
Home » Phase3 drugs (Page 30)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Phase3 Rindopepimut ,CDX-110 Celldex Therapeutics’ brain cancer vaccine

http://clinicaltrials.gov/ct2/show/NCT01480479
MAR 2013
Rindopepimut
Immunotherapeutic vaccine called Rindopepimut showed positive results in prolonging survival in patients with newly diagnosed EGFRvIII-positive glioblastoma (GB), one of the most aggressive forms of brain cancer
Celldex Therapeutics’ brain cancer vaccine, rindopepimut, also known as CDX-110, targets EGFRvIII, an activated mutation of the epidermal growth factor receptor (EGFR). This mutation is found in about 31% of cases of glioblastoma multiforme, a form of fast-growing brain cancer and the most common type of primary brain tumor. It can contribute to tumor growth, and is linked with poor long-term survival. It is not seen in normal tissue.
In the ACT III Phase II trial, which involved people with newly diagnosed EGFRvIII-positive glioblastoma, 65 patients were given rindopepimut in combination with standard-of-care treatment (temozolomide), after having undergone surgery and standard chemotherapy and radiation therapy.
focus on is Rindopepimut, an immunotherapy treatment that targets EGFRvIII. As it’s not found at significant levels in normal tissues but expressed in 30ish% of primary glioblastoma, it’s an ideal target that has produced promising results to date. The drug candidate has shown consistent benefit for patients across three phase 2 studies- that’s no fluke! It’s currently in a global phase 3 trial in patients with newly diagnosed glioblastoma with results due in a couple years
| Phase 3 Study of Rindopepimut in Patients With Newly Diagnosed Glioblastoma (ACT IV) | |
 |
|
| Design: | Phase 3, double-blind, study of rindopepimut compared with KLH control |
| Status: | Currently enrolling at multiple centers in the US; additional centers outside the US planned to be activated in 2012 |
ABOUT THE CLINICAL TRIAL
This 2-arm, randomized, Phase 3 study will investigate the efficacy and safety of the addition of rindopepimut to the current standard of care, temozolomide, in patients with recently diagnosed EGFRvIII positive glioblastoma. All patients will be administered temozolomide. Half the patients will be randomly assigned to receive rindopepimut (given along with GM-CSF as a vaccine adjuvant) and half the patients will be randomly assigned to receive a keyhole limpet hemocyanin (KLH). Patients will be treated in a blinded fashion (neither the patient nor the doctor will know which arm of the study the patient is on). Patients will be treated until disease progression or intolerance to therapy and all patients will be followed for survival.
All patients enrolled in the study will be closely monitored to determine if their cancer is responding to treatment and for any side effects that may occur.
KYTHERA Biopharmaceuticals, Inc. Announces Positive Interim Results from Open-Label Study of ATX-101 in the Reduction of Unwanted Submental Fat (SMF) or “Double Chin”
http://clinicaltrials.gov/ct2/show/NCT01426373
synthesis………..https://newdrugapprovals.org/2014/07/14/some-thing-for-your-chin-fda-accepts-kytheras-atx-101-new-drug-application/
The drug is sodium deoxycholate for injection, code-named ATX-101 was developed for the treatment of lipomas – benign tumors of subcutaneous adipose tissue, as well as other unwanted fatty growths, such as a double chin. This substance, which is a salt of one of the bile acids, emulsifies fats, destroying their excess deposits


ATX-101 (a first-in-class injectable drug being studied for the reduction of localized fat. ATX-101 is a proprietary formulation of deoxycholate a well-studied endogenous compound that is present in the body), a facial injectable drug for the reduction of unwanted fat under the chin, or submental fat. V. Leroy Young, MD, FACS, presented the initial results at the American Society for Aesthetic Plastic Surgery (ASAPS) 45th Annual Aesthetic Meeting in Vancouver, British Columbia, on May 4, 2012.
In August 2010 Bayer Consumer Care AG signed a licensing and development collaboration agreement with KYTHERA, thereby obtaining commercialization rights to ATX-101 outside the US and Canada. KYTHERA and Bayer are collaborating on the development of ATX-101 in Europe.
KYTHERA Biopharmaceuticals Inc. 02 MAR 3013, announced positive interim results from a Phase IIIb multi-center open-label study (ATX-101-11-26) to evaluate the safety and efficacy of ATX-101 an investigational injectable drug for the reduction of unwanted submental fat (SMF) commonly known as double chin. The results presented at the Late Breaking Research Symposium at the 71st American Academy of Dermatology (AAD) Annual Meeting in Miami Beach Fla. found that ATX-101 is well-tolerated and may be effective in reducing SMF by both clinician and patient reported outcome measures. The ATX-101 global clinical development program has enrolled more than 2500 total patients of which more than 1500 have been treated with ATX-101.
“In my practice patients often request a non-surgical way to treat their submental fat or undesirable double chin” said investigator Susan Weinkle MD FAAD a board certified dermatologist and affiliate clinical professor at the University of South Florida. “For these patients double chin is often resistant to diet and exercise. The results of this study suggest that microinjections of ATX-101 can reduce submental fat without worsening skin laxity.”
ATX-101 is a proprietary synthetically-derived formulation of deoxycholic acid (DCA) a naturally-occurring molecule found in the body that aids in fat metabolism. In this open-label Phase IIIb study interim results three months after the last ATX-101 treatment found:
- Reduction of submental fat
- 87 percent of patients achieved at least a one-grade improvement from baseline on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS)
- Similarly 83 percent of patients achieved at least a one-grade improvement on the Patient-Reported Submental Fat Rating Scale (PR-SMFRS)
- 96 percent of patients had unchanged or improved skin laxity based on the clinician rated Submental Skin Laxity Grading Scale (SMSLG)
- 95 percent of patients were satisfied with treatment based on the Global Post Treatment Satisfaction Scale
- Adverse events were of mild to moderate intensity transient and primarily associated with the treatment area

Topline results from this study were announced in November 2012. As previously announced 71.3 percent of subjects had at least a one-grade improvement on the CR-SMFRS / PR-SMFRS composite and 14.0 percent had at least a two-grade improvement on the same composite measure.
These results are based on a multicenter 12-month open-label Phase IIIb study conducted at 21 sites across the United States evaluating 165 adults who received injections of ATX-101 for up to six treatments at four-week intervals. Patients received ATX-101 (2 mg/cm2) by subcutaneous microinjections directly into their SMF and were evaluated three months after their last treatment. The study population includes females (77.6 percent) and males (22.4 percent) with a mean age of 47 who report at least moderate SMF and dissatisfaction with the appearance of their chin. All Fitzpatrick Skin Types an industry standard scale to categorize skin tone are represented.
“We are pleased with these ATX-101 study results” said Patricia S. Walker M.D. Ph.D. chief medical officer KYTHERA Biopharmaceuticals Inc. “These results along with efficacy analyses in double-blind placebo-controlled studies support ATX-101 entering the market as potentially the first medical aesthetic drug approved for the reduction of submental fat.”
About ATX-101
ATX-101 is a potential first-in-class injectable drug candidate under clinical investigation for the reduction of unwanted submental fat. ATX-101 is a proprietary formulation of synthetic deoxycholic acid a well-characterized endogenous compound that is present in the body to promote the natural breakdown of dietary fat. ATX-101 is designed to be a locally-injected drug that causes proximal preferential destruction of adipocytes or fat cells with minimal effect on surrounding tissue. Based on clinical trials conducted to date ATX-101 has exhibited significant meaningful and durable results in the reduction of submental fat which commonly presents as an undesirable “double chin.” These results correspond with subject satisfaction measures demonstrating meaningful improvement in perceived chin appearance.
In August 2010 Bayer signed a licensing and collaboration development agreement with KYTHERA thereby obtaining development and commercialization rights to ATX-101 outside of the U.S. and Canada. Bayer recently completed two pivotal Phase III trials of ATX-101 in Europe for the reduction of submental fat. Topline results from these trials were reported in the second quarter of 2012. KYTHERA completed enrollment in its pivotal Phase III clinical program for ATX-101 in more than 1000 subjects randomized to ATX-101 or placebo in 70 centers across the United States and Canada in August 2012. The Company expects to release topline results in mid-2013.
About KYTHERA Biopharmaceuticals Inc.
KYTHERA Biopharmaceuticals Inc. is a clinical-stage biopharmaceutical company focused on the discovery development and commercialization of novel prescription products for the aesthetic medicine market. KYTHERA initiated its pivotal Phase III clinical program for ATX-101 in March 2012 and completed enrollment of more than 1000 patients randomized to ATX-101 or placebo in 70 centers across the U.S. and Canada in August 2012. KYTHERA also maintains an active research interest in hair and fat biology. Find more information at www.kytherabiopharma.com.
Celgene phase 3 – Oral Apremilast Achieves Statistical Significance for the Primary Endpoint of PASI-75 in the First Phase III Study in Patients with Psoriasis

APREMILAST, N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
mar02,2013
Celgene International Sàrl, a subsidiary of Celgene Corporation (NASDAQ: CELG) today presented the results from ESTEEM 1, the Company’s first phase III study in psoriasis, at the American Academy of Dermatology annual meeting in Miami, Florida.
“I see this as a prime candidate for future management of psoriasis that allows us to treat a range of patients, including more moderate cases earlier on”
The company previously announced statistical significance for the primary and major secondary endpoint of PASI-75 at Week 16 and the Static Physician Global Assessment for patients receiving apremilast in the ESTEEM 1&2 phase III studies. ESTEEM 1&2 are the phase III registrational randomized, placebo-controlled studies evaluating the Company’s oral small-molecule inhibitor of phosphodiesterase-4 (PDE4) in patients with moderate-to-severe chronic plaque psoriasis.
ESTEEM 1, presented today, evaluated efficacy and safety in a range of patients. Approximately one-third of the study population was systemic and/or phototherapy treatment-naïve. Nearly 30 percent of the overall study population had prior biologic therapy, which included biologic-failures.
In the ESTEEM 1 study, a significantly higher percentage of apremilast-treated patients demonstrated PASI-75 at week 16 than did placebo patients (33.1% vs. 5.3%; P<0.0001). Significantly higher PASI-75 scores at week 16 were demonstrated across all patient segments enrolled in this study, including systemic-naïve and biologic-naïve patients receiving apremilast 30 mg BID compared with placebo (38.7% vs. 7.6%; P<0.0001 and 35.8% vs. 5.9%; P<0.0001 respectively). Apremilast demonstrated maintenance of effect over time, as measured by the Mean Percent Change from Baseline in PASI score over 32 weeks, with apremilast demonstrating a 54.9% reduction at week 16 and a 61.9% reduction at week 32.
Statistical significance at week 16 was also demonstrated in the major secondary endpoint, Static Physician Global Assessment (sPGA) of clear or almost clear (P<0.0001), and other key secondary endpoints (change in BSA, Pruritus VAS, DLQI), as well as in assessments of difficult to treat areas (nail and scalp psoriasis).
“I see this as a prime candidate for future management of psoriasis that allows us to treat a range of patients, including more moderate cases earlier on,” said Kristian Reich, M.D., SCIderm Research Institute and Dermatologikum Hamburg, Germany.
The overall safety and tolerability profile was consistent with results from previously reported phase III psoriatic arthritis trials. No cases of tuberculosis or lymphoma were observed through week 16, and there was no increased risk of cardiovascular events or serious opportunistic infection. Apremilast was generally well tolerated. The most common adverse events (AEs) greater than placebo were diarrhea, nausea and headache. Greater than 96% of patients in the study reported no AEs or mild to moderate AEs. A similar percentage of patients reported both serious AEs and severe AEs in the apremilast 30 mg BID treatment group compared to placebo (2.1% vs. 2.8% and 3.6% vs. 3.2%, respectively).
An NDA submission to the U.S. Food and Drug Administration, based on the combined ESTEEM 1&2 studies for psoriasis, is expected in the second half of 2013. The Company previously announced it expects to file a separate NDA for psoriatic arthritis in the first quarter of 2013. A combined PsA/psoriasis MAA submission in Europe is also planned for the second half of 2013.
Top-line positive results from the two pivotal, randomized, placebo-controlled phase III studies of apremilast in psoriasis (ESTEEM 1&2) were released in January 2013. The studies included more than 1,200 patients with moderate-to-severe psoriasis and are ongoing. Results from PSOR-005, a phase IIb dose-range study, were recently published in The Lancet (http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(12)60642-4/fulltext).
About ESTEEM 1 & 2
ESTEEM 1 & 2 are two pivotal phase III randomized, placebo-controlled studies evaluating apremilast in subjects with a diagnosis of moderate-to-severe chronic plaque psoriasis for at least 12 months prior to the screening, and at baseline, and who were also candidates for phototherapy and/or systemic therapy. Approximately 1,250 patients were randomized 2:1 to receive either apremilast 30 mg BID or placebo for the first 16 weeks, followed by a maintenance phase from weeks 16-32 in which placebo subjects were switched to apremilast 30 mg BID through week 32, and a randomized withdrawal phase for responders from Week 32-Week 52 based on their initial apremilast randomization and PASI response.
Apremilast, an oral small-molecule inhibitor of phosphodiesterase 4 (PDE4), works intracellularly to modulate a network of pro-inflammatory and anti-inflammatory mediators. PDE4 is a cyclic adenosine monophosphate (cAMP)-specific PDE and the dominant PDE in inflammatory cells (see http://discoverpde4.com/). PDE4 inhibition elevates intracellular cAMP levels, which in turn down-regulates the inflammatory response by modulating the expression of TNF-α, IL-23, and other inflammatory cytokines. Elevation of cAMP also increases anti-inflammatory cytokines such as IL-10. To learn more go to www.discoverpde4.com/.
Top-line positive results from three pivotal randomized, placebo-controlled phase III studies of apremilast in PsA (PALACE 1, 2 & 3) were released in September 2012. PALACE 1 was also presented as an oral presentation at the ACR annual meeting in November 2012. Taken together, the PALACE program comprises the most comprehensive psoriatic arthritis studies to date intended for regulatory submission.
Results from PSA-001, the phase II study of apremilast in psoriatic arthritis, were recently published online in the journal Arthritis & Rheumatism (http://onlinelibrary.wiley.com/doi/10.1002/art.34627/abstract).
A randomized, placebo-controlled phase III study (POSTURE) of apremilast in ankylosing spondylitis (AS) began enrolling patients in April 2012. AS, a debilitating disease, which may cause fusion of the spine, arthritis, inflammation of the eye and damage to the heart, affects approximately 1.5 million people in the U.S. and Europe. The trial will randomize approximately 450 patients to receive 20 mg or 30 mg apremilast BID, or placebo BID.
Psoriasis is an immune-mediated, non-contagious chronic inflammatory skin disorder of unknown cause. The disorder is a chronic recurring condition that varies in severity from minor localized patches to complete body coverage. Plaque psoriasis is the most common type of psoriasis. About 80 percent of people who develop psoriasis have plaque psoriasis, which appears as patches of raised, reddish skin covered by silvery-white scales. These patches, or plaques, frequently form on the elbows, knees, lower back, and scalp. Psoriasis occurs nearly equally in males and females. Recent studies show that there may be an ethnic link. Psoriasis is believed to be most common in Caucasians and slightly less common in other ethnic groups. Worldwide, psoriasis is most common in Scandinavia and other parts of northern Europe. About 10 percent to 30 percent of patients with psoriasis also develop a condition called psoriatic arthritis, which causes pain, stiffness and swelling in and around the joints.
Celgene International Sàrl, located in Boudry, in the Canton of Neuchâtel, Switzerland, is a wholly owned subsidiary and international headquarters of Celgene Corporation. Celgene Corporation, headquartered in Summit, New Jersey, is an integrated global pharmaceutical company engaged primarily in the discovery, development and commercialization of innovative therapies for the treatment of cancer and inflammatory diseases through gene and protein regulation. For more information, please visit the Company’s website at www.celgene.com.
Apremilast is an orally available small molecule inhibitor of PDE4 being developed by Celgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.”
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06
Taiho Pharma seeks Japanese nod to manufacture,market novel anti-tumour agent TAS-102

TRIFLURIDINE

TIPIRACIL
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
| Combination of | |
|---|---|
| Trifluridine | cytotoxin |
| Tipiracil | thymidine phosphorylase inhibitor |
Clinical trials
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
Mechanism
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
| February 28, 2013, |
|
Taiho Pharmaceutical Co., Ltd. has submitted an application to the Japanese Ministry of Health, Labour and Welfare for approval of the manufacture and marketing of the novel oral nucleoside anti-tumour agent TAS-102 (combination of trifluorothymidine [FTD] and tipiracil hydrochloride [TPI]). Taiho is seeking approval of TAS-102 for the indication of unresectable, advanced, recurrent colorectal cancer. The application for approval is based on the results of a phase II clinical trial (Study 10040030) conducted at 20 facilities throughout Japan. It was a randomized, double-blind comparative study of TAS-102 and a placebo involving 172 patients with unresectable, advanced, recurrent colorectal cancer that was refractory to the standard chemotherapy of at least two or more regimens containing fluoropyrimidine, irinotecan, and oxaliplatin. The results indicated that the group administered TAS-102 had improved overall survival rates (median overall survival: 9.0 months vs. 6.6 months) and a significantly reduced risk of mortality (HR: 0.56, p=0.0011). The most frequently reported adverse drug reaction with a CTCAE grade of 3 or higher was neutropenia. Grade 3 or higher diarrhea, fatigue, nausea, and other adverse reactions were no more than 10 per cent. Taiho Pharmaceutical is currently proceeding with a global phase III clinical trial of TAS-102 in a similar colorectal cancer population (RECOURSE) with the ultimate goal of global registration and commercialization of the agent. Taiho Pharmaceutical believes that TAS-102 will make a significant contribution to cancer patients and will continue its development efforts to broaden its use. TAS-102 is an anti-tumour agent composed of a combination of trifluorothymidine (FTD), a nucleoside that incorporates into DNA and inhibits a variety of genetic functions required for the proliferation of cancer cells, and tipiracil hydrochloride (TPI), an inhibitor of thymidine phosphorylase (which degrades FTD) that maintains an effective blood concentration of FTD. TAS-102 is administered twice daily to achieve a total daily dose of 70mg/m2 for five days followed by two days of rest and then repeated a second time. This is followed by a 14-day rest period to make a 28-day schedule for one course. |
- “New Drug for Colorectal Cancer Shows Promise in Phase II Trial”. 28 Aug 2012.
- “Novel Drug TAS-102 Makes Headway in Refractory Colorectal Cancer”. 4 Oct 2011.
- “Phase II study of TAS-102 for pretreated metastatic colorectal cancer”. 29 Aug 2012.
- “A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA.”. Sept 2004.
|
|
|---|---|
| TRIFLURIDINE | |
| 1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5- (trifluoromethyl) pyrimidine-2,4-dione |
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
TIPIRACIL
| NAME | 5-chloro-6-[(2-iminopyrrolidin-1-yl)methyl]pyrimidine-2,4-(1H,3H)-dione | |||
| CAS | 183204-74-2 | |||
| MOL F | C9H11ClN4O2 | |||
| STR | |
|||
| USE | potentiator of antineoplastics; | |||
Taiho Pharmaceutical, a subsidiary of Otsuka Holdings Co., Ltd., is an R&D-driven specialty pharma focusing on the three fields of oncology, allergies and immunology, and urology.
Phase 3 Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease
 CAS Number:75172-81-5
CAS Number:75172-81-5-
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
- Molecular Structure:

- Formula:C6H14ClNO4
- Molecular Weight:199.63
- Synonyms:3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;Migalastat hydrochloride;Galactostatin hydrochloride;(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
- Melting Point:260 °C
- Boiling Point:382.7 °C at 760 mmHg
- Flash Point:185.2 °C
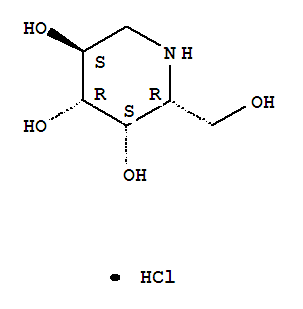
end feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2 Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
http://clinicaltrials.gov/show/NCT01458119
http://www.docstoc.com/docs/129812511/migalastat-hcl
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |

Lumacaftor, VX-809 an experimental drug for the treatment of Late-Stage cystic fibrosis, being developed by Vertex Pharmaceuticals

3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
26,FEB 2013
syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
Vertex Pharmaceuticals announced Tuesday night the design of two phase III studies for its combination therapy to treat the most common form of cystic fibrosis. The studies will each run for six months, so results could be ready as early as the end of 2013 or during first half of 2014.
The studies announced Tuesday will evaluate the two different doses of an experimental medicine VX-809 in combination with Kalydeco. Each study will enroll 500 cystic fibrosis patients randomized to either the VX-809/Kalydeco arms or a placebo for six months of treatment. The studies’ primary endpoint will be the relative improvement in lung function of VX-809/Kalydeco compared to placebo.
Last fall, Vertex presented data from a phase II study demonstrating that a 600 mg dose of VX-809 and Kalydeco worked synergistically to improve lung function in cystic fibrosis patients with the F508del mutation compared to placebo. This same dose combination will be tested in the phase III study along with a higher 800 mg (actually, 400 mg given twice a day) dose of VX-809 plus Kalydeco.
Vertex also announced new data from this phase II study on Tuesday night showing similar lung function improvements between the 800 mg and 600 mg doses of VX-809. For this reason, the higher dose was included in the phase III studies.
Along with the two phase III studies in adult patients, Vertex will also conduct a six-month study of the combination therapy in pediatric patients ages 6 to 11. This study, along with the data from the adult studies, may be used to expand the combination therapy’s approval into younger patients.
In January, FDA anointed Kalydeco and VX-809 with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease. Vertex did not say whether Breakthrough Designation played a specific role in the VX-809/Kalydeco phase III program but the relatively short six-month duration of the studies plus the ability to test the combination in children at the same time does accelerate the development of the combination therapy. If the data from the studies are positive, the drugs could be approved sooner than expected and for more patients.
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis, being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients.[1]
Interim results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis homozygous F508del had an 8.5% increase in lung function (FEV1) after 56 days on a combination of lumacaftor and ivacaftor (Kalydeco).[2]
- Merk; Schubert-Zsilavecz. (in German)Pharmazeutische Zeitung 156 (37): 24–27.
- Vertex Pharmaceuticals. May 29,2012.
- syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
- syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
Phase 3 FDA -Acorafloxacin (Avarofloxacin) Granted QIDP and Fast Track Designation
Avarofloxacin Granted QIDP and Fast Track Designation
JNJ-Q2, JNJ-32729463-AAA
CAS NO 878592-87-1 of base
7-[3-[2-Amino-1(E)-fluoroethylidene]piperidin-1-yl]-1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid

Furiex Pharmaceuticals Inc. announced that the FDA has granted Qualified Infectious Disease Product (QIDP) and Fast Track designation for avarofloxacin (JNJ-Q2). Avarofloxacin is a Phase 3-ready broad-spectrum fluoroquinolone antibiotic for the treatment of acute bacterial skin and skin-structure (ABSSSI) infections, community-acquired pneumonia and has proven to be effective in treating methicillin-resistant Staphylococcus aureus (MRSA) infections.
Avarofloxacin is an investigational novel fluoroquinolone antibiotic that has been shown to be effective in a Phase 2 study of ABSSSI infections. In this study, avarofloxacin demonstrated favorable efficacy for both early clinical response endpoints as well as all clinical cure endpoints for the intent to treat population.

Avarofloxacin has a low tendency for development of drug resistance and exhibits a broad range of antibacterial activities in vitro, including MRSA, fluoroquinolone-resistant Staphylococcus aureus, Streptococcus pneumoniae (including multi-drug resistant strains), gram positive, gram negative, atypical respiratory pathogens (such as legionella and mycoplasma) and anaerobic bacteria, which are often associated with abscesses of the skin and other organs.
The availability of IV and oral formulations for avarofloxacin differentiates it from a number of other products for MRSA infections which are only available for intravenous administration.
For more information call (919) 456-7800or visit http://www.furiex.com/
About Methicillin-Resistant Staphylococcus aureus (MRSA)
MRSA is a strain of the bacteria Staphylococcus aureus (staph) which commonly causes skin and soft tissue infections and is resistant to many antibiotics. Although MRSA had previously been primarily a hospital-acquired pathogen, its incidence has been rising in the community, and it has become the most frequent cause of skin and soft tissue infections presenting to emergency departments in the United States. There are a limited number of antibiotics approved to treat MRSA, and their frequent usage has led to emergence of multi-drug resistant bacteria. Thus, we believe there is significant unmet medical need for new antibiotics such as avarofloxacin that provide flexible (hospital and outpatient) treatment options for MRSA.

WO-2006/101603 describes 7-amino alkylidenyl- heterocyclic quinolones as antimicrobial compounds and the synthesis of 7-[(3E)-3-(2-amino-l-fluoroethylidene)-l- piperidinyl]- 1 -cyclopropyl-6-fluoro- 1 ,4-dihydro-8-methoxy-4-oxo 3-quinolinecarboxylic acid is disclosed as compound (303) in Table 1 on page 20. This compound is conveniently referred to as compound ‘A’ hereafter.
compound ‘A’

7-[(3E)-3-(2-amino-1 -fluoroethylidene)-1 -piperidinyl]-1 -cyclopropyl-6-fluoro- 1 ,4-dihydro-8-methoxy-4-oxo 3-quinolinecarboxylic acid
The in vitro antibacterial properties of compound ‘A’ are described by Morrow B.J. et al. in Antimicrobial Agents and Chemotherapy, vol. 54, pp. 1995 – 1964 (2010).
WO-2008/005670 discloses one-pot methods for the production of substituted allylic alcohols as well as extractive methods for the separation of certain isomeric alcohol products which are useful for preparing quinolones such as the antimicrobial compound 7-[(3E)-3-(2-amino- 1 -fluoroethylidene)- 1 -piperidinyl]- 1 -cyclopropyl-6-fluoro- 1 ,4- dihydro-8-methoxy-4-oxo 3-quinolinecarboxylic acid (i.e. compound ‘Α’). An important intermediate in the overall synthesis route of said antimicrobial compound ‘A’ is 2-[(2E)-2-fluoro-2-(3-piperidinylidene)ethyl]-lH-isoindole-l,3(2H)- dione and its hydrochloric acid salt thereof : compound (1 )

2-[(2E)-2-fluoro-2-(3-piperidinylidene)ethyl]-1 H-isoindole-1 ,3(2H)-dione compound (1 ) .HCI

Compound (1) introduces the desired E- stereochemistry into the overall synthesis route for the antimicrobial compound ‘Α’.
WO-2008/005670 discloses a synthesis route for compound (1) on page 38 as depicted below :

– highly enriched (E)

(Step 3a) O
The detailed reaction procedure for compound (1) is disclosed in WO-2008/005670 in Example 1 on pages 37 to 44 affording compound (1) in Method A with a E:Z ratio of 97:3 in an approximate overall yield of 18 % in Method A (step 1 for the first 3 heptane layers has a yield of 34 % with a ratio E:Z of 71 :29, step 2a has a yield of 53.4% with a ratio E:Z of 97:3, and step 3 has quantitave yield), or affording compound (1) in Method B with an approximate overall yield of 15% with a ratio E:Z of 94.4 : 5.6. WO-2008/005670 discloses a synthesis route for the hydrochloric acid addition salt of compound (1) on page 15 in Scheme 2 as depicted below :

into n-butanol,

1 ) 5/6 N HCl in IPA
2) heat to distill
3) add IPA
enriched E-isomer

compound (1 ) .HCl
The detailed reaction procedure to prepare the HCl salt of compound (1) is disclosed in WO-2008/005670 in Example 4 on pages 49 to 52 affording >95% of desired E-isomer with an overall yield of 18 – 22% starting from N-boc-3-piperidone.
The reaction procedures described in WO-2008/005670 for the preparation of compound (1) or its HCl salt are characterized by lack of selectivity of the Wadsworth- Emmons-Horner reaction which produces the undesired Z-isomer in large quantities. This undesired Z-isomer requires additional time consuming separation steps.
Hence there is a need for a more efficient and less waste-producing procedure for the preparation of compound (1) or its HCl salt. WO-2010/056633 discloses a synthesis scheme XIV on page 87 to prepare tert-butyl 4- (2-ethoxy-2-oxoethylidene)piperidinyl-l-carboxylate and a synthesis scheme XXVI on page 111 to prepare (l-benzyl-piperidin-4-ylidene)bromoacetic acid ethyl ester.
In a first embodiment the present invention relates to an improved process for preparing compounds of formula (III) having an improved ratio of the desired (E)-isomer over the undesired (Z)-isomer.

(I)
In a further embodiment the compound (E)-(III) is then converted in to compound (1) or its hydrochloric acid addition salt thereof.

…………………………..
WO 2008005670
http://www.google.com/patents/WO2008005670A2?cl=en
Scheme 1

3 eq NaBH4 OH
30 – 4O0C

2a 2b 2a

2b shows the preparation of alcohol 2
Scheme 2

1 ) HCI (5 eq )

aqueous layer to enriched E isomer pH 9-10 then extract into n-Butanol, discard aqueous layer
Preparation of
7-[3-(2-Amino-l-fluoroethylidene)piperidin-l-yl]-l-cyclopropyl- 6-fluoro-8-methoxy-4-oxo-l,4-dihydroquinoline-3-carboxylic acid (10) and its HCl salt (12)

7-[3-(2-Amino-1-fluoro-ethyhdene)-piperidin-1-yl]-1-cyclopropyl–fluoro-8-mΘthoxy-4-oxo-1 ,4-dιhydro-quιnolιne-3-carboxylιc acid (10)
Step 1: Preparation of 3-(l-fluoro-2-hydroxyethylidene)piperidine-l-carboxylic acid tert-butyl ester (2a)

A 22-L 4-neck round bottom flask, equipped with a thermocouple controller, overhead mechanical stirrer, condenser, nitrogen inlet adapter, and stopper, was charged with N-Boc-3-piperidone (663.36 g, 3.34 mol), 2-methoxyethanol (6.0 L) and 2-fluorotriethylphosphonoacetate (843.54 g, 3.49 mol). The mixture was stirred to obtain a homogeneous solution and then CS2CO3 was added in portions over 1.5 h. After the CS2CO3 addition was complete, NaBH4 was added in portions over 6 h; during most of this addition the reaction temperature was maintained between 35 0C to 40 0C. After the addition was complete, the reaction was allowed to stir overnight after which time HPLC analysis indicated that the reaction was complete. This run was combined with two additional runs of equal size and transferred to a stirred 100-L Hastalloy® reactor containing water (90 L). The aqueous mixture was extracted with heptane (4 x 20 L) followed by extraction with MTBE (methyl tert-butyl ether) (20 L). The first three heptane extracts provided 842 g of the allylic alcohol as 71:29 (E: Z) mixture (HPLC and NMR). The product mixture from the first three heptane extractions was carried on to the next step without any additional purification. The fourth heptane extract gave 114 g of product that was a 67:33 mixture of is: Z alcohols (NMR). MTBE extraction and concentration gave 1.1 Kg of product as a 33:67 mixture of E:Z alcohols (HPLC). The total overall yield for both isomers was 2.06 Kg (83%). 1H NMR of 2a (400 MHz, CDCl3): £ 1.45 (s, 9 H), 1.52 (m, 2 H), 2.40 (m, 2 H), 3.45 (m, 2 H), 3.90 (s, 2 H), 4.25 (d, 2 H). 1H NMR of 2b (400 MHz, CDCl3): δ 1.46 (s, 9 H), 1.65 (m, 2 H), 2.27 (m, 2 H), 3.45 (m, 2 H), 4.1 (s, 2 H), 4.25 (d, 2 H).
Step 2, Method A: Preparation of 3-is-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester (3-ϋ)

A 22-L 4-neck round bottom flask, equipped with a thermocouple controller, overhead mechanical stirrer, condenser, pressure-equalizing addition funnel, nitrogen inlet adapter, and stopper, was charged with E:Z alcohol mixture 2a and 2b (377.5 g, 1.296 mol corrected), 2-MeTHF (3.31 L), phthalimide (232.8 g, 1.581 mol), and Ph3P (411.3 g, 1.568 mol). The white suspension was stirred under N2 and cooled to -12 0C in an acetone/Dry-Ice bath, DIAD (309 mL, 1.49 mol) was added via the addition funnel over a 36-min period, while the reaction temperature was maintained at -15 0C to -10 0C. After the addition, the reaction was warmed to 20 0C in a water bath and stirred for 2 h. The reaction was cooled to 0 0C in an ice/water bath and quenched with cold 1.0 M HCl (950 mL). The aqueous phase was separated and EtOAc (1.70 L) was added to the organic phase. This phase was washed with cold 1.0 M HCl (0.95 L) (the aqueous phase was pH < 2) and then separated. The organic phase was next washed with cold 4 NNaOH (1.70 L), the alkaline aqueous phase (pH > 13) was separated and the EtOAc layer washed with brine (1.70 L). Concentration of the organic phase at 60 0C under house vacuum (-120 mm Hg) afforded 1,442.0 g of crude 3. This run was repeated on the same scale to provide an additional 1,431.0 g of crude material for a combined yield of 2,873 g (159%). HPLC analysis (area%) indicated crude 3 was a mixture of 3-E (29.4%), 3-Z (10.4 %), Ph3PO (51.0 %), and phthalimide (1.1 %). This was purified by recrystallization as described in step 2a.
Step 2a, Method A: Purification of 3-is-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester

A 22-L 4-neck round bottom flask equipped with a thermocouple controller, overhead mechanical stirrer, condenser, pressure-equalizing addition funnel, nitrogen inlet adapter and stopper was charged with the combined crude 3 (2,873 g) and MeOH (9.0 L). The solution was stirred under nitrogen and heated to 65 0C, while hot (60 0C) D.I. water (7.8 L) was added over a 15-min period. The solution was stirred at 65 0C for 5 min, and then the heating mantle was replaced with a water bath, and the mixture was gradually cooled to 0 0C over a 4-h period, and continued stirring for 1 h at 0 0C. The off-white solid was collected by filtration, and dried by air-suction at 60 0C for 20 h, this provided 1,172.6 g of a mixture of 3-E and 3-Z.
The partially purified product above was recrystallized a second time in the same manner using hot MeOH (7.2 L) and hot water (5.0 L) except that the water was added over a 10-min period to afford 515.6 g (53.4%) of 3-E as a 97:3 mixture of E:Z geometric isomers. This material was used in the next step without additional purification. . 1H NMR of 3-E (400 MHz, CDCl3): δ 1.48 (s, 9 H), 1.52-1.66 (m, 2 H), 2.28-2.38 (m, 2 H), 3.40-3.51 (m, 2 H), 4.18 (s, 2 H), 4.55 (d, J= 21.0 Hz, 2 H), 7.68- 7.77 (m, 2 H), 7.80-7.89 (m, 2 H). MS: 397 (M+Na)+, 771 (2M+Na)+.
3 -E-[2-( 1 ,3 -dioxo- 1 ,3-dihydroisoindol-2-yl)- 1 -fluoroethylidene]-piperidine- 1 – carboxylic acid tert-butyl ester was also prepared with Method B below: Step 2, Method B: Preparation of 3-£-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester (3-E)
Preparation of the methanesulfonate and chloride derivatives

2a
A 12-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with 2a (297.0 g, 1.21 mol) and CH2Cl2 (3.9 L). The solution was cooled to 0 0C under N2 and EtsN (320 mL, 2.30 mol) was added via the addition funnel over a 10- min period. This was followed by methanesulfonyl chloride (115 mL, 1.49 mol) added over a 60-min period then the reaction was stirred for an additional 60-min at 0 0C. The mixture was poured into a mixture of deionized water (4.4 L) and saturated NaHCθ3 (0.78 L), the layers were separated, the aqueous layer was extracted with CH2Cl2 (2 x 2 L). All the CH2Cl2 layers were combined and washed with saturated NaHCθ3 (2 L). The CH2Cl2 was removed under vacuum at 40 0C to afford a mixture of the mesylate and chloride (342.3 g). This mixture was taken on to the next step without any purification.
Conversion of the methanesulfonate/chloride to phthalimide 3

A 5-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with the mixture of the mesylate and chloride from above (342.2 g, 1.21 mol) and DMF (2.0 L) followed by potassium phthalimide (224.9 g, 1.21 mol). The mixture was stirred at 60 0C for 1-h then at 20 0C for 18 h. The mixture was poured into ice- water, allowed to stand for 30-min and filtered. The liquors from the filtration were allowed to stand at 0 0C over the weekend and filtered again. The combined solids were dissolved in acetone (4 L) and concentrated on the rotary evaporator, this process was repeated a second time to give the phthalimide derivative 3 as a mixture oiEIZ (79/31) isomers (263.2 g, 58.1 %).
Step 2a, Method B: Purification of 3-£-[2-(l,3-dioxo-l,3-dihydroisoindol-2-yl)-l- fluoroethylidene]-piperidine-l-carboxylic acid tert-butyl ester

A 12-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with the crude phthalimide derivative 3 (263.1 g) and MeOH (2.74 L). The mixture was heated to 66 – 68 0C while water (2.1 L) was added over 20-min, the mixture was stirred at 68 0C for 5-min, then gradually cooled to 20 0C for 18-h. While the crystallization mixture was cooling it was seeded at 60 0C, 56 0C and 530C. This crystallization gave a white solid that was filtered and dried under vacuum at 50 0C to afford 3-E (118.8 g, 45.2%) as a mixture containing 94.4% E and 5.6% Z isomers (NMR analysis).
Step 3: Preparation of 2-[2-fluoro-2-(3-piperidinylidene)ethyl]-lH-isoindole-l,3)- dione (4)

A 12-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with 3-E (578.0 g, 1.544 mol) and CH2Cl2 (4.5 L). The solution was stirred at 200C under N2 and TFA (476 mL, 6.18 mol) was added via the addition funnel over a 10-min period. The mixture was gently heated to 38 0C and stirred for 3 h. The solvent was removed under vacuum to give the TFA salt of 4 (962.6 g). This material was dissolved in CH2Cl2 (4.0 L) and washed with 2.5 NNa2CO3 (4.6 L)-followed by saturated NaHCO3 (4.6 L). The organic phase was dried (MgSO4), filtered, and condensed in vacuo. The off-white solid was dried at 40 0C under vacuum (20 mm Hg) for 20 h to afford 464.3 g of the free base of 4 as slightly yellowish foamy substance. 1H NMR of 4 TFA salt (400 MHz, CDCl3): δ 1.87-1.98 (m, 2 H), 2.42-2.55 (m, 2 H), 3.38-3.50 (m, 2 H), 4.08-4.18 (br s, 2 H), 4.50 (d, J= 21.0 Hz, 2 H), 7.69-7.78 (m, 2 H), 7.79-7.87 (m, 2 H), 7.98-8.23 (br s, 1 H), 12.48 (s, 1 H). MS: 275 (MH)+, 549 (2M+H)+.
Step 4: Preparation of l-Cyclopropyl-ό^-difluoro-S-methoxy^-oxo-l^- dihydroquinoline-3-carboxylic acid difluoroborate ester (6)

A 22-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure equalizing addition funnel, and a nitrogen inlet adapter was charged with quinoline-3-carboxylic acid 5 (450.0 g, 1.524 mol), THF (5.40 L) and K2CO3 (247.2 g, 1.753 mol). This suspension was first stirred at 20 0C under N2 for 5 min, and BF3 »Et20 (259 mL, 2.04 mol) was added dropwise via the addition funnel to the stirred mixture over a 5-min period. After the addition, the mixture was heated to reflux (66 0C) for 6 h. The reaction was cooled to 10 0C, diluted with Et2O (9.0 L) and stirred for 10 min. The solid was filtered and washed with Et2O (200 mL x 2) and then dried at 50 0C under house vacuum (-160 mm Hg) for 20 h to afford 771.O g of crude difluoroborate ester 6. After this, the crude material was suspended in MeCN (8.0 L) and stirred at 20 0C for 20 min; the solid was collected by filtration. The filter cake was re-suspended and stirred in MeCN four more times (2.0 L x 4), and all filtrates were combined and concentrated at 60 0C under hi-vac (~10 mmHg). The resulting off- white solid was dried at 50 0C under house vacuum (-160 mmHg) for 20 h to afford 508.66 g (97.2% isolated yield, HPLC = 99.2% by area) of pure difluoroborate ester 6. 1H NMR of 6 (400 MHz, CD3CN): «51.17-1.28 (m, 2 H), 1.29-1.40 (m, 2 H), 4.19 (s, 3 H), 4.40-4.52 (m, 1 H), 8.16 (dd, J= 6.9, 7.0 Hz, 1 H), 9.17 (s, 1 H). MS: 344 (MH)+, 667 (2M-F)+.
Step 5: Preparation of intermediate 8


A 5-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure-equalizing addition funnel and a nitrogen inlet adapter was charged with difluoroborate ester 6 (320.0 g, 0.933 mol), DMF (1.10 L) and piperidine 4 (289.0 g, 1.053 mole). This suspension was stirred at 20 0C under N2 for 5 min, EtsN (299 mL, 2.15 mol) was added to the stirred mixture via the addition funnel over an additional 5-min period. After this addition, the mixture was heated to 60 0C and stirred for 3 h, to give crude intermediate 7. HPLC analysis (area%) indicated crude 7 is a mixture of 7 (40.5%), 8 (1.7 %), 6 (24.1%), and the rest of unknowns (33.7%). MS: 598 (MH)+. The coupled crude product 7 was carried on to the next step without isolation.
Removal of the Fluoroborate Ester The above stirred reaction mixture containing 7 was treated in the same flask with EtOH (6.80 L) and Et3N (299 mL, 2.147 mol) under N2 at 60 0C. The amber solution was heated to reflux at 72 0C for 2 h and cooled to 20 0C. The reaction mixture was poured into a rapidly stirred 22-L 4-neck round bottom flask containing a 1 : 1 (v/v) ice-water mixture (8.0 L) over a 10-min period; stirring was continued for -10 min. Cold 1 NHCl (4.0 L) was added to the solution over 20 min to adjust the pH from 9-10 to 3; stirring was continued for an additional 20 min at 0 0C. The yellow solid was isolated by filtration and dried in a filter funnel by air-suction using house vacuum (-160 mm Hg) at 20 0C for 20 h to afford 1,889.0 g of crude 8 as a damp solid (HPLC = 33.6%, area%).
Purification of Intermediate 8
To a 22-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with crude 8 (1889.0 g), MeCN (3.6 L) and EtOH (3.2 L). The suspension was heated to reflux (76 0C), while D.I. H2O (500 mL) was added over 10 min. The solution was stirred at 76 0C for 5 min, and then gradually cooled to 10 0C over 1 h; stirred for an additional hour. The yellow solid was collected by filtration, dried in a vacuum oven under house vacuum (-160 mm Hg) at 60 0C for 20 h to afford 229. Ig (45%) of 8, which was used in next step without further purification. 1H ΝMR of 8 (400 MHz, DMSO-d6): £ 1.02-1.10 (m, 2 H), 1.11-1.19 (m, 2 H), 1.67-1.79 (m, 2 H), 2.34-2.45 (m, 2 H), 3.38-3.49 (m, 2 H), 3.78 (s, 3 H), 4.10 (s, 2 H), 4.15-4.26 (m, 1 H), 4.54 (d, J= 21.0 Hz, 1 H), 7.72 (d, J= 9.1 Hz, 1 H), 7.81 (s, 4 H), 8.71 (s, I H), 14.98 (s, 1 H). MS: 550 (MH)+.
Step 6: Preparation of 7-[3-(2-amino-l-fluoro-ethylidene)-piperidin-l-yl]-l- cyclopropyl-6-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid (10)

8 10 A
22-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure-equalizing addition funnel and a nitrogen inlet adapter was charged with 8 (253.6 g, 0.462 mol) and MeOH (5.10 L). This suspension was stirred at 20 0C under N2 and H2NNH2 (86.9 mL, 2.796 mol) was added over a 5-min period. The yellow suspension was heated to 65 0C and refluxed for 1 h. The reaction was cooled to 60 0C and MeCN (3.84 L) was added. The mixture was heated to reflux for 5 min, and then cooled to 20 0C in a water bath. The light-yellow solid was collected by filtration and the filter cake was washed with MeCN (150 mL x 2). The combined filtrate was concentrated at 60 0C affording 322.0 g of crude product 10. This product was recrystallized from a mixture of MeOH (1.0 L) and water (1.195 L) to give 176.6 g (91.2%) of pure product 10 as a light yellow solid.
BASE FORM
1H NMR of 10 (400 MHz, DMSO- d6): £ 1.0-1.09 (m, 2 H), 1.10-1.19 (m, 2 H), 1.66-1.78 (m, 2 H), 2.30-2.41 (m, 2 H), 3.17 (s, 2 H), 3.35 (s, 1 H), 3.36-3. 47 (m, 2 H), 3.74 (s, 3 H), 3.89 (s, 2 H), 4.13-4.22 (m, 1 H), 5.35-6.18 (br, 2 H), 7.74 (d, J= 8.9 Hz, 1 H), 8.69 (s, 1 H).
MS: 420 (MH)+.
Example 3
Preparation of7-[3-(2-Amino-l-fluoro-ethylidene)-piperidin-l-yl]-l-cyclopropyl-
6-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid hydrogen chloride salt (12)

7-[3-(2-amino-l-fluoro-ethylidene)-piperidin-l-yl]-l-cyclopropyl-6-fluoro-8- methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid (10) was prepared as described in Step 6 of Example 1.
A 5-L 4-neck round bottom flask equipped with an overhead stirrer, thermocouple, condenser, pressure-equalizing addition funnel, and a nitrogen inlet adapter was charged with compound 10 (176.0 g, 0.4196 mol) and EtOH (2.40 L). The suspension was stirred under N2 and cooled to 10 0C with an ice/water bath. A solution of HCl in EtOH (1.25 M, 350 mL) was added via the addition funnel over a 20-min period. After the addition, the reaction was stirred at 10 0C for 5 min. The water bath was replaced with a heating mantle and the solution was heated to 76 0C and stirred for 5 min. The heating mantle was replaced with the water bath, the solution was cooled to 0 0C over 1 h and stirred at this temperature for an additional 1 h. The solid was collected by filtration, washed with ice-cold EtOH (100 mL x 2) and dried at 60 0C under vacuum (~4 mmHg) for 60 h. There was obtained 88.9 g (82%) of HCl salt 12 as an off-white to very light-yellow solid.
HCl SALT
1H NMR of HCl salt 12 (400 MHz, CD3CO2D): £ 1.10-1.19 (m, 2 H), 1.29-1.38 (m, 2 H), 1.81-1.93 (m, 2 H), 2.51-2.60 (m, 2 H), 3.48- 3.60 (m, 2 H), 3.86 (s, 3 H), 4.08 (s, 2 H), 4.18 (s, 1 H), 4.19-4.30 (m, 2 H), 7.92 (d, J= 8.6 Hz, 1 H), 8.98 (s, 1 H) 11.65 (s, 1 H).
MS: 420 (MH)+

………………………………………
http://www.google.com/patents/WO2013045599A1?cl=en
Experiment 6

(VI) compound (1 ) .HCI
Compound (1) .HCI salt from Compound (Via) :
9.8 ml (90.6 mmol) of 1-chloroethyl chloro formate are added slowly to a solution of 30 g (82.3 mmol) of compound (E)-(Va) in 165 ml of toluene kept at 0°C. The reaction mixture is stirred 1 hour at room temperature than 1 hour at 80°C and filtered. 24 ml of ethanol and 15.35 ml (90.6 mmol) of 6M HCI solution in isopropanol are added to the filtrate and the resulting mixture is refluxed for 4 hours then cooled to 0°C. The precipitate is filtered, washed with 16 ml of acetone and 16 ml of toluene and dried under vacuum to give 21.94 g of compound (1) . HCI salt. Yield: 86%>.
NMR and MS data are identical to those of the literature.
-
Avarofloxacin nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/avarofloxacin.pdf
November 28, 2012. N12/130. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN (ZZ-145). AVAROFLOXACIN.
- [PDF]
Avarofloxacin hydrochloride nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/avarofloxacin-hydrochloride.pdf

WO2006101603A1 Feb 2, 2006 Sep 28, 2006 Janssen Pharmaceutica Nv 7-amino alkylidenyl-heterocyclic quinolones and naphthyridones WO2008005670A2 Jun 14, 2007 Jan 10, 2008 Janssen Pharmaceutica Nv One-pot condensation reduction methods for preparing substituted allylic alcohols WO2010056633A2 Nov 10, 2009 May 20, 2010 Janssen Pharmaceutica Nv 7-amino alkylidenyl-heterocyclic quinolones and naphthyridones
Phase3 Ganetespib, a Unique Triazolone-Containing Hsp90 Inhibitor, Exhibits Potent Antitumor Activity and a Superior Safety Profile for Cancer Therapy

Chemical structure of ganetespib and its co-crystal structure with Hsp90 N-terminal.
A, chemical structure of ganetespib.
B, crystallographic complex of ganetespib in the Hsp90 N-terminal.
C, hydrogen bond interactions between ganetespib with amino acid residues in the Hsp90 N-terminal ATP-binding pocket.
Synta Pharmaceuticals has opened a ClinicalTrials.gov listing for their much discussed GALAXY-2 phase 3 trial of their HSP90 inhibitor ganetespib in second-line non-small cell lung cancer (NSCLC), in combination with docetaxel. For the moment at least, the phase 2b GALAXY-1 trial is still listed as enrolling patients, but that would be expected to complete soon.
http://clinicaltrials.gov/ct2/show/NCT01798485
- CAS Number:
- 888216-25-9
-
3H-1,2,4-Triazol-3-one, 5-[2,4-dihydroxy-5-(1-methylethyl)phenyl]-2,4-dihydro-4-(1-methyl-1H-indol-5-yl)-
- Ganetespib
- Molecular Structure:
![Molecular Structure of 888216-25-9 (3H-1,2,4-Triazol-3-one, 5-[2,4-dihydroxy-5-(1-methylethyl)phenyl]-2,4-dihydro-4-(1-methyl-1H-indol-5-yl)-)](https://i0.wp.com/www.lookchem.com/300w/2012-2/e100939d-148c-4f54-9b7c-83632fb0d675.gif)
- Formula: C20H20N4O3
![Molecular Structure of 888216-25-9 (3H-1,2,4-Triazol-3-one, 5-[2,4-dihydroxy-5-(1-methylethyl)phenyl]-2,4-dihydro-4-(1-methyl-1H-indol-5-yl)-)](http://www.lookchem.com/300w/2012-2/e100939d-148c-4f54-9b7c-83632fb0d675.gif)

Hsp90, a chaperone essential for the folding of JAK2, is the target of Synta Pharmaceuticals’ ganetespib.

AstraZeneca ready to file constipation drug naloxegol

4,5α-epoxy-6α-[(3,6,9,12,15,18,21-heptaoxadocosan-1-yl)oxy]-17-(prop-2-en-1-yl)morphinan-3,14-diol
Naloxegol (INN; NKTR-118), or PEGylated naloxol,[1] is a peripherally-selective opioid antagonist under development by AstraZeneca (licensed from Nektar) for the treatment of opioid-induced constipation.[2]
- Roland Seifert; Thomas Wieland; Raimund Mannhold; Hugo Kubinyi, Gerd Folkers (17 July 2006). G Protein-Coupled Receptors as Drug Targets: Analysis of Activation and Constitutive Activity. John Wiley & Sons. p. 227. ISBN 978-3-527-60695-5. Retrieved 14 May 2012.
- “Nektar | R&D Pipeline | Products in Development | CNS/Pain | Oral Naloxegol (NKTR-118) and Oral NKTR-119”. Retrieved 2012-05-14.
phase3
AstraZeneca has presented positive data from a late-stage trial of naloxegol in patients with non-cancer related pain and opioid-induced constipation.
In the fourth trial in a Phase III development programme designed to evaluate long-term safety and adverse event profile , 534 patients received naloxegol once-daily for up to 52 weeks, while 270 were on usual care (laxatives) for OIC. The most commonly-reported AEs occurring more frequently on naloxegol than on usual care included abdominal pain, diarrhoea, nausea and headache but the trial reported no imbalances in serious adverse events.
Briggs Morrison, head of the global medicines development at AstraZeneca, said “these high-level results are similar to the safety results seen in the Phase III studies previously reported and provide further confidence in the data we’ve seen to date for naloxegol”. The programme is now complete and filings in the USA and Europe are planned for the third quarter.
Globally, some 40–50% (28-35 million) of patients taking opioids for long-term pain develop constipation and about 40–50% (11-18 million) of those OIC sufferers achieve the desired outcomes with current options, ie laxatives.
The actual timing of the submissions depend in part on a meeting with the US Food and Drug Administration as naloxegol is currently considered a Schedule II controlled substance across the Atlantic based on its “structural relatedness” to noroxymorphone. AstraZeneca says it has conducted the studies necessary to evaluate the abuse potential and dependence-producing properties of naloxegol and a petition to de-control the drug was submitted in March 2012.
Naloxegol, a peripherally-acting mu-opioid receptor antagonist and formerly known as NKTR-118, was licensed from Nektar Therapeutics in September 2009.
GSK/Isis rare disease drug moves into Phase II/III

feb20,2013
GlaxoSmithKline is paying out $7.5 million to partner Isis Pharmaceuticals as an antisense drug being developed for transthyretin amyloidosis, “a severe and rare genetic disease,” goes into a Phase II/III study.
TTR amyloidosis is characterised by progressive dysfunction of peripheral nerve and/or heart tissues and affects 50,000 patients worldwide and current treatments are limited. The 15-month study of the drug, known as ISIS-TTRRx, will involve some 200 patients with familial amyloid polyneuropathy who experience TTR build-up in their peripheral nerves and experience the loss of motor functions, such as walking.
Lynne Parshall, chief operating officer at Isis, said that “the rapid development of ISIS-TTRRx from a research-stage programme to a drug in late-stage clinical development in just over two years represents the strong commitment of both teams”. She added that the encouraging data from a Phase I study, “in which our drug was well tolerated and produced significant reductions in TTR protein, supported the advancement of ISIS-TTRRx directly into this registration-directed Phase II/III study”.
