
Home » FDA 2018 (Page 5)
Category Archives: FDA 2018
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tildrakizumab-asmn
| Heavy chain: | |
| QVQLVQSGAEVKKPGASVKVSCKASGYIFITYWMTWVRQAPGQGL | |
| EWMGQIFPASGSADYNEKFEGRVTMTTDTSTSTAYMELRSLRSDD | |
| TAVYYCARGGGGFAYWGQGTLVTVSSASTKGPSVFPLAPSSKSTS | |
| GGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS | |
| LSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTC | |
| PPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDP | |
| EVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNG | |
| KEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKN | |
| QVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFL | |
| YSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK | |
| Light chain: | |
| DIQMTQSPSSLSASVGDRVTITCRTSENIYSYLAWYQQKPGKAPK | |
| LLIYNAKTLAEGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQH | |
| HYGIPFTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCL | |
| LNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT | |
| LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC |
Tildrakizumab-asmn
Immunoglobulin G1, anti-(human interleukin 23) (human-Mus musculus monoclonal heavy chain), disulfide with human-Mus musculus monoclonal light chain, dimer
CAS 1326244-10-3, BLA 761067
Tildrakizumab (SCH 900222/MK-3222)
ILUMYA; MK-3222; SCH-900222; SUNPG 1622; SUNPG 1622 I; SUNPG 1623 I; SUNPG 1623 II; SUNPG 1623 III; SUNPG 1623 IV; SUNPG1623; Tildrakizumab-asmn
DRUG BANK https://www.drugbank.ca/drugs/DB14004
Company Sun Pharmaceuticals
Approval Status FDA Approved March 2018 FOR Psoriasis, plaque
Treatments plaque psoriasis
Protein chemical formulaC6426H9918N1698O2000S46
Protein average weight144400.0 DaSequences
>Tildrakizumab Sequence MLGSRAVMLLLLLPWTAQGRAVPGGSSPAWTQCQQLSQKLCTLAWSAHPLVGHMDLREEG DEETTNDVPHIQCGDGCDPQGLRDNSQFCLQRIHQGLIFYEKLLGSDIFTGEPSLLPDSP VGQLHASLLGLSQLLQPEGHHWETQQIPSLSPSQPWQRLLLRFKILRSLQAFVAVAARVF AHGAATLSP
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | IL23 |
| Clinical data | |
| Trade names | Ilumya |
| Synonyms | Tildrakizumab-asmn |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| KEGG | |
| Chemical and physical data | |
| Formula | C6426H9918N1698O2000S46 |
| Molar mass | 144.4 kg/mol |
- Originator Schering-Plough
- Developer Almirall S.A.; Merck & Co; Schering-Plough; Sun Pharmaceutical Industries
- Class Antipsoriatics; Monoclonal antibodies
- Mechanism of Action Interleukin 23 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Registered Plaque psoriasis
- Phase II Ankylosing spondylitis; Psoriatic arthritis
- Discontinued Autoimmune disorders
Most Recent Events
- 21 Mar 2018 Registered for Plaque psoriasis in USA (SC) – First global approval
- 16 Feb 2018 Adverse events data from two phase III trials (reSURFACE 1 and 2) in chronic Plaque psoriasis presented at the 76th Annual Meeting of the American Academy of Dermatology (AAD-2018)
- 16 Feb 2018 Pharmacokinetics data from population PK model in healthy volunteers and patients with psoriasis presented at the 76th Annual Meeting of the American Academy of Dermatology (AAD-2018)
Ilumya (tildrakizumab-asmn) is an interleukin-23 antagonist.
Humanized monoclonal IgG1-kappa antibody against IL-23p19; produced in CHO cells
Immunoglobulin G1, anti-(human interleukin 23) (human-Mus musculus monoclonal heavy chain), disulfide with human-Mus musculus monoclonal light chain, dimer
Ilumya is specifically indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
Ilumya is supplied as a solution for subcutaneous injection. The recommended dose is 100 mg at Weeks 0, 4, and every twelve weeks thereafter.

Tildrakizumab (Ilumya) is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1] In the United States, it is approved for the treatment of moderate-to-severe plaque psoriasis.[2]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system and autoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
Sun Pharmaceutical acquired worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Upon product approval, Sun Pharmaceutical will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. [3]

As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials enrolled nearly 2000 patients. [4][5]
In 2016, tildrakizumab became the first IL-23p19 inhibitor to demonstrate positive results in Phase-3 clinical trials for the treatment of moderate-to-severe plaque psoriasis, further validating the importance of the role of IL-23 in psoriasis. Sun Pharma signed a licensing pact with Spain’s Almirall for marketing tildrakizumab in Europe [6]
In March 2018, it was approved by the Food and Drug Administration for the treatment of moderate-to-severe plaque psoriasis as an injection for subcutaneous use in the United States.[2]
In 2014, Sun Pharma acquired worldwide rights to tildrakizumab from Merck; upon product approval, Sun Pharma is responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the product. In 2016, Almirall sublicensed the product for the development and marketing in Europe for the treatment of psoriasis.
See also
- Ustekinumab, a monoclonal antibody targeting both IL-12 and IL-23 and used to treat plaque psoriasis, launched in the United States under the brand name Stelara
- Guselkumab, another experimental, IL-23-specific monoclonal antibody. (FDA approved in 2017)
- Risankizumab, another experimental, IL-23-specific monoclonal antibody. (In Phase 3 clinical trials for plaque psoriasis as of 2017)
References
- Jump up^ Statement On A Nonproprietary Name Adopted By The USAN Council – Tildrakizumab, American Medical Association.
- ^ Jump up to:a b “FDA approves Ilumya for plaque psoriasis”. National Psoriasis Foundation. March 22, 2018.
- Jump up^ http://www.merck.com/licensing/our-partnership/sunpharma_partnership.html
- Jump up^ http://clinicaltrials.gov/ct2/show/NCT01729754?term=SCH-900222&phase=2&fund=2&rank=1
- Jump up^ http://clinicaltrials.gov/ct2/show/NCT01722331?term=SCH-900222&phase=2&fund=2&rank=2
- Jump up^ http://www.business-standard.com/content/b2b-pharma/sun-pharma-signs-licensing-pact-with-spain-s-almirall-for-tildrakizumab-in-europe-116072800225_1.html
Mechanism of Action
Tildrakizumab is a humanized IgG1/k monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. Tildrakizumab inhibits the release of proinflammatory cytokines and chemokines.
FDA APPROVAL DATA
BLA 761067
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/761067Orig1s000REPLACEMENT_ltr.pdf
Please refer to your Biologics License Application (BLA) dated and received March 23, 2017 and your amendments, submitted under section 351(a) of the Public Health Service Act for ILUMYA (tildrakizumab-asmn) injection. We also refer to our approval letter dated March 20, 2018 which contained the following error: the Final Report Submission date was incorrectly listed for postmarketing requirement 3357-3. This replacement approval letter incorporates the correction of the error. The effective approval date will remain March 20, 2018, the date of the original approval letter.
LICENSING We have approved your BLA for ILUMYA (tildrakizumab-asmn) effective this date. You are hereby authorized to introduce or deliver for introduction into interstate commerce, ILUMYA under your existing Department of Health and Human Services U.S. License No. 0002. ILUMYA is indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
MANUFACTURING LOCATIONS Under this license, you are approved to manufacture ILUMYA drug substance at . The final formulated drug product will be manufactured, filled, labeled, and packaged at MSD Ireland, Carlow, Ireland. You may label your product with the proprietary name, ILUMYA, and market it in 100 mg/1 mL single-dose prefilled syringe
DATING PERIOD The dating period for ILUMYA drug product shall be 36 months from the date of manufacture when stored at 2-8°C. The date of manufacture shall be defined as the date of final sterile filtration of the formulated drug product. The dating period for your drug substance shall be months from the date of manufacture when stored at We have approved the stability protocols in your license application for the purpose of extending the expiration dating period of your drug substance and drug product under 21 CFR 601.12.
PATENTS
WO 2014109927
PAPER
https://www.tandfonline.com/doi/full/10.4161/19420862.2015.988944
Tildrakizumab (SCH 900222/MK-3222) targets the p19 subunit of IL-23. The mAb was developed by Schering-Plough, which was acquired by Merck & Co. in 2009, and it was then licensed by Merck to Sun Pharmaceutical Industries Ltd in September 2014. Clinical development and regulatory activities will be conducted by Merck, but funded by Sun Pharma. As of October 2014, the safety and efficacy of tildrakizumab are being evaluated in 2 Phase 3 studies that are ongoing but not recruiting patients. Both studies include patients with moderate-to-severe chronic plaque psoriasis and subcutaneously administered drug. The 52-week Phase 3 NCT01729754 study has 4 arms (200 mg tildrakizumab; 100 mg tildrakizumab; 50 mg etanercept; and placebo only), and includes an optional long-term safety extension study. The estimated enrollment is 1050, and the estimated primary completion date is October 2019. The 64-week Phase 3 NCT01722331 study is evaluating the effects of either 200 mg or 100 mg tildrakizumab to placebo; it includes an optional long-term safety extension study. The estimated enrollment is 885, and the estimated primary completion date is June 2015.


Mar 21, 2018, 09:04 ET
MUMBAI, India and PRINCETON, N.J., March 21, 2018 /PRNewswire/ — Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715, “Sun Pharma” and includes its subsidiaries and/or associate companies) today announced that the U.S. Food and Drug Administration (FDA) has approved ILUMYA™ (tildrakizumab-asmn) for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. ILUMYA selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor leading to inhibition of the release of pro-inflammatory cytokines and chemokines. ILUMYA is administered at a dose of 100 mg by subcutaneous injection every 12 weeks, after the completion of initial doses at weeks 0 and 4. ILUMYA is contraindicated in patients with a previous serious hypersensitivity reaction to tildrakizumab or to any of the excipients.
“With the approval of ILUMYA and our long-standing commitment in dermatology, we are focused on making a difference for people living with moderate-to-severe plaque psoriasis,” said Abhay Gandhi, President and Chief Executive Officer, North America, Sun Pharma. “We are committed to working with all relevant stakeholders to make ILUMYA available to appropriate people with plaque psoriasis.”
The FDA approval of ILUMYA for the treatment of adults with moderate-to-severe plaque psoriasis was supported by data from the pivotal Phase-3 reSURFACE clinical development program. In the two multicenter, randomized, double-blind, placebo-controlled trials (reSURFACE 1 and reSURFACE 2), 926 adult patients were treated with ILUMYA (N=616) or placebo (N=310). Results from these studies were published in The Lancet in July 2017, with primary endpoints presented at the 25th European Academy of Dermatology and Venereology (EADV) Congress.
Both Phase-3 studies met the primary efficacy endpoints, demonstrating significant clinical improvement with ILUMYA 100 mg compared to placebo when measured by at least 75 percent of skin clearance (Psoriasis Area Sensitivity Index or PASI 75) and Physician’s Global Assessment (PGA) score of “clear” or “minimal” at week 12 after two doses.
|
Efficacy Primary Endpoint at Week 12 in Adults with Plaque Psoriasis (NRI*) |
||||
|
reSURFACE 1 Study (NCT01722331) |
reSURFACE 2 Study (NCT01729754) |
|||
|
ILUMYA 100 mg n=309 |
Placebo n=154 |
ILUMYA 100 mg n=307 |
Placebo n=156 |
|
|
PGA of “clear” (0) or “minimal” (1)† |
179 (58%) |
11 (7%) |
168 (55%) |
7 (4%) |
|
PASI 75† |
197 (64%) |
9 (6%) |
188 (61%) |
9 (6%) |
|
PASI 90 |
107 (35%) |
4 (3%) |
119 (39%) |
2 (1%) |
|
PASI 100 |
43 (14%) |
2 (1%) |
38 (12%) |
0 (0%) |
* NRI = Non-Responder Imputation † Co-Primary Endpoints
Of the patients in the reSURFACE 1 study 74 percent (229 patients) achieved 75 percent skin clearance at week 28 after three doses, and 84 percent of patients who continued receiving ILUMYA 100 mg maintained PASI 75 at week 64 compared to 22 percent of patients who were re-randomized to placebo. In addition, 69 percent of the patients receiving ILUMYA 100 mg who had a PGA score of “clear” or “minimal” at week 28 maintained this response at week 64 compared to 14 percent of patients who were re-randomized to placebo.
Full Prescribing Information and Medication Guide for ILUMYA are attached:
PDF: https://mma.prnewswire.com/media/656994/Sun_Pharma_ILUMYA_US_Prescribing_Information.pdf
PDF: https://mma.prnewswire.com/media/656995/Sun_Pharma_ILUMYA_US_Medication_Guide.pdf
IMPORTANT SAFETY INFORMATION (continued)
Cases of angioedema and urticaria occurred in ILUMYA treated subjects in clinical trial. If a serious hypersensitivity reaction occurs, discontinue ILUMYA immediately and initiate appropriate therapy.
ILUMYA may increase the risk of infection. Treatment with ILUMYA should not be initiated in patients with a clinically important active infection until the infection resolves or is adequately treated. Consider the risks and benefits of treatment prior to prescribing ILUMYA in patients with a chronic infection or a history of recurrent infection. Instruct patients receiving ILUMYA to seek medical help if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a clinically important or serious infection, or is not responding to standard therapy, closely monitor and discontinue ILUMYA until the infection resolves.
Evaluate patients for TB infection prior to initiating treatment with ILUMYA. Initiate treatment of latent TB prior to administering ILUMYA. Monitor patients for signs and symptoms of active TB during and after ILUMYA treatment. Do not administer ILUMYA to patients with active TB infection.
Prior to initiating ILUMYA, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with ILUMYA.
The most common (≥1%) adverse reactions associated with ILUMYA include upper respiratory infections, injection site reactions, and diarrhea. Adverse reactions that occurred at rates less than 1% but greater than 0.1% in the ILUMYA group and at a higher rate than in the placebo group included dizziness and pain in extremity.
About the Phase-3 reSURFACE Trials
The Phase-3 studies (reSURFACE 1 and reSURFACE 2) were randomized, placebo-controlled, multicenter, three-part studies designed to demonstrate efficacy of ILUMYA in moderate-to-severe plaque psoriasis compared to placebo and comparative drug and to assess safety and tolerability. Part one of the studies randomized patients into three or four treatment arms, including ILUMYA 100 mg, ILUMYA 200 mg, placebo and etanercept (reSURFACE 2 only). After Week 12, patients on placebo were then re-randomized into ILUMYA 100 mg and 200 mg treatment arms to proceed into part two of the studies. Finally, in part three of the reSURFACE 1 study, responders (PASI ≥75) and partial responders (PASI ≥50 and PASI <75) to ILUMYA were re-randomized after Week 28 to continue the same treatment, a different dose of ILUMYA or placebo. Partial and non-responders to etanercept were treated with ILUMYA 200 mg in part three of the reSURFACE 2 study. Patients with guttate, erythrodermic, or pustular psoriasis were excluded.
About Psoriasis
Psoriasis is a chronic immune disease that appears on the skin. It is a non-contagious disorder that speeds the growth cycle of skin cells1 and results in thick scaly areas of skin2. The most common form, affecting about 80 to 90 percent of people living with psoriasis, is called plaque psoriasis3. It appears as red, raised areas of skin covered with flaky white scales, which may be itchy and painful and can crack and bleed2. Many people with plaque psoriasis continue to struggle with the ongoing, persistent nature of this chronic disease.
About Sun Dermatology
Sun Dermatology (the branded dermatology division of a wholly owned subsidiary of Sun Pharma) is committed to expanding its dermatology portfolio to bring healthcare providers and patients around the world more treatment options and ongoing support for conditions like moderate-to-severe plaque psoriasis. Sun Pharma, along with its subsidiaries, is ranked fourth in dermatology prescription volume within the U.S. per IMS and is fifth largest specialty generic pharmaceutical company globally. In addition to ILUMYA, Sun Dermatology is comprised of several branded products indicated for the treatment of acne and actinic keratosis with a focus on other dermatologic conditions.
About Sun Pharma, Merck & Co., Inc., Kenilworth, NJ, USA, Agreement
Sun Pharmaceutical Industries Ltd.’s wholly owned subsidiary licensed worldwide rights to ILUMYA from a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, in 2014. Funded by a Sun Pharma subsidiary, Merck & Co., Inc., Kenilworth, NJ, USA was responsible for the completion of Phase-3 trials and submission of a Biologics License Application to the United States Food and Drug Administration (FDA), as well as manufacturing finished goods to support Sun Pharma’s initial product launch. Sun Pharma will be responsible for all post-approval regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. Sun Pharma will also be responsible for all regulatory, pharmacovigilance, post approval studies, manufacturing and commercialization of approved products for all non-U.S. markets. Merck & Co., Inc., Kenilworth, NJ, USA is eligible to receive milestone payments and royalties on sales of ILUMYA.
About Sun Pharma, Almirall S.A, Europe, Agreement
Sun Pharma and its wholly owned subsidiary and Almirall (Spanish Stock Exchange ticker: ALM) closed on July 2016 a licensing agreement on the development and commercialization of tildrakizumab-asmn for psoriasis in Europe. Under the terms of the licensing agreement, Almirall is able to lead European studies, and participate in larger Global clinical studies for plaque psoriasis indication subject to the terms of the Sun Pharma – Merck & Co., Inc., Kenilworth, NJ, USA agreements, as well as certain cost sharing agreements. Sun Pharma will be eligible to receive development and regulatory milestone payments and, additionally, sales milestone payments and royalties on net sales. Sun Pharma will continue to lead development of tildrakizumab-asmn for other indications, where Almirall will have right of first negotiation for certain indications in Europe. The agreement between Sun Pharma and Almirall remains subject to the exclusive licensing agreement between Sun Pharma and Merck & Co., Inc., Kenilworth, NJ, USA.
About Sun Pharmaceutical Industries Ltd. (CIN – L24230GJ1993PLC019050)
Sun Pharma is the world’s fifth largest specialty generic pharmaceutical company and India’s top pharmaceutical company. A vertically integrated business, economies of scale and an extremely skilled team enable us to deliver quality products in a timely manner at affordable prices. It provides high-quality, affordable medicines trusted by customers and patients in over 150 countries across the world. Sun Pharma’s global presence is supported by 41 manufacturing facilities spread across 6 continents, R&D centres across the globe and a multi-cultural workforce comprising over 50 nationalities. In India, the company enjoys leadership across 11 different classes of doctors with 30 brands featuring amongst top 300 pharmaceutical brands in India. Its footprint across emerging markets covers over 100 markets and 6 markets in Western Europe. Its Global Consumer Healthcare business is ranked amongst Top 10 across 3 global markets. Its API business footprint is strengthened through 14 world class API manufacturing facilities across the globe. Sun Pharma fosters excellence through innovation supported by strong R&D capabilities comprising about 2,000 scientists and R&D investments of approximately 8% of annual revenues. For further information, please visit www.sunpharma.com & follow us on Twitter @SunPharma_Live.
References
1. National Psoriasis Foundation. Facts about psoriasis. www.psoriasis.org/sites/default/files/for-media/MediaKit.pdf. Accessed on February 22, 2018.
2. National Psoriasis Foundation. About Psoriasis. www.psoriasis.org/about-psoriasis. Accessed on February 22, 2018.
3. Menter A, Gottlieb A, Feldman SR, Van Voorhees AS et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008 May; 58(5):826-50.
////////////////tildrakizumab-asmn, FDA 2018, MERCK, Schering-Plough, MONOCLONAL ANTIBODY, SCH 900222, MK-3222, Psoriasis, plaque, BLA 761067, SCH-900222, SUNPG 1622, SUNPG 1622 I, SUNPG 1623 I, SUNPG 1623 II, SUNPG 1623 III, SUNPG 1623 IV, SUNPG1623,
Lofexidine, лофексидин , لوفيكسيدين , 洛非西定 ,
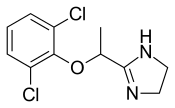
Lofexidine
- Molecular FormulaC11H12Cl2N2O
- Average mass259.132 Da
- (±)-2-[1-(2,6-Dichlorophenoxy)ethyl]-2-imidazoline
FDA Approved May 2018
Lofexidine was developed by US Woldmeds LLC and it got approved by the FDA on May 16, 2018

Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 221-223 | U.S. Patent 3,966,757. |
| boiling point (°C) | 421.5 ºC at 760 mm Hg | ‘MSDS’ |
| water solubility | Soluble | ‘MSDS’ |
| logP | 5.37 | FDA Advisory Committee Briefing Document. |
| pKa | 9.43 | FDA Advisory Committee Briefing Document. |
SYN
Organic Process Research & Development, 13(3), 415-419; 2009


LOFEXIDINE HYDROCHLORIDE
Cas No. 21498-08-8
Lofexidine, sold under the brand name Lucemyra among others,[1] is a medication historically used to treat high blood pressure, but more commonly used to help with the physical symptoms of opioid withdrawal.[2] It is taken by mouth.[3] It is an α2A adrenergic receptoragonist.[3] It was approved for use by the Food and Drug Administration in the United States in 2018.[3]
Medical uses
In the United States, the brand name Lucemyra (lofexidine HCl) is approved for the “mitigation of withdrawal symptoms to facilitate abrupt discontinuation of opioids in adults,” for a treatment duration of 14 days.[1] In the United Kingdom, lofexidine is commonly used in conjunction with the opioid receptor antagonist naltrexone in rapid detoxification cases. When these two drugs are paired, naltrexone is administered to induce an opioid-receptor blockade sending the subject into immediate withdrawal and accelerating the detoxificationprocess, while lofexidine is given to relieve the symptoms associated with the withdrawal including chills, sweating, stomach cramps, muscle pain, and runny nose.[citation needed]
Opioid withdrawal
The United Kingdom’s National Institute for Health and Care Excellence (NICE) guidelines recommend the use of methadone or buprenorphine as first-line agents in the management of opioid use disorder. However, lofexidine is considered an acceptable alternative for people with mild or uncertain opioid dependence in need of short-term detoxification.[4]
Lofexidine is not an opioid.[3] It does not eliminate the symptoms of opioid withdrawal but reduces them.[3] Indeed, one suggested use for lofexidine is to ease withdrawal symptoms of methadone dependence. Its use is approved in the United States for up to 14 days.[3]
Other clinical uses
The possibility of using lofexidine to treat alcohol withdrawal symptoms has been investigated, and has not yet been shown to be an effective treatment.[5] It is also used in treatment of cases suffering from postmenopausal hot flashes.
Special populations
Lofexidine’s safety in pregnancy or in the setting of breastfeeding are unknown.[6] Caution is warranted if chronic kidney impairment is present.[6]
Adverse effects
Adverse effects that have occurred after taking lofexidine include the following:[6]
In addition, people may experience a sudden jump in blood pressure after stopping lofexidine.[1]
Overdose
The LD50 of lofexidine is above 77 mg/kg in animals. Studies of high-dose, single administrations of lofexidine proved tolerable for animals, but repeat administration induced symptoms consistent with toxicity. In studies on mice, rats, and dogs, these included ataxia, somnolence, and tremors. It is expected that an overdose of lofexidine would result in symptoms akin to its pharmacological side effects in humans, such as bradycardia and hypotension.[7]
Interactions
Many drug-drug interactions with lofexidine are possible.[8]
QT prolongation
Lofexidine prolongs the QT interval, which can result in a severe interaction (torsade de pointes) when combined with other drugs that also prolong the QT interval. Patient-specific characteristics that increase the risk for a clinically-significant drug-drug interaction include:[8]
- increasing age
- female sex
- cardiac disease
- electrolyte disturbances (low blood potassium)
As a result, there are many QT-prolonging drugs that may interact with lofexidine. These include medications such as amiodarone, citalopram, and fluconazole. Other medications may increase the risk for a low level of potassium in the blood, thereby indirectly increasing the risk for QT prolongation. For example, dexamethasone, hydrochlorothiazide, and theophylline can lower the level of potassium in the blood.[8]
CNS depression
Lofexidine can depress the central nervous system (CNS), which, in combination with other CNS depressants, may reduce a person’s ability to perform tasks that require skills and attention. For example, clobazam, gabapentin, and levetiracetam all can depress the CNS.[8]
Hypotension
The risk of hypotension (low blood pressure) is increased when lofexidine is combined with other drugs that lower blood pressure. These may include losartan, metoprolol, and pramipexole.[8]
Pharmacology
Lofexidine is an agonist at the α-2A, 2B, and 2C adrenergic receptor subtypes, with the highest activity at the alpha-2A receptor.[9]
-
-
Ki for lofexidine[9] Adrenergic receptor Ki (nM) α-2A 4 α-2B 67 α-2C 69
-
Ki represents the dissociation constant[10] for lofexidine’s binding to a specific subtype of alpha-2 receptor. The smaller the Ki value, the stronger the drug binds to the receptor to exert its activity.
Lofexidine inhibits the release of norepinephrine in the central and peripheral nervous system, thereby reducing some of the symptoms of opioid withdrawal, but it has no documented effect on drug craving and endogenous opioid levels.[2]
Pharmacokinetics
Lofexidine’s oral bioavailability is about 90%, with extensive oral absorption. Peak plasma concentrations occur at 3 hours after a single administration, with a half-life of 11 hours. Lofexidine is extensively metabolized by the liver, and primarily cleared by the kidney. It is 80-90% plasma protein bound.[7]
Chemistry
Lofexidine exists as a solid at room temperature, with a melting point of 127 degrees C.[7] The pair of ortho chlorine (Cl–) atoms on the phenyl ring are necessary for lofexidine’s agonism at the α2a adrenergic receptor subtype; removal of either chlorine atom results in antagonism at the receptor.[9]
Comparison to clonidine

Structure of clonidine and lofexidine
Lofexidine is structurally analogous to clonidine, another α2 adrenergic receptor agonist used for treatment of opioid withdrawal symptoms. A comparison of the two structures is shown at right. Both contain an imidazoline ring and a 2,6-dichlorinated phenyl ring. The differences in structure are shown in red, while the similarities are in black. In addition to the structural differences, administration of lofexidine to people who abuse opioids has been shown to be more effective for a longer duration, with fewer withdrawal symptoms than clonidine even after one day.[11] However, clonidine is often preferred as it is substantially cheaper than lofexidine when purchased with a private (non-NHS) prescription. This factor is exacerbated by the considerable number of and quantities of medications prescribed to alleviate the constellation of withdrawal signs and symptoms. Additionally, clonidine has been shown to significantly lower blood pressure. Therefore, although similar to lofexidine, clonidine is most frequently prescribed to treat high blood pressure.[citation needed]
Society and culture
Britannia Pharmaceuticals has licensed lofexidine to be sold by US WorldMeds for sale in North America.[12] In the United Kingdom, the hydrochloride form, lofexidine HCl, has been licensed and sold since 1992 for opioid withdrawal relief in tablet form as BritLofex by Britannia Pharmaceuticals.[2] BritLofex is only available by prescription. Lofexidine was first approved by the US FDA on May 16, 2018 under the brand name Lucemyra, produced by US WorldMeds.[13] It was noted as the first, non-opioid drug approved in the US for the treatment of opioid withdrawal.[1]
Heroin has been reported to be the most prominent illicit drug of abuse among admissions at public!} -funded substance abuse treatment facilities in the US. At some time in their lives, about 2.4 million people have used heroin; in 1997, there were 81 ,000 new heroin users of whom 87% were less than 26 years of age. In spite of efforts to decrease illicit drug abuse, the problem escalates and the abusing population is increasingly younger. Hospital emergency room episodes from 21 metropolitan areas show that 14% of drug-related emergency room episodes involved heroin, and such episodes increased more than 2-fold from 1991 to 1996. Additionally, prescription opioid abuse escalates; the number of people addicted to prescription pain relievers is 3 -fold higher than those addicted to heroin. For example, from 1999 to 2001, the non-medical use of OxyContin®increased 4-fold, and its use continues to escalate.
[0003] Generally, opioid addiction has been associated with high morbidity and mortality, with a 15-20 fold increase in risk of death for intravenous drug users compared with their same age peers. Clearly, the medical and social importance of the development of effective treatments for opioid addiction is well recognized. Surprisingly, few treatment options for opioid addiction are available.
[0004] Withdrawal, maintenance and relapse are considered the progressive stages for treatment of opioid addiction. There are two predominant management strategies for the treatment of opioid addiction, detoxification and substitution therapy, which are typically combined with medical, social and psychological support. A majority of individuals may benefit from remaining in the maintenance phase for an indefinite period of time, while others may be able to directly undergo medically-supervised detoxification and/or relapse therapy, without the need for maintenance therapy. Methadone and buprenorphine constitute the most commonly used pharmacotherapies. Although patients continue to be successfully treated with methadone, a mμ opioid receptor agonist, several disadvantages of methadone treatment include the length of time for withdrawal, the difficulty of obtaining complete abstinence, and liability for its abuse. Due to the abuse liability of methadone and its consequent Schedule II classification by the Drug Enforcement Administration (DEA), methadone has additional disadvantages with respect to its prescription requirements, the carefully controlled conditions under which it is dispensed, and the annoyance experienced by patients who must frequently visit the dispensing unit to obtain their methadone dosages.
[0005] BritLofex™ (Lofexidine hydrochloride 0.2 mg tablet), an α2-adrenergic agonist, is used as a non-opioid medication for opioid detoxification in the United Kingdom (UK). There is no non-opioid medication approved by the Food and Drug Administration (FDA) for this indication in the US. The only medications currently approved by the FDA for opioid detoxification are methadone and buprenorphine, both opioid receptor agonists and both associated with abuse liability. Clonidine, an 012-adrenergic agonist, is often used “off-label” for this indication in the U.S. However, clonidine has not been approved by the FDA for this indication. However, the use of clonidine is limited by its side-effect profile, i.e., significant hypotension at doses effective in alleviating opioid withdrawal symptoms.
[0006] In contrast, Lofexidine HCl is the only non-opiate, non-addictive treatment approved for use in the UK to manage withdrawal symptoms in patients undergoing opiate detoxification. Lofexidine has been found to be effective in reducing the symptoms associated with heroin withdrawal such as chills, vomiting, sweating, stomach cramps, diarrhea, muscle pain, and runny nose and eyes. In the UK, the treatment is responsible for approximately 20,000 detoxifications per year. The drug’s proven level of safety permits its use in an outpatient situation. This is of great importance to patients in the US who are located in parts of the country where treatment clinics are not readily available.
[0007] Although naltrexone, methadone and more recently buprenorphine are FDA approved in the treatment of opioid addiction, these opioid treatments are associated with high relapse rates. Furthermore, there is currently insufficient availability of methadone and buprenorphine treatment for patients who abuse opioids. A significant number of these patients are undergoing detoxification treatments. However, the great risk of abuse and several other existing restrictions, such as medical prescribing and pharmaceutical dispensing, limit the use of methadone and buprenorphine for outpatient detoxification. In addition, the unapproved status of clonidine, its side effects, such as the lowering of blood pressure, and moderate efficacy limit its use. A substantial amount of research is ongoing to understand the mechanisms that may underline the high rates of relapse associated with opioid addiction. There is growing evidence that chronic drug use results in neuroadaptive changes in brain stress and reward circuits that may be associated with increased drug craving and risk of relapse particularly in the face of environmental triggers such as stressful life events and drug cues.
PATENT
https://patents.google.com/patent/EP2334297A1/en
The lofexidine hydrochloride tablets available in the UK market (BritLofex™) contain the racemic mixture of the drug. However, since lofexidine enantiomers exhibit different affinities for central the nervous system neurotransmitter receptors involved in (±)-lofexidine’s action as a medication for opioid detoxification, each of these enantiomers may have therapeutic benefits in the treatment of opioid addiction.
Experimental
[0028] 1) Resolution of (-)-lofexidine and (+)-lofexidine enantiomers found in the racemic mixture using chiral stationary phases by HPLC method:
[0029] A chiral chromatographic matrix was used to separate a racemic mixture of lofexidine into its component enantiomers by a process of HPLC to obtain optically pure (-)- lofexidine and optically pure (+)-lofexidine. The separation was performed using a chiral stationary phase consisted of D-glucose cyclodextran complex (Cyclobond HP-RSP) from Astec
Company (Whippany, NJ, USA) using a mobile phase consisted of 1OmM ammonium acetate
(88%), acetonitrile (8%), and methanol (8%) at 0.85 ml/min flow rate. Analysis was performed using Agilent series 1100 HPLC system comprising a solvent degasser unit, quaternary pump, autosampler, and DAD detector. Using such chiral stationary phase in a preparative scale enables the yield of gram quantities of desired enantiomers.
[0030] Resolution of (-)-lofexidine and (+)-lofexidine enantiomers found in the racemic mixture using a chiral acid, not only diastereomeric salt formation but also preferential crystallization: [0031] Optical resolution of (±)-lofexidine hydrochloride by using the classical methods of salt formation with a chiral acid such as, [( Di-p-toluoyl-D-tartaric acid [D]D20 +142° (c=l, CH3OH)] as shown in Figure 1, yielded (-)-lofexidine hydrochloride and (+)-lofexidine hydrochloride enantiomers (yield = 87%). The method comprised the following steps: [0032] A racemic form of lofexidine (10 mmol) was placed in ethanol (100 mL), and the chiral acid (+)-Di-p-toluoyl-D-tartaric acid was added in order to form a mixture of the (+)(-) and (+)(+) diastereomeric lofexidine salts. The diastereomeric salts i.e.: (+)(-) lofexidine Di-p- toluoyl-D-tartarate salt was separated from the (+)(+) lofexidine Di-p-toluoyl-D-tartarate salt by a process of fractional crystallization. 10 mL methanol and 1 ml water was added and the mixture was heated for 1 hour at 55-65 0C. After the mixture became clear it was left to cool down at room temperature. The crystals were isolated after two days, dried under vacuum. Recrystallization was performed using ethanol (20 volumes). Final yield was 87%. [0033] Chiral purity of the resulting crystals was tested by the chiral HPLC method. The
(+)(-) lofexidine Di-p-toluoyl-D-tartarate salt or the(+)(+) lofexidine Di-p-toluoyl-D-tartarate salt obtained was treated with a base such as 0.1 N sodium carbonate to liberate (-)-lofexidine and (+)-lofexidine. The resulting enantiomerically pure free base of (-)-lofexidine and (+)-lofexidine was converted to lofexidine hydrochloride salt.
PAPER
A Scalable, Enantioselective Synthesis of the α2-Adrenergic Agonist, Lofexidine

A scalable and high-yielding synthetic route toward pure enantiomers of the α2-adrenergic agonist, lofexidine hydrochloride, is presented. Salient features include a rapid one-pot amide alkylation-imidazoline formation sequence on the carboxamide function of α-(2,6-dichlorophenoxy)propionamide, while preserving the sensitive configuration about the α-carbon of the resulting product. A means to accelerate the sluggish O-alkylation of the carboxamide function of α-(2,6-dichlorophenoxy)propionamide by Me3O+BF4− is also described, which may be of general applicability.
PATENTS
US8101779B2 *2008-10-062012-01-24University Of Kentucky Research FoundationEnantioselective synthesis of (+) and (–)-2-[1-(2,6-dichlorophenoxy)-ethyl]-1,3-diazacyclopent-2-ene
References
- ^ Jump up to:a b c d “Press Announcements – FDA approves the first non-opioid treatment for management of opioid withdrawal symptoms in adults”. http://www.fda.gov. U.S. Food and Drug Administration. Retrieved 16 May 2018.
- ^ Jump up to:a b c Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. p. 330. ISBN 978-0-85711-084-8.
- ^ Jump up to:a b c d e f “Press Announcements – FDA approves the first non-opioid treatment for management of opioid withdrawal symptoms in adults”. http://www.fda.gov. Retrieved 18 May2018.
- Jump up^ “Pharmacological interventions in opioid detoxification for drug misuse in people over 16”. pathways.nice.org.uk. NICE. Retrieved 16 May 2018.
- Jump up^ Keaney F, Strang J, Gossop M, Marshall EJ, Farrell M, Welch S, Hahn B, Gonzalez A. A double-blind randomized placebo-controlled trial of lofexidine in alcohol withdrawal: lofexidine is not a useful adjunct to chlordiazepoxide. Alcohol Alcohol (2001) 36:426–30.
- ^ Jump up to:a b c “LOFEXIDINE HYDROCHLORIDE”. bnf.nice.org.uk. NICE. Retrieved 16 May2018.
- ^ Jump up to:a b c “Lofexidine”. pubchem.ncbi.nlm.nih.gov. National Center for Biotechnology Information. Retrieved 16 May 2018.
- ^ Jump up to:a b c d e “Lofexidine | Interactions | BNF”. bnf.nice.org.uk. NICE. Retrieved 16 May 2018.
- ^ Jump up to:a b c Fulton, Brian (2014). Drug Discovery for the Treatment of Addiction: Medicinal Chemistry Strategies. John Wiley & Sons. p. 151. ISBN 0470614161.
- Jump up^ Neubig, R. R. (1 December 2003). “International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology”. Pharmacological Reviews. 55 (4): 597–606. doi:10.1124/pr.55.4.4.
- Jump up^ G. Gerra, et al., Lofexidine versus clonidine in rapid opioid detoxification, Journal of Substance Abuse TreatmentVolume 21, Issue 1, , July 2001, Pages 11-17.
- Jump up^ Britannia Pharmaceuticals Limited
- Jump up^ “Lucemyra (lofexidine hydrochloride) FDA Approval History – Drugs.com”. Drugs.com. Retrieved 16 May 2018.
 |
|
| Clinical data | |
|---|---|
| Trade names | BritLofex, Lucemyra, Kai Er Ding, others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | >90% |
| Protein binding | 80–90% |
| Metabolism | Liver (glucuronidation) |
| Elimination half-life | 11 hours |
| Excretion | Kidney |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H12Cl2N2O |
| Molar mass | 259.131 g/mol |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
/////////////lofexidine, FDA 2018, лофексидин , لوفيكسيدين , 洛非西定 , Lofetensin, Loxacor
CC(C1=NCCN1)OC2=C(C=CC=C2Cl)Cl
FDA approves new drug Doptelet (avatrombopag) for patients with chronic liver disease who have low blood platelets and are undergoing a medical procedure
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer


FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive). Continue reading.
May 4, 2018
Release
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive).
“This is the first FDA-approved treatment for patients with this aggressive form of thyroid cancer, and the third cancer with this specific gene mutation that this drug combination has been approved to treat,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients.”
Thyroid cancer is a disease in which cancer cells form in the tissues of the thyroid gland. Anaplastic thyroid cancer is a rare, aggressive type of thyroid cancer. The National Institutes of Health estimates there will be 53,990 new cases of thyroid cancer and an estimated 2,060 deaths from the disease in the United States in 2018. Anaplastic thyroid cancer accounts for about 1 to 2 percent of all thyroid cancers.
Both Tafinlar and Mekinist are also approved for use, alone or in combination, to treat BRAF V600 mutation-positive metastatic melanoma. Additionally, Tafinlar and Mekinist are approved for use, in combination, to treat BRAF V600E mutation-positive, metastatic non-small cell lung cancer.
The efficacy of Tafinlar and Mekinist in treating ATC was shown in an open-label clinical trial of patients with rare cancers with the BRAF V600E mutation. Data from trials in BRAF V600E mutation-positive, metastatic melanoma or lung cancer and results in other BRAF V600E mutation-positive rare cancers provided confidence in the results seen in patients with ATC. The trial measured the percent of patients with a complete or partial reduction in tumor size (overall response rate). Of 23 evaluable patients, 57 percent experienced a partial response and 4 percent experienced a complete response; in nine (64 percent) of the 14 patients with responses, there were no significant tumor growths for six months or longer.
The side effects of Tafinlar and Mekinist in patients with ATC are consistent with those seen in other cancers when the two drugs are used together. Common side effects include fever (pyrexia), rash, chills, headache, joint pain (arthralgia), cough, fatigue, nausea, vomiting, diarrhea, myalgia (muscle pain), dry skin, decreased appetite, edema, hemorrhage, high blood pressure (hypertension) and difficulty breathing (dyspnea).
Severe side effects of Tafinlar include the development of new cancers, growth of tumors in patients with BRAF wild-type tumors, serious bleeding problems, heart problems, severe eye problems, fever that may be severe, serious skin reactions, high blood sugar or worsening diabetes, and serious anemia.
Severe side effects of Mekinist include the development of new cancers; serious bleeding problems; inflammation of intestines and perforation of the intestines; blood clots in the arms, legs or lungs; heart problems; severe eye problems; lung or breathing problems; fever that may be severe; serious skin reactions; and high blood sugar or worsening diabetes.
Both Tafinlar and Mekinist can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted Priority Review and Breakthrough Therapy designation for this indication. Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases, was also granted for this indication.
The FDA granted this approval to Novartis Pharmaceuticals Corporation.
///////////////Tafinlar, dabrafenib, Mekinist, trametinib, fda 2018, Priority Review, Breakthrough Therapy designation, Orphan Drug designation, Novartis Pharmaceuticals Corporation,
FDA approves first therapy Crysvita (burosumab) for rare inherited form of rickets, x-linked hypophosphatemia
FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia
The U.S. Food and Drug Administration today approved Crysvita (burosumab), the first drug approved to treat adults and children ages 1 year and older with x-linked hypophosphatemia (XLH), a rare, inherited form of rickets. XLH causes low levels of phosphorus in the blood. It leads to impaired bone growth and development in children and adolescents and problems with bone mineralization throughout a patient’s life.
April 17, 2018
Release
The U.S. Food and Drug Administration today approved Crysvita (burosumab), the first drug approved to treat adults and children ages 1 year and older with x-linked hypophosphatemia (XLH), a rare, inherited form of rickets. XLH causes low levels of phosphorus in the blood. It leads to impaired bone growth and development in children and adolescents and problems with bone mineralization throughout a patient’s life.
“XLH differs from other forms of rickets in that vitamin D therapy is not effective,” stated Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “This is the first FDA-approved medication for the treatment of XLH and a real breakthrough for those living with this serious disease.”
XLH is a serious disease affecting approximately 3,000 children and 12,000 adults in the United States. Most children with XLH experience bowed or bent legs, short stature, bone pain and severe dental pain. Some adults with XLH experience persistent discomfort or complications, such as joint pain, impaired mobility, tooth abscesses and hearing loss.
The safety and efficacy of Crysvita were studied in four clinical trials. In the placebo-controlled trial, 94 percent of adults receiving Crysvita once a month achieved normal phosphorus levels compared to 8 percent of those receiving placebo. In children, 94 to 100 percent of patients treated with Crysvita every two weeks achieved normal phosphorus levels. In both children and adults, X-ray findings associated with XLH improved with Crysvita therapy. Comparison of the results to a natural history cohort also provided support for the effectiveness of Crysvita.
The most common adverse reactions in adults taking Crysvita were back pain, headache, restless leg syndrome, decreased vitamin D, dizziness and constipation. The most common adverse reactions in children were headache, injection site reaction, vomiting, decreased vitamin D and pyrexia (fever).
Crysvita was granted Breakthrough Therapy designation, under which the FDA provides intensive guidance to the company on efficient drug development, and expedites its review of drugs that are intended to treat serious conditions where clinical evidence shows the drug may represent a substantial improvement over other available therapies. Crysvita also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The sponsor is receiving a Rare Pediatric Disease Priority Review Voucher under a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. A voucher can be redeemed at a later date to receive Priority Review of a subsequent marketing application for a different product. This is the 14th Rare Pediatric Disease Priority Review Voucher issued by the FDA since the program began.
The FDA granted approval of Crysvita to Ultragenyx Pharmaceutical Inc.
////////////fda 2018, Crysvita, burosumab, Breakthrough Therapy, priority review. Ultragenyx Pharmaceutical Inc
FDA expands approval of Blincyto (blinatumomab) for treatment of a type of leukemia in patients who have a certain risk factor for relapse
![]()
Blincyto (blinatumomab)
The U.S. Food and Drug Administration granted accelerated approval to Blincyto (blinatumomab) to treat adults and children with B-cell precursor acute lymphoblastic leukemia (ALL) who are in remission but still have minimal residual disease (MRD). MRD refers to the presence of cancer cells below a level that can be seen under the microscope. In patients who have achieved remission after initial treatment for this type of ALL, the presence of MRD means they have an increased risk of relapse.Continue reading.
March 29, 2018
Release
The U.S. Food and Drug Administration granted accelerated approval to Blincyto (blinatumomab) to treat adults and children with B-cell precursor acute lymphoblastic leukemia (ALL) who are in remission but still have minimal residual disease (MRD). MRD refers to the presence of cancer cells below a level that can be seen under the microscope. In patients who have achieved remission after initial treatment for this type of ALL, the presence of MRD means they have an increased risk of relapse.
“This is the first FDA-approved treatment for patients with MRD-positive ALL,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Because patients who have MRD are more likely to relapse, having a treatment option that eliminates even very low amounts of residual leukemia cells may help keep the cancer in remission longer. We look forward to furthering our understanding about the reduction in MRD after treatment with Blincyto. Studies are being conducted to assess how Blincyto affects long-term survival outcomes in patients with MRD.”
B-cell precursor ALL is a rapidly progressing type of cancer in which the bone marrow makes too many B-cell lymphocytes, an immature type of white blood cell. The National Cancer Institute estimates that approximately 5,960 people in the United States will be diagnosed with ALL this year and approximately 1,470 will die from the disease.
Blincyto works by attaching to CD19 protein on the leukemia cells and CD3 protein found on certain immune system cells. Bringing the immune cell close to the leukemia cell allows the immune cells to attack the leukemia cells better. The FDA first approved Blincyto under accelerated approval in December 2014 for the treatment of Philadelphia chromosome (Ph)-negative relapsed or refractory positive B-cell precursor ALL. Full approval for this indication was granted in July 2017, and at that time, the indication was also expanded to include patients with Philadelphia chromosome-positive ALL.
The efficacy of Blincyto in MRD-positive ALL was shown in a single-arm clinical trial that included 86 patients in first or second complete remission who had detectable MRD in at least 1 out of 1,000 cells in their bone marrow. Efficacy was based on achievement of undetectable MRD in an assay that could detect at least one cancer cell in 10,000 cells after one cycle of Blincyto treatment, in addition to the length of time that the patients remained alive and in remission (hematological relapse-free survival). Overall, undetectable MRD was achieved by 70 patients. Over half of the patients remained alive and in remission for at least 22.3 months.
The side effects of Blincyto when used to treat MRD-positive B-cell precursor ALL are consistent with those seen in other uses of the drug. Common side effects include infections (bacterial and pathogen unspecified), fever (pyrexia), headache, infusion related reactions, low levels of certain blood cells (neutropenia, anemia), febrile neutropenia (neutropenia and fever) and low levels of platelets in the blood (thrombocytopenia).
Blincyto carries a boxed warning alerting patients and health care professionals that some clinical trial participants had problems with low blood pressure and difficulty breathing (cytokine release syndrome) at the start of the first treatment, experienced a short period of difficulty with thinking (encephalopathy) or other side effects in the nervous system. Serious risks of Blincyto include infections, effects on the ability to drive and use machines, inflammation in the pancreas (pancreatitis), and preparation and administration errors—instructions for preparation and administration should closely be followed. There is a risk of serious adverse reactions in pediatric patients due to benzyl alcohol preservative; therefore, the drug prepared with preservative free saline should be used for patients weighing less than 22 kilograms.
This new indication for Blincyto was approved under the accelerated approval pathway, under which the FDA may approve drugs for serious conditions where there is unmet medical need and a drug is shown to have certain effects that are reasonably likely to predict a clinical benefit to patients. Further study in randomized controlled trials is required to verify that achieving undetectable MRD with Blincyto improves survival or disease-free survival in patients with ALL.
The FDA granted this application Priority Review and it received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Blincyto to Amgen Inc.
//////amgen, fda 2018, Priority Review m Orphan Drug designation, Blincyto, blinatumomab,
FDA approves new HIV treatment Trogarzo (ibalizumab-uiyk) for patients who have limited treatment options

![]()
Today, the U.S. Food and Drug Administration approved Trogarzo (ibalizumab-uiyk), a new type of antiretroviral medication for adult patients living with HIV who have tried multiple HIV medications in the past (heavily treatment-experienced) and whose HIV infections cannot be successfully treated with other currently available therapies (multidrug resistant HIV, or MDR HIV).Trogarzo is administered intravenously once every 14 days by a trained medical professional and used in combination with other antiretroviral medications. Continue reading.
March 6, 2018
Release
Today, the U.S. Food and Drug Administration approved Trogarzo (ibalizumab-uiyk), a new type of antiretroviral medication for adult patients living with HIV who have tried multiple HIV medications in the past (heavily treatment-experienced) and whose HIV infections cannot be successfully treated with other currently available therapies (multidrug resistant HIV, or MDR HIV).Trogarzo is administered intravenously once every 14 days by a trained medical professional and used in combination with other antiretroviral medications.
“While most patients living with HIV can be successfully treated using a combination of two or more antiretroviral drugs, a small percentage of patients who have taken many HIV drugs in the past have multidrug resistant HIV, limiting their treatment options and putting them at a high risk of HIV-related complications and progression to death,” said Jeff Murray, M.D., deputy director of the Division of Antiviral Products in the FDA’s Center for Drug Evaluation and Research. “Trogarzo is the first drug in a new class of antiretroviral medications that can provide significant benefit to patients who have run out of HIV treatment options. New treatment options may be able to improve their outcomes.”
The safety and efficacy of Trogarzo were evaluated in a clinical trial of 40 heavily treatment-experienced patients with MDR HIV-1 who continued to have high levels of virus (HIV-RNA) in their blood despite being on antiretroviral drugs. Many of the participants had previously been treated with 10 or more antiretroviral drugs. The majority of participants experienced a significant decrease in their HIV-RNA levels one week after Trogarzo was added to their failing antiretroviral regimens. After 24 weeks of Trogarzo plus other antiretroviral drugs, 43 percent of the trial’s participants achieved HIV RNA suppression.
The clinical trial focused on the small patient population with limited treatment options and demonstrated the benefit of Trogarzo in achieving reduction of HIV RNA. The seriousness of the disease, the need to individualize other drugs in the treatment regimen, and safety data from other trials were considered in evaluating the Trogarzo development program.
A total of 292 patients with HIV-1 infection have been exposed to Trogarzo IV infusion. The most common adverse reactions to Trogarzo were diarrhea, dizziness, nausea and rash. Severe side effects included rash and changes in the immune system (immune reconstitution syndrome).
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Trogarzo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Trogarzo to TaiMed Biologics USA Corp.
Theratechnologies Announces FDA Approval of Breakthrough Therapy, Trogarzo™ (ibalizumab-uiyk) Injection, the First HIV-1 Inhibitor and Long-Acting Monoclonal Antibody for Multidrug Resistant HIV-1
- First HIV treatment approved with a new mechanism of action in more than 10 years
- Infused every two weeks, only antiretroviral treatment (ART) that does not require daily dosing
- Trogarzo™ has no drug-drug interactions and no cross-resistance with other ARTs
MONTREAL, March 6, 2018 /PRNewswire/ – Theratechnologies Inc. (Theratechnologies) (TSX: TH) and its partner TaiMed Biologics, Inc. (TaiMed) today announced that the U.S. Food and Drug Administration (FDA) has granted approval of Trogarzo™ (ibalizumab-uiyk) Injection. In combination with other ARTs, Trogarzo™ is indicated for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in heavily treatment-experienced adults with multidrug resistant HIV-1 infection failing their current antiretroviral regimen.1
")
Trogarzo™ represents a critical new treatment advance as the first HIV therapy with a new mechanism of action approved in 10 years and proven effectiveness in difficult-to-treat patients with limited options. Unlike all other classes of ARTs, Trogarzo™ is a CD4-directed post-attachment HIV-1 inhibitor that binds to CD4+ receptors on host cells and blocks the HIV virus from infecting the cells.1
“Today’s approval of Trogarzo™ by the FDA is great news for people infected with difficult-to-treat multidrug resistant HIV. We look forward to bringing this much-needed therapy to patients in the U.S within six weeks,” said Luc Tanguay, President and Chief Executive Officer, Theratechnologies Inc. “We are grateful to the patients, investigators, as well as the FDA who supported the clinical development of Trogarzo™, and are helping address this critical unmet medical need.”
Trogarzo™ previously received Breakthrough Therapy and Orphan Drug designations as well as Priority Review status from the FDA, underscoring the significance of the treatment for this patient population.
“I witnessed some of the earliest cases of HIV and AIDS, at a time when the diagnosis was terrifying to patients because in many cases it was a death sentence,” said David Ho, M.D., chief scientific advisor of TaiMed and scientific director and CEO of the Aaron Diamond AIDS Research Center. “Since then, treatment advances and the discovery that combinations of ARTs was the best way to bring viral load below the level of detection have allowed most people to manage HIV like a chronic condition and live long, healthy lives. However, this is not the reality for people whose HIV is resistant to multiple drugs and whose viral load is not controlled, which is why TaiMed dedicated the past decade to advancing ibalizumab in the clinic. For these patients, it represents the next breakthrough.”
Up to 25,000 Americans with HIV are currently multidrug resistant, of which 12,000 are in urgent need of a new treatment option because their current treatment regimen is failing them and their viral load has risen to detectable levels, jeopardizing their health and making HIV transmittable.2-13 The best way to prevent the transmission of multidrug resistant HIV is to control the virus in those living with it. According to new guidance from the Centers for Disease Control and Prevention (CDC), the HIV virus cannot be transmitted if it is being fully suppressed.13
“I’ve struggled with multidrug resistant HIV for almost 30 years and it was completely debilitating to feel like I had run out of options – I made no long-term plans,” said Nelson Vergel, founder of the Program for Wellness Restoration (PoWeR) and Trogarzo™ patient. “Since starting treatment with Trogarzo™ six years ago and getting my viral load to an undetectable level, I have been my happiest, most productive self. Trogarzo™ is a new source of hope and peace of mind for people whose treatments have failed them, and I feel incredibly lucky to have been able to participate in the clinical trial program.”
TaiMed and Theratechnologies partnered on the development of Trogarzo™ so patients who can benefit from the treatment have access to it. For patients who need assistance accessing Trogarzo™ or who face challenges affording medicines, Theratechnologies has a team of patient care coordinators available to help. Patients can get assistance and expert support by contacting THERA patient support™ at 1-833-23-THERA (84372).
“In Phase 3 ibalizumab trials, we saw marked improvements in patients’ health who not only were heavily treatment-experienced and had limited remaining treatment options, but in cases they also had extremely high viral loads and significantly impaired immune systems,” said Edwin DeJesus, M.D., Medical Director for the Orlando Immunology Center. “As an investigator for ibalizumab clinical trials over nearly 10 years, it was remarkable and inspiring to see the dramatic effect ibalizumab had on such vulnerable patients. As a clinician, I am excited that we will now have another option with a different mechanism of action for our heavily pretreated patients who are struggling to keep their viral load below detection because their HIV is resistant to multiple drugs.”
Clinical Trial Findings
Clinical studies show that Trogarzo™, in combination with other ARTs, significantly reduces viral load and increases CD4+ (T-cell) count among patients with multidrug resistant HIV-1.
The Phase 3 trial showed:1
- Trogarzo™ significantly reduced viral load within seven days after the first dose of functional monotherapy and maintained the treatment response when combined with an optimized background regimen that included at least one other active ART for up to 24 weeks of treatment, while being safe and well tolerated.
- More than 80% of patients achieved the study’s primary endpoint – at least a 0.5 log10 (or 70%) viral load reduction from baseline seven days after receiving a 2,000 mg loading dose of Trogarzo™ and no adjustment to the failing background regimen.
- The average viral load reduction after 24 weeks was 1.6 log10 with 43% of patients achieving undetectable viral loads.
Patients experienced a clinically-significant mean increase in CD4+ T-cells of 44 cells/mm3, and increases varied based on T-cell count at baseline. Rebuilding the immune system by increasing T-cell count is particularly important as people with multidrug resistant HIV-1 often have the most advanced form of HIV.1
The most common drug-related adverse reactions (incidence ≥ 5%) were diarrhea (8%), dizziness (8%), nausea (5%) and rash (5%). No drug-drug interactions were reported with other ARTs or medications, and no cross-resistance with other ARTs were observed.1
About Trogarzo™ (ibalizumab-uiyk) Injection
Trogarzo™ is a humanized monoclonal antibody for the treatment of multidrug resistant HIV-1 infection. Trogarzo™ binds primarily to the second extracellular domain of the CD4+ T receptor, away from major histocompatibility complex II molecule binding sites. It prevents HIV from infecting CD4+ immune cells while preserving normal immunological function.
IMPORTANT SAFETY INFORMATION
Trogarzo™ is a prescription HIV medicine that is used with other antiretroviral medicines to treat human immunodeficiency virus-1 (HIV-1) infections in adults.
Trogarzo™ blocks HIV from infecting certain cells of the immune system. This prevents HIV from multiplying and can reduce the amount of HIV in the body.
Before you receive Trogarzo™, tell your healthcare provider if you:
- are pregnant or plan to become pregnant. It is not known if Trogarzo™ may harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if Trogarzo™ passes into breast milk.
Tell your healthcare provider about all the medicines you take, including all prescription and over-the-counter medicines, vitamins, and herbal supplements.
Trogarzo™ can cause serious side effects, including:
Changes in your immune system (Immune Reconstitution Inflammatory Syndrome) can happen when you start taking HIV-1 medicines. Your immune system might get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your health care provider right away if you start having new symptoms after starting your HIV-1 medicine.
The most common side effects of Trogarzo™ include:
- Diarrhea
- Dizziness
- Nausea
- Rash
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of Trogarzo™. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to at 1-833-23THERA (1-833-238-4372).
About Theratechnologies
Theratechnologies (TSX: TH) is a specialty pharmaceutical company addressing unmet medical needs to promote healthy living and an improved quality of life among HIV patients. Further information about Theratechnologies is available on the Company’s website at www.theratech.com and on SEDAR at www.sedar.com.
/////Trogarzo, ibalizumab-uiyk, fda 2018, Fast Track, Priority Review, Breakthrough Therapy designations, Orphan Drug designation
FDA approves new treatment Erleada (apalutamide) for a certain type of prostate cancer using novel clinical trial endpoint
The U.S. Food and Drug Administration today approved Erleada (apalutamide) for the treatment of patients with prostate cancer that has not spread (non-metastatic), but that continues to grow despite treatment with hormone therapy (castration-resistant). This is the first FDA-approved treatment for non-metastatic, castration-resistant prostate cancer. Continue reading.
February 14, 2018
Release
The U.S. Food and Drug Administration today approved Erleada (apalutamide) for the treatment of patients with prostate cancer that has not spread (non-metastatic), but that continues to grow despite treatment with hormone therapy (castration-resistant). This is the first FDA-approved treatment for non-metastatic, castration-resistant prostate cancer.
“The FDA evaluates a variety of methods that measure a drug’s effect, called endpoints, in the approval of oncology drugs. This approval is the first to use the endpoint of metastasis-free survival, measuring the length of time that tumors did not spread to other parts of the body or that death occurred after starting treatment,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “In the trial supporting approval, Erleada had a robust effect on this endpoint. This demonstrates the agency’s commitment to using novel endpoints to expedite important therapies to the American public.”
According to the National Cancer Institute (NCI) at the National Institutes of Health, prostate cancer is the second most common form of cancer in men in the U.S.. The NCI estimates approximately 161,360 men were diagnosed with prostate cancer in 2017, and 26,730 were expected to die of the disease. Approximately 10 to 20 percent of prostate cancer cases are castration-resistant, and up to 16 percent of these patients show no evidence that the cancer has spread at the time of the castration-resistant diagnosis.
Erleada works by blocking the effect of androgens, a type of hormone, on the tumor. These androgens, such as testosterone, can promote tumor growth.
The safety and efficacy of Erleada was based on a randomized clinical trial of 1,207 patients with non-metastatic, castration-resistant prostate cancer. Patients in the trial either received Erleada or a placebo. All patients were also treated with hormone therapy, either with gonadotropin-releasing hormone (GnRH) analog therapy or with surgery to lower the amount of testosterone in their body (surgical castration). The median metastasis-free survival for patients taking Erleada was 40.5 months compared to 16.2 months for patients taking a placebo.
Common side effects of Erleada include fatigue, high blood pressure (hypertension), rash, diarrhea, nausea, weight loss, joint pain (arthralgia), falls, hot flush, decreased appetite, fractures and swelling in the limbs (peripheral edema).
Severe side effects of Erleada include falls, fractures and seizures.
This application was granted Priority Review, under which the FDA’s goal is to take action on an application within 6 months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The sponsor for Erleada is the first participant in the FDA’s recently-announced Clinical Data Summary Pilot Program, an effort to provide stakeholders with more usable information on the clinical evidence supporting drug product approvals and more transparency into the FDA’s decision-making process. Soon after approval, certain information from the clinical summary report will post with the Erleada entry on Drugs@FDA and on the new pilot program landing page.
The FDA granted the approval of Erleada to Janssen Pharmaceutical Companies.
//////////////fda 2018, Erleada, apalutamide, Priority Review, Janssen
FDA approves new treatment for certain digestive tract cancers Lutathera (lutetium Lu 177 dotatate)

lutetium Lu 177 dotatate
FDA approves new treatment for certain digestive tract cancers
The U.S. Food and Drug Administration today approved Lutathera (lutetium Lu 177 dotatate) for the treatment of a type of cancer that affects the pancreas or gastrointestinal tract called gastroenteropancreatic neuroendocrine tumors (GEP-NETs). This is the first time a radioactive drug, or radiopharmaceutical, has been approved for the treatment of GEP-NETs. Lutathera is indicated for adult patients with somatostatin receptor-positive GEP-NETs. Continue reading.\
January 26, 2018
Release
The U.S. Food and Drug Administration today approved Lutathera (lutetium Lu 177 dotatate) for the treatment of a type of cancer that affects the pancreas or gastrointestinal tract called gastroenteropancreatic neuroendocrine tumors (GEP-NETs). This is the first time a radioactive drug, or radiopharmaceutical, has been approved for the treatment of GEP-NETs. Lutathera is indicated for adult patients with somatostatin receptor-positive GEP-NETs.
“GEP-NETs are a rare group of cancers with limited treatment options after initial therapy fails to keep the cancer from growing,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval provides another treatment choice for patients with these rare cancers. It also demonstrates how the FDA may consider data from therapies that are used in an expanded access program to support approval for a new treatment.”
GEP-NETs can be present in the pancreas and in different parts of the gastrointestinal tract such as the stomach, intestines, colon and rectum. It is estimated that approximately one out of 27,000 people are diagnosed with GEP-NETs per year.
Lutathera is a radioactive drug that works by binding to a part of a cell called a somatostatin receptor, which may be present on certain tumors. After binding to the receptor, the drug enters the cell allowing radiation to cause damage to the tumor cells.
The approval of Lutathera was supported by two studies. The first was a randomized clinical trial in 229 patients with a certain type of advanced somatostatin receptor-positive GEP-NET. Patients in the trial either received Lutathera in combination with the drug octreotide or octreotide alone. The study measured the length of time the tumors did not grow after treatment (progression-free survival). Progression-free survival was longer for patients taking Lutathera with octreotide compared to patients who received octreotide alone. This means the risk of tumor growth or patient death was lower for patients who received Lutathera with octreotide compared to that of patients who received only octreotide.
The second study was based on data from 1,214 patients with somatostatin receptor-positive tumors, including GEP-NETS, who received Lutathera at a single site in the Netherlands. Complete or partial tumor shrinkage was reported in 16 percent of a subset of 360 patients with GEP-NETs who were evaluated for response by the FDA. Patients initially enrolled in the study received Lutathera as part of an expanded access program. Expanded access is a way for patients with serious or immediately life-threatening diseases or conditions who lack therapeutic alternatives to gain access to investigational drugs for treatment use.
Common side effects of Lutathera include low levels of white blood cells (lymphopenia), high levels of enzymes in certain organs (increased GGT, AST and/or ALT), vomiting, nausea, high levels of blood sugar (hyperglycemia) and low levels of potassium in the blood (hypokalemia).
Serious side effects of Lutathera include low levels of blood cells (myelosuppression), development of certain blood or bone marrow cancers (secondary myelodysplastic syndrome and leukemia), kidney damage (renal toxicity), liver damage (hepatotoxicity), abnormal levels of hormones in the body (neuroendocrine hormonal crises) and infertility. Lutathera can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception. Patients taking Lutathera are exposed to radiation. Exposure of other patients, medical personnel, and household members should be limited in accordance with radiation safety practices.
Lutathera was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition. Lutathera also received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Lutathera to Advanced Accelerator Applications.
MORE FROM PUBLIC DOMAIN……………..
WATCH THIS SPACE FOR SYNTHESIS COMING
Dotatate lutenium Lu-177; 437608-50-9; DTXSID20195927
2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(1S,2R)-1-carboxy-2-hydroxypropyl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium(3+)

Lutetium-177
 Lutetium-177 has been quite a late addition as an isotope of significance to the nuclear medicine yet it is making big strides especially as a therapeutic radiopharmaceutical for neuroendocrine tumours in the form of 177Lu-DOTA-TATE on regular basis as described by Das & Pillai (2013). Lutetium-177 a lanthanide is an f block element that has a half-life of 6.7 days and decays mainly by beta emission to Hf-177, is accompanied by two gamma ray emissions. These radionuclide properties are very similar to those of I-131 which has long served as a therapeutic radionuclide, it was therefore not surprising that Lu-177 also emerged as a highly valuable radionuclide for similar applications,
There are several other upcoming applications especially for bone pain palliatiion. As a result of its convenient production logistics Lu-177 as discussed by Pillai et al (2003) is fast emerging a radionuclide of choice in radionuclide therapy (RNT).
Lu-177 can be prepared in a nuclear reactor by one of the two reactions given below :
176Lu(n,gamma)177Lu or
176Yb(n,gamma)177Yb –beta–> 177Lu
The former reaction has a high thermal neutron capture cross section and is presently the method adopted at our reactors in spite of the formation of long lived Lu-177m whose yield is very much low and is considered insignificant to cause any great concern.
Lutetium-177 Impact
 Recently there has been a rush of several research reviews and articles where Lu-177 holds the centre stage, for example, Banerjee et al (2015) have reviewed the chemistry and applications of Lu-177; Dash et al (2015) reviewed its production and available options; Knapp & Pillai (2015) highlighted its usefulness in cancer treatment and chronic diseases and Pillai and Knapp (2015) have discussed the evolving role of Lu-177 in nuclear medicine with this ready availability of Lu-177. Peptide receptor radionuclide therapy is one of the upcoming field of investigation where Lu-177 holds much promise among few other radionuclides. Indeed Lutetium-177 has covered a good distance especially for Therapeutic and as a palliative radiopharmaceutical.
Chemistry
Das et al (2014) have described the preparation of Lu-177 EDTMP kit.
Parus et al (2015) have discussed chemistry of bifunctional chelating agents for binding Lu-177.
Gupta et al (2014) have compiled methods of labelleing antibdoies with radioiodine and radiometals.
Applications
Limouris (2012) has reviewed applications in neuroendocrine tumors with focus on Liver metastasis. Das and Banerjee (2015) described the potential theranostic applications with Lu-177.
Anderson et al (1960) were among the first to use Lutetium (as chloride and citrate) in a clinical trial which were not so successful and did not encourage much promise. Keeling et al (1988) published their results with in vitro uptake of Lutetium hydroxylapatite particles. Lu-EDTMP as bone palliating agent by Ando et al (1998) soon followed, However the greatest impact was seen with the advent of a somatostatin analogue Lu-DOTATATE for targetting neuroendocrine tumors reported by Kwekkeboom et al (2001) and reviewed recently by Bodei et al (2013).
PRRNT – IAEA (2013) has brought out a human health series booklet on the subject with emphasis on neuroendocrine tumors.
177Lu Labelled Peptides in NET Kam et al (2012).
177Lu- DOTATATE – PRRNT – Bakker et al (2006)
177Lu-EDTMP – Bone Pain Palliation – Bahrami-Samani et al (2012)
177Lu-EDTMP – Pharmacokinetics, dosimetry and Therapeutic efficacy – Chakraborty S et al (2015)
177Lu-Hydroxylapatite – Radiosynovectomy – Kamalleshwaran et al. (2014) Shinto et al. (2015)
117Lu- Radioimmunotherapy – Kameshwaran et al (2015)
177Lu – Pretargeted Radioimmunotherapy (PRIT) Frost et al (2015).
More specific applications and additional information about the highly valuable therapeutic isotope would soon be added.
References and Notes
Anderson J, Farmer FT, Haggith JW, Hill M. (1960). The treatment of myelomatosis with Lutetium. Br J Radiol. 33:374-378.
Ando A, Ando L, Tonami N, Kinuya S, Kazuma K, Kataiwa A, Nakagawa M, Fujita N. (1998). 177Lu-EDTMP: a potential therapeutic bone agent. Nucl Med Commun. 19: 587-591.
Bahrami-Samani A, Anvari A, Jalilian AR, Shirvani-Arani S, Yousefnia H, Aghamiri MR, Ghannadi-Maragheh M. (2012). Production, Quality Control and Pharmacokinetic Studies of 177Lu-EDTMP for Human Bone Pain Palliation Therapy Trials. Iran J Pharm Res. 11:137-44.
Bakker WH, Breeman WAP, Kwekkeboom DJ, De Jong LC, Krenning EP. ((2006) Practical aspects of peptide receptor radionuclide therapy with [177Lu][DOTA0, Tyr3]octreotate. Q J Nucl Med Mol Imaging 50: 265-271.
Banerjee S, Pillai MR, Knapp FF (2015). Lutetium-177 Therapeutic Radiopharmaceuticals: Linking Chemistry, Radiochemistry, and Practical Applications. Chem Rev. 115: 2934-2974.
Bodei L, Mueller-Brand J, Baum RP, Pavel ME, Hörsch D, O’Dorisio MS, O’Dorisio TM, Howe JR, Cremonesi M, Kwekkeboom DJ, Zaknun JJ. (2013).The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2013 40:800-16.
Chakraborty S, Balogh L, Das T, Polyák A, Andócs G, Máthé D, Király R, Thuróczy J, Chaudhari PR, Jánoki GA, Jánoki G, Banerjee S, Pillai MR (2015). Evaluation of 177Lu-EDTMP in dogs with spontaneous tumor involving bone: Pharmacokinetics, dosimetry and therapeutic efficacy. Curr Radiopharm (ahead of Pub)
Das T, Banerjee S. (2015). Theranostic Applications of Lutetium-177 in Radionuclide Therapy. Curr Radiopharm. (ahead of print).
Das T , Sarma HD, Shinto A, Kamaleshwaran KK, Banerjee S. (2014). Formulation, Preclinical Evaluation, and Preliminary Clinical Investigation of an In-House Freeze-Dried EDTMP Kit Suitable for the Preparation of Lu-177-EDTMP. Cancer Biotherap Radiopharm. 29: (ahead of publication).
Das T, Pillai M.R.A. (2013).Options to meet the future global demand of radionuclides for radionuclide therapy. Nucl Med Biol. 40: 23-32.
Dash A, Pillai MR, Knapp FF Jr. (2015). Production of 177Lu for targeted radionuclide therapy : Available options. Nucl Med Mol Imaging. 49: 85-107.
Frost SH, Frayo SL, Miller BW, Orozco JJ, Booth GC, Hylarides MD, Lin Y, Green DJ, Gopal AK, Pagel JM, Bäck TA, Fisher DR, Press OW. (2015) Comparative efficacy of 177Lu and 90Y for anti-CD20 pretargeted radioimmunotherapy in murine lymphoma xenograft models. PLoS One. 2015 Mar 18;10(3):e0120561. Gupta S, Batra S, Jain M (2014) Antibody labeling with radioiodine and radiometals. Methods Mol Biol. 2014;1141:147-57.
IAEA (2013). Peptide receptor radionuclide therapy (PRRNT) for neuroendocrine tumors. IAEA Human Health Series No. 20., IAEA, Vienna.
Kam BLR, Teunissen JJM, Krenning EP, de Herder WW, Khan S, van Vliet EI, Kwekkeboom DJ. (2012). Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging 39 (Suppl 1):S103–S112.
Kamaleshwaran KK, Rajamani V, Thirumalaisamy SG, Chakraborty S, Kalarikal R, Mohanan V, Shinto AS.(2014).
Radiosynovectomy of the elbow joint synovitis in rheumatoid arthritis treated with Lutetium – 177 labeled hydroxylapatite (Lu-177HA) particulates; first case report and image of Lu -177 HA in the elbow joint. Indian J Nucl Med. 29:270-2.
Kameshwaran M, Pandey U, Dhakan C, Pathak K, Gota V, Vimalnath KV, Dash A, Samuel G. (2015) .Synthesis and Preclinical Evaluation of (177)Lu-CHX-A”-DTPA-Rituximab as a Radioimmunotherapeutic Agent for Non-Hodgkin’s Lymphoma. Cancer Biother Radiopharm. 2015 Aug;30(6):240-6Kwekkeboom DJ, Bakker WH, Kooij PP, Konijnenberg MW, Srinivasan A, Erion JL, Schmidt MA, Bugaj JL, de Jong M, Krenning EP.. (2001). [177Lu-DOTAOTyr3]octreotate: comparison with [111In-DTPAo]octreotide in patients.Eur J Nucl Med. 28: 1319-1325.
(Russ) Knapp FF, Pillai MR.(2015). Lutetium-177 Labeled Therapeutics: Emerging Importance for Cancer Treatment and Therapy of Chronic Disease. Curr Radiopharm. (ahead of Pub)
Parus JL, Pawlak D, Mikolajczak R, Duatti A. (2015) Chemistry and bifunctional chelating agents for binding 177Lu Curr Radiopharm (Ahead of Pub)
Limouris G. (2012) Neuroendocrine tumors: a focus on liver metastatic lesions. Front Oncol. 2:20 (Ahead of Pub) PMC article
Pillai MR, (Russ) Knapp FF. (2015). Evolving Important Role of Lutetium-177 for Therapeutic Nuclear Medicine Curr Radiopharm (ahead of print).
Pillai MR, Chakraborty S, Das T, Venkatesh M, Ramamoorthy N. (2003). Production logistics of 177Lu for radionuclide therapy. Appl Radiat Isot. 59: 109-118.
Shinto AS, Kamaleshwaran KK, Vyshakh K, Thirumalaisamy SG, Karthik S, Nagaprabhu VN, Vimalnath KV, Das T, Chakraborty S, Banerjee S. (2015) Radiosynovectomy of Painful Synovitis of Knee Joints Due to Rheumatoid Arthritis by Intra‑Articular Administration of 177Lu‑Labeled Hydroxyapatite Particulates: First Human Study and Initial Indian Experience. World J Nucl Med. 14: (ahead of print).
Videos
|
 |
|
| Names | |
|---|---|
| Other names
DOTA-(Tyr3)-octreotate
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| Properties | |
| C65H90N14O19S2 | |
| Molar mass | 1,435.63 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
DOTA-TATE, DOTATATE or DOTA-octreotate is a substance which, when bound to various radionuclides, has been tested for the treatment and diagnosis of certain types of cancer, mainly neuroendocrine tumours.
Chemistry and mechanism of action
DOTA-TATE is an amide of the acid DOTA (top left in the image), which acts as a chelator for a radionuclide, and (Tyr3)-octreotate, a derivative of octreotide. The latter binds to somatostatin receptors, which are found on the cell surfaces of a number of neuroendocrine tumours, and thus directs the radioactivity into the tumour.
Usage examples
Gallium (68Ga) DOTA-TATE (GaTate[1]) is used for tumour diagnosis in positron emission tomography (PET).[2] DOTA-TATE PET/CT has a much higher sensitivitycompared to In-111 octreotide imaging.[1]
Lutetium (177Lu) DOTA-TATE[3] has been tested for the treatment of tumors such as carcinoid and endocrine pancreatic tumor. It is also known as Lutathera.[4]
Patients are typically treated with an intravenous infusion of 7.5 GBq of lutetium-177 octreotate. After about four to six hours, the exposure rate of the patient has fallen to less than 25 microsieverts per hour at one metre and the patients can be discharged from hospital.
A course of therapy consists of four infusions at three monthly intervals.[5]
Availability
Lu177 octreotate therapy is currently available under research protocols in five different medical centers in North America: Los Angeles (CA), Quebec City, (Qc), Birmingham, AL, Edmonton, (Ab), London, (On) as Houston (Tx) on clinical trial.[6] Medical centers in Europe also offer this treatment. For instance: Cerrahpasa Hospital in Turkey, Uppsala Centre of Excellence in Neuroendocrine Tumors in Sweden and Erasmus University in the Netherlands.[7] In Israel, treatment is available at Hadassah Ein Kerem Medical Center. In Australia, treatment is available at St George Hospital and Royal North Shore Hospital, Sydney;[8] the Royal Brisbane and Women’s Hospital in Brisbane [9], the Peter MacCallum Cancer Centre [1] and at the Department of Nuclear Medicine at Fremantle Hospital in Western Australia.[10] In Aarhus universitet hospital in Denmark. In the coming years such therapy will also become commercially available in Latvia, Riga – “Clinic of nuclear medicine”.
See also
- DOTATOC or edotreotide, a similar compound
References
- ^ Jump up to:a b c Hofman, M. S.; Kong, G.; Neels, O. C.; Eu, P.; Hong, E.; Hicks, R. J. (2012). “High management impact of Ga-68 DOTATATE (GaTate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours”. Journal of Medical Imaging and Radiation Oncology. 56 (1): 40–47. doi:10.1111/j.1754-9485.2011.02327.x. PMID 22339744.
- Jump up^ Breeman, W. A. P.; De Blois, E.; Sze Chan, H.; Konijnenberg, M.; Kwekkeboom, D. J.; Krenning, E. P. (2011). “68Ga-labeled DOTA-Peptides and 68Ga-labeled Radiopharmaceuticals for Positron Emission Tomography: Current Status of Research, Clinical Applications, and Future Perspectives”. Seminars in Nuclear Medicine. 41 (4): 314–321. doi:10.1053/j.semnuclmed.2011.02.001. PMID 21624565.
- Jump up^ Bodei, L.; Cremonesi, M.; Grana, C. M.; Fazio, N.; Iodice, S.; Baio, S. M.; Bartolomei, M.; Lombardo, D.; Ferrari, M. E.; Sansovini, M.; Chinol, M.; Paganelli, G. (2011). “Peptide receptor radionuclide therapy with 177Lu-DOTATATE: The IEO phase I-II study”. European Journal of Nuclear Medicine and Molecular Imaging. 38(12): 2125–2135. doi:10.1007/s00259-011-1902-1. PMID 21892623.
- Jump up^ Radiolabeled Peptide Offers PFS Benefit in Midgut NET
- Jump up^ Claringbold, P. G.; Brayshaw, P. A.; Price, R. A.; Turner, J. H. (2010). “Phase II study of radiopeptide 177Lu-octreotate and capecitabine therapy of progressive disseminated neuroendocrine tumours”. European Journal of Nuclear Medicine and Molecular Imaging. 38 (2): 302–311. doi:10.1007/s00259-010-1631-x. PMID 21052661.
- Jump up^ Clinical trial number NCT01237457 for “177Lutetium-DOTA-Octreotate Therapy in Somatostatin Receptor-Expressing Neuroendocrine Neoplasms” at ClinicalTrials.gov
- Jump up^ “PRRT Behandelcentrum Rotterdam”. PRRT Behandelcentrum Rotterdam. Erasmus Universiteit.
- Jump up^ http://www.swslhd.nsw.gov.au/liverpool/pet/PET.html
- Jump up^ https://agitg.org.au/control-nets-study-set-to-commence
- Jump up^ Turner, J. H. (2012). “Outpatient therapeutic nuclear oncology”. Annals of Nuclear Medicine. 26 (4): 289–97. doi:10.1007/s12149-011-0566-z. PMID 22222779.
- Freedman, N; Klein, M; Gross, D; Glasberg, S; Meirovitz, A; Maimon, O; Krausz, Y; Bar-Shalom, R (2014). “Lu177-DOTATATE therapy for NET: Does tumor dose predict response?”. J Nucl Med. 55 (Supplement 1).
//////////////Lutathera, lutetium Lu 177 dotatate, fda 2018, PRIORITY REVIEW, ORPHAN DRUG
CC(C1C(=O)NC(CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCCN)CC2=CNC3=CC=CC=C32)CC4=CC=C(C=C4)O)NC(=O)C(CC5=CC=CC=C5)NC(=O)CN6CCN(CCN(CCN(CC6)CC(=O)[O-])CC(=O)[O-])CC(=O)[O-])C(=O)NC(C(C)O)C(=O)O)O.[Lu+3]
TEZACAFTOR, VX 661 for treatment of cystic fibrosis disease.

TEZACAFTOR, VX 661
CAS : 1152311-62-0;
- Molecular FormulaC26H27F3N2O6
- Average mass520.498 Da
l-(2,2-difluoro-l,3-benzodioxol-5-yl)-N-[l-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-l,l-dimethylethyl)-lH-indol-5-yl]-cyclopropanecarboxamide).
(R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide
Cyclopropanecarboxamide, 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl]-
1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)indol-5-yl]cyclopropane-1-carboxamide
Cyclopropanecarboxamide, 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-(1-((2R)-2,3-dihydroxypropyl)-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl)-
1-(2,2-difluoro-1,3-benzodioxol-5-yl)-N-(1-((2R)-2,3-dihydroxypropyl)-6-fluoro-2-(2-hydroxy-1,1-dimethylethyl)-1H-indol-5-yl)cyclopropanecarboxamide
Vertex (INNOVATOR)
UNII: 8RW88Y506K
![]()
In July 2016, this combination was reported to be in phase 3 clinical development.
Update
Symdeko (tezacaftor/ivacaftor) ; Vertex; For the treatment of cystic fibrosis , Approved February 2018
Urology
Tezacaftor, also known asVX-661, is CFTR modulator. VX-661 is potentially useful for treatment of cystic fibrosis disease. Cystic fibrosis (CF) is a genetic disease caused by defects in the CF transmembrane regulator (CFTR) gene, which encodes an epithelial chloride channel. The most common mutation, Δ508CFTR, produces a protein that is misfolded and does not reach the cell membrane. VX-661 can correct trafficking of Δ508CFTR and partially restore chloride channel activity. VX-661 is currently under Phase III clinical trial.
VX-661 is an orally available deltaF508-CFTR corrector in phase III clinical trials at Vertex for the treatment of cystic fibrosis in patients homozygous to the F508del-CFTR mutation
Novel deuterated analogs of a cyclopropanecarboxamide ie tezacaftor (VX-661), as modulators of cystic fibrosis transmembrane conductance regulator (CFTR) proteins, useful for treating a CFTR-mediated disorder eg cystic fibrosis.
VX-661 (CAS #: 1152311-62-0; l-(2,2-difluoro-l,3-benzodioxol-5-yl)-N-[l-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(2-hydroxy-l,l-dimethylethyl)-lH-indol-5-yl]-cyclopropanecarboxamide). VX-661 is a cystic fibrosis transmembrane conductance regulator modulator. VX-661 is currently under investigation for the treatment of cystic fibrosis. VX-661 has also shown promise in treating sarcoglycanopathies, Brody’s disease, cathecolaminergic polymorphic ventricular tachycardia, limb girdle muscular dystrophy, asthma, smoke induced chronic obstructive pulmonary disorder, chronic bronchitis, rhinosinusitis, constipation, pancreatitis, pancreatic insufficiency, male infertility caused by congenital bilateral absence of the vas deferens (CBAVD), mild pulmonary disease, idiopathic pancreatitis, allergic bronchopulmonary aspergillosis (ABPA), liver disease, hereditary emphysema, hereditary hemochromatosis, coagulation-fibrinolysis deficiencies, such as protein C deficiency, type 1 hereditary angioedema, lipid processing deficiencies, such as familial hypercholesterolemia, type 1 chylomicronemia, abetalipoproteinemia, lysosomal storage diseases, such as I-cell disease/pseudo-Hurler, mucopolysaccharidoses, Sandhof/Tay-Sachs, Crigler-Najjar type II, polyendocrinopathy/hyperinsulinemia, diabetes mellitus, Laron dwarfism, myeloperoxidase deficiency, primary hypoparathyroidism, melanoma, glycanosis CDG type 1, congenital hyperthyroidism, osteogenesis imperfecta, hereditary hypofibrinogenemia, ACT deficiency, diabetes insipidus (DI), neurohypophyseal DI, nephrogenic DI, Charcot-Marie tooth syndrome, Pelizaeus-Merzbacher disease, neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, progressive supranuclear palsy, Pick’s disease, polyglutamine neurological disorders such as Huntington’s, spinocerebellar ataxia type I, spinal and bulbar muscular atrophy, dentatombral pallidoluysian, and myotonic dystrophy, as well as spongifiorm encephalopathies, such as hereditary Creutzfeldt- Jakob disease (due to prion protein processing defect), Fabry disease, Gerstrnarm-Straussler-Scheinker syndrome, chronic obstructive pulmonary disorder, dry-eye disease, or Sjogren’s disease, osteoporosis, osteopenia, bone healing and bone growth (including bone repair, bone regeneration, reducing bone resorption and increasing bone deposition), Gorham’s Syndrome, chloride channelopathies such as myotonia congenita (Thomson and Becker forms), Bartter’s
syndrome type III, Dent’s disease, hyperekplexia, epilepsy, lysosomal storage disease, Angelman syndrome, and primary ciliary dyskinesia (PCD), a term for inherited disorders of the structure and/or function of cilia, including PCD with situs inversus (also known as Kartagener syndrome), PCD without situs inversus, and ciliary aplasia. WO 2014086687; WO2013185112.

VX-661

VX-661 is likely subject to extensive CYP45o-mediated oxidative metabolism. These, as well as other metabolic transformations, occur in part through polymorphically-expressed enzymes, exacerbating interpatient variability. Additionally, some metabolites of VX-661 may have undesirable side effects. In order to overcome its short half-life, the drug likely must be taken several times per day, which increases the probability of patient incompliance and discontinuance.Deuterium Kinetic Isotope Effect
PATENT
Scheme I


EXAMPLE 1
(R)-l-(2,2-difluorobenzo[dl[l,31dioxol-5-vn-N-(l-q,3-dihvdroxypropyn-6-fluoro-2-(l- hvdroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cvclopropanecarboxamide
(VX-661)

![]()
Methyl 2.2-difluorobenzo[dl [1.31dioxole-5-carboxylate: To a 200 mL pressure tank reactor (10 atm. in CO), was placed 5-bromo-2,2-difluoro-2H-l,3-benzodioxole (20.0 g, 84.4 mmol, 1.00 equiv), methanol (40 mL), triethylamine (42.6 g, 5.00 equiv.), Pd2(dba)3 (1.74 g, 1.69 mmol, 0.02 equiv), Pd(dppf)Cl2 (1.4 g, 1.69 mmol, 0.02 equiv.). The resulting solution was stirred at 85 °C under an atmosphere of CO overnight and the reaction progress was monitored by GCMS. The reaction mixture was cooled. The solids were filtered out. The organic phase was concentrated under vacuum to afford 17.5 g of methyl 2,2-difluoro-2H-l,3-benzodioxole-5-carboxylate as a crude solid, which was used directly in the next step. Step 2
![]()
2 step 2 3
(2.2-difluorobenzo[dl [ 1.31 dioxol-5 -vDmethanol : To a 500mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen were placed methyl 2,2-difluoro-2H-l,3-benzodioxole-5-carboxylate (17.5 g, 81.01 mmol, 1.00 equiv.), tetrahydrofuran (200 mL). This was followed by the addition of L1AIH4 (6.81 mg, 162.02 mmol, 2.00 equiv.) at 0 °C. The resulting solution was stirred for 1 h at 25 °C and monitored by GCMS. The reaction mixture was cooled to 0 °C until GCMS indicated the completion of the reaction. The pH value of the solution was adjusted to 8 with sodium hydroxide (1 mol/L). The solids were filtered out. The organic layer combined and concentrated under vacuum to afford 13.25 g (87%) of (2,2-difluoro-2H-l,3-benzodioxol-5-yl)methanol as yellow oil.
Step 3
![]()
step 3
5-(chloromethyl)-2.2-difluorobenzo[diri.31dioxole: (2.2-difluoro-2H-1.3-benzodioxol-5-yl)methanol (13.25 g, 70.4 mmol, 1.00 equiv.) was dissolved in DCM (200 mL). Thionyl chloride (10.02 g, 1.20 equiv.) was added to this solution. The resulting mixture was stirred at room temperature for 4 hours and then concentrated under vacuum. The residue was then diluted with DCM (500 mL) and washed with 2 x 200 mL of sodium bicarbonate and 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate, filtered and evaporated to afford 12.36 g (85%) of 5-(chloromethyl)-2,2-difluoro-2H-l ,3-benzodioxole as yellow oil.
Step 4
![]()
step 4 5
[00160] 2-(2.2-difluorobenzordi ri .31dioxol-5-yl)acetonitrile: 5-(chloromethyl)-2,2-difluoro-2H-l,3-benzodioxole (12.36 g, 60 mmol, 1.00 equiv.) was dissolved in DMSO (120 mL). This was followed by the addition of NaCN (4.41 g, 1.50 equiv.) with the inert temperature below 40 °C. The resulting solution was stirred for 2 hours at room temperature. The reaction progress was monitored by GCMS. The reaction was then quenched by the addition of 300 mL of water/ice. The resulting solution was extracted with 3 x 100 mL of ethyl acetate. The organic layers combined and washed with 3 x 100 mL brine dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.84 g (92%) of 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile as brown oil.
Step 5

l -(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l -carbonitrile: To a 100 mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, were placed 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile (10.84 g, 55 mmol, 1.00 equiv.),
NaOH (50%) in water), 1 -bromo-2-chloroethane (11.92g, 82.5 mmol, 1.50 equiv.), BmNBr
(361 mg, 1.1 mmol, 0.02 equiv.). The resulting solution was stirred for 48 h at 70 °C. The reaction progress was monitored by GCMS. The reaction mixture was cooled. The resulting solution was extracted with 3 x 200 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.12g of 1 -(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile as brown oil.
Step 6

[00162] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carboxylic acid: To a 250-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile (10.12 g, 45.38 mmol, 1.00 equiv), 6 N NaOH (61 mL) and EtOH (60 mL). The resulting solution was stirred for 3 h at 100 °C. The reaction mixture was cooled and the pH value of the solution was adjusted to 2 with hydrogen chloride (1 mol/L) until LCMS indicated the completion of the reaction. The solids were collected by filtration to afford 9.68 g (88%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxylic acid as a light yellow solid.
Step 7

[00163] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carbonyl chloride; To a solution of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxylic acid (687 mg, 2.84 mmol, 1.00 equiv.) in toluene (5 mL) was added thionyl chloride (1.67 g, 5.00 equiv.). The resulting solution was stirred for 3h at 65 °C. The reaction mixture was cooled and concentrated under vacuum to afford 738 mg (99%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonyl chloride as a yellow solid.
Step 8
![]()
9 STEP 8 10
2-methyl-4-(trimethylsilyl)but-3-vn-2-ol: To a solution of ethynyltrimethylsilane (20 g, 203.63 mmol, 1.00 equiv) in THF (100 mL) was added n-BuLi (81 mL, 2.5M in THF)
dropwise with stirring at -78 °C. Then the resulting mixture was warmed to 0 °C for 1 h with stirring and then cooled to -78 °C. Propan-2-one (11.6 g, 199.73 mmol, 1.00 equiv.) was added dropwise with the inert temperature below -78 °C. The resulting solution was stirred at -78 °C for 3 h. The reaction was then quenched by the addition of 100 mL of water and extracted with 3 x 100 mL of MTBE. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 28 g (90%) of 2-methyl-4-(trimethylsilyl)but-3-yn-2-ol as an off-white solid. ¾ NMR (400 MHz, CDCh) δ: 1.50 (s, 6H), 1.16-1.14 (m, 9H).
Step 9
![]()
step 9
10
(3-chloro-3-methylbut-l-vnvntrimethylsilane: To a lOOmL round-bottom flask, was placed 2-methyl-4-(trimethylsilyl) but-3-yn-2-ol (14 g, 89.57 mmol, 1.00 equiv.), cone. HC1 (60 mL, 6.00 equiv.). The resulting solution was stirred for 16 h at 0 °C. The resulting solution was extracted with 3 x 100 mL of hexane. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 8 g (51%) of (3-chloro-3-methylbut-l-yn-l-yl)trimethylsilane as light yellow oil. ¾ NMR (400 MHz, CDCh) δ: 1.84 (s, 6H), 1.18-1.16 (m, 9H).
Step 10
![]()
step 10
11 12
(4-(benzyloxy)-3.3-dimethylbut-l-vnyl)trimethylsilane: Magnesium turnings (1.32 g, 1.20 equiv) were charged to a 250-mL 3-necked round-bottom flask and then suspended in THF (50 mL). The resulting mixture was cooled to 0 °C and maintained with an inert atmosphere of nitrogen. (3-chloro-3-methylbut-l-yn-l-yl)trimethylsilane (8 g, 45.78 mmol, 1.00 equiv.) was dissolved in THF (50 mL) and then added dropwise to this mixture with the inert temperature between 33-37 °C. The resulting solution was stirred at room temperature for an addition 1 h before BnOCH2Cl (6.45 g, 41.33 mmol, 0.90 equiv.) was added dropwise with the temperature below 10 °C. Then the resulting solution was stirred for 16 h at room temperature. The reaction was then quenched by the addition of 50 mL of water and extracted with 3 x 100 mL of hexane. The combined organic layers was dried over
anhydrous sodium sulfate and concentrated under vacuum to afford 10 g (84%) of [4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]trimethylsilane as light yellow oil. ¾ NMR (400 MHz, CDCh) δ: 7.37-7.35 (m, 5H), 4.62 (s, 2H), 3.34 (s, 2H), 1.24 (s, 6H), 0.17-0.14 (m, 9H).
Step 11
![]()
((2.2-dimethylbut-3-vnyloxy)methyl)benzene: To a solution of [4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]trimethylsilane (10 g, 38.40 mmol, 1.00 equiv) in methanol (100 mL) was added potassium hydroxide (2.53 g, 38.33 mmol, 1.30 equiv). The resulting solution was stirred for 16 h at room temperature. The resulting solution was diluted with 200 mL of water and extracted with 3 x 100 mL of hexane. The organic layers combined and washed with 1 x 100 mL of water and then dried over anhydrous sodium sulfate and concentrated under vacuum to afford 5 g (69%) of [[(2,2-dimethylbut-3-yn-l-yl)oxy]methyl]benzene as light yellow oil. ¾ NMR (300 MHz, D20) δ: 7.41-7.28 (m, 5H) , 4.62 (s, 2H), 3.34 (s, 2H), 2.14 (s, 1H), 1.32-1.23 (m, 9H).
Step 12
![]()
14 15
methyl 2.2-difluorobenzo[d1[1.31dioxole-5-carboxylate: To a solution of 3-fluoro-4-nitroaniline (6.5 g, 41.64 mmol, 1.00 equiv) in chloroform (25 mL) and AcOH (80 mL) was added Bn (6.58 g, 41.17 mmol, 1.00 equiv.) dropwise with stirring at 0 °C in 20 min. The resulting solution was stirred for 2 h at room temperature. The reaction was then quenched by the addition of 150 mL of water/ice. The pH value of the solution was adjusted to 9 with sodium hydroxide (10 %). The resulting solution was extracted with 3 x 50 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 50 mL of water and 2 x 50 mL of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was re-crystallized from PE/EA (10: 1) to afford 6 g (61%) of 2-bromo-5-fluoro-4-nitroaniline as a yellow solid.
Step 13

(R)-l-(benzyloxy)-3-(2-bromo-5-fluoro-4-nitrophenylamino)propan-2-ol: 2-bromo-5-fluoro-4-nitroaniline (6.00 g, 25.56 mmol, 1.00 equiv.), Zn(C104)2 (1.90 g, 5.1 mmol, 0.20 equiv.), 4A Molecular Sieves (3 g), toluene (60 mL) was stirred at room temperature for 2 h and maintain with an inert atmosphere of N2 until (2R)-2-[(benzyloxy)methyl]oxirane (1.37 g, 8.34 mmol, 2.00 equiv.) was added. Then the resulting mixture was stirred for 15 h at 85 °C. The reaction progress was monitored by LCMS. The solids were filtered out and the resulting solution was diluted with 20 mL of ethyl acetate. The resulting mixture was washed with 2 x 20 mL of Sat. NH4CI and 1 x 20 mL of brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :5) to afford 7.5 g (70%) of N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluoro-4-nitroaniline as a yellow solid.
Step 14

[00170] (R)-l-(4-amino-2-bromo-5-fluorophenylamino)-3-(benzyloxy)propan-2-ol: To a 250-mL round-bottom flask, was placed N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluoro-4-nitroaniline (7.5 g, 18.84 mmol, 1.00 equiv.), ethanol (80 mL), water (16 mL), NH4CI (10 g, 189 mmol, 10.00 equiv.), Zn (6.11 g, 18.84 mmol, 5.00 equiv.). The resulting solution was stirred for 4 h at 85 °C. The solids were filtered out and the resulting solution was concentrated under vacuum and diluted with 200 mL of ethyl acetate. The resulting mixture was washed with 1 x 50 mL of water and 2 x 50 mL of brine. The organic phase was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :3) to afford 4.16 g (60%) of l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluorobenzene-l ,4-diamine as light yellow oil.
Step 15
TsO

(R)-4-(3-(benzyloxy)-2-hvdroxypropylamino)-5-bromo-2-fluorobenzenaminium 4-methylbenzenesulfonate: l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-bromo-5-fluorobenzene-l ,4-diamine (2 g, 5.42 mmol, 1.00 equiv.) was dissolved in dichloromethane (40 mL) followed by the addition of TsOH (1 g, 5.81 mmol, 1.10 equiv.). The resulting mixture was stirred for 16 h at room temperature and then concentrated under vacuum to afford 2.8 g (95%) of 4-[[(2R)-3-(benzyloxy)-2-hydroxypropyl]amino]-5-bromo-2-fluoroanilinium 4-methylbenzene-l -sulfonate as an off-white solid.
Step 16

(R)-l-(4-amino-2-(4-(benzyloxy)-3.3-dimethylbut-l-vnyl)-5-fluorophenylamino)-3-(benzyloxy)propan-2-ol: To a 100-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed 4-[[(2R)-3-(benzyloxy)-2-hydroxypropyl]amino]-5-bromo-2-fluoroanilinium 4-methylbenzene-l -sulfonate (2.9 g, 5.36 mmol, 1.00 equiv.), [[(2,2-dimethylbut-3-yn-l-yl)oxy]methyl]benzene (1.2 g, 6.37 mmol, 1.20 equiv.), Pd(OAc)2 (48 mg, 0.21 mmol, 0.04 equiv.), dppb (138 mg, 0.32 mmol, 0.06 equiv.), potassium carbonate (2.2 g, 15.92 mmol, 3.00 equiv.) and MeCN (50 mL). The resulting solution was stirred for 16 h at 80 °C. The solids were filtered out and the resulting mixture was concentrated under vacuum until LCMS indicated the completion of the reaction. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1 :4) to afford 2.2 g (86%) of l-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]-5-fluorobenzene-l ,4-diamine as a light brown solid.
Step 17

l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l-carboxylic acid: To a 40-mL vial purged and maintained with an inert atmosphere of nitrogen, was placed 1-N-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[4-(benzyloxy)-3,3-dimethylbut-l-yn-l-yl]-5-fluorobenzene-l,4-diamine (1 g, 2.1 mmol, 1.00 equiv.), MeCN (10 mL), Pd(MeCN)2Cl2 (82 mg, 0.32 mmol, 0.15 equiv.). The resulting solution was stirred for 12 h at 85 °C. The reaction progress was monitored by LCMS. The resulting mixture was concentrated under vacuum to afford 900 mg (crude) of (2R)-l-[5-amino-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-l-yl]-3-(benzyloxy)propan-2-ol as a brown solid, which was used for next step without further purification.
Step 18

(R)-N-(l-(3-(benzyloxy)-2-hvdroxypropyl)-2-(l-(benzyloxy)-2-methylpropan-2-yl)-6-fluoro- lH-indol-5-yl)- 1 -(2.2-difluorobenzo[dl [ 1.31 dioxol-5-vDcvclopropanecarboxamide: To a 40 mL vial purged and maintained with an inert atmosphere of nitrogen, was placed (2R)-l-[5-amino-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-l-yl]-3-(benzyloxy)propan-2-ol (800 mg, 1.68 mmol, 1.00 equiv.), dichloromethane (20 mL), TEA (508 mg, 5.04 mmol, 3.00 equiv.). l-(2,2-difiuoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonyl chloride (524 mg, 2 mmol, 1.20 equiv.) was added to this mixture at 0 °C. The resulting solution was stirred for 2 h at 25 °C. The reaction progress was monitored by LCMS. The resulting solution was diluted with 20 mL of DCM and washed with 3 xlO mL of brine. The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was purified by a silica gel column, eluted with ethyl acetate/petroleum ether (1:5) to afford 400 mg (30%) of N-[l-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-5-yl]-l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxamide as a light yellow solid.
Step 19

(R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide: To a 100-mL 3-necked round-bottom flask purged and maintained with an inert atmosphere of H2, were placed N-[l-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[l-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-lH-indol-5-yl]-l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carboxamide (400 mg, 0.77 mmol, 1.00 equiv.) dry Pd/C (300 mg) and MeOH (5 Ml, 6M HC1). The resulting mixture was stirred at room temperature for 2 h until LCMS indicated the completion of the reaction. The solids were filtered out and the resulting mixture was concentrated under vacuum. The residue was purified by prep-HPLC with the following conditions: Column, XBridge Prep C18 OBD Column 19 x 150 mm, 5um; mobile phase and Gradient, Phase A: Waters (0.1%FA ), Phase B: ACN; Detector, UV 254 nm to afford 126.1 mg (42.4%) of (R)-l-(2,2-difluorobenzo[d] [l,3]dioxol-5-yl)-N-(l-(2,3-dihydroxypropyl)-6-fluoro-2-(l-hydroxy-2-methylpropan-2-yl)-lH-indol-5-yl)cyclopropanecarboxamide as a light yellow solid.
¾ NMR (400 MHz, OMSO-de) δ: 8.32 (s, 1H), 7.54 (s, 1H), 7.41-7.38 (m, 2H), 7.34-7.31 (m, 2H), 6.22 (s, 1H), 5.03-5.02 (m, 1H), 4.93-4.90 (m, 1H), 4.77-4.75 (m, 1H), 4.42-4.39 (m, 1H), 4.14-4.08 (m, 1H), 3.91 (brs, 1H) , 3.64-3.57 (m, 2H), 3.47-3.40 (m, 2H), 1.48-1.46 (m, 2H), 1.36-1.32 (m, 6H), 1.14-1.12 (m, 2H).
LCMS: m/z = 521.2[M+H]+.
PATENT
WO 2015160787
https://www.google.com/patents/WO2015160787A1?cl=en
PATENT
WO 2014014841
https://www.google.com/patents/WO2014014841A1?cl=en
All tautomeric forms of the Compound 1 are included herein. For example, Compound 1 may exist as tautomers, both of which are included herein:

Methods of Preparing Compound 1 Amorphous Form and Compound 1 Form A
Compound 1 is the starting point and in one embodiment can be prepared by coupling an acid chloride moiety with an amine moiety according to Schemes 1-4.
Scheme 1. Synthesis of the acid chloride moiety.

Toluene, H20, 70 °C

Bu4NBr
1. NaOH
2. HC1
Scheme 2. Synthesis of acid chloride moiety – alternative synthesis.

1. NaCN
2. H20

SOC1,

Scheme 3. Synthesis of the amine moiety.



Scheme 4. Formation of Compound 1.

Compound 1
Methods of Preparing Compound 1 Amorphous Form
Starting from Compound 1 , or even a crystalline form of Compound 1 , Compound 1 Amorphous Form may be prepared by rotary evaporation or by spray dry methods.
Dissolving Compound 1 in an appropriate solvent like methanol and rotary evaporating the methanol to leave a foam produces Compound 1 Amorphous Form. In some embodiments, a warm water bath is used to expedite the evaporation.
Compound 1 Amorphous Form may also be prepared from Compound 1 using spray dry methods. Spray drying is a process that converts a liquid feed to a dried particulate form. Optionally, a secondary drying process such as fluidized bed drying or vacuum drying, may be used to reduce residual solvents to pharmaceutically acceptable levels. Typically, spray drying involves contacting a highly dispersed liquid suspension or solution, and a sufficient volume of hot air to produce evaporation and drying of the liquid droplets. The preparation to be spray dried can be any solution, coarse suspension, slurry, colloidal dispersion, or paste that may be atomized using the selected spray drying apparatus. In a standard procedure, the preparation is sprayed into a current of warm filtered air that evaporates the solvent and conveys the dried product to a collector (e.g. a cyclone). The spent air is then exhausted with the solvent, or alternatively the spent air is sent to a condenser to capture and potentially recycle the solvent. Commercially available types of apparatus may be used to conduct the spray drying. For example, commercial spray dryers are manufactured by Buchi Ltd. And Niro (e.g., the PSD line of spray driers manufactured by Niro) (see, US 2004/0105820; US 2003/0144257).
Spray drying typically employs solid loads of material from about 3% to about 30% by weight, (i.e., drug and excipients), for example about 4% to about 20% by weight, preferably at least about 10%. In general, the upper limit of solid loads is governed by the viscosity of (e.g., the ability to pump) the resulting solution and the solubility of the components in the solution. Generally, the viscosity of the solution can determine the size of the particle in the resulting powder product.
Techniques and methods for spray drying may be found in Perry’s Chemical
Engineering Handbook, 6th Ed., R. H. Perry, D. W. Green & J. O. Maloney, eds.), McGraw-Hill book co. (1984); and Marshall “Atomization and Spray-Drying” 50, Chem. Eng. Prog. Monogr. Series 2 (1954). In general, the spray drying is conducted with an inlet temperature of from about 60 °C to about 200 °C, for example, from about 95 °C to about 185 °C, from about 110 °C to about 182 °C, from about 96 °C to about 180 °C, e.g., about 145 °C. The spray drying is generally conducted with an outlet temperature of from about 30 °C to about 90 °C, for example from about 40 °C to about 80 °C, about 45 °C to about 80 °C e.g., about 75 °C. The atomization flow rate is generally from about 4 kg h to about 12 kg/h, for example, from about 4.3 kg/h to about 10.5 kg h, e.g., about 6 kg/h or about 10.5 kg/h. The feed flow rate is generally from about 3 kg/h to about 10 kg/h, for example, from about 3.5 kg/h to about 9.0 kg/h, e.g., about 8 kg/h or about 7.1 kg/h. The atomization ratio is generally from about 0.3 to 1.7, e.g., from about 0.5 to 1.5, e.g., about 0.8 or about 1.5.
Removal of the solvent may require a subsequent drying step, such as tray drying, fluid bed drying (e.g., from about room temperature to about 100 °C), vacuum drying, microwave drying, rotary drum drying or biconical vacuum drying (e.g., from about room temperature to about 200 °C).
Synthesis of Compound 1
Acid Chloride Moiety
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile

ouene, 2 , CN
A reactor was purged with nitrogen and charged with 900 mL of toluene. The solvent was degassed via nitrogen sparge for no less than 16 h. To the reactor was then charged Na3P04 (155.7 g, 949.5 mmol), followed by bis(dibenzylideneacetone) palladium (0) (7.28 g, 12.66 mmol). A 10% w/w solution of tert-butylphosphine in hexanes (51.23 g, 25.32 mmol) was charged over 10 min at 23 °C from a nitrogen purged addition funnel. The mixture was allowed to stir for 50 min, at which time 5-bromo-2,2-difluoro-l,3-benzodioxole (75 g, 316.5 mmol) was added over 1 min. After stirring for an additional 50 min, the mixture was charged with ethyl cyanoacetate (71.6 g, 633.0 mmol) over 5 min followed by water (4.5 mL) in one portion. The mixture was heated to 70 °C over 40 min and analyzed by HPLC every 1 – 2 h for the percent conversion of the reactant to the product. After complete conversion was observed (typically 100% conversion after 5 – 8 h), the mixture was cooled to 20 – 25 °C and filtered through a celite pad. The celite pad was rinsed with toluene (2 X 450 mL) and the combined organics were concentrated to 300 mL under vacuum at 60 – 65 °C. The concentrate was charged with 225mL DMSO and concentrated under vacuum at 70 – 80 °C until active distillation of the solvent ceased. The solution was cooled to 20 – 25 °C and diluted to 900 mL with DMSO in preparation for Step 2. Ή NMR (500 MHz, CDC13) δ 7.16 – 7.10 (m, 2H), 7.03 (d, J = 8.2 Hz, 1H), 4.63 (s, 1H), 4.19 (m, 2H), 1.23 (t, J= 7.1 Hz, 3H).
Synthesis of (2,2-difluoro-l^-benzodioxol-5-yl)-acetonitrile.

[00311] The DMSO solution of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature < 40 °C. The mixture was then heated to 75°C over 1 h and analyzed by HPLC every 1 – 2 h for % conversion. When a conversion of > 99% was observed (typically after 5 – 6 h), the reaction was cooled to 20 – 25 °C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 – 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at < 60°C to remove the solvents. (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 – 130 °C (oven temperature) and 1.5 – 2.0 Torr. (2,2-Difluoro-l,3- benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2- difluoro-l,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). Ή NMR (500 MHz, DMSO) 6 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H). Synthesis of (2,2-difluoro- l,3-benzodioxol-5-yl)-cycIopropanecarbonitrUe.

MTBE
A stock solution of 50% w/w NaOH was degassed via nitrogen sparge for no less than 16 h. An appropriate amount of MTBE was similarly degassed for several hours. To a reactor purged with nitrogen was charged degassed MTBE (143 mL) followed by (2,2-difluoro-l,3- benzodioxol-5-yl)-acetonitrile (40.95 g, 207.7 mmol) and tetrabutylammonium bromide (2.25 g, 10.38 mmol). The volume of the mixture was noted and the mixture was degassed via nitrogen sparge for 30 min. Enough degassed MTBE is charged to return the mixture to the original volume prior to degassing. To the stirring mixture at 23.0 °C was charged degassed 50% w/w NaOH (143 mL) over 10 min followed by l-bromo-2-chloroethane (44.7 g, 311.6 mmol) over 30 min. The reaction was analyzed by HPLC in 1 h intervals for % conversion. Before sampling, stirring was stopped and the phases allowed to separate. The top organic phase was sampled for analysis. When a % conversion > 99 % was observed (typically after 2.5 – 3 h), the reaction mixture was cooled to 10 °C and was charged with water (461 mL) at such a rate as to maintain a temperature < 25 °C. The temperature was adjusted to 20 – 25 °C and the phases separated. Note: sufficient time should be allowed for complete phase separation. The aqueous phase was extracted with MTBE (123 mL), and the combined organic phase was washed with 1 N HC1 (163mL) and 5% NaCl (163 mL). The solution of (2,2-difluoro- 1,3 -benzodioxol-5-yl)- cyclopropanecarbonitrile in MTBE was concentrated to 164 mL under vacuum at 40 – 50 °C. The solution was charged with ethanol (256 mL) and again concentrated to 164 mL under vacuum at 50 – 60 °C. Ethanol (256 mL) was charged and the mixture concentrated to 164 mL under vacuum at 50 – 60 °C. The resulting mixture was cooled to 20 – 25 °C and diluted with ethanol to 266 mL in preparation for the next step. lH NMR (500 MHz, DMSO) 6 7.43 (d, J= 8.4 Hz, 1H), 7.40 (d, J= 1.9 Hz, 1H), 7.30 (dd, J= 8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H). [00314] Synthesis of l-(2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid.

The solution of (2,2-difluoro-l ,3-benzodioxol-5-yl)-cyclopropanecarbonitrile in ethanol from the previous step was charged with 6 N NaOH (277 mL) over 20 min and heated to an internal temperature of 77 – 78 °C over 45 min. The reaction progress was monitored by HPLC after 16 h. Note: the consumption of both (2,2-difluoro-l,3-benzodioxol-5-yl)- cyclopropanecarbonitrile and the primary amide resulting from partial hydrolysis of (2,2-difluoro- l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile were monitored. When a % conversion > 99 % was observed (typically 100% conversion after 16 h), the reaction mixture was cooled to 25 °C and charged with ethanol (41 mL) and DCM (164 mL). The solution was cooled to 10 °C and charged with 6 N HC1 (290 mL) at such a rate as to maintain a temperature < 25 °C. After warming to 20 – 25 °C, the phases were allowed to separate. The bottom organic phase was collected and the top aqueous phase was back extracted with DCM (164 mL). Note: the aqueous phase was somewhat cloudy before and after the extraction due to a high concentration of inorganic salts. The organics were combined and concentrated under vacuum to 164 mL. Toluene (328 mL) was charged and the mixture condensed to 164 mL at 70 – 75 °C. The mixture was cooled to 45 °C, charged with MTBE (364 mL) and stirred at 60 °C for 20 min. The solution was cooled to 25 °C and polish filtered to remove residual inorganic salts. MTBE (123 mL) was used to rinse the reactor and the collected solids. The combined organics were transferred to a clean reactor in preparation for the next step.
Isolation of l-(2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecar boxy lie acid.

The solution of l-(2,2-difluoro- 1 ,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid from the previous step is concentrated under vacuum to 164 mL, charged with toluene (328 mL) and concentrated to 164 mL at 70 – 75 °C. The mixture was then heated to 100 – 105 °C to give a homogeneous solution. After stirring at that temperature for 30 min, the solution was cooled to 5 °C over 2 hours and maintained at 5 °C for 3 hours. The mixture was then filtered and the reactor and collected solid washed with cold 1 :1 toluene/n-heptane (2 X 123 mL). The material was dried under vacuum at 55 °C for 17 hours to provide l-(2,2-difluoro-l,3-benzodioxol-5-yl)- cyclopropanecarboxylic acid as an off-white crystalline solid. l-(2,2-difluoro-l,3-benzodioxol- 5-yl)-cyclopropanecarboxylic acid was isolated in 79% yield from (2,2-difluoro-l,3- benzodioxol-5-yl)-acetonitrile (3 steps including isolation) and with an HPLC purity of 99.0% AUC. ESI-MS m/z calc. 242.04, found 241.58 (M+l)+; Ή NMR (500 MHz, DMSO) δ 12.40 (s, 1H), 7.40 (d, J= 1.6 Hz, 1H), 7.30 (d, J= 8.3 Hz, 1H), 7.17 (dd, J= 8.3, 1.7 Hz, 1H), 1.46 (m, 2H), 1.17 (m, 2H).
Alternative Synthesis of the Acid Chloride Moiety [00319] Synthesis of (2,2-ditluoro-l,3-benzodioxol-5-yl)-methanol.
1. Vitride (2 equiv)
PhCH3 (10 vol)

[00320] Commercially available 2,2-difluoro-l,3-benzodioxole-5-carboxylic acid (1.0 eq) is slurried in toluene (10 vol). Vitride® (2 eq) is added via addition funnel at a rate to maintain the temperature at 15-25 °C. At the end of addition the temperature is increased to 40 °C for 2 h then 10% (w/w) aq. NaOH (4.0 eq) is carefully added via addition funnel maintaining the temperature at 40-50 °C. After stirring for an additional 30 minutes, the layers are allowed to separate at 40 °C. The organic phase is cooled to 20 °C then washed with water (2 x 1.5 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-methanol that is used directly in the next step.
Synthesis of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole.
1. SOCl2 (1.5 equiv)
DMAP (0.01 equiv)

(2,2-difluoro- 1 ,3-benzodioxol-5-yl)-methanol ( 1.0 eq) is dissolved in MTBE (5 vol). A catalytic amount of DMAP (1 mol %) is added and S0C12 (1.2 eq) is added via addition funnel. The S0C12 is added at a rate to maintain the temperature in the reactor at 15-25 °C. The temperature is increased to 30 °C for 1 hour then cooled to 20 °C then water (4 vol) is added via addition funnel maintaining the temperature at less than 30 °C. After stirring for an additional 30 minutes, the layers are allowed to separate. The organic layer is stirred and 10% (w/v) aq. NaOH (4.4 vol) is added. After stirring for 15 to 20 minutes, the layers are allowed to separate. The organic phase is then dried (Na2SO_ , filtered, and concentrated to afford crude 5-chloromethyl- 2,2-difluoro-l,3-benzodioxole that is used directly in the next step.
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile.

A solution of 5-chloromethyl-2,2-difluoro- 1 ,3-benzodioxole ( 1 eq) in DMSO ( 1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 °C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1,8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step.
The remaining steps are the same as described above for the synthesis of the acid moiety.
Amine Moiety
Synthesis of 2-bromo-5-fluoro-4-ntroaniline.

Synthesis of benzyIglycoIated-4-ammonium-2-bromo-5-fluoroaniline tosylate salt.
1) l ^OBn
cat. Zn(C104)2-2H20 ®

DCM
A thoroughly dried flask under N2 was charged with the following: Activated powdered 4A molecular sieves (50 wt% based on 2-bromo-5-fluoro-4-nitroaniline), 2-Bromo-5- fluoro-4-nitroaniline (1.0 equiv), zinc perchlorate dihydrate (20 mol%), and toluene (8 vol). The mixture was stirred at room temperature for NMT 30 min. Lastly, (R)-benzyl glycidyl ether (2.0 equiv) in toluene (2 vol) was added in a steady stream. The reaction was heated to 80 °C (internal temperature) and stirred for approximately 7 hours or until 2-Bromo-5-fluoro-4-nitroaniline was <5%AUC.
The reaction was cooled to room temperature and Celite (50 wt%) was added, followed by ethyl acetate (10 vol). The resulting mixture was filtered to remove Celite and sieves and washed with ethyl acetate (2 vol). The filtrate was washed with ammonium chloride solution (4 vol, 20% w/v). The organic layer was washed with sodium bicarbonate solution (4 vol x 2.5% w/v). The organic layer was concentrated in vacuo on a rotovap. The resulting slurry was dissolved in isopropyl acetate (10 vol) and this solution was transferred to a Buchi hydrogenator.
The hydrogenator was charged with 5wt% Pt(S)/C (1.5 mol%) and the mixture was stirred under N2 at 30 °C (internal temperature). The reaction was flushed with N2 followed by hydrogen. The hydrogenator pressure was adjusted to 1 Bar of hydrogen and the mixture was stirred rapidly (>1200 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with dichloromethane (10 vol). The filtrate was concentrated in vacuo. Any remaining isopropyl acetate was chased with dichloromethane (2 vol) and concentrated on a rotavap to dryness.
The resulting residue was dissolved in dichloromethane (10 vol). jP-Toluenesulfonic acid monohydrate (1.2 equiv) was added and stirred overnight. The product was filtered and washed with dichloromethane (2 vol) and suction dried. The wetcake was transferred to drying trays and into a vacuum oven and dried at 45 °C with N2 bleed until constant weight was achieved. Benzylglycolated-4-ammonium-2-bromo-5-fluoroaniline tosylate salt was isolated as an off-white solid.
Chiral purity was determined to be >97%ee.
[00334] Synthesis of (3-Chloro-3-methylbut-l-ynyl)trimethylsilane.

[00335] Propargyl alcohol (1.0 equiv) was charged to a vessel. Aqueous hydrochloric acid (37%, 3.75 vol) was added and stirring begun. During dissolution of the solid alcohol, a modest endotherm (5-6 °C) is observed. The resulting mixture was stirred overnight (16 h), slowly becoming dark red. A 30 L jacketed vessel is charged with water (5 vol) which is then cooled to 10 °C. The reaction mixture is transferred slowly into the water by vacuum, maintaining the internal temperature of the mixture below 25 °C. Hexanes (3 vol) is added and the resulting mixture is stirred for 0.5 h. The phases were settled and the aqueous phase (pH < 1) was drained off and discarded. The organic phase was concentrated in vacuo using a rotary evaporator, furnishing the product as red oil. [00336] Synthesis of (4-(Benzyloxy)-3,3-dimethylbut-l-yttyl)trimethylsiIane.

[00337] Method A
[00338] All equivalent and volume descriptors in this part are based on a 250g reaction.
Magnesium turnings (69.5 g, 2.86 mol, 2.0 equiv) were charged to a 3 L 4-neck reactor and stirred with a magnetic stirrer under nitrogen for 0.5 h. The reactor was immersed in an ice- water bath. A solution of the propargyl chloride (250 g, 1.43 mol, 1.0 equiv) in THF (1.8 L, 7.2 vol) was added slowly to the reactor, with stirring, until an initial exotherm (-10 °C) was observed. The Grignard reagent formation was confirmed by IPC usingΉ-NMR spectroscopy. Once the exotherm subsided, the remainder of the solution was added slowly, maintaining the batch temperature <15 °C. The addition required ~3.5 h. The resulting dark green mixture was decanted into a 2 L capped bottle.
[00339] All equivalent and volume descriptors in this part are based on a 500g reaction. A 22 L reactor was charged with a solution of benzyl chloromethyl ether (95%, 375 g, 2.31 mol, 0.8 equiv) in THF (1.5 L, 3 vol). The reactor was cooled in an ice-water bath. Two Grignard reagent batches prepared as described above were combined and then added slowly to the benzyl chloromethyl ether solution via an addition funnel, maintaining the batch temperature below 25 °C. The addition required 1.5 h. The reaction mixture was stirred overnight (16 h).
[00340] All equivalent and volume descriptors in this part are based on a 1 kg reaction. A solution of 15%» ammonium chloride was prepared in a 30 L jacketed reactor (1.5 kg in 8.5 kg of water, 10 vol). The solution was cooled to 5 °C. Two Grignard reaction mixtures prepared as described above were combined and then transferred into the ammonium chloride solution via a header vessel. An exotherm was observed in this quench, which was carried out at a rate such as to keep the internal temperature below 25 °C. Once the transfer was complete, the vessel jacket temperature was set to 25 °C. Hexanes (8 L, 8 vol) was added and the mixture was stirred for 0.5 h. After settling the phases, the aqueous phase (pH 9) was drained off and discarded. The remaining organic phase was washed with water (2 L, 2 vol). The organic phase was concentrated in vacuo using a 22 L rotary evaporator, providing the crude product as an orange oil.
[00341] Method B
[00342] Magnesium turnings (106 g, 4.35 mol, 1.0 eq) were charged to a 22 L reactor and then suspended in THF (760 mL, 1 vol). The vessel was cooled in an ice-water bath such that the batch temperature reached 2 °C. A solution of the propargyl chloride (760 g, 4.35 mol, 1.0 equiv) in THF (4.5 L, 6 vol) was added slowly to the reactor. After 100 mL was added, the addition was stopped and the mixture stirred until a 13 °C exotherm was observed, indicating the Grignard reagent initiation. Once the exotherm subsided, another 500 mL of the propargyl chloride solution was added slowly, maintaining the batch temperature <20 °C. The Grignard reagent formation was confirmed by IPC using Ή-NMR spectroscopy. The remainder of the propargyl chloride solution was added slowly, maintaining the batch temperature <20 °C. The addition required -1.5 h. The resulting dark green solution was stirred for 0.5 h. The Grignard reagent formation was confirmed by IPC using Ή-NMR spectroscopy. Neat benzyl
chloromethyl ether was charged to the reactor addition funnel and then added dropwise into the reactor, maintaining the batch temperature below 25 °C. The addition required 1.0 h. The reaction mixture was stirred overnight. The aqueous work-up and concentration was carried out using the same procedure and relative amounts of materials as in Method A to give the product as an orange oil.
[00343] Syntheisis of 4-Benzyloxy-3,3-dimethylbut-l-yne.

2 steps
[00344] A 30 L jacketed reactor was charged with methanol (6 vol) which was then cooled to 5 °C. Potassium hydroxide (85%, 1.3 equiv) was added to the reactor. A 15-20 °C exotherm was observed as the potassium hydroxide dissolved. The jacket temperature was set to 25 °C. A solution of 4-benzyloxy-3,3-dimethyl-l-trimethylsilylbut-l-yne (1.0 equiv) in methanol (2 vol) was added and the resulting mixture was stirred until reaction completion, as monitored by HPLC. Typical reaction time at 25 °C is 3-4 h. The reaction mixture is diluted with water (8 vol) and then stirred for 0.5 h. Hexanes (6 vol) was added and the resulting mixture was stirred for 0.5 h. The phases were allowed to settle and then the aqueous phase (pH 10-11) was drained off and discarded. The organic phase was washed with a solution of KOH (85%, 0.4 equiv) in water (8 vol) followed by water (8 vol). The organic phase was then concentrated down using a rotary evaporator, yielding the title material as a yellow-orange oil. Typical purity of this material is in the 80% range with primarily a single impurity present. Ή NMR (400 MHz, C6D6) δ 7.28 (d, 2 H, J = 7.4 Hz), 7.18 (t, 2 H, J= 7.2 Hz), 7.10 (d, 1H, J= 7.2 Hz), 4.35 (s, 2 H), 3.24 (s, 2 H), 1.91 (s, 1 H), 1.25 (s, 6 H).
[00345] Synthesis of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6- fluoroindole.
[00346] Method A
[00347] Synthesis of Benzylglycolated 4-Amino-2-(4-benzyloxy-3,3-dimethyIbut- l-ynyl)-5- fluoroaniline.

[00348] Benzylglycolated 4-ammonium-2-bromo-5-flouroaniline tosylate salt was freebased by stirring the solid in EtOAc (5 vol) and saturated NaHCC>3 solution (5 vol) until clear organic layer was achieved. The resulting layers were separated and the organic layer was washed with saturated NaHC03 solution (5 vol) followed by brine and concentrated in vacuo to obtain benzylglocolated 4-ammonium-2-bromo-5-flouroaniline tosylate salt as an oil.
[00349] Then, a flask was charged with benzylglycolated 4-ammonium-2-bromo-5- flouroaniline tosylate salt (freebase, 1.0 equiv), Pd(OAc) (4.0 mol%), dppb (6.0 mol%) and powdered K2CO3 (3.0 equiv) and stirred with acetonitrile (6 vol) at room temperature. The resulting reaction mixture was degassed for approximately 30 min by bubbling in N2 with vent. Then 4-benzyloxy-3,3-dimethylbut-l-yne (1.1 equiv) dissolved in acetonitrile (2 vol) was added in a fast stream and heated to 80 °C and stirred until complete consumption of 4-ammonium-2- bromo-5-flouroaniline tosylate salt was achieved. The reaction slurry was cooled to room temperature and filtered through a pad of Celite and washed with acetonitrile (2 vol). Filtrate was concentrated in vacuo and the residue was redissolved in EtOAc (6 vol). The organic layer was washed twice with NH4CI solution (20% w/v, 4 vol) and brine (6 vol). The resulting organic layer was concentrated to yield brown oil and used as is in the next reaction.
[00350] Synthesis of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6- fluoroindole.

[00351] Crude oil of benzylglycolated 4-amino-2-(4-benzyloxy-3,3-dimethylbut-l-ynyl)-5- fluoroaniline was dissolved in acetonitrile (6 vol) and added (MeCN)2PdCl2 (15 mol%) at room temperature. The resulting mixture was degassed using N2 with vent for approximately 30 min. Then the reaction mixture was stirred at 80 °C under N2 blanket overnight. The reaction mixture was cooled to room temperature and filtered through a pad of Celite and washed the cake with acetonitrile (1 vol). The resulting filtrate was concentrated in vacuo and redissolved in EtOAc (5 vol). Deloxane-II THP (5 wt% based on the theoretical yield of N-benzylglycolated-5-amino-2- (2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole) was added and stirred at room temperature overnight. The mixture was then filtered through a pad of silica (2.5 inch depth, 6 inch diameter filter) and washed with EtOAc (4 vol). The filtrate was concentrated down to a dark brown residue, and used as is in the next reaction.
[00352] Repurification of crude N-benzylglycolated-5-amino-2-(2-benzyloxy- 1,1- dimethylethyl)-6-fluoroindole:
[00353] The crude N-benzylglycolated-5-amino-2-(2-benzyloxy- 1 , l-dimethylethyl)-6- fluoroindole was dissolved in dichloromethane (~1.5 vol) and filtered through a pad of silica initially using 30% EtOAc/heptane where impurities were discarded. Then the silica pad was washed with 50% EtO Ac/heptane to isolate N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l- dimethylethyl)-6-fluoroindole until faint color was observed in the filtrate. This filtrate was concentrated in vacuo to afford brown oil which crystallized on standing at room temperature. Ή NMR (400 MHz, DMSO) 6 7.38-7.34 (m, 4 H), 7.32-7.23 (m, 6 H), 7.21 (d, 1 H, J= 12.8 Hz), 6.77 (d, 1H, J= 9.0 Hz), 6.06 (s, 1 H), 5.13 (d, 1H, J = 4.9 Hz), 4.54 (s, 2 H), 4.46 (br. s, 2 H), 4.45 (s, 2 H), 4.33 (d, 1 H, J= 12.4 Hz), 4.09-4.04 (m, 2 H), 3.63 (d, 1H, J= 9.2 Hz), 3.56 (d, 1H, J= 9.2 Hz), 3.49 (dd, 1H, J= 9.8, 4.4 Hz), 3.43 (dd, 1H, J= 9.8, 5.7 Hz), 1.40 (s, 6 H).
[00354] Synthesis of N-benzyIglycolated-5-amino-2-(2-benzyIoxy-l,l-diniethylethyl)-6- fluoroindole.
[00355] Method B

2. (MeCN)2PdCl2
MeCN, 80 <€
3. Silica gel filtration
[00356] Palladium acetate (33 g, 0.04 eq), dppb (94 g, 0.06 eq), and potassium carbonate (1.5 kg, 3.0 eq) are charged to a reactor. The free based oil benzylglocolated 4-ammonium-2-bromo- 5-flouroaniline (1.5 kg, 1.0 eq) was dissolved in acetonitrile (8.2 L, 4.1 vol) and then added to the reactor. The mixture was sparged with nitrogen gas for NLT 1 h. A solution of 4-benzyloxy- 3,3-dimethylbut-l-yne (70%), 1.1 kg, 1.05 eq) in acetonitrile was added to the mixture which was then sparged with nitrogen gas for NLT 1 h. The mixture was heated to 80 °C and then stirred overnight. IPC by HPLC is carried out and the reaction is determined to be complete after 16 h. The mixture was cooled to ambient temperature and then filtered through a pad of Celite (228 g). The reactor and Celite pad were washed with acetonitrile (2 x 2 L, 2 vol). The combined phases are concentrated on a 22 L rotary evaporator until 8 L of solvent have been collected, leaving the crude product in 7 L (3.5 vol) of acetonitrile. [00357] 5 s-acetonitriledichloropalladium ( 144 g, 0.15 eq) was charged to the reactor. The crude solution was transferred back into the reactor and the roto-vap bulb was washed with acetonitrile (4 L, 2 vol). The combined solutions were sparged with nitrogen gas for NLT 1 h. The reaction mixture was heated to 80 °C for NLT 16 h. In process control by HPLC shows complete consumption of starting material. The reaction mixture was filtered through Celite (300 g). The reactor and filter cake were washed with acetonitrile (3 L, 1.5 vol). The combined filtrates were concentrated to an oil by rotary evaporation. The oil was dissolved in ethyl acetate (8.8 L, 4.4 vol). The solution was washed with 20% ammonium chloride (5 L, 2.5 vol) followed by 5% brine (5 L, 2.5 vol). Silica gel (3.5 kg, 1.8 wt. eq.) of silica gel was added to the organic phase, which was stirred overnight. Deloxan THP II metal scavenger (358 g) and heptane (17.6 L) were added and the resulting mixture was stirred for NLT 3 h. The mixture was filtered through a sintered glass funnel. The filter cake was washed with 30% ethyl acetate in heptane (25 L). The combined filtrates were concentrated under reduced pressure to give N- benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole as a brown paste ( 1.4 kgl.Svnthesis of Compound 1
[00358] Synthesis of benzyl protected Compound 1.



[00359] 1 -(2,2-difluoro- 1 ,3 -benzodioxol-5-yl)-cyclopropanecarboxylic acid (1.3 equiv) was slurried in toluene (2.5 vol, based on l-(2,2-difluoro-l,3-benzodioxol-5-yi)- cyclopropanecarboxylic acid) and the mixture was heated to 60 °C. SOCl2 (1.7 equiv) was added via addition runnel. The resulting mixture was stirred for 2 hr. The toluene and the excess
SOCI2 were distilled off using rotavop. Additional toluene (2.5 vol, based on l-(2,2-difluoro- l,3-benzodioxol-5-yl)-cyclopropanecarboxylic acid) was added and distilled again. The crude acid chloride was dissolved in dichloromethane (2 vol) and added via addition funnel to a mixture of N-benzylglycolated-5-amino-2-(2-benzyloxy-l,l-dimethylethyl)-6-fluoroindole (1.0 equiv), and triethylamine (2.0 equiv) in dichloromethane (7 vol) while maintaining 0-3 °C (internal temperature). The resulting mixture was stirred at 0 °C for 4 hrs and then warmed to room temperature overnight. Distilled water (5 vol) was added to the reaction mixture and stirred for NLT 30 min and the layers were separated. The organic phase was washed with 20 wt% K2CO3 (4 vol x 2) followed by a brine wash (4 vol) and concentrated to afford crude benzyl protected Compound 1 as a thick brown oil, which was purified further using silica pad filtration.
[00360] Silica gel pad filtration: Crude benzyl protected Compound 1 was dissolved in ethyl acetate (3 vol) in the presence of activated carbon Darco-G (10wt%, based on theoretical yield of benzyl protected Compound 1) and stirred at room temperature overnight. To this mixture was added heptane (3 vol) and filtered through a pad of silica gel (2x weight of crude benzyl protected Compound 1). The silica pad was washed with ethyl acetate/heptane (1:1, 6 vol) or until little color was detected in the filtrate. The filtrate was concentrated in vacuo to afford benzyl protected Compound 1 as viscous reddish brown oil, and used directly in the next step.
[00361] Repurification: Benzyl protected Compound 1 was redissolved in dichloromethane (1 vol, based on theoretical yield of benzyl protected Compound 1) and loaded onto a silica gel pad (2x weight of crude benzyl protected Compound 1). The silica pad was washed with
dichloromethane (2 vol, based on theoretical yield of benzyl protected Compound 1) and the filtrate was discarded. The silica pad was washed with 30% ethyl acetate/heptane (5 vol) and the filtrate was concentrated in vacuo to afford benzyl protected Compound 1 as viscous reddish orange oil, and used directly in the next step. [00362] Synthesis of Compound 1.

OBn 4 steps

[00363] Method A
[00364] A 20 L autoclave was flushed three times with nitrogen gas and then charged with palladium on carbon (Evonik E 101 NN/W, 5% Pd, 60% wet, 200 g, 0.075 mol, 0.04 equiv). The autoclave was then flushed with nitrogen three times. A solution of crude benzyl protected Compound 1 (1.3 kg, ~ 1.9 mol) in THF (8 L, 6 vol) was added to the autoclave via suction. The vessel was capped and then flushed three times with nitrogen gas. With gentle stirring, the vessel was flushed three times with hydrogen gas, evacuating to atmosphere by diluting with nitrogen. The autoclave was pressurized to 3 Bar with hydrogen and the agitation rate was increased to 800 rpm. Rapid hydrogen uptake was observed (dissolution). Once uptake subsided, the vessel was heated to 50 °C.
[00365] For safety purposes, the thermostat was shut off at the end of every work-day. The vessel was pressurized to 4 Bar with hydrogen and then isolated from the hydrogen tank.
[00366] After 2 full days of reaction, more Pd / C (60 g, 0.023 mol, 0.01 equiv) was added to the mixture. This was done by flushing three times with nitrogen gas and then adding the catalyst through the solids addition port. Resuming the reaction was done as before. After 4 full days, the reaction was deemed complete by HPLC by the disappearance of not only the starting material but also of the peak corresponding to a mono-benzylated intermediate. [00367] The reaction mixture was filtered through a Celite pad. The vessel and filter cake were washed with THF (2 L, 1.5 vol). The Celite pad was then wetted with water and the cake discarded appropriately. The combined filtrate and THF wash were concentrated using a rotary evaporator yielding the crude product as a black oil, 1 kg.
[00368] The equivalents and volumes in the following purification are based on 1 kg of crude material. The crude black oil was dissolved in 1 :1 ethyl acetate-heptane. The mixture was charged to a pad of silica gel (1.5 kg, 1.5 wt. equiv) in a fritted funnel that had been saturated with 1 :1 ethyl acetate-heptane. The silica pad was flushed first with 1 :1 ethyl acetate-heptane (6 L, 6 vol) and then with pure ethyl acetate (14 L, 14 vol). The eluent was collected in 4 fractions which were analyzed by HPLC.
[00369] The equivalents and volumes in the following purification are based on 0.6 kg of crude material. Fraction 3 was concentrated by rotary evaporation to give a brown foam (600 g) and then redissolved in MTBE (1.8 L, 3 vol). The dark brown solution was stirred overnight at ambient temperature, during which time, crystallization occurred. Heptane (55 mL, 0.1 vol) was added and the mixture was stirred overnight. The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (900 mL, 1.5 vol). The filter cake was air-dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 253 g of Compound 1 as an off-white solid.
[00370] The equivalents and volumes for the following purification are based on 1.4 kg of crude material. Fractions 2 and 3 from the above silica gel filtration as well as material from a previous reaction were combined and concentrated to give 1.4 kg of a black oil. The mixture was resubmitted to the silica gel filtration (1.5 kg of silica gel, eluted with 3.5 L, 2.3 vol of 1 :1 ethyl acetate-heptane then 9 L, 6 vol of pure ethyl acetate) described above, which upon concentration gave a tan foamy solid (390 g).
[00371] The equivalents and volumes for the following purification are based on 390 g of crude material. The tan solid was insoluble in MTBE, so was dissolved in methanol (1.2 L, 3 vol). Using a 4 L Morton reactor equipped with a long-path distillation head, the mixture was distilled down to 2 vol. MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 2 vol. A second portion of MTBE (1.6 L, 4 vol) was added and the mixture was distilled back down to 2 vol. A third portion of MTBE (1.2 L, 3 vol) was added and the mixture was distilled back down to 3 vol. Analysis of the distillate by GC revealed it to consist of -6% methanol. The thermostat was set to 48 °C (below the boiling temp of the MTBE-methanol azeotrope, which is 52 °C). The mixture was cooled to 20 °C over 2 h, during which time a relatively fast crystallization occurred. After stirring the mixture for 2 h, heptane (20 mL, 0.05 vol) was added and the mixture was stirred overnight (16 h). The mixture was filtered using a Buchner funnel and the filter cake was washed with 3:1 MTBE-heptane (800 mL, 2 vol). The filter cake was air- dried for 1 h and then vacuum dried at ambient temperature for 16 h, furnishing 130 g of Compound 1 as an off-white solid.
[00372] Method B
[00373] Benzyl protected Compound 1 was dissolved in THF (3 vol) and then stripped to dryness to remove any residual solvent. Benzyl protected Compound 1 was redissolved in THF (4 vol) and added to the hydrogenator containing 5 wt% Pd/C (2.5 mol%, 60% wet, Degussa E5 El 01 N /W). The internal temperature of the reaction was adjusted to 50 °C, and flushed with N2 (x5) followed by hydrogen (x3). The hydrogenator pressure was adjusted to 3 Bar of hydrogen and the mixture was stirred rapidly (>1100 rpm). At the end of the reaction, the catalyst was filtered through a pad of Celite and washed with THF (1 vol). The filtrate was concentrated in vacuo to obtain a brown foamy residue. The resulting residue was dissolved in MTBE (5 vol) and 0.5N HC1 solution (2 vol) and distilled water (1 vol) were added. The mixture was stirred for NLT 30 min and the resulting layers were separated. The organic phase was washed with 10wt% K2CO3 solution (2 vol x2) followed by a brine wash. The organic layer was added to a flask containing silica gel (25 wt%), Deloxan-THP II (5wt%, 75% wet), and
Na2S04 and stirred overnight. The resulting mixture was filtered through a pad of Celite and washed with 10%THF/MTBE (3 vol). The filtrate was concentrated in vacuo to afford crude Compound 1 as pale tan foam.
[00374] Compound 1 recovery from the mother liquor: Option A.
[00375] Silica gel pad filtration: The mother liquor was concentrated in vacuo to obtain a brown foam, dissolved in dichloromethane (2 vol), and filtered through a pad of silica (3x weight of the crude Compound 1). The silica pad was washed with ethyl acetate/heptane (1 :1, 13 vol) and the filtrate was discarded. The silica pad was washed with 10% THF/ethyl acetate (10 vol) and the filtrate was coiicentraied in vacuo to afford Compound 1 as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1.
{00376] Compound 1 recovery from the mother liquor: Option B,
[00377] Silica gel column chromatography: After chromatography on silica gel (50% ethyl acetate/hexaties to 100% ethyl acetate), the desired compound was isolated as pale tan foam. The above crystallization procedure was followed to isolate the remaining Compound 1.
{003781 Additional Recrystaliization of Compound 1
[ 0379‘j Solid Compound 1 (135 kg) was suspended in IPA (5.4 L, 4 vol) and then heated to 82 °C. Upon complete dissolution (visual), heptane (540 mL, 0.4 vol) was added slowly. The mixture was cooled to 58 °C The mixture was then cooled slowly to 51 °C, during which time crystallization occurs. The heat source was shut down and the recrystalfeation mixture was allowed to cool naturally overnight. The mixture was filtered using a benchtop Buclmer funnel and the filter cake was washed with IPA (2.7 L, 2 vol). The filler cake was dried in the tunnel under air flow for 8 h and then was oven-dried in vacuo at 45-50 °C overnight to give 1.02 kg of recrystallized Compound 1 ,
100380] Compound 1 may also be prepared by one of several synthetic routes disclosed in US published patent application U S20090131 92, incorporated herein by reference.
{003811 Table 6 below recites analytical data for Compound 1.
Table 6.

Synthesis of Compound 1 Amorphous Form [00383] Spray-Dried Method
[00384] 9.95g of Hydroxypropylmethylcellulose acetate succinate HG grade (HPMCAS-HG) was weighed into a 500 ml beaker, along with 50 mg of sodium lauryl sulfate (SLS). MeOH (200 ml) was mixed with the solid. The material was allowed to stir for 4 h. To insure maximum dissolution, after 2 h of stirring the solution was sonicated for 5 mins, then allowed to continue stirring for the remaining 2 h. A very fin suspension of HPMCAS remained in solution. However, visual observation determined that no gummy portions remained on the walls of the vessel or stuck to the bottom after tilting the vessel.
[00385] Compound 1 (1 Og) was poured into the 500 ml beaker, and the system was allowed to continue stirring. The solution was spray dried using the following parameters:
Formulation Description: Compound 1 Form A/HPMCAS/SLS (50/49.5/0.5)
Buchi Mini Spray Dryer
T inlet (setpoint) 145 °C
T outlet (start) 75 °C
T outlet (end) 55 °C
Nitrogen Pressure 75 psi
Aspirator 100 %
Pump 35 %
Rotometer 40 mm
Filter Pressure 65 mbar
Condenser Temp -3 °C
Run Time l h
REFERENCES
1: Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS, Lukacs GL. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci Transl Med. 2014 Jul 23;6(246):246ra97. doi: 10.1126/scitranslmed.3008889. PubMed PMID: 25101887.
2: Pettit RS, Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P T. 2014 Jul;39(7):500-11. PubMed PMID: 25083129; PubMed Central PMCID: PMC4103577.
3: Norman P. Novel picolinamide-based cystic fibrosis transmembrane regulator modulators: evaluation of WO2013038373, WO2013038376, WO2013038381, WO2013038386 and WO2013038390. Expert Opin Ther Pat. 2014 Jul;24(7):829-37. doi: 10.1517/13543776.2014.876412. Epub 2014 Jan 7. PubMed PMID: 24392786.
//////TEZACAFTOR, VX 661, PHASE 3, 1152311-62-0, UNII: 8RW88Y506K, deltaF508-CFTR corrector, Vertex, treatment of cystic fibrosis in patients homozygous to the F508del-CFTR mutation
CC(C)(CO)C1=CC2=CC(=C(C=C2N1CC(CO)O)F)NC(=O)C3(CC3)C4=CC5=C(C=C4)OC(O5)(F)F
CC(C)(CO)c1cc2cc(c(cc2n1C[C@H](CO)O)F)NC(=O)C3(CC3)c4ccc5c(c4)OC(O5)(F)F

{kind=link}