-4-carboxamide, Example compound 1A in Scheme 4

To a stirred suspension of 6-fluoro-2-[4-(morpholinomethyl)phenyl]quinoline-4-carboxylic acid (preparation 7) (2.20 g, 6 mmol) in DCM (100 ml) at room temperature, 2-chloro- 4,6-dimethoxy-1 ,3,5-triazine (CDMT) (1.26 g, 7 mmol) and 4-methylmorpholine (NMO) (1.33 ml, 12 mmol) were added. The reaction mixture was stirred at room temperature for 1 h and then 2-pyrrolidin-1-ylethanamine (0.77 ml, 6 mmol) was added and stirred at room temperature for further 3 h. The reaction mixture was washed with NaHC03 saturated aqueous solution (2x 100 ml) and the organic phase was separated, dried over MgS04 and concentrated under reduced pressure. The resulting residue was absorbed on silica gel and purified by flash column chromatography using an 80 g silica gel cartridge and eluting with DCM (Solvent A) and MeOH (Solvent B) and the following gradient: 2 min hold 100% A followed by a 30 min ramp to 10 %B and then 15 min hold at 10%B. The desired fractions were concentrated to dryness under vacuum to obtain the crude product as a yellow solid (95% purity by LCMS). The sample was further purified by a second column chromatography using a 40 g silica gel cartridge, eluting with DCM (Solvent A) and 10% NH3-MeOH in DCM (Solvent B) and the following gradient: 2 min hold 100% A, followed by a 10 min ramp to 23 % B and then 15 min hold at 23% B. The desired fractions were concentrated to dryness under vacuum to obtain product as a white solid (1 g). Re-crystallisation form acetonitrile (18 ml) yielded the title compound as a white solid (625 mg, 1.24 mmol, 20%).
1 H NMR (500 MHz; CDCI3) δ 1.81-1.84 (m, 4H), 2.50-2.52 (m, 4H), 2.63 (brs, 4H), 2.82 (t, 2H, J = 5.9 Hz), 3.61 (s, 2H), 3.71 (dd, 2H, J = 5.4 Hz, J = 1 1.4 Hz), 3.74-3.76 (m, 4H), 6.84 (brs, 1 H), 7.52-7.57 (m, 3H), 7.97-8.00 (m, 2H), 8.13 (d, 2H, J = 8.2 Hz), 8.21 (dd, 1 H, J = 5.5 Hz, J = 9.2 Hz) ppm .
1 H NMR (500 MHz; d6-DMSO) δ 1.72-1.75 (m, 4H), 2.41 (brs, 4H), 2.56 (brs, 4H), 2.67 (t, 2H, J = 6.6 Hz), 3.49-3.52 (m, 2H), 3.56 (s, 2H), 3.60-3.61 (m, 4H), 7.52 (d, 2H, J = 8.3 Hz), 7.73-7.77 (m, 1 H), 8.07 (dd, 1 H, J = 2.9 Hz, J = 10.4 Hz), 8.18-8.21 (m, 2H), 8.26 (d, 2H , J = 8.3 Hz), 8.85 (t, 1 H, J = 6.6 Hz) ppm.
13C NMR (125 MHz; d6-DMS03) 5 23.2, 38.4, 53.2, 53.5, 54.5, 62.1 , 66.2, 109.0, 109.1 , 1 17.3, 120.1 , 120.3, 124.1 , 124.2, 127.1 , 129.4, 132.2, 132.3, 136.8, 139.9, 142.8, 145.2, 155.3, 159.0, 161 .0, 166.1 ppm.
19 F NM R (500 MHz; d6-DMSO) δ -1 12.47 ppm.
Purity by LCMS (UV Chromatogram, 190-450nm) 99 %, rt = 5.0 min, m/z 463 (M+H)+
Angewandte Chemie, International Edition (2015), 54, (46), 13504-13506

Putting a stop to malaria: Phenotypic screening against malaria parasites, hit identification, and efficient lead optimization have delivered the preclinical candidate antimalarial DDD107498. This molecule is distinctive in that it has potential for use as a single-dose cure for malaria and shows a unique broad spectrum of activity against the liver, blood, and mosquito stages of the parasite life cycle.
Professor Ian Gilbert FRSC

Medicinal Chemistry
I am a medicinal chemist and my research interests are in the design and synthesis of potential drugs. The mainstay of my work is synthetic medicinal chemistry as part of the Drug Discovery Unit (DDU). Where possible we make extensive use of molecular modeling to guide our synthetic efforts. I have a particular interests in the following aspects of drug discovery:
Neglected diseases such as human African trypanosomiasis, leishmaniasis and malaria.
Chemical validation of drug targets, including novel targets for which there is little or no precedence for drug discovery.
Novel approaches to and paradigms for drug discovery.
Mode of action studies and target identification.
I am Head of Chemistry in the DDU. Our main focuses are on neglected diseases and novel drug targets. The neglected diseases we are tackling are malaria, tuberculosis and the kinetoplastid diseases. We use both target-based approaches and phenotypic approaches (whole parasite screening). We have had particular success in validating the enzyme N-myristoyltransferase as a drug target in human African trypanosomiasis, and in identifying and optimising phenotypic hits. In our novel targets area, we aim to validate novel areas of biology as potential drug targets.
Dr Neil Norcross


Achim Porzelle
///////////DDD107498, DDD 107498, PRECLINICAL, DUNDEE, MALARIA, DDD 498, Achim Porzelle, Ian Gilbert, MERCK SERENO, Beatriz Baragaña, Medicines for Malaria Venture, University of Dundee, Neil Norcross, 1469439-69-7, 1469439-71-1 , SUCCINATE
Fc1ccc2nc(cc(c2c1)C(=O)NCCN1CCCC1)-c1ccc(cc1)CN1CCOCC1
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone for androgen sensitive prostatic disorders
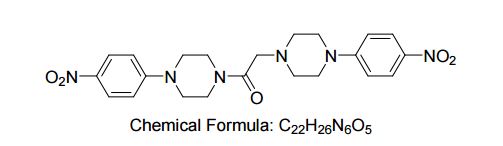
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone
| Molecular Formula: | C22H26N6O5 |
|---|---|
| Molecular Weight: | 454.47904 g/mol |
![]()
CAS 330633-91-5
CDRI-?
For treatment of androgen sensitive prostatic disorders
![1,2-bis[4-(4-nitrophenyl)piperazin-1-yl]ethanone.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=2867018&t=l "A structure of 1,2-bis[4-(4-nitrophenyl)piperazin-1-yl]ethanone")
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone
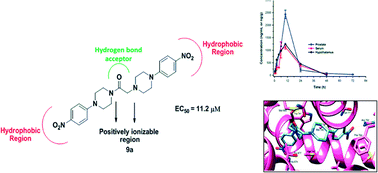
In the quest for novel scaffolds for the management of androgen sensitive prostatic disorders like prostate cancer and benign prostatic hyperplasia, a series of twenty-six aryl/heteroaryl piperazine derivatives have been described. Three compounds, 8a, 8c and 9a, exhibited good activity profiles against an androgen sensitive prostate cancer cell line (LNCaP) with EC50values of 9.8, 7.6 and 11.2 μM, respectively. These compounds caused a decrease in luciferase activity and a decline in PSA and Ca2+ levels, which are indicative of their anti-androgenic and α1A-adrenergic receptor blocking activities, respectively.
Compound 9a reduced the prostate weight of rats (47%) and in pharmacokinetic analysis at 10 mg kg−1 it demonstrated an MRT of ∼14 h post dose, exhibiting high levels in prostate. Compound 9a docked in a similar orientation to hydroxyflutamide on an androgen receptor and showed strong π–π interactions. These findings reveal that compound 9a is a promising candidate for management of prostatic disorders with anti-androgenic and α1A-blocking activities.
Design, synthesis and biological profiling of aryl piperazine based scaffolds for the management of androgen sensitive prostatic disorders
E-mail: vl_sharma@cdri.res.in, vlscdri@gmail.com
Fax: +91 522 2771941
Tel: +91 522 2772450 Ext. 4671
DOI: 10.1039/C6MD00426A, http://pubs.rsc.org/en/Content/ArticleLanding/2016/MD/C6MD00426A?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+rss%2FMD+%28RSC+-+Med.+Chem.+Commun.+latest+articles%29#!divAbstract
1, 2-Bis(4-(4-4-nitrophenyl)piperazin-1-yl)ethanone (9a) To the mixture of 8a (0.3 g, 1.06 mmol) and Et3N (0.3 mL, 2.12 mmol) in CHCl3 (5 mL) was added 1-(4-nitrophenyl)piperazine (7a, 0.320 g, 1.59 mmol) in 5 mL CHCl3 dropwise within 1 h. After complete addition reaction mixture was further stirred in an oil bath at 80-85 °C for 15 h. The reaction mixture was cooled, washed with water (5 mL × 3) and the organic layer was separated. Combined organic layer was dried (anhyd. Na2SO4 and concentrated under reduced pressure in rotavapor. The solid obtained was purified by recrystallization using EtOAc/Hexane which furnished yellow crystals (yield 81%);
mp: 156-157 °C; IR (KBr) (cm-1): 3019, 2399, 1640, 1597, 1506, 1423, 1330;
1H NMR (400 MHz, CDCl3): 8.14-8.09 (4H, m), 6.84-6.81 (4H, m), 3.84-3.83 (4H, m), 3.49-3.44 (8H, m), 3.33 (2H, s), 2.72 (4H, t, J = 5.0 Hz);
13C NMR (75.4 MHz, CDCl3): 167.7, 154.7, 154.3, 138.8, 138.4, 125.9, 125.8, 112.9, 112.7, 60.8, 52.5, 46.9, 46.7, 44.6;
HRMS (ESI positive) m/z calcd. for C22H26N6O5 [M+H]+ : 455.2043, found: 455.2034;
Anal calcd. for C22H26N6O5: C, 58.14; H, 5.77; N, 18.49, found: C, 58.31; H, 5.92; N, 18.66.
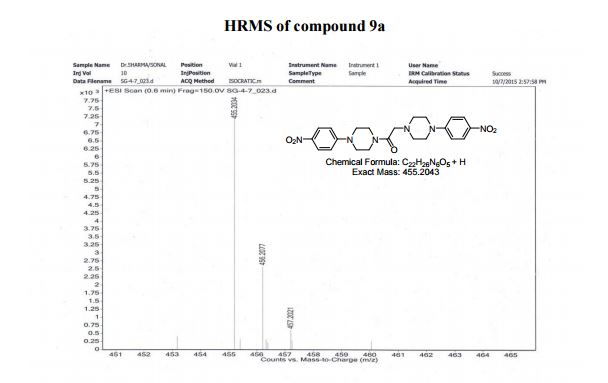



SONAL GUPTA
Medicinal & Process Chemistry Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram ext., Lucknow-226031, India


Dr. VISHNU LAL SHARMA
http://www.cdriindia.org/VL_Sharma.htm
| Dr. VISHNU LAL SHARMA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
Senior Principal Scientist (CSIR-CDRI ) / Professor (AcSIR) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POST-DOCTORAL RESEARCH (ABROAD) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CURRENT AREAS OF INTEREST | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 From left to right upper row: Dr. S.T.V.S. Kiran Kumar, Dr. Lalit Kumar, Dr. V.L. Sharma, Dr. Nand Lal, Dr. Amit Sarswat Lower row: Dhanaraju Mandalapu, Sonal Gupta, Mrs. Tara Rawat (S.T.O.), Dr. Veenu bala, Dr. Santosh Jangir |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| THESIS SUPERVISED | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FORMER Ph.D. STUDENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FORMER PROJECT ASSISTANTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PRESENT Ph.D. STUDENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FORMER POSTGRADUATE STUDENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MEMBERSHIP OF SOCIETES : | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PROJECTS: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PUBLICATIONS & PATENTS- | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SELECTED PUBLICATIONS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LIST OF PATENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
From left to right upper row: Dr. S.T.V.S. Kiran Kumar, Dr. Lalit Kumar, Dr. V.L. Sharma, Dr. Nand Lal, Dr. Amit Sarswat
Lower row: Dhanaraju Mandalapu, Sonal Gupta, Mrs. Tara Rawat (S.T.O.), Dr. Veenu bala, Dr. Santosh Jangir
///////////aryl piperazine, androgen sensitive prostatic disorders, 330633-91-5, CDRI-?
c1(ccc(cc1)[N+]([O-])=O)N2CCN(CC2)C(=O)CN3CCN(CC3)c4ccc(cc4)[N+]([O-])=O
Novel, isoform-selective inhibitor of histone deacetylase 8 (HDAC8)
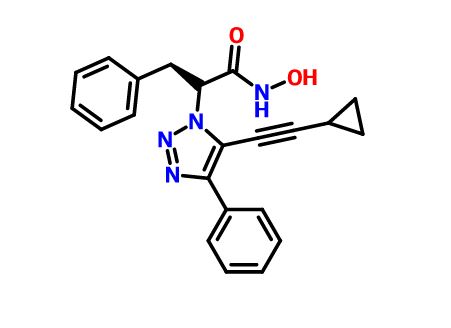

CAS 1620779-53-4
MF C22H20N4O2, MW 372.4
(S)-2-(5-(cyclopropylethynyl)-4-phenyl-1H-1,2,3-triazol-1-yl)-N-hydroxy-3-phenylpropanamide
1H-1,2,3-Triazole-1-acetamide, 5-(2-cyclopropylethynyl)-N-hydroxy-4-phenyl-α-(phenylmethyl)-, (αS)-
| Applicants: | TRUSTEES OF BOSTON UNIVERSITY DANA-FARBER CANCER INSTITUTE, INC. |
| Inventors: | Aaron Beaty BEELER John A. PORCO, JR. Oscar J. INGHAM James E. BRADNER |
|
As histone proteins bind DNA prior to transcription, their biochemical action plays a critical role in the regulation of gene expression and cellular differentiation. Histone deacetylases (HDACs) are an important family of proteins predominantly responsible for specific posttranslational modifications of histone proteins, the chief organizational component of chromatin. HDACs catalyze the removal of acetyl groups from histones and other cellular proteins. HDAC-mediated deacetylation of chromatin-bound histones regulates the expression of a variety of genes throughout the genome. Importantly, HDACs have been linked to cancer, as well as other health conditions. To date, eleven major HDAC isoforms have been described (HDACs 1-11). HDACs are categorized into two classes. Class I HDACs include HDAC1, HDAC2, HDAC3, HDAC8 and HDAC11. Class II HDACs include HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10. HDAC’s are validated targets for a number of disease states, including cancer, neurodegenerative diseases, sickle-cell anemia, muscular dystrophy, and HIV. There are currently two HDAC inhibitors on the market, Vorniostat and Romidepsin. Both are approved for treatment of T-cell lymphoma. However, they are both pan active inhibitors showing very little specificity of binding to HDAC subclasses. Because of this lack of specificity they have a number of side effects.
|
|
Non-selective HDAC inhibitors effect deacetylase activity of most, if not all, of the HDACs. The mechanisms of the anticancer effects of SAHA, a non-selective HDAC inhibitor, are not completely understood, and likely result from both altered gene expression and altered function of proteins regulating cell proliferation and cell death pathways. Non-selective HDAC inhibitors, such as SAHA, induce the accumulation of acetylated histone proteins and non histone proteins.
|
|
Small molecule HDAC inhibitors that are isoform-selective are useful as therapeutic agents with reduced toxicity and as tools for probing the biology of the HDAC isoforms. The present disclosure is related, in part to small molecules that are selective HDAC inhibitors.
|
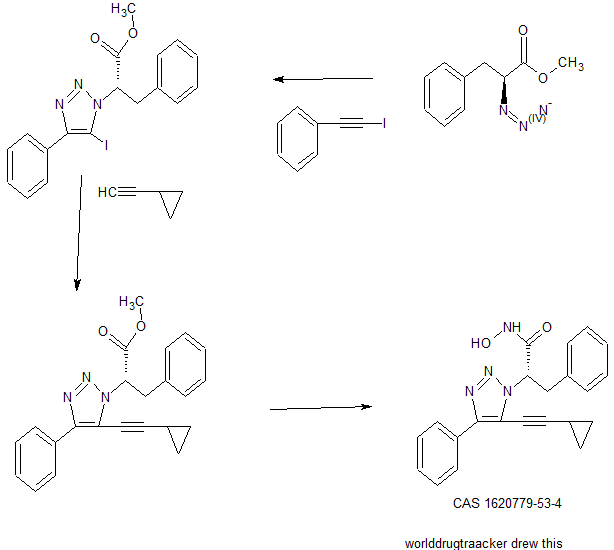

1H NMR (500 MHz, d4-MeOD) 0.80 (2H, m), 0.98 (2H, m), 1.47 (1H, m), 3.51 (1H, dd, J = 11.2, 14.2 Hz), 3.71 (1H, dd, J = 3.9, 14.2 Hz), 5.49 (1H, dd, J = 3.9, 11.2 Hz), 6.96 (2H, m), 7.17-7.20 (3H, m), 7.37 (1H, t, J = 7.3 Hz), 7.43 (2H, t, J = 7.3 Hz), 7.99 (2H, d, J = 8.8 Hz);

13C NMR (100 MHz, d4-MeOD) 0.02, 8.55, 37.07, 60.83, 62.59, 109.09, 118.98, 125.9, 127.16, 128.55, 128.65, 128.71, 129.16, 130.07, 136.09, 147.10, 165.20;
HRMS calculated for C22H21N4O2 + (M+H): 373.1659, found: 373.1665.

PATENT
WO2014116962
https://www.google.com/patents/WO2014116962A1?cl=en
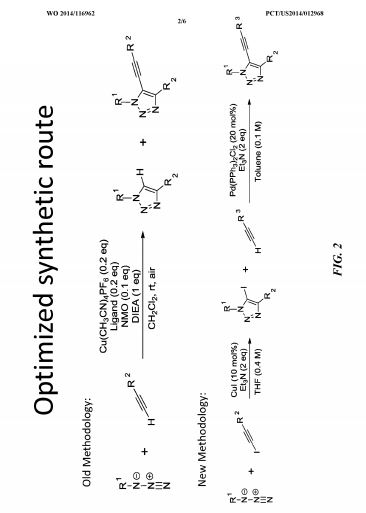
SAR. libraries were synthesized to investigate substitution about the triazole core. In some examples, compounds were synthesized using the synthetic routes shown in Fig. 2.
In one study, compound 
was synthesized as outline in Scheme I.
Scheme I




PATENT
|
SAR libraries were synthesized to investigate substitution about the triazole core. In some examples, compounds were synthesized using the synthetic routes shown in FIG. 2. In one study, compound
|
 was synthesized as outline in Scheme I.
was synthesized as outline in Scheme I.


|
The HDAC assays were carried out as described in Bowers A, West N, Taunton J, Schreiber S L, Bradner J E, Williams R M Total Synthesis and Biological Mode of Action of Largazole: A Potent Class I Histone Deacetylase Inhibitor. J. Am. Chem. Soc. 2008, 130, 11219-11222. Assay results revealed that among the analogues tested a cyclopropane analog was the most active at 0.4 nM (>1000 fold selectivity). These results demonstrated that a small aliphatic group in the 5-position on the triazole can increase potency. Also, compounds with an L-phenylalanine moiety at the 3-position showed significant potency. To expand our understanding of how the molecule interacts with the binding pocket of HDAC 8 and to understand our preliminary SAR, molecular modeling was carried out. The phenyl group from the original amino methyl ester fits snuggly into the Zn binding site and the alkynyl phenyl group sits flat in a hydrophobic groove. In summary, the inventors have developed a potent and highly selective small molecule which inhibits HDAC-8 at approximately 500 pM with over 1000-fold selectivity over HDAC-6 and significantly greater selectivity for all other HDACs. To inventors’ knowledge, to date there are no compounds with this level of potency and selectivity.
|
|
All patents and other publications identified in the specification and examples are expressly incorporated herein by reference for all purposes. These publications are provided solely for their disclosure prior to the filing date of the present application. Nothing in this regard should be construed as an admission that the inventors are not entitled to antedate such disclosure by virtue of prior invention or for any other reason. All statements as to the date or representation as to the contents of these documents is based on the information available to the applicants and does not constitute any admission as to the correctness of the dates or contents of these documents.
|
|
Although preferred embodiments have been depicted and described in detail herein, it will be apparent to those skilled in the relevant art that various modifications, additions, substitutions, and the like can be made without departing from the spirit of the invention and these are therefore considered to be within the scope of the invention as defined in the claims which follow. Further, to the extent not already indicated, it will be understood by those of ordinary skill in the art that any one of the various embodiments herein described and illustrated can be further modified to incorporate features shown in any of the other embodiments disclosed herein.
|
Paper

A novel, isoform-selective inhibitor of histone deacetylase 8 (HDAC8) has been discovered by the repurposing of a diverse compound collection. Medicinal chemistry optimization led to the identification of a highly potent (0.8 nM) and selective inhibitor of HDAC8.
Development of a Potent and Selective HDAC8 Inhibitor
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00239
file:///C:/Users/Inspiron/Downloads/ml6b00239_si_001.pdf
Department of Chemistry and Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States

 Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.
Center for Molecular Discovery (CMD) Director John Porco and members of the CMD lab team.

Aaron Beeler
Aaron Beeler received his Ph.D. in 2002 from Professor John Rimoldi’s laboratory in the Department of Medicinal Chemistry at the University of Mississippi. He then joined the Porco group as a postodoctoral fellow and subsequently the Center for Chemical Methodology and Library Development at Boston University, now the Center for Molecular Discovery. He was promoted to Assistant Director of the CMLD-BU in January 2005. In 2012 Aaron joined the Department of Chemistry as a tenure-track professor in medicinal chemistry.
Degrees and Positions
- B.S. Belmont University, Biology,
- Ph.D. University of Mississippi, Medicinal Chemistry
Research
The Beeler Research Group is truly multidisciplinary, combining organic chemistry, engineering, and biology to solve problems in medicinal chemistry. All of these elements are combined and directed toward significant problems in human health. The Beeler Group is addressing focused disease areas (e.g., schizophrenia, Parkinson’s, cystic fibrosis), as well as project areas with broader impact potential (e.g., new methods for discovery of small molecules with anti-cancer properties).
- Medicinal Chemistry: The goals of medicinal chemistry projects are to optimize small molecules in order to: a) develop a probe that may be utilized as a tool in biological studies; b) develop a lead molecule to facilitate future therapeutics; and c) utilize small molecules to enhance understanding of biological targets that are important for human health. These projects provide students with training in organic chemistry, medicinal chemistry, and focused biology. Projects are selected based on their chemistry and/or biology significance and potential for addressing challenging questions.
- Technology: One of the core components of the research in the Beeler Group is development of technologies and paradigms that facilitate rapid modification of complex scaffolds. These technologies enable optimization of biologically active lead compounds and identification of small molecule leads in biological systems. The projects focus on utilizing automation, miniaturization, and microfluidics to carry out chemical transformations. These projects are highly interdisciplinary with both chemistry and engineering components.
- Photochemistry: This area focuses on photochemical transformations toward the synthesis of natural products, natural product scaffolds, and other complex chemotypes of interest to medicinal chemistry and chemical biology. The foundation of these projects is utilizing microfluidics to enable photochemical reaction development.
Techniques & Resources
Students in the Beeler Research Group will have opportunities to learn a number of exciting research disciplines. Organic synthesis will be at the heart of every project. This will include targeted synthesis, methodology development, and medicinal chemistry. Through collaborations with biological researchers and/or research projects carried out within the Beeler Group, students will learn methods for biological assays, pharmacology, and target identification. Many projects will also include aspects of engineering that will provide opportunities for learning techniques such as microfabrication and microfluidics.
Opportunities
It is becoming evident that successful and impactful science is realized in collaborative interdisciplinary environments. The Beeler Research Group’s multidisciplinary nature and collaborative projects provides opportunities to learn areas of research outside of traditional chemistry.
What’s Next for Graduates of the Beeler Group?
Members of the Beeler Research Group will be positioned for a wide range of future endeavors.
- Undergraduates will be prepared to enter into graduate school for organic chemistry, chemical biology, or chemical engineering or to start careers in industry;
- Graduate students will have the foundation required for postdoctoral studies in organic synthesis or chemical biology as well as an industrial career in biotech or pharma;
- Postdoctoral associates will gain training and experience critical for both academic and industrial careers.
Assistant Professor
Office: SCI 484C
Laboratory: SCI 484A
Phone: 617.358.3487
Fax: 617-358-2847
beelera@bu.edu
Office Hours: by Appointment
Beeler Group Homepage
Google Scholar Page
Oscar J. Ingham below

John A. PORCO, JR below


JAMES E. BRADNER, MD above
Dana-Farber Cancer Institute


 Ron Paranal
Ron Paranal
 Randolph A. Escobar
Randolph A. Escobar
 Han Yueh
Han Yueh
| US20090181943 * | Apr 9, 2008 | Jul 16, 2009 | Methylgene Inc. | Inhibitors of Histone Deacetylase |
| Reference | ||
|---|---|---|
| 1 | * | GERARD, B ET AL.: ‘Synthesis of 1,4,5-trisubstituted-1,2,3-triazoles by copper-catalyzed cycloaddition-coupling of azides and terminal alkynes‘ TETRAHEDRON vol. 62, 12 May 2006, pages 6405 – 6411 |
| 2 | * | VANNINI, A ET AL.: ‘Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor.‘ PNAS, [Online] vol. 101, no. 42, 19 October 2004, pages 15064 – 15069 Retrieved from the Internet: <URL:http://www.pnas.org/content/101/42/15064> |
///////////epigenetic, HDAC, HDAC8, Histone deacetylase, histone deacetylase 8, triazole, PRECLINICAL, Department of Chemistry and Center for Molecular Discovery (BU-CMD), Boston University, 590 Commonwealth Avenue, Boston, Massachusetts 02215, United States, Oscar J. Ingham, Aaron Beeler
n1n(c(c(n1)c2ccccc2)C#CC3CC3)C(C(=O)NO)Cc4ccccc4
PF-05387252
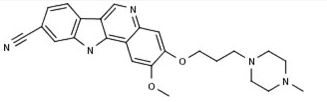
PF-05387252
CAS 1604034-71-0
| C25H27N5O2 | |
| MW | 429.51418 g/mol |
|---|
2-methoxy-3-[3-(4-methylpiperazin-1-yl)propoxy]-11H-indolo[3,2-c]quinoline-9-carbonitrile
IRAK4 inhibitor
Rheumatoid arthritis;
SLE
Preclinical
In the past decade there has been considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFα, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as HMGB1 and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system.
This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
Interleukin-1 receptor associated kinase (IRAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity. IRAK4 is responsible for initiating signaling from TLRs and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice lead to reductions in TLR and IL-1 induced pro-inflammatory cytokines. and 7 IRAK-4 kinase-dead knock-in mice have been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models. Likewise, humans deficient in IRAK4 also display the inability to respond to challenge by TLR ligands and IL-1
However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age implying redundant or compensatory mechanisms for innate immunity in the absence of IRAK4.These data suggest that inhibitors of IRAK4 kinase activity will have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be a viable strategy for the treatment of other inflammatory pathologies such as atherosclerosis.
Indeed, the therapeutic potential of IRAK4 inhibitors has been recognized by others within the drug-discovery community as evidenced by the variety of IRAK4 inhibitors have been reported to-date.12, 13, 14, 15 and 16 However, limited data has been published about these compounds and they appear to suffer from a variety of issues such as poor kinase selectivity and poor whole-blood potency that preclude their advancement into the pre-clinical models. To the best of our knowledge, no in vivo studies of IRAK4 inhibitors have been reported to-date in the literature. Herein we report a new class of IRAK4 inhibitors that are shown to recapitulate the phenotype observed in IRAK4 knockout and kinase-dead mice.
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072.
doi:10.1016/j.bmcl.2014.03.056
http://www.sciencedirect.com/science/article/pii/S0960894X14002832
Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4
- a Pfizer Global R&D, 445 Eastern Point Rd., Groton, CT 06340, USA
- b Pfizer Global R&D, 200 Cambridge Park Dr., Cambridge, MA 02140, USA
- c Pfizer Global R&D, 87 Cambridgepark Dr., Cambridge, MA 02140, USA
- d Pfizer Global R&D, 1 Burtt Rd., Andover, MA 01810, USA
![]()

Abstract
IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis. Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway. A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model. To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.



SYNTHESIS

////////PF-05387252, 1604034-71-0, PF 05387252, TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PRECLINICAL
N1(CCN(CC1)CCCOc3c(cc2c4nc5cc(ccc5c4cnc2c3)C#N)OC)C
OR
CN1CCN(CC1)CCCOC2=C(C=C3C(=C2)N=CC4=C3NC5=C4C=CC(=C5)C#N)OC
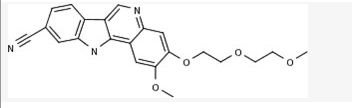
PF-05388169
PF-05388169
CAS 1604034-78-7, MF C22 H21 N3 O4
MW 391.42
- IRAK4 inhibitor
Rheumatoid arthritis;
SLE
Preclinical
![]()
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072.
http://www.sciencedirect.com/science/article/pii/S0960894X14002832
Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4
- a Pfizer Global R&D, 445 Eastern Point Rd., Groton, CT 06340, USA
- b Pfizer Global R&D, 200 Cambridge Park Dr., Cambridge, MA 02140, USA
- c Pfizer Global R&D, 87 Cambridgepark Dr., Cambridge, MA 02140, USA
- d Pfizer Global R&D, 1 Burtt Rd., Andover, MA 01810, USA
IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis. Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway. A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model. To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.
SYNTHESIS
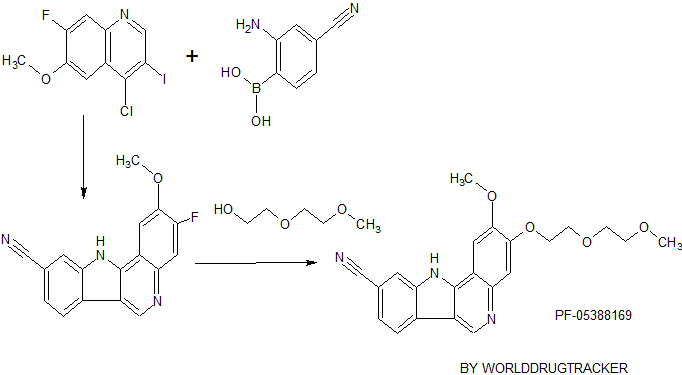
//////////PF-05388169, TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PRECLINICAL, 1604034-78-7
C(COC)OCCOc4c(cc3\C2=N\c1cc(ccc1/C2=C/Nc3c4)C#N)OC
JNJ-54257099
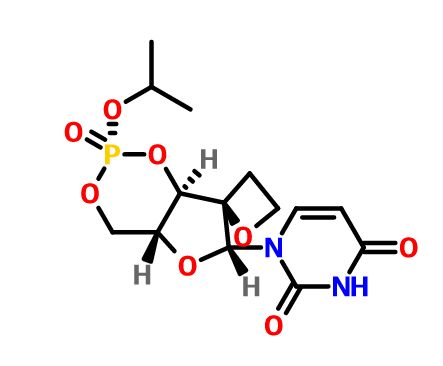

JNJ-54257099,
1-((2R,4aR,6R,7R,7aR)-2-Isopropoxy-2-oxidodihydro-4H,6H-spiro[furo[3,2-d][1,3,2]dioxaphosphinine-7,2′-oxetan]-6-yl)pyrimidine-2,4(1H,3H)-dione
MW 374.28, C14 H19 N2 O8 P
CAS 1491140-67-0
2,4(1H,3H)-Pyrimidinedione, 1-[(2R,2′R,4aR,6R,7aR)-dihydro-2-(1-methylethoxy)-2-oxidospiro[4H-furo[3,2-d]-1,3,2-dioxaphosphorin-7(6H),2′-oxetan]-6-yl]-
1-((2R,4aR,6R,7R,7aR)-2-Isopropoxy-2-oxidodihydro-4H,6H-spiro[furo[3,2-d][1,3,2]dioxaphos-phinine-7,2′-oxetan]-6-yl)pyrimidine-2,4(1H,3H)-dione

Janssen R&D Ireland INNOVATOR
Ioannis Nicolaos Houpis, Tim Hugo Maria Jonckers, Pierre Jean-Marie Bernard Raboisson, Abdellah Tahri,

Tim Jonckers was born in Antwerp in 1974. He studied Chemistry at the University of Antwerp and obtained his Ph.D. in organic chemistry in 2002. His Ph.D. work covered the synthesis of new necryptolepine derivatives which have potential antimalarial activity. Currently he works as a Senior Scientist at Tibotec, a pharmaceutical research and development company based in Mechelen, Belgium, that focuses on viral diseases mainly AIDS and hepatitis. The company was acquired by Johnson & Johnson in April 2002 and recently gained FDA approval for its HIV-protease inhibitor PREZISTA™.

Principal Scientist at Janssen, Pharmaceutical Companies of Johnson and Johnson

DATA
Chiral SFC using the methods described(Method 1, Rt= 5.12 min, >99%; Method 2, Rt = 7.95 min, >99%).
1H NMR (400 MHz, chloroform-d) δ ppm 1.45 (dd, J = 7.53, 6.27 Hz, 6 H), 2.65–2.84 (m, 2 H), 3.98 (td, J = 10.29, 4.77 Hz, 1 H), 4.27 (t,J = 9.66 Hz, 1 H), 4.43 (ddd, J = 8.91, 5.77, 5.65 Hz, 1 H), 4.49–4.61 (m, 1 H), 4.65 (td, J = 7.78, 5.77 Hz, 1 H), 4.73 (d, J = 7.78 Hz, 1 H), 4.87 (dq, J = 12.74, 6.30 Hz, 1 H), 5.55 (br. s., 1 H), 5.82 (d, J = 8.03 Hz, 1 H), 7.20 (d, J = 8.03 Hz, 1 H), 8.78 (br. s., 1 H);
31P NMR (chloroform-d) δ ppm −7.13. LC-MS: 375 (M + H)+.
HCV is a single stranded, positive-sense R A virus belonging to the Flaviviridae family of viruses in the hepacivirus genus. The NS5B region of the RNA polygene encodes a RNA dependent RNA polymerase (RdRp), which is essential to viral replication. Following the initial acute infection, a majority of infected individuals develop chronic hepatitis because HCV replicates preferentially in hepatocytes but is not directly cytopathic. In particular, the lack of a vigorous T-lymphocyte response and the high propensity of the virus to mutate appear to promote a high rate of chronic infection. Chronic hepatitis can progress to liver fibrosis, leading to cirrhosis, end-stage liver disease, and HCC (hepatocellular carcinoma), making it the leading cause of liver transplantations. There are six major HCV genotypes and more than 50 subtypes, which are differently distributed geographically. HCV genotype 1 is the predominant genotype in Europe and in the US. The extensive genetic heterogeneity of HCV has important diagnostic and clinical implications, perhaps explaining difficulties in vaccine development and the lack of response to current therapy.
Transmission of HCV can occur through contact with contaminated blood or blood products, for example following blood transfusion or intravenous drug use. The introduction of diagnostic tests used in blood screening has led to a downward trend in post-transfusion HCV incidence. However, given the slow progression to the end-stage liver disease, the existing infections will continue to present a serious medical and economic burden for decades.
Therapy possibilities have extended towards the combination of a HCV protease inhibitor (e.g. Telaprevir or boceprevir) and (pegylated) interferon-alpha (IFN-a) / ribavirin. This combination therapy has significant side effects and is poorly tolerated in many patients. Major side effects include influenza-like symptoms, hematologic
abnormalities, and neuropsychiatric symptoms. Hence there is a need for more effective, convenient and better-tolerated treatments.
The NS5B RdRp is essential for replication of the single-stranded, positive sense, HCV RNA genome. This enzyme has elicited significant interest among medicinal chemists. Both nucleoside and non-nucleoside inhibitors of NS5B are known. Nucleoside inhibitors can act as a chain terminator or as a competitive inhibitor, or as both. In order to be active, nucleoside inhibitors have to be taken up by the cell and converted in vivo to a triphosphate. This conversion to the triphosphate is commonly mediated by cellular kinases, which imparts additional structural requirements on a potential nucleoside polymerase inhibitor. In addition this limits the direct evaluation of nucleosides as inhibitors of HCV replication to cell-based assays capable of in situ phosphorylation.
Several attempts have been made to develop nucleosides as inhibitors of HCV RdRp, but while a handful of compounds have progressed into clinical development, none have proceeded to registration. Amongst the problems which HCV-targeted
nucleosides have encountered to date are toxicity, mutagenicity, lack of selectivity, poor efficacy, poor bioavailability, sub-optimal dosage regimes and ensuing high pill burden and cost of goods.
Spirooxetane nucleosides, in particular l-(8-hydroxy-7-(hydroxy- methyl)- 1,6-dioxaspiro[3.4]octan-5-yl)pyrimidine-2,4-dione derivatives and their use as HCV inhibitors are known from WO2010/130726, and WO2012/062869, including
CAS-1375074-52-4.
There is a need for HCV inhibitors that may overcome at least one of the disadvantages of current HCV therapy such as side effects, limited efficacy, the emerging of resistance, and compliance failures, or improve the sustained viral response.
The present invention concerns HCV-inhibiting uracyl spirooxetane derivatives with useful properties regarding one or more of the following parameters: antiviral efficacy towards at least one of the following genotypes la, lb, 2a, 2b, 3,4 and 6, favorable
profile of resistance development, lack of toxicity and genotoxicity, favorable pharmacokinetics and pharmacodynamics and ease of formulation and administration.
Such an HCV-inhibiting uracyl spirooxetane derivative is a compound with formula I
including any pharmaceutically acceptable salt or solvate thereof.
PATENT
WO 2015077966
https://www.google.com/patents/WO2015077966A1?cl=en
Synthesis of compound (I)

(5) (6a)

Synthesis of compound (6a)
A solution of isopropyl alcohol (3.86 mL,0.05mol) and triethylamine (6.983 mL, 0.05mol) in dichloromethane (50 mL) was added to a stirred solution of POCI3 (5)
(5.0 mL, 0.055 lmol) in DCM (50 mL) dropwise over a period of 25 min at -5°C. After the mixture stirred for lh, the solvent was evaporated, and the residue was suspended in ether (100 mL). The triethylamine hydrochloride salt was filtered and washed with ether (20 mL). The filtrate was concentrated, and the residue was distilled to give the (6) as a colorless liquid (6.1g, 69 %yield).
Synthesis of compound (4):
CAS 1255860-33-3 is dissolved in pyridine and 1,3-dichloro-l, 1,3,3-tetraisopropyldisiloxane is added. The reaction is stirred at room temperature until complete. The solvent is removed and the product redissolved in CH2CI2 and washed with saturated NaHC03 solution. Drying on MgSC^ and removal of the solvent gives compound (2). Compound (3) is prepared by reacting compound (2) with p-methoxybenzylchloride in the presence of DBU as the base in CH3CN. Compound (4) is prepared by cleavage of the bis-silyl protecting group in compound (3) using TBAF as the fluoride source.
Synthesis of compound (7a)
To a stirred suspension of (4) (2.0 g, 5.13 mmol) in dichloromethane (50 mL) was added triethylamine (2.07 g, 20.46 mmol) at room temperature. The reaction mixture was cooled to -20°C, and then (6a) (1.2 g, 6.78mmol) was added dropwise over a period of lOmin. The mixture was stirred at this temperature for 15min and then NMI was added (0.84 g, 10.23 mmol), dropwise over a period of 15 min. The mixture was stirred at -15°C for lh and then slowly warmed to room temperature in 20 h. The solvent was evaporated, the mixture was concentrated and purified by column chromatography using petroleum ether/EtOAc (10: 1 to 5: 1 as a gradient) to give (7a) as white solid (0.8 g, 32 % yield).
Synthesis of compound (I)
To a solution of (7a) in CH3CN (30 mL) and H20 (7 mL) was add CAN portion wise below 20° C. The mixture was stirred at 15-20° C for 5h under N2. Na2S03 (370 mL) was added dropwise into the reaction mixture below 15°C, and then Na2C03 (370 mL) was added. The mixture was filtered and the filtrate was extracted with CH2C12
(100 mL*3). The organic layer was dried and concentrated to give the residue. The residue was purified by column chromatography to give the target compound (8a) as white solid. (Yield: 55%)
1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.45 (dd, J=7.53, 6.27 Hz, 6 H), 2.65 -2.84 (m, 2 H), 3.98 (td, J=10.29, 4.77 Hz, 1 H), 4.27 (t, J=9.66 Hz, 1 H), 4.43 (ddd, J=8.91, 5.77, 5.65 Hz, 1 H), 4.49 – 4.61 (m, 1 H), 4.65 (td, J=7.78, 5.77 Hz, 1 H), 4.73 (d, J=7.78 Hz, 1 H), 4.87 (dq, J=12.74, 6.30 Hz, 1 H), 5.55 (br. s., 1 H), 5.82 (d, J=8.03 Hz, 1 H), 7.20 (d, J=8.03 Hz, 1 H), 8.78 (br. s., 1 H); 31P NMR (CHLOROFORM-^) δ ppm -7.13; LC-MS: 375 (M+l)+
PATENT
https://www.google.co.in/patents/WO2013174962A1?cl=en
The starting material l-[(4R,5R,7R,8R)-8-hydroxy-7-(hydroxymethyl)-l,6-dioxa- spiro[3.4]octan-5-yl]pyrimidine-2,4(lH,3H)-dione (1) can be prepared as exemplified in WO2010/130726. Compound (1) is converted into compounds of the present invention via a p-methoxybenzyl protected derivative (4) as exemplified in the following Scheme 1. cheme 1

Examples
Scheme 2
Synthesis of compound (8a)

Synthesis of compound (2)
Compound (2) can be prepared by dissolving compound (1) in pyridine and adding l,3-dichloro-l,l,3,3-tetraisopropyldisiloxane. The reaction is stirred at room temperature until complete. The solvent is removed and the product redissolved in CH2CI2and washed with saturated NaHC03 solution. Drying on MgSC^ and removal of the solvent gives compound (2).
Synthesis of compound (3)
Compound (3) is prepared by reacting compound (2) with p-methoxybenzylchloride in the presence of DBU as the base in CH3CN.
Synthesis of compound (4)
Compound (4) is prepared by cleavage of the bis-silyl protecting group in compound (3) using TBAF as the fluoride source.
Synthesis of compound (6a)
A solution of isopropyl alcohol (3.86 mL,0.05mol) and triethylamine (6.983 mL, 0.05mol) in dichloromethane (50 mL) was added to a stirred solution of POCl3 (5) (5.0 mL, 0.055 lmol) in DCM (50 mL) dropwise over a period of 25 min at -5°C. After the mixture stirred for lh, the solvent was evaporated, and the residue was suspended in ether (100 mL). The triethylamine hydrochloride salt was filtered and washed with ether (20 mL). The filtrate was concentrated, and the residue was distilled to give the (6) as a colorless liquid (6.1g, 69 %yield).
Synthesis of compound (7a)
To a stirred suspension of (4) (2.0 g, 5.13 mmol) in dichloromethane (50 mL) was added triethylamine (2.07 g, 20.46 mmol) at room temperature. The reaction mixture was cooled to -20°C, and then (6a) (1.2 g, 6.78mmol) was added dropwise over a period of lOmin. The mixture was stirred at this temperature for 15min and then NMI was added (0.84 g, 10.23 mmol), dropwise over a period of 15 min. The mixture was stirred at -15°C for lh and then slowly warmed to room temperature in 20 h. The solvent was evaporated, the mixture was concentrated and purified by column chromatography using petroleum ether/EtOAc (10:1 to 5: 1 as a gradient) to give (7a) as white solid (0.8 g, 32 % yield).
Synthesis of compound (8a)
To a solution of (7a) in CH3CN (30 mL) and H20 (7 mL) was add CAN portion wise below 20°C. The mixture was stirred at 15-20°C for 5h under N2. Na2S03 (370 mL) was added dropwise into the reaction mixture below 15°C, and then Na2C03 (370 mL) was added. The mixture was filtered and the filtrate was extracted with CH2C12
(100 mL*3). The organic layer was dried and concentrated to give the residue. The residue was purified by column chromatography to give the target compound (8a) as white solid. (Yield: 55%)
1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.45 (dd, J=7.53, 6.27 Hz, 6 H), 2.65 – 2.84 (m, 2 H), 3.98 (td, J=10.29, 4.77 Hz, 1 H), 4.27 (t, J=9.66 Hz, 1 H), 4.43 (ddd, J=8.91, 5.77, 5.65 Hz, 1 H), 4.49 – 4.61 (m, 1 H), 4.65 (td, J=7.78, 5.77 Hz, 1 H), 4.73 (d, J=7.78 Hz, 1 H), 4.87 (dq, J=12.74, 6.30 Hz, 1 H), 5.55 (br. s., 1 H), 5.82 (d, J=8.03 Hz, 1 H), 7.20 (d, J=8.03 Hz, 1 H), 8.78 (br. s., 1 H); 31P NMR (CHLOROFORM-^) δ ppm -7.13; LC-MS: 375 (M+l)+ Scheme 3
Synthesis of compound (VI)

Step 1: Synthesis of compound (9)Compound (1), CAS 1255860-33-3 ( 1200 mg, 4.33 mmol ) and l,8-bis(dimethyl- amino)naphthalene (3707 mg, 17.3 mmol) were dissolved in 24.3 mL of
trimethylphosphate. The solution was cooled to 0°C. Compound (5) (1.21 mL, 12.98 mmol) was added, and the mixture was stirred well maintaining the temperature at 0°C for 5 hours. The reaction was quenched by addition of 120 mL of tetraethyl- ammonium bromide solution (1M) and extracted with CH2CI2 (2×80 mL). Purification was done by preparative HPLC (Stationary phase: RP XBridge Prep CI 8 ΟΒϋ-10μιη, 30x150mm, mobile phase: 0.25% NH4HCO3 solution in water, CH3CN) , yielding two fractions. The purest fraction was dissolved in water (15 mL) and passed through a manually packed Dowex (H+) column by elution with water. The end of the elution was determined by checking UV absorbance of eluting fractions. Combined fractions were frozen at -78°C and lyophilized. Compound (9) was obtained as a white fluffy solid (303 mg, (0.86 mmol, 20%> yield), which was used immediately in the following reaction. Step 2: Preparation of compound (VI)
Compound (9) (303 mg, 0.86 mmol) was dissolved in 8 mL water and to this solution was added N . N’- D ic y c ! he y !-4- mo rph line carboxamidine (253.8 mg, 0.86 mmol) dissolved in pyridine (8.4 mi.). The mixture was kept for 5 minutes and then
evaporated to dryness, dried overnight in vacuo overnight at 37°C. The residu was dissolved in pyridine (80 mL). This solution was added dropwise to vigorously stirred DCC (892.6 mg, 4.326 mmol) in pyridine (80 mL) at reflux temperature. The solution was kept refluxing for 1.5h during which some turbidity was observed in the solution. The reaction mixture was cooled and evaporated to dryness. Diethylether (50 mL) and water (50 mL) were added to the solid residu. N’N-dicyclohexylurea was filtered off, and the aqueous fraction was purified by preparative HPLC (Stationary phase: RP XBridge Prep C18 OBD-ΙΟμιη, 30x150mm, mobile phase: 0.25% NH4HCO3 solution in water, CH3CN) , yielding a white solid which was dried overnight in vacuo at 38°C. (185 mg, 0.56 mmol, 65% yield). LC-MS: (M+H)+: 333.
1H NMR (400 MHz, DMSO-d6) d ppm 2.44 – 2.59 (m, 2 H) signal falls under DMSO signal, 3.51 (td, J=9.90, 5.50 Hz, 1 H), 3.95 – 4.11 (m, 2 H), 4.16 (d, J=10.34 Hz, 1 H), 4.25 – 4.40 (m, 2 H), 5.65 (d, J=8.14 Hz, 1 H), 5.93 (br. s., 1 H), 7.46 (d, J=7.92 Hz, 1 H), 2H’s not observed
Paper
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00382,
Discovery of 1-((2R,4aR,6R,7R,7aR)-2-Isopropoxy-2-oxidodihydro-4H,6H-spiro[furo[3,2-d][1,3,2]dioxaphosphinine-7,2′-oxetan]-6-yl)pyrimidine-2,4(1H,3H)-dione (JNJ-54257099), a 3′-5′-Cyclic Phosphate Ester Prodrug of 2′-Deoxy-2′-Spirooxetane Uridine Triphosphate Useful for HCV Inhibition
JNJ-54257099 (9) is a novel cyclic phosphate ester derivative that belongs to the class of 2′-deoxy-2′-spirooxetane uridine nucleotide prodrugs which are known as inhibitors of the HCV NS5B RNA-dependent RNA polymerase (RdRp). In the Huh-7 HCV genotype (GT) 1b replicon-containing cell line 9 is devoid of any anti-HCV activity, an observation attributable to inefficient prodrug metabolism which was found to be CYP3A4-dependent. In contrast, in vitro incubation of 9 in primary human hepatocytes as well as pharmacokinetic evaluation thereof in different preclinical species reveals the formation of substantial levels of 2′-deoxy-2′-spirooxetane uridine triphosphate (8), a potent inhibitor of the HCV NS5B polymerase. Overall, it was found that 9 displays a superior profile compared to its phosphoramidate prodrug analogues (e.g., 4) described previously. Of particular interest is the in vivo dose dependent reduction of HCV RNA observed in HCV infected (GT1a and GT3a) human hepatocyte chimeric mice after 7 days of oral administration of 9
////////////JNJ-54257099, 1491140-67-0, JNJ54257099, JNJ 54257099
O=C(C=C1)NC(N1[C@H]2[C@]3(OCC3)[C@H](O4)[C@@H](CO[P@@]4(OC(C)C)=O)O2)=O
GSK 6853
GSK 6853
CAS 1910124-24-1
A white solid.
LCMS (high pH): Rt = 0.90 min, [M+H+]+ 410.5.
δΗ NMR (600 MHz, DMSO-d6) ppm 10.74 (s, 1 H), 8.39 (s, 1 H), 8.05 (dd, J = 7.7, 1.8 Hz, 1 H), 7.57 (ddd, J = 8.3, 7.2, 2.0 Hz, 1 H), 7.29 (d, J = 8.1 Hz, 1 H), 7.23 (s, 1 H), 7.17-7.1 1 (m, 1 H), 4.10 (s, 3H), 3.33 (s, 3H), 3.32 (s, 3H), 3.30 (br s, 1 H), 3.07-3.02 (m, 1 H), 3.02-2.99 (m, 1 H), 2.92-2.87 (m, 1 H), 2.80 (td, J = 1 1.3, 2.7 Hz, 1 H), 2.73 (td, J = 1 1 .0, 2.7 Hz, 1 H), 2.68-2.63 (m, 1 H), 2.55 (dd, J = 12.0, 9.8 Hz, 1 H), 0.71 (d, J = 6.1 Hz, 3H).
δ0 NMR (151 MHz, DMSO-d6) ppm 162.1 , 156.8, 154.1 , 134.4, 133.2, 131.5, 130.1 , 126.6, 125.7, 121.9, 121.0, 1 12.5, 103.0, 99.4, 56.8, 55.4, 55.3, 53.3, 46.3, 26.8, 26.6, 16.7.
[aD]25 °c = -50.1 (c = 0.3, MeOH).
Scheme 1



The genomes of eukaryotic organisms are highly organised within the nucleus of the cell. The long strands of duplex DNA are wrapped around an octomer of histone proteins (most usually comprising two copies of histones H2A, H2B, H3 and H4) to form a
nucleosome. This basic unit is then further compressed by the aggregation and folding of nucleosomes to form a highly condensed chromatin structure. A range of different states of condensation are possible, and the tightness of this structure varies during the cell cycle, being most compact during the process of cell division. Chromatin structure plays a critical role in regulating gene transcription, which cannot occur efficiently from highly condensed chromatin. The chromatin structure is controlled by a series of post-translational
modifications to histone proteins, notably histones H3 and H4, and most commonly within the histone tails which extend beyond the core nucleosome structure. These modifications include acetylation, methylation, phosphorylation, ubiquitinylation, SUMOylation and numerous others. These epigenetic marks are written and erased by specific enzymes, which place the tags on specific residues within the histone tail, thereby forming an epigenetic code, which is then interpreted by the cell to allow gene specific regulation of chromatin structure and thereby transcription.
Histone acetylation is usually associated with the activation of gene transcription, as the modification loosens the interaction of the DNA and the histone octomer by changing the electrostatics. In addition to this physical change, specific proteins bind to acetylated lysine residues within histones to read the epigenetic code. Bromodomains are small (=1 10 amino acid) distinct domains within proteins that bind to acetylated lysine residues commonly but not exclusively in the context of histones. There is a family of around 50 proteins known to contain bromodomains, and they have a range of functions within the cell.
BRPF1 (also known as peregrin or Protein Br140) is a bromodomain-containing protein that has been shown to bind to acetylated lysine residues in histone tails, including H2AK5ac, H4K12ac and H3K14ac (Poplawski et al, J. Mol. Biol., 2014 426: 1661-1676). BRPF1 also contains several other domains typically found in chromatin-associated factors, including a double plant homeodomain (PHD) and zinc finger (ZnF) assembly (PZP), and a chromo/Tudor-related Pro-Trp-Trp-Pro (PWWP) domain. BRPF1 forms a tetrameric complex with monocytic leukemia zinc-finger protein (MOZ, also known as KAT6A or MYST3) inhibitor of growth 5 (ING5) and homolog of Esa1 -associated factor (hEAF6). In humans, the t(8;16)(p1 1 ;p13) translocation of MOZ (monocytic leukemia zinc-finger protein, also known as KAT6A or MYST3) is associated with a subtype of acute myeloid leukemia and
contributes to the progression of this disease (Borrow et al, Nat. Genet., 1996 14: 33-41 ). The BRPF1 bromodomain contributes to recruiting the MOZ complex to distinct sites of active chromatin and hence is considered to play a role in the function of MOZ in regulating transcription, hematopoiesis, leukemogenesis, and other developmental processes (Ullah et al, Mol. Cell. Biol., 2008 28: 6828-6843; Perez-Campo et al, Blood, 2009 1 13: 4866-4874). Demont et al, ACS Med. Chem. Lett., (2014) (dx.doi.org/10.1021/ml5002932), discloses certain 1 ,3-dimethyl benzimidazolones as potent, selective inhibitors of the BRPF1 bromodomain.
BRPF1 bromodomain inhibitors, and thus are believed to have potential utility in the treatment of diseases or conditions for which a bromodomain inhibitor is indicated. Bromodomain inhibitors are believed to be useful in the treatment of a variety of diseases or conditions related to systemic or tissue inflammation, inflammatory responses to infection or hypoxia, cellular activation and proliferation, lipid metabolism, fibrosis and in the prevention and treatment of viral infections. Bromodomain inhibitors may be useful in the treatment of a wide variety of chronic autoimmune and inflammatory conditions such as rheumatoid arthritis, osteoarthritis, psoriasis, systemic lupus erythematosus, multiple sclerosis, inflammatory bowel disease (Crohn’s disease and ulcerative colitis), asthma, chronic obstructive airways disease, pneumonitis, myocarditis, pericarditis, myositis, eczema, dermatitis (including atopic dermatitis), alopecia, vitiligo, bullous skin diseases, nephritis, vasculitis, atherosclerosis, Alzheimer’s disease, depression, Sjogren’s syndrome, sialoadenitis, central retinal vein occlusion, branched retinal vein occlusion, Irvine-Gass syndrome (post-cataract and post-surgical), retinitis pigmentosa, pars planitis, birdshot retinochoroidopathy, epiretinal membrane, cystic macular edema, parafoveal telengiectasis, tractional maculopathies, vitreomacular traction syndromes, retinal detachment,
neuroretinitis, idiopathic macular edema, retinitis, dry eye (kerartoconjunctivitis Sicca), vernal keratoconjunctivitis, atopic keratoconjunctivitis, uveitis (such as anterior uveitis, pan uveitis, posterior uveits, uveitis-associated macula edema), scleritis, diabetic retinopathy, diabetic macula edema, age-related macula dystrophy, hepatitis, pancreatitis, primary biliary cirrhosis, sclerosing cholangitis, Addison’s disease, hypophysitis, thyroiditis, type I diabetes, type 2 diabetes and acute rejection of transplanted organs. Bromodomain inhibitors may be useful in the treatment of a wide variety of acute inflammatory conditions such as acute gout, nephritis including lupus nephritis, vasculitis with organ involvement such as
glomerulonephritis, vasculitis including giant cell arteritis, Wegener’s granulomatosis, Polyarteritis nodosa, Behcet’s disease, Kawasaki disease, Takayasu’s Arteritis, pyoderma gangrenosum, vasculitis with organ involvement and acute rejection of transplanted organs. Bromodomain inhibitors may be useful in the treatment of diseases or conditions which involve inflammatory responses to infections with bacteria, viruses, fungi, parasites or their toxins, such as sepsis, sepsis syndrome, septic shock, endotoxaemia, systemic inflammatory response syndrome (SIRS), multi-organ dysfunction syndrome, toxic shock syndrome, acute
lung injury, ARDS (adult respiratory distress syndrome), acute renal failure, fulminant hepatitis, burns, acute pancreatitis, post-surgical syndromes, sarcoidosis, Herxheimer reactions, encephalitis, myelitis, meningitis, malaria and SIRS associated with viral infections such as influenza, herpes zoster, herpes simplex and coronavirus. Bromodomain inhibitors may be useful in the treatment of conditions associated with ischaemia-reperfusion injury such as myocardial infarction, cerebro-vascular ischaemia (stroke), acute coronary syndromes, renal reperfusion injury, organ transplantation, coronary artery bypass grafting, cardio-pulmonary bypass procedures, pulmonary, renal, hepatic, gastro-intestinal or peripheral limb embolism. Bromodomain inhibitors may be useful in the treatment of disorders of lipid metabolism via the regulation of APO-A1 such as hypercholesterolemia, atherosclerosis and Alzheimer’s disease. Bromodomain inhibitors may be useful in the treatment of fibrotic conditions such as idiopathic pulmonary fibrosis, renal fibrosis, postoperative stricture, keloid scar formation, scleroderma (including morphea) and cardiac fibrosis. Bromodomain inhibitors may be useful in the treatment of a variety of diseases associated with bone remodelling such as osteoporosis, osteopetrosis, pycnodysostosis, Paget’s disease of bone, familial expanile osteolysis, expansile skeletal hyperphosphatasia, hyperososis corticalis deformans Juvenilis, juvenile Paget’s disease and Camurati
Engelmann disease. Bromodomain inhibitors may be useful in the treatment of viral infections such as herpes virus, human papilloma virus, adenovirus and poxvirus and other DNA viruses. Bromodomain inhibitors may be useful in the treatment of cancer, including hematological (such as leukaemia, lymphoma and multiple myeloma), epithelial including lung, breast and colon carcinomas, midline carcinomas, mesenchymal, hepatic, renal and neurological tumours. Bromodomain inhibitors may be useful in the treatment of one or more cancers selected from brain cancer (gliomas), glioblastomas, Bannayan-Zonana syndrome, Cowden disease, Lhermitte-Duclos disease, breast cancer, inflammatory breast cancer, colorectal cancer, Wilm’s tumor, Ewing’s sarcoma, rhabdomyosarcoma, ependymoma, medulloblastoma, colon cancer, head and neck cancer, kidney cancer, lung cancer, liver cancer, melanoma, squamous cell carcinoma, ovarian cancer, pancreatic cancer, prostate cancer, sarcoma cancer, osteosarcoma, giant cell tumor of bone, thyroid cancer,
lymphoblastic T-cell leukemia, chronic myelogenous leukemia, chronic lymphocytic leukemia, hairy-cell leukemia, acute lymphoblastic leukemia, acute myelogenous leukemia, chronic neutrophilic leukemia, acute lymphoblastic T-cell leukemia, acute myeloid leukemia, plasmacytoma, immunoblastic large cell leukemia, mantle cell leukemia, multiple myeloma, megakaryoblastic leukemia, acute megakaryocytic leukemia, promyelocytic leukemia, mixed lineage leukaemia, erythroleukemia, malignant lymphoma, Hodgkins lymphoma, non-Hodgkins lymphoma, lymphoblastic T-cell lymphoma, Burkitt’s lymphoma, follicular lymphoma, neuroblastoma, bladder cancer, urothelial cancer, vulval cancer, cervical cancer, endometrial cancer, renal cancer, mesothelioma, esophageal cancer, salivary gland cancer, hepatocellular cancer, gastric cancer, nasopharangeal cancer, buccal cancer, cancer of the mouth, GIST (gastrointestinal stromal tumor) and testicular cancer. In one embodiment the cancer is a leukaemia, for example a leukaemia selected from acute monocytic leukemia, acute myelogenous leukemia, chronic myelogenous leukemia, chronic lymphocytic leukemia,
acute myeloid leukemia and mixed lineage leukaemia (MLL). In another embodiment the cancer is multiple myeloma. In another embodiment the cancer is a lung cancer such as small cell lung cancer (SCLC). In another embodiment the cancer is a neuroblastoma. In another embodiment the cancer is Burkitt’s lymphoma. In another embodiment the cancer is cervical cancer. In another embodiment the cancer is esophageal cancer. In another embodiment the cancer is ovarian cancer. In another embodiment the cancer is breast cancer. In another embodiment the cancer is colarectal cancer. In one embodiment the disease or condition for which a bromodomain inhibitor is indicated is selected from diseases associated with systemic inflammatory response syndrome, such as sepsis, burns, pancreatitis, major trauma, haemorrhage and ischaemia. In this embodiment the
bromodomain inhibitor would be administered at the point of diagnosis to reduce the incidence of: SIRS, the onset of shock, multi-organ dysfunction syndrome, which includes the onset of acute lung injury, ARDS, acute renal, hepatic, cardiac or gastro-intestinal injury and mortality. In another embodiment the bromodomain inhibitor would be administered prior to surgical or other procedures associated with a high risk of sepsis, haemorrhage, extensive tissue damage, SIRS or MODS (multiple organ dysfunction syndrome). In a particular embodiment the disease or condition for which a bromodomain inhibitor is indicated is sepsis, sepsis syndrome, septic shock and endotoxaemia. In another embodiment, the bromodomain inhibitor is indicated for the treatment of acute or chronic pancreatitis. In another embodiment the bromodomain is indicated for the treatment of burns. In one embodiment the disease or condition for which a bromodomain inhibitor is indicated is selected from herpes simplex infections and reactivations, cold sores, herpes zoster infections and reactivations, chickenpox, shingles, human papilloma virus, human immunodeficiency virus (HIV), cervical neoplasia, adenovirus infections, including acute respiratory disease, poxvirus infections such as cowpox and smallpox and African swine fever virus. In one particular embodiment a bromodomain inhibitor is indicated for the treatment of Human papilloma virus infections of skin or cervical epithelia. In one embodiment the bromodomain inhibitor is indicated for the treatment of latent HIV infection.
PATENT
WO 2016062737
http://www.google.com/patents/WO2016062737A1?cl=en
Scheme 1
Example 1
Step 1
5-fluoro-1 H-benzordlimidazol-2(3H)-one

A stirred solution of 4-fluorobenzene-1 ,2-diamine (15.1 g, 120 mmol) in THF (120 mL) under nitrogen was cooled using an ice-bath and then was treated with di(1 -/-imidazol-1 -yl)methanone (23.4 g, 144 mmol) portion-wise over 15 min. The resulting mixture was slowly warmed to room temperature then was concentrated in vacuo after 2.5 h. The residue was suspended in a mixture of water and DCM (250 mL each) and filtered off. This residue was then washed with water (50 mL) and DCM (50 mL), before being dried at 40 °C under vacuum for 16 h to give the title compound (16.0 g, 105 mmol, 88%) as a brown solid.
LCMS (high pH): Rt 0.57 min; [M-H+]“ = 151.1
δΗ NMR (400 MHz, DMSO-d6) ppm 10.73 (br s, 1 H), 10.61 (br s, 1 H), 6.91-6.84 (m, 1 H), 6.78-6.70 (m, 2H).
Step 2
5-fluoro-1 ,3-dimethyl-1 /-/-benzo[dlimidazol-2(3/-/)-one

A solution of 5-fluoro-1 H-benzo[d]imidazol-2(3H)-one (16.0 g, 105 mmol) in DMF (400 mL) under nitrogen was cooled with an ice-bath, using a mechanical stirrer for agitation. It was then treated over 10 min with sodium hydride (60% w/w in mineral oil, 13.1 g, 327 mmol) and the resulting mixture was stirred at this temperature for 30 min before being treated with iodomethane (26.3 mL, 422 mmol) over 30 min. The resulting mixture was then allowed to warm to room temperature and after 1 h was carefully treated with water (500 mL). The aqueous phase was extracted with EtOAc (3 x 800 mL) and the combined organics were washed with brine (1 L), dried over MgS04 and concentrated in vacuo. Purification of the brown residue by flash chromatography on silica gel (SP4, 1.5 kg column, gradient: 0 to 25% (3: 1 EtOAc/EtOH) in cyclohexane) gave the title compound (15.4 g, 86 mmol, 81 %) as a pink solid.
LCMS (high pH): Rt 0.76 min; [M+H+]+ = 181.1
δΗ NMR (400 MHz, CDCI3) ppm 6.86-6.76 (m, 2H), 6.71 (dd, J = 8.3, 2.3 Hz, 1 H), 3.39 (s, 3H), 3.38 (s, 3H).
Step 3
5-fluoro-1 ,3-dimethyl-6-nitro-1 /-/-benzordlimidazol-2(3/-/)-one

A stirred solution of 5-fluoro-1 ,3-dimethyl-1 H-benzo[d]imidazol-2(3/-/)-one (4.55 g, 25.3 mmol) in acetic anhydride (75 mL) under nitrogen was cooled to -30 °C and then was slowly treated with fuming nitric acid (1 .13 mL, 25.3 mmol) making sure that the temperature was kept below -25°C. The solution turned brown once the first drop of acid was added and a thick brown precipitate formed after the addition was complete. The mixture was allowed to slowly warm up to 0 °C then was carefully treated after 1 h with ice-water (100 mL). EtOAc (15 mL) was then added and the resulting mixture was stirred for 20 min. The precipitate formed was filtered off, washed with water (10 mL) and EtOAc (10 mL), and then was dried under vacuum at 40 °C for 16 h to give the title compound (4.82 g, 21 .4mmol, 85%) as a yellow solid.
LCMS (high pH): Rt 0.76 min; [M+H+]+ not detected
δΗ NMR (600 MHz, DMSO-d6) ppm 7.95 (d, J = 6.4 Hz, 1 H, (H-7)), 7.48 (d, J = 1 1.7 Hz, 1 H, (H-4)), 3.38 (s, 3H, (H-10)), 3.37 (s, 3H, (H-12)).
δ0 NMR (151 MHz, DMSO-d6) ppm 154.3 (s, 1 C, (C-2)), 152.3 (d, J = 254.9 Hz, 1 C, (C-5)), 135.5 (d, J = 13.0 Hz, 1 C, (C-9)), 130.1 (d, J = 8.0 Hz, 1 C, (C-6)), 125.7 (s, 1 C, (C-8)), 104.4 (s, 1 C, (C-7)), 97.5 (d, J = 28.5 Hz, 1 C, (C-4)), 27.7 (s, 1 C, (C-12)), 27.4 (s, 1 C, (C-10)).
Step 4
(R)-tert-but \ 4-( 1 ,3-dimethyl-6-nitro-2-oxo-2,3-dihydro-1 H-benzordlimidazol-5-yl)-3-methylpiperazine-1-carboxylate

A stirred suspension of 5-fluoro-1 ,3-dimethyl-6-nitro-1 H-benzo[d]imidazol-2(3/-/)-one (0.924 g, 4.10 mmol), (R)-ie f-butyl 3-methylpiperazine-1 -carboxylate (1.23 g, 6.16 mmol), and DI PEA (1 .43 mL, 8.21 mmol) in DMSO (4 mL) was heated to 120 °C in a Biotage Initiator microwave reactor for 13 h, then to 130 °C for a further 10 h. The reaction mixture was concentrated in vacuo then partitioned between EtOAc and saturated aqueous sodium bicarbonate solution. The aqueous was extracted with EtOAc and the combined organics were dried (Na2S04), filtered, and concentrated in vacuo to give a residue which was purified by silica chromatography (0-100% ethyl acetate in cyclohexane) to give the title compound as an orange/yellow solid (1.542 g, 3.80 mmol, 93%).
LCMS (formate): Rt 1.17 min, [M+H+]+ 406.5.
δΗ NMR (400 MHz, CDCI3) ppm 7.36 (s, 1 H), 6.83 (s, 1 H), 4.04-3.87 (m,1 H), 3.87-3.80 (m, 1 H), 3.43 (s, 6H), 3.35-3.25 (m, 1 H), 3.23-3.08 (m, 2H), 3.00-2.72 (m, 2H), 1.48 (s, 9H), 0.81 (d, J = 6.1 Hz, 3H)
Step 5
(RHerf-butyl 4-(6-amino-1 ,3-dimethyl-2-oxo-2,3-dihydro-1 /-/-benzordlimidazol-5-yl)-3-methylpiperazine-1-carboxylate

To (R)-iert-butyl 4-(1 ,3-dimethyl-6-nitro-2-oxo-2,3-dihydro-1 H-benzo[d]imidazol-5-yl)-3-methylpiperazine-1-carboxylate (1 .542 g) in /‘so-propanol (40 mL) was added 5% palladium on carbon (50% paste) (1.50 g) and the mixture was hydrogenated at room temperature and pressure. After 4 h the mixture was filtered, the residue washed with ethanol and DCM, and the filtrate concentrated in vacuo to give a residue which was purified by silica chromatography (50-100% ethyl acetate in cyclohexane) to afford the title compound (1.220 g, 3.25 mmol, 85%) as a cream solid.
LCMS (high pH): Rt 1 .01 min, [M+H+]+ 376.4.
δΗ NMR (400 MHz, CDCI3) ppm 6.69 (s, 1 H), 6.44 (s, 1 H), 4.33-3.87 (m, 4H), 3.36 (s, 3H), 3.35 (s, 3H), 3.20-2.53 (m, 5H), 1.52 (s, 9H), 0.86 (d, J = 6.1 Hz, 3H).
Step 6
(flVferf-butyl 4-(6-(2-methoxybenzamidoV 1 ,3-dimethyl-2-oxo-2,3-dihvdro-1 H-benzordlimidazol-5-yl)-3-methylpiperazine-1 -carboxylate

A stirred solution of (R)-iert-butyl 4-(6-amino-1 ,3-dimethyl-2-oxo-2,3-dihydro-1 /-/-benzo[d]imidazol-5-yl)-3-methylpiperazine-1 -carboxylate (0.254 g, 0.675 mmol) and pyridine (0.164 ml_, 2.025 mmol) in DCM (2 mL) at room temperature was treated 2-methoxybenzoyl chloride (0.182 mL, 1.35 mmol). After 1 h at room temperature the reaction mixture was concentrated in vacuo to give a residue which was taken up in DMSO:MeOH (1 :1 ) and purified by HPLC (Method C, high pH) to give the title compound (0.302 g, 0.592 mmol, 88%) as a white solid.
LCMS (high pH): Rt 1 .27 min, [M+H+]+ 510.5.
δΗ NMR (400 MHz, CDCI3) ppm 10.67 (s, 1 H), 8.53 (s, 1 H), 8.24 (dd, J = 7.8, 1.7 Hz, 1 H), 7.54-7.48 (m, 1 H), 7.18-7.12 (m, 1 H), 7.07 (d, J = 8.1 Hz, 1 H), 6.82 (s, 1 H), 4.27-3.94 (m, 2H), 4.08 (s, 3H), 3.45 (s, 3H), 3.40 (s, 3H), 3.18-2.99 (m, 2H), 2.92-2.70 (m, 3H), 1.50 (s, 9H), 0.87 (d, J = 6.1 Hz, 3H).
Step 7
(R)-N-( 1 ,3-dimethyl-6-(2-methylpiperazin-1 -yl)-2-oxo-2,3-dihydro-1 H-benzordlimidazol-5-yl)-2-methoxybenzamide

A stirred solution of (R)-ie f-butyl 4-(6-(2-methoxybenzamido)-1 ,3-dimethyl-2-oxo-2,3-dihydro-1 /-/-benzo[d]imidazol-5-yl)-3-methylpiperazine-1-carboxylate (302 mg, 0.592 mmol) in DCM (4 mL) at room temperature was treated with trifluoroacetic acid (3 ml_). After 15 minutes the mixture was concentrated in vacuo to give a residue which was loaded on a solid-phase cation exchange (SCX) cartridge (5 g), washed with MeOH, and then eluted with methanolic ammonia (2 M). The appropriate fractions were combined and concentrated in vacuo to give a white solid (240 mg). Half of this material was taken up in DMSO:MeOH (1 :1 ) and purified by HPLC (Method B, high pH) to give the title compound (101 mg, 0.245 mmol, 41 %) as a white solid.
LCMS (high pH): Rt = 0.90 min, [M+H+]+ 410.5.
δΗ NMR (600 MHz, DMSO-d6) ppm 10.74 (s, 1 H), 8.39 (s, 1 H), 8.05 (dd, J = 7.7, 1.8 Hz, 1 H), 7.57 (ddd, J = 8.3, 7.2, 2.0 Hz, 1 H), 7.29 (d, J = 8.1 Hz, 1 H), 7.23 (s, 1 H), 7.17-7.1 1 (m, 1 H), 4.10 (s, 3H), 3.33 (s, 3H), 3.32 (s, 3H), 3.30 (br s, 1 H), 3.07-3.02 (m, 1 H), 3.02-2.99 (m, 1 H), 2.92-2.87 (m, 1 H), 2.80 (td, J = 1 1.3, 2.7 Hz, 1 H), 2.73 (td, J = 1 1 .0, 2.7 Hz, 1 H), 2.68-2.63 (m, 1 H), 2.55 (dd, J = 12.0, 9.8 Hz, 1 H), 0.71 (d, J = 6.1 Hz, 3H).
δ0 NMR (151 MHz, DMSO-d6) ppm 162.1 , 156.8, 154.1 , 134.4, 133.2, 131.5, 130.1 , 126.6, 125.7, 121.9, 121.0, 1 12.5, 103.0, 99.4, 56.8, 55.4, 55.3, 53.3, 46.3, 26.8, 26.6, 16.7.
[aD]25 °c = -50.1 (c = 0.3, MeOH).
CLIPS
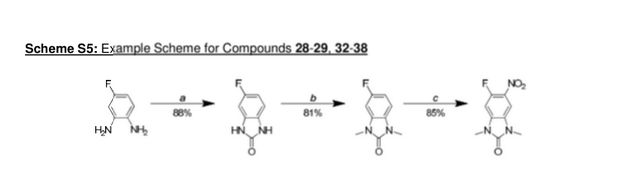
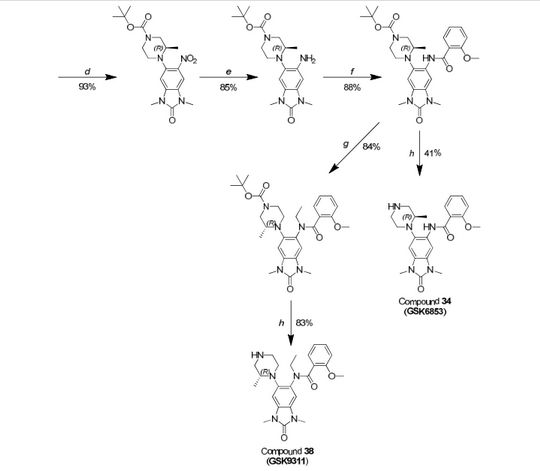

PAPER

The BRPF (Bromodomain and PHD Finger-containing) protein family are important scaffolding proteins for assembly of MYST histone acetyltransferase complexes. A selective benzimidazolone BRPF1 inhibitor showing micromolar activity in a cellular target engagement assay was recently described. Herein, we report the optimization of this series leading to the identification of a superior BRPF1 inhibitor suitable for in vivo studies.
GSK6853, a Chemical Probe for Inhibition of the BRPF1 Bromodomain
//////////////BRPF1, BRPF2, bromodomain, chemical probe, inhibitor, GSK 6853, PRECLINICAL
- Supporting Info SEE NMR COMPD 34, SMILES COc1ccccc1C(=O)Nc2cc4c(cc2N3CCNC[C@H]3C)N(C)C(=O)N4C
CCT 245737

CCT 245737
CAS:1489389-18-5
M.Wt: 379.34
Formula: C16H16F3N7O
2-Pyrazinecarbonitrile, 5-[[4-[[(2R)-2-morpholinylmethyl]amino]-5-(trifluoromethyl)-2-pyridinyl]amino]-
(R)-5-(4-(Morpholin-2-ylmethylamino)-5-(trifluoromethyl)pyridin-2-ylamino)pyrazine-2-carbonitrile
(+)-5-[[4-[[(2R)-Morpholin-2-ylmethyl]amino]-5-(trifluoromethyl)pyridin-2-yl]amino]pyrazine-2-carbonitrile
Cancer Research Technology Limited INNOVATOR
SAREUM
IND Filed, Sareum FOR CANCER
![]()

Synthesis, Exclusive by worlddrugtracker
5-[[4-[[morpholin-2-yl]methylamino]-5- (trifluoromethyl)-2-pyridyl]amino]pyrazine-2-carbonitrile compounds (referred to herein as “TFM compounds”) which, inter alia, inhibit Checkpoint Kinase 1 (CHK1) kinase function. The present invention also pertains to pharmaceutical compositions comprising such compounds, and the use of such compounds and compositions, both in vitro and in vivo, to inhibit CHK1 kinase function, and in the treatment of diseases and conditions that are mediated by CHK1 , that are ameliorated by the inhibition of CHK1 kinase function, etc., including proliferative conditions such as cancer, etc., optionally in combination with another agent, for example, (a) a DNA topoisomerase I or II inhibitor; (b) a DNA damaging agent; (c) an antimetabolite or a thymidylate synthase (TS) inhibitor; (d) a microtubule targeted agent; (e) ionising radiation; (f) an inhibitor of a mitosis regulator or a mitotic checkpoint regulator; (g) an inhibitor of a DNA damage signal transducer; or (h) an inhibitor of a DNA damage repair enzyme.
Checkpoint Kinase 1 (CHK1)
Progression through the cell division cycle is a tightly regulated process and is monitored at several positions known as cell cycle checkpoints (see, e.g., Weinert and Hartwell,
1989; Bartek and Lukas, 2003). These checkpoints are found in all four stages of the cell cycle; G1 , S (DNA replication), G2 and M (Mitosis) and they ensure that key events which control the fidelity of DNA replication and cell division are completed correctly. Cell cycle checkpoints are activated by a number of stimuli, including DNA damage and DNA errors caused by defective replication. When this occurs, the cell cycle will arrest, allowing time for either DNA repair to occur or, if the damage is too severe, for activation of cellular processes leading to controlled cell death.
All cancers, by definition, have some form of aberrant cell division cycle. Frequently, the cancer cells possess one or more defective cell cycle checkpoints, or harbour defects in a particular DNA repair pathway. These cells are therefore often more dependent on the remaining cell cycle checkpoints and repair pathways, compared to non-cancerous cells (where all checkpoints and DNA repair pathways are intact). The response of cancer cells to DNA damage is frequently a critical determinant of whether they continue to proliferate or activate cell death processes and die. For example, tumour cells that contain a mutant form(s) of the tumour suppressor p53 are defective in the G1 DNA damage checkpoint. Thus inhibitors of the G2 or S-phase checkpoints are expected to further impair the ability of the tumour cell to repair damaged DNA. Many known cancer treatments cause DNA damage by either physically modifying the cell’s DNA or disrupting vital cellular processes that can affect the fidelity of DNA replication and cell division, such as DNA metabolism, DNA synthesis, DNA transcription and microtubule spindle formation. Such treatments include for example, radiotherapy, which causes DNA strand breaks, and a variety of chemotherapeutic agents including topoisomerase inhibitors, antimetabolites, DNA-alkylating agents, and platinum- containing cytotoxic drugs. A significant limitation to these genotoxic treatments is drug resistance. One of the most important mechanisms leading to this resistance is attributed to activation of cell cycle checkpoints, giving the tumour cell time to repair damaged DNA. By abrogating a particular cell cycle checkpoint, or inhibiting a particular form of DNA repair, it may therefore be possible to circumvent tumour cell resistance to the genotoxic agents and augment tumour cell death induced by DNA damage, thus increasing the therapeutic index of these cancer treatments.
CHK1 is a serine/threonine kinase involved in regulating cell cycle checkpoint signals that are activated in response to DNA damage and errors in DNA caused by defective replication (see, e.g., Bartek and Lukas, 2003). CHK1 transduces these signals through phosphorylation of substrates involved in a number of cellular activities including cell cycle arrest and DNA repair. Two key substrates of CHK1 are the Cdc25A and Cdc25C phosphatases that dephosphorylate CDK1 leading to its activation, which is a
requirement for exit from G2 into mitosis (M phase) (see, e.g., Sanchez et al., 1997). Phosphorylation of Cdc25C and the related Cdc25A by CHK1 blocks their ability to activate CDK1 , thus preventing the cell from exiting G2 into M phase. The role of CHK1 in the DNA damage-induced G2 cell cycle checkpoint has been demonstrated in a number of studies where CHK1 function has been knocked out (see, e.g., Liu et ai, 2000; Zhao et al., 2002; Zachos et al., 2003).
The reliance of the DNA damage-induced G2 checkpoint upon CHK1 provides one example of a therapeutic strategy for cancer treatment, involving targeted inhibition of CHK1. Upon DNA damage, the p53 tumour suppressor protein is stabilised and activated to give a p53-dependent G1 arrest, leading to apoptosis or DNA repair (Balaint and Vousden, 2001). Over half of all cancers are functionally defective for p53, which can make them resistant to genotoxic cancer treatments such as ionising radiation (IR) and certain forms of chemotherapy (see, e.g., Greenblatt et al., 1994; Carson and Lois, 1995). These p53 deficient cells fail to arrest at the G1 checkpoint or undergo apoptosis or DNA repair, and consequently may be more reliant on the G2 checkpoint for viability and replication fidelity. Therefore abrogation of the G2 checkpoint through inhibition of the CHK1 kinase function may selectively sensitise p53 deficient cancer cells to genotoxic cancer therapies, and this has been demonstrated (see, e.g., Wang et al., 1996; Dixon and Norbury, 2002). In addition, CHK1 has also been shown to be involved in S phase cell cycle checkpoints and DNA repair by homologous recombination. Thus, inhibition of CHK1 kinase in those cancers that are reliant on these processes after DNA damage, may provide additional therapeutic strategies for the treatment of cancers using CHK1 inhibitors (see, e.g., Sorensen et al., 2005). Furthermore, certain cancers may exhibit replicative stress due to high levels of endogenous DNA damage (see, e.g., Cavalier et al., 2009; Brooks et al., 2012) or through elevated replication driven by oncogenes, for example amplified or overexpressed MYC genes (see, e.g., Di Micco et al. 2006; Cole et al., 2011 ; Murga et al. 2011). Such cancers may exhibit elevated signalling through CHK1 kinase (see, e.g., Hoglund et al., 2011). Inhibition of CHK1 kinase in those cancers that are reliant on these processes, may provide additional therapeutic strategies for the treatment of cancers using CHK1 inhibitors (see, e.g., Cole et al., 2011 ; Davies et al., 2011 ; Ferrao et al., 2011).
Several kinase enzymes are important in the control of the cell growth and replication cycle. These enzymes may drive progression through the cell cycle, or alternatively can act as regulators at specific checkpoints that ensure the integrity of DNA replication through sensing DNA-damage and initiating repair, while halting the cell cycle. Many tumours are deficient in early phase DNA-damage checkpoints, due to mutation or deletion in the p53 pathway, and thus become dependent on the later S and G2/M checkpoints for DNA repair. This provides an opportunity to selectively target tumour cells to enhance the efficacy of ionising radiation or widely used DNA-damaging cancer chemotherapies. Inhibitors of the checkpoint kinase CHK1 are of particular interest for combination with genotoxic agents. In collaboration with Professor Michelle Garrett (University of Kent, previously at The Institute of Cancer Research) and Sareum (Cambridge) we used structure-based design to optimise the biological activities and pharmaceutical properties of hits identified through fragment-based screening against the cell cycle kinase CHK1, leading to the oral clinical candidate CCT245737. The candidate potentiates the efficacy of standard chemotherapy in models of non-small cell lung, pancreatic and colon cancer. In collaboration with colleagues at The Institute of Cancer Research (Professor Louis Chesler, Dr Simon Robinson and Professor Sue Eccles) and Newcastle University (Professor Neil Perkins), we have shown that our selective CHK1 inhibitor has efficacy as a single agent in models of tumours with high replication stress, including neuroblastoma and lymphoma.

The checkpoint kinase CHK2 has a distinct but less well characterised biological role to that of CHK1. Selective inhibitors are valuable as pharmacological tools to explore the biological consequences of CHK2 inhibition in cancer cells. In collaboration with Professor Michelle Garrett (University of Kent, previously at The Institute of Cancer Research), we have used structure-based and ligand-based approaches to discover selective inhibitors of CHK2. We showed that selective CHK2 inhibition has a very different outcome to selective CHK1 inhibition. Notably, while CHK2 inhibition did not potentiate the effect of DNA-damaging chemotherapy, it did sensitize cancer cells to the effects of PARP inhibitors that compromise DNA repair.
Synthesis
(R)-5-(4-(Morpholin-2-ylmethylamino)-5-(trifluoromethyl)pyridin-2-ylamino)pyrazine-2-carbonitrile
PATENT
http://www.google.com/patents/WO2013171470A1?cl=enSynthesis 1 D
5-[[4-[[(2R)-Morpholin-2-yl]methylamino]-5-(trifluoromethyl)-2-pyridyl]amino]py
carbonitrile (Compound 1)

A solution of (S)-tert-butyl 2-((2-(5-cyanopyrazin-2-ylamino)-5-(trifluoromethyl)pyridin-4- ylamino)methyl)morpholine-4-carboxylate (1.09 g, 2.273 mmol) in dichloromethane (8 mL) was added dropwise over 10 minutes to a solution of trifluoroacetic acid (52.7 mL, 709 mmol) and tnisopropylsilane (2.61 mL, 12.73 mmol) in dry dichloromethane (227 mL) at room temperature. After stirring for 30 minutes, the mixture was concentrated in vacuo. The concentrate was resuspended in dichloromethane (200 mL) and
concentrated in vacuo, then resuspended in toluene (100 mL) and concentrated.
The above procedure was performed in triplicate (starting each time with 1.09 g (S)-tert- butyl 2-((2-(5-cyanopyrazin-2-ylamino)-5-(trifluoromethyl)pyridin-4- ylamino)methyl)morpholine-4-carboxylate) and the three portions of crude product so generated were combined for purification by ion exchange chromatography on 2 x 20 g Biotage NH2 Isolute columns, eluting with methanol. The eluant was concentrated and 10% methanol in diethyl ether (25 mL) was added. The resulting solid was filtered, washed with diethyl ether (30 mL), and dried in vacuo to give the title compound as a light straw coloured powder (2.30 g, 89%). H NMR (500 MHz, CD3OD) δ 2.62 (1 H, J = 12, 10 Hz), 2.78-2.84 (2H, m), 2.95 (1 H, dd, J = 12, 2 Hz), 3.27-3.38 (2H, m), 3.63 (1 H, ddd, J = 14, 9.5, 3 Hz), 3.73-3.78 (1 H, m), 3.91 (1 H, ddd, J = 11 , 4, 2 Hz), 7.26 (1 H, s), 8.18 (1 H, s), 8.63 (1 H, s), 9.01 (1 H, s).
LC-MS (Agilent 4 min) Rt 1.22 min; m/z (ESI) 380 [M+H+]. Optical rotation [a]D 24 = +7.0 (c 1.0, DMF).
Synthesis 2B
(R)-tert- Butyl 2-((2-chloro-5-(trifluoromethyl)pyridin-4-ylamino)methyl)morpholine-

To a solution of 2-chloro-5-(trifluoromethyl)pyridin-4-amine (1 g, 5.09 mmol) in
dimethylformamide (32.6 mL) was added sodium hydride (60% by wt in oil; 0.407 g, 10.18 mmol) portionwise at room temperature followed by stirring for 10 minutes at 80°C. (S)- tert-Butyl 2-(tosyloxymethyl)morpholine-4-carboxylate (2.268 g, 6.1 1 mmol) was then added portionwise and the reaction mixture was stirred at 80°C for 2.5 hours. After cooling, the mixture was partitioned between saturated aqueous sodium
hydrogencarbonate solution (30 mL), water (100 mL) and ethyl acetate (30 mL). The organic layer was separated and the aqueous layer was further extracted with ethyl acetate (2 x 30 mL). The combined organic layers were washed with brine (2 x 70 mL), dried over magnesium sulfate, filtered, concentrated and dried thoroughly in vacuo. The crude material was purified by column chromatography on a 90 g Thomson SingleStep column, eluting with an isocratic mix of 2.5% diethyl ether / 2.5% ethyl acetate in dichloromethane, to give the title compound as a clear gum that later crystallised to give a white powder (1.47 g, 73%). H NMR (500 MHz, CDCI3) δ 1.48 (9H, s), 2.71-2.83 (1 H, m), 2.92-3.05 (1 H, m), 3.18- 3.23 (1 H, m), 3.33-3.37 (1 H, m), 3.56-3.61 (1 H, m), 3.66-3.71 (1 H, m), 3.80-4.07 (3H, m), 5.32 (1 H, broad s), 6.61 (1 H, s), 8.24 (1 H, s). LC-MS (Agilent 4 min) Rt 3.04 min; m/z (ESI) 396 [MH+]. Svnthesis 2C
(R)-tert-Butyl 2-((2-(5-cyanopyrazin-2-ylamino)-5-(trifluoromethyl)pyridin-4-

(R)-tert-Butyl 2-((2-chloro-5-(trifluoromethyl)pyridin-4-ylamino)methyl)morpholine-4- carboxylate (1.44 g, 3.64 mmol), 2-amino-5-cyanopyrazine (0.612 g, 5.09 mmol, 1.4 eq.), tris(dibenzylideneacetone)dipalladium(0) (0.267 g, 0.291 mmol, 0.08 eq.), rac-2,2′- bis(diphenylphosphino)-1 ,1 ‘-binaphthyl (0.362 g, 0.582 mmol, 0.16 eq.) and caesium carbonate (2.37 g, 7.28 mmol) were suspended in anhydrous dioxane (33 ml_) under argon. Argon was bubbled through the mixture for 30 minutes, after which the mixture was heated to 100°C for 22 hours. The reaction mixture was cooled and diluted with dichloromethane, then absorbed on to silica gel. The pre-absorbed silica gel was added to a 100 g KP-Sil SNAP column which was eluted with 20-50% ethyl acetate in hexanes to give the partially purified product as an orange gum. The crude product was dissolved in dichloromethane and purified by column chromatography on a 90 g SingleStep Thomson column, eluting with 20% ethyl acetate in dichloromethane, to give the title compound (1.19 g, 68%). H NMR (500 MHz, CDCI3) δ 1.50 (9H, s), 2.71-2.88 (1 H, m), 2.93-3.08 (1 H, m), 3.27- 3.32 (1 H, m), 3.40-3.44 (1 H, m), 3.55-3.64 (1 H, m), 3.71-3.77 (1 H, m), 3.82-4.11 (3H, m), 5.33 (1 H, broad s), 7.19 (1 H, s), 8.23 (1 H, s), 8.58 (1 H, s), 8.84 (1 H, s). LC-MS (Agilent 4 min) Rt 2.93 min;m/z (ESI) 480 [MH+].
Paper

Multiparameter optimization of a series of 5-((4-aminopyridin-2-yl)amino)pyrazine-2-carbonitriles resulted in the identification of a potent and selective oral CHK1 preclinical development candidate with in vivo efficacy as a potentiator of deoxyribonucleic acid (DNA) damaging chemotherapy and as a single agent. Cellular mechanism of action assays were used to give an integrated assessment of compound selectivity during optimization resulting in a highly CHK1 selective adenosine triphosphate (ATP) competitive inhibitor. A single substituent vector directed away from the CHK1 kinase active site was unexpectedly found to drive the selective cellular efficacy of the compounds. Both CHK1 potency and off-target human ether-a-go-go-related gene (hERG) ion channel inhibition were dependent on lipophilicity and basicity in this series. Optimization of CHK1 cellular potency and in vivo pharmacokinetic–pharmacodynamic (PK–PD) properties gave a compound with low predicted doses and exposures in humans which mitigated the residual weak in vitro hERG inhibition.
Multiparameter Lead Optimization to Give an Oral Checkpoint Kinase 1 (CHK1) Inhibitor Clinical Candidate: (R)-5-((4-((Morpholin-2-ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-yl)amino)pyrazine-2-carbonitrile (CCT245737)
///////////CCT 245737, IND, PRECLINICAL, Cancer Research Technology Limited, SAREUM
N#CC(C=N1)=NC=C1NC2=NC=C(C(F)(F)F)C(NC[C@@H]3OCCNC3)=C2
- Supporting Info SEE NMR OF COMPD 4
SETIPIPRANT

Setipiprant, KYTH-105
CAS 866460-33-5
2-(2-(1-naphthoyl)-8-fluoro-1,2,3,4-tetrahydropyrido[4,3-b]indol-5-yl)acetic acid
2-[8-fluoro-2-(naphthalene-1-carbonyl)-3,4-dihydro-1H-pyrido[4,3-b]indol-5-yl]acetic acid
5H-Pyrido(4,3-b)indole-5-acetic acid, 8-fluoro-1,2,3,4-tetrahydro-2-(1-naphthalenylcarbonyl)-
MF C24H19FN2O3
MW 402.4176632
IND FILED BY ALLERGAN FOR Alopecia
ACT-129968, a CRTH2 receptor antagonist, had been in phase II clinical trials at Actelion
Setipiprant; UNII-BHF20LA2GM; ACT-129968; 866460-33-5;
Setipiprant is a prostaglandin D2 (PGD2) antagonist. Essentially, it inhibits PGD2 receptor activity
KYTH-105 had previously been studied as a potential allergic inflammation treatment and had undergone eight clinical trials, resulting in a safety database of more than 1,000 patients. Treatment in all studies was well tolerated across all treatment groups.
Intellectual Property
KYTHERA acquired exclusive worldwide rights to KYTH-105, as well as certain patent rights covering the use of PGD2 receptor antagonists for the treatment of hair loss (often presenting as male pattern baldness, or androgenic alopecia).
Next Steps
KYTHERA plans to file an Investigational New Drug (IND) application and initiate a proof-of-concept study to establish the efficacy of KYTH-105 in male subjects with androgenic alopecia (AGA).
In 2015, Allergan acquired Kythera.

![]()
2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic Acid
mp 224.0 °C.
LC(1)/ESI-MS tR = 0.83 min; m/z [M + H+] = 403.09.
1H NMR (DMSO-d6), 65:35 mixture of two rotamers, δ: 8.02 (m, 2 H), 7.76 (d, J = 7.8 Hz, 0.65 H), 7.72 (m, 0.35 H), 7.49–7.64 (m, 3.35 H), 7.35–7.49 (m, 2.35 H), 6.98 (ddd, JH–F = 9.3 Hz, J1 = 9.3 Hz, J2 = 2.4 Hz, 0.65 H), 6.88 (m, 0.65 H), 4.85–5.14 (m, 3.3 H), 4.42 (m, 0.35 H), 4.32 (m, 0.7 H), 4.06 (m, 0.35 H), 3.50 (t, J = 5.5 Hz, 1.3 H), 2.95 (m, 0.70 H), 2.68 (m, 0.65 H), 2.58 (m, 0.65 H).
13C NMR (DMSO-d6) δ: 170.7, 169.2, 157.7 (d, JC–F = 232 Hz), 157.4 (d, JC–F = 233 Hz), 137.1, 136.2, 135.1, 134.9, 134.0, 133.8, 133.5, 129.6, 129.5, 129.4, 129.3, 128.9, 128.8, 127.5, 127.4, 127.0, 126.9, 126.0, 125.9, 125.7 (d, JC–F = 10 Hz), 125.2, 125.1, 125.0, 124.1, 123.9, 110.9 (d, JC–F = 10 Hz), 110.8 (m), 109.3 (d, JC–F = 26 Hz), 109.1 (d, JC–F = 26 Hz), 106.7 (m), 103.3 (d, JC–F = 23 Hz), 103.0 (d, JC–F = 23 Hz), 44.73, 44.70, 44.5, 44.4, 39.5, 39.3, 23.1, 22.3.
HRMS (ESI): m/zcalcd for C24H20N2O3F [M + H+] 403.1458, found 403.1458.
SYNTHERSIS
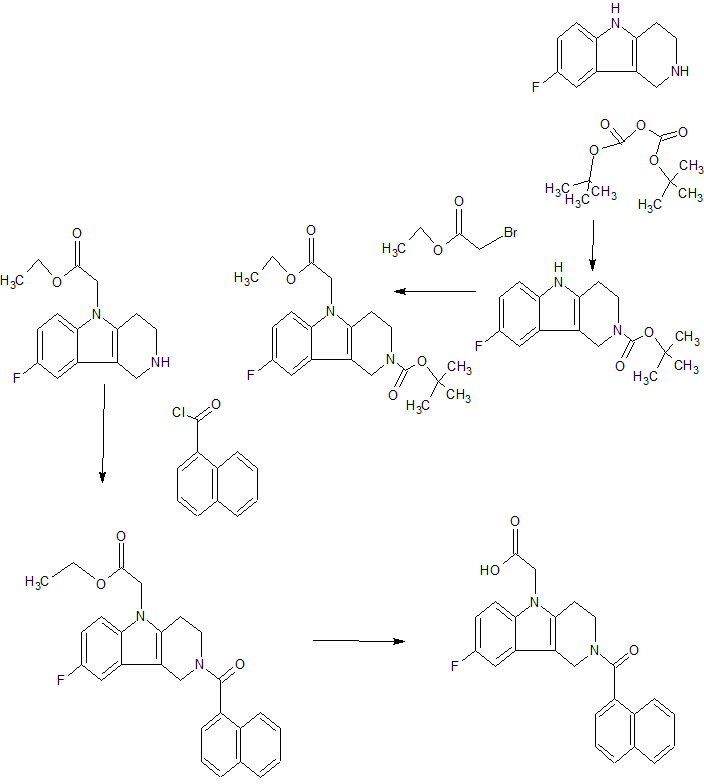

Setipiprant (INN) (developmental code names ACT-129,968, KYTH-105) is a drug originally developed by Actelion which acts as a selective, orally available antagonist of the prostaglandin D2 receptor 2 (DP2).[1] It was initially researched as a treatment for allergies and inflammatory disorders, particularly asthma, but despite being well tolerated in clinical trials and showing reasonable efficacy against allergen-induced airway responses in asthmatic patients,[2][3] it failed to show sufficient advantages over existing drugs and was discontinued from further development in this application.[4]
However, following the discovery in 2012 that the prostaglandin D2 receptor (DP/PGD2) is expressed at high levels in the scalp of men affected by male pattern baldness,[5] the rights to setipiprant were acquired by Kythera with a view to potentially developing this drug as a novel treatment for baldness, with a previously unexploited mechanism of action.[6] While it is too early to tell whether setipiprant will be an effective treatment for this condition, the favorable pharmacokinetics and relative lack of side effects seen in earlier clinical trials mean that fresh clinical trials for this new application can be conducted fairly quickly.[7]
Prostaglandin D2 is a known agonist of the thromboxane A2 (TxA2) receptor, the PGD2 (DP) receptor and the recently identified G-protein-coupled “chemoattractant receptor- homologous molecule expressed on Th2 cells” (CRTH2).
The response to allergen exposure in a previously sensitized host results in a cascade effect involving numerous cell types and release of a number of cytokines, chemokines, and multiple mediators. Among these critical initiators are the cytokines interleukin (IL)-4, IL-13, and IL-5, which play critical roles in Th2 cell differentiation, immunoglobulin (Ig)E synthesis, mast cell growth and differentiation, upregulation of CD23 expression, and the differentiation, recruitment, and activation of eosinophils. The stimulated release of the array of mediators, causes end-organ damage, including constriction and hyperresponsi- veness, vascular permeability, edema, mucous secretion, and further inflammation.
Because of the number of responses targeted, corticosteroids have proven to be the most effective therapy. Rather than antagonizing these specific responses in a directed way, another approach is to alter the immune response, that is, to change the nature of the immunological response to allergen. CRTH2 is preferentially expressed on Th2 cells and is a chemoattractant receptor for PGD2 that mediates PGD2-dependent migration of blood Th2 cells. Chemoattractants are responsible for the recruitment of both Th2 cells and other effector cells of allergic inflammation, which can provide the conceptual basis for the development of new therapeutic strategies in allergic conditions.
So far, few compounds having CRTH2 antagonistic activity have been reported in the patent literature. Bayer AG claims the use of Ramatroban ((3R)-3-(4-fluorobenzene- sulfonamido)-l,2,3,4-tetrahydrocarbazole-9-propionic acid) for the prophylaxis and treatment of allergic diseases, such as asthma, allergic rhinitis or allergic conjuvatitis
(GB 2388540). Further, (2-tert.-butoxycarbonyl-l, 2, 3, 4-tetrahydro-pyrido[4,3-b]indol-5- yl)-acetic acid and (2-ethoxycarbonyl-l, 2, 3, 4-tetrahydro-pyrido[4,3-b]indol~5-yl)-acetic acid are disclosed by Kyle F. et al in two patent applications (US 5817756 and WO 9507294, respectively).
Furthermore, oral bioavailability of the Ramatroban and its ability to inhibit prostaglandin D2-induced eosinophil migration in vitro has been reported (Journal of Pharmacology and Experimental Therapeutics, 305(1), p.347-352 (2003)).
Description of the invention:
It has now been found that compounds of the general Formulae (I) and (II) of the present invention are CRTH2 receptor antagonists. These compounds are useful for the treatment of both chronic and acute allergic/immune disorders such as allergic asthma, rhinitis, chronic obstructive pulmonary disease (COPD), dermatitis, inflammatory bowel disease, rheumatoid arthritis, allergic nephritis, conjunctivitis, atopic dermatitis, bronchial asthma, food allergy, systemic mast cell disorders, anaphylactic shock, urticaria, eczema, itching, inflammation, ischemia-reperfusion injury, cerebrovascular disorders, pleuritis, ulcerative colitis, eosinophil-related diseases, such as Churg-Strauss syndrome and sinusitis, basophil- related diseases, such as basophilic leukemia and basophilic leukocytosis.
The compounds of general Formulae (I) and (II), especially those mentioned as being preferred, display high selectivity towards the CRTH2 receptor. No antagonistic effects (IC50 >10 μM) are observed on e.g. prostaglandin D2 receptor DPI; PGI2 receptor (IP), PGE2 receptors (EPl, EP2, EP3, EP4), PGF2 receptor (FP), thromboxane receptor A2 (TxA2), leukotriene receptors (CysLTl, CysLT2, LTB4), complement receptor (C5a), angiotensin receptors (ATI, AT2) or serotonin receptor 5HT2c.
The solubility of compounds of general Formulae (I) and (II) in buffer at pH 7 is generally >800 μg/ml.
In vitro assays with rat and dog liver microsomes, or with rat and human hepatocytes revealed high metabolic stability for compounds of general Foπnulae (I) and (II), especially for those compounds mentioned as being preferred.
The compounds of general Formulae (I) and (II), especially those mentioned as being preferred, do not interfere with cytochrome P-450 enzymes, e.g. they are neither degraded by, nor do they inhibit such enzymes.
Excellent pharmacokinetic profiles have been observed for compounds of general Formulae (I) and (II), especially for those compounds mentioned as being preferred, after oral administration (10 mg/kg) to rats and dogs (bioavailability 20-80%, Tmax 30 min, Cmax 2000- 6000 ng/ml, low clearance, T] 24-8 h). The compounds of general Formulae (I) and (II), especially those mentioned as being preferred, are efficacious in vitro, inhibiting PGD2-induced migration of eosinophils or other CRTH2 expressing cells in a cell migration assay. A number of techniques have been developed to assay such chemotactic migration (see, e.g., Leonard et al., 1995, “Measurement of α- and β-Chemokines”, in Current Protocols in Immunology, 6.12.1- 6.12.28, Ed. Coligan et al, John Wiley & Sons, Inc. 1995). The compounds of the present invention are tested using a protocol according to H. Sugimoto et al. (J Pharmacol Exp Ther. 2003, 305(1), 347-52), or as described hereinafter: Purified eosinophils are labeled with a fluorescent dye, i.e. Calcein-AM and loaded in BD Falcon FluoroBlock upper inserts. Test compounds are diluted and incubated with eosinophils in the BD Falcon
FluoroBlock upper inserts for 30 min at 37 °C in a humidified CO2 incubator. A constant amount of PGD2 is added to BD Falcon FluoroBlock lower chamber, at a concentration known to have a chemotactic effect on CRTH2 cells. As a control, at least one aliquot in the upper well does not contain test compound. The inserts are combined with the chambers and are incubated for 30 min at 37 °C in a humidified CO2 incubator. After an incubation period, the number of migrating cells on the lower chamber is counted using a fluorescent reader, i.e. an Applied Biosystems Cyto Fluor 4000 plate reader. The contribution of a test compound to the chemotactic activity of PGD2 is measured by comparing the chemotactic activity of the aliquots containing only dilution buffer with the activity of aliquots containing a test compound. If addition of the test compound to the solution results in a decrease in the number of cells detected in the lower chamber relative to the number of cells detected using a solution containing only PGD2, then there is identified an antagonist of PGD2 induction of chemotactic activity of eosinophils.
PAPER
Journal of Medicinal Chemistry (2013), 56(12), 4899-4911
http://pubs.acs.org/doi/abs/10.1021/jm400122f
Identification of 2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic Acid (Setipiprant/ACT-129968), a Potent, Selective, and Orally Bioavailable Chemoattractant Receptor-Homologous Molecule Expressed on Th2 Cells (CRTH2) Antagonist

Herein we describe the discovery of the novel CRTh2 antagonist 2-(2-(1-naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid 28 (setipiprant/ACT-129968), a clinical development candidate for the treatment of asthma and seasonal allergic rhinitis. A lead optimization program was started based on the discovery of the recently disclosed CRTh2 antagonist 2-(2-benzoyl-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid 5. An already favorable and druglike profile could be assessed for lead compound 5. Therefore, the lead optimization program mainly focused on the improvement in potency and oral bioavailability. Data of newly synthesized analogs were collected from in vitro pharmacological, physicochemical, in vitro ADME, and in vivo pharmacokinetic studies in the rat and the dog. The data were then analyzed using a traffic light selection tool as a visualization device in order to evaluate and prioritize candidates displaying a balanced overall profile. This data-driven process and the excellent results of the PK study in the rat (F = 44%) and the dog (F = 55%) facilitated the identification of 28 as a potent (IC50 = 6 nM), selective, and orally available CRTh2 antagonist.
PAtent
WO 2005095397
http://www.google.co.in/patents/WO2005095397A1?cl=en
Formula 6.



Scheme 1
Step a)

Step b)

Scheme 2
Formula (I).

References
- Fretz H, Valdenaire A, Pothier J, Hilpert K, Gnerre C, Peter O, Leroy X, Riederer MA. Identification of 2-(2-(1-naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid (setipiprant/ACT-129968), a potent, selective, and orally bioavailable chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) antagonist. J Med Chem. 2013 Jun 27;56(12):4899-911. doi: 10.1021/jm400122f PMID 23721423
- Sidharta PN, Diamant Z, Dingemanse J. Single- and multiple-dose tolerability and pharmacokinetics of the CRTH2 antagonist setipiprant in healthy male subjects. Fundam Clin Pharmacol. 2014 Dec;28(6):690-9. doi: 10.1111/fcp.12079 PMID 24734908
- Diamant Z, Sidharta PN, Singh D, O’Connor BJ, Zuiker R, Leaker BR, Silkey M, Dingemanse J. Setipiprant, a selective CRTH2 antagonist, reduces allergen-induced airway responses in allergic asthmatics. Clin Exp Allergy. 2014 Aug;44(8):1044-52. doi: 10.1111/cea.12357 PMID 24964348
- Norman P. Update on the status of DP2 receptor antagonists; from proof of concept through clinical failures to promising new drugs. Expert Opin Investig Drugs. 2014 Jan;23(1):55-66. doi: 10.1517/13543784.2013.839658 PMID 24073896
- Garza LA, et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Science Translational Medicine, 21 March 2012; 4(126):126ra34. doi: 10.1126/scitranslmed.3003122
- George Cotsarelis, Garret Fitzgerald, Luis Garza. Compositions and methods for regulating hair growth. US Patent application 2015/0072963
- Pipeline KYTH-105 (setipiprant)
- http://files.shareholder.com/downloads/AMDA-MFNLA/4023632629x0x817836/4E5AC47A-B9EE-4296-9D97-631C0F6B7C97/KYTH-105_setipiprant_.pdf

| Patent ID | Date | Patent Title |
|---|---|---|
| US2015072963 | 2015-03-12 | COMPOSITIONS AND METHODS FOR REGULATING HAIR GROWTH |
| US2014328861 | 2014-11-06 | Combination of CRTH2 Antagonist and a Proton Pump Inhibitor for the Treatment of Eosinophilic Esophagitis |
| US2010234396 | 2010-09-16 | Tetrhydropyridoindole Derivatives |
| US7714132 | 2010-05-11 | Tetrahydropyridoindole derivatives |
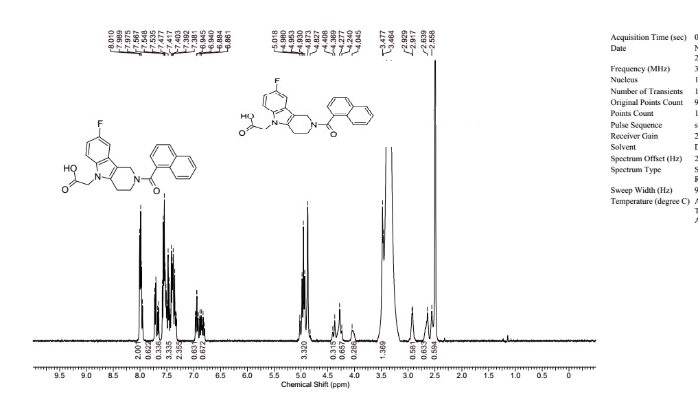
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-[8-fluoro-2-(naphthalene-1-carbonyl)-3,4-dihydro-1H-pyrido[4,3-b]indol-5-yl]acetic acid
|
|
| Clinical data | |
| Administration | Oral |
| Identifiers | |
| CASRN | 866460-33-5 |
| ATC code | none |
| PubChem | CID 49843471 |
| Chemical data | |
| Formula | C24H19FN2O3 |
| Molar mass | 402.417 g/mol |
///////Setipiprant, KYTH-105, 866460-33-5, ALLERGAN, Alopecia, KYTHERA
c15ccccc5cccc1C(=O)N(CC3)Cc2c3n(CC(O)=O)c(cc4)c2cc4F
