
Home » GENERIC DRUG (Page 7)
Category Archives: GENERIC DRUG
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Veterinary- Atipamezole
Atipamezole

4-(2-Ethyl-1,3-dihydroinden-2-yl)-3H-imidazole, Atipamezole, cas 104054-27-5
hydrochloride cas no 104075-48-1
- MPV 1248 (IS: FarmosGroupLt)
- UNII-03N9U5JAF6 (IS)
- UNII-2W4279571X (IS)
Atipamezole is a synthetic alpha2-adrenergic antagonist, indicated for the reversal of the sedative and analgesic effects of dexmedetomidine and medetomidine in dogs. It has also been researched in humans as a potential anti-Parkinsonian drug.Atipamezole is more potent than yohimbine; it is very selective for alpha2-adrenergic vs alpha1sites, but not selelctive for alpha2 – subtypes.
Atipamezole (brand name Antisedan, Pfizer) is a synthetic alpha2–adrenergic antagonist, indicated for the reversal of the sedative and analgesic effects of dexmedetomidine andmedetomidine in dogs.[1] It has also been researched in humans as a potential anti-Parkinsonian drug.[2]
- Pfizer Animal Health ANTISEDAN Product Overview
- Pertovaara A, Haapalinna A, Sirviö J, Virtanen R (2005). “Pharmacological properties, central nervous system effects, and potential therapeutic applications of atipamezole, a selective alpha2-adrenoceptor antagonist”. CNS Drug Reviews 11 (3): 273–88.doi:10.1111/j.1527-3458.2005.tb00047.x. PMID 16389294.

- ANTISEDAN product information page (Pfizer Animal Health)
- Drugs Future, vol. 15, no. 5, 1990, “Atipamezole“, pages 448-452, see page 449
Synonyms
1H-imidazole, 4-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-
1H-imidazole, 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-
5-(2-Ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole
Atipamezole
4-(2-Ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole
4-(2-Ethyl-2-indanyl)imidazole
4-(2-Ethyl-indan-2-yl)-1H-imidazole(Atipamezole)
4-(2-ethylindan-2-yl)imidazole
Antisedan
Antisedan
Atipamezol
Atipamezolum
Atipamezole Hydrochloride CAS 104075-48-1
………………………………
Atipamezole is a selective alpha2 – adrenoceptor antagonist which is currently marketed under the trademark Antisedan® for the reversal of sedative- analgesic veterinary drugs. Atipamezole has been disclosed e.g. in the European Patent EP 183492 as useful for the reversal of detomidine. European Patent EP 0589957 discloses the use of atipamezole for the treatment of male sexual impotence. In US 4698692 the use of atipamezole for the attenuation of ethyl alcohol intoxication is disclosed.
US Patent No. US6543389 discloses insecticidal pet collars for dogs comprising amitraz and atipamezole. Atipamezole in the collar provides amelioration of amitraz toxicosis in combination with the amitraz in case the dogs ingests the collar. The pet collar comprises 0.01 to 1%, preferably 0.1 to 1 %, by weight of atipamezole. Safe, effective ways to eliminate ectoparasites are desired for the companion animal’s well-being, for the well-being and comfort of its human associate and for the prevention of losses in livestock
A substantial amount of work has been devoted to identifying the neurotransmitters involved in the facilitation and inhibition of male sexual behaviour (see e.g. Bitran and Hull 1987, Neuroscience and Behavioral reviews 11 , 365-389). Noradrenergic neuro-transmission seems to have an important role.
Atipamezole is a selective and potent a2*-adrenoceptor antagonist which is currently marketed for the reversal of sedative-analgesic veterinary drugs. Atipamezole has been disclosed e.g. in the European Patent EP 183492 as useful for the reversal of detomidine.
We have now found that this compound is also very effective in increasing male sexual capacity in a monkey model. These findings suggest that atipamezole would be an effective therapy in male impotence in humans as well.
Another a2-adrenoreceptor antagonist, yohimbine, is currently used for the treatment of male impotence. Yohimbine increases noradrenergic neurotransmission and has been reported to facilitate the sexual capacity of male animals, although the results of different studies are conflicting.
Atipamezole is, however clearly advantageous over yohimbine for this use because of its excellent selectivity. The a2/a-|selectivity ratio of atipamezole is
200-300 times higher than that of yohimbine.
- EP 0310745 B (FARMOS OY) 1989.04.12. disclosed preparation of 5-(2-ethyl-2,3-dihydro-1 H-inden-2-yl)-1 H-imidazole salt by two synthetic routes.
-
First synthetic route as starting material was used 2-acetyl-1-indanone, which was alkylated with ethylbromide in acetone in the presence of sodium carbonate to 2-acetyl-2-ethyl-1-indanone. The acetyl group was brominated with bromine in methanol and to imidazole by heating in formamide. Then the intermediate was hydrogenated in 2N hydrochloric acid in the presence of 10% palladium on carbon.
-
Second synthetic route disclosed in the same patent is following, as starting material was used 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester, which was prepared by methylation of 2,3-dihydro-1H-indene-2-carboxylic acid in the presence of sulphuric acid. The 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester was reacted with N-isopropylcyclohexylamide and ethylbromide yielding 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid, then thionyl chloride was added and 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride was obtained. In the next step ethoxymagnesiummalonic acid ethyl ester in dry ether was added to 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride and reaction mixture was treated with sulphuric acid, and 1-(2,3-dihydro-2-ethyl-1H-inden-2-yl)ethanone was obtained, then the intermediate was stirred in methylene chloride and bromine was added by giving a new intermediate 2-bromo-1-(2,3-dihydro-2-methyl-1H-inden-2-yl)ethanone, to which was thereafter added formamide and hydrochloric acid yielding crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole. The last step involved hydrogenation of the crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1 H-imidazole with 10% palladium on carbon.
-
EP 0247764 B (ORION-YHTYMÄ OY) 1987.02.12. disclosed the following process for preparation of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride. The process starts by reaction of alpha, alpha-dibromo-o-xylene with 4-penten-2-one to obtain 1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)ethanone. The obtained intermediate was brominated, e.g. with bromine, methylene chloride was used as solvent and 2-bromo-1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)-ethanone was obtained, which is thereafter reacted with formamide in excess formamide to give a 4(5)-(2,3-dihydro-2-vinyl-1H-inden-2-ylimidazole hydrochloride. As the last step the vinyl group was catalytically hydrogenated to an ethyl group so as to form a product 4(5)-(2,3-dihydro-2-ethyl-1 H-inden-2-yl) imidazole.
-
Another synthetic route for obtaining 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole is disclosed in WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140. The cyclization of alpha, alpha’-dibromo-o-xylene with acetylacetone by means of NaOH and tetrabutylammonium bromide in toluene/water at 80°C under phase-transfer conditions gives the unstable diacetyl derivative, which presumably undergoes cleavage to afford 2-acetylindane. The alkylation of 2-acetylindane with ethyl iodide and potassium tert-butoxide yields 2-acetyl-2-ethylindan, which is brominated with Br2 to give 2-bromoacetyl-2-ethylindan. Finally, this compound is cyclised with formamide at 160°C (some 2-ethyl-2-(4-oxazolyl)indane is also formed but easily eliminated); the cyclization can also be carried out with formamidine in liquid ammonia. Although the substitution of formamide by formamidine acetate eliminates the oxazole formation, it does not increase the yield of Atipamezole (<30%) WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140 in the final step.
The preparation of atipamezole hydrochloride salt is described in U.S. Patent 4,689,339, wherein atipamezole obtained from the hydrogenation step is first recovered from alkaline solution as free base. After the evaporation of methylene chloride solvent to dryness the isolated crystalline product is converted into its hydrochloride salt by treatment with dry hydrogen chloride in ethyl acetate
Other compounds having alpha-2 adrenoceptor antagonist properties which may be useful in accordance with the present invention include idazoxan related compounds [Reckitt & Colman] Doxey, et al., Br. J. Parmacol., Vol. 78, p.489-505 (1983); imiloxan [Syntex] Michel, et al., Br. J. Pharmacol., Vol. 74, p.255-256 (1981); WY 26703 and related compounds [Wyeth] Latimer, et al., Naunvn Schmiedeberg’s Arch. Pharmacol., Vol. 327, p. 312-318 (1984); CH-38083 [Chinoin] Vizi, et a., J. Pharmacol. Exp. Ther., Vol. 238, p. 701-706 (1986); GR 50360A and related compounds [Glaxo] Halliday, et al., Br. J. Pharmacol., Vol. 95, p. 715 (1988); DG 5128 and related compounds of Daiichi Seiyaku Co., Ltd., Tokyo, Japan; and Yohimbine [Sigma].
………………………………….

-
- 1. an essential process for obtaining 5-(2-ethyl-2,3-1H-inden-2-yl)-1H-imidazole, without bromination in any step of process, thus preventing the possibility of brominated by-products;
- 2. This process has given superior yields, compared to patents cited above;
- 3. This process is amenable to large scale production which does not require specialized equipment.
-
The condensing of commercially available 1-trityl-1H-imidazole-4-carboxaldehyde (I) with phtalide to form 2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (II) is performed under the conditions that are similar to those used for synthesis of 4-(indane-1,3-dionyl) pyridine J. Org. Chem. 1971, vol.36, p.1563. surprisingly, the bulky 1-trityl-1H-imidazole-4-carboxaldehyde (I) reacted as expected and produced 2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (II) in over 67% yield. Both ethyl acetate and dioxane can be used as reaction media.
-
The alkylation of (II) by ethyl iodide is performed in boiling acetone with potassium carbonate as basic agent. 2-Ethyl-2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione (III) is formed in over 67% yield and easily isolated from the acetone solution by concentrating it and diluting with water. A high purity (III) is obtained after crystallization from methanol or ethanol.
-
Removing the trityl group of 2-ethyl-2-(1-trityl-1H-imidazole-4-yl)indan-1,3-dione by acid hydrolysis to yield the deprotected 2-ethyl-2-(1H-imidazol-2-yl)indan-1,3-dione.
-
The reduction of (IV) to 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride (V) is performed in hydrogenation apparatus with Pd/C catalyst under hydrogen pressure in HCI solution. The reaction proceeds under variable pressure and temperature conditions, but a pressure of about 3 bar and the temperature of about 80-85°C is preferable. After removing the catalyst the product crystallizes on chilling in over 77% yield. It can be purified by additional crystallization.
-
EP 0310745 B (FARMOS OY) 1989.04.12. disclosed preparation of 5-(2-ethyl-2,3-dihydro-1 H-inden-2-yl)-1 H-imidazole salt by two synthetic routes.
-
First synthetic route as starting material was used 2-acetyl-1-indanone, which was alkylated with ethylbromide in acetone in the presence of sodium carbonate to 2-acetyl-2-ethyl-1-indanone. The acetyl group was brominated with bromine in methanol and to imidazole by heating in formamide. Then the intermediate was hydrogenated in 2N hydrochloric acid in the presence of 10% palladium on carbon.
-
Second synthetic route disclosed in the same patent is following, as starting material was used 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester, which was prepared by methylation of 2,3-dihydro-1H-indene-2-carboxylic acid in the presence of sulphuric acid. The 2,3-dihydro-1H-indene-2-carboxylic acid methyl ester was reacted with N-isopropylcyclohexylamide and ethylbromide yielding 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid, then thionyl chloride was added and 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride was obtained. In the next step ethoxymagnesiummalonic acid ethyl ester in dry ether was added to 2,3-dihydro-2-ethyl-1H-indene-2-carboxylic acid chloride and reaction mixture was treated with sulphuric acid, and 1-(2,3-dihydro-2-ethyl-1H-inden-2-yl)ethanone was obtained, then the intermediate was stirred in methylene chloride and bromine was added by giving a new intermediate 2-bromo-1-(2,3-dihydro-2-methyl-1H-inden-2-yl)ethanone, to which was thereafter added formamide and hydrochloric acid yielding crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole. The last step involved hydrogenation of the crude product of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1 H-imidazole with 10% palladium on carbon.
-
EP 0247764 B (ORION-YHTYMÄ OY) 1987.02.12. disclosed the following process for preparation of 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride. The process starts by reaction of alpha, alpha-dibromo-o-xylene with 4-penten-2-one to obtain 1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)ethanone. The obtained intermediate was brominated, e.g. with bromine, methylene chloride was used as solvent and 2-bromo-1-(2,3-dihydro-2-vinyl-1H-inden-2-yl)-ethanone was obtained, which is thereafter reacted with formamide in excess formamide to give a 4(5)-(2,3-dihydro-2-vinyl-1H-inden-2-ylimidazole hydrochloride. As the last step the vinyl group was catalytically hydrogenated to an ethyl group so as to form a product 4(5)-(2,3-dihydro-2-ethyl-1 H-inden-2-yl) imidazole.
-
Another synthetic route for obtaining 5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole is disclosed in WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140. The cyclization of alpha, alpha’-dibromo-o-xylene with acetylacetone by means of NaOH and tetrabutylammonium bromide in toluene/water at 80°C under phase-transfer conditions gives the unstable diacetyl derivative, which presumably undergoes cleavage to afford 2-acetylindane. The alkylation of 2-acetylindane with ethyl iodide and potassium tert-butoxide yields 2-acetyl-2-ethylindan, which is brominated with Br2 to give 2-bromoacetyl-2-ethylindan. Finally, this compound is cyclised with formamide at 160°C (some 2-ethyl-2-(4-oxazolyl)indane is also formed but easily eliminated); the cyclization can also be carried out with formamidine in liquid ammonia. Although the substitution of formamide by formamidine acetate eliminates the oxazole formation, it does not increase the yield of Atipamezole (<30%) WAI, Wonf, et al. A Concise Synthesis of Atipamezole. Synthesis. 1995, no.2, p.139-140 in the final step.
…………………………….
US Patent 8,431,717
Atipamezole [5-(2-ethyl-2,3-dihydro-1H-inden-2-yl)-1H-imidazole, 1] is a veterinary drug that has been investigated for treating Parkinson’s disease in humans. V. Lusis and co-inventors summarize several ways to synthesize 1. Some routes give a low yield of 1 and produce large quantities of an oxazole byproduct. Other processes involve a sluggish bromination reaction that leads to many byproducts.
The inventors’ process is intended to overcome these problems. In particular, it does not use the bromination reaction and thus avoids forming brominated byproducts. The process, outlined in the figure, begins with the reaction of imidazole 2 with i-PrMgCl to form iodo Grignard reagent 3, which is treated with DMF to give 4. This intermediate is not isolated but is treated with aq NH4Cl to give aldehyde 5, isolated in 73.2% yield. The aldehyde is condensed with phthalide (6) in the presence of NaOMe to produce imidazolylindane 7, recovered in crude form in 67.2% yield.

In the next stage, compound 7 is alkylated with EtI in the presence of K2CO3. Product 8 is isolated in 50.9% yield after being recrystallized from EtOH. Product1 can be produced directly from 8 by making its HCl salt and hydrogenating the salt over Pd/C. Crude atipamezole is isolated as its HCl salt in 26.6% yield.
Alternatively, acid hydrolysis of 8 removes the trityl group to form dione 9, recovered as a white crystalline solid in 76.2% yield. The HCl salt of 9 is then hydrogenated to 1·HCl.
The patent’s claims cover the process to make 1 and new compounds 7 and 8. The overall yield of compound 1 is poor, partly because of the low yield from the hydrogenation step. The inventors claim, however, that the yield is higher than from earlier methods. They point out that the process is amenable to large-scale production without the use of specialized equipment. (JSC Grindeks [Riga, Latvia]. US Patent 8,431,717, April 30, 2013; Keith Turner), View the full-text here.
………………………………
nmr

Atipamezole Hydrochloride CAS 104075-48-1 HNMR
……………………………………………………….
GADODIAMIDE, OMNISCAN Drug Patent Expiration, 1 st oct 2013

GADODIAMIDE
GE HEALTHCARE, OMNISCAN
Drug Patent Expiration
1 st oct 2013, US5560903, CAS 122795-43-1
| GADODIAMIDE | INJECTABLE; INJECTION | 287MG/ML | RX | NDA 020123 |
Gadodiamide is a gadolinium-based MRI contrast agent, used in MR imaging procedures to assist in the visualization of blood vessels. It is commonly marketed under the trade name Omniscan.
For intravenous use in MRI to visualize lesions with abnormal vascularity (or those thought to cause abnormalities in the blood-brain barrier) in the brain (intracranial lesions), spine, and associated tissues.

Gadodiamide is a contrast medium for cranial and spinal magnetic resonance imaging (MRI) and for general MRI of the body after intravenous administration. The product provides contrast enhancement and facilitates visualisation of abnormal structures or lesions in various parts of the body including the central nervous system (CNS). It does not cross an intactblood brain barrier but might give enhancement in pathological conditions.
Based on the behavior of protons when placed in a strong magnetic field, which is interpreted and transformed into images by magnetic resonance (MR) instruments. Paramagnetic agents have unpaired electrons that generate a magnetic field about 700 times larger than the proton’s field, thus disturbing the proton’s local magnetic field. When the local magnetic field around a proton is disturbed, its relaxation process is altered. MR images are based on proton density and proton relaxation dynamics. MR instruments can record 2 different relaxation processes, the T1 (spin-lattice or longitudinal relaxation time) and the T2 (spin-spin or transverse relaxation time). In magnetic resonance imaging (MRI), visualization of normal and pathological brain tissue depends in part on variations in the radiofrequency signal intensity that occur with changes in proton density, alteration of the T1, and variation in the T2. When placed in a magnetic field, gadodiamide shortens both the T1 and the T2 relaxation times in tissues where it accumulates. At clinical doses, gadodiamide primarily affects the T1 relaxation time, thus producing an increase in signal intensity. Gadodiamide does not cross the intact blood-brain barrier; therefore, it does not accumulate in normal brain tissue or in central nervous system (CNS) lesions that have not caused an abnormal blood-brain barrier (e.g., cysts, mature post-operative scars). Abnormal vascularity or disruption of the blood-brain barrier allows accumulation of gadodiamide in lesions such as neoplasms, abscesses, and subacute infarcts.

1.Schenker MP, Solomon JA, Roberts DA. (2001). Gadolinium Arteriography Complicated by Acute Pancreatitis and Acute Renal Failure, Journal of vascular and interventional radiology 12(3):393.[1]
2 Unal O, Arslan H. (1999). Cardiac arrest caused by IV gadopentetate dimeglumine. AJR Am J Roentgenol 172:1141.[2]
3 Cacheris WP, Quay SC, Rocklage SM. (1990). The relationship between thermodynamics and the toxicity of gadolinium complexes, Magn Reson Imaging 8(6):467-81. doi:10.1016/0730-725X(90)90055-7
4 Canavese, C; Mereu, MC; Aime, S; Lazzarich, E; Fenoglio, R; Quaglia, M; Stratta, P (2008). “Gadolinium-associated nephrogenic systemic fibrosis: the need for nephrologists’ awareness”. Journal of nephrology 21 (3): 324–36. PMID 18587720.
COUNTRY PATENT APPROVED, EXPIRY
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5362475 | 1994-11-08 | 2011-11-08 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
Gadolinium contrast agents are used as contrast media to enhance magnetic resonance imaging as they are paramagnetic. This compound has a low incidence of adverse side effects, although there is a rare association with nephrogenic systemic fibrosis (NSF) when given to people with severe renal impairment (ie, GFRglomerular filtration rate <30mL/min/1·73m2).It seems to be related to the liberation of free gadolinium ions, and UK CHM advice is against using the least stable of the agents – Omniscan (gadodiamide) – in patients with severe renal impairment, and carefully considering whether to use others where renal function is impaired.
OMNISCAN (gadodiamide) Injection is the formulation of the gadolinium complex of diethylenetriamine pentaacetic acid bismethylamide, and is an injectable, nonionic extracellular enhancing agent for magnetic resonance imaging. OMNISCAN is administered by intravenous injection. OMNISCAN is provided as a sterile, clear, colorless to slightly yellow, aqueous solution. Each 1 mL contains 287 mg gadodiamide and 12 mg caldiamide sodium in Water for Injection.
The pH is adjusted between 5.5 and 7.0 with hydrochloric acid and/or sodium hydroxide. OMNISCAN contains no antimicrobial preservative. OMNISCAN is a 0.5 mol/L solution of aqua[5,8-bis(carboxymethyl)11-[2-(methylamino)-2-oxoethyl]-3-oxo-2,5,8,11-tetraazatridecan-13-oato (3-)-N5, N8, N11, O3, O5, O8, O11, O13] gadolinium hydrate, with a molecular weight of 573.66 (anhydrous), an empirical formula of C16H28GdN5O9•xH2O, and the following structural formula:
 |
Pertinent physicochemical data for OMNISCAN are noted below:
PARAMETER
| Osmolality (mOsmol/kg water) | @ 37°C | 789 |
| Viscosity (cP) | @ 20°C | 2 |
| @ 37°C | 1.4 | |
| Density (g/mL) | @ 25°C | 1.14 |
| Specific gravity | @ 25°C | 1.15 |
OMNISCAN has an osmolality approximately 2.8 times that of plasma at 37°C and is hypertonic under conditions of use.
gadodiamide, chemical name: [5,8 _ bis (carboxymethyl) -11 – [2_ (methylamino)-2_ ethyl] -3 – O 2 ,5,8, 11 – tetraazacyclododecane-decane -13 – oxo-(3 -)] gadolinium trihydrate. Its structure is shown in formula one.
[0003] Structural Formula:
[0004]

[0005] Magnetic resonance contrast agent gadodiamide resonance than ionic contrast agents safer generation of products, it is non-ionic structure significantly reduces the number of particles in solution, osmotic balance of body fluids is very small.Meanwhile, gadodiamide relatively low viscosity to bring the convenience of nursing staff, making it easier to bolus. In addition, gadodiamide pioneered the use of amide-substituted carboxyl part, not only reduces the toxicity of carboxyl groups and ensure the non-ionic nature of the product solution.
[0006] reported in the literature and their intermediates gadodiamide synthetic route is as follows:
[0007] 1. Compound III synthetic routes for its preparation in U.S. Patent No. US5508388 described as: In the synthesis process, the inventors using acetonitrile as solvent, acetic anhydride as dehydrating agent, pyridine as acid-binding agent, at 55 ~ 60 ° C, the reaction 18h. Anti-
See the reaction should be a process. The disadvantage of this synthesis are acetonitrile toxicity, not widely used.
[0008]

[0009] Reaction a
[0010] (2) Synthesis of Compound III in many articles are reported in the patent and its implementation method similar to the patent US5508388.
[0011] In US3660388, the diethylenetriamine pentaacetic acid (Compound II), pyridine, acetic anhydride, the mixture was reacted at 65 ° C or 20h at 125 ° C the reaction 5min, to give compound III.
[0012] In US4822594, the compounds II, pyridine, acetic anhydride mixture was reacted at 65 ° C 20h, to give compound III.
[0013] In US4698263, the compounds II, pyridine, acetic anhydride heated in a nitrogen or argon atmosphere under reflux for 18h, to give compound III. [0014] In the EPO183760B1, the compounds II, pyridine, acetic anhydride mixture was reacted at 55 ° C 24h, to give compound III.
[0015] In CN1894223A, the compounds II, pyridine, acetic anhydride, the mixture above 65 ° C the reaction mixture, and the pyridine of DTPA feed ratio is: 1: (0.5 to 3).
[0016] The above patents do not provide for the compound III is post-processing method.
[0017] 3 Synthesis of Compound IV.
[0018] In U.S. Patent US4859451, the diethylenetriamine pentaacetic acid dianhydride (compound III) and ammonia, methanol and the reaction of compounds IV, see Reaction Scheme II.
[0019]

[0020] Reaction two
[0021] In the patent US5087439, the compound III with methylamine in aqueous solution for several hours, or overnight reactions, see reaction formula III.
[0022]

[0023] Reactive three
[0024] These two patents using ammonia and methylamine, which can form explosive mixtures with air, in case of fire or high pressure can cause an explosion in the production process of great insecurity. Although raw material prices are lower, but higher production conditions (such as requiring sealed, low temperature, etc.). Compared to this synthesis process,
[0025] 4, gadodiamide (Compound I) synthesis.
[0026] In the patent US4859451, the use of gadolinium chloride with the compound IV is carried out under acidic conditions, complexing. Finally, tune
Section PH neutral, see reaction IV.
[0027]

[0028] Reaction formula tetrakis [0029] in the patent US5087439, the chlorides are used as reactants, and details of the post-processing method of Compound I.
[0030] In the patent US5508388, the use of gadolinium oxide with compound IV in acetonitrile, water with stirring, the resulting compound I.
[0032] The synthetic route is as follows:
[0033]

[0034] 1) Compound II (diethylenetriamine pentaacetic acid) in pyridine, acetic anhydride in the presence of a dehydration reaction into the acid anhydride, and the product was stirred with cold DMF, leaving the solid filtered, washed with ether reagents, drying , to obtain a white powdery solid compound III (diethylenetriamine pentaacetic acid anhydride);
[0035] 2) Compound III in DMF with methylamine hydrochloride, the reaction of the compound IV (5,8 _ bis carboxymethyl methyl-11 – [2 – (dimethylamino) -2 – oxoethyl] – 3 – oxo -2,5,8,11 – tetraazacyclododecane _13_ tridecyl acid); and the control compound III: MeNH2 · HCl molar ratio = 1: (1 to 4), control the temperature between 20 ~ 80 ° C, the reaction time is 4 ~ 6h, after the treatment, the method of distillation under reduced pressure to remove DMF, the product is dissolved in a polar solvent, methanol, and then adding a solvent polarity modulation, so that the target Compound IV from system completely precipitated;
[0036] 3) Compound IV with gadolinium oxide formed in the presence of hydrochloric acid of the complex, after the reaction, filtration and drying, to obtain a white powdery compound I, i.e. gadodiamide.
[0037] Existing gadodiamide Synthesis basically from the synthesis of Compound IV as a starting material, the present invention is first introduced to the compound II as a starting material to synthesize gadodiamide. Synthesis of the conventional method of gadodiamide, the present invention has the advantage of inexpensive starting materials, convenient and easy to get. In addition, the synthetic pathway intermediates are involved in the post-processing is simple, enabling continuous reaction, saving time and cost savings, the reaction becomes controlled step by step, and try to avoid the use of toxic reagents, reducing the possibility of operator injury , while also greatly reducing damage to the environment.
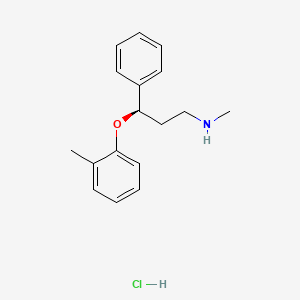
Atomoxetine

Atomoxetine
Atomoxetine hydrochloride (CAS NO.: 82248-59-7)
(R)-(-)-N-Methyl-gamma-(2-methylphenoxy)benzenepropanamine hydrochloride
| Patent No | PatentExpiry Date | |
|---|---|---|
| 5658590 | Nov 26, 2016 | |
| 5658590*PED | May 26, 2017 |
nda 021411 app 2002-11-26
TREATMENT OF ATTENTION-DEFICIT HYPERACTIVITY DISORDER
label
|
Country
|
Patent Number
|
Approved
|
Expires (estimated)
|
|---|---|---|---|
| United States | 5658590 | 1997-05-26 | 2017-05-26 |
The HCl salt of atomoxetine , with the (R)-configuration], which is marketed under the trade name Strattera, is used for treating attention-deficit hyperactivity disorder (ADHD
Atomoxetine is a drug approved for the treatment of attention-deficit hyperactivity disorder(ADHD).[1] It is a selective norepinephrine reuptake inhibitor (NRI),[1] not to be confused with serotonin norepinephrine reuptake inhibitors (SNRIs) or selective serotonin reuptake inhibitors (SSRIs), both of which are currently the most prescribed form of antidepressants.
his compound is manufactured, marketed and sold in theUnited States under the brand name Strattera by Eli Lilly and Company as a hydrochloride salt (atomoxetine HCl), the original patent filing company, and current U.S. patent owner. Generics of atomoxetine are sold in all other countries; they are manufactured by Torrent Pharmaceuticals using the label Tomoxetin, Ranbaxy Laboratories (through its Division: Solus) using the label Attentin, Sun Pharmaceuticals(through its Division: Milmet Pharmaceuticals), and Intas Biopharmaceuticals There is currently no generic manufactured directly in the United States since it is under patent until 2017.[2]
On August 12, 2010, Lilly lost a lawsuit that challenged Lilly’s patent on Strattera, increasing the likelihood of an earlier entry of a generic into the US market.[3] On September 1, 2010, Sun Pharmaceuticals announced it would begin manufacturing a generic in the United States.[4] In a July 29, 2011 conference call, however, Sun Pharmaceutical’s Chairman stated “Lilly won that litigation on appeal so I think [generic Strattera]’s deferred.”[5]
Atomoxetine is designated chemically as (−)-N-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, and has a molecular mass of 291.82.[1] It has a solubility of 27.8 mg/mL in water.[1] Atomoxetine is a white solid that exists as a granular powder inside the capsule, along with pre-gelatinized starch and dimethicone.[1] The capsule shells contain gelatin, sodium lauryl sulfate, FD&C Blue No. 2, synthetic yellow iron oxide, titanium dioxide, red iron oxide, edible black ink, and trace amounts of other inactive ingredients.[1]
The compound (-)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine, or (-)-Λ/-methyl-3-phenyl-3-(o-tolyloxy)-propylamine hydrochloride, is usually known by its adopted name “atomoxetine hydrochloride.” It is represented as shown in Formula 1 and is a selective norepinephrine reuptake inhibitor. A commercialatomoxetine hydrochloride product is sold as STRATTERA™ in the form of capsules containing 10, 18, 25, 40, 60, 80, or 100 mg of atomoxetine, for treating attention-deficit/hyperactivity disorder.
- “STRATTERA® (atomoxetine hydrochloride) CAPSULES for Oral Use. Full Prescribing Information.” Eli Lilly and Company, 2002, 2013. Revised August 5, 2013. [1]
- “Patent and Exclusivity Search Results”. Electronic Orange Book. US Food and Drug Administration. Retrieved 26 April 2009.
- “Drugmaker Eli Lilly loses patent case over ADHD drug, lowers revenue outlook”. Chicago Tribune.
- “Sun Pharma receives USFDA approval for generic Strattera capsules”. International Business Times.
- “Sun Pharma Q1 2011-12 Earnings Call Transcript 10.00 am, July 29, 2011”.
- Strattera by Eli Lilly and Company
- RxList.com – Strattera
- Detailed Strattera Consumer Information: Uses, Precautions, Side Effects
- All disclosed Lilly trials
- MSDS for Atomoxetine HCl
- Strattera Related Published Studies
Synthesis
Also known as: Atomoxetine hydrochloride, Strattera, Atomoxetine HCL, (R)-Tomoxetine hydrochloride, TOMOXETINE HYDROCHLORIDE, Tomoxetine, 82248-59-7
First step appears to be a Mannich reaction between acetophenone, paraformaldehyde and dimethylamine, although not formally written in the scheme.

Foster, B. J.; Lavagnino, E. R.; European Patent, 1982, EP 0052492.

Eli Lilly’s Strattera capsules.


Atomoxetine, designated chemically as (-)-N-methyl-3-phenyl-3-(0-tolyloxy)- propylamine hydrochloride, is structurally represented by the compound of Formula-I and is indicated for the potential treatment of attention-deficit hyperactivity disorder (ADHD). This compound is manufactured, marketed and sold in the United States under the brand name Strattera.

Formula-I Atomoxetine was first disclosed in US Patent No 4314081. The said patent disclosed Atomoxetine, its pharmaceutically acceptable salts and composition containing them.
4-hydroxy Atomoxetine, chemically known as R-(-)-N-methyl-3-(2-methyl-4- hydroxyphenyl)oxy)-3 -phenyl- 1-aminopropane, structurally represented by Formula-II, is a metabolite of Atomoxetine.

Fomnula-ll
4-hydroxy Atomoxetine hydrochloride was first disclosed in US Patent No 7384983, wherein 4-hydroxy Atomoxetine free base was dissolved in ethylacetate, treated the solution with 0.1N HC1; followed by lyophilization yielded a yellow solid which was dissolved in methanol and passed through a short column of activated carbon; the solvent was removed and finally the hydrochloride salt was recrystallized from water to afford 4-hydroxy Atomoxetine hydrochloride. However, this patent does not mention about the nature of the polymorph obtained through this process.
The asymmetric epoxidation of (E)-3-phenyl-2-propen-1-ol (I) by means of titanium tetraisopropoxide, (+)-diethyl tartrate (+)-(DET) and tBu-OOH in dichloromethane gives the chiral epoxide (II), which is opened by means of bis(2-methoxyethoxy)aluminum hydride (Red-Al) in DME to yield the chiral diol (III). The regioselective reaction of (III) with Ms-Cl and TEA in ethyl ether affords the primary mesylate (IV), which is condensed with 2-methylphenol (V) by means of PPh3 and DEAD in ethyl ether to provide the adduct (VI). Finally this compound is treated with methylamine in hot aq. THF to give rise to the target (R)-tomoxetine.

The reduction of omega-chloropropiophenone (I) with NaBH4 in ethanol gives 3-chloro-1-phenyl-1-propanol (II), which is treated with butyric anhydride and pyridine in dichloromethane to yield the corresponding racemic ester (III). The optical resolution of (III) with immobilized lipase B from Candida antarctica (CALB) affords a mixture of unreacted (S)-ester and (R)-alcohol (IV) that are separated by column chromatography. Condensation of th (R)-alcohol (IV) with 2-methylphenol (V) by means of PPh3 and diethyl azodicarboxylate (DEAD) in THF gives the corresponding ether (VI), which is finally treated with methylamine in refluxing ethanol.
more info
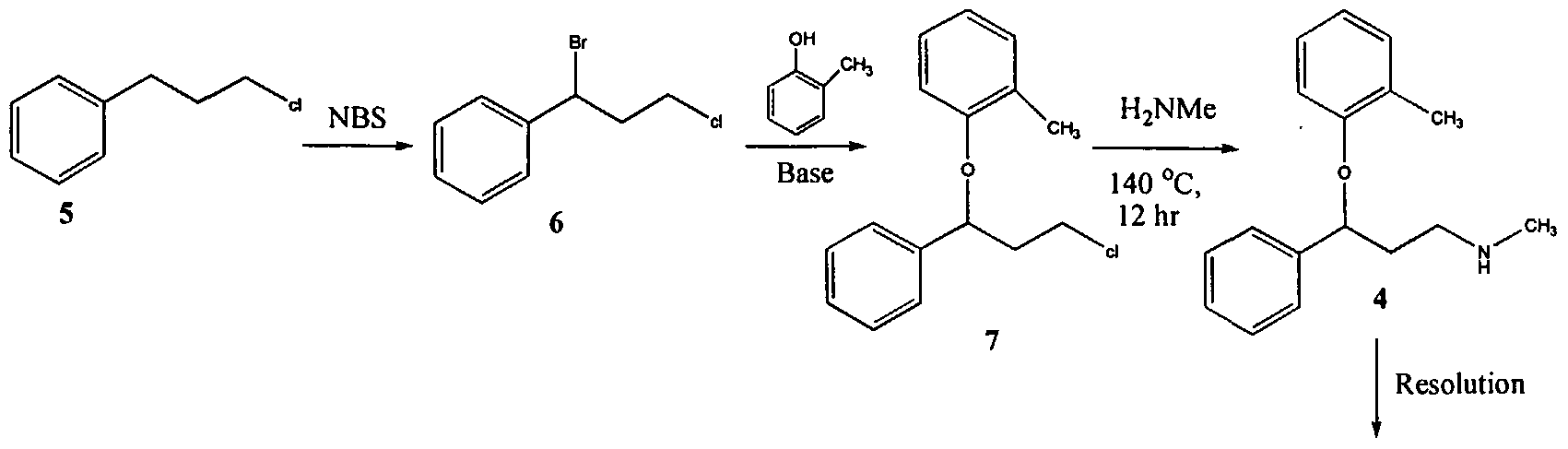
U.S. Patent No. 4,314,081 describes 3-Aryloxy-3-phenyl polyamines, which possess central nervous system activity. Atomoxetine is a member of the above class of compounds, and is a useful drug for the treatment of depression.Atomoxetine was claimed in U. S. Patent No. 4,314,081 and the patent describes a process for the preparation of atomoxetine and related compounds in two different ways as depicted below as Scheme A and Scheme B, respectively.
Scheme A
Atomoxetinc
Scheme B

The process illustrated in Scheme A involves the preparation of the atomoxetineusing 3-phenyl chloropropyl amine (Formula 5) as a starting material. The process involves bromination of said starting compound (Formula 5) by using N-bromosuccinimide. Further the bromo derivative is condensed with o-cresol to result in a compound of Formula 7, which is then subjected to amination using methylamine. Though the process looks very simple, it involves the following disadvantages: i) N-bromosuccinimide being a corrosive and sensitive chemical, its usage demands special care; ii) the workup of the compound formula 7 involves high vacuum (0.03 torr) distillation at 135-1450C, which is a tedious and cumbersome process to carry out at the plant level; and iii) the reaction conditions involved in some of the steps are harsh, for example the amination reaction is conducted at 14O0C. at pressures of 10 kg/cm2 for 12 hours in an autoclave.
All the above points make the process not viable for practicing on a commercial scale. Further, as described in U.S. Patent 4,314,081 , the free base compounds exist as high boiling oils, but form white crystalline salts.
On the other hand, Scheme B describes the preparation of atomoxetine using β-dimethylaminopropiophenone produced by a Mannich reaction; which is reduced to the hydroxy derivative having Formula 9 using diborane; further the hydroxy compound (Formula 9) is converted to the corresponding chloro derivative of Formula 10 using dry HCI gas and thionyl chloride and is followed by condensation with o-cresol.
The said reaction is carried out in methanol at reflux for a duration of five days to achieve the compound of formula 11 and is followed by demethylation using cyanogen bromide to end up with atomoxetine. As can be clearly understood the process is associated with the following problems: i) the use of costly reagents such as diborane makes the process uneconomical; ii) the passage of dry HCI gas followed by thionyl chloride addition is ^ very cumbersome and is not advisable in the plant; iii) this is a time-consuming process, involving a reaction which requires five days for its completion; and iv) use of cyanogen bromide, which is highly toxic, is not desirable.
All of the above-quoted drawbacks make the process unfriendly to practice in a production plant as well as to the environment.
Further, M. Srebnik et al., Journal of Organic Chemistry, Vol. 53, pages2916-2920 (1988); E. Corey et al., Tetrahedron Letters, Vol. 30, pages 5207-5210 (1989);
U.S. Patent No. 4,868,344; Y. Gao et al., Journal of Organic Chemistry, Vol. 53, pages 4081-4084 (1988); J. Deeter et al.,
Tetrahedron Letters, Vol. 31, pages 7101-7104 (1990);
and U.S. Patent No. 4,950,791 disclose stereospecific methods for the preparation of 3-aryloxy-3-phenylpropylamines; the enantiomers of 3-hydroxy-3-phenylpropylamines are prepared by the stereospecific reduction of the corresponding ketones. The thus obtained (S)-3-hydroxy-3-phenyl propylamines are subjected to condensation with aryl alcohols using the Mitsunobo reaction. As can be seen in Scheme C, the reaction involves two critical steps.
Scheme C
Dusopinocampheny) chloroborane


OH
,CH,
DEAD/ tn phenyl phosphine

The first critical step is an asymmetric reduction of the ketone to its corresponding alcohol. The second critical step involves the condensation of the obtained enantiomeric alcohol with the corresponding aryl alcohol. The process suffers from the following disadvantages:
1) the reagent used for the asymmetric reduction of the ketone is highly expensive;
2) the reagent diethyl azodicarboxylate (“DEAD”) is expensive;
3) the DEAD reagent is known to be highly carcinogenic, thus creating problems in handling; and
4) the reaction involves the use of triphenylphosphine and DEAD and the resulting byproducts formed in the reaction, phoshineoxide and a hydrazine derivative, are very difficult to remove.
Therefore, commercial applicability of the said process is limited owing to the above noted disadvantages.
International Patent Publication No. WO 00/58262 relates to a stereo- specific process for the preparation of atomoxetineusing nucleophilic aromatic displacement of an aromatic ring having a functional group, which can be converted to a methyl group. As can be seen, the process is very lengthy and involves many steps and is thus not commercially desirable.
U.S. Patent No. 5,847,214 describes the nucleophilic aromatic displacement reaction of 3-hydroxy-3-arylpropylamines with activated aryl halides, for example the reaction of N-methyl-3-phenyl-3-hydroxypropylamine with 4- triflouromethyl-1-cholro benzene has been reported; the success of this reaction is mainly due to electron withdrawing group on benzene ring of the aryl halides.
U.S. Patent No. 6,541 ,668 describes a process for the preparation of atomoxetine and its pharmaceutically acceptable addition salts which comprises reacting an alkoxide of N-methyl-3-phenyl-3-hydroxy propyl amine or an N protected derivative thereof, with 2-flouro toluene in the presence of 1 ,3-Dimethyl – 2-imidazolidinone (“DMI”) or N-Methyl-3-pyrrolidinone (“NMP”) as the solvent. The process disclosed in the said patent can be shown as Scheme D. Further, the process disclosed in the said patent restricts itself to the solvents DMI and NMP.
Scheme D

Nevertheless, a new crystalline form of N-methyl-3-phenyl-3-(o- tolyloxy)propylamine oxalate and an isolation technique of (±)-atmoxetine free base in a solid form, an intermediate useful in the synthesis of atomoxetine hydrochloride, is desirable.

http://www.sciencedirect.com/science/article/pii/S0040403906025068
There have been several methods reported for preparing (R)-(−)-N-methyl-3-(2-methylphenoxy)-3-phenylpropylamine (Atomoxetine®). For example, U.S. Pat. No. 4,868,344 discloses a process as shown in the following scheme:

In this example, 3-chloropropiophenone is used as the starting material to be asymmetrically reduced with (−)-diisopinocamphenylchloroborane ((−)-IPc2BC1) to give the corresponding chiral alcohol. The resulting chiral alcohol is then reacted with o-cresol via Mitsunobu reaction to form the chiral ether compound. Subsequently, amination of the chiral ether compound with methylamine provided atomoxetine. In this process, the materials such as chiral-borane ((−)-IPc2BC1) and diethyl azodicarboxylate (DEAD) are expensive, and result in high manufacturing cost.
Further, WO 2006/009884 discloses another method for preparing atomoxetine, including the step of reacting N-methyl-3-phenyl-3-hydroxypropylamine with 2-fluorotoluene which is followed by resolution of the resulting product to provide optically pure atomoxetine as shown in the following scheme:

This process involving a chiral resolution step is inefficient due to low product yield, complicated and long time process that renders this process economically less competitive.
………………………………………………………………………………………………

see below
B.-F. Chen and co-inventors describe a synthesis of 5 that avoids costly reagents. It includes the preparation of the chiral amino alcohol 3 as a key intermediate. The route for preparing 5 starts with a Mannich reaction between benzophenone, N,O-dimethylhydroxylamine, and paraformaldehyde to give compound 1, isolated in 87.6% yield.
Ketone 1 is asymmetrically reduced to form alcohol 2 by using the chiral ruthenium catalyst RuCl2-[(S)-DMSEGPHOS)][(S)-DAIPEN]. The hydrogen pressure is described as “predetermined”, but no value is given. Product 2 is recovered as an oily product in 98.7% yield with 98.8% purity and 99% ee. It appears that the catalyst is not removed before the next step in which the oil is hydrogenated over a Raney nickel catalyst to form amino alcohol 3.
Intermediate 3 is also isolated as an oil in 96.4% yield, 96.5% % purity, and 99% ee. After recrystallization from toluene–heptane, the solid product is recovered with 100% ee. In the last step, 3 is treated with fluorotoluene 4 in the presence of t-BuOK to form atomoxetine, isolated as an oil in 91% yield with 97% ee. The purification of 5 and its conversion to the HCl salt are not described.
The inventors provide basic 1H-NMR data for all compounds except 5. The example describing the preparation of 1 lists one of the reactants as 2-acetylthiophene, which is clearly incorrect; and another reagent is called “32% hydrochloride”. These errors should have been spotted by anyone with a fundamental knowledge of chemistry who was involved in writing the patent—perhaps none were. (Sci Pharmtech [Taiwan]. US Patent 8,299,305, Oct. 30, 2012; Keith Turner)
View the full-text patent here.
| Patent Number: | US 8299305 |
| Title: | Process for preparation of atomoxetine |
| Inventor(s): | Chen, Bo-Fong; Li, Yan-Wei; Yeh, Jinun-Ban; Wong, Wei-Chyun |
| Patent Assignee(s): | SCI Pharmtech, Inc., Taiwan |
ATACAND, CANDESARTAN CILEXETIL, ASTRAZENECA.Drug Patent Expiration on 9 th Jan 2014

Candesartan cilexetil Candesartan cilexetil, Candesartan hexetil, H212/91, TCV-116, Kenzen, Blopress 16 mg Plus, Parapres, Ratacand, Blopress, Amias, Atacand
ATACAND
ATACAND (candesartan cilexetil), a prodrug, is hydrolyzed to candesartan during absorption from the gastrointestinal tract. Candesartan is a selective AT1 subtype angiotensin II receptor antagonist. Candesartan cilexetil, a nonpeptide, is chemically described as (±)-1-Hydroxyethyl 2-ethoxy-1-[p-(o-1H-tetrazol-5ylphenyl)benzyl]-7-benzimidazolecarboxylate, cyclohexyl carbonate (ester). Its empirical formula is C33H34N6O6, and its structural formula is:
 |
Candesartan cilexetil is a white to off-white powder with a molecular weight of 610.67. It is practically insoluble in water and sparingly soluble in methanol. Candesartan cilexetil is a racemic mixture containing one chiral center at the cyclohexyloxycarbonyloxy ethyl ester group. Following oral administration, candesartan cilexetil undergoes hydrolysis at the ester link to form the active drug, candesartan, which is achiral. ATACAND is available for oral use as tablets containing either 4 mg, 8 mg, 16 mg, or 32 mg of candesartan cilexetil and the following inactive ingredients: hydroxypropyl cellulose, polyethylene glycol, lactose, corn starch, carboxymethylcellulose calcium, and magnesium stearate. Ferric oxide (reddish brown) is added to the 8-mg, 16-mg, and 32-mg tablets as a colorant.
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| CANDESARTAN CILEXETIL | TABLET; ORAL | 4MG | RX | 020838 | 001 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 8MG | RX | 020838 | 002 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 16MG | RX | 020838 | 003 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 32MG | RX | 020838 | 004 |
Patents
There are 6 patent(s) protecting ASTRAZENECA’s ATACAND. The last patent 5534534*PED expires on 2014-01-09.View patent at USPTO
| Patent US | US | Expiration |
|---|---|---|
| 5534534*PED | 2014-1-9 | |
| 5534534 | Pharmaceutical compositions for oral use and method of preparing them
A pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) having antagonistic action to angiotensin II ##STR1## (wherein the ring W is an optionally substituted N-containing heterocyclic residue; R.sup.3 is a group capable of forming an anion or a group convertible thereinto; X is a direct bond or a spacer having an atomic length of two or less between the phenylene group and the phenyl group; and n is an integer of 1 or 2) and an oily substance having a lower melting point, and a method for preparing a pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) and an oily substance having a lower melting point, which comprises admixing the compound of the formula (I) with an oily substance having a lower melting point and then subjecting the mixture to molding.
|
2013-7-9(expired) |
| 5196444*PED | 2012-12-4(expired) | |
| 5196444 | 1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-c arboxylate and compositions and methods of pharmaceutical use thereof
1-(Cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-car boxylate or a pharmaceutically acceptable salt thereof has potent angiotensin II antihypertensive activity, thus being useful as therapeutic agents for treating circulatory system diseases such as hypertensive diseases, heart diseases (e.g. hypercardia, heart failure, cardiac infarction, etc.), strokes, cerebral apoplexy, nephritis, etc.
|
2012-6-4(expired) |
| 7538133*PED | 2011-10-18(expired) | |
| 5705517*PED | 2011-10-18(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ASTRAZENECA.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 2013-04-26 | 038 | Labeling Revision | |
| 2012-04-27 | 035 | Labeling Revision | |
| 2012-04-13 | 032 | Labeling Revision | |
| 1998-06-04 | 000 | Approval | |
| 2011-06-24 | 033 | Labeling Revision | |
| 2009-10-22 | 031 | Patient Population Altered | |
| 2006-08-17 | 026 | Labeling Revision | |
| 2005-05-18 | 022 | New or Modified Indication | |
| 2005-02-22 | 024 | New or Modified Indication | |
| 2004-12-16 | 023 | Labeling Revision | |
| 2000-06-14 | 008 | Labeling Revision | |
| 2002-09-13 | 015 | Comparative Efficacy Claim | |
| 2003-01-22 | 017 | Labeling Revision | |
| 2003-04-23 | 019 | Labeling Revision | |
| 2013-02-21 | 037 | Manufacturing Change or Addition | |
| 1999-08-11 | 005 | Package Change | |
| 2000-12-27 | 009 | Manufacturing Change or Addition | |
| 2001-05-24 | 012 | Manufacturing Change or Addition | |
| 2001-11-28 | 016 | Labeling Revision | |
| 1999-07-28 | 004 | Control Supplement | |
| 2001-04-02 | 011 | Manufacturing Change or Addition | |
| 2001-10-04 | 014 | Control Supplement | |
| 1998-11-16 | 002 | Manufacturing Change or Addition | |
| 1999-12-08 | 006 | Package Change | |
| 2001-06-07 | 010 | Manufacturing Change or Addition | |
| 2001-03-29 | 013 | Package Change | |
| 1998-12-07 | 001 | Manufacturing Change or Addition |
 Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Synthesis
Candesartan is synthesised as follows:  kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
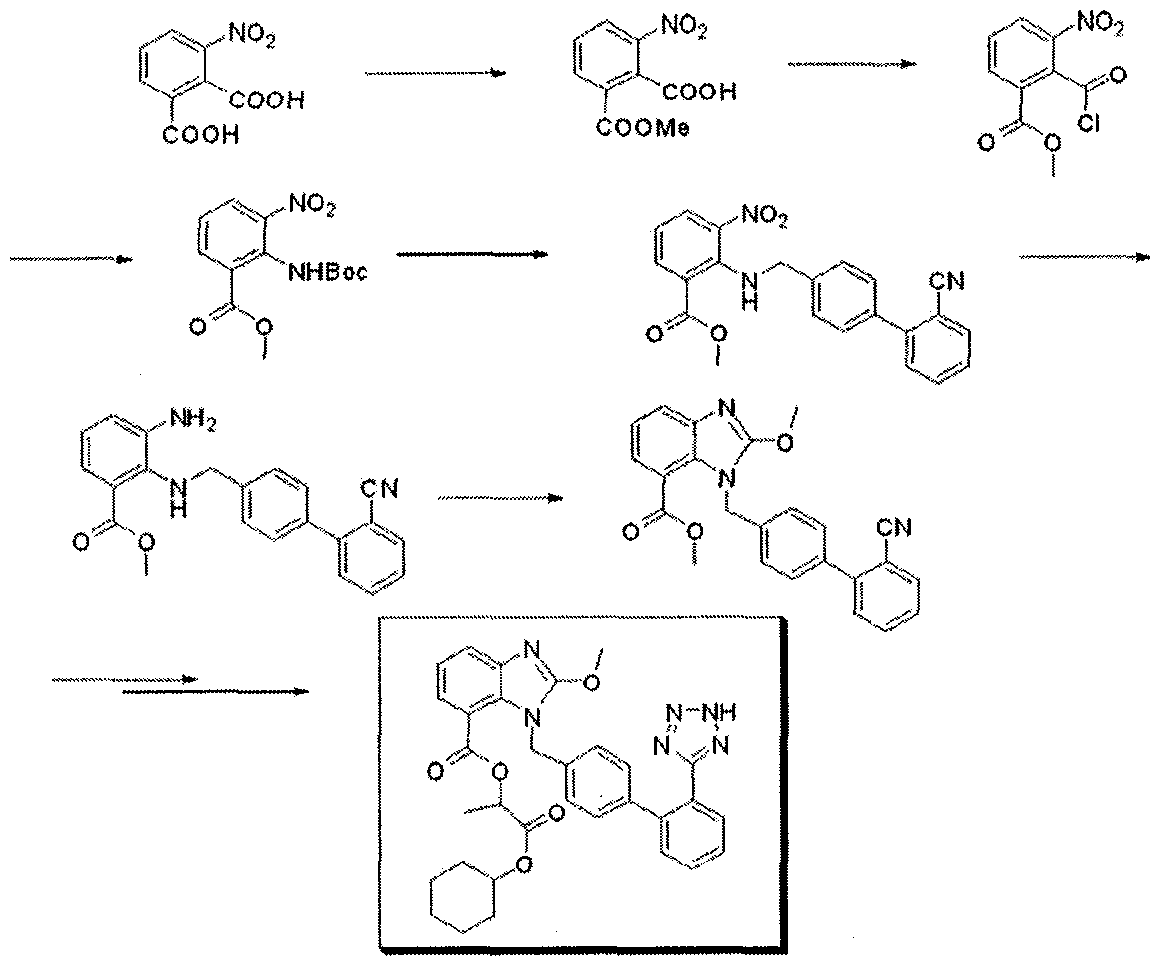
The method has technical problems as follows: a) the starting material is obtained by a minor reaction, b) its yield is relatively low and its industrial applicability is poor (due to N2 gas formation) because the Curtius rearrangement reaction is involved, and c) materials industrially hard to handle such as SOCI2 or NaH are used. In addition, methods for preparing an intermediate of candesartan cilexetil are disclosed in Organic Process Research & Development 11:490-493(2007), as represented by the following Reaction Scheme 2: Reaction Scheme 2

3:1 s

However, the preparation process has also shortcomings of a) undesired byproducts formed by nitrogenation at ortho- or para-position, b) safety problems from strong acids (sulfuric acid and nitric acid) used twice when introducing and rearranging nitrogen groups, and c) utilization of high-flammable Raney Ni.
cut paste
Novel and Practical Synthesis of Candesartan Cilexetil
Yongjun Mao, Ruisheng Xiong, Zheng Liu, Haihong Li, Jingkang Shen, and Jingshan Shen* *Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Rd., Zhangjiang Hi-Tech Park, Shanghai, 201203, China
Abstract
A novel and convergent synthetic route of candesartan cilexetil (API of Atacand), an effective angiotensin II receptor blocker, is described. Cleavage of the N-Boc and N-trityl protective group are implemented simultaneously and formation of the benzimidazole ring is conducted at the last step of this route, which gives candesartan cilexetil in 55% yield over six steps with 99.1% purity (HPLC). Full Text HTMLPDF (567KB)PDF with Links (932KB)
 This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
| Benzimidazole derivs., their production and use | |
| Naka, T.; Nishikawa, K.; Kato, T. (Takeda Chemical Industries, Ltd.) | |
| EP 0459136; EP 0720982; JP 1992364171; JP 1996099960; US 5196444; US 5328919; US 5401764; US 5703110; US 5705517; US 5962491; US 6004989 |
more info Candesartan cilexetil of Formula I, disclosed in U.S. Patent No. 5,196,444 as crystalline form, i.e., Form-I (C-type crystals), is chemically described as 1- cyclohexyloxycarbonyloxyethyl 2-ethoxy-3-[[4-[2-(2H-tetrazol-5- yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate.

H Formula I It is useful in the treatment of cardiovascular complaints such as hypertension and heart failure. Candesartan cilexetil is poorly soluble in water, which is attributed to its hydrophobic nature. Solubility plays an important role in achieving the desired concentration of a drug in systemic circulation for accomplishing the pharmacological response. Various techniques are known in literature to increase the solubility of poorly-soluble drugs, including decreasing the particle size, complexation, changing the surface characteristics of the particles, and incorporation of drug particles into colloidal systems like nanoparticles and liposomes. Among these, the most commonly used technique to increase the solubility is particle size reduction. Sometimes the rate of dissolution of a poorly-soluble drug is the rate limiting factor in its rate of absorption by the body. These drugs may be more readily bioavailable if administered in a finely divided state. Particle size reduction increases the surface area causing an increase in the dissolution rate of the compound, and hence, its bioavailability. There are certain techniques reported in literature to reduce the particle size of such poorly-soluble drugs. PCT Publication No. WO 2006/122254 discloses stable candesartan cilexetil of fine particle size, wherein the stable micronized candesartan cilexetil is prepared by slurrying a sample of candesartan cilexetil of fine particle size in a suitable solvent for a suitable amount of time. In this application, candesartan cilexetil of fine particle size is obtained directly from the synthesis of candesartan cilexetil or by comminuting candesartan cilexetil using milling. PCT Publication No. WO 2005/123720 describes fine particles of candesartan cilexetil having improved pharmacokinetic profile and a process for their production, wherein fine particle size is obtained by a) dissolving candesartan cilexetil in an organic solvent; b) cooling the solution obtained in step a) under stirring to crystallize candesartan cilexetil from the solution; and c) isolating candesartan cilexetil having a particle size of with d90 not more than about 25 μ. U.S. Patent Application No. 2006/0165806 describes compositions comprising a candesartan, such as candesartan cilexitil. The candesartan particles of the composition have an effective average particle size of less than about 2000 nm. U.S. Patent Application No. 2008/0038359 describes a nanoparticle pharmaceutical formulation comprising a poorly soluble drug substance having an average particle size of less than about 1000 nm, a solid or semisolid dispersion vehicle, and optionally a non-surface modifying excipient. U.S. Patent No. 7,828,996 discloses the methods for forming nanoparticles of a material of narrow polydispersity with ultrasonic waves using a partially submersed sonicator that does not touch any part of the apparatus and the point of addition of organic solvent is in the wave funnel produced by sonication and within the selected distance from the wave-source depending on the desired particle size. U.S. Patent No. 7,780,989 discloses the preparation of a dispersion of nanocrystalline particles in an aqueous medium using ultrasound. U.S. Patent No. 5,314,506 describes a crystallization process in which a jet of a solution containing a substance is impinged with a second jet containing an anti-solvent for the substance. The rapid mixing produced by the impinging jets results in a reduction of the crystals so formed compared to conventional slow crystallization processes. The smallest crystals disclosed are about 3 μ and the majority are in the range of from about 3 μ to about 20 μ. PCT Publication No. WO 00/44468 describes a modification to the apparatus described in U.S. Patent No. 5,314,506, wherein ultrasound energy is applied at the point of impingement of the two jets to further enhance localized mixing and is stated to give direct formation of small crystals with a diameter of less than 1 μ. Generally, the crystalline particles described have an average size of 0.5 μ. Conventional particle size reduction methods such as high energy milling may result in loss of yield, noise and dusting, as well as unwanted exposure to highly potent pharmaceutical compounds. Also, in the case of crystalline compounds, stress generated on crystal surfaces during milling can adversely affect labile compounds. Therefore, there is a need for a process for particle size reduction of candesartan cilexetil, which is industrially advantageous, easy to handle and is cost effective.
-
Candesartan is a potent, selective AT1 subtype angiotensin II receptor antagonist and used for treatment of hypertension. Due to poor absorption of Candesartan in body, the prodrug candesartan cilexetilwas developed. The candesartan cilexetil is rapidly and completely hydrolyzed to candesartan in gastrointestinal tract.
-
[0004]U.S. Pat. No. 5,196,444 discloses Candesartan cilexetil and a process for its preparation by the reaction of 2-ethoxy-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid with trityl chloride in presence of triethyl amine in methylene chloride and purification by column chromatography gives 2-ethoxy-1-[[2′-(N-triphenylmethyltetrazol-5-yl)-biphenyl-4-yl]methyl] benzimidazole -7-carboxylic acid, which upon condensation with cyclohexyl 1-iodoethyl carbonate in presence of potassium carbonate in DMF followed by purification with column chromatography gives a colorless powder which is recrystallized in ethanol yields ‘C’ type crystals of Candesartancilexitil.
-
[0005]U.S. Pat. Application No. 2005/131027 discloses a process for preparation of candesartan cilexetil by reaction of trityl candesartanwith cilexetil halide and at least one base in a low boiling solvent in presence of phase transfer catalyst to give Trityl candesartan cilexetil, which upon deprotection with at least one organic acid in at least one organic solvent. U.S. Pat. Application 2005/131027 further discloses the deprotection of Trityl candesartan cilexetil in methanol without an acid.
-
[0006]The PCT publication WO 2005/021535 discloses the deprotection of Trityl candesartan cilexetil with neutral or slightly basic medium in alcohol.
-
[0007]Chem.Pharm.Bull. 47(2), 182-186 (1999) discloses two novel crystalline forms of Candesartan cilexetil, form-I and form-II.
-
[0008]PCT publication WO 04/085426 discloses Candesartan cilexetil 1,4-Dioxane solvate and two more crystalline forms, designated as form-III and form-IV. The disclosed process for preparation of form-III involves crystallization of Candesartan cilexetil in toluene and for form-IV involves crystallization in a mixture of methyl tert-butyl ether and methanol.
-
[0009]PCT publication WO 2005/077941 discloses several crystalline forms, solvates of Candesartan cilexetil along with a process for preparation of form-I (type-C).
-
[0010]The prior art disclosed methods for preparation of Candesartan cilexetilinvolves purification of Trityl candesartan and Candesartan cilexetil by column chromatography or involves the use of strong acids like IN HCl or the use of organic acids or without an acid in methanol for detrytilation of Trityl candesartan cilexetil.
-
[0011]There is a requirement of a process for preparation of Candesartancilexetil which yields a pure Candesartan cilexetil without involving the purification by column chromatography and the usage of strong acids for deprotection.
Candesartan cilexetil of formula (I) shown beiow is chemicaily described as (+/-)-1- [[(cyclohexyloxy)carbonyl]oxy]ethyl-2-ethoxy-1 -[[2′-(1 H-tetrazol-5-yl)-1 , 1 ‘-biphenyl- 4-yl]methyl]-1 H-benzimidazoie-7-carboxylate. An alternative designation is (+-)-1- hydroxyethyf 2~Ethoxy-1 -{p-(o-1 H-tetrazo!-5-yIphenyi)benzyJ)-7-benzϊmidazoie~ carboxyiic acid cyclohexyl carbonate (ester), with candesartan being the underlying carboxylic acid, i.e. 2-Ethoxy-1 -(p-(o-1 H-tetrazol-5-ylphenyl)benzy!)-7-benz- imidazolecarboxylic acid.

Because of its ability to inhibit the angiotensin-converting enzyme it is widely used for the treatment of hypertension and related diseases and conditions. As an angiotensin Ii receptor antagonist, candesartan ciiexetil avoids the side-effects of calcium antagonists, and shows high stability and obvious curative effects. Currently candesartan ciiexetil is soid as racemic mixture, it is produced according to published patents, e.g. EP 0 720 982 B1 and EP 0 459 136. in Chem. Pharm. Bull. 47(2), 182-186 (1999) two crystalline forms (Form I and II), together with an amorphous form, are disclosed and characterized by their DSC thermograms, X-ray diffraction patterns and IR spectra. US 5,196,444 disclosed the C-type crystal (Form I) of candesartan cilexetif, and processes for producing it under acidic conditions. WO 04/085426 discloses the dioxane solvate of candesartan ciiexetil, together with two additional crystalline forms. WO 2005/077941 discloses hydrates and solvates of candesartan ciiexetil, together with processes for their preparation. WO 2006/048237 also describes the preparation of new polymorphic forms ofcandesartan ciiexetil, together with processes for their preparation, including the preparation of amorphous candesartan ciiexetil by precipitating it with a liquid cyclic hydrocarbon from a solution of candesartan ciiexetil in a chlorinated solvent. in WO 2005/123721 processes for the preparation of amorphous candesartanciiexetil are provided, comprised of spray-drying and precipitation. HPLC CUT PASTE , READER TO PICK ONLY REQUIRED INFO Candesartan cilexetil (60 g) is dissolved in isopropanol (900 m!_) at 60-65 0C. Solution is hot filtered into reactor and quickly cooled to 35 0C. At this temperature nucleation is provoked with 300 mg of candesartan cilexetil form I and stirring is enforced. Suspension is cooled to 3O0C in 1 hour and rigorous stirring is continued at this temperature for additional 5 hours. Then stirring power is reduced and the suspension is cooled to 2O0C in 8 hours. The product is filtered, washed with isopropanoi and dried for 2 hours at 38°C. Yield: 48.7 g of candesartan cilexetil form I. Area % HPLC: candesartan cilexetil: 99.73%, alky ester of candesartan cilexetil 0.08%, candesartan cilexetii pyran below 0.05%, tritylcandesartan cϋexetil 0.09% Average particle size: 19 /vm, no agglomerates present (see Figure 2) B) Detection of impurities in candesartan cilexetil Example 6 Detection of candesartan cϊlexetil pyran in candesartan cilexetii by HPLC HPLC (external standard method) was performed using the following specifications : Column: Zorbax Eclipse XDB-C18, 50 mm x 4.6 mm i.d.τ 1.8 μm particles Eluent A: 0.01 M NaH2PO4, pH 2.5 Eluent B: acetonitriie Gradient of Eluent:

Flow rate: about 1.2 ml/min Diluent: acetonitriie : water = 70 : 30 (V/V). Detection: UV, wavelength 225 nm injection volume: 5 μl Column temperature : 500C Autosampler temperature: 7°C Example 7 Detection of cilexetil pyran in 1 -chloroethyl cyclohexylcarbonate by GC GC/FID (area percent method) was performed using the following specifications: Column: capillary (fused-silica) AT-WAX or adequate Length: 30 m ID: 0.32 mm Film thickness: 0.25 μm Carrier gas: helium Carrier gas flow rate: 2.0 ml/mi n Split ratio: 10 : 1 Air flow rate: 400 ml/min Hydrogen flow rate: 40 ml/min Make up gas flow ISb rate: 25 ml/min Column temperature 100°C (0 min) → 10°C/min → 2000C (10 min or prolonged if necessary) Injector temperature: 21 O0C Detector temperature: 250OC Injection volume : 1 μl Diluent: Acetonithle: chromatography grade. Chromatographic system suitability Signal/noise of 1 -chloroethyl cyclohexyl carbonate: not less than 10
………………
Seki M * Mitsubishi Tanabe Pharma Corporation, Osaka, Japan An Efficient C–H Arylation of a 5-Phenyl-1H-tetrazole Derivative: A Practical Synthesis of an Angiotensin II Receptor Blocker. Synthesis 2012; 44: 3231-3237 
Significance
Candesartan cilexetil (Atacand®) is an angiotensin II receptor antagonist that is prescribed for the treatment of hypertension. It is a prodrug that is hydrolyzed to candesartan in the gut. The synthesis depicted, features an efficient protocol for ruthenium-catalyzed C–H arylation of the tetrazole A. Comment A significant challenge in this small-scale synthesis was the final removal of the benzyl protecting group from the tetrazole unit using transfer hydrogenation. Best results were obtained using a ‘thickshell’ Pd/C catalyst from Evonik
RABEPRAZOLE

Pariprazole sodium;Rabeprazole sodium;LY-307640;E-3810;Aciphex;Pariet
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an antiulcer drug in the class of proton pump inhibitors. It was developed by Eisai Co. and is marketed by Janssen-Cilag as the sodium salt under the brand names AcipHex (/ˈæsɨfɛks/, referring to pH) in the US, Pariet in Europe, Brazil, Canada, Japan, Russia and Australia, Acigard, Cyra, Rabium, Esoon,Orporo, Parit, Rabemac, Rabiloz, Razo, Rabifast, Rablet and Rabsiv in India, and Zechin in Pakistan.
Rabeprazole, 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole has the following structural formula

Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H+, K+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. So that it can effectively inhibit the secretion of an acid and is therefore effective in the therapy or prevention of human and animal peptic ulcer.
-
US 5045552 discloses the preparation of Rabeprazole sodium by known traditional procedures, such as dissolution of the product in a mixture of stoichiometric quantity of aqueous sodium hydroxide and ethanol, then removal of water azeotropically, thereafter drying the residue at low pressure and then crystallization of the residue with less polar solvent such as diethyl ether, tert-butyl methyl ether.
The U.S. Pat. No. 5,045,552 discloses the Rabeprazole and many other substituted benzimidazole-type compounds having anti-ulcer activity. This patent further discloses the process for preparation of Rabeprazole by oxidation of Rabeprazole sulfide using 85% m-chloroperbenzoic acid in a mixture of dichloromethane and diethyl ether followed by work up to get product as oil. The obtained oil is crystallized from a mixture of dichloromethane/ether. Optionally the oily crude is dissolved in aqueous solution of sodium hydroxide. The obtained solution is subjected to azeotropic distillation with ethanol to remove water and adding ether to get crystalline Rabeprazole base.

According to the prior art, Rabeprazole base is crystallized using dichloromethane/ether to obtain crystalline off white product. The HPLC purity is less than or equal to 99% and the isolation procedure involves azeotropic distillation of water, during which the product is exposed to high temperature and leads to certain impurities. Repeated crystallization is needed to remove impurities to get desired quality. Using large volumes of chlorinated solvents in the plant leads to environmental hazardous.
Japanese patent application JP2001039975 teaches that the product obtained by example 33 of U.S. Pat. No. 5,045,552 with a melting range of 140-141° C. corresponds to amorphous rabeprazole sodium
The U.S. Pat. No. 6,919,459 patent also discloses the process for the preparation of Rabeprazole by oxidation of Rabeprazole sulfide using m-Chloroperbenzoic acid (m-CPBA) in a suitable solvent. The reaction mass is subjected to repeated washings at different pH levels and isolate the product from aqueous layer.

Rabeprazole is not stable at acidic conditions and decomposes to form unknown impurities. To remove these impurities repeated crystallizations are required to get desire quality of the final product.
The WO2006/117802 PCT application discloses the process for the preparation of Rabeprazole sodium by oxidation of Rabeprazole sulfide with sodium hypo halite solution in water or a mixture of water and water miscible solvent medium using alkali metal hydroxide and catalyst. The reaction mass is saturated by inorganic saturating agents and the Rabeprazole sodium salt is extracted with water immiscible organic solvent. Organic solvent is distilled and the residue is dissolved in second organic solvent to get clear solution, which is precipitated by adding antisolvent.
The WO2006/120701 PCT application discloses process for manufacture of amorphous Rabeprazole sodium by the reaction of Rabeprazole base with aqueous sodium hydroxide. Ethanol is added to the obtained solution. Solvents are distilled from the solution to get thick mass. Organic solvent is added to the obtained residue to get clear solution, to which antisolvent is added to get amorphous Rabeprazole sodium.
The prior art methods cited above have many disadvantages, these methods involve more number of organic solvents and lack successive extractions and washings of the layers during work up procedure. It leads to many impurities that ultimately affect on purity and yield loss of final product.
The U.S. Pat. No. 6,180,652 and WO 2003101452 PCT application discloses the process for the preparation of amorphous rabeprazole sodium, which is obtained by lyophilization of an aqueous solution of rabeprazole sodium acetone complex and an aqueous NaOH solution of Rabeprazole respectively.

Lyophilization technique is not suitable for production at industrial scale and it needs more time cycle and involves the cost.
We observed that rabeprazole is rapidly degraded in chlorinated solvent like dichloromethane to form unknown impurities, due to impurities while distillation gummy material is formed. It leads to yellowish color in final product, finally it leads to yield loss in final product.
According to prior art methods,
-
- (a) Dichloromethane/ether is used for final crystallization gives off white product with HPLC purity less than or equal to 99% and
- (b) Rabeprazole sodium is isolated by using azeotropic distillation. It needs high temperature to remove water and the reaction mass is exposed to high temperature to form unknown impurities, to remove these impurities repeated crystallizations are required to get desire quality of the final product
US 6,313,303 discloses the preparation of sulfoxides by oxidizing thio ether with a peroxoborate salt in the presence of an acid anhydride or a metal catalyst; and the preparation of sulfoxides by oxidizing thio ether with an N- halosuccinimide, l,3-dihalo-5,5-dimethyl-hydantoin or dichloroisocyanuric acid salt in the presence of a base.
IN 192030 discloses the purification process of Rabeprazole, in which sulfone enriched Rabeprazole is treated with an amino alcohol e.g. ethanolamine in the presence of an organic solvent, further the reaction mixture washed with water to remove the sulfone impurities. US 7,439,367 (IN218648, 058/MUM/2003, 193/MUM/2003) discloses the preparation of Rabeprazole by oxidizing its corresponding sulfide compound, where aqueous hypohalite solution is used as an oxidizing agent. The said oxidation is carried out at a controlled temperature and pH. During said oxidation the pH of the reaction mixture is maintained in the range of 9 to 12. This process utilizes catalyst such as pyridine, di-isopropyl ethyl amine and N,N-dimethyl amino pyridine.
US 7,060,837 discloses the purification of lansoprazole using ammonia, ammonium hydroxide, diethylamine, triethylamine and methylamine in the presence of solvent. The said patent utilizes acid for the isolation of lanzoprazole in pure form.
US 2008/0161579 (IN190/MUM/2005) discloses a process for the preparation of Rabeprazole sodium comprising oxidation of Rabeprazole sulfide with sodium hypohalite in water or a mixture of water and water miscible solvent using alkali metal hydroxide and catalyst. It also discloses a process for the preparation of Rabeprazole sulfide.
WO 2008/045777 (1856/CHE/2006) discloses the preparation of
Rabeprazole by oxidizing the corresponding sulfide compound using about 0.8 to 1.25 equivalents of an oxidizing agent in the presence of less than or about 2.25 equivalents of a base where aqueous sodium hypohalite used as an oxidizing agent.
WO 2006/024890 discloses a process for the preparation of Rabeprazole in which the Rabeprazole obtained was treated with the triethylamine in hexane. The use of n-hexane in the final stage is not suitable for manufacturing point of view as it is difficult to remove residual hexane solvent. There are several disadvantages associated with such known processes; all the methods reported in these prior arts leads to the formation of many impurities which ultimately affects the purity of the final product.
US 5,045,552 patent discloses the preparation of Rabeprazole by oxidizing the Rabeprazole sulfide using m-chloroperbenzoic acid as shown in scheme-I. The crude Rabeprazole was dissolved in sodium hydroxide and the resulting solution was azeotropically distilled together with ethanol thrice to remove the water. Finally ether was added to get the crystals of Rabeprazole sodium
WO 03/101452 discloses a method for the preparation of Rabeprazole sodium comprising dissolving Rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization.



Souda, S.; Ueda, N.; Miyazawa, S.; Tagami, K.; Nomoto, S.; Okita, M.; Shimomura, N.; Kaneko, T.; Fujimoto, M.; Murakami, M.; Oketani, K.; Fujisaki, H.; Shibata, H.; Wakabayashi, T. (Eisai Co., Ltd.); Pyridine derivs., pharmaceutical compsns. comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same. AU 8781138; EP 0268956; EP 0475456; EP 0654471; EP 0786461; JP 1989006270; JP 1993247035; JP 1995291967; US 5045552; US 5998445 .
Castaner, J.; Prous, J.; E-3810. Drugs Fut 1991, 16, 1, 19.
Sohda, S.; Tagami, K.; Chiku, S.; Synthesis of 14C-labelled sodium pariprazole (E3810). J Label Compd Radiopharm 1993, 33, 9, 849.

Rabeprazole as “CYRA” (Systopic Labs Pvt Ltd), “Elpizole” (Orchid Chemicals & Pharmaceuticals), Elpizole-20 (Orchid Chemicals & Pharmaceuticals), Rablet (Lupin), Acigard (3D), AcipHex, Rabeloc, Pariet, Rabider (Duta Formulations) Rabsiv 20 (Saharsh Biologicals) is supplied in:
- Tablet, enteric-coated; 10 mg
- Tablet, enteric-coated; 20 mg
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
HPLC METHOD
Rabeprazole with more impurities, particularly at 2.12 RRT (393 mass), 3.51 RRT (491 mass), 4.47 RRT (457 mass), 4.85 RRT (684 mass) and 4.54 RRT (893 mass). The mass (molecular or formula weight) number of the impurities were identified using LCMS. Particularly, the obtained product contains unknown impurities of higher molecular weight in the range of 0.1-1.0 % at relative retention time (RRT) of 2.12, 3.51, 4.47, 4.85, and 4.54 RRT as measured by high performance liquid chromatography (HPLC) method provided below.
The purity of the product obtained is determined by high performance liquid chromatography method under the conditions mentioned below.
Column: Prontosil Kromabond 100-5-C18 (250 x 4.6 mm), 5μ,
Mobile phase A: 1.36g KH2PO4 to 1 litre water, 0.5ml OfEt3N, Mobile phase B: Methanol: ACN (95:5),
Diluent: Mobile phase A and ACN (70:30),
Flow Rate: 1.0 mL/min,
Detection: UV at 280 nm,
Injection Volume: 20 μL, Run Time: 60 min.
Column oven temperature: 3O0C. Surprisingly the applicant identified a method in which, crude Rabeprazole was treated with diethylamine and optionally addition of TBAB (tetrabutylammmonium bromide) as catalyst, where the impurity level reduced. Though the reported amines like triethyl amine, ethanolamine, and ammonia are effectively used to minimize sulfone impurity, those are failed or unsatisfactory to remove the impurities at 2.12 RRT, 3.51 RRT, 4.47 RRT, 4.85 RRT and 4.54 RRT.
SPECTRAL DATA
EP 1869015 B1 FOR RABEPRAZOLE SODIUM
IR Spectra (KBr, cm-1): 3382, 2927, 1583, 1462, 1384, 1298, 1269, 1190, 1157, 1093, 1018, 745.
H NMR Spectra [200 M Hz, CD3OD] δ (ppm): 8.23 – 8.25 (1H, d, ArH); 7.57 – 7.62 (2H, m, ArH); 7.0 – 7.09 (2H, m, ArH); 6.87 – 6.90 (1H, d, ArH); 4.57 – 4.63 (2H, d, O=S-CH2-Ar); 4.0 – 4.1 (2H, t, -O-CH2-CH2-); 3.49 – 3.55 (2H, t, -CH2-O-CH3); 3.31 (3H, s, -OCH3); 2.1 (3H, s, Ar-CH3); 1.96 – 2.0 (2H, t, -CH2-CH2-CH2-).
MP
As per the process described and exemplified in the U. S. Patent No.
5,045,552, rabeprazole sodium is prepared by oxidizing 2-[[4-(3- methoxyporpoxy)-3-methylpyridine-2-yl]rnethylthio]-1 H-benzimidazole with m- chloroperbenzoic acid to afford the rabeprazole base which is further converted to its sodium salt by using 0.1 N aqueous solution of sodium hydroxide, followed by addition of ethanol. The water is removed by azeotropic distillation and the product is precipitated by using ether as solvent such as diethyl ether, tert-butyl methyl ether. The melting point of the disclosed rabeprazole sodium salt is 140- 1410C. The isolation process described in the U. S. Patent No. 5,045,552 has numerous disadvantages such as large volume of solvents is required for azeotropic removal of water during which the product is exposed to high temperature and leads to certain impurities. Based on these drawbacks the isolation process finds to be unsuitable for preparation of amorphous rabeprazole sodium at commercial scale operations.
Japanese patent application JP 2001039975 indicates that the product obtained by example 33 of the U. S. Patent No. 5,045,552 with a melting point of
140-1410C corresponds to amorphous rabeprazole sodium. In this application, the X-ray powder diffraction pattern of the amorphous rabeprazole sodium is shown.
The PCT patent publication No. WO 03/101452 discloses a method for the preparation of rabeprazole sodium comprising dissolving rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization. U.S. Patent No. 6,180,652 B1 (the ‘652 patent) describes acetone complex of rabeprazole sodium, process for its production and characterizes it by powder X-ray diffraction, infra-red spectroscopy and 1H-NMR spectroscopy. The ‘652 patent further reports a process for preparation of amorphous rabeprazole sodium by lyophilizing (freeze-drying) an aqueous solution of rabeprazole sodium acetone complex.
However, lyophilization is a technique, which is not suitable for production at industrial scale because this process presents serious limitations on cost, time, equipment capability and environmental protection.
According to PCT patent publication No. WO 2004/085424A1 , amorphous rabeprazole sodium is obtained by heating the rabeprazole sodium acetone complex at elevated temperature, preferably between 100 and 1100C. It is well known that exposing rabeprazole-type compounds to high temperatures increases the risk of decomposition to form impurities and as such, heat treatment of rabeprazole sodium acetone complex into amorphous rabeprazole sodium is not adequate for the production of a rabeprazole which is suitable for pharmaceutical use.
PCT patent publication No. WO 2007/023393 A2 reports a process for preparation of amorphous rabeprazole sodium, the said process comprises: i) contacting rabeprazole sodium acetone complex with a first solvent system which includes a hydrocarbon solvent or an ether solvent or an alcohol solvent or mixtures thereof; ii) filtering the solid from the solvent system used in step i) or distilling the solvent system used in step i) under reduced or atmospheric pressure, to thereby obtain a residue; iii) contacting the wet solid or the residue of step ii) with a second solvent system which includes a hydrocarbon solvent or an ether solvent; and iv) filtering to obtain a wet solid from the solvent system used in step iii) to obtain a wet solid.
The methods for preparation of amorphous rabeprazole sodium as described in the patents U.S. Patent No. 6,180,652 B1 , PCT patent publication No. WO 2004/085424A1 and PCT patent publication No. WO 2007/023393 A2 involves lengthy process i.e., proceeds via rabeprazole sodium acetone complex intermediate and also the yields obtained in these processes are very low.
U.S. Patent Application No. US2004/0180935A1 teaches a process for production of amorphous rabeprazole sodium by dissolving rabeprazole acid in a mixture of sodium hydroxide and methanol at 25-350C, removing the solvent by evaporation and precipitating the product by adding petroleum ether.
PCT patent publication No. WO 2006/120701 A1 teaches a process for manufacture of amorphous rabeprazole sodium with mean particle diameter between 10 to 55 μm, the said process comprises, addition of rabeprazole to aqueous sodium hydroxide; addition of ethyl alcohol to the solution; distillation of solvents from the solution thus obtained till thick mass is obtained; addition of an organic solvent selected from ethyl acetate, dichloromethane, chloroform, butyl acetate, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, to the residue to obtain a clear solution; addition of this clear solution to an anti-solvent includes diisopropyl ether, diethyl ether, methyl tert-butyl ether, under agitation and isolation of the product.
Since a solvent may play an important role in increasing the yield rate or in determination of physical properties of drug substance such as crystal form, purity, solubility, etc., even if such a solvent is known to be toxic, there may be many cases that the use thereof in the preparation of drug substance cannot be avoided in terms of risk benefits. In such cases, this guideline (ICH guidelines Q3C(R3)) decrees that a concentration of a residual solvent in drug substance should be not more than a specified value, which is toxicologically acceptable. The methods for preparation of amorphous rabeprazole sodium as described in the patents, U.S. Patent Application No. US2004/0180935A1 and PCT patent publication No. WO 2006/120701 A1 suffers with residual solvent problem and thereby commercially not viable. These methods utilize the solvents like diisopropyl ether and petroleum ether as precipitating solvents. These solvents are difficult to remove completely by practical manufacturing techniques. According to the ICH guidelines Q3C(R3), there is no adequate toxicological data for the solvents like diisopropyl ether and petroleum ether on which to base a PDE was found. However, a need still remains for an improved and commercially viable process of preparing pure amorphous rabeprazole sodium that would solve the aforesaid problems associated with processes described in the prior art, which will be suitable for largr-scale preparation, in terms of simplicity, chemical yield and purity of the product, and which would carry out with comparatively smaller volume of solvent
Iroko Pharmaceuticals Receives FDA Approval for ZORVOLEX™
![]()
Philadelphia, Pennsylvania, October 18, 2013 – Iroko Pharmaceuticals, LLC, a global specialty pharmaceutical company dedicated to advancing the science of analgesia, today announced that the U.S. Food and Drug Administration (FDA) has approved ZORVOLEX™ (diclofenac) capsules, a nonsteroidal anti-inflammatory drug (NSAID), for the treatment of mild to moderate acute pain in adults[i]. ZORVOLEX was approved at dosage strengths that are 20 percent lower than currently available diclofenac products. FDA approval of ZORVOLEX was supported by data from a Phase 3 multi-center, randomized study in which patients treated with ZORVOLEX reported significant pain relief compared with patients receiving placebo
FINASTERIDE

(5α, 17β)-N-(1 ,1-dimethylethyl)-3-oxo-4-aza-androst-1-ene-17-carboxamide, finasteride, a 4-aza-steroid compound 5 which exhibits pharmaceutical activity as an inhibitor of the enzyme testosterone 5-α-reductase, and is useful in the treatment of prostate cancer
Finasteride;YM-152;MK-906;Prodel;Propecia;
Chibro-Proscar;Finastid;Prostide;Andozac;Proscar
Finasteride (brand names Proscar and Propecia by Merck, among other generic names) is a synthetic drug for the treatment of benign prostatic hyperplasia (BPH) and male pattern baldness (MPB). It is a type II 5α-reductase inhibitor. 5α-reductase is an enzymethat converts testosterone to dihydrotestosterone (DHT).

Figure . Conversion of testosterone to dihydrotestosterone.
Chemical synthesis

Propecia (finasteride) 1 mg tablets

Propecia 1 mg & Finpecia 1 mg tablets
Finasteride is synthesized fromprogesterone:

History
In 1974, Julianne Imperato-McGinley of Cornell Medical College in New York attended a conference on birth defects. She reported on a group of intersex children in the Caribbean who appeared sexually ambiguous at birth, and were initially raised as girls, but then grew external male genitalia and other masculine characteristic post-onset of puberty. Her research group found that these children shared agenetic mutation, causing deficiency of the 5α-reductase enzyme and male hormone dihydrotestosterone (DHT), which was found to have been the etiology behind abnormalities in male sexual development. Upon maturation, these individuals were observed to have smaller prostates which were underdeveloped, and were also observed to lack incidence of male pattern baldness.
In 1975, copies of Imperato-McGinley’s presentation were seen by P. Roy Vagelos, who was then serving as Merck’s basic-research chief. He was intrigued by the notion that decreased levels of DHT led to the development of smaller prostates. Dr. Vagelos then sought to create a drug which could mimic the condition found in these children in order to treat older men who were suffering from benign prostatic hyperplasia.
In 1992, finasteride (5 mg) was approved by the U.S. Food and Drug Administration (FDA) for treatment of benign prostatic hyperplasia(BPH), which Merck marketed under the brand name Proscar.
In 1997, Merck was successful in obtaining FDA approval for a second indication of finasteride (1 mg) for treatment of male pattern baldness (MPB), which was marketed under the brand name Propecia.
CHEMISTRY
Formerly known as MK-906, finasteride (Figure 1) ([5-![]() , 17-
, 17-![]() -N-(1,1-dimethylethyl) -3-oxo-4-azaandrost- 1-ene-17-carboxamide) belongs to the 4-azasteroid structural class of compounds. (Click on the structure to the right to view a Chime rotatable structure.) Its synthesis, shown in Scheme 1, was published by Rasmusson et al. in 1986.[2] Briefly, beginning with a previously synthesized intermediate, the A-ring of the steroid skeleton was converted from its 3- keto precursor (1) to the required 4-aza system (3) through an open analog (2). Saturation of the B-ring using catalytic hydrogenation gave intermediate 4. Use of the 2-pyridyl thio ester (5) gave a reactive substrate to form the tertiary butyl carboxamide (6). The final step in the synthesis, dehydration of the A-ring with benzeneselenic anhydride, gave the final product, finasteride (7).
-N-(1,1-dimethylethyl) -3-oxo-4-azaandrost- 1-ene-17-carboxamide) belongs to the 4-azasteroid structural class of compounds. (Click on the structure to the right to view a Chime rotatable structure.) Its synthesis, shown in Scheme 1, was published by Rasmusson et al. in 1986.[2] Briefly, beginning with a previously synthesized intermediate, the A-ring of the steroid skeleton was converted from its 3- keto precursor (1) to the required 4-aza system (3) through an open analog (2). Saturation of the B-ring using catalytic hydrogenation gave intermediate 4. Use of the 2-pyridyl thio ester (5) gave a reactive substrate to form the tertiary butyl carboxamide (6). The final step in the synthesis, dehydration of the A-ring with benzeneselenic anhydride, gave the final product, finasteride (7).

Scheme 1. Key intermediates in the synthesis of finasteride by Rasmusson et al. Reagents: a, KMnO4-NaIO4, t-BuOH, reflux; b, NH3, heat; c, H2, Pt, ArOH; d, 2,2′-dipyridyl disulfide, triphenylphosphine, toluene; e, t-butyl amine, THF; f, benzeneselenic anhydride, chlorobenzene.
The preparation of finasteride is described and claimed in U.S. Patent 4.377.584 and further described in U.S. Patent 4.760.071. Other patents which pertain to the preparation of finasteride include Canadian patent application 2.029.859: U.S. patents 5.084.574 and 5.116.983: and Canadian patent applications 2.049.882 and 2.049.881. All these teach the conversion of a final intermediate tofinasteride, which is purified and isolated as a crystalline solid. Althoughfinasteride polymorphs are not mentioned specifically in these items of prior art, the finasteride obtained using them, as a crystalline solid, must be in one or other of the known polymorphic forms, or a mixture of both of them.
Aforementioned Canadian Patent Application 2.103.107 Dolling et a published May 20, 1994, describes preparations of finasteride and the specific polymorphic Form I and Form II thereof. In particular, it teaches that polymorphic Form I can be prepared by crystallization from a mixture of finasteride in an organic solvent and optionally water, such that the amount of organic solvent and water in the mixture is sufficient to cause the solubility of the non-solvated form of finasteride(Form I) to be exceeded and the non- solvated form of finasteride to be less soluble than any other form of finasteride in the mixture. It also teaches that the polymorphic Form I of finasteride can be prepared by heating the polymorphic Form II of finasteride to at least 25°C in water or an organic solvent for a sufficient period of time to effect the conversion. The same reference teaches that polymorphic Form II finasteride can be prepared by crystallization from a mixture of finasteride in an organic solvent and water, such that the amount of organic solvent and water in the mixture is sufficient to cause the solubility of the solvated form of finasteride to be exceeded and the solvated form of finasteride to be less soluble than any other form of finasteride in the mixture, followed by recovery of the solid and removal of the solvent therefrom; or by heating polymorphic Form I finasteride to at least to about 150°C for sufficient time to complete the conversion.
MORE INFO
-
Finasteride, marketed under the tradename of PROSCAR®, by Merck & Co., Inc is 17β-(N-tert-butyl carbamoyl)-4-aza-5α-androst-1-en-3-one and is a 5α-reductase inhibitor for use in treating acne, female hirsutism, and particularly benign prostatic hyperplasia. See US Patent 4,760,071 (1988), the entire disclosure of which is incorporated herein by reference..
-
[0002]The synthesis of finasteride in US Patent 4,760,071 involves reacting the 17β-(2-pyridylthio) carboxylate of 4-aza-5α-androst-1-ene-3-one with t-butylamine. A further synthesis of finasteride is described in Synthetic Communications, 30 (17), p. 2683-2690 (1990). including the reacting of the 17-acylimidazole of 4-aza-5α-androst-1-en-3-one with t-butylamine.
-
[0003]However, both of these reactions require the use of heterocyclic aromatic amines which are expensive and give rise to environmental safety and toxicity considerations. Both of these intermediates are prepared from the 17β-carboxylic acid.
-
[0004]The Bodroux reaction, described by F. Bodroux in the references, Bull. Soc. Chim. France 33, 831 ( 1905); 35, 519 (1906); 1, 912 (1907); Compt. Rend. 138, 1427 (1904); 140, 1108 (1905); 142, 401 (1906) discloses the reaction of the magnesium halide salts of amines with esters. However, there is no description or teaching that the reaction can be applied to the reaction of a sterically hindered amine, e.g. t-butyl amine, with a sterically hindered ester such as 1.

The first method (International Patent: W0200507M97A) is finasteridedihydro as raw materials, benzeneseleninic anhydride synthesis of finasteride, the reaction is as follows:

Used in this reaction toxic and expensive reagents benzeneseleninic anhydride, yield only about 50%, and the product to column chromatography to separate, while the use of certain toxic chlorobenzene as solvent, the cost is very high , environmental hazards large.
[0005] The second method (U.S. Patent No.: US20070167477A1) is finasteridedihydro as raw materials, the use of DDQ / BSTFA (i.e. 3,3 – dichloro-5 ,6 – dicyano-p-benzoquinone / second (third trimethylsilyl) trifluoroacetamide) Oxidation get finasteride, the reaction is as follows:

The reaction yield about 65%, the resulting fluorine-containing wastewater intractable, quinones great harm to the environment.
[0006] The third method (international patent: W02008101308A) is dihydrofinasteride as raw material, the use of phenyl sulfide oxidation get finasteride, the reaction is as follows:

The method steps and more complicated to operate, the total yield of only 60%, the use of expensive lithium diisopropylamide, lithium bis trimethylsilyl test Qi IJ, the cost is higher.
REF
Rasmusson, G.H.; Reynolds, G.F. (Merck & Co., Inc.); 17beta-Substd.-4-aza-5alpha-androstenones and their use as 5alpha-reductase inhibitors. AU 8539167; EP 0155096; EP 0314199; ES 8702430; JP 1985222497; JP 1989093600; US 4760071 .
Rasmusson, G.H.; Reynolds, G.F. (Merck & Co., Inc.); Treatment of prostatic carcinoma with 17beta-N-monosubstd.-carbamoyl-4-aza-5-alpha-androst-1-en-3-ones. EP 0285383 .
Rasmusson, G.H.; Reynolds, G.F.; Steinberg, N.G.; Walton, E.; Patel, G.F.; Liang, T.; Cascieri, M.A.; Cheung, A.H.; Brooks, J.R.; Berman, C.; Azasteroids: structure-activity relationships for inhibition of 5 alpha-reductase and of androgen receptor binding. J Med Chem 1986, 29, 11, 2298.
Castaner, J.; Prous, J.; Finasteride. Drugs Fut 1991, 16, 11, 996.
The oxidative cleavage of N-tert-butyl-3-oxo-5alpha-androst-4-ene-17beta-carboxamide (I) with NaIO4 and KMnO4 in tert butanol – aqueous Na2CO3 gives the seco-ketoacid (II), which is cyclized with liquid ammonia in ethylene glycol at 180 C to afford the DELTA5-azasteroid (III). Hydrogenation of (III) with H2 over PtO2 in acetic acid yields the corresponding saturated aza-steroid (IV), which is finally dehydrogenated with benzeneseleninic anhydride in refluxing chlorobenzene or with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) and bis(trimethylsilyl)trifluoroacetamide (BSTFA) in refluxing dioxane.

Teva: FDA Approves of Generic Tobi in US

TOBRAMYCIN
JERUSALEM–(BUSINESS WIRE)–Oct. 14, 2013– Teva Pharmaceutical Industries Ltd. announces today that the U.S. Food and Drug Administration has granted approval of the generic equivalent to Tobi® (Tobramycin Inhalation Solution USP) in the United States. Pursuant to an agreement with Novartis on this product, Teva expects to launch this product in late November. Marketed by Novartis, Tobi had annual sales of approximately $350 million in the United States, according to IMS data as of June 30, 2013. READ ALL AT………..
http://www.drugs.com/news/teva-fda-approves-tobi-us-48168.html

Tobramycin is an aminoglycoside antibiotic derived from Streptomyces tenebrarius and used to treat various types of bacteria infections, particularly Gram-negative infections. It is especially effective against species of Pseudomonas.[1]

Tobramycin works by binding to a site on the bacterial 30S and 50S ribosome, preventing formation of the 70S complex. As a result, mRNA cannot be translated into protein and cell death ensues. Tobramycin is preferred over gentamicin for Pseudomonas aeruginosapneumonia due to better lung penetration.


Like all aminoglycosides, tobramycin does not pass the gastro-intestinal tract, so forsystemic use it can only be given intravenously or intramuscularly. Ophthalmic (tobramycin only, Tobrex, or combined with dexamethasone, sold as TobraDex) and nebulised formulations both have low systemic absorption. The formulation for injection is branded Nebcin. The nebulised formulation (brand name Tobi) is indicated in the treatment of exacerbations of chronic infection with Pseudomonas aeruginosa in patients diagnosed with cystic fibrosis. A proprietary formulation of micronized, nebulized tobramycin has been tested as a treatment for bacterial sinusitis.[2] Tobrex is a 0.3% tobramycin sterile ophthalmic solution is produced by Bausch & Lomb Pharmaceuticals. Benzalkonium chloride 0.01% is added as a preservative. It is available by prescription only in the United States and Canada. In certain countries, such as Italy, it is available over the counter. Tobrex and TobraDex are indicated in the treatment of superficial infections of the eye, such as bacterial conjunctivitis. Tobramycin (injection) is also indicated for various severe or life-threatening gram-negative infections : meningitis in neonates, brucellosis, pelvic inflammatory disease, Yersinia pestis infection (plague).

Like other aminoglycosides, tobramycin is ototoxic: it can cause hearing loss, or a loss ofequilibrioception, or both in genetically susceptible individuals. These individuals carry a normally harmless genetic mutation that allows aminoglycosides such as tobramycin to affect cochlear cells. Aminoglycoside-induced ototoxicity is generally irreversible.

As with all aminoglycosides, tobramycin is also nephrotoxic, meaning it is toxic to thekidneys. This effect can be particularly worrisome when multiple doses accumulate over the course of a treatment or when the kidney concentrates urine by increasing tubular reabsorption during sleep. Adequate hydration may help prevent excess nephrotoxicity and subsequent loss of renal function. For these reasons parenteral tobramycin needs to be carefully dosed by body weight, and its serum concentration monitored. Tobramycin is thus said to be a drug with a narrow therapeutic index.
|
Mass-spectrum of tobramycin |
|
|

- “Tobramycin” (pdf). Toku-E. 2010-01-12. Retrieved 2012-06-11.
- “Nebulized Tobramycin in treating bacterial Sinusitis” (Press release). July 22, 2008. Retrieved 2009-12-06.

A widely accepted therapy for treating respiratory infections caused by Gram- negative bacteria involves intravenous administration of a single antibiotic or combinations of antibiotics. Gibson et al., 2003 Am. J. Respir. Crit. Care. Med.
168(8):918-951 ; Ramsey, 1996 N. Engl. J. Med. 335(3): 179-188. However, this method of treatment has several significant limitations including: (1 ) narrow spectrum of activity of existing antibiotics, (2) insufficient concentrations of antibiotic reaching the respiratory tract to ensure rapid onset and high rates of bacterial killing, and (3) development of adverse side effects due to high systemic concentrations of drug.
Aerosol administration of antibiotics (Conway, 2005 Chronic Repir. Dis. 2:35- 41 ; O’Riordan, 2000 Respir. Care 45(7):836-845) addresses several of the limitations of parenteral administration (Flume and Klepser, 2002 Pharmacotherapy 22(3 Pt 2):71 S-79S; Kuhn, 2001 Chest. 120:94S-98S). It enables topical delivery of high concentrations of drug to the endobronchial spaces and reduces side effects by lowering systemic exposure to antibiotic. However, patients having chronic respiratory conditions, such as chronic obstructive pulmonary disease, cystic fibrosis and bronchiestasis, may receive prolonged and repeated antibiotic therapies over the entire duration of their adult lives. Gibson et al., 2003 Am. J. Respir. Crit. Care. Med. 168(8):918-951 ; Ramsey, 1996 N. Engi. J. Med. 335(3): 179-188. Therefore, cumulative antibiotic toxicity and development of resistance remains a significant problem. Chronic obstructive pulmonary disease (COPD), a smoking-related condition characterized by progressive and poorly reversible airflow obstruction and airway inflammation, is the fourth most common cause of death in developed countries. COPD is projected to be the third leading cause of global deaths in 2020 and is the only one of the four most common causes of death with an increasing mortality rate. In 2008 in the United States, there were an estimated 10 million patients diagnosed with chronic obstructive pulmonary disease (COPD). SDI COPD Claims Analysis, May 2009. Murray et at., 1997 Lancer 349: 1269-76. Approximately 7 million U.S. patients receive treatment for COPD. Mannino et al; The Epidemiology and
Economics of COPD, Proc Am Tho e Soc 2007. In the US, direct COPD costs in 2002 were approximately $18.0 billion. Statistics from National Center for Health Statistics, National Health Interview Survey: Research for the 1995-2004 redesign, Hyattsville, Maryland: U.S. Department of Health and Human Services, CDC, NCHS. Vital and Health Stat 2( 26), 1999.
The clinical course of COPD is characterized by chronic disability, with intermittent, acute exacerbations which may be triggered by a variety of stimuli including exposure to pathogens, inhaled irritants (e.g., cigarette smoke), allergens, or pollutants. “Acute exacerbation” refers to worsening of a patient’s COPD symptoms from his or her usual state that is beyond normal day-to-day variations, and is acute in onset. See, Rabe et al., 2007 Am J Res Crit Care Med, 176: 532- 555. Acute exacerbations of COPD greatly affect the health and quality of life of patients with COPD. Bathoorn, E, Int J Chron Obstruct Pulmon Dis. 2008 3(2):217- 229. Acute exacerbation of COPD is a key driver of the associated substantial socioeconomic costs of the disease. Approximately 73% ($13 billion) of direct COPD costs in 2002 were due to hospitalizations related to acute exacerbations of COPD. Investigators from the Burden of Obstructive Lung Disease (BOLD) Initiative have estimated the cumulative discounted cost of COPD care in the US to be $880 billion by 2020 – an average of more than $44 billion per year over two decades. Lee et al., 2006 ATS Proceedings, 3:A598. Multiple studies have also shown that prior exacerbation is an independent risk factor for future hospitalization for COPD.
Garcia-Aymerich et al., 2003, Thorax, 58:100-105. Hospitalization consumes roughly 70% of COPD healthcare expenditure in the US. McGhan et al., 2007, Chest, 132(6): 1748-1755. Accordingly, for a new drug therapy to significantly reduce the health and economic costs of COPD, it must address acute exacerbations of COPD.
It is clear that there is a continued need for an improved method of treatment for acute and chronic respiratory infections caused by Gram-negative and Gram- positive bacteria, particularly multidrug resistant bacteria, such as P. aeruginosa. This is particularly evident in patients having chronic respiratory conditions where current therapies are limited by problems with development of resistance and toxicity. Such method of treatment would preferably comprise inhalation of an aerosolized antibiotic composition that delivers a therapeutically effective amount of the active pharmaceutical ingredients directly to the endobronchial space of the airways or to the nasal passages. Such treatment would ideally be efficacious, reduce the frequency of drug resistance, and improve safety.
PCT Publication No. WO2005/1 0022 to Gilead Sciences, Inc. (formerly Corus Pharma) discloses a fosfomycin plus tobramycin combination formulation for delivery by aerosolization. The concentrated fosfomycin/tobramycin combination formulation containing an efficacious amount of fosfomycin and tobramycin is able to inhibit susceptible bacteria. Fosfomycin and tobramycin are formulated in solution such that when reconstituted, the pH is between 4.5 and 8.0 or as a dry powder. Also disclosed is a method for treatment of respiratory tract infections by a formulation delivered as an aerosol having mass medium aerodynamic diameter predominantly from 1 to 5 microns, produced by a jet or ultrasonic nebulizer (or equivalent) or dry powder inhaler.
tobramycin solution for inhalation of various formulations are described in the prior art. 例如,美国专利第5,508,269号公开了一种制剂,该制剂包括在Iml的盐水中40_100mg (毫克)的氨基糖苷被稀释成四分之一生理盐水强度,该制剂的PH值在5. 5到6. 5之间,其中该溶液以5ml浓缩形式气雾给药。 For example, U.S. Patent No. 5,508,269 discloses a preparation which is included in the brine Iml 40_100mg (mg) was diluted into a quarter normal saline aminoglycoside strength, the PH value of the preparation of 5 . 5 to 6.5 between, wherein the solution in concentrated form 5ml aerosol administration. [0011] 美国专利6987094公开了一种气雾制剂,其包括75mg/ml妥布霉素的水性溶液,该溶液包括O. 45%w/v (质量/体积百分比)的氯化钠,该制剂的pH值在4. O到5. 5之间,而渗透压在250到450m0sm/l (毫渗透克分子/毫升)。[0011] U.S. Patent 6,987,094 discloses an aerosol formulation which comprises an aqueous solution of 75mg/ml tobramycin, the solution comprises O. 45% w / v (mass / volume) of sodium chloride, the preparation the pH value of 4. O to 5.5, between the osmotic pressure of 250 to 450m0sm / l (mg penetration mol / ml). [0012] 美国专利申请2007/0116649公开了一种气雾制剂,其包括约100mg/ml到200mg/ ml的抗革兰阴性抗生素。 [0012] U.S. Patent Application 2007/0116649 discloses an aerosol formulation comprising about 100mg/ml to 200mg / ml of antibiotics against gram-negative. 提到了妥布霉素制剂,但是没有公开有妥布霉素的实验。Tobramycin formulations mentioned, but there is no disclosure tobramycin experiment. [0013] 美国专利申请2007/0071686公开了一种妥布霉素组合物,其包括约80mg/ml到120mg/ml的妥布霉素、一酸性辅助剂和低浓度的氯化钠。[0013] U.S. Patent Application 2007/0071686 discloses a tobramycin composition comprises about 80mg/ml to 120mg/ml of tobramycin, an acidic auxiliary agents and a low concentration of sodium chloride. 所述酸性辅助剂可以是硫酸钠或磷酸钠。 The acidic auxiliary agent may be sodium or sodium phosphate. 根据US2007/0071686,活性剂的浓度不超过120mg/ml,这是因为据说妥布霉素的浓度因粘度原因对雾化有负面影响。According to US2007/0071686, the concentration of the active agent is not more than 120mg/ml, this is because the concentration of tobramycin said reasons due to the viscosity of a negative impact on atomization. 而且,根据US2007/0071686的妥布霉素组合物利用喷雾器给药给患者,即活性成分通过潮式呼吸吸入。Moreover, the composition according to US2007/0071686 of tobramycin administered to patients using the spray, that the active ingredient through the tidal breathing inhalation. [0014] 欧洲专利2186508尤其公开了一种少于4ml溶液的组合物,其包括的60 – 200mg/ ml的、在生理上可接受的载体上的氨基糖苷抗生素。 [0014] In particular, in European Patent 2,186,508 discloses a composition of the solution is less than 4ml, comprising of 60 – 200mg / ml, and in a physiologically acceptable carrier aminoglycoside antibiotics. EP2186508中的实验显示,包括120mg/ ml妥布霉素的组合物使用PARI LC PLUS®牌喷雾器(德国Starnberg的Pari Boy N压缩器公司)给药需要约10分钟。EP2186508 The experiments showed that including 120mg / ml tobramycin compositions using PARI LC PLUS ® brand spray (Starnberg, Germany The Pari Boy N compressor company) to about 10 minutes of administration. 尽管这少于商业上可获得的TOBI®给药所需的时间,但从患者配合度和患者友好角度考虑,所需的时间还是太长。 Although this is less than the commercially available TOBI ® dosing time required, but with the degree of the patient, and patient-friendly point of view, the time is too long. EP2186508提到,使用呼吸致动吸入装置对于前面提到的商业上可获得的系统可以获得更快的给药时间。 EP2186508 mentioned breath actuated inhalation device used for the previously mentioned commercially available systems can be obtained faster delivery time. 但是,EP2186508 中使用呼吸致动吸入装置(AcroDose™)所获得的给药时间的实验仅限于低浓度的妥布霉素(60mg/ml)。However, EP2186508 using the breath actuated inhaler device (AcroDose ™) obtained experimental delivery time is limited to low concentrations of tobramycin (60mg/ml). 其还指出,使用AcroDose™系统的60mg/ml组合物给药,还必须给药第二可分量。 It also noted that the use AcroDose ™ system 60mg/ml composition administered may also be administered a second component. 从患者友好和配合角度来看,需要加入和给药第二可分量代表一种缺点。 Friendship and cooperation from the patient’s point of view, and the administration need to add a second component represents a drawback can be. [0015] 妥布霉素溶液也以局部给药出名,例如治疗角膜炎,参见Davis等人在“Canad. J Ophtal. ”(加拿大眼科杂志,1978年第13期273页)的文章,Davis等人在“Arch Opthalmol” (眼科杂志,1978年第96卷123-125页)的文章和Unter man等人在“ J. Cataract Refract. Surg. ”(白内障手术杂志,1988年第14卷500-504页)的文章。[0015] tobramycin solution is also famous for topical administration, such as treatment keratitis, see Davis et al in “Canad. J Ophtal.” (Canadian Journal of Ophthalmology, 1978 13 273) of the article, Davis, etc. in “Arch Opthalmol” (Ophthalmology 1978 Volume 96 pages 123-125) of articles and Unter man and others in “J. Cataract Refract. Surg.” (cataract surgery magazine, Volume 14, 500-504, 1988 pages) of the article. [0016] 现有给药手段和治疗方案的公知缺点是给药所需的时间,尤其影响患者的配合度和患者的生活质量。 Existing methods of administration and treatment programs known

FDA Approves Sanofi’s Nasacort® Allergy 24HR for Over-the-Counter Use – Nasal Spray Treats Adults and Children with Seasonal and Year-Round Nasal Allergies
Nasacort
Paris, France, October 11, 2013 — Sanofi (EURONEXT: SAN and NYSE: SNY) announced today that the U.S. Food and Drug Administration (FDA) approved Nasacort® Allergy 24HR nasal spray as an over-the-counter (OTC) treatment for seasonal and year-round nasal allergies in adults and children 2 years of age and older. Nasacort is the first and only medicine in its class to be available without a prescription and will be marketed by Sanofi’s consumer healthcare division, Chattem, Inc.
read all at
http://www.pharmalive.com/fda-oks-otc-nasacort-allergy-24hr
Triamcinolone acetonide, USP, the active ingredient in NASACORT AQ Nasal Spray, is a corticosteroid with a molecular weight of 434.51 and with the chemical designation 9-Fluoro11β,16α,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone (C24H31FO6).
 |
NASACORT AQ Nasal Spray is a thixotropic, water-based metered-dose pump spray formulation unit containing a microcrystalline suspension of triamcinolone acetonide in an aqueous medium. Microcrystalline cellulose, carboxymethylcellulose sodium, polysorbate 80, dextrose, benzalkonium chloride, and edetate disodium are contained in this aqueous medium; hydrochloric acid or sodium hydroxide may be added to adjust the pH to a target of 5.0 within a range of 4.5 and 6.0.