


















| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 5387603 | 1993-12-01 | 2013-12-01 |
| United States | 5403847 | 1992-11-13 | 2012-11-13 |
Home » GENERIC DRUG (Page 2)
Category Archives: GENERIC DRUG
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
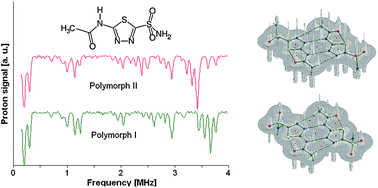
ацетазоламид , أسيتازولاميد [, 乙酰唑胺 , ACETAZOLAMIDE
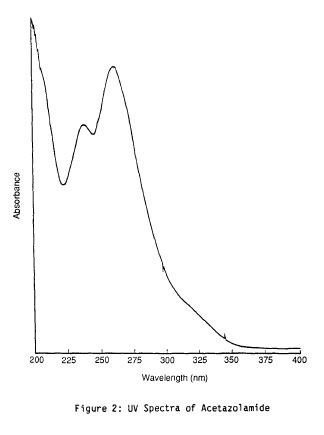
ACETAZOLAMIDE
ацетазоламид , أسيتازولاميد [, 乙酰唑胺 ,
CAS 59-66-5
Acetamide, N-(5-(aminosulfonyl)-1,3,4-thiadiazol-2-yl)-
MW 222.245,MF C4H6N4O3S2
|
Title: Acetazolamide
CAS Registry Number: 59-66-5
CAS Name: N-[5-(Aminosulfonyl)-1,3,4-thiadiazol-2-yl]acetamide
Additional Names: 5-acetamido-1,3,4-thiadiazole-2-sulfonamide; 2-acetylamino-1,3,4-thiadiazole-5-sulfonamide
Manufacturers’ Codes: 6063
Trademarks: Acetamox (Tobishi-Santen); Atenezol (Tsuruhara); Défiltran (Gallier); Diamox (Barr); Didoc (Sawai); Diuriwas (IFI); Donmox (Horita); Edemox (Wassermann); Fonurit (Chinoin); Glaupax (Erco)
Molecular Formula: C4H6N4O3S2
Molecular Weight: 222.25
Percent Composition: C 21.62%, H 2.72%, N 25.21%, O 21.60%, S 28.85%
Literature References: Carbonic anhydrase inhibitor. Prepn: R. O. Roblin, J. W. Clapp, J. Am. Chem. Soc. 72, 4890 (1950); J. W. Clapp, R. O. Roblin, US 2554816 (1951 to Am. Cyanamid). HPLC determn in pharmaceuticals: Z. S. Gomaa, Biomed. Chromatogr. 7, 134 (1993). Effect on retinal circulation: S. M. B. Rassam et al., Eye 7, 697 (1993). Clinical trial in postoperative elevation of intraocular pressure: I. D. Ladas et al., Br. J. Ophthalmol. 77, 136 (1993). Comprehensive description: J. Parasrampuria, Anal. Profiles Drug Subs. Excip. 22, 1-32 (1993). Review of efficacy in acute mountain sickness: L. D. Ried et al.,J. Wilderness Med. 5, 34-48 (1994).
Properties: Crystals from water, mp 258-259° (effervescence). Weak acid. pKa 7.2. Sparingly sol in cold water. Slightly sol in alcohol, acetone. Practically insol in carbon tetrachloride, chloroform, ether. Soly (mg/ml): polyethylene glycol-400 87.81; propylene glycol 7.44; ethanol 3.93; glycerin 3.65; water 0.72.
Melting point: mp 258-259° (effervescence)
pKa: pKa 7.2
Derivative Type: Sodium salt
CAS Registry Number: 1424-27-7
Trademarks: Vetamox (Am. Cyanamid)
Therap-Cat: Antiglaucoma; diuretic; in treatment of acute mountain sickness.
Therap-Cat-Vet: Diuretic.
Keywords: Antiglaucoma; Carbonic Anhydrase Inhibitor; Diuretic; Sulfonamide Derivatives.
|

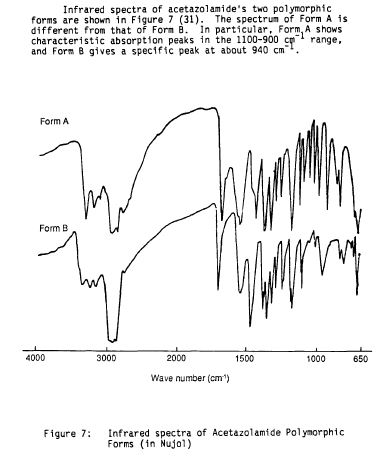
Starting reaction occurs in-between hydrazine hydrate and ammonium thiocyanate that produces 1, 2-bis (thiocarbamoyl) hydrazine which on further treatment with phosgene undergoesrearrangements, particularly molecular rearrangement through loss of ammonia to form 5-amino-2-mercapto-1, 3, 4-thiadiazole. Upon acylation of 5-amino-2-mercapto-1, 3, 4-thiadiazole gives a corresponding amide which on oxidation with aqueous chlorine affords the 2-sulphonyl chloride. The final step essentially consists of amidation by treatment with ammonia.




1H NMR

Paper
14N NQR, 1H NMR and DFT/QTAIM study of hydrogen bonding and polymorphism in selected solid 1,3,4-thiadiazole derivatives
*
Corresponding authors
a»Jozef Stefan« Institute, Jamova 39, 1000 Ljubljana, Slovenia
E-mail: janez.seliger@fmf.uni-lj.si
Fax: +386 1 2517281
Tel: +386 1 4766576
E-mail: janez.seliger@fmf.uni-lj.si
Fax: +386 1 2517281
Tel: +386 1 4766576
bFaculty of Mathematics and Physics, University of Ljubljana, Jadranska 19, 1000 Ljubljana, Slovenia
cFaculty of Physics, Adam Mickiewicz University, Umultowska 85, 61-614 Poznań, Poland
Phys. Chem. Chem. Phys., 2010,12, 13007-13019
DOI: 10.1039/C0CP00195C, http://pubs.rsc.org/en/content/articlelanding/2010/cp/c0cp00195c#!divAbstract

The 1,3,4-thiadiazole derivatives (2-amino-1,3,4-thiadiazole, acetazolamide, sulfamethizole) have been studied experimentally in the solid state by 1H–14N NQDR spectroscopy and theoretically by Density Functional Theory (DFT). The specific pattern of the intra and intermolecular interactions in 1,3,4-thiadiazole derivatives is described within the QTAIM (Quantum Theory of Atoms in Molecules)/DFT formalism. The results obtained in this work suggest that considerable differences in the NQR parameters permit differentiation even between specific pure association polymorphic forms and indicate that the stronger hydrogen bonds are accompanied by the larger η and smaller ν− and e2Qq/h values. The degree of π-electron delocalization within the 1,3,4-thiadiazole ring and hydrogen bonds is a result of the interplay between the substituents and can be easily observed as a change in NQR parameters at N atoms. In the absence of X-ray data NQR parameters can clarify the details of crystallographic structure revealing information on intermolecular interactions.
////////////ацетазоламид , أسيتازولاميد [, 乙酰唑胺 , ACETAZOLAMIDE
CC(=O)NC1=NN=C(S1)S(N)(=O)=O
Febuxostat

Febuxostat
| Febuxostat; 144060-53-7; Uloric; Adenuric; Tei 6720; 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid; | |
| Molecular Formula: | C16H16N2O3S |
|---|---|
| Molecular Weight: | 316.37484 g/mol |
2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-1,3-thiazole-5-carboxylic acid
Febuxostat is a thiazole derivative and inhibitor of XANTHINE OXIDASE that is used for the treatment of HYPERURICEMIA in patients with chronic GOUT.
CAS 144060-53-7
- 2-[3-Cyano-4-(2-methylpropoxy)phenyl]-4-methyl-5-thiazolecarboxylic acid
- 2-(3-Cyano-4-isobutyloxyphenyl)-4-methyl-5-thiazolecarboxylic acid
- FBX
- Febugood
- Feburic
- Febutaz
- TMX 67
- Zurig
Febuxostat (INN; trade names Adenuric in Europe and New Zealand, Uloric in the US, Goturic in Latin America, Feburic in Japan) is a drug that inhibits xanthine oxidase, thus reducing production of uric acid in the body. It is used in the treatment of chronicgout and hyperuricemia.
Febuxostat was discovered by scientists at the Japanese pharmaceutical company Teijin in 1998. Teijin partnered the drug with TAP Pharmaceuticals in the US and Ipsen in Europe. Ipsen obtained marketing approval for febuxostat from the European Medicines Agency in April 2008, Takeda obtained FDA approval in February 2009, and Teijin obtained approval from the Japanese “Pharmaceuticals and Medical Devices Agency” in 2011.
Medical uses
Febuxostat is used to treat chronic gout and hyperuricemia.[2] National Institute for Health and Clinical Excellence concluded that febuxostat is more effective than standard doses of allopurinol, but not more effective than higher doses of allopurinol.[2]

Uloric 40 mg tablet
Febuxostat is in the US pregnancy category C; there are no adequate and well-controlled studies in pregnant women.[3]
Side effects
The adverse effects associated with febuxostat therapy include nausea, diarrhea, arthralgia, headache, increased hepatic serum enzyme levels and rash.[3][4]
Drug interactions
Febuxostat is contraindicated with concomitant use of theophylline and chemotherapeutic agents, namely azathioprine and 6-mercaptopurine, because it could increase blood plasma concentrations of these drugs, and therefore their toxicity.[3][5]
Mechanism of action
Febuxostat is a non-purine-selective inhibitor of xanthine oxidase.[3] It works by non-competitively blocking the molybdenum pterincenter which is the active site on xanthine oxidase. Xanthine oxidase is needed to successively oxidize both hypoxanthine andxanthine to uric acid. Hence, febuxostat inhibits xanthine oxidase, therefore reducing production of uric acid. Febuxostat inhibits both oxidized as well as reduced form of xanthine oxidase because of which febuxostat cannot be easily displaced from the molybdenum pterin site.[4]

History
Febuxostat was discovered by scientists at the Japanese pharmaceutical company Teijin in 1998.[6] Teijin partnered the drug withTAP Pharmaceuticals in the US and Ipsen in Europe.[7][8][9]
Ipsen obtained marketing approval for febuxostat from the European Medicines Agency in April 2008,[10] Takeda obtained FDA approval in February 2009,[11][12] and Teijin obtained approval from the Japanese authorities in 2011.[13] Ipsen exclusively licensed its European rights to Menarini in 2009.[14] Teijin partnered with Astellas for distribution in China and southeast Asia.[15][16]
Society and culture
Cost
In the UK, NICE has found that febuxostat has a higher cost/benefit ratio than allopurinol and on that basis recommended febuxostat as a second-line drug for people who cannot use allopurinol.[2]
Trade names
Febuxostat is marketed as Adenuric in Europe and New Zealand, Uloric in the US, Goturic and Goutex in Latin America, Feburic in Japan, and is generic in several countries and is available by many names in those countries.[1]
Febuxostat (Formula I) is an inhibitor of xanthine oxidase, which was discovered by the Japanese company Teijin Pharma Ltd and it is indicated for use in the treatment of hyperuricemia and chronic gout. Its chemical name is 2-(3-cyano-4-isobutoxyphenyl)-4-methyl- l,3-thiazole-5-carboxylic acid. It is marketed under the brand names Adenuric in Europe, Feburic in Japan and Uloric in USA and Canada.

In EP0513379B1 Febuxostat is prepared from 4-hydroxy-3-nitrobenzaldehyde, according to the following scheme.

This particular process suffers from major drawbacks. Not only it is very long, including seven steps from the starting material to the final product, but, most importantly, it employs the use of cyanides, which are extremely toxic reagents. Cyanide salts are likely to generate hydrocyanide, which sets a high amount of risk in an industrial scale process.
In Japanese patent JP06345724A(JP2706037B) the intermediate ethyl ester of Febuxostat is prepared from p-cyano-nitrobenzene, in three steps. Febuxostat may, then, be prepared by alkaline hydrolysis, according to prior art.
MeCSNH,

The use of extremely toxic potassium cyanide makes this process unsuitable for manufacturing purposes.
Route A

In Japanese patent JP3202607B Febuxostat ethyl ester is prepared, according to the above scheme, through two similar routes. Route A uses flash column chromatography for the purification of the hydroxylamine reaction product, while Route B suffers from low yield and the use of chlorinated solvents for recrystallization. In addition, the reaction solvent is, in both cases, formic acid which causes severe skin burns and eye damage to humans. Formic acid is also corrosive towards metal-based materials of construction (MOC), like stainless steel and nickel alloys, limiting the options, essentially, to glass reactors or vessels. The drawbacks of using this solvent are also related to the high volumes of formic acid required per batch, which hinder the waste treatment.
In CN101723915B focus is made to the improvement of the hydroxylamine reaction. Formic acid is replaced with dimethylformamide (DMF) and other solvents. However, according to widely used organic chemistry textbooks, such as March’s Advanced Organic Chemistry, pi 287, 6th edition, M. B. Smith and J. March, ISBN 0-471-72091-7, the mechanism of the reaction involves the formation of an oxime, upon the action of hydroxylamine, which further dehydrates to form a nitrile, with the aid of a suitable reagent, for example formic acid, or acetic anhydride. In the absence of such a reagent, it is expected that the reaction will, at least, not lead to completion, thereby leading to low yields and undesired impurity levels, namely the intermediate oxime. Such impurities, arising from the reactions of the process and which exhibit similar structure of the desired product, are often difficult to remove with common industrial techniques, e.g. crystallization.
In WO2010142653A1 the intermediate Febuxostat ethyl ester is prepared from 4-cyanophenol, through a five-step process. Febuxostat can be prepared from its respective ethyl ester via alkaline hydrolysis, as in the previous case.
OH

1: patents US5614520 febuxostat synthetic process:

2: Patent JP1994329647 febuxostat synthesis


PATENT
https://www.google.com/patents/CN102936230A?cl=en
Gout occurs because the body produces too much uric acid and renal clearance capacity decreased, uric acid accumulation in the body, leading to urate crystals deposited in the joints and organs. Therefore, it means the treatment of gout usually taken to be: to promote uric acid excretion and suppression of uric acid, and the use of appropriate measures to improve symptoms. Uric acid formation and purine metabolism, the final step in the purine metabolism, hypoxanthine generation xanthine xanthine oxidoreductase (XOR) effect, further generate uric acid, inhibit the activity of the enzyme can effectively reduce uric acid production. Febuxostat is currently the world’s newly developed XOR inhibitors, which act by highly selective to the oxidase, reduce uric acid synthesis, reduce uric acid levels, so as to effectively treat the disease ventilation.
Compared with the traditional treatment of gout drug allopurinol, febuxostat has obvious advantages: (1) allopurinol reduced the XOR only inhibit rather than febuxostat of oxidized and reduced form are XOR significant inhibition, thus reducing the role of uric acid, which is more powerful and lasting; (2) Since allopurinol is a purine analogue, the inevitable result of the purine and other activity related to the impact of pyridine metabolism. So allopurinol treatment should be repeated large doses of the drug to maintain a high level. Which also brought serious or even fatal adverse reactions due to drug accumulation due.Instead of febuxostat non-purine XOR inhibitors, so it has better security.
Document TMX-67. Drugs Fut2001, 26, I, 32, and EP0513379, US5614520, W09209279, public
The detailed preparation febuxostat. Using 3-nitro-4-hydroxybenzaldehyde as the starting material is first reacted with hydroxylamine hydrochloride, to give 3-nitro-4-hydroxybenzonitrile. In effect then HCl, reaction with thioacetamide to give 3-nitro-4-hydroxy-thiobenzamide. Closed loop then reacted with 2-chloro ethyl acetoacetate to give 2- (3_ nitro-4-hydroxyphenyl) methyl-5-thiazolyl -4_ carboxylic acid ethyl ester. Followed by potassium carbonate effect, isobutane is reacted with bromo, to give 2- (3_ nitro-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester. Under the catalytic action of palladium on carbon, hydrogen reduction to give 2- (3-amino-4-isobutyloxyphenyl) -4-methyl-5-thiazole carboxylic acid ethyl ester. Followed by diazotization with sodium nitrite occur, was added cuprous cyanide and potassium cyanide, to give 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl-5-thiazolecarboxylic acid ethyl ester. Finally, under the effect of the hydrolysis of sodium hydroxide, to give the product 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl – thiazole-5-carboxylic acid, to obtain febuxostat.The process route is as follows:

This route in the preparation of febuxostat, there are many disadvantages: raw 3-nitro-4-hydroxybenzaldehyde in the country is difficult to buy; requires the use of palladium-carbon catalytic hydrogenation reaction under the factory equipment higher requirements, there is a certain danger; the cyano preparation, the need to use sodium nitrite diazotization, could easily lead to corrosion of equipment; the cyano preparation, the need to use toxic cyanide copper, potassium cyanide, pollution, higher risk.
Document JP1994329647, JP1998045733, US3518279 reported another synthesis of febuxostat
Methods. From 4-hydroxy-thiobenzamide as a starting material, and the cyclization reaction to give ethyl 2-bromo-acetyl occurred
2- (4_ hydroxyphenyl) -4_ methyl-5-carboxylic acid ethyl ester in polyphosphoric acid effect, HMTA (hexamethylene tetramine) reacts with 2- (3_ aldehyde – 4-hydroxyphenyl) methyl-5-thiazolyl -4_ carboxylic acid ethyl ester. Then two cases: the first case, the effect of potassium carbonate, is reacted with isobutane to give bromo-2- (4-isobutyloxyphenyl 3_ aldehyde) -4_-methyl-5- thiazole carboxylic acid ethyl ester, and then reacted with hydroxylamine hydrochloride to give 2- (3_-cyano-4-isobutyloxyphenyl) -4_-methyl-5-thiazole carboxylic acid ethyl ester; second case is the first with hydroxylamine hydrochloride to give 2- (3_ cyano-4-hydroxyphenyl) methyl-5-thiazolecarboxylic -4_ carboxylic acid ethyl ester, and then under the effect of potassium carbonate, and reacted with isobutane to give bromo-2- (3 _-cyano-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester.
Finally, under the effect of the hydrolysis of sodium hydroxide, to give the product 2- (3_-cyano-4-isobutyloxyphenyl) -4_ methyl – thiazole-5-carboxylic acid, i.e., to obtain febuxostat . The process route is as follows:

This synthesis route febuxostat process, since the introduction of aldehyde HMTA in PPA (polyphosphoric acid) effect. So there are a lot of phosphorus wastewater, serious environmental pollution, but also because PPA has great viscosity, and therefore difficult to stir the production, operation is extremely inconvenient.
Document Heterocyclesl998, 47,2,857 JP1994345724 also reported the synthesis method of febuxostat, using p-nitrophenyl-carbonitrile as a starting material in the reaction with potassium cyanide in DMSO solvent, and then the carbonate lower potassium catalyzed reaction of isobutane and brominated 1,3-cyano-4-diisobutoxybenzene ether. By reaction with thioacetamide to afford
3-cyano-4-isobutyloxyphenyl thiobenzamide. Under heating, and 2-chloro ethyl acetoacetate, ring closure reaction occurs to give 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester, and finally hydrolysis under the effect of sodium hydroxide, to give the product 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl – thiazole-5-carboxylic acid, to obtain febuxostat.
The present invention febuxostat new technology system, comprising the steps of:
(1) 2-hydroxy-5-cyano – NaSH reacted with benzaldehyde to give 4-hydroxy-3- aldehyde thiobenzamide;

(2) the step (I) to give 4-hydroxy-3-aldehyde thiobenzamide reaction with ethyl 2-halo-acetyl, closed
Ring to give 2- (3-aldehyde-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;

X is a halogen, preferably Cl or Br;
(3) the step (2) to give 2- (3-aldehyde-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole with hydroxylamine in formic acid in the reaction solution to give 2- (3- cyano-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;

(4) The step (3) to give 2- (3-cyano-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole isobutane with halo effect in potassium carbonate, to give 2- (3-aldehyde-4-isobutyloxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;
(5) in step (4) to give 2- (3-aldehyde-4-isobutyloxyphenyl) -4-methyl-5-ethoxycarbonyl-thiazol-off hydrolyzable ester group, to obtain a non-Tendon Disposition Tanzania.
[0011] Scheme of the method is as follows:

X is halogen, may be Cl, Br;
Preparation 5 febuxostat Example
To a 500ml reaction flask was added 200ml of absolute ethanol, the product of Step 4 was added with stirring (60g, O. 174mol),
5% sodium hydroxide was added 100ml. Stirring heated to 40 degrees, until it is completely dissolved. 40 degrees heat, reaction 4h. The reaction by TLC tracking. After completion of the reaction, the reaction solution was added 10% hydrochloric acid to adjust the pH to 3, the precipitated solid was filtered. And dried to give a pale yellow solid. Dried over anhydrous recrystallized from methanol to give 31. 2g of white crystals, yield 56.7%.
TLC monitoring of the reaction. Eluent: petroleum ether / ethyl acetate = 3: 1 Melting point:. 201 · 7 ~202 30C (literature value 201 ~202 ° C)
1H-NMR δ:. 1 01 (m, 6H), 2.06 (m, lH), 2.57 (m, 3 H), 3.96 (d, 2H), 7.30 (d, lH), 8.13 (m, 1H), 8. 19 (d, 1H);
MS (m / z):. 316 O (M +)
Infrared detection: 3550-3400cm_1; 2961, 2933,2874; 2227cm_1; 1680U604U511cm_1; 1425cm_1; 1296U283CHT1;
Elemental analysis for C, Η, N, S purified product actual measurement of the content of C, H, N, S content: C:. 60 57%, H:. 5 32%, N:. 8 86%, S: 10. 16%; theoretical value: In C16H16N203S calculated C: 60 74%, H: 510%, N: 885%, S: 1014%..
CLIP
Facile OnePot Transformation of Arenes into Aromatic Nitriles …
Facile OnePot Transformation of Arenes into Aromatic Nitriles under MetalCyanideFree Conditions

Patent
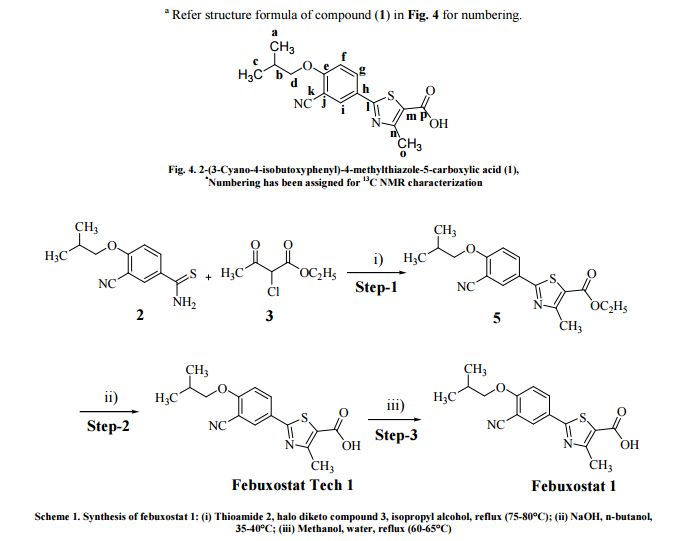
The present invention relates to an improved process for the preparation of anti- hyperuricemia drug 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5- carboxylic acid (commonly known as Febuxostat) and its pharmaceutically acceptable salts, represented by the following structural formula- 1.

Formula- 1
Further, the present invention provides novel amine salts of compound of formula- 1 and its process for the preparation.
The present invention also provides a novel process for the preparation of crystalline forms A, B and G of Febuxostat. Febuxostat is an inhibitor of xanthine oxidase that is indicated for use in the treatment of hyperuricemia and gout.
Febuxostat was approved by the European Medicines and the U.S. Food and Drug Administration. Febuxostat is marketed by Takeda Pharmaceuticals with the brand names Adenuric (EU) and Uloric (US).
Background of Invention:
2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5-carboxylic acid was first disclosed in US 5614520. The disclosed process comprises of reacting the ethyl 2-[4- hydroxy-3-nitrophenyl]-4-methyl-5-thiazolecarboxylate with 1 -bromo-2-methylpropane in the presence of potassium carbonate in dimethyl formamide provides ethyl 2-[4-(2- methylpropoxy)-3-nitrophenyl]-4-methyl-5-thiazole carboxylate which is reduced with Pd/C and the obtained compound is treated with sodium nitrite in water and followed by a mixture of cuprous cyanide and potassium cyanide provides ethyl 2-[4-(2- methylpropoxy)-3-cyanophenyl]-4-methyl-5-thiazolecarboxylate. The obtained ethyl 2- [3-cyano-4-(2-methylpropoxy)phenyl)-4-methyl-5-thiazolecarboxylate was hydrolyzed by treating with aqueous sodium hydroxide in a mixture of ethanol and tetrahydrofuran provides 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5-carboxylic acid having melting range of 238 – 239°C with the yield of 33%. The above process utilizes Pd/C, which is difficult to handle in the laboratory, hence it is not recommended on large scale. However, the yield and purity were also not satisfactory. The said process involves more number of steps, hence there is a need to produce the process for the preparation of 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5-carboxylic acid with less number of steps.
Heterocycles, 1998, Vol. 47, No. 2, 857-864 disclosed a process for the preparation of 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid which comprises of reacting 4-(2-methylpropoxy)-l,3-benzenedicarbonitrile in the presence of hydrochloric acid with thioacetamide in dimethyl formamide provides 4-(2- methylpropoxy)-3-cyanobenzthioarnide. It was reacted with ethyl 2-chloroacetoacetate in ethanol provides ethyl 2-[4-(2-methylpropoxy)-3-nitrophenyl]-4-methyl-5-thiazole carboxylate which is further hydrolyzed with aqueous sodium hydroxide solution in a mixture of tetrahydrofuran and ethanol provides 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid with the yield of 35% which on further recrystallized from acetone provides a colorless crystals having the melting range of 201 – 202°C. However the yield and purity were not satisfactory. JP10-045733 disclosed a process for the preparation of 2-[3-cyano-4-(2- methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid which comprises of reacting ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate with hydroxyl amine in 2011/000328 presence of formic acid and sodium formate provide ethyl 2-(3-cyano-4-hydroxyphenyl)-4- methylthiazole-5-carboxylate. The obtained compound was reacted with l-bromo-2- methylpropane in presence of potassium carbonate provides ethyl 2-[3-cyano-4-(2- methylpropoxy) phenyl]-4-methylthiazole-5-carboxylate, which on further hydrolysis with aq sodium hydroxide provides 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5- carboxylic acid.
2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid produced by the above processes contains 2-[3-carbamoyl-4-(2-methylpropoxy)phenyl]-4- methylthiazole-5-carboxylic acid as an impurity. So there is a need for the purification to get the highly pure 2-[3-cyano-4-(2-methylpropoxy) phenyI]-4-methylthiazole-5-carboxylic acid.WO2011141933
In general, febuxostat prepared by the reported processes contain impurities like amide impurity, ethyl ester impurity and methyl ester impurity etc. It is important that any pharmaceutical compound or intermediate should free of impurities or if present they must be limited to the maximum level set by ICH guidelines. The purification technique of by recrystallisation of the compound from a suitable solvent such as the recrystallisation of febuxostat as disclosed in Heterocycles, Vol. 47, No. 2, 1998 does not yields the desired purity. Hence it is necessary to have an alternate purification technique for the preparation of highly pure febuxostat.
In the aforementioned processes using aqueous sodium hydroxide for hydrolyzation of ethyl 2-[4-(2-methylpropoxy)-3-nitrophenyl]-4-methyl-5-thiazole carboxylate, which lead to the formation of impurities such as ethyl ester impurity, methyl ester impurity, amide impurity and high polar impurities. Hence there is a need to develop a process for the preparation of highly pure febuxostat with high yield.
The crystalline forms A, B, C, D and G of febuxostat and their preparation were first disclosed in US 6225474. However as on date, there is no alternative processes were reported in the prior art for preparing the said polymorphs. Henceforth, there is a need to develop a novel process for the preparation of said crystalline polymorphs.
In view of the foregoing, there is a necessity for the improved process which overcome the problems of prior art and to produce the highly pure 2-[3-cyano-4-(2- methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid with high yield.
Advantages of Present Invention:
• Avoids the usage of Pd/ C .
• Provides the highly pure 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-5- thiazole carboxylic acid with high yield.
• Controls the impurities (such as methyl impurity, ethyl impurity, amide impurity and high polar impurities) levels to meet the ICH guidelines.
• Provides a novel process for the preparation of crystalline forms A, B and G of 2-[3- cyano-4-(2 -methylpropoxy) phenyl]-4-methyl-5-thiazolecarboxylic acid.
· Provides a novel and improved processes for the preparation of 4-(2-methylpropoxy)- 1,3 -benzene dicarbonitrile and also provides a simplest process for the purification of 2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-5-thiazole carboxylic acid.
• Eco-friendly process.
• Uses simple, milder reagents which are easier to handle and use in large scale.

Formula-6 ForrauIa-

Scheme-II:

Scheme-Ill:

Example 8: Preparation of Febuxostat (Formula-1)
A mixture of compound of formula-5 (50 g), sodium hydroxide (23.25 g), tetrahydrofuran (250 ml) and water (13 ml) was heated to 60-65°C and stirred for 8 hours. After completion of the reaction, the reaction mixture was cooled to 25-30°C and quenched it with water. The reaction mixture was stirred for 1 hour at 25-30°C. A solution of hydrose (2.5 g) in water (25 ml) was added to the reaction mixture at 25-30°C and stirred for 30 minutes. The pH of the reaction mixture was adjusted to 1.3 with hydrochloric acid. The reaction mixture was extracted thrice with ethylacetate, washed with water and the organic layer was dried with sodium sulfate. Carbon (2.5 g) was added to the organic layer and stirred for 30 minutes. The reaction mixture was filtered through hyflow bed and washed with ethylacetate. Distilled off the solvent completely from the obtained filtrate to get the solid. Ethyl acetate (200 ml) was added to the obtained solid and heated to reflux temperature and stirred for 30 minutes. The reaction mixture was cooled to 25-30°C, filtered the solid obtained and washed with ethyl acetate. Acetone (250 ml) was added to the wet solid obtained, heated to 55-60°C and stirred for 15 minutes. Water (250 ml) was added to the reaction mixture and stirred for 2 hours. The obtained solid was filtered, washed with aqueous acetone and then dried to get the title compound. Yield: 38 grams.
Example 9: Preparation of Febuxostat (Formula-1)
A mixture of compound of formula-5 (50 g), sodium hydroxide (23.25 g), tetrahydrofuran (250 ml) and water (13 ml) was heated to 60-65°C and stirred for 8 hours. After completion of the reaction, the reaction mixture was cooled to 25-30°C and quenched it with water. The reaction mixture was stirred for 1 hour at 25-30°C. A solution of hydrose (2.5 g) in water (25 ml) was added to the reaction mixture at 25-30°C and stirred for 30 minutes. The pH of the reaction mixture was adjusted to 1.3 with hydrochloric acid. The compound was extracted thrice with ethyl acetate, washed with water and the organic layer was dried with sodium sulfate. Carbon (2.5 g) was added to the reaction mixture and stirred for 30 minutes. The reaction mixture was filtered through hyflow bed and washed with ethylacetate. Distilled off the solvent completely from the obtained filtrate to get the solid. Acetone (250 ml) was added to the solid obtained, heated to 55-60°C and stirred for 15 minutes. Water (250 ml) was added to the reaction mixture and stirred for 45 minutes . Cooled the reaction mixture to 25-30°C and stirred for 30 minutes, further to 0-5 °C and stirred for 2 hours. The obtained solid was filtered, washed with aqueous acetone and then dried. Ethylacetate (200 ml) was added to the obtained solid and heated to reflux temperature and stirred for 30 minutes. The reaction mixture was cooled to 25-30°C. Filtered the solid, washed with ethyl acetate and then dried to get the pure title compound. Yield: 39 grams
Example 10: Preparation of ethyl 2-[3-formyl-4-(2-methyIpropoxy) phenyl]-4- methylthiazole-5-carboxylate compound of formula-12.
To a stirred solution of ethyl 2-[3-formyl-4-hydroxyphenyl]-4-methylthiazole-5- carboxylate (10 gms) in dimethylformamide (40 ml) added potassium carbonate (9.78 gms) and potassium iodide (2.35 gms). Heated the reaction mixture to 70 – 75°C and stirred for 30 minutes. To the above reaction added a solution of l-bromo-2- methylpropane (9.72 gms) in dimethyl formamide (10 ml) and stirred for 5 hours. Cooled the reaction mixture to 25°C, quenched with water and stirred for one hour. Filtered the precipitated solid and dried the material to get the title compound. Yield: 9 gms.
Example 11: Preparation of ethyl 2-[3-cyano-4-(2-methyIpropoxy) phenyl]-4- methyIthiazoIe-5-carboxylate compound of formuIa-5.
To the solution of ethyl 2-[3-formyl-4-(2-methylpropoxy) phenyl]-4- methylthiazole-5-carboxylate compound of formula-12 (10 gms) in formic acid (40 ml) was added hydroxylamine hydrochloride (2.38 gms) and sodium formate (2.35 gms).
Stirred the reaction mixture for 10 minutes. Heated the reaction mixture to 100°C and stirred for four hours at same temperature. Cooled the reaction mixture to 25 °C and quenched with water. Stirred the reaction mixture for 10 hours, filtered the precipitated solid and washed with water. Dried the material to get the title compound. Yield: 10 gms. Take the dry material, added 30 ml of ethyl acetate and heated to reflux temperature. Stirred the reaction mixture for 30 minutes at reflux temperature. Cooled the reaction mixture to 25°C and filtered the precipitated solid. Dry the material to get the title compound as a pure material. Yield: 8.5 gms.
Clip
synthesis describes synthesis of febuxostat (I) from 4-hydroxybenzonitrile (II) in six stages. The synthesis shown is a short, concise route and does not require use of poisonous reagents such as KCN (14). Compound II was converted to 4-hydroxybenzothioamide (III) with 85% yield using NaHS in the presence of hydrated magnesium chloride as Lewis acid. Intermediate III, on cyclization with ethyl-2-chloroacetoacetate, gave thiazole ester (IV) with quantitative yield. In these two stages, the source of potential impurities was identified as an ortho isomer (i.e., 2-hydroxybenzonitrile), which can lead to Impurity VIII and subsequently to Impurity IX . Impurities VIII and IX can be controlled in starting material II with appropriate specification.
|
The ortho formylation of hydroxyl compound IV by using Duff condition (hexamine/TFA) gave aldehyde V (15). The major impurity identified in this reaction was dialdehyde X. Although we have used only 1.0 equivalence of hexamine with respect to Compound IV, the dialdehyde X impurity was formed to a 5-10% ratio in only 2.5 h. It is, therefore, impossible to get rid of this impurity during the reaction, and only effective recrystallization will eliminate it. Impurity X was minimized (≤ 2%) by recrystallization using IPA/H2O (3:5) to get aldehyde V with 50% yield and & #8805; 97% HPLC purity.
Aldehyde V, on alkylation with isobutyl bromide in the presence of potassium carbonate base, gave compound VI with 90% yield. In this stage, Impurities XI and XII were alkylations of carryover Compound IV and dialdehyde, respectively. Two more isomeric impurities n-butyl-aldehyde XIII and 1-methyl propyl-aldehyde XIV were also identified in this stage. Both isomeric impurities can be controlled with appropriate specification for isobutyl bromide. The reaction of Compound VI with hydroxylamine hydrochloride and sodium formate in formic acid at reflux temperature gave Compound VII with 85% yield. Impurities XIII and XIV will also carry forward to impurities n-butyl-nitrile XV and 1-methyl propyl-nitrile XVI, respectively.
In the final step, Compound VII was hydrolyzed using sodium hydroxide in a MeOH:THF:H2O (1:1:1) solvent combination to yield febuxostat (85%). During saponification, methyl ester Impurity XVII was identified via trans-esterification. Its hydrolysis was comparatively slower than its ethyl isomer VII. One way to avoid Impurity XVII is to replace methanol with ethanol. Carryover impurities XI, XV, and XVI were also hydrolyzed to their respective acid derivatives impurities XVIII, XIX, and XX. However, the acid derivatives of impurities X and XII were unexpectedly absent as impurities. It is believed that, because they were present in low concentrations during workup, they were eliminated in the mother liquor. Two additional impurities, amide XXI and diacid XXII, formed by the side reaction of the febuxostat nitrile group with sodium hydroxide, were identified during saponification. The amide XXI and diacid XXII impurities can be controlled by using appropriate equivalence of sodium hydroxide and controlled reaction time. Febuxostat, on acetone recrystallization and seed Crystal A at 45°C, gave pure febuxostat with 75% yield.

http://www.pharmtech.com/investigation-various-impurities-febuxostat
References
- Drugs.com Drugs.com international names for febuxostat Page accessed June 25, 2015
- Febuxostat for the management of hyperuricaemia in people with gout (TA164) Chapter 4. Consideration of the evidence
- Uloric label Updated February, 2009.
- Love BL, Barrons R, Veverka A, Snider KM (2010). “Urate-lowering therapy for gout: focus on febuxostat”. Pharmacotherapy 30 (6): 594–608. doi:10.1592/phco.30.6.594.PMID 20500048.
- Ashraf Mozayani; Lionel Raymon (2011). Handbook of Drug Interactions: A Clinical and Forensic Guide. Springer Science+Business Media.
- Teijin Febuxostat Story Page accessed June 25, 2015
- Tomlinson B. Febuxostat (Teijin/Ipsen/TAP). Curr Opin Investig Drugs. 2005 Nov;6(11):1168-78. PMID 16312139
- Bruce Japsen for the Chicago Tribune. August 17, 2006. FDA puts gout treatment on hold
- Note: TAP Pharmaceuticals was a joint venture between Abbott Laboratories and Takedathat was dissolved in 2008 per this press release: Takeda, Abbott Announce Plans to Conclude TAP Joint Venture
- “Adenuric (febuxostat) receives marketing authorisation in the European Union” (PDF). Retrieved 2008-05-28.
- “Uloric Approved for Gout”. U.S. News and World Report. Retrieved 2009-02-16.
- Teijin and Takeda. February 14, 2009 Press release: ULORIC® (TMX-67, febuxostat) Receives FDA Approval for the Chronic Management of Hyperuricemia in Patients with Gout
- Teijin. January 21, 2011 Press release: TMX-67 (febuxostat) Approved in Japan
- Genetic Engineering News. October 2009. Menarini to Market Takeda/Ipsen Gout Therapy in 41 European Countries
- First Word Pharma. April 1st, 2010 Teijin Pharma and Astellas Pharma enter into agreement for marketing rights of TMX-67 in China and Hong Kong
- Research Views. Aug 11 2011 Teijin Pharma Enters Into Distribution Agreement With Astellas Pharma For Febuxostat
Febuxostat is an inhibitor of xanthine oxidase, and was developed by Teijin pharma. This compound is known as a new drug that is effective against gout and hyperuricemia, and it has been 40 years since the last time a drug of this kind of drug was developed.
Febuxostat has therefore gained a lot of popularity and it has already been accepted as a drug in Europe, USA, Korea and Japan. The synthesis of this molecule have been reported in patents by Teijin pharma as shown below.[1,2]

Recently, Itami group was reported the rapoid synthesis of febxostat by using Ni-catalyzed direct coupling of azoles and arylhalides[3]
References
Sorbera, L.A.; Castaner, J.; Rabasseda, X.; Revel, L.; TMX-67. Drugs Fut 2001, 26, 1, 32
[1] Hasegawa, M.; A facile one-pot synthesis of 4-alkoxy-1,3-benzenedicarbonitrile. Heterocycles 1998, 47, 2, 857. [2] Hasegawa, M.; Hasegawa, M.; Komoriya, K. (Teijin Ltd.); Cyano cpds. and their preparation method. JP 1994345724 . [3] “Nickel-Catalyzed Biaryl Coupling of Heteroarenes and Aryl Halides/Triflates”
Canivet, J.; Yamaguchi, J.; Ban, I.; Itami, K. Org. Lett. 2009, 11, 1733-1736. DOI: 10.1021/ol9001587

Ni-based catalytic systems for the arylation of heteroarenes with aryl halides and triflates have been established. Ni(OAc)2/bipy is a general catalyst for aryl bromides/iodides, and Ni(OAc)2/dppf is effective for aryl chlorides/triflates. Thiazole, benzothiazole, oxazole, benzoxazole, and benzimidazole are applicable as heteroarene coupling partners. A rapid synthesis of febuxostat, a drug for gout and hyperuricemia, is also demonstrated.
A CLIP
A final example of a thiazole containing drug is given in the novel xanthine oxidase inhibitor febuxostat (359, Uloric) which was approved by the FDA in 2009. This inhibitor works by blocking xanthine oxidase in a non-competitive fashion. Consequently, the amount of the oxidation product uric acid is reduced. Thus it is an efficient treatment for hyperuricemia in gout. In order to prepare febuxostat first a synthesis of the noncommercial 4-isobutoxy-1,3-dicyanobenzene building block (363), has to be conducted. An elegant way of achieving this was shown through the reaction of 4-nitrocyanobenzene (360) with potassium cyanide in dry DMSO followed by quenching with isobutyl bromide under basic conditions (Scheme 70). It is suggested that a Meisenheimer-complex intermediate 361 is initially formed, which after rearomatisation, undergoes nucleophilic aromatic substitution of the nitro group by the DMSO solvent [107]. Upon hydrolysis and O-alkylation the desired 4-isobutoxy-1,3-dicyanobenzene (363) is obtained in good overall yield. Subsequently, the less hindered nitrile is converted to the corresponding thioamide 365 in an intriguing reaction using thioacetamide (364). The thiazole ring is then formed by condensation with chloroacetoacetate 366 followed by ester hydrolysis (Scheme 70).
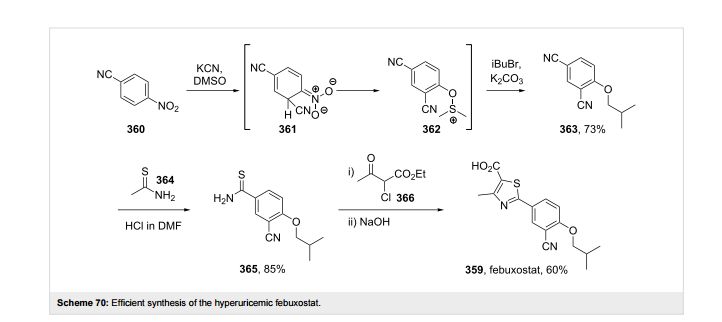
107 Hasegawa, M. Heterocycles 1998, 47, 857–864. doi:10.3987/COM-97-S(N)89
Paper | Special issue | Vol 47, No. 2, 1998, pp.857-864
DOI: 10.3987/COM-97-S(N)89
■ A Facile One-Pot Synthesis of 4-Alkoxy-1,3-benzenedicarbonitrile
Masaichi Hasegawa
*Teijin Institute, Bio-Medical Research, Asahigaoka 4-3-2, Hino, Tokyo 191, Japan
Abstract
2-(3-Cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxlic acid (TEI-6720) was prepared. The introduction of cyano group to 4-nitrobenzonitrile with KCN in dry DMSO followed by quenching with alkyl halide afforded the key intermediates, 4-alkoky-1,3-benzenedicarbonitriles, in good yield. The reaction was completed in dry DMSO, while no reaction occurred in dry DMF. This observation can be suggested by the participation of DMSO in the reaction.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S70
A CLIP
Synthesis and characterization of process-related impurities of an anti-hyperuricemia drug-Febuxostat
Venkateswara Rao Vallu,$ Krunal Girishbhai Desai, Sandip Dhaya Patil, Rajendra Agarwal, Pratap Reddy Padi and Mahesh Reddy Ghanta
*Process Research Laboratory-I, Research & Development Centre, Macleods Pharmaceuticals Ltd, G-2, Mahakali Caves Road, Shantinagar, Andheri (East), Mumbai, Maharastra, India
$Department of Chemistry, Pacific University, Pacific Hills, Airport Road, Pratap Nagar Extension, Debari, Udaipur, Rajasthan, India _____________________________________________________________________
Der Pharma Chemica, 2014, 6(3):300-311 (http://derpharmachemica.com/archive.html)
http://derpharmachemica.com/vol6-iss3/DPC-2014-6-3-300-311.pdf
Synthesis of 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1) [10] A solution of 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1 tech grade, 5.0 g, 0.015 mol.) in methanol (50.0 mL) was heated the reaction mass at 60-65°C till clear solution was obtained. Water (50.0 mL) was added drop wise into reaction mass with in 30.0 min. at 60-65°C. Resultant white crystalline solid was filtrated, Mahesh Reddy Ghanta et al Der Pharma Chemica, 2014, 6 (3):300-311 _____________________________________________________________________________ 302 http://www.scholarsresearchlibrary.com washed with water (10.0 mL) and dried in vacuum tray drier at 50-55°C under vacuum to give
2-(3-cyano-4- isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1). Yield: 95.0 % (4.75 g)
mp 239°C. Purity by HPLC: 99.74 % (10.2 min. retention time),
Anal. Calcd for C16H16N2O3S: C, 60.74; H, 5.10; N, 8.85. Found: C, 60.70; H, 5.11; N, 8.87 %;
IR (KBr) υmax (in cm−1): 3834.61, 3742.03, 3680.30, 3556.85, 3456.55, 2962.76, 2877.89, 2661.85, 2546.12, 2353.23, 2229.79, 2168.06, 2029.18, 1921.16, 1790.00, 1674.27, 1604.83, 1512.24, 1427.37, 1381.08, 1280.78, 1172.76, 1118.75, 1010.73, 918.15, 833.28, 771.55, 725.26, 648.10, 524.66, 462.93; 1H NMR (300 MHz, CDCl3 or DMSO-d6) δH (in ppm): 1.00-1.02 (d, 6H, (CH3)2-CH-), 2.49-2.50 (m, 1H, (CH3)2-CH-), 3.97-3.99 (d, 2H, -CH-CH2−), 7.33–8.25 (d, dd, 3H, Ar-H), 2.64 (s, 3H, -CH3), 13.39 (s, 1H, -COOH);
13C NMR (300 MHz, DMSO–d6) δC (in ppm) (Positiona ): 166.3 (l), 162.9 (p), 162.2 (n), 159.6 (e), 133.1 (g), 131.6 (i), 125.5 (m), 123.0 (h), 115.5 (k), 114.0 (f), 101.7 (j), 75.2 (d), 27.7 (b), 18.8 (a, c), 17.1 (o);
MS m/z (%) (70 eV): m/z =317.0 (100.0 %) [M+1], 318.0 (16.0 %) [M+2], 403.0 (63.0 %), 512.0 (47.0 %), 482.0 (46.0 %), 405.0 (27.0 %), 468.0 (25.0 %), 570.0 (24.0 %).
PATENT
| WO 2012066561 |
As per the present invention, hydroxylamine hydrochloride is added to compound of Formula-Ill in presence of a polar aprotic solvent like DMSO, DMA, ACN or DMF. To this reaction mixture acetyl halides or sulfonyl chlorides are added and temperature raised to 70 -80 °C. Acetyl halides are selected from acetyl bromide or acetyl chloride. Sulfonyl chlorides are selected from methane sulfonyl chloride or para toluene sulfonyl chloride. To this reaction mixture a base selected from alkali metal carbonates like potassium carbonate or sodium carbonate, preferably potassium carbonate and alkyl halide selected from isobutyl bromide is successively added. The reaction mass is washed with water and compound of Formula-II is isolated. In one embodiment the present invention provides, process for the preparation of Febuxostat comprising the steps of:
a) reacting the compound of Formula-III(a) with hydroxylamine hydrochloride in presence of organic solvent;

Formula-III(a)
b) adding acyl halides or sulfonyl chlorides to the reaction mixture;
c) optionally isolating compound of Formula- IV (a)

Formula-IV(a)
d) reacting with isobutyl bromide in presence of base;
e) isolating the compound of Formula-II(a); and

FormuIa-II(a)
f) hydrolyzing the compound of Formula-II(a) to get Febuxostat.
The following examples are provided to illustrate the process of the present invention. They, are however, not intended to limiting the scope of the present invention in any way and several variants of these examples would be evident to person ordinarily skilled in the art. Experimental procedure:
Example – 1: Preparation of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyI thiozole -5-carboxylate
A mixture of 10. Og of Ethyl -2-(3-formyl-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate and 2.85 g of hydroxylamine hydrochloride were stirred for 30 minutes in 40 g of Dimethyl sulfoxide. To this reaction mixture 3.3 grams of acetyl chloride was added and stirred at 70 -80°C for 2-3 hours. Reaction mass was cooled to room temperature and to this 19 g of potassium carbonate and 19 g of isobutyl bromide was added successively. The reaction mass was stirred for 5 hours at 70-80°C. Reaction mass was diluted with 200 ml of purified water. The reaction mass was filtered and washed with purified water to give 10.0 g of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxyltae (yield 84.0%)
Example – 2: Preparation of Ethyl-2-(3-cyano-4-hydroxyphenyl)-4-methyl thiozole – 5-carboxylate
A mixture of 10. Og of Ethyl-2-(3-formyl-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate and 2.85 g of hydroxylamine hydrochloride were stirred for 30 minutes in 30 g of Dimethylformamide. To this reaction mixture 3.3 grams of acetyl chloride was added and stirred at 90°C for 2-3 hours. Reaction mass was cooled to room temperature and diluted with 100 ml of water and stir for 2 hours. The reaction mass was filtered and washed with purified water to give 10.0 g of Ethyl-2-(3-cyano-4-hydroxy phenyl)-4- methyl thiozole -5-carboxyltae (yield 99.0%).
Example – 3: Preparation of Ethyl 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylate
A mixture of 10. Og of Ethyl-2-(3-cyano-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate, 30 g of NMP, 9.6 g of potassium carbonate and 7.2 g of isobutyl bromide were stirred for 3 hours at 90°C. Reaction mass was diluted with 100 ml of purified water. The reaction mass was filtered and washed with purified water and ethanol to give 10.5 g of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxyltae (yield 88.0%). Example – 4: Preparation of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5- carboxylic acid
A mixture of 10. Og of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5- carboxyltae, 2.0g of sodium hydroxide was heated at 45-60°C in 75 ml of aqueous methanol for 1 hour. Reaction mass was cooled to ambient temperature and pH adjusted to 2.0 to 2.5 with dilute hydrochloric acid and precipitated crystal was collected by filtration to give 8.8g of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid (yield 95.8%).
Example – 5-13: Preparation of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole – 5-carboxylic acid
The above compound was prepared by following the procedure as disclosed in Example- 4, using the below listed solvents instead of aqueous methanol.

Example – 14: Preparation of pure 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid
10.0 g of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid was dissolved in 100 ml of ethanol at reflux temperature. After dissolution reaction mass was cooled and precipitated crystal was collected by filtration to give 9.6 g of pure 2-(3- cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid (yield 96%).
PATENT
KR 201603732
PATENT
WO 2015018507
https://www.google.com/patents/WO2015018507A2?cl=en



EXPERIMENTAL
of compound of formula lib

Dissolve 14.14g of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula III) in 55 ml dimethylformamide, at ambient temperature. Add 40g of potassium carbonate, along with 15.9 ml isobutyl bromide. Heat the reaction to 75-80 °C and stir for 4 hours. Cool to 25-30 °C, while 165 ml process water is added. Further cool to 0-5 °C and stir for 30 minutes at this temperature. Filter off the precipitated solid and wash the filter cake with 55 ml process water. The wet cake is dried under vacuum at 40 °C for 7 hours, to furnish 16.43 g of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula lib).
of compound of formula Illb

In a 25 mL round-bottomed flask charge under stirring at 25-30 °C, 1.0 g (2.88 mmol) of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate in 3.0 mL dimethylformamide. Add 34 mg (0.19 mmol) copper acetate under stirring at 25-30 °C. Flush with oxygen (02) and add 0.66 ml (34.92 mmol) 25% aqueous ammonia. Flush again with 02. Heat the reaction mixture to 80-82 °C overnight. Check the progress of the reaction by TLC (cyclohexanerethyl acetate 3:1). Cool reaction mass to 25-30 °C. Add 25mL ethyl acetate and 25mL brine at the reaction mass, separate organic layer and extract aqueous layer twice with 25mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate till dry. The residue is purified with column chromatography (cyclohexane:ethyl acetate 9:1). afforded 0.754g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb) Yield: 75.4%.
EXAMPLE 3: Preparation of compound of formula Illb

In a 25 mL round-bottomed flask charge under stirring at 25-30 °C, 0.17 g (0.49 mmol) of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate in 2.5 mL tetrahydrofuran. Add 2.9 mL (153.43 mmol) 25% aqueous ammonia, under stirring at 25-30 °C. Add 137 mg (0.54 mmol) iodine (I2) to the reaction mass, stir the reaction mixture at 25-30 °C for 15-30 min. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 3:1). Starting material is consumed. Add 2.5 mL 5% w/v aqueous sodium thiosulfate Na2S203 and 15mL ethyl acetate at the reaction mass, separate organic layer and extract twice aqueous layer with 15mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate till dry. 0.158g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb) are collected.
EXAMPLE 4: Preparation of Febuxostat

In a 100 ml 2-neck round-bottomed flask charge 2.407g of ethyl-2-(3-cyano-4-isobutoxyphenyl)-4-methylhiazole-carboxylate in 20ml tetrahydrofuran under stirring, at 25-35 °C, 0.748g of sodium hydroxide and heat reaction mass to 60-65 °C for approximately 8 hrs. Check the progress of the reaction by TLC (cyclohexane:ethyl acetate 3:1). Cool reaction mass to 0-5 °C and add 50 ml process water keeping temperature within 0-5 °C. Adjust pH to 1-2 with 4.5 ml 6 N hydrochloric acid, keeping temperature within 0-5 °C. Warm up reaction mass to 25-30 °C and stir reaction mass at the above temperature for 15 min. Filter off the precipitated solid through Buchner funnel under reduced pressure, spray wash with 2 ml process water and suck dry for 20-30 min. Transfer the crude solid in a 50 ml round-bottomed flask, charge 12 ml process water and 12 ml acetone at 25-30°C. Heat the reaction mass to 50-60 °C for 60 min. Cool down reaction mass to 0-5 °C and stir for 60 min at the above temperature. Filter off the precipitated solid though Buchner funnel under reduced pressure, spray wash with 2 ml of a 1 : 1 mixture of acetone and process water and suck dry for 30-45 min. Dry under vacuum at 60 °C. 1.821g of (compound I) Febuxostat are collected, Purity: 82.6%, Yield: 0.62w/w.
on of compound of formula Ilia

In a 50 mL round-bottomed flask charge under stirring 0.5g (1.72 mmol) of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate in 8.6 mL THF, at 25-30 °C. Add 10.3 mL (544.94 mmol) 25% aqueous ammonia, under stirring at 25-30 °C. Add 480 mg (1.89 mmol) iodine (I2) to the reaction mass, stir the reaction mixture at 25-30 °C for 15-30 min. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 1 :1). Starting material is consumed. Add 8.6 mL 5% w/v aqueous thiosulfate and 40 mL ethyl acetate at the reaction mass, separate organic layer and extract aqueous layer twice with 40 mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate to dryness. Purification of the residue with column chromatography (cyclohexane: ethyl acetate 3: 1) afforded 0.213 g of ethyl 2-(3-cyano-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula Ilia). Yield : 42.6%.
EXAMPLE 6: Preparation of compound Illb
Dissolve 2.2 g of ethyl 2-(3-cyano-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula VI) in 7 ml dimethylformamide and to this mixture add 6.6 g of potassium carbonate and 3.14 g of isobutyl bromide. Stir the reaction at 75 °C for 15 hours and then cool to 40 °C. Add 15 ml process water and cool to 0-5 °C. Filter the precipitated solid off and wash with 15 ml process water, which, after drying, affords 2.28 g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb).
EXAMPLE 7: Preparation of compound I (Febuxostat)
In a 100 ml 2-neck round-bottomed flask charge 2.131 g of ethyl-2(3-cyano-4-isobutoxyphenyl)-4-methylhiazole-carboxylate, 64 ml methanol and 2.5 ml process water are added under stirring at 25-35 °C. Add 1.718 g potassium carbonate and heat reaction mass to reflux for approximately 2-3 hrs. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 3:1). Cool reaction mass to 20-25 °C. Concentrate solvent at below 40 °C. To the residue add 43 ml process water, 21 ml ethyl acetate and stir for 30 min at 25-35 °C. Separate layers and transfer aqueous layer in a 100 ml round-bottomed flask. Adjust pH to 2.3-2.7 with 25 ml 1 N hydrochloric acid, at 25-35 °C. Warm up reaction mass to 40 °C and stir reaction mass at this temperature for 60-90 min. Cool down reaction mass to 25-35 °C. Filter off the precipitated solid through Buchner funnel under reduced pressure, spray wash with 5 ml process water and suck dry for 30-45 min. Dry under vacuum at 60 °C. 1.708g of (compound I) Febuxostat are collected, Purity: 86.7%, Yield: 0.69w/w.
EXAMPLE 8: Preparation of Febuxostat crystalline form III
In a 250 mL round-bottomedflask charge under stirring at 25-30 °C 10 g of crude 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (Febuxostat) in 200 mL ethyl acetate. Heat reaction mass to reflux and stir for 30 min. Cool reaction mass to 25-30°C. Warm again reaction mass and partially distill off solvent from the reaction mass at temperature below 40 °C under reduced pressure. Cool reaction mass to 25-30°C. Filter off the precipitated solid through Buchner funnel under reduced pressure and spray wash with 10 mL ethyl acetate. Dry under vacuum at 60°C. 8.5 g of Febuxostat are collected. Yield: 85 % w/w. XRPD of crystalline compound is in accordance with the one reported in Chinese patent CN101412700B.
PATENT
CN 104418823
https://www.google.com/patents/CN104418823A?cl=zh

PATENT
CN 103588723
https://www.google.com/patents/CN103588723A?cl=zh
Chinese patent CN1275126 described by the Japanese company Teijin invention relates febuxostat Form A, B, C, D, G, and six kinds of amorphous and crystalline preparation method, reported in the literature Form A relatively stable . The method used is a solvent of methanol and water, patent phase diagram (Figure 7 Zone I) can be obtained in anhydrous crystalline Form A (hereinafter referred to as “Form A”), the mixing process by a temperature and the formation of methanol and water to determine the composition of the solvent, and the need to add a certain amount of Form a as a seed crystal to induce precipitation of crystals to control crystallization conditions are very harsh, operable range is very small, easy to form methanol solvate, hydrate or stable crystalline type C, to obtain reproducible single crystal type a low, it is difficult to achieve industrial production, and no mention of the preparation of Form a yield and purity in this patent.
[0011]
[0012] Chinese patent CN102267957A invention discloses a method for preparing febuxostat Form A, the solvent is preferably acetone, dissolved into 25 ~ 40 ° C was allowed to stand, when there began to crystallize when stirred for 20 to 40 minutes, then placed in -15 ~ 0 ° C to continue the crystallization of 8 to 10 hours. The crystallization process need to well below zero, when industrial mass production, resulting in high production costs, is not conducive to industrial production, the process yield up to 95.4%.
[0013] Chinese patent CN101139325 of Example 7 discloses the preparation of Form A with acetone method, although the process is simple, but the yield is low, only 50%.
[0014] Although the Chinese patent CN101684108A isopropyl alcohol as a solvent is disclosed a method for preparing crystalline form, the crystalline form of preparation is used to cool and heat a phased manner was allowed to stand, the crystallization temperature, long crystallization time, about 30 hours, the yield is low, and its products are not crystalline Form A.
[0015] In addition, Chinese patent CN101525319A, CN101805310, CN101926795A, CN101926794, W02012020272A2 are disclosed ethanol as a solvent or aqueous ethanol as a solvent preparation methods, and its products are crystalline ethanol solvate.
[0016] World Patent W02011139886A2 discloses the use of a mixed solvent of alcohol, and its products are not obtained polymorph A0
PAPER
Letters in Organic Chemistry (2015), 12(3), 217-221
Synthesis of the Major Metabolites of Febuxostat
Author(s): Xiao Long Li, Rui Qiu, Wei Li Wan, Xu Cheng, Li Hai and Yong Wu
Affiliation: Key Laboratory of Drug Targeting of Education Ministry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China.
Graphical Abstract:

Abstract:
Total synthesis of three Febuxostat metabolites, named 67M-1, 67M-2, and 67M-4,is described in this article. Through condensation of the key intermediate compound A with different side chains, and then oxidation and hydrolysis, we obtained three target compounds with an overall yield of 19.5%-28.0%.
VOLUME: 12
ISSUE: 3
Page: [217 – 221]
Pages: 5
DOI: 10.2174/1570178612666150108000805, http://www.eurekaselect.com/127479/article

ULORIC (febuxostat) is a xanthine oxidase inhibitor. The active ingredient in ULORIC is 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid, with a molecular weight of 316.38. The empirical formula is C16H16N2O3S.
The chemical structure is:
 |
Febuxostat is a non-hygroscopic, white crystalline powder that is freely soluble in dimethylformamide; soluble in dimethylsulfoxide; sparingly soluble in ethanol; slightly soluble in methanol and acetonitrile; and practically insoluble in water. The melting range is 205°C to 208°C.
LORIC tablets for oral use contain the active ingredient, febuxostat, and are available in two dosage strengths, 40 mg and 80 mg. Inactive ingredients include lactose monohydrate, microcrystalline cellulose, hydroxypropyl cellulose, sodium croscarmellose, silicon dioxide and magnesium stearate. ULORIC tablets are coated with Opadry II, green.
| CN1642546A * | Mar 28, 2003 | Jul 20, 2005 | Teijin Ltd. | Containing a single crystalline solid preparation |
| CN102471295A * | Jul 14, 2010 | May 23, 2012 | Teijin Pharma Ltd. | The method of manufacturing the poor solvent additive method of 2- (3-cyano-4-isobutyl-phenyl) -4-methyl-5-carboxylic acid crystalline polymorph of |
| EP2502920A1 * | Mar 25, 2011 | Sep 26, 2012 | Sandoz Ag | Crystallization process of Febuxostat from A |
| JP2011020950A* | Title not available |
| WO2015018507A3 * | Jul 30, 2014 | Oct 22, 2015 | Pharmathen S.A. | A novel process for the preparation of febuxostat |
| CN103304512A * | Jun 4, 2013 | Sep 18, 2013 | 华南理工大学 | Preparation method for febuxostat |
| WO2011031409A1 * | Aug 12, 2010 | Mar 17, 2011 | Teva Pharmaceutical Industries Ltd. | Processes for preparing febuxostat |
| JP2834971B2 | Title not available | |||
| JP3202607B2 | Title not available | |||
| JPH1045733A * | Title not available | |||
| US5614520 | Jan 30, 1995 | Mar 25, 1997 | Teijin Limited | 2-arylthiazole derivatives and pharmaceutical composition thereof |
| CN102229581A * | Nov 15, 2010 | Nov 2, 2011 | 邹巧根 | Preparation method for febuxostat intermediate |
| JPH1045733A * | Title not available |
| CN101497589A * | Feb 26, 2009 | Aug 5, 2009 | 沈阳药科大学 | Method for synthesizing 2-(3-cyano-4- isobutoxy phenyl)-4-methyl-carboxylate |
| CN101863854A * | Jun 29, 2010 | Oct 20, 2010 | 沈阳药科大学 | Synthesis method of 2-(3-cyan-4-isobutoxy) phenyl-4-methyl-5-thiazole formic acid |
| JP2706037B2 * | Title not available |
| Reference | ||
|---|---|---|
| 1 | * | HASEGAWA, M. ET AL.: ‘A facile one-pot synthesis of 4-alkoxy-1,3-benzenedicarbonitrile‘ HETEROCYCLES vol. 47, no. 2, 1998, pages 857 – 864 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2012131590A1 * | Mar 28, 2012 | Oct 4, 2012 | Sandoz Ag | An improved process for preparation of febuxostat and its polymorphic crystalline form c thereof |
| WO2014009817A1 * | Mar 19, 2013 | Jan 16, 2014 | Alembic Pharmaceuticals Limited | Pharmaceutical composition of febuxostat |
| WO2014057461A1 | Oct 10, 2013 | Apr 17, 2014 | Ranbaxy Laboratories Limited | Process for the preparation of crystalline form g of febuxostat |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(3-cyano-4-isobutoxyphenyl)-4-methyl-
1,3-thiazole-5-carboxylic acid |
|
| Clinical data | |
| Trade names | Uloric, Adenuric, Atenurix, Feburic, Goturic, Goutex. Generic in several countries.[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609020 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | ~49% absorbed |
| Protein binding | ~99% to albumin |
| Metabolism | via CYP1A2, 2C8, 2C9,UGT1A1, 1A3, 1A9, 2B7 |
| Biological half-life | ~5-8 hours |
| Excretion | Urine (~49% mostly as metabolites, 3% as unchanged drug); feces (~45% mostly as metabolites, 12% as unchanged drug) |
| Identifiers | |
| CAS Number | 144060-53-7 |
| ATC code | M04AA03 (WHO) |
| PubChem | CID 134018 |
| IUPHAR/BPS | 6817 |
| DrugBank | DB04854 |
| ChemSpider | 118173 |
| UNII | 101V0R1N2E |
| KEGG | D01206 |
| ChEMBL | CHEMBL1164729 |
| Chemical data | |
| Formula | C16H16N2O3S |
| Molar mass | 316.374 g/mol |
/////////Febuxostat, 144060-53-7, Uloric, Adenuric, Tei 6720, thiazole derivative, inhibitor of XANTHINE OXIDASE, treatment of HYPERURICEMIA, chronic GOUT, FBX, Febugood, Feburic, Febutaz, TMX 67, Zurig
CC1=C(SC(=N1)C2=CC(=C(C=C2)OCC(C)C)C#N)C(=O)O
Title: Febuxostat
CAS Registry Number: 144060-53-7
CAS Name: 2-[3-Cyano-4-(2-methylpropoxy)phenyl]-4-methyl-5-thiazolecarboxylic acid
Additional Names: 2-(3-cyano-4-isobutyloxyphenyl)-4-methyl-5-thiazolecarboxylic acid
Manufacturers’ Codes: TEI-6720; TMX-67
Molecular Formula: C16H16N2O3S
Molecular Weight: 316.37
Percent Composition: C 60.74%, H 5.10%, N 8.85%, O 15.17%, S 10.14%
Literature References: Xanthine oxidase/xanthine dehydrogenase inhibitor. Prepn: S. Kondo et al., EP 513379; eidem, US5614520 (1992, 1997 both to Teijin). Synthesis: M. Hasegawa, Heterocycles 47, 857 (1998). Mechanism of action and crystal structure study: K. Okamoto et al., J. Biol. Chem. 278, 1848 (2003). Pharmacology: Y. Osada et al., Eur. J. Pharmacol. 241, 183 (1993). Clinical pharmacokinetics: M. D. Mayer et al., Am. J. Therap. 12, 22 (2005). Clinical evaluation in hyperuricemia and gout: M. A. Becker et al., N. Engl. J. Med. 353, 2450 (2005). Review of clinical development: B. Tomlinson, Curr. Opin. Invest. Drugs 6,1168-1178 (2005).
Properties: Crystals from ethanol, mp 238-239° (dec). Also reported as crystals from acetone, mp 201-202° (Hasegawa).
Melting point: mp 238-239° (dec); mp 201-202° (Hasegawa)
Therap-Cat: Treatment of hyperuricemia and chronic gout.
Keywords: Antigout; Xanthine Oxidase Inhibitor.
Rifaximin

Rifaximin;
Rifaxidin; Rifacol; Xifaxan; Normix; Rifamycin L 105;L 105 (ansamacrolide antibiotic), L 105SV
(2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-á]-benzimidazole-1,15(2H)-dione,25-acetate
CAS 80621-81-4, 4-Deoxy-4-methylpyrido[1,2-1,2]imidazo[5,4-c]rifamycin SV,
4-Deoxy-4′-methylpyrido[1′,2′-1,2]imidazo[5,4-c]rifamycin SV, Rifacol
| C43H51N3O11 | |
| Molecular Weight: | 785.87854 g/mol |
|---|

XIFAXAN tablets for oral administration are film-coated and contain 200 mg or 550 mg of rifaximin.
Rifaximin is an orally administered, semi-synthetic, nonsystemic antibiotic derived from rifamycin SV with antibacterial activity. Rifaximin binds to the beta-subunit of bacterial DNA-dependent RNA polymerase, inhibiting bacterial RNA synthesis and bacterial cell growth. As rifaximin is not well absorbed, its antibacterial activity is largely localized to the gastrointestinal tract.
Rifaximin (trade names:RCIFAX, Rifagut, Xifaxan, Zaxine) is a semisynthetic antibiotic based on rifamycin. It has poor oral bioavailability, meaning that very little of the drug will be absorbed into the blood stream when it is taken orally. Rifaximin is used in the treatment of traveler’s diarrhea, irritable bowel syndrome, and hepatic encephalopathy, for which it receivedorphan drug status from the U.S. Food and Drug Administration in 1998.
Rifaximin is a rifamycin that was launched in 1988 by Alfa Wasserman for the treatment of bacterial infection, and was commercialized in 2004 by Salix for the treatment of Clostridium difficile-associated diarrhea. In 2008, the product was launched in Germany for the treatment of travelers’ diarrhea caused by non-invasive enteropathogenic bacteria in adults. In 2015, Xifaxan was approved in the U.S. for the treatment of abdominal pain and diarrhea in adult men and women with irritable bowel syndrome with diarrhea. At the same year, Aska filed an application for approval of the product in Japan for the treatment of hepatic encephalopathy.
Rifaximin is licensed by the U.S. Food and Drug Administration to treat traveler’s diarrhea caused by E. coli.[1] Clinical trials have shown that rifaximin is highly effective at preventing and treating traveler’s diarrhea among travelers to Mexico, with fewside effects and low risk of developing antibiotic resistance.[2][3][4] It is not effective against Campylobacter jejuni, and there is no evidence of efficacy against Shigella or Salmonella species.
| Launched – 1988 | Alfa Wassermann | Infection, bacterial |
| Launched – 2004 | Salix | Traveler’s diarrhea |
| Launched – 2010 | Salix | Encephalopathy, hepatic |
| Launched – 2015 | Salix | Irritable bowel syndrome (Diarrhea predominant) |
| Launched | Alfa Wassermann Merck & Co. |
Hyperammonemia |
The drug is also at Salix in phase II trials for the treatment of Crohn’s disease. Alfa Wasserman is also conducting phase II trials for Crohn’s disease. The product was approved and launched in the U.S. for the maintenance of remission of hepatic encephalopathy in 2010. Mayo Clinic is conducting phase II clinical trials in the U.S. for the treatment of primary sclerosing cholangitis and the University of Hong Kong is also conducting Phase II trials for the treatment of functional dyspepsia.
It may be efficacious in relieving chronic functional symptoms of bloating and flatulence that are common in irritable bowel syndrome (IBS),[5][6] especially IBS-D.
In February 1998, Salix was granted orphan drug designation by the FDA for the use of rifaximin to treat hepatic encephalopathy. In 2009, a codevelopment agreement was established between Lupin and Salix in the U.S. for the development of a new formulation using Lupin’s bioadhesive drug delivery technology.
There was recentlya pilot-study done on the efficacy of rifaximin as a means of treatment for rosacea, according to the study, induced by the co-presence of small intestinal bacterial overgrowth.[7]
In the United States, rifaximin has orphan drug status for the treatment of hepatic encephalopathy.[8] Although high-quality evidence is still lacking, rifaximin appears to be as effective as or more effective than other available treatments for hepatic encephalopathy (such as lactulose), is better tolerated, and may work faster.[9] Hepatic encephalopathy is a debilitating condition for those with liver disease. Rifaximin is an oral medication taken twice daily that helps patients to avoid reoccurring hepatic encephalopathy. It has minimal side effects, prevents reoccurring encephalopathy and high patient satisfaction. Patients are more compliant and satisfied to take this medication than any other due to minimal side effects, prolong remission, and overall cost.[10] Rifaximin helps patients avoid multiple readmissions from hospitals along with less time missed from work as well. Rifaximin should be considered a standard prescribed medication for those whom have episodes of hepatic encephalopathy.
The drawbacks to rifaximin are increased cost and lack of robust clinical trials for HE without combination lactulose therapy.
Also treats hyperammonemia by eradicating ammoniagenic bacteria.
Mechanism of action
Rifaximin interferes with transcription by binding to the β-subunit of bacterial RNA polymerase.[11] This results in the blockage of the translocation step that normally follows the formation of the first phosphodiester bond, which occurs in the transcription process.[12]
Efficacy
A 2011 study in patients with IBS (sans constipation) indicated 11% showed benefits over a placebo.[13] The study was supported by Salix Pharmaceuticals, the patent holder.[13] A 2010 study in patients treated for Hepatic Cirrhosis with hospitalization involving Hepatic encephalopathy resulted in 22% of the rifaxmin treated group experiencing a breakthrough episode of Hepatic encephalopathy as compared to 46% of the placebo group. The majority patients were also receivingLactulose therapy for prevention of hepatic encephalopathy in addition to Rifaximin.[14] Rifaximin shows promising results, causing remission in up to 59% of people with Crohn’s disease and up to 76% of people with Ulcerative Colitis.[15]
Availability
In the United States, Salix Pharmaceuticals holds a US Patent for rifaximin and markets the drug under the name Xifaxan, available in tablets of 200 mg and 550 mg.[16][17] In addition to receiving FDA approval for traveler’s diarrhea and (marketing approved for)[17] hepatic encephalopathy, Xifaxan received FDA approval for IBS in May 2015.[18] No generic formulation is available in the US and none has appeared due to the fact that the FDA approval process was ongoing. If Xifaxan receives full FDA approval for hepatic encephalopathy it is likely that Salix will maintain marketing exclusivity and be protected from generic formulations until March 24, 2017.[17] Price quotes received on February 21, 2013 for Xifaxan 550 mg in the Denver Metro area were between $23.57 and $26.72 per tablet. A price quote received on June 24, 2016 for Xifaxan 550 mg was $31.37 per tablet.
Rifaximin is approved in 33 countries for GI disorders.[19][20] On August 13, 2013, Health Canada issued a Notice of Compliance to Salix Pharmaceuticals Inc. for the drug product Zaxine.[21] In India it is available under the brand names Ciboz and Xifapill.[
SPECTRA
LINK IS CLICK
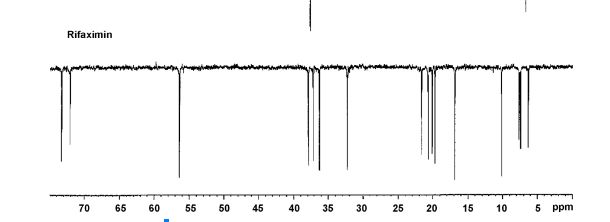
APT 13C NMR RIFAXIMIN
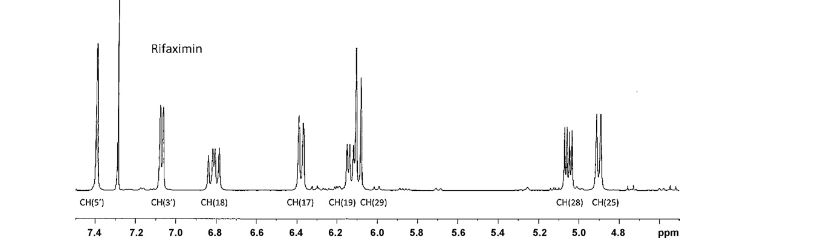
1H NMR PARTIAL
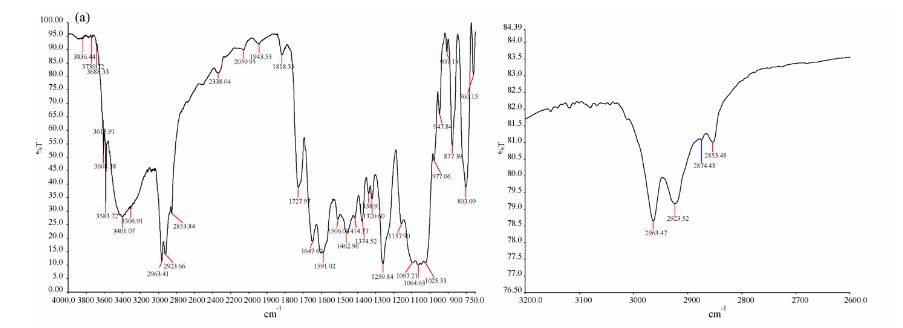
IR
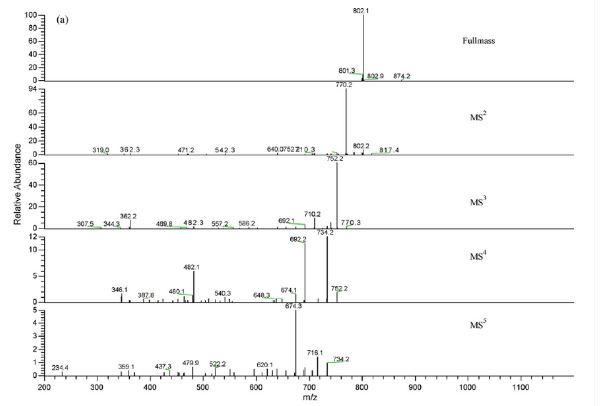
Direct infusion mass analysis ESI (+)
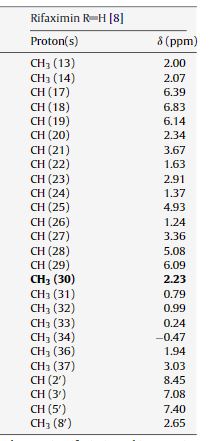
IH NMR
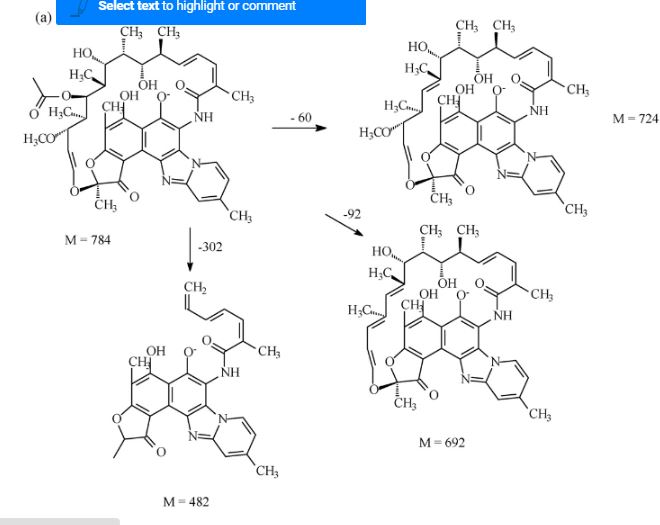
- [-]ESI FRAG PATHWAY
Synthesis
Rifaximin is a broad-spectrum antibiotic belonging to the family of Rifamycins and shows its antibacterial activity, in the gastrointestinal tract against localized bacteria that cause infectious diarrhoea, irritable bowel syndrome, small intestinal bacterial overgrowth, Crohn’s disease, and/or pancreatic insufficiency.
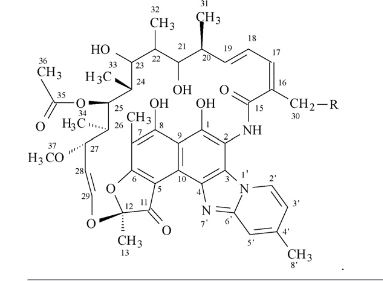
Rifaximin is sold under the brand name Xifaxan® in US for the treatment of Travellers’ diarrhoea and Hepatic Encephalopathy. The chemical name of Rifaximin is (2S , 16Z, 18E,20S ,21 S ,22R,23R,24R,25S ,26S ,27S ,28E)-5,6,21 ,23 ,25-pentahydroxy-27-methoxy-2,4,1 l,16,20,22,24,26-octamethyl-2,7(epoxypentadeca-[l,l l,13]trienimino) benzofuro[4,5-e]pyrido[l,2-a]-benzimidazole-l,15(2H)-dione,25-acetate and the molecular formula is G^HsiNsOn with a molecular weight of 785.9. The structural formula of Rifaximin is:

Formula I
Rifaximin was first described and claimed in Italian patent IT 1154655 and U.S. Pat. No.4,341,785. These patents disclose a process for the preparation of Rifaximin and a method for the crystallisation thereof. The process for the preparation of Rifaximin is as depicted in scheme I given below:

Scheme -I
U.S. Pat. No. 4,179,438 discloses a process for the preparation of 3-bromorifamycin S which comprises reaction of rifamycin S with at least two equivalents of bromine, per one mole of rifamycin S in the presence of at least one mole of pyridine per each equivalent of bromine and in the presence of ethanol, methanol or mixtures thereof with water at a
temperature not above the room temperature. The process is shown in the scheme given below:

Rifamycin S 3-Bromo-Rifamycin-S
U.S. Patent No.4,557, 866 discloses a process for one step synthesis of Rifaximin from Rifamycin O, which is shown in scheme II given below:

Rifamycin O Rifaximin
Scheme -II
US ‘866 patent also discloses purification of Rifaximin by performing crystallization of crude Rifaximin from a 7:3 mixture of ethyl alcohol/water followed by drying both under atmospheric pressure and under vacuum. The crystalline form which is obtained has not been characterized.
U.S. Patent No. 7,045,620 describes three polymorphic forms α, β and γ of Rifaximin. Form a and β show pure crystalline characteristics while the γ form is poorly crystalline. These polymorphic forms are differentiated on the basis of water content and PXRD. This patent also discloses processes for preparation of these polymorphs which involve use of specific reaction conditions during crystallization like dissolving Rifaximin in ethyl alcohol at 45-65°C, precipitation by adding water to form a suspension, filtering suspension and washing the resulted solid with demineralized water, followed by drying at room temperature under vacuum for a period of time between 2 and 72 hours. Crystalline forms a and β are obtained by immediate filtration of suspension when temperature of reaction mixture is brought to 0°C and poorly crystalline form γ is obtained when the reaction mixture is stirred for 5-6 hours at 0°C and then filtered the suspension. In addition to above these forms are also characterized by specific water content. For a form water content should be lower than 4.5%, for β form it should be higher than 4.5% and to obtain γ form, water content should be below 2%.
U.S. Patent No. 7,709,634 describes an amorphous form of Rifaximin which is prepared by dissolving Rifaximin in solvents such as alkyl esters, alkanols and ketones and precipitating by addition of anti-solvents selected from hydrocarbons, ethers or mixtures thereof.
U.S. Patent No. 8,193,196 describes two polymorphic forms of Rifaximin, designated δ and ε respectively. Form δ has water content within the range from 2.5 to 6% by weight (preferably from 3 to 4.5%).
U.S. Patent No 8,067,429 describes a-dry, β-1, β-2, ε-dry and amorphous forms of Rifaximin.
U.S. Patent No. 8,227,482 describes polymorphs Form μ, Form π, Form Omicron, Form Zeta, Form Eta, Form Iota and Form Xi of Rifaximin.
International application publications WO 2008/035109, WO 2008/155728, WO 2012/035544, WO 2012/060675, and WO 2012/156533 describes various amorphous or poorly crystalline forms of Rifaximin.
These polymorphic forms are obtained under different experimental conditions and are characterized by XRPD pattern.
The polymorphic forms of Rifaximin obtained from the prior art methods have specific water content. Transition between different polymorphic forms of Rifaximin occurs by drying or wetting of the synthesized Rifaximin. Hence, it is evident from above that Rifaximin can exist in number of polymorphic forms, formation of these polymorphic forms depends upon specific reaction conditions applied during crystallization and drying.
Rifaximin is a semi-synthetic, rifamycin-based non-systematic antibiotic. It is chemically termed as (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26 S,27S, 28E)-5,6,21,23,255-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-a]-benzimida-zole-1,15(2H)-dione,25-acetate (I).

Rifaximin is used for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli.
Rifaximin was first disclosed in US4341785 which also discloses a process for its preparation and a method for crystallization of rifaximin using suitable solvents or mixture of solvents. However, this patent does not mention the polymorphism of rifaximin.
Canadian patent CA1215976 discloses a process for the synthesis of imidazo rifamycins which comprises reacting rifamycin S with 2-amino-4-methyl pyridine.
US4557866 discloses a process for preparation of rifaximin, but does not mention the polymorphs of rifaximin.
US7045620 discloses crystalline polymorphic forms of rifaximin which are termed as rifaximin α, rifaximin β and rifaximin γ. These polymorphic forms are characterized using X-ray powder diffraction. Further this patent mentions that γ form is poorly crystalline with a high content of amorphous component. This patent also discloses processes for preparation of these polymorphs which involve use of processes of crystallization and drying as disclosed in US4557866along with control of temperature at which the product is crystallized, drying process, water content thereof. Further, according to this patent, crystal formation depends upon the presence of water within the crystallization solvent.
The above patent discloses rifaximin α which is characterized by water content lower than 4.5% & powder X-ray diffractogram having significant peaks are at values of diffraction angles 2θ of 6.6°; 7.4°; 7.9°, 8.8°, 10.5°, 11.1 °, 11.8°, 12.9°, 17.6°, 18.5°, 19.7°, 21.0°, 21.4°, 22.1°; rifaximin β which is characterized by water content higher than 4.5% & powder X-ray diffractogram having significant peaks are at values of diffraction angles 2θ of 5.4°; 6.4°; 7.0°, 7.8°, 9.0°, 10.4°, 13.1°, 14.4°, 17.1°, 17.9°, 18.3°, 20.9° and rifaximin γ which is characterized by poorer powder X-ray diffractogram because of poor crystallinity. The significant peaks are at values of diffraction angles 2θ of 5.0°; 7.1°; 8.4°.
US2005/0272754 also discloses polymorphs of rifaximin namely rifaximin α form, rifaximin β form & rifaximin γ form characterized by powder X-ray diffractogram, intrinsic dissolution rates and processes of preparation of polymorphic forms of rifaximin. However, none of the above patents disclose a wholly amorphous form of rifaximin.
It is a well known fact that different polymorphic forms of the same drug may have substantial differences in certain pharmaceutically important properties. The amorphous form of a drug may exhibit different dissolution characteristics and in some case different bioavailability patterns compared to crystalline forms.
Further, amorphous and crystalline forms of a drug may have different handling properties, dissolution rates, solubility, and stability.
Furthermore, different physical forms may have different particle size, hardness and glass transition temperatures. Amorphous materials do not exhibit the three-dimensional long-range orders found in crystalline materials, but are structurally more similar to liquids where the arrangement of molecules is random.
Amorphous solids do not give a definitive x-ray diffraction pattern (XRD). In addition, amorphous solids do not give rise to a specific melting point and tend to liquefy at some point beyond the glass transition temperature. Because amorphous solids do not have lattice energy, they usually dissolve in a solvent more rapidly and consequently may provide enhanced bioavailability characteristics such as a higher rate and extent of absorption of the compound from the gastrointestinal tract. Also, amorphous forms of a drug may offer significant advantages over crystalline forms of the same drug in the manufacturing process of solid dosage form such as compressibility.
PATENT
https://www.google.com/patents/EP2069363B1?cl=e
The schematic representation for preparation of amorphous rifaximin is as follows :

Amorphous rifaximin according to the present invention can be characterized by various parameters like solubility, intrinsic dissolution, bulk density, tapped density.
Rifaximin is known to exist in 3 polymorphic Forms namely α Form, β Form & γ Form of which the α Form is thermodynamically the most stable. Hence, the amorphous form of rifaximin was studied in comparison with α Form.
Further, when intrinsic dissolution of amorphous rifaximin is carried out against the α Form, it is observed that the amorphous rifaximin has better dissolution profile than α Form which is shown in table below (this data is also shown graphically in Figure 3):
Dissolution medium : 1000 ml of 0.1M Sodium dihydrogen phosphate monohydrate + 4.5g of sodium lauryl sulphate
Temperature : 37±0.5°C
Rotation speed : 100 rpm
Particle size : Amorphous rifaximin – 11 microns
α Form of rifaximin – 13 microns
-
Time in minutes % Release of Amorphous Rifaximin % Release of α Form of Rifaximin 15 1.1 0.8 30 1.9 1.8 45 2.9 3.0 60 3.7 4.4 120 8.1 11.0 180 12.6 18.0 240 16.6 24.6 360 24.7 38.7 480 32.0 47.5 600 39.5 52.7 720 46.4 56.4 960 60.4 62.9 1200 72.9 67.8 1400 83.0 72.7 Amorphous rifaximin exhibits bulk density in the range of 0.3 – 0.4 g/ml and tapped density is in the range of 0.4 – 0.5 g/ml while the α Form rifaximin exhibits bulk density in the range of 0.2 – 0.3 g/ml & tapped density is in the range of 0.3 – 0.4 g/ml. These higher densities of amorphous rifaximin are advantageous in formulation specifically in tablet formulation, for example, it gives better compressibility.
CLIP
Rifaximin (CAS NO.: 80621-81-4), with other name of 4-Deoxy-4-methylpyrido[1,2-1,2]imidazo[5,4-c]rifamycin SV, could be produced through many synthetic methods.
Following is one of the reaction routes:

The reaction of rifamycin S (I) with pyridine perbromide (II) in 2-propanol/chloroform (70/30) mixture at 0 C gives 3-bromorifamicin S (III), which is then condensed with 2-amino-4-methyl-pyridine (IV) at 10 C. The o-quinoniminic compound (V) is then obtained. This compound is finally reduced with ascorbic acid.
POLYMORPHISM
Rifaximin (INN; see The Merck Index, XIII Ed., 8304) is an antibiotic belonging to the rifamycin class, exactly it is a pyrido-imidazo rifamycin described and claimed in Italian Patent IT 1154655, while European Patent EP 0161534 describes and claims a process for its production starting from rifamycin O (The Merck Index, XIII Ed., 8301).
Both these patents describe the purification of rifaximin in a generic way stating that crystallization can be carried out in suitable solvents or solvent systems and summarily showing in some examples that the reaction product can be crystallized from the 7:3 mixture of ethyl alcohol/water and can be dried both under atmospheric pressure and under vacuum without specifying in any way either the experimental conditions of crystallization and drying, or any distinctive crystallographic characteristic of the obtained product.
The presence of different polymorphs had just not been noticed and therefore the experimental conditions described in both patents had been developed with the goal to get a homogeneous product having a suitable purity from the chemical point of view, independent from the crystallographic aspects of the product itself.
It has now been found, unexpectedly, that there are several polymorphous forms whose formation, besides the solvent, depends on time and temperature conditions under which both crystallization and drying are carried out.
In the present application, these orderly polymorphous forms will be, later on, conventionally identified as rifaximin α (FIG. 1) and rifaximin β (FIG. 2) on the basis of their respective specific diffractograms, while the poorly crystalline form with a high content of amorphous component will be identified as rifaximin γ (FIG. 3).
Rifaximin polymorphous forms have been characterized through the technique of the powder X-ray diffraction.
The identification and characterization of these polymorphous forms and, simultaneously, the definition of the experimental conditions for obtaining them is very important for a compound endowed with pharmacological activity which, like rifaximin, is marketed as medicinal preparation, both for human and veterinary use. In fact it is known that the polymorphism of a compound that can be used as active ingredient contained in a medicinal preparation can influence the pharmaco-toxicologic properties of the drug. Different polymorphous forms of an active ingredient administered as drug under oral or topical form can modify many properties thereof like bioavailability, solubility, stability, colour, compressibility, flowability and workability with consequent modification of the profiles of toxicological safety, clinical effectiveness and productive efficiency.
What mentioned above is confirmed by the fact that the authorities that regulate the grant of marketing authorization of the drugs market require that the manufacturing methods of the active ingredients are standardized and controlled in such a way that they give homogeneous and sound results in terms of polymorphism of production batches (CPMP/QWP/96, 2003—Note for Guidance on Chemistry of new Active Substance; CPMP/ICH/367/96—Note for guidance specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances; Date for coming into operation: May 2000).
The need for the above-mentioned standardization has further been strengthened in the field of the rifamycin antibiotics by Henwood S. Q., de Villiers M. M., Liebenberg W. and Lotter A. P., Drug Development and Industrial Pharmacy, 26 (4), 403-408, (2000), who have ascertained that different production batches of the rifampicin (INN) made from different manufacturers differ from each other in that they show different polymorphous characteristics, and as a consequence they show different dissolution profiles, along with a consequent alteration of the respective pharmacological properties.
By applying the crystallization and drying processes generically disclosed in the previous patents IT 1154655 and EP 0161534 it has been found that under some experimental conditions a poorly crystalline form of rifaximin is obtained, while under other experimental conditions other polymorphic crystalline forms of Rifaximin are obtained. Moreover it has been found that some parameters, absolutely not disclosed in the above-mentioned patents, like for instance preservation conditions and the relative ambient humidity, have the surprising effect to determine the polymorph form.
The polymorphous forms of rifaximin object of the present patent application were never seen or hypothesized, while thinking that, whichever method was used within the range of the described condition, a sole homogeneous product would always have been obtained, irrespective of crystallizing, drying and preserving conditions. It has now been found that the formation of α, β and γ forms depends both on the presence of water within the crystallization solvent, on the temperature at which the product is crystallized and on the amount of water present in the product at the end of the drying phase. Form α, form β and form γ of rifaximin have then been synthesized and they are the object of the invention.
Moreover it has been found that the presence of water in rifaximin in the solid state is reversible, so that water absorption and/or release can take place in time in presence of suitable ambient conditions; consequently rifaximin is susceptible of transition from one form to another, also remaining in the solid state, without need to be again dissolved and crystallized. For instance polymorph α, getting water by hydration up to a content higher than 4.5%, turns into polymorph β, which in its turn, losing water by drying up to a content lower than 4.5%, turns into polymorph α.
These results have a remarkable importance as they determine the conditions of industrial manufacturing of some steps of working which could not be considered critical for the determination of the polymorphism of a product, like for instance the washing of a crystallized product, or the preservation conditions of the end product, or the characteristics of the container in which the product is preserved.
The above-mentioned α, β and γ forms can be advantageously used as pure and homogeneous products in the manufacture of medicinal preparations containing rifaximin.
As already said, the process for manufacturing rifaximin from rifamycin O disclosed and claimed in EP 0161534 is deficient from the point of view of the purification and identification of the product obtained; it shows some limits also from the synthetic point of view as regards, for instance, the very long reaction times, from 16 to 72 hours, not very suitable to an industrial use and moreover because it does not provide for the in situ reduction of rifaximin oxidized that may be formed within the reaction mixture.
Therefore, a further object of the present invention is an improved process for the industrial manufacturing of the α, β and γ forms of rifaximin, herein claimed as products and usable as defined and homogeneous active ingredients in the manufacture of the medicinal preparations containing such active ingredient.
PATENT
https://www.google.com/patents/US20090234114
FIG. 1 is a powder X-ray diffractogram of rifaximin polymorphic form α.
FIG. 2 is a powder X-ray diffractogram of rifaximin polymorphic form β.
FIG. 3 is a powder X-ray diffractogram of rifaximin polymorphic form γ.
PATENT
Rifaximin (INN; see The Merck Index, XIII Ed., 8304, CAS no. 80621-81-4), IUPAC nomenclature (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25 pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-(1,11,13)trienimino)benzofuro(4,5-e)pyrido(1,2,-a)benzimidazole-1,15(2H)-dione,25-acetate) is a semi-synthetic antibiotic belonging to the rifamycin class of antibiotics. More precisely rifaximin is a pyrido-imidazo rifamycin described in the Italian patent IT 1154655, whereas the European patent EP 0161534 discloses a process for rifaximin production using rifamycin O as starting material (The Merck Index, XIII Ed., 8301).
U.S. Pat. No. 7,045,620, US 2008/0262220, US 7,612,199, US 2009/0130201 and Cryst. Eng. Comm., 2008, 10 1074-1081 (2008) disclose new forms of rifaximin.
WO 2008/035109 A1 discloses a process to prepare amorphous rifaximin, which comprises reaction of rifamycin S with 2-amino-4 picoline in presence of organic solvent like dichloromethane, ethylacetate, dichloroethylene, chloroform, in an inert atmosphere. When water is added to the reaction mixture, a solid precipitate corresponding to amorphous rifaximin is obtained.
The process described in this document can be assimilated to a crash precipitation, wherein the use of an anti-solvent causes the precipitation of rifaximin without giving any information about the chemical physical and biological characteristics of the rifaximin obtained.
WO 2009/108730 A2 describes different polymorphous forms of rifaximin and also amorphous forms of rifaximin. Amorphous forms are prepared by milling and crash precipitation and with these two different methods the amorphous rifaximin obtained from these two different processes has the same properties.
FIG. 4: 13C-NMR spectrum of rifaximin obtained by spray drying process.

FIG. 5: FT-IR spectrum of rifaximin obtained by spray drying process.

Patent
Rifaximin, lUPAC name:
(2S,16Z,18E,20S,21 S,22H,23H,24H,25S,26S,27S,28£)-5,6,21 ,23,25-pentahydroxy- 27-methoxy-2,4,1 1 ,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1 ,1 1 ,13]-trienimmino)-benzofuro-[4,5-e]-pirido-[1 ,2-oc]-benzimidazol-1 , 15(2 -/)-dione,25-acetate, is the compound of formula (I):

Rifaximin is a broad-spectrum antibiotic belonging to the family of rifamycins, devoid of systemic activity. In view of its physicochemical properties, it is not adsorbed in the gastrointestinal tract and therefore exerts its antimicrobial action inside the gastrointestinal tract. Rifaximin therefore has applications in the treatment of diarrhoea and of microbial infections of the gastrointestinal tract typically caused by E. coli, a microorganism which, being incapable of passing through the mucosa of the gastrointestinal tract, remains in contact with the gastrointestinal fluids. Rifaximin also has applications for the treatment of irritable bowel syndrome, Crohn’s disease, diverticulitis and for antibiotic prophylaxis preceding surgical operations on the intestines.
Rifaximin was obtained and described for the first time in the EP161534 starting from rifamycin O and 2-amino-4-picoline in the presence of ethanol/water and
ascorbic acid/HCI to obtain raw rifaximin which is then treated with Ethanol/water to obtain crystallized rifaximin.
Polymorphic forms of rifaximin, and processes for their synthesis and purification, are described in various documents of the known art.
Rifaximin K was firstly described in WO2012/156951 . Such a crystalline form resulted to be more stable in the presence of humidity than the other known crystalline forms of rifaximin, thus enabling the storage, even for prolonged periods. Such a polymorph was obtained by a process starting from rifaximin comprising the following steps: -suspending or dissolving rifaximin in a 1 ,2-dimethoxyethane based solvent, recovering the product and drying to remove said 1 ,2-dimethoxyethane based solvent. In one of the embodiments of the invention 1 ,2-dimethoxyethane is used as the unique solvent of rifaximin, in other 1 ,2-dimethoxyethane is described as used in combination of n-heptane, methanol, acetonitrile, R-COO-R1 esters wherein R and R1 are independently C3-C6 alkyl radicals, and C3-C7 alkyl ketones, ethanol, isopropanol and water.
Paper
The synthesis of 4-deoxypyrido(1′,2′-1,2)imidazo(5,4-c)rifamycin SV derivatives
J Antibiot 1984, 37(12): 1611
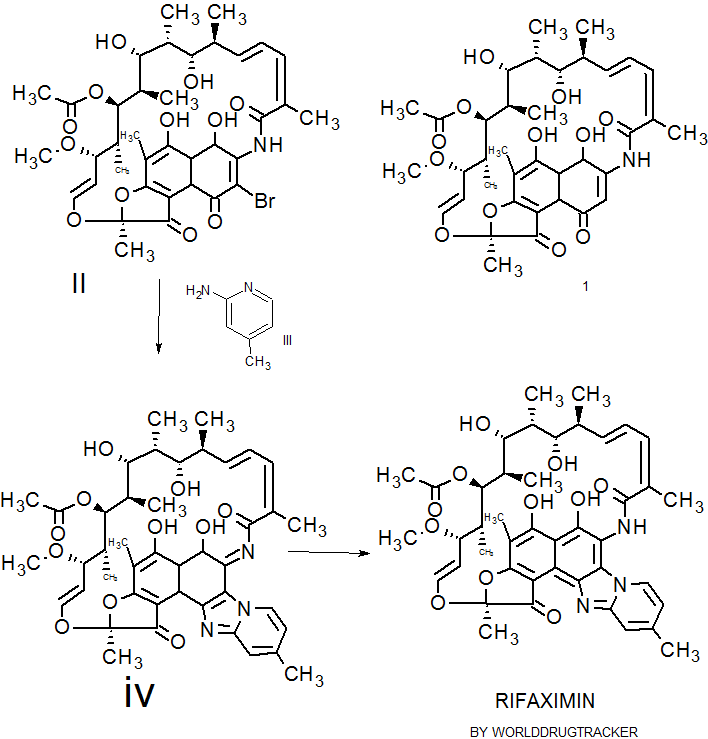
LAST STEP DEPICTED AGAIN
Treatment of rifamycin S (I) with Pyr·Br2 in 2-PrOH/CHCl3 gives 3-bromorifamycin S (II) (1), which upon cyclocondensation with 2-amino-4-methyl-pyridine (III) (1,2,3) in CHCl3 (2) or EtOH (3) yields imine derivative (IV). Finally, reduction of (IV) with L-(+)- ascorbic acid (1,2,3) in MeOH (2) or EtOH (3) provides the target rifaximin (1,2,3).
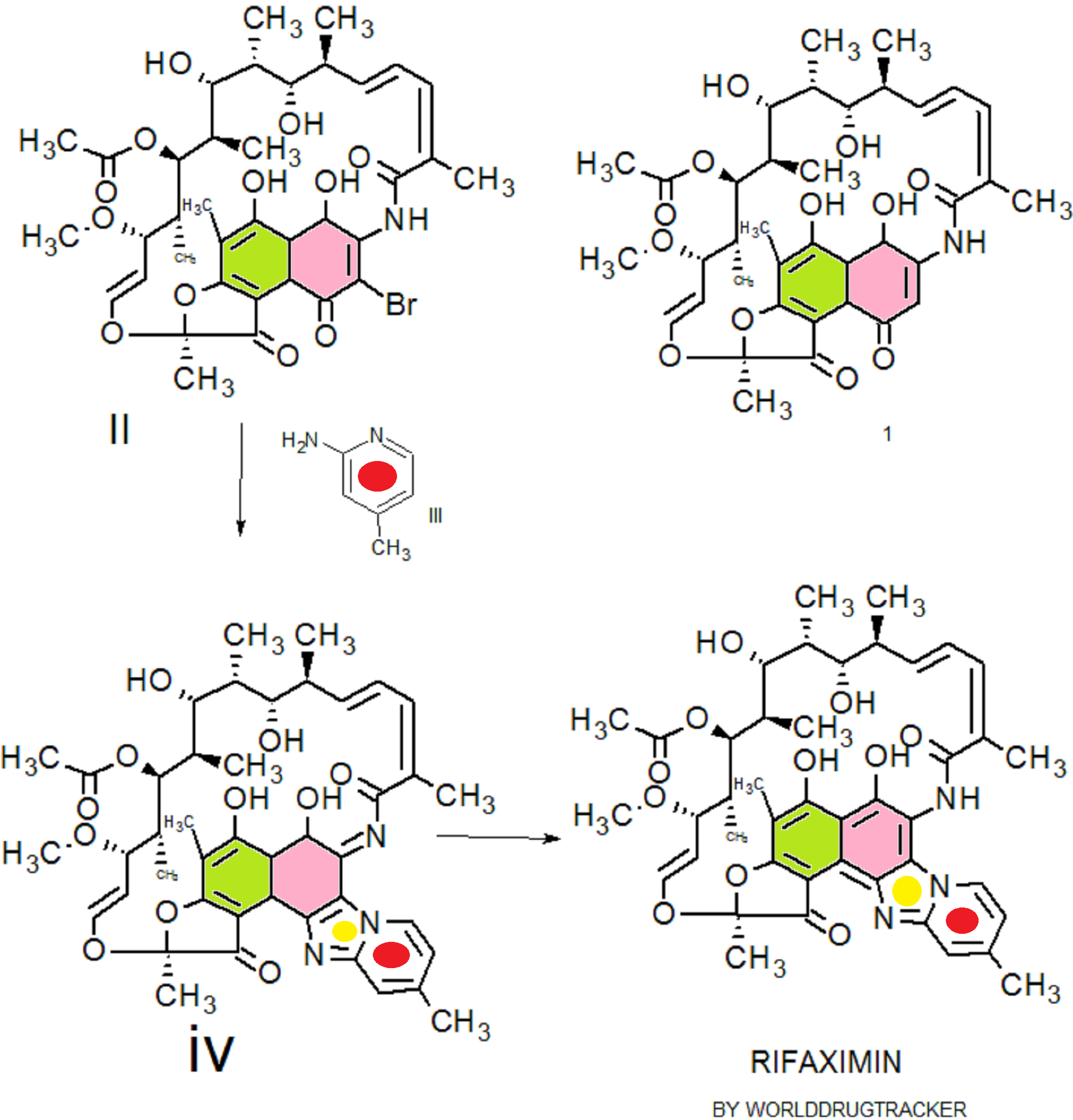
PATENT
WO 2005044823, WO 2012035544, WO 2015014984
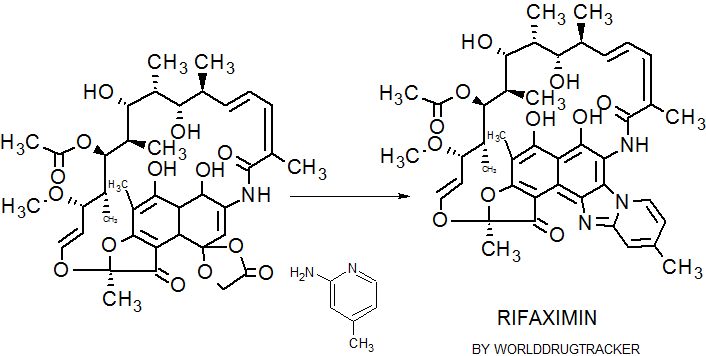
Rifaximin is prepared by the cyclocondensation of rifamycin-O with 2-amino-4-picoline in a solvent mixture such as acetone, acetonitrile, EtOH, MIBK, propylene glycol, i-PrOH or t-BuOH and H2O at 50 °C or EtOH/aceone/H2O or optionally in the presence of I2 in CH2Cl2
PATENT
The process is shown in the scheme given below:

Rifamycin-S
3-halo-Rifamycin-S

Examples
Example 1;
5g of Rifamycin S, 3.1 gms of 2-amino-4-methyl pyridine, 0.45 g of iodine, 1.65 ml of acetic acid and 20ml of acetonitrile were charged in a clean and dry round bottom flask followed by stirring the resultant reaction mixture at about 30°C for about 30 hours. The reaction progress was monitored by TLC, after completion of reaction, the reaction mass was quenched by adding a mixture of 4.0g of ascorbic acid dissolved in 20 ml of water. The resultant reaction suspension was stirred at about 25°C for about 15mins. 25 ml of dichloromethane was charged and stirred for about 15mins. followed by separation of organic and aqueous phases. The aqueous phase was extracted with 25 ml of dichloromethane followed by separation of organic and aqueous phases. The organic phases were combined and distilled at below about 50°C to yield Rifaximin as residue. 11.25ml of purified water and 11.25ml of ethanol were charged to the residue and stirred at about 30°C for about 15 mins. The resultant reaction
suspension was heated to about 75°C and stirred for about 30mins. The resultant reaction solution further cooled to about 25 °C and stirred for about 2 hours followed by further cooling to about 5°C for about 3 hours. The solid precipitated was filtered and the solid was washed with a mixture of 2.5ml of ethanol and 2.5 ml of purified water. The solid obtained was dried at about 50°C for about 10 hours to afford 3 g. of Rifaximin as crystalline form. Purity by HPLC: 99.85 area %.
PAPER
European journal of medicinal chemistry (2015), 103, 551-62
Patent
https://www.google.com/patents/WO2013027227A1?cl=en
Examples
Example 1 : Purification of Rifamycin S
Rifamycin S (500g) and Ethanol (1.5L) were stirred and refluxed for 1 hour. The reaction mixture was then cooled slowly to ambience, stirred at this temperature for 2 hour and filtered. The product dried in vacuum oven at 40 °C to obtain 475g of pure Rifamycin S showing the des acetyl impurity below to 0.6%.
Example 2: Preparation of rifaximin
Rifamycin S (300 g) was stirred in dichloromethane (900 ml) at room temperature for 15 minutes to get a clear solution and then 2-Amino-4-methyl pyridine (139.2g) was added at room temperature under nitrogen atmosphere. Iodine (57. Og) dissolved in dichloromethane (2100ml), was added drop wise in 30-45 minutes at room temperature. The reaction mass was stirred for 22-24 hours at 25-30 °C. After completion of the reaction, a 20% solution of L(-) ascorbic acid in water (300 ml) was added. The reaction mixture was stirred for 45-60 minutes at room temperature and then cooled to 10-15 °C. The pH of the resulting solution was adjusted to 1.5-2.0 with slow addition of dilute hydrochloric acid under stirring. The reaction mass was stirred for 15-20 minutes and layers were separated. The organic layer was washed with demineralized water (1500 ml), 10% sodium thiosulfate solution (1500 ml) and with demineralized water till pH was neutral. The solvent was distilled off under vacuum at 40-45 °C to get a residue which was taken in cyclohexane (1500 ml) and stirred for 1 hour. The resulting solid was filtered, washed with cyclohexane (300 ml) crystallized from a mixture of ethyl alcohol and water (600ml; 420ml ethyl alcohol and 180 ml water) to get 240g of crude rifaximin having purity 99.3% by HPLC.
Example 3: Preparation of rifaximin
Step-1: Preparation of crude rifaximin
Rifamycin S (300 g) was stirred in dichloromethane (900 ml) at room temperature for 15 minutes to get a clear solution and then 2-amino-4-methyl pyridine (139.2g) was added at room temperature under nitrogen atmosphere. Iodine (57. Og) dissolved in dichloromethane (2100ml), was added drop wise in 30-45 minutes at room temperature and was stirred for 22-24 hours. After completion of the reaction, a 20% solution of L (-) ascorbic acid in water (300 ml) was added and stirred for 45-60 minutes. The reaction mass was cooled to 10-15 °C and pH of the resulting solution was adjusted to 1.5-2.0 with slow addition of dilute hydrochloric acid under stirring. The reaction mass was stirred for 15-20 minutes and layers were separated and the organic layer was washed with demineralized water (1500 ml), with 10% sodium thiosulfate solution (1500 ml) and demineralized water till pH was neutral. The solvent was distilled off under vacuum at 40-45 °C to obtain a residue which was crystallized from a mixture of ethyl alcohol and water (378ml ethyl alcohol and 162 ml water) and dried at 35-40 °C to obtain 240g crude rifaximin having purity 98.8% by HPLC. Step-2: Purification of crude rifaximin
Crude rifaximin (240g) was stirred in dichloromethane (2400ml) at room temperature, a neutral alumina (240g) was added, stirred for 1 hour and filtered. The solvent was then distilled off and residue was treated with ethyl acetate (2400ml) and stirred to dissolution. The resulting residue was crystallized from a mixture of ethyl alcohol and water (302ml ethyl alcohol and 130ml water) and dried at 35-40 “C to obtain 192g of rifaximin having purity 99.8% by HPLC.
PATENT
https://www.google.com/patents/US9018225
PAPER

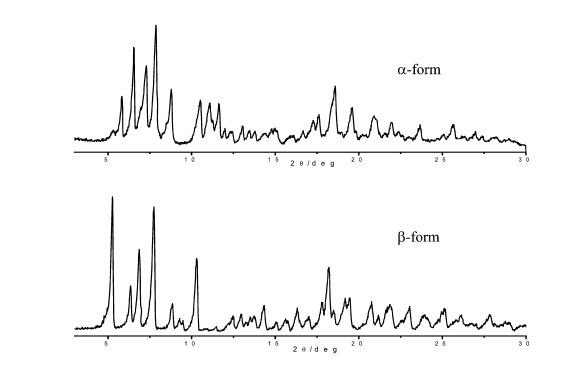
PATENTS
| US4341785 | May 11, 1981 | Jul 27, 1982 | Alfa Farmaceutici S.P.A. | Imidazo-rifamycin derivatives with antibacterial utility |
| US4557866 | Apr 26, 1985 | Dec 10, 1985 | Alfa Farmaceutici S.P.A. | Process for the synthesis of pyrido-imidazo rifamycins |
| US7045620 | Dec 5, 2003 | May 16, 2006 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in medicinal preparations |
| US7612199 | Jun 4, 2009 | Nov 3, 2009 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β, and γ of rifaximin |
| US7902206 | Mar 8, 2011 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β and γ of rifaximin | |
| US7906542 | May 13, 2008 | Mar 15, 2011 | Alfa Wassermann, S.P.A. | Pharmaceutical compositions comprising polymorphic forms α, β, and γ of rifaximin |
| US7915275 | Mar 29, 2011 | Alfa Wassermann, S.P.A. | Use of polymorphic forms of rifaximin for medical preparations | |
| US7923553 | Apr 12, 2011 | Alfa Wassermann, S.P.A. | Processes for the production of polymorphic forms of rifaximin | |
| US7928115 | Apr 19, 2011 | Salix Pharmaceuticals, Ltd. | Methods of treating travelers diarrhea and hepatic encephalopathy | |
| US8158644 | Apr 17, 2012 | Alfa Wassermann, S.P.A. | Pharmaceutical compositions comprising polymorphic forms α, β, and γ of rifaximin | |
| US8158781 | Mar 4, 2011 | Apr 17, 2012 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β and γ of rifaximin |
| US8193196 | Feb 27, 2006 | Jun 5, 2012 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20050272754 * | May 24, 2005 | Dec 8, 2005 | Alfa Wassermann S.P.A. | Polymorphic forms of rifaximin, processes for their production and uses thereof |
| Reference | ||
|---|---|---|
| 1 | Viscomi, G. C., et al., “Crystal forms of rifaximin and their effect on pharmaceutical properties“, Cryst Eng Comm, 2008, 10, 1074-1081, (May 28, 2008), 1074-1081. | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9186355 | Mar 30, 2015 | Nov 17, 2015 | Novel Laboratories | Rifaximin crystalline forms and methods of preparation thereof |
| WO2008035109A1 * | Sep 24, 2007 | Mar 27, 2008 | Cipla Limited | Rifaximin |
| WO2009108730A2 * | Feb 25, 2009 | Sep 3, 2009 | Salix Pharmaceuticals, Ltd. | Forms of rifaximin and uses thereof |
| WO2011080691A1 * | Dec 27, 2010 | Jul 7, 2011 | Silvio Massimo Lavagna | Method for the production of amorphous rifaximin |
| EP1698630A1 * | Mar 3, 2005 | Sep 6, 2006 | ALFA WASSERMANN S.p.A. | New polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20080262220 * | May 13, 2008 | Oct 23, 2008 | Giuseppe Claudio Viscomi | Polymorphic forms alpha, beta and gamma of rifaximin |
| US20090082558 * | Sep 20, 2007 | Mar 26, 2009 | Apotex Pharmachem Inc. | Amorphous form of rifaximin and processes for its preparation |
REFERENCED BY
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015014984A1 * | Aug 1, 2014 | Feb 5, 2015 | Clarochem Ireland Ltd. | A process for preparing rifaximin k |
| CN103360357A * | Aug 7, 2013 | Oct 23, 2013 | 中国药科大学 | A simvastatin-gliclazide co-amorphous compound |
| US9359374 | Jun 13, 2013 | Jun 7, 2016 | Apotex Pharmachem Inc. | Polymorphic forms of rifaximin |
| US4341785 * | May 11, 1981 | Jul 27, 1982 | Alfa Farmaceutici S.P.A. | Imidazo-rifamycin derivatives with antibacterial utility |
| US4557866 * | Apr 26, 1985 | Dec 10, 1985 | Alfa Farmaceutici S.P.A. | Process for the synthesis of pyrido-imidazo rifamycins |
| US7045620 * | Dec 5, 2003 | May 16, 2006 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in medicinal preparations |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8518949 | Jun 4, 2012 | Aug 27, 2013 | Alfa Wassermann S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20140079783 * | Jul 3, 2013 | Mar 20, 2014 | Alfa Wassermann Spa | Pharmaceutical Compositions Comprising Rifaximin and Amino acids, Preparation Methods and Use Thereof |
| CN101836959A * | May 20, 2010 | Sep 22, 2010 | 山东达因海洋生物制药股份有限公司 | Method for preparing almost bitterless rifaximin dry suspension |
| CN103269587A * | Jun 3, 2011 | Aug 28, 2013 | 萨利克斯药品有限公司 | New forms of rifaximin and uses thereof |
| WO2011153444A1 * | Jun 3, 2011 | Dec 8, 2011 | Salix Pharmaceuticals, Ltd | New forms of rifaximin and uses thereof |
References
- Xifaxan label information PDF Retrieved November 15, 2008.
- DuPont, H (2007). “Therapy for and Prevention of Traveler’s Diarrhea”. Clinical Infectious Diseases 45 (45 (Suppl 1)): S78–S84. doi:10.1086/518155. PMID 17582576.
- Ruiz J, Mensa L, Pons MJ, Vila J, Gascon J (May 2008). “Development of Escherichia coli rifaximin-resistant mutants: frequency of selection and stability”. Journal of antimicrobial chemotherapy 61 (5): 1016–9. doi:10.1093/jac/dkn078. PMID 18325895.
- Martinez-Sandoval F, Ericsson CD, Jiang ZD, Okhuysen PC, Romero JH, Hernandez N, Forbes WP, Shaw A, Bortey E, DuPont HL (Mar–Apr 2010). “Prevention of travelers’ diarrhea with rifaximin in US travelers to Mexico.”. J Travel Med. 17 (2): 111–7.doi:10.1111/j.1708-8305.2009.00385.x. PMID 20412178.
- Sharara A, Aoun E, Abdul-Baki H, Mounzer R, Sidani S, ElHajj I (2006). “A randomized double-blind placebo-controlled trial of rifaximin in patients with abdominal bloating and flatulence”. Am J Gastroenterol 101 (2): 326–33. doi:10.1111/j.1572-0241.2006.00458.x.PMID 16454838.
- Antibiotic May Help Ease Irritable Bowel, Businessweek, January 05, 2011
- Small intestinal bacterial overgrowth in rosacea: clinical effectiveness of its eradication. Parodi A, Paolino S, Greco A, Drago F, Mansi C, Rebora A, Parodi A, Savarino V.
- Wolf, David C. (2007-01-09). “Hepatic Encephalopathy”. eMedicine. WebMD. Retrieved 2007-02-15.
- Lawrence KR, Klee JA (2008). “Rifaximin for the treatment of hepatic encephalopathy”.Pharmacotherapy 28 (8): 1019–32. doi:10.1592/phco.28.8.1019. PMID 18657018.Free full text with registration at Medscape.
- Kimer, Nina; Krag, Aleksander; Gluud, Lise L. (March 2014). “Safety, efficacy, and patient acceptability of Rifaximin for hepatic encephalopathy”. Patient Preference and Adherence 8: 331–338. doi:10.2147/PPA.S41565. PMC 3964161. PMID 24672227. Retrieved 14 April 2016.
- http://formularyjournal.modernmedicine.com/formulary-journal/news/clinical/clinical-pharmacology/rifaximin-nonabsorbable-broad-spectrum-antibio?page=full
- http://www.drugbank.ca/drugs/DB01220
- Pimentel, Mark; Lembo, Anthony; Chey, William D.; Zakko, Salam; Ringel, Yehuda; Yu, Jing; Mareya, Shadreck M.; Shaw, Audrey L.; Bortey, Enoch (January 2011). “Rifaximin Therapy for Patients with Irritable Bowel Syndrome without Constipation”. N Engl J Med364 (1): 22–32. doi:10.1056/NEJMoa1004409. PMID 21208106.
- Bass NM, Mullen KD, Sanyal A et al. (March 2010). “Rifaximin treatment in hepatic encephalopathy”. N Engl J Med 362 (12): 1071–1081. doi:10.1056/NEJMoa0907893.PMID 20335583.
- Clark, Brian. “Rifaximin (Xifaxan) is a Promising Drug for the Treatment of Inflammatory Bowel Disease”. Human Data Projct. Human Data Project. Retrieved 28 March 2016.
- http://www.salix.com/products/xifaxan550.aspx
- http://www.accessdata.fda.gov/scripts/cder/ob/docs/obdetail.cfm?Appl_No=022554&TABLE1=OB_Rx
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm448328.htm
- http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM203248.pdf
- http://www.salix.com/news-media/news/previous-years-news/fda-approves-xifaxan%C2%AE-550-mg-tablets-for-reduction-in-risk-of-overt-hepatic-encephalopathy-he-recurrence.aspx
- http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/sbd_smd_2013_zaxine_161256-eng.php
External links
- FDA label approved for Xifaxan (PDF warning)
- Xifaxan200 (manufacturer’s website)
| Patents |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro
[4,5-e]pyrido[1,2-a]-benzimida-zole-1,15(2H)-dione,25-acetate |
|
| Clinical data | |
| Trade names | Xifaxan, Xifaxanta, Normix, Rifagut |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a604027 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | < 0.4% |
| Metabolism | Hepatic |
| Biological half-life | 6 hours |
| Excretion | Fecal (97%) |
| Identifiers | |
| CAS Number | 80621-81-4 |
| ATC code | A07AA11 (WHO) D06AX11(WHO) QG51AA06 (WHO)QJ51XX01 (WHO) |
| PubChem | CID 6436173 |
| DrugBank | DB01220 |
| ChemSpider | 10482302 |
| UNII | L36O5T016N |
| KEGG | D02554 |
| ChEBI | CHEBI:75246 |
| ChEMBL | CHEMBL1617 |
| Chemical data | |
| Formula | C43H51N3O11 |
| Molar mass | 785.879 g/mol |
Giuseppe Viscomi, Manuela Campana, Dario Braga, Donatella Confortini, Vincenzo Cannata, Paolo Righi, Goffredo Rosini, “Polymorphic forms of rifaximin, processes for their production and uses thereof.” U.S. Patent US20050272754, issued December 08, 2005.
///////Rifaximin, Rifaxidin, Rifacol, Xifaxan, Normix, Rifamycin L 105, 80621-81-4
CC1C=CC=C(C(=O)NC2=C(C3=C(C4=C(C(=C3O)C)OC(C4=O)(OC=CC(C(C(C(C(C(C1O)C)O)C)OC(=O)C)C)OC)C)C5=C2N6C=CC(=CC6=N5)C)O)C
TELMISARTAN PART 3/3

CONT…………………….
PAPER
Journal of Organic Chemistry (2015), 80(3), 1915-1919
J. Org. Chem., 2015, 80 (3), pp 1915–1919
DOI: 10.1021/jo5025333

A direct and efficient total synthesis has been developed for telmisartan, a widely prescribed treatment for hypertension. This approach brings together two functionalized benzimidazoles using a high-yielding Suzuki reaction that can be catalyzed by either a homogeneous palladium source or graphene-supported palladium nanoparticles. The ability to perform the cross-coupling reaction was facilitated by the regio-controlled preparation of the 2-bromo-1-methylbenzimidazole precursor. This convergent approach provides telmisartan in an overall yield of 72% while circumventing many issues associated with previously reported processes.
……………………
PAPER
International Journal of Research in Pharmaceutical and Biomedical Sciences (2013), 4(1), 293-295
telmisartan1. [Yield 87%, Purity 99.97% by HPLC.M.P. 260 – 262°C, Sulphated ash < 0.01%].
1H NMR (DMSO-d6): δ 0.98-1.03 (t,3H), 1.73- 1.86 (m, 2H), 2.5 – 2.63 (s, 3H), 2.90-2.95 (s, 2H),3.82 (s, 3H), 5.62 (s, 2H), 7.16-7.34 (m,7H), 7.40-7.59 (m,4H), 7.68-7.70 (m, 3H), 12.86 (s, 1H).
M/Z: 515.50 [M + H]+
………………………..
PATENT
WO 2014027280
http://www.google.com/patents/WO2014027280A1?cl=en
Scheme 1 given below:
Formula .
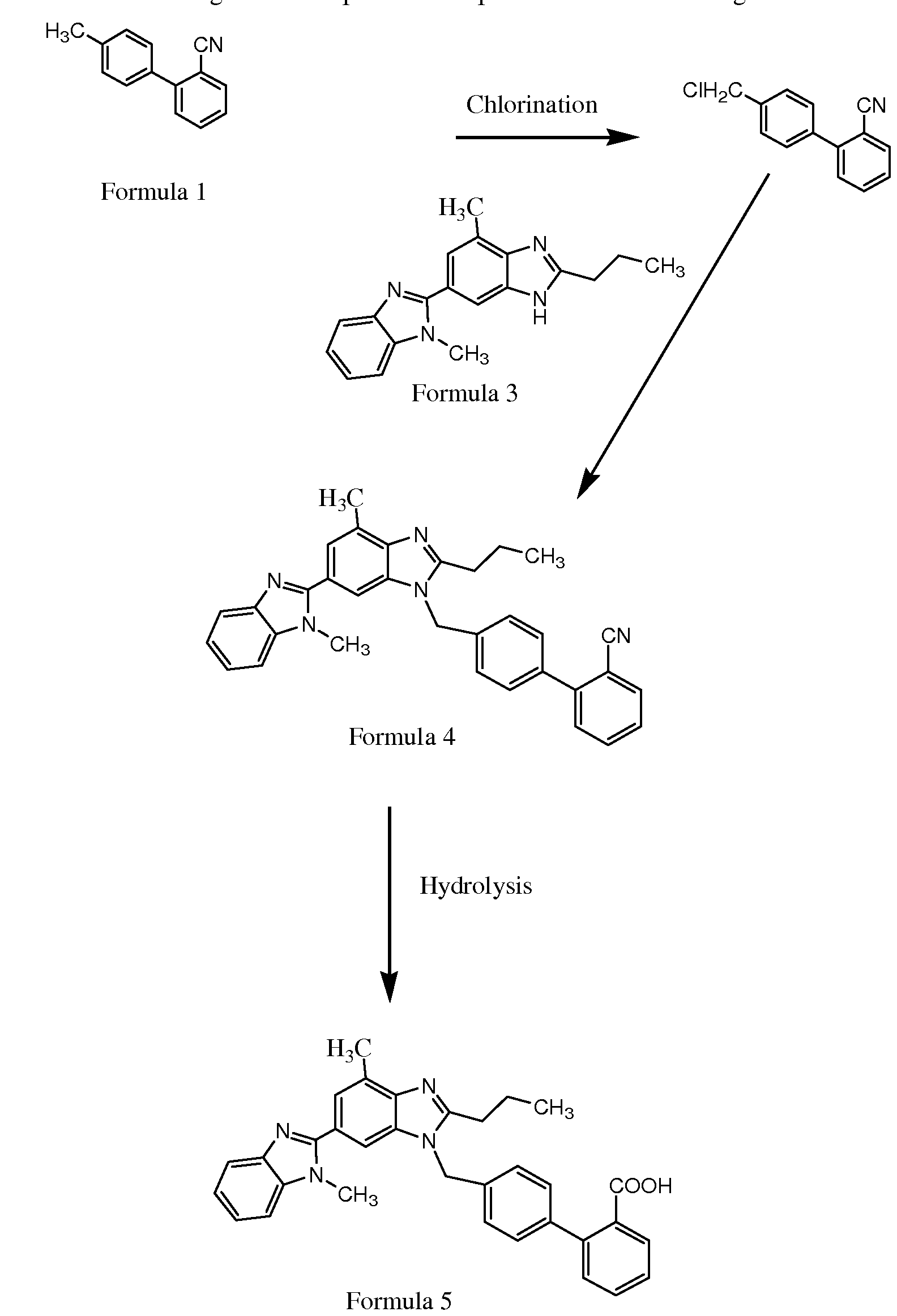
Example 1:
4′-[2-n^ropyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l-ylmeth^
carboxylic acid
In a 2 litre reaction flask was added 400 ml methylene chloride, followed by 100 gm of 2- cyano-4′ -methyl biphenyl. The reaction mass was stirred to get a clear solution and cooled to 20 °C. Chlorine gas was sparged into the reaction mass for a period of 15 hours till completion of the reaction. The reaction was monitored by TLC using mobile phase n-hexane: ethyl acetate (8:2). The excess chlorine from the reaction mass was removed by flushing with nitrogen. The solvent was distilled out completely by distillation at atmospheric pressure and removal of the final traces under vacuum. To the residual mass, 500 ml of methyl isobutyl ketone was added. The reaction mass was stirred and washed with a solution of 300 ml of 5% sodium bicarbonate solution. The lower aqueous layer was separated and the upper organic layer was washed with 300 ml water. The lower aqueous layer was separated. To the organic layer containing 4-chloromethyl-2′-cyanobiphenyl, the compound 2-n-propyl-4-methyl-6-(l’- methylbenzimidazol-2′-yl)benzimidazole was added, followed by a solution of 40 gm sodium hydroxide in 300 ml water. The reaction mass was stirred for 10 minutes and 10 gm of tetrabutyl ammonium hydrogen sulphate was added. The reaction mass was heated to 80 UC and maintained at 80 to 85 °C for 4 hours.
The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). After completion of reaction, the lower aqueous layer was separated. The solvent was distilled out till mass temperature 120 °C and final traces were removed completely under vacuum. To the residual mass, 50 ml of n-butanol was added and the solvent distilled out under vacuum below 100 °C to remove all traces of methyl isobutyl ketone. The residue was dissolved in 750 ml of n-butanol and 83 gm sodium hydroxide added. The reaction mass was heated to reflux and maintained for 24 hours at 123 to 126 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). The solvent was distilled out at atmospheric pressure till the mass temperature reached 140 C. The residual mass was cooled to 100 °C and 300 ml water was added. The solvent was distilled out azeotropically till the mass temperature reached 120 °C. To the reaction mass 750 ml of water was added, the solution warmed to 80 °C. The pH of the reaction mass was adjusted to 8.0 with hydrochloric acid. Finally the pH was adjusted to 6.0 with acetic acid, and the reaction mass maintained at 80 to 85 °C for one hour. The product obtained was filtered, washed with water and dried to yield 120 gm of 4′-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid, which can be purified as per the procedure described mentioned in Example 5. Example 2:
4-chloromethyl-2 ‘-cyanobiphenyl
In a 1 litre reaction flask 400 ml of methylene chloride was added followed by 100 gm of 2- cyano-4′ -methyl biphenyl. The reaction mass was stirred to get a clear solution and cooled to 20 °C. Chlorine gas was sparged into the reaction mass for a period of 15 hours at 20 to 25 °C till completion of the reaction. The reaction was monitored by TLC using mobile phase n- hexane: ethyl acetate (8:2). The excess chlorine from the reaction mass was removed by flushing with nitrogen. The solvent was distilled out completely by distillation at atmospheric pressure and removal of the final traces under vacuum. To the residual mass, 400 ml of n- heptane was added. The reaction mass was stirred and warmed to 60 °C. The clear solution obtained was cooled to 10 °C and the product precipitated was filtered, washed with n-heptane and dried. Further crystallization with n-heptane yielded 80 gm of pure 4-chloromethyl-2’- cyanobiphenyl.
C 73.87%, H 4.41%, N 6.19%; m/z 192.25; 1H NMR DMSO d6400 Mhz : 5ppm 4.84 (s, 2H) 7.32 – 7.66 (aromatic 8H). Example 3:
2-cyano-4,-(2,,-n-propyl-4,,-methyl-6,,-{V”-methylbenzim
ylmethyl) biphenyl
In a 2 litre reaction flask 500 ml of methyl isobutyl ketone was added followed by 100 gm of 2-n-propyl-4-methyl-6-( -methylbenzimidazol-2′-yl)benzimidazole. The reaction mass was stirred and a solution of 40 gm sodium hydroxide in 300 ml water was added. To this solution, 10 gm tetra butyl ammonium hydrogen sulphate and 80 gm of 4-chloromethyl-2′- cyanobiphenyl was added. The reaction mass was warmed to 80 °C and maintained for 4 hours at 80 to 85 °C.
The completion of the reaction was monitored by TLC using mobile phase chloroform : methanol (9:1). After completion of the reaction, the mass was cooled to 20 °C, maintained 3 hours at 15 to 20 °C. The product which precipitated out was filtered, washed with methyl isobutyl ketone, followed by water to yield 126 gm of 2-cyano-4′-(2″-n-propyl-4″-methyl- 6″-(r”-methylbenzimidazol-2″‘-yl)benzimidazol-l”- ylmethyl) biphenyl, melting at 196 – 198 °C. C 80.53%, H 5.70%, N 14.20%; m/z = 496.64 lH NMR DMSO d6 400 Mhz : 5ppm 0.96 – 0.99 (t, 3H) 1.75 – 1.84 (m, 2H) 2.62 (s, 3H) 2.89 – 2.93 (t, 2H) 3.80 (s, 3H) 5.67 (s, 2H) 7.18 – 7.92 (m, 14H)
Example 4:
4′-[2-n^ropyl-4-methyl-6-(l-methylbenzi idazol-2-yl)benzi idazol-ylmethyl]bipheny carboxylic acid
126 gm of 2-cyano-4′-(2″-n-propyl-4″-methyl-6″-(l “‘-methylbenzimidazol-2″‘-yl) benzimidazol-1”- ylmethyl) biphenyl was dissolved in 750 ml of n-butanol and 83 gm sodium hydroxide added. The reaction mass was heated to reflux and maintained for 15 hours at 123 to 126 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1).
The solvent was distilled out at atmospheric pressure till the mass temperature reached 140 °C. The residual mass was cooled to 100 °C and 300 ml water was added. The solvent was distilled out azeotropically till the mass temperature reached 120 °C. To the reaction mass 750 ml of water was added, the solution warmed to 80 °C. The pH of the reaction mass was adjusted to 8.0 with hydrochloric acid. Finally the pH was adjusted to 6.0 with acetic acid, and the reaction mass maintained at 80 to 85 °C for one hour. The product obtained was filtered, washed with water and dried to yield 120 gm of 4’-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid. Example 5:
Purification of 4′-[2-n^ropyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l- ylmethyl]biphenyl-2-carboxytic acid
In a 3 litre reaction flask, 1000 ml of methanol was added followed by the addition of 120 gm of 4′-[2-n-propyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl- 2-carboxylic acid obtained by procedure described in Example 4. The solution was warmed to 50 °C and pH adjusted to 10.0 to 10.5 with 100 ml of a 10% methanolic potassium hydroxide solution. The reaction mass became a clear solution, and 6 gm activated carbon was added. The mass was maintained at 50 to 55 °C for one hour and filtered through hyflo supercel to remove the activated carbon. The clear filtrate obtained was collected and its pH adjusted to 6.0 to 6.5 with 130 ml of acetic acid, maintaining the temperature between 50 to 55 °C. The mass was cooled to 15 °C and maintained one hour at 10 to 15 °C. The product which precipitated out was filtered, washed with 50 ml of methanol followed by 500 ml of water. The wet product was dried to yield 107 gm of 4′-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid.
C 76.49%; H 5.74%, N 11.02%; m/z 515.45.; 1H NMR DMSO d6 400 Mhz : 5ppm 0.97 – 1.01 (t, 3H) 1.76 – 1.85 (m, 2H) 2.62 (s, 3H) 2.90 – 2.94 (t, 3H) 3.81 (s, 3H) 5.61 (s, 2H) 7.15 – 7.71 (14H aromatic);
Melting point of purified telmisartan: 269 °C.
…………………
PAPER
Journal of Organic Chemistry (2014), 79(21), 10568-10580
http://pubs.acs.org/doi/abs/10.1021/jo501665e
J. Org. Chem., 2014, 79 (21), pp 10568–10580
DOI: 10.1021/jo501665e

On the basis of our recently reported aniline aqueous borylation, molecular diversity was achieved in a one-pot process by combining other reactions such as esterification, Suzuki–Miyaura coupling, hydrogenolysis, or Petasis borono-Mannich.
TELMISARTAN IS COMPD 9
……………….
PATENT
US 20150031768
(EN)
Methods of halogenating a carbon containing compound having an sp3 C—H bond are provided. Methods of fluorinating a carbon containing compound comprising halogenation with Cl or Br followed by nucleophilic substitution with F are provided. Methods of direct oxidative C—H fluorination of a carbon containing compound having an sp3 C—H bond are provided. The halogenated products of the methods are provided.
…………………..
PATENT
WO 2014067237
http://www.google.com/patents/WO2014067237A1?cl=en
Telmisartan Preparation: 12 Examples
The title compound (III, R = COOCH 3) (52.8g, O. lmol) of Example 11 with glacial acetic acid
(200ml) and concentrated hydrochloric acid (250ml) mixing, 100 ° C to react for 5 to 6 hours. Evaporated to most mixed acid, residue slowly poured into crushed ice, under ice cooling with saturated K 2 CO ^ solution to adjust the pH to neutral, solid precipitation, filtration, filtrate was washed with water, was for Mischa Tan crude, recrystallization telmisartan (40.1g), liquid purity greater than 99%.
Example 13: Preparation of telmisartan of formula I compound (0.62g, leq) was added to acetonitrile (10ml). After stirring evenly, the KOH (0.14g, 1. leq) was slowly added, after stirring for 10 plus minutes, the title compound of Example 10 of the embodiment (11, R = COOCH 3) (0.5g, leq) was slowly added, stirred for 3-4 hours, TLC the reaction was complete, the direct addition of 50% ethanol (30mL), reflux The reaction for 6 hours. After completion of the reaction by TLC, recovering the organic solvent under reduced pressure, the remaining solution was added dropwise hydrochloric acid (1: 1) to neutral pH. The precipitated solid was filtered, washed with water to give crude telmisartan, telmisartan recrystallized (yield 75.1%), the liquid phase is greater than 98% purity.
Chloromethyl biphenyl -2- (II, R = CN) Preparation of 4′-nitrile:
…………………
Journal of Pharmaceutical and Biomedical Analysis (2015), 108, 86-96.
………………..
IN 262831/EP 1912975
……………………….
JP 2014201585
……………………..
IN 2013KO00463/WO 2014174397
…………………………….
PATENT
http://www.google.com/patents/CN1768044A?cl=en
Example 7: Telmisartan make 5.51 g telmisartan × HCl was dissolved in 50 ml of 40% acetic acid while refluxing. The brown solution was then filtered hot through 1.1 g of carbon, 2.5 ml of 40% acetic acid and washed, and at 80-90 ℃ 2.5 ml of 4N NaOH was added dropwise with stirring to light brown filtrate. Telmisartan crystallization, the suspension was diluted with 30 ml of water, and slowly cooled to ambient temperature. Telmisartan suction filtration, and washed with 50 ml of water. And dried in vacuo at 80 ℃ drying cabinet telmisartan.
Yield: 4.80 g (93.3% of the theoretical yield).
……………………………….
PATENT
http://www.google.com/patents/CN102731407A?cl=en


Example 4 Preparation of telmisartan
[0031] 2-n-propyl group as shown in Formula I-4-methyl-6- (benzimidazol-2-yl-methyl 1’_) benzimidazole (30. 4g, O. 10mol), 4_ bromomethyl-biphenyl-2-carboxylic acid (43. 6g, O. 15mol), three ko amine (12. Ig, O. 15mol) and ko ni ni ether 500ml alcohol were mixed and reacted at 100 ° C for 6 inches The reaction solution was poured into ice water, acidified with dilute hydrochloric acid and slowly adjusted PH2-3, to precipitate a solid. Filtration, 70 ° C drying crude, the resulting crude product ko ko acid ester 300ml heating beating again. Filtered, 70 ° C dry. Recrystallization from DMF telmisartan of formula III as shown in 25. Ig, yield: 50%.
…………………….
PATENT
http://www.google.com/patents/WO2010146187A2?cl=en
For example, WO 2004/087676 describes the hydrolysis of a compound with the chemical name 4 ‘-((1,7’- dimethyl-2 ‘ -propyl-lH, 3 ‘H-2, 5 ‘ -bibenzo [d] imidazol-3 ‘ -yl) – methyl) biphenyl-2-carbonitrile and having formula 2

which is hereinafter referred to as cyanotelmisartan . In par- ticular, the hydrolysis of cyanotelmisartan is carried out at elevated temperatures using strong alkaline conditions. Also, CN 1412183 discloses the hydrolysis of cyanotelmisartan.
US 2006/0264491 Al discloses the hydrolysis of 4′-((l,7′- dimethyl-2 ‘ -propyl-lH, 3 ‘H-2, 5 ‘ -bibenzo [d] imidazol-3 ‘ – yl) methyl) biphenyl-2-carboxamide having formula 3

Example 2: Preparation and isolation of telmisartan
Into a reaction vessel 20.5g (40 mmol) 4 ‘ – ( (1, 7 ‘ -dimethyl-2 ‘ – propyl-IH, 3 ‘ H-2 , 5 ‘ -bibenzo [d] imidazol-3 ‘ -yl) methyl) biphenyl-2- carboxamide and 20 ml (lδOmmol) H2SO4 (1:1) were added. The re- action mixture was heated to about 125°C and stirred at this temperature for 28 h. A sample of the reaction mixture was analyzed by Area% HPLC (starting compound below 0.1%, telmis- artan over 97%) . The reaction mixture was cooled below 800C and 250 ml of water were added. Then, 200 ml of dichloro- methane were added and pH value of mixture was adjusted to 5.4 by addition of 6M NaOH. The mixture was stirred for approximately 5 min and then the phases were separated. The water phase was reextracted by 136 ml of dichloromethane . Collected organic phases were washed with water (2χl36ml) and then treated with activated charcoal (5.3 g) . Subsequently, the organic phase was evaporated an oily residue (26g) . 264 ml of acetone were added. The mixture was stirred at room temperature for at least 6 hours. The precipitated product was sepa- rated and washed with fresh acetone and dried at 65°C under reduced pressure for 3 hours. Yield: 18.3g (89%) Area % HPLC: Telmisartan 99.80%
Example 3: Isolation of telmisartan
Into a reaction vessel 7.5g (15 mmol) of cyanotelmisartan, 30 ml of propylene glycol, 0.8 ml of water and 3g (45 mmol) of 85% KOH were added. The reaction mixture was heated to around 1600C to 170 0C and stirred at this temperature for 24 h. The reaction mixture was cooled below 800C and 75 ml of water were added. Then, pH value of the mixture was adjusted to 4.8 (by addition of 6M HCl) and then 150 ml of dichloromethane were added. The mixture was stirred for approximately 5 min and then the phases were separated. The water phase is reextracted by 50 ml of dichloromethane. Collected organic phases were washed with water (2χ50ml) and then treated with activated charcoal (2 g) . After that the organic phase was evaporated to an oily residue (9.8g) . 100 ml of acetone were added. The mix- ture was stirred at room temperature for at least 6 hours. The precipitated product was separated and washed with fresh acetone and dried at 65°C under reduced pressure for 3 hours. Yield: 6.8g (88%) Area % HPLC: Telmisartan 99.60%
…………………….
PATENT
http://www.google.com/patents/CN1548421A?cl=en
Specific embodiments
14 ‘Example – [(1,4′-dimethyl-2′-propyl [2,6′- two-1H – benzoimidazol] 1′-yl) methyl] – [1, 1’-biphenyl] -2-carboxylic acid sodium salt in 250ml reaction flask, telmisartan 10g (0.0195mol), NaOH0.75g (0.0189mol) and water 100ml, stirred for 1 hour (30 ℃), filtered insoluble materials are removed and concentrated to a small volume, plus ethanol 30ml, concentrated, washed with 30ml of n-hexane, decanted, plus ethanol 30ml, concentrated, and then repeat again, and concentrated to dryness to obtain telmisartan sodium salt 9.9g yield 95.2%. Melting point: 223-225 ℃.
Elemental analysis: C33H29N4O2Na · H2O Calcd: C71.48 H5.10 N10.11 Found: C71.42 H5.08 N10.22 Example 24 ‘- [(1,4′-dimethyl-2′-n propyl [2,6′- two-1H – benzoimidazol] 1′-yl) methyl] – [1,1’-biphenyl] -2-carboxylic acid potassium salt in 250ml reaction flask, Telmisartan 10g (0.0195mol), KOH1.06g (0.0188mol) and water 100ml, stirred for 1 hour (30 ℃), filtered to remove insolubles, and concentrated to a small volume, ethanol 30ml, concentrated, hexane 30ml washed, decanted, plus ethanol 30ml, concentrated, and then repeat again, and concentrated to dryness to obtain telmisartan potassium 10.6g, yield 95.6%. Melting point: 203-205 ℃.
Elemental analysis: C33H29N4O2K · H2O Calcd: C69.04 H5.40 N9.76 Found: C69.01 H5.28 N9.88 Example 3 starting material and the mixed powder was sieved excipients, 5% polyethylene pyrrolidone was granulated and dried. After dried particles were sieved magnesium stearate was added mixed tabletted.
mg / tablet of telmisartan sodium salt 20 Lactose 170 Sodium carboxymethyl starch 10 mg Magnesium stearate 8 meglumine 25% polyvinyl pyrrolidone solution q.s. Example 4 A mixed powder of raw materials and auxiliary materials sieved, added 5 % solution of polyvinylpyrrolidone is granulated and dried. After dried particles were sieved magnesium stearate was added mixed tabletted.
mg / tablet telmisartan sodium Lactose 200 40 140 DCP sodium carboxymethyl starch 16 mg Magnesium stearate 45% povidone solution appropriate amount of Example 5 of this product, according to the dissolution assay (Chinese Pharmacopoeia 2000 edition Appendix II XC second method), phosphate buffer 900ml solvent, the speed of 75 revolutions per minute, operate according to the law, after 30 minutes, take the solution as spectrophotometry (Chinese Pharmacopoeia 2000 edition of the test solution, according to the spectrophotometric two Appendix IVA), absorbance was measured at 295nm wavelength. Another reference standard stock solution 10ml precise amount of determination under set 100ml flask, diluted with phosphate buffer to the mark, then the precise amount of 5ml, set 10ml volumetric flask, dilute to the mark with phosphate buffer , shake, the same method absorption, calculated for each piece of the dissolution of the limits of 80% scalar, should be specified. Dissolution test results in Table.
Table dissolution test results Dissolution (%) telmisartan sodium 97.29 99.65 102.55 95.83 101.10 98.92 99.20 ± 2.45
…………………..
PATENT
http://www.google.com/patents/CN1412183A?cl=en
Example 5 4 ‘- [(1,4′-dimethyl-2′-propyl [2,6′- two -1H- benzimidazol] -1′-yl) methyl] – [1,1’ – biphenyl] -2-carboxylic acid (III) IV (24.8g, 0.05mol) was added ethylene glycol (100ml) and water (150ml) (or other previously described a mixed solvent), sodium ethoxide (or as previously said other alcohols sodium) (13.6g, 0.2mol), was refluxed for 10 hours. After no starting material by TLC was cooled to room temperature, hydrochloric acid was added dropwise (1/1) to pH 5-6, the precipitated solid was filtered, washed with water to give III.
……………………
PATENT
http://www.google.com/patents/CN101550107B?cl=en
Example 3
[0047] 1) Preparation of telmisartan crude methyl ester
Compound II into 50g in 500mL reaction flask, 200mL of methyl isobutyl ketone (MIBK), 25 ° C _30 ° C with stirring until dissolved, was added dropwise 35mL of triethylamine was added 55. Og After the completion of the compound III, 5 (T60 ° C or so for about 4_5 hours, TLC monitoring completion of the reaction, filtered and the filter cake washed with a small amount of MIBK, and then washed with water, dried to give 70. 3g of crude product. 81% yield, purity of about 98%.
(TLC test conditions: ethyl acetate: methanol = 8: 1)
2) preparation of high purity methyl telmisartan
IOOOmL reaction flask, the input step to give the crude methyl ester telmisartan, add 500mL of isopropanol was heated to dissolve, 2gX 2 activated bleaching filtrate was heated to about 90 ° C, added dropwise with stirring 150mL 7jC insulation 0. 5~Ih, cooled slowly to room temperature with stirring. Filtered, and the filter cake washed sequentially with MIBK and water washing, and drying, the yield of about 82%, HPLC purity 99.5%, the single impurities less than 0.1%.
3) Preparation of telmisartan with high purity
[0053] A reaction flask was put in a 500mL high purity 15g telmisartan ester, 3. Og sodium, 200mL of isopropanol, water, 80ml, was heated to reflux for 5 ~ 7 h, TLC monitoring of the reaction was complete, the distillation Isopropanol was removed, and water was added to completely dissolve the solid 40ml, 0. 5g of activated carbon bleaching, the filtrate was added 50ml of water, heated to 80 ° C, lmol / L of acetic acid to adjust the pH to 5. (Γ5. 5, filtered, and the filter cake dried to give 13. 14g of solid, yield 90%, HPLC purity 99.7%, the single impurities less than 0.1%.
(TLC test conditions: ethyl acetate: methanol = 8: 1)
………………………..
http://www.google.com/patents/CN101172968B?cl=en
Example 1
[0023] 1, 100gPPA, 21. 8g (0. Lmol) 2_ n-propyl _4_ _6_ carboxyl methyl benzimidazole and 21. 5gN- methyl-o-phenylenediamine added to the reaction flask in under N2 protection feeding, heated to IO (TC _1601 :, reaction 8-20 hours, down 70-80.C 200ml water was added and the reaction with hydrochloric acid to adjust ffl = 1~2, put charcoal 5_8%,, 8 (TC about 5 to 10 minutes, filtered, and the reaction repeated, the adjustment ra 12-14 with NaOH, for several hours, and filtered to give the crude intermediate 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl ) benzimidazole sodium salt. [0024] 2, the product of the previous step, 2-n-propyl -4-methyl–6_ (methyl benzimidazol-_2_ yl) benzimidazole sodium salt crude product was dissolved into 200 ml of ethanol , and dissolved by heating, cooling to room temperature, 400 ml 1N NaOH, to precipitate the compound 2-n-propyl -4-methyl-6- (methyl benzimidazol-2-yl) benzimidazole .50-8 ( TC dried in vacuo. [0025] 3, product of the previous step -4-methyl-2-n-propyl -6_ (methyl benzimidazol-_2_ yl) benzimidazole into 200 ml of dimethyl sulfoxide was stirred was added at room temperature and 4-bromomethyl – biphenyl-2-carboxylic acid methyl ester 33.55 g, was stirred for 14 hours, extracted with dichloromethane (200, 100, 100), and evaporated to dryness under reduced pressure, 300 ml of methanol and 10% potassium hydroxide (240 ml, 0. 6mo1) mixture was refluxed for 6 hours, cooled, washed with 80 ml of methylene chloride, adjusted with glacial acetic acid ffl = 6, a lot of white floc precipitated precipitate was filtered and dried to give a white Tilmicosin 49.6 g of crude product, the crude product was added 100 ml of chloroform was heated to reflux, activated carbon decolorization, crystallization, filtration, 8 (TC dried in vacuo to give a white pure telmisartan (HLPC> 99. 0%) 41 克, purification yield 82%. mp 261~263.C, H-NMR (d6-DMS0) S 1. 05t, 3H), 1. 83 (m, 2H), 2. 71 (s, 3H), 2. 94 (t, 2H), 3. 81 (s, 3H), 5. 57 (s, 2H), 7. 16-7. 83 (m, 14H) • C33H33N402 [0026] Example 2 Preparation of telmisartan
1, 100gPPA, 21. 8g (0. 1) 2_ [4-methyl-n-propyl-benzimidazole and _6_ 21. 5gN- carboxy-o-phenylenediamine added to the reaction flask in N2 Under the protection of feeding, heated to 100 ° C _160 ° C, the reaction for 8-20 hours, down 70-80. C, the reaction was added 200ml of water, adjusted with hydrochloric acid ffl = 1~2, into charcoal 5_8%, about 8 (TC 5_10 minutes filtered again reacted with K0H ra adjusted to 12-14 for several hours and filtered to give Intermediate crude 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt.
2, the product of the previous step, 2-n-propyl -4-methyl–6_ (methyl benzimidazol-_2_ yl) benzimidazole potassium salt of the crude product into 200 ml of ethanol, and dissolved by heating, cooling to room temperature was added 400 ml 1N K0H, a precipitated compound is 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt. 50-8 (TC dried in vacuo. [0029] 3, 2-n-propyl prepared in the previous step -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt and 27.2 g of 4-bromomethyl-2-cyanobiphenyl, 10.4 g of triethylamine and DMF (DMA, dichloromethane, dichloroethane) were mixed and reacted for 5-10 hours at 35-40 °, TLC detection After no starting material the reaction mixture was poured into 600 g of ice water, extracted with ethyl acetate (300ml * 3), the combined organic phases were washed with water (300ml * 2), dried and desolvation, and then petroleum ether was added and stirred until a solid precipitated was The crude product was 45.6 g.
4, the upper step of the solid 45.6 grams, was added 200ml of ethylene glycol, 150ml water, 12 g of sodium hydroxide, the reaction was refluxed for 10 hours, TLC detected no starting material and then cooled to room temperature, acidified with hydrochloric ra is 5 to 6, there is solid precipitation, filtration, washing, telmisartan was crude, DMF and recrystallized to give 44.5 g of telmisartan pure product (HLPC> 99. 0%) mp261~263 ° C. Force -NMR (de-DMS0) S 1. 05t, 3H), 1. 83 (m, 2H), 2. 71 (s, 3H), 2. 94 (t, 2H), 3. 81 (s, 3H ), 5. 57 (s, 2H), 7. 16-7. 83 (m, 14H) • C33H33N402 [0031]
……………………….
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN100460396C | Mar 8, 2007 | Feb 11, 2009 | 杭州盛美医药科技开发有限公司 | Intermediate of telmisartan, preparation and use thereof |
| CN100506799C | Apr 19, 2007 | Jul 1, 2009 | 北京理工大学 | [(2-n-propyl-4-methyl-1H-Benzimidazole)6-radical] carboxamide-1-radical] methylbiphenyl compound, synthesizing method and usage |
| CN100548293C | Aug 8, 2003 | Oct 14, 2009 | 贝塞斯达药物股份有限公司 | Novel PPAR ligands that do not cause fluid retention, edema or congestive heart failure |
| CN101550107B | Apr 2, 2009 | Jan 12, 2011 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| CN101743228B | Jul 3, 2008 | Jan 29, 2014 | 新梅斯托克尔卡托瓦纳兹德拉韦尔公司 | Process for preparing telmisartan |
| CN101891735B | Nov 25, 2009 | Jul 18, 2012 | 北京理工大学 | Biphenyl sulfafurazole compound, synthesis method and application thereof |
| CN102093297B | Jan 28, 2011 | Aug 1, 2012 | 海南美兰史克制药有限公司 | Telmisartan compound and new preparation method thereof |
| EP1805146A2 * | Oct 18, 2005 | Jul 11, 2007 | Dr. Reddy’s Laboratories Ltd. | Process for preparing telmisartan |
| EP2123648A1 | May 20, 2008 | Nov 25, 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| US7501448 | Oct 13, 2005 | Mar 10, 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7′-dimethyl-2′-propyl-1H,3’H-[2,5′]bibenzoimidazolyl is reacted with 4’bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| US8691999 * | May 10, 2005 | Apr 8, 2014 | Cipla Limited | Process for the preparation of telmisartan |
| WO2006044648A1 * | Oct 13, 2005 | Apr 27, 2006 | Teva Pharma | Process for preparing telmisartan |
| WO2007010558A1 * | Jul 19, 2006 | Jan 25, 2007 | Satyanarayana Chava | A process for the preparation of telmisartan |
| WO2009123483A1 | Mar 30, 2009 | Oct 8, 2009 | Zaklady Farmaceutyczne Polpharma Sa | Process for preparation of telmisartan |
| WO2012028925A2 | Aug 29, 2011 | Mar 8, 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan |
| CN1620437A | Jan 15, 2003 | May 25, 2005 | 贝林格尔英格海姆法玛两合公司 | Method for the production and purification of 1, 7′-dimethyl-2′-propyl-2, 5′-bi-1h-benzimidazole |
| CN1768044A | Mar 26, 2004 | May 3, 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| WO2005108375A1 | May 10, 2005 | Nov 17, 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| CN1344712A | Jul 30, 2001 | Apr 17, 2002 | 中国科学院上海药物研究所 | Synthesis path of Timisatem | |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan | |
| US2006/0094883 | Title not available | ||||
| WO03/059890A1 | Title not available | ||||
| WO2005/108375A1 | Title not available |
| US7501448 | Oct 13, 2005 | Mar 10, 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7′-dimethyl-2′-propyl-1H,3’H-[2,5′]bibenzoimidazolyl is reacted with 4’bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| WO2007147889A2 * | Jun 22, 2007 | Dec 27, 2007 | Krka Tovarna Zdravil D D Novo | Preparation of telmisartan salts |
| WO2010146187A2 | Jun 21, 2010 | Dec 23, 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2012055941A1 | Oct 26, 2011 | May 3, 2012 | Krka,Tovarna Zdravil, D. D., Novo Mesto | Multilayer pharmaceutical composition comprising telmisartan and amlodipine |
| WO2003007876A2 | Jun 25, 2002 | Jan 30, 2003 | Sumner H Burstein | N-fatty acid-amino acid conjugates and therapeutic uses |
| WO2003059327A1 | Jan 16, 2002 | Jul 24, 2003 | Boehringer Ingelheim Pharma | Bilayer pharmaceutical tablet comprising telmisartan and a diuretic and preparation thereof |
| WO2004028505A1 | Sep 18, 2003 | Apr 8, 2004 | Boehringer Ingelheim Int | Solid pharmaceutical formulations comprising telmisartan |
| WO2004087676A1 | Mar 26, 2004 | Oct 14, 2004 | Boehringer Ingelheim Int | Method for the production of telmisartan |
| WO2004096215A1 | Apr 27, 2004 | Nov 11, 2004 | Boehringer Ingelheim Int | Pharmaceutical formulation of the sodium salt of telmisartan |
| WO2005108375A1 * | May 10, 2005 | Nov 17, 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| WO2006044754A2 | Oct 18, 2005 | Apr 27, 2006 | Muthulingam Arunagiri | Process for preparing telmisartan |
| WO2006050509A2 | Nov 3, 2005 | May 11, 2006 | Teva Pharma | Amorphous and polymorphic forms of telmisartan sodium |
| WO2006050921A2 | Nov 9, 2005 | May 18, 2006 | Lek Pharmaceuticals | Preparation of telmisartan salts with improved solubility |
| WO2006063737A1 | Dec 9, 2005 | Jun 22, 2006 | Boehringer Ingelheim Int | Combination therapy comprising telmisartan and hydrochlorothiazide |
| WO2006136916A2 | Jun 20, 2006 | Dec 28, 2006 | Glenmark Pharmaceuticals Ltd | Substantially pure micronized particles of telmisartan and pharmaceutical compositions containing same |
| WO2007010559A2 | Jul 19, 2006 | Jan 25, 2007 | Panacea Biotec Ltd | Novel pharmaceutical modified release dosage form cyclooxygenase enzyme inhibitor |
| WO2007060170A2 | Nov 22, 2006 | May 31, 2007 | Boehringer Ingelheim Int | Bilayer tablet comprising telmisartan and diuretic |
| WO2007144175A2 | Jun 14, 2007 | Dec 21, 2007 | Lek Pharmaceuticals | Pharmaceutical composition comprising hydrochlorothiazide and telmisartan |
| WO2007147889A2 | Jun 22, 2007 | Dec 27, 2007 | Krka Tovarna Zdravil D D Novo | Preparation of telmisartan salts |
| WO2009004064A1 | Jul 3, 2008 | Jan 8, 2009 | Krka Tovarna Zdravil D D Novo | Process for preparing telmisartan |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan |
| CN1548421A | May 22, 2003 | Nov 24, 2004 | 上海医药工业研究院 | Tilmisartan salt and its prepn |
| EP0502314A1 | Jan 31, 1992 | Sep 9, 1992 | Dr. Karl Thomae GmbH | Benzimidazol, medicaments containing them and process for their preparation |
| EP1144386A1 | Jan 7, 2000 | Oct 17, 2001 | Boehringer Ingelheim Pharma KG | Telmisartan polymorphs, methods for producing same and their use in the preparation of a medicament |
| EP1719766A2 | Apr 18, 2006 | Nov 8, 2006 | Dipharma S.p.A. | A process for the preparation of telmisartan |
| US20060264491 | Jun 8, 2006 | Nov 23, 2006 | Chemagis Ltd. | Telmisartan production process |
| CN101172968B | Nov 1, 2006 | May 12, 2010 | 浙江天宇药业有限公司 | 2-propyl-4 methyl-6-(tolimidazole-2group) benzoglioxaline salt and method for producing the same |
| CN101743228B | Jul 3, 2008 | Jan 29, 2014 | 新梅斯托克尔卡托瓦纳兹德拉韦尔公司 | Process for preparing telmisartan |
| CN102015690B | Mar 19, 2009 | Apr 30, 2014 | 力奇制药公司 | Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| CN102036937B | Mar 19, 2009 | Jun 4, 2014 | 力奇制药公司 | 2′-halobiphenyl-4-yl intermediates in the synthesis of angiotensin ii antagonists |
| WO2014067237A1 * | Oct 31, 2013 | May 8, 2014 | Topharman Shanghai Co., Ltd. | Telmisartan preparation method and intermediate thereof |
| WO2010018441A2 * | Aug 10, 2009 | Feb 18, 2010 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2 * | Jun 21, 2010 | Dec 23, 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2011077444A1 * | May 28, 2010 | Jun 30, 2011 | Inogent Laboratories Private Limited | A new process for the preparation of pure telmisartan |
| WO2012028925A2 * | Aug 29, 2011 | Mar 8, 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1768044A * | Mar 26, 2004 | May 3, 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| CN102731407A * | Jul 4, 2012 | Oct 17, 2012 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| EP0627433A1 * | Dec 7, 1993 | Dec 7, 1994 | Eisai Co., Ltd. | Process for producing imidazopyridine derivative and intermediate |
| EP2123648A1 * | May 20, 2008 | Nov 25, 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| EP2305650A1 * | Sep 21, 2009 | Apr 6, 2011 | Chemo Ibérica, S.A. | Novel process for the preparation of telmisartan |
| KR20090000113A * | Title not available | |||
| US20040236113 | Mar 17, 2004 | Nov 25, 2004 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
| US20130137878 | Jan 25, 2013 | May 30, 2013 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
……..
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR
 COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
 amcrasto@gmail.com
amcrasto@gmail.com
LINEZOLID
 LINEZOLID
LINEZOLID
N- [[(5S) – 3 – [3 -Fluoro-4- (4-morpholinyl)phenyl] -2-oxo- 5 -oxazolidinyl] methyl] acetamide and marketed by Pfizer in US under brand name Zyvox. Linezolid is a synthetic antibacterial agent of the oxazolidinone class. It is used for the treatment of infections caused by multi-resistant bacteria including streptococci and methicillin-resistant Staphylococcus aureus.

(S)-N-[[3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl] acetamide.
| N-[[(5s)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide PRODUCT PATENTUS5688792 (1997 to Pharmacia & Upjohn) |
|
| CAS No.: | 165800-03-3 |
|---|---|
| Synonyms: | |
| Formula: | C16H20FN3O4 |
| Exact Mass: | 337.14400 |
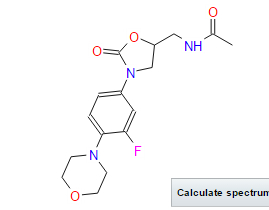

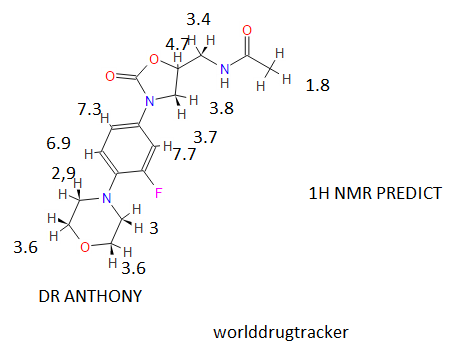
13C
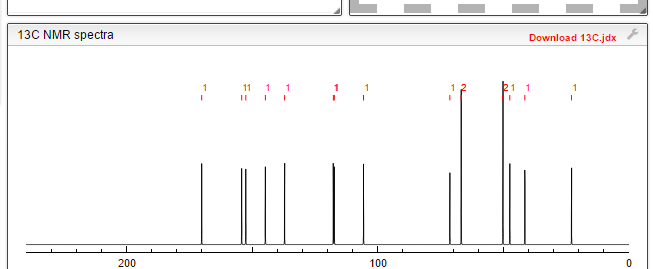
1H NMR AND 13C PREDICT
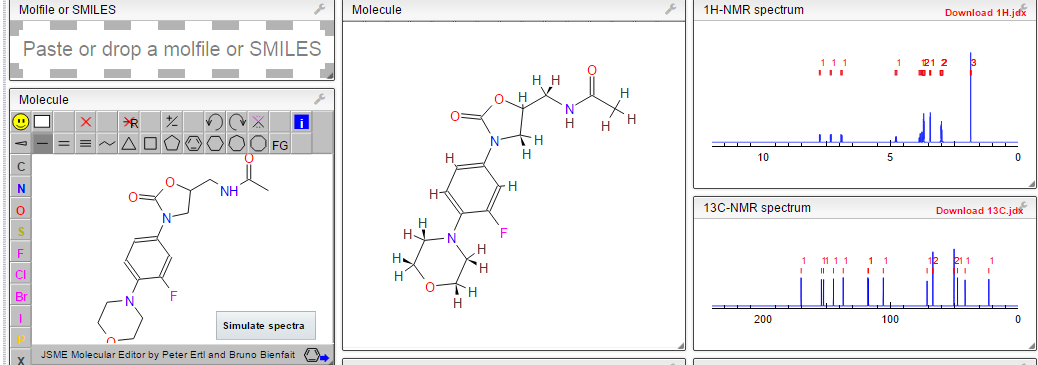

1H NMR PREDICT
![N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide NMR spectra analysis, Chemical CAS NO. 165800-03-3 NMR spectral analysis, N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-19/000/030/236/165800-03-3-1h.png)
13C NMR PREDICT
![N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide NMR spectra analysis, Chemical CAS NO. 165800-03-3 NMR spectral analysis, N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-19/000/030/236/165800-03-3-13c.png)
COSY
PREDICT
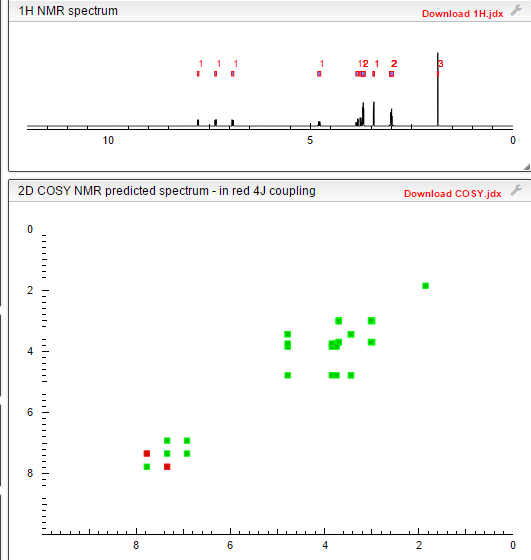
HMBC

ORIGINAL 1H NMR…………...http://www.selleckchem.com/products/Linezolid(Zyvox).html

INTERMEDIATES USED
Arkivoc, , vol. 2012, # 6 p. 45 – 56
WO2011/137222 A1, ;

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 11 ;

THE REGENTS OF THE UNIVERSITY OF CALIFORNIA; GARG, Neil K.; RAMGREN, Stephen D.; SILBERSTEIN, Amanda L.; QUASDORF, Kyle W. Patent: WO2012/94622 A2, 2012 ; Location in patent: Page/Page column 31-32 ;

Lianhe Chemical Technology Co., Ltd. Patent: EP2388251 A1, 2011 ; Location in patent: Page/Page column 11 ;
Tammana, Rajesh; Vemula, Kiran Kumar; Guruvindapalli, Ramadasu; Yanamandr, Ramesh; Gutta, Madhusudhan Arkivoc, 2012 , vol. 2012, # 6 p. 45 – 56

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 10 ;

Song, Lirong; Chen, Xiaobei; Zhang, Shilei; Zhang, Haoyi; Li, Ping; Luo, Guangshun; Liu, Wenjing; Duan, Wenhu; Wang, Wei Organic Letters, 2008 , vol. 10, # 23 p. 5489 – 5492

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 10 ;

JUBILANT LIFE SCIENCES LIMITED; BISWAS, Sujay; PANDA, Atulya, Kumar; GUPTA, Ashish, Kumar; SINGH, Shishupal; TIWARI, Praveen; VIR, Dharam; THOMAS, Saji Patent: WO2013/111048 A1, 2013 ; Location in patent: Page/Page column 24; 25 ;

Perrault, William R.; Pearlman, Bruce A.; Godrej, Delara B.; Jeganathan, Azhwarsamy; Yamagata, Koji; Chen, Jiong J.; Lu, Cuong V.; Herrinton, Paul M.; Gadwood, Robert C.; Chan, Lai; Lyster, Mark A.; Maloney, Mark T.; Moeslein, Jeffery A.; Greene, Meredith L.; Barbachyn, Michael R. Organic Process Research and Development, 2003 , vol. 7, # 4 p. 533 – 546

US6362334 B1, ; Example 13 ;

NMR OF INTERMEDTIATES

………….

…….

………

……………

……………

-
Linezolid is a pharmaceutically active compound useful as an antibacterial agent, e.g. for the treatment of diabetic food infections caused by Gram-positive bacteria. It is represented by the formula (I),

-
[0003]The marketed pharmaceutical compositions are a sterile isotonic solution for an i.v. infusion, a tablet for oral administration and an aqueous suspension for oral administration. They are marketed, i.e., under brand name ZYVOX by Pfizer.
-
[0004]The molecule of linezolid has one asymmetric carbon in the molecule allowing for 2 enantiomers; the marketed compound is the (S)-enantiomer. In the above-marketed compositions, linezolid is present as a free base.
-
[0005]Hereinunder, the name linezolid will be used as the generic name for N-(3-(3-fluoro-4-(morpholin-4-yl)phenyl)-2-oxooxazolidin-5(S)-ylmethyl)acetamide, unless indicated to the contrary.
-
[0006]Linezolid was first disclosed in WO 95/07271 ( EP 0717738 , US 5,688,792 ) of the Upjohn Company.
-
[0007]Various processes for making linezolid are known in the art. In particular, the important ones are these, the final step of which comprises acetylation of an amine precursor of the formula (II) with an acetylhalide or acetic anhydride (see, e.g., WO 2005 099353 ),

-
[0008]This amine precursor (II) may be made from various starting materials, e.g.:
- a) By a reduction of an azide compound of formula (III) by a suitable reductant ( WO2006/091731 , WO 95/07271 , US 5837870 , WO2009/063505 , US 7291614 ),

The starting compound (III) may be made from the corresponding tosylate or chloride of general formula (VII) below ( WO 2005/099353 ).
- b) By a decomposition of a phthalimide compound of formula (IV), e.g. by methylamine ( WO95/07271 ) or by hydrazine ( US 5837870 ),

The starting compound (IV) may be made from the same tosylate or chloride as sub a) ( WO2005/099353 ) or by a cyclization of the oxazolidine ring ( WO 99/24393 , WO2006/008754 ).
- c) From a sulfonate compound of formula (V),

by treatment with ammonium hydroxide in isopropanol or THF ( WO 95/07271 ) or by treatment with ammonia under enhanced pressure ( WO 97/37980 ).
- d) By a reduction of an imine (VI),

wherein R2 is a chlorophenyl, bromophenyl or 2,4,-dichlorophenyl moiety ( WO 2007/116284 ).
- a) By a reduction of an azide compound of formula (III) by a suitable reductant ( WO2006/091731 , WO 95/07271 , US 5837870 , WO2009/063505 , US 7291614 ),
-
[0009]Except of the imine (VI), each of the preceded synthetic approaches is based on a step of converting a starting material of the general formula (VII),

wherein L is a suitable leaving group, for instance a halogen or an alkyl-or aryl sulfonyloxy group,
by a reaction with a nitrogen nucleophile (an azide salt, phthalimide salt, ammonia or ammonium hydroxide), followed, if necessary, by a next step of conversion of the formed reaction intermediate (e.g., compound (III) or compound (IV)) into the amino/compound (II). Apparently, making the starting amine-compound (II) in a good yield and purity is the key aspect of commercial success of any of the above synthetic routes yielding linezolid. However, the known approaches have various drawbacks, for instance serious toxicity and explosion hazard of the azide salts, long reaction times and hazardous agents (hydrazine, methyl amine) in using the phthalimide intermediate, low yields and many side products at the ammonium hydroxide approach, or harsh reaction conditions in reaction with ammonia.
Linezolid [(S)-N-[[3-(3-Fluoro-4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide] is an antimicrobial agent. Linezolid is an oxazolidinone, having the empirical formula C16H20FN3O4 and the following structure (1):

Linezolid (1) is described in The Merck Index (13th edition, Monograph number: 05526, CAS Registry Number: 165800-03-3) as white crystals, with a melting point of 181.5-182.5°. Linezolid (1), as well as a process for its preparation, is disclosed in U.S. Pat. No. 5,688,792 (Example 5), European Patent No. 717738, Israeli Patent No. 110,802, Canadian Patent No. 2,168,560, and International Patent Publication WO 95/07271.
U.S. Pat. No. 5,688,792 discloses the antibacterial agent linezolid as well as a process for its preparation. EXAMPLE 5 reports the linezolid produced had a mp of 181.5-182.5°.
There are many other disclosures of processes to prepare linezolid. J. Med. Chem., 39(3), 673-9 (1996) reports the linezolid was, “recrystallized from ethyl acetate and hexanes . . . white crystals, m.p. 181.5-182.5C.” It also sets forth the IR spectrum as “3284, 3092, 1753, 1728, 1649, 1565, 1519, 1447, 1435”.
Tetrahedron Lett., 40(26), 4855 (1999) discloses linezolid and a process to prepare linezolid. However, this document does not set forth the melting point or IR spectrum of the linezolid prepared.
U.S. Pat. No. 5,837,870 (International Publication WO97/37980 of PCT/US97/03458) discloses a process to prepare linezolid. Linezolid is described in EXAMPLE 18, which does not set forth the melting point or IR spectrum of the linezolid prepared.
International Publication WO99/24393 of PCT/US98/20934 discloses a process to prepare linezolid. Linezolid is described in EXAMPLES 8, 9 and 12 which do no set forth the melting point or IR spectrum of the linezolid prepared.
The form of linezolid being used in the clinical trials to support the filing of the NDA is Form II.
Linezolid (1) is marketed in the United States by Pfizer, Inc. as an injection, tablets, and oral suspension under the name ZYVOX®. Its main indications are nosocomial pneumonia, skin and skin-structure infections, and vancomycin-resistant Enterococcus faecium infections.
U.S. Pat. No. 5,688,792 claims linezolid (1) and its use for the treatment of microbial infections. This patent also discloses, but does not claim, the following method of preparation:

This method of preparation was also disclosed in Bricker, et al., J. Med. Chem., 39 673 -679 (1996), where it was stated that the above route avoids the use of phosgene to make the carbamate precursor of the oxazolidinone ring. The authors also disclose that the use of NaN3 can be avoided by using potassium phthalimide, followed by deblocking of the phthalimide with aqueous methyl amine.
In the above-described synthesis, the intermediate amine, S-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl amine (2)

is reacted without isolation with acetic anhydride as an oily product, or in solution, to produce the acetamide, linezolid (1). This is followed by procedures for isolating the linezolid (1) such as those described in U.S. Pat. No. 5,688,792, at col. 15, 11. 22-28 (chromatography and separation of the desired fraction, followed by evaporation and trituration of the product to obtain pure linezolid (1)).
In the above-described syntheses, the intermediate azide R-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl azide (3)

is reduced to its corresponding amine, S-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl amine (2) in the solvent ethyl acetate by hydrogenation using hydrogen gas and a palladium/carbon catalyst. These reaction conditions lead to the production of an undesirable level of reaction by-products, and, following the acetylation of the intermediate amine (2) to linezolid (1), to undesirably high levels of bis-linezolid (4)

http://www.google.com/patents/US20060252932
FIG. 1 shows the 1H-NMR spectrum of bis-linezolid (4)

FIG. 2 shows the 13C-NMR spectrum of bis-linezolid (4)

FIG. 3 shows the IR spectrum of bis-linezolid (4)

A Novel Synthesis of Oxazolidinone Derivatives (A Key Intermediate of Linezolid)
Pingili Krishna Reddy1,2, K. Mukkanti2 and Dodda Mohan Rao1*
1Symed Research Centre, Plot No. 89/A, Phase-I, Shapoornagar, IDA Jeedimetla, Hyderabad, Andhra Pradesh, India
2Center for Pharmaceutical sciences, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad, India
2Center for Pharmaceutical sciences, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad, India

Reddy P. K, Mukkanti K, Rao D. M. A Novel Synthesis of Oxazolidinone Derivatives (A Key Intermediate of Linezolid). Orient J Chem 2013;29(3). doi : http://dx.doi.org/10.13005/ojc/290322
N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide (7a):
IR (KBr, cm-1): 3338 (N-H stretching), 3117, 3066 (aromatic C-H stretching), 2971, 2863, 2818 (aliphatic C-H stretching), 1738, 1662 (C=O stretching), 1545, 1516,1453 (aromatic C=C stretching), 1425 (C-N stretching), 1381 (aliphatic C-H bending), 1334 (C-F stretching), 1274 (C-O stretching), 1198, 1177 (C-N bending), 1117, 1081 (aromatic C-H bending).
1H NMR (CDCl3) δ ppm: 7.44 (m, 1H), 7.26 (m, 1H), 6.99 (m, 1H), 6.01 (t,1H), 4.76 (m, 1H), 4.02 (m, 2H), 3.80 (m, 4H), 3.61(m, 2H), 3.05 (m, 4H), 2.02 (t, 3H):
C13NMR(CDCl3) δppm: 171.33, 156.87, 154.44, 136.40, 132.84, 118.67, 113.81, 107.52, 71.96, 66.76, 50.79, 47.46, 41.68, 22.81. MS: 338 (M++H);
……………………………………………………………………….
ARKIVOC 2012 (vi) 45-56 Page 45 ©ARKAT-USA, Inc.
An expeditious construction of 3-aryl-5-(substituted methyl)-2- oxazolidinones: a short and efficient synthesis of Linezolid
Rajesh Tammana,a,b Kiran Kumar Vemula,a Ramadasu Guruvindapalli,a Ramesh Yanamandra,c and Madhusudhan Gutta* a
aDepartment of Research & Development, Inogent Laboratories Pvt. Ltd.,
A GVK BIO Company, 28A, IDA, Nacharam, Hyderabad 500 076, Andhra Pradesh, India
bCentre for Pharmaceutical Sciences, Institute of Science and Technology, Jawaharlal Nehru Technological University, Hyderabad 500 072, Andhra Pradesh, India
cDepartment of Analytical Research & Development, GVK Biosciences Pvt. Ltd., 28A, IDA, Nacharam, Hyderabad 500 076, Andhra Pradesh, India
E-mail: madhusudhan.gutta@inogent.com
http://www.arkat-usa.org/get-file/42622/
N-(((S)-3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)acetamide 1 (Linezolid) 1 was prepared according to the method described in literature.12,15
Mp 182-183 °C, (lit.12a 181.5- 182.5 °C); enantiomeric purity 99.9% (by chiral HPLC);
IR (KBr): ν 3343 (NH), 3075 (Ar-H), 2967 (CH), 1741 (C═O), 1660 (C═O) cm-1 ;
1H NMR (CDCl3): δ 2.03 (s, 3H), 3.04-3.07 (t, 4H), 3.56-3.77 (m, 3H), 3.86-3.89 (t, 4H), 4.00-4.06 (t, 1H), 4.74-4.79 (m, 1H), 5.96 (s, 1H), 6.90- 6.96 (t, 1H), 7.06-7.10 (d, 1H), 7.43-7.48 (d, 1H).
13C NMR (DMSO-d6): δ 22.4, 41.4, 47.3, 50.6, 66.1, 71.5, 106.4, 114.0, 119.1, 133.3, 135.5, 154.0, 156.2, 170.0;
ESI-MS (C16H20FN3O4): m/z (%) 338.18 (100, M+ +1).
12. (a) Brickner, S. J.; Hutchinson, D. K.; Barbachyn, M. R.; Manninen, P. R.; Ulanowicz, D. A.;
Garmon, S. A.; Grega, K. C.; Hendges, S. K.; Toops, D. S.; Ford, C. W.; Zurenko, G. E. J.
Med. Chem. 1996, 39, 673. (b) Barbachyn, M. R.; Brickner, S. J.; Hutchinson, D. K. U.S.
patent 5688792; 1997; Chem. Abstr. 1995, 123, 256742. (c) Dhananjay, G. S.; Nandu, B. B.;
Avinash, V. N.; Kamlesh, D. S.; Anindya, S. B.; Tushar, A. N. PCT Int. Appl. 063505, 2009;
Chem. Abstr. 2009, 150, 515152.
13. (a) Imbordino, R. J.; Perrault, W. R.; Reeder, M. R. PCT Int. Appl. 116284, 2007; Chem.
Abstr. 2007, 147, 469356. (b) Pearlman, B. A.; Perrault, W. R.; Barbachyn, M. R.;
Manninen, P. R.; Toops, D. S.; Houser, D. J.; Fleck, T. J. U.S. Patent 5837870, 1998; Chem.
Abstr. 1998, 130, 25061. (c) Perrault, W. R.; Pearlman, B. A.; Godrej, D. B.; Jeganathan, A.;
Yamagata, K.; Chen, J. J.; Lu, C. V.; Herrinton, P. M.; Gadwood, R. C.; Chan, L.; Lyster, M.
A.; Maloney, M. T.; Moeslein, J. A.; Greene, M. L.; Barbachyn, M. R. Org. Proc. Res. Dev.
2003, 7, 533.
14. (a) Yu, D. S.; Huang, L.; Liang, H.; Gong, P. Chin. Chem. Lett. 2005, 16, 875. (b) Pearlman,
B. A. PCT Int. Appl. 9924393, 1999; Chem. Abstr. 1999, 130, 338099. (c) Weigert, F. J. J.
Org. Chem. 1973, 38, 1316.
15. (a) Wang, M.; Tong, H. CN patent 101220001, 2008. (b) Mohan Rao, D.; Krishna Reddy, P.
PCT Int. Appl. 099353, 2005; Chem. Abstr. 2005, 143, 440395. (c) Mohan Rao, D.; Krishna
Reddy, P. PCT Int. Appl. 008754, 2006; Chem. Abstr. 2006, 144, 170978.
……………………………………
Org. Proc. Res. Dev., 2003, 7 (4), pp 533–546
DOI: 10.1021/op034028h
Organic Process Research and Development, 2003 , vol. 7, # 4 p. 533 – 546
http://pubs.acs.org/doi/abs/10.1021/op034028h

Since 1993, a significant process research and development effort directed towards the large-scale synthesis of oxazolidinone antibacterial agents has been ongoing in both Early Chemical Process Research and Development, and Chemical Process Research and Development at Pharmacia. This work has led to the successful development of the current commercial process to produce Zyvox (linezolid), recently approved by the FDA as an antibacterial. While this synthesis is appropriate for the preparation of linezolid in particular, a more convergent and versatile synthesis was developed for the rapid preparation of numerous other oxazolidinone analogues. Toward this end, economical methods for the large-scale preparation of N-[(2S)-2-(acetyloxy)-3-chloropropyl]acetamide 3 and tert-butyl [(2S)-3-chloro-2-hydroxypropyl]carbamate 27 from commercially available (S)-epichlorohydrin via the common intermediate (2S)-1-amino-3-chloro-2-propanol hydrochloride 2a were developed. Also, general methods for coupling these reagents with N-aryl carbamates to giveN-aryl-5(S)-aminomethyl-2-oxazolidinone derivatives in one step were developed. These reagents and procedures have proven widely applicable in the preparation of a diverse array of oxazolidinone analogues such as 23 and 28 in both process and medicinal chemistry research.

(S)-N-[[3-[3-Fluoro-4-(4-morpholinyl)phenyl]-2-oxo- 5-oxazolidinyl]methyl]acetamide: Linezolid: Zyvox
HPLC analyses showed the first and second crops to be 98.9 and 94.6 wt % linezolid, respectively, with <0.2% enantiomer in each; also, an additional 9.7% yield of linezolid was detected in the filtrate by external standard HPLC (total ) 80.6%). Analysis data for 1st crop material: mp ) 73-76 °C;
1 H NMR (CDCl3, 400 MHz)
δ 7.43 (dd, J ) 14.4, 2.4 Hz, 1H), 7.07 (dd, J ) 8.8, 2.0 Hz, 1H), 6.91 (t, J ) 8.8 Hz, 1H), 6.43 (br t, 1H), 4.77 (m, 1H), 4.02 (t, J ) 9.2 Hz, 1H), 3.86 (t, J ) 4.4 Hz, 4H), 3.76 (dd, J ) 8.8, 6.8 Hz, 1H), 3.66 (m, 2H), 3.05 (t, J ) 4.8 Hz, 4H), 2.02 (s, 3H);
13C NMR (CDCl3, 100 MHz)
δ 23.07 (q), 41.93 (t), 47.66 (t), 51.00 (t), 66.95 (t), 71.99 (d), 107.56 (dd, JC-F ) 26.16 Hz), 113.97 (dd, JC-F ) 3.02 Hz), 118.85 (dd, JC-F ) 4.03 Hz), 132.90 (sd, JC-F ) 4.03 Hz), 136.58 (sd, JC-F ) 9.06 Hz), 154.42 (s), 155.50(sd, JC-F ) 246.53 Hz), 171.19 (s)
MS (EI) m/z (relative intensity) 337 (90), 293 (81), 209 (100);
[R]25D ) -16 (c ) 1.05, ethanol).
Anal. Calcd for C16H20FN3O4: C, 56.97; H, 5.97; N, 12.46; found: C, 56.86; H, 6.05; N, 12.44
HPLC (99.0 wt %, 98.9 area % linezolid, tR 1.60 min) conditions: InertsilODS-2 5.0 µm 150 mm × 4.6 mm, flow rate ) 2.0 mL/ min, gradient elution from 40:60 A:B to 80:20 A:B over 10 min; A ) acetonitrile; B ) water. External standard HPLC analysis of the filtrate showed
d 12.9% and 7.6% yield of linezolid and 8, respectively.
SEE HPLC AT http://file.selleckchem.com/downloads/hplc/S140801-Linezolid-Zyvox-HPLC-Selleck.pdf
………………………….
http://www.google.com/patents/WO2007064818A1?cl=en
Linezolid [(S)-N-[[3-(3-Fluoro-4-morpholinyl)phenyl]-2-oxo-5- oxazolidinyljmethyl] acetamide} is an antimicrobial agent. Linezolid is an oxazolidinone, having the empirical formula C16H20FN3O4 and the following structure:

Linezolid
Linezolid is described in The Merck Index (13th edition, Monograph number: 05526, CAS Registry Number: 165800-03-3) as white crystals, with a melting point of 181.5-182.50C. Linezolid, as well as a process for its preparation, is described in U.S. Patent No. 5,688,792 (Example 5), European Patent No. 717738, Israeli Patent No. 110,802, Canadian Patent No. 2,168,560, and International Patent Publication WO 95/07271. Linezolid is marketed in the United States by Pfizer, Inc. as an injection, as tablets, and as an oral suspension under the name ZYVOX®. Its main indications are nosocomial pneumonia, skin and skin-structure infections, and vancomycin-resistant Enterococcus faecium infections.
U.S. Patent No. 5,688,792 describes linezolid and its use for the treatment of microbial infections. This patent also describes the following method for the preparation of linezolid:

This method of preparation was also described in Bricker, et al., J. Med. Chem., 39, 673 — 679 (1996), where it was stated that the above route avoids the use of phosgene to make the carbamate precursor of the oxazolidinone ring. The authors also disclose that the use OfNaN3 can be avoided by using potassium phthalimide, followed by deblocking of the phthalimide with aqueous methyl amine.
An analysis of the commercial tablet ZYVOX® shows the presence of desfluoro linezolid as an impurity of linezolid. An HPLC chromatogram of ZYVOX® is depicted in Figure 1. The desfluoro linezolid haviong a relative retention time (RRT) of 0.69 compared to the retention time of linezolid.
desfluoro linezolid of the following structure:

Desfluoro linezolid
As illustrated in Figure 1, this impurity is ideal for use as a reference standard since it is detectable by HPLC, and yet it is present in much less amounts than linezolid, having a RRT of 0.69 compared to the retention time of linezolid.
The isolated desfluoro linezolid is pure. Preferably it has about 95% purity by weight with respect to other compounds, including linezolid. Preferably, the desfluoro linezolid is isolated in about 99.3% purity by weight. Thus, the isolated desfluoro linezolid contains less than about 5%, preferably less than about 2%, and even more preferably less than about 1%, by weight, linezolid.
The isolated desfluoro linezolid of the present invention can be characterized by data selected from: 1H NMR (400MHz, DMSO-d6) δ (ppm): 1.8a (s), 3.04 (brt), 3.40 (t), 3.68 (m), 3.72 (brt), 4.04 (t), 4.67 (m), 6.95 (d), 6.95 (d), 737 (d), 7.37 (d) and 8.21 (t); 13C NMR (lOOMHz, DMSO-d6) δ (ppm): 22.8, 41.9, 48.0, 49.2, 66.5, 71.7, 115.9, 115.9, 119.9, 119.9, 130.9, 148.0, 154.7, 170.0; EI+m/z (MH+): 319; and IR spectra on KBr at 1523, 1555, 1656, 1731, 2830, 2926, 2968 and 3311 cm‘1.
The isolated desfluoro linezolid of the present invention may be characterized by a 1H NMR, substantially as depicted in figure 2. The isolated desfluoro linezolid of the present invention may be characterized by 13C NMR, substantially as depicted in figure 3. The isolated desfluoro linezolid of the present invention may be characterized by an IR spectrum substantially as depicted in figure 4. The isolated, desfluoro linezolid of the present invention may be characterized by an Mass spectrum substantially as depicted in figure 5. The isolated desfluoro linezolid of the present invention may be prepared by performing the process described in U.S. Patent No. 5,688,792, with l-fluoro-4- nitrobenzene instead of 3,4-difluoronitrobenzene, according to the following scheme:

Desfluoro Linezolid
The desfluoro linezolid of the present invention is isolated by a process comprising the following steps; a) combining (5R)-[[3-[4-(4-morpholinyl)phenyl]-2- oxo-5-oxazolidinyl]methyl]azide with an organic solvent, preferably a C1-C4 alkyl ester or a C6 to C12 aromatic hydrocarbon, more preferably toluene or ethylacetate, most preferably toluene, and hydrogen gas in the presence of a catalyst to obtain a reaction mixture containing (5S)-[[3-[4-(4-morpholinyl)phenyl]-2-oxo-5- oxazolidinyl]methyl] amine; b) filtering the reaction mixture to obtain a solution containing (5S)-[[3-[4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl}methyl]amine; c) adding acetic anhydride to the solution to obtain a precipitate; and d) recovering and drying the precipitate to obtain isolated desfluoro linezolid. Preferably, recovering of the precipitate in step d) is carried out by filtering or decanting. Preferably, the catalyst in step a) is selected from the group consisting of Pd/C, Raney Nickel, and noble metal catalysts, more preferably the catalyst is Pd/C. The isolated desfluoro linezolid of the present invention is useful as a reference marker for linezolid. As such, it may be used in order to detect the desfluoro linezolid impurity in a linezolid sample.
Step 7. Preparation of N-rr(5S)-3-r4-(4-moφholinyl)phenyl1-2-oxo-5- oxazolidinyl]methyl1acetamide (des-fluoro-linezolid). In a IL reactor, 6 g (5R)-[[ 3-(4-morpholinyl)phenyl]-2-oxo-5- oxazolidinyl]methyl]azide were charged with 0.7L toluene followed by 0.6 g Pd/C (10% Pd/C containing 52% water). The system was bubbled with ammonia (gas) during 2 h, and then flushed three times with nitrogen and 3 times with hydrogen. The pressure of hydrogen was set to 1.5 arm. The reaction mixture was stirred at RT and the reaction followed up until completion. The reaction mixture was filtered and the solution was treated with 60 ml acetic anhydride at RT. The precipitate was filtered and dried to obtain 3.3 g of desfluoro linezolid (purity: 99.3%). Desfluorolϊnezolid 1H-NMR and 13C-NMR identification


……………….
HTTP://WWW.GOOGLE.COM/PATENTS/US6559305
Example 1 Preparation of Crystal Form II of Linezolid
Linezolid with better than 99.8% enantiomeric purity, less than 0.2% of the R enantiomer, (1.99 grams) is mixed with ethyl acetate (100 mL). The flask is stoppered and heated to 65° with constant stirring in a temperature controlled oil bath. The linezolid is completely dissolved and the mixture is stirred for an additional 10 minutes. The temperature is maintained at 55° in the flask and one neck of the flask is unstoppered to allow slow evaporation of the solvent. A gentle stream of nitrogen is blown across the open neck to aid in evaporation. Solids spontaneously precipitated from solution and the volume is reduced by about 25% of the initial volume. The flask is sealed and mixed for 90 minutes while maintaining the mixture at 55°. The mixture was then cooled to about 23° while being stirred. The solids are isolated by vacuum filtration using a sintered glass funnel to give linezolid in crystal form. Analysis by powder X-ray diffraction indicates that the solids are linezolid crystal Form II.
……………………
HTTP://WWW.GOOGLE.COM/PATENTS/US7989618
Example 1 Linezolid Dihydrochloride
20 g of linezolid are dissolved in 750 ml of acetone at about 30° C. The solution is kept at about 30° C. and 8 ml of concentrated hydrochloric acid (37% w/w aqueous solution) are added, thus immediately causing linezolid dihydrochloride to precipitate as a white solid. The mixture is kept under stirring at about 30° C. for approximately 30 minutes, then refluxed under stirring for about 2 hours. The mixture is left to cool to room temperature, then cooled on ice-water bath, under stirring, for about 2 hours. A white solid precipitates which is filtered with suction, washed with 30 ml of acetone and dried under vacuum at about 50° C.
A solid water-soluble crystalline product is obtained, characterized by an XRPD spectrum substantially as reported in FIG. 3, wherein the most intense diffraction peaks fall at 13.9; 18.2; 19.1; 19.7; 22.2; 22.9; 23.6; 25.3; 27.1; 28.4±0.2° in 2θ; and by a DSC thermogram substantially as reported in FIG. 4, characterized by an exothermic peak around 178±2° C. The acid-base potentiometric titre is double while the argentimetric one is 17.71% (theor. dihydrochloride 17.77%). Purity 99.8% as determined by HPLC.
1H NMR (300 MHz, DMSO-d6), ppm: 8.37 (bt, 1H), 7.50 (dd, 1H, J=15.3 Hz, J=2.7 Hz), 7.10 (m, 2H), 4.68 (m, 1H), 4.05 (t, 1H, J=9.0 Hz), 3.70 (m, 5H), 3.36 (t, 2H, J=5.1 Hz), 3.07 (t, 4H, J=4.5 Hz), 1.80 (s, 3H).
……………………………………….
http://www.google.com/patents/EP2690100A1?cl=en
Example 3
-
[0034]To a 25 ml, round-bottomed flask equipped with a magnetic stirring bar was charged “amine” (0.49 g) followed by water (8.30 ml). A heterogeneous mixture was stirred and hydrochloric acid (0.12 mL, 35 %) was added. A homogenous solution was obtained. The solution was cooled down in an ice-water bath to 0°C. Acetic anhydride (0.31 mL) was added followed by sodium bicarbonate (0.45 g). Carbon dioxide was immediately released and a formation of white precipitate was observed. The precipitate was filtered off and the filter cake was washed with water (10 ml). The filter cake was collected and dried (100 mbar) at 70°C overnight. An off-white solid linezolid (0.26 g) was isolated.
…………………………..
PATENT
http://www.google.com/patents/WO2007116284A1?cl=en
Example 3 Preparation of (S)~N-[3-(3-fiuoro~4~morpholin-4-yI-ρhenyI)~2-oxo- oxazolidin-5~ylmethyl]-acetarnide (Linezo!id)

Method A
To (S)-5-{[(4-chloro-benzylidene)-amino]-methyl}-3-(3-fluoro-4-morpholin-4-yl- phenyl)-oxazolidin-2-one (129.5g, 31 mmol, 1.0 eq.) is added ethyl acetate (935 mL) and water (935 mL). To the heterogeneous mixture is added 12M aq. HCl (51.58 mL, 620 mmol, 2.0 eq.). Within minutes, the solid went into solution and the reaction mixture is biphasic. After stirring the emulsion at ambient temperature for 2 hours, HPLC assay showed the hydrolysis reaction to be complete (HPLC conditions: YMC 5μ ODS-AM 150 nm X 4.6 nm column, eluting with CH3CN /water + 0.1% TFA from 20% CH3CN to 80% CH3CN in 8 min at 0.5 mL/min, detecting at 254nm, Retention time of (S)-N-[3-(3-fluoro-4-morpholin-4-yl- phenyl)-2-oxo-oxazolidin-5-ylmethyl]-amine is 3.2 min). The phases are separated, the organic layer is discarded, and the aqueous layer is washed with ethyl acetate (500 mL). CH2Cl2 (900 mL) is added and the pH is adjusted to 6.7 with ~ 25 mL aq. 50% aq. NaOH. With constant stirring, Ac2O (58.49 mL, 620 mmol, 2.0 eq.) is added in one portion and the pH dropped to 2. The pH is then readjusted to 6 using 50% aq. NaOH. The pH is adjusted to ca. 7.1 with 50% aq. NaOH and the phases separated. The aqueous phase is extracted with CHiCl2 (800 mL) and the organics are combined and concentrated to ~1L in volume. Ethyl acetate (IL) is added and the volume is reduced to 1.5 L under vacuum. Another IL of ethyl acetate is added and volume is reduced again to IL under vacuum. The resultant slurry is cooled to 00C and the precipitate collected by vacuum filtration. The resulting solid is washed with ethyl acetate (250 mL). The crude product is dried under vacuum at 500C for 2 hours to give the title compound as Hnezolid crystalline Form I.

Following the general procedure of method A and making non-critical variations, but substituting (S)-5- { [2,4-dichloro-benzylidene)-amino]-methyl } -3-(3-fluoro-4-morphoIin-4-yl- phenyl)-oxazolidin-2-one (example 11) for (S)-5-{[(4-chloro-benzylidene)-amino]- methyl}-3-(3-fluoro-4-morρholin-4-yl-phenyl)-oxazolidin-2-one, the title compound is obtained.

Following the general procedure of method B and making non-critical variations, but substituting (S)-5-{ [4-bromo-benzylidene)-amino] -methyl }-3-(3-fluoro-4-morpholin-4-yl~ phenyl)-oxazolidin-2-one (example 9) for (S)-5-{[(4-chloro-benzylidene)-amino]- methyl}-3-(3-fluoro-4-morph.olin-4-yl-phenyl)-oxazoIidin-2-one, the title compound is obtained.
Example 4 Trituration (convert linezolid crystalline Form I to linezolid crystalline Form E) The product from Example (89.18 g) is transferred to a 3L round bottom flask equipped with a mechanical stirrer, thermocouple and heating mantel. Ethyl acetate (2.23 L, 15 mL/g) is added and seeded with Linezolid form II crystals and the slurry is heated to ca. 500C. A slight exotherm of 30C is observed. After 30 minutes of heating the form change is observable as the solid is changing to long needles. Stirring is continued for 2 hours at 500C, at which time the contents are cooled to ambient temperature and stirred for an additional 30 minutes. The contents are then cooled to 30C for 1.5 hours, filtered and washed with cold ethyl acetate (300 mL total). The resultant solids are dried under vacuum at 50°C for 18 hours to give Linezolid (78.12 g) Form II by XRD, 99.8 wt%, 99.9% ee. HPLC conditions: YMC 5μ ODS-AM 150 nm X 4.6 nm column, etuting with CH3CN /water + 0.1% TFA from 20% CH3CN to 80% CH3CN in 8 min at 0.5 mL/min, detecting at 254nm. TR (Linezolid) = 4.4 min; HPLC conditions: Chiralcel OJ-H 250 nm X 4.6 nm column, eluting with 90% CO2/ 10%MeOH at 3.0 mL/min, detecting at 255 nm. TR [title compound] = 3.6 min; TR (enantiomer of title compound) = 4.1 rain
……………………………………..
http://www.google.com/patents/EP2516408A1?cl=en
The polymorphic form obtained by following process disclosed in U.S. Pat. No. 5,688,792 is designated as Form I. Figure- 1 depicts the PXRD graph of Form I obtained by following prior art process. [15] Disadvantage of the process disclosed in U.S. Pat. No. 5,688,792 is that it involves use of n-butyl lithium. Due to its explosive nature it is difficult to handle at plant scale. Also, the said reaction is carried out at temperature of -78°C, which is difficult to attain during commercial production. Further the intermediate obtained requires purification by column chromatography. Column chromatography is a cumbersome technique and difficult to practice during commercial scale production.
The process for the preparation of Linezolid is also disclosed in Journal of Medicinal Chemistry (1996), 39(3), 673-9, U.S. Pat. Nos. 6,492,555, 5,837,870, 6,887,995, 7,307,163, 7,429,661, etc.
Linezolid was first disclosed in U.S. Pat. No. 5,688,792. The process for synthesis is as disclosed in Scheme-I

The synthetic reaction scheme of the present invention is as shown below.

Scheme-ll
Example 6: Synthesis of Linezolid Crude.
[140] Ethyl acetate (3500ml) and 10% palladium on carbon catalyst (6.0g) are added in autoclave having (R)- [N- 3 – (3 -Fluoro-4-morpholinylphenyl) -2-oxo- 5 -oxazolidinyl] methyl azide (lOOg) at 20-30°C. Cool the reaction mass & maintain 2-3kg hydrogen pressure at 15-20°C for 6-7 hrs. Filter it & wash the hyflo bed by Ethyl acetate
(100mlx2). Then add the Triethyl amine (35. lg) & Acetic anhydride (29.9g) slowly at 25-30°C under stirring. Cool the mix, filter it and wash the solid with chilled (0-5°C) Ethyl acetate (100 ml) followed by water (100mlx2). Finally product is dried at 55-60° C. Yield: 0.85.: Percentage 81%w/w.
[141]
[142] Example 7: Synthesis of Linezolid Pure
[143] Reflux the Acetone (1020ml) and Linezolid crude (lOOg) at 55-60°C for the 30
minutes. Filter the hot turbid solution & wash it with hot (55-60°C) acetone (50ml). Cool the reaction mixture at -5 to 0°C for 1 hour, wash the solid with chilled (-5 to 0°C) acetone (50ml). After drying the Linezolid semi pure (77g) add n-Propanol (308ml) reflux it at 95-100°C for 30 min & filter it by hot solution through hyflo bed. Cool the mix to 0-5°C for 1 hour and wash the solid with chilled (0-5°C) n-Propanol (77ml). Dry the material at 55-60°C. Yield: 0.73.: Percentage 73%w/w.
[144]
[145] Example 8: Synthesis of Linezolid
[146] Ethyl acetate (3500ml) and 10% palladium on carbon catalyst (6.0g) are added in autoclave having (R)- [N- 3 – (3 -Fluoro-4-morpholinylphenyl) -2-oxo- 5 -oxazolidinyl] methyl azide (lOOg) at 20-30°C. Cool the reaction mass & maintain 2-3kg hydrogen pressure at 15-20°C for 6-7 hrs. Filter it & wash the hyflo bed by Ethyl acetate. Distill out ethyl acetate at 75-90°C and then cool the reaction mass to 0-5°C. Add acetone (1000ml) & acetic anhydride (29.9g) at 0-5°C. Further, add Triethyl amine (37.8g) slowly at 0-5°C under stirring. Maintain the reaction mass at 0-5°C for 1-2 hrs. Heat the reaction mass to reflux at 65-75°C for 1 hr. Again cool the reaction mass to 0-5°C fori hr. Filter the solid wash it with acetone and water and dry it at 55-60C. Yield: 0.80.: Percentage 80 w/w.
Example 9: Synthesis of Linezolid Form I
[149] Reflux n-propanol (400ml) and Linezolid (lOOg) at 95-100°C till all solid gets
dissolved. Add activated charcoal (2.0g) and heat for 30 mins. Filter thro hyflo bed. Heat the filtrate and concentrate the solution by partially removing n-propanol. Cool to 0-5°C and filter the solid and dry it at 55-60°C under vacuum. Yield: 0.9. : Percentage 90 w/w.
………………………………………..
https://acs.confex.com/acs/green08/techprogram/P52019.HTM
Wednesday, June 25, 2008 – 2:00 PM
New York (Capital Hilton)
128
Convergent Green Synthesis of Linezolid (Zyvox)
Pfizer has developed a novel, convergent, green, second generation synthesis of Linezolid (the active ingredient in ZyvoxTM). The second generation process will replace the launch process after approval by appropriate regulatory agencies and has numerous green chemistry benefits: overall yield is increased by 8%; total waste is reduced by 56%; non-recycled w is eliminated. At current volumes, total waste will be reduced 1.9 million kilograms per year and 1.7 million kg per year non-recyclable waste will be eliminated. The improved process utilizes a highly efficient low dilution convergent synthesis to replace the more dilute linear synthesis utilized in the launch process. The key chlorohydrin imine reagent 1 contains both the chiral center and the key 5-S-aminomethyl moiety of linezolid. In the launch process, S-1-chloro-2,3-propanediol was utilized to install the oxazolidinone functionality. However, this yielded a 5-S-hydroxymethyl group which required activation as the 3-nitrobenzenesulfonate and displacement with excess ammonia to generate the corresponding aminomethyl group of linezolid. The second generation process affords the oxazolidinone imine 3 in the convergent step. The penultimate 5-S-aminomethyl oxazolidinone 4 is then easily formed via hydrolysis with stoichiometric hydrochloric acid. Acylation of this amine with acetic anhydride, utilizing an improved Schotten Baumann reaction, affords high purity linezolid.
………………………………….

……………………………………..

……………………………………….
http://www.google.com/patents/EP2072505A2?cl=en
-
WO 95/07271 , which specifically describes the synthesis of linezolid, namely [(S)-N-[[3-(3-fluoro-4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide], according to the following scheme:

-
[0003]Other synthetic routes for the preparation of linezolid are reported for example in US 6107519 and in Tetrahedron Letters, Vol 37, N° 44, pages 7937-7940, wherein the chiral compound shown below is used instead of glycidyl butyrate as a synthon containing the molecule stereocenter.

-
[0004]It should be appreciated that all of the known approaches to the preparation of linezolid make use of chiral synthons for the construction of the stereocenter. These are small molecules characterized by a high cost, therefore they are not suitable for the production of the compound on an industrial scale.
-
[0005]There is therefore the need for an alternative synthesis which provides oxazolidinone derivatives, linezolid included, from inexpensive starting materials, and which does not require a chiral synthon for the construction of the molecule, so that it can be used for the industrial preparation of such derivatives.
………………………………….
http://pubs.rsc.org/en/content/articlelanding/2010/md/c0md00015a/unauth

………………………………….

…………………………………………

…………………………………
RSC Adv., 2013,3, 24946-24951
DOI: 10.1039/C3RA45186K
http://pubs.rsc.org/en/content/articlelanding/2013/ra/c3ra45186k#!divAbstract


A new asymmetric synthesis of the antibiotic Linezolid was performed through a copper-catalyzed Henry reaction as the key step. The use of camphor-derived aminopyridine ligands helped to improve the yields of the chiral precursor and to obtain Linezolid in good overall yield and enantiomeric excess.
Linezolid 1. Mp: 181–182 C [lit. 181.5–182.5 C];
1 H-NMR (300 MHz; CDCl3) d 2.02 (s, 3H), 3.06 (t, J ¼ 4.7 Hz, 4H), 3.61– 3.78 (m, 3H), 3.87 (t, J ¼ 4.7 Hz, 4H), 4.03 (t, J ¼ 9.0 Hz, 1H), 4.72–4.82 (m, 1H), 6.17 (bt, 1H, exch. with D2O), 6.93 (t, J ¼ 9.0 Hz, 1H), 7.08 (dd, J1 ¼ 9.0 Hz, J2 ¼ 2.5 Hz, 1H), 7.44 (dd, J1 ¼ 14.4 Hz, J2 ¼ 2.5 Hz, 1H); ee ¼ 71%;
HPLC (Daicel CHIRALPAK-IA, hexane/i-PrOH ¼ 70 : 30, ow rate 0.8 mL min 1 , l ¼ 254 nm); tR (major) ¼ 14.1 min; tR (minor) ¼ 16.4 min. A true sample of (S)-Linezolid (ee > 98%) under the same HPLC conditions gave a tR ¼ 14.1 min.
………………………………..
http://www.slideshare.net/vishwajeeta/introduction-new-ppt
………………………………


http://www.slideshare.net/pushechnikov/linezolid-case-study
…………………………………
http://pubs.rsc.org/en/content/articlelanding/2011/cc/c1cc15503b#!divAbstract

…………………………………
http://www.mdpi.com/1424-8247/3/7/1988/htm

………………………………

Numbered structure of linezolid, showing the pharmacophore required for good activity (in blue) and desirable structural features (in orange).
Title: Linezolid
CAS Registry Number: 165800-03-3
CAS Name:N-[[(5S)-3-[3-Fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide
Manufacturers’ Codes: PNU-100766; U-100766
Trademarks: Zyvox (Pharmacia & Upjohn); Zyvoxid (Pharmacia & Upjohn)
Molecular Formula: C16H20FN3O4
Molecular Weight: 337.35
Percent Composition: C 56.96%, H 5.98%, F 5.63%, N 12.46%, O 18.97%
Literature References: Prototype of the oxazolidinone antimicrobials; inhibits bacterial mRNA translation. Prepn: M. R. Barbachyn et al.,WO9507271 (1995 to Upjohn); eidem,US5688792 (1997 to Pharmacia & Upjohn); S. J. Brickner et al.,J. Med. Chem.39, 673 (1996).
Antibacterial spectrum: C. W. Ford et al.,Antimicrob. Agents Chemother.40, 1508 (1996). Mechanism of action study: D. L. Shinabarger et al.,ibid.41, 2132 (1997).
HPLC determn in plasma: C. Buerger et al.,J. Chromatogr. B796, 155 (2003). Clinical comparison with vancomycin, q.v., for MRSA infections: D. L. Stevens et al., Clin. Infect. Dis.34, 1481 (2002).
Review of pharmacology: L. D. Dresser, M. J. Rybak, Pharmacotherapy18, 456-462 (1998); and clinical experience: R. Norrby, Expert Opin. Pharmacother.2, 293-302 (2001).
Properties: White crystals from ethyl acetate and hexanes, mp 181.5-182.5°. [a]D20 -9° (c = 0.919 in chloroform).
Melting point: mp 181.5-182.5°
Optical Rotation: [a]D20 -9° (c = 0.919 in chloroform)
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Oxazolidinones.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (S)-N-({3-[3-fluoro-4-(morpholin-4-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl}methyl)acetamide | |
| Clinical data | |
| Trade names | Zyvox, Zyvoxam, Zyvoxid |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602004 |
| Licence data | US FDA:link |
|
|
|
|
| Intravenous infusion, oral | |
| Pharmacokinetic data | |
| Bioavailability | ~100% (oral) |
| Protein binding | Low (31%) |
| Metabolism | Hepatic (50–70%, CYPnot involved) |
| Half-life | 4.2–5.4 hours (shorter in children) |
| Excretion | Nonrenal, renal, and fecal |
| Identifiers | |
| 165800-03-3 |
|
| J01XX08 | |
| PubChem | CID 441401 |
| DrugBank | DB00601 |
| ChemSpider | 390139 |
| UNII | ISQ9I6J12J |
| KEGG | D00947 |
| ChEMBL | CHEMBL126 |
| NIAID ChemDB | 070944 |
| Chemical data | |
| Formula | C16H20FN3O4 |
| 337.346 g/mol | |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1995007271A1 * | Aug 16, 1994 | Mar 16, 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| AU2001100437A4 * | Title not available | |||
| EP0963980A2 * | Mar 10, 1999 | Dec 15, 1999 | The Wellcome Foundation Limited | 1,2,4-Triazine derivative, its preparation and its use as reference marker for testing purity and stability of “lamotrigine” |
| Reference | ||
|---|---|---|
| 1 | * | [Online] August 2002 (2002-08), XP002388488 Retrieved from the Internet: URL:www.emea.eu.int/pdfs/human/ich/273799e n.pdf> [retrieved on 2006-07-03] |
| 2 | * | [Online] June 1995 (1995-06), XP002388489 Retrieved from the Internet: URL:www.emea.eu.int/pdfs/human/ich/38195en .pdf> [retrieved on 2006-07-03] |
| 3 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JUN ET AL: “Preparation of oxazolidone derivatives as antibacterial agents” XP002429969 retrieved from STN Database accession no. 2003:576097 -& CN 1 355 165 A (INSTITUTE OF MEDICAL AND BIOLOGICAL TECHNOLOGY, CHINESE ACADEMY OF MED) 26 June 2002 (2002-06-26) |
| 4 | * | GLEAVE D M ET AL: “Synthesis and antibacterial activity of [6,5,5] and [6,6,5] tricyclic fused oxazolidinones” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 8, no. 10, 19 May 1998 (1998-05-19), pages 1231-1236, XP004137053 ISSN: 0960-894X |
| 5 | * | REDDY K V S R K ET AL: “Isolation and characterization of process related impurities in linezolid” JOURNAL OF PHARMACEUTICAL AN BIOMEDICAL ANALYSIS, vol. 30, no. 3, 15 October 2003 (2003-10-15), pages 635-642, XP002388486 |
| WO2001057035A1 * | Jan 29, 2001 | Aug 9, 2001 | Upjohn Co | Linezolid-crystal form ii |
| WO2002032857A1 * | Oct 17, 2001 | Apr 25, 2002 | Robert C Gadwood | Methods of producing oxazolidinone compounds |
| WO2002085849A2 * | Apr 15, 2002 | Oct 31, 2002 | Delara B Godrej | Process to prepare oxazolidinones |
| WO2005099353A2 * | Apr 19, 2004 | Oct 27, 2005 | Reddy Pingili Krishna | A novel process for the preparation of linezolid and related compounds |
| WO2006008754A1 | Jul 20, 2004 | Jan 26, 2006 | Reddy Pingili Krishna | Novel intermediates for linezolid and related compounds |
| WO2006031179A1 * | Sep 12, 2005 | Mar 23, 2006 | Astrazeneca Ab | Process for preparation of phtalimide |
| WO2007116284A1 * | Mar 26, 2007 | Oct 18, 2007 | Pfizer Prod Inc | Process for preparing linezolid |
| WO2010081404A1 | Jan 8, 2010 | Jul 22, 2010 | Lianhe Chemical Technology Co., Ltd. | Method for preparing linezolid and intermediates thereof |
| WO2012019632A1 | Aug 11, 2010 | Feb 16, 2012 | Synthon B.V. | Process for making linezolid |
| WO2012019862A1 | Jul 14, 2011 | Feb 16, 2012 | Synthon B.V. | Process for making linezolid |
| WO2012114354A1 | Feb 21, 2012 | Aug 30, 2012 | Lee Pharma Limited | Anhydrous linezolid crystalline form-ii |
| WO2013072923A1 | Sep 18, 2012 | May 23, 2013 | Cadila Healthcare Limited | Process for the preparation of crystalline linezolid |
| WO2013111048A1 | Jan 22, 2013 | Aug 1, 2013 | Jubilant Life Sciences Limited | Improved process for the preparation of stable crystalline form-i of linezolid, substantially free of residual solvent |
| WO2014071990A1 | Nov 9, 2012 | May 15, 2014 | Synthon Bv | Process for making linezolid |
| EP1403267A1 * | Sep 25, 2003 | Mar 31, 2004 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| EP1564215A1 * | Sep 25, 2003 | Aug 17, 2005 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| EP2100884A1 | Oct 16, 2003 | Sep 16, 2009 | Symed Labs Limited | Crystalline form of linezolid |
| EP2690100A1 | Jul 14, 2011 | Jan 29, 2014 | Synhton B.V. | Process for making linezolid |
| US6444813 | Jan 29, 2001 | Sep 3, 2002 | Pharmacia & Upjohn Company | Mixing linezolid of an >98% enantomeric purity in a solvent at >80 degrees; separating a crystal (ii) of >99% purity; analysis by the powder x-ray diffraction spectrum/infrared spectrum as a mineral oil mull; bactericides; stability |
| US6514529 | Mar 15, 2001 | Feb 4, 2003 | Pharmacia & Upjohn Company | A compressed tablet of antibacterial oxazolidinone selected from the group consisting of linezolid, eperezolid and (S)-N-((3-(3-fluoro-4-(tetrahydro-2H-thiopyran-4-yl)phenyl-2-o xo-5-oxazolidinylmethyl)acetamide S,S-dioxide |
| US6544991 | Jun 21, 2001 | Apr 8, 2003 | Pharmacia & Upjohn Company | Compositions and methods for treating bacterial infections |
| US6559305 | May 23, 2002 | May 6, 2003 | Pharmacia & Upjohn Company | Linezolid—crystal form II |
| US6617339 | Jun 3, 1999 | Sep 9, 2003 | Syngenta Limited | Oxazolidinone derivatives, process for their preparation and pharmaceutical compositions containing them |
| US6796975 | Mar 15, 2001 | Sep 28, 2004 | Pharmacia & Upjohn Company | Container for linezolid intravenous solution |
| US6833453 | Oct 17, 2001 | Dec 21, 2004 | Pharmacia & Upjohn Company | As an example, manufacturing a 5-(tert-butylcarbamoyl)-amino-methyl-oxazolidinone by condensing a carbamate with a tert-butylcarbamoyl protected derivative of glycidylamine or a 3-amino-1-halopropanol |
| US6875875 | Sep 25, 2003 | Apr 5, 2005 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| US6887995 | Apr 15, 2002 | May 3, 2005 | Pharmacia & Upjohn Company | Reacting N-aryl-O-alkylcarbamate with an amide derivative in the presence of a lithium cation, a base, and a nucleophile |
| US6989381 | Aug 20, 2001 | Jan 24, 2006 | Pharmacia Corporation | Containing s cyclodextrin compound in a concentration sufficient to maintain the drug in solution at such a drug concentration. |
| US7087784 | Mar 25, 2004 | Aug 8, 2006 | Pharmacia & Upjohn | Process to prepare oxazolidinones |
| US7128928 | Feb 20, 2003 | Oct 31, 2006 | Pharmacia Corporation | Ophthalmic formulation with novel gum composition |
| US7135576 | Jan 7, 2005 | Nov 14, 2006 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| US7307163 | Apr 19, 2004 | Dec 11, 2007 | Symed Labs Limited | Process for the preparation of linezolid and related compounds |
| US7351824 | Oct 8, 2007 | Apr 1, 2008 | Symed Labs Limited | Intermediates for oxazolidinone antibacterials; N-[3-Chloro-2-(R)-hydroxypropyl]-3-fluoro-4-morpholinyl aniline |
| US7429661 | Jul 20, 2004 | Sep 30, 2008 | Symed Labs Limited | Intermediates for linezolid and related compounds |
| US7524954 | Oct 8, 2007 | Apr 28, 2009 | Symed Labs Limited | Reacting 3-fluoro-4-morpholinyl aniline derivative with epichlorohydrin; converting chloromethyl oxazolidinone to aminomethyl oxazolidinone; carbonylation ; reacting with potassium phthalimide, hydrazine hydrate, and acetic anhydride; cyclization, carbamylation |
| US7714128 | Oct 16, 2003 | May 11, 2010 | Symed Labs Limited | crystalline linezolid form III (N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide) an antibacterial agent; thermal stability |
| US7718799 | Sep 26, 2007 | May 18, 2010 | Symed Labs Limited | Crystalline form of linezolid |
| US7718800 | Sep 26, 2007 | May 18, 2010 | Symed Labs Limited | Prepared by mixing linezolid with solvent or mixture of solvents, cooling contents to below 15 degrees C., optionally seeding contents with linezolid form III, stirring, and collecting linezolid form III crystals by filtration or centrifugation; antibacterial agent; thermally stable |
| US7732597 | Sep 26, 2007 | Jun 8, 2010 | Symed Labs Limited | Prepared by acetylating (S)-N-[[3-[3-fluoro-4-[4-morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]amine in a solvent, optionally in presence of an organic base to form linezolid, seeding reaction mixture, and isolating linezolid form III; antibacterial agent; thermally stable |
| US7741480 | Oct 8, 2007 | Jun 22, 2010 | Symed Labs Limited | Process for the preparation of linezolid and related compounds |
| US8658789 | Jan 8, 2010 | Feb 25, 2014 | Lianhe Chemical Technology Co., Ltd. | Method for preparing linezolid and intermediates thereof |
| US5837870 | Mar 28, 1997 | Nov 17, 1998 | Pharmacia & Upjohn Company | Process to prepare oxazolidinones |
| US6107519 | Oct 13, 1998 | Aug 22, 2000 | Pharmacia & Upjohn Company | Amido-substituted secondary alcohol intermediates and preparation thereof |
| US6444813 | Jan 29, 2001 | Sep 3, 2002 | Pharmacia & Upjohn Company | Mixing linezolid of an >98% enantomeric purity in a solvent at >80 degrees; separating a crystal (ii) of >99% purity; analysis by the powder x-ray diffraction spectrum/infrared spectrum as a mineral oil mull; bactericides; stability |
| US6492555 | Jan 15, 2002 | Dec 10, 2002 | Pharmacia & Upjohn Company | Reaction of a carbamate with either a (s)-secondary alcohol or (s)-epoxide or (s)-ester; bactericides |
| US6559305 | May 23, 2002 | May 6, 2003 | Pharmacia & Upjohn Company | Linezolid—crystal form II |
| US6716980 | Jun 27, 2003 | Apr 6, 2004 | Pharmacia & Upjohn Company | Cyclization and acylation of carbamate |
| US6740754 | Apr 24, 2003 | May 25, 2004 | Pharmacia & Upjohn Company | Process to produce oxazolidinones |
| US6833453 | Oct 17, 2001 | Dec 21, 2004 | Pharmacia & Upjohn Company | As an example, manufacturing a 5-(tert-butylcarbamoyl)-amino-methyl-oxazolidinone by condensing a carbamate with a tert-butylcarbamoyl protected derivative of glycidylamine or a 3-amino-1-halopropanol |
| US6887995 | Apr 15, 2002 | May 3, 2005 | Pharmacia & Upjohn Company | Reacting N-aryl-O-alkylcarbamate with an amide derivative in the presence of a lithium cation, a base, and a nucleophile |
| US7649096 * | Jul 17, 2006 | Jan 19, 2010 | Glenmark Pharmaceuticals Limited | crystallization of linezolid antibacterial agent in solvent and antisolvent |
| US20060111350 | Jun 29, 2005 | May 25, 2006 | Judith Aronhime | Solid forms of linezolid and processes for preparation thereof |
| US20060142283 | Jun 29, 2005 | Jun 29, 2006 | Judith Aronhime | Crystalline form IV of linezolid |
| US20090156806 | Dec 11, 2008 | Jun 18, 2009 | Dipharma Francis S.R.I. | Process for the Preparation of Oxazolidinone Derivatives |
| WO1995007271A1 | Aug 16, 1994 | Mar 16, 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO2005035530A1 | Oct 16, 2003 | Apr 21, 2005 | Reddy Pingili Krishna | A novel crystalline form of linezolid |
| WO2007026369A1 | Aug 29, 2005 | Mar 8, 2007 | Reddy Pingili Krishna | A novel amorphous form of linezolid |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009032294A2 * | Sep 5, 2008 | Mar 12, 2009 | Teva Pharma | Processes for the preparation of a linezolid intermediate, linezolid hydroxide |
| WO2011076678A1 * | Dec 17, 2010 | Jun 30, 2011 | F. Hoffmann-La Roche Ag | Substituted benzamide derivatives |
……………
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
 amcrasto@gmail.com
amcrasto@gmail.com
You might enjoy reading:
Isradipine

Isradipine
CAS Registry Number: 75695-93-1
CAS Name: 4-(4-Benzofurazanyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid methyl 1-methylethyl ester
Additional Names: isopropyl 4-(2,1,3-benzoxadiazol-4-yl)-1,4-dihydro-5-methoxycarbonyl-2,6-dimethyl-3-pyridinecarboxylate; 4-(2,1,3-benzoxadiazol-4-yl)-2,6-dimethyl-1,4-dihydro-3-isopropyloxycarbonylpyridine-5-carboxylic acid methyl ester; isrodipine
Manufacturers’ Codes: PN-200-110
Trademarks: Clivoten (Lifepharma); DynaCirc (Novartis); Esradin (Sigma-Tau); Lomir (Novartis); Prescal (Novartis)
Molecular Formula: C19H21N3O5
Molecular Weight: 371.39
Percent Composition: C 61.45%, H 5.70%, N 11.31%, O 21.54%
Properties: mp 168-170°.
Melting point: mp 168-170°
Derivative Type: S(+)-Form
Manufacturers’ Codes: PN-205-033
Properties: Crystals from ether + hexane, mp 142°. [a]D20 +6.7° (c = 1.5 in ethanol).
Melting point: mp 142°
Optical Rotation: [a]D20 +6.7° (c = 1.5 in ethanol)
Derivative Type: R(-)-Form
Manufacturers’ Codes: PN-205-034
Properties: Crystals from ether + hexane, mp 140°. [a]D20 -6.7° (c = 1.67 in ethanol).
Melting point: mp 140°
Optical Rotation: [a]D20 -6.7° (c = 1.67 in ethanol)
Keywords: Antianginal; Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.
Isradipine (tradenames DynaCirc, Prescal) is a calcium channel blocker of the dihydropyridine class. It is usually prescribed for the treatment of high blood pressure in order to reduce the risk of stroke and heart attack. More recent research in animal models suggests that isradipine may have potential uses for treating Parkinson’s disease Chan et al. 2007.
Isradipine is given as either a 2.5mg or 5mg capsule. [1]
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html

Isradipine is a drug used to lower blood pressure but recently it was found by a team from Nortwestern University, that this molecule can also slow the progression of Parkinson’s disease, and restore the dopamine neurons (In animals tests). Isradipine is a calcium channel blocker of the 1,4-dihydropyridine class with a benzoxadiazole moiety in position 4.
The synthesis of the 1,4-dihydropyridine ring is quite classic, the first step consists in a Knoevenagel reaction of methyl acetoacetate on the benzoxadiazole 4-carboxaldehyde using piperidine and acetic acid as catalyst and diisopropylether as solvent in a 61% yield (this is the first time I see a Knoevenagel reaction in an ether!!!???? DCM, Toluene OK, but maybe I am wrong). The second step of this synthesis is the condensation of the acrylate obtained with the isopropyl aminocrotonate in ethanol to give the desire 1,4-dihydropyridine Isradipine in 67% yield after recrystallisation.
(WO/2005/005437) An improved Process for the Manufacture of Isradipine (Shasun Chemical & Drugs Limited)
The synthesis of the 1,4-dihydropyridine ring is quite classic, the first step consists in a Knoevenagel reaction of methyl acetoacetate on the benzoxadiazole 4-carboxaldehyde using piperidine and acetic acid as catalyst and diisopropylether as solvent in a 61% yield (this is the first time I see a Knoevenagel reaction in an ether!!!???? DCM, Toluene OK, but maybe I am wrong). The second step of this synthesis is the condensation of the acrylate obtained with the isopropyl aminocrotonate in ethanol to give the desire 1,4-dihydropyridine Isradipine in 67% yield after recrystallisation.

(WO/2005/005437) An improved Process for the Manufacture of Isradipine (Shasun Chemical & Drugs Limited)
Isradipine is 4-(4-Benzofi–razanyl)-l,4-d–hydro-2,6-dimethyl-3,5- pyiicϊ-nedicarboxylic acid med yl 1-methylethyl ester having die chemical structure of formula (I).

( I ) Isradipine is therapeutically indicated for treating cardiovascular diseases.
The cardiovascular diseases include angina, pectoris, hypertension and congestive heart failure. It is also used to treat high blood pressure. Isradipine was disclosed in the German specification DE 2949491 and US patent Nos. 4466972 and 4567271. DE 2949491 describes the general procedure to prepare 1,4-dihydropyridine derivatives. US 4466972, GBQ2103203A, LU 0088342A9, EP 0000150A1, EP 0000150B1, AU 0538515B2 and od er related patents describe the general mediod for d e preparation of Benzoxadiazoles and dieir derivatives of general formula (EL). These references in its entirety is hereby incorporated by reference into this application.

( II ) Where in Ri is -CH3 and -R2 is — CH(CH3)2 it refers to Isradipine of formula ( I ). When Ri and R2 are not identical the general procedures described in diese patent specifications produces a mixture of isomers of formula ( II ). These procedures for the preparation of Isradipine is characteristic of formation of the ‘ isomeric impurities, 1) 4-(4-Benzoi-urazanyl)-l,4-α^ydro-2,6-climedιyl-3,5- pyridinedicarboxylic acid di-methyl ester of formula ( III ) and 2) 4-(4- Benzofurazanyl)-l,4-α^ydro-2,6-d–methyl-3,5-pyrid–nedicarboxylic acid di-1- med ylethyl ester of formula ( IV) along with Isradipine. The US patent 4466972 describes the preparation of compounds of general formula (II) by refluxing 2, 1, 3-benzoxadiazole-4-carboxaldehyde, keto ester and concentrated ammonia or a β-amino ester in presence of ethanol, followed by evaporation and purification by chromatography.
H

( HI )H

( I V ) Tl ese symmetrical ester isomers ( III ) and ( IN ) are difficult to separate from the Isradipine and the separation is effected only by a chromatographic purifications. The drawback witii the procedures described in these patents is that it is very difficult to produce die product in commercial quantities as it involves d e purification of the product by chromatographic separations. A single step process for the preparation of Isradipine was described in CH 661270. This procedure involves first reacting 2,l,3-benzoxadiazole-4- carboxaldehyde with isopropyl acetoacetate in the presence of catalytic quantities of acetic acid and piperidine in refluxing toluene, and further reacting it widi mediyl-β-aminocrotonate. The Isradipine formed in d e reaction mixture was dien- separated by toluene distillation followed by cyclohexane treatment. The crude product obtained was dien crystallised from etiianol to get Isradipine. When we have repeated this process in our laboratory we got the Isradipine with substantially higher amount of symmetrical ester isomers ( III ) and ( IV ) are present in d e product. Removal of these symmetrical ester isomers is very difficult even after several repurifications from ethanol.

Stap 2

Example 4
Preparation of Isradipine using crude 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester Dissolved the crude 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester ‘ obtained in example – 1 (25 g, 0.10 mol) in absolute edianol (375 ml) and added in to the solution isopropyl-β-aminocrotonate (13.15 ml, 0.09 mol). Stirred the reaction mixture under nitrogen atmosphere at 25-28 °C for 7 hr. Removed sample from d e reaction mixture and analysed die sample by qualitative HPLC. Distilled ethanol from the reaction mixture- under vacuum at 50°C. Dissolved d e residue in ethyl acetate (235 ml) and washed twice widi water (90 ml). Dried the organic layer over sodium sulphate and distillation under vacuum at 50 °C. Dissolved the concentrate in ethanol (65 ml) at 70°C and slowly cooled to 5°C to get die product crystallised. Filtered the product and washed with pre cooled ethanol (25 ml). Recrystauised the product from ethanol (60 ml) and dried at 70°C under vacuum to obtain Isradipine (yield = 20 g, purity = 98.2% and Impurity III = 0.64%, Impurity IV = 0.51% by HPLQ
Example 5 Preparation of Isradipine using purified 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester Dissolved 2-acetyl-3-benzo–urazan-4-yl-acrylic acid methyl ester (25 g, 0.10 mol) in absolute ethanol (375 ml) and added in to the solution isopropyl-β- aminocrotonate (13.15 ml, 0.09 mol). Stirred the reaction mixture under nitrogen atmosphere at 25-28 °C for 5 hr. Removed sample from the reaction mixture and analysed the sample by qualitative HPLC. Distilled ethanol from the reaction mixture under vacuum at 50°C. Dissolved the residue in ethyl acetate (235 ml) and washed twice wid w?ater (90 ml). Dried die organic layer over sodium sulphate and distillation under vacuum at 50 °C. Dissolved the concentrate in ethanol (65 ml) at 70°C and slowly cooled to 5°C to get the product crystallised. Filtered the product. and washed with pre cooled ethanol (25 ml). Recrystallised the product from edianol (60 ml) and dried at 70°C under vacuum to obtain 25 g Isradipine (yield = 67%, purity 99.5%, Impurity III = 0.20%, and Impurity IV = 0.12% by HPLC)
Example 6 Preparation of Isradipine using purified 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester Dissolved the purified 2-acetyl-3-benzofurazan-4-yl-acrylic acid mediyl ester, obtained in example — 3 (25 g, 0.10 mol) in absolute ethanol (375 ml) and added in to the solution isopropyl-β-aminocrotonate (13.15 ml, 0.09 mol). Stirred the reaction mixture under nitrogen atmosphere at 25-28 °C for 5 hr. Removed sample from the reaction mixture and analysed the sample by qualitative HPLC. Distilled ethanol from die reaction mixture under vacuum at 50°C. Dissolved die residue in ethyl acetate (235 ml) and washed twice with water (90 ml). Dried the organic layer over sodium sulphate and distillation under vacuum at 50 °C. Dissolved the concentrate in ethanol (65 ml) at 70°C and slowly cooled to 5°C to get the product crystallised. Filtered the product and washed with pre cooled ethanol (25 ml) and dried at 70°C under vacuum to obtain 30g Isradipine (purity = 99.4%, Impurity III = 0.22%, and Impurity IV = 0.11% by HPLC). Throughout this application, various publications are referenced.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
Side effects
Common side effects include: [2]
- Dizziness
- Warmth, redness, or tingly feeling under your skin
- Headache
- Weakness, tired feeling
- Nausea, vomiting, diarrhea, upset stomach
- Skin rash or itching
Serious side effects include: [2]
- Lightheadedness or fainting
- Shortness of breath, especially from minimal physical activity
- Swelling in the hands and feet
- Rapid and/or heavy heartbeat
- Chest pain
If you experience one or more of these serious side effects, contact your health care provider immediately.
Significant drug interactions
There are other interactions beyond those listed below. Make sure to speak with a Pharmacist or Doctor if you have any concerns.
Three major interactions are listed below.
1. It is advised that those using Isradipine not take Anzemet (Dolasetron), as both agents can cause a dose-dependent PR intervaland QRS complex prolongation. [3]
2. Onmel/Sporanox (Itraconazole) exhibits a negative inotropic effect on the heart and thus could spur an additive effect when used concomitantly with Isradipine. Onmel/Sporanox also inhibits an important cytochrome liver enzyme (CYP 450 3A4) which is needed to metabolize Isradipine and other Calcium Channel Blockers. This will increase plasma levels of Isradipine and could cause an unintentional overdose of the medication. Caution is advised when administering both agents together. [4]
3. Zanaflex (Tizanidine) demonstrates anti-hypertensive effects and should be avoided in patients taking Isradipine due to the possibility of synergism between both medications. [5]
4. The anti-biotic Rifadin (Rifampin) lowered plasma concentrations of Isradipine to below detectable limits. [1]
5. Tagamet (Cimetidine) increased Isradipine mean peak plasma levels. A downward dose adjustment may be necessary with this particular instance of polypharmacy. [1]
6. Severe hypotension was reported with Duragesic (Fentanyl) anesthesia when it was combined with other Calcium Channel Blockers. Even though Isradipine, another Calcium Channel Blocker, has not been used in conjunction with Fentanyl anesthesia in any studies, caution is advised. [1]
Note: There was no significant interaction between Isradipine and Warfarin (Coumadin), Isradipine and Microzide Hydrochlorothiazide, Isradipine and Lanoxin (Digoxin), and Isradipine and Nitrostat (Nitroglycerin).
Overdose
Symptoms of an Isradipine overdose include: [1]
- Lethargy
- Sinus tachycardia
- Transient hypotension
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
…………………
US 4466972
http://www.google.com.na/patents/US4466972
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
………………………………….
http://www.google.com.ar/patents/CN101768153B?cl=en
isradipine is a class Benzofurazan dihydropyridine class of compounds, the synthesis is more complex, especially in the purified material is particularly difficult.
US Patent US4466972 and PCT application W02005 / 00437 respectively disclose two synthetic isradipine
Methods. However, the product prepared by conventional methods contain a certain amount of a formula homologues impurity, the impurity is structured as follows:

wherein, R1, R2 simultaneously or separately as methyl, ethyl and isopropyl. The homologue of the impurities with isradipine extremely difficult to separate, resulting in ineffective purification isradipine.
In addition, conventional processes for preparing key intermediates involved in 4-methyl benzofurazan. However, the preparation method of the key intermediates is still unsatisfactory. For example, the Chinese Patent 200 510 125 267 (Publication No. CN1847233A) in
Discloses an intermediate 4-methyl benzofurazan preparation methods, the preparation process is as follows:

It is clear that the above method steps long, dangerous operation, poor control, and cause a lot of pollution.
Accordingly, there is an urgent need to develop new, efficient and simple isradipine important intermediates for preparing 4-methyl benzofurazan method.
[0019]

[0020] (b) in an inert solvent, such that 4-methyl benzofurazan oxide reduction to form 4-methyl benzofurazan, i.e. a compound of formula 3;
[0021]

[0028]

[0029] to form isradipine.

Example 7 β – amino crotonic acid isopropyl ester (Compound 8)
The isopropyl acetoacetate (72.0g, 0 5mol.), Ammonium acetate (57. 8g, 0 75mol.) And tert-butanol / ethanol (1: 1,600ml) were mixed in IOOOml flask, 300 mesh sieve (50g), heated to reflux, TLC plate monitor. After the reaction is substantially completed, cooled to room temperature, filtered and the filtrate was concentrated until no liquid was distilled off, the residual liquid was distilled under reduced pressure, to collect 110-120 ° C fraction (degree of vacuum of 0. IMPa) to give compound 8 (66. Og). Y = 92. 4%.
1H-WR (CDCl3):…… 5 02-4 95 (1H, m), 4 48 (1H, s), 1 88 (3H, d), 1 23-1 21 (6H , d)
Example 8 isradipine (Isradipine)
In the protection teams, three-necked flask Compound 6 (3.0g, 20mmol), i3- amino crotonic acid isopropyl ester (Compound 8) (2. 7g, 16. 6mmol), methyl acetoacetate (3. 50g, 30mol), Ac2O (2. 05g, 20mmol), conc. H2SO4 (0. 4g, 4mmol) and tert-butanol / ethanol (1: 1,65ml) mixing the liquid phase monitoring, when the remainder is less than 3 Compound 6 When the 7% to terminate the reaction. The reaction was concentrated, the residue was dissolved CH2Cl2 (55ml), washed with water (45ml X Magic, dried, concentrated, drain pump, to give 6. 7g end yellow foam-like solid. Ethanol QOml) dissolved by heating, stirring crystallization (overnight) to give a pale yellow powder isradipine (4. 3g) (HPLC purity> 99.8%, impurity content homologues thereof are less than 0.1), yield 66.8%.
1H-WR (CDCl3):…… 7 62-7 60 (lH, m), 7 31-7 26 (2H, m), 5 46 (lH, s), 4 92-4 . 86 (1H, m), 3. 57 (3H, s), 2. 32-2. 30 (6H, m), 1. 21-1. 19 (3H, d), 0. 95-0. 94 (3H, d)
Comparative Example 1
isradipine (Isradipine) prepared by the United States Patent US4466972:
In the protection teams, three-necked flask Compound 6 (3.0g, 20mmol), i3- amino crotonic acid isopropyl ester (Compound 8) (2. 7g, 16. 6mmol), methyl acetoacetate (3. 50g, 30mol), Ac2O (2. 05g, 20mmol), conc. H2SO4 (0. 4g, 4mmol) and ethanol (65ml) were mixed and stirred, the liquid phase monitoring, when the compound 6 is less than 3.7% remaining, the reaction is stopped. The reaction was concentrated, the residue was dissolved CH2Cl2 (55ml), washed with water (45ml X Magic, dried, concentrated, drain pump, to give 6. 3g end yellow foam-like solid. Ethanol OOml) was dissolved by heating, stirring crystallization (overnight) to give a pale yellow powder isradipine (4. Ig) (HPLC purity: 99.0%, impurity content was homologues greater than 0.3%), a yield of 63.7%.
Compared with Comparative Example 1 (homolog impurity content was greater than 0.3%), was the content of impurities isradipine homologs prepared in Example 1-8 is less than 0.1% by the embodiment of the present invention.
The 10 cases of isradipine
Example 8 was repeated, except that, with t-butanol / ethanol (1: 2,70ml) or t-butanol / ethanol O: 1, 70ml) replaces t-butanol / ethanol (1: 1,65ml) 0
The results showed that isradipine yield of about 62%, the test substance impurity content of less than 0.1% homologous.
Further reading and references
- “”Isradipine: Brands, Medical Use, Clinical Data””.
- “Isradipine Side Effects”.
- “”Isradipine and Anzemet Drug Interactions””.
- “”Isradipine and Onmel Drug Interactions””.
- “”Isradipine and Zanaflex Drug Interactions””.
- Hattori T, Wang P (2006). “Calcium antagonist isradipine-induced calcium influx through nonselective cation channels in human gingival fibroblasts.”. Eur J Med Res 11 (3): 93–6. PMID 16751108.
- Ganz M, Mokabberi R, Sica D (2005). “Comparison of blood pressure control with amlodipine and controlled-release isradipine: an open-label, drug substitution study.”. J Clin Hypertens (Greenwich) 7 (4 Suppl 1): 27–31. doi:10.1111/j.1524-6175.2005.04450.x. PMID 15858400.
- Johnson B, Roache J, Ait-Daoud N, Wallace C, Wells L, Dawes M, Wang Y (2005). “Effects of isradipine, a dihydropyridine-class calcium-channel antagonist, on d-methamphetamine’s subjective and reinforcing effects.”. Int J Neuropsychopharmacol 8 (2): 203–13. doi:10.1017/S1461145704005036. PMID 15850499.
- Fletcher H, Roberts G, Mullings A, Forrester T (1999). “An open trial comparing isradipine with hydralazine and methyl dopa in the treatment of patients with severe pre-eclampsia.”. J Obstet Gynaecol 19 (3): 235–8. doi:10.1080/01443619964977. PMID 15512286.
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ (2007). “‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease.”.Nature 447 (3): 1081–1086. doi:10.1038/nature05865. PMID 17558391.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
External links
- MedlinePlus DrugInfo medmaster-a693048
- DDB 30003
- Drug offers hope for Parkinson’s – BBC News, 11 June 2007.
- [1] – Commentary of Chan et al. publication
- [2] – ArsTechnica – Why neurons die in Parkinson’s patients
…………
CN1847233A21 Nov 200518 Oct 2006圣玛精细化工有限责任公司Method for preparing 4-formoxylbenzofuran
US446697219 Mar 198221 Aug 1984Sandoz Ltd.Hypotensive, antiischemic, antispasmodic agents
WO2005005437A115 Jul 200420 Jan 2005Radhakrishnan Selvar MullaiyurAn improved process for the manufacture of isradipine.
References: Dihydropyridine calcium channel blocker. Prepn: P. Neumann, DE 2949491; idem, US 4466972 (1980, 1984 both to Sandoz). Prepn of enantiomers: A. Vogel, DE 3320616 (1983 to Sandoz), C.A. 101, 7162s (1984). Comparative study of in vitro effects on human and canine cerebral arteries: E. Müller-Schweinitzer, P. Neumann, J. Cereb. Blood Flow Metab.3, 354 (1983). Effect on a-adrenoceptor mediated vasoconstriction in rats: K. Jie et al., Arch. Int. Pharmacodyn. 278, 72 (1985). Pharmacokinetics: F. L. S. Tee, J. M. Jaffe, Eur. J. Clin. Pharmacol. 32, 361 (1987). Clinical evaluation in angina and coronary artery disease: C. E. Handler, E. Sowton, ibid. 27, 415 (1984); in hypertension: E. B. Nelson et al., Clin. Pharmacol. Ther. 40, 694 (1986). Comparison of hemodynamic effects of enantiomers: R. P. Hof et al., J. Cardiovasc. Pharmacol. 8, 221 (1986). Series of articles on pharmacology and clinical use: Am. J. Med. 86, 1-146 (1989).
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 3-methyl 5-propan-2-yl 4-(2,1,3-benzoxadiazol-4-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate | |
| Clinical data | |
| Trade names | DynaCirc |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a693048 |
|
|
| Legal status | |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 15-24% |
| Protein binding | 95% |
| Metabolism | 100% Hepatic |
| Half-life | 8 hours |
| Excretion | 70% Renal, 30% Fecal |
| Identifiers | |
| CAS number | 75695-93-1 |
| ATC code | C08CA03 |
| PubChem | CID 3784 |
| DrugBank | DB00270 |
| ChemSpider | 3652 |
| UNII | YO1UK1S598 |
| KEGG | D00349 |
| ChEMBL | CHEMBL1648 |
| Chemical data | |
| Formula | C19H21N3O5 |
| Molecular mass | 371.387 g/mol |
…….
Alleppey kerala INDIA…..Alappuzha
Alappuzha – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Alappuzha
pronunciation (help·info)), also known as Alleppey, is the administrative headquarters of Alappuzha District of Kerala state of southern India. Alappuzha is the …


Table in restaurant after eating fish, Alleppey, Kerala, India, South Asia,
PAGODA RESORTS ALLEPPEY KERALA INDIA

////////////
CILNIDIPINE 西尼地平

Cilnidipine
西尼地平
CAS 132203-70-4
- (E) – (±) 1 ,4 a dihydro-2 ,6 – dimethyl-4 – (3 – nitrophenyl) -3,5 – pyridinedicarboxylic acid, 2 – methoxy- ethyl butylester 3 – phenyl – 2 – propenyl ester FRC-8653 Cinalong
- More FRC 8653 1,4-Dihydro-2 ,6-dimethyl-4-(3-nitrophenyl) 3 ,5-pyridinedicarboxylic acid 2-methoxyethyl (2E)-3-phenyl-2-propenyl ester
- Molecular formula:C 27 H 28 N 2 O 7
- Molecular Weight:492.52
CAS Name: 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 2-methoxyethyl (2E)-3-phenyl-2-propenyl ester
Additional Names: (±)-(E)-cinnamyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5-pyridinedicarboxylate
Cinnamyl 2-methoxyethyl 4-(3-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate
Manufacturers’ Codes: FRC-8653
Trademarks: Atelec (Morishita); Cinalong (Fujirebio); Siscard (Boehringer, Ing.)
Percent Composition: C 65.84%, H 5.73%, N 5.69%, O 22.74%
Properties: Crystals from methanol, mp 115.5-116.6°. LD50 in male, female mice, rats (mg/kg): ³5000, ³5000, ³5000, 4412 orally;³5000 all species s.c.; 1845, 2353, 441, 426 i.p. (Wada).
Melting point: mp 115.5-116.6°
Toxicity data: LD50 in male, female mice, rats (mg/kg): ³5000, ³5000, ³5000, 4412 orally; ³5000 all species s.c.; 1845, 2353, 441, 426 i.p. (Wada)
Ajinomoto (INNOVATOR)
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.

Cilnidipine (INN) is a calcium channel blocker. It is sold as Atelec in Japan, asCilaheart, Cilacar in India, and under various other trade names in East Asian countries.
Cilnidipine is a dual blocker of L-type voltage-gated calcium channels in vascular smooth muscle and N-type calcium channels in sympathetic nerve terminals that supply blood vessels. However, the clinical benefits of cilnidipine and underlying mechanisms are incompletely understood.
Clinidipine is the novel calcium antagonist accompanied with L-type and N-type calcium channel blocking function. It was jointly developed by Fuji Viscera Pharmaceutical Company, Japan and Ajinomoto, Japan and approved to come into market for the first time and used for high blood pressure treatment in 1995. in india j b chemicals & pharmaceuticals ltd and ncube pharmaceutical develope a market of cilnidipine.
Hypertension is one of the most common cardiovascular disease states, which is defined as a blood pressure greater than or equal to 140/90 mm Hg. Recently, patients with adult disease such as hypertension have rapidly increased. Particularly, since damages due to hypertension may cause acute heart disease or myocardial infarction, etc., there is continued demand for the development of more effective antihypertensive agent.
Meanwhile, antihypertensive agents developed so far can be classified into Angiotensin II Receptor Blocker (ARB), Angiotensin-Converting Enzyme Inhibitor (ACEI) or Calcium Chanel Blocker (CCB) according to the mechanism of actions. Particularly, ARB or CCB drugs manifest more excellent blood pressure lowering effect, and thus they are more frequently used.
However, these drugs have a limit in blood pressure lowering effects, and if each of these drugs is administered in an amount greater than or equal to a specific amount, various side-effects may be caused. Therefore, there have been many attempts in recent years to obtain more excellent blood pressure lowering effect by combination therapy or combined preparation which combines or mixes two or more drugs.
Particularly, since side-effect due to each drug is directly related to the amount or dose of a single drug, there have been active attempts to combine or mix two or more drugs thereby obtaining more excellent blood pressure lowering effect through synergism of the two or more drugs while reducing the amount or dose of each single drug.
For example, US 20040198789 discloses a pharmaceutical composition for lowering blood pressure combining lercanidipine, one of CCB, and valsartan, irbesartan or olmesartan, one of ARB, etc. In addition, a combined preparation composition which combines or mixes various blood pressure lowering drugs or combination therapy thereof has been disclosed.
cilnidipine Compared with other calcium antagonists, clinidipine can act on the N-type calcium-channel that existing sympathetic nerve end besides acting on L-type calcium-channel that similar to most of the calcium antagonists. Due to its N-type calcium-channel blocking properties, it has more advantages compared to conventional calcium-channel blockers. It has lower incidence of Pedal edema, one of the major adverse effects of other calcium channel blockers. Cilnidipine has similar blood pressure lowering efficacy as compared to amlodipine. One of the distinct property of cilnidipine from amlodipine is that it does not cause reflex tachycardia.
In recent years, cardiovascular disease has become common, the incidence increased year by year, about a patient of hypertension in China. 3-1. 500 million, complications caused by hypertension gradually increased, and more and more young patients with hypertension technology. In recent years, antihypertensive drugs also have great development, the main first-line diuretic drug decompression 3 – blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, ar blockers and vascular angiotensin II (Ang II) receptor antagonist.
In the anti-hypertensive drugs, calcium antagonists are following a – blockers after another rapidly developing cardiovascular drugs, has been widely used in clinical hypertension, angina and other diseases, in cardiovascular drugs in the world, ranked first.
Cilnidipine for the long duration of the calcium channel blockers, direct relaxation of vascular smooth muscle, dilation of peripheral arteries, the peripheral resistance decreased, with lower blood pressure, heart rate without causing a reflex effect.
Cilnidipine is a dihydropyridine CCB as well as an antihypertensive. Cilnidipinehas L- and N-calcium channel blocking actions. Though many of the dihydropyridine CCBs may cause an increase in heart rate while being effective for lowering blood pressure, it has been confirmed that cilnidipine does not increase the heart rate and has a stable hypotensive effect. (Takahiro Shiokoshi, “Medical Consultation & New Remedies” vol. 41, No. 6, p. 475-481)
- http://www.mcyy.com.cn/e-product2.asp
- Löhn M, Muzzulini U, Essin K, et al. (May 2002). “Cilnidipine is a novel slow-acting blocker of vascular L-type calcium channels that does not target protein kinase C”. J. Hypertens.20 (5): 885–93. PMID12011649.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
Cilnidipine (CAS NO.: 132203-70-4), with its systematic name of (+-)-(E)-Cinnamyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5-pyridinedicarboxylate, could be produced through many synthetic methods.
Following is one of the synthesis routes: By cyclization of 2-(3-nitrobenzylidene)acetocetic acid cinnamyl ester (I) with 2-aminocrotonic acid 2-methoxyethyl ester (II) by heating at 120 °C.
………………..
http://www.google.com/patents/EP0161877A2?cl=en
AN EXAMPLE
Example 1
-
3.51 g (10 mM) of 2-(3-nitrobenzylidene) acetoacetic acid cinnamyl ester were mixed with 1.38 g (12 mM) of 3-aminocrotonic acid methyl ester, and heated at 120°C for 3 hours. The reaction mixture was separated by silica gel column chromatography, and 3.00 g of cinnamyl methyl 4-(3-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (trans) were obtained (yield 67%). This derivative was recrystallized once from methanol.
-
Elemental Analysis; C25H24N206
- Calcd. (%) C: 66.95, H: 5.39, N: 6.25
- Found (%) C: 67.03, H: 5.31, N: 6.20
(trans)
-
- m.p.; 143.5-144.5°C
- IR (cm-1); vNH 3370, νCO 1700, νNO2 1530, 1350
- NMR δCDCl3; 2.34(s,6H), 3.60(s,3H), 4.69(d,2H), 5.13(s,lH), 6.14(tt,lH), 6.55(d,lH), 7.1-8.1(m,9H)
(cis)
-
- m.p.; 136-137°C
- IR (cm-1); vNH 3360, νCO 1700, 1650, νNO2 1530, 1350
- NMR δCDCl3; 2.30(s,6H), 3,60(s,3H), 4.80(d,lH), 5.10(s,1H), 5.77(tt,lH), 6.56(d,1H), 6.64(bs,1H), 7.1-8.1(m,9H)
EXAMPLE 13
-
Example 13 Cinnamyl 2-methoxyethyl 4-(3-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate
-
Elemental Analysis; C27H28N2O7
- Calcd. (%) C: 65.84, H: 5.73, N: 5.69
- Found (%) C: 65.88, H: 5.70, N: 5.66
- m.p.; 115.5-116.5°C
- IR (cm-1); vNH 3380, νCO 1710, 1680, νNO2 1530, 1350
- NMR δCDCl3; 2.34(s,6H), 3.25(s,3H), 3.50(t,2H), 4.15(t,2H), 4.68(d,2H), 5.15(s,lH), 5.9-6.9(m,3H), 7.1-8.2(m,9H)

cyclization of 2-(3-nitrobenzylidene)acetocetic acid cinnamyl ester (I) with 2-aminocrotonic acid 2-methoxyethyl ester (II) by heating at 120 C.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
MORE
NMR
CARBOHYDRATE POLYMERS 90 PG 1719-1724 , YR2012
Numerous peaks were found in the spectrum of cilnidipine: 2.3555 (3H, s, CH3), 2.3886(3H, s, CH3), 3.2843(CD3OD), 3.3292(3H, s, OCH3), 3.5255–3.5623(2H, m, CH3OCH2CH2 ), 4.1224–4.1597(2H, m, CH3OCH2CH2 ), 4.6695–4.7293(2H, m, CH2 CH CH ), 4.8844(D2O), 5.1576(1H, s, CH), 6.2609(1H, dt, CH2 CH CH ), 6.5518(1H, d, CH2 CH CH ), 7.2488–7.3657(6H, m, ArH), 7.7002(1H, dd, ArH), 7.9805(1H, dd, ArH), 8.1548(1H, s, ArH)
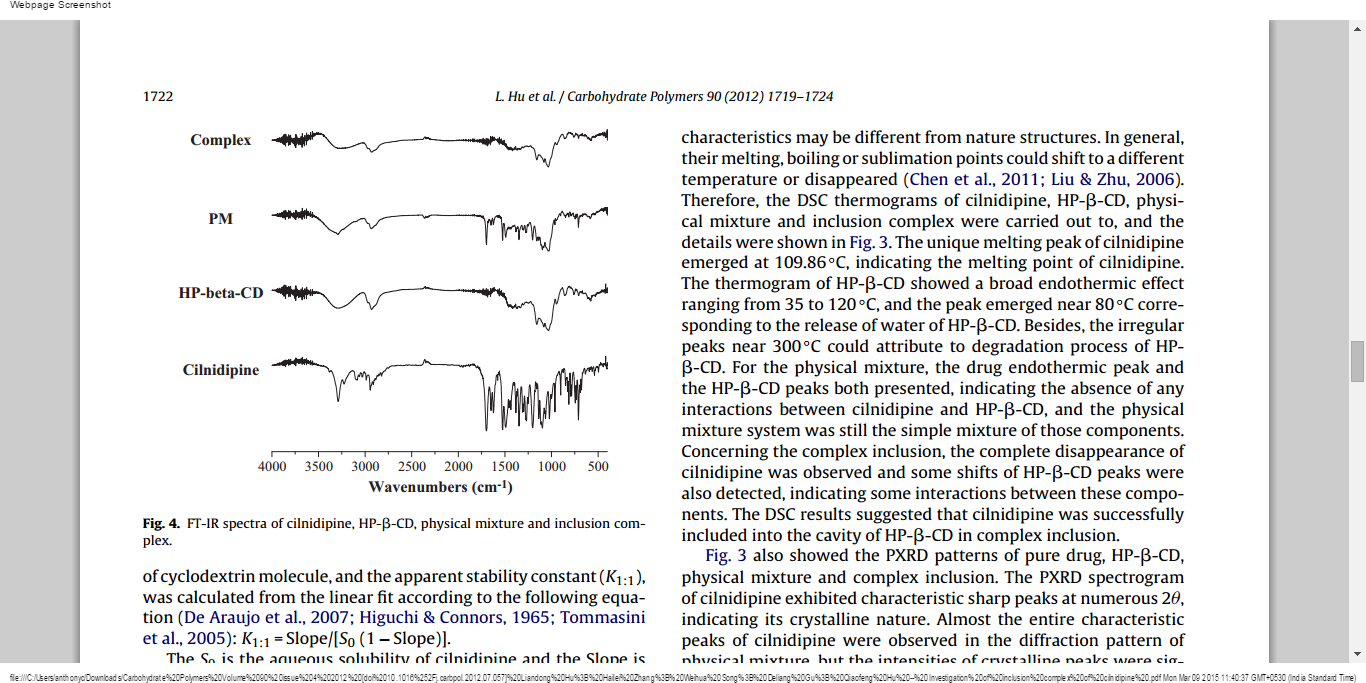
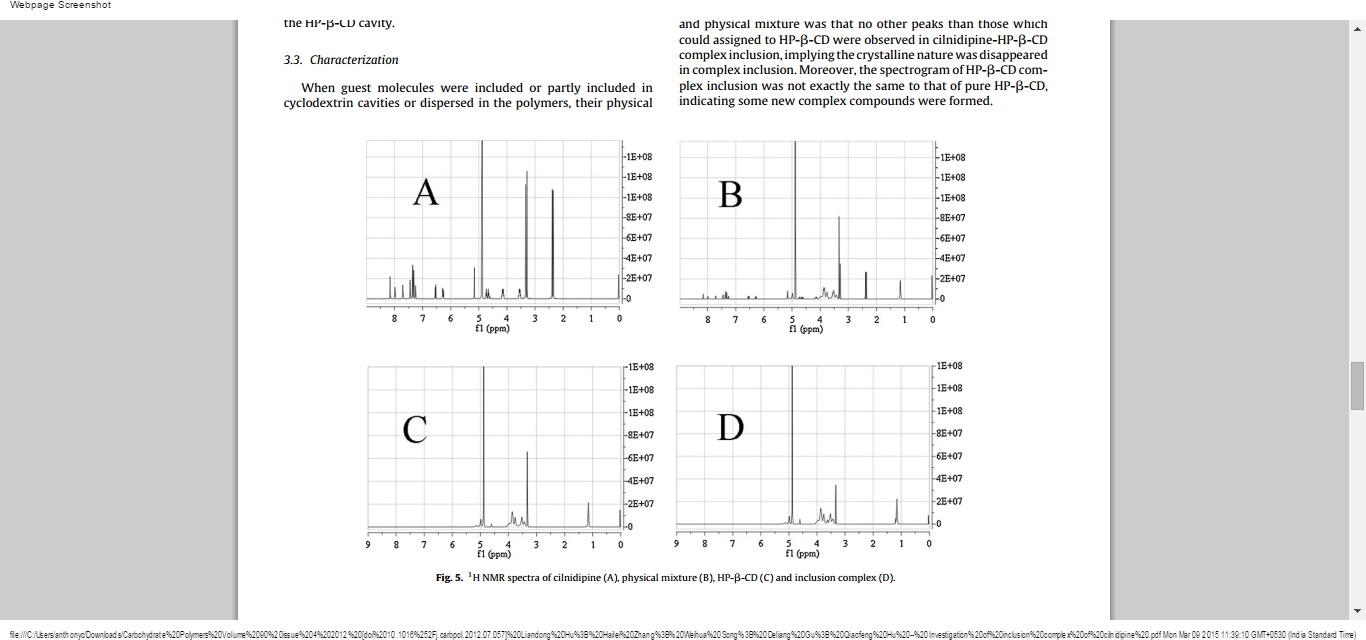
References:
Dihydropyridine calcium channel blocker. Prepn: T. Kutsuma et al., EP 161877; eidem, US 4672068(1985, 1987 both to Fujirebio).
Pharmacology: K. Ikeda et al., Oyo Yakuri 44, 433 (1992).
Mechanism of action study: M. Hosonoet al., J. Pharmacobio-Dyn. 15, 547 (1992).
LC-MS determn in plasma: K. Hatada et al., J. Chromatogr. 583, 116 (1992). Clinical study: M. Ishii, Jpn. Pharmacol. Ther. 21, 59 (1993).
Acute toxicity study: S. Wada et al., Yakuri to Chiryo 20, Suppl. 7, S1683 (1992), C.A. 118, 32711 (1992).
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
U.S Patent No. 4,572,909 discloses amlodipine;
U.S Patent No. 4,446,325 discloses aranidipine;
U.S Patent No. 4,772,596 discloses azelnidipine;
U.S Patent No. 4,220,649 discloses barnidipine;
U.S Patent No. 4,448,964 discloses benidipine;
U.S Patent No. 5,856,346 discloses clevidipine;
U.S Patent No. 4,466,972 discloses isradipine;
U.S Patent No. 4,885,284 discloses efonidipine; and
U.S Patent No. 4,264,61 1 discloses felodipine.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
-
Planar chemical structures of these calcium blockers of formula (I) are shown below.









-
Amlodipine is 2-(2-aminoethoxymethyl)-4-(2-chlorophenyl)-3-ethoxycarbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine disclosed in USP 4,572,909, Japanese patent publication No. Sho 58-167569 and the like.
-
Aranidipine is 3-(2-oxopropoxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(2-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,446,325 and the like.
-
Azelnidipine is 2-amino-3-(1-diphenylmethyl-3-azetidinyloxycarbonyl)-5-isopropoxycarbonyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,772,596, Japanese patent publication No. Sho 63-253082 and the like.
-
Barnidipine is 3-(1-benzyl-3-pyrrolidinyloxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,220,649, Japanese patent publication No. Sho 55-301 and the like.
-
Benidipine is 3-(1-benzyl-3-piperidinyloxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine and is described in the specifications of U.S. Patent No. 4,501,748, Japanese patent publication No. Sho 59-70667 and the like.
-
Cilnidipine is 2,6-dimethyl-5-(2-methoxyethoxycarbonyl)-4-(3-nitrophenyl)-3-(3-phenyl-2-propenyloxycarbonyl)-1,4-dihydropyridine disclosed in USP 4,672,068, Japanese patent publication No. Sho 60-233058 and the like.
-
Efonidipine is 3-[2-(N-benzyl-N-phenylamino)ethoxycarbonyl]-2,6-dimethyl-5-(5,5-dimethyl-1,3,2-dioxa-2-phosphonyl)-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,885,284, Japanese patent publication No. Sho 60-69089 and the like.
-
Elgodipine is 2,6-dimethyl-5-isopropoxycarbonyl-4-(2,3-methylenedioxyphenyl)-3-[2-[N-methyl-N-(4-fluorophenylmethyl)amino]ethoxycarbonyl]-1,4-dihydropyridine disclosed in USP 4,952,592, Japanese patent publication No. Hei 1-294675 and the like.
-
Felodipine is 3-ethoxycarbonyl-4-(2,3-dichlorophenyl)-2,6-dimethyl-5-methoxycarbonyl-1,4-dihydropyridine disclosed in USP 4,264,611, Japanese patent publication No. Sho 55-9083 and the like.
-
Falnidipine is 2,6-dimethyl-5-methoxycarbonyl-4-(2-nitrophenyl)-3-(2-tetrahydrofurylmethoxycarbonyl)-1,4-dihydropyridine disclosed in USP 4,656,181, Japanese patent publication (kohyo) No. Sho 60-500255 and the like.
-
Lemildipine is 2-carbamoyloxymethyl-4-(2,3-dichlorophenyl)-3-isopropoxycarbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine disclosed in Japanese patent publication No. Sho 59-152373 and the like.
-
Manidipine is 2,6-dimethyl-3-[2-(4-diphenylmethyl-1-piperazinyl)ethoxycarbonyl]-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,892,875, Japanese patent publication No. Sho 58-201765 and the like.
-
Nicardipine is 2,6-dimethyl-3-[2-(N-benzyl-N-methylamino)ethoxycarbonyl]-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,985,758, Japanese patent publication No. Sho 49-108082 and the like.
-
Nifedipine is 2,6-dimethyl-3,5-dimethoxycarbonyl-4-(2-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,485,847 and the like.
-
Nilvadipine is 2-cyano-5-isopropoxycarbonyl-3-methoxycarbonyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,338,322, Japanese patent publication No. Sho 52-5777 and the like.
-
Nisoldipine is 2,6-dimethyl-3-isobutoxycarbonyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,154,839, Japanese patent publication No. Sho 52-59161 and the like.
-
Nitrendipine is 3-ethoxycarbonyl-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,799,934, Japanese patent publication (after examination) No. Sho 55-27054 and the like.
-
Pranidipine is 2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-3-(3-phenyl-2-propen-1 -yloxycarbonyl)-1,4-dihydropyridine disclosed in USP 5,034,395, Japanese patent publication No. Sho 60-120861 and the like.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
MAHABALIPURAM, INDIA
Mahabalipuram – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Mahabalipuram
Mahabalipuram, also known as Mamallapuram is a town in Kancheepuram district in the Indian state of Tamil Nadu. It is around 60 km south from the city of …Shore Temple – Seven Pagodas – Pancha Rathas –
.
.



Krishna’s Butter Ball in Mahabalipuram, India. The surface below the rock is …




http://www.weather-forecast.com/locations/Mamallapuram
 Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
 Moonraikers Restaurant, Mamallapuram
Moonraikers Restaurant, Mamallapuram


Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
.
A carving at the Varaha Temple, Mahabalipuram

/////////////
AMPRENAVIR For the treatment of HIV-1 infection in combination with other antiretroviral agents.

Amprenavir
KVX-478, 141W94, VX-478,
(3S)-Tetrahydro-3-furanyl ((1S,2R)-3-(((4-aminophenyl)sulfonyl)(2-methylpropyl)amino)-2-hydroxy-1-(phenylmethyl)propyl)carbamate
(3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate
CAS NO. 161814-49-9, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate
| 161814-49-9 | |
| Weight | 505.224656557 |
|---|---|
| Chemical Formula | C25H35N3O6S |
| Amprenavir is a protease inhibitor used to treat HIV infection. |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.
| Amprenavir is a protease inhibitor with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Protease inhibitors block the part of HIV called protease. HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Amprenavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs. |

| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 5585397 | 1993-12-17 | 2013-12-17 |
| United States | 6730679 | 1997-11-11 | 2017-11-11 |
Background
Research aimed at development of renin inhibitors as potential antihypertensive agents had led to the discovery of compounds that blocked the action of this peptide cleaving enzyme. The amino acid sequence cleaved by renin was found to be fortuitously the same as that required to produce the HIV peptide coat. Structure–activity studies on renin inhibitors proved to be of great value for developing HIV protease inhibitors. Incorporation of an amino alcohol moiety proved crucial to inhibitory activity for many of these agents. This unit is closely related to the one found in the statine, an unusual amino acid that forms part of the pepstatin, a fermentation product that inhibits protease enzymes.
Synthesis
R.D. Tung, M.A. Murcko, G.R. Bhisetti, U.S. Patent 5,558,397 (1996). The scheme shown here is partly based on that used to prepare darunavir and fosamprenavir due to difficulty in deciphering the patent.
AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus (HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
 |
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
………………………………
paper
Org. Biomol. Chem., 2004,2, 2061-2070
DOI: 10.1039/B404071F
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f#!divAbstract

Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.

Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
……………
The reaction of N,N-dibenzyl-L-alaninal (I) with nitromethane, catalyzed by the chiral ammonium salt (II) and KF in THF gives the chiral nitroalcohol (III), which is reduced with NiCl2 and NaBH4 to yield the aminoalcohol (IV). The condensation of (IV) with isobutyraldehyde (V) affords the Schiff base (VI), which is reduced with NaBH4 to provide the secondary amine (VII). The reaction of (VII) with 4-nitrobenzenesulfonyl chloride (VIII) and TEA in dichloromethane furnishes the sulfonamide (IX), which is deprotected by hydrogenation with H2 over Pd/C in methanol, giving the diamino compound (X). Finally, this compound is condensed with 3(S)-tetrahydrofuryl (N-oxysuccinimidyl) carbonate (XI) by means of TEA in dichloromethane to afford the target carbamate.
Angew Chem. Int Ed Engl1999,38,(13-14):1931
……………………………………………………….

The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target compound.
EP 0659181; EP 0885887; JP 1996501299; US 5585397; WO 9405639
……………………………………………………………
Patent
https://www.google.com/patents/WO1999048885A1?cl=ensynthesis of (3S)-tetrahydro-3-furyl N-[(1S,2R)-3(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl]carbamate, hereinafter referred to as the compound of formula (I), and to novel intermediates thereto.The compound of formula (I) has the following structure

and was first described in PCT patent publication number WO94/05639 at Example 168. Currently there is considerable interest in the compound of formula (I) as a new chemotherapeutic compound in the treatment of human immunodeficiency virus (HIV) infection and the associated conditions such as acquired immune deficiency syndrome (AIDS) and AIDS dementia.
There exists at the present time a need to produce large quantities of the compound of formula (I) for clinical investigation into the efficacy and safety of the compound as a chemotherapeutic agent in the treatment of HIV infections.
An ideal route for the synthesis of the compound should produce the compound of formula (I) in high yields at a reasonable speed and at low cost with minimum waste materials and in a manner that is of minimum impact to the environment in terms of disposing of waste-materials and energy consumption.
We have found a new process for the synthesis of the compound of formula (I) with many advantages over previously known routes of synthesis. Such advantages include lower cost, less waste and more efficient use of materials. The new process enables advantageous preparation of the compound of formula (I) on a manufacturing scale.
The route of synthesis of the compound of formula (I) described in the specification of WO94/05639 is specifically described therein in examples 39A, 51A, 51B, 51C, 51D, 167 and 168. The overall yield from these examples is 33.2% of theory.
Generally the route described in WO94/05639 involves protecting the amino alcohol of formula (A) (Ex.39)

wherein P is a protecting group to form a compound of formula (B);

wherein P and P′ are each independently a protecting group;
deprotecting the compound of formula (B) to form a compound of formula (C) (Ex 51A);

wherein P′ is a protecting group;
forming a hydrochloride salt of compound (C) (Ex 51B) then reacting with N-imidazolyl-(S)-tetrahydrofuryl carbamate to form the compound of formula (D) (Ex 51C);

wherein P′ is a protecting group;
deprotecting the compound of formula (D) (Ex 51D) wherein P′ is a protecting group to form the compound of formula (D) wherein P′ is H (Ex 51E); and coupling the resultant secondary amine on the compound of formula (D) to a p-nitrophenylsulphonyl group to form a compound of formula (E) (Ex 167);

the resultant compound of formula (E) is then reduced to form the compound of formula (I) (Ex 168).
In summary, the process disclosed in WO94/05639 for producing the compound of formula (I) from the compound of formula (A) comprises 6 distinct stages:
1) protecting,
2) deprotecting,
3) reacting the resultant compound with an activated tetrahydrofuranol group,
4) deprotecting,
5) coupling with a p-nitrophenylsulfonyl group, and
6) reducing the resultant compound to form a compound of formula (I).
Applicants have now found a process by which the compound of formula (I) may be prepared on a manufacturing scale from the same starting intermediate, the compound of formula (A), in only 4 distinct stages instead of 6. In addition to the associated benefits of fewer stages, such as savings in time and cost, the improved process reduces the number of waste products formed. Furthermore, product may be obtained in a higher yield, of approximately 50% of theory
 .
.
EXAMPLESExample 1
(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(isobutylamino)propyl]carbamate (127.77 g, 379.7 mmol) was heated in toluene (888 ml) to 80° C. and triethylamine (42.6 g, 417.8 mmol) added. The mixture was heated to 90° C. and a solution of p-nitrobenzene sulphonyl chloride (94.3 g, 425.4 mmol) in toluene (250 ml) was added over 30 minutes then stirred for a further 2 hours. The resultant solution of the nosylated intermediate {(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(N-isobutyl- 4-nitrobenzenesulphonamido)propyl]carbamate } was then cooled to 80° C. The solution was maintained at approximately 80° C., and concentrated hydrochloric acid (31.4 ml, 376.8 mmol) was added over 20 minutes. The mixture was heated to reflux (approx 86° C.) and maintained at this temperature for an hour then a further quantity of concentrated hydrochloric acid (26.4 ml, 316.8 mmol) was added. Solvent (water and toluene mixture) was removed from the reaction mixture by azeotropic distillation (total volume of solvent removed approx 600 ml), and the resultant suspension was cooled to 70-75° C. Denatured ethanol (600 ml) was added, and the solution was cooled to 20° C. The mixture was further cooled to approximately −10° C. and the precipitate formed was isolated by filtration, washed with denatured ethanol (50 ml) and dried at approximately 50° C., under vacuum, for approximately 12 hours, to give (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (160 g; 73% of theory yield corrected for assay). NMR: 1H NMR (300Mhz, dmso-d6): 8.37(2H, d, J=9 Hz), 8.16(NH3 +s), 8.06(2H, d, J=9 Hz), 7.31(5H, m), 5.65(1H, d, J=5 Hz), 3.95(1H, m), 3.39(2H, m), 2.95(5H, m), 1.90(1H, m), 0.77(6H, dd, J=21 Hz and 6 Hz).
1,1′-carbonyidiimidazole (27.66 kg, 170.58 mol) was added to ethyl acetate (314.3 kg) with stirring to give 3-(S)-tetrahydrofuryl imidazole-1-carboxylate. (S)-3-hydroxytetrahydrofuran (157 kg, 178.19 mol) was added over 30 minutes, washed in with ethyl acetate (9.95 kg), then the mixture was stirred for a further hour. (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (65.08 kg, 142.10 mol) was added and the mixture heated to reflux for approximately 22 hours. The solution was cooled slightly, and denatured ethanol (98 l) was added. The solution was stirred at 60° C. for 10 minutes then cooled and the product allowed to crystallise. The mixture was cooled to <10° C. and stirred for 2 hours. The product was isolated by filtration, washed with denatured ethanol (33 l) and dried at approximately 50° C., under vacuum to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-1-benzyl-2-hydroxy-3-(N-isobutyl-4-nitrobenzene sulphonamido)propyl]carbamate in a yield of 82% of theory.
NMR: 1H NMR (500 Mhz, dmso-d6): 8.38(2H, d, J=9Hz), 8.06(2H, d, J=9 Hz), 7.20(6H, m), 5.02(1H, d, J=5 Hz), 4.94(1H, m), 4.35(EtOH, broad s), 3.71(EtOH, q), 3.65(1H, m), 3.60(1H, m), 3.51(2H, broad m), 3.40(2H, m), 3.15(1H, dd, J=8 Hz and 14 Hz), 3.07(1H, dd, J=8 Hz and 15 Hz), 2.94(2H, m), 2.48(1H, m), 2.06(1H, m), 1.97(1H, m), 1.78(1H, m), 1.05(EtOH, t), 0.83(6H, dd, J=7 Hz and 16 Hz).
Product from the above stage (80.0 g, 149.4 mmol) was hydrogenated in isopropanol (880 ml) with 5% palladium on carbon (16 g, of a wet paste) and hydrogen pressure (approx 0.5 to 1.5 bar) at 25-50° C. for approximately 5 hours. The mixture was cooled and the catalyst removed by filtration. The solution was distilled to a volume of approximately 320 ml and water (80 ml) was added. This solution was divided into two for the crystallisation step.
To half of the above solution, decolourising charcoal (2 g) was added, the mixture stirred at approximately 32° C. for 4 hours, then filtered. The filtercake was washed with isopropanol (20 ml) then further water (40 ml) was added to the filtrate. The solution was seeded to induce crystallisation and stirred for 5 hours. Water (130 ml) was added slowly over 1 hour then the mixture was stirred for 4 hours. The resultant slurry was cooled to approximately 20° C. and the product was isolated by filtration and washed with a 1:4 mixture of isopropano/water (120 ml). The product was dried at approximately 50° C., under vacuum, for approximately 12 hours to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate (30.3 g; 80% of theory yield).
NMR: 1H NMR (300 Mhz, dmso-d6): 7.39(2H, d, J=9 Hz), 7.18(6H, m), 6.60(2H, d, J=9 Hz), 6.00(2H, s), 4.99(1H, d, J=6 Hz), 4.93(1H, ddt), 3.64(5H, m), 3.34(1H, m), 3.28(1H, dd, J=14 Hz and 3 Hz), 3.01(1H, m, J=14 Hz and 3 Hz), 2.91(1H, m), 2.66(2H, m), 2.50(1H, m), 2.05(1H, m), 1.94(1H, m), 1.78(1H, m), 0.81(6H, dd, J=16 Hz and 7 Hz). m/z: 506.2(M+H+)
…………………………
PATENT
Example 11Synthesis of Amprenavir (1)To a solution of carbamate nitro derivative 15 (0.05 g, 0.09 mmol) in 2 mL of EtOAc was added SnCl2.2H2O (0.1 g, 0.5 mmol) at 70° C. The reaction mixture was heated for 1 h until starting material disappeared and the solution cooled to room temperature. It was then poured into saturated aq. NaHCO3 solution and extracted with EtOAc. The organic extract was dried over anhyd. Na2SO4 and concentrated under reduced pressure. It was purified over chromatography using petroleum ether:EtOAc (3:2) to give amprenavir 1 (0.04 g, 90%).IR: (CHCl3, cm−1): υmax 757, 1090, 1149, 1316, 1504, 1597, 1633, 1705, 2960, 3371; 1H NMR (200 MHz, CDC3): δ 0.86 (d, J=5.7 Hz, 3H), 0.90 (d, J=6.6 Hz, 3H), 1.78-2.21 (m, 3H), 235-3.11 (m, 6H), 3.58-4.11 (m, 7H), 4.25 (s, 2H), 5.01 (br s, 1H), 5.07 (br s, 1H), 6.65 (d, J=8.4 Hz, 2H), 7.20-7.28 (m, 5H), 7.51 (d, J=8.4 Hz, 2H); 13C NMR (50 MHz, CDC3): δ 19.9, 20.2, 27.3, 32.8, 35.4, 35.7, 53.8, 55.0, 58.6, 66.8, 72.6, 73.2, 75.3, 114.0, 125.9, 126.5, 1280.4, 129.5, 137.7, 150.9, 155.9;
Anal. Calcd for C25H35N3O6S: C, 59.39; H, 6.98; N, 8.31; S, 6.34. Found: C, 59.36; H, 6.81; N, 8.25; S, 6.29%.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
……………………..
NMR PREDICTIONS
1H NMR
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-1h.png)
13 C NMR
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-23/000/115/669/161814-49-9-13c.png)
COSY PREDICTION
See also
- Fosamprenavir, a prodrug of amprenavir
External links
- Amprenavir bound to proteins in the PDB
- Shen, C. H.; Wang, Y. F.; Kovalevsky, A. Y.; Harrison, R. W.; Weber, I. T. (2010). “Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters”. FEBS Journal 277 (18): 3699–3714. doi:10.1111/j.1742-4658.2010.07771.x. PMC 2975871. PMID 20695887.
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| Clinical data | |
| Trade names | Agenerase |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699051 |
| Licence data | EMA:Link, US FDA:link |
|
|
| Legal status |
?
|
| Routes | oral |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | hepatic |
| Half-life | 7.1-10.6 hours |
| Excretion | <3% renal |
| Identifiers | |
| CAS number | 161814-49-9 |
| ATC code | J05AE05 |
| PubChem | CID 65016 |
| DrugBank | DB00701 |
| ChemSpider | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| ChEBI | CHEBI:40050 |
| ChEMBL | CHEMBL116 |
| NIAID ChemDB | 006080 |
| Chemical data | |
| Formula | C25H35N3O6S |
| Molecular mass | 505.628 g/mol |
SILODOSIN………For treatment of benign prostatic hypertophy

SILODOSIN
Urief, 160970-54-7, Rapaflo, KMD 3213, Silodyx, KAD 3213, KMD-3213
Molecular Formula: C25H32F3N3O4
Molecular Weight: 495.53449 g/mol
Alpha 1A adrenoceptor antagonist
Prostate hyperplasia
Kissei Pharmaceutical Co Ltd INOVATOR
CAS 160970-54-7
2,3-Dihydro-1-(3-hydroxypropyl)-5-[(2R)-2-[[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl]amino]propyl]-1H-indole-7-carboxamide
160970-64-9 (racemate)
169107-04-4 (diHBr)
Properties: [a]D25 -14.0° (c = 1.01 in methanol).
Optical Rotation: [a]D25 -14.0° (c = 1.01 in methanol)
Therap-Cat: In treatment of benign prostatic hypertophy.
a-Adrenergic Blocker.
In February 2008, the FDA accepted for review an NDA for silodosin for the treatment of dysuria associated with BPH . In October 2008, the FDA approved the drug . In April 2009, Actavis launched silodosin for the treatment of the signs and symptoms of BPH .

1-(3-hydroxypropyl)-5-[(2R)-2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino]propyl]-2,3-dihydroindole-7-carboxamide
Kissei Pharmaceutical, Daiichi Sankyo (formerly Daiichi Seiyaku), Actavis (formerly Watson) and Recordati have developed and launched silodosin (Urief; Trupass; Rapaflo; Thrupas; Silodyx; Urorec; KMD-3213; Youlifu), an oral alpha 1A adrenoceptor antagonist selective for prostatic receptors . The product is comarketed in Europe by several licensees. The drug is indicated for the treatment of the signs and symptoms of benign prostatic hyperplasia (BPH).
Silodosin, a highly selective alpha1A-adrenoceptor antagonist, was launched in May 2006 in Japan for the oral treatment of urinary disturbance associated with benign prostatic hyperplasia (BPH). The product was launched in the U.S. for the treatment of signs and symptoms of benign prostatic hyperplasia in 2009. In 2009, a positive opinion was received in the E.U. for this indication and final approval was obtained in 2010. Launch in the E.U. took place the same year.
In May 2006, silodosin was launched as a capsule formulation in Japan. Actavis launched the drug in the US in April 2009. In June 2010, EU launched began, initially with Germany ; in November 2010, the drug was launched in France; by December 2010, the drug was launched in Spain.
In 2001, Kissei established an agreement with Daiichi Pharmaceutical to codevelop and comarket silodosin. An oral, once-daily formulation of silodosin filed in the U.S. by Watson (now Actavis) was approved in 2008. Watson (now Actavis) obtained exclusive rights in 2004 to develop and market the drug in the U.S.
PRODUCT Was developed and launched byKissei Pharmaceutical, Daiichi Sankyo, Actavis and Recordati. Family members of the product case EP0600675 have SPC protection in most EU states until 2018; while its Orange Book listed equivalent, US5387603, expire in the US in 2018 with US156 extension.
Silodosin (trade names Rapaflo (USA), Silodyx (Europe and South Africa), Rapilif (India), Silodal (India), Urief (Japan), Urorec (Russia)) is a medication for the symptomatic treatment of benign prostatic hyperplasia. It acts as an α1–adrenoceptor antagonist with high uroselectivity (selectivity for the prostate).
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-(3-hydroxypropyl)-5-[(2R)-({2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl}amino)propyl]indoline-7-carboxamide | |
| Clinical data | |
|
|
| Legal status |
|
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 32% |
| Protein binding | 97% |
| Metabolism | Hepatic glucuronidation (UGT2B7-mediated); also minor CYP3A4 involvement |
| Half-life | 13±8 hours |
| Excretion | Renal and fecal |
| Identifiers | |
| CAS number | 160970-54-7 |
| ATC code | G04CA04 |
| PubChem | CID 5312125 |
| IUPHAR ligand | 493 |
| ChemSpider | 4471557 |
| UNII | CUZ39LUY82 |
| ChEMBL | CHEMBL24778 |
| Synonyms | KAD-3213, KMD-3213 |
| Chemical data | |
| Formula | C25H32F3N3O4 |
| Molecular mass | 495.534 g/mol |
History
Silodosin received its first marketing approval in Japan in May 2006 under the tradename Urief, which is jointly marketed by Kissei Pharmaceutical Co., Ltd. and Daiichi Sankyo Pharmaceutical Co., Ltd.
Kissei licensed the US, Canadian, and Mexican rights for silodosin to Watson Pharmaceuticals, Inc. in 2004.
On February 12, 2008, Watson announced that the New Drug Application submitted to the United States Food and Drug Administration for silodosin has been accepted for filing. FDA approved this drug on October 9, 2008.[1] Silodosin is marketed under the trade names Rapaflo in the US and Silodyx in Europe.[2] and Rapilif in India (Ipca Urosciences)
Pharmacology
Since silodosin has high affinity for the α1A adrenergic receptor, it causes practically no orthostatic hypotension (in contrast to other α1 blockers). On the other side, the high selectivity seems to cause more problems with ejaculation.[3]
As α1A adrenoceptor antagonists are being investigated as a means to male birth control due to their ability to inhibit ejaculation but not orgasm, a trial with 15 male volunteers was conducted. While silodosin was completely efficacious in preventing the release of semen in all subjects, 12 out of the 15 patients reported mild discomfort upon orgasm. The men also reported the psychosexual side effect of being strongly dissatisfied by their lack of ejaculation.[4]

///////////////////////////////
CN 103848772
http://www.google.com/patents/CN103848772A?cl=en
silodosin (Silodosin) is 〃 2 Japanese orange Johnson invented – receptor antagonist, for the treatment of benign prostatic hyperplasia or hypertrophy, and other related symptoms. Clinical trials showed that 25% of patients with benign prostatic hyperplasia need for drugs or surgery. Although prostatectomy is better, the mortality rate is not high, but patients bring varying degrees of damage. So look for an effective and safe non-surgical treatment, not only can control the further development of the disease, while relieving the symptoms of the patient.
benign prostatic hyperplasia in older male patients have a higher prevalence, and clinical alternative drugs rarely, so the development of a benign prostatic hyperplasia drug treatment, not only has good social benefits, but also to bring good economic benefits. The study confirmed that silodosin is the treatment of benign prostatic hyperplasia in an important class of drugs.

Currently, the research reported in the published literature on the preparation of compounds of silodosin, are:


Early 1995, Kitazawa M et al patent US5387603, the reporter silodosin total synthesis method, but the method reaction step is long, the yield is not too high, not suitable for our industrial production.

In 2009, 翟富民 et al patent CN102115455A, which reported a method for preparing Sailuoduoxin key intermediates. The appropriate method for improving existing methods, although shorter than the previous method step in the step, but low synthesis yield of the process, we can not meet the needs of industrial production.
In summary, the compounds prepared silodosin more synthetic methods are constantly improved, but there are still a lot of flaws. Therefore, there is need for further research on the preparation of compounds of silodosin to get simple process, product yield, product easy separation of the new preparation. SUMMARY
The present invention is to overcome the above problems of the prior art, there is provided a method for preparing important intermediates silodosin, the present invention is simple process, high yield, easy separation of the product, the method suitable for industrial production .
To achieve the above technical object, to achieve the above technical result, the present invention is realized by the following technical scheme:
One kind of silodosin preparation of important intermediate, comprising the steps of:
Step I) in a flask, 282g of raw materials 1-acetyl-5- (2-bromo-propyl) indoline, 222g phthalimide potassium salt and 700mL DMF, was heated at 110 ° C for 2h; After completion of the reaction, to which was added the right amount of water to wash away the excess solvent DMF and salt extraction desolventizing after EA, was 296g crude;
Step 2) In a flask was added 296g crude product obtained in step I, dissolved 800mL ethanol, was added 165mL of hydrazine hydrate, 50 ° C is heated to precipitate a white solid; After completion of the reaction, cooling suction filtered, the filter cake washed with ethanol, and then the mother liquor removing solvent under reduced pressure; After dissolving EA, washed with water to wash away the excess hydrazine, and finally the organic phase the solvent was removed to give 165g intermediate, i.e. 1-acetyl-5- (2-aminopropyl) indoline;
Step 3) In the three-necked flask, 165g of Intermediate 1-acetyl-5- (2-aminopropyl) indoline, dissolved 600mL methanol, stirred at room temperature, and thereto was slowly added dropwise bromine; the addition was complete After stirring at room temperature 5-6h; After completion of the reaction, slowly poured into saturated NaHSO3, and wash away excess bromine; extracted with ethyl acetate, washed with water and saturated brine, dried over anhydrous sodium sulfate; After filtration, the solvent was removed in vacuo to give the crude product recrystallized from toluene to give 177g pure product;
Step 4) In a flask was added 177g of pure product obtained in Step 3 and 65g CuCN, after use 700mL DMF, was heated at 110 ° C for 3 to 5h; After completion of the reaction, the amount of water was added thereto, washing off excess solvent DMF and salt, EA desolventizing crude extract, after recrystallization 121g pure 1-acetyl-5- (2-bromo-propyl) -7-cyano-indoline that silodosin important intermediates;
Step (1), (2), (3), (4) synthesis reaction is:

Further, the step I) to step 4) by TLC plate tracking point detection reaction.
The beneficial effects of the present invention are:
Preparation silodosin important intermediate of the present invention, mention of the method is simple, high reaction yield, product easily separated, suitable for industrial production and so on.
Preparation Method A silodosin important intermediate, comprising the following steps: Step I) in a flask, 282g of raw materials 1-acetyl-5- (2-bromo-propyl) indoline, 222g o phthalimide potassium and 700mL DMF, heated at 110 ° C for 2h; After completion of the reaction, to which was added the right amount of water to wash away the excess solvent DMF and salt extraction desolventizing after EA, was 296g crude;
Step 2) In a flask was added 296g crude product obtained in step I, dissolved 800mL ethanol, was added 165mL of hydrazine hydrate, 50 ° C is heated to precipitate a white solid; After completion of the reaction, cooling suction filtered, the filter cake washed with ethanol, and then the mother liquor removing solvent under reduced pressure; After dissolving EA, washed with water to wash away the excess hydrazine, and finally the organic phase the solvent was removed to give 165g intermediate, i.e. 1-acetyl-5- (2-aminopropyl) indoline;
Step 3) In the three-necked flask, 165g of Intermediate 1-acetyl-5- (2-aminopropyl) indoline, dissolved 600mL methanol, stirred at room temperature, and thereto was slowly added dropwise bromine; the addition was complete After stirring at room temperature 5-6h; After completion of the reaction, slowly poured into saturated NaHSO3, and wash away excess bromine; extracted with ethyl acetate, washed with water and saturated brine, dried over anhydrous sodium sulfate; After filtration, the solvent was removed in vacuo to give the crude product recrystallized from toluene to give 177g pure product;
Step 4) In a flask was added 177g of pure product obtained in Step 3 and 65g CuCN, after use 700mL DMF, was heated at 110 ° C for 3 to 5h; After completion of the reaction, the amount of water was added thereto, washing off excess solvent DMF and salt, EA desolventizing crude extract, after recrystallization 121g pure 1-acetyl-5- (2-bromo-propyl) -7-cyano-indoline that silodosin important intermediates;
Step (1), (2), (3), (4) synthesis reaction is:

Further, the step I) to step 4) by TLC plate tracking point detection reaction.
…………………………………………………
WO2013056842
http://www.google.com/patents/WO2013056842A1?cl=en
Silodosin is commercially available under the tradenames RAPAFLO® or
UROPvEC as a capsule formulation for oral use containing 4 mg or 8 mg of the drug. The capsules are to be taken orally once daily for the treatment of the signs and symptoms of benign prostatic hyperplasia. US 5,387,603 and EP 0 600 675 disclose silodosin as a therapeutic agent for the treatment for dysurea associated with benign prostatic hyperplasia. The molecular structure of silodosin (XXV) is shown below.
(XXV)

The synthesis of silodosin is relatively complex and requires a sequence of multiple steps. A key intermediate compound in the synthesis of silodosin is the optically active amine compound represented by the general formula R-Y:

1
wherein, R represents a protecting group and R represents a cyano (CN) or carbamoyl (CONH2) group. The intermediate compound R-Y bears the asymmetric carbon atom that imparts the optical activity to silodosin. Therefore, it is important to obtain the compound R-Y with high optical purity, because according to the methods reported in the state of the art the optical purity of the compound R-Y determines the optical purity of the final product silodosin.
JP 2001-199956 discloses a process for the preparation of a compound of formula R-Y, wherein l-(3-benzoyloxypropyl)-7-cyano-5-(2-oxopropyl)-2,3- dihydroindole or the corresponding 7-carbamoyl derivative is reacted with an optically active amine, namely L-2-phenylglycinol or L-l-phenylethanamine, to afford an imine compound of formula III as depicted in the below scheme 1. Scheme l . JP 2001-199956

R1 = COPh; R2 = CN or CONH2; R3 = H or OH a = 1. cat. deprotection
2. frational crystallization with L-tartaric acid
b = 1. chromatographic separation
2. cat. deprotection
The optically active imine III is subjected to catalytic hydrogenation using platinum(IV) oxide as a catalyst affording the diastereomers IV in a ratio of 3.8:1. The chiral auxiliary II is subsequently removed by catalytic hydrogenation using 10% palladium on carbon, i. e. under the typical conditions which lead to the cleavage and removal of benzylic protecting groups from nitrogen or oxygen atoms. The catalytic deprotection reaction affords the desired intermediate compound R-Y with an optical purity corresponding to the ratio of the diasteromers obtained in the previous step, i. e. the ratio of compound R-Y to S-Y is approximately 3.8: 1, which corresponds to an optical purity of approximately 58.3% enantiomeric excess (e.e.).
In order to increase the optical purity of the intermediate R-Y JP 2001-199956 suggests to conduct a fractional crystallization of the desired enantiomer with L-tartaric acid. After a series of fractional crystallizations the compound R-Y is obtained with an optical purity of 97.6% enantiomeric excess. Alternatively, the diastereomers of the compound of formula IV are separated using chromatographic techniques as column chromatography on silicagel. The pure diastereomer R-TV affords the desired enantiomer R-Y with an optical purity of 100% e.e. after removal of the chiral auxiliary II with hydrogen using 10% palladium on carbon as catalyst.
Another approach for the synthesis of the key intermediate compound R-Y is reported in JP 2002-265444. The route of synthesis disclosed in said document is depicted in the below scheme 2.
Scheme 2. JP 2002-265444


R1 = CH2Ph (Bn); R2 = CN The process involves the reaction of an enantiomeric mixture of the compound of formula VI with (I S, 2R)-2-benzylaminocyclohexane methanol (VII) to obtain a diastereomeric mixture containing the salt VIII. After a series of crystallizations the diastereomer VIII was obtained with an optical purity of 92.8% diastereomeric excess (d.e.). Subsequently, the salt VIII was treated with an acidic aqueous solution to release the acid R-Vl from the salt. After extraction from the aqueous solution with ethyl acetate the acid R-Vl is converted into its amide IX. The compound IX is finally subjected to a Hofmann type rearrangement reaction to obtain the desired intermediate compound R-V.
WO 201 1/030356 discloses a process for the preparation of the intermediate compound R-V, which avoids the resolution of the enantiomers of specific intermediate compounds using chiral auxiliaries or optically active bases. The route of synthesis described in WO 201 1/030356 starts from L-alanine (X), which is a naturally occurring optically active amino acid. The process described in
WO 2011/030356 is depicted in the below scheme 3.

R1 = trimethylsilyl (TMS), tert-butyl dimethylsilyl (TBDMS), allyl, benzyl, propargyl R2 = CN or CONH2 The amino acid is protected by the addition of ethyl chloroformate and subsequently activated by the addition of oxalyl chloride to afford i?-(N-ethoxycarbonyl)alanine as an acyl chloride (XI). Said acyl chloride is reacted with hydroxy protected l-(3- hydroxypropyl)-7-cyano-2,3-dihydroindole of formula XII in a Friedel-Crafts acylation reaction, which gives a compound of formula XIII. The oxo group in compound XIII is reduced to afford a compound of formula XIV that is subsequently subjected to a hydrolysis reaction to yield the key intermediate compound R-Y. It is an object of the present invention to provide a process for preparing silodosin or a pharmaceutically acceptable salt thereof, which process affords the drug with high optical purity and with better yield compared to the prior art processes. This object is solved by the subject matter as defined in the claims.
Scheme 5. Conversion of com ound V to silodosin

R = protecting group
R2 = CN or CONH2
X = leaving group
Example 11. Silodosin (XXV)
A. The compound XXIV (18.0 g) was dissolved in methanol (150 ml) and 5% aqueous sodium hydroxide solution (50 ml). The reaction mixture was stirred at room temperature for 2 h. The deprotected compound XXIV, i. e. a compound of formula XXIV with R = hydrogen and R = cyano, was extracted with toluene. Subsequently, a 10% lactic acid solution (25 ml) was added to the toluene phase in order to extract the product in the aqueous phase. The aqueous solution was separated and then basified. The deprotected product was finally extracted with ethyl acetate. Removal of the solvent gives the deprotected compound to XXIV (R1 = H and R2 = CN; 1 1.0 g) as an oily mass.
B. A mixture of compound XXIV (R1 = H and R2 – CN; 10.0 g), DMSO (80 ml) and 5N NaOH solution (9.0 ml) was stirred for 15 min. at room temperature. An aqueous H202 (30%) solution (1 1.0 ml) was added to the reaction mixture, which was stirred at room temperature for additional 2 h after completion of the addition. Water was added to the reaction mixture, the product was extracted with ethyl acetate, and the solvent was subsequently evaporated to afford 9.0 g crude silodosin.
Example 12. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 85.0% e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 97.5% e.e.
Example 13. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 98.5% e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25 °C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 99.9% e.e.
Example 14. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 90.0 %e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 97.0% e.e.
Example 15. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 92.0% e.e.) was dissolved in isopropyl acetate (160 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 8.2 g of XXV with an optical purity of 98.0% e.e. Example 16. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 98.0% e.e.) was dissolved in isopropyl acetate (160 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 8.0 g of XXV with an optical purity of 99.5% e.e.
………………………………
EP2475634
http://www.google.com/patents/EP2475634A2?cl=en
Scheme- 1.

Scheme-2.

Scheme-3.




Scheme-4.

Scheme-5.

Example-14
Preparation of Preparation of l-(3-Hydroxy-propyl)-5-(2(R)-{2-[2-(2, 2, 2-trifluoro- ethoxy)-phenoxy]-ethyIamino}-propyl)-2,3-dihydro-lH-indol-7-carboxylic acid amide (I)(Silodosin)
To a solution of Benzoic acid 3-[5(R)-(2-amino-propyl)-7-cyano-2, 3-dihydro-indol-l- yl]-propyl ester (XV) (3.5 g, 10 mmole) in Dimethyl sulphoxide (60 ml), charged Hydrogen peroxide (10% w/w) (11 ml). Then added 5 N sodium hydroxide solution (12.3 ml) and reaction mass was stirred for 2 hours. After completion of reaction water was added and extracted the product in ethyl acetate. Organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated below 40°C under reduced pressure and added methanol (25 ml). To this solution charged glacial acetic acid (0.25 g, 4mmole) and [2-(2, 2, 2-Trifluoro-ethoxy)-phenoxy]-acetaldehyde (VIII) (3 g, 0.0125 mole). Reaction mixture was stirred at 25-30°C for 1 hour. Then reacted with sodium cyanoborohydride (0.15 g, 2.8 mmoles) and heated at 40-45°C for 2 hours. After the completion of reaction solvent was distilled off below 40°C under reduced pressure and added water to the residue. Reaction mass was then acidified with aqueous mineral acid. The aqueous layer was then basified and product was extracted in ethyl acetate. Organic layer was washed with water and dried over sodium sulphate. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using a mixture of ethyl acetate and hexane (5/95) as eluent to give 0.8g of (I) as yellow solid. Purity (by HPLC) = 98%
Example 15
Preparation of l-(3-hydroxypropyl)-5-[(2R)-({2-[2-(2, 2, 2-trifIuoroethoxy) phenoxy]-ethyl} amino) propyl]-2, 3-dihydro-lH-indole-7-carbonitriIe (XVII) A mixture of 3-[7-Cyano-5 (R)-[-2-{2-[2-(2,2,2-trifluoroethoxy)-phenoxy] ethyl} amino) propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (XVI) (6.0 g , 0.010 mole), methanol (30 ml) and aqueous solution of Sodium hydroxide ( 1.6 g in 8 ml of water) was stirred at ambient temperature for 6 hours. To the reaction mixture water (90ml) was added and product was extracted with ethyl acetate (90 ml). The organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give 3.85 g of (XVII). Example 16
Preparation of l-(3-Hydroxy-propyl)-5(R)-(2-{2-[2-(2, 2, 2-trifluoro-ethoxy)- phenoxy]-ethylamino}-propyl)-2, 3-dihydro-lH-indol-7-carboxylic acid amide (I) (Silodosin)
To a solution of l-(3-hydroxypropyl)-5(R)-[2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy]- ethyl}amino)propyl]-2,3-dihydro-lH-indole-7-carbonitrile (XVII) (6.0 g , 0.013 mole) in dimethylsulfoxide (75 ml) was added 5 N sodium hydroxide solution (4.5 ml). To this reaction mixture, 30 % hydrogen peroxide (2.63 ml) was added slowly below 25°C. Reaction mixture was stirred at ambient temperature for 6 hours. Aqueous solution of sodium sulfite (2.1 in 150 ml water) was added to the reaction mixture. The reaction mixture was extracted with ethyl acetate. The combined ethyl acetate layer was extracted 2N hydrochloric acid. The aqueous layer was neutralized with sodium bicarbonate and extracted the product in ethyl acetate. The organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate. The resulting solution was cooled to 5°C and filtered to get 4.51 g of (I) as solid.
…………………………………………………
WO2012147019
http://www.google.com/patents/WO2012147019A1?cl=en
The present invention provides a process for the preparation of Silodosin of formula (I). More particularly, the present invention provides the process for preparation of tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy ] ethyl } amino)propyl] -2, 3 -dihydro- 1 H-indol- 1 -y 1 } propyl benzoate of formula (IV), which is a precursor in the preparation of Silodosin.

Background of the Invention:
A compound of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (IV) is a key intermediate for preparation of Silodosin. The chemical name of Silodosin is l-(3- hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl} amino) propyl]-2,3-dihydro-lH-indole-7-carboxamide and structurally represented as
(IV)

(I)
U.S.Pat. No. 5,387,603 discloses Silodosin as therapeutic agents for the treatment of dysuria, urinary disturbance associated with benign prostatic hyperplasia.
U.S.Pat. No. 6,310,086 discloses a process for preparing a Silodosin analogue compound from reaction of (R)-3-{5-(2-aminopropyl)-7-cyano-2,3- dihydro- 1 H-indol- 1 -yl jpropylbenzoate with 2-(2-Ethoxyphenoxy)ethyl methane sulfonate and finally isolated as residue and purified by column chromatography on silicagel. The said literature process has certain drawbacks like use of column chromatography.
U.S.Pat. No. 7,834,193 (IN 3178/DELNP/2007) discloses the process for preparation of monooxalate salt of 3-{7-cyano-5-[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy ] ethyl } amino)propyl] -2, 3 -dihydro- 1 H-indol- 1 -y 1 } propyl benzoate (IV). This patent specifically discloses the preparation of monooxalate salt of formula (IV) helps to remove N,N-dialkyl impurity to certain extend. CN 101993405 A discloses the reaction of (R)-5-(2-aminopropyl)-l-(3-(4- fluorobenzoyloxy)propyl)-7-cyanoindoline with 2-(2-(2,2,2-trifluoroethoxy) phenoxy)ethyl methane sulfonate followed by oxalic acid salt preparation.
The main drawback in the prior art process, the formation of N,N-dialkyl impurity compound of formula (VI), as disclosed in detailed description, in the preparation of Silodosin, during condensation of compound of formula (II) with compound of formula (III), the impurity which is not removable by crystallization method or precipitation technique and column chromatography purification is not suitable for commercial purpose. So considering the commercial importance of Silodosin, the present invention focus on the preparation of pure Silodosin, and surprisingly found that the isolation of formula (IV) as tartrate salt helps to prepare Silodosin having less than 0.2 % of N,N dialkyl impurity and with good yield. None of the prior arts teaches or motivates isolation of tartaric acid addition salt of formula (IV). The preparation of Silodosin from tartrate salt of 3-{7-cyano-5-[(2R)- 2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH- indol-l-yl} propyl benzoate (IV) or its freebase of the present invention has purity of greater than 99.6 %.

Example 3
Preparation of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl-} amino) propyl]-2,3-dihydro-lH-indole-7-carboxamide (Silodosin)
Method A: The compound of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy] ethyl- } amino)propyl] -2,3 -dihydro- 1 H-indole-7- carbonitrile of formula (V) in dimethylsulfoxide was treated with 48% hydrogen peroxide and 20% sodium hydroxide solution and stirred at room temperature till completion of reaction. After completion of reaction, reaction mass quenched with 5% sodium bisulphite solution and ethylacetate was added over it. The ethylacetate layer was separated and treated with 20 % aqueous hydrochloric acid. The aqueous layer separated, neutralized with sodium bicarbonate solution and extracted with ethylacetate. The separated organic layer was washed with 10% sodium bicarbonate solution, brine solution and dried under vacuum. The organic layer distilled upto residue under vacuum at 50-55°C. The obtained residue was crystallized in ethylacetate.
Method B: To the tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (IV) (100 grams) in methanol, aqueous potassium hydroxide solution (38.38 grams) was added and stirred at room temperature till reaction completion. After completion of reaction, DM water and dichloromethane was added over it under stirring. Organic layer separated, washed with brine solution distilled under vacuum upto less than 1 volume. To the solution, dimethyl sulphoxide, 20% sodium hydroxide and hydrogen peroxide was added and stirred till completion of reaction. After completion of reaction, water containing sodium bisulfite was added to the reaction mass. The pH of the reaction mixture adjusted to about 8.5 using 10% sodium hydroxide and extracted in dichloromethane twice, washed with water, dried and concentrated upto 1-2 volume under vacuum. To the obtained solution, toluene was added over it at room temperature under stirring. The reaction mixture maintained for complete solid formation, filtered and dried under vacuum. Yield 58 grams. Example 4
Purification of Silodosin:
Method A: To the mixture of toluene and acetonitrile solvent, Silodosin was added over it and heated to 50° – 55 °C for complete dissolution. The reaction mass gradually cooled to room temperature and maintained for completion of solid formation. The obtained solid is filtered, washed with toluene and dried under vacuum. Method B: To the mixture of ethyl acetate and toluene solvent, Silodosin was added over it and heated to 60° – 65 °C for complete dissolution. The reaction mass gradually cooled to room temperature and maintained for completion of solid formation. The obtained solid is filtered, washed with toluene and dried under vacuum.
//////////////////////////////////////////
CN101993407
http://www.google.com/patents/CN101993407B?cl=en
silodosin for selective inhibition of urethral smooth muscle contraction and reduce the pressure within the urethra, but no significant impact on blood pressure, for the treatment of benign prostatic hyperplasia. At present, the method of synthesis Silodosin many reports, but the lack of high yield method for industrial production.
JP200199956 reported that benzoic acid as a starting material, 1_ (3_ benzoyloxy-propyl) indoline hydrochloride (structural formula (1), R is a hydrogen atom) in 60% yield, then through the multi-step reaction was further prepared silodosin intermediate 1- (3-benzoyloxy-propyl) -5- (2-nitro-propyl) -7-cyano-indoline (structural formula VIII ), the total yield is low, and only 20 percent. Compound (VIII) with potassium carbonate, the reaction of hydrogen peroxide to yield compound (IX), impurities, and purified by column chromatography to be not suitable for industrial production. Compound (IX) under catalysis of molybdenum oxide, and L- (S) – benzyl glycyl alcohol asymmetric reactions, protecting groups may be due to steric hindrance is small, low chiral induction, is 3.8: I.




Silodosin Preparation: 12 Example
Example 11 to give 8 g solid, dissolved in DMSO 100ml, was added 5mol / L NaOH 12ml, 18 ~ 20 ° C was added dropwise slowly with 30% H2027 grams, then 30 ° C, the reaction ended 4h. Extracted with ethyl acetate, the combined organic layer was washed 2N HCl and then the organic layer, the aqueous layer was neutralized with sodium hydroxide, and then extracted with ethyl acetate, washed with saturated sodium bicarbonate, dried over anhydrous sodium sulfate, and evaporated concentrated and then dissolved in ethyl acetate, natural cooling crystallization, filtration, drying 5 g (87%), purity> 99%.
Mp 105 ~ 108 ° C
[a] 20d = -16.2 C = I, MeOH
1NMR spectrum (DMS0-d6): δ ppm 0.9-1.0 (3H, d), 1.5-1.6 (1H, s), 1.6-1.7 (2H, m),
2.3-2.4 (1H, dd), 2.6-2.7 (1H, dd), 2.8-3.0 (5H, m), 3.1-3.2 (2H, m), 3.3-3.4 (2H, m),
3.4-3.5 (2H, t), 4.0-4.1 (2H, t), 4.2-4.3 (1H, s), 4.6-4.8 (2H, t), 6.9-7.15 (6H, m),
7.2-7.3 (1H, s), 7.5-7.6 (1H, s)
…………………………………………………..
see
Silodosin is (I) (formula 1, claim 1, page 31).
Process for the prepartion of pure polymorphic gamma form of silodosin – comprising dissolving any polymorphic form of silodosin in a solvent and seeding gamma form of silodosin.
Crude (I) (50 g) was dissolved in methanol, filtered and solvent was distilled under vacuum. The residue was dissolved in isopropanol at 50 degreeC, cooled and seed of (I) gamma form was added and further cooled and cyclohexane (500 mL) was added, solid was filtered, washed and dried to obtain pure polymorphic form gamma of (I) having a toluene content of 12 ppm (example 10, pages 29-30).
A process for the preparation of silodosin and/or its salt is claimed, comprising the reaction of 3-[5-((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-1H-indol-1-yl]propyl benzoate(2R,3R)-monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate to form a cyano benzyloxy intermediate, followed by hydrolysis to form a cyano hydroxy intermediate, which is then reacted with tartaric acid and hydrolyzed in the presence of an oxidizing agent to obtain the product. An alternate method of preparation of silodosin comprising the hydrolyses of tartrate salt of cyano hydroxy intermediate in the presence of an oxidizing agent, pure polymorphic form gamma of silodosin, and the cyano hydroxy intermediate are also claimed. Further processes for the prepartion of the pure polymorphic form gamma of silodosin are claimed, wherein the process involves the dissolution of of any polymorphic form of silodosin in a solvent by heating at 30-100 degree C, cooling before and after seeding with gamma form of silodosin, adding an antisolven, isolating the polymorph and optionally micronizing.
The present invention provides an improved and efficient process for the preparation of

It acts as an selective ai -adrenoceptor antagonist and is useful in the symptomatic treatment of benign prostatic hyperplasia (BPH). Chemically it is known as l-(3-hydroxypropyl)-5-[(2R)- ( { 2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino) propyl] indoline-7-carboxamide.
Silodosin and its pharmaceutically acceptable salts are first disclosed in US patent 5,387,603. Synthetic approach for the production of silodosin, is described in patent ‘603 can be represented as shown below in scheme 1.
l

Scheme 1
As represented in scheme 1, silodosin is prepared by the reaction of l-acetyl-5-(2r aminopropyl)indoline-7-carbonitrile with 2-[2-(2,2,2-trifiuoroethoxy)phenoxy] ethyl methanesulfonate in the presence of sodium bicarbonate in ethanol to give l-acetyl-5-[2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino]propyl]indoline-7-carbonitrile, which upon reaction with di-tert-butyldicarbonate in methylene chloride produces protected acetyl indoline carbonitrile compound. Further deacetylation with sodium hydroxide in ethanol followed by treatment with acetic acid provides protected indoline carbonitrile compound, which upon hydrolysis using dimethyl sulfoxide, 30% hydrogen peroxide, sodium hydroxide and acetic acid gives protected indoline carboxamide, which upon further reaction with 2-tert-butyldimethylsiloxy)ethyl-4-nitrobenzene sulfonate in the presence of cis-dicyclohexano-18 crown-6 and potassium carbonate in dioxane gives protected (tert-butyl-dimethylsiloxy) ethyl indoline carbonitrile. Further treatment with tetrabutylammonium fluoride in tetrahydrofuran produces N-boc protected hydroxy deprotected propyl indoline carbonitrile, which under goes facile deprotection of boc group upon treatment with trifluoroacetic acid, in methylene chloride to yield silodosin. The complete process is very complex, make use of pyrophoric reagents
which are very difficult to handle in large scale and have many extra steps involving protection and depfotection. Further in US patent ‘603, concrete detail of preparation and purification of silodosin have not been reported. Furthermore, isolated silodosin is characterized using IR, NMR and specific rotation but the patent is silent on product appearance and crystalline nature. There are several processes known for the preparation of silodosin and its intermediates viz; in JP 4634560; JP 4921646; JP-2006- 188470; WO2011/124704 and WO2011/101864. In most of the inventions, silodosin is prepared by following reaction as shown in scheme 2. Major disadvantages of these processes are the formation of N,N dialkyl impurity, and other impurities which forms during the condensation of 3-[5-((2/?)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]propyl benzoate or its salts like monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate. N,N dialkyl impurity forms in about 12-15% and may form due to reaction of one molecule of benzoate compound with two molecules of methanesulfonate compound. Removal of this impurity is not possible by simple purification

wherein R is benzoyl, benzyl, tetrahydropyranyl, 2-trimethylsilylethyl, dinitrophenyl, diphenyl methyl and the like
Scheme 2
US patent 7,834,193 discloses a process for preparation of silodosin with similar condensation of 3-[5-((2R)-2-arriinopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]pfopyl benzoate or its salts like monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate, but 3-{7-cyano-5-[(2R)-2-({2-(2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propylbenzoate is purified by preparing monooxalate salt as shown below in
scheme 3. This patent specifically prepares monooxalate salt of 3- {7-cyano-5-[(2R)-2-({ 2- (2,2,2-trifluoroethoxy)-phenoxy]ethyl }amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propyl benzoate to remove N,N÷dialkyl impurity, but impurity has not been removed completely, only a certain % of it, has been removed.

Scheme 3
In PCT publication WO2012/131710, preparation of silodosin is described wherein improved processes for preparation of 3-[5-((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l- yl]propyl benzoate have been disclosed which is then converted to silodosin by condensation with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate. In exemplified process, 3-[5- ((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]propyl benzoate is condensed with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate and the resulting benzoate compound is hydrolyzed to give l-(3-hydroxy propyl)-5-[(2R)-2-({ 2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyr]-2,3-dihydro-lH-indol-7-carbonitrile.The carbonitrile compound is treated with oxalic acid to prepare its oxalate salt having purity greater than 99%, which is then hydrolyzed using a base to prepare free carbonitrile compound having purity greater than 99%, but this patent is silent about N, N- dialkyl impurity or its removal.
In PCT publication WO2012/147019, preparation of silodosin using 3-{ 7-cyano-5-[(2R)-2-({2- (2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propyl benzoate tartrate salt has been described as shown below in scheme 4.

Scheme 4
One other PCT publication WO2012/147107 describes preparation of silodosin by preparing hydrochloride and acetic acid salts of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyl]-2,3-dihydro-lH-indol-7-carbonitrile to remove N,N dialkyl impurity. It has been observed that in exemplified process, wherein hydroxy compound namely l-(3-hydroxy propyl)-5-[(2R)-2-({2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyl]-2,3-dihydro-lH-indol-7-carbonitrile is purified by preparing its acetate salt to, remove the impurities but still N, N-dialkyl impurity remains in an amount of 0.6%, which is difficult to remove in next stage or require extra purifications.
Beside to use highly pure silodosin, use of a pure polymorphic form of API is an essential requirement of drug formulation, these both aspects when address jointly, and obtained silodosin can be converted to pure polymorph then only a complete solution of prior art problems can be achieved. Apart from above mentioned process patents/publications which aimed to prepare the pure silodosin, there are exist some polymorph patents/publications which also aims to prepare pure polymorphic form of silodosin.
Polymorphism is considered as one of the- most important solid-state property of drug substance, since different polymorph have different physiochemical and biological properties and in pharmaceutical chemistry it is often desired to obtain one particular form that is biologically active and also offers ease of handling during formulation. The available literature references related to polymorph of silodosin are incorporated herein.
Japanese patent 3331048 (publication No.H07-330726), discloses a process for purification of silodosin wherein silodosin is dissolved in ethyl acetate, dried over anhydrous magnesium sulfate, solvent is distilled off and again dissolved in ethyl acetate at 70°C and crystallizes below room temperature. The resulting product is characterized by melting point, IR, NMR and specific rotation. Here also disclosure is silent about polymorphic form of product.
US patent publication US2006/0142374A1 (equivalent European patent EP1541554B 1) discloses polymorphic forms of silodosin including three crystalline polymorphic form of silodosin which are named as alpha (a), beta (β) and gamma (γ) and one amorphous form. These polymorphic forms have been characterized by X-ray powder diffraction pattern. In the patent publication, processes for the preparation of all these three crystalline forms have been disclosed. In. a given process, form alpha is prepared by dissolving crude silodosin in appropriate amount of ethyl acetate, ethyl formate, acetone, methyl ethyl ketone, acetonitrile, tetrahydrofuran or mixture of acetone and acetonitrile (1: 1), preferably ethyl acetate under heating, allowing to stand at room temperature to precipitate the crystal gradually. Similarly, form beta is prepared by dissolving crude silodosin in appropriate amount of methanol under heating, adding petroleum ether as a anti-solvent, crystal precipitation is ensured using vigorous stirring.
In a second process, to prepare the form beta, crude silodosin is dissolved in ethanol or 1-propanol and the reaction mass is cooled quickly. The crystalline form gamma is prepared by dissolving crude silodosin in appropriate amount of toluene or a mixture of acetonitrile and toluene (1:4) or ethyl acetate and toluene (1: 19), preferably in toluene, under heating, cooling to room temperature and allowing to precipitate gradually upon standing. In a second process to prepare form gamma, crude silodosin is dissolved in 2-propanol and the crystals are precipitated by adding an appropriate amount of toluene. In spite of disclosing three crystalline polymorphic forms, the patent publication prefers preparation and use of form alpha by highlighting the problems faced for preparation and use of other forms. It is disclosed that crystal form beta has manufacturing difficulties at industrial scale since precipitation occurs only when the nonpolar antisolvent is added to warm solution which leads to inconsistency in quality of crystals.
With the second process for preparation of form beta, desired level of yield and purity has not been achieved. Further, according to this publication, preparation of gamma form involves use of toluene which can not be removed completely from final product, because of its high boiling point and raises the problem of residual solvent. In the case of toluene, a class 2 solvent, its limits should not be more than 890 ppm. In the exemplified process, toluene content has not been disclosed, which clearly reflects that product was not suitable for pharmaceutical composition having problem of high residual content of toluene. Furthermore patent publication also states that all the three crystal forms donot have any difference in hygroscopicity and stabilities.
Thereafter, several patents/publications disclose preparation of polymorphic forms alpha and beta. For example a PCT publication WO2012/147107 discloses a process for preparation of beta form using isopropyl acetate and methyl isobutylketone. In another PCT publication WO2012/077138, preparation of alpha and beta forms are disclosed using various solvent , system. Similarly, in a Chinese patent CN102010359, crystalline form beta is prepared by dissolving the crude silodosin in alcoholic solvent by heating and the product is crystallized by cooling or by adding an antisolvent such as ketone or ether.
European patent EP2474529 discloses new polymorphic forms delta (δ) and eta (ε) of silodosin by using a solvent (tetrahydrofuran) and antisolvent (n-heptane, n-hexane, cyclohexane, tert butylmethyl ether).Further it discloses conversion of delta form to beta form by just heating the delta form at a particular temperature. The form delta can also be transformed into form eta by. slurrying in aqueous methanol. One new crystalline form designated as delta has also been disclosed in a Chinese patent publication CN102229558. An Indian patent application 478/MUM/2010, also discloses a new polymorphic form Zy-S which is prepared by using solvent such as esters, aromatic hydrocarbons, ketones, and alcohols.
All the above disclosures are silent about the preparation of gamma form of silodosin and only available disclosure reports that gamma form have problem of residual solvent, as impurity and is not suitable for pharmaceutical compositions.
Method C: l-(3-HydroxypropyI)-5-[(2R)-2-({2-[2,2,2-trifIuoroethoxy)phenoxy] ethyl} amino) propyl]-2,3-dihydro-lH-indol-7-carbonitrile tartrate (lOg) dissolved in dimethylsulfoxide (120 ml) and to this solution, was added 5 mol/L aqueous sodium hydroxide solution (15ml). To the reaction mixture, 30% hydrogen peroxide (5ml) was added and keeping the temperature below 25°C. The reaction mixture was stirred at 20-25°C, for 5 hours. To the reaction mixture, sodium sulfite (5g) dissolved in water (100ml) was added slowly. The reaction mixture was extracted with ethyl acetate (1x200ml) and ethyl acetate layer was concentrated under reduced pressure. The resulting product was dissolved in methanol and clear solution was filtered through micron filter paper of size 0.22 micron two times and filtrate was concentrated.The resulting compound was dissolved in toliiene (70ml) and isopropyl alcohol (7ml) at 50-55°C and the solution was cooled to 20-25°C, cyclohexane was added and stirred for further 4 hours, filtered and dried to give title compound having purity 99.86% and N,N-dialkyl impurity not detected by HPLC. Example 5: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin (15g) having toluene content 1872 ppm, was micronized under air pressure. The micronized product was dried under vacuum at 55°C-60°C for 23.0 hours to afford pure polymorphic form gamma of silodosin having toluene content 460 ppm.
Example 6: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin [having toluene content 1327 ppm] was micronized under air pressure. The micronized product was dried under vacuum at 55°C-60°C for 16 hours to afford pure polymorphic form gamma of silodosin having toluene content 350 ppm.
Example 7: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin crude (3.0g) was dissolved in isopropanol (12ml) at 50°C and reaction mass was cooled to 35°C and seed of silodosin gamma form (O.lg) was added. Thereafter reaction mass was again cooled to 15-20°C and cyclohexane (30ml) was added to the reaction mass and stirred for further 0.5 hour. The resulting solid, thus obtained, was filtered, washed with cyclohexane and dried to afford pure polymorphic form gamma of silodosin having toluene content 34 ppm.
References
- “Drugs.com, Watson Announces Silodosin NDA Accepted for Filing by FDA for the Treatment of Benign Prostatic Hyperplasia”. Retrieved 2008-02-13.
- European Medicines Agency: Assessment report for Silodyx
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Kobayashi K, Masumori N, Kato R, Hisasue S, Furuya R, Tsukamoto T. (December 2009). “Orgasm is preserved regardless of ejaculatory dysfunction with selective alpha1A-blocker administration.”. Int J Impot Res. 21 (5): 306–10. doi:10.1038/ijir.2009.27. PMC 2834370. PMID 19536124.
External links
a1a-Adrenoceptor antagonist. Prepn: M. Kitazawa et al., EP 600675; eidem, US 5387603 (1994, 1995 both to Kissei).PRODUCT PATENT
Adrenoceptor binding study: K. Shibata et al., Mol. Pharmacol. 48, 250 (1995); and tissue selectivity: S. Murata et al., J. Urol. 164, 578 (2000).
Pharmacology: K. Akiyama et al., Pharmacology 64, 140 (2002).
Series of articles on pharmacology, pharmacokinetcs and toxicology: Yakugaku Zasshi 126, 187-263 (2006).
Review of development and therapeutic potential: F. Kamali, Curr. Opin. Cent. Peripher. Nerv. Syst. Invest. Drugs 1, 248-252 (1999)
| CN101993405A * | Aug 27, 2009 | Mar 30, 2011 | 浙江华海药业股份有限公司;上海医药工业研究院 | Indoline derivative as well as preparation method and application thereof |
| JP2006188470A * | Title not available | |||
| US7834193 * | Apr 16, 2007 | Nov 16, 2010 | Kissei Pharmaceutical Co., Ltd. | industrial production of silodosin (for treating dysuria associated with benign prostatic hyperplasia) via mixing 3-{7-cyano-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino]propyl]-2,3-dihydro-1H-indol-1-yl}-propyl benzoate and oxalic acid, nitrilizing, hydrolyzing |
| WO2011030356A2 * | Sep 13, 2010 | Mar 17, 2011 | Sandoz Ag | Process for the preparation of indoline derivatives and their intermediates thereof |
| WO2011124704A1 * | Apr 8, 2011 | Oct 13, 2011 | Ratiopharm Gmbh | Process for preparing an intermediate for silodosin |
| WO2012131710A2 * | Mar 27, 2012 | Oct 4, 2012 | Panacea Biotec Ltd | Novel process for the synthesis of indoline derivatives |
| JP2006188470A * | Title not available |
| Patent | Submitted | Granted |
|---|---|---|
| Combination therapy for the treatment of benign prostatic hyperplasia [US6410554] | 2002-06-25 | |
| Indoline compound and process for producing the same [US7834193] | 2007-08-23 | 2010-11-16 |
| Agents and crystals for improving excretory potency of urinary bladder [US8252814] | 2009-10-22 | 2012-08-28 |
| METHODS FOR TREATING BENIGN PROSTATIC HYPERPLASIA [US2011319464] | 2011-12-29 | |
| PREVENTIVE AND/OR THERAPEUTIC AGENT FOR URINE COLLECTION DISORDER ACCOMPANYING LOWER URINARY TRACT OBSTRUCTION [US2009227651] | 2009-09-10 | |
| PREVENTIVE AND/OR THERAPEUTIC AGENT FOR URINE COLLECTION DISORDER ACCOMPANYING LOWER URINARY TRACT OBSTRUCTION [US2010137399] | 2010-06-03 | |
| Agents for improving excretory potency of urinary bladder [US2004116457] | 2004-06-17 | |
| Medicinal Composition for Prevention of Transition to Operative Treatment for Prostatic Hypertrophy [US2008090893] | 2008-04-17 | |
| METHODS FOR TREATING BENIGN PROSTATIC HYPERPLASIA [US2008242717] | 2008-10-02 | |
| Agents and crystals for improving excretory potency of urinary bladder [US2006281725] | 2006-12-14 |
Synthesis of Ibuprofen Using Silica-Supported Preyssler Nanoparticles as an Eco-Friendly, Inexpensive, and Efficient Catalyst,
 |
|
|
Synthesis of Ibuprofen Using Silica-Supported Preyssler Nanoparticles as an Eco-Friendly, Inexpensive, and Efficient Catalyst,
Organic Chemistry International
Volume 2014 (2014), Article ID 906801, 6 pages
http://dx.doi.org/10.1155/2014/906801
http://www.hindawi.com/journals/oci/2014/906801/
Ali Gharib,1,2 Nader Noroozi Pesyan,3 Leila Vojdani Fard,4 and Mina Roshani1
1Department of Chemistry, Islamic Azad University, Mashhad, Iran
2Agricultural Researches and Services Center, Mashhad, Iran
3Department of Chemistry, Faculty of Science, Urmia University, Urmia 57159, Iran
4Education Organization of Razavi Khorasan, Education Ministry, Mashhad, Iran
Received 5 January 2014; Revised 15 February 2014; Accepted 31 March 2014; Published 6 May 2014
Academic Editor: Jonathan White
Copyright © 2014 Ali Gharib et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
This paper describes an alternative and simple procedure for the synthesis of Ibuprofen using Silica-Supported Preyssler Nanoparticles (H14[NaP5W30O110]/SiO2) (SPNPs), as an eco-friendly, inexpensive, and efficient catalyst. High yields, simplicity of operation, and easy work-up procedure are some advantages of this protocol. Silica-Supported Preyssler Nanoparticles (H14[NaP5W30O110]/SiO2) (SPNPs) offer the advantages of a higher hydrolytic and thermal stability. The salient features of Preyssler’s anion are availability, nontoxicity and reusability. We believe this methodology can find usefulness in organic synthesis.
Synthesis of Ibuprofen (6)
To a solution of ethyl-2-(4-isobutylphenyl) propanoate (1 g, 4.27 mmol) in 6 mL of CH3OH a solution of KOH was added (479 mg, 8.55 mmol) in 5 mL of H2O. The resultant solution was stirred at room temperature for 4 h. Methanol was removed under reduced pressure and the resulting solution was extracted with ethyl acetate and the organic extracts were washed with H2O, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give compound 6.
M.P (°C) 130-133,
IR (KBr, cm−1): 3100, 2920, 2870, 1716, 1408, 1419, 1321, 1230, 1184, 935, 779, 668, 583. 1H NMR (400 MHz, CDCl3) 7.15 (d, J = 8.1 Hz, 2H), 7.02 (d, J = 8.1 Hz, 2H), 3.64 (q, J = 7.2 Hz, 1H), 2.37 (d, J = 7.1 Hz, 2H), 1.75 (m, 1H), 1.43 (d, J = 7.1 Hz, 3H), 0.82 (d, J = 6.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): 22.81, 22.82, 29.07, 42.64, 44.50, 128.80, 128.93, 128.95, 132.22, 140.23, 181.26. Anal. Calcd. for C13H18O2: C, 75.69; H, 8.80%. Found: C, 75.61; H, 8.70%.
HRMS (EI) Calcd. for C26H25FN4O6 [M]+, 206.1600, Found 206.1009.
