Graphical abstract

PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Phase2 drugs (Page 23)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

Radezolid
869884-78-6 cas no
http://www.ama-assn.org/resources/doc/usan/radezolid.pdf
Rib-X Pharmaceuticals
Phase II completed
N-{[(5S)-3-(2-fluoro-4′-{[(1H-1,2,3-triazol-5-ylmethyl)amino]methyl}biphenyl-4-yl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide
Rib-X Pharmaceuticals has completed two Phase II clinical trials of radezolid for the treatment of pneumonia and uncomplicated skin infections. The trial completion dates were in 2008 and 2009, but to date the Phase III trials have not been initiated [1-6].
Radezolid (INN, codenamed RX-1741) is a novel oxazolidinone antibiotic being developed by Rib-X Pharmaceuticals, Inc. for the treatment of serious multi-drug–resistant infections. Radezolid has completed two phase-II clinical trials. One of these clinical trials was for uncomplicated skin and skin-structure infections (uSSSI) and the other clinical trial was for community acquired pneumonia (CAP).
Oxazolidinone antibiotics are a relatively new class of antibacterial agents with activity against a broad spectrum of gram-positive pathogens. The first member of this new class to be commercialized, linezolid, was approved in 2000. Since that time the development of linezolid resistant organisms has prompted efforts to discover more effective members of the oxazolidinone class.
A new family of biaryl oxazolidinone antibacterials with activity against both linezolid-susceptible and -resistant Gram-positive bacteria, as well as certain Gram-negative bacteria has been reported (see Bioorganic & Medicinal Chemistry Letters, 2008, 18, 6175-6178, and PCT Patent Publication WO 2005/019211).
Among the known biaryloxazolidinones is N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-bipheny- l-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide, more commonly known as radezolid (RX-1741), currently being developed for multi-drug-resistant infections.
Although a monohydrochloride salt of radezolid was disclosed in PCT Patent Publication WO 2006/133397, there is a continuing need for new salts and polymorphs thereof having improved properties such as solubility to optimize bioavailability on therapeutic administration.
Synthesis 1
http://www.google.co.il/patents/WO2005019211A2?hl=iw&cl=en
Scheme A

Scheme B

Scheme C

Scheme D

Scheme E


Scheme G


Scheme I

Scheme J

producing compounds of the present invention. Known iodoaryl oxazolidinone intermediate 50 (see U.S. Patent Nos. 5,523,403 and 5,565,571) is coupled to a substituted aryl boronic acid (the Suzuki reaction) to produce biaryl alcohol 51. Mesylate 52, azide 53, and amine 54 are then synthesized using chemistry well known to those skilled in the art. Scheme 1

NaN3, DMF, 70 °C


NO 2
http://www.google.com/patents/US20100234615
| TABLE 1 | |
| Compound | |
| Number | Structure |
| 1 |
 |
Example 1 Synthesis of Compound 1
Compound 1 and its hydrochloride salt are synthesized according to the following Scheme:


4-Methoxybenzyl Azide
1001.
A solution of 4-methoxybenzyl chloride 1000 (51.8 g, 331.0 mmol) in anhydrous DMF (200 mL) was treated with solid sodium azide (21.5 g, 331.0 mmol, 1.0 equiv) at 25° C., and the resulting mixture was stirred at 25° C. for 24 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was quenched with H2O (400 mL) and ethyl acetate (EtOAc, 400 mL) at room temperature.
The two layers were separated, and the aqueous layer was extracted with EtOAc (200 mL). The combined organic extracts were washed with H2O (2×200 mL) and saturated NaCl aqueous solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude 4-methoxybenzyl azide (51.2 g, 53.95 g theoretical, 94.9% yield) was obtained as colorless oil, which by HPLC and 1H NMR was found to be essentially pure and was directly used in the subsequent reaction without further purifications. For 4-methoxybenzyl azide 1001:
1H NMR (300 MHz, CDCl3) δ 3.84 (s, 3H, ArOCH3), 4.29 (s, 2H, Ar—CH2), 6.96 (d, 2H, J=8.7 Hz), 7.28 (d, 2H, J=7.8 Hz).
C-[1-(4-Methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-Methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine
(1003 and 1004).
A solution of 4-methoxybenzyl azide 1001 (61.2 g, 375.5 mmol) in toluene (188 mL) was heated with propargylamine 1002 (commercially available, 30.97 g, 38.6 mL, 563.0 mmol, 1.5 equiv) at 25° C., and the resulting reaction mixture was warmed up to gentle reflux at 100-110° C. for 21 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was cooled down to room temperature before being concentrated in vacuo to remove the excess amount of propargylamine and solvent.
The oily residue was then treated with 30% ethyl acetate-hexane (v/v, 260 mL), and the resulting mixture was warmed up to reflux and stirred at reflux for 30 min before being cooled down to room temperature for 1 h. The pale-yellow solids were then collected by filtration, washed with 30% ethyl acetate-hexane (v/v, 2×100 mL), and dried in vacuo at 40° C. for overnight to afford the crude, cycloaddition product (78.8 g, 81.75 g theoretical, 96.4%) as a mixture of two regioisomers, C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004), in a ratio of 1.2 to 1 by 1H NMR.
The crude cycloaddition product was found to be essentially pure and the two regioisomers were not separated before being used directly in the subsequent reaction without further purification. For 1003 and 1004:
1H NMR (300 MHz, DMSO-d6) δ 1.82 (br. s, 2H, NH2), 3.72 and 3.73 (two s, 3H, Ar—OCH3), 5.47 and 5.53 (two s, 2H, ArCH2), 6.89 and 6.94 (two d, 2H, J=8.7 Hz, Ar—H), 7.17 and 7.29 (two d, 2H, J=8.7 Hz, Ar—H), 7.58 and 7.87 (two br. s, 1H, triazole-CH); C11H14N4O, LCMS (EI) m/e 219 (M++H) and 241 (M++Na).
4-({tert-Butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-Butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009).
Method A. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.0 g, 91.74 mmol) in 1,2-dichloroethane (DCE, 280 mL) was treated with 4-formylphenylboronic acid 1005 (commercially available, 12.39 g, 82.57 mmol, 0.9 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.2 g, 137.6 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture was concentrated in vacuo. The residue, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with tetrahydrofuran (THF, 100 mL) and water (H2O, 100 mL).
The resulting solution was subsequently treated with solid potassium carbonate (K2CO3, 37.98 g, 275.2 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.02 g, 91.74 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo.
The crude, regioisomeric 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 35.98 g, 37.32 g, 96.4%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo.
This crude material was directly used in the subsequent reaction without further purification. For 1008 and 1009:
1H NMR (300 MHz, DMSO-d6) δ 1.32 and 1.37 (two br. s, 9H, COOC(CH3)3), 3.70, 3.73 and 3.74 (three s, 3H, Ar—OCH3), 4.07-4.39 (m, 4H), 5.49 and 5.52 (two s, 2H), 6.70-8.04 (m, 9H, Ar—H and triazole-CH); C23H29BN4O5, LCMS (EI) m/e 453 (M++H) and 475 (M++Na).
Method B. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.06 g, 92.0 mmol) in tetrahydrofuran (THF, 300 mL) was treated with 4-formylphenylboronic acid (13.11 g, 87.4 mmol, 0.95 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.25 g, 138.0 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with water (H2O, 200 mL).
The resulting aqueous solution was subsequently heated with solid potassium carbonate (K2CO3, 38.0 g, 276 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.08 g, 92 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude, 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 38.45 g, 39.50 g, 97.3%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo. This crude material was found to be essentially identical in every comparable aspect as the material obtained from Method A and was directly used in the subsequent reaction without further purification.
(5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester
(1011 and 1012).
A suspension of the crude regioisomeric mixture of 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 37.62 g, 83.23 mmol) and N-[3-(3-fluoro-4-iodo-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1010, 28.32 g, 74.9 mmol, 0.90 equiv) in toluene (150 mL) was treated with powder K2CO3 (34.45 g, 249.7 mol, 3.0 equiv), EtOH (50 mL), and H2O (50 mL) at 25° C.,
and the resulting mixture was degassed three times under a steady stream of Argon at 25° C. Pd(PPh3)4 (866 mg, 0.749 mmol, 0.01 equiv) was subsequently added to the reaction mixture, and the resulting reaction mixture was degassed three times again under a stead stream of Argon at 25° C. before being warmed up to gentle reflux for 18 h. When TLC and HPLC/MS showed the coupling reaction was complete, the reaction mixture was cooled down to room temperature before being treated with H2O (100 mL) and ethyl acetate (100 mL). The two layers were then separated, and the aqueous layer was extracted with EtOAc (100 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×150 mL), H2O (100 mL), and the saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The residual oil was solidified upon standing at room temperature in vacuo to afford the crude, (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-y]methyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester (1011) and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1012) as a regioisomeric mixture.
This crude product (43.36 g, 49.28 g theoretical, 88%) was used directly in the subsequent reaction without further purification. For the mixture of 1011 and 1012: 1H NMR (300 MHz, DMSO-d6) δ 1.35 and 1.38 (two br. s, 9H, COO(CH3)3), 1.85 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.73 and 3.76 (two s, 3H, Ar—OCH3), 3.79 (dd, 1H, J=6.6, 9.1 Hz), 4.18 (t, 1H, J=9.1 Hz), 4.35-4.43 (m, 4H), 4.73-4.81 (m, 1H), 5.50 (br. s, 2H), 6.90 and 6.98 (two d, 2H, J=8.7 Hz), 7.28 and 7.32 (two d, 2H, J=8.7 Hz), 7.35 (dd, 2H, J=2.2, 8.6 Hz), 7.42 (dd, 1H, J=2.2, 8.6 Hz), 7.49-7.63 (m, 4H, aromatic-H), 7.90 and 7.99 (two br. s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3); C35H39FN6O6, LCMS (EI) m/e 659 (M++H) and 681 (M++Na).
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1013)
and
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H–[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1014).
A solution of a regioisomeric mixture of (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1011 and 1012, 37.28 g, 56.65 mmol) in ethyl acetate (EtOAc, 150 mL) and methanol (MeOH, 30 mL) was treated with a solution of 4 N hydrogen chloride in 1,4-dioxane (113.3 mL, 453.2 mmol, 8.0 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 12 h. When TLC and HPLC/MS showed that the N-BOC deprotection reaction was complete,
the solvents were removed in vacuo. The residue was then suspended in 250 mL of 5% methanol (MeOH) in acetonitrile (CH3CN), and the resulting slurry was stirred at room temperature for 1 h. The solids were then collected by filtration, washed with toluene (2×100 mL) and 5% methanol in acetonitrile (2×50 mL), and dried in vacuo to afford a regioisomeric mixture of the crude, (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 30.0 g, 33.68 g theoretical, 89.1% yield) as off-white crystals in a ratio of 1.2 to 1.
This material was found by 1H NMR and HPLC/MS to be essentially pure and was directly used in the subsequent reactions without further purification. For the regioisomeric mixture of 1013 and 1014:
1H NMR (300 MHz, DMSO-d6) δ 1.84 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.71 and 3.74 (two s, 3H, Ar—OCH3), 3.80 (dd, 1H, J=6.6, 9.1 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.23-4.30 (m, 4H), 4.73-4.80 (m, 1H), 5.58 and 5.70 (two s, 2H), 6.88 and 6.93 (two d, 2H, J=8.7 Hz), 7.15 and 7.32 (two d, 2H, J=8.7 Hz), 7.43 (dd, 2H, J=2.2, 8.6 Hz), 7.52-7.62 (m, 6H, aromatic-H), 8.28 (s, 1H, triazole-CH), 8.32 (t, 1H, J=5.8 Hz, NHCOCH3), 9.91 and 10.32 (two br. s, 2H, ArCH2N+H2); C30H31FN6O4, LCMS (EI) m/e 559 (M++H) and 581 (M++Na).
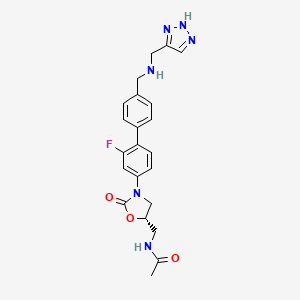
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide hydrochloride (1 hydrochloride salt).
A solution of the crude regioisomeric mixture of (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-1H-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 29.17 g, 49.07 mmol) in trifluoroacetic acid(TFA, 150 mL) was warmed up to 65-70° C., and the resulting reaction mixture was stirred at 65-70° C. for 12 h. When TLC and HPLC/MS showed that the deprotection reaction was complete, the solvents were removed in vacuo.
The residual solids were then treated with ethyl acetate (EtOAc, 100 mL) and H2O (150 mL) before being treated with a saturated aqueous solution of sodium carbonate (30 mL) at room temperature. The resulting mixture was then stirred at room temperature for 1 h before the solids were collected by filtration, washed with EtOAc (2×50 mL) and H2O (2×50 mL), and dried in vacuo at 40-45° C. to afford the crude, (5S)-N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl)-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1 as the free base, 18.9 g, 21.49 g theoretical, 87.9%) as off-white powders, which by HPLC/MS and 1H NMR was found to be one pure regioisomer and this regioisomer was found to be identical as the material obtained from deprotection of 1013 alone by the same method.
For 1 as the free base: 1H NMR (300 MHz, DMSO-d6) δ 1.85 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.74 (s, 2H), 3.77 (s, 2H), 3.79 (dd, 1H, J=6.4, 9.2 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.72-4.81 (m, 1H), 7.39-7.62 (m, 7H, aromatic-H), 7.73 (s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH); C22H23FN6O3, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
A suspension of 1 free base (18.0 g, 41.1 mmol) in ethyl acetate (EtOAc, 80 mL), and methanol (MeOH, 20 mL) was treated with a solution of 4.0 N hydrogen chloride in 1,4-dioxane (41.1 mL, 164.4 mmol, 4.0 equiv) at room temperature, and the resulting mixture was stirred at room temperature for 8 h. The solvents were then removed in vacuo, and the residue was further dried in vacuo before being treated with a mixture of 10% methanol in acetonitrile (80 mL). The solids were collected by filtration, washed with 10% MeOH/acetonitrile (2×40 mL), and dried in vacuo to afford 1 hydrochloride salt (18.13 g, 19.50 g theoretical, 93% yield) as off-white crystals.
The crude 1 hydrochloride salt can be recrystallized from acetonitrile and water, if necessary, according to the following procedure: A suspension of the crude 1 hydrochloride salt (50.0 g) in acetonitrile (1250 mL) was warmed up to reflux before the distilled water (H2O, 280 mL) was gradually introduced to the mixture. The resulting clear yellow to light brown solution was then stirred at reflux for 10 min before being cooled down to 45-55° C. The solution was then filtered through a Celite bed at 45-55° C., and the filtrates were gradually cooled down to room temperature before being further cooled down to 0-5° C. in an ice bath for 1 h. The solids were then collected by filtration, washed with acetonitrile (2×50 mL), and dried in vacuo at 40° C. for 24 h to afford the recrystallized 1 hydrochloride salt (42.5 g, 50.0 g theoretical, 85% recovery) as off-white crystals.
For 1: 1H NMR (300 MHz, DMSO-d6) δ 1.86 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.84 (dd, 1H, J=6.4, 9.2 Hz), 4.19 (t, 1H, J=9.1 Hz), 4.24 (br. s, 2H), 4.31 (br. s, 2H), 4.74-4.79 (m, 1H), 7.44 (dd, 1H, J=2.2, 8.6 Hz), 7.57-7.66 (m, 6H, aromatic-H), 8.17 (s, 1H, triazole-CH), 8.30 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH);
13C NMR (75 MHz, DMSO-d6) δ 22.57, 40.69, 41.50, 47.36, 49.23, 71.85, 105.70 (d, J=28.5 Hz), 114.14 (d, J=2.9 Hz), 122.29 (d, J=13.3 Hz), 128.82 (d, J=3.0 Hz), 130.70, 130.94, 131.0, 131.22, 135.30, 137.92 (br. s), 139.66 (d, J=11.2 Hz), 154.11, 159.13 (d, J=243.5 Hz), 170.19;
C22H23FN6O3—HCl, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
……………………………..
http://www.sciencedirect.com/science/article/pii/S0960894X0801192X

| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6969726 * | Jun 2, 2004 | Nov 29, 2005 | Rib X Pharmaceuticals Inc | Biaryl heterocyclic compounds and methods of making and using the same |
| US20050043317 * | Jun 2, 2004 | Feb 24, 2005 | Jiacheng Zhou | Biaryl heterocyclic compounds and methods of making and using the same |
|
9-17-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
9-17-2010
|
Process for the synthesis of triazoles
|
|
|
4-28-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
11-26-2008
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
10-26-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
12-13-2006
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
11-30-2005
|
Biaryl heterocyclic compounds and methods of making and using the same
|

October 10, 2012
Rib-X Pharmaceuticals announced that the FDA designated radezolid as a Qualified Infectious Disease Product (QIDP) for the indications of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CABP).
The QIDP designation will enable Rib-X to benefit from certain incentives for the development of new antibiotics, including an additional five years of market exclusivity, priority review and eligibility for fast-track status, provided under the new Generating Antibiotic Incentives Now (GAIN) program. GAIN was included in the FDA Safety and Innovation Act (FDASIA), formerly known as PDUFA V, which received bipartisan Congressional support and was signed into law by President Obama in July 2012.
Radezolid has completed two Phase 2 clinical trials with an oral formulation in uncomplicated skin and skin structure infections (uSSSI) and in CABP. A Phase 1 study with an IV formulation was recently completed in healthy subjects. Rib-X recently announced data from a positive Phase 1 IV dosing study conducted in healthy subjects and an in vivo long-term safety study vs. linezolid (Zyvox; Pfizer).
Radezolid is a next-generation oxazolidinone with a safety profile permitting long-term treatment of resistant infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
For more information call (203) 624-5606 or visit www.rib-x.com



BC-3781
Topical pleuromutilin antibiotic agent
Gram-positive, including MRSA, PHASE 2 COMPLETED
Nabriva (Austria)
SEE UPDATED POST AT https://newdrugapprovals.org/2014/10/10/nabrivas-lefamulin-bc-3781-receives-fda-fast-track-status-to-treat-cabp-and-absssi/ ………….C0NTAINS SYNTHESIS
BC-3781
The pleuromutilin BC-3781 belongs to the first generation of pleuromutilins to combine excellent oral
bioavailability with substantial activity against Gram-positive pathogens and atypicals as well as some
Gram-negative pathogens. In particular, BC-3781 is highly active against multi-drug resistant (MDR)
pathogens including methicillin resistant Staphylococcus aureus (MRSA), MDR Streptococcus pneumonia
(i.e. macrolide and quinolone resistance), and vancomycin resistant Enterococcus faecium. It is
characterized by excellent in vivo activities (e.g. pneumonia model), outstanding PK/PD parameters,
allowing once a day dosing, and a novel mode of action. BC-3781 is being developed for both oral and IV
administration and is intended for the treatment of serious multi-drug resistant skin & skin structure
infections (CSSI) and moderate to severe pneumonia (CAP, HAP etc).
Pleuromutilins have been known since 1951, but only entered the market![]() in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
Nabriva is currently working on the development of new compounds is this class. The lead compound, BC-3781, if approved, will be the first pleuromutilin for systemic use in humans.
The compound shows potent in vitro activity against a large collection of staphylococci, streptococci, andE. faecium. When compared to linezolid and vancomycin, the compound shows greater overall potency againstS. aureus [121]. BC-3781 shows improved activity against most bacteria commonly associated with community-acquired respiratory tract infections, the compound is especially potent against S. pneumoniaincluding penicillin resistant strains. It also shows improved activity against H. influenza, M. catarrhalis, M. pneumoniae and C. pneumoniae.
BC-3781 is undergoing Phase I clinical trials for CAP and in March of 2011 has completed a Phase II clinical study comparing it to vancomycin for treatment of aBSSSI [119,120,121,122,123]. Nabriva Therapeutics AG announced that the cooperation with Forest Laboratories to develop the compound had elapsed, and that Nabriva retained all rights in BC-3781. The company informed that the product was Phase III ready and that it was seeking partners to continue further development [203].
Nabriva is also developing BC-7013 for topical use against Gram-positive infections and working on the discovery of new pleuromutilins [119,124].
Dr William Prince, CMO Nabriva Therapeutics commented:
“This is the first patient study with a systemic pleuromutilin. It will be an important proof of concept
for an exciting new class of antibiotics. The phase II study builds on our extensive preclinical and
phase I data which have demonstrated that BC-3781 can achieve therapeutically relevant blood and
tissue levels in man with excellent tolerability when administered by either oral or intravenous
routes.”
Dr. David Chiswell, CEO Nabriva Therapeutics commented:
“With a worldwide problem due to antibiotic resistant bacteria, there is a very significant need for
new classes of antibiotics with unique modes of action such as the pleuromutilins. The commercial
prospects for BC-3781 as the leading compound of an exciting new class are excellent, especially as it
has an ideal anti-bacterial spectrum for both skin and respiratory infections and is being developed
with both oral and intravenous formulations”
BC-3781 is highly active against key pathogens, including MRSA, associated with skin infections and
community and hospital acquired pneumonia and is more potent than Linezolid and vancomycin. The
compound’s novel mode of action ensures that it overcomes resistance mechanisms affecting all
approved classes of antibiotics. BC-378
About Nabriva Therapeutics
Nabriva Therapeutics is a biotechnology company focused on developing a new class of antibiotics for
the treatment of serious infections caused by resistant pathogens. Nabriva’s lead systemic product,
BC-3781, is being developed for the treatment of serious skin infections and bacterial pneumonia
caused by S. aureus, , S. pneumoniae, H. influenza, Mycoplasma, Legionella and other bacteria,
including drug resistant strains such as MRSA and vancomycin resistant E. faecium. In addition,
Nabriva Therapeutics’ topical pleuromutilin product candidate, BC-7013, is in clinical phase I. Nabriva
Therapeutics has a proven track record in world-class medicinal chemistry, clinical expertise, a
seasoned management team and solid IP. Nabriva Therapeutics is located in Vienna, Austria.
For more information on Nabriva please visit http://www.nabriva.com.
REF
http://www.phase4-partners.com/wp-content/uploads/2013/09/100412.pdf
http://www.glsv-vc.com/downloads/2010-06-02_First%20Patient_PressRelease.pdf
119
Nabriva. Pleuromutilins. Available online: http://www.nabriva.com/programs/pleuromutilins/ (accessed on 7 December 2012).
120
Forest Laboratories. Our pipeline: Solid, and set for further growth. Available online: http://www.frx.com/research/pipeline.aspx (accessed on 13 April 2013).
121
Sader, H.S.; Biedenbach, D.J.; Paukner, S.; Ivezic-Schoenfeld, Z.; Jones, R.N. Antimicrobial activity of the investigational pleuromutilin compound BC-3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2012,56, 1619–1623, doi:10.1128/AAC.05789-11.
122
Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175, doi:10.1093/jac/dks001.
123
Nabriva Therapeutics AG. Study comparing the safety and efficacy of two doses of BC-3781 vs. vancomycin in patients with acute bacterial skin and skin structure infection (ABSSSI). Available online: http://www.clinicaltrials.gov/ct2/show/NCT01119105 (accessed on 13 April 2013).
124
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. NY Acad. Sci. 2011, 1241, 71–81, doi:10.1111/j.1749-6632.2011.06219.x.
 valnemulin
valnemulin
 retapamulin
retapamulin

FINAFLOXACIN
(S-cyano-1-cyclopropyl-ό-fluoro-T-^aS, 7aS)-hexahydropyrrolo [3,4- b]-1,4-oxazin-6(2H)-yl]-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid)
7-[(4aS,7aS)-3,4,4a,5,7,7a-hexahydro-2H-pyrrolo[3,4-b][1,4]oxazin-6-yl]-8-cyano-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid |
BAY-35-3377
BY-377
CAS Registry Number: 209342-40-5
HYD SALT
(-)-(4aS,7aS)-8-Cyano-1-cyclopropyl-6-fluoro-4-oxo-7-(perhydropyrrolo[3,4-b]-1,4-oxazin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid hydrochloride
209342-41-6,
| C20 H19 F N4 O4 . Cl H | |
| MW | 434.849 |
Synonyms: Finafloxacin, UNII-D26OSN9Q4R,
MerLion Pharmaceuticals (Singapore)…POSTER…….http://www.merlionpharma.com/sites/default/files/file/PPS/F1-2036_Wohlert.pdf
H. pylori, Broad-Spectrum
Finafloxacin is a novel fluoroquinolone being developed by MerLion Pharmaceuticals. Under neutral pH conditions (pH 7.2–7.4), the compound has shown in vitro activity equivalent to that of ciprofloxacin. However, under slightly acidic pH5.8 the compound shows enhanced potency.
Other marketed fluoroquinolones, such as ciprofloxacin, levofloxacin and moxifloxacin, exhibit reduced activity at slightly acidic pH 5.0–6.5. This feature of finafloxacin makes the compound suitable for use in the treatment of infections in acidic foci of infections such as urinary tract infections
Finafloxacin hydrochloride, a novel highly potent antibiotic, is in phase III clinical trials at Alcon for the treatment of ear infections. MerLion Pharmaceuticals is evaluating the product in phase II clinical trials at for the treatment of Helicobacter pylori infection and for the treatment of lower uncomplicated urinary tract infections in females.
A quinolone, finafloxacin holds potential for the treatment of Helicobacter pylori infection and urinary tract infection. Unlike existing antibiotics, finafloxacin demonstrates a unique acid activated activity whereby it becomes increasingly active under acidic conditions.
In 2009, a codevelopment agreement was signed between Chaperone Technologies and MerLion Pharmaceuticals. In 2011, finafloxacin hydrochloride was licensed to Alcon by MerLion Pharmaceuticals in North America for the treatment of ear infections.
MerLion Pharmaceuticals has announced that the FDA has granted a Qualified Infectious Disease Product Designation and Fast Track Status for finafloxacin. The company is currently recruiting patients for the Phase II clinical trial of the compound for the treatment of complicated urinary tract infections (cUTI) and/or acute pyelonephritis compared to ciprofloxacin
Finafloxacin and derivatives thereof can be synthesized according to the methods described in U.S. Patent No. 6,133,260 to Matzke et al., the contents of which are herein incorporated by reference in their entirety. The compositions of the invention are particularly directed toward treating mammalian and human subjects having or at risk of having a microbial tissue infection. Microbial tissue infections that may be treated or prevented in accord with the method of the present invention are referred to in J. P. Sanford et al., “The Sanford Guide to Antimicrobial Therapy 2007” 37 Edition (Antimicrobial Therapy, Inc.). Particular microbial tissue infections that may be treatable by embodiments of the present invention include those infections caused by bacteria, protozoa, fungi, yeast, spores, and parasites.
SYNTHESIS
WO1998026779A1
http://www.google.sc/patents/WO1998026779A1 COPY PASTE ON BROWSER
8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-di-azabicyclo [4.3.0] non-8-yl)-l, 4-dihydro-4-oxo-3-quinolinecarboxylic acid.
The compounds, which are suitable for use in the invention are known already to some extent in EP-A-0350733, EP-A-0550903 as well as from DE-A-4329600 or can be prepared according to the processes described in .
If, for example 9,10-difluoro-3 ,8-dimethyl-7-oxo-2 ,3-dihydro-7H-pyrido [l ,2,3-d, e] [l, 3,4] benzoxadiazine-6 -carboxylic acid and 2-oxa-5 ,8-diazabicyclo [4.3.0] nonane, the reaction can be represented by the following equation:

The 7-halo-quinolonecarboxylic acid derivatives used for preparing the compounds of Fomel (I) of the invention are known or can be prepared by known methods. Thus, the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid, or of the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro- l been ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester described in EP-A-0 276 700th The corresponding 7-fluoro derivatives can be, for example, via the following reaction sequence to build:

An alternative process for preparing the intermediate compound 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride as the starting material for the preparation of 7-chloro-
8-cyano-1-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is used (EP-A-0276700) and in the 3-cyano-2 ,4,5-trifluoro- benzoyl can be converted, is based on 5-fluoro-l ,3-xylene, 5-fluoro-l ,3-xylene, in the presence of a catalyst under ionic conditions in the nucleus disubstituted to 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene, and this is subsequently chlorinated chlorinated under free radical conditions in the side chains of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methylbenzene. This is the 2,4-dichloro-5-fluoro-3-dichloromethyl-benzoic acid to give 2,4-dichloro-5-fluoro-3-formyl-benzoic acid, and then hydrolyzed to 2,4-dichloro-5-fluoro-3 N-hydroxyiminomethyl acid implemented. By treatment with thionyl chloride, 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride is obtained, which can still be ,4,5-trifluoro-ben-zoylfluorid converted by a chlorine / fluorine exchange on-3-cyano-2 .



The amines used for the preparation of compounds of formula (I) according to the invention are known from EP-A-0550903, EP-A-0551653 as well as from DE-A-4 309 964th
An alternative to the synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-dihydro-drobromid or the free base 1 S, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0 ] nonane and the corresponding IR, 6R enantiomer provides the following path represents:
Starting material for this synthesis is the cis-l ,4-dihydroxy-2-butene, which is converted to the bis-mesylate with mesylation tosylamide for 1-tosylpyrrolidine. This is converted into the epoxide m-chloroperbenzoic. The ring opening of the epoxide by heating in isopropanol with ethanolamine to trans-3-hydroxy-4 – (2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl)-pyrrolidine in 80% yield. Tetrahydrofuran is then in pyridine / reacted with tosyl chloride, with cooling to Tris-tosylate, which as a crude product in a mixture with some tetra-tosyl derivative with basichen reaction conditions to give the racemic trans-5 ,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane is cylisiert. At this stage occurs with high selectivity of a chromatographic resolution kieselgelgebundenem poly (N-methacryloyl-L-leucine-d menthylamide) as the stationary phase. The desired enantiomer, (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane, is of a purity of
> 99% ee. Cleavage of the p-tosyl protecting groups is carried out with HBr-acetic acid to the lS, 6S-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide, the one with a base such as sodium or potassium hydroxide or with the aid of ion exchanger can be converted into the free base. The analogous sequence may be used for the preparation of lR, 6R-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide.
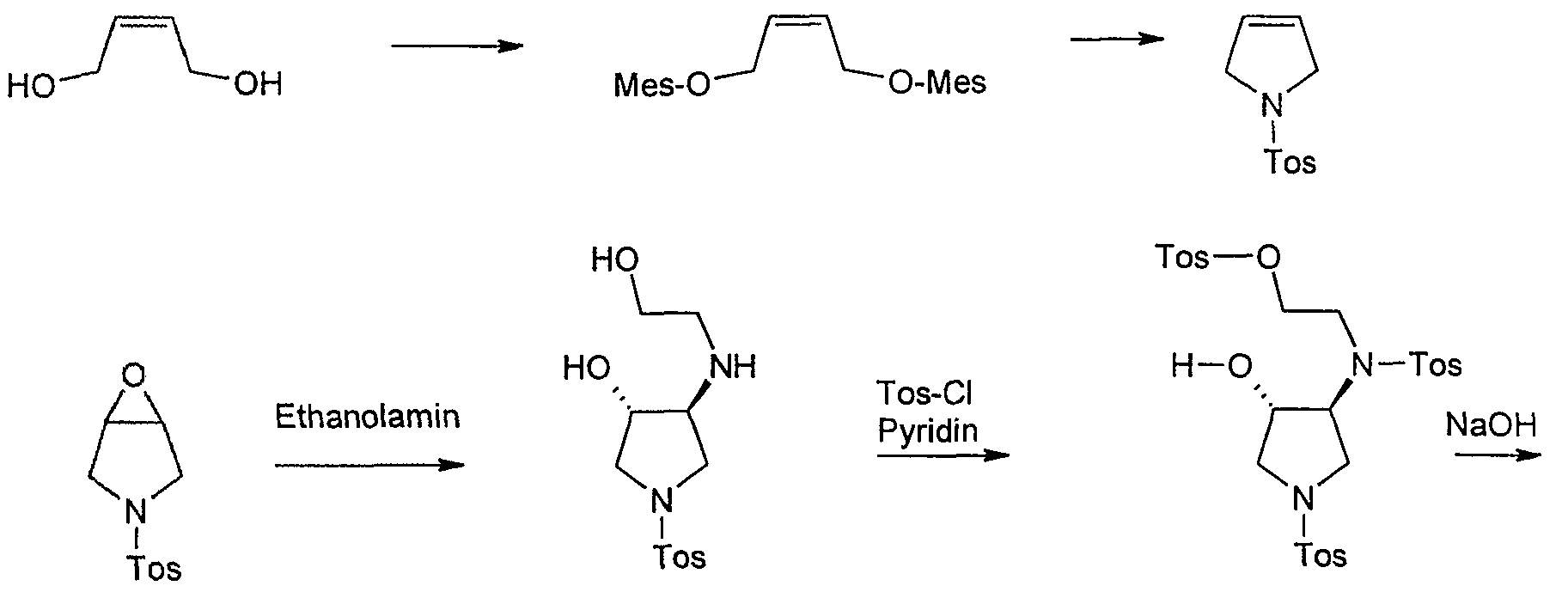

HBr / AcOH

Synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Examples of compounds of the invention are mentioned in addition to the compounds listed in the preparation examples, the compounds listed in Table 1 below, which can be used both in racemic form as well as enantiomerically pure or diastereomerically pure compounds. Table 1:


Example 1 Z
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester

a 3-bromo-2 ,4,5-trifluoro-benzoate
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 – trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 – cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene

a solvent-free
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 – dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 – 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene

In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid

In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid

In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 – 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 – 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor

In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl

50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 – dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 –
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride

76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 – cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 – (toluene-4-sulfonyl-pyrroline

In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 – (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o “|” h “CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-(‘toluene-4-sulfonyl’) pyrrolidine

643.7 g (2.65 mol) 3 – (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] – ftoluol-4-sulfonyl)-amino]-ethyl ester)

552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) – pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane

1092 g of crude trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl) – pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l – (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide

38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid

Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan

1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 – oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + – form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan

Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Example Z 19
2 – (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester

To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
Example Z 20
2 – (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate

To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester

To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid

33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
Example 1

A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl – 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-(‘(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] non-8-yl) – l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
……………….
Tetrahedron Lett 2009, 50(21): 2525
A novel approach to Finafloxacin hydrochloride (BAY35-3377)Pages 2525-2528 |
||
Finafloxacin hydrochloride, an important clinical compound was synthesized by a novel synthetic approach. An active intermediate ethyl 7-chloro-8-cyano-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate 19 was prepared by a new route. The chiral (S,S′)-N-Boc 10 was derived from protected pyrrolidine and the absolute stereochemistry was established by X-ray analysis.
http://www.sciencedirect.com/science/article/pii/S0040403909005875
……………….



| WO2011003091A1 * | 2 Jul 2010 | 6 Jan 2011 | Alcon Research, Ltd. | Compositions comprising finafloxacin and methods for treating ophthalmic, otic, or nasal infections |
| US7723524 | 29 Sep 2004 | 25 May 2010 | Daiichi Pharmaceutical Co., Ltd. | 8-cyanoquinolonecarboxylic acid derivative |
| US8536167 | 2 Jul 2010 | 17 Sep 2013 | Alcon Research, Ltd. | Methods for treating ophthalmic, otic, or nasal infections |
| DE4329600A1 * | 2 Sep 1993 | 9 Mar 1995 | Bayer Ag | Pyrido [1,2,3-d,e] [1,3,4] benzoxadiazinderivate |
| EP0276700A1 * | 15 Jan 1988 | 3 Aug 1988 | Bayer Ag | 8-Cyano-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids, process for their preparation, and antibacterial agents containing them |
| EP0350733A2 * | 30 Jun 1989 | 17 Jan 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 * | 28 Dec 1992 | 14 Jul 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0603887A2 * | 23 Dec 1993 | 29 Jun 1994 | Daiichi Pharmaceutical Co., Ltd. | Bicyclic amine derivatives |
| EP0676199A1 * | 23 Mar 1995 | 11 Oct 1995 | Pfizer Inc. | Use of trovafloxacin or derivatives thereof for the manufacture of a medicament for the treatment of H. pylori infections |
| GB2289674A * | Title not available |


PICLAMILAST
An antiasthmatic agent and phosphodiesterase 4 inhibitor.
144035-83-6
SANOFI
Piclamilast (RP 73401), is a selective PDE4 inhibitor.[1] It is comparable to other PDE4 inhibitors for its anti-inflammatory effects. It has been investigated for its applications to the treatment of conditions such as chronic obstructive pulmonary disease, bronchopulmonary dysplasia andasthma. It is a second generation compound that exhibits structural functionalities of the PDE4 inhibitors cilomilast and roflumilast. The structure for piclamilast was first elucidated in a 1995 European patent application.[2] The earliest mention of the name “piclamilast” was used in a 1997 publication.[3]
Piclamilast functions through the selective inhibition of the four PDE4 isoforms (PDE4A-D). It shows no inhibition of the other PDEs. The PDE4 isoforms are especially important to inflammatory and immunomodulatory cells. They are the most common PDE in inflammatory cells such as mast cells, neutrophils, basophils, eosinophils, T lymphocytes, macrophages, and structural cells such as sensory nerves and epithelial cells. PDE4hydrolyzes cyclic adenosine monophosphate (cAMP) to inactive adenosine monophosphate (AMP). Inhibition of PDE4 blocks hydrolysis of cAMP thereby increasing levels of cAMP within cells. cAMP suppresses the activity of immune and inflammatory cells. PDE4 inhibition in an induced chronic lung disease murine model was shown to have anti-inflammatory properties, attenuate pulmonary fibrin deposition and vascular alveolar leakage, and prolong survival in hyperoxia-induced neonatal lung injury. A study of PDE4 inhibition in a murine model of allergic asthma showed that piclamilast significantly improves the pulmonary function, airway inflammation and goblet cell hyperplasia.[4][5]
Emesis is the most commonly cited side effect of piclamilast. It has proven difficult to separate the emetic side effects from the therapeutic benefits of several PDE4 inhibitors, including piclamilast.[6]
The preparation steps for synthesis of piclamilast are as follows (both discovery[7] and production[8] routes have been documented)

SEE
J Med Chem 1994, 37(11): 1696
http://pubs.acs.org/doi/abs/10.1021/jm00037a021
AND
Org Process Res Dev 1998, 2(3): 157
http://pubs.acs.org/doi/full/10.1021/op9700385

3-(cyclopentyloxy)-N-(3,5-dichloropyrid-4-yl)-4-methoxybenzamide (1) (26.4 g, 69%) as an off-white solid, mp 155−157 °C (lit.1 mp 155−157 °C). 1H NMR: δ 1.55−2.05 (m, 8H), 3.93 (s, 3H), 4.87 (m, 1H), 6.95 (d, 1H, J = 8 Hz), 6.98−7.53 (m, 2H), 7.65 (s, 1H), 8.56 (s, 2H). Anal. Calcd for C18H18Cl2N2O3: C, 56.7; H, 4.76; Cl, 18.6; N, 7.35. Found: C, 56.3; H, 4.7; Cl, 18.4; N, 7.2.


Vatiquinone
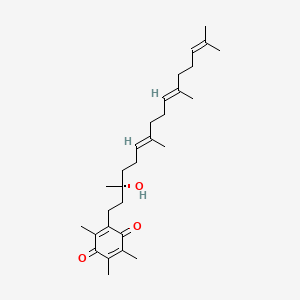
バチキノン
Vatiquinone; Alpha-Tocotrienol quinone; EPI-743; UNII-6O85FK9I0X; 1213269-98-7; Vincerenone
| Molecular Formula: | C29H44O3 |
|---|---|
| Molecular Weight: | 440.668 g/mol |
2-[(3R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trienyl]-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
2-((R,6E,10E)-3-hydroxy-3,7,11,15-tetramethylhexadeca-6,10,14-trien-1-yl)-3,5,6-trimethylcyclohexa-2,5-diene-1,4-dione
Vatiquinone is in phase II/III clinical trials for the treatment of leigh syndrome in JP. Phase II clinical trials is also ongoing for Friedreich’s ataxia, Parkinson’s disease, Pearson syndrome, cobalamin C deficiency syndrome, hearing loss and Rett’s syndrome.
Vatiquinone was originally developed by Edison Pharmaceuticals, then licensed to Sumitomo Dainippon Pharma in Japan in 2013.
Orphan drug designations for the treatment of Friedreich’s, Leigh syndrome and Rett’s syndrome were granted to the compound by FDA in 2014.
In 2013, the compound was licensed to Sumitomo Dainippon Pharma by Edison Pharmaceuticals in Japan for development and commercialization for the treatment of pediatric orphan inherited mitochondrial and adult central nervous system diseases.
EU
On 17 January 2018, orphan designation (EU/3/17/1971) was granted by the European Commission to Edison Orphan Pharma BV, The Netherlands, for vatiquinone (also known as alpha-tocotrienol quinone) for the treatment of RARS2 syndrome.
Vatiquinone, also known as EPI 743, is an orally bioavailable para-benzoquinone being developed for inherited mitochondrial diseases. The mechanism of action of EPI-743 involves augmenting the synthesis of glutathione, optimizing metabolic control, enhancing the expression of genetic elements critical for cellular management of oxidative stress, and acting at the mitochondria to regulate electron transport.
Vatiquinone has been investigated for the treatment and prevention of Retinopathy, Rett Syndrome, Genetic Disease, Noise-induced Hearing Loss, and Methylmalonic Aciduria and Homocystinuria,Cblc Type.
EPI-743 (vatiquinone) is a compound being developed by BioElectron (previously known as Edison Pharmaceuticals) to treat Friedreich’s ataxia (FA), a rare, autosomal recessive genetic disorder. The disorder is caused by mutations in the FXN gene, which encodes for a protein called frataxin. Frataxin is required for the normal functioning of mitochondria, or the energy factories of the cells. Decreased levels of frataxin, as observed in patients with FA, disrupts the normal function of mitochondria and leads to the gradual development of symptoms associated with the disease: impairment of muscle coordination, loss of muscle strength and sensation, and impaired speech, vision, and hearing.
Currently, there are no drugs available that could cure or help to effectively manage the condition, although a large number of potential treatments are in the pipeline.
EPI-743 is a drug belonging to the class of para-benzoquinones, a group of potent antioxidants. The regulation of oxidative stress is disturbed in people with FA. EPI-743 targets an enzyme called NADPH quinone oxidoreductase 1 (NQO1), helping to increase the biosynthesis of glutathione, a compound essential for the control of oxidative stress. The drug does not target any FA-specific biochemical pathways directly, but helps to improve the regulation of cellular energy metabolism in general. Due to its non-specific mechanism, the drug can be used in a variety of disorders where mitochondrial function is affected.
In December 2012, Edison Pharmaceuticals started a placebo-controlled Phase 2 study (NCT01728064) to examine the safety and efficacy of EPI-743 on visual and neurological function in FA patients. The study was completed in February 2016. The results indicated no significant differences in visual function at six months between patients treated with EPI-743 and those who received a placebo. However, researchers reported a trend toward improvement in neurological function.
In October 2013, the University of South Florida started a small Phase 2 study (NCT01962363) to evaluate the effects of EPI-743 in patients with rare point mutations leading to FA. The study investigated whether treatment with EPI-743 has a discernible impact on neurological function. The results announced in April 2016 demonstrated significant improvements in neurological functions over 18 months. However, the trial only included three participants.
Currently, no further trials testing EPI-743 in FA patients is taking place. However, the drug is in clinical trials for several other disorders that affect the functions of mitochondria, including Leigh syndrome, mitochondrial respiratory chain disease, Pearson syndrome, and others.
In February 2014, the U.S. Food and Drug Administration (FDA) granted orphan drug status to EPI-743, which allows a more expedited drug approval process. The FDA also granted fast track status to EPI-743 for the treatment of FA in March 2014.
Edison Pharmaceuticals is developing vatiquinone, which was awarded Fast Track status for Friedreich’s ataxia in March 2014.
Reference
Bioorg. Med. Chem. Lett. 2011, 21, 3693-3698.
https://www.sciencedirect.com/science/article/pii/S0960894X11005440

Reference
WO2013041676A1 / US9045402B2.

It is known that a-tocotrienol quinones are pharmaceutically active.
US 201 1 /0172312 A1 discloses that tocotrienol quinones are used in treating Leight Syndrome. WO 2010/126909 A1 and US 2006/0281809 A1 disclose that tocotrienol quinones can be used for treating ophthalmic diseases and mitochondrial diseases. US 5,318,993 discloses the activity of tocotrienol quinones as cholesterol suppression. W.D. Shrader et al., Bioorganic & Medical Chemistry Letters 21 (201 1 ), 3693-3698 disclose that the R-isomer of a-tocotrienol quinone is a metabolite of α-tocotrienol and is a potent cellular protectant against oxidative stress and ageing. The R-isomer of α-tocotrienol used for this study has been extracted from Elaeis guineensis. All these documents either use tocotrienol from natural sources or do not disclose the source of tocotrienol respectively tocotrienol quinones or disclose very specific complex synthesis thereof. These methods are very expensive and limited in producing industrial amounts of the desired products.
It is well known that from vitamin E the tocopherols and tocotrienols having the R-configuration have a significantly higher bioactivity (biopotency) than the corresponding S-isomer. This is also the case for the corresponding R-isomers of tocotrienol quinones.
Synthetic pathways to produce the R-isomer of tocotrienol quinones in a stereospecific way are very expensive and therefore only of limited interest.
The synthesis of a-tocotrienol is known from Kabbe and Heitzer, Synthesis 1978, 888-889, however, no indication of chirality whatsoever is indicated.
The synthesis of tocotrienol from the corresponding 4-oxo-chromanol-derivative is known from US 6,096,907, however, no indication of chirality is indicated.
J. Org. Chem. 1981 , 46, 2445-2450 and CH 356754 disclose the chemical transformation of a-tocopherol to a-tocopheryl quinone and to a-tocopherylhydro-quinone, however, neither tocotrienols nor tocotrienol quinones are mentioned.
Separation of chiral compounds by chromatography is principally known. However, it is also known that the quantitative separation is very often very difficult to achieve.
Due to the importance of these substances, there exists a high interest in a process which would produce R-tocotrienol quinones in a large scale in an easy and economic way.
Examples
The present invention is further illustrated by the following experiments.
1 . Chromatographic separation
Starting materials:
Solvents and reagents used as received were heptane (Fluka, 51750), ethanol (Merck, 1 .00983), isopropanol (Sigma-Aldrich, 59300) and acetic acid (Fluka, 45730).
Chromatography:
Preparative separations were performed on an Agilent 1 100 series hplc system consisting of an Agilent 1 100 degasser, Agilent 1 100 preparative pump, Agilent 1 100 diode array detector, Agilent 1 100 MPS G2250A autosampler/fraction collector controlled by chemstation/CC-mode software package.
HPLC conditions for preparative separation:
Column: Daicel Chiracel® OD-H, 250 mm x 20 mm; eluent 0.5% isopropanol, 0.2 % acetic acid in n-heptane; flow 13 ml/min; detection 220 nm, 400 μΙ injection.
Separation of (R)-6-hydroxy-2,5,7,8-tetramethyl-2-((3E,7E)-4,8, 12-trimethyl-trideca-3,7, 11-trienyl) chroman-4-one and (S)-6-hydroxy-2,5,7,8-tetramethyl-2-((3E, 7E)-4,8, 12-trimethyltrideca-3, 7, 11-trienyl) chroman-4-one
Example 1 :
6-Hydroxy-2,5,7,8-tetramethyl-2-((3E,7E)-4,8,12-trimethyltrideca-3,7,1 1 -trienyl) chroman-4-one was prepared according to the example 6a in Kabbe and Heitzer, Synthesis 1978, 888-889.
The product was analyzed by HPLC (Column: Daicel Chiracel® OD-H, 250 mm x 4.6 mm; eluent 1 % ethanol in n-hexane; flow 1 ml/min; detection 220 nm, 2 μΙ injection). Figure 9 b) shows this chromatogram. It shows that the product is a 49.5 : 50.5 mixture (Retention time 13.2 and 14.2 min.)
87.5 mg of this product in heptane was injected and the two peaks with retention time at maximum 35.4 min. (1 ) (50.9%) resp. 43.5 min. (2) (49.1 %) were se-parated by the preparative HPLC separation. Figure 9 a) shows the chromatogram of the preparative HPLC separation.
After evaporation to dryness and dissolution the two collected fractions have been reanalysis on an analytical column (Daicel Chiracel® OD-H, 250 mm x 4.6 mm; eluent 1 % ethanol in n-hexane; flow 1 ml/min; detection 220 nm, 2 μΙ injection). Figure 9 c), respectively Figure 9 d), show the chromatogram of the first fraction, respectively the second fraction. The separation of the two isomers (Retention time 13.2 min, resp. 14.2 min) in the two fraction shows to be 94.9 : 5.1 (Figure 9 c)) resp. 7.1 : 92.9 (Figure 9 d)). Hence, the two isomers have been separation by preparative chromatography almost completely.
Patent
The active component of the formulation of the present invention is selected from alpha- tocotrienol quinone, beta-tocotrienol quinone, gamma-tocotrienol quinone, delta-tocotrienol quinone, and mixtures thereof. In one embodiment, the formulation of the present invention comprises alpha-tocotrienol quinone as the active component. In other embodiments, the formulations of the present invention comprise one or more tocotrienol quinones of Formula I or mixtures thereof, in a pharmaceutically acceptable vehicle, and in other embodiments, the formulations of the present invention comprise alpha-tocotrienol quinone in a pharmaceutically acceptable vehicle. In other particular embodiments, the formulations are administered orally. In other embodiments, the formulations of the present invention comprise one or more tocotrienol quinones of Formula I or mixtures thereof, in an ophthalmically acceptable vehicle for topical, periocular, or intraocular administration, and in other embodiments, the formulations of the present invention comprise alpha-tocotrienol quinone in an ophthalmically acceptable vehicle.
[0120] The formulations of the present invention comprise tocotrienol quinones which can be produced synthetically from the respective tocotrienol by oxidation with suitable oxidizing agents, as for example eerie ammonium nitrate (CAN). Particularly, the formulations of the present invention comprise alpha-tocotrienol quinone (CAS Reg. No. 1401-66-7) produced by oxidation of alpha-tocotrienol. A preferred process for the production of alpha-tocotrienol has been described in co-owned US provisional application USAN 61/197,585 titled “Process for Enrichment and Isolation of alpha-Tocotrienol from Natural Extracts”.
[0121] Syntheses of various members of the tocotrienol family in the d,l- or (RS)-form have been published, see for example Schudel et al, HeIv. Chim. Acta (1963) 46, 2517-2526; H. Mayer et al, HeIv. Chim. Acta (1967) 50, 1376-11393; H.-J. Kabbe et al, Synthesis (1978), 888-889; M. Kajiwara et al, Heterocycles (1980) 14, 1995-1998; S. Urano et al, Chem. Pharm. Bull. (1983) 31, 4341-4345, Pearce et al, J. Med Chem. (1992), 35, 3595-3606 and Pearce et al, J. Med. Chem. (1994). 37, 526-541. None of these reported processes lead to the natural form of the tocotrienols, but rather produces racemic mixtures. Syntheses of natural form d-tocotrienols have been published. See for example. J. Scott et al, HeIv. CMm. Acta (1976) 59, 290-306, Sato et al. (Japanese Patent 63063674); Sato et al. (Japanese Patent NoJP 01233278) and Couladouros et al. (US Patent No. 7,038,067).
[0122] While synthetic and natural tocopherols are readily available in the market, the natural tocotrienol supply is limited, and generally comprises a mixture of tocotrienols. Crude palm oil which is rich in tocotrienols (800-1500 ppm) offers a potential source of natural tocotrienols. Carotech, Malaysia is able to extract and concentrate tocotrienols from crude palm oil, by a process patented in U.S. Pat. No. 5,157,132. Tocomin®-50 typically comprises about 25.32% mixed tocotrienols (7.00% alpha-tocotrienol, 14.42% gamma-tocotrienol, 3.30% delta-tocotrienol and 0.6% beta-tocotrienol ), 6.90% alpha-tocopherol and other phytonutrients such as plant squalene, phytosterols, co-enzyme QlO and mixed carotenoids.
[0123] Other methods for isolation or enrichment of tocotrienol from certain plant oils and plant oil by-products have been described in the literature. For some examples of such isolation and purification processes, see for instance Top A. G. et al, U.S. Pat. No. 5,190,618; Lane R et al, U.S. Pat No. 6,239,171; Bellafiore, L. et al. U.S. Pat. No.6,395,915; May, CY et al, U.S. Pat. No.6,656,358; Jacobs, L et al, U.S. Pat. No. 6,838,104; Sumner, C et al. Int. Pat. Pub. WO 99/38860, or Jacobs, L, Int. Pat. Pub. WO 02/500054. The compounds for use in the present invention and the other therapeutically active agents can be administered at the recommended maximum clinical dosage or at lower doses. Dosage levels of the active compounds in the compositions for use in the present invention may be varied so as to obtain a desired therapeutic response depending on the route of administration, severity of the disease and the response of the patient. When administered in combination with other therapeutic agents, the therapeutic agents can be formulated as separate compositions that are given at the same time or different times, or the therapeutic agents can be given as a single composition.
1: Peragallo JH, Newman NJ. Is there treatment for Leber hereditary optic neuropathy? Curr Opin Ophthalmol. 2015 Nov;26(6):450-7. doi: 10.1097/ICU.0000000000000212. PubMed PMID: 26448041; PubMed Central PMCID: PMC4618295.
2: Miller DK, Menezes MJ, Simons C, Riley LG, Cooper ST, Grimmond SM, Thorburn DR, Christodoulou J, Taft RJ. Rapid identification of a novel complex I MT-ND3 m.10134C>A mutation in a Leigh syndrome patient. PLoS One. 2014 Aug 12;9(8):e104879. doi: 10.1371/journal.pone.0104879. eCollection 2014. PubMed PMID: 25118196; PubMed Central PMCID: PMC4130626.
3: Strawser CJ, Schadt KA, Lynch DR. Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev Neurother. 2014 Aug;14(8):949-57. doi: 10.1586/14737175.2014.939173. Epub 2014 Jul 18. PubMed PMID: 25034024.
4: Enns GM. Treatment of mitochondrial disorders: antioxidants and beyond. J Child Neurol. 2014 Sep;29(9):1235-40. doi: 10.1177/0883073814538509. Epub 2014 Jun 30. PubMed PMID: 24985754.
5: Avula S, Parikh S, Demarest S, Kurz J, Gropman A. Treatment of mitochondrial disorders. Curr Treat Options Neurol. 2014 Jun;16(6):292. doi: 10.1007/s11940-014-0292-7. PubMed PMID: 24700433; PubMed Central PMCID: PMC4067597.
6: Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014 Apr;49:105-11. doi: 10.1016/j.biocel.2014.01.020. Epub 2014 Feb 2. Review. PubMed PMID: 24495877.
7: Chicani CF, Chu ER, Miller G, Kelman SE, Sadun AA. Comparing EPI-743 treatment in siblings with Leber’s hereditary optic neuropathy mt14484 mutation. Can J Ophthalmol. 2013 Oct;48(5):e130-3. doi: 10.1016/j.jcjo.2013.05.011. PubMed PMID: 24093206.
8: Pastore A, Petrillo S, Tozzi G, Carrozzo R, Martinelli D, Dionisi-Vici C, Di Giovamberardino G, Ceravolo F, Klein MB, Miller G, Enns GM, Bertini E, Piemonte F. Glutathione: a redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol Genet Metab. 2013 Jun;109(2):208-14. doi: 10.1016/j.ymgme.2013.03.011. Epub 2013 Mar 24. PubMed PMID: 23583222.
9: Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Experiment Ophthalmol. 2013 Sep-Oct;41(7):702-12. doi: 10.1111/ceo.12086. Epub 2013 Apr 11. Review. PubMed PMID: 23433229.
10: Kerr DS. Review of clinical trials for mitochondrial disorders: 1997-2012. Neurotherapeutics. 2013 Apr;10(2):307-19. doi: 10.1007/s13311-013-0176-7. Review. PubMed PMID: 23361264; PubMed Central PMCID: PMC3625388.
11: Blankenberg FG, Kinsman SL, Cohen BH, Goris ML, Spicer KM, Perlman SL, Krane EJ, Kheifets V, Thoolen M, Miller G, Enns GM. Brain uptake of Tc99m-HMPAO correlates with clinical response to the novel redox modulating agent EPI-743 in patients with mitochondrial disease. Mol Genet Metab. 2012 Dec;107(4):690-9. doi: 10.1016/j.ymgme.2012.09.023. Epub 2012 Sep 28. PubMed PMID: 23084792.
12: Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, Hinman A, Shrader WD, Thoolen M, Klein MB, Bertini E, Miller G. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh Syndrome. Mol Genet Metab. 2012 Nov;107(3):383-8. doi: 10.1016/j.ymgme.2012.09.007. Epub 2012 Sep 10. PubMed PMID: 23010433.
13: Büsing A, Drotleff AM, Ternes W. Identification of α-tocotrienolquinone epoxides and development of an efficient molecular distillation procedure for quantitation of α-tocotrienol oxidation products in food matrices by high-performance liquid chromatography with diode array and fluorescence detection. J Agric Food Chem. 2012 Aug 29;60(34):8302-13. doi: 10.1021/jf301137b. Epub 2012 Aug 16. PubMed PMID: 22747466.
14: Sadun AA, Chicani CF, Ross-Cisneros FN, Barboni P, Thoolen M, Shrader WD, Kubis K, Carelli V, Miller G. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012 Mar;69(3):331-8. doi: 10.1001/archneurol.2011.2972. PubMed PMID: 22410442.
15: Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012 Jan;105(1):91-102. doi: 10.1016/j.ymgme.2011.10.009. Epub 2011 Oct 21. PubMed PMID: 22115768.
16: Shrader WD, Amagata A, Barnes A, Enns GM, Hinman A, Jankowski O, Kheifets V, Komatsuzaki R, Lee E, Mollard P, Murase K, Sadun AA, Thoolen M, Wesson K, Miller G. α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg Med Chem Lett. 2011 Jun 15;21(12):3693-8. doi: 10.1016/j.bmcl.2011.04.085. Epub 2011 Apr 24. PubMed PMID: 21600768.
17: Gagnon KT. HD Therapeutics – CHDI Fifth Annual Conference. IDrugs. 2010 Apr;13(4):219-23. PubMed PMID: 20373247.
18: Bidichandani SI, Delatycki MB. Friedreich Ataxia. 1998 Dec 18 [updated 2014 Jul 24]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1281/ PubMed PMID: 20301458.
19: Yu-Wai-Man P, Chinnery PF. Leber Hereditary Optic Neuropathy. 2000 Oct 26 [updated 2013 Sep 19]. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1174/ PubMed PMID: 20301353.

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9162957 | METHODS FOR SELECTIVE OXIDATION OF ALPHA TOCOTRIENOL IN THE PRESENCE OF NON-ALPHA TOCOTRIENOLS |
2012-07-19
|
2014-09-04
|
| US9670545 | METHODS AND KITS FOR TREATING AND CLASSIFYING INDIVIDUALS AT RISK OF OR SUFFERING FROM TRAP1 CHANGE-OF-FUNCTION |
2014-06-11
|
2016-06-30
|
| US2017297991 | METHODS FOR SELECTIVE OXIDATION OF ALPHA TOCOTRIENOL IN THE PRESENCE OF NON-ALPHA TOCOTRIENOLS |
2017-01-20
|
|
| US2014221674 | PROCESS FOR THE PRODUCTION OF ALPHA-TOCOTRIENOL AND DERIVATIVES |
2013-09-26
|
2014-08-07
|
| US8575369 | Process for the production of alpha-tocotrienol and derivatives |
2012-01-25
|
2013-11-05
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017037023 | PROCESS FOR THE PRODUCTION OF ALPHA-TOCOTRIENOL AND DERIVATIVES |
2016-03-11
|
|
| US9670170 | RESORUFIN DERIVATIVES FOR TREATMENT OF OXIDATIVE STRESS DISORDERS |
2014-03-14
|
2016-02-11
|
| US9296712 | RESORUFIN DERIVATIVES FOR TREATMENT OF OXIDATIVE STRESS DISORDERS |
2013-03-15
|
2014-09-18
|
| US8106223 | PROCESS FOR THE PRODUCTION OF ALPHA-TOCOTRIENOL AND DERIVATIVES |
2010-04-29
|
2012-01-31
|
| US9567279 | METHODS FOR SELECTIVE OXIDATION OF ALPHA TOCOTRIENOL IN THE PRESENCE OF NON-ALPHA TOCOTRIENOLS |
2015-09-10
|
2016-01-07
|
////////////orphan drug status, EPI-743, fast track, EPI743, EPI-743, EPI 743, Vatiquinone; alpha-Tocotrienol quinone, Vincerenone, バチキノン , BioE-743
CC1=C(C(=O)C(=C(C1=O)C)CCC(C)(CCC=C(C)CCC=C(C)CCC=C(C)C)O)C
19 February 2013 EPI-743 Vatiquinone is a new drug that is based on vitamin E. Tests have shown that it can help improve the function of cells with mitochondrial problems. It may be able to treat people with genetic disorders that affect metabolism and mitochondria Edison Pharmaceuticals and Bambino Gesu Children’s Hospital have announced the commencement of EPI-743 Phase 2 cobalamin C deficiency syndrome trial. EPI-743 is an orally bioavailable small molecule and a member of the para-benzoquinone class of drugs. The trial’s principal investigator, Bambino Gesu Children’s Hospital, division of metabolism Professor Carlo Dionisi-Vici said, “Given the central role of glutathione in cellular redox balance and antioxidant defense systems, we are eager to explore whether a therapeutic that increases glutathione such as EPI-743 will provide clinical benefit.” Improvement in visual function is the primary endpoint of the placebo-controlled study while secondary outcome measurements assess neurologic and neuromuscular function, glutathione biomarkers, quality of life, in addition to safety parameters. The investigation is aimed at assessing the efficacy of EPI-743 in disorders of intermediary metabolism that also result in redox disturbances. EPI-743 is an orally absorbed small molecule that readily crosses into the central nervous system. It works by targeting the enzyme NADPH quinone oxidoreductase 1 (NQO1). Its mode of action is to synchronize energy generation in mitochondria with the need to counter cellular redox stress Friedreich’s ataxia (FRDA) is an autosomal recessive neurodegenerative and cardiodegenerative disorder caused by decreased levels of the protein frataxin. The disease causes the progressive loss of voluntary motor coordination (ataxia) and cardiac complications. Symptoms typically begin in childhood, and the disease progressively worsens as the patient grows older; patients eventually become wheelchair-bound due to motor disabilities. Patients with Friedreich’s ataxia develop loss of visual acuity or changes in color vision. Most have jerky eye movements (nystagmus), but these movements by themselves do not necessarily interfere with vision. ……………… Bioorg Med Chem Lett 2011, 21(12): 3693 http://www.sciencedirect.com/science/article/pii/S0960894X11005440We report that α-tocotrienol quinone (ATQ3) is a metabolite of α-tocotrienol, and that ATQ3 is a potent cellular protectant against oxidative stress and aging. ATQ3 is orally bioavailable, crosses the blood–brain barrier, and has demonstrated clinical response in inherited mitochondrial disease in open label studies. ATQ3 activity is dependent upon reversible 2e-redox-cycling. ATQ3 may represent a broader class of unappreciated dietary-derived phytomolecular redox motifs that digitally encode biochemical data using redox state as a means to sense and transfer information essential for cellular function. 
Figure 1.
The conversion of α-tocotrienol to α-tocotrienol quinone.

Figure 1.
The conversion of α-tocotrienol to α-tocotrienol quinone.


Sonidegib/Erismodegib
CODE DESIGNATION ..LDE225, NVP-LDE-225
Treatment of medulloblastoma PHASE3 2014 FDA FILING
Treatment of advanced basal cell carcinoma PHASE3 2014 FDA FILING
Treatment of SOLID TUMORS..PHASE1 2017 FDA FILING
READMalignant Solid Tumors of Childhood
THERAPEUTIC CLAIM Oncology, Antineoplastics & Adjunctive Therapies

CHEMICAL NAMES
1. [1,1′-Biphenyl]-3-carboxamide, N-[6-[(2R,6S)-2,6-dimethyl-4-morpholinyl]-3-pyridinyl]-2-
methyl-4′-(trifluoromethoxy)-, rel-
2. N-{6-[(2R,6S)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl}-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide
N-[6-[(2S,6R)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl]-2-methyl-3-[4-(trifluoromethoxy)phenyl]benzamide
N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide
MOLECULAR FORMULA C26H26F3N3O3
MOLECULAR WEIGHT 485.5
SPONSOR Novartis Pharma AG
CAS REGISTRY NUMBER 956697-53-3 free form
NOTE… DIPHOSPHATE SALT IS THE DRUG WITH CAS 1218778-77-8
About the Study
The Phase II, randomized, double-blind BOLT (Basal cell carcinoma Outcomes in LDE225 Trial) study was designed to assess the safety and efficacy of two oral dose levels of LDE225 (200 mg and 800 mg) in patients with locally advanced or metastatic basal cell carcinoma[4], which are subtypes of advanced basal cell carcinoma.
The primary endpoint was the proportion of patients achieving an objective response rate, defined as a confirmed complete response and partial response as their best overall response per modified RECIST criteria, within six months of starting treatment with LDE225. Key secondary endpoints of the study included assessing the duration of tumor responseand the rate of complete response. Other secondary endpoints included progression-free survival, time to tumor response and overall surviva

Sonidegib (INN) or Erismodegib (USAN), also known as LDE225 is a Hedgehog signalling pathway inhibitor (via smoothened antagonism) being developed as an anticancer agent by Novartis.[1][2] It has been investigated as a potential treatment for:
NVP-LDE-225, a product candidate developed by Novartis, is in phase III clinical trials for the treatment of medulloblastoma and basal cell carcinoma. Phase II trials are in progress for the treatment of adult patients with relapsed or refractory or untreated elderly patients with acute leukemia.
Early clinical trials are ongoing for the oral treatment of advanced solid tumors, for the treatment of myelofibrosis in combination with ruxolitinib and for the treatment of small cell lung cancer. A phase II clinical trial for the treatment of basal cell carcinomas in Gorlin’s syndrome patients with a cream formulation of NVP-LDE-225 was discontinued in 2011 since the formulation did not demonstrate tumor clearance rate sufficient to support further development.
Dana-Farber Cancer Institute and the Massachusetts General Hospital are conducting phase I clinical trials for the treatment of locally advanced or metastatic pancreatic cancer in combination with chemotherapy. In 2009, orphan drug designation was assigned in the E.U. for the treatment of Gorlin syndrome.
It has demonstrated significant efficacy against melanoma in vitro and in vivo.[21] It also demonstrated efficacy in a mouse model of pancreatic cancer.[22]
NVP-LDE225 Diphosphate salt (Erismodegib, Sonidegib)

About LDE225
LDE225 (sonidegib) is an oral, investigational, selective smoothened inhibitor being studied in a variety of cancers. Smoothened (SMO) is a molecule that regulates the hedgehog (Hh) signaling pathway, which plays a critical role in stem cell maintenance and tissue repair. LDE225 is currently in clinical development for a variety of diseases including myelofibrosis, leukemia and solid tumors.
Given that LDE225 is an investigational compound, the safety and efficacy profile has not yet been fully established. Access to this investigational compound is available only through carefully controlled and monitored clinical trials. These trials are designed to better understand the potential benefits and risks of the compound. Given the uncertainty of clinical trials, there is no guarantee that LDE225 will ever be commercially available anywhere in the world.
Possibility (LDE225) is effective in medulloblastoma relapsed or refractory hedgehog pathway inhibitor sonidegib has been revealed. That the anti-tumor effect was observed in some patients and tolerability in 1/2 test phase.
4th Quadrennial Meeting of the World Federation of Neuro-Oncology in conjunction with the 18th Annual Meeting of the Society for Neuro-Oncology, which was held in San Francisco November 21 to 24 in (WFNO-SNO2013), rice Dana-Farber It was announced by Mark Kieran Mr. Children’s Hospital Cancer Center.
The research group, announced the final results of the Phase 1 trial that target advanced solid cancer in children of sonidegib. 1 dose increased multi-test phase, was initiated from 372mg/m2 once-daily dosing to target children under the age of 18 more than 12 months. (233mg/m2 group 11 people, 16 people 372mg/m2 group, 11 people group 425mg/m2, 680mg/m2 group 21 women) who participated 59 people, including medulloblastoma 38 patients. 12 median age was (2-17).
Creatine phosphokinase elevation of grade 4 only were seen at 372mg/m2 as dose-limiting toxicity only, and became two recommended dose phase and 680mg/m2. Nausea muscle pain creatine kinase rise malaise (22.0%) (15.3%) (15.3%), (13.6%), vomiting side effects were many, was (13.6%). Hypersensitivity vomiting creatine kinase increased (3.4%) (1.7%) (1.7%), rhabdomyolysis side effects of grade 3/4 was (1.7%). (One group 372mg/m2, 425mg/m2 group one) complete response was obtained in two people, a strong correlation was found between the activation of the hedgehog pathway and effect.
Phase III clinical trials that target medulloblastoma the activated hedgehog pathway currently are underway.
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and diagnostic tools, over-the-counter and animal health products. Novartis is the only global company with leading positions in these areas. In 2013, the Group achieved net sales of USD 57.9 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 136,000 full-time-equivalent associates and operate in more than 140 countries around the world.

The following Examples serve to illustrate the invention without limiting the scope thereof, it is understood that the invention is not limited to the embodiments set forth herein, but embraces ali such forms thereof as come within the scope of the disclosure,

Step 1:
To a solution of 2-chloro-5-nitro-pyridine 1 (5.58 g, 35.2 mmoL) and c/s-2,6- dimethylmorpholine (4.05 g, 35.2 mmoL) in anhydrous DMF (30 mi.) was added K2CO3 (9.71 g, 70.4 mnrtoL). The mixture was heated at 50ºC overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 3 as a yellow solid, after purification by silica gel chromatography, obtained pure product (7.80 g, 93.2%). LC-MS m/z: 238.2 (M+ 1).
Step 2:
The above material 3 (7.3Og. 30.8 mmoL) was hydrogenated in the presence of 10% Pd-C (1.0 g) in MeOH (120 ml) under hydrogen overnight. The suspension was filtered through celite and the filtrate was concentrated to give the crude product 4 (5.92 g) as a dark brown oil which was used directly in the next step without further purification. LC-MS m/z. 208.2 (M+1).
Step 3:
To a solution of 3-bromo-2-methyl benzoic acid (2.71 g, 12.6 mmoL), 6-((2S,6R)-2,6- dimethylmorpholino)pyridin-3-arnine 4 (2.61 g, 12.6 mmoL), and HATU (4.80 g, 12.6 mmoL) in anhydrous DMF (30 mL) was added diisopropylethylamine (6.58 mL, 37.8 mmoL) dropwise. The resulting mixture was stirred overnight at room temperature. The reaction mixture was diluted with water (50 mL), and then extracted with EtOAc (3×120 mL). The organic layer was dried and concentrated to give the crude product. This crude product was then purified by flash column chromatography using 30% EtOAc in hexane as eiuent to give 5 as a white solid (4.23 g, 83.0%). LC-MS m/z: 404.1 (M+1).
Step 4:
A mixture of 4-(trif!uoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo- N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-ylJ-4-methyl-benzamide 5 (250 mg, 0.62mmol), Pd(PPh3)4 (36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL) in a sealed tube was heated at 130ºC overnight. The reaction mixture was diluted with EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and concentrated to give the crude product which was then purified by preparative mass triggered HPLC (C18 column, etuted with CH3CN-H2O containing 0.05% TFA) to give N-(6-((2S,6R)-2,6-dimethyfmorpholino)pyridin-3-yl)-2-rnethyl- 4′-(trifluoromethoxy)biphenyi-3-carboxamide (183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
The resultant crystalline product (Form A) was converted to the amorphous form by dissolving in 3% w/w aqueous ethanol, and the resultant solution spray dried at about 150ºC.
Form B was prepared by heating the amorphous form in an oven at 110ºC for 2 hours. In a further embodiment, the invention relates to a process step or steps, or an intermediate as described herein.

…………………………..
SYNTHESIS

| LC-MS m/z 486.2 (M + 1) |

Step 1: To a solution of 3-iodo-4-methyl-benzoic acid (10.0 g, 38.2 mmol) in methanol (70 ml) is added concentrated sulfuric acid (0.5 ml). The reaction mixture is heated at 70° C. for 48 hours, cooled to room ambient temperature and then concentrated. After that, ethyl acetate (100 ml) and aqueous NaHCO3 (saturated, 100 ml) solution are added to the residue. The organic layer is separated and washed again with aqueous NaHCO3 (saturated, 100 ml) solution. The organic layer is separated, dried over anhydrous Na2SO4 and concentrated to yield 3-iodo-4-methyl-benzoic acid methyl ester 1. It is used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.87 (d, 1H, J=8.4 Hz), 7.48 (d, 1H, J=8.4 Hz), 3.85 (s, 3H), 3.35 (s, 3H); LC-MS m/z: 277.0 (M+1).
Step 2: To a round-bottom flask containing 3-iodo-4-methyl-benzoic acid methyl ester (1.38 g, 5.00 mmol), 4-cyanophenylboronic acid (1.10 g, 7.48 mmol), palladium acetate (168 mg, 0.748 mmol), 2-(dicyclohexylphosphino)biphenyl (0.526 g, 1.50 mmol) and potassium fluoride (0.870 g, 15.0 mmol) is added anhydrous 1,4-dioxane (15 ml). The flask is purged with argon and sealed. The mixture is stirred at 130° C. for 18 hours, cooled to ambient temperature and then water (20 ml) and ethyl acetate (20 ml) are added. Solid is removed under vacuum filtration. The filtrate is extracted with EtOAc (20 ml×2). The organic layers are combined, washed with aqueous HCl (5%, 20 ml) and saturated NaHCO3 (20 ml). It is dried over MgSO4, and concentrated. The residue is purified by silica gel column chromatography (EtOAc/Hexane, gradient) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2; LC-MS m/z: 252.1 (M+1).
Step 3: To a solution of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2 (2.56 g, 10.3 mmol) in 1,4-dioxane-H2O (1:1 mixture, 20 ml) is added NaOH (1.22 g, 30.2 mmol)). The reaction is stirred at ambient temperature for 24 hours. To this mixture is added aqueous HCl (1 N, 36 ml) and it is then extracted with ethyl acetate (40 ml×3). The organic layers are combined, dried over anhydrous Na2SO4. The solver is removed. The solid obtained is washed with small amount of acetonitrile and air dried to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3: 1H NMR (DMSO-d6) δ 7.94 (d, 2H, J=8.0 Hz), 7.84 (dd, 1H, J1=8.4 Hz, J2=1.2 Hz), 7.75 (d, 1H, J=1.2 Hz), 7.61 (d, 2H, J=8.0 Hz), 7.48 (d, 1H, J=8.4 Hz), 2.29 (s, 3 H); LC-MS m/z 238.1 (M+1).
Step 4: To a suspension of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3 (40 mg, 0.17 mmol) in anhydrous methylene chloride (5 ml) is added 2 drops of DMF. Then oxalyl chloride (32 mg, 22 μl, 0.25 mmol) is added. The mixture is stirred at ambient temperature until it turns clear. After that, it is concentrated, re-dissolved in anhydrous methylene chloride (3 ml), and added to a solution of 4-(morpholine-4-sulfonyl)-phenylamine (61 mg, 0.25 mmol) and triethylamine (34 mg, 47 μl, 0.33 mmol) in methylene chloride (2 ml). The mixture is stirred for 2 hours, concentrated and the residue is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [4-(morpholine-4-sulfonyl)-phenyl]-amide: 1H NMR (DMSO-d6) δ 10.64 (s, 1H), 8.07 (d, 2H, J=8.8 Hz), 7.97 (d, 2H, J=8.4 Hz), 7.95 (d, 1H, J=8.8 Hz), 7.89 (s, 1H), 7.43 (d, 2H, J=8.4 Hz), 7.67 (d, 2H, J=8.8 Hz), 7.53 (d, 2H, J=8.8 Hz), 3.63 (m, 4H), 2.84 (m, 4H) 2.32 (s, 3H); LC-MS m/z: 462.1 (M+1).
Example 2 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide

Step 1: To a solution of 2-chloro-5-nitro-pyridine 4 (2.38 g, 15 mmol.) and cis-2,6-dimethylmorpholine (1.73 g, 15 mmol.) is added K2CO3 (4.14 g, 30 mmol.). The mixture was heated at 50° C. overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 6 as a yellow solid. The crude product is used directly in next step without further purification. LC-MS m/z: 238.1 (M+1).
Step 2: The above crude material 6 is hydrogenated in the presence of Pd—C (0.2 g) in MeOH (100 mL) under hydrogen over 10 h. The suspension is filtered through celite and the filtrate is concentrated to give the crude product 7 as a dark brown oil which is used directly in the next step without further purification. LC-MS m/z: 208.1 (M+1).
Step 3: To a solution of 3-bromo-4-methyl benzoic acid (108 mg, 0.5 mmol.), 6-(2,6-Dimethyl-morpholin-4-yl)-pyridin-3-ylamine 7 (104 mg, 0.5 mmol.), amd HATU (190 mg, 0.5 mmol.) in dry DMF (5 mL) is added triethylamine (139 uL, 1.0 mmol.) dropwise. The resulting mixture is stirred at room temperature for 2 h. After concentration, the residue is partitioned between EtOAc and water. The organic layer is dried and concentrated to give the crude product. The final compound is purified by flash column chromatography using 50% EtOAc in hexane as eluent to give 8 as a white solid. LC-MS m/z: 404.1 (M+1).
Step 4: A mixture of 4-cyanophenyl boronic acid (18 mg, 0.12 mmol), 3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide 8 (40 mg, 0.1 mmol), Pd(PPh3)4 (11 mg, 0.01 mmol), and Na2CO3 (42 mg, 0.4 mmol) in a combined solvent system of toluene (0.2 mL) and water (0.2 mL) and ethanol (0.05 mL) is heated at 140° C. under microwave irradiation for 30 min. The reaction mixture is diluted with EtOAc and water. The aqueous layer is extracted with EtOAc. The combined organic layer is washed with brine and concentrated to give the crude product which is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide. LC-MS m/z: 427.2 (M+1).

CONDENSE WITH …3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamideACS Medicinal Chemistry Letters, 2010 , vol. 1, 3 p. 130 – 134

A mixture of 4-(trifluoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo-N-[6-(2,6-
dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide E (250 mg, 0.62mmol), Pd(PPh3)4
(36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL)
in a sealed tube was heated at 1300C overnight. The reaction mixture was diluted with EtOAc
and water. The aqueous layer was extracted with EtOAc. The combined organic layer was
washed with brine and concentrated to give the crude product which was then purified by
preparative mass triggered HPLC (C18 column, eluted with CH3CN-H2O containing 0.05% TFA)
to give N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide (5m, 183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
HRMS (m/z): [M+H]+
calcd for C26H27N3O3F3 486.2005; found 486.1986,
1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.15 (s, 1H), 8.43 (d, 1H), 7.94 (dd, 1H), 7.52-7.43
(m, 5H), 7.38 (m, 1H), 7.33 (m, 1H), 6.86 (d, 1H), 4.06 (d, 2H), 3.62 (m, 2H), 2,34 (m, 2H), 2.22
(s, 3H), 1.16 (d, 6H).
http://pubs.acs.org/doi/suppl/10.1021/ml1000307/suppl_file/ml1000307_si_001.pdf
sonidegib
Skin Cancer Foundation. “Skin Cancer Facts.” Available at:http://www.skincancer.org/skin-cancer-information/skin-cancer-facts . Accessed on February 14, 2014.
Rubin AI, Chen EH, Ratner D (2005). Current Concepts: Basal-Cell Carcinoma. N Engl J Med; 353:2262-9.
ClinicalTrials.gov. “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)” Available at:http://clinicaltrials.gov/ct2/show/NCT01327053?term=%22LDE225%22+and+%22BOLT%22&rank=1. Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Complete Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45652 . Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Partial Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45819 . Accessed on February 14, 2014.
Wong C S M, Strange R C, Lear J T (2003). Basal cell carcinoma. BMJ; 327:794-798.
Copcu E, Aktas A. Simultaneous two organ metastases of the giant basal cell carcinoma of the skin. Int Semin Surg Oncol. 2005;2:1-6. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC544837/ . Accessed on February 14, 2014.
Skin Cancer Foundation. “Basal Cell Carcinoma Treatment Options.” Available athttp://www.skincancer.org/skin-cancer-information/basal-cell-carcinoma/bcc-treatment-options . Accessed on February 14, 2014.
Stuetz A, et al. LDE225, a specific smoothened inhibitor, for the topical treatment of nevoid basal cell carcinoma syndrome (Gorlin’s syndrome). Melanoma Research. 2010; 20:e40. Available at:http://journals.lww.com/melanomaresearch/Fulltext/2010/06001/FC24_LDE225,_a_specific_smoothened_inhibitor,_for.87.aspx#FC24_LDE225%2C_a_specific_smoothened_inhibitor%2C_for.87.aspx?s=2&_suid=139234380607909969110518506816.
Novartis.com. “The Pipeline of Novartis Oncology: LDE225.” Available at:http://www.novartisoncology.com/research-innovation/pipeline.jsp #. Accessed on February 14, 2014.
Children’s Medical Research Center, Children’s Memorial Hospital/Northwestern University Feinberg School of Medicine. “The Sonic hedgehog/patched/gli signal transduction pathway.” Available at http://www.childrensmrc.org/iannaccone/gli/ . Accessed on February 14, 2014.
Gupta S, Takebe N, LoRusso P. Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol. 2010 July; 2(4): 237-250. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3126020/ . Accessed on February 14, 2014.
| WO2004078163A2 | Feb 26, 2004 | Sep 16, 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007113120A1 | Mar 22, 2007 | Oct 11, 2007 | Frank Hoffmann | Stamping apparatus with feed device |
| WO2007131201A2 * | May 4, 2007 | Nov 15, 2007 | Irm Llc | Compounds and compositions as hedgehog pathway modulators |
| WO2008154259A1 | Jun 4, 2008 | Dec 18, 2008 | Irm Llc | Biphenylcarboxamide derivatives as hedgehog pathway modulators |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ



K salt monohydrate, N-[[2-[2-(methylamino)-4-pyrimidinyl]-1H-indol-5-yl]carbonyl]-3-(phenyl-2-pyridinylamino)- L-Alanine,
2-{[2-(2-methylamino-pyrimidin-4-yl)-lH-indole-5- carbonyl]-amino}-3-(phenylpyridin-2-yl-amino)-propionic acid, as the monopotassium monohydrate salt., 899418-66-7 , C28 H25 N7 O3 . H2 O . K
IC 50= 0.4 nm
K SALT
L-Alanine, N-[[2-[2-(methylamino)-4-pyrimidinyl]-1H-indol-5-yl]carbonyl]-3-(phenyl-2-pyridinylamino)-, monopotassium salt , 899418-65-6, C28 H25 N7 O3 . K
Free acid
As an inhibitor of IKB kinase, the compound of the invention, functions via the selective inhibition of IKK, particularly an IKK-2 inhibitor; as well as exhibiting localized activity, as opposed to a systemic activity. Such an inhibitor is particularly useful for treating a patient suffering from or subject to IKK- 2 mediated pathological diseases or conditions, e.g., asthma, rhinitis, chronic obstructive pulmonary disorder (COPD), or COPD exacerbations, that could be ameliorated by the targeted administering of the inhibitor.
Sanofi.. INNOVATOR
SANOFI LISTS http://clinicaltrials.gov/show/NCT01463488 SAR113945 AS IkB kinase inhibitors IN PHASE II…. BUT I AM NOT SURE OF THIS….Protein Kinases as Small Molecule Inhibitor Targets … – ResearchGa click here to see see table 7 (cont)……2227
EMAIL ME amcrasto@gmail.com
WO 2005113554
………………….
Synthesis
EXAMPLES
Example 1, Step 1
Synthesis of 2-{[2-(2-Methylamino-pyrimidin-4-yl)-lH-indole-5-carbonyl]amino}-3-(phenyl-pyridin-
2-yl-amino)-propionic acid

6.04 mmol of the 2-{[2-(2-methylamino-pyrimidin-4-yl)-lH-indole-5-carbonyl]-amino}-3-(phenyl- ρyridin-2-yl-amino)-propionic acid, methyl ester prepared essentially as described in patent application WO2005/113544, is dissolved in 70 mL of ethanol. 24.2 mL of 0.5 N aqueous ΝaOΗ is added and the mixture is stirred at room temperature for 2 h. After the reaction is complete, the pH is adjusted to ~5 using 1 N HCl. Water is added slowly and the resulting precipitate is filtered off and washed with water. After drying under reduced pressure of about 1 mbar at 400C, 2.49 g of 2-{[2-(2-methylamino- pyrimidin-4-yl)-lH-indole-5-carbonyl]-arnino}-3-(phenyl-pyridin-2-yl-amino)-propionic acid is isolated. Empirical formula C28H25N7O3; M. W. = 507.56; MS (M+H) 508.3. 1H NMR (DMSO-^6) 2.95 (s, 3 H), 4.32-4.50 (m, 2 H), 4.65-4.72 (m, 1 H), 6.29-6.36 (d? 1 H), 6.70- 6.79 (m, 1 H), 6.90-7.10 (sb, 1 H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 4 H), 7.40-7.48 (m, 3 H), 7.50-7.55 (m, 1 H), 7.57-7.60 (m, 1 H), 7.96 (bs, 1 H), 8.34-8.40(m, 2 H), 8.80-8.90 (d, 1 H), 11.80 (s, 1 H) 12.8 (bs, IH). Chiral HPLC shows 94% ee.
Example 1, Step 2
Enantiomeric Purification of 2-{[2-(2-Methylaminopyrimidin-4-yl)-lH-indole-5-carbonyl]amino}-3-
(phenylpyridin-2-yl-amino)-propionic acid

2- { [2-(2-methylaminopyrimidin-4-yl)- lH-indole-5-carbonyl]amino} -3-(phenylpyridin-2-yl-amino)- propionic acid, prepared essentially according to Example 1, Step 1 above, is heated under reflux for 15 minutes. The insoluble racemic compound is removed by hot filtration. The TΗF of the resulting filtrate is removed by distillation and the residue is precipitated by the addition of isopropanol. After drying under reduced pressure of about 1 mbar at 400C, the desired 2-{[2-(2-methylaminopyrimidin-4- yl)-lH-indole-5-carbonyl]amino}-3-(phenylpyridin-2-yl-amino)-propionic acid is isolated with an ee = 98.5%.
Example 1, Step 3
Synthesis of 2-{[2-(2-Methylamino-pyrimidin-4-yl)-lH-indole-5-carbonyl]-amino}-3-(phenyl-pjτidin- 2-yl-amino)-propionic acid monopotassium monohydrate salt

To a slurry of 2-{[2-(2-methylaminopyrimidin-4-yl)-lH-indole-5-carbonyl]amino}-3-(phenylpyridin- 2-yl-amino)-propionic acid (50.8 mmol from Example 1, Step 2 above) in H2O and EtOH is added 1.02 M KOH (2.00 equiv) with vigorous swirling. The mixture is heated to 670C with swirling on a steam bath to dissolve the starting material, while braking up any remaining clumps. After several minutes the clear orange solution is filtered and the flask containing the filtrate is wrapped in aluminum foil and allowed to cool slowly to room temperature in the hot water remaining in the steam bath. After 17 hours, the mixture is cooled in an ice-bath and the salt is collected by filtration and washed 4 times with ice-cold H2O. The last two washes have a pH of 8. The salt is dried in a vacuum oven at 45 0C with an N2 bleed to yield the desired compound as fine needles:1H NMR (DMSO-«k) 2.95 (s,3 H)5 3.95-4.05 (m, 1 H), 4.35-4.40 (m, IH), 4.55-4.62 (m, 1 H), 6.35-6.39 (d, 1 H), 6.58-6.60 (m, IH), 6.90-7.10 (sb, 1 H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 6 H), 7.40-7.48 (m, 3 H), 7.57-7.60 (m,l H), 7.70 (s, 1 H), 8.10-8.15(d, 1 H), 8.30 (bs, 1 H), 11.80 (s, 1 H); LC-MS m/z 509 (M+ + 2), 508 (M+ H- I), 275, 254 (100). Anal. Calcd for C28H24KN7O3-H2O (563.66): C, 59.67; H, 4.65; N, 17.39; K. 6.94; H2O (Karl Fischer), 3.20. Found: C, 59.59; H, 4.66; N, 17.39; K5 6.44; H2O (Karl Fischer), 3.16. Chiral HPLC showed 99.5% S-enantiomer.
Example 2 Synthesis of 2-{[2-(2-Methylammo-pyrimidin-4-yl)-lH-indole-5-carbonyl]-amino}-3-(phenyl-pyridin-
2-yl-amino)-propionic acid monopotassium monohydrate salt

As an alternative procedure for preparing the compound of formula Ha3 (3.8 mmol) of methyl ester 1 is dissolved in ethanol and water and 2 N aqueous KOH is added and the mixture is stirred at room temperature for 4 h. The product starts to crystallize and the mixture is diluted with additional water. The resulting crystalline precipitate is filtered off and washed with water. After drying under reduced pressure of about 1 mbar at 400C, the monopotassium monohydrate salt π is isolated. Empirical formula C28H24KN7O3-H2O M.W. = 563.65; MS (free acid, M+H) 508.3. 1H ΝMR (DMSO-J6) 2.95 (s, 3 H), 3.95-4.05 (m, 1 H), 4.35-4.40 (m, IH), 4.55-4.62 (m, 1 H), 6.35-6.39 (d, 1 H), 6.58-6.60 (m, 1 H), 6.90-7.10 (sb, 1 H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 6 H), 7.40-7.48 (m, 3 H), 7.57-7.60 (m, 1 H), 7.70 (s, 1 H), 8.10-8.15(d, 1 H), 8.30 (bs, 1 H), 11.80 (s, 1 H). Water (Karl-Fischer): 3.2% (Monohydrate). XRPD (2 theta): 5.28, 6.45, 7.97, 9.46, 10.18, 10.93, 13.23, 13.66, 14.94, 15.94, 16.71, 18.15, 19.49, 20.38, 21.04, 21.42, 23.76, 24.38, 25.36, 25.71, 26.19, 27.13, 27.67, 28.13, 28.61, 29.12, 29.75, 30.95, 31.37, 32.94. ee: 99.8% (Chiralpak AD-H, 250 x 4.6mm, Heptane : EtOH : MeOH 5 : 1 : 1, RT).
It is known that indole derivatives are used as units for the synthesis of active pharmaceutical ingredients. For example, 2-(2-aminopyrimidin-4-yl)-1H-indole-5-carboxylic acids or their salts are important units for the preparation of IkB kinase inhibitors (see WO 01/30774 A1):

2-(2-Aminopyrimidin-4-yl)-1H-indole-5-carboxylic acids can be prepared by classical Fischer indole synthesis starting from the corresponding 4-acetylpyrimidines (III) and 4-hydrazinobenzoic acid (II) (see scheme 1):

One disadvantage here is the severe reaction conditions which are required for a full conversion. Secondly, the products of this reaction are obtained in a mixture with the corresponding oligomers, which leads to a poor isolability, especially with regard to the filtration times. Moreover, these oligomers, owing to the low solubility of 2-(2-aminopyrimidin-4-yl)-1H-indole-5-carboxylic acids in organic solvents, can only be removed with difficulty and are entrained as an impurity in the further reactions, in some cases up to the active ingredient.
Here are two ways to make a kinase inhibitor intermediates.
http://www.google.com/patents/US8232395
J. Graeser and co-inventors describe indole derivatives such as 4 and 12 as intermediates for preparingIκB kinase inhibitors. Although indoles can be prepared by the classical Fisher synthesis, the inventors state that this method is not satisfactory when it is used for making the desired compounds. Severe reaction conditions are needed, and oligomeric compounds are formed that are difficult to remove.
The inventors describe two routes for preparing the desired compounds. The first route (Figure 1, top) begins with the reaction of indoleboronic acid 1 and chloropyrimidine 2in the presence of (Ph3P)4Pd to form 3, which is isolated in 93% yield and 96% purity. Compound 3 is converted to amine derivative 4 by treating it with MeNH2. The product was isolated in quantitative yield and with 97.6% purity. If desired, the ester group in 4can be hydrolyzed with NaOH to produce sodium salt 5.

Indoleboronic acid 1 is obtained by treating tert-butoxycarbonyl (Boc)–protected indole6 with B(O-i-Pr)3 in the presence of LiN-i-Pr2 (Figure 1, bottom) The reaction initially forms Boc-protected compound 7. After acid hydrolysis, 1 is isolated in 61% yield with 92.7% purity.
The inventors mention the advantage of using unprotected indole 1 in the reaction with2 rather than the N-protected compound. Their explanation is that although some 6 is formed by the loss of the boronate group from 1 during the coupling reaction with 2, 6does not subsequently react with 2. Hence the yield of 3 in the coupling step is not reduced.
The second route to the desired compound is quite different from the first. Figure 2 outlines the process for preparing 12, the methyl ester analogue of 4. This route starts with the preparation of silylated acetylene compound 8, isolated in 90% yield with 99% purity after what is described as an aqueous workup. In the next step, the silyl group is removed, and primary alkyne 9 is isolated in quantitative yield. Alkyne 9 is treated with chloropyrimidine 10 in the presence of CuI and a palladium catalyst in DMF to give 11, which is isolated after aqueous workup in 85% yield and 99.7% purity. The cyclization of 11 to form 12 is carried out with a strong base such as KO-t-Bu. The product is isolated after an aqueous workup in 58% yield and 92.3% purity.
Although the inventors do not provide details for preparing 10, they state that it can be synthesized by the route shown at the bottom of Figure 2. The reaction produces isomers 10 and 13, which can be separated by chromatographic methods or steam distillation.
The inventors describe an alternative route to 4 in which 1 reacts with 10 in place of 2. They point out that 1 reacts with a mixture of 10 and 13 to give 4. Although it may be expected that 13 would react to give an isomer of 4, they claim that this reaction does not take place. No examples of the reaction of 1 and 10 with or without 13 are given Also, the inventors mention “aqueous workup” several times but do not explain what this means.
These processes provide alternative routes to a drug intermediate that overcome product isolation problems. (Sanofi [Paris]. US Patent 8,232,395, July 31, 2012;

EXAMPLE 1 Synthesis of ethyl 2-(2-chloropyrimidin-4-yl)-1H-indole-5-carboxylate

28 g (114 mmol) of 2-borono-5-ethoxycarbonylindole, 12 g (113 mmol) of sodium carbonate and 17.2 g of 2,4-(113 mmol) dichloropyrimidine were initially charged in 412 ml of ethanol. The clear solution was freed of oxygen by vigorous stirring and passing argon through (20 minutes). At RT, 2.67 g of tetrakis(triphenylphosphine)palladium(0) were added. The mixture was heated to from 65° C. to 70° C. for 2 hours (h). Subsequently, 112 ml of water and 112 ml of 30% hydrochloric acid were added and the mixture was cooled to 0° C. After filtration and drying under reduced pressure, 37.3 g (93% of theory) of ethyl 2-(2-chloropyrimidin-4-yl)-1H-indole-5-carboxylate were obtained (HPLC >96%).
The purity was determined by high-pressure liquid chromatography (HPLC):
| Column: | Waters Symetry Shield RP8 3.9 * 150 | ||||
| Temperature: | 40° C. | ||||
| Flow rate: | 1 ml/min | Injection volume: | 10 μl | ||
| Pressure: | 90 bar | UV: | 254 nm | ||
| Eluent: | A: Water/trifluoroacetic acid (0.05%) | ||||
| B: Acetonitrile/trifluoroacetic acid (0.05%) | |||||
| Time (min) | 0 | 15 | 20 | 25 | 30 |
| A (%) | 80 | 25 | 25 | 80 | 80 |
| B (%) | 20 | 75 | 75 | 20 | 20 |
| Retention time of | 12.6 min | ||||
| title compound: | |||||
EXAMPLE 2 Synthesis of ethyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate

30 g (95.4 mmol) of ethyl 2-(2-chloropyrimidin-4-yl)-1H-indole-5-carboxylate were initially charged and suspended in 150 ml of ethanol. 53.9 g of methylamine solution in ethanol (8 M) were added to this suspension which was heated to from 75° C. to 80° C. in an autoclave for 4 h. After concentration and washing with ethanol, 29.7 g of ethyl 2-(2-methylamino-pyrimidin-4-yl)-1H-indole-5-carboxylate were obtained (97.6 HPLC area %). LCMS: [M+H]⊕ 297.12
HPLC method as in example 1; retention time of title compound: 5.8 min
EXAMPLE 3 Synthesis of 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylic acid sodium salt

25 g of ethyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate were admixed with 200 ml of ethanol and 24.5 g of 33% sodium hydroxide solution, and heated to from 65° C. to 70° C. for 4 h. After cooling, the mixture was filtered with suction and the precipitate was washed with 15 ml of ethanol/water (9:1). 24.5 g (87.6% of theory) of 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylic acid sodium salt were obtained (98.1 HPLC area %). LCMS: [M+H]⊕ 269.10
HPLC method as in example 1; retention time of title compound: 3.3 min
EXAMPLE 4 Synthesis of methyl 4-amino-3-trimethylsilylethynylbenzoate

5.83 g (20 mmol) of methyl 4-aminobenzoate, 20.2 g (198 mmol) of triethylamine and 80 ml of toluene were initially charged. The clear solution was freed of oxygen by vigorous stirring and passing argon through (20 minutes). At an internal temperature of 20° C., 3.2 g (33 mmol) of trimethylsilylacetylene, 76 mg of copper(I) iodide and 52 mg of triphenylphosphine were added. After aqueous workup, 5.45 g of 4-amino-3-trimethylsilylethynylbenzoate were obtained (HPLC: >99 area %). HPLC method as in example 1.
EXAMPLE 5 Synthesis of methyl 4-amino-3-ethynylbenzoate

1.9 g (7.7 mmol) of methyl 4-amino-3-trimethylsilylethynylbenzoate were initially charged in 20 ml of tetrahydrofuran (THF). At from 5° C. to 8° C., 8.45 ml (8.5 mmol) of tetrabutylammonium fluoride solution (1 M in THF) were added dropwise within 5 minutes. After 25 min at 2° C., 438 ml of acetic acid were added. After addition of water and extraction with dichloromethane, and after removal of the solvent, 1.35 g of methyl 4-amino-3-ethynylbenzoate were obtained. HPLC method as in example 1.
EXAMPLE 6 Synthesis of methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)-ethynylbenzoate

3.0 g (17 mmol) of methyl 4-amino-3-ethynylbenzoate and 2.6 g (19 mmol) of 4-chloro-2-methylaminopyrimidine were initially charged in 20 ml of dimethylformamide (DMF) and 8.7 g (85 mmol) of triethylamine, and degassed with argon while stirring for 5 min. Subsequently, 65 mg of copper(I) iodide and 20 mg of tetrakis(triphenylamine)palladium(0) were added and the mixture was heated to 71° C. for 3 h. After aqueous workup, 4.1 g of methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)ethynylbenzoate were obtained. (HPLC: 99.7 area %) HPLC method as in example 1.
EXAMPLE 7 Synthesis of methyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate by cyclizing methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)ethynylbenzoate

73 mg (0.7 mmol) of potassium tert-butoxide were dissolved in 1 ml of NMP and admixed with a solution of 140 mg (0.5 mmol) of methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)ethynylbenzoate in 1 ml of NMP. Subsequently, stirring was continued at RT for 24 h. Aqueous workup afforded 115 mg of methyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate (HPLC: 92.3 area %).
EXAMPLE 8 Synthesis of 2-borono-5-ethoxycarbonylindole

150 g (519 mmol) of N-Boc-5-ethoxycarbonylindole and 192 ml (833 mmol) of triisopropyl borate in 350 ml of toluene were admixed at from 5° C. to 10° C. with 350 ml of a 1.8 molar solution of LDA in THF. The mixture was stirred for a further 5 min and the reaction mixture was added to a solution of 278 g of 30% hydrochloric acid and 940 ml of water. Subsequently, the mixture was stirred at from 5° C. to 10° C. for 30 min. Thereafter, the mixture was filtered and the filtercake was suspended in 530 ml of ethanol. This suspension was added at 40° C. to a solution of 500 ml of 30% hydrochloric acid and 224 ml of ethanol. Subsequently, the mixture was stirred at from 40° C. to 45° C. for 2.5 h and admixed at 30° C. with 380 ml of water. The mixture was then cooled to from 10° C. to 15° C., stirred at this temperature for 30 min and filtered. Drying under reduced pressure afforded 79.5 g (61% of theory) of 2-borono-5-ethoxycarbonylindole (HPLC: 92.7 area %).
…………………………………………….
C) Synthesis of the heterocyclic base

C.1) indole base synthesis. Of 2 – (2-methylamino-pyrimidin-4-yl) -1 H-indole-5-carboxylic acid (20) C.1.1) 1-Dimethylamino-4 ,4-dimethoxy-pent. -1-en-3-one (18)
100 g (0.76 mol) of 3,3-dimethoxy-2-butanone (16) of (17) (0.76 mol) at 120 ° C with stirring 90.2 g of 48 N, N-dimethylformamide dimethyl acetal h. The methanol formed during the reaction was continuously removed from the reaction solution by distillation. On cooling, the solution became a spontaneous crystallization, which was brought by adding a little heptane to completion. This gave 128.24 g of crude 18 (90% yield), which was reacted without further purification. Molecular formula C 9 Hι 7 N0 3, MW = 187.24, MS (M + H) 188.2 i H NMR (DMSO-de) 1.22 (s, 3H), 2.80 (s, 3H), 3.10 (s, 9H), 5.39. (d, J = 15 Hz, 1 H), 7:59 (d, J = 15 Hz, 1 H). . . . . . . .
C.1.2). [4 – (1,1-Dimethoxy-ethyl)-pyrimidin-2-yl]-methyl-amine (19)
1:22 g (53 mmol) of sodium were dissolved in 100 ml absolute ethanol. This was
Stirring 5.8 g (53 mmol) Methylguanidinhydrochlorid and 10 g (53 mmol) of 1-dimethylamino-4,4-dimethoxy-penM-en-3-one (18) and heated to boiling for 4 h. To stop the reaction, the ethanol was evaporated. The product 19 thus obtained was used without further purification for the subsequent reaction. Yield 11.5 g (58 mmol, quantitative) Molecular Formula C9H15N3O2, MW = 197.24, MS (M + H) 198.2 1 H NMR (DMSO-de) 1.45 (s, 3H), 2.78 (s, 3H), 3.10 (s,. 6H), 6.75 (d, J = .3 Hz, 1 H), 7.0 – 7.1 (s (b), 1 H), 8.30 (d, J = 3 Hz, 1 H).
C.1.3) 2 -. (2-methylamino-pyrimidin-4-yl) -1 H-indole-5-carboxylic acid (20) Into 150 ml of 50% sulfuric acid at room temperature 5 g (25 mmol) [4 – ( 1, 1 – dimethoxy-ethyl)-pyrimidin-2-yl]-methyl-amine (19) and, 3.85 g of 4-hydrazinobenzoic acid with stirring and heated 4 h at 130 ° C. The methanol formed during the reaction was continuously removed from the reaction solution by distillation. After cooling to 10 ° C the reaction mixture was poured into 200 mL of ice and adjusted to a pH of about 5.5 with concentrated sodium hydroxide solution. The precipitate formed from sodium sulfate, and the product mixture was filtered and the filter residue was extracted several times with methanol. The combined methanol extracts were concentrated and the product 20 by flash chromatography (DCM / methanol 9:1). Yield: 0.76 g (11%) Molecular formula Oι Hι3 N 4 4 0 2, MW = 268.28, MS (M + H) 269.1.
1 H NMR (DMSO-de) 2.95 (s, 3H), 6.90 – 7.10 (s (b), 1 H), 7.18 (d, J = 3 Hz, 1H), 7.4 (s, 1 H), 7:58 (d, J = 4.5 Hz, 1H), 7.80 (d, J = 4.5 Hz, 1H), 8.30 (s, 1H), 7.80 (d, J = 4.5 Hz, 1H), 8:38 (d, J = 3 Hz, 1H), 11.85 (s, 1H), 12:40 – 12.60 (s (b), 1 H).
| US7285560 | Aug 18, 2003 | Oct 23, 2007 | Sanofi-Aventis Deutschland Gmbh | Indole derivatives or benzimidazole derivatives for modulating IκB kinase |
| US7342029 | Jul 22, 2005 | Mar 11, 2008 | Sanofi-Aventis Deutschland Gmbh | Substituted indoles |
| US7462638 | Aug 18, 2003 | Dec 9, 2008 | Sanofi-Aventis Deutschland Gmbh | Use of IκB-kinase inhibitors in pain therapy |
| US20030119820 | Oct 4, 2002 | Jun 26, 2003 | Aventis Pharma Deutschland Gmbh | Substituted indoles |
| US20040116494 | Aug 18, 2003 | Jun 17, 2004 | Aventis Pharma Deutschland Gmbh | Use of IkappaB-kinase inhibitors in pain therapy |
| US20040209868 | May 11, 2004 | Oct 21, 2004 | Aventis Pharma Deutschland Gmbh | Substituted indoles |
| US20070244139 | Jun 6, 2007 | Oct 18, 2007 | Sanofi-Aventis Deutschland Gmbh | Indole Derivatives or Benzimidazole Derivatives for Modulating IkB Kinase |
| US20090069358 | Nov 6, 2008 | Mar 12, 2009 | Sanofi-Aventis Deutschland Gmbh | Use of IKappaB-Kinase Inhibitors in Pain Therapy |
| JP2003519101A | Title not available | |||
| WO1998040380A1 | Feb 27, 1998 | Sep 17, 1998 | Alessio Roberto D | Indolyl-pyrrolydenemethylpyrrole derivatives and process for their preparation |
| WO2001030774A1 | Oct 17, 2000 | May 3, 2001 | Aventis Pharma Gmbh | Substituted indoles for modulating nfkb activity |
| WO2003066629A2 | Feb 6, 2003 | Aug 14, 2003 | Michael J Arnost | Heteroaryl compounds useful as inhibitors of gsk-3 |
| WO2004022057A1 | Aug 5, 2003 | Mar 18, 2004 | Aventis Pharma Gmbh | USE OF IκB KINASE INHIBITORS FOR THE TREATMENT OF PAIN |
| WO2004022553A1 | Aug 5, 2003 | Mar 18, 2004 | Aventis Pharma Gmbh | INDOLE OR BENZIMIDAZOLE DERIVATIVES FOR MODULATING IκB KINASE |
| WO2004089913A1 | Apr 8, 2004 | Oct 21, 2004 | Novartis Ag | Aminopyrimidine derivatives and their medical use |
| WO2005040133A1 | Oct 11, 2004 | May 6, 2005 | Michael Clare | Pyrimidine compounds for the treatment of inflammation |
| WO2004022553A1 * | Aug 5, 2003 | Mar 18, 2004 | Aventis Pharma Gmbh | INDOLE OR BENZIMIDAZOLE DERIVATIVES FOR MODULATING IκB KINASE |
Fiduxosin hydrochloride, 208992-74-9, NCGC00162178-02, AC1L58WW,
 Fiduxosin
Fiduxosin







3-[4-((3aR,9bR)- cis -9-Methoxy-1,2,3,3a,4,9b-hexahydro-[1]-benzopyrano[3,4-c]pyrrol-2-yl)butyl]-8-chloro-pyrazino[2′,3′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione hydrochloride

Fiduxosin (ABT-980), α1a-adrenoreceptor antagonist, a development compound at Abbot for the treatment of benign prostate hyperplasia, is disclosed in Organic Process Research & Development 2004, 8, 897-902 and references cited therein.
The synthetic route for preparation of Fiduxosin is as follows:


Fiduxosin (1) has been under development at Abbott Laboratories for the treatment of benign prostatic hyperplasia. A convergent strategy required methodologies for preparation of an enantiomerically pure 3,4-cis-disubstituted pyrrolidine and a 2,3,5-trisubstituted thienopyrazine in a regiospecific manner.
A [3+2] cycloaddition of an enantiopure azomethine ylide followed by a diastereoselective crystallization was employed to prepare the benzopyranopyrrolidine in high diastereomeric and enantiomeric purity. Conditions for reduction of an O-aryl lactone susceptible to epimerization were developed, and cyclization of the alcohol/phenol to the ether was accomplished in high yield.
The thienopyrazine was prepared by condensation of methyl thioglycolate and a regiospecifically prepared 2-bromo-3-cyano-5-phenylpyrazine. Conditions for effective halogen substitutive deamination to prepare regiospecific trisubstituted pyrazines will be described.
The mixture of 5 – and 6-phenyl regioisomers of 2-hydroxy-3-carboxamidopyrazine (IX) and (X), prepared by a known method, was treated with POCl3 and Et3N to produce the corresponding chloro nitriles (XI) and (XII ). Condensation of this mixture with methyl thioglycolate in the presence of NaOMe, followed by chromatographic separation of isomers furnished the desired thienopyrazine intermediate (XIII).
http://pubs.acs.org/doi/suppl/10.1021%2Fop049889k
…………………………………………………..
 Fiduxosin
Fiduxosin
……………………………………………………….
SYNTHESIS

Cycloaddition of the azomethine ylide resulting from N-trimethylsilylmethyl-N-methoxymethyl-(R)-alpha-methylbenzylamine (II) to 5-methoxycoumarin (I) produced the chiral cis-benzopyranopyrrole system (III). Lactone reduction by means of LiAlH4 or LiBH4 afforded diol (IV). After conversion of the primary alcohol of (IV) to either the corresponding chloride or the mesylate, cyclization in the presence of potassium tert-butoxide generated the tricyclic compound (V).
The alpha-methylbenzyl group of ( V) was removed by catalytic hydrogenation to give amine (VI), which was alkylated with 4-bromobutyronitrile yielding (VII). Reduction of the cyano group of (VII) using LiAlH4 in the presence AlCl3 or by catalytic hydrogenation in the presence of Raney -Ni produced the primary amine (VIII).
…………………………………………………

The mixture of 5 – and 6-phenyl regioisomers of 2-hydroxy-3-carboxamidopyrazine (IX) and (X), prepared by a known method, was treated with POCl3 and Et3N to produce the corresponding chloro nitriles (XI) and (XII ). Condensation of this mixture with methyl thioglycolate in the presence of NaOMe, followed by chromatographic separation of isomers furnished the desired thienopyrazine intermediate (XIII).
………………………………………………………….

In a regioselective synthetic method, phenyl glyoxime (XIV) was condensed with aminomalononitrile to produce the pyrazine N-oxide (XV). Reduction of the N-oxide of (XV) with triethyl phosphite yielded (XVI). Diazotization of the amino group of (XVI), followed by diazo displacement with CuBr2, furnished bromo pyrazine (XVII). This was then cyclized with methyl thioglycolate as above to yield the desired thienopyrazine intermediate (XIII).
………………………………………………….

In an alternative synthesis, phenylacetaldehyde (XVIII) was condensed with pyrrolidine (XIX) to give enamine (XX). Nitrosation of malononitrile (XXI), followed by treatment with tosyl chloride, produced the O-tosyl oxime (XXII). This was condensed with enamine (XX), and to the intermediate adduct (XXIII) was added thiophenol producing the phenylthiopyrazine (XXIV). Subsequent oxidation of the sulfide group of (XXIV) to sulfone (XXV), followed by condensation with methyl thioglycolate, gave the desired thienopyrazine (XIII).
……………………………………………………………..

The amino ester intermediate (XIII) was treated with phosgene and Et3N, and to the resulting isocyanate (XXVI) was added the primary amine (VIII), producing urea (XXVII). Then, cyclization of (XXVII) in refluxing toluene generated the desired compound,
fiduxosin
|
2-1-2002
|
Effect of fiduxosin, an antagonist selective for alpha(1A)- and alpha(1D)-adrenoceptors, on intraurethral and arterial pressure responses in conscious dogs.
|
The Journal of pharmacology and experimental therapeutics
|
|
|
2-1-2002
|
Modeling of relationships between pharmacokinetics and blockade of agonist-induced elevation of intraurethral pressure and mean arterial pressure in conscious dogs treated with alpha(1)-adrenoceptor antagonists.
|
The Journal of pharmacology and experimental therapeutics
|
|
|
1-1-2002
|
Effect of food on the pharmacokinetics of fiduxosin in healthy male subjects.
|
European journal of drug metabolism and pharmacokinetics
|
|
9-1-2012
|
Identification and analysis of hepatitis C virus NS3 helicase inhibitors using nucleic acid binding assays.
|
Nucleic acids research
|
|
|
3-1-2012
|
Small molecule screening identifies targetable zebrafish pigmentation pathways.
|
Pigment cell & melanoma research
|
|
|
7-1-2010
|
A small molecule inverse agonist for the human thyroid-stimulating hormone receptor.
|
Endocrinology
|
|
|
11-1-2009
|
A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel.
|
Analytical biochemistry
|
|
|
10-1-2009
|
Genetic mapping of targets mediating differential chemical phenotypes in Plasmodium falciparum.
|
Nature chemical biology
|
|
|
5-1-2007
|
Chemical genetics reveals a complex functional ground state of neural stem cells.
|
Nature chemical biology
|
|
|
5-1-2006
|
Microsphere-based protease assays and screening application for lethal factor and factor Xa.
|
Cytometry. Part A : the journal of the International Society for Analytical Cytology
|
|
|
5-1-2002
|
Single- and multiple-dose pharmacokinetics of fiduxosin under nonfasting conditions in healthy male subjects.
|
Journal of clinical pharmacology
|
|
|
5-1-2002
|
Multiple dose pharmacokinetics of fiduxosin under fasting conditions in healthy elderly male subjects.
|
The Journal of pharmacy and pharmacology
|
|
|
2-1-2002
|
Preclinical pharmacology of fiduxosin, a novel alpha(1)-adrenoceptor antagonist with uroselective properties.
|
The Journal of pharmacology and experimental therapeutics
|
 Dasantafil
Dasantafil

569351-91-3 CAS NO
405214-79-1 (racemate)
THERAPEUTIC CLAIM treatment of erectile dysfunction (phosphodiesterase (PDE) 5 isoenzyme inhibitor)
read all at
ALL ABOUT DRUGS
CLICK BELOW
http://www.allfordrugs.com/2014/01/29/dasantafil-for-treatment-of-erectile-dysfunction/

GISEDENAFIL
GISEDENAFIL BESYLATE
334826-98-1 free form
334827-98-4 (as besylate)
THERAPEUTIC CLAIM Treatment of lower urinary tract
symptoms associated with BPH
LEARN SPECTROSCOPY USING GISADENAFIL INTERMEDIATES
CHEMICAL NAMES FREE FORM
1. ……..7H-Pyrazolo[4,3-d]pyrimidin-7-one, 5-[2-ethoxy-5-[(4-ethyl-1-
piperazinyl)sulfonyl]-3-pyridinyl]-3-ethyl-2,6-dihydro-2-(2-methoxyethyl)-
2. …….5-{2-ethoxy-5-[(4-ethylpiperazin-1-yl)sulfonyl]pyridin-3-yl}-3-ethyl-2-(2-
methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one
3………1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine
MOLECULAR FORMULA C23H33N7O5S
MOLECULAR WEIGHT 519.6
CODE DESIGNATION UK-369,003
CAS REGISTRY NUMBER 334826-98-1
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-6,7-dihydro-2H-pyrazolo[4,3-d]pyrimidin-7-one
Phosphodiesterase PDE5A Inhibitors , Treatment of Erectile Dysfunction
Pfizer (Originator)
UK-369003 is a phosphodiesterase V (PDE V) inhibitor which had been under development for the treatment of erectile dysfunction, pulmonary hypertension and for the treatment of lower urinary tract symptoms, but no recent development has been reported for these indications. Trials for the treatment of benign prostatic hyperplasia were discontinued.
D09622, 334827-98-4
M.Wt:677.79
5-(2-ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl)-3-ethyl-2-(2-methoxyethyl)-2H-pyrazolo[4,3-d]pyrimidin-7(6H)-one benzenesulfonate
1-[[6-Ethoxy-5-[3-ethyl-4,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridinyl]sulfonyl]-4-ethylpiperazine Monobenzenesulfonate
Formula:C23H33N7O5S.C6H6O3S
| Certificate of Analysis |
|
| Biological Activity:Potent and selective PDE5 inhibitor (IC50: 1.23 nM) with improved selectivity over PDE6(PDE5/6 selectivity value 117 and >3000-fold selectivity over other PDEs).Gisadenafil has the potential for oral bioavailability and dose-proportional pharmacokinetics. Close analogue of Sildenafil (Viagra; Axon 2046) |
Gisadenafil besylate is a PDE5 inhibitor. Inhibition of PDE5 prevents the breakdown of cyclic phosphodiester secondary messenger molecules. This has the effect of prolonging and enhancing signal transduction.
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=UK-369,003
………………………….
PAPERS
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 1 p. 498 – 509
http://www.sciencedirect.com/science/article/pii/S0968089611008303
Scheme 1.
Reagents and conditions: (i) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, hydroxybenzotriazole, di-isopropylethylamine, THF, 20 °C, 20 h; (ii) caesium carbonate, alkyl mesylate or alkyl chloride, DMF, 20 °C, 20 h; (iii) KHMDS, R1OH, 120 °C, 20 h.
Scheme 2.
Reagents and conditions: (i) KHMDS, nBuOH, 120–130 °C, pressure vessel (ii) TFA, CH2Cl2; (iii) methanesulphonyl chloride, NEt3, CH2Cl2; (iv) HOAc, NaCNBH3, CH2O (v) KHMDS, nBuOH, reflux.
Scheme 3.
Reagents and conditions: (i) caesium carbonate, RCl, DMF; (ii) 50 psi H2, 10% Pd/C (iii) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, HOBT, di-isopropylethylamine, THF, 20 °C, 20 h; (iv) KHMDS, ethanol, 120 °C, pressure vessel; (v) TFA, CH2Cl2; (vi) CH2O, HOAc, NaCNBH3; (vii) R1OH, KHMDS, 120 °C.
Scheme 4.
Reagents and conditions: (i) NaNO2, HCl, H2O; (ii) TFAA, Et2O; (iii) ethyl propynoate, xylene, reflux, 2 h; (iv) NaOH, H2O, dioxan; (v) HNO3/H2SO4, 40–55 °C; (vi) (COCl)2, CH2Cl2, DMF; (vii) NH3, THF; (viii) 10% Pd/C, EtOH, 60 psi H2, 20 °C, 14 h; (ix) acid chloride of 3, NEt3, CH2Cl2; (x) KHMDS, EtOH, 130 °C, 14 h, pressure vessel; (xi) methoxyethanol, KHMDS, reflux, 14 h.
……………………………
PAPERS
http://pubs.acs.org/doi/abs/10.1021/op0300241

………………………….

PAPERS

UK-369,003 was nominated for development as the lead candidate for treatment of benign prostatic hyperplasia (BPH). The free base was found to be moderately crystalline with a melting point of 168 °C. Solubility of the free base at physiological pH was found to be poor hence necessitating a comprehensive screen for a suitable salt form of the API. Benzenesulfonic acid was found to form the most suitable counterion for the API with a melting point of 248 °C and satisfied all our requirements for primary and secondary processing. The process for the formation of the benzenesulfonic acid salt involved the use of water/methyl ethyl ketone (4% water by volume) as the reaction medium. The water level at 4% ensured an optimum balance between product quality (purging of impurities) and the reaction yield. The cyclisation reaction (step 2/Scheme 01) involves the use of ethanol as the reaction media. Any residual amount of ethanol in the isolated step 2 product was therefore considered to be a considerable risk factor in the potential formation of ethyl besylate during the final step processing (step 3/Scheme 01).

aCDI = carbonyl diimidazole; MEK = methyl ethyl ketone; EtOAc = ethyl acetate; KOtBu = potassium tertiary butoxide; EtOH = ethanol.
……………………
SYNTHESIS
Compound 1E is also known as 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, or alternatively as 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridyl sulphonyl}-4-ethylpiperazine (the compound of Example 103 of WO 01/27113 and exemplified hereinafter as Example 1).
Preparation 1
2,2-dimethoxybutane:
Methyl ethyl ketone (672 mL) was charged to a 2 L round bottomed flask and stirred at room temperature before being treated with, trimethylorthoformate (763 mL) and para-toluenesulphonic acid (6.65 g, 0.5 mol %). Over a 15 min period the internal temperature rose to 46° C., so the reaction was cooled to 0° C. for 30 min. The reaction was then stirred at room temperature for 2 h. The reaction was then neutralised by pouring onto sodium carbonate (ca. 750 g) with constant stirring. The resultant slurry was filtered under vacuum and the resultant filtrate was distilled at atmospheric pressure. The fraction boiling in the range 118° C.-124° C. was collected as a colourless liquid, 582 g, 70%.
1H NMR (CDCl3): δ=0.88 (3H, t), 1.24 (3H, s), 1.61 (2H, q), 3.17 (6H, s).
Example 1 N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide
(a) Ethyl 3-ethyl-1H-pyrazole-5-carboxylate (IIA) from (IlI) and (V)

To a stirred solution of 2,2-dimethoxybutane (10 g, 84.7 mMol) in CH2Cl2 (50 mL) under a nitrogen atmosphere at 0° C. was added pyridine (13.7 mL, 169.5 mMol). The reaction mixture was maintained at 0° C. and a solution of trichloroacetyl chloride (18.9 mL, 169.5 mMol) in CH2Cl2 (35 mL) was added over 1 hour with constant stirring. The yellow-orange solution begins to precipitate a white solid as the reaction progresses. The reaction mixture is allowed to warm to room temperature over 20 h. The reaction mixture was diluted with ethanol (150 mL) and re-cooled to 0° C. before treatment with hydrazine hydrate (8.2 mL, 169.5 mMol) as a solution in ethanol (35 mL) over 30 min. The reaction was heated to 50° C. and solvent was distilled at atmospheric pressure. The temperature was increased until the head temperature reached 78° C. Reflux was maintained for a further 2 h, before cooling to room temperature. The reaction mixture was diluted with water (250 mL) and ethanol was removed by evaporation at reduced pressure. The resultant mixture was extracted with CH2Cl2 (3×200 mL). The combined organics were dried (MgSO4), filtered and evaporated at reduced pressure to afford the title compound as a brown oil, 12.05 g, 85%.
1H NMR (300 MHz, CDCl3): δ=1.20 (3H, t), 1.28 (3H, t), 2.67 (2H, q), 4.29 (2H, q), 6.55 (1H, s), 12.56 (1H, s).
LRMS m/z=167.1 [M-H]+, C8H12N2O2 requires 168.2.
(b) Ethyl 3-ethyl-1H-pyrazole-5-carboxylic acid (IIA) from (IIA) via route 1

Aqueous sodium hydroxide solution (10M; 100 ml, 1.0 mol) was added dropwise to a stirred suspension of the title compound of Example (a) (66.0 g, 0.39 mol) in methanol and the resulting solution heated under reflux for 4 hours. The cool reaction mixture was concentrated under reduced pressure to ca. 200 ml, diluted with water (200 ml) and this mixture washed with toluene (3×100 ml). The resulting aqueous phase was acidified with concentrated hydrochloric acid to pH 4 and the white precipitate collected and dried by suction to provide the title compound (34.1 g). δ (DMSOd6): 1.13 (3H,t), 2.56 (2H,q), 6.42 (1H,s).
(c) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxylic acid
Fuming sulphuric acid (17.8 ml) was added dropwise to stirred, ice-cooled fuming nitric acid (16.0 ml), the resulting solution heated to 50° C., then 3-n-propyl-1H-pyrazole-5-carboxylic acid (Chem. Pharm. Bull., 1984, 32,1568; 16.4 g, 0.106 mol) added portionwise over 30 minutes whilst maintaining the reaction temperature below 60° C. The resulting solution was heated for 18 hours at 60° C., allowed to cool, then poured onto ice. The white precipitate was collected, washed with water and dried by suction to yield the title compound (15.4 g), m.p. 170-172° C. Found: C, 42.35; H, 4.56; N, 21.07. C7H9N3O4requires C, 42.21; H, 4.55; N, 21.10%. δ (DMSOd6): 0.90 (3H,t), 1.64 (2H,m), 2.83 (2H,m), 14.00 (1 H,s).
(d) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxylic acid (IIA) to (AA) via route 2

Obtained from the title compound of Example (b), by analogy with the process of Example (c), as a brown solid (64%). δ (DMSOd6): 1.18 (3H,t), 2.84 (2H,m), 13.72 (1 H,s).
(e) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxamide
A solution of the title compound of Example (c) (15.4 g, 0.077 mol) in thionyl chloride (75 ml) was heated under reflux for 3 hours and then the cool reaction mixture evaporated under reduced pressure. The residue was azeotroped with tetrahydrofuran (2×50 ml) and subsequently suspended in tetrahydrofuran (50 ml), then the stirred suspension ice-cooled and treated with gaseous ammonia for 1 hour. Water (50 ml) was added and the resulting mixture evaporated under reduced pressure to give a solid which, after trituration with water and drying by suction, furnished the title compound (14.3 g).
m.p. 197-199° C. Found: C, 42.35; H, 5.07; N, 28.38. C7H10N4O3 requires C, 42.42; H, 5.09; N, 28.27%. δ (DMSOd6): 0.90 (3H,t), 1.68 (2H,m), 2.86 (2H,t), 7.68 (1 H,s), 8.00 (1 H,s).
(f) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxamide BA from AA via route 3

Obtained from the title compound of Example (d), by analogy with Example (e), as a white solid (90%). δ (DMSOd6): 1.17 (3H,t), 2.87 (2H,m), 7.40 (1H,s), 7.60 (1H,s), 7.90 (1H,s). LRMS: m/z 185 (M+l)+.
(g)(i) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide CA from BA via route 4

A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (2.5 kg, 13.6 Mol), sodium carbonate (1.8 Kg, 17.0 Mol) and 2-bromoethyl methyl ether (1.98 kg, 14.2 Mol) in THF (22.5 L) and water (2.5 L) was heated under reflux and stirred for 20 hours. The mixture was cooled to ambient temperature and CH2Cl2 (67.5 L) and water (22.5 L) were added. The resultant organic and aqueous layers were separated. The aqueous phase was extracted with CH2Cl2 (22.5 L) and the combined organic solution was distilled under atmospheric pressure and replaced with ethyl acetate (33 L) to a final volume of 17 L. The cooled mixture was granulated at ambient temperature for 2 hours, filtered and washed with ethyl acetate (2.5 L). This afforded 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a white crystalline solid, 2.10 kg, 57%. m.p.=140° C. Found: C, 44.46; H, 5.79; N, 23.01. C9H14N4O4 requires C, 44.63; H, 5.79; N, 23.14%.
δ (CDCl3): 1.18 (3H, t), 2.98 (2H, q), 3.22 (3H, s), 3.77 (2H, t), 4.28 (2H, q), 6.03 (1H, s), 7.36 (1H, s).
LRMS: m/z=243 (M+1)+
(g)(ii) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide.
A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (25 g, 0.136 Mol), sodium carbonate (18 g, 0.17 Mol) and sodium iodide (20.4 g, 0.136 Mol) were suspended in ethyl methyl ketone (125 mL) at room temperature. 2-bromoethyl methyl ether (12.8 mL, 0.142 Mol) was added and the mixture was heated to reflux and stirred for 70 hours. The mixture was cooled to ambient temperature and water (250 mL) was added. The resultant slurry was warmed to reflux and held at that temperature for 30 min before cooling to room temperature. The resultant precipitate was granulated at room temperature for 3 h, filtered and vacuum dried to afford 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a yellow crystalline solid 24.3 g, 74%. Data as reported for Example (g)(i).
(h) 4-Amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (IA) from CA via route 5

A mixture of 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide (20 g, 82.6 mMol) and 5% Pd/C (1 g) in methanol (200 mL) was pressurised at 50psi/25° C. in a sealed vessel and stirred for 15 hours. At the end of the reaction the mixture was filtered through arbocel and the filter cake was washed with methanol. The methanolic solution was distilled at atmospheric pressure and replaced with ethyl acetate to a final volume of 100 mL. The cooled mixture was granulated at ambient temperature for 2 h filtered and washed with ethyl acetate (20 mL) to afford 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide as a white crystalline solid, 15 g, 88%. m.p.=131° C. Found: C, 50.75; H, 7.62; N, 26.38. C9H16N4O2 requires C, 50.94; H, 7.55; N, 26.42%. δ (CDCl3): 1.20 (3H, t), 2.63 (2H, q), 3.32 (3H, s), 3.74 (2H, t), 3.95 (2H, s), 4.15 (2H, t), 5.27 (1H, s), 6.59 (1H, s).
LRMS: m/z=213 (M+1)+
(i) N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide.

2-ethoxy-5-(4-ethyl-1-piperazinylsulfonyl)nicotinic acid (2.31 kg, 6.73 Mol) was suspended in ethyl acetate (16.2 L) and 1,1-carbonyldimidazole (1.09 kg, 6.73 Mol) was added at room temperature. The reaction mixture was heated at 45° C. for 40 minutes and then the reaction was stirred for a further 40 minutes at reflux. After cooling to ambient temperature 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (1.5 kg, 7.06 Mol) was added to the cooled mixture, and the reaction stirred for a further 15 hours under reflux. The mixture was cooled filtered and the filter cake was washed with 90% water/10% ethyl acetate, (2 mL /g) to afford N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide as an off white crystalline solid, 3.16 kg, 88%. m.p.=156° C. Found: C, 51.33; H, 6.56; N, 18.36. C23H35N7O6S requires C, 51.40; H, 6.53; N, 18.25%.
δ (CDCl3): 1.04 (3H, t), 1.22 (3H, t), 1.60 (3H, t), 2.44 (2H, q), 2.54 (4H, m), 2.96 (2H, q), 3.12 (4H, m), 3.36 (3H, s), 3.81 (2H, t), 4.27 (2H, t), 4.80(2H, q), 5.35(1H, s), 6.68 (1H, s), 8.66 (1H, d), 8.86 (1H, d), 10.51 (1H, s).
LRMS: m/z=539 (M+1)+
(i) 1-(6-Ethoxy-5-[3-ethyll-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine•ethyl acetate solvate.

GISADENAFIL
A mixture of N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120° C. for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55° C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4 L. The precipitated solids were granulated at 5° C. for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p.=157° C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5CO2CH3 requires C, 53.21; H, 6.49; N, 18.25%.
δ (CDCl3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1H, d), 9.06 (1H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
LRMS: m/z=520 (M+1)+
……………..
Example 102
1-(6-Ethoxy-5-f3-ethyll-6,7-dihvdro-2-(2-methoxyethvn-7-oxo-2r7-pyrazoler4.3- cf1pyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine»ethyl acetate solvate.

To prepare the compound of Example 8 a mixture of Λ/-[3-carbamoyl-5-ethyl- 1 -(2-methoxyethyl)-1 /-/-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1 -piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120°C for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55°C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4L. The precipitated solids were granulated at 5°C for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1 -(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo- 2H-pyrazole[4,3-o pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p. = 157°C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5C02CH3 requires C, 53.21 ; H, 6.49; N, 18.25%.
δ(CDCI3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1 H, d), 9.06 (1 H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
LRMS: m/z = 520 (M+1)+
Example 103
1-(6-ethoxy-5-r3-ethyl-6.7-dihvdro-2-(2-methoxyethvn-7-oxo-2H-pyrazolor4.3- dlpyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine

GISADENAFIL
10g (0.019 mol) of the compound of Example 8 and Example 102, 1-{6- ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine ethyl acetate solvate, was charged followed by 12ml/g (120mls) of 16% water in ethyl alcohol. The slurry was heated to reflux to yield a solution and 6ml/g (60mls) distilled off at atmospheric pressure. The solution was then cooled to room temperature with crystallisation occurring at 40°C. The slurry was then cooled to 5-10°C and granulated for 30 minutes following which it was filtered and washed with 2ml/g ethyl alcohol (20 mis). The damp solid was dried in vacuo overnight at 55-60 °C to yield a white crystalline solid. (Yield 7.6g, 76%). Melting Point 162- 165°C.
δ (CDCI3): 1.05 (3H,t), 1.42 (3H,t), 1.58 (3H,t), 2.43 (2H,q), 2.57 (4H,t), 3.09 (2H, t), 3.15 (4H,t), 3.30 (3H,s), 3.93 (2H,t), 4.48 (2H,t), 4.90 (2H,q), 8.65 (1 H,d), 9.05 (1 H,d), 10.65 (1 H,s).
In the process of Example 103, water and pharmaceutically acceptable alcohols such as methanol, ethanol, propanol, butanol and mixtures thereof can be used to prepare the compound of Examples 8 and 102.
BESYLATE SALT
Example 104 1-(6-ethoxy-5-r3-ethyl-6,7-dihvdro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolor4.3- d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine benzene-sulfonate salt.

170g (0.33 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7- dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3- pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a water/ 2- butanone (4% v/v) at 10 ml/g (1.7 litres) and warmed to reflux. 53g (0.33 mol) of benzene sulfonic acid dissolved in water (23mls, resulting in 70 % w/w solution) was added to the refluxing solution over 30 minutes.5.3ml/g (0.9 litres) of 2-butanone were striped and replaced and the slurry cooled. The slurry was cooled to 5-10°C and granulated for 2 hours after which it was filtered and washed with 2ml/g (0.3 litres) of 2-butanone. The salt was dried overnight in vacuo at 55-60°C to yield a white crystalline solid. Yield 215g, 96.4%. Mpt 242-244°C. δ (DMSO): 1.17 (3H, t), 1.28 (3H, t), 1.35 (3H, t), 2.73 (2H, q), 2.97 (2H, q), 3.2 (3H, s), 3.58 (2H, t), 3.78 (3H, t), 3.81 (2H, t), 4.49 (2H, t) 4.51 (2H, q), 7.29-7.33 (3H, m), 7.57-7.60 (2H, m), 8.28 (1 H, d), 8.73 (1 H, d), 9.13 (1 H,s), 11.90(1 H,s).
The powder X-ray diffraction (PXRD) pattern for this salt, having Mpt 242- 244°C, was determined using a Siemens D5000 powder X-ray diffractometer fitted with a theta-theta goniometer, automatic beam divergence slits, a secondary monochromator and a scintillation counter. The specimen was rotated whilst being irradiated with copper K-alpha1 X-rays (Wavelength = 1.5046 Angstroms) filtered with a graphite monochromator (λ = 0.15405nm) with the X-ray tube operated at 40 kV/mA. The main peaks (in degrees θ) of the PXRD pattern are illustrated in Table I.
Table


The same besylate salt, as defined by the XRD pattern described in Table 1 , when made via alternative routes can have a melting point in the range of from 235-246°C (measured using a Perkin Elmer DSC7 at a heating rate of 20°C/minute).

References
1 The discovery of UK-369003, a novel PDE5 inhibitor with the potential for oral bioavailability and dose-proportional pharmacokinetics
Bioorg Med Chem 2012, 20(1): 498………….MP 161 – 162 °C
2. Hajikarimian, Y.; Yeo, S.; Ryan, R.W.; Levett, P.; Stoneley, C.; Singh, P.
Investigation into the formation of the genotoxic impurity ethyl besylate in the final step manufacturing process of UK-369,003-26, a novel PDE5 inhibitor
Org Process Res Dev 2010, 14(4): 1027
3. Bentham; Dawson; Dunn; Papadopoulos; Taylor; Mitchell; Snowden; Taylor
Organic Process Research and Development, 2004 , vol. 8, 4 PG. 674 – 679 ………….AS ENTRY B
PATENTS
1. WO 2010062366
2. WO 2007072156
3 WO 2007072156
4.US2002/22732 A1,
5.US2002/28799 A1,
6.
| WO1998049166A1 * | Apr 10, 1998 | Nov 5, 1998 | Mark Edward Bunnage | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| WO1999054333A1 * | Mar 25, 1999 | Oct 28, 1999 | Mark Edward Bunnage | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction |
| US4666921 * | 15 окт 1985 | 19 май 1987 | Ludwig Heumann & Co. Gmbh | Pyrazole derivatives, processes for their preparation and pharmaceutical preparations containing these compounds |
| US5808092 * | 15 окт 1997 | 15 сен 1998 | Ube Industries, Ltd. | Process for preparing-1-ethyl-5-hydroxypyrazole |
| US6015911 * | 24 мар 1998 | 18 янв 2000 | Dow Agrosciences Llc | Process for preparing 1-alkyl-4-(2-chloro-3-alkoxy-4-alkylsulfonylbenzoyl)-5-hydroxypyrazole and related compounds |
| EP0463756A1 | 7 июн 1991 | 2 янв 1992 | Pfizer Limited | Pyrazolopyrimidinone antianginal agents |
| EP0812845A1 | 4 июн 1997 | 17 дек 1997 | Pfizer Limited | Process for preparing sildenafil |
| EP0994115A2 | 11 окт 1999 | 19 апр 2000 | Pfizer Limited | Process for preparation of pyrazolo-(4,3-d)pyrimidin-7-ones and intermediates thereof |
| EP0995750A1 | 15 окт 1999 | 26 апр 2000 | Pfizer Inc. | Pyrazolopyrimidinone cGMP PDE5 inhibitors for the treatment of sexual dysfunction |
| WO1998049166A1 | 10 апр 1998 | 5 ноя 1998 | Mark Edward Bunnage | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| WO1999054333A1 | 25 мар 1999 | 28 окт 1999 | Mark Edward Bunnage | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction |
| WO2001027112A1 | 4 окт 2000 | 19 апр 2001 | Charlotte Moira Norfo Allerton | 5-(2-substituted-5-heterocyclylsulphonylpyrid-3-yl)-dihydropyrazolo[4,3-d]pyrimidin-7-ones as phosphodiesterase inhibitors |
| WO2001027113A2 | 11 окт 2000 | 19 апр 2001 | Mark Edward Bunnage | PYRAZOLO `4,3-d! PYRIMIDINE DERIVATIVES |
PDE5 inhibitors mirodenafil

sildenafil

tadalafil

udenafil 3-(l-methyl-7-oxo-3-propyl-4H-pyrazolo[5,4-e]pyrimidin-5-yl)-N- [2-(l -methylpyrrolidin-2-yl)ethyl] -4-propoxybenzenesulfonamide

vardenafil 4-[2-ethoxy-5-(4-ethylpiperazin-l-yl)sulfonyl-phenyl]-9-methyl-7- propyl- 3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one

avanafil 4-[(3-chloro-4-methoxy-phenyl)methylamino]-2-[(2S)-2- (hydroxymethyl)pyrrolidin- 1 -yl] -N-(pyrimidin-2- ylmethyl)pyrimidine-5-carboxamide

dasantafil 7-[(3-bromo-4-methoxyphenyl)methyl]-l-ethyl-8-[[(lR,2R)-2- hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)purine-2,6-dione
NM 702 (Nissan Chemical Industries)

SLX 101 (Surface Logix) – Structure Not Available
UK 369003 (Pfizer) – Gisadenafil besylate


334827-98-4 (as besylate)
symptoms associated with BPH

 1…………..
1…………..
















1.07 (3H, t), METHYL OF -N CH2-CH3 ON PIPERAZINE RING
1.42 (3H, t), METHYL OF -CH2-CH3 ON PYRAZOLE SIDE CHAIN
1.61 (3H, t), METHYL OF -O-CH2-CH3 ON PYRIMIDINE RING
2.44 (2H, q), CH2 OF -N CH2-CH3 ON PIPERAZINE RING
2.57 (4H, m),4H OF –NCH2 ON PIPERAZINE RING BOTH SIDE OF N ATOM
3.08 (2H, q), CH2 OF –CH2-CH3 ON PYRAZOLE SIDE CHAIN
3.15 (4H, m),4H OF –NCH2 ON PIPERAZINE RING BOTH SIDE OF N ATOM CLOSE TO SO2 GP
3.32 (3H, s),METHYL OF -OCH3 ON PYRAZOLE SIDE CHAIN
3.92 (2H, q), CH2 OF NCH2-CH2-O-CH3 ON PYRAZOLE SIDECHAIN
4.48 (2H, q), CH2 OF NCH2 –CH2-O-CH3 ON PYRAZOLE SIDECHAIN
4.77 (2H, q), CH2 OF O-CH2 CH3 ON PYRIMIDINE RING
8.65 (1H, d), PYRIMIDINE AROM H …..AWAY/PARA TO C=O-NH -PYRAZOLE GP
9.06 (1H, d). PYRIMIDINE AROM H …..CLOSER/ORTHO TO C=O-NH -PYRAZOLE GP, reason this signal will shift from 8,86 delta to 9.06 after cyclization in this step ie formation of GISADENAFIL
The spectrum also has signals that correspond to a solvate with ethyl acetate.