
Home » Phase3 drugs (Page 25)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Baricitinib

Baricitinib
NDA submitted jan 2016
2-[1-ethylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile,
3-Azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1Hpyrazol-1-yl]-
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
3-Azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-
2-(3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(ethylsulfonyl)azetidin-3-yl)acetonitrile
For the treatment of rheumatoid arthritis and diabetic kidney disease
Incyte Corporation INNOVATOR
http://www.ama-assn.org/resources/doc/usan/baricitinib.pdf
MF C16H17N7O2S
MW 371.4
SPONSOR Eli Lilly and Company
CODE LY3009104, INCB028050
CAS 1187594-09-7
UPDATE……..APPROVED PMDA 2017
WO 2009114512
2-[3-(4-{7H-pyrrolo[2,3-d]pyrimidin-4-yl}-1H-pyrazol- 1-yl)-1-(ethylsulfonyl)azetidin-3-yl]acetonitrile (baricitinib) FREE FORM
m.p. 193–195 °C;
IR: 3203, 3113, 2998, 2847, 2363, 1584, 1328, 1137 cm–1.
Anal. calcd for C16H17N7 O2 S: C, 51.74; H, 4.61; N, 26.40; found: C, 51.91; H, 4.49; N, 26.57%. MS (m/z): 372 [M + H]+;
1 H NMR (300 MHz, DMSO-d6 ): δ 1.25 (t, J = 7.3 Hz, 3H), 3.23 (m, J = 7.3 Hz, 2H), 3.69 (s, 2H), 4.24 (d, J = 9.0 Hz, 2H), 4.61 (d, J = 9.0 Hz, 2H), 7.08 (s, 1H), 7.62 (s, 1H), 8.47 (s, 1H), 8.71 (s, 1H), 8.92 (s, 1H), 12.12 (s, 1H);
13C NMR (125 MHz, DMSO-d6 ): δ 7.4, 24.9, 39.3, 43.4, 58.5, 99.9, 113.0, 116.6, 126.9, 129.5, 139.9, 149.3, 150.9, 152.2.
REF Journal of Chemical Research, Volume 40, Number 4, April 2016, pp. 205-208(4)

Baricitinib phosphate
{1-(Ethylsulfonyl)-3-[4-(1H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-3-azetidinyl}acetonitrile phosphate (1:1)
Cas 1187595-84-1, C16H20N7O6PS, 469.41
- Originator Incyte Corporation
- Developer Eli Lilly; Incyte Corporation
- Class Acetonitriles; Antipsoriatics; Antirheumatics; Azetidines; Pyrazoles; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors; Janus kinase-2 inhibitors
Highest Development Phases
- Preregistration Rheumatoid arthritis
- Phase II Atopic dermatitis; Diabetic nephropathies; Psoriasis; Systemic lupus erythematosus
Most Recent Events
- 01 Jul 2016 Eli Lilly completes a phase I trial in Healthy volunteers in China (PO) (NCT02758613)
- 09 Jun 2016 Efficacy and adverse events data from the RA-BEYOND phase III trial in Rheumatoid arthritis presented at the 17thAnnual Congress of the European League Against Rheumatism (EULAR-2016)
- 09 Jun 2016 Final efficacy and safety data from the phase III trials, RA-BEAM and RA-BEGIN in Rheumatoid arthritis were presented at the 17th Annual Congress of the European League Against Rheumatism (EULAR – 2016)
The Janus kinase (JAK) is a family of four tyrosine receptor kinases that play a pivotal role in cytokine receptor signalling pathways via their interaction with signal transducers and
activators of transcription proteins. The four JAK family members are Janus kinase 1 (JAK1), Janus kinase 2 (JAK2), Janus kinase 3 (JAK3) and tyrosine kinase (TYK2), whose
lengths range from 120 to 140 kDa. It has been shown that JAK2 activation may be critical for tumour growth and progression,indicating its selection as a therapeutic target. Moreover, since JAK3 is required for immune cell development, targeting JAK3 could be a useful strategy for generating a novel class of immunosuppressant drugs. JAK1 and TYK2 have been implicated in disease and immune suppression.
Over the past decade there have been extensive efforts to identify and design novel small-molecule JAK inhibitors with varied profiles of subtype selectivity to address unmet medical needs Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes JAK1 and JAK2. It was approved by the U.S. Food and Drug Administration (FDA) for the treatment
of intermediate or high-risk myelofibrosis in November 2011.
Selective inhibitors of JAK are viewed as having considerable potential as disease-modifying anti-inflammatory drugs for the treatment of rheumatoid arthritis. Tofacitinib, which was the first oral non-biological disease-modifying antirheumatic drug, was approved for the management of rheumatoid arthritis (RA) at the end of 2012. Baricitinib and filgotinib are beingmevaluated in phase III and phase II clinical trials respectively for
the treatment of rheumatoid arthritis. Baricitinib (also known as LY3009104 or INCB028050) is a novel and potent small molecule inhibitor of the Janus kinase family of enzymes with selectivity for JAK1 and JAK2. In in vitro studies baricitinib inhibited JAK1 and JAK2 in the low nanomolar range, while it demonstrated low inhibitory activity for JAK3 and moderate activity for TYK2.9–13 The data from two phase III studies showed that baricitinib can achieve impressive responses in RA patients who have not responded well to established therapies.
Therefore, improvement in the preparation of baricitinib is of practical significance.
1 L. Tan, K. Akahane, R. McNally, K.M.S.E. Reyskens, S.B. Ficarro, S. Liu,
G.S. Herter-Sprie, S. Koyama, M.J. Pattison, K. Labella, L. Johannessen,
E.A. Akbay, K. Wong, D.A. Frank, J.A. Marto, T.A. Look, J.S.C. Arthur,
M.J. Eck and N.S. Gray, J. Med. Chem., 2015, 58, 6589.
2 J.J. Kulagowski, W. Blair, R.J. Bull, C. Chang, G. Deshmukh, H.J. Dyke,
C. Eigenbrot, N. Ghilardi, P. Gibbons, T.K. Harrison, P.R. Hewitt, M.
Liimatta, C.A. Hurley, A. Johnson, T. Johnson, J.R. Kenny, P.B. Kohli, R.J.
Maxey, R. Mendonca, K. Mortara, J. Murray, R. Narukulla, S. Shia, M.
Steffek, S. Ubhayakar, M. Ultsch, A. Abbema, S.I. Ward, B. Waszkowycz
and M. Zak, J. Med. Chem., 2012, 55, 5901.
3 J.F. Kadow, Y. Ueda, N.A. Meanwell, T.P. Connolly, T. Wang, C. Chen, K.
Yeung, J. Zhu, J.A. Bender, Z. Yang, D. Parker, P. Lin, R.J. Colonno, M.
Mathew, D. Morgan, M. Zheng, C. Chien and D. Grasela, J. Med. Chem.,
2012, 55, 2048.
4 Q. Su, S. Ioannidis, C. Chuaqui, L. Almeida, M. Alimzhanov, G. Bebernitz,
K. Bell, M. Block, T. Howard, S. Huang, D. Huszar, J.A. Read, C.R. Costa,
J. Shi, M. Su, M. Ye and M. Zinda, J. Med. Chem., 2014, 57, 144.
5 J.D. Clark, M.E. Flanagan and J. Telliez, J. Med. Chem., 2014, 57, 5023.
6 P. Norman, Expert. Opin. Investig. Drugs, 2014, 23, 1067.
7 L.J. Farmer, M.W. Ledeboer, T. Hoock, M.J. Arnost, R.S. Bethiel, Y.L.
Bennani, J.J. Black, C.L. Brummel, A. Chakilam, W.A. Dorsch, B. Fan,
J.E. Cochran, S. Halas, E.M. Harrington, J.K. Hogan, D. Howe, H. Huang,
D.H. Jacobs, L.M. Laitinen, S. Liao, S. Mahajan, V. Marone, G. Martinez-
Botella, P. McCarthy, D. Messersmith, M. Namchuk, L. Oh, M.S. Penney,
A.C. Pierce, S.A. Raybuck, A. Rugg, F.G. Salituro, K. Saxena, D. Shannon,
D. Shlyakter, L. Swenson, S. Tian, C. Town, J. Wang, T. Wang, M.W.
Wannamaker, R.J. Winquist and H.J. Zuccola, J. Med. Chem., 2015, 58,
7195.
8 S.C. Meyer and R.L. Levine, Clin. Cancer. Res., 2014, 20, 2051.
Baricitinib (formerly INCB28050, LY3009104)[1] is an oral JAK1 and JAK2 inhibitor.
Baricitinib is in Phase III development by Eli Lilly and Incyte as a potential treatment for rheumatoid arthritis.[2] It is in Phase II development as a potential treatment for psoriasis and diabetic nephropathy. The related compound in JAK inhibitor is Tofacitinib, currently approved for the treatment of rheumatoid arthritis (RA) in the United States.
The companies announced 52-week data from a Phase IIb study of baricitinib at the European Congress of Rheumatology meeting in Madrid which showed that clinical improvements previously observed at week 24 were sustained for the full year in RA patients. Specifically, 49% of patients were ACR50 responders (ie a 50% improvement in their condition) after 52 weeks compared to 41% at week 24. For the full year, 21% reached ACR70 compared with 27% after 24 weeks.
To date, baricitinib, an orally administered selective JAK1 and JAK2 inhibitor, has demonstrated “an acceptable safety profile and side effects have generally been straightforward to manage”, said Oxford University’s Peter Taylor. He added that “these encouraging findings support further investigation of this new drug in RA”.
Baricitinib is already in Phase III for RA and in Phase II for psoriasis and diabetic nephropathy.
WO2009114512, also to Incyte, discloses azetidine and cyclobutane derivatives of the general structure shown below as JAK inhibitors.

Baricitinib (also known as LY3009104 or INCB28050) is in phase II clinical trials for the treatment of rheumatoid arthritis and diabetic kidney disease. Baricitinib is shown below.

About Baricitinib
Baricitinib is a once-daily, oral, selective JAK1 and JAK2 inhibitor. There are four known JAK enzymes: JAK1, JAK2, JAK3 and TYK2. JAK-dependent cytokines have been implicated in the pathogenesis of a number of inflammatory and autoimmune diseases, suggesting that JAK inhibitors may be useful for the treatment of a broad range of inflammatory conditions. Baricitinib demonstrates approximately 100-fold greater potency of inhibition against JAK1 and JAK2 than JAK 3 in kinase assays.
In December 2009, Lilly and Incyte announced an exclusive worldwide license and collaboration agreement for the development and commercialization of baricitinib and certain follow-on compounds for patients with inflammatory and autoimmune diseases. Baricitinib is currently in Phase 3 clinical development for rheumatoid arthritis and Phase 2 development for psoriasis and diabetic nephropathy.
About Rheumatoid Arthritis
Rheumatoid arthritis is an autoimmune diseasei characterized by inflammation and progressive destruction of joints.ii More than 23 million people worldwide suffer from RA.iii Approximately three times as many women as men have the disease. Patients and physicians indicate there remains an important opportunity to improve patient care. Current treatment of RA includes the use of non-steroidal anti-inflammatory drugs, oral disease-modifying anti-rheumatic drugs such as methotrexate, and injectable biological response modifiers that target selected mediators implicated in the pathogenesis of RA.iv
About Baricitinib Phase 3 Trials
Lilly and Incyte have conducted four pivotal Phase 3 clinical trials of baricitinib in patients with moderately-to-severely active rheumatoid arthritis to support regulatory submission in most countries. An additional Phase 3 study was initiated to support clinical development in China and remains ongoing. The clinical trial program includes a wide range of patients including those who are methotrexate naïve, inadequate responders to methotrexate, inadequate responders to conventional disease-modifying anti-rheumatic drugs, or inadequate responders to TNF inhibitors. Patients completing any of the five Phase 3 studies can enroll in a long-term extension study. For additional information on this clinical trial program, please visit http://www.clinicaltrials.gov.
About Incyte
Incyte Corporation is a Wilmington, Delaware-based biopharmaceutical company focused on the discovery, development and commercialization of proprietary therapeutics for oncology and inflammation. For additional information on Incyte, please visit the Company’s web site at http://www.incyte.com.
About Eli Lilly and Company
Lilly is a global healthcare leader that unites caring with discovery to make life better for people around the world. We were founded more than a century ago by a man committed to creating high-quality medicines that meet real needs, and today we remain true to that mission in all our work. Across the globe, Lilly employees work to discover and bring life-changing medicines to those who need them, improve the understanding and management of disease, and give back to communities through philanthropy and volunteerism. To learn more about Lilly, please visit us at http://www.lilly.com and newsroom.lilly.com/social-channels.
SOURCE: Eli Lilly
Biological Activity
Baricitinib (formerly INCB28050, LY3009104) is a selective orally bioavailable JAK1/JAK2 inhibitor. Baricitinib preferentially inhibits JAK1 and JAK2, with 10-fold selectivity over Tyk2 and 100-fold over JAK3. INCB-28050 (baricitinib) inhibits intracellular signaling of multiple proinflammatory cytokines including IL-6 at concentrations <50 nM. Baricitinib also inhibits pSTAT3 stimulated by IL-23 with IC50 of 20 nM in isolated naive T-cells. Baricitinib (INCB028050) was also effective in multiple murine models of arthritis, with no evidence of suppression of humoral immunity or adverse hematologic effects. Baricitinib reduces levels of pSTAT3 in a dose- and time-dependent manner in the peripheral blood of rAIA animals. INCB28050 (Baricitinib) (10 mg/mL, p.o.) improves a composite score of joint damage by 47% in the murine CIA model.
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.


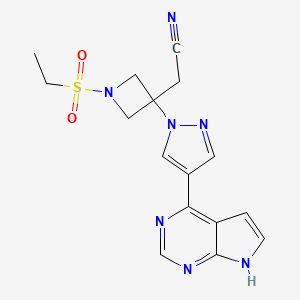
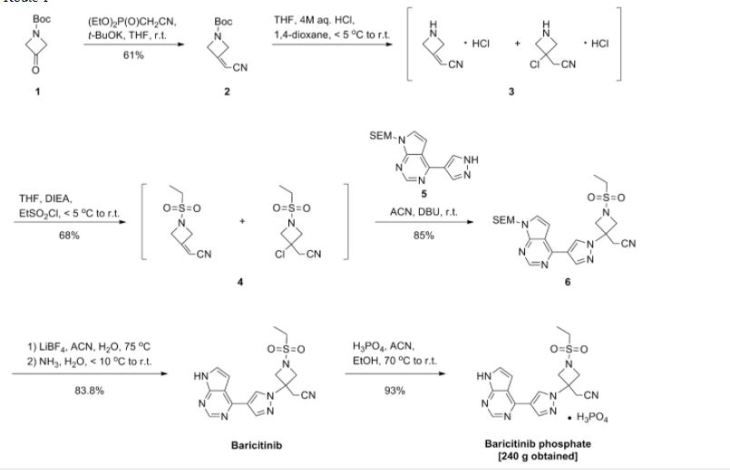
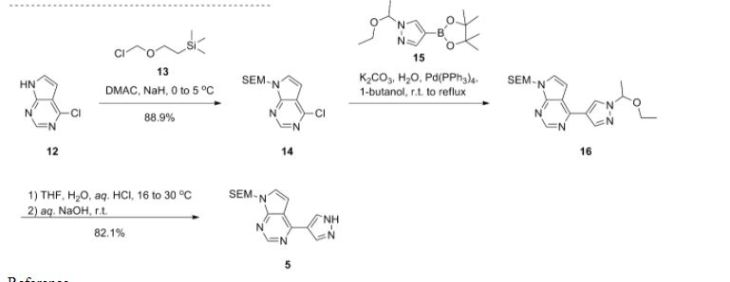
As shown in Scheme 1, Rodgers et al. have reported the first synthetic route to baricitinib.
ref J.D. Rodgers, S. Shepard, T.P. Maduskuie, H. Wang, N. Falahatpisheh, M. Rafalski, A.G. Arvanitis, L. Sorace, R.K. Jalluri, J.S. Fridman and K. Vaddi, 2007, US20070135461.
tert-Butyl 3-oxoazetidine- 1-carboxylate (1) was employed as the starting material. This was transformed to compound 2 by a Horner–Emmons reaction, followed by deprotection of the N-Boc group in acidic conditions. The intermediate 4 was obtained by the sulfonamidation reaction of compound 3 with ethanesulfonyl chloride. The other part of baricitinib was acquired by utilising 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (5) as the starting material. Compound 5 reacted with [2-(chloromethoxy)ethyl] trimethylsilane (SEM-Cl) to afford the intermediate 6, which was converted by reaction with 7 via the intermediate 8 to 4-(1H-pyrazol-4-yl)-7-{[2-(trimethylsilyl)ethoxy]methyl}-7Hpyrrolo[2,3-d]pyrimidine (9) via a Suzuki coupling reaction and a hydrolysis reaction. After the nucleophilic addition reaction and deprotection of the SEM group, baricitinib was obtained through eight steps. This synthetic route had drawbacks of high cost, low overall yield and the requirement of strict operating conditions.
PATENT
WO2009114512
EXAMPLES
Example 1. {l-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l- yl]azetidin-3-yl}acetonitrile trifluoroacetic acid salt

Step 1. tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate
0I \t
To a suspension of sodium hydride (60% dispersion in mineral oil, 0.257 g, 6.42 mmol) in tetrahydrofuran (32 mL) at 0 0C under a nitrogen atmosphere was added diethyl cyanomethylphosphonate (1.19 g, 6.72 mmol) (purchased from Aldrich). The reaction was then stirred for 45 minutes at room temperature. A solution of tert-butyl 3-oxoazetidine-l- carboxylate (1.00 g, 5.84 mmol) (purchased from Alfa Aesar) in tetrahydrofuran (8.8 mL) was introduced dropwise and the mixture was stirred for 16 hours. Brine and ethyl acetate were added and the layers separated. The aqueous layer was extracted with three portions of ethyl acetate. The combined extracts were dried over sodium sulfate, filtered and concentrated to afford product, used without further purification in Step 2 (1.12 g, 99%). 1H NMR (300 MHz, CDCl3): δ 5.38 (p, IH), 4.73-4.68 (m, 2H), 4.64-4.59 (m, 2H), 1.46 (s, 9H).
Step 2. tert-butyl 3-(cyanomethyl)’3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- djpyrim idin-4-yl) – 1 H-pyrazol-1 -yl]azetidine-l -carboxylate

To a solution of 4-(lH-pyrazol-4-yl)-7-[2-(trimethylsilyl)ethoxy]methyl-7H- pyrrolo[2,3-d]pyrimidine (4.61 g, 14.6 mmol) (prepared according to the method of WO 2007/070514 in Example 65, Step 2) and tert-butyl 3-(cyanomethylene)azetidine-l- carboxylate (2.84 g, 14.6 mmol) in acetonitrile (100 mL) was added 1,8- diazabicyclo[5.4.0]undec-7-ene (2.19 mL, 14.6 mmol). The reaction was stirred at room temperature for 16 hours. The acetonitrile was removed in vacuo and the residue was dissolved in ethyl acetate. This solution was sequentially washed with IN HCl and brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography, eluting with 80% ethyl acetate/hexanes to afford desired product (5.36 g, 72%).
1H NMR (300 MHz, CDCl3): δ 8.86 (s, IH), 8.44 (s, IH), 8.34 (s, IH), 7.42 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.54 (d, 2H), 4.29 (d, 2H), 3.59-3.51 (m, 2H), 3.33 (s, 2H), 1.47 (s, 9H), 0.96-0.89 (m, 2H), -0.06 (s, 9H); LCMS (M+H)+: 510.2.
Step 3. 3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH- pyrazol- 1 -yl] azetidin-3-ylacetonitrile

To a solution of tert-butyl 3-(cyanomethyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidine-l-carboxylate (5.36 g, 10.5 mmol) in 1,4-dioxane (100 mL) was added 4.00 M of hydrogen chloride in 1,4-dioxane (40 mL, 160 mmol) and the mixture was stirred at room temperature for 16 hours. The reaction was poured into saturated sodium bicarbonate solution sufficient to neutralize. The product was extracted with three portions of ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, filtered and concentrated to afford product which was used without further purification (3.0 g, 69%). 1H NMR (400 MHz, CDCl3): δ 8.85 (s, IH), 8.42 (s, IH), 8.32 (s, IH), 7.41 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.30 (d, 2H), 3.88 (d, 2H), 3.58-3.51 (m, 2H), 3.42 (s, 2H), 0.96-0.89 (m, 2H), -0.06 (s, 9H); LCMS (M+H)+: 410.2. Step 4. l-(ethylsulfonyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- d]pyritnidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile

To a solution of 3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H-pyrrolo[2,3- d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile (0.100 g, 0.244 mmol) in tetrahydrofuran (2 mL) containing N,N-diisopropylethylamine (0.085 mL, 0.49 mmol) was added ethanesulfonyl chloride (0.023 mL, 0.24 mmol). After stirring for 1.5 hours, the reaction mixture was poured into dilute HCl and extracted with three portions of ethyl acetate. The combined extracts were washed with brine, dried over sodium sulfate, decanted and concentrated to afford product, used without further purification in Step 5 (111 mg, 91%).
1H NMR (300 MHz, CDCl3): δ 8.86 (s, IH), 8.63 (s, IH), 8.35 (s, IH), 7.45 (d, IH), 6.83 (d, IH), 5.68 (s, 2H), 4.63 (d, 2H), 4.26 (d, 2H), 3.54 (t, 2H), 3.42 (s, 2H), 3.09 (q, 2H), 1.41 (t, 3H), 0.92 (t, 2H), -0.06 (s, 9H); LCMS (M+H)+: 502.1.
Step 5. l-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3- ylacetonitrile trifluoroacetate salt
To a solution of l-(ethylsulfonyl)-3-[4-(7-[2-(trimethylsilyl)ethoxy]methyl-7H- pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-ylacetonitrile (0.111 g, 0.22 mmol) in methylene chloride (3 mL) was added trifluoroacetic acid (2 mL) and the solution was stirred for 1.5 hours. The solvents were removed in vacuo and the residue was dissolved in methanol (3 mL) and ethylenediamine (0.1 mL) was added. After stirring for 3 hours, the volume was reduced in vacuo and the product was purified by preparative-HPLC/MS, (SunFire Cl 8 column, eluting with a gradient Of MeCNZH2O containing 0.1% TFA) to afford the product as the trifluoroacetic acid salt (50 mg, 47%). 1H NMR (400 MHz, d6-dmso): δ 12.55 (br d, IH), 9.03 (s, IH), 8.83 (s, IH), 8.56 (s, IH), 7.79-7.75 (m, IH), 7.24-7.19 (m, IH), 4.59 (d, 2H), 4.26 (d, 2H), 3.71 (s, 2H), 3.25 (q, 2H), 1.24 (t, 3H); LCMS (M+H)+: 372.1.
Alternatively, the deprotection and sulfonylation steps could be performed in the reverse order, as in Example 2.
Example 66. tert-Butyl 3-oxoazetidine-l-carboxylate (7).
A solution of tert-buty\ 3-hydroxyazetidine-l-carboxylate (24, 50 g, 289 mmol) in ethyl acetate (400 mL) was cooled to 0 0C. The resulting solution was then treated with solid TEMPO (0.5 g, 3.2 mmol, 0.011 equiv) and a solution of potassium bromide (KBr, 3.9 g, 33.2 mmol, 0.115 equiv) in water (60 mL) at 0 – 5 0C. While keeping the reaction temperature between 0 – 5 0C a solution of saturated aqueous sodium bicarbonate (NaHCO3, 450 mL) and an aqueous sodium hypochlorite solution (NaClO, 10 – 13 % available chlorine, 450 mL) were added. Once the solution of sodium hypochlorite was added, the color of the reaction mixture was changed immediately. When additional amount of sodium hypochlorite solution was added, the color of the reaction mixture was gradually faded. When TLC showed that all of the starting material was consumed, the color of the reaction mixture was no longer changed. The reaction mixture was then diluted with ethyl acetate (EtOAc, 500 mL) and two layers were separated. The organic layer was washed with water (500 rnL) and the saturated aqueous sodium chloride solution (500 mL) and dried over sodium sulfate (Na2SO4). The solvent was then removed under reduced pressure to give the crude product, tert-butyl 3-oxoazetidine-l-carboxylate (7, 48 g, 49.47 g theoretical, 97% yield), which was found to be sufficiently pure and was used directly in the subsequent reaction without further purification. For crude 7: 1H NMR (CDCl3, 300 MHz), δ 4.65 (s, 4H), 1.42 (s, 9H) ppm.
Example 67. tert-Buty\ 3-(cyanomethylene)azetidine-l-carboxylate (9). Diethyl cyanomethyl phosphonate (8, 745 g, 4.20 mol, 1.20 equiv) and anhydrous tetrahydrofuran (THF, 9 L) was added to a four-neck flask equipped with a thermowell, an addition funnel and the nitrogen protection tube at room temperature. The solution was cooled with an ice-methanol bath to -14 0C and a 1.0 M solution of potassium tert-butoxide (^-BuOK) in anhydrous tetrahydrofuran (THF, 3.85 L, 3.85 mol, 1.1 equiv) was added over 20 min while keeping the reaction temperature below -5 0C. The resulting reaction mixture was stirred for 3 h at -10 0C and a solution of l-terf-butoxycarbonyl-3-azetidinone (7, 600 g, 3.50 mol) in anhydrous tetrahydrofuran (THF, 2 L) was added over 2 h while keeping the internal temperature below -5 0C. The reaction mixture was stirred at -5 to -10 0C over 1 h and then slowly warmed up to room temperature and stirred at room temperature for overnight. The reaction mixture was then diluted with water (4.5 L) and saturated aqueous sodium chloride solution (NaCl, 4.5 L) and extracted with ethyl acetate (EtOAc, 2 x 9 L). The combined organic layers were washed with brine (6 L) and dried over anhydrous sodium sulfate (Na2SO4). The organic solvent was removed under reduced pressure and the residue was diluted with dichloromethane (CH2Cl2, 4 L) before being absorbed onto silica gel (Siθ2, 1.5 Kg). The crude product, which was absorbed on silica gel, was purified by flash column chromatography (SiO2, 3.5 Kg, 0 – 25% EtOAc/hexanes gradient elution) to afford tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (9, 414.7 g, 679.8 g theoretical, 61% yield) as white solid. For 9: 1H NMR (CDCl3, 300MHz), δ 5.40 (m, IH), 4.70 (m, 2H), 4.61 (m, 2H), 1.46 (s, 9H) ppm; Ci0H14N2O2 (MW, 194.23), LCMS (EI) mle 217 (M+ + Na).
C8H13NO3 MoI Wt 171 19

2

11 step 3 10
C7H10N2O2S C5H7CIN2 MoI Wt 186 23 MoI Wt 130 58
Example 68. 2-(l-(Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11).
A solution of tert-buty\ 3-(cyanomethylene)azetidine-l-carboxylate (9, 100Og, 5.2 mol) in acetonitrile (7 L) and a 3 N aqueous HCl solution (7 L) was stirred at room temperature for 18 h. When HPLC showed that all the starting material (9) was consumed, the reaction mixture was concentrated under reduced pressure to dryness. The residue, which contains the crude desired deprotection product (10), was then suspended in acetonitrile (12 L) and the resulting suspension was cooled to O – 5 0C. Diisopropyethylamine (DIEA, 3.14 L, 18.03 mol, 3.5 equiv) was then slowly added while keeping the internal temperature below 5 0C. The resulting homogeneous solution was allowed to cool down to O 0C and ethane sulfonyl chloride (EtSO2Cl, 730 mL, 7.73 mol, 1.5 equiv) was added over 1 h while keeping the internal temperature below 5 0C. The resulting reaction mixture was allowed to gradually warm to room temperature and stirred at room temperature for overnight. When HPLC showed that the reaction was complete, the reaction mixture was concentrated under reduced pressure to a volume of approximately 2 L. The bath temperature of the rotary evaporator is set to not exceed 45 0C. The concentrated residue was then diluted with dichloromethane (CH2CI2, 10 L) and the resulting dichloromethane solution was washed with aqueous sodium chloride solution (10 L). The aqueous phase was back extracted with dichloromethane (CH2CI2, 5 L). The combined organic layers were dried over anhydrous sodium sulfate
(Na2SO^ and the residue was absorbed onto silica gel (SiO2, 1 Kg) under reduced pressure. The bath temperature of the rotary evaporator was set to not exceed 45 0C. The material was then loaded onto a silica gel column (SiO2, 2.5 Kg) and eluted with 20 – 60 % ethyl acetate in heptane to afford 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 882 g, 968.4 g theoretical, 91 % yield) as off-white solids. For 11: 1H NMR (CDCl3, 300 MHz) δ 5.46 (m, IH), 4.77 (m, 2H), 4.70 (m, 2H), 3.05 (q, 2H), 1.39 (t, 3H) ppm; C7Hi0N2O2S (MW, 186.23), LCMS (EI) mle 187 (M+ + H).
Example 69. 2-(l-(Ethylsulfonyl)-3-(4-(7-((2-(trimethyIsiIyl)ethoxy)methyl)-7H- pyrrolo[2,3-</]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (12).
Method A. To a suspension of 4-(lH-pyrazol-4-yl)-7-((2-
(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-^pyrimidine (5, 440 g, 1.395 mol) and 2-(l- (ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 312.4 g, 1.68 mol, 1.2 equiv) in acetonitrile (4.4 L) was added DBU (249.8 mL, 1.67 mol, 1.2 equiv) drop wise to keep the reaction temperature between 15 – 25 0C. After adding DBU, the reaction mixture became homogeneous, but a precipitate appeared in 30 min. The reaction mixture was stirred for 3 h at room temperature. When ΗPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (11 L). The resulting mixture was stirred at room temperature for additional 30 min and then filtered. The solid cake was washed with water (4 L), MTBE (2 L) and dried in vacuum oven at 35 0C for 24 h to afford crude 2-(l –
(ethylsulfonyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-(i]pyrimidin-4-yl)- lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (12, 681 g, 699.8 g theoretical, 97.3 % yield) as white solids, which was found to be sufficiently pure for the subsequent reaction without further purification. For 12: 1HNMR (CDCl3, 300 MHz), δ 8.86 (s, IH), 8.45 (s, IH), 8.35 (s, IH), 7.43 (d, IH), 6.80 (d, IH), 5.68 (s, 2H), 4.65 (d, 2H), 4.27 (d, 2H), 3.55 (s, 2H), 3.4 (t, 2H), 3.07 (m, 2H), 1.42 (m, 3H), 0.92 (m, 2H), -0.05 (s, 9H) ppm; C22H3IN7O3SSi (MW, 501.68), LCMS (EI) mle 502 (M+ + H).

12
C15H21N5OSi C22H31N7O3SSi MoI Wt 315 45 MoI Wt 501 68

Phosphate salt
C16H17N7O2S C16H20N7O6PS
MoI Wt 371 42 MoI Wt 469 41
Example 72. tert-Butyl 3-(cyanomethyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrroIo[2,3-rf]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidine-l-carboxyIate (15).
To a suspension of tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (9, 417.2 g, 2.15 mol, 1.05 equiv) and 4-(lH-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrrolo[2,3-<f]pyrimidine (5, 645 g, 2.04 mol) in acetonitrile (4.9 L) was added DBU (30.5 mL, 0.204 mol, 0.1 equiv) drop wise at room temperature. The resulting reaction mixture was then stirred at room temperature for 3 h. After about 1 h, a clear, brown solution was obtained. When LCMS showed that no starting material remained, silica gel (SiO2, 1 Kg) was added and the mixture was concentrated to dryness under reduced pressure. This material, which contains the crude desired product (15), was then loaded onto a pre-packed silica column (Siθ2, 2.5 Kg) and the column was eluted with 60 – 80% of ethyl acetate/heptane. The fractions containing the pure desired product (15) were combined and concentrated under reduced pressure to give the desired product as thick oil which was then stirred in heptane at room temperature until crystallization occurred. The solids were collected by filtration and washed with heptane to afford tert-buty\ 3-(cyanomethyl)-3-(4-(7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-ύ(]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidine- 1-carboxylate (15, 1014.9 g, 1039.7 g theoretical, 97.6% yield) as white solids. For 15: 1H NMR (DMSO-^6, 300 MHz) δ 8.93 (s, IH), 8.77 (s, IH), 8.47 (s, IH), 7.80 (d, IH, J= 3.8 Hz), 7.20 (d, IH, J = 3.7 Hz), 5.63 (s, 2H), 4.50 (d, 2H, J= 9.3 Hz), 4.21 (d, 2H, J= 9.3 Hz), 3.66 (s, 2H), 3.52 (t, 2H, J= 7.8 Hz), 1.40 (s, 9H), 0.82 (t, 2H, J= 8.1 Hz), -0.12 (s, 9H) ppm; C25H35N7O3Si (MW, 509.68), LCMS (EI) m/e 510 (M+ + H) and 532 (M+ + Na).

15
C15H21N5OSi C25H35N7O3Si MoI Wt 31545 MoI Wt 509 68

16 12
C20H27N7OSi C22H31N7O3SSi MoI Wt 409 56 MoI Wt 501 68

14 phosphate
C16H17N7O2S C16H20N7O6PS

MoI Wt 371 42 MoI Wt 46941
Example 77. (4-(l-(3-(Cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)- 7H-pyrrolo[2,3-</]pyrimidin-7-yl)methyl pivalate (20).
To a suspension of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-</|pyrimidin-7-yl]methyl pivalate (19, 10.0 g, 33.4 mmol) and 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (11, 6.22 g, 33.4 mmol, 1.0 equiv) in N,N-dimethylformamide (DMF, 20 mL) was added DBU (254 mg, 1.67 mmol, 0.05 equiv) drop wise to keep the reaction temperature between 15 – 25 0C. After adding DBU, the reaction mixture became homogeneous within 90 min. The reaction mixture was stirred for 3 h at room temperature. When ΗPLC showed that the reaction was deemed complete, the reaction mixture was quenched with water (120 mL) and acetonitrile (80 mL). The resulting mixture was stirred at room temperature for an additional 30 min. The solids were collected by filtration, washed with a mixture of acetonitrile and water (2/3 by volume, 2 x 20 mL), and dried in vacuum oven at 40 – 45 0C for 24 h to afford crude (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H- pyrrolo[2,3-</)pyrimidin-7-yl)methyl pivalate (20, 14.5 g, 16.2 g theoretical, 89.5 % yield) as white solids, which was found to be sufficiently pure (> 98.0% by ΗPLC) for the subsequent reaction without further purification. For 20: 1FTNMR (CDCl3, 300 MHz), δ 8.87 (s, IH), 8.43 (s, IH), 8.37 (s, IH), 7.51 (d, IH, J= 3.6 Hz), 6.76 (d, IH, J= 3.6 Hz), 6.26 (s, 2H),
4.64 (d, 2H, J = 9.6 Hz), 4.25 (d, 2H, J = 9.6 Hz), 3.41 (s, 2H), 3.09 (q, 2H, J= 7.6 Hz), 1.42 (t, 3H, J= 7.6 Hz), 1.17 (s, 9H) ppm; C22H27N7O4S (MW, 485.56), LCMS (EI) mle 486 (M+ + H).

C15H17N5O2 C22H27N7O4S MoI Wl 299 33 MoI Wt 48556

14 phosphate
C16H17N7O2S C16H20N7O6PS MoI Wt 371 42 MoI Wt 46941
PAPER
Authors: Xu, Jiaojiao; Cai, Jin; Chen, Junqing; Zong, Xi; Wu, Xuan; Ji, Min; Wang, Peng
Source: Journal of Chemical Research, Volume 40, Number 4, April 2016, pp. 205-208(4), http://dx.doi.org/10.3184/174751916X14569294811333
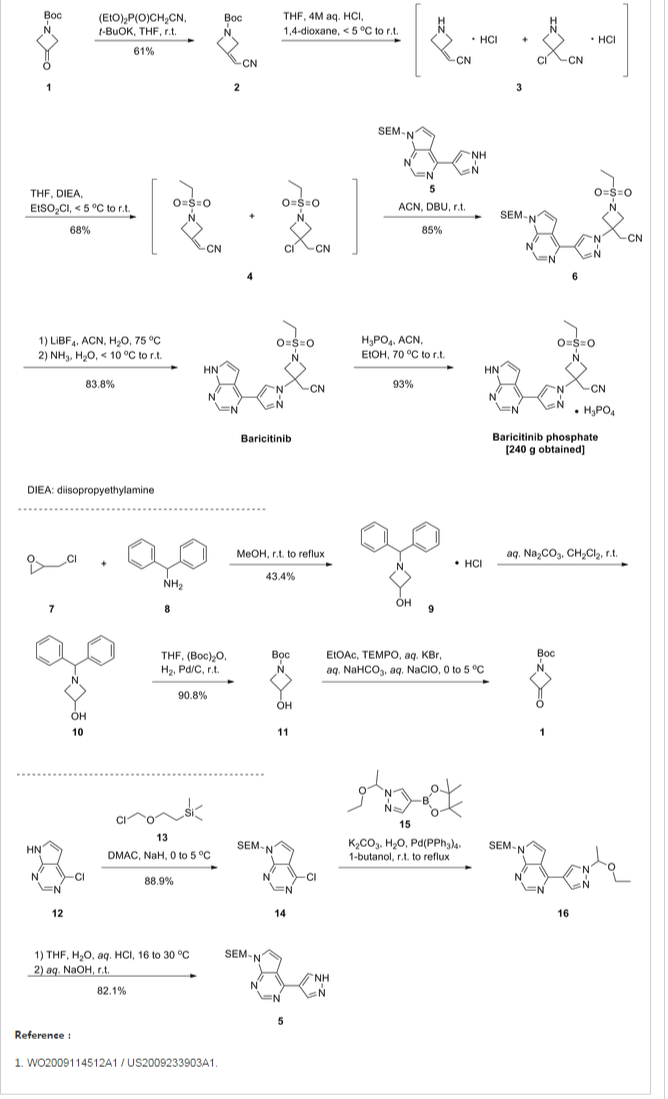
A highly efficient method for the synthesis of baricitinib was developed. The starting material tert-butyl 3-oxoazetidine-1-carboxylate was converted to intermediate 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile via the Horner–Emmons reaction, deprotection of the N-Boc-group and a final sulfonamidation reaction. Then the nucleophilic addition reaction was carried out smoothly to afford the borate intermediate in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene under reflux. Finally, the desired compound baricitinib was obtained by the Suzuki coupling reaction of 4-chloro-7-H-pyrrolo[2,3-d]pyrimidine with the above borate intermediate. All compounds were characterised by IR, MS, 1H NMR and 13C NMR. The overall yield in this synthetic route was as high as 49%. Moreover, this procedure is straightforward to carry out, has low cost and is suitable for industrial production.

In order to improve the procedure, we designed a novel synthetic route for the synthesis of baricitinib (Scheme 2). Similarly, we also applied tert-butyl 3-oxoazetidine-1-carboxylate (1) as the starting material. First, we optimised the preparation of compound 4. In the Horner–Emmons reaction, NaH was used as the base instead of t-BuOK, which led to a yield as high as 84%. Then the deprotection of the N-Boc group was carried out smoothly under trifluoroacetic acid (TFA) cleavage conditions to afford compound 3, which was reacted with ethanesulfonyl chloride without further purification. Next, the nucleophilic addition reaction between compound 4 and 4-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-lH-pyrazole (11) proceeded successfully in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). With the intermediate compound 12 in hand, we optimised the conditions of the Suzuki coupling reaction with compound 5. Several coupling systems were evaluated, such as Pd(PPh3 )4 – K2 CO3 –t-butanol/H2 O, Pd(PPh3 )4 –Na2 CO3 –t-butanol/H2 O and Pd(OAc)2 –K2 CO3 –dioxane/H2 O. The Pd(PPh3 )4 –CsF–t-butanol/ toluene/H2 O system afforded the most satisfactory yield. Finally, baricitinib was obtained efficiently and the overall yield was as high as 49% based on tert-butyl 3-oxoazetidine-1-carboxylate (1)
PATENT
Baricitinib is a Janus kinase (JAK) inhibitor. It is chemically designated as { 1 (ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile, having the structure as depicted in Formula I.

Formula I
U.S. Patent No. 8,158,616 discloses processes for the preparation of baricitinib of Formula I and [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.

Formula II
U.S. Patent No. 8, 158,616 involves a three-step process for the preparation of [4- (lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II as depicted in Scheme 1 below:
Scheme 1

Formula V

Formula VI

Formula II Formula VII
The process disclosed in U.S. Patent No. 8, 158,616 involves the use of sodium hydride as a base for reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III with chloromethyl pivalate of Formula IV, and the use of a protected pyrazole borolane derivative of Formula VI for the conversion of (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7- yl)methyl 2,2-dimethylpropanoate of Formula V into [4-(lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
The use of sodium hydride is not suitable on an industrial scale due to its inflammable and hazardous nature. The use of a protected pyrazole borolane derivative of Formula VI increases the cost of the manufacturing process, as an additional deprotection step is required for obtaining [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
Thus, there exists a need for the development of an economical and industrially advantageous process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II that avoids the use of sodium hydride and involves a lesser number of steps.
The present invention provides a convenient, economical, and industrially advantageous two-step process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II. The process of the present invention involves the use of an alkali or alkaline earth metal hydroxide, carbonate, or bicarbonate as a base for reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III with chloromethyl pivalate of Formula IV, and the use of an unprotected pyrazole borolane of Formula VIII for the conversion of (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate of Formula V into [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II. The process of the present invention provides [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II in high yield.
A first aspect of the present invention provides a process for the preparation of [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II,

Formula II
comprising the steps of:
i) reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III

Formula III
with chloromethyl pivalate of Formula IV

Formula IV
in the presence of an alkali or alkaline earth metal hydroxide, carbonate, bicarbonate as a base to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7- yl)methyl 2,2-dimethylpropanoate of Formula V; and

ii) reacting the (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2- dimethylpropanoate of Formula V with 4-(4,4,5,5-tetramethyl-l,3,2 dioxaborolan-2-yl)-lH-pyrazole of Formula VIII

Formula VIII
to obtain the [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II.
A second aspect of the present invention provides a process for the preparation of baricitinib of Formula I,

Formula I
comprising the steps of:
i) reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine of Formula III

Formula III
with chloromethyl pivalate of Formula IV

Formula IV
in the presence of an alkali or alkaline earth metal hydroxide, carbonate, or bicarbonate base to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate of Formula V;

Formula V
ii) reacting the (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2- dimethylpropanoate of Formula V with 4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazole of Formula VIII

Formula VIII
to obtain [4-( lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II; and

Formula II
iii) reacting the [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate of Formula II with [l-(ethylsulfonyl)azetidin-3-ylidene]acetonitrile of Formula IX

Formula IX
to obtain baricitinib of Formula I.
EXAMPLES
Example 1 : Preparation of (4-chloro-7H-pyrrolor2.3-dlpyrimidin-7-yl)methyl 2.2-dimethylpropanoate (Formula V)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (25 g; Formula III), potassium carbonate (27 g), and chloromethyl pivalate (27 g; Formula IV) were added to a reaction vessel containing N,N-dimethylformamide (100 mL) at ambient temperature. The reaction mixture was stirred for 14 hours. The progress of the reaction was monitored by thin layer chromatography. Water (250 mL) was added to the reaction mixture, and then the mixture was stirred for 2 hours. The reaction mixture was filtered, then washed with water (50 mL), and then dried under reduced pressure at 40°C to 45°C for 12 hours to obtain (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate.
Yield: 98.85%
Example 2: Preparation of r4-(lH-pyrazol-4-yl)-7H-pyrrolor2.3-dlpyrimidin-7-yllmethyl pivalate (Formula II)
(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methyl 2,2-dimethylpropanoate (10 g; Formula V), water (50 mL), and potassium carbonate (15.5 g) were added into a reaction vessel at ambient temperature. 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (8.7 g; Formula VIII), 1,4-dioxane (100 mL), and
tetrakis(triphenylphosphine)palladium(0) (0.08 g) were added to the reaction mixture. The reaction mixture was heated to a temperature of 80°C to 85°C, and then stirred at the same temperature for 14 hours. The progress of the reaction was monitored by thin layer chromatography. On completion, ethyl acetate (100 mL) was added to the reaction mixture. The contents were stirred for 1 hour, then filtered through a Hyflo®, and then washed with ethyl acetate (40 mL). The organic layer was separated, and then concentrated under reduced pressure to obtain [4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl]methyl pivalate.
Yield: 82.27%
PATENT
PATENT
https://www.google.com/patents/CN105566332A?cl=en
PATENT
The present invention provides processes for the preparation of baricitinib of Formula I and an intermediate of Formula V. The present invention also provides the of the intermediate of Formula V for the preparation of baricitinib.

Formula V
Background of the Invention
Baricitinib is a Janus kinase (JAK) inhibitor. It is chemically designated as (ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-lH-pyrazol-l-yl]azetidin-3 yl}acetonitrile, having the structure as depicted in Formula I.

Formula I
U.S. Patent No. 8,158,616 discloses a process for the preparation of baricitinib comprising the reaction of 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile of Formula II with 4-(lH-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-7-yl methyl pivalate of Formula
III to provide an intermediate of Formula IV, followed by deprotection of the intermediate of Formula IV to obtain baricitinib of Formula I, as depicted in Scheme I below:
Scheme I

Formula IV
The process disclosed in U.S. Patent No. 8, 158,616 requires a deprotection step in the last stage of the synthesis, which adds to the cost of the overall synthesis.
Thus, there exists a need for an alternate, cost-effective, and industrially advantageous process for the preparation of baricitinib.
EXAMPLES
Example 1 : Preparation of 3-(cvanomethylene)azetidine hydrochloride (Formula VIP
Aqueous hydrochloric acid (6N, 10 mL) and montmorillonite K-10 (2 g) were added into a reaction vessel at ambient temperature. The contents were stirred for 1 hour, and then filtered under reduced pressure to obtain activated montmorillonite K-10. The activated montmorillonite K-10 was added into another reaction vessel containing tert-butyl 3-(cyanomethylidene)azetidine-l-carboxylate (2 g; Formula VI) and methanol (20
mL) at ambient temperature. The reaction mixture was refluxed for about 12 hours to about 15 hours. On completion, the reaction mixture was filtered under reduced pressure followed by recovery of methanol under reduced pressure at about 40°C to about 45°C to obtain 3-(cyanomethylene)azetidine hydrochloride.
Yield: 75%
Example 2: Preparation of 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile (Formula ID
N,N-Diisopropylethylamine (4.5 mL) was added into a reaction vessel containing acetonitrile (50 mL) and 3-(cyanomethylene)azetidine hydrochloride (1.5 g; Formula VII) at about 0°C to about 10°C. The reaction mixture was stirred for about 10 minutes.
Ethanesulfonyl chloride (2.22 g) was added into the reaction mixture at about 0°C to about 5°C over about 5 minutes. The temperature of the reaction mixture was raised to about 20°C to about 25 °C, and then the reaction mixture was stirred for about 16 hours. On completion of the reaction, acetonitrile was recovered from the reaction mixture under reduced pressure at about 40°C to about 45°C to obtain an oily residue. Dichloromethane (50 mL) was added into the residue. The contents were washed with a saturated sodium chloride solution (30 mL), followed by complete recovery of dichloromethane under reduced pressure at about 40°C to obtain 2-(l-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile .
Yield: 98.59%
Example 3: Preparation of { l-(ethylsulfonyl)-3-[4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-yl)-lH-pyrazol-l-yllazetidin-3-yl}acetonitrile (Formula V)
1,4-Dioxane (20 mL) was added into a reaction vessel containing a solution of potassium carbonate (4.5 g) in water (30 mL) at about 20°C to about 25 °C. 2-(l-(Ethylsulfonyl)azetidin-3-ylidene)acetonitrile (2 g; Formula II) and 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (2.30 g; Formula VIII) were added into the reaction mixture at about 20°C to about 25 °C. The reaction mixture was stirred at about 20°C to about 25 °C for about 16 hours to about 18 hours. On completion of the reaction, 1,4-dioxane was recovered from the reaction mixture under reduced pressure at about 45 °C to obtain a residue. Ethyl acetate (20 mL) was added into the residue, and the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45 °C to obtain { l-(ethylsulfonyl)- 3-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile.
Yield: 85.78%
Mass: 381.4 [M + H]+
Example 4: Preparation of baricitinib (Formula I)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine (0.8 g; Formula IX) was added into a reaction vessel containing a solution of potassium carbonate (2.1 g) in water (30 mL) at about 20°C to about 25°C. A solution of { l-(ethylsulfonyl)-3-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile (2.0 g; Formula V) in 1,4-dioxane (30 mL) was added into the reaction mixture at about 20°C to about 25 °C, followed by the addition of tetrakis(triphenylphosphine)palladium(0) (0.1 g). The reaction mixture was stirred at about 80°C to about 85°C for about 5 hours. On completion of the reaction, 1,4-dioxane was recovered from the reaction mixture under reduced pressure at about 45°C to obtain a residue. Ethyl acetate (50 mL) was added into the residue, and then the contents were stirred for about 5 minutes. The organic and aqueous layers were separated. The organic layer was concentrated under reduced pressure at about 45°C to obtain baricitinib.
Yield: 99.0%
Patent
PATENT
Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
Example: Preparation of crystalline form of baricitinib
(4-( 1 -(3-(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3-yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate (8 g), methanol (40 mL), tetrahydrofuran (160 mL), and 1M sodium hydroxide (18.4 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched with water (80 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid. Half of the solvent was recovered at a temperature of 40°C to 50°C. The reaction mixture was stirred at 20°C to 25°C for 18 hours, and then cooled to 5°C to 10°C. The solids were filtered, washed with a mixture of acetonitrile (50 mL) and water (100 mL), and then dried at 40°C to 50°C under reduced pressure for 24 hours to obtain the crystalline form of baricitinib.
Yield: 70%
PATENT
Figure 1 : X-ray Powder Diffraction (XRPD) pattern of the crystalline form of baricitinib. BELOW

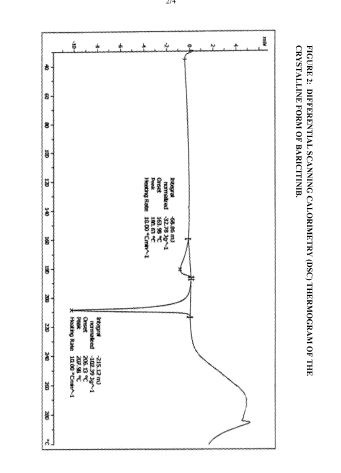
Figure 2: Differential Scanning Calorimetry (DSC) thermogram of the crystalline form of baricitinib.
Figure 3 : Thermogravimetric Analysis (TGA) of the crystalline form of baricitinib.
Figure 4: Infra-red (IR) spectrum of the crystalline form of baricitinib.
The crystalline form of baricitinib is further characterized by a DSC having endotherms at about 180.63°C and about 207.98°C.
The crystalline form of baricitinib has a water content of about 3%, as determined by TGA.
The crystalline form of baricitinib is also characterized by an XRPD pattern as depicted in Figure 1, a DSC thermogram as depicted in Figure 2, a TGA as depicted in Figure 3, and an IR spectrum as depicted in Figure 4.
The preparation of the crystalline form of baricitinib is carried out by reacting (4-(l-(3-(cyanomethyl)-l-(ethylsulfonyl)azetidin-3-yl)-lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate with a base in the presence of one or more solvents at a temperature of about 15°C to 50°C, stirring the reaction mixture for about 30 minutes to about 10 hours, partially recovering the solvent(s) from the reaction mixture at a temperature of about 35°C to about 60°C under reduced pressure, stirring the contents at about 15°C to 35°C for about 5 hours to about 24 hours, filtering the solid, washing the solid with a mixture of acetonitrile and water, and drying.
The (4-( 1 -(3 -(cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate may be obtained by following the process disclosed in U.S. Patent No. 8, 158,616.
Example: Preparation of crystalline form of baricitinib
(4-( 1 -(3-(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3-yl)- lH-pyrazol-4-yl)-7H-pyrrolo [2,3-d]pyrimidin-7-yl)methyl pivalate (8 g), methanol (40 mL), tetrahydrofuran (160 mL), and 1M sodium hydroxide (18.4 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched with water (80 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid. Half of the solvent was recovered at a temperature of 40°C to 50°C. The reaction mixture was stirred at 20°C to 25°C for 18 hours, and then cooled to 5°C to 10°C. The solids were filtered, washed with a mixture of acetonitrile (50 mL) and water (100 mL), and then dried at 40°C to 50°C under reduced pressure for 24 hours to obtain the crystalline form of baricitinib.
Yield: 70%
PATENT
https://www.google.com/patents/WO2015145286A1?cl=en
EXAMPLES
Comparative Examples
Example 1 : Repetition of the process according to Example 78. Method B of U.S. Patent No. 8.158.616
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1 g), methanol (5 mL), tetrahydrofuran (20 mL), and 1M sodium hydroxide (2.3 mL) were added into a reaction vessel at 20°C to 25 °C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched by adding water (20 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid, and the contents were stirred for 1.5 hours. No solid material was obtained. Example 2: Repetition of the process according to Example 78. Method C of U.S. Patent No. 8.158.616
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (2 g), lithium hydroxide monohydrate (0.51 g), acetonitrile (8 mL), and 2-propanol (2 mL) were added into a reaction vessel at 20°C to 25°C. The reaction mixture was stirred at 45°C to 50°C for 6 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was cooled to 20°C to 25°C. The pH was adjusted to 6.0 to 7.0 by adding IN hydrochloric acid, and the contents were stirred overnight. No solid material was obtained.
Working Example:
Preparation of an amorphous form of baricitinib
4-( 1 -(3 -(Cyanomethyl)- 1 -(ethylsulfonyl)azetidin-3 -yl)- lH-pyrazol-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-7-yl)methyl pivalate (1 g), methanol (5 mL), tetrahydrofuran (20 mL), and 1M sodium hydroxide (2.3 mL) were added into a reaction vessel at 20°C to 25 °C. The reaction mixture was stirred for 3 hours. Progress of the reaction was monitored by thin layer chromatography. On completion, the reaction mixture was quenched by adding water (20 mL). The pH was adjusted to 7.0 to 7.5 by adding IN hydrochloric acid, followed by completely recovering the solvent under reduced pressure at 40°C to 50°C. A sticky material was obtained. Water (10 mL) was added to the sticky material at 20°C to 25°C. The contents were stirred for 10 minutes. A solid material was precipitated out. The solid material was filtered, washed with water (20 mL), and then dried under reduced pressure at 40°C to 45°C for 24 hours to obtain the amorphous form of baricitinib.
Yield: 81%.
The amorphous form of baricitinib may be used in a pharmaceutical composition with one or more pharmaceutically acceptable carriers, diluents, or excipients, and optionally other therapeutic ingredients. The pharmaceutical composition may be used for the treatment of JAK-associated diseases.
PATENT
CN 105693731
https://www.google.com/patents/CN105693731A?cl=en
Baricitinib crystal form A and preparation method
Crystalline forms of 1 ethylsulfonyl 3 4 7H pyrrolo 2 3 d pyrimidin 4 yl …
priorart.ip.com/IPCOM/000244270
Nov 27, 2015 – Crystalline forms of baricitinib were found and are described … on their appearance temperature As follows the polymorph observed at room …
IR (KBr): 3203, 3116, 2256, 1583, 1328, 1138 cm-1.
(d, J=3.2 Hz, 1H), 7.10 (d, J=3.4 Hz, 1H), 4.62 (d, J=9.0 Hz, 2H), 4.26 (d, J=9.1 Hz,
2H), 3.72 (s, 2H), 3.26 (q, J=7.3 Hz, 2H), 1.26 (t, J=7.3 Hz, 3H) ppm.
116.86, 113.25, 100.14, 58.74, 56.26, 43.50, 27.03, 7.63 ppm.
7.10-100.14, 4.62-58.74, 4.26-58.74, 3.72-27.03, 3.26-43.50, 1.26-7.63.
56.26), 8.73-(152.39, 149.55, 113.25), 8.50-(129.80, 122.42), 7.64-(152.39, 113.25,
100.14), 7.10-(152.39, 127.13, 113.25), (4.62, 4.26)-(58.74, 56.26, 27.03), 3.72-
(116.86, 58.74, 56.26), 3.26-7.63, 1.26-43.50.
C 51.74%; H 4.61%; N 26.40%; S 8.63%.
Found C 51.62%; H 4.59%; N 26.28%; S 8.78%.
References
- “Baricitinib” (pdf). Statement on a nonproprietary name adopted by the USAN council. American Medical Association.
- “Lilly, Incyte Treatment Shows Positive Results”. http://www.insideindianabusiness.com. 9 Dec 2014. Retrieved 2 Mar 2015.
| Patent | Submitted | Granted |
|---|---|---|
| AZETIDINE AND CYCLOBUTANE DERIVATIVES AS JAK INHIBITORS [US8158616] | 2009-09-17 | 2012-04-17 |
| AZETIDINE AND CYCLOBUTANE DERIVATIVES AS JAK INHIBITORS [US2013225556] | 2013-03-29 | 2013-08-29 |
| JANUS KINASE INHIBITORS FOR TREATMENT OF DRY EYE AND OTHER EYE RELATED DISEASES [US2010113416] | 2010-05-06 | |
| METHOD OF TREATING MUSCULAR DEGRADATION [US2013310340] | 2013-05-15 | 2013-11-21 |
| METHOD OF SELECTING THERAPEUTIC INDICATIONS [US2014170157] | 2012-06-15 | 2014-06-19 |
| CYCLODEXTRIN-BASED POLYMERS FOR THERAPEUTIC DELIVERY [US2014357557] | 2014-05-30 | 2014-12-04 |
| Azetidine and cyclobutane derivatives as JAK inhibitors [US8420629] | 2011-12-09 | 2013-04-16 |
| BIOMARKERS AND COMBINATION THERAPIES USING ONCOLYTIC VIRUS AND IMMUNOMODULATION [US2014377221] | 2013-01-25 | 2014-12-25 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010039939A1 * | Oct 1, 2009 | Apr 8, 2010 | Incyte Corporation | Janus kinase inhibitors for treatment of dry eye and other eye related diseases |
| WO2011028685A1 | Aug 31, 2010 | Mar 10, 2011 | Incyte Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| WO2011103423A1 * | Feb 18, 2011 | Aug 25, 2011 | Incyte Corporation | Cyclobutane and methylcyclobutane derivatives as janus kinase inhibitors |
| WO2012068450A1 * | Nov 18, 2011 | May 24, 2012 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as jak inhibitors |
| WO2012177606A1 | Jun 19, 2012 | Dec 27, 2012 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as jak inhibitors |
| WO2013026025A1 | Aug 17, 2012 | Feb 21, 2013 | Incyte Corporation | Cyclohexyl azetidine derivatives as jak inhibitors |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2014028756A1 * | Aug 15, 2013 | Feb 20, 2014 | Concert Pharmaceuticals, Inc. | Deuterated baricitinib |
| WO2014138168A1 * | Mar 5, 2014 | Sep 12, 2014 | Incyte Corporation | Processes and intermediates for making a jak inhibitor |
| WO2015095492A1 | Dec 18, 2014 | Jun 25, 2015 | Incyte Corporation | Tricyclic heterocycles as bet protein inhibitors |
| WO2015123424A1 | Feb 12, 2015 | Aug 20, 2015 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| WO2015131031A1 | Feb 27, 2015 | Sep 3, 2015 | Incyte Corporation | Jak1 inhibitors for the treatment of myelodysplastic syndromes |
| CN102844317B * | Feb 18, 2011 | Jun 3, 2015 | 因西特公司 | Cyclobutane and methylcyclobutane derivatives as janus kinase inhibitors |
| CN103415515B * | Nov 18, 2011 | Aug 26, 2015 | 因塞特公司 | 作为jak抑制剂的环丁基取代的吡咯并吡啶和吡咯并嘧啶衍生物 |
| CN103797010A * | Jun 19, 2012 | May 14, 2014 | 因塞特公司 | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| CN104024256A * | Sep 6, 2012 | Sep 3, 2014 | 因塞特公司 | Processes and intermediates for making a JAK inhibitor |
| EP2647629A1 * | Dec 1, 2011 | Oct 9, 2013 | Nissan Chemical Industries, Ltd. | Pyrazole compound having therapeutic effect on multiple myeloma |
| EP2647629A4 * | Dec 1, 2011 | Apr 23, 2014 | Nissan Chemical Ind Ltd | Pyrazole compound having therapeutic effect on multiple myeloma |
| US8415362 | Jun 12, 2008 | Apr 9, 2013 | Incyte Corporation | Pyrazolyl substituted pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8722693 | Dec 5, 2013 | May 13, 2014 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8822481 | Apr 18, 2014 | Sep 2, 2014 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8829013 | Apr 18, 2014 | Sep 9, 2014 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8933085 | Nov 18, 2011 | Jan 13, 2015 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US8933086 | Sep 20, 2013 | Jan 13, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B]pyridines and pyrrolo[2,3-B]pyrimidines as Janus kinase inhibitors |
| US8946245 | Mar 30, 2011 | Feb 3, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8987443 | Mar 5, 2014 | Mar 24, 2015 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9023840 | Feb 21, 2014 | May 5, 2015 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9034884 | Nov 18, 2011 | May 19, 2015 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US9079912 | May 12, 2014 | Jul 14, 2015 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase inhibitors |
| US9193733 | May 17, 2013 | Nov 24, 2015 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9206187 | Sep 6, 2013 | Dec 8, 2015 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase |
| US9216984 | Nov 8, 2013 | Dec 22, 2015 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9221845 | Mar 11, 2015 | Dec 29, 2015 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |

PHOSHATE SEE……http://www.medchemexpress.com/product_pdf/HY-15315A/Baricitinib%20phosphate-NMR-HY-15315A-08874-2013.pdf
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-[1-ethylsulfonyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1187594-09-7 |
| ATC code | None |
| PubChem | CID: 44205240 |
| ChemSpider | 26373084 |
| ChEMBL | CHEMBL2105759 |
| PDB ligand ID | 3JW (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C16H17N7O2S |
| Molecular mass | 371.42 g/mol |
SEE………http://apisynthesisint.blogspot.in/2016/01/baricitinib.html
//////////LY3009104, INCB028050, LY 3009104, INCB 028050, nda, baricitinib
CCS(=O)(=O)N1CC(C1)(CC#N)N2C=C(C=N2)C3=C4C=CNC4=NC=N3
Review of literature
Almost all the synthetic methods (WO2009114512A1, CN201510880931.X, CN201610080433.1, WO2016088094A1, WO2016125080A2, WO2016205487A1, CN201610903498.1, WO2017109524A1, CN201710181322.4, CN201710165830.3) reported for the preparation of baricitinib employed important intermediates 2-(1-(ethylsulfonyl)azetidin-3-ylidene)acetonitrile(2) and tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate(3), for which the development of a green and facile synthetic method for intermediates 2 and 3 has a strong demand. However, several reported research-scale synthetic methods for the preparation of intermediates 2 and 3
1
In Scheme 1, compounds 2-(chloromethyl)oxirane (I-1) and diphenylmethanamine (I-2) were used as the starting material (WO2009114512A1). Intermediate 2 was obtained through reduction reaction, boc-protecting reaction, oxidizing reaction, and wittig reaction, which was then employed to afford intermediate 3 by deprotect and hinsber reactions
Synthesis of intermediate 2 and 3 using 2-(chloromethyl)oxirane (I-1) and diphenylmethanamine (I-2) as starting material
2
In Scheme 2, compound azetidin-3-ol hydrochloride (II-1) was used as start material, which was employed to afford intermediate 3 through hinsber reaction, oxidizing reaction, and wittig reaction (WO2016205487A1) Besides, another patent reported that the start material 1-amino-3-chloropropan-2-ol hydrochloride (III-1) was first reacted with ethanesulfonyl chloride to afford compound N-(3-chloro-2-hydroxypropyl)ethanesulfonamide (III-2), which was then converted to the same intermediate 1-(ethylsulfonyl)azetidin-3-ol (III-3, II-2) after cyclization
Synthesis of intermediate 3 with II-1 as starting material
3
Key intermediate 3 was obtained by the same method as that of Scheme 2 (Scheme 3, CN201710165830.3)
Synthesis of intermediate 3 with III-1 as starting material
4
In Scheme 4, compound azetidin-3-one hydrochloride (IV-1) was used as raw start material, which was converted to intermediate 3 through hinsber reaction and aldol condensation reaction (CN201610903498.1).
Synthesis of intermediate 3 with IV-1 as starting material
van Vollenhoven R, Helt C, Arora V, Zhong J, Correia AP, de la Torre I, Muram D (2018) Safety and efficacy of baricitinib in patients receiving conventional synthetic disease-modifying antirheumatic drugs or corticosteroids. Rheumatol Ther 5(2):525–536
https://www.mdpi.com/1424-8247/12/1/37/htm
Baricitinib (2, Figure 2) is the active ingredient of Olumiant®, commercialized by Eli Lilly and Co. Its IUPAC name is: 2-[1-(ethanesulfonyl)-3-(4-{7H-pyrrolo[2,3-d]pyrimidin-4-yl}-1H-pyrazol-1-yl)azetidin-3-yl] acetonitrile, CAS 1187594-09-7.
After a rejection in April 2017, baricitinib (2 mg tablets) has been approved on May 31, 2018 for treatment of rheumatoid arthritis. [3] Noticeably, it had been approved, for the same purpose, in the European Union (EU) in February 2017. [4]
There are essentially two routes for the preparation of baricitinib 2. As depicted in Scheme 1, they can be distinguished by introducing central pyrazole ring in the molecule. In the original procedure [20,21], the pyrazole ring was linked to the pyrrolo[2,3-d]pyrimidine system (to afford 6) and then coupled to the azetidine moiety 7 to give the intermediate 8. In an alternative route [22,23], the bound between the pyrazole and the azetidine was formed (to yield 10) before reaction with the fused system 9.
Scheme 2. Preparation of baricitinib 2 from a 4-pyrazolyl-7H-pyrrolo[2,3-d]pyrimidine
Thus (Scheme 2), 4-chloro-7H– pyrrolo[2,3-d]pyrimidine was protected on position 7 by reaction with 2-(trimethylsilyl)ethoxymethyl chloride. The protected fused system was then coupled with 4-pyrazoleboronic acid pinacol ester 12 by a Suzuli-Miyaura reaction, giving 6. Parallelly, 7 was obtained from 1-Boc-3-azetidinone 13 and diethyl cyanomethylphosphonate. Reaction between 6 and 7 in the presence of DBU afforded the ester 8. Subsequent hydrolysis, decarboxylation, sulfonation, and finally deprotection of the pyrrolopyrimidine moiety yielded the targeted derivative 2. In a variant [21], also used to prepare deuterated samples of 2 [24], the azetidine derivative 7 has been deprotected and sulfonated before coupling with 6.
Scheme 3. Preparation of baricitinib 2 from a 4-chloro-7H-pyrrolo[2,3-d]pyrimidine.
In a more recent patent [22], the sulfonated azetidine 14 (Scheme 3) was prepared from azetidine-3-ol by a sequence including a sulfonation, an oxidation, and introduction of the cyanomethylene moiety. Interestingly, there is no need to protect any position in that sequence. Additionally, let us emphasize that the oxidation step could be performed both in batch or under flow conditions. [22,25]. Then, 14 was reacted with 4-pyrazoleboronic acid pinacol ester 12 to yield 10. The bound between the azetidinylpyrazole group and the pyrrolo[2,3-d]pyrimidine system was then created through a Suzuki-Miyaura reaction involving 7-Boc-4-chloro-7H-pyrrolo[2,3-d]pyrimidine 9 or even the unprotected 4-chloro-7H-pyrrolo[2,3-d]pyrimidine.
- 20 Rodgers, J.; Shepard, S.; Maduskuie, T.; Wang, H.; Falahatpisheh, N.; Rafalski, M.; Arvanitis, A.; Storace, L.; Jalluri, R.; Fridman, J.; et al. Heteroaryl Substituted Pyrrolo[2,3-b]pyridines and Pyrrolo[2,3-b]pyrimidines as Janus Kinase Inhibitors. US20070135461A1, 14 June 2007.
- 21 Rodgers, J.D.; Shepard, S.; Li, Y.-L.; Zhou, J.; Liu, P.; Meloni, D.; Xia, M. Azetidine and Cyclobutane Derivatives as JAK Inhibitors. U.S. Patent US20090233903A1, 17 September 2009.
- 22 Kobierski, M.E.; Kopach, M.E.; Martinelli, J.R.; Varie, D.L.; Wilson, T.M. Processes and Intermediates for the Preparation of {1(Ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1yl]azetidin-3-yl}acetonitrile. WO 2016/ 205487, 22 December 2016]
- 23 Xu, J.; Cai, J.; Chen, J.; Zong, X.; Wu, X.; Ji, M.; Wang, P. An Efficient Synthesis of Baricitinib. Chem. Res.2016, 40, 205–208.
- 24 Tung, R.D. Deuterated Baricitinib. U.S. Patent US20180221374A1, 9 August 201
- 25 Hughes, D.L. Applications of Flow Chemistry in Drug Development: Highlights of Recent Patent Literature. Process Res. Dev.2018, 22, 13–20

Reference:1. WO2009114512A1 / US2009233903A1.



: J. Med. Chem. 2019, 62, 7340−7382
Baricitinib (Olumiant). Baricitinib is an inhibitor of Janus family tyrosine kinase (JAK)-1 and -2 approved by the USFDA in 2017 as monotherapy or in combination with methotrexate for the treatment of adults with moderate to severe active rheumatoid arthritis.83 Baricitinib was discovered by Incyte and codeveloped with Eli Lilly. It is also in clinical trials for the treatment of atopic dermatitis, systemic lupus erythematosus, and giant cell arteritis. Numerous synthetic routes to baricitinib have been reported in the patent literature.28 The largest scale synthesis was reported by Incyte and is described in Schemes 35−37. 28a Horner−Emmons reaction between tert-butyl 3-oxoazetidine1-carboxylate (156) and diethyl cyanomethyl phosphonate (157) gave cyanomethylene azetidine 158 in 61% yield (Scheme 35). Acidic removal of the Boc protecting group was followed by treatment with ethanesulfonyl chloride to give the sulfonamide subunit of baricitinib 159 in 91% yield. The synthesis of baricitinib was completed as described in Scheme 36. Deprotonation of chloropyrrolopyrimidine 160 with sodium hydride followed by treatment with trimethylsilylethoxymethyl chloride gave the SEM protected chloropyrrolopyrimidine 161 in 89% yield. Suzuki coupling with commercially available boronic ester 162 followed by acidic removal of the ethoxyethyl protecting group gave pyrazole 163 in 87% yield. 1,4-Addition of pyrazole 163 to cyanomethylazetidine 159 was accomplished in the presence of DBU to give SEM-protected baricitinib 164 in high yield. Finally, treatment of 164 with lithium tetrafluoroborate followed by ammonium hydroxide gave baricitinib (XVII) in 81% yield. An alternative synthesis of baricitinib that avoids the use of the ethoxylethyl and SEM protecting groups and changes the order of the 1,4- addition and Suzuki coupling steps has also been reported on large scale and is described in Scheme 37. 28b,84
(28) (a) Rodgers, J. D.; Shepard, S.; Li, Y.-L.; Zhou, J.; Liu, P.; Meloni, D.; Xia, M. Preparation of Azetidine and Cyclobutane Derivatives as Jak Inhibitors. WO 2009114512, 2009. (b) Kobierski, M. E.; Kopach, M. E.; Martinelli, J. R.; Varie, D. L.; Wilson, T. M. Processes and Intermediates for the Preparation of {1-(Ethylsulfonyl)- 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3- yl}acetonitrile. WO 2016205487, 2016.
(84) Xu, J.; Cai, J.; Chen, J.; Zong, X.; Wu, X.; Ji, M.; Wang, P. An Efficient Synthesis of Baricitinib. J. Chem. Res. 2016, 40, 205−208.
///////////////





Formulations f.c. tablet 2 mg, 4 mg (as phosphate) References Jiaojiao, X. et al., Journal of Chem. Research, (2016) 40 (4), 205. a WO 2009 114512 (Incyte; 17.09.2009; US-prior. 11.03.2008). US 8 158 616 (Incyte; 17.04.2012; US-prior. 11.03.2008). b WO 2016 205487 (Eli Lilly & Co.; 22.12.2016; US-prior. 19.06.2015). Preparation of I Qiyan, L. et al., Organic Letters, (2009) 11(9), 1999-2002
Ledipasvir (formerly GS-5885), Treatment of chronic Hepatitis C infection

Ledipasvir (formerly GS-5885), Treatment of chronic Hepatitis C infection
Ledipasvir nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/ledipasvir.pdf
November 28, 2012. N12/139. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN ZZ-132. LEDIPASVIR.
MOLECULAR FORMULA C49H54F2N8O6
MOLECULAR WEIGHT 889
Gilead Sciences
CODE DESIGNATION GS-5885
CAS REGISTRY NUMBER 1256388-51-8
Ledipasvir (formerly GS-5885) is an experimental drug for the treatment of hepatitis C being developed by Gilead Sciences.[1] It is currently in Phase III clinical trials.[2] It is being studied in combination with other direct-acting antiviral agents that interfere with HCV replication.
Ledipasvir is an inhibitor of the hepatitis C virus HCV NS5A protein.
Ledipasvir is being tested in interferon-free regimens with other direct-acting antiviral agents for hepatitis C.
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of the HCV protease inhibitor sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[3][4] Gilead is developing a sofosbuvir + ledipasvir coformulation that is being tested with and without ribavirin.
- “Ledipasvir”. United States Adopted Name.
- “GS-5885”. Gilead Sciences.
- ELECTRON: 100% Suppression of Viral Load through 4 Weeks’ Post-treatment for Sofosbuvir + Ledipasvir (GS-5885) + Ribavirin for 12 Weeks in Treatment-naïve and -experienced Hepatitis C Virus GT 1 Patients. Gane, Edward et al. 20th Conference on Retroviruses and Opportunistic Infections. March 3–6, 2013. Abstract 41LB.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, Liz. HIVandHepatitis.com. 4 March 2013.
Cempra Provides Guidance on the Clinical Program Required for Regulatory Approval for Solithromycin for Community-Acquired Bacterial Pneumonia (CABP)

solithromycin
(3aS,4R,7S,9R,10R,11R,13R,15R,15aR)-1-[4-[4-(3-aminophenyl)-1H-1,2,3-triazol-1-yl]butyl]-4-ethyl-7-fluorooctahydro-11-methoxy-3a,7,9,11,13,15-hexamethyl-10-{[3,4,6-trideoxy-3-(dimethylamino)-β-D–xylo-hexopyranosyl]oxy}-2H-Oxacyclotetradecino[4,3-d]oxazole-2,6,8,14(1H,7H,9H)-tetrone
| Legal status | Phase III clinical trials, North America, South America, Europe |
|---|---|
| Routes | oral, intravenous |
| Identifiers | |
| CAS number | 760981-83-7 |
Cempra Provides Guidance on the Clinical Program Required for Regulatory …
The Herald | HeraldOnline.com
The Phase 3 solithromycin clinical program in CABP will be planned to consist of an oral trial and an intravenous (IV)-to-oral clinical trial. Cempra followed the CABP guidance that the FDA proposed in a November, 2011, meeting of the Anti-Infective …
READ ALL AT
http://www.heraldonline.com/2013/06/13/4944834/cempra-provides-guidance-on-the.html
Solithromycin (formerly known as CEM-101 and OP-1068) is a novel ketolide antibiotic undergoing clinical development for the treatment of community-acquired pneumonia (CAP) and other infections.It is expected to be the first macrolide antibiotic available in intravenous, oral, and pediatric suspension formulations in over 20 years.
Solithromycin exhibits excellent in vitro activity against a broad spectrum of Gram-positive respiratory tract pathogens, including macrolide-resistant strains. Solithromycin has activity against a wide variety of pathogens, and further research is being conducted for other infections.
- May 2011: Solithromycin is in a Phase 2 clinical trial for serious community-acquired bacterial pneumonia (CABP) and in a Phase 1 clinical trial with an intravenous formulation.
- September 2011 : Encouraging results from the phase 2 clinical trial versus levofloxacin were reported.
Cadila banks on diabetes drug, Lipaglyn, Saroglitazar

(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
- saroglitazar
- ZYH1 compound
-
- E0YMX3S4JD
- cas no 495399-09-2
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |


 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn — to treat “diabetic dyslipidemia”.
Diabetic dyslipidemia is a condition where a person is diabetic and has elevated levels of total cholesterol. Over 80 per cent of diabetic patients are dyslipidemic.
http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Zydus Cadila said it is looking for partnership to market its new chemical entity (NCE) Lipaglyn, to be used for treating a type of diabetes in developed and developing markets. “Lipaglyn is the first glitazar to be approved in the world and the first NCE discovered and developed indigenously by an Indian pharma company.
The new drug is expected to be launched in Q3 of this fiscal in the country,” Zydus Cadila Chairman and Manging Director Pankaj Patel told reporters.
The company has spent USD 250 million in developing Lipaglyn and aims to spend another USD 150-200 million to launch the drug in overseas markets in next 3-5 years period, Patel said, adding that the company is looking for marketing partnerships.
“We expect this to be a blockbuster drug, which means over USD 1 billion sales a year, when the drug is sold globally, he said. The market for this drug is estimated at Rs 100 crore in the local market over the next three years and having market potential size of over USD 30 billion in the world market, he said.
Zydus Cadila took about eight years to develop the molecule and conducted clinical trials on more than 1,000 patients in India, Patel said, adding that the company is yet to finalise the price, but believes that it will be reasonably priced in the local market.
The company said that the Indian drug regulator Drug Controller General of India (DCGI) has approved Lipaglyn to be used for treating ‘diabetic dyslipidemia’.
| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid | |
| Clinical data | |
| Trade names | Lipaglyn |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 495399-09-2 |
| ATC code | None |
| PubChem | CID 60151560 |
| ChemSpider | 32079086 |
| Chemical data | |
| Formula | C25H29NO4S |
| Mol. mass | 439.56 g/mol |
MORE DETAILS
Saroglitazar (INN, trade name Lipaglyn) is a drug for the treatment of type 2 diabetes mellitus and dyslipidemia. It is approved for use in India by the Drug Controller General of India.[1] Saroglitazar is indicated for the treatment of diabetic dyslipidemia andhypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy. In clinical studies, saroglitazar has demonstrated reduction of triglycerides (TG), LDL cholesterol, VLDL cholesterol, non-HDL cholesterol and an increase in HDL cholesterol a characteristic hallmark of atherogenic diabetic dyslipidemia (ADD). It has also shown favorable glycemic control by reducing the fasting plasma glucose and HBA1c in diabetes patients.
Zydus-Cadila has developed and launched saroglitazar (ZYH-1; Lipaglyn; structure shown), a lipid metabolism modulator, a potent PPAR-alpha agonist with relatively weak PPAR-gamma activity, an insulin sensitizer (glucose-lowering agent), for the once-daily oral treatment of metabolic disorders, including diabetic dyslipidemia and hypertriglyceridemia
In June 2013, the Drug Controller General of India (DCGI) approved the drug for launch in India ; in September 2013, the drug was launched . The company is also developing the drug for the potential treatment of lipodystrophy. In May 2014, a phase III trial was initiated . In June 2012, the company was seeking to outlicense the drug for regional/global partnerships
By June 2012, an NDA filing had been made for dyslipidemia. In June 2013, the DCGI approved the drug for launch in India . By September 2013, the drug was launched for dyslipidemia and hypertriglyceridemia .
Mechanism of action
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]
_MoA.png)
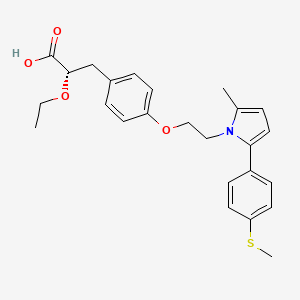
Clinical trials
The approval for saroglitazar was based on the results obtained from clinical studies, which were conducted for more than eight years.
The studies evaluated the efficacy, safety, pharmacokinetics and pharmacodynamics of the drug. Phase I clinical trials on saroglitazar were conducted in 2005. The highest dose of saroglitazar evaluated in a Phase I trial was 128 mg, several times the estimated therapeutic doses (1–4 mg). The pharmacokinetics of saroglitazar support a once daily dosage schedule. No serious adverse events were reported.[3] Phase II studies were completed in 2006.
The Phase III clinical trials were conducted between 2008 and 2011. The first Phase III clinical trials on saroglitazar compared saroglitazar 4 mg dose with pioglitazone 45 mg. The results of the study demonstrated that patients who were administered with saroglitazar 4 mg dose showed reduction in LDL cholesterol and triglycerides, and increase in HDL cholesterol. The study also showed that saroglitazar administered patients showed a reduction in fasting plasma glucose and glycosylated hemoglobin.
Saroglitazar 2 mg and 4 mg significantly reduced (P < 0.001) plasma triglycerides from baseline by 26.4% (absolute change ± SD: −78.2 ± 81.98 mg/dL) and 45% (absolute change ± SD −115.4 ± 68.11 mg/dL), respectively, as compared to pioglitazone -15.5% (absolute change ± SD: −33.3 ± 162.41 mg/dL) at week 24. Saroglitazar 4 mg treatment also demonstrated marked decrease in low-density lipoprotein (5%), very-low-density lipoprotein (45.5%), total cholesterol (7.7%), and apolipoprotein-B (10.9%).[4]
The second Phase III clinical trials on saroglitazar were conducted to evaluate the diabetic dyslipidemic patients insufficiently controlled with statin therapy. The second Phase III study results showed that patients treated with saroglitazar showed pronounced beneficial effect on both the lipid and glycaemic parameters.
At Week 12, saroglitazar 2-mg and 4-mg tablets significantly reduced mean plasma triglyceride levels by -45.5±3.03% and -46.7±3.02% (mean±SE), respectively, and the difference was significant (P<0.001) compared with placebo. Saroglitazar 2 mg demonstrated significant decrease in levels of non-HDL-C, very LDL-C, total cholesterol, and fasting plasma glucose. Additionally, saroglitazar 4 mg also significantly reduced LDL-C and apolipoprotein B levels. Saroglitazar was found to be safe and well tolerated by patients.[5]
Safety
Saroglitazar was found to be safe and well tolerated during the clinical program. In Phase III trials, There was no edema or weight gain reported in any of the study arms. During this study, subjects were monitored for cardiac events, ECG abnormalities, and cardiac function by 2-D ECHO at the start of the study, at the end of 12 weeks, and at 24 weeks after the last dose of the study drug. There were no adverse events reported as far as cardiac safety is concerned.
After 12 weeks of treatment, there were a no significant changes in hemoglobin, liver enzymes (alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, andγ-glutamyl transferase), renal function (creatinine, enhanced glomerular filtration rate, and blood urea nitrogen), CPK, and high-sensitivity C-reactive protein in the saroglitazar and placebo arms.[6][7]
In Phase I clinical trials saroglitazar was used up to 128 mg and found well tolerated. No serious adverse events were reported. Adverse events were generally mild and moderate in nature and did not show any clinically relevant findings in clinical laboratory investigations, physical examinations, vital signs and electrocardiograms.[8]
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
http://www.sciencedirect.com/science/article/pii/S0040403914006200

PATENT
WO 2003009841
http://www.google.co.in/patents/WO2003009841A1?cl=en
PATENT
US 20030236254
http://www.google.com/patents/US20030236254
PATENT
US 20140099333
http://www.google.com/patents/US20140099333
PATENT
http://patentscope.wipo.int/search/en/WO2014174524
 (I)
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
PATENT
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.

Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.

wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1

In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)

wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2

Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).

Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3

Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
PATENT
saroglitazar magnesium alongwith its intermediates may be prepared by the reaction scheme- 1, scheme-2 and scheme-3 as shown below, which is also the scope of the present invention.

Scheme-1

EXAMPLES
Example-l:
Preparation of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, sodium methoxide (165 g) and toluene (1000.0 ml) were added under nitrogen environment and cooled to 8°C to 12°C. Methyl acetoacetate (331.55 g) was added dropwise and stirred for 1 hour. 2-bromo-l-(4-methyl sulfonyl phenyl) ethanone (500.0 g) compound (El) in toluene (1500.0 ml) and sodium sulfate
(75.0 g) mixture was stirred for 10 min and filtered at 25° to 35°C. The filtrate as obtained was added dropwise into the previous reaction mixture and stirred at 30°C to 35°C for 30 min. The organic layer was collected and washed with 10% sodium bicarbonate solution. The separated organic layer was collected and washed with water. 2-[2-(4-Methyl sulfanyl-phenyl)-2-oxo-ethyl]-3-oxo-butynic acid methyl ester as obtained in toluene layer is diluted with methanol (2500 ml) and sodium hydroxide solution (89.75 g) in water (2500 ml) was added and heated to 50° to 55°C for 1 hour. The layers were separated and the toluene layer was collected and heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (57.3 g) and ethanol amine (143.9 g) were added and heated to 105° to 1 15°C for removing water azeotropically. The toluene layer was separated and triethyl amine (271.85 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (282.5 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum to obtain the residue. The residue was dissolved in toluene (1500 mL) and used for further process.
ExampIe-2:
Preparation of methanesulfonic acid 2-f2-methyl-5-(4-methylsulfanyl-pheny0-pyrrol- 1-viyethyl ester (Al)

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 4-(methylthio)benzaldehyde (10 g), methyl vinyl ketone (3.63 g), triethylamine (9.95 g) and 3-methyl-5-(2-hydroxyethyl)-4-methyI thiazolium iodide (stetter
catalyst) (2.8 g) were heated to 70°C to 80°C and maintained overnight. The reaction mixture was cooled to room temperature and ethanol (100 mL) was added. The reaction mixture was stirred for 30 min and filtered. The product was washed with ethanol and dried to obtain 1 ,4-diketo compound (CI).
1 ,4-diketo compound (CI) obtained above and toluene (50 mL) were heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (5.7 g) and ethanol amine (14.4 g) were added and heated to 105° to 1 15°C and cooled to 25°C. Triethyl amine (27.2 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (28.3 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum, methanol (2500 ml) was added and heated to 55° to 65 °C and charcoalized for 30 min. The reaction mixture was filtered and washed with methanol. The reaction mixture was cooled to 25° to 35°C and stirred for 30 min. Reaction mass was further cooled to -5° to 5°C and filtered. The wet-cake was washed with methanol and dried to obtain compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.2).
Example-3:
Purification of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 70 g methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l -yl]-ethyl ester (Al) and 420 mL ethyl acetate were added at 25°C. The reaction mixture was stirred for 30 min to obtain clear solution. 3.5 g charcoal was added and stirred for 30 min. The reaction mixture was filtered and washed with ethyl acetate. The filtrate was concentrated and 315 mL methanol was added. The reaction mixture was stirred for 2 hours at 25°C and cooled to 0°C. The product precipitated was filtered and washed with methanol to obtain crystalline
compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.3).
Example-4:
Preparation of saroglitazar magnesium (T)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) (100.0 g) and toluene (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. Toluene solution of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol- 1 -yl]-ethyl ester (Al) (150.24 g) obtained in example- 1, 18-Crown-6 (5.0 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour, The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with toluene (200.0 ml) and stirred for 15 min. The organic, layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with HP-120 (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was added sodium hydroxide 20.14 g solution in water (200.0 ml) and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for 1 hour. The aqueous layer was extracted with methylene dichloride (2000 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove toluene completely. The residue was diluted with toluene (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
The reaction of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al) and 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) may also be performed in similar manner as above in absence of phase transfer catalyst 18-Crown-6.
ExampIe-5:
Preparation of saroglitazar (S)-(-)-phenyl ethylamine salt:

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, residue-A obtained in example- 1 and ethanol (400 mL) were stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with isopropyl acetate (400 mL). The separated aqueous layer was diluted with isopropyl acetate (500 mL) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with (S)-(-)-phenyl ethylamine (55.94 g) and stirred for 2 hours at 25°C and 30 min at 45°C. The reaction mixture was cooled to 0°C and stirred for 2 hours, filtered and washed with isopropyl acetate. The wet-cake was dried to obtain saroglitazar phenyl ethylamine salt.
ExampIe-6:
Preparation of saroglitazar magnesium from saroglitazar (SH-)-phenyl ethylamine salt:
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, saroglitazar phenyl ethylamine wet-cake obtained in example-7 and isopropyl acetate (800 mL) were added at 25°C. The reaction mixture was diluted with water (400.0 ml) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with sodium hydroxide solution (20.14 g) in water (200 mL) and stirred for 30 min. The separated aqueous layer was treated with magnesium acetate tetrahydrate (2.29 g) in water (5 mL) solution and stirred for 60 min. The reaction mixture was extracted with methylene dichloride (800 mL). The methylene dichloride was complete removed by distillation under vacuum below 40°C to obtain the residue. The residue was diluted with methylene dichloride (50 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
References
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
- “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
-
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- 7 “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- 8 “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
by WORLD DRUG TRACKER
DR ANTHONY
do not miss out on updates
see my update at https://newdrugapprovals.org/2015/03/09/saroglitazar-magnesium-new-patent-wo-2015029066-cadila-healthcare-ltd/ 9 may 2015
////////////
CCO[C@@H](Cc1ccc(cc1)OCCn2c(ccc2c3ccc(cc3)SC)C)C(=O)O
Novartis Japan Achieves Primary Endpoint In HER2 Positive Advanced Breast Cancer Phase III Afinitor Trials

Everolimus
Novartis announced it achieved its primary endpoint of significantly extending progression-free survival with Afinitor (everolimus) in Phase III trials of patients with HER2 positive advanced breast cancer.
read all at
Everolimus (RAD-001) is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors for use in a number of cancers.
It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
GSK’s Votrient meets primary objective in Phase III ovarian cancer trial

pazopanib
GlaxoSmithKline’s (GSK) Votrient (pazopanib) has met the primary objective of a statistically significant improvement in the time to disease progression or death that is the progression-free survival (PFS) against placebo in Phase III ovarian cancer..
Pazopanib (trade name Votrient) is a potent and selective multi-targeted receptortyrosine kinase inhibitor of VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-a/β, and c-kit that blocks tumor growth and inhibits angiogenesis. It has been approved for renal cell carcinoma and soft tissue sarcoma by the U.S. Food and Drug Administration.Pazopanib may also be active in ovarian cancer Pazopanib also appears effective in the treatment of non-small cell lung carcinoma.
Merck Provides Update on Phase III Clinical Program for Preladenant, the Company’s Investigational Parkinson’s Disease Medicine

preladenant
Merck , known as MSD outside the United States and Canada, today provided an update on the clinical program for preladenant, Merck’s investigational adenosine A2A receptor antagonist for the treatment of Parkinson’s disease (PD). An initial review of data from three separate Phase III trials did not provide evidence of efficacy for preladenant compared with placebo…..read more at

Preladenant (SCH 420814) was a drug that was developed by Schering-Plough which acted as a potent and selective antagonist at the adenosine A2A receptor. It was being researched as a potential treatment for Parkinson’s disease.Positive results were reported in Phase II clinical trials in humans, but it did not prove itself to be more effective than a placebo during Phase III trials, and so was discontinued in May 2013
Ferring Presents Phase III Data for a Controlled Release Misoprostol Vaginal Delivery System for Labour Induction at the First European Congress on Intrapartum Care
MISOPROSTOL
READ AT
Misoprostol is a synthetic prostaglandin E1 (PGE1) analog that is used for the prevention of nonsteroidal anti-inflammatory drug (NSAID) induced gastric ulcers, to treat missed miscarriage, to induce labor, and as an abortifacient. The latter use is controversial in theUnited States. Misoprostol was invented and marketed by G.D. Searle & Company (nowPfizer) under the trade name Cytotec, but other brand-name and generic formulations are now available as well.
Misoprostol is approved for use in the prevention of NSAID induced gastric ulcers. It acts upon gastric parietal cells, inhibiting the secretion of gastric acid via G-protein coupled receptor mediated inhibition of adenylate cyclase, which leads to decreased intracellularcyclic AMP levels and decreased proton pump activity at the apical surface of the parietal cell. Because
Novo Nordisk Says Will Seek Approval of Obesity Drug Liraglutide Next Year

liraglutide
read all at
http://www.pharmalive.com/novo-nordisk-says-will-seek-approval-of-obesity-drug-next-year
Systematic (IUPAC) name
L-histidyl-L-alanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-valyl-L-seryl-L-seryl-L-tyrosyl-L-leucyl-L-α-glutamylglycyl-L-glutaminyl-L-alanyl-L-alanyl-N6-[N-(1-oxohexadecyl)-L-γ-glutamyl]-L-lysyl-L-α-glutamyl-L-phenylalanyl-L-isoleucyl-L-alanyl-L-tryptophyl-L-leucyl-L-valyl-L-arginylglycyl-L-arginyl-glycine
Liraglutide (NN2211), marketed under the brand name Victoza, is a long-acting glucagon-like peptide-1 agonist (GLP-1 agonist) developed by Novo Nordisk for the treatment of type 2 diabetes. The product was approved by the European Medicines Agency (EMA) on July 3, 2009, and by the U.S. Food and Drug Administration (FDA) on January 25, 2010.
Liraglutide is marketed under the brandname Victoza in the U.S., India, Canada, Europe and Japan. It has been launched in Germany, Denmark, the Netherlands, the United Kingdom, Ireland, Sweden, Japan, Canada, the United States, France, Malaysia and Singapore.
Phase I trials of an oral variant of Victoza (NN9924) started in 2010.
Takeda’s Ixazomib, Multiple Myeloma Drug

CAS#: 1201902-80-8
Synonym: Ixazomib; MLN-9708.
IUPAC/Chemical name:
4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid
UPDATES AT THE BOTTOM OF PAGE
CAMBRIDGE, Mass., May 23, 2013 – Takeda Pharmaceutical Company Limited (TSE:4502) today announced the initiation of an international phase 3 clinical trial evaluating once a week MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma who are not candidates for transplant. The multi-center study with MLN9708, an investigational, oral proteasome inhibitor, will be conducted in Europe and North America.———————-READ MORE AT
http://www.pharmalive.com/takeda-begins-phase-iii-trial-of-multiple-myeloma-drug
Description of Ixazomib: ixazomib is an orally bioavailable second generation proteasome inhibitor (PI) with potential antineoplastic activity. Ixazomib inhibits the activity of the proteasome, blocking the targeted proteolysis normally performed by the proteasome, which results in an accumulation of unwanted or misfolded proteins; disruption of various cell signaling pathways may follow, resulting in the induction of apoptosis. Compared to first generation PIs, second generation PIs may have an improved pharmacokinetic profile with increased potency and less toxicity. Proteasomes are large protease complexes that degrade unneeded or damaged proteins that have been ubiquinated
MLN9708 is an investigational proteasome inhibitor that, compared with bortezomib, has improved pharmacokinetics, pharmacodynamics, and antitumor activity in preclinical studies. MLN9708 rapidly hydrolyzes to MLN2238, the biologically active form. MLN9708 has a shorter proteasome dissociation half-life and improved pharmacokinetics, pharmacodynamics, and antitumor activity compared with bortezomib.MLN9708 has a larger blood volume distribution at steady state, and analysis of 20S proteasome inhibition and markers of the unfolded protein response confirmed that MLN9708 has greater pharmacodynamic effects in tissues than bortezomib. MLN9708 showed activity in both solid tumor and hematologic preclinical xenograft models, and we found a correlation between greater pharmacodynamic responses and improved antitumor activity. Moreover, antitumor activity was shown via multiple dosing routes, including oral gavage. Taken together, these data support the clinical development of MLN9708 for both hematologic and solid tumor indications. (source: Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16.).
| References |
1: Mullard A. Next-generation proteasome blockers promise safer cancer therapy. Nat Med. 2012 Jan 6;18(1):7. doi: 10.1038/nm0112-7a. PubMed PMID: 22227650.
2: Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445-52. Epub 2012 Jan 3. PubMed PMID: 22215754.
3: Appel A. Drugs: More shots on target. Nature. 2011 Dec 14;480(7377):S40-2. doi: 10.1038/480S40a. PubMed PMID: 22169800.
4: Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011 Dec 1;17(23):7313-23. Epub 2011 Sep 8. PubMed PMID: 21903769.
5: Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011 Aug 15;17(16):5311-21. doi: 10.1158/1078-0432.CCR-11-0476. Epub 2011 Jun 30. PubMed PMID: 21724551; PubMed Central PMCID: PMC3156932.
6: Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16. Erratum in: Cancer Res. 2010 May 1;70(9):3853. Hales, Paul [added]. PubMed PMID: 20160034.
7: Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 Mar;15(5-6):243-9. Epub 2010 Jan 29. Review. PubMed PMID: 20116451.8: Marblestone JG. Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs. 2009 Dec;12(12):750-3. PubMed PMID: 19943215.

http://www.cancernetwork.com/conference-reports/ash2012/content/article/10165/2119611
Nasopharyngeal cancer is a sub-type of head and neck cancer that arises from the epithelial cells that cover the surface and line the nasopharynx. The incidence of nasopharyngeal cancer has been reported at approximately 0.5 to 2 new cases per year per 100,000 in Europe and the USA. Rottey et ah, Curr. Opin. Oncol., 23(3): 254-258 (201 1). There are three subtypes of nasopharyngeal cancer recognized in the World Health Organization (WHO) classification: (i) Type 1 – squamous cell carcinoma, typically found in the older adult population; (ii) Type 2 non-keratinizing carcinoma; and (iii) Type 3 – undifferentiated carcinoma. Treatment for nasopharyngeal cancer often involves radiotherapy and/or chemotherapy. There remains a continuing need for new and improved treatments for patients with nasopharyngeal cancer. There remains a further need to identify nasopharyngeal patients most likely to benefit from treatment with a proteasome inhibitor.
Proteasome inhibition represents an important new strategy in cancer treatment. King et al. , Science 274: 1652-1659 ( 1996), describes an essential role for the ubiquitin-proteasome pathway in regulating cell cycle, neoplastic growth and metastasis. The authors teach that a number of key regulatory proteins, including cyclins, and the cyclin-dependent kinases p21 and p27K,P ! , are temporally degraded during the cell cycle by the ubiquitin-proteasome pathway. The ordered degradation of these proteins is required for the cell to progress through the cell cycle and to undergo mitosis.
The proteasome inhibitor VELCADE© (bortezomib; N-2-pyrazinecarbonyl-L -phenylalanine -L- leucineboronic acid) is the first proteasome inhibitor to achieve regulatory approval. Mitsiades et ai, Current Drug Targets, 7: 1341 (2006), reviews the clinical studies leading to the approval of bortezomib for the treatment of multiple myeloma patients who have received at least one prior therapy. Fisher et ai , J. Clin. Oncol, 30:4867, describes an international multi-center Phase II study confirming the activity of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Ishii et al, Anti-Cancer Agents in Medicinal Chemistry, 7:359 (2007), and Roccaro et al., Curr. Pharm. Biotech., 7: 1341 (2006), discuss a number of molecular mechanisms that may contribute to the antitumor activities of bortezomib. The proteasome inhibitor MLN9708 [2,2′-{2-[(lR)- l -( {[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3- methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid] is currently undergoing clinical evaluation for hematological and solid cancers. MLN9708 is a citrate ester which rapidly hydrolyzes to the active form [(lR)-l -({[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3-methylbutyl]boronic acid (MLN2238) on exposure to aqueous solution or plasma. MLN9708 has demonstrated anti-tumor activity in a range of hematological and solid tumor xenograft models (Kupperman et al. (2010) Cancer Res. 70: 1970- 1980),
Summary
The invention relates to the discovery that patients with nasopharyngeal cancer respond to treatment with MLN9708. In one aspect, the invention relates to the discovery of the increased expression of Nuclear Factor Kappa-B RelA 65,000 dalton subunit (NFKB p65) in biological samples comprising cells obtained from patients with nasopharyngeal cancer and responsive to MLN9708.
Accordingly, the invention features treating nasopharyngeal cancer patients withMLN9708 if a sample from the patient demonstrates an elevated expression of NFKB p65.
PATENT

or a pharmaceutically acceptable salt or a pharmaceutical composition or a boronic acid anhydride thereof.
[048| The compound of formula (II), [( l R)-l -( } [(2,5-dichlorobenzoyl)amino]acetyl} amino)-3- methylbutyljboronic acid (MLN2238) is disclosed in Olhava and Danca, U .S. Patent No. 7,442,830, herein incorporated by reference in its entirety. [049] In some other embodiments, Z and Z together form a moiety derived from a compound having at least two hydroxyl groups separated by at least two connecting atoms in a chain or ring, said chain or ring comprising carbon atoms and, optionally, a heteroatom or heteroatoms which can be N, S, or O, wherein the atom attached to boron in each case is an oxygen atom.
In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A) or (III-B):

(III-B), or a mixture thereof or a pharmaceutical composition thereof.
[054] In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A):

or a pharmaceutical composition thereof.
[055] The compound of formula (III-A), 2,2′- {2-[( l i?)- l -( { [(2,5-dichlorobenzoyl)amino]acetyl } amino)- 3-methylbutyl]-5-oxo- l ,3,2-dioxaborolane-4,4-diyl} diacetic acid (MLN9708) is disclosed in Elliott et al. , WO 09/ 154737, herein incorporated by reference in its entirety
PATENT
http://www.google.com/patents/WO2009154737A1?cl=en
Example 1: Synthesis of 4-(/?,S)-(carboxymethyl)-2-( (R)-I -(2-(2,5- dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-l,3,2-dioxaborinane-4- carboxylic acid (1-1)

Step l: 2,5-r(dichlorobenzoyI)aminolacetic acid
[0310] To a mixture of NaOH (12 g, 300 mmol) and glycine (18 g, 239 mmol) in water (120 mL) was added dropwise over 45 min a solution of 2,5-dichlorobenzoyl chloride (10 g, 48 mmol) in THF (15 mL) keeping the internal temperature below about 25 0C. After 1 h, the mixture was acidified with 2.0 M HCl (125 mL) keeping the internal temperature below about 5 0C. The resulting precipitate was collected by vacuum filtration. The crude product was recrystallized from water to give 2,5-[(dichlorobenzoyl)amino]acetic acid as a white, crystalline solid (6.1 g, 52%). mp 173.3 0C. 1H NMR (300 MHz, DMSOd6, δ): 12.72 (bs, IH), 8.89 (t, J = 6.0 Hz, IH), 7.54 (m, 2H), 7.48 (m, IH), 3.93 (d, J = 6.0 Hz). 13C NMR (75 MHz, DMSO-Ci6, δ): 41.6, 129.3, 129.6, 131.4, 132.2, 138.2, 171.4, 165.9. MS (ni/z): [M+H] calculated for C9H8Cl2NO3, 248.0; found, 248.0; [M+Na] calculated for C9H7Cl2NNaO3, 270.0; found 270.2.
2,5-[(dichlorobenzoyl)amino]acetic acid was also be prepared via the following procedure: To a mixture of glycine (21.5 g, 286 mmol) in water (437 mL), was added 2.0 M NaOH (130 mL) and the resulting solution was cooled to 0 0C. A solution of 2,5-dichlorobenzoyl chloride (50.0 g, 239 mmol) in THF (75 mL) was added dropwise at such a rate that the internal temperature was maintained at 0 ± 1 0C. During the addition, the pH was controlled at 11.0 ± 0.2 using a pH controller titrated with 2.0 M NaOH. After complete addition, the mixture was stirred at 0 ± 1 0C for an additional 2 h. The mixture was then acidified with 2.0 M HCl (176 mL) to a final pH of 2.5. The resulting precipitate was collected by filtration, washed with cold water (125 mL), and dried at 45 0C in a vacuum oven to afford 2,5-[(dichlorobenzoyl)amino]acetic acid as a white solid (57.6 g, 97.3%). Step 2: 2,5-dichloro-N-f2-(( (lR’)-3-niethyl-l-r(3aS,4S.6S.7aR)-3a,5,5-trimethylhexahvdro-
4,6-methano-l,3,2-benzodioxaborol-2-yllbutyl }amino)-2-oxoethvπbenzamide
To a solution of 2,5-[(dichlorobenzoyl)amino]acetic acid (6.10 g, 24.6 mmol) and TBTU (8.34 g, 26.0 mmol) in DMF (40 mL) with an internal temperature below about 5 0C was added (IR)- 3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5-trimethylhexahydro-4,6-methano-l,3,2-benodioxaborol-2- yl]butan-l-amine»TFA (9.35 g, 24.7 mmol). DIPEA (13 mL, 75 mmol) was then added dropwise over 2 h keeping the internal temperature below about 5 0C. After 40 min, the mixture was diluted with EtOAc (90 mL), washed with 5% NaCl (150 mL), twice with 10% NaCl (2 x 40 mL), once with 2% K2CO3 (1 x 40 mL), once with 1% H3PO4 (1 x 40 mL), and once with 10% NaCl (1 x 40 mL). The resulting organic layer was concentrated to a thick oil, diluted with heptane (40 mL) and evaporated to yield 2,5-dichloro-N-[2-({ (lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l ,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide as a white solid which was used in the next step without purification.
Step 3: N,N\N’Wboroxin-2A6-triyltrisir(lR)-3-methylbutane-l J-diyllimino(2-oxoethane- 2,l-diyl)^ ^tris(2,5-dichlorobenzamide)
To a solution of 2,5-dichloro-N-[2-({(lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide (12.2 g, 24.6 mmol) in methanol/hexane (1 :1) (250 mL) were added IN HCl (30 mL, 30 mmol) and (2-methylpropyl)boronic acid (6.5 g, 64 mmol). The reaction mixture was allowed to stir overnight. The phases were separated and the methanol layer was washed twice with additional heptane (2 x 55 mL). The resulting organic layer was concentrated to about 10 mL and partitioned between 2.0M NaOH (30 mL) and DCM (25 mL). The DCM layer was washed once with additional 2.0M NaOH (5 mL). The basic aqueous layers were then combined, washed twice with DCM (2 x 25 mL) and acidified with IM HCl (60 mL). The resulting mixture was diluted with DCM (40 mL), the layers were separated, and the resulting aqueous layer was washed three times with DCM (3 x 10 mL). The combined DCM extracts were dried over MgSO4 (25 g) and evaporated to a thick oil. The product was precipitated with heptane (50 mL) and collected by filtration to yield N,N’,N”-{boroxin-2,4,6- -riyltris[[(lR)-3-methylbutane-l,l-diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) as a white solid (6.6 g, 74%). 1H NMR (300 MHz, DMSO-Cl6, δ): 8.93 (t, J – 6.0 Hz, IH), 8.68 (bs, IH), 7.63 (m, IH), 7.52 (m, 2H), 4.00 (d, J = 6.0 Hz, 2H), 2.62 (m, IH), 1.59 (m, IH), 1.33 (m, IH), 1.24 (m, IH), 0.81 (d, / = 5.9 Hz, 6H). 13C NMR (125 MHz, DMSO-Cl6, δ): 23.2, 25.8, 40.1, 40.7, 43.0, 129.0, 130.0, 131.0, 137.5, 165.0, 172.5. MS (m/z) in CH3CN: [M+H] calculated for C42H52B3Cl6N6O9, 1027.2; found, 1027.3; [M+Na] calculated for C42H51B3Cl6N6NaO9, 1049.2; found 1049.5.
Step 4: 4-(/?.S)-(carboxymethyl)-2-((/?)-l-(2-(2,5-dichlorobenzamido)acetamido)-3- methylbutyl)-6-oxo-l,3,2-dioxaborinane-4-carboxylic acid (1-1)
Form 1: To a solution of citric acid (2.75 g, 14.3 mmol) in EtOAc (85 mL) with an internal temperature of about 74 0C was added N,N’,N”-{boroxin-2,4,6-triyltris[[(lR)-3-methylbutane-l,l- diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) (5.00 g, 4.87 mmol) as a solid. The solution was cooled uncontrolled until the internal temperature was about 25 0C and the mixture was stirred overnight. The resulting precipitate was collected by filtration to yield 2,2′-{2-[(lR)-l-({ [(2,5- dichlorobenzoyl)amino]acetyl }amino)-3-methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid Form 1 as a crystalline solid (6.65 g, 88 %). 1H NMR (500 MHz, DMSOd6, δ 110 0C): 10.08 (s, IH), 8.69 (s, IH), 7.61 (s, IH), 7.52 (d, J = 1.3 Hz, 2H), 4.26 (d, J = 5.5 Hz, 2H), 2.70 (q, J = 14.5 Hz, 4H), 2.70 (bs, IH), 1.72 (sept, J – 6.5 Hz, IH), 1.42 (ddd, J = 5.2 Hz, J = 8.6 Hz, J = 13.9 Hz, IH), 1.28 (ddd, J = 5.3, J = 9.4 Hz, J = 14.3 Hz, IH), 0.91 (dd, J = 3.3 Hz, J = 6.6 Hz, 6H). MS (m/z) in CH3CN: [M+Na] calculated for C20H23BCl2N2NaO9, 539.1; found, 539.1.
Ixazomib citrate [USAN]
1,3,2-Dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- [ACD/Index Name]
1,3,2-Dioxaborolane-4,4-diacetic acid,2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-
1239908-20-3 [RN]
2,2′-{2-[(1R)-1-{[N-(2,5-Dichlorbenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4,4-diyl}diessigsäure [German] [ACD/IUPAC Name]
2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacetic acid [ACD/IUPAC Name]
2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diacetic acid
2-[4-(carboxymethyl)-2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methyl-butyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
Acide 2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-méthylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacétique [French] [ACD/IUPAC Name]
MLN9708
UPDATES………..
Ixazomib (trade name Ninlaro) is a drug for the treatment of multiple myeloma, developed by Takeda Pharma. It acts as aproteasome inhibitor and has orphan drug status in the US. In November 2015, the U.S. Food and Drug Administration approved ixazomib for use in combination with lenalidomide and dexamethasone for the treatment of multiple myeloma after at least one prior therapy.[2]
Mechanism
Ixazomib is a peptide analogue that reversibly inhibits the protein proteasome subunit beta type-5 (PSMB5), which is part of the 20Sproteasome complex.[3]
Chemistry

Ixazomib citrate—a prodrug for ixazomib
U.S. FDA Approves Takeda’s NINLARO® (ixazomib), the First and Only Oral Proteasome Inhibitor to Treat Multiple Myeloma
NINLARO Provides a New Option for Patients Living with Multiple Myeloma Who Have Received at Least One Prior Therapy
Cambridge, Mass. and Osaka, Japan, November 20, 2015 – Takeda Pharmaceutical Company Limited (TSE: 4502) today announced that the U.S. Food and Drug Administration (FDA) has approved NINLARO®(ixazomib) capsules, the first and only oral proteasome inhibitor, indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is a once-weekly pill. More information is available at www.NINLARO.com.
Takeda submitted a New Drug Application for NINLARO to the FDA in July 2015, and in September NINLARO was granted Priority Review status with a PDUFA date of March 10, 2016, reflecting the profound and continuing unmet need for new treatments for multiple myeloma, a devastating, relapsing and incurable rare cancer.
“With the approval of NINLARO, we can now offer patients a once-weekly oral proteasome inhibitor as part of a highly active triplet therapy,” said Paul Richardson, M.D., Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center Institute Physician at Dana-Farber Cancer Institute, and investigator for TOURMALINE-MM1, the pivotal Phase 3 trial on which today’s approval is based. “We, as investigators of the TOURMALINE-MM1 trial, felt it was vital to conduct a comprehensive ‘real world’ evaluation of this combination that included some of the most common patient types in the relapsed/refractory multiple myeloma setting, such as older patients, patients with moderate renal impairment, light chain disease, and high risk cytogenetics. Further, we treated patients until disease progression to determine the sustainability of NINLARO in treating their relapsed/refractory disease. The TOURMALINE-MM1 data demonstrate convincingly that oral NINLARO-based triplet treatment is effective at extending progression-free survival, over and above the clinical benefit seen with lenalidomide and dexamethasone, with a tolerable safety profile.”
“We introduced the first proteasome inhibitor for multiple myeloma, VELCADE, into clinical research approximately 20 years ago. Since that time, we’ve significantly advanced scientific understanding of this rare cancer, culminating in the introduction of NINLARO,” said Andy Plump, M.D., Ph.D, Takeda Chief Medical and Scientific Officer. “NINLARO is an entirely new molecule that offers the efficacy of this proteasome inhibitor in a convenient once-weekly pill with a tolerable safety profile. Takeda is delighted to bring this significant innovation to multiple myeloma patients today, and we continue to examine the potential of NINLARO through a robust clinical development program.”
Dr. Brian Durie, Chairman of the International Myeloma Foundation, said, “The IMF is pleased by the approval of ixazomib. This opens the door for a fully oral proteasome inhibitor-based triplet combination therapy. Having worked in multiple myeloma for decades, I’ve seen notable progress, yet significant unmet needs remain. With today’s approval, we now have another attractive option for many patients living with multiple myeloma.”
The FDA approval of NINLARO is based on results from the TOURMALINE-MM1 Phase 3 clinical trial, the first double-blind, placebo-controlled trial with a proteasome inhibitor. TOURMALINE-MM1 is the first of five ongoing Phase 3 clinical trials with study results available. The TOURMALINE program has enrolled approximately 3,000 patients to date in 40 countries. Data from the NINLARO Phase 3 TOURMALINE-MM1 pivotal trial will be presented at the upcoming 57th Annual Meeting of the American Society of Hematology on December 7, 2015.
“The approval of ixazomib offers a much-needed additional option in the multiple myeloma treatment landscape. It is developments such as these that help us to better understand the disease and provide continued hope for patients,” said Kathy Giusti, Founder and Executive Chairman of the Multiple Myeloma Research Foundation (MMRF). “A cancer diagnosis today is different from what it was just a few years ago and it’s exciting to see continued progress. As a patient, I understand the urgent need for advancing research through partnerships that bring new treatment options, as we’ve done with Takeda.”
“NINLARO is a first-of-its-kind innovation that is supported by a global development program, unprecedented for us at Takeda Oncology, and we would like to express our immense appreciation for all patients involved for their incredible strength and invaluable participation. The introduction of NINLARO marks an important step forward, as its efficacy and safety profile – coupled with its completely oral administration – potentially can reduce some logistical burdens, and help enable patients to reap the full benefits of this sustainable therapy,” explained Christophe Bianchi, M.D., President, Takeda Oncology. “As part of our unwavering 20-year commitment, Takeda will continue to pursue advances for these patients, and we look forward to introducing and expanding access to NINLARO in other markets around the world.”
About the TOURMALINE-MM1 Trial
TOURMALINE-MM1 is an international, randomized, double-blind, placebo-controlled clinical trial of 722 patients, designed to evaluate NINLARO plus lenalidomide and dexamethasone compared to placebo plus lenalidomide and dexamethasone in adult patients with relapsed and/or refractory multiple myeloma. Results showed NINLARO is effective in extending Progression Free Survival (PFS) and has a manageable safety profile. The trial achieved its primary endpoint and demonstrated a clinically meaningful and statistically significant prolongation in PFS at this analysis, which showed that patients treated in the NINLARO arm lived without their disease worsening for a significantly longer time compared to patients in the control arm. Patients continue to be treated to progression in this trial and will be evaluated for long term outcomes.
In the TOURMALINE-MM1 trial, the most common adverse reactions (≥20%) in patients receiving NINLARO included diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting and back pain. Serious adverse reactions reported in ≥2% patients included thrombocytopenia (2%) and diarrhea (2%).
Efficacy and safety data were reviewed by an Independent Data Monitoring Committee (IDMC), who recommended the study be continued in blinded fashion to allow further maturation of long term outcomes, including overall survival (OS) and long-term safety.
About NINLARO (ixazomib) capsules
NINLARO (ixazomib) is the first and only oral proteasome inhibitor indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is administered orally, once-weekly on days 1, 8, and 15 of a 28-day treatment cycle. NINLARO is currently under review by the European Medicines Agency (EMA) and was granted an accelerated assessment by the Committee for Medicinal Products for Human Use (CHMP). NINLARO also received Breakthrough Therapy status by the U.S. FDA for relapsed or refractory systemic light-chain (AL) amyloidosis, a related ultra orphan disease, in 2014.
The TOURMALINE clinical development program further reinforces Takeda’s ongoing commitment to developing innovative therapies for people living with multiple myeloma worldwide and the healthcare professionals who treat them. Five global Phase 3 trials are ongoing:
- TOURMALINE-MM1, investigating ixazomib vs. placebo, in combination with lenalidomide and dexamethasone in relapsed and/or refractory multiple myeloma
- TOURMALINE-MM2, investigating ixazomib vs. placebo, in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma
- TOURMALINE-MM3, investigating ixazomib vs. placebo as maintenance therapy in patients with newly diagnosed multiple myeloma following induction therapy and autologous stem cell transplant (ASCT)
- TOURMALINE-MM4, investigating ixazomib vs. placebo as maintenance therapy in patients with newly diagnosed multiple myeloma who have not undergone ASCT
- TOURMALINE-AL1, investigating ixazomib plus dexamethasone vs. physician choice of selected regimens in patients with relapsed or refractory AL amyloidosis
In addition to the TOURMALINE program, a large number of investigator initiated studies are evaluating ixazomib for patients globally.
For additional information on the ongoing Phase 3 studies please visit www.clinicaltrials.gov. To learn more about NINLARO, please visit www.NINLARO.com or call 1-844-N1POINT (1-844-617-6468).
![]()
References
- “Ninlaro (ixazomib) Capsules, for Oral Use. Full Prescribing Information” (PDF). NINLARO (ixazomib) For Healthcare Professionals. Takeda Pharmaceutical Company Limited Cambridge, MA 02139. Retrieved 21 November 2015.
- “FDA Okays Ixazomib, Another Multiple Myeloma Drug”. November 20, 2015.
- KEGG: Ixazomib
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N2-(2,5-Dichlorobenzoyl)-N-[(1R)-1-(dihydroxyboryl)-3-methylbutyl]glycinamide
|
|
| Clinical data | |
| Trade names | Ninlaro |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 58%[1] |
| Protein binding | 99% |
| Metabolism | hepatic, CYP3A4 (42%),CYP1A2 (26%) and others |
| Biological half-life | 9.5 days |
| Excretion | urine (62%), feces (22%) |
| Identifiers | |
| CAS Number | 1072833-77-2 |
| ATC code | L01XX50 |
| PubChem | CID 25183872 |
| ChemSpider | 25027391 |
| UNII | 71050168A2 |
| KEGG | D10130 |
| ChEBI | CHEBI:90942 |
| Synonyms | MLN2238 |
| Chemical data | |
| Formula | C14H19BCl2N2O4 |
| Molar mass | 361.03 g·mol−1 |

//////////
see….http://apisynthesisint.blogspot.in/2016/02/takedas-ixazomib-multiple-myeloma-drug.html
