
Home » Peptide drugs (Page 3)
Category Archives: Peptide drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Imdevimab
| (Heavy chain) QVQLVESGGG VVQPGRSLRL SCAASGFTFS NYAMYWVRQA PGKGLEWVAV ISYDGSNKYY ADSVKGRFTI SRDNSKNTLY LQMNSLRTED TAVYYCASGS DYGDYLLVYW GQGTLVTVSS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW QQGNVFSCSV MHEALHNHYT QKSLSLSPGK (Light chain) QSALTQPASV SGSPGQSITI SCTGTSSDVG GYNYVSWYQQ HPGKAPKLMI YDVSKRPSGV SNRFSGSKSG NTASLTISGL QSEDEADYYC NSLTSISTWV FGGGTKLTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADSSPVK AGVETTTPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H96, H147-H203, H223-L215, H229-H’229, H264-H324-H370-H428, H’22-H’96, H’147-H’203, H’223-L’215, H’264-H’324, H’370-H’428, L22-L90, L138-L197, L’22-L’90, L’138-L’197) |
イムデビマブ;
- Immunoglobulin G1, anti-(severe acute respiratory syndrome coronavirus 2 spike glycoprotein) (human monoclonal REGN10987 γ1-chain), disulfide with human monoclonal REGN10987 λ-chain, dimer
| Formula | C6396H9882N1694O2018S42 |
|---|---|
| CAS | 2415933-40-1 |
| Mol weight | 144141.7693 |
Monoclonal antibody
Treatment and prophylaxis of SARS-CoV-2 infection
ANTIVIRAL
SARS-CoV-2 spike glycoprotein
- REGN 10987
- RG 6412
Fact Sheet – US Food and Drug Administration
https://www.fda.gov › media › download
PDFBenefit of treatment with casirivimab and imdevimab has not been observed in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab.
Casirivimab/imdevimab, sold under the brand name REGEN-COV,[1] is an experimental medicine developed by the American biotechnology company Regeneron Pharmaceuticals. It is an artificial “antibody cocktail” designed to produce resistance against the SARS-CoV-2 coronavirus responsible for the COVID-19 pandemic.[3][4] It consists of two monoclonal antibodies, casirivimab (REGN10933) and imdevimab (REGN10987) that must be mixed together.[1][5][6] The combination of two antibodies is intended to prevent mutational escape.[7]
Trials
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
References
- ^ Jump up to:a b c “REGEN-COV- casirivimab and imdevimab kit”. DailyMed. Retrieved 18 March 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19”. U.S. Food and Drug Administration (FDA) (Press release). 21 November 2020. Retrieved 21 November 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Kelland K (14 September 2020). “Regeneron’s antibody drug added to UK Recovery trial of COVID treatments”. Reuters. Retrieved 14 September 2020.
- ^ “Regeneron’s COVID-19 Response Efforts”. Regeneron Pharmaceuticals. Retrieved 14 September 2020.
- ^ Morelle R (14 September 2020). “Antibody treatment to be given to Covid patients”. BBC News Online. Retrieved 14 September2020.
- ^ “Safety, Tolerability, and Efficacy of Anti-Spike (S) SARS-CoV-2 Monoclonal Antibodies for Hospitalized Adult Patients With COVID-19”. ClinicalTrials. 3 September 2020. Retrieved 14 September2020.
- ^ Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. (August 2020). “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies”. Science. 369 (6506): 1014–1018. Bibcode:2020Sci…369.1014B. doi:10.1126/science.abd0831. PMC 7299283. PMID 32540904.
- ^ “RECOVERY COVID-19 phase 3 trial to evaluate Regeneron’s REGN-COV2 investigational antibody cocktail in the UK”. Recovery Trial. Retrieved 14 September 2020.
- ^ “Phase III prevention trial showed subcutaneous administration of investigational antibody cocktail casirivimab and imdevimab reduced risk of symptomatic COVID-19 infections by 81%”. streetinsider.com. Archived from the original on 2021-04-12. Retrieved 2021-04-12.
- ^ Jump up to:a b c “Regeneron Reports Positive Interim Data with REGEN-COV Antibody Cocktail used as Passive Vaccine to Prevent COVID-19”(Press release). Regeneron Pharmaceuticals. 26 January 2021. Retrieved 19 March 2021 – via PR Newswire.
- ^ “Fact Sheet For Health Care Providers Emergency Use Authorization (EUA) Of Casirivimab And Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
- ^ “Casirivimab and Imdevimab”. Regeneron Pharmaceuticals. Retrieved 19 March 2021.
- ^ “EMA starts rolling review of REGN‑COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 1 February 2021. Retrieved 1 February 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “EMA reviewing data on monoclonal antibody use for COVID-19” (Press release). European Medicines Agency (EMA). 4 February 2021. Retrieved 4 March 2021.
- ^ “EMA issues advice on use of REGN-COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 26 February 2021. Retrieved 5 March 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ https://timesofindia.indiatimes.com/india/india-approves-roche/regeneron-antibody-cocktail-to-treat-covid-19/articleshow/82407551.cms
- ^ “Roche receives Emergency Use Authorisation in India for its investigational Antibody Cocktail (Casirivimab and Imdevimab) used in the treatment of Covid-19 | Cipla”. http://www.cipla.com. Retrieved 2021-05-06.
- ^ Williams, Stephen (3 October 2020). “Experimental drug given to President made locally”. The Daily Gazette.
- ^ Stanton, Dan (11 September 2020). “Manufacturing shift to Ireland frees up US capacity for Regeneron’s COVID antibodies”. BioProcess International.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ “Roche and Regeneron link up on a coronavirus antibody cocktail”. CNBC. 19 August 2020. Retrieved 14 September 2020.
- ^ Jump up to:a b Thomas K (2 October 2020). “President Trump Received Experimental Antibody Treatment”. The New York Times. ISSN 0362-4331. Retrieved 2 October 2020.
- ^ Hackett DW (3 October 2020). “8-Gram Dose of COVID-19 Antibody Cocktail Provided to President Trump”. http://www.precisionvaccinations.com. Archived from the original on 3 October 2020.
External links
- “Casirivimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Imdevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Casirivimab and Imdevimab EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA).
- “Frequently Asked Questions on the Emergency Use Authorization of Casirivimab + Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
| REGN10933 (blue) and REGN10987 (orange) bound to SARS-CoV-2 spike protein (pink). From PDB: 6VSB, 6XDG. | |
| Combination of | |
|---|---|
| Casirivimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Imdevimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | REGEN-COV |
| Other names | REGN-COV2 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Casirivimab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: Unapproved (Emergency Use Authorization)[1][2] |
| Identifiers | |
| DrugBank | DB15691 |
| KEGG | D11938D11939 |
////////Imdevimab, ANTI VIRAL, PEPTIDE, CORONA VIRUS, COVID19, APPROVALS 2020, FDA 2020, イムデビマブ, REGN 10987, RG 6412,
NEW DRUG APPROVALS
one time
$10.00
Casirivimab
(Heavy chain)
QVQLVESGGG LVKPGGSLRL SCAASGFTFS DYYMSWIRQA PGKGLEWVSY ITYSGSTIYY
ADSVKGRFTI SRDNAKSSLY LQMNSLRAED TAVYYCARDR GTTMVPFDYW GQGTLVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(Light chain)
DIQMTQSPSS LSASVGDRVT ITCQASQDIT NYLNWYQQKP GKAPKLLIYA ASNLETGVPS
RFSGSGSGTD FTFTISGLQP EDIATYYCQQ YDNLPLTFGG GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H147-H203, H223-L214, H229-H’229, H232-H’232, H264-H324, H370-H428, H’22-H’96, H’147-H’203, H’223-L’214, H’264-H’324, H’370-H’428, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Casirivimab
カシリビマブ;
- Immunoglobulin G1, anti-(severe acute respiratory syndrome coronavirus 2 spike glycoprotein) (human monoclonal REGN10933 γ1-chain), disulfide with human monoclonal REGN10933 κ-chain, dimer
| Formula | C6454H9976N1704O2024S44 |
|---|---|
| CAS | 2415933-42-3 |
| Mol weight | 145233.3296 |
Monoclonal antibody
Treatment and prophylaxis of SARS-CoV-2 infection (COVID-19)
SARS-CoV-2 spike glycoprotein
- Protein Sequence
- Sequence Length: 1328, 450, 450, 214, 214
- REGN 10933
- RG 6413
https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19 November 21, 2020
Today, the U.S. Food and Drug Administration issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age or older weighing at least 40 kilograms [about 88 pounds]) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19. This includes those who are 65 years of age or older or who have certain chronic medical conditions.
In a clinical trial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.
Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.
Casirivimab and imdevimab are not authorized for patients who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19. A benefit of casirivimab and imdevimab treatment has not been shown in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation.
“The FDA remains committed to advancing the nation’s public health during this unprecedented pandemic. Authorizing these monoclonal antibody therapies may help outpatients avoid hospitalization and alleviate the burden on our health care system,” said FDA Commissioner Stephen M. Hahn, M.D. “As part of our Coronavirus Treatment Acceleration Program, the FDA uses every possible pathway to make new treatments available to patients as quickly as possible while continuing to study the safety and effectiveness of these treatments.”
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens such as viruses. Casirivimab and imdevimab are monoclonal antibodies that are specifically directed against the spike protein of SARS-CoV-2, designed to block the virus’ attachment and entry into human cells.
“The emergency authorization of these monoclonal antibodies administered together offers health care providers another tool in combating the pandemic,” said Patrizia Cavazzoni, M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “We will continue to facilitate the development, evaluation and availability of COVID-19 therapies.”
The issuance of an EUA is different than an FDA approval. In determining whether to issue an EUA, the FDA evaluates the totality of available scientific evidence and carefully balances any known or potential risks with any known or potential benefits of the product for use during an emergency. Based on the FDA’s review of the totality of the scientific evidence available, the agency has determined that it is reasonable to believe that casirivimab and imdevimab administered together may be effective in treating patients with mild or moderate COVID-19. When used to treat COVID-19 for the authorized population, the known and potential benefits of these antibodies outweigh the known and potential risks. There are no adequate, approved and available alternative treatments to casirivimab and imdevimab administered together for the authorized population.
The data supporting this EUA for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms. Of these patients, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline. Viral load reduction in patients treated with casirivimab and imdevimab was larger than in patients treated with placebo at day seven. However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment. For patients at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated patients on average compared to 9% in placebo-treated patients. The effects on viral load, reduction in hospitalizations and ER visits were similar in patients receiving either of the two casirivimab and imdevimab doses.
Under the EUA, fact sheets that provide important information about using casirivimab and imdevimab administered together in treating COVID-19 as authorized must be made available to health care providers and to patients and caregivers. These fact sheets include dosing instructions, potential side effects and drug interactions. Possible side effects of casirivimab and imdevimab include: anaphylaxis and infusion-related reactions, fever, chills, hives, itching and flushing.
The EUA was issued to Regeneron Pharmaceuticals Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Related Information
- Casirivimab and Imdevimab EUA Letter of Authorization
- Frequently Asked Questions on the Emergency Use Authorization for Casirivimab and Imdevimab
- Emergency Use Authorization: Therapeutics
- Coronavirus Disease (COVID-19)
Casirivimab/imdevimab, sold under the brand name REGEN-COV,[1] is an experimental medicine developed by the American biotechnology company Regeneron Pharmaceuticals. It is an artificial “antibody cocktail” designed to produce resistance against the SARS-CoV-2 coronavirus responsible for the COVID-19 pandemic.[3][4] It consists of two monoclonal antibodies, casirivimab (REGN10933) and imdevimab (REGN10987) that must be mixed together.[1][5][6] The combination of two antibodies is intended to prevent mutational escape.[7]
Trials
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
References
- ^ Jump up to:a b c “REGEN-COV- casirivimab and imdevimab kit”. DailyMed. Retrieved 18 March 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19”. U.S. Food and Drug Administration (FDA) (Press release). 21 November 2020. Retrieved 21 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Kelland K (14 September 2020). “Regeneron’s antibody drug added to UK Recovery trial of COVID treatments”. Reuters. Retrieved 14 September 2020.
- ^ “Regeneron’s COVID-19 Response Efforts”. Regeneron Pharmaceuticals. Retrieved 14 September 2020.
- ^ Morelle R (14 September 2020). “Antibody treatment to be given to Covid patients”. BBC News Online. Retrieved 14 September2020.
- ^ “Safety, Tolerability, and Efficacy of Anti-Spike (S) SARS-CoV-2 Monoclonal Antibodies for Hospitalized Adult Patients With COVID-19”. ClinicalTrials. 3 September 2020. Retrieved 14 September2020.
- ^ Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. (August 2020). “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies”. Science. 369 (6506): 1014–1018. Bibcode:2020Sci…369.1014B. doi:10.1126/science.abd0831. PMC 7299283. PMID 32540904.
- ^ “RECOVERY COVID-19 phase 3 trial to evaluate Regeneron’s REGN-COV2 investigational antibody cocktail in the UK”. Recovery Trial. Retrieved 14 September 2020.
- ^ “Phase III prevention trial showed subcutaneous administration of investigational antibody cocktail casirivimab and imdevimab reduced risk of symptomatic COVID-19 infections by 81%”. streetinsider.com. Archived from the original on 2021-04-12. Retrieved 2021-04-12.
- ^ Jump up to:a b c “Regeneron Reports Positive Interim Data with REGEN-COV Antibody Cocktail used as Passive Vaccine to Prevent COVID-19”(Press release). Regeneron Pharmaceuticals. 26 January 2021. Retrieved 19 March 2021 – via PR Newswire.
- ^ “Fact Sheet For Health Care Providers Emergency Use Authorization (EUA) Of Casirivimab And Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
- ^ “Casirivimab and Imdevimab”. Regeneron Pharmaceuticals. Retrieved 19 March 2021.
- ^ “EMA starts rolling review of REGN‑COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 1 February 2021. Retrieved 1 February 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “EMA reviewing data on monoclonal antibody use for COVID-19” (Press release). European Medicines Agency (EMA). 4 February 2021. Retrieved 4 March 2021.
- ^ “EMA issues advice on use of REGN-COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 26 February 2021. Retrieved 5 March 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ https://timesofindia.indiatimes.com/india/india-approves-roche/regeneron-antibody-cocktail-to-treat-covid-19/articleshow/82407551.cms
- ^ “Roche receives Emergency Use Authorisation in India for its investigational Antibody Cocktail (Casirivimab and Imdevimab) used in the treatment of Covid-19 | Cipla”. http://www.cipla.com. Retrieved 2021-05-06.
- ^ Williams, Stephen (3 October 2020). “Experimental drug given to President made locally”. The Daily Gazette.
- ^ Stanton, Dan (11 September 2020). “Manufacturing shift to Ireland frees up US capacity for Regeneron’s COVID antibodies”. BioProcess International.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ “Roche and Regeneron link up on a coronavirus antibody cocktail”. CNBC. 19 August 2020. Retrieved 14 September 2020.
- ^ Jump up to:a b Thomas K (2 October 2020). “President Trump Received Experimental Antibody Treatment”. The New York Times. ISSN 0362-4331. Retrieved 2 October 2020.
- ^ Hackett DW (3 October 2020). “8-Gram Dose of COVID-19 Antibody Cocktail Provided to President Trump”. http://www.precisionvaccinations.com. Archived from the original on 3 October 2020.
External links
- “Casirivimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Imdevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Casirivimab and Imdevimab EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA).
- “Frequently Asked Questions on the Emergency Use Authorization of Casirivimab + Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
| REGN10933 (blue) and REGN10987 (orange) bound to SARS-CoV-2 spike protein (pink). From PDB: 6VSB, 6XDG. | |
| Combination of | |
|---|---|
| Casirivimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Imdevimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | REGEN-COV |
| Other names | REGN-COV2 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Casirivimab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: Unapproved (Emergency Use Authorization)[1][2] |
| Identifiers | |
| DrugBank | DB15691 |
| KEGG | D11938 |
//////////// Casirivimab, ANTI VIRAL, PEPTIDE, SARS-CoV-2, MONOCLONAL ANTIBODY, FDA 2020, 2020APPROVALS, CORONA VIRUS, COVID 19, カシリビマブ, REGN-COV2, REGN10933+REGN10987 combination therapy, REGN 10933, RG 6413
NEW DRUG APPROVALS
ONE TIME
$10.00
Evinacumab
(Heavy chain)
EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA ISGDGGSTYY
ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL RNTIFGVVIP DAFDIWGQGT
MVTVSSASTK GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF
LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE
QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS
QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK
SRWQEGNVFS CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK ASSLESGVPS
RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H140-L214, H153-H209, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’140-L’214, H’153-H’209, H’267-H’327, H’373-H’431, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Evinacumab
エビナクマブ (遺伝子組換え)
Immunoglobulin G4, anti-(human protein ANGPTL3 (angiopoietin-like 3)) (human monoclonal REGN1500 heavy chain), disulfide with human monoclonal REGN1500 light chain, dimer
| Formula | C6480H9992N1716O2042S46 |
|---|---|
| CAS | 1446419-85-7 |
| Mol weight | 146081.9345 |
Protein Sequence
Sequence Length: 1334, 453, 453, 214, 214multichain; modified (modifications unspecified)
FDA APPROVED, 2021/2/11, EVKEEZA
Antihyperlipidemic, Anti-angiopietin like 3
Monoclonal antibody
Treatment of dyslipidemia
- REGN 1500
- REGN-1500
- REGN1500
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-22 – Cys-96 | disulfide bridge |
| bridge | Cys-140 – Cys-214” | disulfide bridge |
| bridge | Cys-153 – Cys-209 | disulfide bridge |
| bridge | Cys-232 – Cys-232′ | disulfide bridge |
| bridge | Cys-235 – Cys-235′ | disulfide bridge |
| bridge | Cys-267 – Cys-327 | disulfide bridge |
| bridge | Cys-373 – Cys-431 | disulfide bridge |
| bridge | Cys-22′ – Cys-96′ | disulfide bridge |
| bridge | Cys-140′ – Cys-214”’ | disulfide bridge |
| bridge | Cys-153′ – Cys-209′ | disulfide bridge |
| bridge | Cys-267′ – Cys-327′ | disulfide bridge |
| bridge | Cys-373′ – Cys-431′ | disulfide bridge |
| bridge | Cys-23” – Cys-88” | disulfide bridge |
| bridge | Cys-134” – Cys-194” | disulfide bridge |
| bridge | Cys-23”’ – Cys-88”’ | disulfide bridge |
| bridge | Cys-134”’ – Cys-194”’ | disulfide bridge |
PATENTS
WO 2017024062
US 20170305999
Evinacumab, sold under the brand name Evkeeza, is a monoclonal antibody medication for the treatment of homozygous familial hypercholesterolemia (HoFH).[1][2]
Evinacumab is a recombinant human IgG4 monoclonal antibody targeted against angiopoietin-like protein 3 (ANGPTL3) and the first drug of its kind. The ANGPTL family of proteins serve a number of physiologic functions – including involvement in the regulation of lipid metabolism – which have made them desirable therapeutic targets in recent years.2 Loss-of-function mutations in ANGPTL3 have been noted to result in hypolipidemia and subsequent reductions in cardiovascular risk, whereas increases in function appear to be associated with cardiovascular risk, and it was these observations that provided a rationale for the development of a therapy targeted against ANGPTL3.3
In February 2021, evinacumab became the first-and-only inhibitor of ANGPTL3 to receive FDA approval after it was granted approval for the adjunctive treatment of homozygous familial hypercholesterolemia (HoFH) under the brand name “Evkeeza”.8 Evinacumab is novel in its mechanism of action compared with other lipid-lowering therapies and therefore provides a unique and synergistic therapeutic option in the treatment of HoFH.
Common side effects include nasopharyngitis (cold), influenza-like illness, dizziness, rhinorrhea (runny nose), and nausea. Serious hypersensitivity (allergic) reactions have occurred in the Evkeeza clinical trials.[2]
Evinacumab binds to the angiopoietin-like protein 3 (ANGPTL3).[2] ANGPTL3 slows the function of certain enzymes that break down fats in the body.[2] Evinacumab blocks ANGPTL3, allowing faster break down of fats that lead to high cholesterol.[2] Evinacumab was approved for medical use in the United States in February 2021.[2][3]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable |  | |
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable | |
History
The effectiveness and safety of evinacumab were evaluated in a double-blind, randomized, placebo-controlled, 24-week trial enrolling 65 participants with homozygous familial hypercholesterolemia (HoFH).[2] In the trial, 43 participants received 15 mg/kg of evinacumab every four weeks and 22 participants received the placebo.[2] Participants were taking other lipid-lowering therapies as well.[2]
The primary measure of effectiveness was the percent change in low-density lipoprotein (LDL-C) from the beginning of treatment to week 24.[2] At week 24, participants receiving evinacumab had an average 47% decrease in LDL-C while participants on the placebo had an average 2% increase.[2]
The U.S. Food and Drug Administration (FDA) granted the application for evinacumab orphan drug, breakthrough therapy, and priority review designations.[2] The FDA granted approval of Evkeeza to Regeneron Pharmaceuticals, Inc.[2]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves add-on therapy for patients with genetic form of severely”. U.S. Food and Drug Administration (FDA). 11 February 2021. Retrieved 12 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA Approves First-in-class Evkeeza (evinacumab-dgnb) for Patients with Ultra-rare Inherited Form of High Cholesterol” (Press release). Regeneron Pharmaceuticals. 11 February 2021. Retrieved 12 February 2021 – via PR Newswire.
Further reading
- Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. (July 2017). “Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease”. N Engl J Med. 377 (3): 211–221. doi:10.1056/NEJMoa1612790. PMC 5800308. PMID 28538136.
External links
- “Evinacumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Angiopoietin-like 3 (ANGPTL3) |
| Clinical data | |
| Trade names | Evkeeza |
| Other names | REGN1500, evinacumab-dgnb |
| License data | US DailyMed: Evinacumab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1446419-85-7 |
| DrugBank | DB15354 |
| ChemSpider | none |
| UNII | T8B2ORP1DW |
| KEGG | D11753 |
| Chemical and physical data | |
| Formula | C6480H9992N1716O2042S46 |
| Molar mass | 146083.95 g·mol−1 |
//////////////
#Evinacumab, #Peptide, #APPROVALS 2021, #FDA 2021, #Monoclonal antibody, #dyslipidemia, #エビナクマブ (遺伝子組換え) , #REGN 1500, #REGN-1500, #REGN1500, #Anthony melvin crasto, #world drug tracker. # new drug approvals, #pharma
TROFINETIDE

Trofinetide
- Molecular FormulaC13H21N3O6
- Average mass315.322 Da
Tofinetide , NNZ-256610076853400-76-7[RN]
glycyl-2-methyl-L-prolyl-L-glutamic acid
H-Gly-PMe-Glu-OHL-Glutamic acid, glycyl-2-methyl-L-prolyl-UNII-Z2ME8F52QLZ2ME8F52QLтрофинетид [Russian] [INN]تروفينيتيد [Arabic] [INN]曲非奈肽 [Chinese] [INN]
| IUPAC Condensed | H-Gly-aMePro-Glu-OH |
|---|---|
| Sequence | GXE |
| HELM | PEPTIDE1{G.[*C(=O)[C@@]1(CCCN1*)C |$_R2;;;;;;;;_R1;$|].E}$$$$ |
| IUPAC | glycyl-alpha-methyl-L-prolyl-L-glutamic acid |
An (1-3) IGF-1 analog with neuroprotective activity.
OPTICAL ROT; -52.4 ° Conc: 0.19 g/100mL; water ; 589.3 nm; Temp: 20 °C; Len: 1.0 dm…Tetrahedron 2005, V61(42), P10018-10035
EU Customs Code CN, 29339980
Harmonized Tariff Code, 293399
- L-Glutamic acid, glycyl-2-methyl-L-prolyl-
- glycyl-2-methyl-L-prolyl-L-glutamic acid
- Glycyl-L-2-methylprolyl-L-glutamic acid
FDA APPROVED 2023/3/10, Daybue
853400-76-7 CAS
トロフィネチド;
Trofinetide (NNZ-2566) is a drug developed by Neuren Pharmaceuticals that acts as an analogue of the neuropeptide (1-3) IGF-1, which is a simple tripeptide with sequence Gly–Pro–Glu formed by enzymatic cleavage of the growth factor IGF-1 within the brain. Trofinetide has anti-inflammatory properties and was originally developed as a potential treatment for stroke,[1][2] but has subsequently been developed for other applications and is now in Phase II clinical trials against Fragile X syndrome and Rett syndrome.[3][4][5]
Trofinetide (NNZ-2566), a neuroprotective analogue of glypromate, is a novel molecule that has a profile suitable for both intravenous infusion and chronic oral delivery. It is currently in development to treat traumatic brain injury.
In February 2021, Neuren is developing trofinetide (NNZ-2566, phase 2 clinical ), a small-molecule analog of the naturally occurring neuroprotectant and N-terminus IGF-1 tripeptide Glypromate (glycine-proline-glutamate), for intravenous infusion treatment of various neurological conditions, including moderate to severe traumatic brain injury (TBI), stroke, chronic neurodegenerative disorders and peripheral neuropathies. At the same time, Neuren is also investigating an oral formulation of trofinetide (phase 3 clinical) for similar neurological indications, including mild TBI.
Autism Spectrum Disorders and neurodevelopment disorders (NDDs) are becoming increasingly diagnosed. According to the fourth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-4), Autism spectrum disorders (ASD) are a collection of linked developmental disorders, characterized by abnormalities in social interaction and communication, restricted interests and repetitive behaviours. Current classification of ASD according to the DSM-4 recognises five distinct forms: classical autism or Autistic Disorder, Asperger syndrome, Rett syndrome, childhood disintegrative disorder and pervasive developmental disorder not otherwise specified (PDD-NOS). A sixth syndrome, pathological demand avoidance (PDA), is a further specific pervasive developmental disorder.
More recently, the fifth edition of the American Psychiatric Association’s (APA) Diagnostic and Statistical Manual oƒ Mental Disorders (DSM-5) recognizes recognises Asperger syndrome, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified (PDD-NOS) as ASDs.
This invention applies to treatment of disorders, regardless of their classification as either DSM-4 or DSM-5.
Neurodevelopment Disorders (NDDs) include Fragile X Syndrome (FXS), Angelman Syndrome, Tuberous Sclerosis Complex, Phelan McDermid Syndrome, Rett Syndrome, CDKL5 mutations (which also are associated with Rett Syndrome and X-Linked Infantile Spasm Disorder) and others. Many but not all NDDs are caused by genetic mutations and, as such, are sometimes referred to as monogenic disorders. Some patients with NDDs exhibit behaviors and symptoms of autism.
As an example of a NDD, Fragile X Syndrome is an X-linked genetic disorder in which affected individuals are intellectually handicapped to varying degrees and display a variety of associated psychiatric symptoms. Clinically, Fragile X Syndrome is characterized by intellectual handicap, hyperactivity and attentional problems, autism spectrum symptoms, emotional lability and epilepsy (Hagerman, 1997a). The epilepsy seen in Fragile X Syndrome is most commonly present in childhood, but then gradually remits towards adulthood. Hyperactivity is present in approximately 80 percent of affected males (Hagerman, 1997b). Physical features such as prominent ears and jaw and hyper-extensibility of joints are frequently present but are not diagnostic. Intellectual handicap is the most common feature defining the phenotype. Generally, males are more severely affected than females. Early impressions that females are unaffected have been replaced by an understanding of the presence of specific learning difficulties and other neuropsychiatric features in females. The learning disability present in males becomes more defined with age, although this longitudinal effect is more likely a reflection of a flattening of developmental trajectories rather than an explicit neurodegenerative process.
The compromise of brain function seen in Fragile X Syndrome is paralleled by changes in brain structure in humans. MRI scanning studies reveal that Fragile X Syndrome is associated with larger brain volumes than would be expected in matched controls and that this change correlates with trinucleotide expansion in the FMRP promoter region (Jakala et al, 1997). At the microscopic level, humans with Fragile X Syndrome show abnormalities of neuronal dendritic structure, in particular, an abnormally high number of immature dendritic spines (Irwin et al, , 2000).
Currently available treatments for NDDs are symptomatic – focusing on the management of symptoms – and supportive, requiring a multidisciplinary approach. Educational and social skills training and therapies are implemented early to address core issues of learning delay and social impairments. Special academic, social, vocational, and support services are often required. Medication, psychotherapy or behavioral therapy may be used for management of co-occurring anxiety, ADHD, depression, maladaptive behaviors (such as aggression) and sleep issues, Antiepileptic drugs may be used to control seizures.
Patent
WO 2014085480,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014085480

EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro-Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
WO95/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS to increase TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses certain GPE analogs having amino acid substitutions and certain other modification that are capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including injury or disease in the CNS.
EXAMPLES
The following examples are intended to illustrate embodiments of this invention, and are not intended to limit the scope to these specific examples. Persons of ordinary skill in the art can apply the disclosures and teachings presented herein to develop other embodiments without undue experimentation and with a likelihood of success. All such embodiments are considered part of this invention.
Example 1: Synthesis of N,N-Dimethylglycyl-L-prolyl)-L-glutamic acid
The following non-limiting example illustrates the synthesis of a compound of the invention, N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
All starting materials and other reagents were purchased from Aldrich; BOC=tert-butoxycarbonyl; Bn=benzyl.
BOC-L-proline-(P-benzyl)-L-glutamic acid benzyl ester
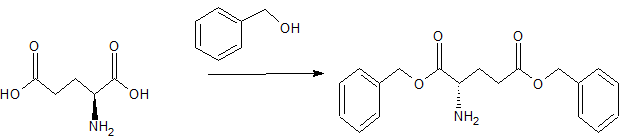
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem. Soc: 79, 6810, 1994] (10 mmol) in dichloromethane (50 mi), cooled to 0°C, was added triethylamine (1 .39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl-L-glutamate (10 mmol) was then added and the mixture stirred at 0° C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol 1-1) then dried (MgSO4) and concentrated at reduced pressure to give BOC-L-proline-L-glutamic acid dibenzyl ester (5.0 g, 95%).
L-proline-L-glutamic acid dibenzyl ester
A solution of BOC-L-glutamyl-L-proline dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 h. at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give L-proline-L-glutamic acid dibenzyl ester.
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of L-proline-L-glutamic acid dibenzyl ester (10 mmol), N,N-dimethylglycine (10 mmol) and triethylamine ( 10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0°C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallised from ethyl acetate to yield the tripeptide derivative.
It can be appreciated that following the method of the Examples, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Eample 2: Synthesis of Glycyl-L-2-Methyl-L-Prolyl-L-Glutamate
L-2-Methylproline and L-glutamic acid dibenzyl ester p-toluenesulphonate were purchased from Bachem, N-benzyloxycarbonyl-glycine from Acros Organics and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BoPCl, 97%) from Aldrich Chem. Co.
Methyl L-2-methylprolinate hydrochloride 2
Thionyl chloride (5.84 cm3, 80.1 mmol) was cautiously added dropwise to a stirred solution of (L)-2-methylproline 1 (0.43 g, 3.33 mmol) in anhydrous methanol (30 cm3) at -5 °C under an atmosphere of nitrogen. The reaction mixture was heated under reflux for 24 h, and the resultant pale yellow-coloured solution was. concentrated to dryness in vacuo. The residue was dissolved in a 1 : 1 mixture of methanol and toluene (30 cm3) then concentrated to dryness to remove residual thionyl chloride. This procedure was repeated twice more, yielding hydrochloride 2 (0.62 g, 104%) as an hygroscopic, spectroscopically pure, off-white solid: mp 127- 131 °C; [α]D -59.8 (c 0.24 in CH2Cl2); vmax (film)/cm-1 3579, 3398 br, 2885, 2717, 2681 , 2623, 2507, 1743, 1584, 1447, 1432, 1374, 1317, 1294, 1237, 1212, 1172, 1123, 981 , 894, 861 and 764; δH (300 MHz; CDCl3; Me4Si) 1.88 (3H, s, Proα-CH3), 1 .70-2.30 (3H, br m, Proβ-HAΗΒ and Proγ-H2), 2.30-2.60 (1H, br m, Proβ-HAΗΒ), 3.40-3.84 (2H, br m, Proδ-H2), 3.87 (3H, s, CO2CH3), 9.43 (1H, br s, NH) and 10.49 ( 1H, br s, HCl); δC (75 MHz; CDCl3) 21.1 (CH3, Proα-CH3), 22.4 (CH2, Proγ-C), 35.6 (CH2, Proβ-C), 45.2 (CH2, Proδ-C), 53.7 (CH3, CO2CH3), 68.4 (quat., Proα-C) and 170.7 (quat, CO); m/z (FAB+) 323.1745 [M2.H35Cl.H+: (C7H13NO2)2. H35Cl.H requires 323.1738] and 325.1718 [M2.H37Cl.H+: (C7H13NOz)2. H37Cl.H requires 325.1708],
N-Benxyloxycarbonyl-glycyl-L-2-methylproline 5
Anhydrous triethylamine (0.45 cm3, 3.23 mmol) was added dropwise to a mixture of methyl L-2-methylprolinate hydrochloride 2 (0.42 g, 2.34 mmol) and N-benzyloxycarbonyl-glycine (98.5%) 3 (0.52 g, 2.45 mmol) in methylene chloride (16 cm3), at 0 °C, under an atmosphere of nitrogen. The resultant solution was stirred for 20 min and a solution of 1 ,3-dicyclohexylcarbodiimide (0.56 g, 2.71 mmol) in methylene chloride (8 cm3) at 0 °C was added dropwise and the reaction mixture was warmed to room temperature and stirred for a further 20 h. The resultant white mixture was filtered through a Celite™ pad to partially remove 1 ,3-dicyclohexylurea, and the pad was washed with methylene chloride (50 cm3). The filtrate was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and concentrated to dryness in vacuo. Further purification of the residue by flash column chromatography (35 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) afforded tentatively methyl N-benzyloxycarbonyl-glycyl-L-2-methylprolinate 4 (0.56 g), containing 1 ,3-dicyclohexylurea, as a white semi-solid: Rf 0.65 (EtOAc); m/z (ΕI+) 334.1534 (M+. C17H22N2O5 requires 334.1529) and 224 ( 1 ,3-dicyclohexylurea).
To a solution of impure prolinate 4 (0.56 g, ca. 1.67 mmol) in 1,4-dioxane (33 cm3) was added dropwise 1 M aqueous sodium hydroxide (10 cm3, 10 mmol) and the mixture was stirred for 19 h at room temperature. Methylene chloride ( 100 cm3) was then added and the organic layer extracted with saturated aqueous sodium hydrogen carbonate (2 x 100 cm3). The combined aqueous layers were carefully acidified with hydrochloric acid (32%), extracted with methylene chloride (2 x 100 cm3), and the combined organic layers dried (MgSO4), filtered, and
concentrated to dryness in vacuo. Purification of the ensuing residue (0.47 g) by flash column chromatography ( 17 g SiO2; 50% ethyl acetate – hexane to 30% methanol – dichloromethane; gradient elution) gave N-protected dipeptide 5 (0.45 g, 60%) as a white foam in two steps from hydrochloride 2. Dipeptide 5 was shown to be exclusively the frafw-orientated conformer by NMR analysis: Rf 0.50 (20% MeOH – CH2Cl2); [α]D -62.3 (c 0.20 in CH2Cl2); vmax (film)/cm-1 3583, 3324 br, 2980, 2942, 1722, 1649, 1529, 1454, 1432, 1373, 1337, 1251 , 1219, 1179, 1053, 1027, 965, 912, 735 and 698; δH (300 MHz; CDCl3; Me4Si) 1.59 (3H, s, Proα-CH3), 1 .89 (1H, 6 lines, J 18.8, 6.2 and 6.2, Proβ-HAHB), 2.01 (2H, dtt, J 18.7, 6.2 and 6.2, Proγ-H2), 2.25-2.40 (1H, m, Proβ-HAΗΒ), 3.54 (2H, t, J 6.6, Proδ-H2), 3.89 (1H, dd, J 17.1 and 3.9, Glyα-HAHB), 4.04 (1H, dd, J 17.2 and 5.3, Glyα-HAΗΒ), 5.11 (2H, s, OCH2Ph), 5.84 (I H, br t, J 4.2, N-H), 7.22-7.43 (5H, m, Ph) and 7.89 (1 H, br s, -COOH); δC (75 MHz; CDCl3) 21.3 (CH3, Proα-CH3), 23.8 (CH2, Proγ-C), 38.2 (CH2, Proβ-C), 43.6 (CH2, Glyα-C), 47.2 (CH2, Proδ-C), 66.7 (quat, Proα-C), 66.8 (CH2, OCH2Ph), 127.9 (CH, Ph), 127.9 (CH, Ph), 128.4, (CH, Ph), 136.4 (quat., Ph), 156.4 (quat., NCO2), 167.5 (quat., Gly-CON) and 176.7 (quat., CO); m/z (EI+) 320.1368 (M+. C16Η20Ν2Ο5 requires 320.1372).
Dibenzyl N-benzyloxycarbonyl-glycyl-L-2-methylprolyl-L-glutamate 7
Triethylamine (0.50 cm3, 3.59 mmol) was added dropwise to a solution of dipeptide 5 (0.36 g, 1.12 mmol) and L-glutamic acid dibenzyl ester /Moluenesulphonate 6 (0.73 g, 1.46 mmol) in methylene chloride (60 cm3) under nitrogen at room temperature, and the reaction mixture stirred for 10 min. Bis(2-oxo-3-oxazoIidinyl)phosphinic chloride (BoPCl, 97%) (0.37 g, 1.41 mmol) was added and the colourless solution stirred for 17 h. The methylene chloride solution was washed successively with 10% aqueous hydrochloric acid (50 cm3) and saturated aqueous sodium hydrogen carbonate (50 cm3), dried (MgSO4), filtered, and evaporated to dryness in vacuo. Purification of the resultant residue by repeated (2x) flash column chromatography (24 g SiO2; 30-70% ethyl acetate – hexane; gradient elution) yielded ƒully protected tripeptide 7 (0.63 g, 89%) as a colourless oil. Tripeptide 7 was shown to be exclusively the trans-orientated conformer by NMR analysis: Rf 0.55 (EtOAc); [α]D -41.9 (c 0.29 in CH2Cl2); vmax (film)/cm-1 3583, 3353 br, 2950, 1734, 1660, 1521, 1499, 1454, 1429, 1257, 1214, 1188, 1166, 1051, 911, 737 and 697; δH (400 MHz; CDCl3; Me4Si) 1.64 (3H, s, Proot-CH3), 1.72 (1H, dt, J 12.8, 7.6 and 7.6, Proβ-HAHB), 1.92 (2H, 5 lines, J 6.7, Proγ-H2), 2.04 (1H, 6 lines, J 7.3 Gluβ-HAHB), 2.17-2.27 (1H, m, Gluβ-HAΗΒ), 2.35-2.51 (3H, m, Proβ-HAΗΒ and Gluγ-H2), 3.37-3.57 (2H, m, Proδ-H2), 3.90 (1 H, dd, J 17.0 and 3.6, Glyα-HAHB), 4.00 (1H, dd, J 17.1 and 5.1, Glyα-HAΗΒ), 4.56 (1H, td, J 7.7 and 4.9, Glyα-H), 5.05-5.20 (6H, m, 3 x OCH2Ph), 5.66-5.72 (1H, br m, Gly-NH), 7.26-7.37 (15H, m, 3 x Ph) and 7.44 (1H, d, J 7.2, Glu-NH); δC (100 MHz; CDCl3) 21.9 (CH3, Proα-CH3), 23.4 (CH2, Proγ-C), 26.6 (CH2, Gluβ-C), 30.1 (CH2, Gluγ-C), 38.3 (CH2, Proβ-C),
43.9 (CH2, Glyα-C), 47.6 (CH2, Proδ-C), 52.2 (CH, Glua-C), 66.4 (CH2, OCH2Ph), 66.8 (CH2, OCH2Ph), 67.1 (CH2, OCH2Ph), 68.2 (quat, Proα-C), 127.9 (CH, Ph), 128.0 (CH, Ph), 128.1, (CH, Ph), 128.2, (CH, Ph), 128.2, (CH, Ph), 128.3, (CH, Ph), 128.4, (CH, Ph), 128.5, (CH, Ph), 128.5, (CH, Ph), 135.2 (quat., Ph), 135.7 (quat., Ph), 136.4 (quat, Ph), 156.1 (quat, NCO2), 167.3 (quat., Gly-CO), 171.4 (quat., CO), 172.9 (quat., CO) and 173.4 (quat., CO); m/z (FAB+) 630.2809 (MH+. C35H40N3O8 requires 630.2815).
Glycyl-L-2-methylprolyl-L-glutamic acid (G-2-MePE)
A mixture of the protected tripeptide 7 (0.63 g, 1.00 mmol) and 10 wt % palladium on activated carbon (0.32 g, 0.30 mmol) in 91 :9 methanol – water (22 cm3) was stirred under an atmosphere of hydrogen at room temperature, protected from light, for 23 h. The reaction mixture was filtered through a Celite™ pad and the pad washed with 75 :25 methanol – water (200 cm3). The filtrate was concentrated to dryness under reduced pressure and the residue triturated with anhydrous diethyl ether to afford a 38: 1 mixture of G-2-MePE and tentatively methylamine 8 (0.27 g, 86%) as an extremely hygroscopic white solid. Analytical reverse-phase HPLC studies on the mixture [Altech Econosphere C 18 Si column, 150 x 4.6 mm, 5 ☐m; 5 min flush with H2O (0.05% TFA) then steady gradient over 25 min to MeCN as eluent at flow rate of 1 ml/min; detection using diode array] indicated it was a 38: 1 mixture of two eluting peaks with retention times of 13.64 and 14.44 min at 207 and 197 nm, respectively. G-2-MePE was shown to be a 73 :27 trans:cis mixture of conformers by 1H NMR analysis (the ratio was estimated from the relative intensities of the double doublet and triplet at δ 4.18 and 3.71 , assigned to the Gluα-H protons of the major and minor conformers, respectively):
mp 144 °Cɸ;
[ α]D -52.4 (c 0.19 in H2O);
δα (300 MHz; D2O; internal MeOH) 1.52 (3H, s, Proα-CH3), 1.81-2.21 (6H, m, Proβ-H2, Proγ-H, and Gluβ-H2), 2.34 (1.46H, t, J 7.2, Gluy-H2), 2.42* (0.54H, t, 77.3, Gluγ-H2), 3.50-3.66 (2H, m, Pro6-H2), 3.71 * (0.27H, t, J 6.2, Gluoc-H), 3.85 (1H, d, J 16.6, Glyα-HAHB), 3.92 (1H, d, J 16.6, Glyα-HAΗΒ) and 4.18 (0.73H, dd, J 8.4 and 4.7, Glua-H);
δC (75 MHz; D2O; internal MeOH) 21.8 (CH3, Proα-CH3), 25.0 (CH2, Proγ-C), 27.8* (CH2: Gluβ-C), 28.8 (CH2, Gluβ-C), 32.9 (CH2, Gluγ-C), 40.8 (CH2, Proβ-C), 42.7 (CH2, Glyα-C), 49.5 (CH2, Proδ-C), 56.0* (CH, Gluα-C), 56.4 (CH, Gluα-C), 69.8 (quat, Proα-C), 166.5 (quat., Gly-CO), 177.3 (quat., Pro-CON), 179.2 (quat., Gluα-CO), 180.2* (quat., Gluγ-CO) and 180.6 (quat., Gluγ-CO);
m/z (FAB+) 3 16.1508 (MH+. C13H22N3O6 requires 316.1509).
PATENT
WO02094856
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
Testing; Material and Methods
The following experimental protocol followed guidelines approved by the
University of Auckland animal ethics committee.
Preparation of cortical astrocyte cultures for harvest of metabolised cell culture supernatant
One cortical hemisphere from a postnatal day 1 rat was used and collected into
4ml of DMEM. Trituration was done with a 5ml glass pipette and subsequently through an 18 gauge needle. Afterwards, the cell suspension was sieved through a lOOμm cell strainer and washed in 50ml DMEM (centrifugation for 5min at 250g). The sediment was resuspended into 20ml DMEM+10% fetal calf serum. 10 Milliliters of suspension was added into each of two 25cm3 flasks and cultivated at 37°C in the presence of 10% C02, with a medium change twice weekly. After cells reached confluence, they were washed three times with PBS and adjusted to Neurobasal/B27 and incubated for another 3 days. This supernatant was frozen for transient storage until usage at -80°C.
Preparation of striatal and cortical tissue from rat E18/E19 embryos
A dam was sacrificed by C02-treatment in a chamber for up to 4 minutes and was prepared then for cesarean section. After surgery, the embryos were removed from their amniotic sacs, decapitated and the heads put on ice in DMEM/F12 medium for striatum and PBS + 0.65% D(+)-glucose for cortex.
Striatal tissue extraction procedure and preparation of cells
Whole brain was removed from the skull with the ventral side facing upside in DMEM/F12 medium. The striatum was dissected out from both hemispheres under a stereomicroscope and the striatal tissue was placed into the Falcon tube on ice.
The collected striatal tissue was triturated by using a PI 000 pipettor in 1ml of volume. The tissue was triturated by gently pipetting the solution up and down into the pipette tip about 15 times, using shearing force on alternate outflows. The tissue pieces settled to the bottom of the Falcon tube within 30 seconds, subsequently the supernatant was transferred to a new sterile Falcon tube on ice. The supernatant contained a suspension of dissociated single cells. The tissue pieces underwent a second trituration to avoid excessively damaging cells already dissociated by over triturating them. 1 Milliliter of ice-cold DMEM/F12 medium was added to the tissue pieces in the first tube and triturated as before. The tissue pieces were allowed to settle and the supernatant was removed to a new sterile Falcon tube on ice. The cells were centrifuged at 250g for 5 minutes at 4°C. The resuspended cell pellet was ready for cell counting.
Plating and cultivation of striatal cells
Striatal cells were plated into Poly-L-Lysine (O.lmg/ml) coated 96-well plates (the inner 60 wells only) at a density of 200,000 cells /cm2 in Neurobasal/B27 medium (Invitrogen). The cells were cultivated in the presence of 5% C02 at 37°C under 100% humidity. Complete medium was changed on days 1, 3 and 6.
Cortical tissue extraction procedure and preparation of cells
The two cortical hemispheres were carefully removed by a spatula from the whole brain with the ventral side facing upside into a PBS +0.65% D(+)-glucose containing petri dish. Forcips were put into the rostral part (near B. olfactorius) of the cortex for fixing the tissue and two lateral – sagittal oriented cuttings were done to remove the paraform and entorhinal cortices. The next cut involved a frontal oriented cut at the posterior end to remove the hippocampal formation. A final frontal cut was done a few millimeters away from the last cut in order to get hold of area 17/18 of the visual cortex.
The collected cortices on ice in PBS+0.65% D(+)-glucose were centrifuged at 350g for 5min. The supernatant was removed and trypsin/EDTA (0.05%/0.53mM) was added for 8min at 37°C. The reaction was stopped by adding an equal amount of DMEM+10%) fetal calf serum. The supernatant was removed by centrifugation followed by two subsequent washes in Neurobasal/B27 medium.
The cells were triturated once with a glass Pasteur pipette in 1 ml of
Neurobasal/B27 medium and subsequently twice by using a 1ml insulin syringe with a 22 gauge needle. The cell suspension was passed through a lOOμm cell strainer and subsequently rinsed by 1ml of Neurobasal B27 medium. Cells were counted and adjusted to 50,000 cells per 60μl.
Plating and cultivation of cortical cells
96-well plates were coated with 0.2mg/ml Poly-L-Lysine and subsequently coated with 2μg/ml laminin in PBS, after which 60μl of cortical astrocyte-conditioned medium was added to each well. Subsequently, 60μl of cortical cell suspension was added. The cells were cultivated in the presence of 10% C02 at 37°C under 100%) humidity. At day 1, there was a complete medium change (1:1- Neurobasal/B27 and astrocyte-conditioned medium) with addition of lμM cytosine-β-D-arabino-furanoside (mitosis inhibitor). On the second day, 2/3 of medium was changed. On day 5, 2/3 of the medium was changed again.
Cerebellar microexplants from P8 animals: preparation, cultivation and fixation
The laminated cerebellar cortices of the two hemispheres were explanted from a P8 rat, cut into small pieces in PBS + 0.65% D(+)glucose solution and triturated by a 23gauge needle and subsequently pressed through a 125 μm pore size sieve. The microexplants that were obtained were centrifuged (60 g) twice (media exchange) into serum-free BSA-supplemented START V-medium (Biochrom). Finally, the
microexplants were reconstituted in 1500 μl STARTV-medium (Biochrom). For cultivation, 40μl of cell suspension was adhered for 3 hours on a Poly-D-Lysine
(O.lmg/ml) coated cover slip placed in 35mm sized 6-well plates in the presence of 5% C02 under 100% humidity at 34°C. Subsequently, 1ml of STARTV-medium was added together with the toxins and drugs. The cultures were monitored (evaluated) after 2-3 days of cultivation in the presence of 5% C02 under 100% humidity. For cell counting analysis, the cultures were fixed in rising concentrations of paraformaldehyde (0.4%, 1.2%, 3% and 4% for 3min each) followed by a wash in PBS.
Toxin and drug administration for cerebellar, cortical and striatal cells: analysis
All toxin and drug administration experiments were designed that 1/100 parts of okadaic acid (30nM and lOOnM concentration and 0.5mM 3-nitropropionic acid for cerebellar microexplants only), GPE (InM -ImM) and G-2Methyl-PE (InM-lmM) were used respectively at 8DIV for cortical cultures and 9DIV for striatal cultures. The incubation time was 24hrs. The survival rate was determined by a colorimetric end-point MTT-assay at 595nm in a multi-well plate reader. For the cerebellar microexplants four windows (field of 0.65 mm2) with highest cell density were chosen and cells displaying neurite outgrowth were counted.
Results
The GPE analogue G-2Methyl-PE exhibited comparable neuroprotective capabilities within all three tested in vitro systems (Figures 12-15).
The cortical cultures responded to higher concentrations of GPE (Figure 12) /or
G-2Methyl-PE (lOμM, Figure 13) with 64% and 59% neuroprotection, respectively.
Whereas the other 2 types of cultures demonstrated neuroprotection at lower doses of G-2Methyl-PE (Figures 14 and 15). The striatal cells demonstrated
neuroprotection within the range of InM to ImM of G-2Methyl-PE (Figure 15) while the postnatal cerebellar microexplants demonstrated neuroprotection with G-2Methyl-PE in the dose range between InM and lOOnM (Figure 14).
While this invention has been described in terms of certain preferred embodiments, it will be apparent to a person of ordinary skill in the art having regard to that knowledge and this disclosure that equivalents of the compounds of this invention may be prepared and administered for the conditions described in this application, and all such equivalents are intended to be included within the claims of this application.
PATENT
WO-2021026066
Composition and kits comprising trofinetide and other related substances. Also claims a process for preparing trofinetide and the dosage form comprising the same. Disclosed to be useful in treating neurodegenerative conditions, autism spectrum disorders and neurodevelopmental disorders.
Trofinetide is a synthetic compound, having a similar core structure to Glycyl-Prolyl-Glutamic acid (or “GPE”). Trofinetide has been found to be useful in treating neurodegenerative conditions and recently has been found to be effective in treating Autism Spectrum disorders and Neurodevelopmental disorders.
Formula (Ila),
Example 1: Trofinetide Manufacturing Process
In general, trofinetide and related compounds can be manufactured from a precursor peptide or amino acid reacted with a silylating or persilylating agent at one or more steps. In the present invention, one can use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group.
Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
Step 1: Preparation of Z-Gly-OSu
Several alternative procedures can be used for this step.
Procedure 1A
One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 27 eq of iPrOH and 4 eq of CH2Cl2 at 21 °C. The mixture was cooled and when the temperature reached -4 °C, 1.1 eq of EDC.HCl was added gradually, keeping the temperature below 10 °C. During the reaction a dense solid appeared. After addition of EDC.HCl, the mixture was allowed to warm to 20 °C. The suspension was cooled to 11 °C and filtered. The cake was washed with 4.9 eq of cold iPrOH and 11 eq of IPE before drying at 34 °C (Z-Gly-OSu dried product -Purity: 99.5%; NMR assay: 96%; Yield: 84%).
Procedure 1B
This Procedure is for a variant of Procedure 1A, and differs by replacing iPrOH with ACN. One (1) eq of Z-Gly-OH and 1.1 eq of Suc-OH were solubilized in 22 eq of ACN at 35 °C. The mixture was cooled in an ice bath. When the temperature reached 1 °C, 0.9 eq of DCC in 5.5 eq of ACN was added gradually to keep the temperature below 5 °C. The coupling reaction took about 20 hrs. During the reaction, DCU precipitated and was removed by filtration at the end of the coupling. After filtration, DCU was washed with ACN to recover the product. The mixture of Z-Gly-OSu was then concentrated to reach 60% by weight. iPrOH (17 eq) was added to initiate the crystallization. Quickly after iPrOH addition a dense solid appeared. An additional 17 eq of iPrOH was needed to liquify the suspension. The suspension was cooled in an ice bath and filtered. The solid was washed with 9 eq of iPrOH before drying at 45 °C (Z-Gly-OSu dried product – Purity: 99.2%; HPLC assay: 99.6%; Yield: 71%).
Step 2: Preparation of Z-Gly-MePro-OH
Several alternative procedures can be used for this step.
Procedure 2A
One (1) eq of MePro.HCl was partially solubilized in 29 eq of CH2Cl2 at 35 °C with 1.04 eq of TEA and 1.6 eq of TMA. The mixture was heated at 35 °C for 2 hrs to perform the silylation. Then 1.02 eq of Z-Gly-OSu was added to the mixture. The mixture was kept at 35 °C for 3 hrs and then 0.075 eq of butylamine was added to quench the reaction. The mixture was allowed to return to room temperature and mixed for at least 15 min. The Z-Gly-MePro-OH was extracted once with 5% w/w NaHCO3 in 186 eq of water, then three times successively with 5% w/w NaHCO3 in 62 eq of water. The aqueous layers were pooled and the pH was brought to 2.2 by addition of 34 eq of HCl as 12N HCl at room temperature. At this pH, Z-Gly-MePro-OH formed a sticky solid that was solubilized at 45 °C with approximately 33 eq of EtOAc and 2.3 eq of iButOH. Z-Gly-MePro-OH was extracted into the organic layer and washed with 62 eq of demineralized water. The organic layer was then dried by azeotropic distillation with 11.5 eq of EtOAc until the peptide began to precipitate. Cyclohexane (12 eq) was added to the mixture to complete the precipitation. The suspension was cooled at 5 °C for 2 hrs and filtered. The solid was washed with 10 eq of cyclohexane before drying at 45 °C (Z-Gly-MePro-OH dried product – Purity: 100%; HPLC assay: 100%; Yield 79%).
Procedure 2B
This Procedure is for a variant of Procedure 2A. One (1) eq of MePro.HCl was partially solubilized in 36.6 eq of CH2Cl2 at 34 °C with 1.01 eq of TEA and 0.1 eq of TMA. Then 1.05 eq of Z-Gly-OSu was added to the mixture, followed by 1.0 eq of TEA. The mixture was maintained at 35 °C for approximately 1 hr, cooled to 25 to 30 °C and 0.075 eq of DMAPA was added to stop the reaction. One hundred (100) eq of water, 8.6 eq of HCl as 12N HCl and 0.3 eq of KHSO4 were added to the mixture (no precipitation was observed, pH=1.7). Z-Gly-MePro-OH was extracted into the organic layer and washed twice with 97 eq of demineralized water with 0.3 eq of KHSO4, then 100 eq of demineralized water, respectively. EtOAc (23 eq) was added to the mixture and CH2Cl2 was removed by distillation until the peptide began to precipitate. Cyclohexane (25 eq) was added to the mixture to complete the precipitation. The suspension was cooled at -2 °C overnight and filtered. The solid was washed with 21 eq of cyclohexane before drying at 39 °C (Z-Gly-MePro-OH dried product – Purity: 98.7%; NMR assay: 98%; Yield 86%).
Procedure 2C
In reactor 1, MePro.HCl (1 eq) was suspended in EtOAc (about 7 eq). DIPEA (1 eq) and TMA (2 eq) were added, and the mixture heated to dissolve solids. After dissolution, the solution was cooled to 0 °C. In reactor 2, Z-Gly-OH (1 eq) was suspended in EtOAc (about 15 eq). DIPEA (1 eq), and pyridine (1 eq) were added. After mixing, a solution was obtained, and cooled to -5 °C. Piv-Cl (1 eq) was added to reactor 2, and the contents of reactor 1 added to reactor 2. Upon completed addition, the contents of reactor 2 were taken to room temperature. The conversion from Z-Gly-OH to Z-Gly-MePro-OH was monitored by HPLC. When the reaction was complete, the reaction mixture was quenched with DMAPA (0.1 eq), and washed with an aqueous solution comprised of KHSO4, (about 2.5 wt%), NaCl (about 4 wt%), and conc. HCl (about 6 wt%) in 100 eq H2O. The aqueous layer was re-extracted with EtOAc, and the combined organic layers washed with an aqueous solution comprised of KHSO4 (about 2.5 wt%) and NaCl (about 2.5 wt%) in 100 eq H2O, and then with water (100 eq). Residual water was removed from the organic solution of Z-Gly-MePro-OH by vacuum distillation with EtOAc. The resulting suspension was diluted with heptane (about 15 eq) and cooled to 0 °C. The product was isolated by filtration, washed with cold heptane (about 7 eq), and dried under vacuum at 45 °C. Z-Gly-MePro-OH (85% yield) was obtained.
Step 3: Preparation of Z-Gly-MePro-Glu-OH
Several alternative procedures can be used in this step.
Procedure 3A
H-Glu-OH (1.05 eq) was silylated in 2 eq of CH2Cl2 with 3.5 eq of TMA at 65 °C. Silylation was completed after 2 hrs. While the silylation was ongoing, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 24 eq of CH2Cl2 and 1.0 eq of DMA at room temperature in another reactor. EDC.HCl (1.0 eq.) was added. The activation rate reached 97% after 15 min. The activated Oxyma Pure solution, was then added to silylated H-Glu-OH at 40 °C and cooled at room temperature. Coupling duration was approximately 15 min, with a coupling rate of 97%. Addition of 8.2% w/w NaHCO3 in 156 eq of water to the mixture at room temperature (with the emission of CO2) was performed to reach pH 8. Z-Gly-MePro-Glu-OH was extracted in water. The aqueous layer was washed twice with 29 eq of CH2Cl2. Residual CH2Cl2 was removed by concentration. The pH was brought to 2.5 with 2.5N HCl, followed by 1.4 eq of solid KHSO4 to precipitate Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 3 x 52 eq of water. The filtered solid was added to 311 eq of demineralized water and heated to 55-60 °C. iPrOH (29 eq) was added gradually until total solubilization of the product. The mixture was slowly cooled to 10 °C under moderate mixing during 40 min to initiate the crystallization. The peptide was filtered and washed with 2 x 52 eq of water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.5%; NMR assay: 96%; Yield 74%).
Procedure 3B
One (1) eq of Z-Gly-MePro-OH and 1.05 eq of Suc-OH were solubilized in 40 eq of ACN and 30 eq of CH2Cl2 at room temperature. The mixture was cooled in an ice bath, and when the temperature was near 0 °C, 1.05 eq of DCC dissolved in 8 eq of ACN was added gradually, keeping the temperature below 5 °C. After addition of DCC, the mixture was progressively heated from 0 °C to 5 °C over 1 hr, then to 20 °C between 1 to 2 hrs and then to 45 °C between 2 to 5 hrs. After 5 hrs, the mixture was cooled to 5 °C and maintained overnight. The activation rate reached 98% after approximately 24 hrs. DCU was removed by filtration and washed with 13.5 eq of ACN. During the activation step, 1.1 eq of H-Glu-OH was silylated in 30 eq of ACN with 2.64 eq of TMA at 65 °C. Silylation was completed after 2 hrs. Z-Gly-MePro-OSu was then added gradually to the silylated H-Glu-OH at room temperature, with 0.4 eq of TMA added to maintain the solubility of the H-Glu-OH. The mixture was heated to 45 °C and 0.7 eq of TMA was added if precipitation occurred. The coupling duration was about 24 hrs to achieve a coupling rate of approximately 91%. The reaction was quenched by addition of 0.15 eq of butylamine and 2.0 eq of TEA. Water (233 eq) was added and the mixture concentrated until gelation occurred. Z-Gly-MePro-Glu-OH was extracted in water by addition of 5% w/w NaHCO3 in 233 eq of water and 132 eq of CH2Cl2. The aqueous layer was washed twice with 44 eq of CH2Cl2. Residual CH2Cl2 was removed by distillation. The pH was brought to 2.0 with 24 eq of HCl as 12N HCl followed by 75 eq of HCl as 4N HCl. At this pH, Z-Gly-MePro-Glu-OH precipitated. The mixture was cooled in an ice bath over 1 hr and filtered. The solid was washed with 186 eq of cold water before drying at 45 °C (Z-Gly-MePro-Glu-OH dried product – HPLC Purity: 98.4%; NMR assay: 100%; Yield 55%).
Procedure 3C
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq) was silylated in 3.7 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after approximately 1.5 to 2 hrs, as evidenced by solubilization. During the silylation step, 1.0 eq of Z-Gly-MePro-OH and 1.0 eq of Oxyma Pure were solubilized in 31.5 eq of CH2Cl2 at 22 °C. One (1.06) eq of EDC.HCl was added to complete the activation. The silylated H-Glu-OH was then added to the activated Oxyma Pure solution. The temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 153 eq of water and 9 eq of iPrOH to reach a pH of 1.65. Residual CH2Cl2 was removed by concentration. The mixture was cooled to 12 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 90 eq of water before drying at 36 °C.
Procedure 3D
This Procedure is for a variant of Procedure 3A. H-Glu-OH (1.05 eq.) was silylated in 3.9 eq of CH2Cl2 with 3.5 eq of TMA at 62 °C. Silylation was completed after 2 hrs, as evidenced by Solubilization. During the silylation step, 1 eq of Z-Gly-MePro-OH and 1 eq of Oxyma Pure were solubilized in 25 eq of CH2Cl2 at 23 °C. One (1) eq of EDC.HCl was added. To complete the activation, an additional 0.07 eq of EDC. HCl was added. Silylated H-Glu-OH was then added to the activated Oxyma Pure solution. Temperature was controlled during the addition to stay below 45 °C. Desilylation was performed by addition of a mixture of 2.5% w/w KHSO4 in 160 eq of water and 9.6 eq of iPrOH to reach pH 1.63.
Residual CH2Cl2 was removed by concentration. The mixture was cooled to 20 °C to precipitate the Z-Gly-MePro-Glu-OH. The mixture was filtered and the solid was washed with 192 eq of water before drying at about 25 °C for 2.5 days. The solid was then solubilized at 64 °C by addition of 55 eq of water and 31 eq of iPrOH. After solubilization, the mixture was diluted with 275 eq of water and cooled to 10 °C for crystallization. The mixture was filtered and the solid was washed with 60 eq of water before drying at 27 °C (Z-Gly-MePro-Glu-OH dried product – Purity: 99.6%; NMR assay: 98%; Yield 74%).
Procedure 3E
In reactor 1, H-Glu-OH (1.05 eq) was suspended in ACN (about 2.2 eq). TMA (about 3.5 eq) added, and the mixture was heated to dissolve solids. After dissolution, the solution was cooled to room temperature. In reactor 2, Z-Gly-MePro-OH (1 eq) was suspended in ACN (14 eq). Oxyma Pure (1 eq) and EDC.HCl (1 eq) were added. The mixture was stirred at room temperature until the solids dissolved. The contents of reactor 2 were added to reactor 1. The conversion from Z-Gly-MePro-OH to Z-Gly-MePro-Glu-OH was monitored by HPLC. Upon completion the reaction mixture was added to an aqueous solution comprised of KHSO4 (about 2.5 wt%) dissolved in about 100 eq H2O. ACN was removed from the aqueous suspension of Z-Gly-MePro-Glu-OH by vacuum distillation with H2O. After stirring at room temperature, the product in the resulting suspension was isolated by filtration and washed with water. The solid obtained was dissolved in an aqueous solution comprised of NaHCO3 (about 5 wt%) in 110 eq H2O, and recrystallized by addition of an aqueous solution comprised of KHSO4 (about 10 wt%) in 90 eq H2O. The product was isolated by filtration, washed with water, and dried under vacuum at 45 °C. Z-Gly-MePro-Glu-OH (75% yield) was obtained.
Step 4: Deprotection and Isolation of Trofinetide
Several alternative procedures can be used in this step.
Procedure 4A
Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 25 eq) and EtOAc (about 15 eq). Pd/C (0.025 eq by weight and containing 10% Pd by weight) was added, and the reaction mixture hydrogenated by bubbling hydrogen through the reaction mixture at room temperature. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration, and the layers separated. Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained. Alternatively, deprotection can be accomplished using MeOH only, or a combination of iPrOH and MeOH, or by use of ethyl acetate in water.
Procedure 4B
This Procedure is for a variant of Procedure 4A, excluding EtOAc. Z-Gly-MePro-Glu-OH (1 eq) was suspended in water (about 50 eq). Pd/C (0.05 eq, 5% Pd by weight) was added, and the reaction mixture hydrogenated at room temperature with a pressure of 5 bar. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon
reaction completion the catalyst was removed by filtration, and the aqueous layer washed with EtOAc (about 5 eq). Residual EtOAc was removed from the aqueous solution containing trofinetide by sparging with nitrogen or washing with heptane. The aqueous solution was spray-dried to isolate the product. Trofinetide (90% yield) was obtained.
Procedure 4C
This Procedure is for a variant of Procedure 4A, replacing EtOAc with MeOH. Z-Gly-MePro-Glu-OH (1 eq) was suspended in MeOH (100 eq) and water (12 eq). Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC, and upon reaction completion the catalyst was removed by filtration and the layers were washed with MeOH and iPrOH. The solvents were concentrated under vacuum at 45 °C, and trofinetide precipitated. The precipitate was filtered and dried at 45 °C to provide trofinetide.
Procedure 4D
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 105 eq of MeOH and 12 eq of water. Pd/Si (0.02 eq by weight) was added and the mixture was heated at 23 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate approximately 99% after 1 hr), the catalyst was filtered off and washed with 20-30 eq of MeOH. iPrOH (93 eq) was added and MeOH was replaced by iPrOH by concentration at 45 °C under vacuum. The peptide was concentrated until it began to precipitate. The peptide was filtered and dried at 45 °C (H-Gly-MePro-Glu-OH dried product: Purity: 98.1%; NMR assay: 90%; Yield 81%).
Procedure 4E
This Procedure is for a variant of Procedure 4A, removing H2O and replacing Pd/C with Pd/Si. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 44 eq of MeOH. Pd/Si type 340 (0.02 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 99.9%, after 3-3.5 hrs), the catalyst was filtered off and washed with 8 eq of MeOH. Deprotected peptide was then precipitated in 56 eq of iPrOH. After 30 min at 5 °C, the peptide was filtered and washed with three times with 11 eq of iPrOH before drying at 25 °C (H-Gly-MePro-Glu-OH dried product: Purity: 99.4%; HPLC assay: ~98%; Yield: 81%).
Procedure 4F
This Procedure is for a variant of Procedure 4A. One (1) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 14 eq of EtOAc and 25 eq of water. Pd/C (0.01 eq by weight) was added and the mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (conversion rate about 100%, after about 3.5 hrs), the catalyst was filtered off and washed with a mixture of 3.5 eq of EtOAc and 6 eq of water. The aqueous layer was then ready for spray-drying (Aqueous H-Gly-MePro-Glu-OH peptide solution: Purity: 98.6%; Yield: ~95%).
Procedure 4G
This Procedure is for a variant of Procedure 4A, replacing Pd/C with Pd/Si, EtOAc with MeOH, and removing H2O. Pd/Si type 340 (0.02 eq by weight) was added to 2.9 vols of MeOH for pre-reduction during 30 min. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 34 eq of MeOH. The reduced palladium was then transferred to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. Pd/C type 39 (0.007 eq by weight) was added to the mixture to increase reaction kinetics. At the end of the deprotection, the catalyst was filtered off and washed with 13.6 eq of MeOH. The deprotected peptide was then precipitated in 71 eq of iPrOH. After about 40 min, the peptide was filtered and washed with 35 eq of iPrOH. The peptide was dried below 20 °C and was then ready for solubilization in water and spray-drying.
Procedure 4H
This Procedure is for a variant of Procedure 4A. One (1.0) eq of Z-Gly-MePro-Glu-OH was partially solubilized in 24.8 eq of water and 13.6 eq of EtOAc. Pd/C type 39 (0.025 eq by weight) was added to the peptide mixture. The mixture was kept at 20 °C for the hydrogenolysis. Solubilization of the peptide occurred during the deprotection. At the end of the deprotection (19 hrs), the catalyst was removed by filtration and washed with 5.3 eq of water and 2.9 eq of EtOAc. The biphasic mixture was then decanted to remove the upper organic layer. The aqueous layer was diluted with water to reach an H-Gly-MePro-Glu-OH concentration suitable for spray-drying the solution.
Example 2: Alternative Trofinetide Manufacturing Process
An alternative method for synthesis of Trofinetide is based on U.S. Patent No.
8,546,530 adapted for a tripeptide as follows.
The persilylated compounds used to synthesis Formula (Ia) (trofinetide) are obtained by silylating a corresponding peptide or amino acid by reaction with a silylating agent, optionally in an organic solvent. The persilylated peptide or amino acid can be isolated and purified if desired. One can use the persilylated peptide or amino acid in situ, e.g. by combining a solution containing persilylated peptide or amino acid with a solution containing, optionally activated, peptide or amino acid.
In step 2, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-MePro-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid can be isolated and purified if desired. One can use the persilylated amino acid in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated, amino acid (for example, Z-Gly-OH).
In step 3, the persilylated compound of an amino acid is obtained by silylating a corresponding amino acid (for example, H-Glu-OH) by reaction with a silylating agent, optionally in an organic solvent. The persilylated amino acid or peptide can be isolated and purified if desired. It is however useful to use the persilylated amino acid or peptide in situ, e.g. by combining a solution containing the persilylated amino acid with a solution containing, optionally activated (for example, by using EDC.HCl and Oxyma Pure), peptide (for example, Z-Gly-MePro-OH).
In the present invention, it is useful to use silylating agents, such as N-trialkylsilyl amines or N-trialkylsilyl amides, not containing a cyano group. Examples of such silylating reagents include N,O-bis(trimethylsilyl)acetamide (BSA), N,O-bis(trimethylsilyl)trifluoroacetamide, hexamethyldisilazane, N-methyl-N-(trimethylsilyl)acetamide (TMA), N-methyl-N-(trimethylsilyl)trifluoroacetamide, N-(trimethylsilyl)acetamide, N-(trimethylsilyl)diethylamine, N-(trimethylsilyl)dimethylamine, 1-(trimethylsilyl)imidazole, 3-(trimethylsilyl)-2-oxazolidone.
The reaction of step 2 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 40 °C, and optionally from 15 °C to 30 °C.
The reaction of step 3 is generally carried out at a temperature from 0 °C to 100 °C, optionally from 10 °C to 60 °C, optionally from 15 °C to 50 °C.
In the reaction of step 2, generally 0.5 to 5 equivalents, optionally 1 to 3 equivalents, optionally about 1.5 to 2.5 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2 to 4 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible. “Functional groups to be silylated” means particular groups having an active hydrogen atom that can react with the silylating agent such as amino, hydroxyl, mercapto or carboxyl groups.
In the reaction of step 3, generally 0.5 to 5 equivalents, optionally 2 to 4.5 equivalents, optionally about 3 to 4 equivalents of silylating agent are used relative to the molar amount of functional groups to be silylated. Use of 2.5 to 4.5 equivalents of silylating agent relative to the molar amount of functional groups to be silylated is also possible.
It is understood that “persilylated” means an amino acid or peptide or amino acid analogue or peptide analogue in which the groups having an active hydrogen atom that can react with the silylating agent are sufficiently silylated to ensure that a homogeneous reaction medium for a coupling step is obtained.
In the process according to the invention, the reaction between the amino acid or peptide and the persilylated amino acid or peptide is often carried out in the presence of a carboxyl group activating agent. In that case the carboxylic activating reagent is suitably selected from carbodiimides, acyl halides, phosphonium salts and uronium or guanidinium salts. More optionally, the carboxylic activating agent is an acyl halide, such as isobutyl chloroformate or pivaloyl chloride or a carbodiimide, such as EDC.HC1 or DCC.
Good results are often obtained when using additional carboxylic activating reagents which reduce side reactions and/or increase reaction efficiency. For example, phosphonium and uronium salts can, in the presence of a tertiary base, for example, N,N-diisopropylethylamine (DIPEA) and triethylamine (TEA), convert protected amino acids into activated species. Other reagents help prevent racemization by providing a protecting reagent. These reagents include carbodiimides (for example, DCC) with an added auxiliary nucleophile (for example, 1-hydroxy-benzo triazole (HOBt), 1-hydroxy-azabenzotriazole (HOAt), or Suc-OH) or derivatives thereof. Another reagent that can be utilized is TBTU. The mixed anhydride method, using isobutyl chloroformate, with or without an added auxiliary nucleophile, is also used, as is the azide method, due to the low racemization associated with it. These types of compounds can also increase the rate of carbodiimide-mediated couplings. Typical additional reagents include also bases such as N,N-diisopropylethylamine (DIPEA), triethylamine (TEA) or N-methylmorpholine (NMM).
When the silylation is carried out in the presence of a solvent, said solvent is optionally a polar organic solvent, more optionally a polar aprotic organic solvent. An amide type solvent such as N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAC)
can be used. In the present invention for step 2, one can use an alkyl acetate solvent, in particular ethyl acetate is more particularly optional.
In the present invention for step 3, one can use a chlorinated hydrocarbon solvent or alkyl cyanide solvent, in particular dichloromethane or acetonitrile are more particularly optional.
In another embodiment, silylation is carried out in a liquid silylation medium consisting essentially of silylating agent and amino acid or peptide.
In the present invention, amino acid or peptide is understood to denote in particular an amino acid or peptide or amino acid analogue or peptide analogue which is bonded at its N-terminus or optionally another position, to a carboxylic group of an amino protected amino acid or peptide.
Example 3: Specifications for Compositions Containing Compounds of Formula (I)
1 ICH guideline Q3C on impurities: guideline for residual solvents
Example 4: Alternative Manufacturing of Trofinetide Example 1, Step 4, Procedure 4B
This Procedure is for a variant of Step 4, Procedure 4B. Z-Gly-MePro-Glu-OH (1 eq) was added in portions to Pd/C (0.027 eq by weight and containing 5% Pd by weight) in about 50 eq of water. The reaction mixture was hydrogenated at 20 °C at a pressure of 5 bar for at least 4 cycles of 4 hrs each. Pd/C (0.0027 eq by weight) was charged between cycles, as needed, to speed up the reaction. The conversion from Z-Gly-MePro-Glu-OH to trofinetide was monitored by HPLC. Upon reaction completion the catalyst was removed by filtration, washed with water (12.5 eq) and the aqueous layer washed with EtOAc (about 14 eq). After phase separation, residual EtOAc was removed from the aqueous solution containing
trofinetide by sparging with nitrogen under vacuum at 20 °C for about 3 hrs. The aqueous solution was filtered. The final concentration of trofinetide was about 25 wt% and the solution was then ready for spray-drying to isolate the product.
Example 5: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (I)
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (I) on an anhydrous basis.
Example 6: Alternative Composition of Trofinetide
A composition comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (II):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, and/or a compound of Formula (III):
or a stereoisomer, hydrate, or pharmaceutically acceptable salt thereof, wherein R1, R2, R3 and R4 independently are selected from the group consisting of hydrogen and C1-4 alkyl, provided that least one of R1, R2, R3 and R4 is C1-4 alkyl, and wherein the composition comprises at least 90 wt%, such as 91 wt%, 92 wt%, 93 wt%, 94 wt%, 95 wt%, 96 wt%, or 97 wt% of the compound of Formula (Ia) on an anhydrous basis.
Example 7: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, wherein the product comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101
wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 8: A Product of Trofinetide
A product, including a kit containing a dosage form with instructions for use, comprising a compound of Formula (Ia)
or a hydrate, or pharmaceutically acceptable salt thereof, and a compound of Formula (IIa)
or a hydrate, or pharmaceutically acceptable salt thereof, and additionally comprising one or more compounds selected from the group consisting of Formula (III), Formula (IIIa), Formula (IV), Formula (V), Formula (VI), Formula (VII), Formula (VIII), and Formula (IX), wherein the composition comprises between 95 wt% and 105 wt%, such as 96 wt%, 97 wt%, 98 wt%, 99 wt%, 100 wt%, 101 wt%, 102 wt%, 103 wt%, or 104 wt% of the specified amount of the compound of Formula (Ia) in the product.
Example 9: Analysis of Products and Compositions
The products and compositions disclosed herein may be analyzed by liquid chromatography, a suitable chromatographic method using UPLC, e.g. using materials and conditions such as Waters Acquity CSH C18, 1.7 µm, 150 x 2.1 mm column, water with 0.1 % TFA (mobile phase A), and water/ACN 70/30 + 0.1 % TFA (mobile phase B), ranging from (4% phase A/6% phase B to 100% phase B and flushed with 4% phase A/6% phase B).
Flow rate: 0.35 ml/min, Column temperature: 40 °C, autosampler temperature: 4 °C, injection volume: 4 ml (e.g. prepared by weighing about 10 mg of powder in a 10 ml volumetric flask and diluted to volume with water). Examples of detectors are UV (ultraviolet, UV 220 nm) and MS (mass spectrometry).
INDUSTRIAL APPLICABILITY
This invention finds use in the pharmaceutical, medical, and other health care fields.
PATENT
WO2014085480 ,
claiming use of trofinetide for treating autism spectrum disorders including autism, Fragile X Syndrome or Rett Syndrome.
EP 0 366 638 discloses GPE (a tri-peptide consisting of the amino acids Gly-Pro- Glu) and its di-peptide derivatives Gly-Pro and Pro-Glu. EP 0 366 638 discloses that GPE is effective as a neuromodulator and is able to affect the electrical properties of neurons.
W095/172904 discloses that GPE has neuroprotective properties and that administration of GPE can reduce damage to the central nervous system (CNS) by the prevention or inhibition of neuronal and glial cell death.
WO 98/14202 discloses that administration of GPE can increase the effective amount of choline acetyltransferase (ChAT), glutamic acid decarboxylase (GAD), and nitric oxide synthase (NOS) in the central nervous system (CNS).
WO99/65509 discloses that increasing the effective amount of GPE in the CNS, such as by administration of GPE, can increase the effective amount of tyrosine hydroxylase (TH) in the CNS for increasing TH-mediated dopamine production in the treatment of diseases such as Parkinson’s disease.
WO02/16408 discloses GPE analogs capable of inducing a physiological effect equivalent to GPE within a patient. The applications of the GPE analogs include the treatment of acute brain injury and neurodegenerative diseases, including but not limited to, injury or disease in the CNS.
Example
The following non-limiting example illustrates the synthesis of a compound of the invention, NN-dimethylglycyl-L-prolyl-L-glutamic acid.
All starting materials and other reagents were purchased from Aldrich;
BOC = tert-butoxycarbonyl; Bn = benzyl.
BOC-(γ-benzyl)-L-prolyl-L-glutamic acid benzyl ester
To a solution of BOC-proline [Anderson GW and McGregor AC: J. Amer. Chem.
Soc: 79, 6180, 1957] (10 mmol) in dichloromethane (50 ml), cooled to 0 °C, was added triethylamine (1.39 ml, 10 mmol) and ethyl chloroformate (0.96 ml, 10 mmol). The resultant mixture was stirred at 0 °C for 30 minutes. A solution of dibenzyl L-glutamate (10 mmol) was then added and the mixture stirred at 0 °C for 2 hours then warmed to room temperature and stirred overnight. The reaction mixture was washed with aqueous sodium bicarbonate and citric acid (2 mol l“1) then dried (MgS04) and concentrated at reduced pressure to give BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (5.0 g, 95%).
(7-Benzyl)-L-prolyl-L-glutamic acid dibenzyl ester
A solution of BOC-(γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (3.4 g, 10 mmol), cooled to 0 °C, was treated with trifluoroacetic acid (25 ml) for 2 hr at room temperature. After removal of the volatiles at reduced pressure the residue was triturated with ether to give (γ-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (I).
N,N-Dimethylglycyl-L-prolyl-L-glutamic acid
A solution of dicyclohexylcarbodiimide (10.3 mmol) in dichloromethane (10 ml) was added to a stirred and cooled (0 °C) solution of (7-benzyl)-L-prolyl-L-glutamic acid dibenzyl ester (10 mmol), TVN-dimethylglycine (10 mmol) and triethylamine
(10.3 mmol) in dichloromethane (30 ml). The mixture was stirred at 0 °C overnight and then at room temperature for 3 h. After filtration, the filtrate was evaporated at reduced pressure. The resulting crude dibenzyl ester was dissolved in a mixture of ethyl acetate (30 ml) and methanol (30 ml) containing 10% palladium on charcoal (0.5 g) then hydrogenated at room temperature and pressure until the uptake of hydrogen ceased. The filtered solution was evaporated and the residue recrystallized from ethyl acetate to yield the tri-peptide derivative.
It will be evident that following the method of the Example, and using alternative amino acids or their amides or esters, will yield other compounds of Formula 1.
PAPER
Tetrahedron (2005), 61(42), 10018-10035. (CLICK HERE)
The synthesis of ten proline-modified analogues of the neuroprotective tripeptide GPE is described. Five of the analogues incorporate a proline residue with a hydrophobic group at C-2 and two further analogues have this side chain locked into a spirolactam ring system. The pyrrolidine ring was also modified by replacing the γ-CH2 group with sulfur and/or incorporation of two methyl groups at C-5.
Graphical Abstract

PAPER
Bioorganic & Medicinal Chemistry Letters (2005), 15(9), 2279-2283
A series of GPE analogues, including modifications at the Pro and/or Glu residues, was prepared and evaluated for their NMDA binding and neuroprotective effects. Main results suggest that the pyrrolidine ring puckering of the Pro residue plays a key role in the biological responses, while the preference for cis or trans rotamers around the Gly-Pro peptide bond is not important.
Graphical abstract
A series of Pro and/or Glu modified GPE analogues is described. Compounds incorporating PMe and dmP showed higher affinity for glutamate receptors than GPE and neuroprotective effects similar to those of this endogenous tripeptide in culture hippocampal neurons exposed to NMDA.

PATENT
US 20060251649
WO 2006127702
US 20070004641
US 20080145335
WO 2012102832
WO 2014085480
US 20140147491
References
- ^ Bickerdike MJ, Thomas GB, Batchelor DC, Sirimanne ES, Leong W, Lin H, et al. (March 2009). “NNZ-2566: a Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke”. Journal of the Neurological Sciences. 278 (1–2): 85–90. doi:10.1016/j.jns.2008.12.003. PMID 19157421. S2CID 7789415.
- ^ Cartagena CM, Phillips KL, Williams GL, Konopko M, Tortella FC, Dave JR, Schmid KE (September 2013). “Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3”. Neuromolecular Medicine. 15 (3): 504–14. doi:10.1007/s12017-013-8236-z. PMID 23765588. S2CID 12522580.
- ^ Deacon RM, Glass L, Snape M, Hurley MJ, Altimiras FJ, Biekofsky RR, Cogram P (March 2015). “NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome”. Neuromolecular Medicine. 17 (1): 71–82. doi:10.1007/s12017-015-8341-2. PMID 25613838. S2CID 11964380.
- ^ Study Details – Rett Syndrome Study
- ^ Neuren’s trofinetide successful in Phase 2 clinical trial in Fragile X
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 3 | Enrolling by Invitation | Treatment | Rett’s Syndrome | 1 |
| 3 | Recruiting | Treatment | Rett’s Syndrome | 1 |
| 2 | Completed | Supportive Care | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Fragile X Syndrome (FXS) | 1 |
| 2 | Completed | Treatment | Injuries, Brain | 1 |
| 2 | Completed | Treatment | Rett’s Syndrome | 2 |
| 2 | Terminated | Treatment | Concussions | 1 |
| 1 | Completed | Treatment | Brain Injuries,Traumatic | 2 |
| Legal status | |
|---|---|
| Legal status | US: Investigational New Drug |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 853400-76-7 |
| PubChem CID | 11318905 |
| ChemSpider | 9493869 |
| UNII | Z2ME8F52QL |
| Chemical and physical data | |
| Formula | C13H21N3O6 |
| Molar mass | 315.322 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@]1(CCCN1C(=O)CN)C(=O)N[C@@H](CCC(=O)O)C(=O)O | |
| InChI[hide]InChI=1S/C13H21N3O6/c1-13(5-2-6-16(13)9(17)7-14)12(22)15-8(11(20)21)3-4-10(18)19/h8H,2-7,14H2,1H3,(H,15,22)(H,18,19)(H,20,21)/t8-,13-/m0/s1Key:BUSXWGRAOZQTEY-SDBXPKJASA-N |
////////////Tofinetide , NNZ 2566, PHASE 2, PHASE 3. NEUREN, Amino Acids, Peptides, Proteins,
CC1(CCCN1C(=O)CN)C(=O)NC(CCC(=O)O)C(=O)O
J. Med. Chem. 2025, 68, 2147−2182
Trofinetide (Daybue). Trofinetide (8) was developed by Neuren Pharmaceuticals and Acadia Pharmaceuticals for the treatment of rare childhood neurodevelopmental disorders and
was approved by the USFDA in March 2023 for adults and pediatric patients two years of age or older with Rett syndrome. 59 In most cases, Rett syndrome is caused by loss of-function mutations in the X-linked gene that encodes methyl CpG-binding protein 2 (MeCP2). 60,61 MeCP2 is a critical transcriptional regulator required for normal neurological development. In the past, treatment of Rett syndrome has
been limited to symptom management based on knowledge from treating other conditions, 62
but new research has focused on targeting the underlying genetic cause and finding agents to
restore MeCP2 function. Trofinetide is an orally available synthetic analog of glycine-proline-glutamate (GPE), the Nterminal tripeptide metabolite of insulin like growth factor-1 (IGF-1). GPE has been shown to partially reverse Rett-like symptoms in MeCP2 deficient mouse models and trofenitide was developed to have an improved pharmakokinetic profile to GPE. 64 Its use has shown significant improvement over placebo in clinical trials.
Anumberofaccountsrelated to the preparation of trofinetide have been reported with various protecting group strategies, but they are moreamenabletosmall-scaleproductionduetoreagent selection and challenging isolations requiring column chromatography.65−67 A commercially viable synthesis of the drug has been described by researchers at Neuren Pharmaceuticals and is depicted in Scheme 12.68
This synthesis takes advantage of in situ silyl protection/deprotection during its amidation steps to
avoid lengthy protecting group manipulations. Activation of Cbz protected glycine 8.1 with hydroxysuccinimide 8.2 using EDCI provided 8.3 in 84% yield as a direct drop crystallization
(isolated by crystallization directly from the reaction mixture). This activated ester was then coupled with commercially available methyl proline 8.4 69 by first silylation of the carboxylic acid of the proline analog in situ with 8.5, then adding 8.3 to couple with the amine. Deprotection of the silyl ester during the
workup provided amide 8.6 in 79% yield. In a similar sequence, 8.6 was activated with Oxyma Pure and EDCI while in a second vessel 8.7 was silyl protected using 8.5. Subsequent combination of these streams followed by workup and crystallization gave amide 8.8 in good yield. Finally, Cbz deprotection was
accomplished using Pd/C and hydrogen. Trofinetide (8) was extracted into the aqueous layer and isolated by spray drying in 90% yield.
(60) Kyle, S. M.; Vashi, N.; Justice, M. J. Rett syndrome: a
neurological disorder with metabolic components. Open Biol. 2018,
8, No. 170216.
(61) Collins, B. E.; Neul, J. L. Rett syndrome and MECP2 duplication
syndrome: disorders of MeCP2 dosage. Neuropsychiatr. Dis. Treat.
2022, 18, 2813−2835.
(62) Fu, C.; Armstrong, D.; Marsh, E.; Lieberman, D.; Motil, K.; Witt,
R.; Standridge, S.; Nues, P.; Lane, J.; Dinkel, T.; et al. Consensus
guidelines on managing Rett syndrome across the lifespan. BMJ
Paediatr. Open 2020, 4, No. e000717.
(63) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D.G.;Marsh,E.D.;Lin,T.;Stankovic,S.;Bishop,K. M.;Youakim,J.M.
Trofinetide for the treatment of Rett syndrome: a randomized phase 3
study. Nat. Med. 2023, 29, 1468−1475.
(64) Neul, J. L.; Percy, A. K.; Benke, T. A.; Berry-Kravis, E. M.; Glaze,
D. G.; Peters, S. U.; Jones, N. E.; Youakim, J. M. Design and outcome
measures of LAVENDER, a phase 3 study of trofinetide for Rett
syndrome. Contemp. Clin. Trials 2022, 114, No. 106704.
(65) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl-l-2-methylprolyl-l-glutamic acid. US
20140147491, 2014.
(66) Glass, L.; Bickerdike, M. J.; Snape, M. F. Treatment of autism
spectrum disorders using glycyl L2012102832, 2012.-2-methylprolyl
L-glutamic acid. WO 2012102832, 2012
(67) Brimble, M.A.;Harris, P. W.R.;Sieg, F. Preparation of analogsof
glycyl-prolyl-glutamate as neuroprotective agents. US 20080145335,
2008
(68) Blower, C.; Peterson, M.; Shaw, J. M.; Bonnar, J. A.; Moniotte, E.
D. F. P.; Bousmanne, M. B. C.; Betti, C.; Decroos, K. W. L.; Ayoub, M.
Compositions of trofinetide. WO 2021026066, 2021.
(69) Beck, A. K.; Blank, S.; Job, K.; Seebach, D.; Sommerfeld, T.
Synthesis of (S)-2-methylproline: a general method for the preparation
of α-branched amino acids (L.-proline, 2-methyl-). Org. Synth. 1995, 72, 62−73

.
GLUCAGON
glucagon
EMA……Ogluo (glucagon), a hybrid medicine for the treatment of severe hypoglycaemia in diabetes mellitus. Hybrid applications rely in part on the results of pre-clinical tests and clinical trials of an already authorised reference product and in part on new data.
On 10 December 2020, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product Ogluo, intended for the treatment of severe hypoglycaemia in diabetes mellitus. The applicant for this medicinal product is Xeris Pharmaceuticals Ireland Limited.
Ogluo will be available as 0.5 and 1 mg solution for injection. The active substance of Ogluo is glucagon, a pancreatic hormone (ATC code: H04AA01); glucagon increases blood glucose concentration by stimulating glycogen breakdown and release of glucose from the liver.
The benefits with Ogluo are its ability to restore blood glucose levels in hypoglycaemic subjects. The most common side effects are nausea and vomiting.
Ogluo is a hybrid medicine1 of GlucaGen/GlucaGen Hypokit; GlucaGen has been authorised in the EU since October 1962. Ogluo contains the same active substance as GlucaGen but is available as a ready-to-use formulation intended for subcutaneous injection.
The full indication is:
Ogluo is indicated for the treatment of severe hypoglycaemia in adults, adolescents, and children aged 2 years and over with diabetes mellitus.
Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
1 Hybrid applications rely in part on the results of pre-clinical tests and clinical trials for a reference product and in part on new data.
Glucagon is a peptide hormone, produced by alpha cells of the pancreas. It works to raise the concentration of glucose and fatty acids in the bloodstream, and is considered to be the main catabolic hormone of the body.[3] It is also used as a medication to treat a number of health conditions. Its effect is opposite to that of insulin, which lowers extracellular glucose.[4] It is produced from proglucagon, encoded by the GCG gene.
The pancreas releases glucagon when the amount of glucose in the bloodstream is too low. Glucagon causes the liver to engage in glycogenolysis: converting stored glycogen into glucose, which is released into the bloodstream.[5] High blood-glucose levels, on the other hand, stimulate the release of insulin. Insulin allows glucose to be taken up and used by insulin-dependent tissues. Thus, glucagon and insulin are part of a feedback system that keeps blood glucose levels stable. Glucagon increases energy expenditure and is elevated under conditions of stress.[6] Glucagon belongs to the secretin family of hormones.
Function
Glucagon generally elevates the concentration of glucose in the blood by promoting gluconeogenesis and glycogenolysis.[7] Glucagon also decreases fatty acid synthesis in adipose tissue and the liver, as well as promoting lipolysis in these tissues, which causes them to release fatty acids into circulation where they can be catabolised to generate energy in tissues such as skeletal muscle when required.[8]
Glucose is stored in the liver in the form of the polysaccharide glycogen, which is a glucan (a polymer made up of glucose molecules). Liver cells (hepatocytes) have glucagon receptors. When glucagon binds to the glucagon receptors, the liver cells convert the glycogen into individual glucose molecules and release them into the bloodstream, in a process known as glycogenolysis. As these stores become depleted, glucagon then encourages the liver and kidney to synthesize additional glucose by gluconeogenesis. Glucagon turns off glycolysis in the liver, causing glycolytic intermediates to be shuttled to gluconeogenesis.
Glucagon also regulates the rate of glucose production through lipolysis. Glucagon induces lipolysis in humans under conditions of insulin suppression (such as diabetes mellitus type 1).[9]
Glucagon production appears to be dependent on the central nervous system through pathways yet to be defined. In invertebrate animals, eyestalk removal has been reported to affect glucagon production. Excising the eyestalk in young crayfish produces glucagon-induced hyperglycemia.[10]
Mechanism of action
Metabolic regulation of glycogen by glucagon.
Glucagon binds to the glucagon receptor, a G protein-coupled receptor, located in the plasma membrane of the cell. The conformation change in the receptor activates G proteins, a heterotrimeric protein with α, β, and γ subunits. When the G protein interacts with the receptor, it undergoes a conformational change that results in the replacement of the GDP molecule that was bound to the α subunit with a GTP molecule. This substitution results in the releasing of the α subunit from the β and γ subunits. The alpha subunit specifically activates the next enzyme in the cascade, adenylate cyclase.
Adenylate cyclase manufactures cyclic adenosine monophosphate (cyclic AMP or cAMP), which activates protein kinase A (cAMP-dependent protein kinase). This enzyme, in turn, activates phosphorylase kinase, which then phosphorylates glycogen phosphorylase b (PYG b), converting it into the active form called phosphorylase a (PYG a). Phosphorylase a is the enzyme responsible for the release of glucose 1-phosphate from glycogen polymers. An example of the pathway would be when glucagon binds to a transmembrane protein. The transmembrane proteins interacts with Gɑβ𝛾. Gɑ separates from Gβ𝛾 and interacts with the transmembrane protein adenylyl cyclase. Adenylyl cyclase catalyzes the conversion of ATP to cAMP. cAMP binds to protein kinase A, and the complex phosphorylates phosphorylase kinase.[11] Phosphorylated phosphorylase kinase phosphorylates phosphorylase. Phosphorylated phosphorylase clips glucose units from glycogen as glucose 1-phosphate. Additionally, the coordinated control of glycolysis and gluconeogenesis in the liver is adjusted by the phosphorylation state of the enzymes that catalyze the formation of a potent activator of glycolysis called fructose 2,6-bisphosphate.[12] The enzyme protein kinase A (PKA) that was stimulated by the cascade initiated by glucagon will also phosphorylate a single serine residue of the bifunctional polypeptide chain containing both the enzymes fructose 2,6-bisphosphatase and phosphofructokinase-2. This covalent phosphorylation initiated by glucagon activates the former and inhibits the latter. This regulates the reaction catalyzing fructose 2,6-bisphosphate (a potent activator of phosphofructokinase-1, the enzyme that is the primary regulatory step of glycolysis)[13] by slowing the rate of its formation, thereby inhibiting the flux of the glycolysis pathway and allowing gluconeogenesis to predominate. This process is reversible in the absence of glucagon (and thus, the presence of insulin).
Glucagon stimulation of PKA also inactivates the glycolytic enzyme pyruvate kinase in hepatocytes.[14]
Physiology
Production
A microscopic image stained for glucagon
The hormone is synthesized and secreted from alpha cells (α-cells) of the islets of Langerhans, which are located in the endocrine portion of the pancreas. Production, which is otherwise freerunning, is suppressed/regulated by amylin, a peptide hormone co-secreted with insulin from the pancreatic β cells.[15] As plasma glucose levels recede, the subsequent reduction in amylin secretion alleviates its suppression of the α cells, allowing for glucagon secretion.
In rodents, the alpha cells are located in the outer rim of the islet. Human islet structure is much less segregated, and alpha cells are distributed throughout the islet in close proximity to beta cells. Glucagon is also produced by alpha cells in the stomach.[16]
Recent research has demonstrated that glucagon production may also take place outside the pancreas, with the gut being the most likely site of extrapancreatic glucagon synthesis.[17]
Regulation
Secretion of glucagon is stimulated by:
- Hypoglycemia
- Epinephrine (via β2, α2,[18] and α1[19] adrenergic receptors)
- Arginine
- Alanine (often from muscle-derived pyruvate/glutamate transamination (see alanine transaminase reaction).
- Acetylcholine[20]
- Cholecystokinin
- Gastric inhibitory polypeptide
Secretion of glucagon is inhibited by:
- Somatostatin
- Amylin [21]
- Insulin (via GABA)[22]
- PPARγ/retinoid X receptor heterodimer.[23]
- Increased free fatty acids and keto acids into the blood.[24]
- Increased urea production
- Glucagon-like peptide-1
Structure
Glucagon is a 29-amino acid polypeptide. Its primary structure in humans is: NH2–His–Ser–Gln–Gly–Thr–Phe–Thr–Ser–Asp–Tyr–Ser–Lys–Tyr–Leu–Asp–Ser–Arg–Arg–Ala–Gln–Asp–Phe–Val–Gln–Trp–Leu–Met–Asn–Thr–COOH.
The polypeptide has a molecular mass of 3485 daltons.[25] Glucagon is a peptide (nonsteroid) hormone.
Glucagon is generated from the cleavage of proglucagon by proprotein convertase 2 in pancreatic islet α cells. In intestinal L cells, proglucagon is cleaved to the alternate products glicentin, GLP-1 (an incretin), IP-2, and GLP-2 (promotes intestinal growth).[26]
Pathology
Abnormally elevated levels of glucagon may be caused by pancreatic tumors, such as glucagonoma, symptoms of which include necrolytic migratory erythema,[27] reduced amino acids, and hyperglycemia. It may occur alone or in the context of multiple endocrine neoplasia type 1[28]
Elevated glucagon is the main contributor to hyperglycemic ketoacidosis in undiagnosed or poorly treated type 1 diabetes. As the beta cells cease to function, insulin and pancreatic GABA are no longer present to suppress the freerunning output of glucagon. As a result, glucagon is released from the alpha cells at a maximum, causing rapid breakdown of glycogen to glucose and fast ketogenesis.[29] It was found that a subset of adults with type 1 diabetes took 4 times longer on average to approach ketoacidosis when given somatostatin (inhibits glucagon production) with no insulin. Inhibiting glucagon has been a popular idea of diabetes treatment, however some have warned that doing so will give rise to brittle diabetes in patients with adequately stable blood glucose.[citation needed]
The absence of alpha cells (and hence glucagon) is thought to be one of the main influences in the extreme volatility of blood glucose in the setting of a total pancreatectomy.
History
In the 1920s, Kimball and Murlin studied pancreatic extracts, and found an additional substance with hyperglycemic properties. They described glucagon in 1923.[30] The amino acid sequence of glucagon was described in the late 1950s.[31] A more complete understanding of its role in physiology and disease was not established until the 1970s, when a specific radioimmunoassay was developed.[citation needed]
Etymology
Kimball and Murlin coined the term glucagon in 1923 when they initially named the substance the glucose agonist.[32]
References
- ^ Jump up to:a b c GRCh38: Ensembl release 89: ENSG00000115263 – Ensembl, May 2017
- ^ “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Voet D, Voet JG (2011). Biochemistry (4th ed.). New York: Wiley.
- ^ Reece J, Campbell N (2002). Biology. San Francisco: Benjamin Cummings. ISBN 978-0-8053-6624-2.
- ^ Orsay J (2014). Biology 1: Molecules. Examkrackers Inc. p. 77. ISBN 978-1-893858-70-1.
- ^ Jones BJ, Tan T, Bloom SR (March 2012). “Minireview: Glucagon in stress and energy homeostasis”. Endocrinology. 153 (3): 1049–54. doi:10.1210/en.2011-1979. PMC 3281544. PMID 22294753.
- ^ Voet D, Voet JG (2011). Biochemistry (4th ed.). New York: Wiley.
- ^ HABEGGER, K. M., HEPPNER, K. M., GEARY, N., BARTNESS, T. J., DIMARCHI, R. & TSCHÖP, M. H. (2010). “The metabolic actions of glucagon revisited”. Nature Reviews. Endocrinology. 6 (12): 689–697. doi:10.1038/nrendo.2010.187. PMC 3563428. PMID 20957001.
- ^ Liljenquist JE, Bomboy JD, Lewis SB, Sinclair-Smith BC, Felts PW, Lacy WW, Crofford OB, Liddle GW (January 1974). “Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men”. The Journal of Clinical Investigation. 53 (1): 190–7. doi:10.1172/JCI107537. PMC 301453. PMID 4808635.
- ^ Leinen RL, Giannini AJ (1983). “Effect of eyestalk removal on glucagon induced hyperglycemia in crayfish”. Society for Neuroscience Abstracts. 9: 604.
- ^ Yu Q, Shuai H, Ahooghalandari P, Gylfe E, Tengholm A (July 2019). “Glucose controls glucagon secretion by directly modulating cAMP in alpha cells”. Diabetologia. 62 (7): 1212–1224. doi:10.1007/s00125-019-4857-6. PMC 6560012. PMID 30953108.
- ^ Hue L, Rider MH (July 1987). “Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues”. The Biochemical Journal. 245 (2): 313–24. doi:10.1042/bj2450313. PMC 1148124. PMID 2822019.
- ^ Claus TH, El-Maghrabi MR, Regen DM, Stewart HB, McGrane M, Kountz PD, Nyfeler F, Pilkis J, Pilkis SJ (1984). “The role of fructose 2,6-bisphosphate in the regulation of carbohydrate metabolism”. Current Topics in Cellular Regulation. 23: 57–86. doi:10.1016/b978-0-12-152823-2.50006-4. ISBN 9780121528232. PMID 6327193.
- ^ Feliú JE, Hue L, Hers HG (August 1976). “Hormonal control of pyruvate kinase activity and of gluconeogenesis in isolated hepatocytes”. Proceedings of the National Academy of Sciences of the United States of America. 73 (8): 2762–6. Bibcode:1976PNAS…73.2762F. doi:10.1073/pnas.73.8.2762. PMC 430732. PMID 183209.
- ^ Zhang, Xiao-Xi (2016). “Neuroendocrine Hormone Amylin in Diabetes”. World J Diabetes. 7 (9): 189–197. doi:10.4239/wjd.v7.i9.189. PMC 4856891. PMID 27162583.
- ^ Unger RH, Cherrington AD (January 2012). “Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover”. The Journal of Clinical Investigation. 122(1): 4–12. doi:10.1172/JCI60016. PMC 3248306. PMID 22214853.
- ^ Holst JJ, Holland W, Gromada J, Lee Y, Unger RH, Yan H, Sloop KW, Kieffer TJ, Damond N, Herrera PL (April 2017). “Insulin and Glucagon: Partners for Life”. Endocrinology. 158(4): 696–701. doi:10.1210/en.2016-1748. PMC 6061217. PMID 28323959.
- ^ Layden BT, Durai V, Lowe WL (2010). “G-Protein-Coupled Receptors, Pancreatic Islets, and Diabetes”. Nature Education. 3 (9): 13.
- ^ Skoglund G, Lundquist I, Ahrén B (November 1987). “Alpha 1- and alpha 2-adrenoceptor activation increases plasma glucagon levels in the mouse”. European Journal of Pharmacology. 143 (1): 83–8. doi:10.1016/0014-2999(87)90737-0. PMID 2891547.
- ^ Honey RN, Weir GC (October 1980). “Acetylcholine stimulates insulin, glucagon, and somatostatin release in the perfused chicken pancreas”. Endocrinology. 107 (4): 1065–8. doi:10.1210/endo-107-4-1065. PMID 6105951.
- ^ Zhang, Xiao-Xi (2016). “Neuroendocrine Hormone Amylin in Diabetes”. World J Diabetes. 7 (9): 189–197. doi:10.4239/wjd.v7.i9.189. PMC 4856891. PMID 27162583.
- ^ Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, Liu S, Wendt A, Deng S, Ebina Y, Wheeler MB, Braun M, Wang Q (January 2006). “Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system”. Cell Metabolism. 3 (1): 47–58. doi:10.1016/j.cmet.2005.11.015. PMID 16399504.
- ^ Krätzner R, Fröhlich F, Lepler K, Schröder M, Röher K, Dickel C, Tzvetkov MV, Quentin T, Oetjen E, Knepel W (February 2008). “A peroxisome proliferator-activated receptor gamma-retinoid X receptor heterodimer physically interacts with the transcriptional activator PAX6 to inhibit glucagon gene transcription”. Molecular Pharmacology. 73 (2): 509–17. doi:10.1124/mol.107.035568. PMID 17962386. S2CID 10108970.
- ^ Johnson LR (2003). Essential Medical Physiology. Academic Press. pp. 643–. ISBN 978-0-12-387584-6.
- ^ Unger RH, Orci L (June 1981). “Glucagon and the A cell: physiology and pathophysiology (first two parts)”. The New England Journal of Medicine. 304 (25): 1518–24. doi:10.1056/NEJM198106183042504. PMID 7015132.
- ^ Orskov C, Holst JJ, Poulsen SS, Kirkegaard P (November 1987). “Pancreatic and intestinal processing of proglucagon in man”. Diabetologia. 30 (11): 874–81. doi:10.1007/BF00274797 (inactive 2020-10-11). PMID 3446554.
- ^ John AM, Schwartz RA (December 2016). “Glucagonoma syndrome: a review and update on treatment”. Journal of the European Academy of Dermatology and Venereology. 30 (12): 2016–2022. doi:10.1111/jdv.13752. PMID 27422767. S2CID 1228654.
- ^ Oberg K (December 2010). “Pancreatic endocrine tumors”. Seminars in Oncology. 37 (6): 594–618. doi:10.1053/j.seminoncol.2010.10.014. PMID 21167379.
- ^ Fasanmade OA, Odeniyi IA, Ogbera AO (June 2008). “Diabetic ketoacidosis: diagnosis and management”. African Journal of Medicine and Medical Sciences. 37 (2): 99–105. PMID 18939392.
- ^ Kimball C, Murlin J (1923). “Aqueous extracts of pancreas III. Some precipitation reactions of insulin”. J. Biol. Chem. 58 (1): 337–348.
- ^ Bromer W, Winn L, Behrens O (1957). “The amino acid sequence of glucagon V. Location of amide groups, acid degradation studies and summary of sequential evidence”. J. Am. Chem. Soc. 79 (11): 2807–2810. doi:10.1021/ja01568a038.
- ^ “History of glucagon – Metabolism, insulin and other hormones – Diapedia, The Living Textbook of Diabetes”. http://www.diapedia.org. Archived from the original on 2017-03-27. Retrieved 2017-03-26.
External links
- PDBe-KB provides an overview of all the structure information available in the PDB for Human Glucagon
| GCG | ||
|---|---|---|
| Available structuresPDBHuman UniProt search: PDBe RCSBshowList of PDB id codes | ||
| Identifiers | ||
| Aliases | GCG, GLP1, glucagon, GRPP, GLP-1, GLP2 | |
| External IDs | OMIM: 138030 HomoloGene: 136497 GeneCards: GCG | |
| hideGene location (Human)Chr.Chromosome 2 (human)[1]Band2q24.2Start162,142,882 bp[1]End162,152,404 bp[1] | ||
| hideRNA expression patternMore reference expression data | ||
| showGene ontology | ||
| Orthologs | ||
| Species | Human | Mouse |
| Entrez | 2641 | n/a |
| Ensembl | ENSG00000115263 | n/a |
| UniProt | P01275 | n/a |
| RefSeq (mRNA) | NM_002054 | n/a |
| RefSeq (protein) | NP_002045 | n/a |
| Location (UCSC) | Chr 2: 162.14 – 162.15 Mb | n/a |
| PubMed search | [2] | n/a |
| Wikidata | ||
| View/Edit Human |
///////////GLUCAGON, DIABETES, PEPTIDE, HORMONE
Naxitamab

(Heavy chain)
QVQLVESGPG VVQPGRSLRI SCAVSGFSVT NYGVHWVRQP PGKGLEWLGV IWAGGITNYN
SAFMSRLTIS KDNSKNTVYL QMNSLRAEDT AMYYCASRGG HYGYALDYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPELLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSRDEL
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
EIVMTQTPAT LSVSAGERVT ITCKASQSVS NDVTWYQQKP GQAPRLLIYS ASNRYSGVPA
RFSGSGYGTE FTFTISSVQS EDFAVYFCQQ DYSSFGQGTK LEIKRTVAAP SVFIFPPSDE
QLKSGTASVV CLLNNFYPRE AKVQWKVDNA LQSGNSQESV TEQDSKDSTY SLSSTLTLSK
ADYEKHKVYA CEVTHQGLSS PVTKSFNRGE C
(Disulfide bridge: H22-H95, H146-H202, H222-L211, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’95, H’146-H’202, H’222-L’211, H’263-H’323, H’369-H’427, L23-L88, L131-L191, L’23-L’88, L’131-L’191)
Naxitamab
ナキシタマブ;
Antineoplastic, Anti-GD2 antibody
| Formula | C6414H9910N1718O1996S44 |
|---|---|
| CAS | 1879925-92-4 |
| Mol weight | 144434.4882 |
FDA APPROVED 2020/11/25, Danyelza
FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow
On November 25, 2020, the Food and Drug Administration granted accelerated approval to naxitamab (DANYELZA, Y-mAbs Therapeutics, Inc.) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) for pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy.
Efficacy was evaluated in patients with relapsed or refractory neuroblastoma in the bone or bone marrow enrolled in two single-arm, open-label trials: Study 201 (NCT 03363373) and Study 12-230 (NCT 01757626). Patients with progressive disease following their most recent therapy were excluded. Patients received 3 mg/kg naxitamab administered as an intravenous infusion on days 1, 3, and 5 of each 4-week cycle in combination with GM-CSF subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. At the investigator’s discretion, patients were permitted to receive pre-planned radiation to the primary disease site in Study 201 and radiation therapy to non-target bony lesions or soft tissue disease in Study 12-230.
The main efficacy outcome measures were confirmed overall response rate (ORR) per the revised International Neuroblastoma Response Criteria (INRC) and duration of response (DOR). Among 22 patients treated in the multicenter Study 201, the ORR was 45% (95% CI: 24%, 68%) and 30% of responders had a DOR greater or equal to 6 months. Among 38 patients treated in the single-center Study 12-230, the ORR was 34% (95% CI: 20%, 51%) with 23% of patients having a DOR greater or equal to 6 months. For both trials, responses were observed in either the bone, bone marrow or both.
The prescribing information contains a Boxed Warning stating that naxitamab can cause serious infusion-related reactions and neurotoxicity, including severe neuropathic pain, transverse myelitis and reversible posterior leukoencephalopathy syndrome (RPLS). To mitigate these risks, patients should receive premedication prior to each naxitamab infusion and be closely monitored during and for at least two hours following completion of each infusion.
The most common adverse reactions (incidence ≥25% in either trial) in patients receiving naxitamab were infusion-related reactions, pain, tachycardia, vomiting, cough, nausea, diarrhea, decreased appetite, hypertension, fatigue, erythema multiforme, peripheral neuropathy, urticaria, pyrexia, headache, injection site reaction, edema, anxiety, localized edema, and irritability. The most common Grade 3 or 4 laboratory abnormalities (≥5% in either trial) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased platelet count, decreased potassium, increased alanine aminotransferase, decreased glucose, decreased calcium, decreased albumin, decreased sodium and decreased phosphate.
The recommended naxitamab dose is 3 mg/kg/day (up to 150 mg/day) on days 1, 3, and 5 of each treatment cycle, administered after dilution as an intravenous infusion in combination with GM-CSF, subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. Treatment cycles are repeated every 4 to 8 weeks.
View full prescribing information for DANYELZA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761171lbl.pdf
This review used the Real-Time Oncology Review (RTOR) pilot program and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted accelerated approval based on overall response rate and duration of response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.
This application was granted priority review, breakthrough therapy, and orphan drug designation. A priority review voucher was issued for this rare pediatric disease product application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
////////////Naxitamab, priority review, breakthrough therapy, orphan drug, FDA 2020, 2020 APPROVALS, Danyelza, MONOCLONAL ANTIBODY, PEPTIDE, ナキシタマブ,
Setmelanotide


Setmelanotide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
- Molecular FormulaC49H68N18O9S2
- Average mass1117.309 Da
- N-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide (2->8)-disulfide
1,2-Dithia-5,8,11,14,17,20-hexaazacyclotricosane-4-carboxamide, 22-[[(2S)-2-(acetylamino)-5-[(diaminomethylene)amino]-1-oxopentyl]amino]-10-[3-[(diaminomethylene)amino]propyl]-16-(1H-imidazol-5-ylmeth yl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-13-(phenylmethyl)-, (4R,7S,10S,13R,16S,19R,22R)- [ACD/Index Name]10011920014-72-8[RN]Imcivree [Trade name]N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-Ltryptophyl- L-cysteinamide, cyclic (2-8)-disulfideN7T15V1FUYRM-493, BIM-22493UNII-N7T15V1FUYсетмеланотид [Russian] [INN]سيتميلانوتيد [Arabic] [INN]司美诺肽 [Chinese] [INN](4R,7S,10S,13R,16S,19R,22R)-22-[[(2S)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-13-benzyl-10-[3-(diaminomethylideneamino)propyl]-16-(1H-imidazol-5-ylmethyl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-1,2-dithia-5,8,11,14,17,20-hexazacyclotricosane-4-carboxamide
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age
Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan


update Imcivree EMA APPROVED 2021/7/16
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
 |
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355



Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects.[2] In addition to reducing appetite, setmelanotide increases resting energy expenditure in both obese animals and humans.[3] Importantly, unlike certain other MC4 receptor agonists, such as LY-2112688, setmelanotide has not been found to produce increases in heart rate or blood pressure.[4]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Research
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor.[6][4] Its peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It was first discovered at Ipsen and is being developed by Rhythm Pharmaceuticals for the treatment of obesity and diabetes.[6] In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity.[7] As of December 2014, the drug is in phase II clinical trials for obesity and PWS.[6][8][9][needs update] So far, preliminary data has shown no benefit of Setmelanotide in Prader-Willi syndrome.[10]
PATENT
WO 2007008704
WO 2011060355
WO 2011060352
US 20120225816
PAPER
Journal of Medicinal Chemistry, 61(8), 3674-3684; 2018
PATENT
https://patents.google.com/patent/US9314509
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2

The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
TABLE 1 Example Mol. Wt. Mol. Wt. Purity Number (calculated) (ES-MS) (HPLC) 1 1117.3 1116.9 95.1% 2 1117.3 1116.8 99.2% 3 1280.5 1280.6 98.0% 5 1216.37 1216.20 99.9%
Preparation of Pamoate Salt of Example 1
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment for weight management for people with certain rare genetic conditions”. U.S. Food and Drug Administration (FDA) (Press release). 27 November 2020. Retrieved 27 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Kim GW, Lin JE, Blomain ES, Waldman SA (January 2014). “Antiobesity pharmacotherapy: new drugs and emerging targets”. Clinical Pharmacology and Therapeutics. 95 (1): 53–66. doi:10.1038/clpt.2013.204. PMC 4054704. PMID 24105257.
- ^ Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. (April 2015). “RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals”. The Journal of Clinical Endocrinology and Metabolism. 100 (4): 1639–45. doi:10.1210/jc.2014-4024. PMC 4399297. PMID 25675384.
- ^ Jump up to:a b Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, et al. (February 2013). “Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques”. Diabetes. 62 (2): 490–7. doi:10.2337/db12-0598. PMC 3554387. PMID 23048186.
- ^ Muniyappa R, Chen K, Brychta R, Abel B, Mullins K, Staker P, et al. (June 2014). “A Randomized, Double-Blind, Placebo-Controlled, Crossover Study to Evaluate the Effect of a Melanocortin Receptor 4 (MC4R) Agonist, RM-493, on Resting Energy Expenditure (REE) in Obese Subjects” (PDF). Endocrine Reviews. Rhythm Pharmaceuticals. 35 (3). Retrieved 2015-05-21.
- ^ Jump up to:a b c Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID 25853469.
- ^ “Obesity and Diabetes Caused by Genetic Deficiencies in the MC4 Pathway”. Rhythm Pharmaceuticals. Retrieved 2015-05-21.
- ^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID 25000213. S2CID 23198484.
- ^ “RM-493: A First-in-Class, Phase 2-Ready MC4 Agonist: A New Drug Class for the Treatment of Obesity and Diabetes”. Rhythm Pharmaceuticals. Archived from the original on 2015-06-14. Retrieved 2015-05-21.
- ^ Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother L, et al. (March 2019). “A multidisciplinary approach to the clinical management of Prader-Willi syndrome”. Molecular Genetics & Genomic Medicine. 7 (3): e514. doi:10.1002/mgg3.514. PMC 6418440. PMID 30697974.
ADDITIONAL INFORMATION
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
L-Cysteinamide, N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-, cyclic (2->8)-disulfide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
REFERENCES
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
- “Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Imcivree |
| Other names | RM-493; BIM-22493; IRC-022493; N2-Acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide, cyclic (2-8)-disulfide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 920014-72-8 |
| PubChem CID | 11993702 |
| ChemSpider | 10166169 |
| UNII | N7T15V1FUY |
| KEGG | D11927 |
| Chemical and physical data | |
| Formula | C49H68N18O9S2 |
| Molar mass | 1117.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@@H]1C(=O)N[C@H](C(=O)N[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)C)C(=O)N)Cc2c[nH]c3c2cccc3)CCCN=C(N)N)Cc4ccccc4)Cc5cnc[nH]5 | |
| InChI[hide]InChI=1S/C49H68N18O9S2/c1-26-41(70)63-37(20-30-22-55-25-59-30)46(75)64-35(18-28-10-4-3-5-11-28)44(73)62-34(15-9-17-57-49(53)54)43(72)65-36(19-29-21-58-32-13-7-6-12-31(29)32)45(74)66-38(40(50)69)23-77-78-24-39(47(76)60-26)67-42(71)33(61-27(2)68)14-8-16-56-48(51)52/h3-7,10-13,21-22,25-26,33-39,58H,8-9,14-20,23-24H2,1-2H3,(H2,50,69)(H,55,59)(H,60,76)(H,61,68)(H,62,73)(H,63,70)(H,64,75)(H,65,72)(H,66,74)(H,67,71)(H4,51,52,56)(H4,53,54,57)/t26-,33+,34+,35-,36+,37+,38+,39+/m1/s1Key:HDHDTKMUACZDAA-PHNIDTBTSA-N |
///////////Setmelanotide, FDA 2020, 2020 APPROVALS, Imcivree, Orphan, PEPTIDE, ANTIOBESITY, UNII-N7T15V1FUY, сетмеланотид , سيتميلانوتيد , 司美诺肽 , BIM 22493, RM 493
CC1C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(CSSCC(C(=O)N1)NC(=O)C(CCCN=C(N)N)NC(=O)C)C(=O)N)CC2=CNC3=CC=CC=C32)CCCN=C(N)N)CC4=CC=CC=C4)CC5=CN=CN5
Isatuximab
(A chain)
QVQLVQSGAE VAKPGTSVKL SCKASGYTFT DYWMQWVKQR PGQGLEWIGT IYPGDGDTGY
AQKFQGKATL TADKSSKTVY MHLSSLASED SAVYYCARGD YYGSNSLDYW GQGTSVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(B chain)
QVQLVQSGAE VAKPGTSVKL SCKASGYTFT DYWMQWVKQR PGQGLEWIGT IYPGDGDTGY
AQKFQGKATL TADKSSKTVY MHLSSLASED SAVYYCARGD YYGSNSLDYW GQGTSVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(C chain)
DIVMTQSHLS MSTSLGDPVS ITCKASQDVS TVVAWYQQKP GQSPRRLIYS ASYRYIGVPD
RFTGSGAGTD FTFTISSVQA EDLAVYYCQQ HYSPPYTFGG GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(D chain)
DIVMTQSHLS MSTSLGDPVS ITCKASQDVS TVVAWYQQKP GQSPRRLIYS ASYRYIGVPD
RFTGSGAGTD FTFTISSVQA EDLAVYYCQQ HYSPPYTFGG GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: A22-A96, A147-A203, A223-C214, A229-B229, A232-B232, A264-A324, A370-A428, B22-B96, B147-B203, B223-D214, B264-B324, B370-B428, C23-C88, C134-C194, D23-D88, D134-D194)
Isatuximab
イサツキシマブ (遺伝子組換え)
APPROVED USFDA 2020/3/2, Sarclisa
EU APPROVED 2020/5/30
JAPAN APPROVED 2020/6/29
CAS 1461640-62-9
| Antineoplastic, Anti-CD38 antibody | |
| Disease | Multiple myeloma |
|---|
Isatuximab, sold under the brand name Sarclisa, is a monoclonal antibody (mAb) medication for the treatment of multiple myeloma.[4][3]
The most common side effects include neutropenia (low levels of neutrophils, a type of white blood cell), infusion reactions, pneumonia (infection of the lungs), upper respiratory tract infection (such as nose and throat infections), diarrhoea and bronchitis (inflammation of the airways in the lungs).[3]
Isatuximab is an anti-CD38 mAb intended to treat relapsed or refractory multiple myeloma.[5] It entered in Phase II trials for multiple myeloma[6] and T-cell leukemia in 2015.[7]
Medical uses
In the United States it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with multiple myeloma who have received at least two prior therapies including lenalidomide and a proteasome inhibitor.[8][9][10]
In the European Union it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with relapsed and refractory multiple myeloma (MM) who have received at least two prior therapies including lenalidomide and a proteasome inhibitor (PI) and have demonstrated disease progression on the last therapy.[3]
History
It was granted orphan drug designation for multiple myeloma by the European Medicines Agency (EMA) in April 2014, and by the U.S. Food and Drug Administration (FDA) in December 2016.[3][11]
Researchers started a Phase I study with isatuximab in combination with pomalidomide and dexamethasone for the treatment of patients with multiple myeloma (MM). The results during the Phase I trial showed that 26 out of the 45 patients discontinued the treatment due to progression of the disease. The patients had already been heavily pretreated. The latter lead to a manageable safety profile where the dose of isatuximab in combination with pomalidomide and dexamethasone would be capped to the maximum of 10 mg/kg weekly every two weeks for future studies.[12]
Based on the remarkable findings during the Phase I trial, a Phase II trial was launched where researchers investigated isatuximab as a single agent in patients with MM. The heavily pretreated patients reacted well to the single administration of isatuximab during Phase II of the trial.[13]
A Phase III combination trial for plasma cell myeloma is comparing pomalidomide and dexamethasone with and without isatuximab is in progress with an estimated completion date of 2021.[medical citation needed]
Additionally, two Phase III trials were added in 2017. The first trial highlights whether there is an added value in the combination of isatuximab with bortezomib, lenalidomide and dexamethasone. The latter will be tested in patients with newly diagnosed MM who are not qualified for a transplant (IMROZ trial). The second trial evaluates the combinations of isatuximab with carfilzomib and dexamethasone compared to carfilzomib with dexamethasone. The second trial was designed for patients who were previously treated with one to three prior lines (IKEMA). There is currently[when?] no treatment for MM, however promising improvements have been made and the study is still ongoing.[14][15]
In March 2020, it was approved for medical use in the United States.[8][9][10]
The U.S. Food and Drug Administration (FDA) approved isatuximab-irfc in March 2020, based on evidence from a clinical trial (NCT02990338) of 307 subjects with previously treated multiple myeloma.[10] The trial was conducted at 102 sites in Europe, North America, Asia, Australia and New Zealand.[10]
The trial evaluated the efficacy and side effects of isatuximab-irfc in subjects with previously treated multiple myeloma.[10] Subjects were randomly assigned to receive either isatuximab-irfc (in combination with pomalidomide and low-dose dexamethasone) or active comparator (pomalidomide and low-dose dexamethasone).[10] Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity.[10] Both subjects and health care providers knew which treatment was given.[10] The trial measured the time patients lived without the cancer growing (progression-free survival or PFS).[10]
It was approved for medical use in the European Union in May 2020.[3]
Structure and reactivity
The structure of isatuximab consists of two identical immunoglobulin kappa light chains and also two equal immunoglobulin gamma heavy chains. Chemically, isatuximab is similar to the structure and reactivity of daratumumab, hence both drugs show the same CD38 targeting. However, isatuximab shows a more potent inhibition of its ectozyme function. The latter gives potential for some non-cross reactivity. Isatuximab shows action of an allosteric antagonist with the inhibition of the CD38 enzymatic activity. Additionally, isatuximab shows potential where it can induce apoptosis without cross linking.[16] Lastly, Isatuximab reveals direct killing activity when a larger increase in apoptosis is detected in CD38 expressing cancer cells. Furthermore, isatuximab demonstrated a dose dependent inhibition of CD38 enzymatic activity. However, daratumumab with the same experimental conditions shows a more limited inhibition without a dose response.[17]
Reactions
Isatuximab binds uniquely to an epitope on the CD38 receptor and is the only CD38 antibody which can start apoptosis directly.[18] Isatuximab binds to a different CD38 epitope amino-acid sequence than does the anti-CD38 monoclonal antibody daratumumab.[19] The binding with the CD38 receptor is mainly via the gamma heavy chains and are more potent than other CD38 antibodies such as daratumumab which can inhibit the enzymatic activity of CD38. Moreover, isatuximab inhibits the hydrolase activity of CD38.[medical citation needed]
The antibodies show signs of improving antitumor immunity by eliminating regulatory T cells, B cells and myeloid-derived suppressor cells. The difference in binding between isatuximab and daratumumab is in the recognition of the different amino acid groups. Isatuximab identifies 23 amino acids of CD38 to the contrary with daratumumab who has 27. The residue of Glu233 has a flexible sidechain and faces the N-terminal of Asp1 residue in the isatuximab light chain. The latter light chain of isatuximab is also flexible which makes the interaction between CD38/Glu233 and the Asp1 weaker than the other interactions between CD38 and isatuximab. The caspase-dependent apoptotic pathway and the lysosomal mediated cell death pathway in MM cells is induced by isatuximab. The MM cell death follows the downstream reactions of the lysosomal activation. The latter also activates the production of reactive oxygen species.[20]
Available forms
Isatuximab or isatuximab-irfc is available as a drug in an intravenous infusion form. Injection doses are 100 mg/5 mL (20 mg/mL) solution in single-dose vial or 500 mg/25 mL (20 mg/mL) solution in single-dose vial.[4]
Mechanism of action
Cancer of the blood that is distinguished by an overproduction of malignant plasma cells in the bone marrow is called multiple myeloma. The myeloma cells are marked with uniformed overexpression of CD38 surface glycoproteins. Although these proteins are also expressed on other myeloid and lymphoid cells, the extent is relatively minor compared to myeloma cells. The fact that CD38 glycoproteins carry out various important cellular functions, and that they are plentiful on the surface of myeloma cells, has made them an appealing target for multiple myeloma treatment.[21] CD38 was first described as an activation marker, but later the molecule displayed functions in adhesion to endothelial CD31 proteins, e.g. as an aiding component of the synapse complex, as well as an ectoenzyme implicated in the metabolism of extracellular NAD+ and cytoplasmic NADP. The tumour cells can evade the immune system, possibly due to adenosine, an immunosuppressive molecule that arises as a product of the ectoenzymatic activity of CD38.[22]
Isatuximab-irfc is an IgG1-derived monoclonal antibody that selectively binds to the CD38 that exists on the exterior of hematopoietic and multiple myeloma cells (as well as other tumor cells). This drug induces apoptosis of tumor cells and activates immune effector mechanisms such as complement dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC). Isatuximab-irfc is able to stimulate natural killer (NK) cells in the absence of CD38-positive target tumor cells and blocks CD38-positive T-regulatory cells.[4] Furthermore, the NADase activity of CD38 is adjusted by isatuximab, similarly to other CD38 antibodies. Contrarily to daratumumab however, isatuximab can incite apoptosis directly without cross-linking, and in its binding epitope.[23] According to the FDA, isatuximab-irfc alone has reduced ADCC and direct tumor cell killing activity in vitro in comparison to when it is combined with pomalidomide. As well as increased anti-tumor activity as opposed to isatuximab-irfc or pomalidomide only in a human multiple myeloma xenograft model.[4]
Metabolism and toxicity
Metabolism
Isatuximab-irfc is likely to be metabolized through catabolic pathways into smaller peptides. When isatuximab is at a constant state it is expected that the ≥99% elimination will occur approximately two months after the last dose was administered. The clearance percentage diminished when the dosages were increased over time, as well as when multiple doses were administered. However, the elimination of isatuximab-irfc did not differ when applied as a single agent or as a combination therapy.[4]
Toxicity
A dose-limiting toxicity (DLT) has characterized been characterized as the development of any of the following: grade ≥ 3 non-hematologic toxicity; grade 4 neutropenia or grade 4 thrombocytopenia lasting more than 5 days; grade ≥ 2 allergic reactions or hypersensitivity (i.e., infusion reactions); or any other toxicity considered to be dose-limiting by the investigators or sponsor. Grade ≤ 2 infusion reactions were excluded from the DLT definition, because, with suitable care, patients that suffered a grade 2 infusion reaction prior to completion of the infusion were able to finalize isatuximab administration.[23]
There is no recommended reduced dose of isatuximab-irfc. In the eventuality of hematological toxicity it may be necessary to delay administration so that the blood count may be recovered.[4] Although there is no counteracting agent for isatuximab, clinical experience with overdoses is seemingly nonexistent as well. Overdose symptoms will probably be in line with the side effects attached to isatuximab. Therefore, infusion reactions, gastrointestinal disturbances and an elevated risk of infections may occur. It is necessary to carefully monitor the patient in case of an overdose and to employ clinically indicated symptomatic and supportive procedures.[21]
No studies have been conducted with isatuximab concerning carcinogenicity, genotoxicity or fertility.[4]
Pregnancy
When given to pregnant women isatuximab-irfc can cause fetal injury, due to the mechanism of action. It can precipitate depletion of immune cells as well as decreased bone density in the fetus. Pregnant women are therefore notified of the potential risks to a fetus, and women that are able to reproduce are advised to use effective contraceptives during treatment and at least five months subsequent to the last dose of isatuximab-irfc.
Furthermore, it is not recommended to combine isatuximab-irfc with pomalidomide in women that are carrying a child, because pomalidomide may cause birth defects and death of the unborn child.[4]
Indications
Isatuximab is indicated as a CD38-directed cytolytic antibody. By inhibiting the enzymatic activity of CD38.
The binding of isatuximab to CD38 on multiple myeloma (MM) cells leads to a trigger to several mechanisms leading to direct apoptosis of target cancer cells. The triggered pathways are the caspase-dependent apoptotic and the lysosome-mediated cell death pathway in MM cells.[24]
It is used in a combination with dexamethasone and pomalidomide. The drug is thus to treat patients with multiple myeloma. Restrictions for the use of isatuximab is that the patients have to be adults who have at least received two previous treatments with lenalidomide and a proteasome inhibitor.[4]
Isatuximab is currently[when?] also being tested in a Phase II trial as a monotherapy against refractory/recurrent systemic light-chain amyloidosis.[24]
Efficacy and side effects
Efficacy
A Phase III study of patients with refractory and relapsed MM, who were resistant to lenalidomide and a proteasome inhibitor, and could not have received daratumumab, another anti-CD38 monoclonal antibody was published in 2019 (ICARIA-MM). The addition of isatuximab to pomalidomide and dexamethasone improved progression free survival to 11.5 months compared to 6.5 months, with an overall response rate of 63%.[25]
Side effects
Adverse reactions to isatuximab-irfc may include neutropenia, infusion-related reactions and/or secondary primary malignancies.[4] Of these three the most commonly occurring ones are the infusion-related reactions.[24] Examples of the most frequent symptoms of infusion-related reactions are dyspnea, cough, chills, and nausea, while the severest signs and symptoms included hypertension and dyspnea.[4]
Effects on animals
The activity of isatuximab has been researched in mouse tumor models. It has been proven that isatuximab leads to antitumor activity in MM cells. Furthermore, the combination of isatuximab and pomalidomide will lead to an extra enhanced antitumor activity in MM cells. Thus, pomalidomide in vivo and in vitro leads to an increase of the activity of isatuximab.[24]
Animal studies in reproduction toxicity have not yet been carried out. So, the risks of birth defects and miscarriage risks are unknown.[4]
Names
Isatuximab is the United States Adopted Name (USAN).[26]
It was developed by ImmunoGen and Sanofi-Aventis with the development name SAR-650984.
References
- ^ Jump up to:a b “Sarclisa Australian prescription medicine decision summary”. Therapeutic Goods Administration (TGA). 14 May 2020. Retrieved 16 August 2020.
- ^ “Isatuximab (Sarclisa) Use During Pregnancy”. Drugs.com. 25 March 2020. Retrieved 25 June 2020.
- ^ Jump up to:a b c d e f “Sarclisa EPAR”. European Medicines Agency (EMA). 24 March 2020. Retrieved 25 June 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h i j k l “Sarclisa- isatuximab injection, solution, concentrate”. DailyMed. 2 March 2020. Retrieved 26 March 2020.
- ^ ImmunoGen, Inc. Announces Data Presentations at Upcoming 57th ASH Annual Meeting and Exposition
- ^ Martin T (2015). “A Dose Finding Phase II Trial of Isatuximab (SAR650984, Anti-CD38 mAb) As a Single Agent in Relapsed/Refractory Multiple Myeloma”. Blood. 126 (23): 509. doi:10.1182/blood.V126.23.509.509.
- ^ “Safety and Efficacy of Isatuximab in Lymphoblastic Leukemia”. ClinicalTrials.gov. Retrieved 4 March 2020.
- ^ Jump up to:a b “FDA approves isatuximab-irfc for multiple myeloma”. U.S. Food and Drug Administration (FDA). 2 March 2020. Retrieved 2 March 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “FDA Approves New Therapy for Patients with Previously Treated Multiple Myeloma”. U.S. Food and Drug Administration (FDA) (Press release). 2 March 2020. Retrieved 4 March 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h i “Drug Trials Snapshots: Sarclisa”. U.S. Food and Drug Administration(FDA). 2 March 2020. Retrieved 25 March 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Isatuximab Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 4 March 2020.
- ^ Mikhael J, Richardson P, Usmani SZ, Raje N, Bensinger W, Karanes C, et al. (July 2019). “A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma”. Blood. 134 (2): 123–133. doi:10.1182/blood-2019-02-895193. PMC 6659612. PMID 30862646.
- ^ Martin T (7 December 2015). “A Dose Finding Phase II Trial of Isatuximab (SAR650984, Anti-CD38 mAb) As a Single Agent in Relapsed/Refractory Multiple Myeloma”. ASH.
- ^ Orlowski RZ, Goldschmidt H, Cavo M, Martin TG, Paux G, Oprea C, Facon T (20 May 2018). “Phase III (IMROZ) study design: Isatuximab plus bortezomib (V), lenalidomide (R), and dexamethasone (d) vs VRd in transplant-ineligible patients (pts) with newly diagnosed multiple myeloma (NDMM)”. Journal of Clinical Oncology. 36 (15_suppl): TPS8055. doi:10.1200/JCO.2018.36.15_suppl.TPS8055.
- ^ Moreau P, Dimopoulos MA, Yong K, Mikhael J, Risse ML, Asset G, Martin T (January 2020). “Isatuximab plus carfilzomib/dexamethasone versus carfilzomib/dexamethasone in patients with relapsed/refractory multiple myeloma: IKEMA Phase III study design”. Future Oncology. 16 (2): 4347–4358. doi:10.2217/fon-2019-0431. PMID 31833394.
- ^ Rajan AM, Kumar S (July 2016). “New investigational drugs with single-agent activity in multiple myeloma”. Blood Cancer Journal. 6 (7): e451. doi:10.1038/bcj.2016.53. PMC 5030378. PMID 27471867.
- ^ Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. (June 2017). “A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma”. Blood. 129 (25): 3294–3303. doi:10.1182/blood-2016-09-740787. PMC 5482100. PMID 28483761.
- ^ Martin TG, Corzo K, Chiron M (2019). “Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab”. Cells. 8 (12): 1522. doi:10.3390/cells8121522. PMC 6953105. PMID 31779273.
- ^ Dhillon S (2020). “Isatuximab: First Approval”. Drugs. 80 (9): 905–912. doi:10.1007/s40265-020-01311-1. PMID 32347476. S2CID 216597315.
- ^ Martin TG, Corzo K, Chiron M, Velde HV, Abbadessa G, Campana F, et al. (November 2019). “Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab”. Cells. 8 (12): 1522. doi:10.3390/cells8121522. PMC 6953105. PMID 31779273.
- ^ Jump up to:a b “Isatuximab”. Drugbank. 20 May 2019.
- ^ Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V, Malavasi F (28 November 2018). “CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma”. Frontiers in Immunology. 9: 2722. doi:10.3389/fimmu.2018.02722. PMC 6279879. PMID 30546360.
- ^ Jump up to:a b Martin T, Strickland S, Glenn M, Charpentier E, Guillemin H, Hsu K, Mikhael J (March 2019). “Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma”. Blood Cancer Journal. 9 (4): 41. doi:10.1038/s41408-019-0198-4. PMC 6440961. PMID 30926770.
- ^ Jump up to:a b c d Martin TG, Corzo K, Chiron M, Velde HV, Abbadessa G, Campana F, et al. (November 2019). “Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab”. Cells. 8 (12): 1522. doi:10.3390/cells8121522. PMC 6953105. PMID 31779273.
- ^ Attal, Michel; Richardson, Paul G; Rajkumar, S Vincent; San-Miguel, Jesus; Beksac, Meral; Spicka, Ivan; Leleu, Xavier; Schjesvold, Fredrik; Moreau, Philippe; Dimopoulos, Meletios A; Huang, Jeffrey Shang-Yi (2019). “Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study”. The Lancet. 394 (10214): 2096–2107. doi:10.1016/s0140-6736(19)32556-5. ISSN 0140-6736. PMID 31735560. S2CID 208049235.
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Isatuximab, American Medical Association
External links
- “Isatuximab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02990338 for “Multinational Clinical Study Comparing Isatuximab, Pomalidomide, and Dexamethasone to Pomalidomide and Dexamethasone in Refractory or Relapsed and Refractory Multiple Myeloma Patients (ICARIA-MM)” at ClinicalTrials.gov
| Isatuximab (pale blue) binding CD38 (purple). PDB: 4CMH | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Chimeric (mouse/human) |
| Target | CD38 |
| Clinical data | |
| Trade names | Sarclisa |
| Other names | SAR-650984, isatuximab-irfc |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620023 |
| License data | US DailyMed: Sarclisa |
| Pregnancy category | AU: C[1]US: N (Not classified yet)[2] |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-onlyEU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1461640-62-9 |
| DrugBank | DB14811 |
| ChemSpider | none |
| UNII | R30772KCU0 |
| KEGG | D11050 |
| Chemical and physical data | |
| Formula | C6456H9932N1700O2026S44 |
| Molar mass | 145190.99 g·mol−1 |
////////Isatuximab, Sarclisa, 2020APPROVALS, JAPAN 2020, US 2020, EU 2020, PEPTIDE, SANOFI , イサツキシマブ (遺伝子組換え) ,
Bulevirtide acetate

Bulevirtide acetate
(N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt.
molecular formula C248H355N65O72,
molecular mass is 5398.9 g/mol
ブレビルチド酢酸塩;
APROVED 2020/7/31, EU, Hepcludex
MYR GmbH
|
Antiviral, Entry inhibitor
|
|
| Disease |
Hepatitis delta virus infection
|
|---|
Bulevirtide is a 47-amino acid peptide with a fatty acid, a myristoyl residue, at the N-terminus and an amidated C-terminus. The active substance is available as acetate salt. The counter ion acetate is bound in ionic form to basic groups of the peptide molecule and is present in a non-stoichiometric ratio. The chemical name of bulevirtide is (N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt. It corresponds to the molecular formula C248H355N65O72, its relative molecular mass is 5398.9 g/mol
Bulevirtide appears as a white or off-white hygroscopic powder. It is practically insoluble in water and soluble at concentrations of 1 mg/ml in 50% acetic acid and about 7 mg/ml in carbonate buffer solution at pH 8.8, respectively. The structure of the active substance (AS) was elucidated by a combination of infrared spectroscopy (IR), mass spectrometry (MS), amino acid analysis and sequence analysis Other characteristics studied included ultraviolet (UV) spectrum, higher order structure (1D- and 2D- nuclear magnetic resonance spectroscopy (NMR)) and aggregation (Dynamic Light Scattering). Neither tertiary structure nor aggregation states of bulevirtide have been identified. With regard to enantiomeric purity, all amino acids are used in L-configuration except glycine, which is achiral by nature. Two batches of bulevirtide acetate were evaluated for enanatiomeric purity and no relevant change in configuration during synthesis was detected.
Bulevirtide is manufactured by a single manufacturer. It is a chemically synthesised linear peptide containing only naturally occurring amino acids. The manufacturing of this peptide is achieved using standard solidphase peptide synthesis (SPPS) on a 4-methylbenzhydrylamine resin (MBHA resin) derivatised with Rink amide linker in order to obtain a crude peptide mixture. This crude mixture is purified through a series of washing and preparative chromatography steps. Finally, the purified peptide is freeze-dried prior to final packaging and storage. The process involves further four main steps: synthesis of the protected peptide on the resin while side-chain functional groups are protected as applicable; cleavage of the peptide from the resin, together with the removal of the side chain protecting groups to obtain the crude peptide; purification; and lyophilisation. Two chromatographic systems are used for purification. No design space is claimed. Resin, Linker Fmoc protected amino acids and myristic acid are starting materials in line with ICH Q11. Sufficient information is provided on the source and the synthetic route of the starting materials. The active substance is obtained as a nonsterile, lyophilised powder. All critical steps and parameters were presented and clearly indicated in the description of the manufacturing process. The process description includes also sufficient information on the type of equipment for the SPPS, in-process controls (IPCs). The circumstances under which reprocessing might be performed were clearly presented. No holding times are proposed. Overall the process is sufficiently described.
The finished product is a white to off white lyophilised powder for solution for injection supplied in single-use vials. Each vial contains bulevirtide acetate equivalent to 2 mg bulevirtide. The composition of the finished product was presented. The powder is intended to be dissolved in 1 ml of water for injection per vial. After reconstitution the concentration of bulevirtide net peptide solution in the vial is 2 mg/ml. The components of the formulation were selected by literature review and knowledge of compositions of similar products available on the market at that time, containing HCl, water, mannitol, sodium carbonate, sodium hydrogen carbonate and sodium hydroxide. All excipients are normally used in the manufacture of lyophilisates. The quality of the excipients complies with their respective Ph. Eur monographs. The intrinsic properties of the active substance and the compounding formulation do not support microbiological growth as demonstrated by the stability data. No additional preservatives are therefore needed.
Hepcludex is an antiviral medicine used to treat chronic (long-term) hepatitis delta virus (HDV) infection in adults with compensated liver disease (when the liver is damaged but is still able to work), when the presence of viral RNA (genetic material) has been confirmed by blood tests.
HDV is an ‘incomplete’ virus, because it cannot replicate in cells without the help of another virus, the hepatitis B virus. Because of this, patients infected with the virus always also have hepatitis B.
HDV infection is rare, and Hepcludex was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 19 June 2015. For further information on the orphan designation, see EU/3/15/1500.
Hepcludex contains the active substance bulevirtide.
Bulevirtide, sold under the brand name Hepcludex, is an antiviral medication for the treatment of chronic hepatitis D (in the presence of hepatitis B).[2]
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[2]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[2] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[2]
Bulevirtide was approved for medical use in the European Union in July 2020.[2]
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[2][3]
Pharmacology
Mechanism of action
Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking both viruses from entering hepatocytes.[4]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[5][6] that can also dock to NTCP, blocking the virus’s entry mechanism.[7]
The drug is also effective against hepatitis D because the hepatitis D virus is only infective in the presence of a hepatitis B virus infection.[7]
References
- ^ Deterding, K.; Wedemeyer, H. (2019). “Beyond Pegylated Interferon-Alpha: New Treatments for Hepatitis Delta”. Aids Reviews. 21 (3): 126–134. doi:10.24875/AIDSRev.19000080. PMID 31532397.
- ^ Jump up to:a b c d e f g “Hepcludex EPAR”. European Medicines Agency (EMA). 26 May 2020. Retrieved 12 August 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Summary of opinion: Hepcludex” (PDF). European Medicines Agency. 28 May 2020.
- ^ Francisco, Estela Miranda (29 May 2020). “Hepcludex”. European Medicines Agency. Retrieved 6 August 2020.
- ^ Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, et al. (May 2013). “The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus”. Journal of Hepatology. 58 (5): 861–7. doi:10.1016/j.jhep.2012.12.008. PMID 23246506.
- ^ Abbas Z, Abbas M (August 2015). “Management of hepatitis delta: Need for novel therapeutic options”. World Journal of Gastroenterology. 21 (32): 9461–5. doi:10.3748/wjg.v21.i32.9461. PMC 4548107. PMID 26327754.
- ^ Jump up to:a b Spreitzer H (14 September 2015). “Neue Wirkstoffe – Myrcludex B”. Österreichische Apothekerzeitung (in German) (19/2015): 12.
External links
- “Bulevirtide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Hepcludex |
| Other names | MyrB, Myrcludex-B[1] |
| License data | |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
/////////Bulevirtide acetate, ブレビルチド酢酸塩 , orphan designation, MYR GmbH, PEPTIDE, EU 2020, 2020 APPROVALS
Imlifidase
MDSFSANQEI RYSEVTPYHV TSVWTKGVTP PANFTQGEDV FHAPYVANQG WYDITKTFNG
KDDLLCGAAT AGNMLHWWFD QNKDQIKRYL EEHPEKQKIN FNGEQMFDVK EAIDTKNHQL
DSKLFEYFKE KAFPYLSTKH LGVFPDHVID MFINGYRLSL TNHGPTPVKE GSKDPRGGIF
DAVFTRGDQS KLLTSRHDFK EKNLKEISDL IKKELTEGKA LGLSHTYANV RINHVINLWG
ADFDSNGNLK AIYVTDSDSN ASIGMKKYFV GVNSAGKVAI SAKEIKEDNI GAQVLGLFTL
STGQDSWNQT N
Imlifidase
イムリフィダーゼ;
| Formula |
C1575H2400N422O477S6
|
|---|---|
| CAS |
1947415-68-0
|
| Mol weight |
35070.8397
|
EMA APPROVED, 2020/8/25, Idefirix
Pre-transplant treatment to make patients with donor specific IgG eligible for kidney transplantation
Immunosuppressant, Immunoglobulin modulator (enzyme)
Imlifidase is under investigation in clinical trial NCT02854059 (IdeS in Asymptomatic Asymptomatic Antibody-Mediated Thrombotic Thrombocytopenic Purpura (TTP) Patients).
Imlifidase, brand name Idefirix, is a medication for the desensitization of highly sensitized adults needing kidney transplantation, but unlikely to receive a compatible transplant.[1]
Imlifidase is a cysteine protease derived from the immunoglobulin G (IgG)‑degrading enzyme of Streptococcus pyogenes.[1] It cleaves the heavy chains of all human IgG subclasses (but no other immunoglobulins), eliminating Fc-dependent effector functions, including CDC and antibody-dependent cell-mediated cytotoxicity (ADCC).[1] Thus, imlifidase reduces the level of donor specific antibodies, enabling transplantation.[1]
The benefits with imlifidase are its ability to convert a positive crossmatch to a negative one in highly sensitized people to allow renal transplantation.[1] The most common side effects are infections and infusion related reactions.[1]
In June 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended the approval of Imlifidase.[1][2]
Medical uses
Per the CHMP recommendation, imlifidase will be indicated for desensitization treatment of highly sensitized adult kidney transplant people with positive crossmatch against an available deceased donor.[1] The use of imlifidase should be reserved for people unlikely to be transplanted under the available kidney allocation system including prioritization programmes for highly sensitized people.[1]
History
Imlifidase was granted orphan drug designations by the European Commission in January 2017, and November 2018,[3][4] and by the U.S. Food and Drug Administration (FDA) in both February and July 2018.[5][6]
In February 2019, Hansa Medical AB changed its name to Hansa Biopharma AB.[4]
| PHASE | STATUS | PURPOSE | CONDITIONS | COUNT |
|---|---|---|---|---|
| 2 | Recruiting | Treatment | Anti-Glomerular Basement Membrane Disease | 1 |
| 2 | Recruiting | Treatment | Guillain-Barré Syndrome (GBS) | 1 |
| 2 | Recruiting | Treatment | Kidney Transplant Rejection | 1 |
| 2 | Terminated | Treatment | Thrombotic Thrombocytopenic Purpura (TTP) | 1 |
| Not Available | Recruiting | Not Available | Kidney Transplant Failure and Rejection | 1 |
References
- ^ Jump up to:a b c d e f g h i “Imlifidase: Pending EC decision”. European Medicines Agency (EMA). 25 June 2020. Retrieved 26 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “New treatment to enable kidney transplant in highly sensitised patients”. European Medicines Agency (Press release). 26 June 2020. Retrieved 26 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “EU/3/16/1826”. European Medicines Agency (EMA). 12 January 2017. Retrieved 27 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “EU/3/18/2096”. European Medicines Agency (EMA). 13 February 2019. Retrieved 27 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Imlifidase Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 3 July 2018. Retrieved 27 June 2020.
- ^ “Imlifidase Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 14 February 2018. Retrieved 27 June 2020.
Further reading
- Ge S, Chu M, Choi J, Louie S, Vo A, Jordan SC, et al. (October 2019). “Imlifidase Inhibits HLA Antibody-Mediated NK Cell Activation and Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) In Vitro”. Transplantation. doi:10.1097/TP.0000000000003023. PMID 31644495.
- Lin J, Boon L, Bockermann R, Robertson AK, Kjellman C, Anderson CC (March 2020). “Desensitization using imlifidase and EndoS enables chimerism induction in allosensitized recipient mice”. Am. J. Transplant. doi:10.1111/ajt.15851. PMID 32185855.
- Lonze BE, Tatapudi VS, Weldon EP, Min ES, Ali NM, Deterville CL, et al. (September 2018). “IdeS (Imlifidase): A Novel Agent That Cleaves Human IgG and Permits Successful Kidney Transplantation Across High-strength Donor-specific Antibody”. Ann. Surg. 268 (3): 488–496. doi:10.1097/SLA.0000000000002924. PMID 30004918.
- Lorant T, Bengtsson M, Eich T, Eriksson BM, Winstedt L, Järnum S, et al. (November 2018). “Safety, immunogenicity, pharmacokinetics, and efficacy of degradation of anti-HLA antibodies by IdeS (imlifidase) in chronic kidney disease patients”. Am. J. Transplant. 18 (11): 2752–2762. doi:10.1111/ajt.14733. PMC 6221156. PMID 29561066.
External links
- “Imlifidase”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | im lif’ i dase |
| Trade names | Idefirix |
| Other names | HMED-IdeS |
| Routes of administration |
Intravenous |
| ATC code | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C1575H2400N422O477S6 |
| Molar mass | 35071.36 g·mol−1 |
//////////Imlifidase, Idefirix, PEPTIDE, イムリフィダーゼ , 2020 APPROVALS, EMA 2020, EU 2020

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}