
Home » Phase3 drugs (Page 16)
Category Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cortendo AB: First Patient Enrolled into NormoCort Phase 3 SONICS Trial Following a Successful EU Investigator Meeting
KETOCONAZOLE 2S 4R
ALSO
142128-57-2
228850-16-6 (tartrate)
228850-16-6 (tartrate)
(-)-cis-1-Acetyl-4-[4-[2(S)-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4(R)-ylmethoxy]phenyl]piperazine
531.431, C26 H28 Cl2 N4 O4
COR-003
DIO-902
LDKTZ
DIO-902
LDKTZ
CORTENDO
licensee DiObex
| Biological Role(s): | antifungal agent
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
|
| Application(s): | antifungal agent
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
|
Ketoconazole, 1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3– dioxolan-4-yl]methoxy]phenyl]piperazine, is a racemic mixture of the cis enantiomers (-)-(2S,4R) and (+)-(2R,4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis.(-)-Ketoconazole, the (2S,4R) enantiomer contained in the racemate of ketoconazole, is in phase III clinical trials at Cortendo for the treatment of endogenous Cushing’s syndrome. The company and licensee DiObex had also been developing the drug candidate for the treatment of type 2 diabetes; however, no recent development has been reported for this research.Preclinical studies have demonstrated the drug candidate’s ability to inhibit the synthesis of cortisol, resulting in substantial clinical benefits including lowering both blood pressure and cholesterol in addition to controlling glucose levels. It has also been shown that (-)-ketoconazole is responsible for virtually all of the cortisol synthesis inhibitory activity present in the racemate. Rights to the compound are shared with Cortendo.In 2012, orphan drug designation was assigned in the U.S. for the treatment of endogenous Cushing’s syndrome.
August 12, 2014 02:30 AM Eastern Daylight Time
GÖTEBORG, Sweden.–(BUSINESS WIRE)–Cortendo AB (OSE:CORT) today announced that the first patient has been enrolled into the Phase 3 SONICS trial, i.e., “Study Of NormoCort In Cushing’s Syndrome.”
“The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”
The patient was enrolled by one of the trial’s lead principal investigators at a Pituitary Center from a prestigious institution in Baltimore, Maryland. “The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”, said Dr. Theodore R Koziol. ”The SONICS clinical trial team is acutely focused on the implementation of the trial following a successful EU Investigator’s meeting in Barcelona in July, which we believe further solidified the foundation for the trial.”
Cortendo successfully completed its European Investigator meeting supporting SONICS held in Barcelona, Spain on July 17-18. More than 35 investigators/study coordinators, including many of the world’s leading Cushing’s experts from 24 study sites, were in attendance and received training for the trial. Based on the positive feedback from the meeting, Cortendo has gained further confidence that NormoCort (COR-003) has the potential to be an important future treatment option for patients afflicted with Cushing’s Syndrome. A second US Investigator meeting is also being planned for later this year.
”It was gratifying to participate in the NormoCort SONICS trial investigator meeting in my home town of Barcelona with so many esteemed colleagues dedicated to treating patients with Cushing’s Syndrome”, said Susan Webb M.D. Ph.D. Professor of Medicine Universitat Autonoma de Barcelona. ”There remains a significant unmet medical need for patients, and I am delighted to be part of the development of this new therapy”.
Cortendo has also further strengthened its internal as well as external teams to support the study and to position the trial for an increased recruitment rate. In July, Cortendo added both an experienced physician and internal Clinical Operations Director to the NormoCort development team. Cortendo, working in concert with its CROs supporting the SONICS trial, now has a team of approximately 20 personnel on the NormoCort development program.
Cortendo has previously communicated its plan to meet the recruitment goal by increasing the number of study sites from 38 to 45 worldwide. The company is at various levels of activation with more than 30 study sites to date. Therein, Cortendo expects a large proportion of the sites to be activated by the end of the third quarter this year and remains confident that essentially all sites will be open by the end of 2014.
Risk and uncertainty
The development of pharmaceuticals carries significant risk. Failure may occur at any stage during development and commercialization due to safety or clinical efficacy issues. Delays may occur due to requirements from regulatory authorities not anticipated by the company.
About Cortendo
Cortendo AB is a biopharmaceutical company headquartered in Göteborg, Sweden. Its stock is publicly traded on the NOTC-A-list (OTC) in Norway. Cortendo is a pioneer in the field of cortisol inhibition and has completed early clinical trials in patients with Type 2 diabetes. The lead drug candidate NormoCort, the 2S, 4R-enantiomer of ketoconazole, has been re-focused to Cushing’s Syndrome, and has entered Phase 3 development. The company’s strategy is to primarily focus its resources within orphan drugs and metabolic diseases and to seek opportunities where the path to commercialization or partnership is clear and relatively near-term. Cortendo’s business model is to commercialize orphan and specialist product opportunities in key markets, and to partner non-specialist product opportunities such as diabetes at relevant development stages.
Cortendo AB (publ)
Sweden: Box 47 SE-433 21 Partille Tel. / Fax: +46 (0)31-263010
USA: 555 East Lancaster Ave Suite 510 Radnor, PA 19087 Tel: +1 610-254-9200 Fax: +1 610-254-9245
This information was brought to you by Cision http://news.cision.com
Contacts
Alexander Lindström
Chief Financial Officer Office
+1 610 254 9200
Mobile : +1 917 349 7210
E-mail : alindstrom@cortendo.com
-
Ketoconazole, 1-acetyl-4- [4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolan-4-yl] methoxy] phenyl] piperazine, is a racemic mixture of the cis enantiomers (-)-(2S, 4R) and (+)-(2R, 4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis. Ergosterol is a key component of fungal cell walls.
-
More recently, ketoconazole was found to decrease plasma cortisol and to be useful, alone and in combination with other agents, in the treatment of a variety of diseases and conditions, including type 2 diabetes, Metabolic Syndrome (also known as the Insulin Resistance Syndrome, Dysmetabolic Syndrome or Syndrome X), and other medical conditions that are associated with elevated cortisol levels. SeeU.S. Patent Nos. 5,584,790 ; 6,166,017 ; and 6,642,236 , each of which is incorporated herein by reference. Cortisol is a stress-related hormone secreted from the cortex of the adrenal glands. ACTH (adenocorticotropic hormone) increases cortisol secretion. ACTH is secreted by the pituitary gland, a process activated by secretion of corticotropin releasing hormone (CRH) from the hypothalamus.
-
Cortisol circulates in the bloodstream and activates specific intracellular receptors, such as the glucocorticoid receptor (GR). Disturbances in cortisol levels, synthetic rates or activity have been shown to be associated with numerous metabolic complications, including insulin resistance, obesity, diabetes and Metabolic Syndrome. Additionally, these metabolic abnormalities are associated with substantially increased risk of cardiovascular disease, a major cause of death in industrialized countries. See Mårin P et al., “Cortisol secretion in relation to body fat distribution in obese premenopausal women.” Metabolism 1992; 41:882-886, Bjorntorp, “Neuroendocrine perturbations as a cause of insulin resistance.” Diabetes Metab Res Rev 1999; 15(6): 427-41, and Rosmond, “Role of stress in the pathogenesis of the metabolic syndrome.” Psychoneuroendocrinology 2005; 30(1): 1-10, each of which is incorporated herein by reference.
-
While ketoconazole is known to inhibit some of the enzymatic steps in cortisol synthesis, such as, for example, 17α hydroxylase (Wachall et al., “Imidazole substituted biphenyls: a new class of highly potent and in vivo active inhibitors of P450 17 as potential therapeutics for treatment of prostate cancer.” Bioorg Med Chem 1999; 7(9): 1913-24, incorporated herein by reference) and 11b-hydroxylase (Rotstein et al., “Stereoisomers of ketoconazole: preparation and biological activity.” J Med Chem 1992; 35(15): 2818-25) and 11β-hydroxy steroid dehydrogenase (11β-HSD) (Diederich et al., “In the search for specific inhibitors of human 11β-hydroxysteroid-dehydrogenases (11β-HSDs): chenodeoxycholic acid selectively inhibits 11β-HSD-L” Eur J Endocrinol 2000; 142(2): 200-7, incorporated herein by reference) the mechanisms by which ketoconazole decreases cortisol levels in the plasma have not been reported. For example, there is uncertainty regarding the effect of ketoconazole on the 11β-hydroxy steroid dehydrogenase (11β-HSD) enzymes. There are two 11β-HSD enzymes. One of these, 11β-HSD-I, is primarily a reductase that is highly expressed in the liver and can convert the inactive 11-keto glucocorticoid to the active glucocorticoid (cortisol in humans and corticosterone in rats). In contrast, the other, 11β-HSD-II, is primarily expressed in the kidney and acts primarily as an oxidase that converts active glucocorticoid (cortisol in humans and corticosterone in rats) to inactive 11-keto glucocorticoids. Thus, the plasma concentration of active glucocorticoid is influenced by the rate of synthesis, controlled in part by the activity of adrenal 11β-hydroxylase and by the rate of interconversion, controlled in part by the relative activities of the two 11β-HSD enzymes. Ketoconazole is known to inhibit these three enzymes (Diederich et al., supra) and the 2S,4R enantiomer is more active against the adrenal 11β-hydroxylase enzyme than is the 2R,4S enantiomer (Rotstein et al., supra). However, there are no reports describing the effect of the two ketoconazole enantiomers on either of 11β-HSD-I or 11β-HSD-II, so it is not possible to predict what effects, if any, the two different ketoconazole enantiomers will each have on plasma levels of the active glucocorticoid levels in a mammal.
-
Ketoconazole has also been reported to lower cholesterol levels in humans (Sonino et al. (1991). “Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients.” Clin Endocrinol (Oxf). 35(4): 347-52; Gylling et al. (1993). “Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia.” J Lipid Res. 34(1): 59-67) each of which is incorporated herein by reference). The 2S,4R enantiomer is more active against the cholesterol synthetic enzyme 14 αlanosterol demethylase than is the other (2R,4S) enantiomer (Rotstein et al infra). However, because cholesterol level in a human patient is controlled by the rate of metabolism and excretion as well as by the rate of synthesis it is not possible to predict from this whether the 2S,4R enantiomer of ketoconazole will be more effective at lowering cholesterol levels.
-
The use of ketoconazole as a therapeutic is complicated by the effect of ketoconazole on the P450 enzymes responsible for drug metabolism. Several of these P450 enzymes are inhibited by ketoconazole (Rotsteinet al., supra). This inhibition leads to an alteration in the clearance of ketoconazole itself (Brass et al., “Disposition of ketoconazole, an oral antifungal, in humans.” Antimicrob Agents Chemother 1982; 21(1): 151-8, incorporated herein by reference) and several other important drugs such as Glivec (Dutreix et al., “Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects.” Cancer Chemother Pharmacol 2004; 54(4): 290-4) and methylprednisolone (Glynn et al., “Effects of ketoconazole on methylprednisolone pharmacokinetics and cortisol secretion.” Clin Pharmacol Ther 1986; 39(6): 654-9). As a result, the exposure of a patient to ketoconazole increases with repeated dosing, despite no increase in the amount of drug administered to the patient. This exposure and increase in exposure can be measured and demonstrated using the “Area under the Curve” (AUC) or the product of the concentration of the drug found in the plasma and the time period over which the measurements are made. The AUC for ketoconazole following the first exposure is significantly less than the AUC for ketoconazole after repeated exposures. This increase in drug exposure means that it is difficult to provide an accurate and consistent dose of the drug to a patient. Further, the increase in drug exposure increases the likelihood of adverse side effects associated with ketoconazole use.
-
[0008]Rotstein et al. (Rotstein et al., supra) have examined the effects of the two ketoconazole cis enantiomers on the principal P450 enzymes responsible for drug metabolism and reported “…almost no selectivity was observed for the ketoconazole isomers” and, referring to drug metabolizing P450 enzymes: “[t]he IC50 values for the cis enantiomers were similar to those previously reported for racemic ketoconazole”. This report indicated that both of the cis enantiomers could contribute significantly to the AUC problem observed with the ketoconazole racemate.
-
One of the adverse side effects of ketoconazole administration exacerbated by this AUC problem is liver reactions. Asymptomatic liver reactions can be measured by an increase in the level of liver specific enzymes found in the serum and an increase in these enzymes has been noted in ketoconazole treated patients (Sohn, “Evaluation of ketoconazole.” Clin Pharm 1982; 1(3): 217-24, and Janssen and Symoens, “Hepatic reactions during ketoconazole treatment.” Am J Med 1983; 74(1B): 80-5, each of which is incorporated herein by reference). In addition 1:12,000 patients will have more severe liver failure (Smith and Henry, “Ketoconazole: an orally effective antifungal agent. Mechanism of action, pharmacology, clinical efficacy and adverse effects.” Pharmacotherapy 1984; 4(4): 199-204, incorporated herein by reference). As noted above, the amount of ketoconazole that a patient is exposed to increases with repeated dosing even though the amount of drug taken per day does not increase (the “AUC problem”). The AUC correlates with liver damage in rabbits (Ma et al., “Hepatotoxicity and toxicokinetics of ketoconazole in rabbits.” Acta Pharmacol Sin 2003; 24(8): 778-782 incorporated herein by reference) and increased exposure to the drug is believed to increase the frequency of liver damage reported in ketoconazole treated patients.
-
Additionally, U.S. Patent No. 6,040,307 , incorporated herein by reference, reports that the 2S,4R enantiomer is efficacious in treating fungal infections. This same patent application also reports studies on isolated guinea pig hearts that show that the administration of racemic ketoconazole may be associated with an increased risk of cardiac arrhythmia, but provides no data in support of that assertion. However, as disclosed in that patent, arrhythmia had not been previously reported as a side effect of systemic racemic ketoconazole, although a particular subtype of arrhythmia, torsades de pointes, has been reported when racemic ketoconazole was administered concurrently with terfenadine. Furthermore several published reports (for example, Morganroth et al. (1997). “Lack of effect of azelastine and ketoconazole coadministration on electrocardiographic parameters in healthy volunteers.” J Clin Pharmacol. 37(11): 1065-72) have demonstrated that ketoconazole does not increase the QTc interval. This interval is used as a surrogate marker to determine whether drugs have the potential for inducing arrhythmia. US Patent Number 6,040,307 also makes reference to diminished hepatoxicity associated with the 2S,4R enantiomer but provides no data in support of that assertion. The method provided in US Patent Number 6,040,307 does not allow for the assessment of hepatoxicity as the method uses microsomes isolated from frozen tissue.
…………………………
http://www.google.com/patents/EP1853266B1?cl=en
-
DIO-902 is the single enantiomer 2S,4R ketoconazole and is derived from racemic ketoconazole. It is formulated using cellulose, lactose, cornstarch, colloidal silicon dioxide and magnesium stearate as an immediate release 200 mg strength tablet. The chemical name is 2S,4R cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl] methoxyl]phenyl] piperazine, the formula is C26H28Cl2N4O4, and the molecular weight is 531.44. The CAS number is 65277-42-1, and the structural formula is provided below. The chiral centers are at the carbon atoms 2 and 4 as marked.

-
[0132]Ketoconazole is an imidazole-containing fungistatic compound. DIO-902 is an immediate release tablet to be taken orally and formulated as shown in the table below.
Component Percentage 2S,4R ketoconazole;
DIO-90250% Silicified Microcrystalline Cellulose, NF
(Prosolv HD 90)16.5 Lactose Monohydrate, NF (316 Fast-Flo) 22.4 Corn Starch, NF (STA-Rx) 10 Colloidal Silicon Dioxide, NF (Cab-O-Sil M5P) 0.5 Magnesium Stearate, NF 0.6 The drug product may be stored at room temperature and is anticipated to be stable for at least 2 years at 25° C and 50% RH. The drug is packaged in blister packs.

ketoconazole 2S,4R enantiomer
ketoconazole 2S,4S enantiomer
-
ketoconazole 2R,4R enantiomer
ketoconazole 2R,4S enantiomer
……………………..
Journal of Medicinal Chemistry (Impact Factor: 5.61). 08/1992; 35(15):2818-25. DOI: 10.1021/jm00093a015
http://pubs.acs.org/doi/abs/10.1021/jm00093a015
…………………….
Enantioselective separation of ketoconazole enantiomers by membrane extraction
http://www.sciencedirect.com/science/article/pii/S1383586611001638
A new process has been developed to separate ketoconazole (KTZ) enantiomers by membrane extraction, with the oppositely preferential recognition of hydrophobic and hydrophilic chiral selectors in organic and aqueous phases, respectively. This system is established by adding hydrophobic l-isopentyl tartrate (l-IPT) in organic strip phase (shell side) and hydrophilic sulfobutylether-β-cyclodextrin (SBE-β-CD) in aqueous feed phase (lumen side), which preferentially recognizes (+)-2R,4S-ketoconazole and (−)-2S,4R-ketoconazole, respectively. The studies performed involve two enantioselective extractions in a biphasic system, where KTZ enantiomers form four complexes with SBE-β-CD in aqueous phase and l-IPT in organic phase, respectively. The membrane is permeable to the KTZ enantiomers but non-permeable to the chiral selector molecules. Fractional chiral extraction theory, mass transfer performance of hollow fiber membrane, enantioselectivity and some experimental conditions are investigated to optimize the separation system. Mathematical model of I/II = 0.893e0.039NTU for racemic KTZ separation by hollow fiber extraction, is established. The optical purity for KTZ enantiomers is up to 90% when 9 hollow fiber membrane modules of 30 cm in length in series are used.

- I, (−)-2S,4R-ketoconazole;
- II, (+)-2R,4S-ketoconazole;
- CDs, cyclodextrin derivatives;
- l-IPT, l-isopentyl tartrate;
- d-IPT, d-isopentyl tartrate;
- HP-β-CD, hydroxypropyl-β-cyclodextrin;
- Me-β-CD, methyl-β-cyclodextrin;
- β-CD, β-cyclodextrin;
- NTU, number of transfer units;
- HTU, height of a transfer unit;
- PVDF,polyvinylidene fluoride
…………………….
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-
-
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-epichlorohydrin
Original Research Article
- Pages 1283-1294
- Pelayo Camps, Xavier Farrés, Ma Luisa García, Joan Ginesta, Jaume Pascual, David Mauleón, Germano Carganico
-
Bromobenzoates (2R,4R)- and (2S,4S)-18, prepared stereoselectively from (R)- and (S)-epichlorohydrin, were transformed into (2R,4S)-(+)- and (2S,4R)-(−)-Ketoconazole, respectively, following the known synthetic protocols for the racemic mixture.

Tetrahedron Asymmetry 1995, 6(6): 1283
Stereoselective syntheses of both enantiomers of ketoconazole (1) from commercially available (R)- or (S)-epichlorohydrin has been developed. The key-step of these syntheses involves the selective substitution of the methylene chlorine atom by benzoate on a mixture of ![]() and
and ![]() or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
………………..
WO 1996029325
http://www.google.com/patents/WO1996029325A1?cl=en
The incidence of fungal infections has considerably increased over the last decades. Notwithstanding the utility of the antifungal compounds commercialized in the last 15 years, the investigation in this field is however very extensive. During this time, compounds belonging to the azole class have beer, commercialized for both the topical and oral administrations, such a class including imidazoles as well as 1,2,4-triazoles. Some of these compounds car. show m some degree a low gastrointestinal tolerance as well as hepatotoxycity.
A large number of pharmaceutically active compounds are commercialized as stereoisomeric mixtures. On the other hand, the case in which only one of said stereoisomers is pharmaceutically active is frequent.
The undesired enantiomer has a lower activity and it sometimes may cause undesired side-effects.
Ketoconazole (1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine), terconazole (1-[4-[[2(2,4-dichlorophenyl)-2-[(1H-1 , 2 ,4-triazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine) and other related azole antifungal drugs contain in their structure a substituted 1,3-dioxolane ring, in which carbon atoms C2 and C4 are stereogenic centres, therefore four possible stereoisomers are possible. These compounds are commercialized in the form or cis racemates which show a higher antifungal activity than the corresponding trans racemates.
The cis homochiral compounds of the present invention, which are intermediates for the preparation of enantiomerically pure antifungal drugs, have been prepared previously in the racemic form and transformed into the different azole antifungal drugs in the racemic form [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979). J . Med . Chem . , 26, 611 (1983), J . Med . Chem . , 27 , 894 (1984) and US 4,144,346, 4,223,036, 4,358,449 and 4,335,125].
Scheme 1 shows the synthesis described for racemic ketoconazole [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979)]. Scheme 1
)

The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.

The preparation of racemic itraconazole [J. Heeres et al., J. Med . Chem. , 27 , 894 (1984)] is similar to that of terconazole, differing only in the nature of the phenol used in the last step of the synthetic sequence.

In the class of azoles containing a 1,3-dioxolane ring and a piperazine ring and moreover they are pure enantiomers, only the preparation of (+)- and (-)-ketoconazole has been described [D. M. Rotstein et al., J. Med . Chem . , 35, 2818 (1992)] (Scheme 2) starting from the tosylate of (+)- and (-) 2,2-dimethyl-1,3-dioxolane-4-methanol.
Scheme 2

This synthesis suffers from a series of drawbacks, namely: a) the use of expensive, high molecular weight starting products which are available only on a laboratory scale, and b) the need for several chromatographies during the process in order to obtain products of suitable purity, which maKes said synthesis economically unattractive and difficult to apply industrially.
Recently (N. M. Gray, WO 94/14447 and WO 94/14446) the use of (-)-ketoconazole and (+)-ketoconazole as antifungal drugs causing less side-effects than (±)-ketoconazole has been claimed.
The industrial preparation of enantiomerically pure antifungal drugs with a high antifungal activity and less side-effects is however a problem in therapy. The present invention provides novel homochiral compounds which are intermediates for the industrial preparation of already known, enantiomerically pure antifungal drugs such as ketoconazole enantiomers, or of others which have not yet been reported in literature, which are described first in the present invention, such as (+)-terconazole and (-)-terconazoie, which show the cited antifungal action, allowing to attain the same therapeutical effectiveness using lower dosages than those required for racemic terconazole
Example 14 : (2S,4R)-(-)-1-acetyl-4-[4-[ [2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine, (2S,4R) -(- )-ketoconazole.
This compound is prepared following the process described above for (2R,4S)-(+)-ketoconazole. Starting from HNa (60-65% dispersion in paraffin, 32 mg, 0.80 mmol), 1-acetyl-4-(4-hydroxyphenyl)piperazine (153 mg, 0.69 mol) and (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (250 mg, 0.61 mmol), upon crystallization from an acetone:ethyl acetate mixture, (2S,4R) -(-)-ketoconazole is obtained [(2S,4R)-V Ar = 2,4-dichlorophenyl, Y = CH, Z = COCH3] (196 mg, 61% yield) as a solid, m.p. 153-155ºC (lit. 155-157ºC); [α]D 20 = -10.50 (c = 0.4, CHCl3) (lit. [α]D 25 = -10.58. c = 0.4, CHCl3) with e.e. > 99% (determined by HPLC using the chiral stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1 % diethylamine as the eluent).

+ KETOCONAZOLE…. UNDESIRED
Example 7: (2 R ,4S)-(+)-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine (22, 4 S)-(+)-ketoconazole.
To a suspension of NaH (dispersed in 60-65% paraffin, 19.2 mg, 0.48 mmol) in anhydrous DMSO (3 ml),
1-acetyl-4-(hydroxyphenyl)piperazine (102 mg, 0.46 mmol) is added and the mixture is stirred for 1 hour at room temperature. Then, a solution of (2R,4R) – (+)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (160 mg, 0.39 mmol) in anhydrous DMSO (5 ml) is added, and the mixture is heated at 80ºC for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water
(20 ml) and extracted with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with water (3 × 25), dried with Na2SO4 and the solvent is evaporated off under vacuum. The oily residue thus obtained is crystallized from an acetone:ethyl acetate mixture to give (2R,4S)-(+)-ketoconazole ( (2R, 4 S) -V , Ar 2,4-dichlorophenyl, Y = CH , Z = COCH3 ) ( 110 mg , 5 3 % yie ld ) as a white solid, m.p. 155-156°C (lit. 154-156ºC), [α]D 20 = + 8.99 (c = 0.4, CHCl3) (lit. [α]D 25 = + 8.22, c = 0.4, CHCl3), with e.e. > 99% (determined by HPLC using the chirai stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1% of diethylamine, as the eluent; (+)-Ketoconazole retention time 73,28 min. (-)-Ketoconazole, retention time 79.06 min).
IR (KBr), ʋ : 2875, 1645, 1584, 1511, 1462, 1425, 1250, 103S, 313 cm-1.
1H NMR (500 MHz, CDCl3), δ : 2.12 (s, 3H, COCH3),
3.02 (m, 2H, 3-H2), 3.05 (m, 2H, 5-H2), 3.27 (dd, J= 9.5
Hz, J’=7.0 Hz, 1H) and 3.70 (dd, J=9.5 Hz, J’=5.0 Hz, 1 H) (4″-CH2), 3.60 (m, 2H, 6-H2), 3.76 (m, 2H, 2-H2), 3.73 (dd, J=8.0 Hz, J’=5.0 Hz, 1H) and 3.86 (dd, J=8.0 Hz, J’=6.5 Hz, 1H) (5″-H2), 4.34 (m, 1H, 4″-H), 4.40 (d, J=15.0 Hz, 1H) and 5.00 (d, J=15.0 Hz, 1H) (CH2-N), 4.34
(m, 1H, 4″-H), 6.76 [d, J = 9.0 Hz, 2H, 2′(C6′ )-H], 6.88
[d, J=9.0 Hz, 2H, C3′(C5)-H], 6.96 (s, 1H, imidazole 5- H), 6.99 (s, 1H, imidazole 4-H), 7.25 (dd, J=8.5 Hz, J’=2.0 Hz, 1H, 5″‘-H), 7.46 (d, J=2.0 Hz, 1H, 3″‘-H),
7.53 (s, 1H, imidazole 2-H), 7.57 (d, J=8.5 Hz, 1H,
6″‘-H).
13C NMR (75.4 MHz, CDCI3), δ : 21.3 (CH3, COCH3), 41.4 (CH2, C2), 46.3 (CH2, C6), 50.6 (CH2, C3), 51.0 (CH2, C5), 51.2 (CH2, CH2-N), 67.6 [CH2, C5″ and 4″-CH2), 74.7 (CH, C4″), 108.0 (C, C2″), 115.2 [CH, C2′(6′)], 118.8 [CH, C3′(5′)], 121.2 (CH, imidazole C5), 127.2 (CH, C5″‘), 128.5 (CH, imidazole C4), 129.5 (CH, C6′”), 131.3 (CH, C3″‘), 133.0 (C, C2″‘), 134.6 (C, C1′”), 135.8 (C, C4″‘), 138.8 (CH, imidazole C2), 145.6 (C, C1′), 152.8 (C, C4’), 168.9 (C, CO).
…………………………
Experimental and theoretical analysis of the interaction of (+/-)-cis-ketoconazole with beta-cyclodextrin in the presence of (+)-L-tartaric acid
J Pharm Sci 1999, 88(6): 599
Enrico Redenti, Paolo Ventura, Giovanni Fronza, Antonio Selva, Silvia Rivara, Pier Vincenzo Plazzi and Marco Mor
Article first published online: 12 JUN 2000 | DOI: 10.1021/js980468o
http://onlinelibrary.wiley.com/doi/10.1021/js980468o/pdf
1H NMR spectroscopy was used for determining the optical purity of cis-ketoconazole enantiomers obtained by fractional crystallization. The chiral analysis was carried out using β-cyclodextrin in the presence of (+)-l-tartaric acid. The mechanism of the chiral discrimination process, the stability of the complexes formed, and their structure in aqueous solution were also investigated by 1H and 13C chemical shift analysis, two-dimensional NOE experiments, relaxation time measurements, and mass spectrometry experiments. Theoretical models of the three-component interaction were built up on the basis of the available NMR data, by performing a conformational analysis on the relevant fragments on ketoconazole and docking studies on the components of the complex. The model derived from a folded conformation of ketoconazole turned out to be fully consistent with the molecular assembly found in aqueous solution, as inferred from NOE experiments. An explanation of the different association constants for the complexes of the two enantiomers is also provided on the basis of the interaction energies.
| WO1993019061A1 * | Mar 10, 1993 | Sep 30, 1993 | Janssen Pharmaceutica Nv | Itraconazole and saperconazole stereoisomers |
| WO1994025452A1 * | Apr 28, 1994 | Nov 10, 1994 | Ashit K Ganguly | Process for preparing intermediates for the synthesis of antifungal agents |
| EP0050298A2 * | Oct 13, 1981 | Apr 28, 1982 | Hoechst Aktiengesellschaft | 1-(1,3-Dioxolan-2-ylmethyl) azoles, process for their preparation and their use |
| EP0052905A1 * | Nov 19, 1981 | Jun 2, 1982 | Janssen Pharmaceutica N.V. | Novel (2-aryl-4-phenylthioalkyl-1,3-dioxolan-2-yl-methyl)azole derivatives |
| US5208331 * | Jun 18, 1992 | May 4, 1993 | Syntex (U.S.A.) Inc. | Process for preparing 1,3-dioxolane derivatives |
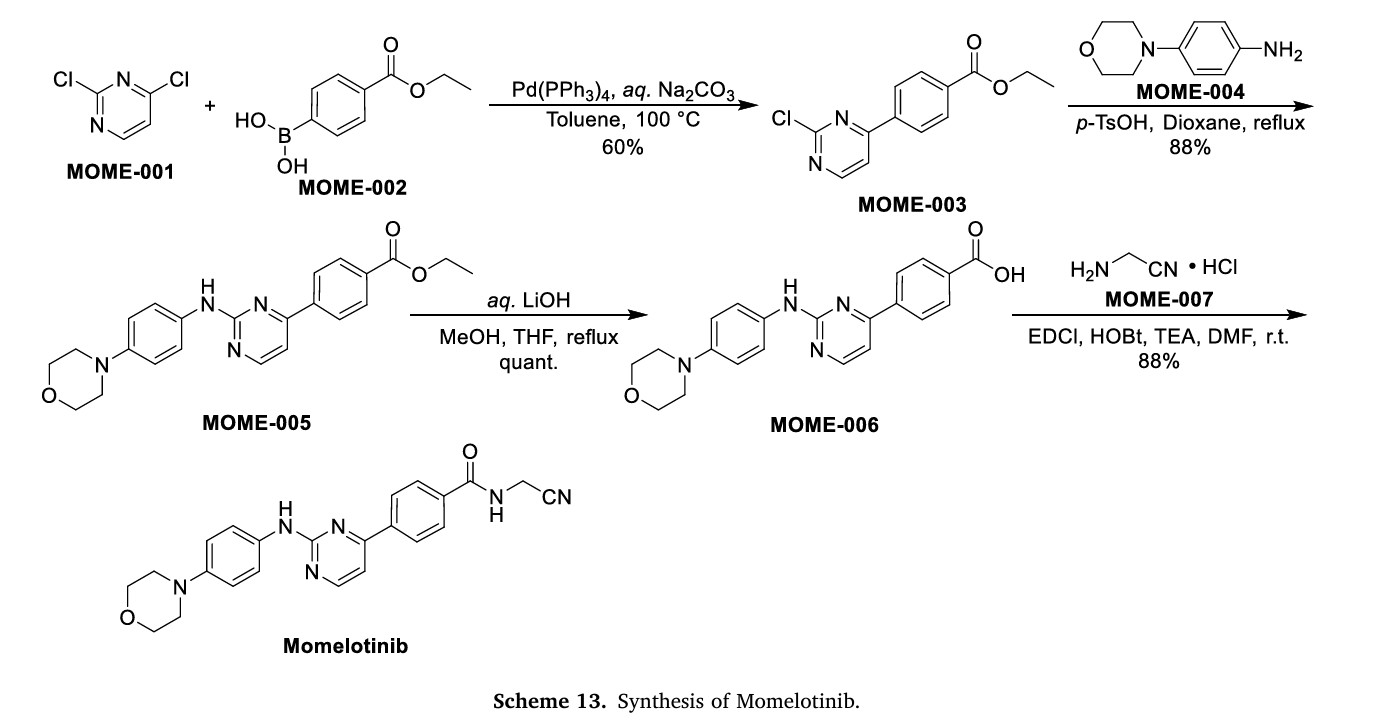
Momelotinib


Momelotinib
414.47, C23H22N6O2,
1056634-68-4
FDA 2023, Ojjaara,
| To treat intermediate or high-risk myelofibrosis in adults with anemia Drug Trials Snapshot |
N-(Cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide
N-(Cyanomethyl)-4-[2-[4-(4-morpholinyl)phenylamino]pyrimidin-4-yl]benzamide
Jak2 tyrosine kinase inhibitor; Jak1 tyrosine kinase inhibitor
Inflammatory disease; Myelofibrosis; Myeloproliferative disorder; Pancreatic ductal adenocarcinoma; Polycythemia vera
CYT 387; CYT-387; momelotinib)
GS-0387
CYT387 sulfate saltCAS No: 1056636-06-6
 CYT387 Mesylate CAS No: 1056636-07-7
CYT387 Mesylate CAS No: 1056636-07-7
DI HCL SALT 1380317-28-1
Momelotinib, sold under the brand name Ojjaara among others, is an anticancer medication used for the treatment of myelofibrosis.[5] It is a Janus kinase inhibitor and it is taken by mouth.[5]
The most common adverse reactions include dizziness, fatigue, bacterial infection, hemorrhage, thrombocytopenia, diarrhea, and nausea.[8]
Momelotinib was approved for medical use in the United States in September 2023,[5][8][9] and in the European Union in January 2024.[6][10]
CYT387 is an ATP-competitive small molecule JAK1 / JAK2 inhibitor with IC50 of 11 and 18 nM for JAK1 and JAK2, respectively. CYT387 is useful for treatment of myeloproliferative disorders and anti-cancer.
CYT-387 is a potent, orally administered JAK1/JAK2/ Tyk2 inhibitor in phase III clinical studiest at Gilead for the treatment of post-polycythemia vera, for the treatment of primary myelofibrosis and for the treatment of post-essential thrombocythemia. Phase II studies are also ongoing, in combination with gemcitabine and nab-paclitaxel, in adults with untreated metastatic pancreatic ductal adenocarcinoma.
The compound possesses an excellent selectivity and safety profile. In 2010 and 2011, orphan drug designation was assigned by the FDA and the EMA, respectively, for the treatment of myelofibrosis. In 2011, orphan drug designation was assigned by the EMA for the treatment of post-essential thrombocythemia myelofibrosis and for the treatment of post-polycythemia vera myelofibrosis.
PAT
http://www.google.com.ar/patents/US8486941?cl=ja
N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide
| 3 |
 |
414.18 | 1H NMR (300 MHz, d6-DMSO): δ 9.47 (1 H, s), 9.32 (1 H, t, J = 5.5 Hz), 8.54 (1 H, d, J = 5.0 Hz), 8.27 (2 H, d, J = 8.7 Hz), 8.02 (2 H, d, J = 8.2 Hz), 7.67 (2 H, d, J = 9.1 Hz), 7.41 (1 H, d, J = 5.5 Hz), 6.93 (2 H, d, J = 9.1 Hz), 4.36 (2 H, d, J = 5.5 Hz), 3.75 (4 H, m), 3.05 (4 H, m). | m/z 415.3 [M + H]+ | N-(cyanomethyl)-4-(2-(4- morpholinophenylamino)pyrimidin- 4-yl)benzamide |
Example 1Synthesis of Compound 3
A mixture of 4-ethoxycarbonylphenyl boronic acid (23.11 g, 119 mmol), 2,4-dichloropyrimidine (16.90 g, 113 mmol), toluene (230 mL) and aqueous sodium carbonate (2 M, 56 mL) was stirred vigorously and nitrogen was bubbled through the suspension for 15 minutes. Tetrakis(triphenylphosphine)palladium[0] (2.61 g, 2.26 mmol) was added. Nitrogen was bubbled through for another 10 min., the mixture was heated to 100° C., then at 75° C. overnight. The mixture was cooled, diluted with ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. The aqueous layer was extracted with ethyl acetate (100 ml) and the two organic extracts were combined. The organics were washed with brine, filtered through sodium sulfate, concentrated, and the resultant solid was triturated with methanol (100 mL) and filtered. The solids were washed with methanol (2×30 mL) and air dried. This material was dissolved in acetonitrile (150 mL) and dichloromethane (200 mL), stirred with MP.TMT Pd-scavenging resin (Agronaut part number 800471) (7.5 g) over 2 days. The solution was filtered, the solids were washed with dichloromethane (2×100 mL), and the filtrate concentrated to give ethyl 4-(2-chloropyrimidin-4-yl)benzoate as an off-white solid (17.73 g, 60%)—additional washing with dichloromethane yielded a further 1.38 g and 0.5 g of product. 1H NMR (300 MHz, d6-DMSO) δ 8.89 (1H, d, J=5.0 Hz); 8.32 (2H, d, J=8.7 Hz); 8.22 (1H, d, J=5.5 Hz); 8.12 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=7.1 Hz); 1.34 (3H, t, J=7.1 Hz); LC-ESI-MS (method B): rt 7.3 min.; m/z 263.0/265.0 [M+H]+.
A mixture of ethyl 4-(2-chloropyrimidin-4-yl)benzoate (26.15 g, 99.7 mmol) and 4-morpholinoaniline (23.10 g, 129.6 mmol) was suspended in 1,4-dioxane (250 mL). p-Toluenesulfonic acid monohydrate (17.07 g, 89.73 mmol) was added. The mixture was heated at reflux for 40 h., cooled to ambient temperature, concentrated then the residue was partitioned between ethyl acetate and 1:1 saturated sodium bicarbonate/water (1 L total). The organic phase was washed with water (2×100 mL) and concentrated. The aqueous phase was extracted with dichloromethane (3×200 mL). The material which precipitated during this workup was collected by filtration and set aside. The liquid organics were combined, concentrated, triturated with methanol (200 mL) and filtered to yield additional yellow solid. The solids were combined, suspended in methanol (500 mL), allowed to stand overnight then sonicated and filtered. The solids were washed with methanol (2×50 mL) to give, after drying, ethyl 4-(2-(4-morphonlinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 88%). 1H NMR (300 MHz, d6-DMSO) δ 9.49 (1H, s); 8.54 (1H, d, J=5.0 Hz); 8.27 (2H, d, J=8.7 Hz); 8.10 (2H, d, J=8.7 Hz), 7.66 (2H, d, J=9.1 Hz); 7.38 (1H, d, J=5.0 Hz); 6.93 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=6.9 Hz), 3.73 (4H, m); 3.04 (4H, m); 1.34 (3H, t, J=6.9 Hz); LC-ESI-MS (method B): rt 7.5 min.; m/z 404.1 [M+H].
A solution of ethyl 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 87.6 mmol) in 3:1 methanol/tetrahydrofuran (350 mL) was treated with lithium hydroxide (4.41 g, 183.9 mmol) in water (90 mL). The mixture was heated at reflux for 2 h., cooled, concentrated and acidified with hydrochloric acid (2M, 92.5 mL, 185 mmol). The dark precipitate was filtered, washed with water, and dried under vacuum. The solid was ground to a powder with a mortar and pestle, triturated with methanol (500 mL) then filtered again to yield 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid as a muddy solid. This material was washed with ether, air dried overnight, and ground to a fine powder with mortar and pestle. On the basis of mass recovery (34.49 g) the yield was assumed to be quantitative. 1H NMR (300 MHz, d6-DMSO) δ 9.47 (1H, s); 8.53 (1H, d, J=5.2 Hz); 8.24 (2H, d, J=8.5 Hz); 8.08 (2H, d, J=8.8 Hz), 7.66 (2H, d, J=9.1 Hz); 7.37 (1H, d, J=5.2 Hz); 6.93 (2H, d, J=9.1 Hz); 3.73 (4H, m); 3.04 (4H, m). LC-ESI-MS (method C): rt 7.3 min.; m/z 377.1 [M+H]+.
To a suspension of 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid (theoretically 32.59 g, 86.6 mmol) in DMF (400 mL) was added triethylamine (72.4 mL, 519.6 mmol, 6 eq.) The mixture was sonicated to ensure dissolution. Aminoacetonitrile hydrochloride (16.02 g, 173.2 mmol) was added followed by N-hydroxybenzotriazole (anhydrous, 14.04 g, 103.8 mmol) and 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (19.92 g, 103.8 mmol). The suspension was stirred vigorously overnight. The solvent was evaporated under reduced pressure, the residue was diluted with 5% sodium bicarbonate (400 mL) and water (300 mL), giving a yellow solid, which was broken up and filtered. The solids were washed several times with 100 mL portions of water, triturated with hot methanol/dichloromethane (500 mL, 1:1), concentrated to a volume of approximately 300 mL), cooled and filtered. The solids were washed with cold methanol (3×100 mL), ether (200 mL) and hexane (200 mL) prior to drying to afford
Compound 3 (31.69 g, 88%). M.p. 238-243° C.
Microanalysis: Found C, 66.52; H, 5.41; N, 20.21. C23H26N6O10S2 requires C, 66.65; H, 5.35; N, 20.28%.
13C NMR (75.5 MHz, d6-DMSO) δ 166.04, 162.34, 160.26, 159.14, 146.14, 139.87, 134.44, 132.73, 127.80, 126.84, 120.29, 117.49, 115.50, 107.51, 66.06, 49.16, 27.68.
1H NMR GIVEN ABOVE
Example 6Salt Formation from Compound 3
Compound 3 (10.0 g) was suspended in methanol (1 L). Concentrated sulfuric acid (10.52 g, 90% w/w) was added dropwise to the stirring solution. A clear brown solution resulted and a solid lump formed. The solution was filtered quickly then allowed to continue stirring for 3 h (a second precipitate appeared within minutes). After this time the pale yellow precipitate was collected by filtration, washed with methanol (10 mL) then dried under vacuum overnight to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium hydrogensulfate, as a pale yellow solid (10.20 g, 69%). m.p. 205° C. Microanalysis: Found C, 45.18; H, 4.36; N, 13.84; S, 10.24. C23H26N6O10S2 requires C, 45.24; H, 4.29; N, 13.76; S 10.50%. 1H NMR (300 MHz, d6-DMSO) δ 9.85 (br. s, 1H), 9.34 (t, J=5.4 Hz, 1H), 8.59 (d, J=5.2 Hz, 1H), 8.27 (d, J=8.5 Hz, 2H), 8.03 (d, J=8.5 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 7.50 (d, J=5.2 Hz, 1H), 7.34 (br. s, 2H), 4.36 (d, J=5.4 Hz, 2H), 3.89 (br. s, 4H), 3.37 (br. s, 4H); 13C NMR (75.5 MHz, d6-DMSO) δ 166.07, 163.36, 159.20, 158.48, 140.19, 139.34, 136.45, 134.89, 128.00, 127.22, 121.13, 119.89, 117.59, 109.05, 64.02, 54.04, 27.82. LC-ESI-MS (method D): rt 10.0 min.; m/z 415.1 [M+H]+.
Compound 3 (0.25 g) was suspended in methanol (25 ml). Methane sulfonic acid (0.255 g) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 3 h, after which the volume was reduced to 9 ml. The resultant precipitate was collected and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium methanesulfonate as a pale yellow solid (0.22 g). m.p. 208° C. 1H NMR (300 MHz, d6-DMSO) δ 9.83 (br. s, 1H), 9.35 (t, J=5.3 Hz, 1H), 8.59 (d, J=5.1 Hz, 1H), 8.28 (d, J=8.5 Hz, 2H), 8.04 (d, J=8.5 Hz, 2H), 7.83 (d, J=9.0 Hz, 2H), 7.50 (d, J=5.5 Hz, 1H), 7.31 (d, J=9.0 Hz, 2H), 4.36 (d, J=5.5 Hz, 2H), 3.88 (m, 4H), 3.35 (br. s, 4H), 2.36 (s, 6H); LC-ESI-MS (method D): rt 10.2 min.; m/z 415.3 [M+H]+.
Compound 3 (0.50 g) was suspended in methanol (45 ml). A freshly prepared solution of hydrochloric acid in methanol (2.6 ml, HCl conc. 40 mg/ml) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 2 h, then the resultant precipitate was collected, washed with methanol (5 ml) and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium chloride a pale yellow solid (0.30 g). m.p. 210° C. 1H NMR (300 MHz, d6-DMSO) 1H NMR (300 MHz, DMSO) δ 9.92 (br. s, 1H), 9.42 (t, J=5.3, 1H), 8.62 (d, J=4.8, 1H), 8.29 (d, J=8.1, 2H), 8.06 (d, J=8.1, 2H), 7.89 (d, J=9.0, 2H), 7.53 (br. s, 3H), 4.36 (d, J=5.4, 2H), 3.82 (br. s, 4H), 3.43 (br. s, 4H)
LC-ESI-MS (method D): rt 10.3 min.; m/z 415.3 [M+H]+.
PAT
WO 2014114274
References on CYT387
. [1] A Pardanani et al CYT387, a Selective JAK1 / JAK2 inhibitor: in vitroassessment of kinase selectivity and preclinical s using Cell lines and Primary cells from polycythemia vera Patients Leukemia (2009) 23, 1441-1445
Abstract
Somatic mutations in Janus kinase 2 (JAK2), including JAK2V617F, result in dysregulated JAK-signal transducer and activator transcription (STAT) signaling, which is implicated in myeloproliferative neoplasm (MPN) pathogenesis. CYT387 is an ATP-competitive small molecule that potently inhibits JAK1 / JAK2 kinases ( IC (50) = 11 and 18 nM, respectively), with significantly less activity against other kinases, including JAK3 (IC (50) = 155 nM). CYT387 inhibits growth of Ba / F3-JAK2V617F and human erythroleukemia (HEL) cells ( IC (50) approximately 1500 nM) or Ba / F3-MPLW515L cells (IC (50) = 200 nM), but has considerably less activity against BCR-ABL harboring K562 cells (IC = 58 000 nM). Cell lines harboring mutated JAK2 alleles (CHRF-288-11 or Ba / F3-TEL-JAK2) were inhibited more potently than the corresponding pair harboring mutated JAK3 alleles (CMK or Ba / F3-TEL-JAK3), and STAT-5 phosphorylation was inhibited in HEL cells with an IC (50) = 400 nM. …
[2]. Tyner Jeffrey W. et al CYT387, a novel JAK2 inhibitor, induces Hematologic Responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms Blood June 24, 2010vol. no 115. 255232-5240
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. IDENTIFIED We have an aminopyrimidine derivative ( CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting JAK2 inhibitors That May be Unable to Eliminate JAK2 (V617F) cells, Consistent with Preliminary results from Clinical Trials of JAK2 inhibitors in myelofibrosis. …
[3]. Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH.Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma.J Chromatogr B Analyt Technol Biomed Life Sci 2012 May 1; 895-896:. 174-7 Epub 2012 Mar 23..
abstract
A quantitative bioanalytical Liquid Chromatography-Tandem Mass spectrometric (LC-MS / MS) assay for the JAK2 inhibitor CYT387 WAS Developed and validated. Plasma samples Were Treated using pre-Protein precipitation with acetonitrile containing cediranib as Internal Standard. The extract WAS Directly Injected into the chromatographic system after dilution with water. This system consisted of a sub-2 μm particle, trifunctional bonded octadecyl silica column with a gradient using 0.005% (v / v) of formic acid in a mixture of water and methanol. The eluate was transferred into the electrospray interface with positive ionization and the analyte was detected in the selected reaction monitoring mode of a triple quadrupole mass spectrometer. The assay was validated in a 0.25-1000 ng / ml calibration range. Within day precisions were 3.0-13.5%, BETWEEN Day Precisions 5.7% and 14.5%. Accuracies Were BETWEEN 96% and 113% for the Whole Calibration range. The Drug WAS stable under All Relevant Analytical Conditions. Finally, the assay successfully WAS Used to ASSESS Drug Levels in mice.
[4] . Monaghan KA, Khong T, Burns CJ, Spencer A.The novel JAK inhibitor CYT387 suppresses Multiple Signalling pathways, and induces apoptosis in Prevents Proliferation phenotypically Diverse myeloma cells.Leukemia 2011 Dec; 25 (12):. 1891-9.
Abstract
Janus kinases (JAKs) are involved in various signalling pathways exploited by malignant cells. In multiple myeloma (MM), the interleukin-6 / JAK / signal transducers and activators of transcription (IL-6 / JAK / STAT) pathway has been the focus of research for a number of years and IL-6 has an established role in MM drug resistance. JAKs therefore make a rational drug target for anti-MM therapy. CYT387 is a novel, orally bioavailable JAK1 / 2 inhibitor, which has recently been described. This preclinical evaluation of CYT387 for treatment of MM demonstrated that CYT387 was able to prevent IL-6-induced phosphorylation of STAT3 and greatly decrease IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in human myeloma cell lines (HMCL). CYT387 inhibited MM proliferation in a time- and dose-dependent manner in 6/8 HMCL, and this was not abrogated by the addition of exogenous IL-6 (3/3 HMCL). Cell cycling was inhibited with a G (2) / M accumulation of cells, and apoptosis was induced by CYT387 in all HMCL tested (3/3). CYT387 synergised in killing HMCL when used in combination with the conventional anti-MM therapies melphalan and bortezomib. Importantly, WAS Also apoptosis induced in Primary Patient MM cells (N = 6) with CYT387 as a single agent, and synergy WAS Seen Again when Combined with Conventional therapies.
[5]. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW.CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms.Blood 2010 Jun 24; 115 (25):. 5232- 40. Epub 2010 Apr 12.
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. We have IDENTIFIED an aminopyrimidine derivative (CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting that JAK2 inhibitors may be unable to eliminate JAK2 (V617F) cells, consistent with preliminary results from clinical trials of JAK2 inhibitors in myelofibrosis. While the clinical benefit of JAK2 inhibitors may be substantial, not the least due to reduction of inflammatory cytokines and symptomatic improvement, our data add to increasing evidence that kinase inhibitor monotherapy of malignant disease is not curative, suggesting a need for drug combinations to optimally target the malignant cells.
JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to, amongst other things, cellular proliferation.
The central role played by the JAK family of protein tyrosine kinases in the cytokine dependent regulation of both proliferation and end function of several important cell types indicates that agents capable of inhibiting the JAK kinases are useful in the prevention and chemotherapeutic treatment of disease states dependent on these enzymes. Potent and specific inhibitors of each of the currently known four JAK family members will provide a means of inhibiting the action of the cytokines that drive immunological and inflammatory diseases.
Myeloproliferative disorders (MPD) include, among others, polycythemia vera (PV), primary myelofibrosis, thrombocythemia, essential thrombocythemia (ET), idiopathic myelofibrosis (IMF), chronic myelogenous leukemia (CML), systemic mastocystosis (SM), chronic neutrophilic leukemia (CNL), myelodisplastic syndrome (MDS) and systemic mast cell disease (SMCD). JAK2 is a member of the JAK family of kinases in which a specific mutation (JAK2V617F) has been found in 99% of polycythemia vera (PV) patients and 50% of essential thrombocytopenia (ET) and idiopathic myelofibrosis (MF). This mutation is thought to activate JAK2, giving weight to the proposition that a JAK2 inhibitor will be useful in treating these types of diseases.
Asthma is a complex disorder characterized by local and systemic allergic inflammation and reversible airway obstruction. Asthma symptoms, especially shortness of breath, are a consequence to airway obstruction, and death is almost invariably due to asphyxiation. Airway Hyper Responsiveness (AHR), and mucus hyper secretion by goblet cells are two of the principle causes of airway obstruction in asthma patients. Intriguingly recent work in animal experimental models of asthma has underscored the importance of IL-13 as a key player in the pathology of asthma. Using a specific IL-13 blocker, it has been demonstrated that IL-13 acts independently of IL-4 and may be capable of inducing the entire allergic asthma phenotype, without the induction of IgE (i.e. in a non-atopic fashion). This and other models have pointed to an important second tier mechanism for elicitating the pathophysiology of asthma, that is not dependent on the production of IgE by resident B-cells or the presence of eonisophils. A direct induction of AHR by IL-13, represents an important process that is likely to be an excellent target for intervention by new therapies. A contemplated effect of a JAK2 inhibitor to the lungs would result in the suppression of the local release of IL-13 mediated IgE production, and therefore reduction in histaminine release by mast cells and eosinophils. This and other consequences of the absence of IL-13 indicate that many of the effects of asthma may be alleviated through administration of a JAK2 inhibitor to the lungs.
Chronic Obstructive Pulmonary Disease (COPD) is a term which refers to a large group of lung diseases which can interfere with normal breathing. Current clinical guidelines define COPD as a disease state characterized by airflow limitation which is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles and gases, particularly cigarette smoke and pollution. Several studies have pointed to an association between increased production of IL-13 and COPD, lending support to the proposition that the potential alleviation of asthma symptoms by use of a JAK2 inhibitor, may also be achieved in COPD. COPD patients have a variety of symptoms including cough, shortness of breath, and excessive production of sputum. COPD includes several clinical respiratory syndromes including chronic bronchitis and emphysema.
Chronic bronchitis is a long standing inflammation of the bronchi which causes increased production of mucus and other changes. The patient’s symptoms are cough and expectoration of sputum. Chronic bronchitis can lead to more frequent and severe respiratory infections, narrowing and plugging of the bronchi, difficult breathing and disability.
Emphysema is a chronic lung disease which affects the alveoli and/or the ends of the smallest bronchi. The lung loses its elasticity and therefore these areas of the lungs become enlarged. These enlarged areas trap stale air and do not effectively exchange it with fresh air. This results in difficult breathing and may result in insufficient oxygen being delivered to the blood. The predominant symptom in patients with emphysema is shortness of breath.
Additionally, there is evidence of STAT activation in malignant tumors, among them lung, breast, colon, ovarian, prostate and liver cancer, as well as Hodgkins lymphoma, multiple myeloma and hepatocellular carcinoma. Chromosomal translocations involving JAK2 fusions to Tel, Bcr and PCM1 have been described in a number of hematopoietic malignancies including chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), chronic eosinophilic leukemia (CEL), myelodisplastic syndrome (MDS), myeloproliferative disease (MPD) and acute lymphocytic leukemia (ALL). This suggests treatment of hyperproliferative disorders such as cancers including multiple myeloma; prostate, breast and lung cancer; Hodgkin’s Lymphoma; CML; AML; CEL; MDS; ALL; B-cell Chronic Lymphocytic Leukemia; metastatic melanoma; glioma; and hepatoma, by JAK inhibitors is indicated.
Potent inhibitors of JAK2, in addition to the above, will also be useful in vascular disease such as hypertension, hypertrophy, cardiac ischemia, heart failure (including systolic heart failure and diastolic heart failure), migraine and related cerebrovascular disorders, stroke, Raynaud’s phenomenon, POEMS syndrome, Prinzmetal’s angina, vasculitides, such as Takayasu’s arteritis and Wegener’s granulomatosis, peripheral arterial disease, heart disease and pulmonary arterial hypertension.
Pulmonary arterial hypertension (PAH) is a pulmonary vascular disease affecting the pulmonary arterioles resulting in an elevation in pulmonary artery pressure and pulmonary vascular resistance but with normal or only mildly elevated left-sided filling pressures. PAH is caused by a constellation of diseases that affect the pulmonary vasculature. PAH can be caused by or associated with collagen vascular disorders such as systemic sclerosis (scleroderma), uncorrected congenital heart disease, liver disease, portal hypertension, HIV infection, Hepatitis C, certain toxins, splenectomy, hereditary hemorrhagic teleangiectasia, and primary genetic abnormalities. In particular, a mutation in the bone morphogenetic protein type 2 receptor (a TGF-b receptor) has been identified as a cause of familial primary pulmonary hypertension (PPH). It is estimated that 6% of cases of PPH are familial, and that the rest are “sporadic.” The incidence of PPH is estimated to be approximately 1 case per 1 million population. Secondary causes of PAH have a much higher incidence. The pathologic signature of PAH is the plexiform lesion of the lung which consists of obliterative endothelial cell proliferation and vascular smooth muscle cell hypertrophy in small precapillary pulmonary arterioles. PAH is a progressive disease associated with a high mortality. Patients with PAH may develop right ventricular (RV) failure. The extent of RV failure predicts outcome. The JAK/STAT pathway has recently been implicated in the pathophysiology of PAH. JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to proliferation of endothelial cells and smooth muscle cells, and cause hypertrophy of cardiac myocytes. There are three different isoforms of JAK: JAK1, JAK2, and JAK3. Another protein with high homology to JAKs is designated Tyk2. An emerging body of data has shown that the phosphorylation of STAT3, a substrate for JAK2, is increased in animal models of PAH. In the rat monocrotaline model, there was increased phosphorylation of the promitogenic transcription factor STAT3. In this same study pulmonary arterial endothelial cells (PAECs) treated with monocrotaline developed hyperactivation of STAT3. A promitogenic agent or protein is an agent or protein that induces or contributes to the induction of cellular proliferation. Therefore, one effect of JAK2 inhibition would be to decrease proliferation of endothelial cells or other cells, such as smooth muscle cells. A contemplated effect of a JAK2 inhibitor would be to decrease the proliferation of endothelial cells or other cells which obstruct the pulmonary arteriolar lumen. By decreasing the obstructive proliferation of cells, a JAK2 inhibitor could be an effective treatment of PAH.
Additionally the use of JAK kinase inhibitors for the treatment of viral diseases and metabolic diseases is indicated.
Although the other members of the JAK family are expressed by essentially all tissues, JAK3 expression appears to be limited to hematopoetic cells. This is consistent with its essential role in signalling through the receptors for IL-2, IL4, IL-7, IL-9 and IL-15 by non-covalent association of JAK3 with the gamma chain common to these multichain receptors. Males with X-linked severe combined immunodeficiency (XSCID) have defects in the common cytokine receptor gamma chain (gamma c) gene that encodes a shared, essential component of the receptors of interleukin-2 (IL-2), IL-4, IL-7, IL-9, and IL-15. An XSCID syndrome in which patients with either mutated or severely reduced levels of JAK3 protein has been identified, suggesting that immunosuppression should result from blocking signalling through the JAK3 pathway. Gene Knock out studies in mice have suggested that JAK3 not only plays a critical role in B and T lymphocyte maturation, but that JAK3 is constitutively required to maintain T cell function. Taken together with the biochemical evidence for the involvement of JAK3 in signalling events downstream of the IL-2 and IL-4 receptor, these human and mouse mutation studies suggest that modulation of immune activity through the inhibition of JAK3 could prove useful in the treatment of T-cell and B-cell proliferative disorders such as transplant rejection and autoimmune diseases. Conversely undesired inhibition of JAK3 could have a devastating effect on the immune status of an individual treated with drug.
Although the inhibition of various types of protein kinases, targeting a range of disease states, is clearly beneficial, it has been to date demonstrated that the identification of a compound which is selective for a protein kinase of interest, and has good “drug like” properties such as high oral bioavailability, is a challenging goal. In addition, it is well established that the predictability of inhibition, or selectivity, in the development of kinase inhibitors is quite low, regardless of the level sequence similarity between the enzymes being targeted.
The challenges in developing therapeutically appropriate JAK2 inhibitors for use in treatment kinase associated diseases such as immunological and inflammatory diseases including organ transplants; hyperproliferative diseases including cancer and myeloproliferative diseases; viral diseases; metabolic diseases; and vascular diseases include designing a compound with appropriate specificity which also has good drug-likeliness.
There is therefore a continuing need to design and/or identify compounds which specifically inhibit the JAK family of kinases, and particularly compounds which may preferentially inhibit one of the JAK kinases relative to the other JAK kinases, particularly JAK2. There is a need for such compounds for the treatment of a range of diseases.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Omjjara (GlaxoSmithKline Australia Pty Ltd)”. Therapeutic Goods Administration (TGA). 14 January 2025. Retrieved 20 January 2025.
- https://www.tga.gov.au/resources/artg/442230 [bare URL]
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-12-20]”. Health Canada. 20 December 2024. Retrieved 21 December 2024.
- “Ojjaara product information”. Health Canada. 8 November 2024. Retrieved 27 December 2024.
- “Ojjaara- momelotinib tablet”. DailyMed. U.S. National Library of Medicine. 15 September 2023. Archived from the original on 30 November 2023. Retrieved 20 September 2023.
- “Omjjara EPAR”. European Medicines Agency. 5 August 2011. Retrieved 18 March 2024.
- “Omjjara Product information”. Union Register of medicinal products. 26 January 2024. Retrieved 18 March 2024.
- “FDA Roundup: September 19, 2023”. U.S. Food and Drug Administration (FDA) (Press release). 19 September 2023. Archived from the original on 21 September 2023. Retrieved 20 September 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 15 September 2023. Archived from the original on 21 January 2023. Retrieved 20 September 2023. This article incorporates text from this source, which is in the public domain.
- “GSK’s Omjjara Authorized in EU for Treating Myelofibrosis With Anemia”. MarketWatch. Retrieved 30 January 2024.
- Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A (August 2009). “CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients”. Leukemia. 23 (8): 1441–1445. doi:10.1038/leu.2009.50. PMID 19295546. S2CID 26947444.
- “Omjjara: Pending EC decision”. European Medicines Agency (EMA). 10 November 2023. Archived from the original on 29 November 2023. Retrieved 5 December 2023.
External links
- Clinical trial number NCT04173494 for “A Study of Momelotinib Versus Danazol in Symptomatic and Anemic Myelofibrosis Patients (MOMENTUM)” at ClinicalTrials.gov
- Clinical trial number NCT01969838 for “Momelotinib Versus Ruxolitinib in Subjects With Myelofibrosis (Simplify 1)” at ClinicalTrials.gov
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
N-(Cyanomethyl)-4-{2-[4-(morpholin-4-yl)anilino]pyrimidin-4-yl}benzamide
|
|
Other names
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank |
|
| KEGG | |
|
PubChem CID
|
|
| UNII |
|
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C23H22N6O2 | |
| Molar mass | 414.469 g·mol−1 |
| Pharmacology | |
| L01EJ04 (WHO) | |
| By mouth | |
| Legal status | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Clinical data | |
|---|---|
| Other names | Momelotinib hydrochloride hydrate (JAN JP), Momelotinib dihydrochloride (USAN US) |
| License data |
|
| Identifiers | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
//////////Momelotinib, APPROVALS 2023, FDA 2023, Ojjaara, high-risk myelofibrosis, anemia, APPROVALS 2024, EU 2024, EMA 2024
REF
European Journal of Medicinal Chemistry 265 (2024) 116124
Scheme 13 illustrates the synthesis of Momelotinib Dihydrochloride [48]. The Pd(PPh3) 4-catalyzed Suzuki coupling reaction between 2,4-dichloropyrimidine (MOME-001) and boronic acid MOME-002
resulted in the formation of MOME-003. Subsequently, MOME-003 underwent a substitution reaction with aniline MOME-004 in the presence of p-toluenesulfonic acid (TsOH), yielding MOME-005.
MOME-005 was hydrolyzed by lithium hydroxide, leading to the formation of carboxylic acid MOME-006. MOME-006 underwent amidation with 2-aminoacetonitrile hydrochloride (MOME-007) to produce
Momelotinib.
[48] G.D. Smith, R. Fida, M.M. Kowalski, N-(cyanomethyl)-4-[2-[[4-(4-morpholinyl)
phenyl]amino]-4-pyrimidinyl]-benzamide [CYT387] or a Related Compound,
2012. WO2012071612A1.
.
Decernotinib … JAK inhibitor for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.

Decernotinib
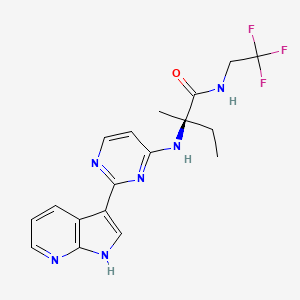
Decernotinib
N2-[2-(1H-Pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]-N-(2,2,2-trifluoroethyl)-D-isovalinamide
(R)-2-(2-(lH-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2- trifluoroethyl)butanamide
Vertex Pharmaceuticals Inc
UNII-MZK2GP0RHK, VX-509, VRT-831509, cas 944842-54-0
Molecular Formula: C18H19F3N6O
Molecular Weight: 392.37827
In phase 3 for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.
 DECERNOTINIB
DECERNOTINIB
The Janus kinases (JAK) are a family of tyrosine kinases consisting of JAK1, JAK2, JAK3, and TYK2. The JAKs play a critical role in cytokine signaling. The down-stream substrates of the JAK family of kinases include the signal transducer and activator of transcription (STAT) proteins. JAK/STAT signaling has been implicated in the mediation of many abnormal immune responses such as psoriasis. Moreover, JAK kinases represent an established therapeutic target for this disease.
For example, JAK kinases are an established therapeutic target for treating psoriasis. Stump K. L., et al., Arthritis Res. Ther. (201 1) 13:R68; Fridman J.S., et al., J Immunol. (2010) 184:5298-5307; West K., Curr. Op. Investig. Drugs (2009) 10:491-504; Kremer J. M. et al., Arthritis Rheumatism (2009) 60(7):1895- 1905; Xiong, W. et al., Ther Adv Musculoskelet Dis. (201 1) 3(5): 255-266; Panes, J. et al. 19th Ann. Eur. Gastroenterology Week (Oct 22-26, 2011) Stockholm, SE, PI 456; and Drugs in R & D “Tofacitinib” (2010) 10(4):271-84.
Compounds described as kinase inhibitors, particularly the JAK family kinases, are disclosed in WO 2005/095400 and WO 2007/084557. Also disclosed in these publications are processes and intermediates for preparing these compounds
Decernotinib ( VX-509 ) is an oral selective JAK3 inhibitor being evaluated for the treatment of rheumatoid arthritis ( RA ). This was a 24-week, randomized, placebo-controlled, double-blind, phase 2 study of four dosing regimens of Decernotinib, administered to patients with RA with inadequate response to Methotrexate ( MTX ).
The aim of the study was to assess the efficacy and safety of four dosing regimens of VX-509 administered to patients with rheumatoid arthritis on stable background Methotrexate therapy.
Patients with active rheumatoid arthritis ( C-reactive protein [ CRP ] greater than ULN, greater than or equal to 6 swollen joints [ of 66 ], and greater than or equal to 6 tender joints [ of 68 ] ) taking stable doses of MTX were randomized 1:1:1:1:1 to receive placebo or one of four dosing regimens of Decernotinib ( 100 mg QD, 150 mg QD, 200 mg QD, or 100 mg BID ) for a duration of 24 weeks.
The primary efficacy endpoints at week 12 were met and have previously been reported; 24-week efficacy and safety results are now reported.
A total of 358 patients were randomized and received greater than or equal to 1 dose of study drug; 81% of patients were female, with a mean age of 53 years.
At baseline, the mean tender joint count was 23.8, the mean swollen joint count was 16.1, and the average disease duration was 7.3 years.
After 24 weeks of treatment the proportion of patients achieving ACR20, ACR50, ACR70, DAS28 ( CRP ) less than 2.6 and DAS28 ( ESR ) less than 2.6 and the decrease from baseline in DAS28 ( CRP ) were statistically significantly greater in each of the Decernotinib dose groups than in the placebo group.
Over 24 weeks, the percentage of patients with any adverse event was higher in the Decernotinib group ( all Decernotinib dose groups combined ) ( 59.9% ) relative to placebo ( 42.3% ) and led to study discontinuation in 9.1% and 8.5% of patients in the Decernotinib and placebo groups, respectively.
The most common adverse reactions in the Decernotinib group were headache ( 8.7% ), hypercholesterolemia ( 5.2% ), and diarrhea ( 4.5% ).
Serious adverse reactions occurred in similar proportions of patients receiving Decernotinib ( 7.3% ) or placebo ( 5.6% ), but there were more serious infections in the Decernotinib group ( 3.5% ) compared with placebo ( 1.4% ).
Through 24 weeks there were two serious adverse effects that resulted in death; one was cardiac failure in the Decernotinib 100 mg BID group ( previously reported ) and one was pancytopenia in a patient with pneumonia in the Decernotinib 200 mg QD group.
Elevations in transaminase levels and decreases in median neutrophil and lymphocyte counts were observed in the Decernotinib groups and were generally mild.
Safety profiles were comparable across groups receiving Decernotinib.
In conclusion, all tested doses of Decernotinib significantly improved signs and symptoms of rheumatoid arthritis versus placebo when administered in combination with stable background Methotrexate therapy for 24 weeks.
Decernotinib was associated with small increases in adverse reactions rates, serious infections, and mostly minor laboratory abnormalities. ( Xagena )
Source: EULAR Meeting – van Vollenhoven R et al, Ann Rheum Dis 2014;73(Suppl2)
see
WO 2007084557
http://www.google.com/patents/WO2007084557A2?cl=en
………………………………………
WO 2013006634
http://www.google.com/patents/WO2013006634A2?cl=en

Formula I is:
The present invention provides a process for preparing (R)-2-(2-(lH-pyrrolo[2,3- b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2-trifluoroethyl)butanamide of Formula la:

la
comprising the steps of:
ivb) reacting lH-pyrrolo[2,3-b]pyridine (5a) with p-toluenesulfonyl chloride in the presence of an organic solvent to generate l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a)

5a 9a
vb) reacting l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a) in an organic solvent with N-bromosuccinimide to generate 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a)

vi) reacting 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a) with triisopropyl borate in the presence of a strong lithium base in an organic solvent to generate
l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) 0H

8a
vii) esterifying l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) with pinacolate alcohol in an organic solvent to generate
3 -(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -tosyl- 1 H-pyrrolo[2,3 -bjpyridine (la) :

viiib) reacting 2,4-dichloropyrimidine (11a) with a hydrochloride salt of D-isovaline (15a) under coupling condition to generate a compound of Formula 2a

11a 2a
ixb) reacting the compound of Formula 2a with HC1 to generate the hydrochloride salt of the compound of Formula 2a;
i) reacting the compound of Formula la with the compound of Formula 2a with in the presence of water, an organic solvent, an inorganic base, and a transition metal catalyst to generate a compound of Formula 3a,

ii) deprotecting the compound of Formula 3a under basic conditions to generate a compound of Formula 4a

4a ; and iii) reacting the compound of Formula 4a with 2,2,2-trifluoroethylamine in the presence of a coupling agent and an organic solvent to generate the compound of Formula la.


– l13C4, 15N2]


……………………………………………………………….
WO 2013070606
http://www.google.com/patents/WO2013070606A1?cl=en
………………………………………………….
patent WO2014074471
WO2014074471 claiming use of heterocyclic compound (preferably decernotinib) for treating psoriasis. Vertex is developing decernotinib, an oral JAK 3 inhibitor, for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis. As of July 2014, the drug is Phase 3 trials.
http://www.google.com/patents/WO2014074471A1?cl=en
Table 1:
COMPD 1 IS DECERNOTINIB


Example 1: Analytical Methods Used
[0260] (A) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (60:40:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm. Run time was 25-26 minutes.
[0261] (B) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (90: 10:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm.
[0262] (C) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9:1). Gradient 5 % to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 245 nm.
T = 25 °C.
[0263] (D) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9: 1). Gradient 15% to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 220 nm.
T = 35 °C.
[0264] Example 2: Preparation of Compounds of Formula I [0265] General Synthetic Scheme

[0266] The Boc-protected amino acid starting material (1) undergoes amidation in the presence of an activating agent, a coupling reagent, and the acid salt of the amine HNR7R17 to generate the Boc-protected amide intermediate (2). The amide intermediate (2) is
deprotected under acidic conditions and reacted with the halogenated heteroaryl (3) to generate the aminoheteroaryl intermediate (4). Boronated azaindole (5) is coupled with the aminoheteroaryl intermediate (4) under cross-coupling condition to generate the compound of Formula I.
………………………………………………………………………….
Patent
http://www.google.com/patents/US8163917
| 346 | M+H393.20 | RT 1.60 | (DMSO-d6, 300 MHz) 11.95 (bs, 1H), 8.7 (d, |
| 1H), 8.25 (m, 2H), 8.12 (d, 1H), 8.02 (d, 1H), | |||
| 7.28 (s, 1H), 7.13 (dd, 1H), 6.38 (bd, 1H), 3.75 | |||
| (m, 2H), 2.06 (m, 1H), 1.83 (m, 1H), 1.46 (s, | |||
| 3H), 0.8 (t, 3H); |
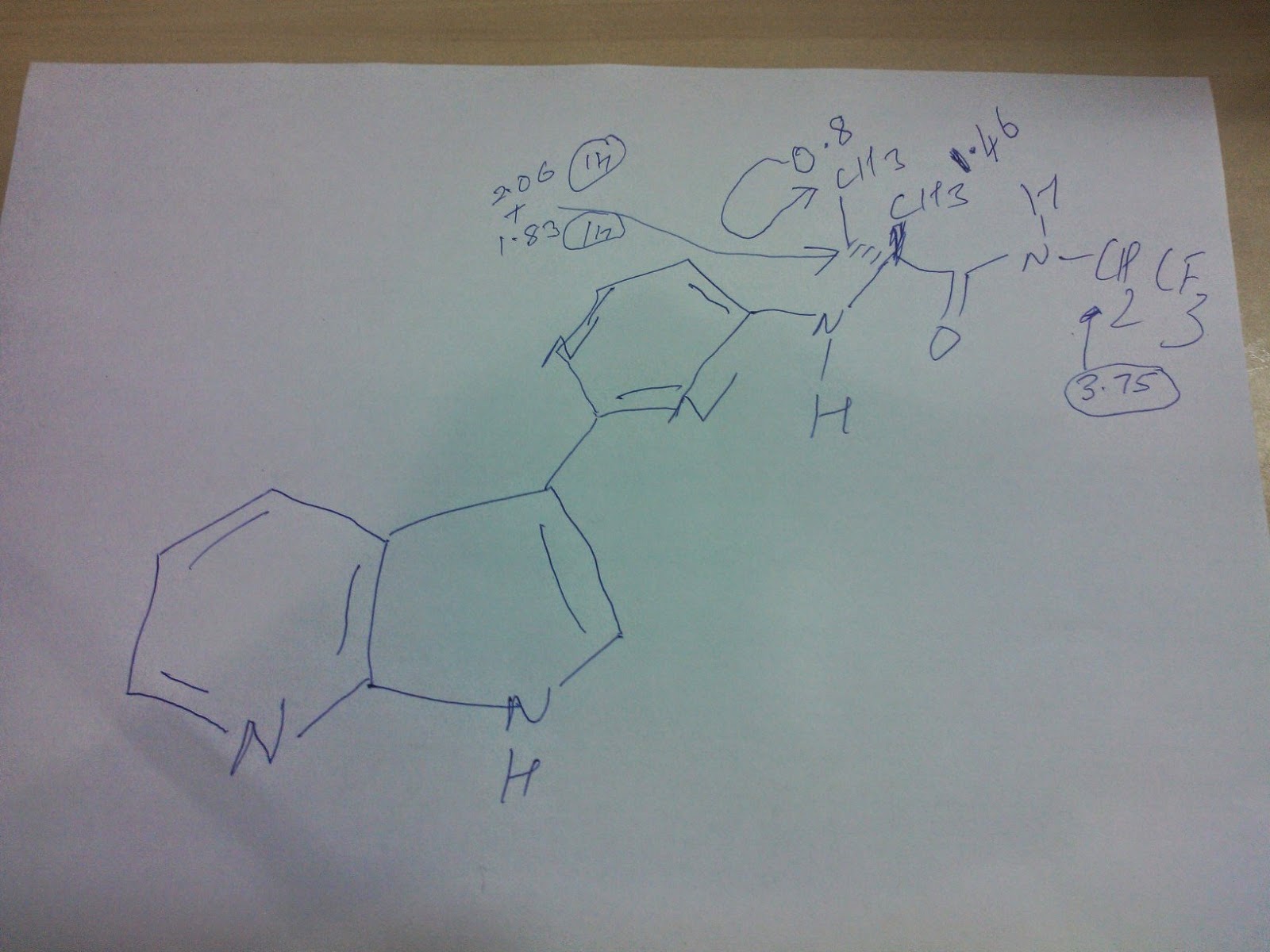
| 346 |
|
|
Example 1 Preparation of Compounds of the Invention
General Synthetic Scheme

Step 1
To a stirred solution of Boc-valine (1; R1 is Me; 3.8 g, 0.02 mol), EDC (4.63 g, 0.024 mol), HOBt (4.0 g, 0.026 mol), DIEA (10.5 mL, 0.06 mol) in 100 mL of DCM is added trifluoroethylamine HCl (2.92 g, 0.022 mol). The reaction mixture is stirred for 16 h. It is concentrated to dryness and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give 5.4 g (98%) of 2 as a white solid.
Step 2
Compound 2 (5.32 g, 0.0197 mol) is deprotected with a 1:1 mixture of DCM/TFA at rt for 45 min. Concentration to dryness gives the intermediate amine that is used directly for the next step. A mixture of 5-fluoro-2,4-dichloropyrimidine (3; R is F; 3.28 g, 0.0197 mol), the crude amine TFA salt (5.25 g, 0.0197 mol) and DIEA (10.27 mL, 0.059 mol) are stirred in isopropanol at rt for 16 h. The reaction mixture is concentrated in vacuo and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give a crude oil that is subjected to chromatography (50% EtOAc/50% hexanes) to yield the desired compound 4.
Step 3
A mixture of 5 (30 mg, 0.075 mmol; prepared according to WO 2005/095400), 4 (23 mg, 0.075 mmol), Pd (Ph3P)4 (9 mg, 0.0078 mmol) and sodium carbonate 2M (115 uL, 0.23 mmol) in 1 mL of DME is microwaved at 150° C. for 10 minutes. The reaction mixture is filtered through a short pad of silica gel with 30% EtOAc-70% hexanes as eluent to provide, after concentration to dryness, the crude intermediate that is used directly for the next step.
The crude intermediate is dissolved in 1 mL of dry methanol and 200 uL of sodium methoxide in methanol 25% was added. The reaction mixture is stirred at 60° C. for 1 h and quenched with 6N HCl (154 uL). The mixture is dried under a flow of nitrogen and purified by reverse phase HPLC (10-60 MeCN/water w/0.5% TFA) to provide the desired material of formula 6a.
Compounds of formulae 6b and 6c may be prepared in an analogous manner using the appropriate starting reagents. For instance, a compound of formula 6b may generally be made by substituting Cert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)pyrrolidine-1-carboxylate for compound 1, while a compound of formula 6c may generally be made by substituting tert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)propan-2-ylcarbamate for compound 1.
Example 2 Analytical Results
Tables 4, 5 and 6 below depicts exemplary 1H-NMR data (NMR) and liquid chromatographic mass spectral data, reported as mass plus proton (M+H), as determined by electrospray, and retention time (RT) for certain compounds of the present invention, wherein compound numbers in Tables 4, 5 and 6 correspond to the compounds depicted in Tables 1, 2 and 3, respectively (empty cells indicate that the test was not performed):
PATENTS
|
4-25-2012
|
Azaindoles Useful as Inhibitors of Janus Kinases
|
|
|
8-4-2010
|
Azaindoles useful as inhibitors of janus kinases
|
new patent
WO-2014110259
| US8450489 * | Mar 1, 2012 | May 28, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US8530489 * | May 22, 2012 | Sep 10, 2013 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| US8686143 * | Oct 25, 2011 | Apr 1, 2014 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of Janus kinases |
| US20120157429 * | Oct 25, 2011 | Jun 21, 2012 | Wannamaker Marion W | Compounds useful as inhibitors of janus kinases |
| US20120165307 * | Mar 1, 2012 | Jun 28, 2012 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US20120309963 * | May 22, 2012 | Dec 6, 2012 | Vertex Pharmaceuticals Incorporated | 5-cyano-4- (pyrrolo [2,3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| US20130237516 * | Apr 25, 2013 | Sep 12, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2013070606A1 * | Nov 6, 2012 | May 16, 2013 | Vertex Pharmaceuticals Incorporated | Methods for treating inflammatory diseases and pharmaceutical combinations useful therefor |
PARP Inhibitor.. Veliparib (ABT-888) 维利帕尼

Veliparib
2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide
CAS number: 912444-00-9 (Veliparib),
912445-05-7 (Veliparib dihydrochloride)
Mechanism of Action:poly (adenosine diphosphate [ADP]–ribose) polymerase (PARP) inhibitor
Indiction:cancer treatment
Development Status:Phase III
Drug Company: AbbVie
PARP Inhibitor Veliparib (ABT-888)
Also known as: ABT-888, 912444-00-9, ABT 888, carboxamide, CHEBI:62880, 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, ABT888, Veliparib
Molecular Formula: C13H16N4O Molecular Weight: 244.29234
| Systematic (IUPAC) name | |
|---|---|
| 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide | |
| Clinical data | |
| Legal status | experimental |
| Identifiers | |
| ATC code | None |
| PubChem | CID 11960529 |
| DrugBank | DB07232 |
| ChemSpider | 10134775 |
| UNII | 01O4K0631N |
| ChEMBL | CHEMBL506871 |
| Chemical data | |
| Formula | C13H16N4O |
| Mol. mass | 244.29 g/mol |
|
2-10-2012
|
PARP1 TARGETED THERAPY
|
|
|
4-17-2009
|
2-{(R)-2-METHYLPYRROLIDIN-2-YL)-1H-BENZIMIDAZOLE-4-CARBOXAMIDE CRYSTALLINE FORM 1
|
Veliparib (ABT-888)[1] is a potential anti-cancer drug acting as a PARP inhibitor. It kills cancer cells by blocking a protein called PARP, thereby preventing the repair of DNA or genetic damage in cancer cells and possibly making them more susceptible to anticancer treatments. Veliparib may make whole brain radiation treatment work more effectively against brain metastases from NSCLC.
It inhibits both PARP1 and PARP2.[2][3]
AbbVie’s Veliparib (ABT-888,), an inhibitor of poly ADP-ribose polymerase 1 and 2 (PARP 1 and PARP 2), is being investigated in multiple tumor types, including 3 phase III studies, all initiated this year, in neoadjuvant treatment of triple-negative breast cancer (clinical trial number:NCT02032277), non-small cell lung cancer (NSCLC, clinical trial number:NCT02106546) and HER2-negative, BRCA1 and/or BRCA2-positive breast cancer (clinical trial number:NCT02163694).
AbbVie, which was spun off from Abbott Laboratories in early 2013, is currently looking to buy Irish drug maker Shire for $46 billion. The proposed deal follows Pfizer’s failed $120 billion attempt to buy AstraZeneca. Humira, AbbVie’s rheumatoid arthritis drug and the world’s top-selling drug last year, accounts for 60% of company revenue and is going off-patent in at the end of 2016. The threat of growing competition for Humira may be a major motivation for AbbVie.


Clinical trials
Numerous phase I clinical trials are in progress.[4]
A phase I/II clinical trial for use with/out doxorubicin (for Metastatic or Unresectable Solid Tumors or Non-Hodgkin Lymphoma) started in 2008 and is due to complete in 2010.[5] Results (inc MTD) with topotecan.[6]
A phase II clinical trial for metastatic melanoma has started recruiting.[7] Due to end Dec 2011.
A phase II clinical trial for metastatic breast cancer has started recruiting.[8] Due to end Nov 2011.
A phase II clinical trial for add-on to Radiation Therapy for Patients with Brain Metastases from Non-Small Cell Lung Cancer.
It was included in the I-SPY2 breast cancer trial,[9] and there are encouraging data from that study [10]
A phase I clinical trial for prostate cancer in men who carry the BRCA mutation is underway and is now recruiting (as of May 2013).[11]


……………….
http://www.google.com/patents/US20060229289
EXAMPLE 1
2-(2-methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide EXAMPLE 1A 1-benzyl 2-methyl 2-methylpyrrolidine-1,2-dicarboxylate
A solution of 1-benzyl 2-methyl pyrrolidine-1,2-dicarboxylate (15.0 g, 57 mmol) and iodomethane (7.11 ml, 114 mmol) in THF (100 mL) was treated with NaN(TMS)2 (1.0 M solution in THF, 114 mL, 114 mmol) at −75° C. under nitrogen. The temperature of the cooling bath was then slowly raised to −20° C. within 1 h and the mixture was stirred at the same temperature for another 3 h. After quenching with water, the mixture was acidified with 2 N HCl (˜100 mL) and was partitioned between water (400 mL) and EtOAc (400 mL). The organic phase was washed with brine and concentrated. The residue was purified by flash column chromatography (silica gel, EtOAc/hexane) to give Example 1A (15.15 g, Yield: 96%). MS (DCI/NH3) m/z 278 (M+H)+.
EXAMPLE 1B
1-[(benzyloxy)carbonyl]-2-methylpyrrolidine-2-carboxylic acid
A solution of Example 1A (15.15 g, 54.63 mmol) in a mixture of THF (100 mL) and water (50 mL) was treated with LiOH.H2O (4.58 g, 109.26 mmol) in water (50 mL). Methanol was added until a transparent solution formed (60 mL). This solution was heated at 60° C. for overnight and the organic solvents were removed under vacuum. The residual aqueous solution was acidified with 2 N HCl to pH 2 and was partitioned between ethyl acetate and water. The organic phase was washed with water, dried (MgSO4), filtered and concentrated to give Example 1B as a white solid (13.72 g, 95.4% yield). MS (DCI/NH3) m/z 264 (M+H)+.
EXAMPLE 1C
benzyl 2-({[2-amino-3-(aminocarbonyl)phenyl]amino}carbonyl)-2-methylpyrrolidine-1-carboxylate
A solution of Example 1B (13.7 g, 52 mmol) in a mixture of pyridine (60 mL) and DMF (60 mL) was treated with 1,1′-carbonyldiimidazole (9.27 g, 57.2 mmol) at 45° C. for 2 h. 2,3-Diamino-benzamide dihydrochloride (11.66 g, 52 mmol), which was synthesized as described in previous patent application WO0026192, was added and the mixture was stirred at rt overnight. After concentration under vacuum, the residue was partitioned between ethyl acetate and diluted sodium bicarbonate aqueous solution. The slightly yellow solid material was collected by filtration, washed with water and ethyl acetate, and dried to give Example 1C (16.26 g). Extraction of the aqueous phase with ethyl acetate followed by concentration, filtration and water-EtOAc wash, provided additional 1.03 g of Example 1C. Combined yield: 84%. MS (APCI) m/z 397 (M+H)+.
EXAMPLE 1D
benzyl 2-[4-(aminocarbonyl)-1H-benzimidazol-2-yl]-2-methylpyrrolidine-1-carboxylate
A suspension of Example 1C (17.28 g, 43.6 mmol) in acetic acid (180 mL) was heated under reflux for 2 h. After cooling, the solution was concentrated and the residual oil was partitioned between ethyl acetate and sodium bicarbonate aqueous solution. The organic phase was washed with water and concentrated. The residue was purified by flash column chromatography (silica gel, 3-15% CH3OH in 2:1 EtOAc/hexane) to provide Example 1D (16.42 g, Yield: 99%).
MS (APCI) m/z 379 (M+H)+.
EXAMPLE 1E 2-(2-methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide
A solution of Example 1D (15.0 g, 40 mmol) in methanol (250 ml) was treated with 10% Pd/C (2.8 g) under 60 psi of hydrogen for overnight. Solid material was filtered off and the filtrate was concentrated. The residual solid was recrystallized in methanol to give 7.768 g of Example 1E as free base. The bis-HCl salt was prepared by dissolving the free base in warm methanol and treating with 2 equivalents of HCl in ether (10.09 g). MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): δ 1.92 (s, 3 H), 2.00-2.09 (m, 1 H), 2.21-2.29 (m, 1 H), 2.35-2.41 (m, 1 H), 2.52-2.57 (m, 1 H), 3.54-3.65 (m, 2 H), 7.31 (t, J=7.93 Hz, 1 H), 7.68 (dd, J=8.24, 0.92 Hz, 1 H), 7.72 (dd, J=7.63, 0.92 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72N, 17.66. Found: C, 49.30; H, 5.60; N, 17.39.
EXAMPLE 3 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide EXAMPLE 3A benzyl(2R)-2-[4-(aminocarbonyl)-1H-benzimidazol-2-yl]-2-methylpyrrolidine-1-carboxylate
Example 1D (1.05 g, 2.8 mmol) was resolved on chiral HPLC (Chiralcel OD, 80/10/10 hexane/EtOH/MeOH). The faster eluting peak was collected and concentrated to provide Example 3A (99.4% e.e., 500 mg). MS (APCI) m/z 379 (M+H)+.
EXAMPLE 3B 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide
A solution of Example 3A (500 mg, 1.32 mmol) in methanol (10 ml) was treated with 10% Pd/C (150 mg) under hydrogen for overnight (balloon). Solid material was filtered off and the filtrate was concentrated. The residual solid was further purified by HPLC (Zorbax C-18, CH3CN/H2O/0.1%TFA) and was converted to bis-HCl salt to provide Example 4 as white solid (254 mg). Co-crystallization of the free base with 1 equivalent of L-tartaric acid in methanol gave a single crystal that was suitable for X-ray study. The X-ray structure with L-tartaric acid was assigned the R-configuration. MS (APCI) m/z 245 (M+H)+; 1H NMR (500 MHz, D2O): δ 2.00 (s, 3 H), 2.10-2.19 (m, 1 H), 2.30-2.39 (m, 1 H), 2.45-2.51 (m, 1 H), 2.61-2.66 (m, 1 H), 3.64-3.73 (m, 2 H), 7.40 (t, J=7.95 Hz, 1 H), 7.77 (d, J=8.11 Hz, 1 H), 7.80 (d, J=7.49 Hz, 1 H); Anal. Calcd for C13H16N4O.2 HCl: C, 49.22; H, 5.72; N, 17.66. Found: C, 49.10; H, 5.52; N, 17.61.
……………….
WO2009049111
http://www.google.com/patents/WO2009049111A1?cl=en
EXAMPLE 1 Preparation of ABT-888 Crystalline Form 1 A mixture of ABT-888 dihydrochloride (10 g) was stirred in saturated potassium bicarbonate (50 mL) and n-butanol (50 mL) until the ABT-888 dihydrochloride completely dissolved. The aqueous layer was extracted with a second portion of n-butanol then discarded. The extracts were combined, washed with 15% sodium chloride solution (50 mL) and concentrated. The concentrate was chase distilled three times with heptane (50 mL),dissolved in refluxing 2-propanol (45 mL) and filtered hot. The filtrate was cooled to ambient temperature with stirring over 18 hours, cooled to 0-50C, stirred for 1 hour, and filtered. The filtrant was washed with 2-propanol and dried in a vacuum oven at 45-500C with a slight nitrogen purge.
EXAMPLE 2
Preparation of ABT-888 Crystalline Form 2
A mixture of ABT-888 in methanol, in which the ABT-888 was completely dissolved, was concentrated at about 35 0C, and the concentrate was dried to a constant weight.
EXAMPLE 3 Preparation of ABT-888 Crystalline Form 1

15 16
Step 1 : 2-(2-methyl-2-pyrrolidino)-benzimidazole-4-carboxamide 2 HCl (15) is dissolved in water (3.5 kg / kg 15) at 20 + 5 0C. Dissolution of 15 in water results in a solution of pH 0 – 1.
Step 2: The reaction is run at 20 – 25 0C. One equivalent of sodium hydroxide is added, raising the pH to 2 – 3 with only a mild exotherm (100C observed with rapid addition of 1.0 equiv.). This generates a solution that remains clear for several days even when seeded with free base crystals. 3N NaOH (1.0 equiv., 1.25 kg / kg 15) is charged and the solution polish filtered into the crystallizer/ reactor.
Step 3: 5% Na2CO3 (1.5 equiv., 10.08 kg / kg 15) is then filtered into the crystallizer over 2 hours. Nucleation occurs after approximately l/6th of the Na2CO3 solution is added (-0.25 equiv.)
Step 4: The slurry is mixed for NLT 15 min before sampling (typically 1 to 4 hours (2.5 mg/mL product in the supernatant)). The slurry is filtered at 200C and washed with 6 portions of water (1.0 kg / kg 15 each). Each wash was applied to the top of the cake and then pressured through. No mixing of the wetcake was done.
Step 5 : The solids are then dried. Drying was performed at 500C keeping the Cogeim under vacuum while applying a slight nitrogen bleed. The agitator blade was left in the cake to improve heat transfer to the cake. It was rotated and lifted out of the cake once per hour of drying to speed the drying process while minimizing potential crystal attrition that occurs with continuous agitator use. In one embodiment of Step 1, the volume of water for dissolution of the Dihydrochloride (15) is about 1.3 g water/g 15. In another embodiment of Step 1,, the volume of water for dissolution is about 1.3 g to about 4 g water/g 15. In another embodiment of Step 1, the volume of water for dissolution is 1.3 g to 3.5 g water/g 15. In another embodiment of Step 1, the volume of water for dissolution is 3.5 g water/g 15.
……………………
J. Med. Chem., 2009, 52 (2), pp 514–523
DOI: 10.1021/jm801171j

(2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide
excellent PARP enzyme potency as well as single-digit nanomolar cellular potency. These efforts led to the identification of 3a (2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide, ABT-888), currently in human phase I clinical trials. Compound 3a displayed excellent potency against both the PARP-1 and PARP-2 enzymes with a Ki of 5 nM and in a C41 whole cell assay with an EC50 of 2 nM. In addition, 3a is aqueous soluble, orally bioavailable across multiple species, and demonstrated good in vivo efficacy in a B16F10 subcutaneous murine melanoma model in combination with temozolomide (TMZ) and in an MX-1 breast cancer xenograft model in combination with either carboplatin or cyclophosphamide.
References
- “ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models” May 2007
- http://www.cancer.gov/drugdictionary/?CdrID=496464
- “ABT-888, an Orally Active Poly(ADP-Ribose) Polymerase Inhibitor that Potentiates DNA-Damaging Agents in Preclinical Tumor Models”, 2007
- http://clinicaltrialsfeeds.org/clinical-trials/results/term=Drug:+ABT-888
- “ABT-888 and Cyclophosphamide With Versus Without Doxorubicin in Treating Patients With Metastatic or Unresectable Solid Tumors or Non-Hodgkin Lymphoma”
- Phase I Study of ABT-888, a PARP Inhibitor, in Combination with Topotecan Hydrochloride in Adults with Refractory Solid Tumors and Lymphomas.. July 2011. doi:10.1158/0008-5472.CAN-11-1227.
- “A Study Evaluating Efficacy of ABT-888 in Combination With Temozolomide in Metastatic Melanoma”
- “ABT-888 and Temozolomide for Metastatic Breast Cancer”
- “Breast cancer study aims to speed drugs, cooperation”, March 2010
- http://www.centerwatch.com/news-online/article/5737/new-presurgery-combination-therapy-for-triple-negative-breast-cancer
- “Veliparib in Treating Patients With Malignant Solid Tumors That Did Not Respond to Previous Therapy. Clinical Trial NCT00892736”
|
4-1-2013
|
Design, synthesis and biological evaluation of novel imidazo[4,5-c]pyridinecarboxamide derivatives as PARP-1 inhibitors.
|
Bioorganic & medicinal chemistry letters
|
|
8-15-2013
|
Discovery of novel benzo[b][1,4]oxazin-3(4H)-ones as poly(ADP-ribose)polymerase inhibitors.
|
Bioorganic & medicinal chemistry letters
|
|
|
8-1-2013
|
Identification of potent Yes1 kinase inhibitors using a library screening approach.
|
Bioorganic & medicinal chemistry letters
|
|
5-1-2010
|
A rapid and sensitive method for determination of veliparib (ABT-888), in human plasma, bone marrow cells and supernatant by using LC/MS/MS.
|
Journal of pharmaceutical and biomedical analysis
|
|
1-22-2009
|
Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer.
|
Journal of medicinal chemistry
|
External links
http://kdwn.com/2013/12/16/new-drug-study-method-show-breast-cancer-promise/
| US8013168 | Oct 10, 2008 | Sep 6, 2011 | Abbott Laboratories | Veliparib crystal structure; an anticancer PARP inhibitor |
| US8372987 | Oct 10, 2008 | Feb 12, 2013 | Abbvie Inc. | Title compound is Veliparib, a Poly(ADP-ribose) polymerase i.e. PARP inhibitor; anticancer agent |
| US20060229289 * | Apr 11, 2006 | Oct 12, 2006 | Gui-Dong Zhu | 2-(2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide, aka veliparib, for example; poly(ADP-ribose)polymerase inhibitors; antiinflammatory, antitumor agents; Parkinson’s disease |
Penning, Thomas D. et al. Discovery of the Poly(ADP-ribose) Polymerase (PARP) Inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the Treatment of Cancer. Journal of Medicinal Chemistry, 52(2), 514-523; 2009
Zhu, Guidong. 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide crystalline form 2 compositions and preparation for treating cancer. PCT Int. Appl. (2009), WO2009049109 A1 20090416
Kolaczkowski, Lawrence . 2-((R)-2-Methylpyrrolidin-2-yl)-1H-benzimidazole-4-carboxamide (ABT-888) crystalline form I and its pharmaceutical composition for cancer treatment. PCT Int. Appl. (2009), WO2009049111 A1 20090416.
Zhu, Gui-Dong; Gong, Jianchun; Gandhi, Virajkumar B.; Penning, Thomas D.; Giranda, Vincent L. Preparation of 1H-benzimidazole-4-carboxamides as poly(ADP-ribose)polymerase (PARP) inhibitors. U.S. Pat. Appl. Publ. (2006), US20060229289 A1 20061012.

Cebranopadol GRT 6005 セブラノパドール a Potent Analgesic NOP and Opioid Receptor Agonist

Cebranopadol
(GRT-6005; GRT 6005; GRT6005)
CAS: 863513-91-1
(1r,4r)-6′-Fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4-b]indol]-4-amine
Spiro[cyclohexane-1,1′(3’H)-pyrano[3,4-b]indol]-4-amine, 6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-
- C24H27FN2O
- Average mass: 378.482391 Da
Grünenthal GmbH innovator
Cebranopadol(GRT-6005) is a novel first in class compounds with potent agonist activity on ORL-1 (opioid receptor like -1) and the well established mu opioid receptor.
Cebranopadol exhibits highly potent and efficacious antinociceptive and antihypersensitive effects in several experimental model models of acute and chronic pain (tail–flick, rheumatoid arthritis, bone cancer, spinal nerve ligation, diabetic neuropathy) with ED50 values of 0.5–5.6 μg/kg after intravenous and 25.1 μg/kg after oral administration. Unlike morphine, cebranopadol did not disrupt motor coordination and respiration at doses within and exceeding the analgesic dose range. Cebranopadol, by its combination of agonism at NOP and opioid receptors, affords highly potent and efficacious analgesia in various pain models with a favorable side–effect profile.
GRT-6005 is a centrally active analgesic in phase II clinical development for the oral treatment of neuropathic pain in patients with painful diabetic polyneuropathy and for the treatment of pain due to osteoarthritis of the knee. It is being developed by Grüenenthal and Forest. No recent development has been reported for research into the treatment of moderate to severe pain following bunionectomy. In 2010, GRT-6005 was licensed to Forest and Grünenthal in Canada and the U.S. for the treatment of moderate to severe chronic pain.

Description: IC50 Value: N/A Cebranopadol and GRT 6006 are novel first-in-class compounds with unique pharmacological and pharmacokinetic profiles that may enhance their effect in certain pain conditions. The unique mode of action of these compounds builds on the ORL-1 receptor and, supported by the established mu opioid receptor, is particularly suitable for the treatment of moderate to severe chronic pain [1]. in vitro: N/A in vivo: N/A Clinical trial: Cebranopadol has successfully completed initial proof-of-concept studies in nociceptive and neuropathic pain with further Phase II studies planned prior to initiation of Phase III studies.
Neuropathic pain
Neuropathic pain is caused when peripheral nerves are damaged by mechanical, metabolic or inflammatory way. The pain occurring images are mainly due to the occurrence of spontaneous pain, hyperalgesia and allodynia (pain is already triggered by non-noxious stimuli) in. As a result, the lesions to increased expression of Na + channels and thus to spontaneous activity in the damaged axons and their Nachbaraxonen (England et al., Neurology, 1996, 47, 272-276).The excitability of the neurons is increased and they react to incoming stimuli with an increased discharge frequency. This results in an increased sensitivity to pain, which contributes to the development of hyperalgesia and spontaneous pain (Baron, Clin J Pain 2000;. 16 (2 Suppl), 12-20). The causes and manifestations, and therefore the treatment needs of neuropathischerm pain are varied. They arise as a result of injury or disease of the brain, spinal cord or peripheral nerves.Causes may be operations, such as phantom pain after amputation, stroke, multiple sclerosis, spinal cord injury, alcohol or drug abuse or other toxins, cancers but also
Metabolic diseases such as diabetes, gout, kidney failure or liver cirrhosis, or infectious diseases such as mononucleosis, ehrlichiosis, typhoid, diphtheria, HIV, syphilis or Lyme disease. The pain experience is very different signs and symptoms that can change over time in number and intensity. Paradoxically, patients with neuropathic pain outline a slowdown or failure of acute pain perception and the simultaneous increase of neuropathic pain. The typical symptoms of neuropathic pain as tingling, burning, shooting or described, or radiating electrifying. Pharmacological basis for treatment of neuropathic pain include tricyclic antidepressants and anticonvulsants, which are used as monotherapy or in combination with opioids. These drugs usually provide only a certain pain relief during a pain-free but is often not achieved. The often-adjusting side effects are dose increases while the drug to achieve adequate pain relief often in the way. In fact, a higher dosage of a μ-opioid is often required as the treatment of acute pain, thereby reducing the side effects get even more important for satisfactory treatment of neuropathic pain. By the occurrence of typical μ-opioid tolerance development and the concomitant need for dose escalation of this problem is exacerbated. In summary it can be stated that neuropathic pain is difficult to treat and today is alleviated by high doses of μ-opioids only partially (Saudi Pharm J. 2002, 10 (3), 73-85). There is therefore an urgent need for medicines for the treatment of chronic pain, the dose should not be increased until the occurrence of intolerable side effects to ensure a satisfactory pain treatment.
……………
http://www.google.com/patents/US7547707
Example 24 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Non-polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (651 mg, 3 mmoles) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 537 mg, 3 mmoles) were initially introduced into abs. MC (20 ml) under argon. Trifluoromethanesulfonic acid trimethylsilyl ester (0.6 ml, 3.1 mmoles) was then added very rapidly. The mixture was stirred at RT for 20 h. For working up, 1 M NaOH (30 ml) was added to the reaction mixture and the mixture was stirred for 30 min. The organic phase was separated, and the aqueous phase which remained was extracted with MC (3×60 ml). The combined organic phases were washed with water (2×30 ml) and dried over sodium sulfate. Methanol (30 ml) was added to the solid residue obtained after the solvent had been distilled off, and the mixture was heated, and stirred for 15 hours. The solid contained in the suspension was filtered off with suction and dried. 955 mg of the more non-polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole were obtained (m.p. 284-292° C.). 850 mg of this were dissolved in hot ethanol (900 ml), and a similarly hot solution of citric acid (1 g, 5.2 mmoles) in ethanol (20 ml) was added. After approx. 15 minutes, crystals precipitated out at the boiling point. After cooling to approx. 5° C., the mixture was left to stand for 2 h. The solid formed was filtered off with suction. 640 mg of the hemicitrate were obtained as a white solid (m.p. 258-282° C.).
Example 25 1,1-(3-Dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole hemicitrate, More Polar diastereoisomer
4-Dimethylamino-4-phenylcyclohexanone (217 mg, 1 mmole) and 2-(5-fluoro-1H-indol-3-yl)-ethanol (“5-fluorotryptophol”, 179 mg, 1 mmole) were dissolved in conc. acetic acid (4 ml). Phosphoric acid (1 ml, 85 wt. %) was slowly added dropwise to this mixture. The mixture was stirred at RT for 16 h. For working up, the mixture was diluted with water (20 ml), brought to pH 11 with 5 M NaOH and extracted with MC (3×20 ml). The combined organic phases were dried with sodium sulfate and evaporated. The residue (364 mg of white solid) was suspended in hot ethanol (20 ml), and a similarly hot solution of citric acid (185 mg, 0.96 mmole) in ethanol (5 ml) was added. The residue thereby dissolved completely and no longer precipitated out even on cooling to approx. 5° C. Ethanol was removed on a rotary evaporator and the hemicitrate of the more polar diastereoisomer of 1,1-(3-dimethylamino-3-phenylpentamethylene)-6-fluoro-1,3,4,9-tetrahydropyrano[3,4-b]indole was obtained in this way in a yield of 548 mg as a white solid (m.p. 148-155° C.).
| 24 |
 |
hemicitrate | more non-polar diastereomer |
| 25 |
 |
hemicitrate | more polar diastereomer |
………………..
WO 2013113690
(1 r,4r)-6′-fluoro-N,N- dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1 ,1 ‘-pyrano[3,4-b]indol]-4-amine (free base), has the following structural formula (I):
http://www.google.com/patents/WO2013113690A1?cl=en

…………………

see A4
…………………………
One particular drug that is of great interest for use in treating cancer pain (and other acute, visceral, neuropathic and chronic pain pain disorders) is (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine. This drug is depicted below as the compound of formula (I).

The solid forms of (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4b]indol]-4-amine that are known so far are not satisfactory in every respect and there is a demand for advantageous solid forms
A) Synthesis of Crystalline Form A100 mg (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine [crystalline form D according to D)] was suspended in 0.5 mL TBME. The suspension was stirred at RT for six days. The resulting solid was filtered out and dried in air. A crystalline solid of crystalline form A was obtained and characterized by FT Raman, TG-FTIR and PXRD.
……………………

In a previous communication, our efforts leading from 1 to the identification of spiro[cyclohexane-dihydropyrano[3,4-b]indole]-amine 2a as analgesic NOP and opioid receptor agonist were disclosed and their favorable in vitro and in vivo pharmacological properties revealed. We herein report our efforts to further optimize lead 2a, toward trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine (cebranopadol, 3a), which is currently in clinical development for the treatment of severe chronic nociceptive and neuropathic pain.
Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol
http://pubs.acs.org/doi/full/10.1021/ml500117c
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/ml500117c
6′-Fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3’H)-pyrano[3,4-
b]indol]-4-amine, trans-, 2-hydroxy-1,2,3-propanetricarboxylate (2:1)
b]indol]-4-amine, trans-, 2-hydroxy-1,2,3-propanetricarboxylate (2:1)
hemicitrate were obtained as a white solid (mp 258-282 °C).1H-NMR (300 MHz; DMSO-d6): 1.75-1.87 (m, 4 H); 2.14 (s, 6 H); 2.27 (t, 2 H); 2.61-
2.76 (m,6 H); 3.88 (t, 2 H); 6.86 (dt, 1 H); 7.10 (dd, 1 H); 7.30-7.43 (m, 6 H); 10.91 (br
s, 1 H).
2.76 (m,6 H); 3.88 (t, 2 H); 6.86 (dt, 1 H); 7.10 (dd, 1 H); 7.30-7.43 (m, 6 H); 10.91 (br
s, 1 H).
13C-NMR (75.47 MHz; DMSO-d6): 22.1; 27.6; 30.2 (2 C); 38.0 (2 C); 43.1; 58.8 (2 C,
overlap); 71.5; 72.2; 102.3 (2JC,F = 23 Hz); 105.6 (3JC,F = 4 Hz); 108.3 (2JC,F = 26 Hz);
overlap); 71.5; 72.2; 102.3 (2JC,F = 23 Hz); 105.6 (3JC,F = 4 Hz); 108.3 (2JC,F = 26 Hz);
112.0 (3JC,F = 10 Hz); 126.5; 126.6; 126.7 (2 C); 127.4 (2 C); 132.4; 138.7; 141.5;
156,7 (1JC,F = 231 Hz); 171.3 (2 C), 175.3.HPLC-MS: m/z 378.9 [M + H]+
156,7 (1JC,F = 231 Hz); 171.3 (2 C), 175.3.HPLC-MS: m/z 378.9 [M + H]+
…………………………..
| US20120034297 * | Aug 4, 2011 | Feb 9, 2012 | Gruenenthal Gmbh | Pharmaceutical dosage forms comprising 6′-fluoro-(N-methyl- or N,N-dimethyl-)-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3’h-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| WO2004043967A1 | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2008040481A1 | Sep 26, 2007 | Apr 10, 2008 | Gruenenthal Gmbh | MIXED ORL 1/μ AGONISTS FOR TREATING PAIN |
-
CORAL – Cebranopadol Versus Morphine Prolonged-release in Patients With Chronic Moderate to Severe Pain Related to Cancer
Efficacy, Safety, and Tolerability of Oral Cebranopadol Versus Morphine Sulfate PR in Subjects With Chronic Moderate to Severe Pain Related to Cancer.Average amount of daily rescue medication at the end of the maintenance period.
UK Clinical Trials Gateway, 07 October 2013
-
CORAL XT – Open-label Extension Trial of the CORAL Trial
An Open-label, Multi-site Trial to Describe the Safety and Tolerability of Oral Cebranopadol Administered for 26 Weeks in Subjects With Cancer-related Pain Who Have Completed Treatment in the KF6005/07 Trial.Absolute…
UK Clinical Trials Gateway, 12 December 2013
| WO2004043967A1 * | Nov 5, 2003 | May 27, 2004 | Otto Aulenbacher | Spirocyclic cyclohexane derivatives |
| WO2005066183A1 * | Dec 21, 2004 | Jul 21, 2005 | Gruenenthal Gmbh | Spirocyclic cyclohexane derivatives with affinity for the orl1-receptor |
| US20050153998 * | Aug 19, 2004 | Jul 14, 2005 | Fumitaka Ito | Tetrahydroisoquinoline or isochroman compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7799931 * | Feb 17, 2009 | Sep 21, 2010 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7951948 * | Apr 19, 2010 | May 31, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US7960404 | Aug 21, 2009 | Jun 14, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8034936 | Nov 4, 2010 | Oct 11, 2011 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds useful to treat substance dependency |
| US8053576 | Feb 17, 2009 | Nov 8, 2011 | Gruenenthal Gmbh | Treating conditions associated with the nociceptin/ORL1 receptor system, e.g. pain, drug withdrawal, anxiety, muscle relaxants, anxiolytic agents; e.g. 1,1-[3-dimethylamino-3-(pyridin-2-yl)pentamethylene]-3,4-dihydro-1H-2,9-diazafluorene |
| US8288406 | Sep 22, 2010 | Oct 16, 2012 | Gruenenthal Gmbh | Hydroxymethylcyclohexylamines |
| US8288430 | Mar 25, 2009 | Oct 16, 2012 | Grunenthal Gmbh | Spiro(5.5)undecane derivatives |
| US8293758 * | Mar 25, 2009 | Oct 23, 2012 | Grunenthal Gmbh | Substituted spirocyclic cyclohexane derivatives |
| US8357705 | Mar 25, 2009 | Jan 22, 2013 | Gruenenthal Gmbh | Substituted cyclohexyldiamines |
| US8404740 | Aug 21, 2009 | Mar 26, 2013 | Gruenenthal Gmbh | Spirocyclic cyclohexane compounds |
| US8614245 * | Jan 8, 2013 | Dec 24, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3′H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US8618156 * | Jul 6, 2012 | Dec 31, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-N,N-dimethyl-4-phenyl-4′,9′-dihydro-3’H-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |
| US20130012563 * | Jul 6, 2012 | Jan 10, 2013 | Gruenenthal Gmbh | Crystalline (1r,4r)-6′-fluoro-n,n-dimethyl-4-phenyl-4′,9′-dihydro-3’h-spiro[cyclohexane-1,1′-pyrano[3,4,b]indol]-4-amine |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
http://anthonycrasto.jimdo.com/
blogs are


Congratulations! Your presentation titled “Anthony Crasto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
join my process development group on google
you can post articles and will be administered by me on the google group which is very popular across the world
LinkedIn group
blogs are
MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ
http://www.stumbleupon.com/stumbler/amcrasto
MY CHINA AND JAPAN BLOGS
VIETNAM
ICELAND
RUSSIA
========================

Luseogliflozin, TS 071…………. strongly inhibited SGLT2 activity,

LUSEOGLIFLOZIN, CAS 898537-18-3
An antidiabetic agent that inhibits sodium-dependent glucose cotransporter 2 (SGLT2).
(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol
(1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
Taisho Pharmaceutical Co., Ltd
Taisho (Originator), PHASE 3
Click to access 2013041801-e.pdf
TS-071
| Taisho Pharmaceutical Holdings Co. Ltd. | |
| Description | Oral sodium-glucose cotransporter 2 (SGLT2) inhibitor |

TS-071, an SGLT-2 inhibitor, is in phase III clinical development at Taisho for the oral treatment of type 1 and type 2 diabetes
In 2012, the product was licensed to Novartis and Taisho Toyama Pharmaceutical by Taisho in Japan for comarketing for the treatment of type 2 diabetes.
Diabetes is a metabolic disorder which is rapidly emerging as a global health care problem that threatens to reach pandemic levels. The number of people with diabetes worldwide is expected to rise from 285 million in 2010 to 438 million by 2030. Diabetes results from deficiency in insulin because of impaired pancreatic β-cell function or from resistance to insulin in body, thus leading to abnormally high levels of blood glucose.
Diabetes which results from complete deficiency in insulin secretion is Type 1 diabetes and the diabetes due to resistance to insulin activity together with an inadequate insulin secretion is Type 2 diabetes. Type 2 diabetes (Non insulin dependent diabetes) accounts for 90-95 % of all diabetes. An early defect in Type 2 diabetes mellitus is insulin resistance which is a state of reduced responsiveness to circulating concentrations of insulin and is often present years before clinical diagnosis of diabetes. A key component of the pathophysiology of Type 2 diabetes mellitus involves an impaired pancreatic β-cell function which eventually contributes to decreased insulin secretion in response to elevated plasma glucose. The β-cell compensates for insulin resistance by increasing the insulin secretion, eventually resulting in reduced β-cell mass. Consequently, blood glucose levels stay at abnormally high levels (hyperglycemia).
Hyperglycemia is central to both the vascular consequences of diabetes and the progressive nature of the disease itself. Chronic hyperglycemia leads to decrease in insulin secretion and further to decrease in insulin sensitivity. As a result, the blood glucose concentration is increased, leading to diabetes, which is self-exacerbated. Chronic hyperglycemia has been shown to result in higher protein glycation, cell apoptosis and increased oxidative stress; leading to complications such as cardiovascular disease, stroke, nephropathy, retinopathy (leading to visual impairment or blindness), neuropathy, hypertension, dyslipidemia, premature atherosclerosis, diabetic foot ulcer and obesity. So, when a person suffers from diabetes, it becomes important to control the blood glucose level. Normalization of plasma glucose in Type 2 diabetes patients improves insulin action and may offset the development of beta cell failure and diabetic complications in the advanced stages of the disease.
Diabetes is basically treated by diet and exercise therapies. However, when sufficient relief is not obtained by these therapies, medicament is prescribed alongwith. Various antidiabetic agents being currently used include biguanides (decrease glucose production in the liver and increase sensitivity to insulin), sulfonylureas and meglitinides (stimulate insulin production), a-glucosidase inhibitors (slow down starch absorption and glucose production) and thiazolidinediones (increase insulin sensitivity). These therapies have various side effects: biguanides cause lactic acidosis, sulfonylurea compounds cause significant hypoglycemia, a-glucosidase inhibitors cause abdominal bloating and diarrhea, and thiazolidinediones cause edema and weight gain. Recently introduced line of therapy includes inhibitors of dipeptidyl peptidase-IV (DPP-IV) enzyme, which may be useful in the treatment of diabetes, particularly in Type 2 diabetes. DPP-IV inhibitors lead to decrease in inactivation of incretins glucagon like peptide- 1 (GLP-1) and gastric inhibitory peptide (GIP), thus leading to increased production of insulin by the pancreas in a glucose dependent manner. All of these therapies discussed, have an insulin dependent mechanism.
Another mechanism which offers insulin independent means of reducing glycemic levels, is the inhibition of sodium glucose co-transporters (SGLTs). In healthy individuals, almost 99% of the plasma glucose filtered in the kidneys is reabsorbed, thus leading to only less than 1% of the total filtered glucose being excreted in urine. Two types of SGLTs, SGLT-1 and SGLT-2, enable the kidneys to recover filtered glucose. SGLT-1 is a low capacity, high-affinity transporter expressed in the gut (small intestine epithelium), heart, and kidney (S3 segment of the renal proximal tubule), whereas SGLT-2 (a 672 amino acid protein containing 14 membrane-spanning segments), is a low affinity, high capacity glucose ” transporter, located mainly in the S 1 segment of the proximal tubule of the kidney. SGLT-2 facilitates approximately 90% of glucose reabsorption and the rate of glucose filtration increases proportionally as the glycemic level increases. The inhibition of SGLT-2 should be highly selective, because non-selective inhibition leads to complications such as severe, sometimes fatal diarrhea, dehydration, peripheral insulin resistance, hypoglycemia in CNS and an impaired glucose uptake in the intestine.
Humans lacking a functional SGLT-2 gene appear to live normal lives, other than exhibiting copious glucose excretion with no adverse effects on carbohydrate metabolism. However, humans with SGLT-1 gene mutations are unable to transport glucose or galactose normally across the intestinal wall, resulting in condition known as glucose-galactose malabsorption syndrome.
Hence, competitive inhibition of SGLT-2, leading to renal excretion of glucose represents an attractive approach to normalize the high blood glucose associated with diabetes. Lower blood glucose levels would, in turn, lead to reduced rates of protein glycation, improved insulin sensitivity in liver and peripheral tissues, and improved cell function. As a consequence of progressive reduction in hepatic insulin resistance, the elevated hepatic glucose output which is characteristic of Type 2 diabetes would be expected to gradually diminish to normal values. In addition, excretion of glucose may reduce overall caloric load and lead to weight loss. Risk of hypoglycemia associated with SGLT-2 inhibition mechanism is low, because there is no interference with the normal counter regulatory mechanisms for glucose.
The first known non-selective SGLT-2 inhibitor was the natural product phlorizin
(glucose, 1 -[2-P-D-glucopyranosyloxy)-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)- 1 – propanone). Subsequently, several other synthetic analogues were derived based on the structure of phlorizin. Optimisation of the scaffolds to achieve selective SGLT-2 inhibitors led to the discovery of several considerably different scaffolds.
C-glycoside derivatives have been disclosed, for example, in PCT publications
W.O20040131 18, WO2005085265, WO2006008038, WO2006034489, WO2006037537, WO2006010557, WO2006089872, WO2006002912, WO2006054629, WO2006064033, WO2007136116, WO2007000445, WO2007093610, WO2008069327, WO2008020011, WO2008013321, WO2008013277, WO2008042688, WO2008122014, WO2008116195, WO2008042688, WO2009026537, WO2010147430, WO2010095768, WO2010023594, WO2010022313, WO2011051864, WO201 1048148 and WO2012019496 US patents US65151 17B2, US6936590B2 and US7202350B2 and Japanese patent application JP2004359630. The compounds shown below are the SGLT-2 inhibitors which have reached advanced stages of human clinical trials: Bristol-Myers Squibb’s “Dapagliflozin” with Formula A, Mitsubishi Tanabe and Johnson & Johnson’s “Canagliflozin” with Formula B, Lexicon’s “Lx-421 1″ with Formula C, Boehringer Ingelheim and Eli Lilly’s “Empagliflozin” with Formula D, Roche and Chugai’s “Tofogliflozin” with Formula E, Taisho’s “Luseogliflozin” with Formula F, Pfizer’ s “Ertugliflozin” with Formula G and Astellas and Kotobuki’s “Ipragliflozin” with Formula H.

Formula G Formula H
In spite of all these molecules in advanced stages of human clinical trials, there is still no drug available in the market as SGLT-2 inhibitor. Out of the potential candidates entering the clinical stages, many have been discontinued, emphasizing the unmet need. Thus there is an ongoing requirement to screen more scaffolds useful as SGLT-2 inhibitors that can have advantageous potency, stability, selectivity, better half-life, and/ or better pharmacodynamic properties. In this regard, a novel class of SGLT-2 inhibitors is provided herein
………………………
SYNTHESIS
- Example 5

Synthesis of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose
-
Five drops of 1,2-dibromoethane were added to a mixture of magnesium (41 mg, 1.67 mmol), 1-bromo-3-(4-ethoxybenzyl)-6-methoxy-4-methylbenzene (0.51 g, 1.51 mmol) and tetrahydrofuran (2 mL). After heated to reflux for one hour, this mixture was allowed to stand still to room temperature to prepare a Grignard reagent. A tetrahydrofuran solution (1.40 mL) of 1.0 M i-propyl magnesium chloride and the prepared Grignard reagent were added dropwise sequentially to a tetrahydrofuran (5 mL) solution of 2,3,4,6-tetra-O-benzyl-5-thio-D-glucono-1,5-lactone (0.76 g, 1.38 mmol) while cooled on ice and the mixture was stirred for 30 minutes. After the reaction mixture was added with a saturated ammonium chloride aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate =4:1) to obtain (0.76 g, 68%) a yellow oily title compound.
1H NMR (300 MHz, CHLOROFORM-d) δ ppm 1.37 (t, J=6.92 Hz, 3 H) 2.21 (s, 3 H) 3.51 – 4.20 (m, 12 H) 3.85 – 3.89 (m, 3 H) 4.51 (s, 2 H) 4.65 (d, J=10.72 Hz, 1 H) 4.71 (d, J=5.75 Hz, 1 H) 4.78 – 4.99 (m, 3 H) 6.59 – 7.43 (m, 26 H)
Example 6
-
[0315]


Synthesis of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol
-
An acetonitrile (18 mL) solution of 2,3,4,6-tetra-O-benzyl-1-C-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-5-thio-D-glucopyranose (840 mg, 1.04 mmol) was added sequentially with Et3SiH (0.415 mL, 2.60 mmol) and BF3·Et2O (0.198 mL, 1.56 mmol) at -18°C and stirred for an hour. After the reaction mixture was added with a saturated sodium bicarbonate aqueous solution and extracted with ethyl acetate, the organic phase was washed with brine and then dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate=4:1) to obtain the title compound (640 mg, 77%).
1H NMR (600 MHz, CHLOROFORM-d) δ ppm 1.35 (t, J=6.88 Hz, 3 H) 2.21 (s, 3 H) 3.02 – 3.21 (m, 1 H) 3.55 (t,J=9.40 Hz, 1 H) 3.71 (s, 1 H) 3.74 – 3.97 (m, 10 H) 4.01 (s, 1 H) 4.45 – 4.56 (m, 3 H) 4.60 (d, J=10.55 Hz, 2 H) 4.86 (s, 2 H) 4.90 (d, J=10.55 Hz, 1H) 6.58 – 6.76 (m, 5 H) 6.90 (d, J=7.34 Hz, 1 H) 7.09 – 7.19 (m, 5 H) 7.23 – 7.35 (m, 15 H).
ESI m/z = 812 (M+NH4).
Example 7

Synthesis of (1S)-1,5-anhydro-1-[3-(4-ethoxybenzyl)-6-methoxy-4-methylphenyl]-1-thio-D-glucitol
-
A mixture of (1S)-1,5-anhydro-2,3,4,6-tetra-O-benzyl-1-[2-methoxy-4-methyl-5-(4-ethoxybenzyl)phenyl]-1-thio-D-glucitol (630 mg, 0.792 mmol), 20% palladium hydroxide on activated carbon (650 mg) and ethyl acetate (10 mL) – ethanol (10 mL) was stirred under hydrogen atmosphere at room temperature for 66 hours. The insolubles in the reaction mixture were filtered off with celite and the filtrate was concentrated. The obtained residue was purified by silica gel column chromatography (chloroform:methanol =10:1) to obtain a colorless powdery title compound (280 mg, 81%) as 0.5 hydrate. 1H NMR (600 MHz, METHANOL- d4) δ ppm 1.35 (t, J=6.9 Hz, 3 H) 2.17 (s, 3 H) 2.92 – 3.01 (m, 1 H) 3.24 (t, J=8.71 Hz, 1 H) 3.54 – 3.60 (m, 1 H) 3.72 (dd, J=11.5, 6.4 Hz, 1 H) 3.81 (s, 3 H) 3.83 (s, 2 H) 3.94 (dd, J=11.5, 3.7 Hz, 1 H) 3.97 (q, J=6.9 Hz, 2 H) 4.33 (s, 1 H) 6.77 (d, J=8.3 Hz, 2 H) 6.76 (s, 1 H) 6.99 (d, J=8.3 Hz, 2 H) 7.10 (s, 1 H). ESI m/z = 452 (M+NH4+), 493 (M+CH3CO2-). mp 155.0-157.0°C. Anal. Calcd for C23H30O6S·0.5H2O: C, 62.28; H, 7.06. Found: C, 62.39; H, 7.10.
………………………………..
PAPER
(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (TS-071) is a Potent, Selective Sodium-Dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for Type 2 Diabetes Treatment
(Journal of Medicinal Chemistry) Saturday March 20th 2010
Author(s): ,
DOI:10.1021/jm901893x
GO TO: [Article]
http://pubs.acs.org/doi/abs/10.1021/jm901893x

(1S)-1,5-Anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-d-glucitol (3p)
Compound 3p (0.281 g, 81%) was prepared as a colorless powder from 21p (0.630 g, 0.792 mmol) according to the method described for the synthesis of 3a. (Method A)
mp 155.0−157.0 °C.
1H NMR (600 MHz, MeOH-d4) δ 1.35 (t, J = 6.9 Hz, 3 H), 2.17 (s, 3 H), 2.92−3.01 (m, 1 H), 3.24 (t, J = 8.7 Hz, 1 H), 3.54−3.60 (m, 1 H), 3.72 (dd, J = 6.4, 11.5, Hz, 1 H), 3.81 (s, 3 H), 3.83 (s, 2 H), 3.94 (dd, J = 3.7, 11.5 Hz, 1 H), 3.97 (q, J = 6.9 Hz, 2 H), 4.33 (brs, 1 H), 6.77 (d, J = 8.3 Hz, 2 H), 6.76 (s, 1 H), 6.99 (d, J = 8.3 Hz, 2 H), 7.10 (s, 1 H).
MS (ESI) m/z 452 (M+NH4).
Anal. Calcd for (C23H30O6S·0.5H2O) C, 62.28; H, 7.06. Found C, 62.39; H, 7.10.
 3p is compd
3p is compd
| cmpds | R1 | R2 | R3 | SGLT2 (nM) mean (95% CI) | SGLT1 (nM) mean (95% CI) | T1/T2 selectivity |
|---|---|---|---|---|---|---|
| 1 | 27.8 (21.8−35.3) | 246 (162−374) | 8.8 | |||
| 3a | H | H | OEt | 73.6 (51.4−105) | 26100 (20300−33700) | 355 |
| 3b | H | OH | OEt | 283 (268−298) | 14600 (11500−18500) | 51.6 |
| 3c | H | OMe | OEt | 13.4 (11.3−15.8) | 565 (510−627) | 42.2 |
| 3d | H | F | OEt | 9.40 (5.87−15.0) | 7960 (7180−8820) | 847 |
| 3e | H | Me | OEt | 2.29 (1.76−2.99) | 671 (230−1960) | 293 |
| 3f | H | Cl | OEt | 1.77 (0.95−3.30) | 1210 (798−1840) | 684 |
| 3g | OH | H | OEt | 17.4 (15.9−19.0) | 4040 (1200−13600) | 232 |
| 3h | OMe | H | OEt | 37.9 (26.4−54.4) | 100000 (66500−151000) | 2640 |
| 3i | OMe | OMe | OEt | 10.8 (6.84−17.1) | 4270 (1560−11600) | 395 |
| 3j | H | Cl | OMe | 1.68 (1.08−2.60) | 260 (72.5−931) | 155 |
| 3k | H | Cl | Me | 1.37 (0.97−1.95) | 209 (80.2−545) | 153 |
| 3l | H | Cl | Et | 1.78 (0.88−3.63) | 602 (473−767) | 338 |
| 3m | H | Cl | iPr | 4.01 (1.75−9.17) | 8160 (4860−13700) | 2040 |
| 3n | H | Cl | tBu | 18.8 (11.0−32.1) | 35600 (31900−39800) | 1890 |
| 3o | H | Cl | SMe | 1.16 (0.73−1.85) | 391 (239−641) | 337 |
| 3p | OMe | Me | OEt | 2.26 (1.48−3.43) | 3990 (2690−5920) | 1770 |
| 3q | OMe | Me | Et | 1.71 (1.19−2.46) | 2830 (1540−5200) | 1650 |
| 3r | OMe | Me | iPr | 2.68 (2.15−3.34) | 17300 (14100−21100) | 6400 |
| 3s | OMe | Cl | Et | 1.51 (0.75−3.04) | 3340 (2710−4110) | 2210 |
PATENT
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2004014930A1 * | Aug 8, 2003 | Feb 19, 2004 | Asanuma Hajime | PROCESS FOR SELECTIVE PRODUCTION OF ARYL 5-THIO-β-D- ALDOHEXOPYRANOSIDES |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | AL-MASOUDI, NAJIM A. ET AL: “Synthesis of some novel 1-(5-thio-.beta.-D-glucopyranosyl)-6-azaur acil derivatives. Thio sugar nucleosides” NUCLEOSIDES & NUCLEOTIDES , 12(7), 687-99 CODEN: NUNUD5; ISSN: 0732-8311, 1993, XP008091463 |
| 2 | * | See also references of WO2006073197A1 |
| EP2419097A1 * | Apr 16, 2010 | Feb 22, 2012 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| EP2455374A1 * | Oct 15, 2009 | May 23, 2012 | Janssen Pharmaceutica N.V. | Process for the Preparation of Compounds useful as inhibitors of SGLT |
| EP2601949A2 * | Apr 16, 2010 | Jun 12, 2013 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| EP2668953A1 * | May 15, 2009 | Dec 4, 2013 | Bristol-Myers Squibb Company | Pharmaceutical compositions comprising an SGLT2 inhibitor with a supply of carbohydrate and/or an inhibitor of uric acid synthesis |
| WO2009143020A1 | May 15, 2009 | Nov 26, 2009 | Bristol-Myers Squibb Company | Method for treating hyperuricemia employing an sglt2 inhibitor and composition containing same |
| WO2010043682A2 * | Oct 15, 2009 | Apr 22, 2010 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of sglt |
| WO2010119990A1 | Apr 16, 2010 | Oct 21, 2010 | Taisho Pharmaceutical Co., Ltd. | Pharmaceutical compositions |
| WO2013152654A1 * | Mar 14, 2013 | Oct 17, 2013 | Theracos, Inc. | Process for preparation of benzylbenzene sodium-dependent glucose cotransporter 2 (sglt2) inhibitors |
-
Luseogliflozin: Phase III data 10/21/2013
Week in Review, Clinical ResultsTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Molecular target: Sodium-glucose cotransporter 2 (SGLT2) Description: Oral sodium-glucose… -
Luseogliflozin: Phase III data 10/21/2013
Week in Review, Clinical ResultsTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Molecular target: Sodium-glucose cotransporter 2 (SGLT2) Description: Oral sodium-glucose… -
Luseogliflozin regulatory update 05/13/2013
Week in Review, RegulatoryTaisho Pharmaceutical Holdings Co. Ltd. (Tokyo:4581), Tokyo, Japan Product: Luseogliflozin (TS-071) Business: Endocrine/Metabolic Last month, Taisho’s Taisho Pharmaceutical Co. Ltd. subsidiary submitted a regulatory … -
Strategy: Doubling down in diabetes 05/06/2013
Merck shoring up slowing diabetes franchise with Pfizer, Abide dealsBioCentury on BioBusiness, StrategyAs sales flatten for Merck’s sitagliptin franchise and a new class of oral diabetes drugs comes to market, the pharma has tapped Pfizer and Abide to shore up its position.
see
SEE
http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-132352


PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF)

ATALUREN
PTC 124
3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
| MF C15H9FN2O3 | ||
| Molecular Weight | 284.24 | |
| CAS Registry Number | 775304-57-9 |
PTC Therapeutics Initiates Confirmatory Phase 3 Clinical Trial of Translarna™ (ataluren) in Patients with Nonsense Mutation Cystic Fibrosis (nmCF) – MarketWatch

Ataluren, formerly known as PTC124, is a small-molecular agent designed by PTC Therapeutics and sold under the trade nameTranslarna. It makes ribosomes less sensitive to premature stop codons (referred to as “read-through”). This may be beneficial in diseases such as Duchenne muscular dystrophy where the mRNA contains a mutation causing premature stop codons or nonsense codons. There is ongoing debate over whether Ataluren is truly a functional drug (inducing codon read-through), or if it is nonfunctional, and the result was a false-positive hit from a biochemical screen based on luciferase.[1]
Ataluren has been tested on healthy humans and humans carrying genetic disorders caused by nonsense mutations,[2][3] such as some people with cystic fibrosis and Duchenne muscular dystrophy. In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[4] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug. However, other phase 2 clinical trials were successful for cystic fibrosis in Israel, France and Belgium.[5] Multicountry phase 3 clinical trials are currently in progress for cystic fibrosis in Europe and the USA.[6]
In cystic fibrosis, early studies of ataluren show that it improves nasal potential difference.[7]
Ataluren appears to be most effective for the stop codon ‘UGA’.[2]
On 23 May 2014 ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA).[8]

It is not that ataluren is a complex molecule. To judge from one of the patents, synthesis is straightforward starting from 2-cyanobenoic acid and 2-fluorobenzoyl chloride, both commercially available. The synthetic steps are methylation of 2-cyanobenoic acid (iodomethane), nitrile hydrolysis with hydroxylamine, esterification with the fluoro acid chloride using DIPEA, high-temperature dehydration to the oxadiazole and finally ester hydrolysis (NaOH).


References
- Derek (2013-09-18). “The Arguing Over PTC124 and Duchenne Muscular Dystrophy. In the Pipeline:”. Pipeline.corante.com. Retrieved 2013-11-28.
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (May 2007). “PTC124 targets genetic disorders caused by nonsense mutations”. Nature 447 (7140): 87–91.doi:10.1038/nature05756. PMID 17450125.
- Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL (Apr 2007). “Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers”. Journal of clinical pharmacology 47 (4): 430–444. doi:10.1177/0091270006297140. PMID 17389552.
- “PTC THERAPEUTICS AND GENZYME CORPORATION ANNOUNCE PRELIMINARY RESULTS FROM THE PHASE 2B CLINICAL TRIAL OF ATALUREN FOR NONSENSE MUTATION DUCHENNE/BECKER MUSCULAR DYSTROPHY (NASDAQ:PTCT)”. Ptct.client.shareholder.com. Retrieved 2013-11-28.
- Wilschanski, M.; Miller, L. L.; Shoseyov, D.; Blau, H.; Rivlin, J.; Aviram, M.; Cohen, M.; Armoni, S.; Yaakov, Y.; Pugatsch, T.; Cohen-Cymberknoh, M.; Miller, N. L.; Reha, A.; Northcutt, V. J.; Hirawat, S.; Donnelly, K.; Elfring, G. L.; Ajayi, T.; Kerem, E. (2011). “Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis”. European Respiratory Journal 38 (1): 59–69. doi:10.1183/09031936.00120910. PMID 21233271. Sermet-Gaudelus, I.; Boeck, K. D.; Casimir, G. J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; Miller, N. L.; Constantine, S.; Reha, A.; Ajayi, T.; Elfring, G. L.; Miller, L. L. (November 2010). “Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis”. American Journal of Respiratory and Critical Care Medicine 182 (10): 1262–1272.doi:10.1164/rccm.201001-0137OC. PMID 20622033.
- “PTC Therapeutics Completes Enrollment of Phase 3 Trial of Ataluren in Patients with Cystic Fibrosis (NASDAQ:PTCT)”. Ptct.client.shareholder.com. 2010-12-21. Retrieved 2013-11-28.
- Wilschanski, M. (2013). “Novel therapeutic approaches for cystic fibrosis”. Discovery medicine 15 (81): 127–133. PMID 23449115.
- http://www.marketwatch.com/story/ptc-therapeutics-receives-positive-opinion-from-chmp-for-translarna-ataluren-2014-05-23
External links
other sources
rINN: Ataluren
Other Names
PTC124®, 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid
Pharmacological Information
Pharmacology Images

Ataluren Molecule

Web information on Ataluren
Relevant Clinical Literature
UK Guidance
Regulatory Literature
Other Literature
Orphan drug under investigation for treatment of genetic conditions where nonsense mutations result in premature termination of polypeptides. This drug, which is convenient to deliver orally, appears to allow ribosomal transcription ofRNA to continue past premature termination codon mutations with correct reading of the full normal transcript which then terminates at the proper stop codon. Problematically it has been postulated that assay artifact may have complicated evaluation of its efficacy which appears to be less than gentamicin.[1] Faults of this class in the transcription process are involved in several inherited diseases.
Some forms of cystic fibrosis and Duchenne muscular dystrophy are being targeted in the development stage of the drug.[2] Phase I and II trials are promising for cystic fibrosis.[3][4] In a mouse model of Duchenne muscular dystrophy, restoration of muscle function occurred.[5]
A potential issue is that there may be parts of the human genome whose optimal gene function through evolution has resulted from relatively recent in evolutionary terms insertion of a premature termination codon and so functional suboptimal transcripts of other proteins or functional RNAs might result.
References
- ↑ Roberts RG. A read-through drug put through its paces. PLoS biology. 2013; 11(6):e1001458.(Link to article – subscription may be required.)
- ↑ Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, Miller LL. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. Journal of clinical pharmacology. 2007 Apr; 47(4):430-44.(Link to article– subscription may be required.)
- ↑ Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, Elfring GL, Northcutt VJ, Miller LL, Kerem B, Wilschanski M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008 Aug 30; 372(9640):719-27.(Link to article – subscription may be required.)
- ↑ Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL. Ataluren (PTC124) Induces Cystic Fibrosis Transmembrane Conductance Regulator Protein Expression and Activity in Children with Nonsense Mutation Cystic Fibrosis. American journal of respiratory and critical care medicine. 2010 Nov 15; 182(10):1262-72.(Link to article – subscription may be required.)
- ↑ Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007 May 3; 447(7140):87-91.(Link to article – subscription may be required.)
old cut paste
A large-scale, multinational, phase 3 trial of the experimental drug ataluren has opened its first trial site, in Cincinnati, Ohio.
The trial is recruiting boys with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) caused by anonsense mutation — also known as a premature stop codon — in the dystrophin gene. This type of mutation causes cells to stop synthesizing a protein before the process is complete, resulting in a short, nonfunctional protein. Nonsense mutations are believed to cause DMD or BMD in approximately 10 to 15 percent of boys with these disorders.
Ataluren — sometimes referred to as a stop codon read-through drug — has the potential to overcome the effects of a nonsense mutation and allow functional dystrophin — the muscle protein that’s missing in Duchenne MD and deficient in Becker MD — to be produced.
The orally delivered drug is being developed by PTC Therapeutics, a South Plainfield, N.J., biotechnology company, to whichMDA gave a $1.5 million grant in 2005.
PTC124 has been developed by PTC Therapeutics.
Mast Therapeutics’ MST-188 Would Fit Well In Merck’s Drug Development Pipeline

MST-188 (purified poloxamer 188)
MST-188 is an investigational agent, formulated using a purified form of poloxamer 188. Substantial research has demonstrated that poloxamer 188 has cytoprotective and hemorrheologic properties and inhibits inflammatory processes and thrombosis. We believe the pharmacologic effects of poloxamer 188 support the development of MST-188 in multiple clinical indications for diseases and conditions characterized by microcirculatory insufficiency (endothelial dysfunction and/or impaired blood flow). We are enrolling patients in EPIC, a pivotal phase 3 study of MST-188 in sickle cell disease. In addition, our MST-188 pipeline includes development programs in adjunctive thrombolytic therapy (e.g., acute limb ischemia, stroke), heart failure, and resuscitation (i.e., restoration of circulating blood volume and pressure) following major trauma.

POTENTIAL APPLICATIONS OF MST-188
We believe the pharmacodynamic properties of MST-188 (cytoprotective, hemorheologic, anti-inflammatory, antithrombotic/pro-fibrinolytic) enable it simultaneously to address, or prevent activation of, multiple biochemical pathways that can result in microcirculatory insufficiency, a multifaceted condition principally characterized by endothelial dysfunction and impaired blood flow. The microcirculation is responsible for the delivery of blood through the smallest blood vessels (arterioles and capillaries) embedded within tissues. A healthy endothelium is critical to a functional microcirculation. Without the regular delivery of blood and transfer of oxygen to tissue from the microcirculation, individual cells (in both the endothelium and tissue) are unable to maintain aerobic metabolism and, through a series of complex and interrelated events, eventually die. If microcirculatory insufficiency continues, the patient will suffer tissue necrosis, organ damage and, eventually, death.


EPIC’s study drug, MST-188, is a new class of drug that acts by attaching to the damaged surfaces of the cell membranes, potentially improving blood flow and oxygen delivery.
Improving blood flow and oxygen delivery may reduce the duration and severity of pain crises faced by sickle cell patients.





Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): a randomised, multicentre, double-blind, placebo-controlled phase 3 trial.
Angiogenesis is a valid target in the treatment of epithelial ovarian cancer. Trebananib inhibits the binding of angiopoietins 1 and 2 to the Tie2 receptor, and thereby inhibits angiogenesis. We aimed to assess whether the addition of trebananib to single-agent weekly paclitaxel in patients with recurrent epithelial ovarian cancer improved progression-free survival.
Lancet Oncol. 2014 Jun 17. pii: S1470-2045(14)70244-X. doi: 10.1016/S1470-2045(14)70244-X.
http://www.ncbi.nlm.nih.gov/pubmed/24950985
old cut paste
Amgen’s Experimental Ovarian Cancer Drug, Trebananib, Shows Positive Results In Late Stage Clinical Trials
STRUCTURAL FORMULA ,Trebananib, AMG-386
Monomer
MDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE 50
DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY 100
KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV 150
KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ 200
GNVFSCSVMH EALHNHYTQK SLSLSPGKGG GGGAQQEECE WDPWTCEHMG 250
SGSATGGSGS TASSGSGSAT HQEECEWDPW TCEHMLE 287
Disulfide bridges location
7-7′ 10-10′ 42-102 42′-102′ 148-206
148′-206′ 239-246 239′-246′ 275-282 275′-282′
CAS REGISTRY NUMBER 894356-79-7
MOLECULAR FORMULA C2794H4248N752O886S30
Trebananib
Immunoglobulin G1 (synthetic human Fc domain fragment) fusion protein with
angiopoietin 1/angiopoietin 2-binding peptide (synthetic)
http://www.ama-assn.org/resources/doc/usan/trebananib.pdf
http://www.genome.jp/dbget-bin/www_bget?dr:D10177
Amgen’s Experimental Ovarian Cancer Drug, Trebananib, Shows Positive …
Medical Daily
Amgen, a large biotechnology company out of Thousand Oaks, Calif. has announced that its drug for reoccurring ovarian cancer has shown positive results in Phase III clinical trials. The trials sought to stop the progression of ovarian cancer and extend …
read all at

Selexipag Meets Primary Endpoint in Pivitol Phase III Griphon Outcome Study in Patients with Pulmonary Arterial Hypertension
June 16, 2014
Actelion Ltd today announced the top-line results of the pivotal Phase III GRIPHON study in 1,156 patients with pulmonary arterial hypertension (PAH) with selexipag, the first selective oral prostacyclin IP receptor agonist. Initial analysis shows that the event-driven outcome study has met its primary efficacy endpoint with high statistical significance.
June 16, 2014
Actelion Ltd today announced the top-line results of the pivotal Phase III GRIPHON study in 1,156 patients with pulmonary arterial hypertension (PAH) with selexipag, the first selective oral prostacyclin IP receptor agonist. Initial analysis shows that the event-driven outcome study has met its primary efficacy endpoint with high statistical significance.

Selexipag
N-[2-[4-[N-(5,6-Diphenylpyrazin-2-yl)-N-isopropylamino]butoxy]acetyl]methanesulfonamide
2-[4-[N-(5,6-Diphenylpyrazin-2-yl)-N-isopropylamino]butoxy]-N-(methylsulfonyl)acetamide
phase 3 pulmonary hypertention
Selexipag (ACT-293987, NS-304) is a drug currently in development by Actelion as a treatment of pulmonary arterial hypertension. Selexipag and its active metabolite, ACT-333679, are agonists at the PGI2 prostaglandin receptor, which leads to vasodilation in the pulmonary circulation
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of
5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors.
In April 2008, Actelion and Nippon Shinyaku signed a licensing agreement, under which Actelion will be responsible for the global development and commercialization of selexipag outside Japan, and the two companies will co-develop and co-commercialize the drug in Japan.
http://www1.actelion.com/sites/en/scientists/development-pipeline/phase-3/selexipag.page
ABOUT THE ACTELION / NIPPON SHINYAKU ALLIANCE
Actelion and Nippon Shinyaku entered into an exclusive worldwide alliance in April 2008 to collaborate on selexipag, a first-in-class orally-available, selective IP receptor agonist for patients suffering from pulmonary arterial hypertension (PAH). This compound was originally discovered and synthesized by Nippon Shinyaku. Phase II evaluation has been completed, and a Phase III program in PAH patients has been initiated. Actelion is responsible for global development and commercialization of selexipag outside Japan, while the two companies will co-develop and co-commercialize in Japan. Nippon Shinyaku will receive milestone payments based on development stage and sales milestones as well as royalties on any sales of selexipag.
| Selexipag | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 475086-01-2 |
| PubChem | 9913767 |
| ChemSpider | 8089417 |
| UNII | 5EXC0E384L |
| KEGG | D09994 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C26H32N4O4S |
| Molar mass | 496.6 g·mol−1 |
NS-304 (ACT-293987), an orally available long acting non-prostanoid prostaglandin I2 (PGI-2) receptor agonist, is in phase III clinical trials at Actelion for the oral treatment of pulmonary hypertension. Nippon Shinyaku is conducting phase III clinical trials with NS-304 for this indication in Europe. In Japan, phase II clinical trials are ongoing for the treatment of pulmonary hypertension and chronic thromboembolic pulmonary hypertension.
Originally discovered and synthesized by Nippon Shinyaku, NS-304 stimulates PGI-2 receptors in blood vessels and exerts vasodilating effects.
In 2008, the compound was licensed to Actelion by Nippon Shinyaku on a worldwide basis with the exception of Japan for the oral treatment of pulmonary arterial hypertension (PAH). According to the final licensing agreement, Actelion will be responsible for global development and commercialization of NS-304 outside Japan, while the two companies will codevelop and co-commercialize the product candidate in Japan. In 2005, orphan drug designation was assigned in the E.U. by Nippon Shinyaku for the treatment of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension.
…………………….
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
…………………………………
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
………………………
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
……………………
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a isthe drug
2a isthe drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
References
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
ABOUT SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
SELEXIPAG
Selexipag, originally discovered and synthesized by Nippon Shinyaku, is a potent, orally available, selective prostacyclin IP receptor agonist.
Selexipag selectively targets the prostacyclin receptor (also called IP-receptor). The IP receptor is one of 5 types of prostanoid receptor. Prostacyclin activates the IP receptor inducing vasodilation and inhibiting proliferation of vascular smooth muscle cells. Selexipag, unlike prostacyclin analogs, is selective for the IP receptor over other prostanoid receptors. In preclinical models selective IP receptor agonism has shown to maintain efficacy and reduce the risk of side effects mediated by activation of other prostanoid receptors, such as EP1 and EP3 receptors. [2,4,5]
Selexipag was previously evaluated in a Phase II, 43-patient, placebo-controlled, double-blind study, where patients were randomized in a 3:1 ratio receiving selexipag or placebo on top of PDE-5 inhibitor and/or ERA [6]
