WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
update………APPROVED JAPAN 2021, 2021/9/27, Raiatt MIBG-I 131
July 30, 2018
Release
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
“Many patients with these ultra-rare cancers can be treated with surgery or local therapies, but there are no effective systemic treatments for patients who experience tumor-related symptoms such as high blood pressure,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Patients will now have an approved therapy that has been shown to decrease the need for blood pressure medication and reduce tumor size in some patients.”
Pheochromocytomas are rare tumors of the adrenal glands. These glands are located right above the kidneys and make hormones including stress hormones called epinephrines and norepinephrines. Pheochromocytomas increase the production of these hormones, leading to hypertension (high blood pressure) and symptoms such as headaches, irritability, sweating, rapid heart rate, nausea, vomiting, weight loss, weakness, chest pain or anxiety. When this type of tumor occurs outside the adrenal gland, it is called a paraganglioma.
The efficacy of Azedra was shown in a single-arm, open-label, clinical trial in 68 patients that measured the number of patients who experienced a 50 percent or greater reduction of all antihypertensive medications lasting for at least six months. This endpoint was supported by the secondary endpoint, overall tumor response measured by traditional imaging criteria. The study met the primary endpoint, with 17 (25 percent) of the 68 evaluable patients experiencing a 50 percent or greater reduction of all antihypertensive medication for at least six months. Overall tumor response was achieved in 15 (22 percent) of the patients studied.
The most common severe side effects reported by patients receiving Azedra in clinical trials included low levels of white blood cells (lymphopenia), abnormally low count of a type of white blood cells (neutropenia), low blood platelet count (thrombocytopenia), fatigue, anemia, increased international normalized ratio (a laboratory test which measures blood clotting), nausea, dizziness, hypertension and vomiting.
As it is a radioactive therapeutic agent, Azedra includes a warning about radiation exposure to patients and family members, which should be minimized while the patient is receiving Azedra. The risk of radiation exposure is greater in pediatric patients. Other warnings and precautions include a risk of lower levels of blood cells (myelosuppression), underactive thyroid, elevations in blood pressure, renal failure or kidney injury and inflammation of lung tissue (pneumonitis). Myelodysplastic syndrome and acute leukemias, which are cancers of the blood and bone marrow, were observed in patients who received Azedra, and the magnitude of this risk will continue to be studied. Azedra can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception after receiving Azedra. Radiation exposure associated with Azedra may cause infertility in males and females.
The FDA granted this application Fast Track, Breakthrough Therapy and Priority Review designations. Azedra also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Azedra to Progenics Pharmaceuticals, Inc.
AdreView (iobenguane I 123 Injection) is a sterile, pyrogen-free radiopharmaceutical for intravenous injection. Each mL contains 0.08 mg iobenguane sulfate, 74 MBq (2 mCi) of I 123 (as iobenguane sulfate I 123) at calibration date and time on the label, 23 mg sodium dihydrogen phosphate dihydrate, 2.8 mg disodium hydrogen phosphate dihydrate and 10.3 mg (1% v/v) benzyl alcohol with a pH of 5.0 – 6.5. Iobenguane sulfate I 123 is also known as I 123 meta-iodobenzlyguanidine sulfate and has the following structural formula:
Physical Characteristics
Iodine 123 is a cyclotron-produced radionuclide that decays to Te 123 by electron capture and has a physical half-life of 13.2 hours.
Iobenguane I-131 is a guanidine analog with specific affinity for tissues of the sympathetic nervous system and related tumors. The radiolabeled forms are used as antineoplastic agents and radioactive imaging agents. (Merck Index, 12th ed) MIBG serves as a neuron-blocking agent which has a strong affinity for, and retention in, the adrenal medulla and also inhibits ADP-ribosyltransferase.
Iobenguane i-131 is a Radioactive Diagnostic Agent. The mechanism of action of iobenguane i-131 is as a Radiopharmaceutical Activity.
Iobenguane I-131 is an I 131 radioiodinated synthetic analogue of the neurotransmitter norepinephrine. Iobenguane localizes to adrenergic tissue and, in radioiodinated forms, may be used to image or eradicate tumor cells that take up and metabolize norepinephrine.
The radioisotope of iodine used for the label can be iodine-123 (for imaging purposes only) or iodine-131 (which must be used when tissue destruction is desired, but is sometimes used for imaging also).
Pheochromocytoma seen as dark sphere in center of the body (it is in the left adrenal gland). Image is by MIBG scintigraphy, with radiation from radioiodine in the MIBG. Two images are seen of the same patient from front and back. Note dark image of the thyroid due to unwanted uptake of iodide radioiodine from breakdown of the pharmaceutical, by the thyroid gland in the neck. Uptake at the side of the head are from the salivary glands. Radioactivity is also seen in the bladder, from normal renal excretion of iodide.
It localizes to adrenergic tissue and thus can be used to identify the location of tumors[2] such as pheochromocytomas and neuroblastomas. With I-131 it can also be used to eradicate tumor cells that take up and metabolize norepinephrine.
Thyroid precautions
Thyroid blockade with (nonradioactive) potassium iodide is indicated for nuclear medicine scintigraphy with iobenguane/mIBG. This competitively inhibits radioiodine uptake, preventing excessive radioiodine levels in the thyroid and minimizing the risk of thyroid ablation ( in the case of I-131). The minimal risk of thyroid carcinogenesis is also reduced as a result.
The FDA-approved dosing of potassium iodide for this purpose are as follows: infants less than 1 month old, 16 mg; children 1 month to 3 years, 32 mg; children 3 years to 18 years, 65 mg; adults 130 mg.[3] However, some sources recommend alternative dosing regimens.[4]
Not all sources are in agreement on the necessary duration of thyroid blockade, although agreement appears to have been reached about the necessity of blockade for both scintigraphic and therapeutic applications of iobenguane. Commercially available iobenguane is labeled with iodine-123, and product labeling recommends administration of potassium iodide 1 hour prior to administration of the radiopharmaceutical for all age groups,[5] while the European Associated of Nuclear Medicine recommends (for iobenguane labeled with either I-131 or I-123,) that potassium iodide administration begin one day prior to radiopharmaceutical administration, and continue until the day following the injection, with the exception of newborns, who do not require potassium iodide doses following radiopharmaceutical injection.[4]
Product labeling for diagnostic iodine-131 iobenguane recommends potassium iodide administration one day before injection and continuing 5 to 7 days following.[6] Iodine-131 iobenguane used for therapeutic purposes requires a different pre-medication duration, beginning 24–48 hours prior to iobenguane injection and continuing 10–15 days following injection.[7]
Alternative imaging modality for pheochromocytoma
The FDOPAPET/CT scan has proven to be nearly 100% sensitive for detection of pheochromocytomas, vs. 90% for MIBG scans.[8][9][10] Centers which offer FDOPA PET/CT, however, are rare.
Clinical trials
Iobenguane I 131 for cancers
Iobenguane I 131 (as Azedra) has had a clinical trial as a treatment for malignant, recurrent or unresectable pheochromocytoma and paraganglioma, and the US FDA has granted it a Priority Review.[11]
Percent Composition: C 34.93%, H 3.66%, I 46.13%, N 15.28%
Literature References: Norepinephrine analog with specific affinity for tissues of sympathetic nervous system and related tumors; prepd as 123I and 131I labeled forms. Prepn and imaging studies: D. M. Wieland et al.,J. Nucl. Med.21, 349 (1980); eidem,US4584187 (1986). Improved synthesis: P. A. P. M. van Doremalen, A. G. M. Janssen, J. Radioanal. Nucl. Chem. Lett.96, 97 (1985). Metabolism in man: T. J. Mangner et al.,J. Nucl. Med.27, 37 (1986). HPLC determn in serum and urine: D. Schwabe et al.,J. Chromatogr.487, 177 (1989). Radiopharmacokinetics: S. Ertl et al.,Nucl. Med. Commun.8, 643 (1987). Clinical evaluation of myocardial imaging: D. Fagret et al.,Eur. J. Nucl. Med.15, 624 (1989). Diagnostic use in pheochromocytoma: B. Shapiro et al.,J. Nucl. Med.26, 576 (1985); therapeutic use: M. Krempf et al.,J. Clin. Endocrinol. Metab.72, 455 (1991). Symposia on therapeutic and diagnostic use in neuroblastoma: Advances in Neuroblastoma Research2, A. E. Evans et al., Eds. (Alan R. Liss, Inc., New York, 1988) p 643-726; Med. Pediatr. Oncol.15, 157-228 (1987). Review of pharmacology: J. C. Sisson, D. M. Weiland, Am. J. Physiol. Imaging1, 96-103 (1986); of biodistribution and clinical studies: A. R. Wafelman et al.,Eur. J. Nucl. Med.21, 545-559 (1994); of therapeutic use in neural crest tumors: L. Troncone, V. Rufini, Anticancer Res.17, 1823-1832 (1997).
Derivative Type: Sulfate
Molecular Formula: (C8H10IN3)2.H2SO4
Molecular Weight: 648.26
Percent Composition: C 29.64%, H 3.42%, I 39.15%, N 12.96%, S 4.95%, O 9.87%
Properties: Colorless crystals from water + ethanol, mp 166-167°.
Melting point: mp 166-167°
Therap-Cat: Radiolabeled forms as antineoplastic; diagnostic aid (radioactive imaging agent).
Jump up^Scarsbrook AF, Ganeshan A, Statham J, et al. (2007). “Anatomic and functional imaging of metastatic carcinoid tumors”. Radiographics. 27 (2): 455–77. doi:10.1148/rg.272065058. PMID17374863.
Jump up^Kowalsky RJ, Falen, SW. Radiopharmaceuticals in Nuclear Pharmacy and Nuclear Medicine. 2nd ed. Washington DC: American Pharmacists Association; 2004.
Jump up^6-[18FFluorodopamine Positron Emission Tomographic (PET) Scanning for Diagnostic Localization of Pheochromocytoma. Pacek et al. 2001] full text
Jump up^Luster M, Karges W, Zeich K, Pauls S, Verburg FA, Dralle H; et al. (2010). “Clinical value of (18)F-fluorodihydroxyphenylalanine positron emission tomography/computed tomography ((18)F-DOPA PET/CT) for detecting pheochromocytoma”. European journal of nuclear medicine and molecular imaging. 37 (3): 484–93. doi:10.1007/s00259-009-1294-7. PMID19862519.
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
“Many patients with these ultra-rare cancers can be treated with surgery or local therapies, but there are no effective systemic treatments for patients who experience tumor-related symptoms such as high blood pressure,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Patients will now have an approved therapy that has been shown to decrease the need for blood pressure medication and reduce tumor size in some patients.”
Pheochromocytomas are rare tumors of the adrenal glands. These glands are located right above the kidneys and make hormones including stress hormones called epinephrines and norepinephrines. Pheochromocytomas increase the production of these hormones, leading to hypertension (high blood pressure) and symptoms such as headaches, irritability, sweating, rapid heart rate, nausea, vomiting, weight loss, weakness, chest pain or anxiety. When this type of tumor occurs outside the adrenal gland, it is called a paraganglioma.
The efficacy of Azedra was shown in a single-arm, open-label, clinical trial in 68 patients that measured the number of patients who experienced a 50 percent or greater reduction of all antihypertensive medications lasting for at least six months. This endpoint was supported by the secondary endpoint, overall tumor response measured by traditional imaging criteria. The study met the primary endpoint, with 17 (25 percent) of the 68 evaluable patients experiencing a 50 percent or greater reduction of all antihypertensive medication for at least six months. Overall tumor response was achieved in 15 (22 percent) of the patients studied.
The most common severe side effects reported by patients receiving Azedra in clinical trials included low levels of white blood cells (lymphopenia), abnormally low count of a type of white blood cells (neutropenia), low blood platelet count (thrombocytopenia), fatigue, anemia, increased international normalized ratio (a laboratory test which measures blood clotting), nausea, dizziness, hypertension and vomiting.
As it is a radioactive therapeutic agent, Azedra includes a warning about radiation exposure to patients and family members, which should be minimized while the patient is receiving Azedra. The risk of radiation exposure is greater in pediatric patients. Other warnings and precautions include a risk of lower levels of blood cells (myelosuppression), underactive thyroid, elevations in blood pressure, renal failure or kidney injury and inflammation of lung tissue (pneumonitis). Myelodysplastic syndrome and acute leukemias, which are cancers of the blood and bone marrow, were observed in patients who received Azedra, and the magnitude of this risk will continue to be studied. Azedra can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception after receiving Azedra. Radiation exposure associated with Azedra may cause infertility in males and females.
The FDA granted this application Fast Track, Breakthrough Therapy and Priority Review designations. Azedra also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Azedra to Progenics Pharmaceuticals, Inc.
TAVALISSE™
(fostamatinib disodium hexahydrate) Tablets, for Oral Use
DESCRIPTION
Fostamatinib is a tyrosine kinase inhibitor. TAVALISSE is formulated with the disodium hexahydrate salt of fostamatinib, a phosphate prodrug that converts to its pharmacologically active metabolite, R406, in vivo.
The chemical name for fostamatinib disodium hexahydrate is disodium (6-[[5-fluoro-2-(3,4,5trimethoxyanilino) pyrimidin-4-yl]amino]-2,2-dimethyl-3-oxo-pyrido[3,2-b][1,4]oxazin-4-yl)methyl phosphate hexahydrate. The molecular formula is C23H24FN6Na2O9P·6H2O, and the molecular weight is 732.52. The structural formula is:
Fostamatinib disodium is a white to off-white powder that is practically insoluble in pH 1.2 aqueous buffer, slightly soluble in water, and soluble in methanol.
Each TAVALISSE oral tablet contains 100 mg or 150 mg fostamatinib, equivalent to 126.2 mg or 189.3 mg fostamatinib disodium hexahydrate, respectively.
The inactive ingredients in the tablet core are mannitol, sodium bicarbonate, sodium starch glycolate, povidone, and magnesium stearate. The inactive ingredients in the film coating are polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350, talc, iron oxide yellow, and iron oxide red.
Fostamatinib has been investigated for the treatment and basic science of Rheumatoid Arthritis and Immune Thrombocytopenic Purpura (ITP). It was approved on April 17, 2018 under the trade name Tavalisse for use in ITP [8]. Fostamatinib has also been granted orphan drug status by the FDA [8].
Fostamatinib is indicated for use in the treatment of chronic immune thrombocytopenia (ITP) in patients who have had insufficient response to previous therapy [Label].
Syk is a protein tyrosine kinase associated with various inflammatory cells, including macrophages, which are presumed to be the cells responsible for ITP platelet clearance.[3] When FcγRs I, IIA, and IIIA bind to their ligands, the receptor complex becomes activated and triggers the phosphorylation of the immunoreceptor-activating motifs (ITAMs). This leads to various genes becoming activated, which causes a cytoskeletal rearrangement that mediates phagocytosis in cells of the monocyte/macrophage lineage. Because Syk plays an important role in FcγR-mediated signal transduction and inflammatory propagation, it is considered a good target for the inhibition of various autoimmune conditions, including rheumatoid arthritis and lymphoma.
The investigation of fostamatinib began with studies involving the treatment of mouse models with cytopenia. Mice were used to measure the effectiveness of R788, a small molecule prodrug of the biologically active R406, a Syk inhibitor. In animal models, treatment with R406/R788 was shown to be safe and effective in reducing inflammation and joint damage in immune-mediated rheumatoid arthritis. The models responded favorably to treatment so the study progressed to Phase 2 trials involving humans. Human studies have shown that R788 has good oral bioavailability, biologic activity, is well tolerated, and does not exhibit collagen or ADP-induced platelet aggregation. In NCT00706342, 16 adults with chronic ITP were entered into an open-label, single-arm cohort dose-escalation trials beginning with 75 mg and rising to 175 mg twice a day. The dose was increased until a persistent response was evident, toxicity was reached, or 175 mg twice a day was met. 8 patients achieved persistent responses with platelet counts greater than 50,000 mm3/L on more than 67% of their visits. 3 of these patients had not persistently responded to thrombopoietic agents. 4 others had nonsustained responses. Mean peak platelet count exceeded 100,000 mm3/L in these 12 patients. Toxicity was evidenced primarily in GI-related side effects, notable diarrhea, urgency, and vomiting. 2 patients developed transaminitis.[5]
Rheumatoid arthritis
A phase II study of rheumatoid arthritis patients failing to respond to a biologic agent showed little efficacy as compared to placebo, but the drug was well tolerated. In patients with high inflammatory burden, measured by levels of C-reactive protein, ACR20 was achieved by a significantly higher portion of those in the fostamatinib group (42%) versus the placebo group (26%).[6]
Autoimmune thrombocytopenia
Immune thrombocytopenic purpura (ITP) is an autoimmune disease where the immune system attacks and destroys platelets in the blood, causing abnormally low platelet counts. It is characterized by the antibody-mediated destruction of platelets. Patients with ITP have accelerated clearance of circulating IgG-coated platelets via Fcγ receptor-bearing macrophages in the spleen and liver, leading to different levels of thrombocytopenia and variable degrees of mucocutaneous bleeding.[7] Recent studies of ITP pathophysiology suggest decreased platelet production may also be an important component of the thrombocytopenia. Many patients exhibit responses to established therapies, including corticosteroids, IV immunoglobulin, anti-D, splenectomy, and rituximab. However, there are a significant minority of patients who retain persistently low platelet counts despite treatment. These patients are consistently at risk of intracranial hemorrhage and other bleeding complications. Several thrombopoiesis-stimulating therapies including eltrombopag and AMG 531 are being investigated to help combat low platelet counts in ITP patients. Rigel reported results from two Phase III clinical trials for fostamatinib as an ITP treatment in August and October 2016. The study is the second Phase 3, multi-center, randomized, double-blind, placebo controlled, study of fostamatinib disodium in the treatment of persistent/chronic immune thrombocytopenic purpura that Rigel has conducted. Primary outcome measures are defined as a stable platelet response by the end of the study (week 24) of at least 50,000/µL on at least 4 of the 6 visits between weeks 14-24. Participants received either a placebo, 100 mg, or 150 mg of the drug in the morning and evening for 24 full weeks. The first study, FIT 1 (047) met the primary endpoint in a statistically significant manner, with 18% of patients hitting the 50,000 platelets/µL of blood and no patients receiving the placebo meeting that criteria. As of June 2016, the open-label, long term extension study (049) is currently tracking 118 patients who opted to receive fostamatinib after completing either study 047 or 048.[8]
Autoimmune hemolytic anemia
Approval for treatment of autoimmune hemolytic anemia (AIHA) is in Stage 1 of Phase II trials. This study is a Phase 2, multi-center, open label, Simon two-stage study to evaluate the safety and efficacy of fostamatinib disodium in the treatment of warm antibody autoimmune hemolytic anemia. Primary outcome measures examined include a hemoglobin response measured by levels higher than 10 g/dL and 2 g/dL higher than the baseline hemoglobin. Responses were studied for a period of 12 weeks and for a dose of 150 mg in the morning and evening. The study began in April 2016 and is estimated to conclude in September 2017. The study is currently recruiting participants from U.S. states including Arizona, California, D.C., Massachusetts, New York, North Carolina, and Texas. Subjects must have had a diagnosis of primary or secondary warm antibody AIHA, and must have failed at least 1 prior treatment regimen for AIHA. Subjects cannot have a platelet count less than 30,000/µL, have AIHA secondary to autoimmune disease, have uncontrolled or poorly controlled hypertension, or have cold antibody AIHA, cold agglutinin syndrome, mixed type AIHA, or paroxysmal cold hemoglobinuria.[9]
Immunoglobulin A nephropathy
Fostamatinib as a treatment for IgA nephropathy (IgAN) is in Phase II trials, which will conclude at the end of 2016. IgAN is a chronic autoimmune disease associated with inflammation in the kidneys that reduces their ability to successfully filter blood. There are currently no disease-targeted therapies for IgAN. Participants are currently being recruited from the US, Austria, Germany, Hong Kong, Taiwan, and the UK. Patients must be between 18 and 70 years old, have renal biopsy findings consistent with IgA nephropathy, have been treated with an Angiotensin Converting Enzyme inhibitor (ACEi) and/or an Angiotensin II Receptor Blocker (ARB) for at least 90 days at the maximum approved dose, have a proteinuria > 1 gm/day at diagnosis of IgA nephropathy and a level > 0.5 gm/day at the second screening visit, and a blood pressure controlled to ≤ 1302/80 with angiotensin blockade. Eligible candidates cannot have recently used cyclophosphamide, mycophenolate mofetil, azathioprine, Rituximab, or > 15 mg/day of prednisone or any other corticosteroid equivalent. The study investigates whether fostamatinib is a safe and effective treatment for IgAN. It is a Phase 2, multi-center, randomized, double-blind, ascending-dose, placebo-controlled clinical study. Primary outcome measures include the mean change in proteinuria as measured by spot urine protein/creatinine ratio (sPCR). Effects were evaluated for 100 mg, 150 mg, and placebo formulations taken twice daily by mouth for 24 weeks. The study began in October 2014 and is expected to complete by June 2017.[10]
Suitable active 2,4-pyrimidinediamine compounds are described, for example, in U.S. application Serial No. 10/355,543 filed January 31 , 2003 (US2004/0029902A1), international application Serial No. PCT/US03/03022 filed January 31, 2003 (WO 03/063794), U.S. application Serial No. 10/631,029 filed July 29, 2003 (US 2005/0028212), international application Serial No. PCT/US03/24087 (WO2004/014382), U.S. application Serial No. 10/903,263 filed July 30, 2004 (US2005/0234049), and international application Serial No.
PCT/US2004/24716 (WO 2005/016893), the disclosures of which are incorporated herein by reference. In such 2,4-pyrimidinediamine compounds, the progroup(s) Rp can be attached to any available primary or secondary amine, including, for example, the N2 nitrogen atom of the 2,4-pyrimidinediamine moiety, the N4 nitrogen atom of the 2,4-pyrimidinediamine moiety, and/or a primary or secondary nitrogen atom included in a substituent on the 2,4-pyrimidinediamine compound. The use of phosphate-containing progroups Rp is especially useful for 2,4-pyrimidinediamine compounds that exhibit poor water solubility under physiological conditions (for example, solubilities of less than about 10 μg/ml). While not intending to be bound by any theory of operation, it is believed that the phosphate-containing progroups aid the solubility of the underlying active 2,4-pyrimidinediamine compound, which in turn increases its bioavailability when administered orally. It is believed that the phosphate progroups Rp are metabolized by phosphatase enzymes found in the digestive tract, permitting uptake of the underlying active drug.
[0024] It has been discovered that the water solubility and oral bioavailability of a particular biologically active 2,4-pyrimidinediamine compound, illustrated below (Compound 1), increased dramatically when formulated to include a progroup Rp of the formula -CH2-O-P(O)(OH)2 at the ring nitrogen atom highlighted with the asterisk (Compound 4):
[0260] N4-(2,2-dimethyl-3-oxo-4H-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (1, 1.0 g, 2.12 mmol), Cs2CO3 (1.0 g, 3.07 mmol) and di-tert-butyl chloromethyl phosphate (2, 0.67 g, 2.59 mmol) in acetone (20 mL) was stirred at room temperature under nitrogen atmosphere. Progress of the reaction was monitored by LC/MS. Crude reaction mixture displayed three product peaks with close retention times with M++H 693 (minor-1), 693 (major; 3) and 477 (minor-2) besides starting material (Compound 1). Upon stirring the contents for 4 days (70% consumption), the reaction mixture was concentrated and diluted with water. The resultant pale yellow precipitate formed was collected by filtration and dried. The crude solid was purified by silica gel (pretreated with 10%NEt3/CH2Cl2 followed by eluting with hexanes) column chromatography by gradient elution with 70% EtOAc / hexanes-100% EtOAc). The fractions containing Compound 1 and M++H 693 were collected and concentrated. The resulting crude white solid was subjected to repurifϊcation in the similar manner as described previously but by eluting with 30%-50%-75%-100% EtOAc/hexanes. The major product peak with M++H 693 was collected as a white solid (270 mg, 18%) and was characterized as N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3). 1H NMR (DMSO-d6): δ 9.21 (s, IH), 9.17 (s, IH), 8.16 (d, IH, J = 2.6 Hz), 7.76 (d, IH, J = 8.5 Hz), 7.44 (d, IH, J = 8.5 Hz), 7.02 (s, 2H), 5.78 (d, IH, J3PH = 6.1 Hz), 3.64 (s, 6H), 3.58 (s, 3H), 1.45 (s, 6H), 1.33 (s, 9H). LCMS: ret. time: 14.70 min.; purity: 95%; MS (m/e): 693 (MH+). 31P NMR (DMSO-d6): -11.36.
[0261] Trifluoroacetic acid (1.5 mL) was added dropwise as a neat for 5 min to N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3, 120 mg, 0.173 mmol ) dissolved in CH2Cl2 (10 mL) at 00C under nitrogen atmosphere. The contents were allowed to stir for 1.5 h. Progress of the reaction mixture was monitored by LC/MS. After complete consumption of the starting material, reaction mixture was concentrated, dried and triturated with ether. The ethereal layer was decanted and dried to provide the crude solid. LC/MS analysis of the crude displayed three peaks with M++H 581, 471 and 501. The peak corresponding to M++H 581 was collected by preparative HPLC chromatographic purification. The fractions were lyophilised and dried to provide 53 mg (52%) of off white fluffy solid and characterized as N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[ 1 ,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4). 1H NMR (DMSO-d6): δ 9.21 (br s, 2H), 8.16 (d, IH, J = 2.6 Hz), 7.93 (d, IH, J = 8.5 Hz), 7.39 (d, IH, J = 8.5 Hz), 7.05 (s, 2H), 5.79 (d, IH, J3PH = 6.6 Hz), 3.67 (s, 6H), 3.59 (s, 3H), 1.44 (s, 6H). LCMS: ret. time: 8.52 min.; purity: 95%; MS (m/e): 581 (MH+). 31P NMR (DMSO-d6): -2.17.
2. Alternative Synthesis of Prodrug Compound 4
[0262] An alternative method of synthesizing prodrug Compound 4 which alleviates the need for column chromatography and HPLC purification is provided below.
[0263] N4-(2,2-dimethyl-3-oxo-4H-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 1, 19.73 g, 41.97 mmol),
Cs2CO3 (15.04 g, 46.16 mmol) and di-tert-butyl chloromethyl phosphate (13.0 g, 50.38 mmol) in DMF (100 mL) was stirred at room temperature under nitrogen atmosphere. Progress of the reaction was monitored by in process LC/MS. Crude reaction mixture displayed two product peaks (ratio 1 :6.5) with close retention times displaying M++H 693 (minor) and 693 (major) besides starting material (Compound 1). Initial yellow reaction mixture turned to olive green as the reaction progressed. Workup was carried out as follows 1). Upon stirring the contents for 30 h (92% consumption), reaction mixture was poured onto ice-water (400 mL) and stirred the contents by adding brine solution (200 mL). Fine yellow tan solid formed was filtered, washed with water and dried overnight.
2). The solid (35 g) was dissolved in MTBE (500 mL) and washed with water (40OmL). Aqueous layer was extracted with MTBE (2 X 350 mL) till the absence of UV on TLC. Combined organic layers were dried over anhydrous Na2SO4 and decanted.
Note: step 2 can be done directly, however, DMF extraction back into solution leads to difficulty in the crystallization step.
3). The dark red clear solution was subjected to 10 g of activated charcoal treatment, heated to boil and filtered.
4). The dark red clear solution was concentrated by normal heating to 400 mL of its volume and left for crystallization. The solid crystallized as granules was filtered, crushed the granules to powder, washed with MTBE (400 mL) and dried under high vacuum. See step 7 for the workup of mother liquor. Weight of the solid: 17 g; purity: 90% (Compound 3), 6.26% (Compound 1), 1.8% (minor M+ 693).
5). At this stage solid was taken in 500 ml of ethyl ether and heated to boil. Cooled and filtered to remove undissolved material. Filtrate was concentrated.
6). Above concentrate was subjected to crystallization in MTBE (300 mL).
The white solid formed was filtered, washed with MTBE (100 mL) and dried under high vacuum to provide the desired N4-(2,2-dimethyl-4-[(di-tert-butyl
phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3) in 97% purity. 1H NMR (DMSO-d6): δ 9.21 (s, IH), 9.17 (s, IH), 8.16 (d, IH, J = 2.6 Hz), 7.76 (d, IH, J = 8.5 Hz), 7.44 (d, IH, J = 8.5 Hz), 7.02 (s, 2H), 5.78 (d, IH, J3PH = 6.1 Hz), 3.64 (s, 6H), 3.58 (s, 3H), 1.45 (s, 6H), 1.33 (s, 9H). LCMS: ret. time: 14.70 min.; purity: 95%; MS (m/e): 693 (MH+). 31P NMR (DMSO-d6): -11.36. Weight of the solid: 15.64 g (yield: 55%); purity: 97% (Compound 3), 3% (Compound 1).
7). The mother liquor was concentrated and steps 5 and 6 were repeated to provide Compound 3.
2.2. Synthesis of N4-(2,2-dimethyl-4-[(dihydrogen
phosphonoxy)methyl] -3-oxo-5-pyrido [ 1 ,4] oxazin-6-yl)-5- fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine
(Compound 4)
[0264] N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3); (15.0 g, 21.67 mmol) dissolved in AcOH:H20 (225 niL, 4:1) was heated at 65 0C (oil bath temp). The progress of the reaction was monitored by in process LC/MS. The reaction mixture transformed to faint tan white solid after Ih of heating. At this point most of Compound 3 converted to mono des t-butyl product. After 3h of heating, consumption of SM and complete conversion of intermediate (mono des t-butylated) to product was observed.
[0265] Reaction mixture was cooled, poured onto ice-water (200 mL), stirred for 20 min and filtered. The clear white filter cake was washed with water (600 ml) and acetone (200 mL) successively, dried for 2h followed by drying under high vacuum over P2O5 in a desiccator. Weight of the solid: 12.70 g; purity: 97% (Compound 3) and 3% (Compound 1) 1H NMR indicated acetic acid presence (1 :1)
[0266] To remove acetic acid, the solid was taken in acetonitrile (300 mL) and concentrated by rotovap vacuum. This process was repeated 2 times with acetonitrile and toluene (3 X 300 mL). The solid obtained was dried under high vacuum at 50 OC. [0267] Finally, the solid was taken in acetone (400 mL), filtered and dried to provide solid N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4). 1H NMR (DMSO-d6): δ 9.21 (br s, 2H), 8.16 (d, IH, J = 2.6 Hz), 7.93 (d, IH, J = 8.5 Hz), 7.39 (d, IH, J = 8.5 Hz), 7.05 (s, 2H), 5.79 (d, IH, J3PH = 6.6 Hz), 3.67 (s, 6H), 3.59 (s, 3H), 1.44 (s, 6H). LCMS: ret. time: 8.52 min.; purity: 95%; MS (m/e): 581 (MH+). 31P NMR (DMSO-d6): -2.17.
3. Synthesis of N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo- 5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4- pyrimidinediamine mono calcium salt (Prodrug Salt 6)
[0268] Aqueous (10 niL) NaHCO3 (0.17 g, 2.02 mmol) solution was added dropwise to a suspension of N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (0.5 g, 0.86 mmol) in water (5 mL) at room temperature while stirring the contents. The clear solution formed was treated with aqueous (10 mL) CaCl2 (0.11 g in 10 mL water, 0.99 mmol) n a dropwise manner at room temperature. The addition resulted in the precipitation of a white solid from reaction mixture. Upon completion of addition, the contents were stirred for a period of 30 min, filtered, washed with water (40 mL) and dried. The clear white solid was taken in water (30 mL) and heated on a stir plate to boil. The solution was cooled, filtered and dried. The white solid collected and further dried under high vacuo at 80 0C for 32 h to provide 0.41 g (83%) of solid N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[ 1 ,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine mono calcium salt (Prodrug Salt 6).
[0269] Ca(OAc)2 may also used in place Of CaCl2 in this preparation.
4. Synthesis of N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo- 5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4- pyrimidinediamine disodium salt hexahydrate and monosodium salt
hydrate
[0270] A round-bottomed flask was charged with 10.00 g N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[ 1 ,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4) and 140 mL water into a round bottom flask to form a slurry having a pH between 3.6 and 3.7. The pH was adjusted to in the range of 9.3 to 10.3 by addition of 1 M aqueous NaOH, initially forming a turbid solution, which returned to a suspension upon prolonged stirring. The mixture was heated at reflux, then the turbid solution was hot filtered through filter paper. The solid collected in the filter paper was rinsed with 10 mL hot water.
Isopropanol (75 mL) was added to the filtrate, yielding a clear solution, which was allowed to cool to room temperature over about 1.5 hours with stirring, during which time a solid precipitated. The precipitate was collected by filtration, rinsed with 47 mL isopropanol, and taken up in 73 mL acetone to form a slurry, which was stirred for 1.5 hours at room temperature. The solid was again collected by filtration and rinsed with 18 mL acetone, then dried at about 40 0C under vacuum until substantially all isopropanol and acetone was removed (i.e., below 0.5 wt% each). The product was exposed to air at about 40% relative humidity and room temperature until the water content stabilized at about 15% by Karl Fisher titration, yielding 8.18 g of the title compound. 1H NMR (D2O): δ 7.67 (d, IH, J = 3.8 Hz), 7.49 (d, IH, J = 8.8 Hz), 6.87 (d, IH, J = 8.8 Hz), 6.50 (s, 2H), 5.52 (d, IH, J3PH = 2.0 Hz), 3.53 (s, 3H), 3.47 (s, 6H), 1.32 (s, 6H). 31P NMR (D2O): 2.75. The prodrug salt hydrate was obtained as a pure-white, highly crystalline material. Microscopic investigation indicated that the crystallites are plate-like with a particle size of less than 10 μm. Polarized light microscopy revealed birefringence corroborating the crystalline nature of the hydrate. [0271] The monosodium salt can be prepared from N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fiuoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine and sodium hydroxide by a proper pH control; pH of 5-5.5 results in predominantly the formation of monosodium salt.
References
^ Jump up to:abS.P. McAdoo; F.W.K. Tam (2011). “Fostamatinib Disodium”. Drugs of the Future. 36 (4): 273–280.
Jump up^Braselmann, S; Taylor, V; Zhao, H; Wang, S; Sylvain, C; Baluom, M; Qu, K; Herlaar, E; et al. (2006). “R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation”. The Journal of Pharmacology and Experimental Therapeutics. 319 (3): 998–1008. doi:10.1124/jpet.106.109058. PMID16946104.
Jump up^Morales-Torres, Jorge (2010). “R788 (fostamatinib disodium): a novel approach for the treatment of rheumatoid arthritis”. International Journal of Clinical Rheumatology. 5 (1): 9–15. doi:10.2217/ijr.09.69.
This new drug application provides for the use of KRINTAFEL (tafenoquine) tablets for the radical cure (prevention of relapse) of Plasmodium vivax malaria in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute P. vivax infection….https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/210795Orig1s000Ltr.pdf
Tafenoquine under the commercial name of Krintafel is an 8-aminoquinoline drug manufactured by GlaxoSmithKline that is being investigated as a potential treatment for malaria, as well as for malaria prevention.[2][3]
The proposed indication for tafenoquine is for treatment of the hypnozoite stages of Plasmodium vivax and Plasmodium ovale that are responsible for relapse of these malaria species even when the blood stages are successfully cleared. This is only now achieved by administration of daily primaquine for 14 days. The main advantage of tafenoquine is that it has a long half-life (2–3 weeks) and therefore a single treatment may be sufficient to clear hypnozoites. The shorter regimen has been described as an advantage.[4]
Like primaquine, tafenoquine causes hemolysis in people with G6PD deficiency.[2] Indeed, the long half-life of tafenoquine suggests that particular care should be taken to ensure that individuals with severe G6PD deficiency do not receive the drug.
The dose of tafenoquine has not been firmly established, but for the treatment of Plasmodium vivax malaria, a dose of 800 mg over three days has been used.[5]
In 2018 United States Food and Drug Administration (FDA) approved single dose tafenoquine for the radical cure (prevention of relapse) of Plasmodium vivax malaria[6].
Tafenoquine is used for the treatment and prevention of relapse of Vivax malaria in patients 16 years and older. Tafenoquine is not indicated to treat acute vivax malaria.[1]
Malaria is a disease that remains to occur in many tropical countries. Vivax malaria, caused by Plasmodium vivax, is known to be less virulent and seldom causes death. However, it causes a substantive illness-related burden in endemic areas and it is known to present dormant forms in the hepatocytes named hypnozoites which can remain dormant for weeks or even months. This dormant form produces ongoing relapses
FDA Approves Tafenoquine, First New P VivaxMalaria Treatment in 60 Years
JUL 23, 2018
The US Food and Drug Administration (FDA) has approved, under Priority Review, GlaxoSmithKline (GSK)’s tafenoquine (Krintafel), which is the first single-dose medicine for the prevention of Plasmodium vivax (P vivax) malaria relapse in patients over the age of 16 years who are receiving antimalarial therapy. This is the first drug to be approved for the treatment of P vivax in over 60 years.
“[The] approval of Krintafel, the first new treatment for Plasmodium vivax malaria in over 60 years, is a significant milestone for people living with this type of relapsing malaria.” Hal Barron, MD, chief scientific officer and president of research and development of GSK, said in the announcement, “Together with our partner, Medicines for Malaria Venture (MMV), we believe Krintafel will be an important medicine for patients with malaria and contribute to the ongoing effort to eradicate this disease.”
Tafenoquine is an 8-aminoquinoline derivative with activity against all stages of the P vivax lifecycle, including hypnozoites. It was first synthesized by scientists at the Walter Reed Army Institute of Research in 1978, and in 2008, GSK entered into a collaboration with MMV, to develop tafenoquine as an anti-relapse medicine.
After an infected mosquito bite, the P vivax parasite infects the blood and causes an acute malaria episode and can also lie dormant in the liver (in a form known as hypnozoite) from where it periodically reactivates to cause relapses, which can occur weeks, months, or years after the onset of the initial infection. The dormant liver forms cannot be readily treated with most anti-malarial treatments. Primaquine, an 8-aminoquinolone, has been the only FDA-approved medicine that targeted the dormant liver stage to prevent relapse; however, effectiveness only occurs after 14 days and the treatment has shown to have poor compliance.
“The US FDA’s approval of Krintafel is a major milestone and a significant contribution towards global efforts to eradicate malaria,” commented David Reddy, PhD, chief executive officer of MMV in a recent statement, “The world has waited decades for a new medicine to counter P vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance.”
Approval for tafenoquine was granted based on the efficacy and safety data gleaned from a comprehensive global clinical development program for P vivaxprevention of relapse which has been designed by GSK and MMV in agreement with the FDA. The program consisted of 13 studies assessing the safety of a 300 mg single-dose of tafenoquine, including 3 double-blind studies referred to as DETECTIVE Parts 1 and 2 and GATHER.
With the approval of tafenoquine, GSK has also been awarded a tropical disease priority review voucher by the FDA. Additionally, GSK is waiting for a decision from Australian Therapeutics Good Administration regarding the regulatory submission for the drug.
P vivax malaria has caused around 8.5 million clinical infections each year, primarily in South Asia, South-East Asia, Latin America, and the Horn of Africa, a peninsula in East Africa. Symptoms include fever, chills, vomiting, malaise, headache and muscle pain, and can lead to death in severe cases.
Tafenoquine should not be administered to: patients who have glucose-6-phosphate dehydrogenase (G6PD) deficiency or have not been tested for G6PD deficiency, patients who are breastfeeding a child known to have G6PD deficiency or one that has not been tested for G6PD deficiency, or patients who are allergic to tafenoquine or any of the ingredients in tafenoquine or who have had an allergic reaction to similar medicines containing 8-aminoquinolines
Stereochemistry
Tafenoquine contains a stereocenter and consists of two enantiomers. This is a mixture of (R) – and the (S) – Form:
Enantiomers of tafenoquine
(R)-Form
(S)-Form
CLIP
US 4431807
Nitration of 1,2-dimethoxybenzene (XXIX) with HNO3/AcOH gives 4,5-dimethoxy-1,2-dinitrobenzene (XXX), which is treated with ammonia in hot methanol to yield 4,5-dimethoxy-2-nitroaniline (XXXI). Cyclization of compound (XXXI) with buten-2-one (XXXII) by means of H3PO4 and H3AsO4 affords 5,6-dimethoxy-4-methyl-8-nitroquinoline (XXXIII), which is selectively mono-demethylated by means of HCl in ethanol to provide 5-hydroxy-6-methoxy-4-methyl-8-nitroquinoline (XXXIV). Reaction of quinoline (XXXIV) with POCl3 gives the corresponding 5-chloro derivative (XXXV), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH to yield the diaryl ether (XXXVI). Finally, the nitro group of (XXXVI) is reduced by means of H2 over PtO2 in THF or H2 over Raney nickel.
Nitration of 2-fluoroanisole (XXXVII) with HNO3/Ac2O gives 3-fluoro-4-methoxynitrobenzene (XXXVIII), which is reduced to the corresponding aniline (XXXIX) with SnCl2/HCl. Reaction of compound (XXXIX) with Ac2O yields the acetanilide (XL), which is nitrated with HNO3 to afford 5-fluoro-4-methoxy-2-nitroacetanilide (XLI). Hydrolysis of (XLI) with NaOH provides 5-fluoro-4-methoxy-2-nitroaniline (XLII), which is cyclized with buten-2-one (XXXII) by means of As2O5 and H3PO4 to furnish 5-fluoro-6-methoxy-4-methyl-8-nitroquinoline (XLIII). Condensation of quinoline (XLIII) with 3-(trifluoromethyl)phenol (IV) by means of K2CO3 gives the diaryl ether (XXXIV), which is finally reduced by means of H2 over PtO2 in THF.
CLIP
US 4617394
Reaction of 8-amino-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline (XIV) with phthalic anhydride (XV) affords the phthalimido derivative (XVI), which is oxidized with MCPBA to yield the quinoline N-oxide (XVII). Treatment of compound (XVII) with neutral alumina gives the quinolone derivative (XVIII), which by reaction with POCl3 in refluxing CHCl3 provides the 2-chloroquinoline derivative (XIX). Alternatively, reaction of the quinoline N-oxide (XVII) with POCl3 as before also gives the 2-chloroquinoline derivative (XIX) The removal of the phthalimido group of compound (XIX) by means of hydrazine in refluxing ethanol gives the chlorinated aminoquinoline (XX), which is finally treated with MeONa in hot DMF.
CLIP
US 6479660; WO 9713753
Chlorination of 6-methoxy-4-methylquinolin-2(1H)-one (I) with SO2Cl2 in hot acetic acid gives the 5-chloro derivative (II), which is nitrated with HNO3 in H2SO4 to yield the 8-nitroquinolinone (III). Condensation of compound (III) with 3-(trifluoromethyl)phenol (IV) by means of KOH in NMP provides the diaryl ether (V), which is treated with refluxing POCl3 to afford the 2-chloroquinoline (VI). Reaction of compound (VI) with MeONa in refluxing methanol results in the 2,6-dimethoxyquinoline derivative (VII), which is reduced with hydrazine over Pd/C to give the 8-aminoquinoline derivative (VIII). Condensation of aminoquinoline (VIII) with N-(4-iodopentyl)phthalimide (IX) by means of diisopropylamine in hot NMP yields the phthalimido precursor (X), which is finally cleaved with hydrazine in refluxing ethanol.
Reaction of 1,4-dibromopentane (XI) with potassium phthalimide (XII) gives N-(4-bromopentyl)phthalimide (XIII), which is then treated with NaI in refluxing acetone.
Reaction of 4-methoxyaniline (XXI) with ethyl acetoacetate (XXII) by means of triethanolamine in refluxing xylene gives the acetoacetanilide (XXIII), which is cyclized by means of hot triethanolamine and H2SO4 to yield 6-methoxy-4-methylquinolin-2(1H)-one (I), which is treated with refluxing POCl3 to provide 2-chloro-6-methoxy-4-methylquinoline (XXIV). Reaction of compound (XXIV) with SO2Cl2 in hot AcOH affords 2,5-dichloro-6-methoxy-4-methylquinoline (XXV), which is treated with MeONa in refluxing methanol to furnish 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Alternatively, the reaction of compound (XXIV) with MeONa as before gives 2,6-dimethoxy-4-methylquinoline (XXVII), which is treated with SO2Cl2 in hot AcOH to give the already described 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Nitration of compound (XXVI) with KNO3 and P2O5 gives the 8-nitroquinoline derivative (XXVIII), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH in hot NMP to yield the diaryl ether (VII). Finally, the nitro group of compound (VII) is reduced with hydrazine over Pd/C.
aResearch Center for Solar Energy Chemistry, Division of Chemical Engineering, Graduate School of Engineering Science, Osaka University, Toyonaka 560-8531, Japan E-mail:shiraish@cheng.es.osaka-u.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Saitama 332-0012, Japan
Abstract
Tafenoquine (TQ), a fluorescent antimalarial drug, was used as a receptor for the fluorometric detection of hypochlorite (OCl−). TQ itself exhibits a strong fluorescence at 476 nm, but OCl−-selective cyclization of its pentan-1,4-diamine moiety creates a blue-shifted fluorescence at 361 nm. This ratiometric response facilitates rapid, selective, and sensitive detection of OCl− in aqueous media with physiological pH. This response is also applicable to a simple test kit analysis and allows fluorometric OCl− imaging in living cells.
Synthesis of the intermediate diazepinone (IV) is accomplished by a one-pot synthesis. Condensation of 2-chloro-3-aminopyridine (I) with the anthranilic ester (II) is effected in the presence of potassium tert-butoxide as a catalyst. The resulting anthranilic amide (III) is cyclized under the influence of catalytic amounts of sulfuric acid. Treatment of (IV) with chloroacetylchloride in toluene yields the corresponding choroacetamide (V). The side chain of AQ-RA 741 is prepared starting from 4-picoline, which is alkylated by reaction with 3-(diethylamino)propylchloride in the presence of n-butyllithium. Hydrogenation of (VIII) using platinum dioxide as a catalyst furnishes the diamine (IX), which is coupled with (V) in the presence of catalytic amounts of sodium iodide in acetone leading to AQ-RA 741 as its free base.
Shanks GD, Oloo AJ, Aleman GM et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis33 (12): 1968–74. doi:10.1086/324081. JSTOR4482936.PMID11700577.
Lell B, Faucher JF, Missinou MA et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”.Lancet355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID10885356.
Percent Composition: C 62.19%, H 6.09%, F 12.30%, N 9.07%, O 10.36%
Literature References: Analog of primaquine, q.v. Prepn: P. Blumbergs, M. P. LaMontagne, US4617394 (1986 to U.S. Sec. Army); M. P. LaMontagne et al.,J. Med. Chem.32, 1728 (1989). HPLC determn in blood and plasma: D. A. Kocisko et al.,Ther. Drug Monit.22, 184 (2000). Metabolism: O. R. Idowu et al.,Drug Metab. Dispos.23, 1 (1995). Clinical pharmacokinetics: M. D. Edstein et al.,Br. J. Pharmacol.52, 663 (2001). Clinical evaluation in prevention of malaria relapse: D. S. Walsh et al.,J. Infect. Dis.180, 1282 (1999); in malaria prophylaxis: B. Lell et al.,Lancet355, 2041 (2000); B. R. Hale et al.,Clin. Infect. Dis.36, 541 (2003).
Derivative Type: Succinate
CAS Registry Number: 106635-81-8
Trademarks: Etaquine (GSK)
Molecular Formula: C24H28F3N3O3.C4H6O4
Molecular Weight: 581.58
Percent Composition: C 57.83%, H 5.89%, F 9.80%, N 7.23%, O 19.26%
Properties: Crystals from acetonitrile, mp 146-149°. LD50 in male, female rats (mg/kg): 102, 71 i.p.; 429, 416 orally (LaMontagne).
April 28, 2014
GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV) announced the start of a Phase 3 global program to evaluate the efficacy and safety of tafenoquine, an investigational medicine which is being developed for the treatment and relapse prevention (radical cure) of Plasmodium vivax (P. vivax) malaria.
P. vivax malaria, a form of the disease caused by one of several species of Plasmodium parasites known to infect humans, occurs primarily in South and South East Asia, Latin America and the horn of Africa. Severe anemia, malnutrition and respiratory distress are among the most serious consequences described to be caused by the infection.
The Phase 3 program includes two randomized, double-blind treatment studies to investigate tafenoquine in adult patients with P. vivax malaria. The DETECTIVE study (TAF112582) aims to evaluate the efficacy, safety and tolerability of tafenoquine as a radical cure for P. vivax malaria, co-administered with chloroquine, a blood stage anti-malarial treatment. The GATHER study (TAF116564) aims to assess the incidence of hemolysis and safety and efficacy of tafenoquine compared to primaquine, the only approved treatment currently available for the radical cure of P. vivax malaria.
Tafenoquine is not yet approved or licensed for use anywhere in the world.
“P. vivax malaria can affect people of all ages and is particularly insidious because it has the potential to remain dormant within the body in excess of a year, and causes some patients to experience repeated episodes of illness after the first mosquito bite,” said Nicholas Cammack, head, Tres Cantos Medicines Development Center for Diseases of the Developing World. “Our investigation of tafenoquine for the treatment of P. vivax malaria is part of GSK’s efforts to tackle the global burden of malaria. Working with our partners, including MMV, we are determined to stop malaria in all its forms.”
“One of the big challenges we face in tackling malaria is to have new medicines to prevent relapse, caused by dormant forms of P. vivax,” said Dr. Timothy Wells, MMV’s chief scientific officer. “The Phase 3 program is designed to build upon the promising results of the Phase 2b study which showed that treatment with tafenoquine prevented relapses. If successful, tafenoquine has the potential to become a major contributor to malaria elimination. It’s a great privilege to be working with GSK on this project; they have a clear commitment to changing the face of public health in the countries in which we are working.”
/////////////Tafenoquine, タフェノキン , Orphan, FDA 2018, KRINTAFEL, Priority Review, GlaxoSmithKline
FDA approves first cancer drug through new oncology review pilot that enables greater development efficiency FDA expands the use of breast cancer drug
The U.S. Food and Drug Administration today approved Kisqali (ribociclib) in combination with an aromatase inhibitor for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, as initial endocrine-based therapy. The FDA also approved Kisqali in combination with fulvestrant for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer, as initial endocrine based therapy or following disease progression on endocrine therapy.
The U.S. Food and Drug Administration today approved Kisqali (ribociclib) in combination with an aromatase inhibitor for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, as initial endocrine-based therapy. The FDA also approved Kisqali in combination with fulvestrant for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer, as initial endocrine based therapy or following disease progression on endocrine therapy.
This is the first approval that FDA has granted as a part of two new pilot programs announced earlier this year that collectively aim to make the development and review of cancer drugs more efficient, while improving FDA’s rigorous standard for evaluating efficacy and safety. With this real-time review, the FDA was able to start evaluating the clinical data as soon as the trial results become available, enabling FDA to be ready to approve the new indication upon filing of a formal application with the Agency.
The first new program, called Real-Time Oncology Review, allows for the FDA to review much of the data earlier, after the clinical trial results become available and the database is locked, before the information is formally submitted to the FDA. This allows the FDA to begin its analysis of the data earlier, and provide feedback to the sponsor on how they can most effectively analyze the data to answer key regulatory questions. The pilot focuses on early submission of data that are the most relevant to assessing safety and effectiveness of the product. Then, when the sponsor submits the application with the FDA, the review team will already be familiar with the data and in a better position to conduct a more efficient, timely, and thorough review.
The second program is a new templated Assessment Aid that the applicant uses to organize its submission into a structured format to facilitate FDA’s review of the application. By using a structured template, the FDA is able to layer its assessment into the same file submitted by the sponsor, allowing this annotated application to serve as the document that contains the FDA review. This voluntary submission form provides for a more streamlined approach to reviewing data and illustrating FDA’s analysis. It allows for drug reviewers to focus on the key benefit-risk and labeling issues rather than administrative issues.
“With this approval, we’ve demonstrated some of the benefits of the new programs that we’re piloting for our review of cancer drugs, to improve regulatory efficiency while enhancing the process for evaluating the data submitted to us. This shows that, with smart policy approaches, we can gain efficiency while also improving the rigor of our process. These new programs were designed to reduce some of the administrative issues that can add to the time and cost of the review process, including the staffing burdens on the FDA. For example, by analyzing data earlier in the process, before formal submission to the FDA, and evaluating submissions in a structured template, we can make it easier to identify earlier when applications are missing key analysis or information that can delay reviews,” said FDA Commissioner Scott Gottlieb, M.D. “With today’s approval, the FDA used these new approaches to allow the review team to start analyzing data before the actual submission of the application and help guide the sponsor’s analysis of the top-line data to tease out the most relevant information. This enabled our approval less than one month after the June 28 submission date and several months ahead of the goal date.”
These new processes are good for patients, good for health care providers, good for product developers, and good for the FDA, by allowing our staff to have more time to engage with product developers and focus on the key aspects of drug reviews. We can improve efficiency and solidify our gold standard for review.”
Currently the two pilot programs are being used for supplemental applications for already-approved cancer drugs and could later be expanded to original drugs and biologics.
Kisqali was first approved in March 2017 for use with an AI to treat HR-positive, HER2-negative breast cancer in post-menopausal women whose cancer is advanced or has spread to other parts of the body.
“The approval adds a new treatment choice for patients with breast cancer,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to continuing to bring more treatment options to patients.”
Breast cancer is the most common form of cancer in the United States. The National Cancer Institute at the National Institutes of Health estimates approximately 266,120 women will be diagnosed with breast cancer this year and 40,920 will die of the disease. Approximately 72 percent of patients with breast cancer have tumors that are HR-positive and HER2-negative.
The efficacy of Kisqali in combination with an AI for pre/perimenopausal women was demonstrated in a clinical trial of 495 participants who received either Kisqali and an AI or placebo and an AI. All pre- or peri-menopausal patients on this study received ovarian suppression with goserelin. The trial measured progression-free survival (PFS), which is generally the amount of time after the start of this treatment during which the cancer does not substantially grow and the patient is alive. PFS was longer for patients taking Kisqali plus an AI (median PFS of 27.5 months) compared to patients who received placebo plus an AI (median PFS of 13.8 months).
The efficacy of Kisqali in combination with fulvestrant in treating advanced or metastatic breast cancer was demonstrated in a clinical trial that included 726 participants who received either Kisqali and fulvestrant or placebo and fulvestrant. The trial measured PFS, which was longer for patients taking Kisqali plus fulvestrant (median PFS of 20.5 months) compared to patients who received placebo plus fulvestrant (median PFS of 12.8 months).
The common side effects of Kisqali are infections, abnormally low count of a type of white blood cell (neutropenia), a reduction in the number of white cells in the blood (leukopenia), headache, cough, nausea, fatigue, diarrhea, vomiting, constipation, hair loss and rash.
Warnings include the risk of a heart problem known as QT prolongation that can cause an abnormal heartbeat and may lead to death, serious liver problems, low white blood cell counts that may result in infections that may be severe, and fetal harm.
Tecovirimat, sold under the brand name Tpoxx among others,[6] is an antiviral medication with activity against orthopoxviruses such as smallpox and monkeypox.[4][7][8] It is the first antipoxviral drug approved in the United States.[9][10] It is an inhibitor of the orthopoxvirus VP37 envelope wrapping protein.[4]
The drug works by blocking cellular transmission of the virus, thus preventing the disease.[11] Tecovirimat has been effective in laboratory testing; it has been shown to protect animals from monkeypox and rabbitpox and causes no serious side effects in humans.[6] Tecovirimat was first used for treatment in December 2018, after a laboratory-acquired vaccinia virus infection.[12]
The World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980. However, there have been longstanding concerns that smallpox may be used as a bioweapon.2,5 Tecovirimat is an antiviral drug that was identified via a high-throughput screen in 2002.2 It is effective against all orthopoxviruses, including vaccinia, cowpox, ectromelia, rabbitpox, monkeypox, and Variola (smallpox) virus.1,4
Tecovirimat was approved by the FDA in July 2018 as the first drug ever approved to treat smallpox.6,5 Tecovirimat was later approved by Health Canada in December 2021,7 followed by the approval from the European Commission in January 2022.9 Other than smallpox, tecovirimat is also indicated to treat complications due to replication of the vaccinia virus following vaccination against smallpox, and to treat monkeypox and cowpox in adults and children.8 Tecovirimat is available as both oral and intravenous formulations.10
Medical uses
In the United States, tecovirimat is indicated for the treatment of human smallpox disease.[4] In the European Union it is indicated for the treatment of smallpox, monkeypox, and cowpox.[5]
Mechanism of action
Tecovirimat inhibits the function of a major envelope protein required for the production of extracellular virus. The drug prevents the virus from leaving an infected cell, hindering the spread of the virus within the body.[16]
Chemistry
The first synthesis of tecovirimat was published in a patent filed by scientists at Siga Technologies in 2004. It is made in two steps from cycloheptatriene.[17]
The scheme has taken from SmartChem a knowledgebase by ROW2 Technologies, Inc. (www.row2technologies.com)
A perfect amalgamation of information on chemicals and global suppliers. A database where you can search for information on more than 150,000 chemicals and around 15,000 Global chemicals suppliers, including routes of synthesis, Applications, end uses, and validated contact details of global suppliers. For more information, please visit www.row2technologies.com or contact,
Reference: Dai, Dongcheng. Process for the preparation of tecovirimat. Assignee Siga Technologies, Inc., USA. WO 2014028545. (2014).
SYN 3
Synthetic Description
Reference: Medical composition containing ST-246, its preparation and anti-poxvirus application. Assignee Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences, PLA, Peop. Rep. China. CN 101912389. (2010).
The present invention provides a process for making ST-246 outlined in Scheme 1
The present invention also provides a process for making ST-246 outlined in Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3
The present invention also provides a process for making ST-246 outlined in Scheme 4
The present invention further provides a process for making ST-246 outlined in Scheme 5
The present invention also provides the following compounds useful in the synthesis of ST-246:
EXAMPLE 1Synthetic Route I
Step A. Synthesis of Compound 6 (P=Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO04112718) in EtOH (80 mL, EMD, AX0441-3) was added tert-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc-hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCl3: δ 6.30 (br s, 1H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1.46 (s, 9H), 1.06-1.16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCl Salt)
Compound 6 (3.6 g, 11.83 mmol) was dissolved in i-PrOAc (65 mL, Aldrich, 99.6%). 4M HCl in dioxane (10.4 mL, 41.4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (15 mL) and dried under vacuum to yield HCl salt of compound 7 (1.9 g, 67% yield) as a white solid. The filtrate was concentrated to ⅓ its volume and stirred at 10-15° C. for 30 min. The solid was filtered, washed with minimal volume of i-PrOAc and dried to afford additional 0.6 g (21% yield) of compound 7. Total yield: 2.5 g (88% yield). 1H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1.07-1.17 (m, 2H), 0.18-0.29 (m, 1H), −0.01-0.07 (m, 1H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1.17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20° C. The resulting solution was stirred for 5 minutes at 15-20° C., to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH4Cl (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent.
EXAMPLE 2Synthetic Route II
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (11.6%).
The reaction mixture was cooled to 45° C. and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1.5 g, 54% yield) as an off-white solid. 1H NMR in CDCl3: δ 8.44 (s, 1H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 1.5 h at 95° C., LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo=94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95° C., LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 110° C. and the reaction was monitored. After heating at 110° C. for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo=94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo=97:3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+
EXAMPLE 3Synthetic Route III
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and tert-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine by-product (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1H NMR in DMSO-d6: δ 9.61 (s, 1H), 7.16 (s, 2H), 1.42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
Step B. Synthesis of Compound 11 (HCl salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in i-PrOAc (57 mL, Aldrich, 99.6%). 4M HCl in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (10 mL) and dried at 45° C. under vacuum for 1 h to afford HCl salt of compound 11 (2.39 g, 89% yield) as a white solid. 1H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 113.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1.19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylamine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20° C. The resulting solution was stirred for 5 minute at 15-20° C. and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1.31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4Cl (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1.76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 110-115° C. under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo=94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo=93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo=99:1) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO04112718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo=91:9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
EXAMPLE 4Synthetic Route IV
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and tert-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 mL, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1.0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31.1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 15 h at 95° C., LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105° C. overnight. After total 40 h at 95-105° C., LC-MS analysis at 254 nm showed ˜99% conversion to the desired product (endo:exo=93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo=91:9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCl salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in i-PrOAc (26 mL, Aldrich, 99.6%). 4M HCl in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (5 mL) and dried under vacuum to yield HCl salt of compound 7 (1.57 g, 97% yield) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1.34 mL, 7.7 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20° C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4Cl (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
EXAMPLE 5Synthetic Route V
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1.6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 mL,) was added triethylamine (2.04 mL, 14.63 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minute. 4-Iodobenzoyl chloride 12 (1.95 g, 7.31 mmol, 1.1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20° C. After 24 h, additional 0.18 g (0.1 equiv, used total 1.6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and ˜5% of compound 7. The reaction was diluted with dichloromethane (100 mL). The organic phase was washed with saturated aqueous NH4Cl (100 mL), water (100 mL) and saturated aqueous NaHCO3 (100 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 13 (1.63 g, 57% yield, HPLC area 93% pure) as a white solid. 1H NMR in DMSO-d6: δ 11.19 and 10.93 (two singlets with integration ratio of 1.73:1, total of 1H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1.18 (s, 2H), 0.27 (q, 1H), 0.06 (s, 1H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 mL) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (0.44 mL, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at −90° C. for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45° C. and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt) Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. Example 2: Synthetic Route II
Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co- injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III
ST-246 9
P = Boc Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+. duct
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+ Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:
P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III. Im ine by-product
Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV) To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:
Scheme 5 Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
As of 2009, the results of clinical trials support its use against smallpox and other related orthopoxviruses. It shows potential for a variety of uses including preventive healthcare, as a post-exposure therapeutic, as a therapeutic, and an adjunct to vaccination.[21][
Tecovirimat can be taken by mouth and as of 2008, was permitted for phase II trials by the U.S. Food and Drug Administration (FDA). In phase I trials, tecovirimat was generally well tolerated with no serious adverse events.[22] Due to its importance for biodefense, the FDA designated tecovirimat for fast-track status, creating a path for expedited FDA review and eventual regulatory approval. On 13 July 2018, the FDA announced approval of tecovirimat.[23]
Society and culture
Legal status
In November 2021, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization under exceptional circumstances for the medicinal product tecovirimat siga, intended for the treatment of orthopoxvirus disease (smallpox, monkeypox, cowpox, and vaccinia complications) in adults and in children who weigh at least 13 kilograms (29 lb)[24] The applicant for this medicinal product is Siga Technologies Netherlands B.V.[24] Tecovirimat was approved for medical use in the European Union in January 2022.[5][25]
In December 2021, Health Canada approved oral tecovirimat for the treatment of smallpox in people weighing at least 13 kilograms (29 lb).[26][27]
^ Jump up to:abc“Tecovirimat Siga EPAR”. European Medicines Agency. 10 November 2021. Archived from the original on 16 May 2022. Retrieved 23 April 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^ Jump up to:abAU patent 2004249250, Bailey, Thomas R.; Jordan, Robert & Rippin, Susan R., “Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases”, published 2004-12-29, assigned to Siga Pharmaceuticals Inc
^Hughes, David L. (2019). “Review of the Patent Literature: Synthesis and Final Forms of Antiviral Drugs Tecovirimat and Baloxavir Marboxil”. Organic Process Research & Development. 23 (7): 1298–1307. doi:10.1021/acs.oprd.9b00144. S2CID197172102.
^ Jump up to:ab“Tecovirimat Siga: Pending EC decision”. European Medicines Agency. 11 November 2021. Archived from the original on 13 November 2021. Retrieved 13 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
Prior to its eradication in 1980, variola virus, the virus that causes smallpox, was mainly spread by direct contact between people. Symptoms typically began 10 to 14 days after infection and included fever, exhaustion, headache and backache. A rash initially consisting of small, pink bumps progressed to pus-filled sores before finally crusting over and scarring. Complications of smallpox could include encephalitis (inflammation of the brain), corneal ulcerations (an open sore on the clear, front surface of the eye) and blindness.
TPOXX’s effectiveness against smallpox was established by studies conducted in animals infected with viruses that are closely related to the virus that causes smallpox, and was based on measuring survival at the end of the studies. More animals treated with TPOXX lived compared to the animals treated with placebo. TPOXX was approved under the FDA’s Animal Rule, which allows efficacy findings from adequate and well-controlled animal studies to support an FDA approval when it is not feasible or ethical to conduct efficacy trials in humans.
The safety of TPOXX was evaluated in 359 healthy human volunteers without a smallpox infection. The most frequently reported side effects were headache, nausea and abdominal pain.
The FDA granted this application Fast Track and Priority Review designations. TPOXX also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases and a Material Threat Medical Countermeasure Priority Review Voucher, which provides additional incentives for certain medical products intended to treat or prevent harm from specific chemical, biological, radiological and nuclear threats.
The FDA granted approval of TPOXX to SIGA Technologies Inc.
TPOXX was developed in conjunction with the U.S. Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA).
Tecovirimat (Tpoxx) Tecovirimat is a drug used for the treatment or prophylaxis of viral infections, particularly those caused by the orthopoxvirus (Figure 12). In 2015, Dai described a procedure for the preparation of tecovirimat in a US patent (Scheme 33).[57 ] The developed method started with a cycloaddition reaction of cycloheptatriene with maleic anhydride in xylene to yield adduct 192, which after reaction with tert-butyl carbazate provided compound 193. Deprotection in acidic media gave rise to hydrazine derivative 194 and subsequent reaction with p-trifluoromethylbenzoyl chloride afforded tecovirimat (191).
57 [57] D. Dai, US Patent 0322010, 2015.
Synthesis
RAW MATERIAL
Key RM is, 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 3a,4,4a,5,5a,6-hexahydro-, (3aR,4R,4aR,5aS,6S,6aS)-rel–
The present invention provides a process for making ST-246 outlined in Scheme 1
P = Boc
Scheme 1
The present invention also provides a process for making ST-246 outlined in, Scheme 2
Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3
ST-246
P = Boc
Scheme 3
P = Boc
Scheme 4
The present invention further provides a process for making ST-246 outlined in
Scheme 5
Scheme 5
Example 1 : Synthetic Route I:
P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt)
Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 -50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent.
Example 2: Synthetic Route II
Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III
ST-246 9
P = Boc
Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
duct
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:
P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Im ine by-product
Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:
Scheme 5
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55
mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
PAPER
N-(3,3a,4,4a,5,5a,6,6a-Octahydro-1,3-dioxo-4,6- ethenocycloprop[f]isoindol-2-(1H)-yl)carboxamides: Identification of Novel Orthopoxvirus Egress Inhibitors
ViroPharma Incorporated, 397 Eagleview Boulevard, Exton, Pennsylvania 19341, United States Army Medical Research Institute of Infectious Diseases, 1425 Porter Street, Frederick, Maryland 21702, University of Alabama, Birmingham, Alabama 35294, and SIGA Technologies, Inc., 4575 SW Research Way, Corvallis, Oregon 97333
J. Med. Chem., 2007, 50 (7), pp 1442–1444
DOI: 10.1021/jm061484y
A series of novel, potent orthopoxvirus egress inhibitors was identified during high-throughput screening of the ViroPharma small molecule collection. Using structure−activity relationship information inferred from early hits, several compounds were synthesized, and compound 14was identified as a potent, orally bioavailable first-in-class inhibitor of orthopoxvirus egress from infected cells. Compound 14 has shown comparable efficaciousness in three murine orthopoxvirus models and has entered Phase I clinical trials.
A mixture of 2.00 g (9.8 mmol) of 4-(trifluoromethyl) benzoic acid hydrazide, 1.86 g (9.8 mmol) of 4,4a,5,5a,6,6a-hexahydro-4,6-etheno-1Hcycloprop[f]isobenzofuran-1,3(3aH)-dione, and one drop of diisopropylethylamine in 40 mL of absolute ethanol was refluxed for 4.5 h. Upon cooling to rt, 4 mL of water was added, and the product began to crystallize. The suspension was cooled in an ice bath, and the precipitate collected by filtration. The crystalline solid was air-dried affording 3.20 g (87%) of the product as a white solid;
Preparation of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide
a. Preparation of Compounds 1(a) and 1(b).
A mixture of cycloheptatriene (5 g, 54.26 mmol) and maleic anhydride (6.13 g, 62.40 mmol) in xylenes (35 mL) was heated at reflux under argon overnight. The reaction was cooled to room temperature and a tan precipitate was collected by filtration and dried to give 2.94 grams (28%) of the desired product, which is a mixture of compounds 1(a) and 1(b). Compound 1(a) is normally predominant in this mixture and is at least 80% by weight. The purity of Compound 1(a) may be further enhanced by recrystallization if necessary. Compound 1(b), an isomer of compound 1(a) is normally less than 20% by weight and varies depending on the conditions of the reaction. Pure Compound 1(b) was obtained by concentrating the mother liquid to dryness and then subjecting the residue to column chromatography. Further purification can be carried out by recrystallization if necessary. 1H NMR (500 MHz) in CDCl3: δ 5.95 (m, 2H), 3.42 (m, 2H), 3.09 (m, 2H), 1.12 (m, 2H), 0.22 (m, 1H), 0.14 (m, 1H).
b. Preparation of N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. desired
A mixture of compound 1(a) (150 mg, 0.788 mmol) and 4-trifluoromethylbenzhydrazide (169 mg, 0.827 mmol) in ethanol (10 mL) was heated under argon overnight. The solvent was removed by rotary evaporation. Purification by column chromatography on silica gel using 1/1 hexane/ethyl acetate provided 152 mg (51%) of the product as a white solid.
c. Preparation of N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. UNWANTED
N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]4-(trifluoromethyl)-benzamide was prepared and purified in the same fashion as for N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide by replacing 1(a) with 1(b) and was obtained as a white solid. 1H NMR (300 MHz) in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+.
EXAMPLE 42 Characterization of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (“ ”)
In the present application, ST-246 refers to: N-[(3aR,4R,4aR,5aS,65,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide.
Physico-Chemical Properties
Appearance: ST-246 is a white to off-white powder.
Melting Point: Approximately 196° C. by DSC.
Permeability: The calculated log P is 2.94. Based on the partition coefficient, ST-246 is expected to have good permeability.
Particle Size: The drug substance is micronized to improve its dissolution in the gastrointestinal fluids. The typical particle size of the micronized material is 50% less than 5 microns.
Solubility: The solubility of ST-246 is low in water (0.026 mg/mL) and buffers of the gastric pH range. Surfactant increases its solubility slightly. ST-246 is very soluble in organic solvents. The solubility data are given in Table 5.
Damon, Inger K.; Damaso, Clarissa R.; McFadden, Grant (2014). “Are We There Yet? The Smallpox Research Agenda Using Variola Virus”. PLoS Pathogens10 (5): e1004108.doi:10.1371/journal.ppat.1004108. PMID24789223.
Referenced by Citing Patent Filing date Publication date Applicant Title CN101912389A * Aug 9, 2010 Dec 15, 2010 中国人民解放军军事医学科学院微生物流行病研究所 Pharmaceutical composition containing ST-246 and preparation method and application thereof CN102406617A * Nov 30, 2011 Apr 11, 2012 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof CN102406617B Nov 30, 2011 Aug 28, 2013 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof CN103068232B * Mar 23, 2011 Aug 26, 2015 西佳科技股份有限公司 多晶型物形式st-246和制备方法 US8530509 Jul 29, 2011 Sep 10, 2013 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases US8802714 Aug 14, 2013 Aug 12, 2014 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases US9045418 Jul 3, 2014 Jun 2, 2015 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases
“Treatment options are limited for people with emphysema who have severe symptoms that have not improved from taking medicines. These have included lung surgery, such as lung volume reduction or lung transplants, which may not be suitable or appropriate for all patients,” said Tina Kiang, Ph.D., acting director, Division of Anesthesiology, General Hospital, Respiratory, Infection Control and Dental Devices, in the FDA’s Center for Devices and Radiological Health. “This novel device is a less invasive treatment that expands the options available to patients.”
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
“Treatment options are limited for people with emphysema who have severe symptoms that have not improved from taking medicines. These have included lung surgery, such as lung volume reduction or lung transplants, which may not be suitable or appropriate for all patients,” said Tina Kiang, Ph.D., acting director, Division of Anesthesiology, General Hospital, Respiratory, Infection Control and Dental Devices, in the FDA’s Center for Devices and Radiological Health. “This novel device is a less invasive treatment that expands the options available to patients.”
The Centers for Disease Control and Prevention estimates that 3.5 million American adults have been diagnosed with emphysema. Emphysema, including severe emphysema, is a type of chronic obstructive pulmonary disease (COPD) due to damage to the air sacs (alveoli) in the lungs. Lung damage from emphysema is irreversible. The damaged alveoli can cause used air to become trapped in the lungs during exhalation. This can cause the diseased parts of the lung to get larger and put pressure on the healthy part of the lung, which makes it difficult to breathe. As a result, the body may not get the oxygen it needs.
Using a flexible bronchoscope, a doctor places Zephyr Valves, similar in size to pencil erasers, into the diseased areas of the lung airways during a procedure in a hospital setting. Design of the device is intended to prevent air from entering the damaged parts of the lung and allow trapped air and fluids to escape. During inhalation, the valves close, preventing air from entering the damaged part of the lung and during exhalation, the valves open, letting out trapped air, which is intended to relieve pressure.
The FDA reviewed data from a multi-center study of 190 patients with severe emphysema. In this study, 128 patients were treated with Zephyr Valves and medical management according to current clinical guidelines, including medications (bronchodilators, corticosteroids, antibiotics or anti-inflammatory maintenance medications) and pulmonary rehabilitation, while 62 patients (the control group) received medical management only. Results of treatment were measured by how many patients in each arm of the study had at least a 15 percent improvement in pulmonary function scores (the volume of air that can forcibly be blown out in one second after full inhalation). At one year, 47.7 percent of patients treated with Zephyr Valves experienced at least a 15 percent improvement in their pulmonary function scores, compared with 16.8 percent of patients in the control group. Adverse events observed in the study include death, air leak (pneumothorax), pneumonia, worsening of emphysema, coughing up blood, shortness of breath and chest pain.
The Zephyr Valve device is contraindicated for patients with active lung infections; those who are allergic to nitinol, nickel, titanium or silicone; active smokers and those who are not able to tolerate the bronchoscopic procedure. Patients who have had major lung procedures, heart disease, large bubbles of air trapped in the lung or who have not responded to other treatments should talk with their providers to determine if the Zephyr Valve device is appropriate for them.
The Zephyr Valve was granted Breakthrough Device designation, meaning the FDA provided intensive interaction and guidance to the company on efficient device development, to expedite evidence generation and the agency’s review of the device. To qualify for such designation, a device must provide for more effective treatment or diagnosis of a life-threatening or irreversibly debilitating disease or condition, and meet one of the following criteria: the device must represent a breakthrough technology; there must be no approved or cleared alternatives; the device must offer significant advantages over existing approved or cleared alternatives; or the availability of the device is in the best interest of patients.
The FDA reviewed the Zephyr Valve device through the premarket approval review pathway, a regulatory pathway for the highest risk class of devices.
The FDA granted approval of the Zephyr Valve device to Pulmonx Inc.
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
CBD is a chemical component of the Cannabis sativa plant, more commonly known as marijuana. However, CBD does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC).
It is THC (and not CBD) that is the primary psychoactive component of marijuana.
“This approval serves as a reminder that advancing sound development programs that properly evaluate active ingredients contained in marijuana can lead to important medical therapies. And, the FDA is committed to this kind of careful scientific research and drug development,” said FDA Commissioner Scott Gottlieb, M.D. “Controlled clinical trials testing the safety and efficacy of a drug, along with careful review through the FDA’s drug approval process, is the most appropriate way to bring marijuana-derived treatments to patients. Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes. We’ll continue to support rigorous scientific research on the potential medical uses of marijuana-derived products and work with product developers who are interested in bringing patients safe and effective, high quality products. But, at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims. Marketing unapproved products, with uncertain dosages and formulations can keep patients from accessing appropriate, recognized therapies to treat serious and even fatal diseases.”
Dravet syndrome is a rare genetic condition that appears during the first year of life with frequent fever-related seizures (febrile seizures). Later, other types of seizures typically arise, including myoclonic seizures (involuntary muscle spasms). Additionally, status epilepticus, a potentially life-threatening state of continuous seizure activity requiring emergency medical care, may occur. Children with Dravet syndrome typically experience poor development of language and motor skills, hyperactivity and difficulty relating to others.
Lennox-Gastaut syndrome begins in childhood. It is characterized by multiple types of seizures. People with Lennox-Gastaut syndrome begin having frequent seizures in early childhood, usually between ages 3 and 5. More than three-quarters of affected individuals have tonic seizures, which cause the muscles to contract uncontrollably. Almost all children with Lennox-Gastaut syndrome develop learning problems and intellectual disability. Many also have delayed development of motor skills such as sitting and crawling. Most people with Lennox-Gastaut syndrome require help with usual activities of daily living.
“The difficult-to-control seizures that patients with Dravet syndrome and Lennox-Gastaut syndrome experience have a profound impact on these patients’ quality of life,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition.”
Epidiolex’s effectiveness was studied in three randomized, double-blind, placebo-controlled clinical trials involving 516 patients with either Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex, taken along with other medications, was shown to be effective in reducing the frequency of seizures when compared with placebo.
The most common side effects that occurred in Epidiolex-treated patients in the clinical trials were: sleepiness, sedation and lethargy; elevated liver enzymes; decreased appetite; diarrhea; rash; fatigue, malaise and weakness; insomnia, sleep disorder and poor quality sleep; and infections.
Epidiolex must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. As is true for all drugs that treat epilepsy, the most serious risks include thoughts about suicide, attempts to commit suicide, feelings of agitation, new or worsening depression, aggression and panic attacks. Epidiolex also caused liver injury, generally mild, but raising the possibility of rare, but more severe injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice and/or dark urine.
Under the Controlled Substances Act (CSA), CBD is currently a Schedule I substance because it is a chemical component of the cannabis plant. In support of this application, the company conducted nonclinical and clinical studies to assess the abuse potential of CBD.
The FDA prepares and transmits, through the U.S. Department of Health and Human Services, a medical and scientific analysis of substances subject to scheduling, like CBD, and provides recommendations to the Drug Enforcement Administration (DEA) regarding controls under the CSA. DEA is required to make a scheduling determination.
The FDA granted Priority Review designation for this application. Fast-Track designation was granted for Dravet syndrome. Orphan Drug designation was granted for both the Dravet syndrome and Lennox-Gastaut syndrome indications.
The FDA granted approval of Epidiolex to GW Research Ltd.
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
The U.S. Food and Drug Administration today approved Fulphila (pegfilgrastim-jmdb) as the first biosimilar to Neulasta (pegfilgrastim) to decrease the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells), in patients with non-myeloid (non-bone marrow) cancer who are receiving myelosuppressive chemotherapy that has a clinically significant incidence of febrile neutropenia.
“Bringing new biosimilars to patients is a top priority for the FDA, and a key part of our efforts to help promote competition that can reduce drug costs and promote access,” said FDA Commissioner Scott Gottlieb, M.D. “We’ll continue to prioritize reviews of these products to help ensure that biosimilar medications are brought to the market efficiently and through a process that makes certain that these new medicines meet the FDA’s rigorous standard for approval. This summer, we’ll release a comprehensive new plan to advance new policy efforts that promote biosimilar product development. Biologics represent some of the most clinically important, but also costliest products that patients use to promote their health. We want to make sure that the pathway for developing biosimilar versions of approved biologics is efficient and effective, so that patients benefit from competition to existing biologics once lawful intellectual property has lapsed on these products.”
Biological products are generally derived from a living organism and can come from many sources, such as humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on data showing that it is highly similar to a biological product already approved by the FDA (reference product) and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Fulphila is based on review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrates Fulphila is biosimilar to Neulasta. Fulphila has been approved as a biosimilar, not as an interchangeable product.
The most common side effects of Fulphila are bone pain and pain in extremities. Patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as pegfilgrastim or filgrastim products should not take Fulphila.
Serious side effects from treatment with Fulphila include rupture of the spleen, acute respiratory distress syndrome, serious allergic reactions including anaphylaxis, acute inflammation of the kidney (glomerulonephritis), an abnormally high level of white blood cells (leukocytosis), capillary leak syndrome and the potential for tumor growth. Fatal sickle cell crises have occurred.
The FDA granted approval of Fulphila to Mylan GmbH.
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody.
This drug is indicated for the treatment of X-linked hypophosphatemia with radiological evidence of bone disease in children of 1 year of age and older and adolescents with growing skeletons [4].
Burosumab (INN, trade name Crysvita) known as KRN23 is a human monoclonal antibody designed for the treatment of X-linked hypophosphatemia.[1][2][3] Burosumab was approved by the FDA for its intended purpose, in patients aged 1 year and older, on 17 April 2018.[4] The FDA approval fell under both the breakthrough therapy and orphan drug designations.[4]
This drug was developed by Ultragenyx and is in a collaborative license agreement with Kyowa Hakko Kirin.[5]
Burosumab (KRN23) is an entirely human monoclonal IgG1 antibody that binds excess fibroblast growth factor 23 (FGF23) and has been successfully tested in clinical trials in children with X-linked hypophosphatemic rickets [1].
The U.S. Food and Drug Administration approved Crysvita (burosumab) in April 2018. This is the first drug approved to treat adults and children ages 1 year and older with X-linked hypophosphatemia (XLH), which is a rare, inherited form of rickets. X-linked hypophosphatemia causes low circulating levels of phosphorus in the blood. It causes impaired bone growth and development in children and adolescents and issues with bone mineralization throughout a patient’s life [3].
XLH is a serious disease which affects about 3,000 children and 12,000 adults in the United States. Most children with XLH suffer from bowed or bent legs, short stature, bone pain and severe dental pain. Some adults with this condition suffer from persistent, unrelenting discomfort and complications, such as joint pain, impaired mobility, tooth abscesses and hearing loss [3]
Crysvita is specifically indicated for the treatment of X-linked hypophosphatemia (XLH) in adult and pediatric patients 1 year of age and older.
Crysvita is supplied as a subcutaneous injection. The recommended starting dose for pediatrics is 0.8 mg/kg of body weight, rounded to the nearest 10 mg, administered every two weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg. After initiation of treatment with Crysvita, measure fasting serum phosphorus every 4 weeks for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is above the lower limit of the reference range for age and below 5 mg/dL, continue treatment with the same dose. Follow dose adjustment schedule per the drug label. The recommended dose regimen in adults is 1 mg/kg body weight, rounded to the nearest 10 mg up to a maximum dose of 90 mg, administered every four weeks. After initiation of treatment with Crysvita, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the normal range, continue with the same dose. See drug label for specific dose adjustments.
Mechanism of Action
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody. X-linked hypophosphatemia is caused by excess fibroblast growth factor 23 (FGF23) which suppresses renal tubular phosphate reabsorption and the renal production of 1,25 dihydroxy vitamin D. Burosumab-twza binds to and inhibits the biological activity of FGF23 restoring renal phosphate reabsorption and increasing the serum concentration of 1,25 dihydroxy vitamin D.
Kutilek S: Burosumab: A new drug to treat hypophosphatemic rickets. Sudan J Paediatr. 2017;17(2):71-73. doi: 10.24911/SJP.2017.2.11. [PubMed:29545670]
Kinoshita Y, Fukumoto S: X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases -Prospect for new treatment. Endocr Rev. 2018 Jan 26. pii: 4825438. doi: 10.1210/er.2017-00220. [PubMed:29381780]
FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia [Link]
//////////////Burosumab-twza, Crysvita FDA 2018, BLA 761068, Protein Based Therapies, Monoclonal antibody, mAb, KRN 23, breakthrough therapy, orphan drug designations, Peptide, ブロスマブ

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











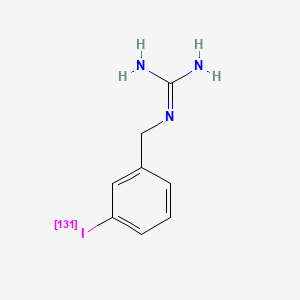

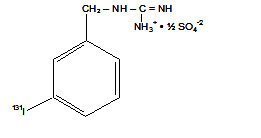










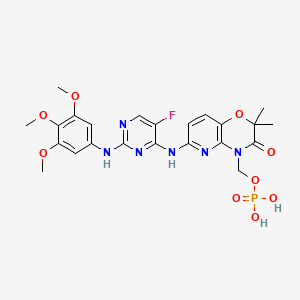

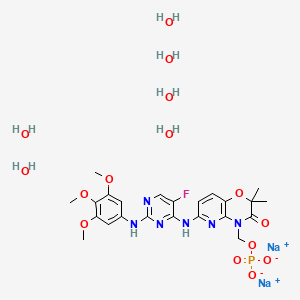








-Tafenoquin_Structural_Formula_V1.svg)
-Tafenoquin_Structural_Formula_V1.svg)
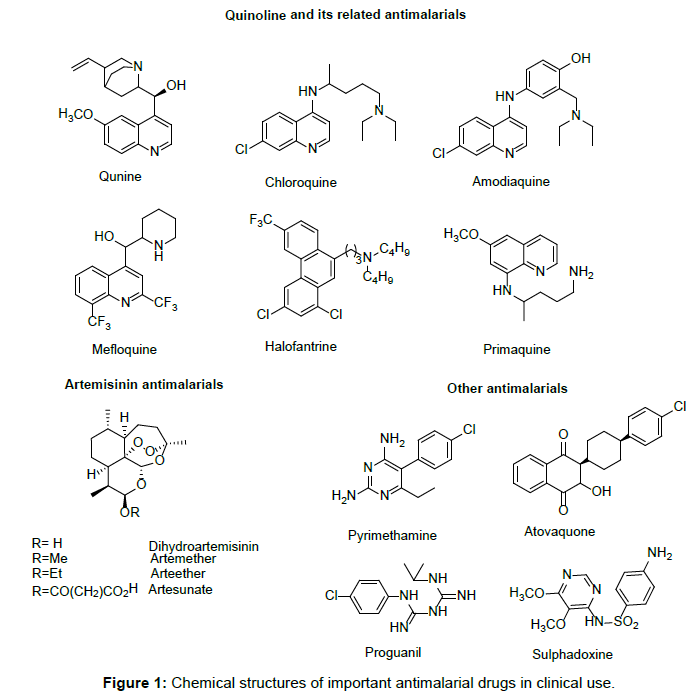
 CLIP
CLIP











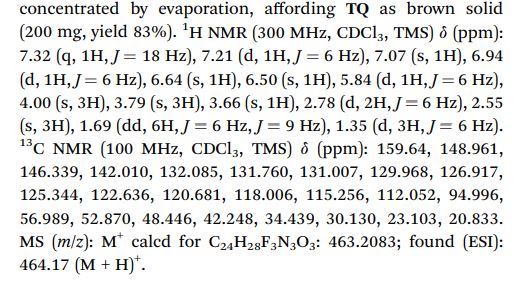





-Tafenoquin_Structural_Formula_V1.svg)







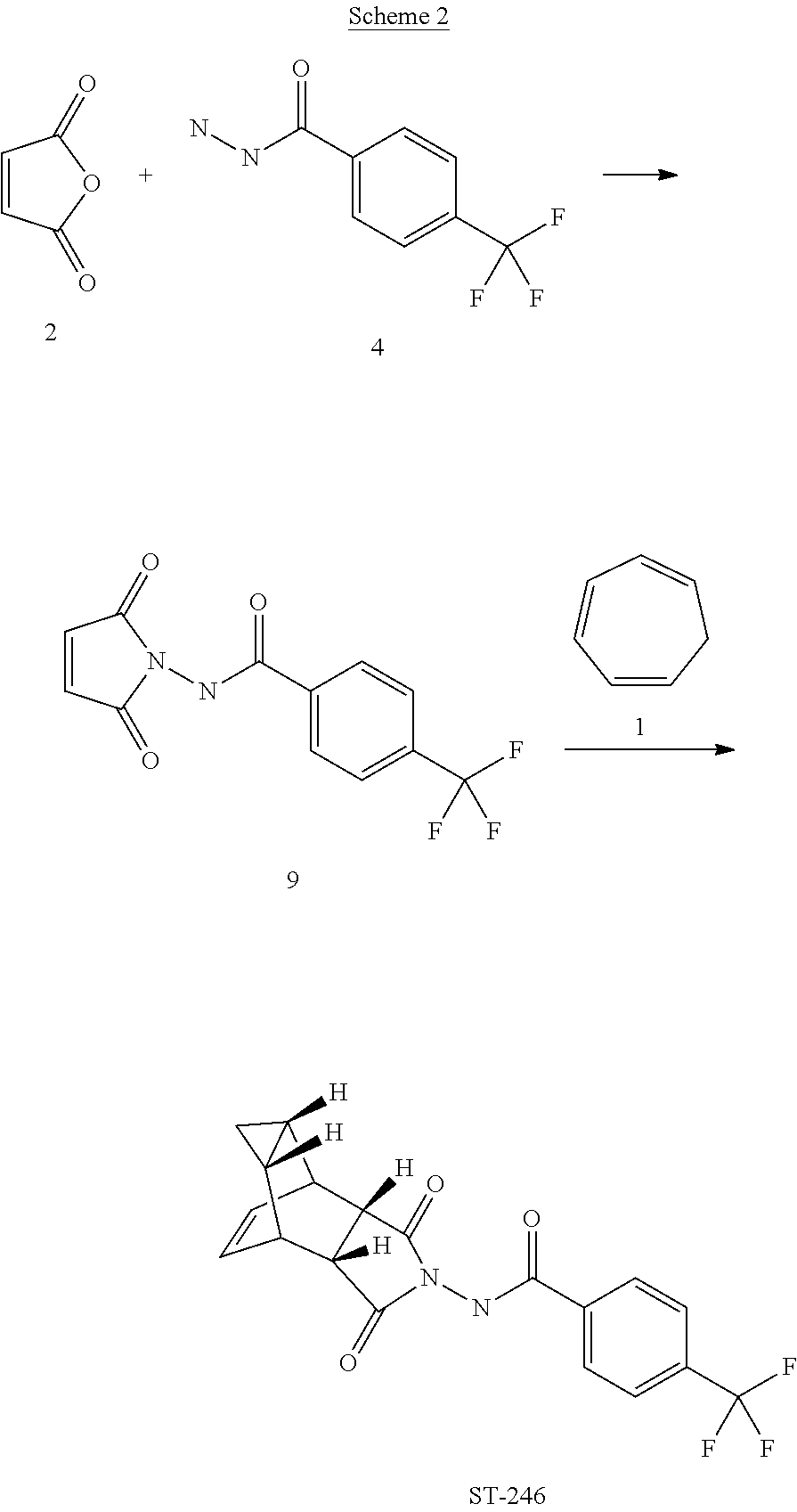
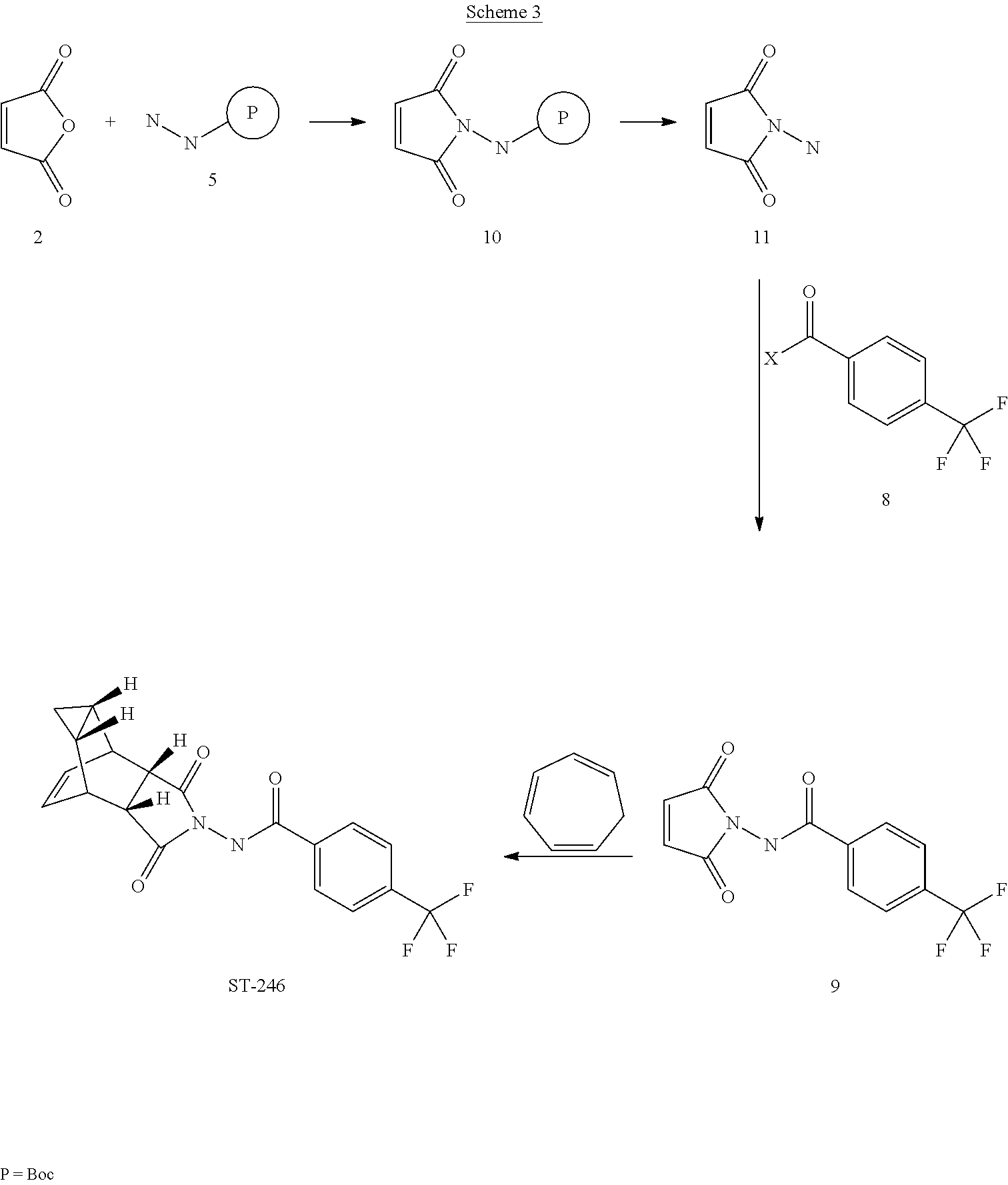























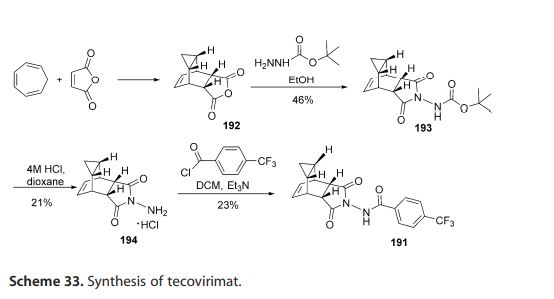
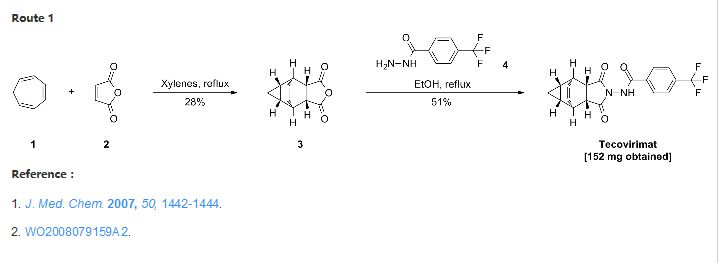












{kind=link}