

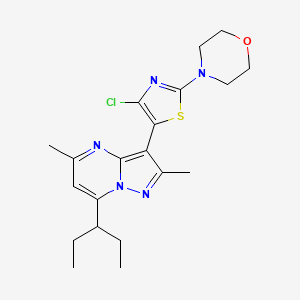

![4-[(2S,4S)-4-Ethoxy-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl]benzoic acid.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=90467622&t=l)
Making small-molecule drugs usually goes something like this: set up a reaction, purify the intermediate, change a solvent, and repeat, repeat, repeat to get the final product. But there’s a lot of waste involved, which is why chemists stress the environmental benefits of an alternate approach: biocatalysis. Engineering enzymes to make reactions happen saves a lot of materials, minimizes chemical and hazardous waste, and even uses less plasticware and glassware. And not having to isolate intermediates saves time.
Home » Phase2 drugs (Page 4)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Catequentinib, Anlotinib
Catequentinib
C23H22FN3O3 407.4 g/mol
1-[[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxyquinolin-7-yl]oxymethyl]cyclopropan-1-amine
1058156-90-3
CAS No. 1360460-82-7 DI HCL
| Molecular Weight | 480.36 |
| Formula | C23H22FN3O3 • 2HCl |
Anlotinib
AL3818
UNII-GKF8S4C432
Chia Tai Tianqing Pharmaceutical Group Co Ltd
Launched (Metastatic non small cell lung cancer – China – May-2018)
Orphan Drug; Priority Review
MOA:VEGFR inhibitor
Indication:advanced gastric adenocarcinoma; Advanced renal cell carcinoma (RCC); Medullary thyroid cancer (MTC); Metastatic colorectal cancer (CRC); Non small cell lung cancer (NSCLC); Soft tissue sarcoma; Ovarian cancerStatus:Phase III (Active)
AL-3818 ; AL-3818, Jiangsu Chia-tai Tianqing Pharmaceutical ; FOCUS-V ; FuKeWei ; VEGFR2/VEGFR3 inhibitor (capsule, cancer), Jiangsu Chia Tai Tianqing Pharmaceutical ; anlotinib ; anlotinib dihydrochloride ; catequentinib ; catequentinib ; catequentinib dihydrochloride
NMR https://file.selleckchem.com/downloads/nmr/S872601-Anlotinib-AL3818-hnmr-selleck.pdf
Anlotinib (AL3818) is a highly potent and selective VEGFR2 inhibitor with IC50 less than 1 nM. It has broad-spectrum antitumor potential in clinical trials.
Anlotinib dihydrochloride is in phase II/III clinical trials for the treatment of metastatic colorectal cancer and advanced gastric adenocarcinoma. The compound was co-developed by CTTQ Pharmaceutical (正大天晴) and Advenchen Laboratory.
It is also in phase II clinical trials for the treatment of ovarian cancer, endometrial cancer, non small cell lung cancer (NSCLC), medullary thyroid cancer (MTC), soft tissue sarcoma and advanced renal cell carcinoma (RCC).
In 2015, orphan drug designation was received in the U.S. for the treatment of ovarian cancer.
PATENT
WO 2016179123
https://patents.google.com/patent/WO2016179123A1/en
new process to synthesize l-((4-(4-Fluoro-2-methyl- lH- indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by condensing intermediate (XI) with (Yl) in a solvent at the presence of KI or Nal, or intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) in Scheme I. A stable crystalline form of l-((4-(4-Fluoro-2 -methyl- lH-indol-5-yloxy)-6- methoxyquinolin-7-yloxy)-methyl)cyclopropanamine and its salts as well as crystalline forms of salts have also been prepared.

Wherein, R is selected from H and Ci-Cealkoxy.
Process A

R is selected from H and C1 -C6 alkoxy
The final compound (AL3818) was prepared according to Process Al when R is H by deprotecting intermediate (Z-l) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25°C-80°C for 0.1-4 hours. (Z-l) was prepared by reacting intermediate (XI) with (Yl-1) at the presence of KI or Nal with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60°C-160°C for 2-24 hours.
Process Al (R=H)

The final compound (AL3818) was prepared according to Process A2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0°C-30°C for 1-24 hours. (Z-2) was prepared by reacting intermediate (XI) with (Y 1-2) at the presence of KI or Nal with K2C03 in a solvent, such as acetone or DMF, at a temperature of 60°C -160°C for 2-24 hours.
Process A2 (R=4-OMe)

The present invention relates a new process to synthesize l-((4-(4-Fluoro-2 -methyl- 1H- indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by reacting intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process B. Proce B

R is selected from H and C1-C6 alkoxy
The final compound (AL3818) was prepared according to Process Bl when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25°C-80°C for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X2-1) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60°C – 160°C for 1-12 hours.
Process Bl R=H)

The final compound (AL3818) was prepared according to Process B2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0°C-30°C for 1-24 hours. (Z-2) was prepared by reacting intermediate (X2-2) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60°C -160°C for 1-12 hours.
Process B2 (R=4-OMe)

The following examples further illustrate the present invention, but should not be construed as in any way to limit its scope.
Example 1
Representation of Process A, Process Al
Process for preparation of l-((4-(4-Fluoro-2 -methyl- lH-indol-5-yloxy)-6-methoxy- quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
To a stirred mixture of benzyl l-(hydroxymethyl)cyclopropylcarbamate (50 g) and DCM (200 ml) was added DIPEA (39g). The result solution was cooled to 0-5 °C with ice/water and further stirred under this temperature for 15 min. MsCl (30g) was added via an addition funnel dropwise keeping temperature below 5°C for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5°C for 30 min and quenched with saturated NaHC03 (150 ml). The solution was extracted with 150 ml DCM twice. The combined DCM layer was washed with 0.1 N HCl (400 ml) followed by brine. It was dried over Na2S04 and concentrated to obtain an off-white solid 60 gram as (l-(benzyloxycarbonylamino)cyclopropyl)methyl methanesulfonate (Yl-1), MS: (M+l) 300.
To a stirred mixture of (Yl-1) (16 g), XI [(4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6- methoxy-7-hydroxyquinoline, 12 g] , K2CO3 (21 g) and KI (21 g) was added DMF (100 ml), the reaction suspension was heated at 80°C for 10 hours and (Yl-l) (10 g) was added to continuously heated 80°C for 10 hours. The reaction then was quenched with water (150 ml) and extracted with 150 ml DCM twice. The combined DCM layer was washed with 2 N NaOH (100 ml) followed by water and brine. It was dried over Na2SC>4 and concentrated, further recrystallized from EtOH to obtain a yellow solid as benzyl l-((4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6-methoxyquinolin- 7-yloxy)methyl)cyclopropylcarbamate (Z-l) 9.5 g. MS: (M+l) 542.
To a stirred mixture of (Z-l) (9.5 g), HCOONH4 (4.7 g) and Pd/C (10%, wet 50%, 4.7g) was added MeOH, the reaction mixture was heated at 45°C for 1.5 hours. It was then cooled and filtered through Celite, further evaporated. 2N HCl (200 ml) was added and extracted with DCM/MeOH (10/1, 100 ml) twice. The aqueous layer was basified with 3N NaOH to adjust pH 11-12 to generate a solid precipitation. The solid was filtered and washed with water to neutral, further suction dry. The solid was dissolved into a mixture of DCM/MeOH (250 ml, 10/1) and further washed with water and brine. It was dried with MgS04 and filtered, further evaporated to give a light yellow solid 5.5 g crude product. Further purification was conducted by dissolving the crude product into DCM/MeOH (40 ml, 10/1) to triturate with petroleum ether (40 ml) for 2 hours slow stirring. The precipitate was filtered and dried in an oven to give the final crystalline product 4.4 g (MP: 203-208 C) and it can be further purified by recrystallizing from EtOH to give purer final product as a same crystalline form. MS: (M+l) 408; ¾ NMR(DMSO-dg) δ 0.60- 0.63(d, 4H), 2.41(s, 1H), 2.42-2.5 l(t, 2H), 3.3 l(s, 2H), 3.96(s, 3H), 4.04(s, 2H), 6.27(s, 1H), 6.31-6.32(m, 1H), 6.97-7.02(t, 1H), 7.20-7.22(d, 1H), 7.36(s, 1H), 7.60(s, 1H), 8.40-8.42(d, 1H), 1 1.41(s, 1H). MP: 208-210°C; DSC Melting Range (Endo): 207-220°C with Peak Temp=216°CPATENTWO 2019154273https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=11C1DF5485B11ADA40E45C9488AB5679.wapp1nB?docId=WO2019154273&tab=FULLTEXT
Tyrosine kinases are a group of enzymes that catalyze the phosphorylation of protein tyrosine residues. They play an important role in intracellular signal transduction. They are involved in the regulation, signal transmission and development of normal cells, and are also related to tumor cells. Proliferation, differentiation, migration and apoptosis are closely related. Many receptor tyrosine kinases are related to the formation of tumors, and can be divided into epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial cell growth factor receptor according to the structure of their extracellular region. Body (VEGFR), Fibroblast Growth Factor Receptor (FGFR), etc.[0003]WO2008112407 discloses the compound 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy in Example 24 )Methyl)cyclopropylamine and its preparation method, its structural formula is shown in formula I:[0004]
[0005]It is a multi-target receptor tyrosine kinase inhibitor that can inhibit the activity of vascular endothelial cell growth factor receptors (VEGFR1, VEGFR2/KDR and VEGFR3), stem cell factor receptors, platelet-derived growth factor receptors and other kinase activities. Inhibit the downstream signal transduction mediated by VEGFR2, thereby inhibiting tumor angiogenesis.[0006]Solid drugs generally have multiple crystal forms, such as polymorphs, solvates (hydrates), salts, and co-crystals. The change in the crystal form of the same drug usually results in different melting points, solubility, stability, biological activity, etc., which are important factors that affect the difficulty of drug preparation, storage stability, preparation difficulty, and bioavailability. . When the compound has multiple crystal forms, due to the specific thermodynamic properties and stability of the specific crystal form of the drug, it is important to understand the crystal form of the compound used in each dosage form during the preparation process to ensure the production process Use the same form of medicine. Therefore, it is necessary to ensure that the compound is a single crystal form or a known mixture of some crystal forms.[0007]WO2016179123 discloses the crystalline form 1 of the free base anhydrate of the compound of formula I and a preparation method thereof. CN201010245688.1 discloses the anhydrate and dihydrate crystals of quinoline derivative dihydrochloride and the preparation method thereof.[0008]The discovery of a variety of new crystal forms of medicinal compounds provides an opportunity to improve the physical properties of the drug, that is, to expand all the properties of the substance, which can better guide the research of the compound and its preparation. Therefore, the quinoline derivative provided in this application The crystals and pharmaceutical compositions containing the crystals have commercial value in the manufacture of medicines and other applications.Example 1 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropylamine (Formula I compound) preparation[0081]
[0082]Put intermediate 1 (its chemical name is (1-((4-(4-fluoro-2-methyl-1H-indol-5-yl)oxy-6-methoxy Quinolin-7-yl)oxy)methyl)cyclopropyl)benzyl carbamate) 100g, 10% palladium on carbon 30g, ammonium formate 50g and methanol 800ml. Incubate the reaction at 45-55°C, TLC tracking showed that the reaction was complete, filtered, the filter cake was washed with a small amount of methanol, the filtrate was concentrated to dryness under reduced pressure, ethyl acetate and 2mol/L hydrochloric acid were added, stirred for 10 minutes, and then stood for 10 minutes. Separate the aqueous phase, adjust the pH to above 12 with 4N sodium hydroxide, and a large amount of solids will precipitate out. After washing with water until neutral, the aqueous phase is filtered to obtain the crude product of the title compound.[0083]Example 2 Preparation of amorphous compound of formula I[0084]According to the preparation method disclosed in Example 24 of WO2008112407, 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yl (Oxy)methyl)cyclopropylamine is composed of (1-(((4-(4-fluoro-2-methyl-1H-indol-5-yl)oxy-6-methoxyquinolin-7-yl )Oxy)methyl)cyclopropyl)benzyl carbamate (Intermediate 1) was prepared according to the following methods 2.1 and 2.2.[0085]2.1 Take 100 mg of Intermediate 1 and Pd/C (10%, 40 mg) into ethanol (20 ml), and hydrogenate at 50 psi for 12 hours. The reaction solution was filtered with diatomaceous earth, and evaporated to obtain an amorphous compound of formula I, and its X-ray powder diffraction (XRD) pattern was obtained as shown in FIG. 11.[0086]
2.2 Take 100 mg of Intermediate 1, acetic acid (1ml) and 33% hydrobromic acid/acetic acid (1ml) and mix. The reaction was stirred for 1 hour at room temperature, diluted with ethyl acetate/water, and then basified with sodium carbonate. The organic layer is dried, concentrated, and purified by silica gel column to obtain the amorphous compound of formula I.PATENTUS 20160326138https://patents.google.com/patent/US20160326138A1/enNew process has been outlined in Scheme I.

- The present invention relates a new process to synthesize 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by condensing intermediate (X1) with (Y1) in a solvent at the presence of KI or NaI to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process A.
- [0040]
The final compound (AL3818) was prepared according to Process A1 when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25° C.-80° C. for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X1) with (Y1-1) at the presence of KI or NaI with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60° C.-160° C. for 2-24 hours. - [0041]
The final compound (AL3818) was prepared according to Process A2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0° C.-30° C. for 1-24 hours. (Z-2) was prepared by reacting intermediate (X1) with (Y1-2) at the presence of KI or NaI with K2CO3 in a solvent, such as acetone or DMF, at a temperature of 60° C.-160° C. for 2-24 hours. - [0042]
The present invention relates a new process to synthesize 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropanamine (AL3818) by reacting intermediate (X2) with (Y2) in a solvent to form intermediate (Z) which is deprotected to give the final compound (AL3818) according to Process B. - [0043]
The final compound (AL3818) was prepared according to Process B1 when R is H by deprotecting intermediate (Z-1) with HCOONH4 (ammonium formate) and Pd/C in an alcoholic solvent, such as MeOH, at 25° C.-80° C. for 0.1-4 hours. (Z-1) was prepared by reacting intermediate (X2-1) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60° C.-160° C. for 1-12 hours. - [0044]
The final compound (AL3818) was prepared according to Process B2 when R is 4-OMe by deprotecting intermediate (Z-2) with TFA in DCM at 0° C.-30° C. for 1-24 hours. (Z-2) was prepared by reacting intermediate (X2-2) with (Y2) in a solvent, such as pyridine or lutidine, at a temperature of 60° C.-160° C. for 1-12 hours. - [0045]
The following examples further illustrate the present invention, but should not be construed as in any way to limit its scope.
Example 1Representation of Process A, Process A1Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
- [0046]
To a stirred mixture of benzyl 1-(hydroxymethyl)cyclopropylcarbamate (50 g) and DCM (200 ml) was added DIPEA (39 g). The result solution was cooled to 0-5° C. with ice/water and further stirred under this temperature for 15 min. MsCl (30 g) was added via an addition funnel dropwise keeping temperature below 5° C. for about 1.5 hours. After completion of addition, the reaction mixture was allowed stirring at 0-5° C. for 30 min and quenched with saturated NaHCO3 (150 ml). The solution was extracted with 150 ml DCM twice. The combined DCM layer was washed with 0.1 N HCl (400 ml) followed by brine. It was dried over Na2SO4 and concentrated to obtain an off-white solid 60 gram as (1-(benzyloxycarbonylamino)cyclopropyl)methyl methanesulfonate (Y1-1), MS: (M+1) 300. - [0047]
To a stirred mixture of (Y1-1) (16 g), X1 [(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-7-hydroxyquinoline, 12 g], K2CO3 (21 g) and KI (21 g) was added DMF (100 ml), the reaction suspension was heated at 80° C. for 10 hours and (Y1-1) (10 g) was added to continuously heated 80° C. for 10 hours. The reaction then was quenched with water (150 ml) and extracted with 150 ml DCM twice. The combined DCM layer was washed with 2 N NaOH (100 ml) followed by water and brine. It was dried over Na2SO4 and concentrated, further recrystallized from EtOH to obtain a yellow solid as benzyl 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)methyl)cyclopropylcarbamate (Z-1) 9.5 g. MS: (M+1) 542. - [0048]
To a stirred mixture of (Z-1) (9.5 g), HCOONH4 (4.7 g) and Pd/C (10%, wet 50%, 4.7 g) was added MeOH, the reaction mixture was heated at 45° C. for 1.5 hours. It was then cooled and filtered through Celite, further evaporated. 2N HCl (200 ml) was added and extracted with DCM/MeOH (10/1, 100 ml) twice. The aqueous layer was basified with 3N NaOH to adjust pH 11-12 to generate a solid precipitation. The solid was filtered and washed with water to neutral, further suction dry. The solid was dissolved into a mixture of DCM/MeOH (250 ml, 10/1) and further washed with water and brine. It was dried with MgSO4 and filtered, further evaporated to give a light yellow solid 5.5 g crude product. Further purification was conducted by dissolving the crude product into DCM/MeOH (40 ml, 10/1) to triturate with petroleum ether (40 ml) for 2 hours slow stirring. The precipitate was filtered and dried in an oven to give the final crystalline product 4.4 g (MP: 203-208° C.) and it can be further purified by recrystallizing from EtOH to give purer final product as a same crystalline form. MS: (M+1) 408; 1H NMR (DMSO-d6) δ 0.60-0.63 (d, 4H), 2.41 (s, 1H), 2.42-2.51 (t, 2H), 3.31 (s, 2H), 3.96 (s, 3H), 4.04 (s, 2H), 6.27 (s, 1H), 6.31-6.32 (m, 1H), 6.97-7.02 (t, 1H), 7.20-7.22 (d, 1H), 7.36 (s, 1H), 7.60 (s, 1H), 8.40-8.42 (d, 1H), 11.41 (s, 1H). MP: 208-210° C.; DSC Melting Range (Endo): 207-220° C. with Peak Temp=216° C. TGA demonstrating as an unsolvated material with weight loss at about 210° C. (between 205-215° C.). XRPD having pattern comprising characteristic 10 peaks with intensity % greater than 10% expressed in d values and angles as follows: - Angle d value 13.344 6.62986 15.858 5.58405 16.799 5.27326 17.640 5.02377 18.770 4.72373 20.650 4.29771 21.633 4.10463 23.087 3.84934 25.128 3.54112 26.607 3.34755
- [0049]
It was similar prepared according to the preparation procedures of (Z-1) described in Example 1 by using 4-methoxybenzyl 1-(hydroxymethyl)cyclopropylcarbamate to first generate (1-((4-methoxybenzyloxy)carbonylamino)cyclopropyl)methyl methanesulfonate (Y1-2) then to give 4-methoxybenzyl 1-((4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)-methyl)cyclopropylcarbamate (Z-2), MS: (M+1) 572 - [0050]
To a stirred mixture of (Z-2) (1.5 g) in DCM (15 ml) at 0° C. was added TFA (1.5 ml) for about 30 min and warmed up to RT. The reaction was stirred at RT for 2 hours and added into water (30 ml). The aqueous layer was extracted with DCM twice (100 ml×2) and basified with 2N NaOH to adjust pH 11-12. The mixture was extracted with DCM (100 ml×3) and further washed with brine (100 ml). It was dried with MgSO4 and filtered. The solution was evaporated to give 1.05 g crude final product. Further purification was conducted to dissolve the crude product into DCM/MeOH and triturated with petroleum ether and dried in an oven to give the final pure product 0.8 g AL3818 with the same crystalline form.
Example 3Representation of Process A, Process B1Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
- [0051]
To a mixture of benzyl 1-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropyl-carbamate (X2-1) (5 g), 4-fluoro-2-methyl-1H-indol-5-ol (Y2) (5 g) and DMAP (4 g) was added 1,6-lutidine (15 ml). The reaction was stirred and heated at 135° C. for 5 hours and was cooled followed by adding IPA with slow stirring for 2 hours at RT. The solid was filtered and further washed with IPA, dried to give (Z-1) 5.2 g as a solid. It was then similarly prepared according to deprotection procedures described of (Z-1) in Example 1 to give the final compound AL3818 with the same crystalline form.
Example 4Representation of Process A, Process B2Process for preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)methyl)cyclopropanamine (AL3818)
- [0052]
(Z-2) was similarly prepared according to the procedures described in Example 3 by using 4-methoxybenzyl 1-((4-chloro-6-methoxyquinolin-7-yloxy)methyl)cyclopropylcarbamate (X2-2) and (Y2). It was then similarly prepared according to deprotection procedures of (Z-2) described in Example 2 to give the final compound AL3818 with the same crystalline form.
Example 5
- [0053]
Preparation of 1-((4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-quinolin-7-yloxy)-methyl)cyclopropanamine bishydrochloride acid salt and its crystalline - [0054]
To a 25 ml flask was added 250 mg free base (AL3818), 4N HCl in dioxane 0.625 mL (2.5 mmol, 4 eq.) in 10 ml EtOH, the reaction was heated at 75° C. for 30 minutes, cooled to RT and stirred for O.N. The solid was filtered and rinsed with acetone twice. It was dried in oven at 50° C. for 4 hours to give 126 mg white solid as the bishydrochloride salt as a crystalline and further recrystallized from EtOH to give a purer product as a same crystalline form. 1H NMR (DMSO-d6) δ 1.09-1.24 (m, 4H), 2.43 (s, 3H), 4.08 (s, 3H), 4.40 (s, 2H), 6.32 (s, 1H), 6.76 (s, 1H), 7.05-7.11 (t, 1H), 7.27-7.30 (d, 1H), 7.65 (s, 1H), 7.82 (s, 1H), 8.64 (s, 2H), 8.70-8.73 (m, 1H), 11.51 (s, 1H). Chloride ion chromatography showed 2 molecular ratio ions (16.1%). DSC Melting Range (Exo): 249-280 with Peak Temp=268° C. - [0055]
To a 10 mL flask, charged 140 mg of 3818-2HCl salt from above Example 4 and 0.7 mL (×5 with salt volume) of 80% MeOH in H2O. The result suspension was heated to 70° C. to form a solution and cooled to RT and further stirred for O.N. The solid was filtered and rinsed with acetone twice. It was dried in oven at 50° C. for 4 hours to obtain off-white solid 110 mg as the crystalline bishydrochloride hydrate salt. 1H NMR (DMSO-d6) δ 1.09 (s, 2H), 1.22 (s, 2H), 2.44 (s, 1H), 2.52 (s, 2H), 4.09 (s, 3H), 4.44 (s, 2H), 6.32 (s, 1H), 6.81-6.82 (d, 1H), 7.08-7.14 (t, 1H), 7.29-7.32 (d, 1H), 7.79 (s, 1H), 7.85 (s, 1H), 8.75-8.78 (d, 1H), 8.85 (s, 2H), 11.66 (s. 1H). Chloride ion chromatography showed 2 molecular ratio ions (17.8%). DSC Melting Range (Exo): 207-260° C. with Peak Temp=226° C. TGA demonstrating 2.68% (˜3%, 1 water) weight loss till 120° C. (between 115-125° C.) and further weight loss at about 170° C. (between 165-175° C.).
US8148532B2.
https://patents.google.com/patent/US8148532B2/en
Patent
2. US20080227811A1.

/////////////catequentinib, ANLOTINIB, AL3818, AL 3818, PHASE 2, CHINA 2018
NC1(CC1)COc1cc2nccc(Oc3ccc4[NH]c(C)cc4c3F)c2cc1OC
ETRUMADENANT
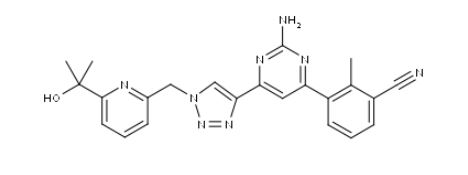
ETRUMADENANT
CAS 2239273-34-6
C23 H22 N8 O, 426.47
Benzonitrile, 3-[2-amino-6-[1-[[6-(1-hydroxy-1-methylethyl)-2-pyridinyl]methyl]-1H-1,2,3-triazol-4-yl]-4-pyrimidinyl]-2-methyl-
- 3-[2-Amino-6-[1-[[6-(1-hydroxy-1-methylethyl)-2-pyridinyl]methyl]-1H-1,2,3-triazol-4-yl]-4-pyrimidinyl]-2-methylbenzonitrile
- AB 928
Arcus Biosciences is developing etrumadenant, the lead from the small molecule adenosine (A2a/A2b) dual receptor antagonist program, for treating cancer. In November 2020, preliminary data from ARC-7 in metastatic NSCLC were expected to report in the first half of 2021.
- OriginatorArcus Biosciences
- ClassAmines; Antineoplastics; Nitriles; Pyridines; Pyrimidines; Small molecules; Triazoles
- Mechanism of ActionAdenosine A2A receptor antagonists; Adenosine A2B receptor antagonists
- Phase IINon-small cell lung cancer
- Phase I/IIProstate cancer
- Phase IBladder cancer; Breast cancer; Cancer; Colorectal cancer; Endometrial cancer; Gastrointestinal cancer; Head and neck cancer; Malignant melanoma; Merkel cell carcinoma; Oesophageal cancer; Ovarian cancer; Renal cancer
- 19 Sep 2020Updated efficacy and adverse events data from a phase I/Ib trial in Non-small cell lung cancer presented at the 45th European Society for Medical Oncology Congress (ESMO-2020)
- 06 Aug 2020Efficacy data from a phase I trial in Colorectal cancer presented at the American Association for Cancer Research Meeting (AACR-2020)
- 13 Jul 2020Arcus Biosciences and Gilead Sciences complete closing of partnership agreement to co-develop and co-promote AB 928 in USA
PAPER
Organic Process Research & Development (2020), 24(7), 1254-1261.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00124
AB928 is a potent and selective dual antagonist of the A2a and A2b receptors, which is currently in clinical trials. Here, we report the development of two scalable and practical syntheses of AB928. The first-generation synthesis was used to successfully obtain AB928 in excellent yield and purity to support our preclinical and initial clinical studies. Recently, we have developed a second-generation synthesis of AB928 featuring a palladium-free protocol to access 3-(2-amino-6-chloropyrimidin-4-yl)-2-methylbenzonitrile, a key intermediate in the AB928 synthesis. The new method is scalable, practical, and significantly more cost-effective.



PAPER
Tetrahedron Letters (2020), 61(20), 151855.
PAPENT
WO 2020018680
Example 1: Synthesis of 3-[2-amino-6-(l-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-lH-l,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile (Compound I)
[0208] Step 1 : In a 250mL round bottom flask equipped with a magnetic stir bar was successively charged the boronic ester (3.89 g, 16 mmol) and the 2-amino-4,6-dichloropyrimidine (3.67 g, 22,4 mmol). Absolute ethanol (100 mL) was added followed by a solution of KHCO3 (4.81 g, 48 mmol) in deionized water (19 mL). The resulting suspension was degassed with nitrogen for 5 minutes. PdChiPPluk (112 mg, 1 mol%) was then added and the mixture was heated to 78 °C for 3 hours under a nitrogen atmosphere. Ethanol was evaporated under reduced pressure and deionized water (150 mL) was added. The suspension was filtered and the solid was washed with additional water (100 mL). The solid was then dissolved in acetone (220 mL) and collected in a 500 mL round bottom flask. A mixture of silica and celite (1 : 1, 150 g) was added and the solvent was removed under reduced pressure. The resulting crude material was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate gradient 0% to 15%). The desired product was obtained as a white solid (1.91 g, 49%). LCMS: Method A, retention time = 2.93 min, ESI MS [M+H]+ for C12H9CIN4, calcd 245.7, found 245.2
[0209] Step 2 : In a round-bottom flask 5.1 g (20.8 mmol) of chloro-pyrimidine was suspended in 42 mL of degassed THF. To this suspension was added 8.68 mL (62.4 mmol) of Et3N and 5.95 mL (25.0 mmol) of TIPS-acetylene. The reaction mixture was stirred for 5 min, followed by addition of 219 mg (0.312 mmol) of PdCl2(PPh3)2 and 119 mg (0.624 mmol) of Cul. The reaction mixture was stirred at 50 °C for 5h under N2. After cooling the reaction to room temp., solvent was removed and the crude material was resuspended in 100 mL EtOAc from which insoluble solid was filtered off. The filtrate was washed with (1 : 1) NH4CI/NH4OH (2 x 100 mL) and 10% Na2S204 (1 x 100 mL). The organic layer was dried using Na2S04, concentrated and taken to next step without further purification.
[0210] Step 3 : In a round-bottom flask the crude TIPS product from previous step was dissolved in 42 mL dry THF and cooled to 0 °C. To this was added 25 mL (25.0 mmol) of TBAF (1.0 M in THF). The reaction was stirred at 0 °C for 15 min. Saturated NH4CI (100 mL) was added to quench the reaction. The organics were extracted from the aqueous layer with EtOAc (2 x 100 mL). The combined organic layer was washed with (1 : 1) NH4CI/NH4OH (2 x 100 mL) and 10% Na2S204 (1 x 100 mL). The organic layer was dried using Na2S04, concentrated and the pure product 5 was obtained by triturating with 40% CH2Cl2/Hexane as a light brown solid. Yield: 3.71 g (76%, 2-steps).
[0211] Step 4 : To a solution of methylmagnesium bromide (3 M in Et20, 40 mL, 120 mmol, 4.0 equiv) at 0 °C under N2 was added a solution of methyl 2-(hydroxymethyl)pyridine-2-carboxylate (5.0 g, 29.9 mmol) in THF (70 mL, 0.4 M) over the course of 30 minutes. The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was quenched with NH4CI aq (55 mL) and EtOAc (50 mL) was added. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 x 40 mL). The combined organic extracts were washed with saturated aqueous sodium bisulfite (7 x 20 mL), then dried (Ni^SCh), filtered and concentrated in vacuo to give the title compound (3.45 g, 69% yield; 96% purity as judged by LCMS) as a pale yellow liquid. LCMS: Method A, retention time = 0.722 and 1.06 min, ESI MS [M+H]+ for C9H13NO2, calcd 167.09, found 167.2
[0212] Step 5 : To a solution of 2-hydroxymethyl-6-(l -hydroxy- 1 -methyl ethyljpyri dine (5 g,
29.9 mmol, 1.0 equiv) in PhMe (33 mL, 0.9 M) at 0 °C under N2 was added diphenylphosphoryl azide (7.73 mL, 35.9 mmol, 1.2 equiv.), followed by l,8-diazabicyclo[5.4.0]undec-7-ene (5.37 mL, 35.9 mmol, 1.2 equiv.). The resulting mixture was to warm to room temperature and stirred for 14 h. Upon completion, diluted with ethyl acetate and washed with water, the organic layer was dried (Na2S04), filtered and concentrated. The residue was dissolved in 1N aq HC1 (2 eq, 60 mmol) and extracted with MTBE in hexanes (3:7, 100 mL), the organic layer was washed with water (50 mL) and the combined aqueous layer was neutralized with 2N aqueous NaOH and extracted with ethyl acetate (3X75 mL), dried the organic layer (Na2S04), filtered through a plug of cotton and concentrated the filtrate to afford the pure compound as pale yellow color liquid (3.75 g, 75%). LCMS: Method A, retention time = 2.67 min, ESI MS [M+H]+ for C9H12N4O, calcd 193.1, found 193.2
[0213] Step 6: A mixture of azide (3.34 g, 17.4 mmol), alkyne (3.71 g, 15.8 mmol), copper(II) sulfate (39 mg; 0.158 mmol), and sodium ascorbate (156 mg, 0.790 mmol) in 2: 1 /-BuOH/EbO (158 mL) was heated at 60 °C for 13 h. The solvent was removed in vacuo, the residue dry loaded onto silica gel, and purified by silica gel chromatography (0-100% EtOAc in hexanes) to afford the desired product as an off-white solid (6.08 g, 90%). ‘H NMR (400 MHz, DMSO-cfc) d 8.69 (s, 1H), 7.90 (d, J= 7.8 Hz, 1H), 7.80 (t, J= 7.8 Hz, 1H), 7.76 (d, J= 7.8 Hz, 1H), 7.61 (d, J= 8.0 Hz, 1H), 7.51 (t, /= 7.8 Hz, 1H), 7.28 (s, 1H), 7.10 (d, J= 7.6 Hz, 2H), 6.90 (s, 2H), 5.81 (s, 2H), 5.23 (s, 1H), 2.55 (s, 3H), 1.38 (s, 6H). ESI MS [M+H]+ for C23H23N8O, calcd 427.2, found 427.3.
Example 2: Preparation of Crystalline Solid Form of 3-[2-amino-6-(l-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-lH-l,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile
[0214] The product from Example 1, Step 6 (7.53 g) was dissolved in acetone (109 mL) by heating to reflux at which point water (218 mL) was added at a rate of 10 mL/min to initiate crystallization. The mixture was cooled and the solids were collected by filtration, washed with 1 :2 acetone/water (109 mL), and dried under vacuum to afford Form I of Compound I as a white solid (7.08 g; 94%).
PATENT
WO 2019161054
PATENT
WO2020185859 , claiming method for treating a subject identified as having an oncogene driven cancer comprising an agent (eg AB-928) targeting the extracellular production of adenosine and/or antagonizing the activation by adenosine of one of its receptors.
PATENT
WO-2020247789
Processes for preparing aminopyrimidine compounds, particularly etrumadenant (AB-928).
Example 1: Trifluoroethanol Assisted Condensation of B-Ketoesters to Provide a
Hydroxypyrimidine (and Chloropyrimidine).
bromo-2-methylaniline (18.6 g, 100 mmol) dropwise so that a fine white suspension forms. The mixture was cooled to 0 °C and a solution of sodium nitrite (7.31 g, 106 mmol) in water (15.1 mL) was added dropwise. The mixture was stirred at 0 °C for 30 minutes. To the resultant homogeneous mixture at 0 °C was added sodium bicarbonate (17.8 g, 212 mmol) at such a rate to avoid excessive gas evolution. The aqueous phase of the resultant brown suspension was found to have pH ~7. This suspension was maintained at 0 °C.
[0070] In a separate flask, copper cyanide (9.85 g, 110 mmol), potassium cyanide (13.0 g, 200 mmol), and water (31 mL) were heated to 60 °C to form a homogeneous solution. To this solution at 60 °C with stirring was added the above suspension dropwise to avoid excessive gas evolution. After addition, the mixture was stirred at 100 °C for 30 minutes. The mixture was cooled, MTBE (200 mL) was added, the mixture agitated, and filtered to remove any solids, washing with MTBE. The organic phase was dried over Na2SO4 and concentrated. The resultant crude product was purified by vacuum distillation to afford the desired product as a light orange solid (13.6 g, 69%).
[0071] Step 2: In a two liter two-necked flask, aryl bromide (101.9 g, 520 mmol, 1.0 equiv.) was dissolved in THF (520 mL) under an atmosphere of N2, and the mixture was cooled in an
ice-water bath. iPrMgClLiCl (400 mL, 1.3 M in THF, 520 mmol, 1.0 equiv.) was added by cannula. Upon completion of the addition, the ice bath was removed. After four hours, the flask was cooled in an ice-water bath and dry ice (~ 230 g, 5.2 mol, 10 equiv.) was added portionwise to prevent overheating or bubbling over (note: CO2 gas can be bubbled through the solution in place of solid dry ice). When bubbling from the addition was complete, the mixture was diluted with MTBE (500 mL) and 2M HC1 (250 mL). The layers were separated, and the aqueous layer was washed with additional MTBE (500 mL). The organic layer was extracted with 10% NaOH (190 mL x 2), and the combined aqueous layers were cooled in an ice-water bath and acidified with concentrated HC1 until a white precipitate formed. The precipitate was isolated by filtration and washed with water before being dried overnight in a vacuum oven at 80° C to afford the benzoic acid as a white solid (64.1 g, 76% yield).
[0072] Step 3: The benzoic acid (50 g, 311 mmol, 1.0 equiv.) was suspended in CH2CI2, and oxalyl chloride (40 mL, 466 mmol, 1.5 equiv.) was added, followed by DMF (~ 30 drops). Off gassing was observed immediately, and the reaction flask was open to the atmosphere under positive pressure of N2. Upon complete consumption of the starting acid as determined by LCMS and visual inspection (complete dissolution of starting material), the reaction mixture was concentrated. Excess oxalyl chloride was removed by azeotropic distillation with toluene to afford the corresponding acid chloride as a tannish-brown solid.
[0073] In a separate two-necked flask equipped with an overhead stirrer, potassium ethyl malonate (66.1 g, 388 mmol, 1.25 equiv.), triethylamine (108 mL, 777 mmol, 2.5 equiv.) and MeCN (777 mL) were cooled in a salt/ice-brine bath. Solid MgCl2 (74 g, 777 mmol, 2.5 equiv.) was added, and the resulting suspension was vigorously stirred at ~ -10° C. After one hour, the solid acid chloride was added at a rate to ensure dissolution into the thick suspension. The suspension rapidly became homogenous, and the stirring rate was reduced to avoid splashing.
The ice bath was removed. Upon complete consumption of the starting material as determined by TLC analysis, the reaction mixture was cooled in an ice-water bath, and 2M HC1 (971 mL, 1.9 mol, 6.25 equiv.) was added, and the ice bath was removed. After 30 minutes, the layers were separated, and the aqueous layer was extracted with MTBE. The combined organic layers were washed with saturated NaHCO3 and brine, dried over sodium sulfate, filtered, and concentrated to afford the keto-ester as a tannish-brown solid (67 g, 93% yield).
[0074] Step 4: A round-bottom flask was charged with 42.0 g (181.8 mmol) of the b-keto-ester, 32.7 g (181.8 mmol) of guanidinium carbonate and 227 mL of trifluoroethanol. The suspension was then heated to reflux under N2 for 16 h.
[0075] Work-up: The reaction was cooled to room temperature and solvent was evaporated under reduced pressure to obtain a viscus red oil. The oil was re-dissolved in 250 mL H2O and the aqueous solution was extracted with dichloromethane (2 x 250 mL). The aqueous phase is then acidified to pH ~2-3 using 1.0 M HCl(aq ). The precipitated product was collected by filtration, washed thoroughly with H2O and dried in a vacuum oven at 70 °C. Yield 30.81 g (75%), Purity >99%.
[0076] Step 5: A round-bottom flask was charged with 50.0 g (221.2 mmol) pyrimidone from step 4 and 100.8 g (442.2 mmol) of benzyltriethylammonium chloride. The mixture was suspended in 442.2 mL of dry acetonitrile and 31.0 mL (331.8 mmol) of POCI3 was added. The suspension thus obtained was then heated to reflux under N2 for 4 h.
[0077] Work-up: The reaction was cooled to room temperature and ~200 g crushed ice was added. The mixture was then stirred for 30 min flowed by dropwise addition of ice-cold 15% aqueous NH4OH to ~ pH 10 -11. {Note: Slow addition of cold NH4OH is recommended to avoid sudden exotherm due to quenching of excess POCI3). The suspension was then stirred at room temperature for an additional 1.5 h. The precipitated product was collected by filtration, washed thoroughly with H2O and dried in a vacuum oven at 70 °C. Yield 48.2 g (89%), Purity >99%.
HPLC conditions
HPLC: Agilent 1 100
Column: YMC-HPLC Column; 250 x 4.6; S-5 pm, 20 nm; AQ20S05-2546WT; No.0425058945
Solvent: H2O / MeCN with 0.1% HCO2H
Flow Rate: 0.8 mL/min
Column Temperature: 30 °C
Method:
Example 2: Comparative Pyrimidine Coupling
[0078] The synthetic route for preparing 3-[2-amino-6-(l- {[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-1H-1 ,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile utilizing boronic ester benzonitrile to linked the phenyl and pyrimidine rings is shown below and is also provided in WO2018/136700.
[0079] The scheme below displays the synthetic route used to prepare the boronic ester benzonitrile used in the process above and subsequent reaction with pyrimidine to form a compound of Formula (I). Notably, the desired linkage between the pyrimidine and the phenyl provides a yield of less than 50%.
[0080] The below scheme displays the synthetic route used to prepare a compound of Formula (I) that utilized a conversion of a b-diketoester to a pyrimidine using guanidine. The route provides a 75% yield.
PATENT
WO 2018136700
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018136700
Example 1: Synthesis of 3-[2-amino-6-(1-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-1H-1,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile
[0269] Step 1: In a 250mL round bottom flask equipped with a magnetic stir bar was successively charged the boronic ester (3.89 g, 16 mmol) and the 2-amino-4,6- dichloropyrimidine (3.67 g, 22,4 mmol). Absolute ethanol (100 mL) was added followed by a solution of KHCO3 (4.81 g, 48 mmol) in deionized water (19 mL). The resulting suspension was degassed with nitrogen for 5 minutes. PdCl2(PPh3)2 (112 mg, 1 mol%) was then added and the mixture was heated to 78 °C for 3 hours under a nitrogen atmosphere. Ethanol was evaporated under reduced pressure and deionized water (150 mL) was added. The suspension was filtered and the solid was washed with additional water (100 mL). The solid was then dissolved in acetone (220 mL) and collected in a 500 mL round bottom flask. A mixture of silica and celite (1:1, 150 g) was added and the solvent was removed under reduced pressure. The resulting crude material was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate gradient 0% to 15%). The desired product was obtained as a white solid (1.91 g, 49%). LCMS: Method A, retention time = 2.93 mm, ESI MS [M+H]+ for C12H9ClN4, calcd 245.7, found 245.2
[0270] Step 2: In a round-bottom flask 5.1 g (20.8 mmol) of chloro-pyrimidine was suspended in 42 mL of degassed THF. To this suspension was added 8.68 mL (62.4 mmol) of Et3Ν and 5.95 mL (25.0 mmol) of TIPS -acetylene. The reaction mixture was stirred for 5 min, followed by addition of 219 mg (0.312 mmol) of PdCl2(PPh3)2 and 119 mg (0.624 mmol) of Cul. The reaction mixture was stirred at 50 °C for 5h under N2. After cooling the reaction to room temp., solvent was removed and the crude material was resuspended in 100 mL EtOAc from which insoluble solid was filtered off. The filtrate was washed with (1:1) NH4C1/NH4OH (2 × 100 mL) and 10% Na2S2O4 (1 × 100 mL). The organic layer was dried using Na2SO4, concentrated and taken to next step without further purification.
[0271] Step 3: In a round-bottom flask the crude TIPS product from previous step was dissolved in 42 mL dry THF and cooled to 0 °C. To this was added 25 mL (25.0 mmol) of TBAF (1.0 M in THF). The reaction was stirred at 0 °C for 15 mm. Saturated NH4Cl (100 mL) was added to quench the reaction. The organics were extracted from the aqueous layer with EtOAc (2 x 100 mL). The combined organic layer was washed with (1:1) NH4Cl/NH4OH (2 x 100 mL) and 10% Na2S2O4 (1 x 100 mL). The organic layer was dried using Na2SO4, concentrated and the pure product 5 was obtained by triturating with 40% CH2Cl2/Hexane as a light brown solid. Yield: 3.71 g (76%, 2-steps).
[0272] Step 4: To a solution of methylmagnesium bromide (3 M in Et2O, 40 mL, 120 mmol, 4.0 equiv) at 0 °C under N2 was added a solution of methyl 2-(hydroxymethyl)pyridine-2-carboxylate (5.0 g, 29.9 mmol) in THF (70 mL, 0.4 M) over the course of 30 minutes. The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was quenched with NH4Cl aq (55 mL) and EtOAc (50 mL) was added. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 x 40 mL). The combined organic extracts were washed with saturated aqueous sodium bisulfite (7 x 20 mL), then dried (Na2SO4), filtered and concentrated in vacuo to give the title compound (3.45 g, 69% yield; 96% purity as judged by LCMS) as a pale yellow liquid. LCMS: Method A, retention time = 0.722 and 1.06 mm, ESI MS [M+H]+ for C9H13NO2, calcd 167.09, found 167.2
[0273] Step 5: To a solution of 2-hydroxymethyl-6-(1-hydroxy-1-methylethyl)pyridine (5 g, 29.9 mmol, 1.0 equiv) in PhMe (33 mL, 0.9 M) at 0 °C under N2 was added diphenylphosphoryl azide (7.73 mL, 35.9 mmol, 1.2 equiv.), followed by l,8-diazabicyclo[5.4.0]undec-7-ene (5.37 mL, 35.9 mmol, 1.2 equiv.). The resulting mixture was to warm to room temperature and stirred for 14 h. Upon completion, diluted with ethyl acetate and washed with water, the organic layer was dried (Na2SO4), filtered and concentrated. The residue was dissolved in 1N aq HCl (2 eq, 60 mmol) and extracted with MTBE in hexanes (3:7, 100 mL), the organic layer was washed with water (50 mL) and the combined aqueous layer was neutralized with 2N aqueous NaOH and extracted with ethyl acetate (3×75 mL), dried the organic layer (Na2SO4), filtered through a plug of cotton and concentrated the filtrate to afford the pure compound as pale yellow color liquid (3.75 g, 75%). LCMS: Method A, retention time = 2.67 mm, ESI MS [M+H]+ for C9H12N4O, calcd 193.1, found 193.2
[0274] Step 6: A mixture of azide (3.34 g, 17.4 mmol), alkyne (3.71 g, 15.8 mmol), copper(II) sulfate (39 mg; 0.158 mmol), and sodium ascorbate (156 mg, 0.790 mmol) in 2:1 t-BuOH/H2O (158 mL) was heated at 60 °C for 13 h. The solvent was removed in vacuo, the residue dry loaded onto silica gel, and purified by silica gel chromatography (0-100% EtOAc in hexanes) to afford the desired product as an off-white solid (6.08 g, 90%). 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.61 (d, J= 8.0 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.28 (s, 1H), 7.10 (d, J = 7.6 Hz, 2H), 6.90 (s, 2H), 5.81 (s, 2H), 5.23 (s, 1H), 2.55 (s, 3H), 1.38 (s, 6H). ESI MS [M+H]+ for C23H23N8O, calcd 427.2, found 427.3.
/////////ETRUMADENANT, AB-928, AB 928, PHASE 2
TILDACERFONT
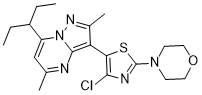
TILDACERFONT
| Synonyms: |
Tildacerfont 1014983-00-6 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(1-ethyl-propyl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine 7-(1-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine |
|---|---|
| MW/ MF | 420 g/mol/ C20H26ClN5OS |
- Originator Spruce Biosciences
- Class2 ring heterocyclic compounds; Morpholines; Pyrazoles; Pyrimidines; Small molecules; Thiazoles
- Mechanism of Action Corticotropin receptor antagonists
- Orphan Drug Status Yes – Congenital adrenal hyperplasia
- New Molecular Entity Yes
- Phase II Congenital adrenal hyperplasia
- 09 Jul 2020 Spruce Biosciences initiates a phase II trial in Congenital adrenal hyperplasia in USA (PO) (NCT04457336)
- 24 Sep 2019 Spruce Biosciences completes a phase II trial in Congenital adrenal hyperplasia in USA (NCT03687242)
- 19 Sep 2019 Updated safety and efficacy data from a phase II trial in Congenital adrenal hyperplasia release by Spruce Biosciences
Deuterated pyrazolo[1,5-a]pyrimidine derivatives, particularly tildacerfont (SPR-001), useful as CRF antagonists for treating congenital adrenal hyperplasia. Spruce Bioscience is developing tildacerfont under license from Lilly as an oral capsule formulation for the treatment of congenital adrenal hyperplasia; in July 2017, a phase II trial for CAH was initiated.
Corticotropin releasing factor (CRF) is a 41 amino acid peptide that is the primary physiological regulator of proopiomelanocortin (POMC) derived peptide secretion from the anterior pituitary gland. In addition to its endocrine role at the pituitary gland, immunohistochemical localization of CRF has demonstrated that the hormone has a broad extrahypothalamic distribution in the central nervous system and produces a wide spectrum of autonomic, electrophysiological and behavioral effects consistent with a neurotransmitter or neuromodulator role in the brain. There is also evidence that CRF plays a significant role in integrating the response in the immune system to physiological, psychological, and immunological stressors.
PATENT
Product case, WO2008036579 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008036579
Example 16
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo [ 1 ,5 -α]pyrimidine
Under a nitrogen atmosphere dissolve 3-(4-bromo-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (116 mg, 0.25 mmol) in THF (1.5 mL) and chill to -78 0C. Add n-butyl lithium (0.1 mL. 2.5 M in hexane, 0.25 mmol) and stir at -78 0C for 30 min. Add N-chlorosuccinimide (33.4 mg, 0.25 mmol) and stir for another 30 min, slowly warming to room temperature. After stirring overnight, quench the reaction by adding a solution of saturated ammonia chloride and extract with ethyl acetate. Wash the organic layer with brine, dry over sodium sulfate, filter, and concentrate to a residue. Purify the crude material by flash chromatography, eluting with hexanes:dichloromethane: ethyl acetate (5:5:2) to provide the title compound (54 mg). MS (APCI) m/z (35Cl) 420.6 (M+l)+; 1H NMR (400 MHz, CDCl3): 6.44 (s, IH), 3.79 (t, 4H, J=4.8 Hz), 3.63-3.56 (m, IH), 3.47 (t, 4H, J=4.8 Hz), 2.55 (s, 3H), 2.45 (s, 3H), 1.88-1.75 (m, 4H), 0.87 (t, 6H, J=7.5 Hz).
Alternate Preparation from Preparation 6:
Combine 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine, (9 g,
26.2 mmol) and 4-chloro-2-morpholino-thiazole (7.5 g, 36.7 mmol) in
dimethylformamide (90 mL) previously degassed with nitrogen. Add cesium carbonate (17.8 g, 55 mmol), copper iodide (250 mg, 1.31 mmol), triphenylphosphine (550 mg, 2.09 mmol) and palladium acetate (117 mg, 0.52 mmol). Heat the mixture to 125 0C for 16 h and then cool to 22 0C. Add water (900 mL) and extract with methyl-?-butyl ether (3 x 200 mL). Combine the organic portions and evaporate the solvent. Purify by silica gel chromatography eluting with hexanes/ethyl acetate (4/1) to afford the title compound (6.4 g, 62%). ES/MS m/z (35Cl) 420 (M+l)+.
Example 16a
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo[l,5-α]pyrimidine, hydrochloride
Dissolve 3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (1.40 g, 3.33 mmol) in acetone (10 mL) at 50 0C and cool to room temperature. Add hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and stir well in a sonicator. Concentrate the solution a little and add a minimal amount of diethyl ether to crystallize the HCl salt. Cool the mixture in a refrigerator overnight. Add additional hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and cool in a refrigerator. Filter the crystalline material and dry to obtain the title compound (1.15 g, 75%). ES/MS m/z (35Cl) 420 (M+l)+; 1H NMR(CDCO): 9.18 (br, IH), 6.86 (s, IH), 3.72 ( m, 4H), 3.49(m, IH), 3.39 (m, 4H), 2.48 (s, 3H), 2.38(s, 3H), 1.79 (m, 4H), 0.79 (m, 6H).
PATENT
US-20200255436
PATENT
WO2019210266
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019210266
claiming the use of CRF-1 antagonists (eg tildacerfont).
PATENT
WO 2010039678
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010039678
EXAMPLES
Example 1 : 7-(l-ethyl-propyl)-3-(‘2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine nthroline
Charge 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.03 g, 3.00 mmoles), K3PO4 (1.95 g, 9.00 mmoles), 2,4-dichlorothiazole (0.58 g, 3.75 mmoles), 1,10 phenanthroline (0.05 g, 0.30 mmoles) and anhydrous DMAC (5 mL) to a round bottom flask equipped with a magnetic stir bar, thermal couple and N2 inlet. Degas the yellow heterogeneous reaction mixture with N2 (gas) for 30 min. and then add CuI (0.06 g, 0.30 mmoles) in one portion followed by additional 30 min. degassing with N2 (gas). Stir the reaction mixture at 120 0C for about 6 hr. Cool the reaction mixture to room temperature overnight, add toluene (10 mL) and stir for 1 hr. Purify the mixture through silica gel eluting with toluene (10ml). Extract with 1 M HCl (10 mL), water (10 mL), brine (10 mL) and concentrate under reduced pressure to give a yellow solid. Recrystallize the solid from methanol (5ml) to yield the title compound as a yellow crystalline solid. (0.78 g, 70% yield, >99% pure by LC) MS(ES) = 369 (M+ 1). 1H NMR (CDCl3)= 6.5 (IH, s); 3.6 (IH, m); 2.6 (3H, s); 2.5 (3H, s); 1.9 (4H, m); 0.9 (6H, t).
Example 2: 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolol“! ,5-aipyrimidine
Charge 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (0.37 g, 1.00 mmoles), K2CO3 (0.28 g, 2.00 mmoles) and anhydrous morpholine (3 mL) to a round bottom flask equipped with a magnetic stir bar and N2 inlet. Stir the yellow mixture at 100 0C for about 4 hr., during which time the reaction becomes homogeneous. Cool the reaction mixture to room temperature, add H2O (10 mL) and stir the heterogeneous reaction mixture overnight at room temperature. Collected the yellow solid by filtration, wash with H2O and allowed to air dry overnight to give the crude title compound (391mg). Recrystallize from isopropyl alcohol (3 mL) to yield the title compound as a light yellow crystalline solid (380 mg, 90.6% yield, >99% by LC). MS(ES) = 420 (M+l). 1H NMR (CDCl3)= 6.45 (IH, s); 3.81 (m, 4H); 3.62 (IH, m); 3.50 (m, 4H); 2.6 (3H, s); 2.45 (3 H, s); 1.85 (4H, m); 0.9 (6H, t).
Example 3 :
The reactions of Example 1 are run with various other catalysts, ligands, bases and solvents, which are found to have the following effects on yield of 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. (See Tables 1 – 4).
Table 1 : Evaluation of different li ands
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole, 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.5 mmol CuI, 0.5 mmol ligand and 2.1 mmol Cs2CO3 in 4 mL DMAC. The reactions are degassed under N2 for 30 min. and then heated at between 80 and
1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak. Longer reaction times are shown in parenthesis) Table 2: Evaluation of various solvents
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.25 mmol CuI, 0.25 mmol 1,10-phenanthroline and 2.1 mmol Cs2CO3 in 3 mL specified solvent. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 3 : Evaluation of different copper sources
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.05 mmol CuX, 0.01 mmol 1,10-phenanthroline and 3 equivalents K3PO4 in 3 mL DMAC. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 4: Evaluation of various inorganic bases
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.1 mmol CuI, 0.1 mmol 1,10-phenanthroline and 2.1 mmol base and degassed for 30 minutes prior to the addition of 3 mL DMAC. The reactions are degassed under N2 for 10 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Example 4. Use of morpholine both as a reactant and base in 2-MeTHF as solvent.
solvent
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (15.2 g, 41.16 mmoles) is charged into a 250 mL 3-necked round bottomed flask, followed by addition of 2-MeTHF (61 mL, 4.0 volumes), the yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (19 g, 218.18 mmoles) is added over 2-5 minutes. Contents are heated to reflux and maintained at reflux for 12 hr. The slurry is cooled to 25 0C, followed by addition of 2-MeTHF (53 mL, 3.5 volumes) and water ( 38 mL 2.5 volumes). The reaction mixture is warmed to 40 0C, where upon a homogenous solution with two distinct layers formed. The layers are separated, the organic layer is filtered and concentrated to ~3 volumes at atmospheric pressure. Four volumes 2-propanol (61 mL) are added. The solution is concentrated to ~3 volumes followed by addition of 4 volumes 2-propanol (61 mL), re-concentrated to ~3 volumes, followed by addition of another 6 volumes 2-propanol (91 mL), and refluxed for 15 min. The clear solution is gradually cooled to 75 0C, seeded with 0.45 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 2 mL 2-propanol, rinsed with an additional 2 mL 2-propanol and transferred to a crystallization flask. The slurry is cooled to between 0-5 0C, maintained for 1 hr, filtered and the product rinsed with 2-propanol (30 mL, 2 volumes). The solid is dried at 60 0C in a vacuum oven to afford 16.92 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. Purity of product by HPLC assay is 100.00 %. XRPD and DSC data of product is consistant with reference sample. MS(ES) = 420 (M+ 1).
Example 5. Use of morpholine as both reactant and base in 2-propanol as solvent.
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (11.64 mmoles) is charged into a 100 mL 3 -necked round bottomed flask followed by addition of 2-propanol ( 16 mL, 3.72 volumes). The yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (3.3 g, 37.84 mmoles) is added over 2-5 minutes. Contents are refluxed for 6 hr. The slurry is cooled to 25 0C. 2-Propanol ( 32 mL, 7.44 volumes) and water ( 8.6 mL, 2.0 volumes) are added and the mixture warmed to 70-75 0C, filtered and concentrated to ~ 9 volumes at atmospheric pressure. The clear solution is gradually cooled to 55 0C, seeded with 0.06 g of crystalline 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 0.5 mL 2-propanol, rinsed with additional 0.5 mL 2-propanol and added to crystallization flask. The slurry is cooled to 0-5 0C, maintained for 1 hr., filtered and the product rinsed with 2-propanol ( 9 mL, 2.1 volumes). Suctioned dried under vacuum at 60 0C to afford 4.6 g of dry 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (88.8 % yield, purity by HPLC assay is 99.88 % ). MS(ES) = 420 (M+ 1).
Example 6: 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine
7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (10 g, 29.17 mmoles), 2, 4-dichlorothiazole (5.2 g , 33.76 mmoles), cesium carbonate(19.9g, 61.07 mmoles) and 1,10-phenanthroline (1 g, 5.5 mmoles) are charged into a 250 mL 3-necked round bottomed flask, followed by 2-MeTHF (36 mL, 3.6 volumes). The reaction mixture is degassed with nitrogen and then evacuated. Cuprous chloride (0.57 g, 5.7 mmoles), DMAC (10 mL, 1 volume) and 2-MeTHF (4 mL, 0.4 volumes) are added in succession. The reaction mixture is degassed with nitrogen and then evacuated. The contents are refluxed for 20 hr. The reaction mixture is cooled to -70 0C and 2-MeTHF (100 mL, 10 volumes) is added. The contents are filtered at ~70 0C and the residual cake is washed with 2-MeTHF (80 mL, 8 volumes) at about 65-72°C. The filtrate is transferred into a separatory funnel and extracted with water. The organic layer is separated and washed with dilute HCl. The resulting organic layer is treated with Darco G60, filtered hot (600C). The filtrate is concentrated at atmospheric pressure to -2.8 volumes. 25 mL 2-propanol is added, followed by re-concentration to -2.8 volumes. An additional 25 mL 2-propanol is added, followed again by re-concentration to -2.8 volumes. Finally, 48 mL 2-propanol is added. The contents are cooled to -7 0C, maintained at -7 0C for 1 hr., filtered and rinsed with 20 mL chilled 2-propanol. Product is suction dried and then vacuum dried at 60 0C to afford 9.41 g 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (purity of product by HPLC assay is 95.88 %). MS(ES) = 369 (M+ 1).
Example 7. Synthesis of 7-(l-ethyl-propyl)-3-(2, 4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori,5-a1pyrimidine using 1,4-Dioxane solvent and CuCl catalyst
Add dioxane (9.06X), Cs2CO3 (2.00X), 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.0 equivalent), 2,4-dichlorothiazole (0.54 equivalent) to a reactor under N2. Purge the reactor with N2 three times, degas with N2 for 0.5-1 hr., and then add 1,10-phenanthroline (0.3 eq) and CuCl (0.3eq) under N2 , degassing with N2 for 0.5-1 hr. Heat the reactor to 1000C -1100C under N2 . Stir the mixture for 22-24 hr. at 100 0C -1100C. Cool to 10~20°C and add water (10V) and CH3OH (5V), stir the mixture for 1-1.5 hr. at 10~20°C. Filter the suspension, resuspend the wet cake in water, stirr for 1-1.5 hr. at 10~20°C, and filter the suspension again. Charge the wet cake to n-heptane (16V) and EtOAc (2V) under N2. Heat the reactor to 40 °C~500C under N2.
Active carbon (0. IX) is added at 40 °C~500C. The reactor is heated to 55°C~650C under N2 and stirred at 55 °C~650C for 1-1.5 hr. The suspension is filtered at 40~55°C through diatomite (0.4 X). The cake is washed with n-heptane (2.5V). The filtrate is transferred to another reactor. EtOAc (10V) is added and the the organic layer washed with 2 N HCl (10V) three times, followed by washing two times with water (10X, 10V). The organic layer is concentrated to 3-4V below 500C. The mixture is heated to 80-90 0C. The mixture is stirred at this temperature for 40-60 min. The mixture is cooled to 0~5°C, stirred for 1-1.5 hr. at 0~5°C and filtered. The cake is washed with n-heptane (IV) and vacuum dried at 45-500C for 8-10 hr. The crude product is dissolved in 2-propanol (7.5V) under N2, and re-crystallized with 2-propanol. The cake is dried in a vacuum oven at 45°C~50°C for 10-12 hr. (55-80% yield). 1H NMR56.537 (s, IH) 3.591-3.659 (m, IH, J=6.8Hz), 2.593 (s, 3H), 2.512 (s, 3H), 1.793-1.921(m, 4H), 0.885-0.903 (m, 6H).
REFERENCES
1: Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol. 2014 Apr;35(2):234-44. doi: 10.1016/j.yfrne.2014.01.001. Epub 2014 Jan 20. Review. PubMed PMID: 24456850; PubMed Central PMCID: PMC4213066.
/////////////tildacerfont, SPR 001, Orphan Drug Status, Congenital adrenal hyperplasia, SPRUCE BIOSCIENCES, PHASE 2
CCC(CC)C1=CC(=NC2=C(C(=NN12)C)C3=C(N=C(S3)N4CCOCC4)Cl)C
SULCARDINE SULPHATE
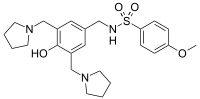
sulcardine, HBI-3000
B 87823
- Molecular FormulaC24H33N3O4S
- Average mass459.602 Da
N-[[4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)phenyl]methyl]-4-methoxybenzenesulfonamide
Benzenesulfonamide, N-[[4-hydroxy-3,5-bis(1-pyrrolidinylmethyl)phenyl]methyl]-4-methoxy-
N-[4-Hydroxy-3,5-bis(1-pyrrolidinylmethyl)benzyl]-4-methoxybenzenesulfonamide
343935-60-4 [RN]
heart arrhythmia
.gif)
CAS No. : 343935-61-5 (Sulcardine sulfate)
| Synonyms: | B-87823; HBI-3000; B87823; HBI3000; B 87823; HBI 3000;N-(4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)benzyl)-4-methoxybenzenesulfonamide sulfate |
| Molecular Formula: | C24H35N3O8S2 |
| Molecular Weight: | 557.67 |
- Originator Jiangsu Furui Pharmaceuticals; Shanghai Institute of Materia Medica
- Developer HUYA Bioscience International; Jiangsu Furui Pharmaceuticals
- Class Antiarrhythmics; Small molecules
- Mechanism of ActionIon channel antagonists
- Phase I Atrial fibrillation
- No development reported Arrhythmias
- 13 Mar 2020 Chemical structure information added
- 28 Feb 2020 No recent reports of development identified for preclinical development in Arrhythmias in USA (IV)
- 16 Dec 2019 Adverse events data from a phase I trial in Atrial fibrillation (In volunteers) presented at the American Heart Association Scientific Sessions 2019 (AHA-2019)
HUYA Bioscience , under license from Shanghai Institute of Materia Medica (SIMM), is developing sulcardine (HBI-3000, oral, i.v, heart arrhythmia), a myocardial ion channel inhibitory compound, for the treatment of arrhythmia; In September 2016, the drug was still in phase II development, as of August 2020, the company website states that a phase II trial was pending in China.
HBI-3000 (sulcardine sulfate) is an experimental drug candidate that is currently in phase II of human clinical trials as an antiarrhythmic agent.[1][needs update] Clinical investigation will test the safety and efficacy of HBI-3000 as a treatment for both atrial and ventricular arrhythmias.[2]
The molecular problem
Anti-arrhythmic medication is taken to treat irregular beating of the heart. This irregular beating results from a deregulation of the initiation or propagation of the electrical stimulus of the heart. The most common chronic arrhythmia is atrial fibrillation.[3] There is an increased incidence of atrial fibrillation in the elderly and some examples of complications include heart failure exacerbation, hypotension and thrombembolic events.[3]
Most anti-arrhythmic medications exert their effects by decreasing the permeability of potassium ion channels (IKr) in heart cells. These potassium channel blockers delay ventricular repolarization and prolong action potential duration (APD; the prolongation of the electrical stimulus within heart cells). These changes can lower heart rate, eliminate atrial fibrillation, and ultimately sudden cardiac death.[4][5]
Mechanism of action in ventricular myocytes
Ventricular myocytes are heart muscle cells found in the lower chambers of the heart. Heart rate is dependent on the movement of an electrical stimulus through the individual heart cells. This is mediated by the opening of ion channels on cell surfaces. HBI-3000 exerts its effects on the heart by inhibiting multiple ion channels (INa-F, INa-L, ICa-L and IKr), but predominantly the INa-L ion channel . By decreasing the ion permeability of these channels, HBI-3000 slightly prolongs APD (due to IKr); however, unlike pure IKr channel blockers, it is self-limited (due to the decreased permeability of INa-L and ICa-L). This is similar to the medications ranolazine and amiodarone.[5] HBI-3000 suppresses early afterdepolarizations (EADs; a change in the normal net flow of ions during repolarization), does not produce any electrical abnormalities, and displays minimally pronounced prolongation of APD during a slow heart rate (i.e. stimulated at a slower frequency). Pronounced prolongation of APD during a slow heart rate can lead to proarrythmias. Overall, HBI-3000 seems to have a low proarrhythmic risk. The effect of HBI-3000 on contractility and cardiac conduction requires further investigation.[5]
Studies
Animal model
In a canine model, the intravenous injection of HBI-3000 demonstrated to be an effective anti-arrhythmic and anti-fribrillatory agent.[6]
Cellular isolation
The administration of HBI-3000 to isolated heart muscle cells demonstrated the potential to improve arrhythmias while having low proarrhythmic risk.[5]
Human studies
Jiangsu Furui Pharmaceuticals Co., Ltd is currently recruiting participants in their study.[1][
PAPER
http://www.simm.cas.cn/wyp/wyp_lw/201804/W020180420480084769998.pdf

N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate) (1) To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensul-fonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1 H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+ ), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
(2) A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ~2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (~300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0°C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1 H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+ ), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
(3) Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0°C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140°C. 1 H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45– 3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S∙H2SO4∙3H2O) C, H, N, S.
PATENT
Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
https://patents.google.com/patent/US6605635
Synthesis and antiarrhythmic activities of changrolin (1) have been reported (Liangquan Li, et al., Scientia Sinica, 1979, 7, 723; Weizhou Chen, et al., Acta Pharmaceutica Sinica, 1979, 14, 710). Thereafter, investigations of the chemical structural modifications and the physiological activities have successively been carried out by domestic and foreign scientists (Cunji Sun, et al., Acta Pharmaceutica Sinica, 1981, 16, 564; 1986, 21, 692; Mulan Lin, et al., ibid., 1982, 17, 212; D. M. Stout, et al. J. Med. Chem., 1983, 26, 808; 1984, 27, 1347; 1985, 28, 295; 1989, 32, 1910; R. J. Chorvat, et al., ibid., 1993, 36, 2494).

Changrolin is an effective antiarrhythmic agent. Ventricular premature beats disappear 2-3 days after oral administration of changrolin to patients suffering from arrhythmia; I.v. injection or instillaton may result in significant reduction or even disappearence of ventricular premature beats and ventricular tachycardia. However, oral administration of changrolin for a period of over one month may cause a reversible pigmentation on the skin of patients, which gradually retrogresses after ceasing the administration. This pigmentation is associated to the subcutaneous oxidation of certain structural moieties in changrolin molecule or to its instability in solution.
EXAMPLE 1N-[3,5-bis(1-Piperidinomethyl)-4-hydroxy]phenyl-1-naphthalenesulfonamide (B-87836)
(1) To a solution of 4-aminophenol (4.5 g) in dioxane (20 ml) was added dropwise a solution of 1-naphthalenesulfonyl chloride (4.4 g) in dioxane (20 ml). The mixture was further stirred at room temperatue for 4.5 hours and poured into water. The precipitate was collected by filtration, recrystallized from ethanol and decolored with activated carbon to give N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (4.2 g), mp 195-196° C.
(2) A mixture of N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g), 37% aqueous formaldehyde (4.5 g) and piperidine (5.6 g) in ethanol (100 ml) was heated to reflux for 50 hours. The ethanol was removed by evaporation in vacuo and chloroform was added to the residue. The organic layer was washed with water then dried over anhydrous Na2SO4. Then the chloroform was removed in vacuo and the residue was triturated in water to give a solid, which was then recrystallized from ethanol to give the titled product (1.4 g), mp 197-198° C.
1HNMR(CDCl3): 1.30-1.50(m, 12H), 2.10-2.21(m, 8H), 3.28(s, 4H), 6.45(s, 2H), 7.24-8.04(m, 6H), 8.56(m, 1H). Elemental analysis (C28H35N3O3S ): Calcd. (%): C, 68.12; H, 7.15; N, 8.51. Found (%): C, 67.96; H, 7.16; N, 8.56.
PATENT
WO-2020159959
Novel crystalline forms of acid salts of sulcardine useful for treating arrhythmia and atrial fibrillation.
4-Methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)-4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), also known as sulcardine, and its salts, such as sulcardine sulfate, constitute a group of compounds with potent anti -arrhythmic activity. Sulcardine is a multi-ion channel blocker that specifically inhibits iNa-Peak, iNa-Late, Ica,L, and Ixrwith similar in vitro potencies (and Ito and IKUT to a lesser degree) in human atrial cardiomyocytes and represents what may be the sole example of a substituted sulfonamide class of anti-arrhythmic. Sulcardine salts can be used as an intravenous injectable or as oral doses for the treatment of arrhythmias, including supraventricular tachyarrhythmia, premature ventricular contractions, ventricular tachycardia, ventricular fibrillation, and atrial fibrillation. See, e.g ., U.S. Patent Nos. 8,541,464 and 8,637,566. Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
[0004] In addition, the evidence to date suggests that one advantage of sulcardine and its salts is that they lack significant pro-arrhythmic activity, as demonstrated in rigorous preclinical safety models, including a post-MI sudden-death conscious canine model and the validated rabbit ventricular wedge model. Additionally, it has been shown that they do not significantly increase defibrillation threshold, nor increase defibrillation failure risk in a post-MI canine model as was seen with flecainide. On the basis of these data, sulcardine and salts, with their very low apparent pro-arrhythmic potential, could potentially be used to treat acute and recurrent atrial fibrillation in the presence of organic heart disease, prolonged QR syndrome, and ventricular arrhythmias, including premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), in either acute- or chronic-administration settings owing to their ability to be formulated into intravenous and oral dosing formulations.
Sulcardine has a chemical name of 4-methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)- 4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), and has the following structure:
[0062] Sulcardine sulfate has the following structure:
[0063] Sulcardine sulfate can exist in a hydrated form. One such form is a trihydrate.
HPLC analysis was performed on a Dionex Ultimate 3000 instrument with the following parameters:
Column: Phenomenex Luna C18, 150×4.6mm, 5pm
Column Temperature: 30°C
Mobile Phase A: 0.2% Phosphoric Acid
Mobile Phase B: Methanol
Diluent: 50:50 MeOH:H20
Runtime: 12 minutes
Flow Rate: l.OmL/min
Injection Volume: 5pL
Detection: 237 nm
Gradient:
EXAMPLE 2 – PREPARATION OF FREE BASE AND SCREENING
[00348] Sulcardine sulfate trihydrate was dissolved in ethyl acetate (16 vol.) and saturated sodium bicarbonate solution (16 vol.). The biphasic solution was transferred to a separating funnel and the layers separated. The organic layer was dried over sodium sulfate and then the solvent was removed by rotary evaporation and the resulting oil dried under vacuum at ambient temperature for ca. 3 hr. FIG. 4 is an XRPD pattern of the resulted amorphous sulcardine free base. In all cases, the initial screening work detailed below was performed on 10 mg of sulcardine free base. All XRPD diffractograms were compared with sulcardine sulfate trihydrate, sulcardine free base and relevant counterions and found to be distinct.
Patent
WO2020123824
claiming treatment of atrial fibrillation (AF) by intravenously administering sulcardine sulfate .
PATENT
References
- ^ Jump up to:a b Jiangsu Furui Pharmaceuticals (November 5, 2010). “Efficacy and safety of sulcardine sulfate tablets in patients with premature ventricular contractions”. ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 2019-12-20.
- ^ “HUYA Bioscience Int’l announces clinical trial milestones in China for promising new anti-arrhythmic compound; Data supports desirable safety profile” (Press release). San Francisco, California: HUYA Bioscience International. Retrieved 2019-12-20.
- ^ Jump up to:a b Mashal, Abdallah; Katz, Amos; Shvartzman, Pesach (2011). “Atrial fibrillation: A primary care cross-sectional study”. Israel Medical Association Journal. 13 (11): 666–671. PMID 22279699.
- ^ Farkas, András; Leprán, István; Papp, Julius Gy. (1998). “Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits”. European Journal of Pharmacology. 346 (2–3): 245–253. doi:10.1016/S0014-2999(98)00067-3. PMID 9652366.
- ^ Jump up to:a b c d Guo, Donglin; Liu, Que; Liu, Tengxian; Elliott, Gary; Gingras, Mireille; Kowey, Peter R.; Yan, Gan-Xin (2011). “Electrophysiological properties of HBI-3000: A new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes”. Journal of Cardiovascular Pharmacology. 57 (1): 79–85. doi:10.1097/FJC.0b013e3181ffe8b3. PMID 20980921.
- ^ Lee, Julia Y.; Gingras, Mireille; Lucchesi, Benedict R. (2010). “HBI-3000 prevents sudden cardiac death in a conscious canine model”. Heart Rhythm. 7 (11): 1712. doi:10.1016/j.hrthm.2010.09.028.
 |
|
| Names | |
|---|---|
| IUPAC name
N-({4-Hydroxy-3,5-bis[(pyrrolidin-1-yl)methyl]phenyl}methyl)-4-methoxybenzene-1-sulfonamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H33N3O4S | |
| Molar mass | 459.61 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////sulcardine sulfate, phase 2, china, HBI 3000, atrial fibrillation, B 87823,
COC1=CC=C(C=C1)S(=O)(=O)NCC2=CC(=C(C(=C2)CN3CCCC3)O)CN4CCCC4
NARONAPRIDE


NARONAPRIDE
860174-12-5
Average: 537.1
C27H41ClN4O5
ATI 7505 / ATI-7505
(3R)-1-azabicyclo[2.2.2]octan-3-yl 6-[(3S,4R)-4-(4-amino-5-chloro-2-methoxybenzamido)-3-methoxypiperidin-1-yl]hexanoate
| INGREDIENT | UNII | CAS | |
|---|---|---|---|
| Naronapride dihydrochloride | 898PE2W8US | 860169-57-9 |
860174-12-5 (free base) 860169-57-9 (HCl)
Naronapride (free base), also known as ATI-7505, is a highly selective, high-affinity 5-HT(4) receptor agonist for gastrointestinal motility disorders. ATI-7505 accelerates overall colonic transit and tends to accelerate GE and AC emptying and loosen stool consistency.
Investigated for use/treatment in gastroesophageal reflux disease (GERD) and gastroparesis.
Renexxion , presumed to have been spun-out from Armetheon , under license from ARYx Therapeutics is developing naronapride (ATI-7505; phase 2 clinical in February 2020), an analog of the gastroprokinetic 5-HT 4 agonist cisapride identified using ARYx’s RetroMetabolic platform technology (ARM), for the oral treatment of upper GI disorders. In September 2018, this was still the case . PATENT
WO2005068461
NEW PATENT
WO-2020096911
Process for preparing trihydrate salt of naronapride hydrochloride as 5-HT 4 receptor agonist useful for treating gastrointestinal disorders such as dyspepsia, gastroparesis, constipation, post-operative ileus. Appears to be the first filing from the assignee and the inventors on this compound,
In some aspects, provided herein is a method of making a trihydrate form of (3S, 4R, 3’R)-6-[4-(4-amino-5-chloro-2-methoxy-benzoylamino)-3-methoxy-piperidin-l-yl]-hexanoic acid l-azabicyclo[2.2.2]oct-3’-yl ester di-hydrochloride salt, which has the following formula:
Example 5: NMR Characterization of the Trihydrate
[0282] ^-Nuclear Magnetic Resonance Spectroscopy (‘H-NMR) : Approximately 6 mg of the trihydrate was dissolved in in 1 g of deuterated solvent (dimethylsulfoxide (DMSO)-C45 99.9% d, with 0.05% v/v tetramethyl silane (TMS)). A Varian Gemini 300 MHz FT-NMR spectrometer was used to obtain the ¾-NMK spectrum. A list of the peaks is provided in Table 1 below. A representative ‘H-NMR spectrum is provided in FIG. 6.
Table 1. ‘H-NMR peak list for trihydrate
[0283] 13 C-Nuclear Magnetic Resonance Spectroscopy ( 13C-NMR ): Approximately 46 mg of the trihydrate was dissolved in 1 mL of deuterated solvent (deuterium oxide, Aldrich, 99.9% D, TPAS 0.75%). The 13C-NMR spectrum was obtained using a Varian Gemini 300 MHz FT-NMR spectrometer. A list of the peaks is provided in Table 2 below. A representative 13C-NMR spectrum is provided in FIG. 7.
Table 2. 13C-NMR peak list for trihydrate

PATENT
US10570127 claiming composition (eg tablet) comprising a trihydrate form of naronapride.
patent
ARYX THERAPEUTICS, WO2005/68461, A1, (2005)
Methods
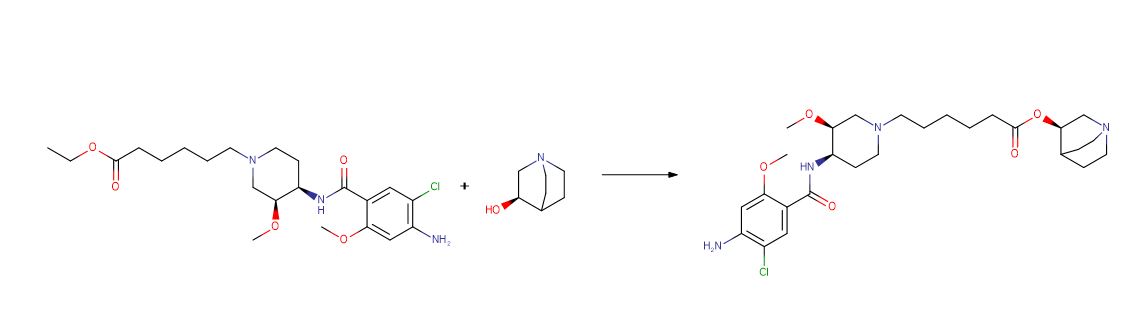
titanium tetraethoxide; toluene;
Reactants can be synthesized in 1 step.
ARYX THERAPEUTICS, WO2005/68461, A1, (2005) The ester (1 part by weight) and (R)-3-Quinuclidinol (about 1.12 part by weight) were suspended in toluene before slowly adding titanium (IV) ethoxide (about 0.5 part by weight) to the stirred suspens ion. The mixture was heated to about 91 °C under a stream of nitrogen, and partial vacuum was applie d to the flask through a distillation apparatus in order to azeotropically remove the ethanol. Addit ional toluene was added as needed to maintain a minimum solvent volume in the flask. The reaction was considered complete after about 33 hours. The mixture was cooled to about room temperature and ext racted five times with water. The organic layer was concentrated under reduced pressure and the resulting residue was redissolved in EtOH/iPrOH (about 1: 1 v/v) and then filtered through a 0.45 micron membrane filter to remove any particulates. Concentrated hydrochloric acid was added slowly to the stirred filtrate to precipitate out the desired product as the dihydrochloride salt. The resulting s uspension was stirred for several hours at room temperature and collected under vacuum filtration and rinsed with EtOH/tPrOH (1: 1; v/v) to provide 0.53 part by weight of the crude product salt. Crude dihydrochloride salt was resuspended in ethanol and heated to reflux before cooling to room temperature over about 1 hour. The product was collected under vacuum filtration and rinsed with ethanol an d then air-dried. The solids were resuspended in ethanol and warmed to about 55 °C to give a clear s olution before adding warm isopropanol and the product was allowed to precipitate by slow cooling to room temperature. The resulting suspension was stirred for several hours before vacuum filtering and rinsing with, e. g., isopropanol. The product was vacuum dried, initially at room temperature for several hours and then at about 55 °C until a constant weight was achieved.
Patent
Methods
dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; DMFA;
Reactants can be synthesized in 2 steps.
ARYX THERAPEUTICS, WO2007/28073, A2, (2007) Production of Compound IV and Compound VI[0394] A mixture of (+)-Comrhoound II (1 eq.), (R)-(-)-3-quinuclidinol HCl salt (1 eq.), EDAC (1 eq.) and DMAP (1 eq.) in DMF is heated at around 5OC overnight . After cooling and diluting with water, the mixture is purified by chromatography or by crystallization to provide Compound IV. Similarly, using (S)-(+)-quinuclidinol, Compound VI is obtained
REFERENCES
1: Jiang C, Xu Q, Wen X, Sun H. Current developments in pharmacological therapeutics for chronic constipation. Acta Pharm Sin B. 2015 Jul;5(4):300-9. doi: 10.1016/j.apsb.2015.05.006. Epub 2015 Jun 6. Review. PubMed PMID: 26579459; PubMed Central PMCID: PMC4629408.
2: Buchwald P, Bodor N. Recent advances in the design and development of soft drugs. Pharmazie. 2014 Jun;69(6):403-13. Review. PubMed PMID: 24974571.
3: Mozaffari S, Didari T, Nikfar S, Abdollahi M. Phase II drugs under clinical investigation for the treatment of chronic constipation. Expert Opin Investig Drugs. 2014 Nov;23(11):1485-97. doi: 10.1517/13543784.2014.932770. Epub 2014 Jun 24. Review. PubMed PMID: 24960333.
4: Shin A, Camilleri M, Kolar G, Erwin P, West CP, Murad MH. Systematic review with meta-analysis: highly selective 5-HT4 agonists (prucalopride, velusetrag or naronapride) in chronic constipation. Aliment Pharmacol Ther. 2014 Feb;39(3):239-53. doi: 10.1111/apt.12571. Epub 2013 Dec 5. Review. PubMed PMID: 24308797.
5: Stevens JE, Jones KL, Rayner CK, Horowitz M. Pathophysiology and pharmacotherapy of gastroparesis: current and future perspectives. Expert Opin Pharmacother. 2013 Jun;14(9):1171-86. doi: 10.1517/14656566.2013.795948. Epub 2013 May 11. Review. PubMed PMID: 23663133.
6: Tack J, Camilleri M, Chang L, Chey WD, Galligan JJ, Lacy BE, Müller-Lissner S, Quigley EM, Schuurkes J, De Maeyer JH, Stanghellini V. Systematic review: cardiovascular safety profile of 5-HT(4) agonists developed for gastrointestinal disorders. Aliment Pharmacol Ther. 2012 Apr;35(7):745-67. doi: 10.1111/j.1365-2036.2012.05011.x. Epub 2012 Feb 22. Review. PubMed PMID: 22356640; PubMed Central PMCID: PMC3491670.
7: Hoffman JM, Tyler K, MacEachern SJ, Balemba OB, Johnson AC, Brooks EM, Zhao H, Swain GM, Moses PL, Galligan JJ, Sharkey KA, Greenwood-Van Meerveld B, Mawe GM. Activation of colonic mucosal 5-HT(4) receptors accelerates propulsive motility and inhibits visceral hypersensitivity. Gastroenterology. 2012 Apr;142(4):844-854.e4. doi: 10.1053/j.gastro.2011.12.041. Epub 2012 Jan 4. PubMed PMID: 22226658; PubMed Central PMCID: PMC3477545.
8: Bowersox SS, Lightning LK, Rao S, Palme M, Ellis D, Coleman R, Davies AM, Kumaraswamy P, Druzgala P. Metabolism and pharmacokinetics of naronapride (ATI-7505), a serotonin 5-HT(4) receptor agonist for gastrointestinal motility disorders. Drug Metab Dispos. 2011 Jul;39(7):1170-80. doi: 10.1124/dmd.110.037564. Epub 2011 Mar 29. PubMed PMID: 21447732.
9: Tack J. Current and future therapies for chronic constipation. Best Pract Res Clin Gastroenterol. 2011 Feb;25(1):151-8. doi: 10.1016/j.bpg.2011.01.005. Review. PubMed PMID: 21382586.
10: Manabe N, Wong BS, Camilleri M. New-generation 5-HT4 receptor agonists: potential for treatment of gastrointestinal motility disorders. Expert Opin Investig Drugs. 2010 Jun;19(6):765-75. doi: 10.1517/13543784.2010.482927. Review. PubMed PMID: 20408739.
11: Sanger GJ. Translating 5-HT receptor pharmacology. Neurogastroenterol Motil. 2009 Dec;21(12):1235-8. doi: 10.1111/j.1365-2982.2009.01425.x. Review. PubMed PMID: 19906028.
12: Vakil N. New pharmacological agents for the treatment of gastroesophageal reflux disease. Rev Gastroenterol Disord. 2008 Spring;8(2):117-22. Review. PubMed PMID: 18641594.
13: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2007 Jun;29(5):359-73. PubMed PMID: 17805439.
14: Camilleri M, Vazquez-Roque MI, Burton D, Ford T, McKinzie S, Zinsmeister AR, Druzgala P. Pharmacodynamic effects of a novel prokinetic 5-HT receptor agonist, ATI-7505, in humans. Neurogastroenterol Motil. 2007 Jan;19(1):30-8. PubMed PMID: 17187586.
////////////NARONAPRIDE, ATI 7505, ATI 7505,PHASE 2
CO[C@H]1CN(CCCCCC(=O)O[C@H]2CN3CCC2CC3)CC[C@H]1NC(=O)C1=C(OC)C=C(N)C(Cl)=C1
NIDUFEXOR
NIDUFEXOR
LMB763
4-[[benzyl-(8-chloro-1-methyl-4H-chromeno[4,3-c]pyrazole-3-carbonyl)amino]methyl]benzoic acid
Nidufexor is a farnesoid X receptor (FXR) agonist.
| Molecular Weight |
487.93 |
|---|---|
| Formula |
C₂₇H₂₂ClN₃O₄ |
| CAS No. |
1773489-72-7 |
PHASE 2 Treatment of Liver and Biliary Tract Disorders,
Agents for Diabetic Nephropathy, NOVARTIS
1773489-72-7, LMB-763, UNII-CJ1PL0TE6J, CJ1PL0TE6J, BCP28929, EX-A1854
Nidufexor pound LMB-763 pound(c)
4-((N-benzyl-8-chloro-1-methyl-1,4-dihydrochromeno[4,3-c]pyrazole-3-carboxamido)methyl)benzoic acid

https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.9b01621
1 (7.6 g, 89% yield) as a white solid. Melting point: 232.6 °C.
1 H NMR (400 MHz, DMSO): δ 12.93 (s, 1H), 7.96−7.85 (m, 2H), 7.71 (dd, J = 7.1, 2.5 Hz, 1H), 7.42−7.20 (m, 8H), 7.06 (dd, J = 8.7, 1.9 Hz, 1H), 5.45 (d, J = 3.9 Hz, 2H), 5.25 (d, J = 9.2 Hz, 2H), 4.58 (d, J = 12.1 Hz, 2H), 4.12 (d, J = 16.6 Hz, 3H).
13C NMR (101 MHz, DMSO-d6): δ 167.07, 162.21, 151.98, 142.65, 139.18, 132.20, 132.67, 129.70, 129.50, 129.50, 128.53, 128.53, 127.43, 127.43, 127.43, 127.43, 127.43, 125.53, 122.24, 119.0, 117.09, 116.64, 64.51, 50.68, 48.24. LC-MS m/z: 488.2/490.2 (M +H)+ ; chlorine pattern; method 3; RT = 1.41 min.
Elemental Analysis calcd for C27H22ClN3O4: C 66.46, H 4.54, N 8.61; found: C 66.43, H 4.56, N 8.62.
TRIS Salt Formation. Methanol (400 mL) was added to a mixture of 1 (4.0 g, 8.2 mmol) and 2-amino-2-hydroxymethylpropane-1,3-diol (TRIS, 1.0 g, 8.2 mmol). The mixture was heated to 70 °C for 0.5 h. After cooling to room temperature, the solvent was removed in vacuum. The residue was sonicated in dichloromethane (10 mL) and concentrated again. The resulting white solid was dried under vacuum overnight. The crude material was crystallized by slurring the solid residue in a 4:1 mixture of acetonitrile and methanol (5 mL). The mixture was stirred at room temperature for 24 h to give 4-((N-benzyl-8-chloro-1-methyl-1,4-dihydrochromeno- [4,3-c]pyrazole-3-carboxamido)methyl)benzoic acid TRIS salt as a white salt (3.7 g, 73% yield). Melting point: 195.6 °C. 1 H NMR (400 MHz, DMSO): δ 7.92−7.80 (m, 2H), 7.78−7.64 (m, 1H), 7.41− 7.19 (m, 8H), 7.13−7.00 (m, 1H), 5.44 (s, 2H), 5.25−5.14 (m, 2H), 4.61−4.48 (m, 2H), 4.18−4.03 (m, 3H), 3.39 (s, 7H). TRIS OH masked by water peak. LC-MS m/z: 488.0/490.0 (M+H)+ ; chlorine pattern, method 3. RT = 1.58 min. Elemental Analysis calc for C31H33ClN4O7: C 61.00, H 5.36, N 9.15; found: C 60.84, H 5.34, N 9.13.






https://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.9b01621/suppl_file/jm9b01621_si_001.pdf
Patent
WO 2015069666
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015069666&tab=PCTDESCRIPTION
///////NIDUFEXOR, LMB 763, Phase II, PHASE 2, Liver and Biliary Tract Disorders, Diabetic Nephropathy, NOVARTIS
CN1C(C2=CC(Cl)=CC=C2OC3)=C3C(C(N(CC4=CC=CC=C4)CC5=CC=C(C(O)=O)C=C5)=O)=N1
LNP 023

LNP 023
CAS 1644670-37-0
ROTATION +
4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid
| M.Wt | 422.525 | |
| Formula | C25H30N2O4 | |
4-[(2S,4S)-4-ethoxy-1-[(5-methoxy-7-methyl-1H-indol-4-yl)methyl]piperidin-2-yl]benzoic acid
LNP023
RENRQMCACQEWFC-UGKGYDQZSA-N
PATENT US9682968, Example-26a
BDBM160475
4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid
4-[(2~{S},4~{S})-4-ethoxy-1-[(5-methoxy-7-methyl-1~{H}-indol-4-yl)methyl]piperidin-2-yl]benzoic acid
LNP023 (LNP-023) is a highly potent, reversible, selective inhibitor of factor B (IC50=10 nM), the proteolytically active component of the C3 and C5 convertases.
LNP023 (LNP-023) is a highly potent, reversible, selective inhibitor of factor B (IC50=10 nM), the proteolytically active component of the C3 and C5 convertases; shows direct, reversible, and high-affinity binding to human FB with Kd of 7.9 nM in SPR assays, demonstrates potent inhibition of AP-induced MAC formation in 50% human serum with IC50 of 0.13 uM; shows no inhibition of factor D (FD), as well as classical or lectin complement pathway activation (up to 100 uM), and no significant effects (up to 10 μM) in a broad assay panel of receptors, ion channels, kinases, and proteases; blocks zymosan-induced MAC formation membrane attack complex (MAC) with IC50 of 0.15 uM, prevents KRN-induced arthritis in mice and is effective upon prophylactic and therapeutic dosing in an experimental model of membranous nephropathy in rats afer oral adminstration; also prevents complement activation in sera from C3 glomerulopathy patients and the hemolysis of human PNH erythrocytes.
Other Indication
Phase 2 Clinical

PATENT
WO 2015009616
https://patents.google.com/patent/WO2015009616A1/en
PATENT
https://patents.google.com/patent/US9682968B2/en
Example-26Example-26a4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid ((+) as TFA Salt)

A mixture of methyl 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoate, Intermediate 6-2b peak-1 (tr=1.9 min), (84 mg, 0.192 mmol) and LiOH in H2O (1 mL, 1 mmol) in THF (1 mL)/MeOH (2 mL) was stirred at room temperature for 16 h, and then concentrated. The resulting residue was purified by RP-HPLC (HC-A) to afford the title compound. Absolute stereochemistry was determined by comparison with enantiopure synthesis in Example-26c. 1H NMR (TFA salt, 400 MHz, D2O) δ 8.12 (d, J=8.19 Hz, 2H), 7.66 (br. d, J=8.20 Hz, 2H), 7.35 (d, J=3.06 Hz, 1H), 6.67 (s, 1H), 6.25 (d, J=3.06 Hz, 1H), 4.65 (dd, J=4.28, 11.49 Hz, 1H), 4.04 (d, J=13.00 Hz, 1H), 3.87-3.98 (m, 2H), 3.53-3.69 (m, 5H), 3.38-3.50 (m, 1H), 3.20-3.35 (m, 1H), 2.40 (s, 3H), 2.17-2.33 (m, 2H), 2.08 (br. d, J=15.70 Hz, 1H), 1.82-1.99 (m, 1H), 1.28 (t, J=7.03 Hz, 3H); HRMS calcd. for C26H31N2O3 (M+H)+ 423.2284, found 423.2263.
PATENT
WO 2020016749
The present invention relates to a process for the preparation of phenylpiperidinyl indole derivatives. More particularly, the present invention relates to a process for the preparation of the compound of formula (I)
also referred to as 4-((2S,4S)-(4-ethoxy-1 -((5-methoxy-7-methyl-1 /-/-indol-4-yl)methyl)piperidin-2-yl))benzoic acid, or a pharmaceutically acceptable salt thereof, which is capable of inhibiting the activation of the alternative pathway of the complement system. The complement system plays a major role in the innate and adaptive immunity system and comprises a group of proteins that are normally present in an inactive state. These proteins are organized in three activation pathways: the classical, the lectin, and the alternative pathways (Holers, In Clinical Immunology: Principles and practice, ed. R.R. Rich, Mosby Press; 1996, 363-391 ). Molecules from microorganisms, antibodies or cellular components can activate these pathways resulting in the formation of protease complexes known as the C3-convertase and the C5-convertase. The classical pathway is a calcium / magnesium-dependent cascade, which is normally activated by the formation of antigen-antibody complexes. It can also be activated in an antibody-independent manner by the binding of C-reactive protein complexed to
ligand and by many pathogens including gram-negative bacteria. The alternative pathway is a magnesium-dependent cascade, which is activated by deposition and activation of C3 on certain susceptible surfaces (e.g. cell wall polysaccharides of yeast and bacteria, and certain biopolymer materials). The alternative pathway (AP) utilizes C3 fragments (C3b) to opsonize the pathogens hence targeting them for phagocytosis without the need for antibodies. Hyperactivity of the complement system, and in particular in its AP, plays a role in a large number of complement-driven diseases, such as C3 glomerulopathy (C3G), paroxysmal nocturnal hemoglobinuria (PNH) and IgA nephropathy (IgAN). Phenylpiperidinyl indole derivatives, such as compound of formula (I), or a pharmaceutically acceptable salt thereof, play a role in the inhibition of complement factor B, a known critical enzyme for activation of the alternative complement pathway (Lesavre et al J. Exp. Med. 1978, 148, 1498-1510; Volanakis et al New Eng. J. Med. 1985, 312, 395-401 ), which may also be a suitable target for the inhibition of the amplification of the complement pathways. The phenylpiperidinyl indole derivatives, such as compound of formula (I), or a pharmaceutically acceptable salt thereof, and a method for preparing such derivatives, are described in WO2015/009616. In particular, compound of formula (I) is described in example 26, of WO2015/009616. One of the drawbacks of the synthesis was the use of hazardous chemicals (such as sodium hydride, or dimethylacetamide, which represent safety concerns on a larger scale) and the poor enantio- and diastereo-selectivity of the steps, leading to unwanted stereoisomers.
Thus, there is a need to provide an alternative reaction route in a process for producing compound of formula (I), or a pharmaceutically acceptable salt thereof, generating less by products, and easier to handle on a large scale.
Scheme 1 , vide infra.
Compound of fformula (II)
ormu a ( )
formula (1)
Scheme 1
1. Asymmetric synthesis of compound of formula (II): .
One aspect of the present invention relates to an asymmetric process for preparing a compound of formula (II), or salt thereof, as outlined in Scheme 2 below, wherein the stereocenters in position 2 and in position 4 on the piperidine are obtained in high enantio- and diastereo-selectivity.
formula (ii)
Scheme 2
Example 1 : Synthesis of Benzyl-2-r4-(methoxycarbonyl)phenyl1-4-oxopiperidine-1 -carboxylate according to the following sequence:
Y
R = Methyl R = Methyl R =: Methyl
Step 1 : Synthesis of Benzyl-2-[4-(methoxycarbonyl)phenyl]-4-oxo-3, 4-dihydro pyridine-1(2W)-carboxylate (C3, wherein Pi = Cbz and R = methyl)
iPrMgCI (2N THF, 109.96 g, 54.98 ml_, 2.0 eq) was charged in a reactor. A solution of bis[2 -(N,N-dimethylaminoethyl)] ether (2.5 eq, 22.03 g, 137.46 mmol) in THF (24 ml.) was added at 15 – 25 °C. The mixture was stirred for 1 hour. A solution of C1 (20.17 g, 76.98 mmol, 1 .4 eq) in THF (102 ml.) was added slowly at 15 – 25 °C. The mixture was heated to 25 – 30 °C, stirred for more than 1 hour, and checked by HPLC. The mixture was cooled to -30 °C. A solution of C2 (methyl 4-iodobenzoate, 6.0 g, 54.98 mmol, 1 .0 eq) in THF (20 ml.) was added, followed by a solution of benzyl chloroformate (1 .15 eq, 10.79 g, 63.23 mmol) in THF (36 ml_). The mixture was stirred for 2 hours and quenched with AcOH (6.60 g, 109.96 mmol, 2 eq). Isopropyl acetate (60 ml.) was added. Hydrogen chloride (15%, 90 g) was added to adjust the pH = 1 – 2. The organic layer was separated and washed with brine (15%, 100 g), and concentrated. Isopropyl acetate (160 ml.) was added and concentrated to remove the THF. The crude product was recrystallized in Isopropyl
acetate (1 14 ml.) and n-heptane (120 ml_). The product was dried at 60 °C to provide C3 as light yellow solid (16.0 g, 79.65 % yield). 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 8.1 1 (dd, J=8.39, 1.01 Hz, 1 H), 7.91 (d, J=8.39 Hz, 2H), 7.33 – 7.37 (m, 6H), 5.82 (d, J= 7.20 Hz, 1 H), 5.20 – 5.35 (m, 3H) , 3.83 (s, 3H), 3.41 (br. s, 1 H), 3.31 (dd, J=16.64, 7.52 Hz, 1 H), 2.66 (br. d, J=16.55 Hz, 1 H).
Step 2: Synthesis of Benzyl-2-[4-(methoxycarbonyl)phenyl]-4-oxopiperidine-1 -carboxylate (C4, wherein Pi = Cbz and R = methyl)
A solution of C3 (25 g, 68.42 mmol, 1 .0 eq) in AcOH (200 ml.) was heated to 50 – 60 °C to form a clear solution. The solution was then cooled to 35 °C. Zn powder (13.42 g, 205.26 mmol, 3.0 eq) was added portionwise while keeping the inner temperature at 35 – 40 °C. After addition, the mixture was stirred for more than 8 hours and checked by HPLC. THF (250 ml.) was added. The mixture was cooled to 25 °C, filtered, and the filter cake was washed with THF (125 volume). The filtrate was concentrated to dryness. Isopropanol (375 ml.) was added. The solution was cooled to 0 – 5 °C. EDTA-4Na.2H20 (40 g) in water (200 ml.) was added. The mixture was neutralized to pH = 9 – 10 with 30% sodium hydroxide solution and stirred for 2 hours. The organic layer was collected, washed with brine (15%, 250 g) and concentrated to about 50 ml_. MTBE (100 ml.) was added and concentrated to about 50 ml_. MTBE (80 ml.) was added followed by n-heptane (20 ml.) dropwise. Then the mixture was cooled to 0 °C gradually. The mixture was filtered and the filter cake was dried to afford C4 as a light yellow solid (20.1 1 g, 80.0 % yield). 1 H NMR (400 MHz, CDCIs) d (ppm)= 7.99 (d, J=8.31 Hz, 2H), 7.27 – 7.39 (m, 7H), 5.83 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.20 – 4.42 (m, 1 H), 3.92 (s, 3H), 3.12 – 3.33 (m, 1 H), 2.84 – 3.04 (m, 2H), 2.46 – 2.65 (m, 1 H), 2.23 – 2.45 (m, 1 H).
Example 2: Synthesis of Benzyl -4-hvdroxy-2-(4-(methoxycarbonyl)phenyl)piperidine-1-
carboxylate (C5. wherein Pi = Cbz and R = methyl)
P1 = Cbz P i = Cbz
R = Methyl R = Methyl
A 0.1 M pH = 7.0 PBS was prepared with disodium phosphate dodecahydrate (22.2 g), sodium dihydrogen phosphate dihydrate (6.2 g) and purified water (999 g). To a reactor equipped with a pH meter 0.1 M pH = 7.0 PBS (499 g), D-glucose (40.2 g, 233.14 mmol, 2.0 eq), NADP (EnzymeWorks, 0.72 g), GDH (EnzymeWorks, 0.41 g) and KRED-EW124 (EnzymeWorks, 2.05 g)
were added, followed by addition of emulsion of C4 (41 g, 1 1 1 .60 mmol, 1 .0 eq) in DMSO (102.5 ml_). The mixture was heated to JT < 45 °C, IT 41 ± 3 °C and stirred at IT 41 ± 3 °C for > 16 h while controlling pH 6.9-7.2 by adding 1 M sodium hydroxide solution. A mixture of NADP (0.29 g), GDH (0.16 g) and KRED-EW124 (0.82 g, #Enzyme Works Inc. China) in 0.1 M pH = 7.0 PBS (1 1 g) were charged and stirred at IT 41 ± 3 °C for > 20 hours. The reaction was monitored by HPLC.
The reaction was filtered to afford white wet cake. To a 1 .0 L Radleys reactor equipped with anchor agitator crude C5 wet cake (80 g) and acetonitrile (500 ml.) were charged. The mixture was stirred to form a light yellow suspension (700 RPM). The suspension was heated to IT = 70 ± 5 °C and stirred for 4 hours, and then cooled to IT = 25 ± 5 °C. The suspension was filtered and the cake was washed with acetonitrile (75 ml_). To a clean 500 ml. Radleys reactor equipped with anchor agitator the resulting mother liquor was charged. The mother liquid was concentrated to about 95 g, solvent exchanged with three portions of toluene (105 g) to 95 g residue. Toluene (170 g) was charged and the reaction was checked by GC (acetonitrile / (toluene + acetonitrile) < 1 .2%). The suspension was heated to IT = 80 ± 5 °C, held for 1 hour, cooled to IT = 45 ± 3 °C and adjusted the agitation speed to low mode. Sequential operations of seeding and aging for 2 hours, charging n-heptane (10.2 g) in 0.5 hours and aging for 1 hour, charging n-heptane (34 g) over 1 .5 hours and aging for 0.5 hours were carried out. The mixture was cooled to IT = 10 ± 3 °C over 7 hours and maintained at 10 ± 3 °C for 2 hours. The mixture was filtered and the cake was washed with cold mixed solvents of toluene (50 ml.) and n-heptane (10 ml.) to afford a light yellow solution of C5 (330 g, trans/cis = 90/10, assay 6.8%, yield 52%). The mother liquor was telescoped to the next step. 1 H-NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets): d (ppm) = 7.99 (d, J=8.44 Hz, 2H) [7.92 (d, J=8.44 Hz, 0.04H)], 7.23 – 7.39 (m, 7H) [7.10 – 7.18 (m, 0.21 H)], 5.69 (br. s, 1 H) [5.40-5.42 (m, 0.1 1 H)], 5.19 (s, 2H) [5.14 (s, 0.23H)], 4.26 (br. d, J=13.33 Hz, 1 H) [4.18-4.20(m, 0.13H)], 3.91 (s, 3H) [3.90 (s, 0.4H)], 3.67 – 3.79 (m, 1 H) [3.38-3.45 (m, 0.1 1 H)], 2.83 (td, J=13.51 , 2.81 Hz, 1 H), 2.64 (br. d, J=13.33 Hz, 1 H) [2.41 -2.47 (m, 0.12H)], 1 .81-1 .91 (m, 2H) [2.17-2.22 (m, 0.12H)], 1 .72 – 1 .77 (m, 1 H), 1 .45 – 1 .56 (m, 1 H). HRMS: Calcd for C21 H24NO5 (M+H): 370.1654m, found 370.1662.
Example 3: Synthesis of Methyl 4-r(2S,4S)-4-ethoxypiperidin-2-yl1benzoate (Compound of formula according to the following sequence:
R = Methyl R = Methyl R = Methyl
Step 1 : Synthesis of Benzyl (4S)-4-((tert-butyldimethylsilyl)oxy)-2-(4-(methoxycarbonyl) phenyl)piperidine-1 -carboxylate (C8, wherein Pi = Cbz, P2 = TBS and R = methyl).
To a 500 ml. Radleys Reactor charged with C5 in a toluene/heptane solution (1 .0 eq, 145.67 g from previous step, assay 6.07%, 23.94 mmol). The solution was concentrated to about 25 g. Then dichloromethane (1 17.1 g) was charged and the solution was cooled to 23 ± 4 °C. To the clear solution, imidazole (3.42 g, 50.26 mmol, 2.1 eq) and TBS-CI (6.13 g, 40.69 mmol, 1 .7 eq) were introduced. The yellow suspension was stirred at 23 ± 4 °C for 10 hours. The reaction was monitored by HPLC. Then 10% Na2CC>3 (70.7 g) was charged and the mixture was stirred for 1 hours. The organic phase was washed with 5% brine (53 g) and concentrated to about 30 g. Then the solvent was exchange with toluene (45 g) to about 25 g. The residue was diluted with dichloromethane (66 g) and the mixture was filtered through a pad of 200-300 mesh silica gel (1 .66 g). The silica gel was eluted with another portion of dichloromethane (17.5 g). The eluent was concentrated and the residue was subjected to solvent exchange with acetonitrile (71 .1 g + 98.2 g) to 90 g (yield 100%). C8 in acetonitrile solution was used in the next step. 1 H-NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets): d (ppm) = 8.01 (d, J=8.44 Hz, 2H) [7.94 (d, J=8.44 Hz, 0.17H)], 7.26 – 7.34 (m, 7H) [7.09 – 7.18 (m, 0.13H)], 5.65 (br. d, J=2.04 Hz, 1 H) [5.41 (br. d, J=2.04 Hz, 0.08H)], 5.19 (s, 2H) [5.13 (s, 0.16H)], 4.22 (br. d, J=13.69 Hz, 1 H) [4.10-4.14(m, 0.19H)], 3.92 (s, 3H) [3.90 (s, 0.3H)], 3.62 – 3.69 (m, 1 H) [3.43-3.50 (m, 0.08H)], 2.81 (td, J=13.54, 2.87 Hz, 1 H), 2.49 (br. d, J=13.57 Hz, 1 H) [2.31 -2.35 (m, 0.1 OH)], 1.84-1 .92 (m, 1 H) [2.08-2.14 (m, 0.07H)], 1 .74 – 1 .75 (m, 1 H), 1 .48 – 1 .59 (m, 1 H), 0.86 (s, 9H) [0.56 (s, 0.65H)], 0.03 (s, 3H) [0.09 (s, 0.27H)].
Step 2: Synthesis of Benzyl (4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)piperidine-1 -carboxylate (C9, wherein Pi = Cbz, R = methyl)
To a 250 ml. Radleys Reactor equipped with impeller agitator C8 in acetonitrile solution (135.5 g, assay 12.53%, 35.10 mmol) was charged and rinsed with acetonitrile (with 8.5 g). Et3SiH (12.25 g, 105.31 mmol, 3.0 eq) was charged. The reactor was cooled to IT = 4 ± 5 °C. TESOTf (1 .392 g,
5.265 mmol, 0.15 eq) was charged. A solution of 2,4,6-trimethyM ,3,5-trioxane (4.64 g, 35.10 mmol, 1 .0 eq) in acetonitrile (7.9 g) was added to the mixture in 60 min at IT = 4 ± 5 °C. After addition, the mixture was stirred for 15 min and followed by HPLC. To the reaction mixture was charged 5% aqueous Na2CC>3 (21 .22 g) and water (30 g). Followed by n-heptane (20.4 g) and the mixture was stirred at 25 ± 5 °C for 30 min. Phase cut and the bottom acetonitrile phase was collected. The acetonitrile phase was concentrated to about 65 g. MTBE (100.6 g) and 5% aqueous Na2CC>3 (43.44 g) were charged to the residual acetonitrile solution. The mixture was stirred for 30 min. The upper MTBE phase was collected and filtered via Charcoal film. The charcoal film was washed with MTBE (7.4 g). The mother liquor was concentrated to about 35 g. To the residue methanol (79.2 g) was charged and the solution was concentrated to 70 g. The solution was telescoped to the next step. 1 H NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets) d (ppm) = 8.01 (d, J= 8.31 Hz, 2H) [7.96 (d, J= 8.31 Hz, 0.21 H)], 7.29 – 7.32 (m, 7H) [7.07 – 7.22 (m, 0.40H)], 5.68 (br. s, 1 H) [5.32 – 5.34 (m, 0.10H)], 5.19 (s, 2H) [5.1 1 (s, 0.19H)], 4.27 (br. d, J=13.08 Hz, 1 H) [4.05 – 4.14 (m, 0.15H)], 3.91 (s, 3H) [3.89 (s, 0.15H)], 3.41 – 3.54 (m, 2H) [3.14 – 3.25 (m, 0.21 )], 3.30 – 3.40 (m, 1 H) [3.86 – 3.75 (m, 0.13H)], 2.84 (td, J=13.51 , 2.81 Hz, 1 H), 2.66 (br. d, J=13.20 Hz, 1 H), 1 .62 – 1 .95 (m, 2H), 1 .40 – 1 .53 (m, 1 H), 1 .18 (t, J= 6.97 Hz, 3H).
Step3: Synthesis of Methyl 4-((4S)-4-ethoxypiperidin-2-yl)benzoate (removal of the protecting group Pi = Cbz – R = methyl)
To a 500 ml. autoclave charged with 10% Pd/C (50% wet, 3.83 g), C9 solution in methanol (assay 19.97%, 192 g, 96.46 mmol) and methanol (28 g). The reactor was purged with vacuum/H2, three times. The mixture was hydrogenated at 3 bar and at a temperature of 25 ± 4 °C for 4 hours. The mixture was filtered and the Pd/C cake was washed with methanol (20 g). The mother liquor was concentrated to 48 g, solvent swapped twice with 142 g isopropyl acetate to 106 g, cooled to 8 ± 5 °C, and 3% hydrogen chloride solution (90.2 g) was added. After phase separation, the aqueous phase was collected and washed with isopropyl acetate (86.4 g). To the aqueous phase MTBE (72 g) and 10% Na2C03 (99.2 g) were added. After phase separation, the aqueous phase was extracted with MTBE (72 g). The combined MTBE phase was washed with water (40 g). The MTBE solution was introduced into the next step. 1 H NMR (400 MHz, CDCI3, mixture of two isomers, data for the minor isomer is shown in brackets) d (ppm) = 7.96 (m, J= 8.31 Hz, 2H), 7.40 – 7.46 (m, 2H), 4.06 (dd, J=1 1 .62, 2.45 Hz, 1 H), 3.88 (s, 3H), 3.70 – 3.79 (m, 1 H) [3.64 – 3.69 (m, 0.12H)], 3.48 -3.56 (m, 2H) [3.38 – 3.45(m, 0.1 1 H)], 3.1 1 – 3.18 (m, 1 H) [3.21 – 3.26 (m, 0.1 1 H)], 2.88 – 2.97 (m, 1 H) [2.73 – 2.80 (m, 0.12H )], 1 .94 – 2.00 (m, 1 H) [ 2.14 – 2.19 (m, 0.10H)], 1.84 – 1 .89 (m, 1 H) [2.02 – 2.07 (m, 0.12H)], 1 .75 (S, 1 H), 1 .65 – 1 .70 (m, 1 H) [1 .45 – 1 .49 (m, 0.10H)], 1 .59 – 1 .64 (m, 1 H) [1 .36 – 1 .42 (m, 0.1 1 H)], 1 .22 – 1 .25 (t, 3H) [1 .17 – 1 .20 (t, J= 6.97, 0.24H)].
Step 4: Synthesis of Methyl 4-[(2S,4S)-4-ethoxypiperidin-2-yl]benzoate (Compound of formula (II) – R = methyl).
To a 500 ml. one neck flask was added the crude solution of step 3 (above) in MTBE (telescoped from last step, 1 10 g, assay 10.52%, light yellow solution, 43.95 mmol). The solution was concentrated to 18.4 g and the solvent was exchanged (JT = 60 °C) with 55 g of n-heptane twice to get 35 g yellow solution. The solution was transferred to 100 ml. Easy Max equipped with impeller agitator. The solution was heated to 50 °C with 300 RPM , aged for 30 min, cooled to 41 ± 2 °C and seed was added. The agitation was adjusted to low speed. The mixture was aged at 41 ± 2 °C for 2 hours, cooled to 35 ± 2 °C in 8 – 10 hours and then aged at 35 ± 2 °C for 1 – 2 hours n-heptane (7.9 g) was added dropwise. The agitation was adjusted to medium speed. The mixture was cooled to IT = 25 ± 2 °C in 1 hour and aged at 25 ± 2 °C for 10 – 20 minutes. The mixture was filtered. The filtrate was re-charged to the reactor for rinsing the solid on the reactor wall. The mixture was filtered and the filter cake was washed with pre-cooled (-5 °C) n-heptane (7.9g). The cake was dried at 40 °C for > 10 hours to afford 6.4 g of white solid (50% yield). 1H NMR (400 MHz, CDCIs) d (ppm) = 7.99 (m, J=8.31 Hz, 2H), 7.45 (m, J=8.19 Hz, 2H), 4.09 (dd, J=1 1 .62, 2.20 Hz, 1 H), 3.90 (s, 3H), 3.75 (t, J=2.81 Hz, 1 H), 3.53 (q, J= 6.97 Hz, 2H), 3.17 (td, J=12.13, 2.63 Hz, 1 H), 2.91 – 2.99 (m, 1 H), 1.99 (dd, J=13.57, 2.69 Hz, 1 H), 1 .88 (dt, J=13.79, 2.58 Hz, 1 H), 1 .69 – 1 .79 (m, 1 H), 1 .57 – 1 .68 (m, 2H), 1 .25 (t, J= 7.03 Hz, 3H).
Example 4: Enantioselective synthesis of compound according to the following
sequence:
Step 1 : Synthesis of Benzyl 4-oxo-3,4-dihydropyridine-1 (2H)-carboxylate (C6, wherein Pi = Cbz and R = methyl)
To a 2.0 L reactor, 4-methoxypyridine (C1 , 45.0 g, 412.39 mmol, 1 .0 eq) and methanol (900 ml.) were added. The mixture was cooled to -75 °C with dry ice/acetone bath. A solution of benzyl
chloroformate (73.86 g, 432.99 mmol, 1 .05 eq) in THF (90 ml.) was charged dropwise while keeping IT < -70 °C. The reaction was stirred for 1 hour to afford a white suspension at -70 °C. Sodium borohydride (16.38 g, 432.99 mmol, 1 .05 eq) was added in portions while keeping IT < -70 °C. The reaction was stirred at -70 °C for 2 hours. Water (200 g) was added and the cooling bath was removed. A solution of 36% hydrogen chloride (16.72 g, 164.95 mmol, 0.4 eq) in water (50 ml.) was added in 10 min at 0 – 5 °C and stirred for 1 hour. Then 20% Na2CC>3 (85.5 g) was added to adjust pH = 7 while maintained IT < 5 °C. Organic solvents were removed under vacuum. The resulting residue was extracted with dichloromethane (450 ml_). The dichloromethane phase was washed with 3wt% hydrogen chloride (151 ml.) and 3 wt% Na2C03 (151 ml_). After solvent exchange with MTBE, about 4 volume (180 ml) of the MTBE mixture was obtained. The mixture was heated to 50 °C to afford a solution and then cooled to 45 °C. Crystal seed of C6 was charged and the mixture was aged at 40 – 45 °C for 7 hours. The mixture was cooled to 10 – 15 °C in 3 hours. The white suspension was filtered and the wet cake was rinsed with cold MTBE (45 ml_). The cake was dried under vacuum at 40 – 50 °C for 2 hours to afford C6 as a white powder (91.56 g, 60% yield). 1H NMR (400 MHz, CDCI3): d (ppm) = 7.85 (br. s, 1 H), 7.37 – 7.43 (m, 5H), 5.43 (br. s, 1 H), 5.26 (s, 2H), 4.05 (t, J=7.34 Hz, 2H), 2.54 – 2.58 (m, 2H).
Step 2: Synthesis of Benzyl (S)-2-(4-(methoxycarbonyl)phenyl)-4-oxopiperidine-1 -carboxylate ((S)-C4, wherein Pi = Cbz and R = methyl)
Method 1 : A 500 ml Radleys reactor was purged 3 times with vacuum/N2. C6 (8 g, 34.60 mmol, 1.0 eq), C7 (9.34 g, 51.89 mmol, 1 .5 eq), tert- Amyl alcohol (160 ml.) and deionized water (16 ml.) were added. The mixture was stirred for > 40 minutes to give a clear colorless solution. The solution was purged 4 times with vacuum / N2 and bubbled with N2 via a syringe needle for 1 hour. To the colorless solution was charged the mixed solid of (S)-XylBINAP (0.381 g, 0.519 mmol, 0.015 eq) and Rh(Acac)(C2H4)2 (0.134 g, 0.519 mmol, 0.015 eq). The mixture was continued to bubble with N2 for 15 minutes and purged 4 times with vacuum / N2. The suspension was stirred for another 2 hours to dissolve (S)-XylBINAP. The reaction mixture was stirred at 55 ± 4 °C for 15 hours. The reaction was followed by HPLC. The mixture was cooled and treated with 7.7% sodium hypochlorite (1 g, 1 .04 mmol, 0.03 eq) for 1 .5 hours at 40 ± 4 °C. tert- Amyl alcohol was distilled off. The residue was extracted with isopropyl acetate (64 ml.) and ethyl acetate (8 ml.) and filtered. The organic phase was washed with 5% NaHC03 (50 g) then with 15% brine (40 g) at 50 ± 5 °C. Some solvents were removed and ethyl acetate (21 .6 g) was added. The solution was treated with Smopex-234 (1 .2 g) at IT =55 ± 5 °C for 2 hours then filtered via 200 – 300 mesh silica gel (1 .6 g). After solvent exchange with n-heptane, MTBE (44.4 g) was added. The mixture was cooled to IT = 42 ± 3 °C. (S)-C4 seed (10 mg) was added. The mixture was aged for 2 hours and cooled to IT =
31 ± 3 °C in 3 hours n-heptane (23.2 g) was then charged in 1 – 2 hours. The mixture was aged for 2 hours and cooled to IT = 20 ± 3 °C in 2 hours. The mixture was filtered and the cake was washed with a mixed solvent of MTBE (4.4 g) and n-heptane (4.1 g). Dried the wet cake at 60 °C for > 5 hours to afford (S)-C4 (7.63 g, 60% yield) as yellow powder. 1H NMR (400 MHz, CDCI3): d (ppm) = 7.99 (d, J=8.44 Hz, 2 H), 7.28 – 7.37 (m, 7H), 5.82 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.30 (br. s, 1 H), 3.91 (s, 3H), 3.22 (br. d, J=8.31 Hz, 1 H), 2.84 – 3.03 (m, 2H), 2.46 – 2.64 (m, 1 H), 2.38 (br. d, J=16.26 Hz, 1 H).
Method 2: To a 500 ml Radleys reactor purged 3 times with vacuum/N2, C6 (8 g, 34.60 mmol, 1 .0 eq), C7 (9.34 g, 51 .89 mmol, 1 .5 eq), fe/f-Amyl alcohol (160 ml.) and deionized water (16 ml.) were added. The mixture was stirred for roughly 40 minutes to give a clear colorless solution. The solution was purged 4 times with vacuum / N2 and bubbled with N2 via a syringe needle for 1 hour. To the colorless solution, was charged the mixed solid of (R, R)-Ph-BPE-Rh(Acac) (0.005 eq., 0.122 g, 0.173 mmol). The mixture was continued to bubble with N2 for 15 minutes and purged with vacuum / N2. The reaction mixture was stirred at 55 ± 4 °C for 15 hours. The reaction was followed by HPLC. Tert- amyl alcohol was distilled off. The residue was extracted with isopropyl acetate (64 ml.) and ethyl acetate (8 ml_), and then filtered. The organic phase was washed with 5% NaHC03 (50 g), then with 15% brine (40 g) at 50 ± 5 °C. Some solvents were removed and ethyl acetate (21 .6 g) was added. The solution was treated with Smopex-234 (1 .2 g) at IT = 55 ± 5 °C for 2 hours then filtered via 200 – 300 mesh silica gel (1 .6 g). After solvent exchange with n-heptane, MTBE (44.4 g) was added. The mixture was cooled to IT = 42 ± 3 °C. (S)-C4 seed (10 mg) was added. The mixture was aged for 2 hours and cooled to IT = 31 ± 3 °C in 3 hours n-heptane (23.2 g) was then charged in 1 – 2 hours. The mixture was aged for 2 hours and cooled to IT = 20 ± 3 °C in 2 hours. The mixture was filtered and the cake was washed with a mixed solvent of MTBE (4.4 g) and n-heptane (4.1 g). The wet cake was dried at 60 °C for roughly 5 hours to afford (S)-C4 (10.17 g, 80% yield) as yellow powder. 1 H NMR (400 MHz, CDCI3) d (ppm) = 7.99 (d, J=8.44 Hz, 2 H), 7.28 – 7.37 (m, 7H), 5.82 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.30 (br. s, 1 H), 3.91 (s, 3H), 3.22 (br. d, J=8.31 Hz, 1 H), 2.84 – 3.03 (m, 2H), 2.46 – 2.64 (m, 1 H), 2.38 (br. d, J=16.26 Hz, 1 H).
Method 3: To a 500 ml Radleys reactor purged 3 times with vacuum/N2. C6 (8 g, 34.60 mmol, 1 .0 eq), C7 (9.34 g, 51 .89 mmol, 1 .5 eq), tert- amyl alcohol (160 ml.) and deionized water (16 ml.) were added. The mixture was stirred for roughly 40 minutes to give a clear colorless solution. The solution was purged 4 times with vacuum / N2, and bubbled with N2 via a syringe needle for 1 hour. To the colorless solution was charged the mixed solid of (S)-XylBINAP-Rh(Acac) (0.01 eq., 0.324
g, 0.346 mmol). The mixture was continued to bubble with N2 for 15 minutes and purged with vacuum / N2. The reaction mixture was stirred at 55 ± 4 °C for 15 hours. The reaction was followed by HPLC. Tert- amyl alcohol was distilled off. The residue was extracted with isopropyl acetate (64 mL) and ethyl acetate (8 mL), and then filtered. The organic phase was washed with 5% NaHC03 (50 g), then with 15% brine (40 g) at 50 ± 5 °C. Some solvents were removed and ethyl acetate (21 .6 g) was added. The solution was treated with Smopex-234 (1 .2 g) at IT =55 ± 5 °C for 2 hours then filtered via 200 – 300 mesh silica gel (1 .6 g). After solvent exchange with n-heptane, MTBE (44.4 g) was added. The mixture was cooled to IT = 42 ± 3 °C. (S)-C4 seed (10 mg) was added. The mixture was aged for 2 hours and cooled to IT = 31 ± 3 °C in 3 hours n-heptane (23.2 g) was then charged in 1 – 2 hours. The mixture was aged for 2 hours and cooled to IT = 20 ± 3 °C in 2 hours. The mixture was filtered, and the cake was washed with a mixed solvent of MTBE (4.4 g) and n-heptane (4.1 g). The wet cake was dried at 60 °C for roughly 5 hours to afford (S)-C4 (10.30 g, 81 % yield) as yellow powder. 1H NMR (400 MHz, CDCI3) d (ppm) = 7.99 (d, J=8.44 Hz, 2 H), 7.28 – 7.37 (m, 7H), 5.82 (br. s, 1 H), 5.14 – 5.28 (m, 2H), 4.30 (br. s, 1 H), 3.91 (s, 3H), 3.22 (br. d, J=8.31 Hz, 1 H), 2.84 – 3.03 (m, 2H), 2.46 – 2.64 (m, 1 H), 2.38 (br. d, J=16.26 Hz, 1 H).
Example 5: Synthesis of Benzyl -4-hvdroxy-2-(4-(methoxycarbonyl)phenyl)piperidine-
1-carboxylate f(S)-C5, wherein Pi = Cbz and R = methyl)
R = Methyl R = Methyl
Preparation of 0.1 M PBS, pH 7.0, with 0.1 % TPGS buffer solution: To a 500 ml. Radleys reactor equipped with impeller agitator was charged Na2HP04.12H20 (8.63 g), NaH2P04.2H20 (2.41 g), Tap Water (388.6 g) and TPGS-750-M.001 (0.388 g). The mixture was stirred for > 3 hours at IT = 60 ± 5 °C and then cooled to IT = 51 ± 3 °C. 80 g of the buffer solution was taken from the reactor to a flask and cooled to < 35 °C. Check pH value of the buffer solution (7.0 ± 0.5). To the above Radleys reactor (S)-C4 (20.0 g, 54.4 mmol, 1 .0 eq), Isopropanol (16.36 g, 272.2 mmol, 5.0 eq) and 0.1 % TPGS buffer solution (60 g) were added. To a 25 mL flask was charged KRED-P3-G09 (0.4 g, #Codexis), NADP+ (0.1 g) and 0.1 % TPGS buffer solution (60 g) from the above flask. All the solid was dissolved. The solution of enzyme was charged to the 500 mL Reactor at IT =50 ± 5 °C. Rinsed the 25 mL flask with 0.1 % TPGS buffer (10 g) and transferred the solution to the 500 mL reactor at IT =50 ± 5 °C. The mixture was stirred with agitation speed > 500 RPM at 51 ± 3 °C for >
8 hours. The reaction was followed by HPLC. To the reactor 2-MeTHF (200 mL) was added and the mixture was stirred for > 60 minutes at 50 ± 5 °C. The mixture was held for > 50 minutes without agitation and the bottom aqueous phase was separated. The organic phase was washed twice with another 200 g of water at 50 ± 5 °C. The organic phase was concentrated to about 70 g. After solvent exchange with twice 158 g acetonitrile to give about 80 g solution, which was cooled to < 30 °C then filtered via MCC. MCC cake was washed with isopropyl acetate (40 mL/35.5 g) to afford (S)-C5 in a light color solution (1 14.3 g, assay 16.95% 96.34% yield). The acetonitrile / isopropyl acetate solution was telescoped to the next step directly. 1 H NMR (400 MHz, CDCI3): d (ppm) = 7.98 (d, J=8.44 Hz, 2H), 7.23 – 7.38 (m, 7H), 5.61 – 5.72 (m, 1 H), 5.18 (s, 2H), 4.23 (br. d, J=13.33 Hz, 1 H), 3.90 (s, 3H), 3.62 – 3.75 (m, 1 H), 2.81 (td, J=13.51 , 2.81 Hz, 1 H), 2.62 (br. d, J=13.33 Hz, 1 H), 2.45 (br. s, 1 H), 1 .79 – 1 .91 (m, 2H), 1 .41 – 1 .56 (m, 1 H).
Example 6: Asymmetric synthesis of Methyl 4-r(2S.4S)-4-ethoxypiperidin-2-yl1benzoate
(Compound of formula . or a salt thereof. – R= methyl) according to the following
sequence:
(S)-C5 (S)-C9 Compound of (Pi = Cbz) (Pi = Cbz, P2 = TBS) (Pi = Cbz) formula (II) R = Methvl R = Methyl R = Methyl R = Methyl
Step 1 : Synthesis of Benzyl (2S,4S)-4-{[tert-butyl(dimethyl)silyl]oxy}-2-[4-(methoxy carbonyl) phenyl]piperidine-1 -carboxylate ((S)-(C8), wherein Pi = Cbz, P2 = TBS, and R = methyl).
To a 500 ml Radleys Reactor was charged with (S)-C5 solution (in acetonitrile / isopropyl acetate, 271 .8 g, assay 14.72%, contained 40.0 g of (S)-C5, 108.31 mmol, 1 .0 eq) from the previous step. After solvent exchange with isopropyl acetate (159.8 g / 180 ml_), 100 g clear solution was obtained. Isopropyl acetate (176 g /198 ml_), imidazole (26.54 g, 389.90 mmol, 3.6 eq) and TBS-CI (27.75 g, 184.12 mmol, 1 .7 eq) were added. The yellow suspension was stirred at 55 ± 4 °C for 7 hours. The reaction was followed by HPLC. The reaction mixture was cooled to 23 ± 4 °C and filtered through MCC (2 g). The cake was washed with isopropyl acetate (88.8 g / 100 ml_). 6% NaHC03 (240 g) was added and the mixture was stirred for 20 minutes. The organic phase was washed with 5% brine (2×240 g) and concentrated to about 105 g. After solvent exchange with toluene (120 g / 135.4 ml_), 105 g solution was obtained. Dichloromethane (298 g / 224.5 ml.) was added and the solution was filtered via 200-300 mesh silica gel (4.4 g). The silica gel was eluted with another portion of dichloromethane (44 g / 33 ml_). The mother liquor was concentrated and the solvent was exchanged with acetonitrile (2×280 ml_, 442.4 g in total) to 100 g. The residue was diluted with acetonitrile (105 g / 132.9 ml.) to afford a light yellow solution (205 g, assay 25.55%, 100% yield), which was used for the next step directly. 1 H NMR (400 MHz, CDCI3) d (ppm) = 8.01 (d, J=8.44 Hz, 2 H), 7.23 – 7.37 (m, 7 H), 5.60 – 5.70 (m, 1 H), 5.18 (s, 2H), 4.22 (br. d, J=13.45 Hz, 1 H), 3.90 (s, 3H), 3.62 – 3.71 (m, 1 H), 2.82 (td, J=13.51 , 2.81 Hz, 1 H), 2.49 (br. d, J=13.45 Hz, 1 H), 1.83 – 1 .96 (m, 1 H), 1 .75 – 1 .80 (m, 1 H), 1 .47 – 1.60 (m, 1 H), 0.86 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H).
Step 2: Synthesis of Benzyl (2S, 4S)-4-ethoxy-2-[4-(methoxycarbonyl)phenyl]piperidine-1 -carboxylate ((S)-C9, wherein Pi = Cbz amd R = methyl)
To a 500 ml. Radleys Reactor equipped with impeller agitator (S)-C8 in an acetonitrile solution (170.8 g, assay 29.28%, 103.38 mmol, 1 .0 eq) and fresh acetonitrile (220 g) were charged, followed by Et3SiH (36.06 g, 310.13 mmol, 3.0 eq). The mixture was cooled to IT =4 ± 5 °C and TESOTf (5.47 g, 20.68 mmol, 0.2 eq) was charged. To the mixture was charged a solution of 2,4,6-trimethyl-1 ,3,5-trioxane (13.66 g, 103.38 mmol, 1 .0 eq) in acetonitrile (23 g) over 60 minutes at IT =4 ± 5 °C. Upon addition, the mixture was stirred for 15 minutes. The reaction was followed by HPLC. To the reaction mixture was charged 5% aqueous sodium hydroxide (16.54 g, 20.68 mmol, 0.2 eq) and 20 g water, followed by n-heptane (60 g). The mixture was stirred for 30 minutes at 20 ± 5 °C. The bottom acetonitrile phase was collected. To the acetonitrile phase was charged with MTBE (1 1 1 g) and 10% brine (300 g). The mixture was stirred for 30 minutes. The upper MTBE phase was washed with 10% brine (2×300 g), concentrated to 90 g. MTBE (185 g) and water (150 g) were charged. After phase separation at 38 ± 4 °C and solvent exchange of the organic layer with isopropyl acetate (2×266.4 g), 205 g solution was obtained, which was filtered through Charcoal film slowly. The charcoal film was washed with isopropyl acetate (22.2 g) to afford as a light yellow solution (223 g, 100% yield). The solution was telescoped to the next step directly. 1 H NMR (400 MHz, CDCI3) d (ppm) = 8.01 (d, J=8.44 Hz, 2H), 7.25 – 7.38 (m, 7H), 5.68 (br. s, 1 H), 5.19 (s, 2H), 4.27 (br. d, J=13.33 Hz, 1 H), 3.92 (s, 3H), 3.42 – 3.54 (m, 2H), 3.34 (ddd, J=10.88, 6.91 , 4.22 Hz, 1 H), 2.84 (td, J=13.51 , 2.81 Hz, 1 H), 2.66 (br. d, J=13.20 Hz, 1 H), 1 .96 (br. d, J=10.51 Hz, 1 H), 1 .75 – 1 .90 (m, 1 H), 1 .33 – 1 .53 (m, 1 H), 1 .18 (t, J= 6.97 Hz, 3H).
Step 3: Synthesis of Methyl 4-((2S,4S)-4-ethoxypiperidin-2-yl)benzoate (compound of Formula (II), or a salt thereof – R= methyl)
To a 500 ml. autoclave which was purged with vacuum / N2 (S)-C9 in an isopropyl acetate solution (278.4 g, assay 17.96%, 50 g of (S)-C9, 125.80 mmol) and 10% Pd/C (5.0 g, 50% wet) were
charged. The reactor was purged with vacuum / H2 and stirred for > 7 hours at 25 ± 5 °C. The reaction was followed by HPLC analysis. Filtered the reaction mixture via MCC (7.7 g) which was pre-washed with isopropyl acetate . Rinsed the reactor and MCC with isopropyl acetate (39 g). The mother liquor was combined to afford compound of formula (II) as a light yellow solution (315 g, assay 10.0%, 95.1 % yield). 1 H NMR (400 MHz, CDCI3) d (ppm) = 7.99 (m, J=8.31 Hz, 2H), 7.45 (m, J=8.19 Hz, 2H), 4.09 (dd, J=1 1 .62, 2.20 Hz, 1 H), 3.90 (s, 3H), 3.75 (t, J=2.81 Hz, 1 H), 3.53 (q, J= 6.97 Hz, 2H), 3.17 (td, J=12.13, 2.63 Hz, 1 H), 2.91 – 2.99 (m, 1 H), 1 .99 (dd, J=13.57, 2.69 Hz, 1 H), 1 .88 (dt, J=13.79, 2.58 Hz, 1 H), 1 .69 – 1 .79 (m, 1 H), 1 .57 – 1 .68 (m, 2H), 1 .25 (t, J= 7.03 Hz, 3H).
Step 4: Synthesis of the maleic salt of compound of formula (II) (R = methyl)
To a 500 ml. Radleys Reactor equipped with impeller agitator a solution of methyl 4-((2S,4S)-4-ethoxypiperidin-2-yl)benzoate (381 g, assay 10.03%, 145.12 mmol, 1 .0 eq) from the previous step was charged. The solution was concentrated to 281 g and fresh isopropyl acetate (28.6 g) was added. Then a solution of maleic acid (8.45 g, 72.56 mmol, 0.5 eq) in acetone (30.5 ml.) was added at 51 ± 3 °C in 30 minutes. After stirring for 15 minutes, a seed of the maleic salt of compound of formula (II) was added and the mixture was aged for 2 hours. A solution of maleic acid (8.45 g, 72.56 mmol, 0.5 eq) in acetone (30.5 ml.) was charged at 51 ± 3 °C in 60 minutes and the mixture was aged for 2 hours. The mixture was cooled to IT = 10 ± 3 °C in 6 hours and stirred for > 120 minutes. The mixture was filtered and the filter cake was washed with pre-cooled isopropyl acetate (44.4 g). The cake was dried under high vacuum at 55 °C for 5 – 12 hours to afford maleic salt of compound of formula (II) as white solid (49.8 g, Yield 90.4%). 1 H NMR (400 MHz, CDCIs) d (ppm) 9.35 – 9.78 (m, 2H), 8.02 (m, J=8.31 Hz, 2H), 7.58 (m, J=8.31 Hz, 2H), 6.17 (s, 2H), 4.56 (br. d, J=1 1.13 Hz, 1 H), 3.90 (s, 3H), 3.86 (s, 1 H), 3.48 – 3.57 (m, 2H), 3.38 – 3.44 (m, 2H), 2.42 (br. t, J=13.57 Hz, 1 H), 1 .98 – 2.20 (m, 3H), 1 .24 (t, J= 6.97 Hz, 3H).
The maleic salt of compound of formula (II) may be characterized by a x-ray powder diffraction pattern (XRPD) comprising four or more 2Q values (CuKa l=1 .5418 A) selected from the group consisting of 5.893, 6.209, 1 1 .704, 13.014, 16.403, 17.295, 17.592, 18.629, 18.942, 21 .044, 21 .733, 21 .737, 22.380, 23.528, 24.195, 26.013, 26.825, 29.017, 29.515, 32.250, 35.069, 35.590, and 37.932, measured at a temperature of about 22 °C and an x-ray wavelength, l, of 1 .5418 A.
Example 7: Synthesis of fert-butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate
(Compound of formula (III), or a salt thereof) according to the following seguence:
Step 1 : Synthesis of 7-methyl-1 H-indol-5-ol (C11 )
To a 250 ml. flask equipped with a thermometer 3.4% Na2HP04 (100 g, pH = 8.91 ) was charged, followed by addition of Fremy’s salt (4.84 g, 2.4 eq). The mixture was stirred at 20 ± 5 °C until a clear solution was formed. A solution of 7-methylindoline in acetone (9.1 g, 1 1 %) was added in one portion. The mixture was stirred at 20 ± 5 °C for 1 .5 hours. Then sodium sulfite (0.38 g) was added. The mixture was extracted with ethyl acetate (100 ml. x 2) The combined organic extracts were dried over anhydrous sodium sulfate, filtered and concentrated. To the residue 20ml_ acetonitrile was added. The solution was used directly in the next step.
Step 2: Synthesis of fert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12, wherein P3 = Boc)
The above as prepared solution was cooled to 0 ± 5 °C. DMAP (0.34 g, 0.4 eq) was charged followed by addition of (Boc)20 (4.9 g, 3.0 eq). The mixture was warmed to 20 ± 5 °C, stirred at 20 ± 5 °C for 30 minutes and concentrated. To the residue was added methanol (40 ml_). The mixture was cooled to 0 ± 5 °C. Potassium carbonate (5.1 g, 5.0 eq) was added. The mixture was stirred at 0 ± 5 °C for 4 hours, warmed to 20 ± 5 °C and stirred for additional 2 hours. The mixture was cooled to 0 ± 5 °C. Acetic acid (2 g) was added. pH was 7-8. The mixture was filtered and the filter cake was washed with methanol (10 mL x 2). The filtrate was concentrated and ethyl acetate (30 ml.) was added. The mixture was washed with water (20 ml.) and 5% brine (20 ml_). The organic layer was concentrated to afford a dark oil, which was slurried with (3:2) n-heptane: Ethyl acetate (5 g) to afford a yellow solid. The solid was collected by filtration and dried to give C12 as yellow solid. 27.4% isolate yield from C10. 1 H-NMR (400 MHz, DMSO-d6): d (ppm) = 9.13 (s, 1 H), 7.52 (d, J= 3.67 Hz, 1 H), 6.74 (d, J= 2.2 Hz, 1 H), 6.56 (m, 1 H), 6.50 (d, J= 3.67 Hz, 1 H), 2.45 (s, 3 H), 1.57 (s, 9 H). LCMS (m/z): positive mode 248.1 [M]+, LCMS (m/z): negative mode 246.1 [M-1 ]-.
Step 3: Synthesis of fert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13, wherein P3 = Boc)
To a solution of fe/f-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12) (53.8% assay, 1 .0 g, 2.2 mmol) in THF (20 ml.) was added dropwise the solution of CH3MgBr in THF (1 N, 2.2 ml_, 2.2 mmol). The resulting mixture was stirred at 20 – 25 °C for 10 minutes. (CHO)n (0.2 g, 6.53 mmol)
was added to the mixture. The reaction mixture was heated to 65 – 70 °C and stirred for 1 hours. The reaction mixture was cooled to 20 – 25 °C. Saturated NH4CI (20 ml.) and MTBE (20 ml.) were added. The mixture was separated and the aqueous layer was extracted with MTBE (20 ml_). The organic layers were combined and concentrated to give compound C13 as yellow solid (0.7 g, 79% assay, 92% yield). 1H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.74 (s, 1 H), 10.54 (s, 1 H), 7.82 (d, J= 4.0 Hz, 1 H), 7.34 (d, J= 4.0 Hz, 1 H), 6.81 (s, 1 H), 2.59 (s, 3H), 1 .65 (s, 9H). LCMS (m/z): positive mode 290.1 [M]+.
Step 4: Synthesis of fert-Butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III)).
To a solution of compound C13 (50 mg, 0.182 mmol) in dry DMF (3 ml.) was added K2CO3 (50.2 mg, 0.363 mmol). The mixture was stirred for 10 minutes and then dimethyl sulfate (25.2 mg, 0.20 mmol) was added. The reaction mixture was stirred for 1 hours and poured into ice-water (12 ml_). The mixture was filtered and the filter cake was washed with water. The cake was dried under vacuum to give tert- Butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III)) as pale solid (48 mg, 91 % yield). 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.51 (s, 1 H), 7.80 (d, J= 4.0 Hz, 1 H), 7.31 (d, J= 4.0 Hz, 1 H), 6.81 (s, 1 H), 3.95 (s, 3H), 2.61 (s, 3H), 1 .59 (s, 9H). LCMS (m/z): negative mode 274.1 [M-1 ]-.
Example 8: Synthesis of fert-butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate
(Compound of formula (III), or a salt thereof) according to the following sequence:
f available (P3 = Boc)
formula (ill)
Step 1 : Synthesis of 5-(benzyloxy)-1 ,3-dimethyl-2 -nitrobenzene
To a solution of commercially available 3,5-dimethyl-4-nitrophenol (100.0 g, 590.4 mmol) in DMF (500 ml_), CS2CO3 (230.8 g, 708.5 mmol) was added and the resulting mixture was stirred for 10 minutes. Then, (bromomethyl)benzene (104.1 g, 590.4 mmol) was added dropwise to the mixture within 30 minutes. The reaction mixture was stirred at 20-25 °C for 1 hour, and then poured into ice-water (1800 ml_). The solid separated out was collected by filtration and washed with water (500 ml_). The cake was dissolved in ethyl acetate (500 ml.) and the solution was washed with a saturated solution of NaCI (50 ml_), was separated, and the solution was concentrated to give 5-(benzyloxy)-l ,3-dimethyl-2-nitrobenzene 2 (147 g, 97.8% yield) as brown solid. HPLC purity
99.7%. 1H-NMR (400 MHz, DMSO-d6) d (ppm) = 7.42 (m, 5 H), 6.94 (s, 2H), 5.16 (s, 2 H), 2.25 (s, 6 H); LCMS (m/z): negative mode 256.2 [M-1 ]-
Step 2: Synthesis of fert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12, wherein P3 = Boc)
To a solution of 5-(benzyloxy)-1 ,3-dimethyl-2-nitrobenzene (60.0 g, 233.2 mmol, from Step 1) in DMF (300 ml.) were added DMF-DMA (87.8 g, 699.6 mmol) and pyrrolidine (50.3 g, 699.6 mmol). The solution was heated to 85-90 °C and stirred for 19 hours under nitrogen, then the mixture was cooled to 20-25 °C. The volatile components (DMF-DMA, pyrrolidine and DMF) were removed at 65-70 °C on a rotary evaporator. The crude mixture was dissolved in ethyl acetate (300 ml_), and Raney Nickel (6.0 g) was added. The reaction mixture was subjected to catalytic hydrogenation under atmospheric pressure, overnight. Then, the reaction mixture was put under nitrogen. The mixture was filtrated and the filtrate was concentrated to provide 5-(benzyloxy)-7-methyM H-indole as a black oil. 5-(benzyloxy)-7-methyl-1 H-indole was used without further purification into the next step.
5-(benzyloxy)-7-methyl-1 H-indole was dissolved in acetonitrile (300 ml_), (Boc)20 (53.6 g, 233.2 mmol) and DMAP (5.7 g, 46.6 mmol) were added. The reaction mixture was stirred at 20-25 °C for 1 hour. Acetonitrile was removed on a rotary evaporator, and the residual mixture was dissolved in ethyl acetate (300 ml_). The solution was washed with a saturated aqueous solution of NaHC03 and then concentrated to give a crude oil which was purified by column chromatography (Si02, 500 g) using a mixture of heptane / MTBE (1 :10) to provide the intermediate tert-butyl 5-(benzyloxy)-7-methyl-1 H-indole-1 -carboxylate as a brown oil (42.1 g, 49.2% yield). HPLC purity 93.5%. 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 7.59 (d, J= 3.67 Hz, 1 H), 7.40 (m, 5 H), 7.04 (d, J= 2.45 Hz, 1 H), 6.81 (d, J= 2.2 Hz, 1 H), 6.57 (d, J= 3.67 Hz, 1 H), 5.1 1 (s, 2 H), 2.51 (s, 3 H), 1.58 (s, 9 H). LCMS (m/z): negative mode 336.2 [M-1 ]- To a solution of intermediate tert-butyl 5-(benzyloxy)-7-methyl-1 H-indole-1-carboxylate (36.7 g, 100 mmol) in ethanol (250 mL), under nitrogen, 10% Pd/C (10.6 g, 10 mmol) and ammonium formate (6.8 g, 105 mmol) were added. The solution was heated to 45-50 °C and stirred for 5 hours under nitrogen. Then the mixture was cooled to room temperature, filtered, and the filtrate was concentrated to give a residue oil. The residual oil was dissolved in ethyl acetate (250 mL), the solution was washed with a saturated aqueous solution of NaCI (100 mL), the phases were separated. The organic layers were collected and concentrated. The obtained crude mixtures was slurried with a (1 :15) mixture of MTBE / Heptane (160 mL) for 2 hours. The precipitate was filtered and washed with heptane (50 mL). The cake was dried under vacuum to give tert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12) as a tawny solid (21 .8 g, 87.2% yield). HPLC purity 97.7%. 1 H-NMR (400 MHz, DMS0-d6) d (ppm) = 9.13 (s, 1 H), 7.52 (d, J= 3.67 Hz, 1 H), 6.74 (d, J= 2.2 Hz, 1 H), 6.56 (m, 1 H), 6.50 (d, J= 3.67 Hz, 1 H), 2.45 (s, 3 H), 1 .57 (s, 9 H). LCMS (m/z): negative mode 246.2 [M-1 ]-
Step 3: Synthesis of fert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13, wherein P3 = Boc)
To a mixture of MgCI2 (1 1 .6 g, 1 19.7 mmol) and (CHO)n (5.0 g, 159.6 mmol), in THF (150 ml), under nitrogen, triethylamine (17.8 ml_, 127.7 mmol) was added dropwise and the resulting mixture was stirred at 20-25 °C for 10 minutes. Then, tert-butyl 5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C12) (10.0 g, 39.9 mmol) was added to the mixture. The reaction mixture was heated to 65-70 °C and stirred for 3 hours. The reaction mixture was cooled to 20-25 °C, followed by addition of 2N HCI (70 ml) and isopropyl acetate (150 ml). The mixture was separated and the organic layer was washed with a 5% NaCI solution. Then, the solution was concentrated to give a crude solid. The solid was slurried with ethanol (100 ml.) for 1 hour. The solid precipitate was filtrated, and washed with ethanol (20 ml_). The cake was dried under vacuum to give tert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13) as a tawny solid (7.2 g, 63.9% yield). HPLC purity 96.5%. The filtrate solution was concentrated to 20 mL, then stirred for 1 hour. The solid was filtrated, and washed with ethanol (5 mL). The cake was dried by vacuum to give an additional amount of tert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13) as a tawny solid (1 .1 g, 95.3% assay, 9.5% yield.). HPLC purity 90.5%. 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.69 (s, 1 H), 10.47 (s, 1 H), 7.75 (d, J= 3.35 Hz, 1 H), 7.27 (d, J= 3.55 Hz, 1 H), 6.74 (s, 1 H), 2.51 (s, 3 H), 1 .59 (s, 9 H); LCMS (m/z): negative mode 274.2 [M-1 ]-.
Step 4: Synthesis of fert-Butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III)).
To a suspension of tert-butyl 4-formyl-5-hydroxy-7-methyl-1 H-indole-1 -carboxylate (C13) (6.0 g, 21 .3 mmol) in MeCN (60 mL), 50% K2C03 solution (20 mL) and dimethyl sulfate (2.26 mL, 23.4 mmol) were added. The resulting mixture was stirred at 35-40 °C for 3 hours. The reaction mixture was cooled to 20-25 °C and isopropyl acetate (30 mL) was added. The mixture was then extracted; the water layer was extracted with isopropyl acetate (15 mL), the organic layers were combined and concentrated to give a crude residual. The crude residual was dissolved in isopropyl acetate (60 mL), the solution was washed with a statured NH4CI solution, and then concentrated to give a crude product (6.6 g). The crude was slurried with ethyl acetate / Heptane (100 mL, 1/50) for 3 hours. The solid was filtrated, washed with heptane (20 mL). The cake was dried under vacuum to give tert-butyl 4-formyl-5-methoxy-7-methyl-1 H-indole-1 -carboxylate (Compound of formula (III))
as a pink solid (5.5 g, 87.8% yield). HPLC purity 99.3%. 1 H-NMR (400 MHz, DMSO-d6) d (ppm) = 10.52 (s, 1 H), 7.79 (d, J= 3.67 Hz, 1 H), 7.31 (d, J= 3.67 Hz, 1 H), 7.02 (s, 1 H) , 3.95 (s, 3 H), 2.61 (s, 3 H), 1 .60 (s, 9 H); LCMS (m/z): positive mode 290 [M]+.
Example 9: Synthesis of Compound of formula , or salt thereof (R = methyl).
Method 1 (Pa = Boc and R = methyl): To a vessel were added lr(CO)2acac (1 mg, 0.1 mol%), compound of formula (II) (maleic salt, 3 mmol, 1 .137g), compound of formula (III) (3 mmol, 0.867g) in 9 ml. of degassed ethanol. The autoclave was purged 3 times with nitrogen and 3 times with H2 under stirring (250 RPM). The reactions were run for 24 hours at 75 °C under 20 bar of H2 at 700 RPM. An aliquot of the reaction was diluted in methanol and was analyzed by HPLC. Compound of formula (C15) was obtained after 24 hours in 88% conversion.
Method 2 (Pa = Boc and R = methyl): To a vessel were added lrCI3, xH20 (0.05 mol%, 0.9 mg, anhydrous), compound of formula (II) (maleic salt, 6 mmol, 2.274 g ), compound of formula (III) (6 mmol, 1 .735g) in 12 ml. of degassed ethanol. The autoclave was purged 3 times with nitrogen and 3 times with carbon monoxide (CO) (250 RPM). The autoclave was pressurized with 1 bar of CO and 19 bar of H2 and run for 24 hours at 75 °C under 20 bar of H2 / CO at 700 RPM. An aliquot of the reaction was diluted in methanol and was analyzed by HPLC. Compound of formula (C15) was obtained after 24 hours in 62% conversion.
1H NMR (400 MHz, DMSO-d6) d ppm 8.13 (d, J=8.16 Hz, 2H), 7.77 (br. d, J=7.84 Hz, 2H), 7.62 -7.68 (m, 1 H), 6.85 (s, 1 H), 6.80 (d, J= 3.76 Hz, 1 H), 4.01 (s, 3H), 3.92 (s, 3H), 3.73 (br. s, 1 H), 3.55 – 3.67 (m, 4H), 3.39 – 3.42 (m, 1 H), 2.60 – 2.70 (m, 5H), 1 .99 – 2.02(br. d, 1 H), 1 .82 – 1.90 (m, 2H), 1.74 (s, 9H), 1 .64 – 1 70(m, 1 H), 1 .35 (t, J= 6.97 Hz, 3H).
1. Schubart A, et al. Proc Natl Acad Sci U S A. 2019 Mar 29. pii: 201820892.
Proceedings of the National Academy of Sciences of the United States of America (2019), 116(16), 7926-7931.
//////LNP 023, BDBM160475, ZINC223246892, HY-127105, CS-0093107, LNP023
O=C(O)c1ccc(cc1)[C@@H]4C[C@H](CCN4Cc2c(OC)cc(C)c3nccc23)OCC
LYS 228

LYS228
BOS-228
LYS-228
Molecular Formula, C16-H18-N6-O10-S2
Molecular Weight, 518.4783
(3S,4R)-3-((Z)-2-(2-Ammoniothiazol-4-yl)-2-((1-carboxycyclopropoxy)imino)acetamido)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)azetidine-1-sulfonate
RN: 1810051-96-7
UNII: 29H7N9XI1B
UNII-005B24W9YP
005B24W9YP
Lys-228 trihydrate
2091840-43-4
Yclopropanecarboxylic acid, 1-(((Z)-(1-(2-amino-4-thiazolyl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxo-3-oxazolidinyl)methyl)-1-sulfo-3-azetidinyl)amino)ethylidene)amino)oxy)-, hydrate (1:3)
1-[(Z)-[1-(2-amino-1,3-thiazol-4-yl)-2-oxo-2-[[(3S,4R)-2-oxo-4-[(2-oxo-1,3-oxazolidin-3-yl)methyl]-1-sulfoazetidin-3-yl]amino]ethylidene]amino]oxycyclopropane-1-carboxylic acid;trihydrate
BOS-228 (LYS-228) is a monobactam discovered at Novartis and currently in phase II clinical development at Boston Pharmaceuticals for the treatment of complicated urinary tract infection and complicated intraabdominal infections in adult patients.
The compound has been granted fast track and Qualified Infectious Disease Product (QIDP) designation from the FDA.
In October 2018, Novartis licensed to Boston Pharmaceuticals worldwide rights to the product.
Paper
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00330
Patent
US 20150266867
PATENT
WO 2017050218
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017050218&tab=FULLTEXT
Compound X: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0126]
Step 1: Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate. To a solution of (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetic acid (854 mg, 1.59 mmol) prepared according to published patent application US2011/0190254, Intermediate B (324 mg, 1.75 mmol) and HATU (785 mg, 2.07 mmol) in DMF (7.9 mL) , DIPEA was added (832 μL, 4.77 mmol) . After 1 h of stirring, it was poured into water and extracted with EtOAc. Brine was added to the aqueous layer, and it was further extracted with ethyl acetate (EtOAc) (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo. The crude residue was purified via silica gel chromatography (0-10%MeOH-DCM) to afford the title compound (1.09 g, 97%) as a beige foam. LCMS: R t = 0.97 min, m/z =705.3 (M+1) Method 2m_acidic.
[0127]
Instead of HATU, a variety of other coupling reagents can be used, such as any of the typical carbodiimides, or CDMT (2-chloro-4, 6-dimethoxy-1, 3, 5-triazine) and N-methylmorpholine to form the amide bond generated in Step 1.
[0128]
Step 2: (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid. Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate (1.00 g, 1.42 mmol) in DMF (7.0 mL) at 0 ℃ was treated with SO 3·DMF (448 mg, 2.84 mmol) . After 2 h of stirring at rt, the solution was poured into ice-cold brine and extracted with EtOAc (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo, affording the title compound (assumed quantitative) as a white solid. LCMS: Rt =0.90 min, m/z = 785.2 (M+1) Method 2m_acidic.
[0129]
Step 3: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0130]
[0131]
To a solution of (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid (1.10 g, 1.40 mmol) in DCM (1.5 mL) at 0℃, TFA (5.39 mL, 70.0 mmol) was added, and after 10 minutes, the ice bath was removed. Additional TFA (3.24 mL, 42.0 mmol) was added after 1 hr at rt and the solution was diluted with DCM and concentrated in vacuo after an additional 30 min. Optionally, anisole may be added to the TFA reaction to help reduce by-product formation, which may increase the yield of desired product in this step. The crude residue was purified by reverse phase prep HPLC (XSelect CSH, 30 x 100 mm, 5 μm, C18 column; ACN-water with 0.1%formic acid modifier, 60 mL/min) , affording the title compound (178 mg, 23%) as a white powder. LCMS: R t = 0.30 min, m/z = 518.9 (M+1) Method 2m_acidic; 1H NMR (400 MHz, DMSO-d 6) δ 9.27 (d, J = 9.0 Hz, 1H) 6.92 (s, 1H) 5.23 (dd, J = 9.1, 5.7 Hz, 1H) 4.12-4.23 (m, 3H) 3.72-3.62 (m, 2H assumed; obscured by water) 3.61-3.52 (m, 1H assumed; obscured by water) 3.26 (dd, J = 14.5, 5.9 Hz, 1H) 1.36 (s, 4H) . 1H NMR (400 MHz, D 2O) δ 7.23 (s, 1H) , 5.48 (d, J = 5.8 Hz, 1H) , 4.71-4.65 (m, 1H) , 4.44 (t, J = 8.2 Hz, 2H) , 3.89-3.73 (m, 3H) , 3.54 (dd, J = 14.9, 4.9 Hz, 1H) , 1.65-1.56 (m, 2H) , 1.56-1.46 (m, 2H) . The product of this process is amorphous. Compound X can be crystallized from acetone, ethanol, citrate buffer at pH 3 (50 mM) , or acetate buffer at pH 4.5 (50 mM) , in addition to solvents discussed below.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(4), 748-755.
https://www.sciencedirect.com/science/article/pii/S0960894X18300064
PATENT
WO 2019026004
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019026004&tab=PCTDESCRIPTION
Over the past several decades, the frequency of antimicrobial resistance and its association with serious infectious diseases have increased at alarming rates. The increasing prevalence of resistance among nosocomial pathogens is particularly disconcerting. Of the over 2 million (hospital-acquired) infections occurring each year in the United States, 50 to 60% are caused by antimicrobial-resistant strains of bacteria. The high rate of resistance to commonly used antibacterial agents increases the morbidity, mortality, and costs associated with nosocomial infections. In the United States, nosocomial infections are thought to contribute to or cause more than 77,000 deaths per year and cost approximately $5 to $10 billion annually.
Important causes of Gram-negative resistance include extended-spectrum 13- lactamases (ESBLs), serine carbapenemases (KPCs) and metallo-13-lactamases (for example NDM-1 ) in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis, high-level third-generation cephalosporin (AmpC) 13-lactamase resistance among Enterobacter species and Citrobacter freundii, and multidrug-resistance genes observed in Pseudomonas, Acinetobacter, and Stenotrophomonas. The problem of antibacterial resistance is compounded by the existence of bacterial strains resistant to multiple antibacterials. For example, Klebsiella pneumonia harboring NDM-1 metallo-13- lactamase carries frequently additional serine-13-lactamases on the same plasmid that carries the NDM-1 .
Thus there is a need for new antibacterials, particularly antibacterial compounds that are effective against existing drug-resistant microbes, or are less susceptible to development of new bacterial resistance. Monobactam antibiotic, which is referred to herein as Compound X, is primarily effective against Gram-negative bacteria, including strains that show resistance to other monobactams.
The present invention relates to a process for the preparation of monobactam antibiotic Compound X and intermediates thereof.
More particularly, the present invention relates to a process for the preparation of Compound X
Compound X
also referred to as 1 -(((Z)-(1 -(2-aminothiazol-4-yl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)-1 -sulfoazetidin-3-yl)amino)ethylidene)amino)oxy)cyclopropanecarboxylic acid, or a salt thereof, or a solvate including hydrate thereof.
Patent application number PCT/US2015/02201 1 describes certain monobactam antibiotics. Compound X may be prepared using the method disclosed in PCT/US2015/02201 1 , in particular example 22, and in PCT/CN2016/099482.
A drawback from these processes is that they exhibit a large number of process steps and intermediate nitrogen protection/deprotection steps, reducing the overall yield and efficiency. Furthermore, these processes require several chromatographic purification steps to be carried out in course of the processes. We have found that the preparation of Compound X, as previously prepared on a manufacturing scale, possesses a number of disadvantages, in particular poor handling characteristics.
It would thus be beneficial to develop alternative or improved processes for the production of Compound X that do not suffer from some or all of these disadvantages.
Compound x Compound x
Scheme 1
Preparation of Compound X from Intermediates 22 and 2A
Scheme 3
Examples
The Following examples are merely illustrative of the present disclosure and they should not be considered as limiting the scope of the disclosure in any way, as these examples and other equivalents thereof will become apparent to those skilled in the art in the light of the present disclosure, and the accompanying claims.
Synthesis of Compound 8 (R = benzyl)
1 .50kg oxazolidin-2-one (7b) was charged into the reactor. 7.50kg THF was charged and the stirring started. The mixture was cooled to 10~20°C. 2.18kg potassium fert-butoxide was charged intol 2.00kg THF and stirred to dissolve.
The potassium fert-butoxide solution was added dropwise into the reactor while maintaining the temperature at 10-20 °C. The reaction was stirred for 1 ~2hrs at 10-20 °C after the addition. The solution of 2.36kg methyl-2-chloroacetate (7a) in 3.00kg of THF was added to the reactor while maintaining the temperature at 10-20 °C. The reaction mixture was stirred for 16-18 h at 20-25 °C. The IPC (in process control) showed completion of the reaction. The mixture was centrifuged and the wet cake was washed with 7.50kg THF. The filtrate was concentrated and the crude 7 was provided as reddish brown liquid, which was used for the next step without further purification,
1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.65 – 3.71 (m, 2 H) 3.74 (s, 3 H) 4.02 (s, 2 H) 4.34 – 4.45 (m, 2 H).
The dried reactor was exchanged with N2 three times. 3.71 kg LiHMDS solution in THF/Hep (1 M) and 1 .30kg THF were charged under nitrogen protection. The stirring was started and the solution was cooled to -70—60 °C. The solution of 0.71 kg benzyl acetate (6) in 5.20 kg THF was added dropwisely at -70— 60 °C, and the resulted mixture was stirred for 1 -1 .5 h after the addition. The solution of 0.65kg 7 in 3.90kg THF was added dropwise while maintaining the temperature at -70—60 °C, then stirred for 30-40 minutes. The reaction mixture was warmed to 20-25 °C and stirring was continued for 0.5-1 .0 h. IPC showed 6 was less than1 .0% (Otherwise, continue the reaction till IPC passes). The reaction mixture was poured into 13.65 kg aqueous citric acid below 10 °C. The mixture was stirred for 15-20 minutes after the addition. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (6.50kg * 2). The organic layer was combined, washed by 6.50 kg 28% NaCI solution and dried with 0.65
kg anhydrous MgSC . The mixture was filtered and the wet cake was washed with 1 .30kg EA. The filtrate was concentrated under vacuum to provide crude 8. The crude 8 was stirred in 2.60 kg MTBE at 20-25 °C for 1 -1 .5 h. The mixture was cooled to 0-10 °C and stirred for 1 .5-2.0 h and filtered. The filter cake was washed with 0.65kg pre-cooled MTBE and dried under vacuum (<-0.096Mpa) at 20-25 °C for 12~16hrs till a constant weight to give 513 g of 8 as a white solid, Yield: 45%, HPLC purity 96.4%,1 H NMR (400 MHz, CHLOROFORM-c δ ppm 3.48 – 3.55 (m, 1 H) 3.56 – 3.63 (m, 2 H) 3.66 – 3.74 (m, 1 H) 4.17 – 4.26 (m, 2 H) 4.31 – 4.44 (m, 2H) 5.12 – 5.24 (m, 2 H) 7.30 – 7.44 (m, 5 H).
Synthesis of Compound 9 (R = benzyl)
The dried reactor was charged with 3.75kg HOAc and 1 .50 kg 8. The stirring was started and the reaction mixture was cooled to 0-5 °C. 3.53kg aqueous NaN02 was added dropwise at 0-10 °C, and the reaction mixture was stirred for 15-30 minutes after the addition. IPC showed 8 was less than 0.2%. The reaction mixture was treated with 7.50kg EA and 7.50 kg water. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (7.50kg * 2). The organic layers were combined, washed with 7.50 kg 28% NaCI solution, and concentrated under vacuum to provide crude 9. The crude 9 was slurried with 5.25 kg water at 10-20 °C for 3~4hrs, and filtered. The wet cake was washed with 1 .50kg water. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .44 Kg of 9, yield: 86.9%, HPLC purity 92.9%,1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.60 – 3.76 (m, 2 H) 4.44 (t, J=8.07 Hz, 2 H) 4.60 (s, 2 H) 5.25 – 5.41 (m, 2 H) 7.30 – 7.43 (m, 5 H) 1 1 .62 (br s, 1 H).
Synthesis of Compound 9a (R = benzyl)
9
The dried reactor was charged with 0.58 kg Zn, 4.72kg (Βο Ο, 6.00 kg water, 1 .20 kg NH4CI and 6.00kg THF. The reaction mixture was stirred and heated to 50-55 °C. The solution of 0.60 kg 9 in 4.20kg THF
was added dropwisely while maintaining the temperature at 50-55 °C. The reaction mixture was stirred for 0.5-1 .Ohrs after the addition. IPC showed 9 was less than 0.1 %. The reaction mixture was treated withl .50 kg ethyl acetate and stirred for 15-20 minutes. Phase was separated and the water layer was extracted by1 .50 kg ethyl acetate. The organic layers were combined, washed with 6.00 kg 28% NaCI solution and concentrated under vacuum to provide crude 9a. The crude 9a was stirred with 3.60kg*2 n-heptane to remove excess (Βο Ο. The residue was purified by silica gel chromatography column eluted with ethyl acetate: Heptane= 1 :1 to provide crude 9a solution. The solution was concentrated under reduced pressure to obtain crude 9a. The crude 9a was slurried with 1 .80 kg MTBE for 2.0-3. Ohrs, filtered, and the wet cake was washed with MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 50-55 °C for 16-18 h till a constant weight to give 392 g of 9a as a white solid, Yield: 51 %, HPLC purity 98.1 %,1H NMR (400 MHz, DMSO-cfe) δ ppm 1.17 – 1 .57 (m, 9 H) 3.39 – 3.61 (m, 2 H) 4.20 – 4.45 (m, 3 H) 5.10 – 5.32 (m, 3 H) 5.75 (s, 1 H) 7.38 (br s, 5 H) 7.75 – 7.99 (m, 1 H).
Synthesis of compound (VII) (R = benzyl, X = CI)
9a VII
The dried reactor was charged with 13.0kg HCI in IPA and the stirring was started. 1 .33 kg 9a was charged in portions at 20-25 °C. The mixture was stirred at 20-25 °C for 3-4 h. IPC showed 9a was less than 0.1 %. The reaction solution was concentrated under vacuum 40-45 °C. The residue was treated with 21 .58kg MTBE at 20-25 °C for 3-4 h. The mixture was filtered and the wet cake was washed with 2.60kg MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .045 Kg of compound VII (R = benzyl, X = CI) as a yellow solid, Yield: 93.7%, HPLC purity 99.2%,1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.16 – 3.74 (m, 3 H) 4.10 – 4.35 (m, 4 H) 5.09 – 5.39 (m, 2 H) 7.27 – 7.60 (m, 5 H) 8.72 (br s, 2 H).
Synthesis of compound (Vile) (R = benzyl)
VII Vile
To an autoclave (3L) were added VII (R = benzyl, X = CI) (100 g, 304.2 mmol, 1 .0 equiv.), DCM (2650 g, 26.5 equiv., w/w) and (S-BINAP)RuCl2 (2.4 g, 3.04 mmol, 0.01 equiv.), successively. Air in the autoclave was replaced with N2 5 times. N2 in the autoclave was was replaced with H2 5 times. The solution was stirred with 250-260 r/min and H2 (2.1 ±0.1 MPa) at 40±5°C for 24 h. The reaction mixture was filtered, and the filter cake was washed with DCM (400 g, 4.0 equiv., w/w). The filter cake was slurried with IPA (785 g, 7.85 equiv., w/w) and H2O (40 g, 0.4 equiv., w/w) overnight (18-20 h). The mixture was filtered. The filter cake was washed with IPA (200 g, 2.0 equiv., w/w) and dried at 45±5°C overnight (18-20 h). Vile (R = benzyl) was obtained as off-white solid, 80.4 g, 79.9% yield, 95.5% purity, 97.6% de, >99.5% ee. 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.34-3.38 (m, 2 H) 3.50-3.52 (m, 1 H) 3.60-3.62 (m, 1 H) 4.18-4.24 (m, 4 H) 5.23 (s, 2H) 6.16 (s, 1 H) 7.32 (m, 5H) 8.74 (s, 1 H).
Alternative synthesis of compound 9a (R = benzyl)
5b
Mg(OtBu)2
To a flask was added 5a (1 .88 g, 12.93 mmol), THF (40 mL), and CDI (2.20 g, 13.58 mmol) at 25 °C. The mixture was stirred for 3 h. To the reaction mixture was added 5b (2.00 g, 6.47 mmol), and Mg(OfBu)2 (2.21 g, 12.93 mmol). The reaction mixture was stirred at 25 °C for 24 h. The reaction mixture was concentrated under vacuum to remove most of the THF solvent. To the concentrated solution was added MTBE (40 mL), followed by addition of an aqueous solution of HCI (1 M, 60mL) to adjust to pH = 2-3. Two phases were separated, and the water phase was extracted with MTBE (20 mL). The combined organic phase was washed with aqueous NaHCC (5%, 50 mL) and brine (20%, 40 mL). The organic phase was concentrated to a weight of -19 g, and a lot of white solid was obtained in the concentration process. The suspension was cooled to 0 °C, and filtered. The filter cake was washed with cold MTBE (5 mL) and dried under vacuum to obtain product 9a (1 .6g, 63% yield).
Synthesis of compound (Vile) (R = benzyl, PG = Cbz)
Vile Vile
To a flask (5 L) were added Vile (R = benzyl) (140 g, 423.2 mmol, LOequiv.), H20 (1273 g, 9.09 equiv., w/w) and toluene (2206 g, 15.76 equiv., w/w). The solution was stirred and cooled to 0-5 °C with ice bath. Then NaHCOa (78.4 g, 933 mmol, 2.22 equiv.) was added and CbzCI (89.6 g, 527 mmol, 1 .24 equiv.) was dropped into the stirring solution, respectively. The solution was stirred at 30±5 °C overnight (18-20 h). Heptane (3612 g, 25.8 equiv., w/w) was added dropwise to the stirring solution over 1 h at 20-30 °C. The mixture was filtered. The filter cake was washed with heptane (280 g, 2.00 equiv., w/w) and MTBE (377 g, 2.69 equiv., w/w), respectively. The filter cake was dried at 45±5°C overnight (18-20 h). Vile (R = benzyl, PG = Cbz) was obtained as an off-white solid, 169.4 g, 93% yield, 96.7% purity, 98% de, >99.5% ee, 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.23-3.24 (m, 1 H) 3.30 (m, 1 H) 3.51 -3.55 (m, 2 H) 3.99 (s, 1 H) 4.17-4.21 (m, 3 H) 5.02-5.03 (m, 2H) 5.12 (s, 2H) 5.46-5.48 (d, 1 H) 7.33-7.36 (m, 10H) 7.75-7.73 (d, 1 H).
Synthesis of compound (IV) (PG = Cbz)
Vile IV
Vile (R = benzyl) (220 g, 513.5 mmol, 1 .0 equiv.) was dissolved in THF (1464g, 6.65 equiv., w/w). The solution was filtered. The filter cake was washed with THF (488g, 2.22 equiv., w/w). The filtrate (Vile) was collected. To an autoclave (3L) were added the filtrate (Vile). The reactor was cooled down to -75 – -65 °C with dry-ice/EtOH bath, and bubbled with NH3 for not less than 4 h. Then the solution was stirred at 25±5 °C with NH3 (0.5-0.6 MPa) for 24 h. The autoclave was deflated to release NH3. The reaction solution was concentrated with a rotary evaporator to remove THF until the residue was around 440 g. The residue was slurried with EA (2200 g, 10 equiv., w/w) at 70±2 °C, then cooled to 25±5 °C and stirred for 16-18 h. The mixture was filtered. The filter cake was washed with EA (440 g). The filter cake was slurried with EA (1320 g, 6.00 equiv. w/w), and the temperature was raised to 70±2 °C, then cooled to 25±5 °C and stirred for 16-20 h. The mixture was filtered. The filter cake was washed with EA, and dried at 50±5 °C overnight (18-20 h). IV (PG = Cbz) was obtained as off-white solid, 141 g, 81 .5% yield, 99.1 % purity, >99.5% assay, 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.12 – 3.23 (m, 2 H) 3.31 (br s, 1 H) 3.56 (t, J=8.01 Hz, 2 H) 3.88 (quin, J=6.02 Hz, 1 H) 3.93 – 4.03 (m, 1 H) 4.20 (t, J=8.01 Hz, 2 H) 5.02 (s, 2 H) 5.27 (d, J=5.87 Hz, 1 H) 7.12 (s, 1 H) 7.22 – 7.45 (m, 5 H).
Synthesis of compound (III) (PG = Cbz, LG = S02CH3)
IV III
To a flask was added IV (PG = Cbz) (14.00 g, 41 .50 mmol, 1 .00 equiv), and dry 1 , 2-dimethoxyethane (300 mL) under N2. The mixture was stirred at -5°C ~ 0°C for 1 h to obtain a good suspension. MsCI (7.89 g, 68.89 mmol, 5.33 mL, 1 .66 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min, and Et3N (12.60 g, 124.50 mmol, 17.26 mL, 3.00 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min side to side. The reaction mixture was stirred for additional 5 min at -5°C ~ 0°C, and was quenched with water (6 mL). The reaction mixture was concentrated to remove DME. The solid was slurried in water (250 mL) and MTBE (125 mL) for 1 h. The solid was collected by filtration, and then slurried in water (250 mL) for 1 hr. The solid was collected by filtration, and washed with water (25 mL) to give white solid. The solid was slurried in EA (150 mL) and dried in vacuum at 60°C for 24 h to give III (PG = Cbz, LG = SO2CH3) (15.00 g, 36.1 1 mmol, 87.01 % yield), 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.17 (s, 3 H) 3.26 (br d, J=15.04 Hz, 1 H) 3.47 – 3.57 (m, 1 H) 3.64 (br d, J=6.36 Hz, 2 H) 4.22 (br dd, J=17.79, 8.50 Hz, 2 H) 4.50 (br s, 1 H) 4.95 – 5.17 (m, 3 H) 7.21 – 7.56 (m, 5H) 7.43 (s, 1 H) 7.63 – 7.89 (m, 2 H).
Synthesis of compound II (PG = Cbz, LG = SO2CH3, M+ = NBu4+)
O OMs o CISO3H, 2-picoline – ° O ?yO
HN Bu4NHS04< NHCbz
“Cbz
III II
To a flask was added 2-picoline (1 1 .50 g, 12.23 mL) and DMF (10 mL). The solution was cooled to 5 SC, followed by slow addition of chlorosulfonic acid (7.20 g, 4.14 mL). The temperature was increased to 20 SC. Ill (PG = Cbz, LG = SO2CH3) (5.13 g, 12.35 mmol) was added to the reaction mixture. The reaction mixture was heated to 42 SC for 18h. IPC (in process control) showed complete conversion of starting material. The reaction was cooled to 20 SC and dropwise added to a solution of tetrabutylammonium hydrogen sulfate (4.6 g, 13.6 mmol) in the mixed solvents of dichloromethane (100 mL) and water (100 mL) at 5SC. The phases were separated and the water phase was extracted with dichloromethane (2*50mL). The combined organic phase was washed with water (5*100mL). The organic phase was concentrated to dryness and purified by column chromatography (dichloromethane/methanol = 15/1 v/v) to afford II (PG = Cbz, LG = SO2CH3, M+ = NBii4+) (8.4 g, 92.30%), 1 H NMR (400 MHz, CHLOROFORM-c/) δ ppm 0.99 (t, J=7.34 Hz, 12 H) 1 .36 – 1 .50 (m, 8 H) 1 .54 – 1 .76 (m, 8 H) 3.15 (br d, J=8.31 Hz, 2 H) 3.21 – 3.35 (m, 8 H) 3.47 (br dd, J=14.73, 7.27 Hz, 1 H) 3.54 – 3.65 (m, 1 H) 3.67 – 3.81 (m, 2 H) 4.17 – 4.32 (m, 1 H) 4.39 – 4.62 (m, 1 H) 4.74 (br s, 1 H) 5.1 1 (s, 3 H) 5.32 – 5.50 (m, 1 H) 6.47 (br s, 1 H) 7.29 – 7.47 (m, 5 H) 8.69 – 8.94 (m, 1 H).
Synthesis of compound (IA)
A solution of II (PG = Cbz, LG = SO2CH3, M+ = NBu4+) (4.0 g) in dichloromethane (38 mL) was pumped to tube A at rate of 2.0844 mL/min, and a solution of KHCO3 (3.0 g) in water (100 mL) was pumped to tube B at a rate of 1 .4156 mL/min side to side. These two streams were mixed in a cross-mixer then flowed to a tube coil that was placed in an oil bath at 100 °C. The residence time of the mixed stream in the coil was 2 min. The reaction mixture flowed through a back-pressure regulator that was set at ~ 7 bars, and was collected to a beaker. After completion of the collection, two phases was separated. The organic phase was concentrated to dryness. The residue was slurried in ethyl acetate (5 mL). The solid was filtered and the filter cake was dried to give IA (2.6 g, 75%),
1H NMR (400 MHz, CHLOROFORM-c/) δ ppm 1.00 (t, J=7.27 Hz, 12 H) 1 .42 (sxt, J=7.31 Hz, 8 H) 1 .62 (quin, J=7.83 Hz, 8 H) 3.13 – 3.39 (m, 8 H) 3.54 – 3.69 (m, 2 H) 3.81 (dd, J=14.98, 2.51 Hz, 1 H) 3.96 – 4.13 (m, 1 H) 4.22 – 4.47 (m, 3 H) 4.99 – 5.23 (m, 3 H) 6.42 (br d, J=9.29 Hz, 1 H) 7.26 – 7.44 (m, 5 H).
Synthesis of compound 2A
Step 1
To a stirring solution of compound 16b (2 g, 10.14mmol, 1 .0 eq) in DMF (20 ml_) was added CS2CO3 (5.29g, 16.22 mmol, 1 .6 eq), then the resulting solution was stirred at room temperature for 10mins, then compound 16a (5.27g, 20.28mmol, 2eq) was added dropwise to the mixture for 2 minutes, then the resulting solution was stirred for another 2 hours. TLC showed the starting material was consumed completely. The mixture was added with water (60mL) and extracted with MTBE (20mL*3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The crude was slurried in heptane to give 1 .65 g 16 as a white solid (Yield: 57%), 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.48-7.28 (m, 10 H), 5.00-4.96 (t, J=6.0 Hz, 1 H), 3.81 (s, 3H), 3.44-3.42 (m, 2H), 2.40-2.37 (m, 2H).
Compound 16 (1 g, 2.66mmol, 1 eq) was dissolved in THF (20mL) under Nitrogen, and cooled to -40 °C. NaHMDS (1 .6mL, 2.0M THF solution, 1 .2 eq) was added dropwise. The reaction was stirred for 1 h at -40 °C. HPLC indicated the reaction was finished. The reaction was quenched with 10% Citric acid, extracted with MTBE (25 ml_ x 2). The combined organic layers were washed with brine (30 ml_), dried with Na2S04, filtered and concentrated to give 17 as a yellow solid, which was used for the next step without purification (assay yield: 65%); 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.27-7.13 (m, 10 H), 3.46 (s, 3H), 1 .21 -1 .17(dd, J=7.2, 10.4 Hz, 2H ); 1 .14-1 .1 1 (dd, J=7.2, 10.4 Hz, 2H).
Step 3
Compound 17 (100 mg) was dissolved in methanol (5 mL) and 2.0 M HCI IPAC solution (5 mL). The solution was heated at 45 °C for 3 days. HPLC indicated the reaction was finished. The reaction was cooled to room temperature and was diluted with 10 mL water. The reaction mixture was washed with MTBE (10 mL x 2), organic layer was discarded and the aqueous layer was concentrated to give compound 2A HCI (32 mg, 62% yield), 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.80-3.44 (br, 4H), 1 .56 (s, 2H), 1 .38 (s, 2H).
Step 4
To a solution of 2A HCI (0.70 g, 4.57 mmol) in methanol (5 mL) was added triethylamine (1 .26 mL, 9.14 mmol) at room temperature. The solution was stirred for 20 min, and the solvent was removed under vacuum. To the residue was added IPAC (10 mL) leading to precipitation. The solid was filtered, and the filtrate was concentrated to provide 2A (0.50g, 94% yield) containing ca. 6 wt% Et3N-HCI.
Synthesis of Compound X from compound of formula (I), (IA)
Compound x
To a flask was charged 21 (1 .00 g, 68.43 wt%, 2.50 mmol) and DMF (10 mL). The suspension was cooled to -20 °C, to which was added diphenylphosphinic chloride (0.52 mL, 2.75 mmol). The solution was stirred at -20 °C for 30 min, followed by addition of a mixed solution of (IA) (1 .52g, 3.00 mmol) and triethylamine (0.52 mL, 3.76 mmol) in DMF (2mL). The reaction mixture was stirred at 20 °C for 20 h, followed by addition of MTBE (20 mL). The reaction mixture was adjusted to pH = 2-3 using aqueous HCI solution (37%). To the mixture was added isopropanol (100 mL). The resulting mixture was stirred for 4 h to obtain a suspension. The suspension was filtered and the filter cake was dried under vacuum to afford crude 22 (1 .17 g). The crude 22 was slurried in a combined solvent of THF/H2O (= 12 mL / 3mL), and filtered to afford 22 (0.744 g, 75 wt% by Q-NMR, 53.3% yield). 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.47 – 3.55 (m, 2 H) 3.59 – 3.63 (m, 2 H) 4.13 – 4.21 (m, 3 H ) 5.05 (dd, J=8.8, 5.6 Hz, 1 H) 8.22 (s, 1 H) 9.73 (d, J=8.7 Hz, 1 H).
To a suspension of 22 (580 mg, 75 wt%, 1 .037 mmol) in DMAC (1 .5 mL) was added 2A (214.3 mg, 85 wt%, 1 .556 mmol). The reaction was stirred at 25 °C for 3 days, and in process control showed 22, Compound X = 4/96, and Z/E = 91 /9. the mixture was slowly added into 15ml acetone to precipitate yellowish solid. The reaction mixture was filtered to afford Compound X (0.7 g, 34 wt% by QNMR, 44% yield).
Synthesis of compound 3 (R2 = CH(Ph)2)
R2 = CH(Ph)2
2-(2-aminothiazol-4-yl)-2-oxoacetic acid (Y) (10.00 g, 47.93 mmol) and compound W (R2 = CH(Ph)2) (13.31 g, 46.98 mmol) were suspended in DMAC (40 mL), followed by addition of triethylamine (5.01 mL, 35.95 mmol). The reaction mixture was stirred at 20 °C for 5 h. HPLC showed completion of the reaction, and Z/E
= 97/3. To the reaction mixture was added water (120 mL) with stirring. The mixture was stirred for 20 min to obtain a suspension. The suspension was filtered and the filter cake was washed with water (50 mL).
The filter cake was slurried in a combined solvent of THF/ethyl acetate (50 mL / 50 mL) at 60 °C and cooled to 20 °C. The solid was filtered and dried at 50 °C for 3 h to get 3 (R2 = CH(Ph)2) (19.5 g, 88% yield). 1H
NMR (400 MHz, DMSO-cfe) δ ppm 1.37 -1 .42 (m, 2 H) 1 .44 – 1 .49 (m, 2 H) 6.87 (s, 1 H) 6.94 (s, 1 H) 7.22
– 7.30 (m, 6 H) 7.45 – 7.49 (m, 4 H).
Alternative Synthesis of Compound X from compound of formula (I), (IA)
Compound x
IA (40.14 g, 62.63 mmol) was dissolved in methanol (200 ml_), followed by addition of Pd/C (10%, 1 .1 g). The reaction mixture was maintained under hydrogen atmosphere (1 -2 bar) at 20 °C for 24 h. In process control showed completion of the reaction. The reaction mixture was filtered. The filtrate was concentrated to give an oil of IB (M+ = NBu4+) (58.20 g, 55 wt% by Q-NMR, 100% yield). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 0.93 (t, J=7.3 Hz, 12 H) 1 .23 – 1 .36 (m, 8 H) 1 .57 (m, 8 H) 2.99 – 3.28 (m, 8 H) 3.37 (dd, J=14.3, 7.5 Hz, 1 H) 3.65 – 3.70 (m, 3 H) 3.84 – 3.88 (m, 1 H) 4.08 (d, J=5.6 Hz, 1 H) 4.18 – 4.22 (m, 2 H).
3 (R2 = CH(Ph)2) (0.95 g, 2.17 mmol) was dissolved in THF (20 ml_). To the solution was added /V-methyl morpholine (0.77 g, 7.60 mmol) and 2-chloro-4,6-dimethoxy-1 ,3,5-triazine (0.57 g, 3.26 mmol). The reaction mixture was stirred at 20 °C for 1 h followed by addition of IB (M+ = NBu +) (2.70 g, 48.98 wt%, 2.61 mmol). The reaction was stirred at 20 °C for 5 h. In process control showed completion of the reaction. To the reaction mixture was added ethyl acetate (20 ml_). The organic phase was washed with brine (10 ml_). Solvent was removed. Acetone (40ml) was added to dissolve residue. TFA (1 .24 g, 10.86 mmol) dissolved in acetone (3 ml) was added slowly. The white solid was filtered and washed by acetone (10 ml) two times. Dried at 40 °C for 5h to get compound 4 (R2 = CH(Ph)2). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 1 .49 – 1 .55 (m, 4 H) 3.27 (dd, J=14.4, 6.2 Hz, 1 H) 3.49 – 3.65 (m, 2 H) 3.71 (dd, J=14.4, 6.2 Hz, 1 H) 4.04 – 4.10 (m, 1 H) 4.07 (dd, J=16.0, 8.6 Hz, 1 H) 4.17 (dd, J=1 1 .8, 6.0 Hz, 1 H) 5.28 (dd, J=9.0, 5.7 Hz, 1 H) 6.88 (s, 1 H) 7.03 (s, 1 H) 7.18 – 7.32 (m, 6 H) 7.43 (m, 4 H) 9.45 (d, J=9.0 Hz, 1 H).
Crude 4 (R2 = CH(Ph)2) (2.13 g) was dissolved in dichloromethane (20 ml_). The solution was cooled to 0 °C. To the solution was added anisole (0.68 ml_, 6.24 mmol) and trifluoroacetic acid (2.16 ml_, 28.08 mmol). The reaction was warmed to 20 °C, and stirred for 15 h. In process control showed completion of the
reaction. The aqueous phase was separated and added to acetone (40 mL) to obtain a suspension. The suspension was filtered to afford Compound X (0.98 g, 54.5% yield over two steps). 1 H NMR (400 MHz, DMSO-c/e) δ ppm 1.40 (m, 4 H) 3.26 (dd, J=14.4, 6.0 Hz, 1 H) 3.54 – 3.69 (m, 3 H) 4.14 – 4.21 (m, 3 H) 5.25 (dd, J= 8.9, 5.7 Hz, 1 H) 7.02 (s, 1 H) 9.38 (d, J=9.0 Hz, 1 H).
REF
Synthesis and optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae – Identification of LYS228
57th Intersci Conf Antimicrob Agents Chemother (ICAAC) (June 1-5, New Orleans) 2017, Abst SATURDAY-297
//////////////LYS228, LYS 228, BOS-228, LYS-228, monobactam, Novartis, phase II, Boston Pharmaceuticals, complicated urinary tract infection, complicated intraabdominal infections, fast track, Qualified Infectious Disease Product, QIDP,
Nc1nc(cs1)\C(=N\OC2(CC2)C(=O)O)\C(=O)N[C@H]3[C@@H](CN4CCOC4=O)N(C3=O)S(=O)(=O)O
Islatravir (MK-8591, EFdA)
Islatravir (MK-8591, EFdA)
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
- Molecular FormulaC12H12FN5O3
- Average mass293.254 Da
- 865363-93-5
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine
Adenosine, 2′-deoxy-4′-C-ethynyl-2-fluoro-
(2R,3S,5R)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol
2′-deoxy-4′-c-ethynyl-2-fluoroadenosine
865363-93-5 [RN]
9H-Purin-6-amine, 9-(2-deoxy-4-C-ethynyl-β-D-erythro-pentofuranosyl)-2-fluoro-
E2FDA
(2R,3S,5R)-5-(6-amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)oxolan-3-ol
MK-8591
UNII:QPQ082R25D
FDA APPROVED 2026, 4/20/2026, doravirine and islatravir, Idvynso
To treat HIV-1 infection (as a complete regimen) in adults to replace the current antiretroviral regimen in those who are virologically-suppressed on a stable antiretroviral regimen with no history of virologic treatment failure and no known substitutions associated with resistance to doravirine
Islatravir is known to be a nucleoside reverse transcriptase inhibitor, useful for treating HIV-1 and -2 infection and AIDS.
Islatravir (MK-8591, EFdA), useful for the treatment of eg HIV, AIDS and related diseases.
Merck & Co and Idenix , under license from Yamasa Shoyu , are developing islatravir, a nucleoside reverse transcriptase inhibitor, for the oral prevention and treatment of HIV-1 and HIV-2 infection; in July 2019, data from a phase IIb trial in patients with HIV-1 infection were presented.In August 2015, Merck licensed Codexis ‘ CodeEvolver® protein engineering platform technology to develop enzymes for use in the manufacture of the pharmaceutical products such as islatravir.
Islatravir (4′-ethynyl-2-fluoro-2′-deoxyadenosine, EFdA, or MK-8591) is an investigational drug for the treatment of HIV infection.[1]It is classified as a nucleoside reverse transcriptase translocation inhibitor (NRTTI).[2] Merck is developing a subdermal drug-eluting implant to administer islatravir.[3][4]
Biological activity
Islatravir has activity against HIV in animal models,[5] and is being studied clinically for HIV treatment and prophylaxis.[6] Islatravir is a nucleoside analog reverse transcriptase translocation inhibitor that unlike other such inhibitors, inhibits HIV through multiple mechanisms,[5] providing rapid suppression of the virus, when tested in macaques and mice.[7] Nevertheless, there are HIV strains resistant to islatravir and research is ongoing.[8]
PATENTS
WO2020014046 ,
PATENT
WO2020014047
PATENT
WO2020014050 (assigned to Codexis ), covering engineered phosphopentomutase (PPM) enzymes, useful in the synthesis of pharmaceutical compounds including islatravir.
PATENT
WO-2020014041
4’-Ethynyl-2’-deoxy nucleoside analogs are known for activity against HIV, AIDS and related diseases.
One example of a 4’-ethynyl nucleoside analog is 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA, also known as MK-8591) which is a nucleoside reverse transcriptase translocation inhibitor that blocks HIV-l and SIV viral replication in vitro (Kawamoto, A., Kodama, E., Sarafianos S. F. et al, Int. J. Biochem. Cell Biol.; 40(l l):24lO-2O [2008]; Ohrui, H., Kohgo, S., Hayakawa, H. et al, Nucleosides, Nucleotides & Nucleic Acids, 26, 1543-1546
[2007]) and in vivo (Hattori, S., Ide, K., Nakata, H. et al. Antimicrobial. Agents and
Chemotherapy, 53, 3887-3893 [2009]). EFdA is claimed in US Patent No. 7,339,053 (referred to in the‘053 patent as 2,-deoxy-4’-C-ethynyl-2-fluoroadenosine). EFdA has the following chemical structure:
EFdA is metabolized in cells to its active triphosphate anabolite which inhibits HIV reverse transcriptase. In contrast to nucleoside reverse transcriptase inhibitors (NsRTIs) and nucleotide reverse transcriptase inhibitors (NtRTIs) currently available for the treatment of HIV infection which lack a 3′-OH group to block incorporation of incoming nucleotide, EFdA retains a 3′ OH group and acts as a chain terminator by preventing translocation of the primer template in the reverse transcriptase (RT) active site and preventing binding of incoming
deoxyribonucleotide triphosphates (dNTPs). In addition, the pucker of the modified ribose ring of EFdA is believed to contribute to inhibition of reverse transcriptase by placing the 3′-OH in a vector in which phosphotransfer from the incoming nucleotide is inefficient. (Michailidis E, et ak, Mechanism of inhibition of HIV-l reverse transcriptase by 4’-ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, J Biol Chem 284:35681-35691 [2009]; Michailidis E, et ak, 4’-Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) inhibits HIV-l reverse transcriptase with multiple mechanisms, J Biol Chem 289:24533-24548 [2014] ).
In in-vitro HIV replication assays, EFdA is a potent antiretroviral and exhibits comparable antiviral activity against clinical isolates across all subtypes that have been evaluated. It is rapidly anabolized in both lymphoid derived cell lines and in peripheral blood mononuclear cells to the active triphosphate in vitro, and the intracellular half-life of EFdA Triphosphate (EFdA- TP) exceeds 72 hrs. (Stoddart, C. A., Galkina, et ak, Oral Administration of the Nucleoside EFdA (4’-Ethynyl-2-Fluoro-2’-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque, Antimicrob Agents Chemother, 2015 Jul; 59(7): 4190-4198, Published online 2015 May 4).
EFdA has been shown to have efficacy in animal models of HIV infection including humanized mouse models and an SIV infected rhesus macaque model. Pharmacokinetic studies of orally administered EFdA in mouse and rhesus monkey have demonstrated rapid absorption and high plasma concentrations. A long intracellular half-life was demonstrated by the fact that isolated peripheral blood mononuclear cells from the rhesus macaque were refractory to SIV infection 24 hr after drug administration. (Ibid.)
Previous syntheses of 4’-ethynyl nucleoside analogs including EFdA suffer from modest stereoselectivity in the formation of the C-N bond between the ethynyl-deoxyribose sugar and the 2-fluoroadenine (also referred to as 2-fluoro-9H-purin-6-amine) nucleobase. The previous syntheses also require protecting groups to carry out the glycosylation reaction which reduces the efficiency of the syntheses.
The synthesis described in Kei Fukuyama, et ak, Synthesis of EFdA via a
Diastereoselective Aldol Reaction of a Protected 3-Keto Furanose, Organic Letters 2015, 17(4), pp. 828-831; DOI: 10.102 l/ol5036535) is a l4-step synthesis from D-glucose diacetonide that uses diastereoselective reactions to set the three stereocenters. The stereochemistry of the anomeric center is controlled by having a 2′-acetoxy directing group that is subsequently removed by hydrolysis and deoxygenation. This route requires 4 chromatographic purifications, and the stoichiometric use of a toxic organotin reagent for late-stage deoxygenation.
In another route (see Mark McLaughlin, et al., Enantioselective Synthesis of 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) via Enzymatic Desymmetrization, Organic Letters 2017, 19 (4), pp. 926-929), the fully-substituted 4′- carbinol is generated stereoselectively with an enzymatic desymmetrization. The 3 ‘-stereocenter is set with a catalytic asymmetric transfer hydrogenation, and the anomeric 1 ‘-linkage is established in modest stereoselectivity using substrate control, with an upgrade in stereochemical purity achieved by crystallization of an intermediate. This process requires 15 steps, requires the use of several protecting groups and generates the glycosyl linkage between the nucleobase and sugar fragments in low
stereoselectivity (1.8: 1).
A l2-step synthesis for making EFdA from R-glyceraldehyde acetonide is described in Kageyama, M., et al., Concise Synthesis of the Anti-HIV Nucleoside EFdA, Biosci. Biotechnol. Biochem, 2012 , 76, pp. 1219 -1225; and Enantioselective Total Synthesis of the Potent Anti-HIV Nucleoside EFdA, Masayuki Kageyama, et al., Organic Letters 2011 13 (19), pp. 5264-5266 [DOL 10.1021 / ol202116k] . The syntheses use the chiral starting material to set the 3′-stereocenter with moderate diastereoselectivity. After chromatographic separation of stereoisomers, the new stereocenter is used to guide a diastereoselective alkyne addition to set the fully-substituted 4′-stereocenter. The anomeric 1 ‘-position is established with little stereocontrol and requires chromatography to separate the anomers. This route requires chromatographic separation of diastereoisomers at two different stages and starts from an expensive chiral starting material.
Kohgo, S., et al., Design, Efficient Synthesis, and Anti-HIV Activity of 4′-C-Cyano- and 4′-C-Ethynyl-2′-deoxy Purine Nucleosides, Nucleosides, Nucleotides and Nucleic Acids, 2004, 23, pp. 671-690 [ DOL 10.1081/NCN-120037508] describes a synthetic route that starts from an existing nucleoside and modifies both the sugar and nucleobase portions. It is an 18-step synthesis starting from 2-amino-2′-deoxy adenosine with a low 2.5% overall yield.
It is known that enzymes such as purine nucleoside phosphorylase (PNP, EC 2.4.2.1) can form the glycosyl linkage in nucleosides and nucleoside analogs in high stereoselectivity and without the use of protecting groups. See for example the review: New Trends in Nucleoside Biotechnology, Mikhailopulo, I. A., Miroshnikov, A.I,. Acta Naturae 2010, 2, pp. 36-58.
However, the current scope of the sugar fragments capable of undergoing reaction catalyzed by PNP has been limited to the a- 1 -phosphates of natural ribose and deoxyribose along with a small number of analogs with small H, NH2, or F substituents at the C2’ and C3’ positions and replacements of the C5’ OH group. There have been no reports of successful glycosylation catalyzed by PNP using sugars with carbon substituents on the ring or any substitution at the C4’ position.
Access to the ribose and deoxyribose a- 1 -phosphate substrates for the PNP-catalyzed glycosylation has been demonstrated by translocation of the phosphate group from the 5’-hydroxyl to G -hydroxyl position with the enzyme phosphopentomutase (PPM, EC 5.4.2.7) (see Mikhailopulo, I. A., et al. supra). However, the scope of the sugars for which PPM is capable of catalyzing this reaction has been limited to ribose, arabinose, 2-deoxyribose, and 2,3-dideoxyribose. No examples have been reported of successful reaction with sugar phosphates containing any additional substituents.
Deoxyribose phosphate aldolase (DERA, EC 4.1.2.4) enzymes are known to catalyze the aldol addition of acetaldehyde to other short-chain aldehydes (see review: Stephen M. Dean, et al., Recent Advances in Aldolase-Catalyzed Asymmetric Synthesis, Adv. Synth. Catal. 2007, 349, pp. 1308 – 1320; DOI: 10. l002/adsc.200700115). However, no examples have been reported with aldehydes bearing a fully substituted carbon a to the aldehyde.
ETS Patent 7,229, 797 describes the formation of deoxyribonucleosides from the natural unsubstituted deoxyribose 1 -phosphate by use of purine nucleoside phosphorylase (PNP) and additionally using enzymes such as sucrose phosphorylase to remove the inorganic phosphate byproduct and drive the equilibrium. It does not disclose enzyme engineering for the creation of PNP enzymes that can generate nucleosides from the unnatural 4-ethynyl-D-2-deoxyribose 1-phosphate, nor that through engineering of PPM and DERA enzymes to act on unnatural substrates, 4-ethynyl-D-2-deoxyribose 1 -phosphate can be generated.
In view of the difficult and lengthy synthetic options developed to date for producing 4’-ethynyl nucleoside analogs, it would be desirable to develop an improved enzymatic synthesis for 4’-ethynyl nucleoside analogs such as EFdA that reduces the number of process steps, minimizes the use of protecting groups, improves the stereoselectivity of glycosylation and avoids the use of toxic materials.
Surprisingly, it has been found that PPM enzymes have some activity with the 3-atom ethynyl substituent at the 4’ position on ribose and that the PPM enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a reaction for
isomerization of
4-ethynyl-D-2-deoxyribose 5-phosphate (6) to 4-ethynyl-D-2-deoxyribose 1 -phosphate (6.5) catalyzed by PPM to enable a more efficient method for production of 4’-ethynyl-2’-deoxy nucleosides.
Additionally, PNP enzymes have also been found to have some activity with the 3-atom ethynyl substituent at the 4 position on deoxyribose and that the PNP enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a glycosylation reaction catalyzed by PNP to enable a more efficient method for production of 4’ -ethynyl -2’-deoxy nucleosides.
Even further improvement to the overall synthetic method came from the finding that
DERA enzymes, particularly the DERA from Shewanella halifaxensis, have activity for aldol reaction with 2-ethynyl-glyceraldehyde 3-phosphate which has a fully substituted a-carbon. This discovery allowed for the efficient synthesis of 4-ethynyl-D-2-deoxyribose 5-phosphate, a precursor to 4’-ethynyl-2’-deoxy nucleoside analogs, e.g., including EFdA.
SUMMARY OF THE INVENTION
The present invention involves the use of engineered enzymes in a novel enzymatic synthesis of 4’-ethynyl-2’-deoxy nucleoside analogs, including EFdA, that eliminates the use of protecting groups on intermediates, improves the stereoselectivity of glycosylation and greatly reduces the number of process steps needed to make said compounds compared to prior methods, among other process improvements. It further relates to novel intermediates which are an integral part of the enzymatic process.
The overall process is summarized in the following Scheme 1 and Scheme 2; the latter scheme provides an alternative method for making compound 5:
Scheme 1
kinase
p p y
Scheme 1A
kinase galactose oxidase
3 2X+ 9
2
p p y
It has been discovered that 4’-ethynyl-2’-deoxy nucleoside analogs such as EFdA can be synthesized employing a final step one-pot process by combining 4-ethynyl-D-2-deoxyribose 5-phosphate (6) with two enzymes, phosphopentomutase (PPM) [for example but not limited to SEQ ID NO.: 8] and purine nucleoside phosphorylase (PNP) [for example but not limited to SEQ ID NO.: 9, SEQ ID NO.: 15], as shown in Scheme 2.
Scheme 2
Scheme 2A
Several upstream intermediates used in the present process for the synthesis of the final product 4’-ethynyl-2’-deoxy nucleosides and analogs thereof are also made using enzymatic reaction methods as shown in Scheme 3; Scheme 3 A and Scheme 3B
Scheme 3
Scheme 3A
o2
pTsOH
deoxyribose
aldolase
Scheme 3B
Experimental Procedures
Preparation of 2-ethynyl-2-hvdroxypropane-l,3-diyl diacetate 12)
Method A:
To a -35 °C solution of diacetoxyacetone (1) (159 g, 914.0 mmol) in THF (1000 mL) was added 1600 mL of a 0.5 M solution of ethynyl magnesium chloride in THF maintaining the temperature below -20 °C. After the reaction reached completion, acetic acid (78 mL) in 400 mL methyl tert-butyl ether (MTBE) was added dropwise keeping the temperature below -20 °C. MTBE (800 mL) was then added and the mixture was warmed to room temp. Saturated NaCl in water (1000 mL) was added followed by saturated NH4CI solution in water (1050 mL). The organic layer was separated, dried over Na2SC>4 and evaporated to give compound (2) as an oil (160 g, 88%). 1H NMR (CDCI3, 500 MHz): d 4.26 (dd, 4 H), 2.55 (s, 1H), 2.14 (s, 6H).
Preparation of 2-ethynyl-propane-l,2,3-triol 13)
Method B:
To a solution of 2-ethynyl-2-hydroxypropane-l,3-diyl diacetate (2) (70 g, 350 mmol) in ethanol was added a 0.5M solution of sodium methoxylate in methanol (69.9 mL, 35.0 mmol) at room temperature (rt). The reaction was stirred at rt for 2 hours (h) to reach completion. The solvents were evaporated and the residue was re-dissolved in 100 mL water and extracted with 3 x 50 mL MTBE. The aqueous layer was sparged with nitrogen to remove residual solvents to give a 40.9% solution of 2-ethynyl-propane-l,2,3-triol (3) (108 g , 100% yield) as determined by nuclear magnetic resonance (NMR) (maleic acid as internal standard). lH NMR (D2O, 500 MHz): d 3.60 (dd, 4 H), 2.85 (s, 1H).
Alternate Preparations o ethynyl-glvcer aldehyde 14)
Method Cl:
In a stirred reactor, 2-ethynyl-propane-l,2,3-triol (3) (1.1 g, 9.47 mmol) in sodium phosphate buffer (30 mL, 100 mM, pH 7.0) containing antifoam 204 (Sigma A6426, 1 drop ~ 20 pL) was warmed to 30 °C with air sparging at 12.5 seem. Galactose oxidase (GOase, SEQ ID NO.: 1) (250 mg), Horseradish Peroxidase* (Type I, 5 mg) and bovine catalase** (5 mg) dissolved in sodium phosphate buffer (5 mL 100 mM, pH 7.0) were added to the reactor, followed by the addition of CuS04 aq. solution (100 mM, 150 pL). The reaction mixture was stirred at 600 rpm with air sparging for 47h to give (f?)-2-ethynyl-glyceraldehyde (4) in 47% conversion (by NMR) and 72% e.e. . (The product was not isolated). lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish Type I, commercially available from SIGMA (P8125), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345)
Method C2:
In a stirred 100 L jacketed reactor charged with deionized water (56.2 kg), sodium dihydrogen phosphate (1.212 kg, 10 moles) was added. The pH was adjusted to 7.02 using 10 N sodium hydroxide solution (852.6 g) at 25 °C. The reactor was charged with Antifoam 204 (A6426, 10 mL), followed CuS04*5H20 (6.5 g). Galactose oxidase (451.2 g) (SEQ ID NO.: 10) was added and stirred for 15 min while sparged with air. Horseradish peroxidase* (200.2 g) and catalase** (502.6 g) were added and the reactor was rinsed with water (2.0 kg). Next 2-ethynyl-propane- 1,2, 3 -triol (3) solution in water (9.48%, 30.34 kg, 24.72 mol) was added followed by an additional portion of Antifoam 204 (A6426, 10 mL). The reaction was sparged with air and
stirred overnight to give 94.0 kg of (A)-2-ethynyl-glyceraldehyde (4) in 66% conversion (by NMR) and 84% e.e. Assay yield 60%: 1H NMR (D20, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish purified, commercially available from Toyobo (PEO-301), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345).
The above reaction was also performed using the galactose oxidase (SEQ ID NO.: 11) and the product (4) was obtained in 67% conversion (by NMR) and 88% e.e. and assay yield 59%: 1H NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
Method C3:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 0.5 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase (SEQ ID NO.: 17, 450 mg enzyme powder) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (Cl 345, Sigma-Aldrich, 150 mg, 2000-5000 U/mg, 0.75 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg,
130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (25 wt%, 12 mL, 25.8 mmol). The reaction mixture was stirred at 30 °C with aeration at 125 seem and sampled using EasySampler over 20h to give 70% conversion and form compound (4) ((A)- 2-ethynyl-glyceraldehyde) in 58% assay yield and 99% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C4: Oxidation with immobilized galactose oxidase
Galactose
Oxidase
immobilized
3
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (16 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 160 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution. In a vessel evolved galactose oxidase (SEQ ID NO.: 17, 2.00 g) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (50 mL) and the resin. The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 160 mL) and potassium PIPES buffer (10 column volumes, 160 mL; 50 mM, pH 7.5) and it was used directly in a reaction. Reaction procedure:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 1 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase immobilized on the resin (SEQ ID NO.: 17, 750 mg enzyme powder per 6 mL resin) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (C1345, Sigma-Aldrich, 210 mg, 2000-5000 U/mg, 1.05 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg, 130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane- 1,2, 3 -triol (3) (25 wt%, 13 mL, 29.4 mmol). The reaction mixture was stirred at 25 °C with aeration at 125 seem. After 22h the reaction reached 91% conversion to give 200 mM (//)-2-ethynyl-glyceraldehyde (4) solution (100 mL, 68% assay yield, 97% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C5: Optional Isolation of aldehyde via formation of aminal (8)
Step 1: Preparation of (S)-2-( \ .3-dibenzylimidazolidin-2-yl )but-3-yne- l 2-diol
A 100 L jacketed cylindrical vessel equipped with nitrogen bubbler, mechanical stirrer and thermocouple was charged with crude oxidase reaction stream containing (f?)-2-ethynyl-glyceraldehyde ((4), 26.0 kg, 1.85 wt% aldehyde, 3.64 mol) and inerted with N2 atmosphere. The aqueous solution was warmed to 20 °C and Af,A-di methyl dodecan- 1 -ami ne oxide (DDAO) (30 wt% in water, 798 g, 0.96 mol;) was added, followed by MTBE (55.3 kg, 76 L) and N,N -dibenzylethane-l, 2-diamine (1.55 kg, 6.43 mol). The brown, biphasic mixture was stirred overnight at 20 °C under nitrogen atmosphere. After 17 hours the stirring was stopped and the organic phase was removed and discarded. A light brown MTBE solution of fV)-2-( l ,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (56.5 kg, 2.02 wt% aminal, 3.39 mmol, 93% assay yield) was obtained.
Six similar MTBE solutions were processed together in a single distillation and crystallization step (in total 374.4 kg of solution, containing 7.91 kg aminal).
A 50 L jacketed cylindrical vessel equipped with mechanical stirrer, distillation head (condenser at -20 °C) and thermocouple was charged with aminal solution (45 L). Vacuum was applied to the vessel (65-95 torr) and the jacket was set to 40 °C. Solvent was removed by distillation until a volume of 35 L had been reached. At this point, the internal temperature was 6.1 °C and an off-white solid had begun to crystallize. The remaining MTBE solution was slowly added, maintaining a constant volume of 35-40 L and an internal temperature of 0-10 °C. Once all the MTBE solution had been added the volume was decreased to 25 L. Distillation was halted, the vessel was inerted with nitrogen and the jacket temperature was decreased to 10 °C. The resulting pale yellow suspension was aged at this temperature for 2 hours and the solids were collected by filtration. The filter cake was washed with cold (-2 °C) MTBE (12.7 kg) and then dried under nitrogen flow for 7 hours. (5)-2-(l,3-dibenzylimidazolidin-2-yl)-but-3-yne-l,2-diol was obtained as an off-white crystalline solid (5.75 kg) lff NMR (500 MHz, DMSO-i¾) d 7.42 – 7.35 (m, 4H), 7.32 (td, J= 7.5, 1.6 Hz, 4H), 7.27 – 7.21 (m, 2H), 5.10 (t, J= 5.6 Hz, 1H), 5.03 (s, 1H), 4.28 (d, J= l3.3Hz, 1H), 4.16 (d, J= 13.3 Hz, 1H), 3.76 (s, 1H), 3.70 – 3.58 (m, 4H), 3.21 (d, J= 0.9 Hz, 1H), 2.90 – 2.80 (m, 2H), 2.60 – 2.51 (m, 2H).13C NMR (126 MHz, DMSO-i¾) d 140.0, 140.0, 128.5, 128.3, 128.2, 128.1, 126.8, 126.8, 88.6, 86.9, 75.0, 74.0, 66.4, 60.7, 60.5, 50.4, 50.3, 39.5. HR-MS (ESI) Aminal (M + H+) C21H25N202+ calculated 337.1911; found 337.1922.
Step 2: Prep l (8)
A 4 L jacketed cylindrical vessel equipped with nitrogen bubbler and mechanical stirrer was charged with of TsOH»H20 (12.0 g, 63.1 mmol), water (60 mL), (ri)-2-(l,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (110 g, 327 mmol) and MTBE (1700 mL). The biphasic mixture was placed under nitrogen and the jacket temperature was set to 15 °C. A solution of TsOH»H20 (114 g, 599.3 mmol) in water (600 mL) was added dropwise over 1.5 hours with overhead stirring (200 rpm). After addition had completed, the jacket temperature was lowered to 5 °C and the resulting slurry was aged for 1 hour. The solids were removed by filtration and washed with cold water (270 mL). The biphasic solution was transferred to a separating funnel and the organic phase was removed and discarded. The aqueous phase was treated with DOWEX™ MARATHON™ A resin (hydroxide form, 11.0 g) and AMBERLYST® 15 resin (hydrogen form, 11.0 g) while sparging with N2 at a rate of 200 seem for 24 hours to remove residual MTBE. The resins were removed by filtration to give a colorless aqueous solution of (f?)-2-hydroxy-2-(hydroxymethyl)but-3-ynal (774 g, 4.6 wt% aldehyde, 82% yield). lH MR (500 MHz, D2O) d 5.01 (s, 1H), 3.77 (d, J= 11.7 Hz, 1H), 3.73 (d, J= 11.7 Hz, 1H), 2.92 (s, 1H). 13C NMR (126 MHz, D2O) d 129.4, 125.4, 90.3, 81.0, 76.0, 73.9, 65.3. HRMS
(ESI) Aldehyde dimer (2M + Na+) CioHi2Na06+ calculated 251.0526; found 251.0530.
Alternate Preparations o ethvnyl-glvceraldehvde 3-phosphate (5):
Method Dl: Acetate kinase: ATP -regeneration system
Pantothenate kinase PanK
ATP
Acetate kinase
4 Acetate phosphate
5
In a stirred reactor, to a solution of adenosine diphosphate disodium salt (40 mg, 0.087 mmol) and magnesium chloride (38 mg, 0.400 mmol) in HEPES buffer (66 mM, pH 7.5, 30 mL) was added (i?)-2-ethynyl-glyceraldehyde (4) (1.9 mL, 210 g/L solution in water, 3.51 mmol), followed by acetate kinase (SEQ ID NO.: 3) (40 mg), and pantothenate kinase (SEQ ID NO.: 2) (120 mg). The reaction mixture was warmed to 25 °C and a solution of acetyl phosphate lithium potassium salt (1.3 g, 7.01 mmol) in HEPES buffer (50 mM, pH 7.5, 10 mL) was added dropwise over 4 hours, with pH maintained at 7.5 using 5M sodium hydroxide. The reaction was stirred for 18 hours to give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 85% conversion (by HPLC) (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method D2: Pyruvate oxidase ATP -regeneration system
Pan
Pyruvate oxidase
Pyruvate
Phosphate
02
In a stirred reactor, a solution of sodium pyruvate (3.11 g, 28 mmol) and phosphoric acid (0.523 mL, 7.71 mmol) in 76 mL water pH 7.5 was charged with (i?)-2-ethynyl-glyceraldehyde (4) (3.8 mL, 210 g/L solution in water, 7.01 mmol), adenosine diphosphate disodium salt (80 mg, 0.174 mmol), thiamine pyrophosphate (40 mg, 0.086 mmol), flavin adenine dinucleotide disodium salt hydrate (64 mg, 0.077 mmol), and magnesium chloride (400 pL, 1 M solution in water, 0.4 mmol). The pH was re-adjusted to 7.5 with 5M aq sodium hydroxide and the reaction volume was re-adjusted to 80 mL with water. Acetate kinase (SEQ ID NO.: 3) (80 mg), pyruvate oxidase (SEQ ID NO.: 4) (80 mg, lyophilized cell free extract), pantothenate kinase (SEQ ID NO.: 2) (400 mg), and catalase (800 pL, ammonium sulfate suspension CAT-101, Biocatalytics) were added. The reaction was stirred at 500 rpm and 30 °C with air sparging for 72 hours to give (//)-2-ethynyl-glyceraldehyde 3 -phosphate 5 in 95% conversion (by HPLC) (The product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
The above reaction was also performed using the pantothenate kinase (SEQ ID NO.: 13) and the product 5 was obtained in 66% conversion. (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H).
Method D3: Acetate kinase: ATP -regeneration system using immobilized enzymes
Panth
Acetate phosphate
Enzyme immobilization procedure:
NUVIA™ Immobilized Metal-ion Affinity Chromatography (IMAC) nickel-charged resin (168 mL based on settled volume) was added to a filter funnel and washed with binding buffer (1.6 L; 500 mM sodium chloride, 50 mM sodium phosphate, pH 8.0). In a vessel, pantothenate kinase
(8.4 g) (SEQ ID NO.: 12) and acetate kinase (2.8 g) (SEQ ID NO.: 3) were dissolved in binding buffer (500 mL). The washed resin was charged to the vessel and the solution was stirred for 4 hours at 20 °C. The resin was filtered and washed first with binding buffer (1.6 L) followed by piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (840 mL; 50 mM, pH 6.5). The washed resin was used directly in the next step.
Reaction procedure:
To a 1 L reactor, a solution of (f?)-2-ethynyl-glyceraldehyde (4) in water (608.7 g, 4.6 wt%, 212 mmol) was charged and cooled to 5 °C. To the cooled solution piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (32.7 mL, 1 M, pH 6.5, 32.7 mmol), magnesium chloride (9.33 mL, 1 M, 9.33 mmol), acetyl phosphate diammonium salt (51.8 g, 265 mmol), adenosine diphosphate disodium salt hydrate (1.17 g, 2.12 mmol), and water (192 mL) were added. The solution was allowed to stir and pH was adjusted to 6.4 using 5 N KOH. The reaction was warmed to 20 °C and 168 mL of resin with co-immobilized pantothenate kinase (SEQ ID NO.: 12) and acetate kinase (SEQ ID NO.: 3) was added. The reaction was stirred for 10 hours with 5 N KOH used to maintain a pH of 6.4 to give (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in
92% conversion (by HPLC) and 91% yield (by 3 lp NMR with tetraphenylphosphonium chloride as internal standard) (the product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Preparation of 4-ethynyl-D-2-deoxyribose 5-phosphate 16)
Method E:
To a solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (5, 20 mL, 5.3 mmol) in water, a solution of acetaldehyde in water (40 wt.%, 2.02 mL, 15.9 mmol) was added at room
temperature, followed by the addition of Deoxyribose-phosphate aldolase (DERA) (SEQ ID NO. : 6), 25 mg solution in triethanolamine hydrochloride buffer (1 mL, 1 M, pH 7.0). The reactor was sealed and the mixture was stirred overnight at 30 °C and 600 rpm to give 4-ethynyl-D-2-deoxyribose 5-phosphate (6) in 99% conv. and 99% e.e., 99% d.e. as a 1 : 1 anomer mixture (The product was not isolated) a-anomer: lH NMR (D2O, 600 MHz) 5 5.31 (t, 1H), 4.13 (t, 1H), 3.81-3.72 (m, 2H), 2.89 (s, 1H), 2.42-2.34 (m, 1H), 1.87-1.79 (m, 1H); 13c NMR (D2O, 151 MHz) 5 97.7 (s), 81.4 (d), 79.4 (s), 78.9 (s), 71.1 (s), 67.7 (d), 39.6 (s). b-anomer: 1H NMR
(D2O, 600 MHz) 5 5.40 (dd, 1H), 4.28 (t, 1H), 3.88-3.80 (m, 2H), 2.87 (s, 1H), 2.13-2.06 (m,
1H), 2.04-1.97 (m, 1H); 13C NMR (D20, 151 MHz) 5 97.3 (s), 82.2 (d), 78.7 (s), 78.5 (s), 71.3 (s), 68.4 (d), 39.6 (s). LC-MS: (ES, m/z): calculated for C7H10O7P (M-H): 237.0; found 237.0
Alternate Preparations of (2 ?,3A,5 ?)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol monohydrate (7) [alternative name 4’-ethynyl-2-fluoro- 2’-deoxvadenosine or EFdAI
Method FI:
Ammonium ((2f?,3ri)-2-ethynyl-3,5-dihydroxytetrahydrofuran-2-yl)m ethyl hydrogen phosphate (1.00 g, 3.91 mmol) was dissolved in 10 mL of pH 7.5 buffer (100 mM triethanolamine ΉO containing 5 mM MnCl2). The solution pH was adjusted to 7.3 with 5 N NaOH. To the solution was added 2-fluoroadenine (0.599 g, 3.91 mmol) and sucrose (2.68 g, 7.82 mmol). The enzyme solution was prepared by dissolving phosphopentomutase (SEQ ID NO. : 8) (100 mg), purine nucleoside phosphorylase (SEQ ID NO.: 9) (50 mg), and sucrose phosphorylase (SEQ ID NO. :
7) (10 mg) in 10 mL of the pH 7.5 buffer. The enzyme solution was added to the reagent mixture and the resulting suspension was shaken at 40 °C. After 20 h, the suspension was cooled to 0 °C and filtered, rinsing with cold water. The solid was suction dried to give the title compound (1.12 g, 92%) as a single isomer.
iH NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J =
5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13c NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
The PPM and PNP enzymes used in this step were each derived from mutations starting from the enzymes from E. coli ( Escherichia coli). The sucrose phosphorylase (SP) used in this step was derived from Alloscardovia omnicolens ; SP derived from other organisms could also be used.
Method F2:
To an aqueous solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (950 mL, 157 mmol) containing piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer at a pH from about 5.5 to 6.0 was added triethanolamine (7.09 g, 47.5 mmol). The pH of the solution was adjusted from 7.1 to 7.6 using potassium hydroxide (8 mL, 8M). Manganese(II) chloride hydrate (0.592 g, 4.70 mmol) was added followed by sucrose (161 g, 470 mmol), giving a pH of 7.5 To the solution
was added the following enzymes: deoxyribose-phosphate aldolase (SEQ ID NO. : 14) (461 mg), sucrose phosphorylase (SEQ ID NO. : 7) (494 mg), phosphopentomutase (SEQ ID NO.: 8)(2.63 g), and purine nucleoside phosphorylase (SEQ ID NO. : 15) (659 mg). Once the enzymes were dissolved, 2-fluoroadenine (19.80 g, 125 mmol) was added. The reaction was heated to 35 °C and acetaldehyde was added (40 wt% in isopropyl alcohol, 29.8 mL, 235 mmol). After reacting for 2h, the mixture was seeded with EFdA crystalline product (0.96 g, 2 mol%). After reacting over 26 h at 35 °C, the slurry was cooled to 0 °C, and the solids were collected by filtration, washing with water two times (40 mL ea.). The solids were dried under a nitrogen sweep. Yield 43.2 g, 92 wt%, 96.2% corrected. ¾ NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J = 5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13C NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
Alternate Preparations of -2-ethvnyl-propane-l,2,3-triol 1 1-phosphate 19) :
Method Gl: Acetate kinase: ATP-regeneration system using enzymes SEQ. ID No.: 2 and SEQ. ID No.: 3
Panthotenate kinase PanK
ATP
Acetate kinase
Acetate phosphate
A 50 mL reactor was charged with a solution of 2-ethynyl-propane-l,2,3-triol (3) in water (9.29 g, 9.46 wt%, 7.57 mmol) potassium PIPES buffer (1.02 mL, 1 M, pH 6.5, 1.02 mmol), magnesium chloride (292 pL, 1 M, 0.292 mmol), acetyl phosphate diammonium salt (1.851 g, 89 wt%, 9.46 mmol), adenosine diphosphate disodium salt hydrate (ADP, 42 mg, 0.076 mmol, 0.01 eq), and water (28 mL). The pH was adjusted to 6.4 using 5 M KOH, the solution was warmed to 20 °C and evolved pantothenate kinase PanK SEQ. ID No.: 2 (264 mg) and acetate kinase AcK SEQ. ID No. : 3 (88 mg) were added. The reaction was stirred for 16 hours with pH maintained at 6.4 using 5 N KOH. The final reaction contents provided C.V)-2-ethynyl -propane- 1 ,2,3-triol 1-phosphate (9) in >95% e.e. and 99% conversion (by 31P NMR). The product was not isolated. ¾ NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H). 13C NMR (D2O, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s). 31P NMR (D2O, 202 MHz) d 3.39. HRMS: (ESI, m/z): calculated for [M-l] CsHsOeP: 195.0058; Found 195.0068 [M-H] : 195.0058.
Method G2: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
To a jacketed reactor aqueous solution 2-ethynyl-propane-l,2,3-triol (3) (11.47 kg, 8.7% wt, 8.61 mol) and water (7.5kg) was charged, followed by 1M BIS-TRIS methane buffer pH 6.5 (1L) and magnesium chloride (41.4 g). ATP (48g, 0.086 mol, 0.01 equivalent) and diammonium acetyl phosphate (2.021 kg, 89%, 10.33 mmol) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using KOH (270.4 g). Evolved pantothenate kinase SEQ. ID No.: 20 (20.4 g) and evolved acetate kinase SEQ. ID No.: 21 (3 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which pH dropped to 5.5.
Quantitative conversion of 2-ethynyl-propane-l,2,3-triol (3) was obtained as judged by ‘H and 31P NMR. Such prepared (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) solution (397 mM, 22.5 kg, 98% yield) was used in subsequent oxidation step without any further purification. ‘H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H).
Method G3: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21 with deuterated compound (3) to assign absolute stereochemistry and demonstrate desymmetrizing phosphorylation.
Acetate phosphate
Z-d2, 95:5 er
Evolved pantothenate kinase SEQ. ID No. : 20 (100 pL of 10 g/L solution in water ) and evolved acetate kinase SEQ. ID No. : 21 (100 pL of 2g/L solution in water) were added to a solution containing diammonium acetyl phosphate (41 mg), 2-ethynyl-propane-l, l-72-l,2,3-triol ((A)- 3-d2, 20 mg, 170 pmol), magnesium chloride (10 pL of 1 M solution in water), ADP (10 pL of 100 g/L solution in water), and sodium phosphate buffer (10 pL of 1 M solution in water) in water (800 pL) at pH 6.5. The reaction was incubated for 24h at rt to give deuterated 2-ethynyl-propane-l,2,3-triol l-phosphate analogs (S)-9-(3,3-d2) and (S)-9-(l,l-d2) in 95:5 ratio and 99% overall yield. The ratio of phosphorylated compounds was determined by 31P NMR to be -95:5, confirming stereoselective phosphorylation of the 2-ethynyl-propane-l,2,3-triol (3) at the pro-(S) hydroxyl group (i.e. a desymmetrizing phosphorylation). 1H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H), 2.93 (s, 1H). 13C NMR (D20, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s).
Method G4: Acetate kinase: ATP-regeneration system using immobilized enzymes SEQ. ID No. : 20 and enzyme SEQ. ID No. : 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (75 mL based on settled volume) was added to a filter funnel and washed with water (9 column volumes, 3 x 225 mL) and binding buffer (1 column volume, 75mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0). In a vessel pantothenate kinase (SEQ ID NO. : 20, 6.0 g) lyophilized powder was resuspended in binding buffer (200 mL) and the washed resin was added. The solution was mixed using rotating mixer at 25 °C for 6h. The resin was filtered and washed with binding buffer (6 column volumes, 6 x 225 mL) and BIS-TRIS buffer (8 column volumes, 600 mL; 50 mM, pH 6.2).
Reaction procedure:
An aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (574 g, 8.7% wt, 0.430 mol) and water (350 mL) was charged to a jacketed reactor, followed by 1M BIS-TRIS methane buffer pH 6.5 (50 mL) and magnesium chloride (2.033 g, 0.01 mol). ATP (2.37g, 0.0043 mol, 0.01 equivalent) and diammonium acetyl phosphate (101 g, 89%, 0.530 mmol, 1.2 eq) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using 5 M KOH.
Resin with immobilized pantothenate kinase SEQ. ID No. : 20 (25 mL) and evolved acetate kinase SEQ. ID No. : 21 (0.15 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which the pH dropped to 5.5. Quantitative conversion of 2-ethynyl-propane- I,2,3-triol (3) to (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) was obtained as judged by ¾ and 31P NMR. ¾ NMR (D20, 500 MHz) d 3.89 (m, 2H), 3.72 (d, J= 11.6 Hz, 1 H), 3.65 (d, J =
I I .6 Hz, 1H), 2.93 (s, 1H).
Alternate Preparations of (i?V2-ethvnyl-glvceraldehvde 3-phosphate 15):
Method HI: Immobilized galactose oxidases SEP ID No.: 16
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 16 g of washed resin. In a vessel evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the glycosylation reaction.
Reaction procedure:
The resin with immobilized galactose oxidase SEQ ID NO.: 16 (3.0 g) was added to a solution of S)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (f?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the(f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used
directly in the glycosylation reaction. iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H2: Immobilized galactose oxidases SEP ID No.: 17
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give l6g of washed resin. In a vessel, evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS methane buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the reaction.
Reaction procedure:
The resin with immobilized evolved galactose oxidase SEQ ID NO.: 17 (3.0 g) was added to a solution of (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was used directly in the glycosylation reaction. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H3: Immobilized galactose oxidases SEQ ID No.: 18
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 18, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution ((9), 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 90% conversion and the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H),
4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (MΉ): 193.1; found 193.0.
Method H4: Immobilized galactose oxidases SEP ID No.: 19
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 19, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution (9, 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 100% conversion and (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was obtained in >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
PATENT
CA 2502109
WO 2017053216
US 20200010834
US 20200010868
PAPER
Organic letters (2017), 19(4), 926-929.
Organic Letters (2017), 19(4), 926-929.
Journal of medicinal chemistry (2018), 61(20), 9218-9228.
Bioscience, Biotechnology, and Biochemistry (2020), 84(2), 217-227.
PAPER
Organic letters (2011), 13(19), 5264-6.
A concise enantioselective total synthesis of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), an extremely potent anti-HIV agent, has been accomplished from (R)-glyceraldehyde acetonide in 18% overall yield by a 12-step sequence involving a highly diastereoselective ethynylation of an α-alkoxy ketone intermediate.

Processes for preparing islatravir and its analogs comprising the reaction of a substituted tetrahydrofuran compound with purine nucleoside phosphorylase and a nucleobase, followed by stereochemical synthesis, glycosylation, reduction, oxidation and isolation are claimed. Also claimed are novel intermediates of islatravir and processes for their preparation and their use for the preparation of islatravir.
(2R,3S,5R)-5-(6-Amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)- tetrahydrofuran-3-ol (1). To a stirred solution of 16 (66.2 mg, 0.115 mmol) in MeOH/CH2Cl2 (2:1, 1.5 mL) was added NH4F (85.1 mg, 2.30 mmol) at room temperature. After 16 h, MeOH (0.5 mL) was added, and the resulting mixture was stirred for an additional 27 h. To the mixture was added 10% NaOH in MeOH (1.5 mL) to adjust the pH of the mixture to ca. 10. After 10 min, Dowex 50W×8 (200– 400 mesh (H)) was added until the pH of the mixture reached ca. 4. To the resulting mixture was added CaCO3 (259 mg, 2.59 mmol), and the mixture was stirred for 30 min. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 10:1) to give 29.3 mg (87%) of 1. Mp: 220.0–221.4 °C (dec.); [α] 25 D +12.4 (c 0.97, MeOH); IR: νmax 3315 (br m), 3179 (br m), 1690 (vs), 1356 (vs); 1 H NMR (600 MHz, DMSO-d6): δ 2.43 (1H, ddd, J = 13.2, 7.3, 7.3 Hz), 2.70 (1H, ddd, J = 13.2, 6.8, 5.1 Hz), 3.52 (1H, s), 3.54 (1H, dd, J = 11.7, 6.4 Hz), 3.65 (1H, dd, J = 11.7, 5.0 Hz), 4.57 (1H, m), 5.32 (1H, m), 5.60 (1H, m), 6.24 (1H, dd, J = 7.2, 5.1 Hz), 7.82 (1H, br s), 7.92 (1H, br s), 8.31 (1H, s); 13C NMR (150 MHz): δ 38.3, 64.4, 70.3, 79.2, 81.7, 82.2, 85.4, 117.6, 140.0, 150.4 (d, JCF = 20.7 Hz), 157.8 (d, JCF = 21.2 Hz), 158.8 (d, JCF = 203.4 Hz); HRMS (FAB): m/z calcd for C12H13FN5 O3, 294.1002; found, 294.1000 ([M+H]+ ).


PAPER
Organic Letters (2011), 13(19), 5264-5266.
PAPER
Bioscience, biotechnology, and biochemistry (2012), 76(6), 1219-25.
EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine), a nucleoside reverse transcriptase inhibitor with extremely potent anti-HIV activity, was concisely synthesized from (R)-glyceraldehyde acetonide in an 18% overall yield by a 12-step sequence involving highly diastereoselective ethynylation of an α-alkoxy ketone intermediate. The present synthesis is superior, both in overall yield and in the number of steps, to the previous one which required 18 steps from an expensive starting material and resulted in a modest overall yield of 2.5%.
PAPER
Bioscience, Biotechnology, and Biochemistry (2012), 76(6), 1219-1225.
Organic letters (2015), 17(4), 828-31.
Organic Letters (2015), 17(4), 828-831.
PAPER

Some pharmaceutical companies are investigating biocatalysis at different points in their drug development pipelines, but mostly at one or two steps into the making of a small molecule. Scientists at Merck & Co. have taken this further—they are reporting an entire drug synthesis using a chain of nine enzymes, five of which had been engineered, to produce an experimental HIV drug at high yield in just a few steps (Science 2019, DOI: 10.1126/science.aay8484).
This biocatalytic cascade is turning heads. For the most part, scientists aren’t using biocatalysis to manufacture a compound so much as to develop it, says Princeton University chemist Todd Hyster. The Merck process stitches together nine enzymes to get good yields of the final product, which Hyster says is no small feat.
It literally took my breath away.
Alison Narayan, assistant professor, University of Michigan
“I was blown away,” Hyster says of the first time he saw Merck scientists talk about this work. “It’s something that was very complicated.”
Mark Huffman, a chemist who led the work at Merck with Anna Fryszkowska, says they turned to biocatalysis in order to overcome a couple of key hurdles in synthesizing some molecules. One is stereochemistry. Islatravir is a nucleoside that blocks the HIV enzyme reverse transciptase and traditionally, in medicinal chemistry, it’s been hard to get the stereochemistry of nucleosides right, Huffman says. But this is something enzymes are designed by nature to do. The other is preventing unwanted side reactions. A number of steps in the traditional chemical synthesis of islatravir put the compound’s functional groups at risk of being lopped off, so they must be protected. Huffman says enzymes are specific in the types of reactions they catalyze, so there’s little to no risk of an unwanted side reaction.
Scientists at Merck & Co. use nine enzymes, five of which are engineered, to biocatalytically make an HIV drug called islatravir without having to purify intermediates. Enzymes not engineered by Merck are not pictured.
On top of that, Huffman says, they are doing these reactions at neutral pH, in aqueous solvents, and at room temperature, which cuts down on electricity and the need for multiple bioreactors running under different conditions. Islatravir normally takes between 12 and 18 steps to make. With biocatalysis, the team has cut this down to three.
“You don’t have rigorous equipment requirements,” he says. “You’re usually running [these reactions] under much milder conditions.”
To run the cascade, the team started with 2-ethynylglycerol, and added a mixture of three enzymes to run one group of reactions. They then added more enzymes to drive a second set of reactions. Then, they remove the enzymes from the solution, which are immobilized and easy to filter out, and use four more enzymes to drive the final reactions that lead to islatravir. There are no intermediate purification steps. The overall yield is about 51% using biocatalysis, compared to yields of 7% and 15% using two more traditional syntheses.
To make their biocatalysts, the team surveyed natural enzymes, mostly from microbes, that interacted with the different intermediates in islatravir production. One of the reasons why Huffman says islatravir is an ideal small molecule to produce using biocatalysis is that most organisms have to make and break down nucleosides, so there are several natural enzymes found across multiple species. This gave the team a lot of starting material from which to alter amino acids and build the enzymes they needed to do their syntheses. By making adjustments to active sites and other areas of the enzymes, the team built five of the nine enzymes needed to make islatravir biochemically.
Huffman says that while islatravir is a good molecule to show that scientists can build large biocatalytic cascades, Merck is also looking at biocatalysis to make other small molecules and biologic drugs.
Alison Narayan, a biocatalysis chemist at the University of Michigan, calls Merck “bold” for putting the time, money, and people behind this change in production—it takes a lot of resources to try an entire synthesis via biocatalysis. And, she says, they’ve succeeded spectacularly. “It literally took my breath away,” Narayan says of her first exposure to this project in 2018. “I think it’s a huge accomplishment.”
She says that Merck’s islatravir work shows that industry is starting to appreciate what biocatalysis can do for their drug pipelines and their financial bottom lines. Alongside Merck, companies like GlaxoSmithKline and Pfizer are also exploring biocatalysis at different points in drug development and manufacturing.
“It’s an important proof of concept,” Narayan says. “This is a practical way to build molecules, and this will be the way that people will build molecules when you take into consideration efficiency, green-ness, and constructing an effective synthesis. Biocatalysis has a lot to offer.”
PAPER
Biocatalytic cascades go viral
IN SECTION: PERSPECTIVE | BIOCATALYSIS
An investigational drug targeting the HIV virus is synthesized with nine enzymes
Natural biosynthesis assembles a vast array of complex natural products starting from a limited set of building blocks, under physiological conditions, and in the presence of numerous other biomolecules. Organisms rely on the extraordinary selectivity of enzymes and their ability to operate under similar reaction conditions, meaning that these catalysts are perfectly adapted to mediate cascade reactions. In these multistep processes, the product of one biocatalytic step becomes the substrate for the next transformation (Display footnote number:1-3). On page 1255 of this issue, Huffman et al. (Display footnote number:4) report the development of an impressive nine-enzyme biocatalytic cascade for the synthesis of the investigational drug islatravir for the treatment of human HIV.
This study represents a partnership between scientists from Merck and Codexis. These two companies have a history of successfully collaborating to develop biocatalysts for the synthesis of important pharmaceuticals. Almost a decade ago, they developed a chemoenzymatic route for the synthesis of the type 2 diabetes drug sitagliptin (Januvia), relying on a key enzyme-catalyzed transamination with a highly engineered (R)-selective transaminase (Display footnote number:5). The work was considered a landmark example of directed evolution and functioned to highlight the potential application of biocatalysis to revolutionize industrial chemical processes.
The cascade for synthesizing islatravir was inspired by the bacterial nucleoside salvage pathway, which recycles precious nucleosides by using three key enzymes: a purine nucleoside phosphorylase (PNP), a phosphopentomutase (PPM), and a deoxyribose-5-phosphate aldolase (DERA) (see the figure). However, to achieve the synthesis of the target molecule, Huffman et al. required the natural nucleoside degradative cascade to run in reverse. The reversible nature of enzymes is central to the design of this cascade and is one of the important features that sets biocatalysts apart from the majority of traditional chemical catalysts.
The success of the cascade developed by the team also relied on all three enzymes accepting non-natural substrates bearing a fully substituted carbon at the C-4 position of the 2-deoxyribose ring. The authors reconstructed the reverse nucleoside salvage pathway from a PNP and PPM found in Escherichia coli and a DERA from Shewanella halifaxensis. The native E. coli enzymes required engineering to improve their activity. The DERA displayed existing high activity and stereoselectivity for the formation of the desired sugar phosphate enantiomer, but it required engineering to improve its ability to operate at high substrate concentration.
One of the many advantages of performing biocatalytic cascade reactions is the effective displacement of unfavorable reaction equilibria that can be achieved through product removal. However, despite performing the PNP and PPM steps in tandem, the reaction proceeded with poor conversion, and the inorganic phosphate by-product inhibits the enzymes. An elegant solution to these issues was the inclusion of an auxiliary sucrose phosphorylase, along with its sugar substrate, which removed free phosphate and effectively displaced the reaction equilibrium toward product formation.
Having assembled enzymes for the three key steps in the cascade, Huffman et al. sought to develop a biocatalytic route for the synthesis of the DERA substrate 2-ethynylglyceraldehyde 3-phosphate. Extensive screening of a broad range of kinases resulted in the identification of pantothenate kinase (PanK) from E. coli, which displayed low levels of activity (∼1% conversion) toward the (R)-enantiomer of the target aldehyde. Despite the modest initial activity, directed evolution was successfully used to substantially improve the productivity and stability of this enzyme. Finally, after 12 rounds of evolution, the authors reversed the enantioselectivity and improved the activity, stability, and expression of a galactose oxidase variant for the desymmetrization of the starting substrate, 2-ethynylglycerol.
Opens in modal lightbox
Viewable Image – engineering a biocatalytic cascade
Image Caption
GRAPHIC: A. KITTERMAN/SCIENCE
Advancements in protein engineering, rapid gene sequencing, and the availability of low-cost DNA synthesis have made it possible to alter the properties of enzymes and fine-tune them for biocatalytic applications (Display footnote number:6-8). The work by Huffman et al. is a milestone in cascade design, largely because of the number of biocatalysts operating in tandem and the engineering feat required to optimize five of the nine enzymes involved in the synthesis. It also highlights how biosynthetic or degradative pathways can be a source of inspiration for the design of efficient biocatalytic cascades and how sequences can be reconstituted using enzymes recruited from multiple sources—in this case, of bacterial, fungal, plant, and mammalian origin. The diverse role that biocatalysts can play is also exemplified in this work, where five engineered enzymes are directly involved in the synthesis of the target molecule, and four additional enzymes function to recycle coenzyme, remove inhibitory by-products, and maintain the correct oxidation state of the copper cofactor.
Previous approaches reported for the synthesis of islatravir relied on multistep syntheses and require protecting group manipulations and intermediate purification steps (Display footnote number:9, 10). The incorporation of a key biocatalytic step or steps has the potential to revolutionize synthetic design strategies by making possible transformations that are not accessible using solely chemical approaches (Display footnote number:11, 12). The application of enzymes in industry and the development of chemoenzymatic routes to complex molecules is now well established. However, multistep syntheses exclusively comprising biocatalytic transformations are rare (Display footnote number:13), and this contribution sets a new standard for the synthesis of complex molecules with enzymatic cascades.
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. Email: elaine.oreilly@ucd.ie.
REFERENCES AND NOTES
1. S. P. France, L. J. Hepworth, N. J. Turner, S. L. Flitsch, ACS Catal. 7, 710 (2017).
2. S. Gandomkar, A. Żadło-Dobrowolska, W. Kroutil, ChemCatChem 11, 225 (2019).
3. P. Both et al., Angew. Chem. Int. Ed. 55, 1511 (2016).
4. M. A. Huffman et al., Science 366, 1255 (2019).
5. C. K. Savile et al., Science 329, 305 (2010).
6. F. H. Arnold, Angew. Chem. Int. Ed. 57, 4143 (2018).
7. C. Zeymer, D. Hilvert, Annu. Rev. Biochem. 87, 131 (2018).
8. C. A. Denard, H. Ren, H. Zhao, Curr. Opin. Chem. Biol. 25, 55 (2015).
9. M. McLaughlin et al., Org. Lett. 19, 926 (2017).
10. M. Kageyama, T. Nagasawa, M. Yoshida, H. Ohrui, S. Kuwahara, Org. Lett. 13, 5264 (2011).
11. N. J. Turner, E. O’Reilly, Nat. Chem. Biol. 9, 285 (2013).
12. M. Hönig, P. Sondermann, N. J. Turner, E. M. Carreira, Angew. Chem. Int. Ed. 56, 8942 (2017).
13. S. Wu et al., Nat. Commun. 7, 11917 (2016).
ACKNOWLEDGMENTS
J.R. acknowledges the School of Chemistry, University College Dublin, for support.
References
- ^ Kawamoto, A; Kodama, E; Sarafianos, SG; Sakagami, Y; Kohgo, S; Kitano, K; Ashida, N; Iwai, Y; Hayakawa, H; Nakata, H; Mitsuya, H; Arnold, E; Matsuoka, M (2008). “2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants”. The International Journal of Biochemistry & Cell Biology. 40 (11): 2410–20. doi:10.1016/j.biocel.2008.04.007. PMID 18487070.
- ^ Roy M. Gulick (2018). “Investigational Antiretroviral Drugs: What is Coming Down the Pipeline”. Top Antivir Med. 25 (4): 127–132. PMC 5935216. PMID 29689540.
- ^ “Someday, an Arm Implant May Prevent H.I.V. Infection for a Year”. New York Times. July 23, 2019.
- ^ “Merck Presents Early Evidence on Extended Delivery of Investigational Anti-HIV-1 Agent Islatravir (MK-8591) via Subdermal Implant”(Press release). July 23, 2019.
- ^ Jump up to:a b Michailidis, Eleftherios; Huber, Andrew D.; Ryan, Emily M.; Ong, Yee T.; Leslie, Maxwell D.; Matzek, Kayla B.; Singh, Kamalendra; Marchand, Bruno; Hagedorn, Ariel N.; Kirby, Karen A.; Rohan, Lisa C.; Kodama, Eiichi N.; Mitsuya, Hiroaki; Parniak, Michael A.; Sarafianos, Stefan G. (2014). “4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) Inhibits HIV-1 Reverse Transcriptase with Multiple Mechanisms”. Journal of Biological Chemistry. 289 (35): 24533–48. doi:10.1074/jbc.M114.562694. PMC 4148878. PMID 24970894.
- ^ Grobler, Jay (February 22–25, 2016). Long-Acting Oral and Parenteral Dosing of MK-8591 for HIV Treatment or Prophylaxis. Boston, Massachusetts. Conference on Retroviruses and Opportunistic Infections. 98.
- ^ Stoddart, Cheryl A.; Galkina, Sofiya A.; Joshi, Pheroze; Kosikova, Galina; Moreno, Mary E.; Rivera, Jose M.; Sloan, Barbara; Reeve, Aaron B.; Sarafianos, Stefan G.; Murphey-Corb, Michael; Parniak, Michael A. (2015). “Oral Administration of the Nucleoside EFdA (4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque”. Antimicrobial Agents and Chemotherapy. 59 (7): 4190–8. doi:10.1128/AAC.05036-14. PMC 4468726. PMID 25941222.
- ^ Bruno Marchand. “The Crystal Structure of EFdA‐Resistant HIV‐1 Reverse Transcriptase Reveals Structural Changes in the Polymerase Active Site” (PDF).
 | |
| Names | |
|---|---|
| IUPAC name
2′-Deoxy-4′-ethynyl-2-fluoroadenosine
| |
| Other names
EFdA; MK-8591
| |
| Identifiers | |
|
3D model (JSmol)
| |
| ChemSpider | |
|
PubChem CID
| |
| UNII | |
| Properties | |
| C12H12FN5O3 | |
| Molar mass | 293.258 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |
////////////////Islatravir, MK-8591, EFdA, PHASE 2, HIV-1 , HIV-2, FDA 2026, APPROVALS 2026, Idvynso
C#CC1(C(CC(O1)N2C=NC3=C(N=C(N=C32)F)N)O)CO
GRAPIPRANT

{kind=link}
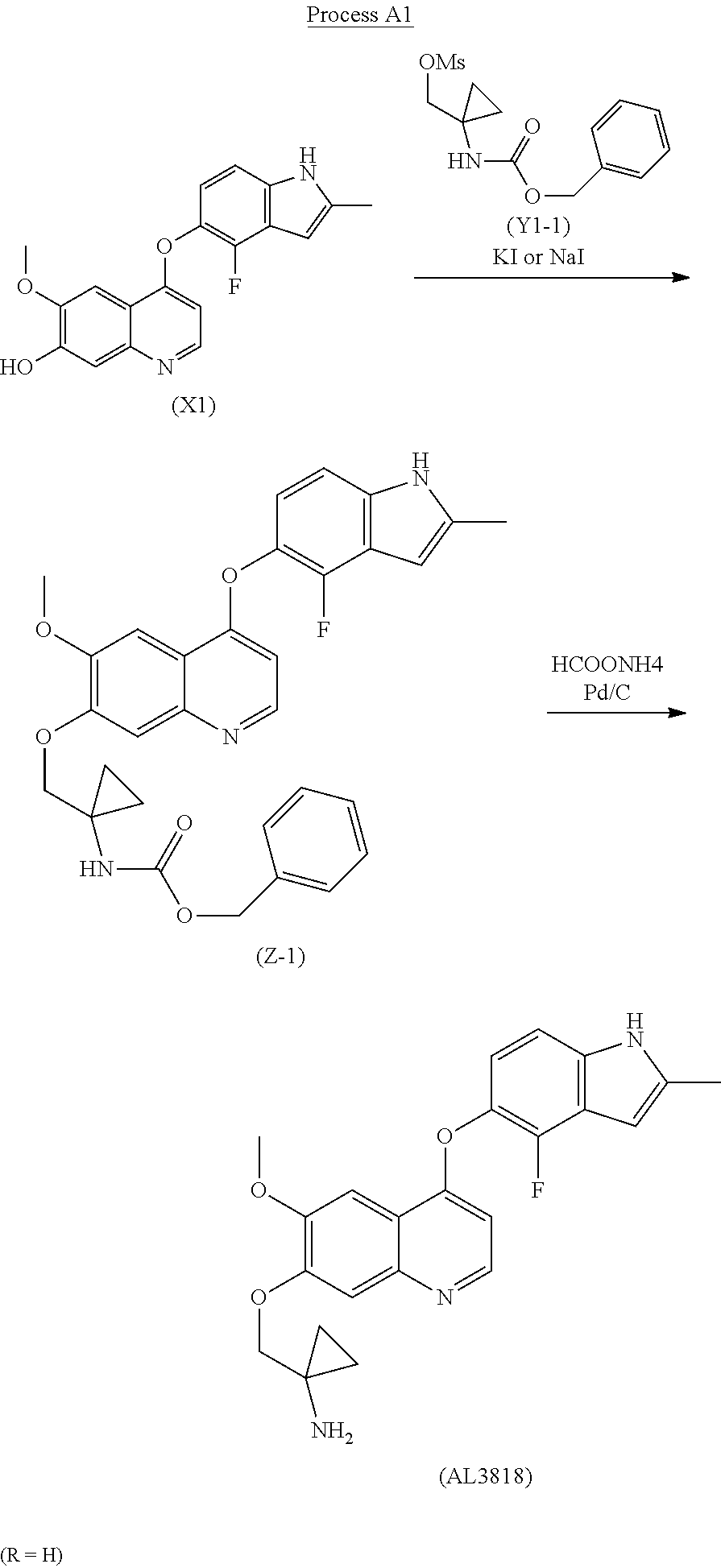{kind=link}
{kind=link}
{kind=link}
{kind=link}
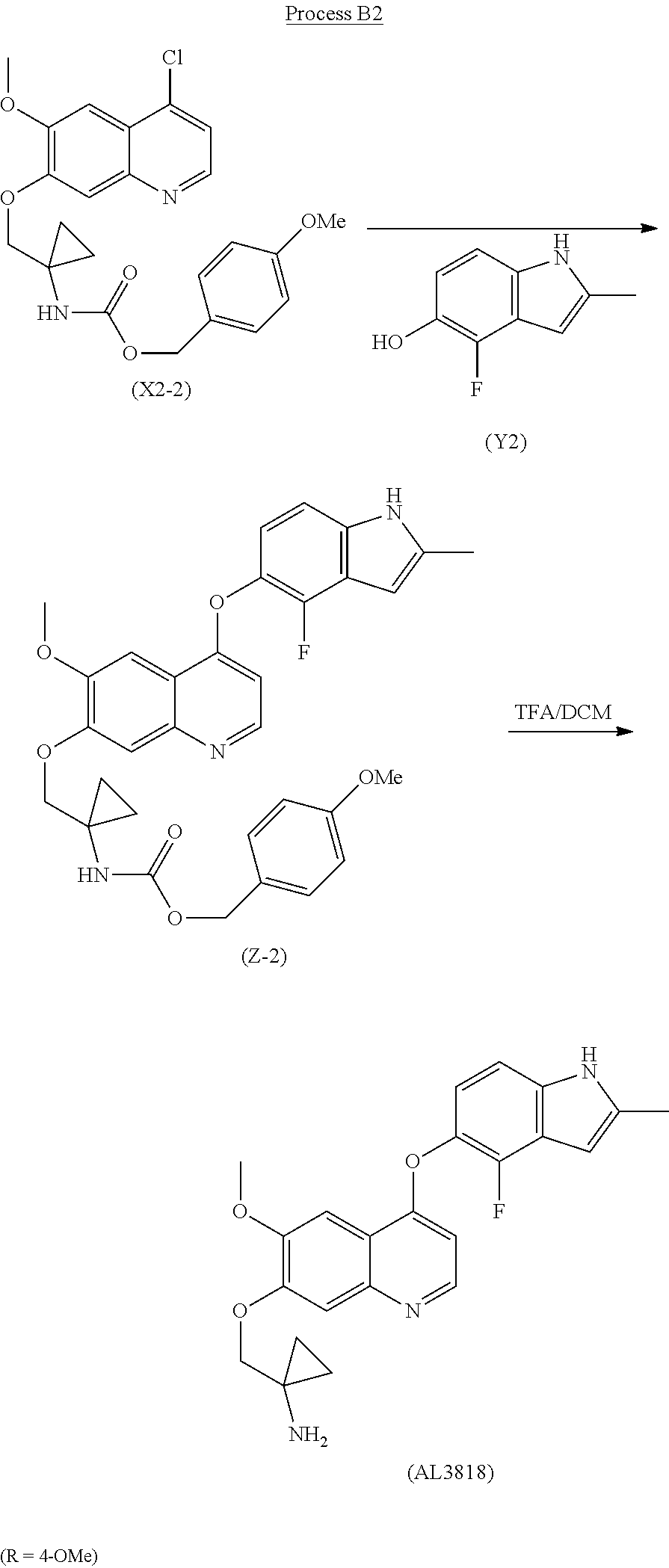{kind=link}
{kind=link}

GRAPIPRANT
- Molecular FormulaC26H29N5O3S
- Average mass491.605 Da
CAS 415903-37-6
UNII-J9F5ZPH7NB, CJ 023423, CJ-023423,
Phase II, Arrys Therapeutics, CANCER,
PAIN, AskAt Phase II,
N-{2-[4-(2-ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl}-N’-[(4-methylphenyl)sulfonyl]urea
RQ-00000007, MR10A7
9763
AAT-007
Benzenesulfonamide, N-[[[2-[4-(2-ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]amino]carbonyl]-4-methyl-
CJ-023,423
- N-[[[2-[4-(2-Ethyl-4,6-dimethyl-1H-imidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]amino]carbonyl]-4-methylbenzenesulfonamide
- 1-[2-[4-(2-Ethyl-4,6-dimethylimidazo[4,5-c]pyridin-1-yl)phenyl]ethyl]-3-(4-methylphenyl)sulfonylurea
- 2-Ethyl-4,6-dimethyl-1-[4-[2-[[[[(4-methylphenyl)sulfonyl]amino]carbonyl]amino]ethyl]phenyl]-1H-imidazo[4,5-c]pyridine
- AAT 007
- CJ 023423
- Grapiprant
- MR 10A7
- RQ 00000007
- RQ 7
Synonyms and Mappings
- 415903-37-6
- GRAPIPRANT [GREEN BOOK]
- CJ-023
- GRAPIPRANT [INN]
- GRAPIPRANT [WHO-DD]
- MR-10A7
- AAT-007
- MR10A7
- RQ-00000007
- RQ-7
- GRAPIPRANT [USAN]
- GRAPIPRANT
- 2-ETHYL-4,6-DIMETHYL-1-(4-(2-(((((4-METHYLPHENYL)SULFONYL)AMINO)CARBONYL)AMINO)ETHYL)PHENYL)-1H-IMIDAZO(4,5-C)PYRIDINE
- N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYLBENZENESULFONAMIDE
- CJ 023423
- BENZENESULFONAMIDE, N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYL-
- CJ-023,423
- N-(((2-(4-(2-ETHYL-4,6-DIMETHYL-1H-IMIDAZO(4,5-C)PYRIDIN-1-YL)PHENYL)ETHYL)AMINO)CARBONYL)-4-METHYL-BENZENESULFONAMIDE
- CJ-023423
SYN

Arrys Therapeutics (under license from AskAt ) and affiliate Ikena Oncology (formerly known as Kyn Therapeutics ) are developing ARY-007 , an oral formulation of grapiprant, for treating cancers; in December 2019, preliminary data were expected in 2020
Grapiprant (trade name Galliprant) is a small molecule drug that belongs in the piprant class. This analgesic and anti-inflammatory drug is primarily used as a pain relief for mild to moderate inflammation related to osteoarthritis in dogs. Grapiprant has been approved by the FDA’s Center for Veterinary Medicine and was categorized as a non-cyclooxygenase inhibiting non-steroidal anti-inflammatory drug (NSAID) in March 2016.[1]
Preclinical studies also indicate that grapiprant is not only efficacious as a acute pain but also in chronic pain relief and inflammation drug. The effect of the drug is directly proportional to the dosage and its effects were comparable to human medication such as rofecoxib and piroxicam.[2]
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Grapiprant is also used in humans, and was researched to be used as a pain control and inflammation associated with osteoarthritis. The effect of grapiprant could be explained through the function of prostaglandin E2, in which acts as a pro-inflammatory mediator of redness of the skin, edema and pain which are the typical signs of inflammation. The effect of PGE2 stems from its action through the four prostaglandin receptor subgroups EP1, EP2, EP3 and EP4, in which the prostaglandin EP4 receptor acts as the main intermediary of the prostaglandin-E2-driven inflammation. Grapiprant is widely accepted in veterinary medicine due to its specific and targeted approach to pain management in dogs. The serum concentration of grapiprant is increased when used in conjunction with other drugs such as acetaminophen, albendazole, and alitretinoin.
Common side effects are intestinal related effects such as mild diarrhea, appetite loss, and vomiting.[3] Additionally, it is found that it might lead to reduced tear production due to it being a sulfa-based medication and also reduced albumin levels.
Grapiprant, a prostanoid EP4 receptor antagonist, is in phase II clinical trials at AskAt for the treatment of chronic pain. Phase I/II clinical trials are ongoing at Arrys Therapeutics in combination with pembrolizumab for the treatment of patients with microsatellite stable colorectal cancer and in patients with advanced or metastatic PD-1/L1 refractory non-small cell lung cancer (NSCLC).
Medical uses
Grapiprant is used once a day as an oral pain relief for dogs with inflammation-related osteoarthritis. It is a non-steroidal anti-inflammatory (NSAID) that functions as a targeted action to treat osteoarthritis pain and inflammation in dogs.
Mechanism of action
Grapiprant acts as a specific antagonist that binds and blocks the prostaglandin EP4 receptor, one out of the four prostaglandin E2 (PGE2) receptor subgroups. The EP4 receptor then mediates the prostaglandin-E2-elicited response to pain, and hence grapiprant was proven to be effective in the decrease of pain in several inflammatory pain models of rats. It was also proven to be effective in reducing osteoarthritis-related pain in humans, which serves as a proof for its mechanism of action. The approximate calculation for canine efficacy dose is between the range of 1.3 and 1.7 mg/kg, in conjunction with a methylcellulose suspending agent. Based on the calculations from the comparisons of binding affinity of grapiprant to the EP4 receptors of dogs, rats, and humans, the study of plasma and serum protein binding determinations, the effective doses determined in inflammation pain models of rats, and human-related clinical studies, it is evaluated that Grapiprant should be administered just once a day. The approved dose of the commercial Grapiprant tablet by the FDA for the pain relief and inflammation associated with osteoarthritis to dogs is reported to be 2 mg/kg a day.[4]
Absorption
Studies in animals such as horses have shown the presence of Grapiprant in serum 72 hours with a concentration >0.005 ng/ml after the initial administration of a dose of 2 mg/kg. Grapiprant is expeditiously absorbed and the reported serum concentration was reported to be 31.9 ng/ml in an amount of time of 1.5 hours. The actual body exposure to grapiprant after administration of one dose was shown to be 2000 ng.hr/ml. The degree and rate at which grapiprant is absorbed into the body, presents a mean bioavailability of 39%. A significant reduction in the bioavailability, concentration time and maximal concentration were reported to have occurred after food intake.[1] And thus, grapiprant is usually not administered with food as it will not be as efficient.[5]
Distribution
The volume of distribution in cat studies was reported to be 918 ml/kg.[1]
Route of elimination
Following an oral administration, the majority of the dose was metabolized within the first 72 hours. Equine studies have shown that grapiprant is present in urine 96 hours after the first administration of a dose of 2 mg/kg and has a concentration >0.005 ng/ml. From the excreted dose conducted in horses, it is found that 55%, 15% and 19% of the orally-administered dose was excreted in bile, urine, and faeces respectively.[1]
Toxicity
Safety studies conducted on grapiprant have demonstrated that it generally possesses an exceptional safety profile and a wide safety margin in veterinary studies.[6] In animal studies, a research on 2.5-12 times overdose was conducted for grapiprant and the study resulted in soft-blobs and mucous-filled faeces, occasional bloody stools and emesis.
PATENT
WO-2020014465
Novel crystalline forms of grapiprant and their salts eg HCl (designated as Form A), useful for inhibiting prostaglandin EP4 receptor activity and treating cancers.
Prostaglandins are mediators of pain, fever and other symptoms associated with inflammation. Prostaglandin E2 (PGE2) is the predominant eicosanoid detected in inflammation conditions. In addition, it is also involved in various physiological and/or pathological conditions such as hyperalgesia, uterine contraction, digestive peristalsis, awakeness, suppression of gastric acid secretion, blood pressure, platelet function, bone metabolism, angiogenesis or the like.
[0003] Four PGE2 receptor subtypes (EP1, EP2, EP3 and EP4) displaying different pharmacological properties exist. The EP4 subtype, a Gs-coupled receptor, stimulates cAMP production as well as PI3K and GSK3P signaling, and is distributed in a wide variety of tissue suggesting a major role in PGE2-mediated biological events. Various EP4 inhibitors have been described previously, for example, in WO 2002/032900, WO 2005/021508, EiS 6,710,054, and US 7,238,714, the contents of which are incorporated herein by reference in their entireties.
[0004] Accordingly, there is a need for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. The present invention addresses such a need.
It has now been found that compounds of the present invention, and compositions thereof, are useful for treating, preventing, and/or reducing severity of a proliferative disorder associated with prostaglandin EP4 receptor activity. In general, salt forms and co-crystal forms, and pharmaceutically acceptable compositions thereof, are useful for treating or lessening the severity of proliferative disorders associated with prostaglandin EP4 receptor activity, as described in detail herein. Such compounds are represented by the chemical structure below, denoted as compound A (also known as grapiprant):
A
or a pharmaceutically acceptable salt thereof.
United States Patent 7,960,407, filed March 1, 2006 and issued June 14, 2011 (“the ‘407 patent,” the entirety of which is hereby incorporated herein by reference), describes certain EP4 inhibitor compounds. Such compounds include compound A:
or a pharmaceutically acceptable salt thereof.
[0037] Compound A, N-[({2-[4-(2-Ethyl-4,6-dimethyl-lH-imidazo[4,5-c]pyridin-l-yl) phenyl]ethyl}amino)carbonyl]-4-methylbenzenesulfonamide, is described in detail in the ‘407
patent, including its synthetic route. The ‘407 patent also discloses a variety of physical forms of compound A.
[0038] It would be desirable to provide a solid form of compound A (e.g., as a co-crystal thereof or salt thereof) that imparts characteristics such as improved aqueous solubility, stability and ease of formulation. Accordingly, the present invention provides both co-crystal forms and salt forms of compound A:
A.
PATENT
WO 2002032900
PATENT
WO 2002032422
Family members of the product case ( WO0232422 ) of grapiprant have protection in most of the EU states until October 2021 and expire in the US in October 15, 2021.
PATENT
WO 2003086371
PATENT
WO2020014445 covering combinations of grapiprant and an immuno-oncology agent.
WO 2005102389
WO 2006095268
US 7960407
US 20190314390
References
- ^ Jump up to:a b c d “Grapiprant”. http://www.drugbank.ca. Retrieved 2019-05-15.
- ^ PubChem. “Grapiprant”. pubchem.ncbi.nlm.nih.gov. Retrieved 2019-05-15.
- ^ Paul Pion, D. V. M.; Spadafori, Gina (2017-08-08). “Veterinary Partner”. VIN.com.
- ^ Nagahisa, A.; Okumura, T. (2017). “Pharmacology of grapiprant, a novel EP4 antagonist: receptor binding, efficacy in a rodent postoperative pain model, and a dose estimation for controlling pain in dogs”. Journal of Veterinary Pharmacology and Therapeutics. 40 (3): 285–292. doi:10.1111/jvp.12349. ISSN 1365-2885. PMID 27597397.
- ^ Paul Pion, D. V. M.; Spadafori, Gina (2017-08-08). “Veterinary Partner”. VIN.com.
- ^ Kirkby Shaw, Kristin; Rausch-Derra, Lesley C.; Rhodes, Linda (February 2016). “Grapiprant: an EP4 prostaglandin receptor antagonist and novel therapy for pain and inflammation”. Veterinary Medicine and Science. 2 (1): 3–9. doi:10.1002/vms3.13. ISSN 2053-1095. PMC 5645826. PMID 29067176.
 |
|
| Clinical data | |
|---|---|
| Trade names | Galliprant |
| Routes of administration |
Oral |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | 6.6 L/kg, high volume of distribution |
| Elimination half-life | 5.86 hours in horses |
| Excretion | Urine |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C26H29N5O3S |
| Molar mass | 491.61 g·mol−1 |
| 3D model (JSmol) | |
//////////////GRAPIPRANT, 415903-37-6, UNII-J9F5ZPH7NB, CJ 023423, CJ-023423, RQ-00000007, MR10A7, Galliprant, Phase II, Arrys Therapeutics, CANCER, PAIN, AskAt
CCC1=NC2=C(N1C3=CC=C(C=C3)CCNC(=O)NS(=O)(=O)C4=CC=C(C=C4)C)C=C(N=C2C)C