Teriflunomide,
Teriflunomide, HMR-1726, 1726, A-771726, RS-61980, SU-0020,
(Z)-2-Cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide
108605-62-5, 282716-73-8 (monosodium salt)
C12-H9-F3-N2-O2
270.2091
Sugen (Licensee)

Antiarthritic Drugs, Disease-Modifying Drugs, Immunologic Neuromuscular Disorders, Treatment of, IMMUNOMODULATING AGENTS, Immunosuppressants, Multiple Sclerosis, Agents for, NEUROLOGIC DRUGS, TREATMENT OF MUSCULOSKELETAL & CONNECTIVE TISSUE DISEASES, Dihydroorotate Dehydrogenase Inhibitors
CAMBRIDGE, Mass.–Aug. 30, 2013–(BUSINESS WIRE)–Genzyme, a Sanofi company (EURONEXT: SAN and NYSE: SNY), announced today that the European Commission has granted marketing authorization for Aubagio® (teriflunomide) 14 mg, a once-daily, oral therapy indicated for the treatment of adult patients with relapsing remitting multiple sclerosis (RRMS).
read all at
http://www.pharmalive.com/ec-approves-genzyme-s-aubagio-for-ms

Teriflunomide (trade name Aubagio, marketed by Sanofi, also known as A77 1726) is the active metabolite of leflunomide.[1]Teriflunomide was investigated in the Phase III clinical trial TEMSO as a medication for multiple sclerosis (MS). The study was completed in July 2010.[2] 2-year results were positive.[3] However, the subsequent TENERE head-to-head superiority trial reported that “although permanent discontinuations [of therapy] were substantially less common among MS patients who received teriflunomide compared with interferon beta-1a, relapses were more common with teriflunomide.”[4] The drug was approved by the FDA on September 13, 2012.[5]

Mechanisms of action
Teriflunomide is an immunomodulatory drug inhibiting pyrimidine de novo synthesis by blocking the enzyme dihydroorotate dehydrogenase. It is uncertain whether this explains its effect on MS lesions.[6]
Teriflunomide inhibits rapidly dividing cells, including activated T cells, which are thought to drive the disease process in MS. Teriflunomide may decrease the risk of infections compared to chemotherapy-like drugs because of its more-limited effects on the immune system.[7]
It has been found that teriflunomide blocks the transcription factor NF-κB. It also inhibits tyrosine kinase enzymes, but only in high doses not clinically used.[8]
Activation of leflunomide to teriflunomide
 →
→ ↔
↔
The structure which results from ring opening can interconvert between the E and Z enolic forms (and the corresponding keto-amide), with the Z enol being the most stable and therefore most predominant form.

Space filling model of the E isomer of teriflunomide
^ Magne D, Mézin F, Palmer G, Guerne PA (2006). “The active metabolite of leflunomide, A77 1726, increases proliferation of human synovial fibroblasts in presence of IL-1beta and TNF-alpha”. Inflamm. Res. 55 (11): 469–75. doi:10.1007/s00011-006-5196-x. PMID 17122964.- ^ ClinicalTrials.gov Phase III Study of Teriflunomide in Reducing the Frequency of Relapses and Accumulation of Disability in Patients With Multiple Sclerosis (TEMSO)
- “Sanofi-Aventis’ Teriflunomide Comes Up Trumps in Two-Year Phase III MS Trial”. 15 Oct 2010.
- Gever, John (June 4, 2012). “Teriflunomide Modest Help but Safe for MS”. medpage. Retrieved June 04, 2012. Unknown parameter
|source= ignored (help)
- ^ “FDA approves new multiple sclerosis treatment Aubagio” (Press release). US FDA. Retrieved 2012-09-14.
- ^ H. Spreitzer (March 13, 2006). “Neue Wirkstoffe – Teriflunomid”. Österreichische Apothekerzeitung (in German) (6/2006).
- Dr. Timothy Vollmer (May 28, 2009). “MS Therapies in the Pipeline: Teriflunomide”. EMS News (in English) (May 28, 2009).
- ^ Breedveld, FC; Dayer, J-M (November 2000). “Leflunomide: mode of action in the treatment of rheumatoid arthritis”. Ann Rheum Dis 59 (11): 841–849. doi:10.1136/ard.59.11.841.PMC 1753034. PMID 11053058.


………………………
http://www.google.com/patents/WO2014177978A3?cl=en

Formula i
Teriflunomide is an immunosuppressant, acting as a tyrosine kinase inhibitor. It is also evaluated in the treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis. An oral film coated tablet containing teriflunomide as the active ingredient is marked in the United States by Sanofi Aventis US using brand AUBAGIO™. AUBAGIO is indicated for the treatment of patients with relapsing forms of multiple sclerosis.
U.S. Patent No. 5,679,709 appears to claim teriflunomide and its pharmaceutically acceptable salts, the same patent also further covers pharmaceutical composition and method of administering top a patients suffering from autoimmune disease.
U.S. Patent No. 5,494,91 I disclosesthe process for the preparation of teriflunomide by reacting 5-methylisoxazole-4-carbonyl chloride with trifluoromethyl aniline in the presence of acetonitrile to yield Leflunomide with on further hydrolysis with aqueous sodium hydroxide solution in methanol gives teriflunomide of formula I.
U.S. Patent No. 5,990,141 discloses the process for the preparation of teriflunomide by reacting 4-trifluoromethyl aniline with cyano acetic acid ethyl ester to yield cyanoaceto-(4-trifluromethyl)-aniline, with on further reacted with acetyl chloride in the presence of sodium hydride base and THF and acetonitrile solvent to give teriflunomide of formula I.
U.S. patent No. 6,365,626 discloses the process for the preparation of teriflunomide by reacting 4-trifluromethylaniline with cyanoacetic acid to give cyanoacet-(4- trifluoromethyl)anilide which on further reacted with acetyl chloride in the presence of sodium hydride to give teriflunomide of formula I.
U.S. Patent No. 6,894,184 discloses the process for the preparation of teriflunomide involves reacting 4-trifluromethylaniline with cyanoacetic acid to give cyanoacet-(4- trifluoromethyl)anilide which on further reacted with acetic anhydride in the presence of base to give teriflunomide of formula I.
International PCT application No. WO 2009/147624 discloses the process for the preparation of teriflunomide involves condensation of ethyl-2-cyano-3-hydroxybut-2-enoate and 4-(trifluoromethyl) aniline in presence of xylene solvent at reflux temperatures for 16 hours to give teriflunomide of formula I.
preparation of teriflunomide (I) comprises steps of;
1 ) condensation of cyanoacetic acid of formula (II) with 4-trifluoromethyl aniline of formula (III) in the presence of chlorinating agent to give 2-cyano-N-[4-(trifluromethyl)phenyl]acetamide of formula (IV);

(II I) (IV)
2) acetylation of 2-cyano-N-[4-(trifluromethyl)phenyl] acetamide of
formula (IV) with an acetylating agent in the presence of base and suitable solvents to yield teriflunomide of formula (I).
EXAMPLE 1 : Preparation of 2-cvano-N-f4-(trifluoromethyl> phenyl! acetamide (IV)
A round bottom flask is charged with cyanoacetic acid (100 g) and phosphorous pentachloride and tetrahydrofuran (300 ml) and the reaction mixture is stirred at room temperature for 4 hours. 4-trifluoromethyl aniline (161 g) dissolved in tetrahydrofuran (100 ml) is slowly added to the reaction mixture and stirred for completion of reaction. The resultant reaction mass is cooled and separated solid is filtered and washed with slurry of Isoproapnol and cyclohexane and dried under reduced pressure to afford the title compound. Weight: 196 gm.
Purity by HPLC: 98%
EXAMPLE 2: preparation of 2-cyano-3-hvdroxy-N-f4-( trifluoromethyl) phenyl] but-2-enamide (Teriflunomide crude)
A round bottom flask is charged with 2-cyano-N-[4-{trifluromethyl} phenyl] acetamide (100g), sodium hydroxide (70 gm) and dimethyl formamide is added and the reaction mixture is stirred for 30 minutes. Isopropenyl acetate (60 ml) is added slowly and the resultant mixture is stirred for about 4-5 hours at room temperature. After completion of the reaction, the resulting reaction mixture is diluted with water and acidified with Cone. HCI solution and stirred for solid separation. The separated solid is filtered and washed with water and dried under reduced pressure to afford Teriflunomide.
The obtained teriflunomide is charged in round bottom flask and aqueous solution of sodium hydroxide solution (29.6 g in 300 ml water) is added slowly at 25-35°C and stirred for 1 to 2 hours. The mixture is brought to 5 to 10°C and dichloromethane is added, the mixture is stirred for 15 minutes. The organic and the aqueous layer are separated, and the resultant aqueous layer is acidified with aq. Hcl and stirred. The separated solid is filtered and washed with water and dried under vacuum at 65-70°C for 10-12 hours to afford teriflunomide.
Weight: 101 gm
Purity by HPLC: 95%
EXAMPLE 3; Purification of Teriflunomide:
Teriflunomide (5 g) is charged into a flask followed by addition of acetonitrile (125 ml) and heated to reflux and stirred for 2 hours. The resultant reaction solution is filtered through highflow bed to obtain a clear solution and cooled to room temperature and stirred for solid separation. The separated solid is filtered, washed with Isopropanol (50 ml) and dried under vacuum to afford pure teriflunomide.
Weight: 3.8 gm
Purity by HPLC: 99.7%
…………………………………………………………………………………………………………

EP 0527736; JP 1993506425; JP 1999322700; JP 1999343285; US 5494911; US 5532259; WO 9117748
5-Methylisoxazole-4-carboxylic acid (I) was converted to the corresponding acid chloride (II) upon refluxing with SOCl2. Coupling of acid chloride (II) with 4-(trifluoromethyl)aniline (III) produced anilide (IV). Finally, isoxazole ring opening in the presence of NaOH gave rise to the title cyano amide.
- Magne D, Mézin F, Palmer G, Guerne PA (2006). “The active metabolite of leflunomide, A77 1726, increases proliferation of human synovial fibroblasts in presence of IL-1beta and TNF-alpha”. Inflamm. Res. 55 (11): 469–75. doi:10.1007/s00011-006-5196-x. PMID 17122964.
- ClinicalTrials.gov Phase III Study of Teriflunomide in Reducing the Frequency of Relapses and Accumulation of Disability in Patients With Multiple Sclerosis (TEMSO)
- “Sanofi-Aventis’ Teriflunomide Comes Up Trumps in Two-Year Phase III MS Trial”. 15 Oct 2010.
- ^ Gever, John (June 4, 2012). “Teriflunomide Modest Help but Safe for MS”. medpage. Retrieved June 04, 2012. Unknown parameter
|source= ignored (help)
- “FDA approves new multiple sclerosis treatment Aubagio” (Press release). US FDA. Retrieved 2012-09-14.
- ^ H. Spreitzer (March 13, 2006). “Neue Wirkstoffe – Teriflunomid”. Österreichische Apothekerzeitung (in German) (6/2006).
- ^ Dr. Timothy Vollmer (May 28, 2009). “MS Therapies in the Pipeline: Teriflunomide”. EMS News (in English) (May 28, 2009).
- Breedveld, FC; Dayer, J-M (November 2000). “Leflunomide: mode of action in the treatment of rheumatoid arthritis”. Ann Rheum Dis 59 (11): 841–849. doi:10.1136/ard.59.11.841.PMC 1753034. PMID 11053058.
Teriflunomide, a dihydroorotate dehydrogenase (DHODH) inhibitor, is the active metabolite of leflunomide a synthetic, low-molecular-weight drug currently used in the treatment of rheumatoid arthritis. The mechanisms by which teriflunomide exerts its antiinflammatory, antiproliferative and immunosuppressive effects are not yet completely understood, although inhibition of pyrimidine biosynthesis (via suppression of DHODH) and interference with tyrosine kinase activity both appear to be involved. Based on its efficacy shown in animal models of experimental allergic encephalomyelitis, teriflunomide was tested in a phase II study in patients with multiple sclerosis with relapses. Recruitment is ongoing for a phase III study to determine the efficacy of teriflunomide in reducing the frequency of relapses and accumulation of disability in multiple sclerosis patients.
The chemical name of Teriflunomide is 2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide and formula is C12H9F3N2O2 and molecular weight is 270.207.
Teriflunomide is used as Immunosupressant. It acts as tyrosine kinase inhibitor. It is used in treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis.
Teriflunomide was first disclosed and claimed in U.S. Pat. No. 5,679,709 but this patent does not mention any process of preparation for salt formation.
U.S. Pat. No. 5,494,911, U.S. Pat. No. 5,990,141 disclose various processes for preparing Teriflunomide. These patents do not disclose process for preparation Teriflunomide salts or mention any its polymorphic form.

EP 2280938 A2
HISTORY OF SYNTHESIS
The chemical name of Teriflunomide is
2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide and formula is Ci2H9 F3N2O2 and molecular weight is 270.207.
Teriflunomide is used as Immunosupressant. It acts as tyrosine kinase inhibitor. It is used in treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis.
Teriflunomide was first disclosed and claimed in US patent no. 5,679,709 but this application does not mention the process of preparation.
US patent no. 5,494,911 discloses a process for preparation of Teriflunomide as shown in given below
4-trifluoromethylaniline (IV) in acetonitrile to give leflunomide (VI). The subsequent hydrolysis with aqueous sodium hydroxide solution in methanol gives Teriflunomide (I). US patent 5,990,141 discloses a process for preparation of Teriflunomide as shown in given below
Teriflunomide (I)
The process involves reacting 4-trifluorometyl aniline (IV) with cyanoacetic acid ethyl ester (II) to give cyanoacet-(4-trifluoromethyl)-anilide (VII). This compound is further reacted first with sodium hydride in acetonitrile and then with acetylchloride in THF to give Teriflunomide (I).
US patent no. 6,365,626 discloses a process for preparation of Teriflunomide which is as given in below
Teriflunomide
ONE MORE

http://pubs.rsc.org/en/content/articlelanding/2012/cc/c2cc36352f GET ABOVE DETAILS HERE
Teriflunomide is used as Immunosupressant. It acts as tyrosine kinase inhibitor. It is used in treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis.
Teriflunomide was first disclosed and claimed in US patent no. 5,679,709 but this application does not mention the process of preparation.
[H] US patent no. 5,494,911 discloses a process for preparation of Teriflunomide in Example-4 as shown in given below scheme-I
(V) (IV) (VI) (D
Scheme-I
The proces; 5 involves re acting 5-metlr
4-trifluoromethylaniline (IV) in acetonitrile to give leflunomide (VI). The subsequent hydrolysis with aqueous sodium hydroxide solution in methanol gives Teriflunomide (I). US patent 5,990,141 discloses a process for preparation of Teriflunomide as shown in given below scheme-II.
Teriflunomide (I)
Scheme-II The process involves reacting 4-trifluorometyl aniline (IV) with cyanoacetic acid ethyl ester (II) to give cyanoacet-(4-trifluoromethyl)-anilide (VII). This compound is further reacted first with sodium hydride in acetonitrile and then with acetylchloride in THF to give Teriflunomide (I).
US patent no. 6,365,626 discloses a process for preparation of Teriflunomide in Fig. 19 which is as given in below scheme-Ill.
Teriflunomide
(I)
Scheme-Ill The process involves reacting 4-trifluoromethyl aniline (IV) with cyanoacetic acid (Ha) to give compound of formula (VII). This compound is further reacted first with sodium hydride and then with acetylchloride to give Teriflunomide (I)
………………………….

Example-1 Preparation of Ethyl-2-cyano-3-hydroxy-but-2-enoate (III) [77] Potassium carbonate (73.3 g) was added to the well stirred solution of Ethylcy- anoacetate (50 g) in Dimethylformamide (250 ml) and stirred for 15 minute at ambient temperature. Acetic anhydride (90.25 g) was added drop wise to the above well stirred solution during 2 to 3 hours at ambient temperature. Reaction mixture was stirred at ambient temperature for 15 to 20 hours. Reaction mixture was diluted with water (500 ml) and extracted with dichloromethane (3 xlOO ml). Combined organic layer was washed with saturated sodium carbonate solution (3x100ml). Aqueous carbonate layer was separated and acidified with 50% HCl solution and extracted with dichloromethane (3x100ml). Combined organic layer was washed with brine solution (100 ml), dried over sodium sulfate and evaporated to yield Ethyl 2-cyano-3-hydroxy-but-2-enoate (58 g).
Yield: 84.6%Example-2 ] Preparation of Teriflunomide (I) [82] Ethyl 2-cyano-3-hydroxybut-2-enoate (III) (50 g) and 4-(trifluoromethyl) aniline (51.9 g) in xylene (1000 ml) was refluxed for 48 hours. The reaction mixture was allowed to cool at room temperature. Separated solid was filtered and washed with xylene (2×100 ml). Solid was dried under vacuum at 700C to yield (62 g) of Teri- flunomide.
Yield: 71.0%
Purity: 99.4%
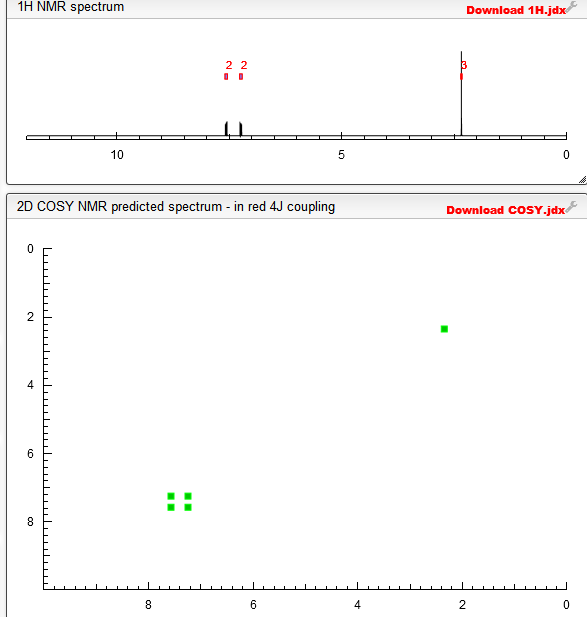
! HNMR (DMSO, 300MHz) :δ 2.24(s, 3H); 5.36(bs, IH); 7.65(d, J=8.7Hz, 2H);
7.76(d, J=8.6Hz, 2H); 10.89(s, IH) ppm.
13 CNMR (DMSO, 75MHz) :δ 23.5, 82.1, 118.3, 122.2, 126.5, 126.9, 142.1, 167.4,
187.8 ppm.
MS(FD) : m/e 269(M”, 100). [88] IR : 3305, 2220, 1633, 1596, 1554, 1418, 1405, 1325, 1247, 1114, 1157, 1073, 971,
842, 684 cm-1.
…………………
see
http://pubs.rsc.org/en/Content/ArticleLanding/2004/OB/b312682j#!divAbstract
………………………………
http://www.google.com/patents/CN103848756A?cl=en
Currently, for the preparation of teriflunomide mainly in the following three categories:
The first synthetic methods: mainly 5-methyl-isoxazole-4-carboxylic acid starting materials or by Synthesis of 5-methyl-isoxazole-4-carboxylic acid intermediate, then reacted with 4- trifluoromethyl base – aniline was synthesized teriflunomide, specific synthetic route is as follows:
[0007]
The general reaction step above normal class methods, not easy to intermediate purification, total yield is low, and the synthesis process using a large number of chloride corrosion of equipment can easily produce large amounts of acid mist and acidic water, thus polluting the environment .
The second class of methods: 2-cyano-acetic acid derivatives and 4-trifluoromethyl aniline. Such methods will be first prepared as a 2-cyano acetic acid chloride, and then 4-trifluoromethyl-aniline to give the corresponding amide, and then acetyl chloride for
With, the condensation reaction between the molecules to give the desired product, the synthesis route is as follows:
This class methods used in the reaction process large amounts of chloride reagent for large equipment and environmental damage.
The third method: This method is quite similar to the second type of method, mainly in the 2-cyano-acetic acid derivatives and 4-trifluoromethyl-aniline; The method of the second type is different, In the last step with 1-methyl-2-chloro-propylene oxide as raw materials to build α, β-unsaturated nitrile of the enol structure, i.e., to give the desired product, the synthesis route is as follows:

Teriflunomide Preparation Example 18 [0185] Implementation
Example 17 was obtained as a pale yellow solid of 61.2g crude compound was used directly in the synthesis of teriflunomide. In a 2L round bottom flask was added compound 27.2g (0.32mol) having the structure shown in formula IV, dry dioxane (620mL), sodium hydride 4g (0.16mol, in g / mL count, mass volume ratio 60% saving in kerosene), calcium hydride
6.7g (0.16mol), 15 ° C was stirred for I h, then slowly added dropwise in Example 17 was obtained as a pale yellow solid compound 61.2g (0.32mol) embodiment of dioxane 200mL, approximately I hour addition was complete, After the addition was complete the reaction was heated to reflux, the reaction at 80 ° C for 24 hours, the reaction process using a nitrogen blanket. After completion of the reaction was added 500mL of ice water to quench the reaction, with 2mol / L of HCl (aq.) And the reaction solution was adjusted to neutral pH, and extracted with EtOAc three times each in an amount of 500mL, and the combined organic phase was washed with saturated aqueous NaCl solution 800mL, dried over anhydrous Na2SO4, concentrated under reduced pressure, the mixed solution was twice recrystallized from methanol i_PrOH, the volume ratio of 1-PrOH and methanol is 2: 1, by volume of each recrystallized with a mixed solution of methanol with i_PrOH for 600mL, the crystallization temperature of 10 ° C, to give 58.8g of white solid compound in a yield of 66%, the total yield of 54% ο

using mass spectrometry, nuclear magnetic resonance spectroscopy and NMR spectra of the resulting white solid carbon compound structures were identified. MS data [M-H +] = 269.1, H NMR data = 1H-NMR (DMSO-Cie) δ the white solid compound: 10.88 (s, 1Η), 10.07 (br, s, 1H), 7.79 ( d, 2H), 7.66 (d, 2H), 2.26 (s, 3H), carbon NMR spectral data for: 13C-NMR (DMS0-d6) δ: 23.5,80.2,119.1,119.9,120.3,122.4,122.0, 123.5,125.3,126.2,141.8,166.2,186.0. Structural analysis by a white solid compound obtained in the present embodiment example for teriflunomide. Cases detected by HPLC obtained teriflunomide the embodiment of purity, calculated based on the peak area normalization method available, the present embodiment obtained teriflunomide a purity of 99.9%.
………………………
http://www.google.com/patents/WO2015029063A2?cl=en

Scheme-A
Scheme-A

Pure Teriflunomide ………………………………………….Crude Teriflunomide
xamples
Example- 1: Preparation of N-(4′-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide (Formula-2)
Methylene chloride (125 ml) and dimethyl formamide (2.87 gms) were added to 5-methylisoxazole-4-carboxylic acid (25 gms) at 25-30°C. Heated the reaction mixture to 35-40°C and thionyl chloride (47.59 gms) was slowly added and stirred for 4 hours at the same temperature. After completion of the reaction, distilled off the solvent completely from the reaction mixture. To the obtained compound, dichloromethane was added at 25-30°C. Distilled off the solvent completely from the reaction mixture. Acetonitrile (50 ml) was added to the obtained compound at 25-30°C and slowly added to a mixture of acetonitrile (300 ml) and 4-(trifluoromethyl)aniline (64.45 gms) at 25-30°C and stirred the reaction mixture for 5 hours at the same temperature. Filtered the reaction mixture and distilled off the solvent completely from the filtrate. Methanol (225 ml), followed by activated carbon (2.5 gms) were added to the obtained compound at 25-30°C and stirred for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with methanol. Water (250 ml) was slowly added to the obtained filtrate at 25-30°C and stirred the reaction mixture for 2 hours. Filtered the precipitated solid, washed with water and dried to get the title compound. Yield: 39.8 gms; Melting point: 165-168°C. Purity by HPLC: 99.63%.
Example-2: Preparation of N-(4′-trifluoromethylphenyl)-5-methylisoxazoIe-4-carboxamide (FormuIa-2)
Methylene chloride (15 Its) and dimethyl formamide (40 ml) were added to 5-methylisoxazole-4-carboxylic acid (3 kgs) at 25-30°C. Thionyl chloride (5.70 kgs) was slowly added to the reaction mixture at 25-30°C. Heated the reaction mixture to 40-45°C and stirred for 4 hours at the same temperature. After completion of the reaction, distilled off the solvent completely from the reaction mixture. Cooled the reaction mixture to 25-30°C and dichloromethane was added at the same temperature. Distilled off the solvent completely from the reaction mixture. Cooled the reaction mixture to 25-30°C and dissolved the obtained compound in acetonitrile (6.0 Its) at the same temperature. Slowly added to a mixture of acetonitrile (36 Its) and 4-(trifluoromethyl)aniline (7.70 kgs) at 25-30°C and stirred the reaction mixture for 5 hours at the same temperature. After completion of the reaction, filtered the reaction mixture and distilled off the solvent completely from the filtrate. Methanol (27 Its), followed by activated carbon (30 gms) was added to obtained compound at 25-30°C and stirred for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with methanol. Water (30 Its) was slowly added to the obtained filtrate at 25-30°C and stirred the reaction mixture for 2 hours. Filtered the precipitated solid, washed with water. To the obtained wet compound, toluene (9 Its) was added at 25-30°C. Heated the reaction mixture to 55-60°C and stirred for 30 minutes at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 3 hours at the same temperature. Filtered the solid, washed with toluene and dried to get the title compound. Yield: 4.7 kg.
Example-3: Preparation of (Z)-2-cyano-3-hydroxy-but-2-enoic acid-(4-trifluoromethyl phenyl)-amide (Formula-l)
Methanol (150 ml) was added to N-(4′-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide (50 gms) at 25-30°C. Cooled the reaction mixture to 0-5°C and aqueous sodium hydroxide solution was slowly added to the reaction mixture at the same temperature. Stirred the reaction mixture for 2 hours at 0-5°C. Water was added to the reaction mixture. Adjust the pH of the reaction mixture to 7.5 by using dilute hydrochloric acid at 25-30°C. Filtered the precipitated solid, washed with water and dried to get the title compound. Yield: 46.0 gms;
Example-4: Preparation of crystalline form-M of (Z)-2-cyano-3-hydroxy-but-2-enoic acid-(4-trifluoromethyl phenyl)-amide (Formula-1)
Dimethylformamide (300 ml) was added to (Z)-2-cyano-3-hydroxy-but-2-enoic acid-(4-trifluoromethylphenyl)-amide (50 gms) at 25-30°C. Heated the reaction mixture to 55-60°C and stirred for 30 minutes at the same temperature. Filtered the reaction mixture and washed with dimethyl formamide. To the obtained filtrate, methanol (350 ml) was added at 25-30°C. Cooled the reaction mixture to 10-15°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with chilled methanol and dried to get the title compound. Yield: 41 gms;
Melting point: 228-231°C; Water content: 0.06% w/w; Phenyl isoxazole impurity: 0.004%; Purity by HPLC: 99.97%.
Particle size distribution before micronisation: D10: 6.71 μιτι; D50: 34.4 μπι; D90: 109.8 μηι; Particle size distribution after micronisation: DIO: 1.35 μητ, D50: 4.52 μητ, D90: 10.26 μιη.
The P-XRD of the obtained compound is shown in figure- 1.
The DSC thermogram of the obtained compound is shown in figure-2.
Reference Example- 1: Preparation of (Z)-2-cyano-3-hydroxy-but-2-enoicacid-(4-trifluoromethylphenyl)-amide according to US5494911 (Formula-1)
Methanol (74 ml) was added to N-(4′-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide (20 gms) at 25-30°C. Cooled the reaction mixture to 0-5°C and aqueous sodium hydroxide solution {prepared by dissolving sodium hydroxide (3.26 gms) in water (74 ml)} was slowly added to the reaction mixture at the same temperature. Stirred the reaction mixture for 1 hour at 0-5°C. After completion of the reaction, 20% aqueous hydrochloric acid solution was added to the reaction mixture at 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and dried to get the title compound. Yield: 8.7 gms.
The P-XRD pattern of the obtained compound is shown in figure-3.
The DSC thermogram of the obtained compound is shown in figure-4.
………………….



…………….



Teriflunomide,
HMR-1726, 1726, A-771726, RS-61980, SU-0020,
(Z)-2-Cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide
108605-62-5, 282716-73-8 (monosodium salt)
C12-H9-F3-N2-O2 270.2091
17= US2011/0105795A1

1H NMR AND 13C NMR


above 13C NMR
! HNMR (DMSO, 300MHz) :δ 2.24(s, 3H); 5.36(bs, IH); 7.65(d, J=8.7Hz, 2H);
7.76(d, J=8.6Hz, 2H); 10.89(s, IH) ppm.
13 CNMR (DMSO, 75MHz) :δ 23.5, 82.1, 118.3, 122.2, 126.5, 126.9, 142.1, 167.4,
187.8 ppm.
MS(FD) : m/e 269(M”, 100).
IR : 3305, 2220, 1633, 1596, 1554, 1418, 1405, 1325, 1247, 1114, 1157, 1073, 971,
842, 684 cm-1.
REF EP 2280938 A2
Example-1 Preparation of Ethyl-2-cyano-3-hydroxy-but-2-enoate (III) [77] Potassium carbonate (73.3 g) was added to the well stirred solution of Ethylcy- anoacetate (50 g) in Dimethylformamide (250 ml) and stirred for 15 minute at ambient temperature. Acetic anhydride (90.25 g) was added drop wise to the above well stirred solution during 2 to 3 hours at ambient temperature. Reaction mixture was stirred at ambient temperature for 15 to 20 hours. Reaction mixture was diluted with water (500 ml) and extracted with dichloromethane (3 xlOO ml). Combined organic layer was washed with saturated sodium carbonate solution (3x100ml). Aqueous carbonate layer was separated and acidified with 50% HCl solution and extracted with dichloromethane (3x100ml). Combined organic layer was washed with brine solution (100 ml), dried over sodium sulfate and evaporated to yield Ethyl 2-cyano-3-hydroxy-but-2-enoate (58 g).
Yield: 84.6% Example-2 Preparation of Teriflunomide (I) [82] Ethyl 2-cyano-3-hydroxybut-2-enoate (III) (50 g) and 4-(trifluoromethyl) aniline (51.9 g) in xylene (1000 ml) was refluxed for 48 hours. The reaction mixture was allowed to cool at room temperature. Separated solid was filtered and washed with xylene (2×100 ml). Solid was dried under vacuum at 700C to yield (62 g) of Teri- flunomide.
Yield: 71.0%
Purity: 99.4%
! HNMR (DMSO, 300MHz) :δ 2.24(s, 3H); 5.36(bs, IH); 7.65(d, J=8.7Hz, 2H);
7.76(d, J=8.6Hz, 2H); 10.89(s, IH) ppm.
13 CNMR (DMSO, 75MHz) :δ 23.5, 82.1, 118.3,
122.2, 126.5,
126.9, 142.1, 167.4,
187.8 ppm.
MS(FD) : m/e 269(M”, 100).
IR : 3305, 2220, 1633, 1596, 1554, 1418, 1405, 1325, 1247, 1114, 1157, 1073, 971,
842, 684 cm-1.
1H NMR PREDICT


COSY

HPLC
HPLC method of analysis:
N-(4′-trifluoromethylphenyI)-5-methylisoxazole-4-carboxamide of formula-2:
Apparatus: A liquid chromatographic system equipped with variable wavelength UV- detector; Column: Cosmicsil APT CI 8, 100 x 4.6 mm, 3 μιη (or) equivalent; Flow rate: 1.5 ml/min; Wavelength: 210 nm; Column Temperature: 25°C; Injection volume: 20 μί; Run time: 40 min; Diluent: Mobile phase; Needle wash: Tetrahydrofuran; Elution: Isocratic; Mobile phase: 5 ml of triethyl amine into a 650 ml of water. Adjusted the pH to 3.4 with dil. Orthophosphoric acid and filter this solution through 0.22 μπι nylon membrane filter paper and sonicate to degas it. (Z)-2-cyano-3-hydroxy-but-2-enoicacid-(4-trifluoromethyl phenyl)-amide compound of formula- 1:
Apparatus: A liquid chromatographic system equipped with variable wavelength UV- detector; Column: Kromasil 100 C18, 250 x 4.6 mm, 5 μηι (or) equivalent; Flow rate: 1.0 ml/min; Wavelength: 250 nm; Column Temperature: 35°C; Injection volume: 5 μί; Run time: 37 min; Diluent: 0.01 M dipotassium hydrogen orthophosphate in 1000 ml of water; Elution: Gradient; Mobile phase-A: Buffer (100%); Mobile phase-B: Acetonitrile : Buffer (70:30 v/v); Buffer: 1 ml of ortho phosphoric acid into a 1000 ml of water and 3.0 grams of 1 -octane sulfonic acid sodium salt anhydrous. Adjust pH to 6.0 with potassium hydroxide solution and filtered through 0.22μηι Nylon membrane filter paper and sonicate to degas it……..http://www.google.com/patents/WO2015029063A2?cl=en
| WO2009147624A2 * |
3 Jun 2009 |
10 Dec 2009 |
Alembic Limited |
A process for preparing teriflunomide |
| WO2011004282A2 * |
22 Jun 2010 |
13 Jan 2011 |
Alembic Limited |
Novel polymorphic form of teriflunomide salts |
| US5494911 |
24 Oct 1990 |
27 Feb 1996 |
Hoechst Aktiengesellschaft |
Isoxazole-4-carboxamides and hydroxyalkylidenecyanoacetamides, pharmaceuticals containing these compounds and their use |
| US5679709 |
7 Jun 1995 |
21 Oct 1997 |
Hoechst Aktiengesellschaft |
N-(4-trifluoromethylphenyl)-2-cyano-3-hydroxycrotonamide or salts, used for reduction of b-cell produced self-antibodies |
| US5990141 |
6 Jan 1995 |
23 Nov 1999 |
Sugen Inc. |
Administering 5-methyl-isoxazole-4-carboxylic acid-n-(4-trifluoromethyl)anilide or 2-cyano-3-hydroxy-n-(4-trifluoro-methyl)phenyl-2-butenamide; antitumor,-carcinogenic and proliferative agents; kinase inhibitors |

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....