
Home » APPROVALS 2022 (Page 3)
Category Archives: APPROVALS 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GIMERACIL
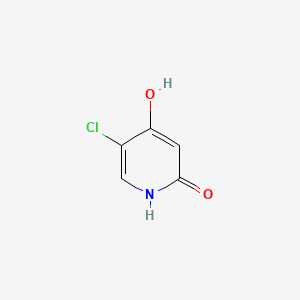

GIMERACIL
C5H4ClNO2, 145.54
5-chloro-4-hydroxy-1H-pyridin-2-one
5-Chloro-2,4-dihydroxypyridine
5-Chloro-4-hydroxy-2(1H)-pyridone
CDSCO APPROVED,01.02.2022

Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
| Combination of | |
|---|---|
| Tegafur | Antineoplastic drug |
| Gimeracil | Enzyme inhibitor |
| Oteracil | Enzyme inhibitor |
| Clinical data | |
| Trade names | Teysuno, TS-1 |
| Other names | S-1[1] |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by Tegafur |
| Pregnancy category | Contraindicated |
| Routes of administration | By mouth |
| ATC code | L01BC53 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) [2]EU: Rx-only [3]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 150863-82-4 |
| PubChem CID | 54715158 |
Tegafur/gimeracil/oteracil, sold under the brand names Teysuno and TS-1,[3][4] is a fixed-dose combination medication used for the treatment of advanced gastric cancer when used in combination with cisplatin,[3] and also for the treatment of head and neck cancer, colorectal cancer, non–small-cell lung, breast, pancreatic, and biliary tract cancers.[5]: 213
The most common severe side effects when used in combination with cisplatin include neutropenia (low levels of neutrophils, a type of white blood cell), anaemia (low red blood cell counts) and fatigue (tiredness).[3]
Tegafur/gimeracil/oteracil (Teysuno) was approved for medical use in the European Union in March 2011.[3] It has not been approved by the U.S. Food and Drug Administration (FDA).[5]: 213
Medical uses
In the European Union tegafur/gimeracil/oteracil is indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.[3]
Contraindications
In the European Union, tegafur/gimeracil/oteracil must not be used in the following groups:
- people receiving another fluoropyrimidine (a group of anticancer medicines that includes tegafur/gimeracil/oteracil) or who have had severe and unexpected reactions to fluoropyrimidine therapy;[3]
- people known to have no DPD enzyme activity, as well as people who, within the previous four weeks, have been treated with a medicine that blocks this enzyme;[3]
- pregnant or breastfeeding women;[3]
- people with severe leucopenia, neutropenia, or thrombocytopenia (low levels of white cells or platelets in the blood);[3]
- people with severe kidney problems requiring dialysis;[3]
- people who should not be receiving cisplatin.[3]
Mechanism of action
Tegafur is the actual chemotherapeutic agent. It is a prodrug of the active substance fluorouracil (5-FU).[3] Tegafur, is a cytotoxic medicine (a medicine that kills rapidly dividing cells, such as cancer cells) that belongs to the ‘anti-metabolites’ group. Tegafur is converted to the medicine fluorouracil in the body, but more is converted in tumor cells than in normal tissues.[3] Fluorouracil is very similar to pyrimidine.[3] Pyrimidine is part of the genetic material of cells (DNA and RNA).[3] In the body, fluorouracil takes the place of pyrimidine and interferes with the enzymes involved in making new DNA.[3] As a result, it prevents the growth of tumor cells and eventually kills them.[3]
Gimeracil inhibits the degradation of fluorouracil by reversibly blocking the dehydrogenase enzyme dihydropyrimidine dehydrogenase (DPD). This results in higher 5-FU levels and a prolonged half-life of the substance.[6]
Oteracil mainly stays in the gut because of its low permeability, where it reduces the production of 5-FU by blocking the enzyme orotate phosphoribosyltransferase. Lower 5-FU levels in the gut result in a lower gastrointestinal toxicity.[6]
Within the medication, the molar ratio of the three components (tegafur:gimeracil:oteracil) is 1:1:0.4.[7]
The maximum tolerated dose differed between Asian and Caucasian populations (80 mg/m2 and 25 mg/m2 respectively), perhaps due to differences in CYP2A6 genotype.[5]: 213
Research
It is being developed for the treatment of hepatocellular carcinoma.[8] and has activity in esophageal,(Perry Chapter 33) breast,[citation needed] cervical,[citation needed] and colorectal cancer.[9]
References
- ^ Liu TW, Chen LT (201). “S-1 with leucovorin for gastric cancer: how far can it go?”. Lancet Oncol. 17 (1): 12–4. doi:10.1016/S1470-2045(15)00478-7. PMID 26640038.
- ^ “Teysuno 20mg/5.8mg/15.8mg hard capsules – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 30 July 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Teysuno EPAR”. European Medicines Agency (EMA). Retrieved 30 July 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “ティーエスワン 患者さん・ご家族向け総合情報サイト | 大鵬薬品工業株式会社”.
- ^ Jump up to:a b c DeVita, DeVita; Lawrence, TS; Rosenberg, SA (2015). DeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology (10th ed.). LWW. ISBN 978-1451192940.
- ^ Jump up to:a b A. Klement (22 July 2013). “Dreier-Kombination gegen Magenkrebs: Teysuno”. Österreichische Apothekerzeitung (in German) (15/2013): 23.
- ^ Peters GJ, Noordhuis P, Van Kuilenburg AB et al. (2003). “Pharmacokinetics of S-1, an oral formulation of ftorafur, oxonic acid and 5-chloro-2,4-dihydroxypyridine (molar ratio 1:0.4:1) in patients with solid tumors”. Cancer Chemother. Pharmacol. 52 (1): 1–12. doi:10.1007/s00280-003-0617-9. PMID 12739060. S2CID 10858817.
- ^ “BCIQ”.
- ^ Miyamoto Y, Sakamoto Y, Yoshida N, Baba H (2014). “Efficacy of S-1 in colorectal cancer”. Expert Opin Pharmacother. 15 (12): 1761–70. doi:10.1517/14656566.2014.937706. PMID 25032886. S2CID 23637808.
External links
- “Tegafur”. Drug Information Portal. U.S. National Library of Medicine.
- “Gimeracil”. Drug Information Portal. U.S. National Library of Medicine.
- “Oteracil”. Drug Information Portal. U.S. National Library of Medicine.
Gimeracil is an adjunct to antineoplastic therapy, used to increase the concentration and effect of the main active componets within chemotherapy regimens. Approved by the European Medicines Agency (EMA) in March 2011, Gimeracil is available in combination with Oteracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Gimeracil’s main role within Teysuno is to prevent the breakdown of Fluorouracil (5-FU), which helps to maintin high enough concentrations for sustained effect against cancer cells 2. It functions by reversibly and selectively blocking the enzyme dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU 1. This allows higher concentrations of 5-FU to be achieved with a lower dose of tegafur, thereby also reducing toxic side effects.
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////


SYNTHESIS

SYN
https://europepmc.org/article/pmc/pmc7717319
Synthesis of Gimeracil 20a
aReagents and conditions: (a) CH3C(OCH3)3, MeOH, then (CH3)2NHCH(OCH3)2, reflux, 92%; (b) aq AcOH, 130 °C, 2 h, 95%; (c) SO2Cl2, HOAc, 50 °C, 0.5 h, 91%; (d) 40% H2SO4, 130 °C, 4 h, 91%; (e) SO2Cl2, HOAc, 50 °C, 45 min, 86%; (f) 75% H2 SO4, 140 °C, 3 h, then NaOH, then pH 4–4.5, 89%


In 1953, Kolder and Hertog reported a synthesis of the TS-1 additive gimeracil 20, which was completed in seven steps using 4-nitropyridine N-oxide as starting material.222 Later, Yano et al. reported an alternative gram-scale synthesis (Scheme 15).223 The one-pot, three component condensation of malononitrile 111, 1,1,1-trimethoxyethane, and 1,1-dimethyoxytrimethylamine generated the dicyano intermediate 112, which was into 2(1H)-pyridinone 113.224 Selective chlorination of 113 was followed by acid-mediated demethylation, hydrolysis, and decarboxylation, to afford gimeracil 20. Interestingly, Xu et al. found that treatment of intermediate 113 with sulfuryl chloride resulted in dichloro 115 formation, which could still be converted to gimeracil 20 by treatment with sulfuric acid.225
(222) Kolder CR; den Hertog HJ Synthesis and reactivity of 5-chloro-2,4-dihydroxypyridine. Rec. Trav. Chim 1953, 72, 285–295. [Google Scholar]
(223) Yano S; Ohno T; Ogawa K Convenient and practical synthesis of 5-chloro-4-hydroxy-2(1H)-pyridinone. Heterocycles 1993, 36, 145–148. [Google Scholar]
(224) Mittelbach M; Kastner G; Junek H Synthesen mit Nitrilen, 71. Mitt. Zur Synthese von 4-Hydroxynicotinsaure aus Butadiendicarbonitrilen. Arch. Pharm 1985, 318 (6), 481–486. [Google Scholar]
(225) Xu Y; Mao D; Zhang F CN Patent 1915976, 2007.

NEW DRUG APPROVALS
THIS MAY NOT RUN WITHOUT SUBSCRIPTION HELP. AVOID CLOSURE OF THIS BLOG
$10.00
//////////GIMERACIL, APPROVALS 2022, INDIA 2022
OC1=CC(=O)NC=C1Cl
Darinaparsin
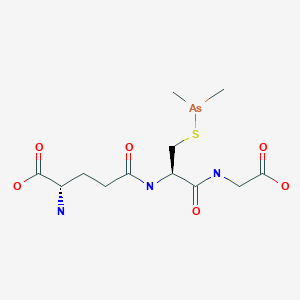
Darinaparsin
ダリナパルシン , Darvias
JAPAN 2022 APPROVED, PMDA 2022/6/20
(2S)-2-amino-5-[[(2R)-1-(carboxymethylamino)-3-dimethylarsanylsulfanyl-1-oxopropan-2-yl]amino]-5-oxopentanoic acid
Glycine, L-gamma-glutaMyl-S-(diMethylarsino)-L-cysteinyl-
| Formula | C12H22AsN3O6S |
|---|---|
| CAS | 69819-86-9 |
| Mol weight | 411.3062 |
| Efficacy | Antineoplastic |
|---|---|
| Comment | organic arsenical |
Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L
- OriginatorTexas A&M University; University of Texas M. D. Anderson Cancer Center
- DeveloperSolasia Pharma; ZIOPHARM Oncology
- ClassAmines; Antineoplastics; Arsenicals; Oligopeptides; Pentanoic acids; Small molecules; Sulfides
- Mechanism of ActionApoptosis stimulants; Cell cycle inhibitors; Reactive oxygen species stimulants
- Orphan Drug StatusYes – Peripheral T-cell lymphoma
- PreregistrationPeripheral T-cell lymphoma
- DiscontinuedLiver cancer; Lymphoma; Multiple myeloma; Non-Hodgkin’s lymphoma; Solid tumours
- 28 Mar 2022No recent reports of development identified for phase-I development in Peripheral-T-cell-lymphoma in China (IV, Injection)
- 26 Jan 2022ZIOPHARM Oncology is now called Alaunos Therapeutics
- 11 Dec 2021Safety and efficacy data from a phase II trial in Peripheral T-cell lymphoma presented at the 63rd American Society of Hematology Annual Meeting and Exposition (ASH-2021)
Darinaparsin is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects. Compared to inorganic arsenic compounds such as arsenic trioxide (As2O3), darinaparsin appears to exhibit a wide therapeutic window.
Darinaparsin, also know as ZIO-101 and SP-02, is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects.
Darinaparsin is an organic arsenical composed of dimethylated arsenic linked to glutathione, and is being investigated for antitumor properties in vitro and in vivo. While other arsenicals, including arsenic trioxide, have been used clinically, none have shown significant activity in malignancies outside of acute promyelocytic leukemia. Darinaparsin has significant activity in a broad spectrum of hematologic and solid tumors in preclinical models. Here, we review the literature describing the signaling pathways and mechanisms of action of darinaparsin and compare them to mechanisms of cell death induced by arsenic trioxide. Darinaparsin has overlapping, but distinct, signaling mechanisms. We also review the current results of clinical trials with darinaparsin (both intravenous and oral formulations) that demonstrate significant antitumor activity.
PAPER
Biochemical Pharmacology (Amsterdam, Netherlands), 126, 79-86; 2017



PATENT
WO 2015085208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015085208
Preparation of Darinaparsin
[0071] Sterile water (15.5 L) and ethyl alcohol (200 proof, 15.5 L) were charged in a reaction flask prior to the addition of L-glutathione (3.10 kg). While being stirred, the reaction mixture was cooled to 0-5 °C prior to the addition of triethylamine (1.71 L). Stirring was continued until most of the solids were dissolved and the solution was filtered. After filtration, the reaction mixture was cooled to 0-5 °C prior to the addition of chlorodimethylarsine (1.89 kg) over 115 minutes while maintaining the temperature at 0-5 °C. Stirring continued at 0-5 °C for 4 hours before acetone (30.6 L) was added over 54 minutes while maintaining the temperature at 0-5 °C. The suspension was stored at 0-5°C overnight prior to filtration. The solid was collected in a filter funnel, washed successively with ethyl alcohol (200 proof, 13.5 L) and acetone (13.5 L) and dried in suction for 23 minutes. A second similar run was performed and the collected solids from both runs were combined. Ethyl alcohol (200 proof, 124 L) and the combined solids (11.08 kg) were charged in a vessel. The slurry was stirred at ambient temperature for 2 hours before filtration, washing successively with ethyl alcohol (200 proof, 27 L) and acetone (27 L) and dried in suction for 60 minutes. The resulting solid was transferred to drying trays and dried in a vacuum oven at ambient temperature for 66 hours to provide darinaparsin as a solid with the differential scanning calorimetry (DSC) thermogram of Figure 1, with an extrapolated onset temperature at about 191.36° C and a peak temperature at about 195.65° C.
PATENT
WO 2010021928
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109°C was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
SGLU-1: Glutathione (14.0 g, 45.6 mmol) was stirred rapidly in glyme while dimethylchoroarsine (6.5 g, 45.6 mmol) was added dropwise. Pyridine (6.9 g, 91.2 mmol) was then added to the slurry and the mixture was subsequently heated to reflux. The heat was removed immediately and the mixture stirred at room temperature for 4 h. Isolation of the resultant insoluble solid and recrystallization from ethanol afforded 4 as the pyridine hydrochloride complex (75% yield). mp 115-118°C; NMR (D20) δ1.35 (s, 6H), 1.9-4.1 (m’s, 10H), 7.8-9.0 (m, 5H); mass spectrum (m/e) 140, 125, 110, 105, 79, 52, 45, 36.
PATENT
WO 2009075870
Step 1
Example 1. Preparation of Dimethylchloroarsine (DMCA). A 3-neck round-bottom flask (500 mL) equipped with mechanical stirrer, inlet for nitrogen, thermometer, and an ice bath was charged with cacodylic acid (33 g, 0.23 mol) and cone. hydrochloric acid (67 mL). In a separate flask, a solution of SnCl2·2H2O (54 g, 0.239 mol) in cone. hydrochloric acid (10 mL) was prepared. The SnCl2·2 H2O solution was added to the cacodylic acid in HCl solution under nitrogen while maintaining the temperature between 5 °C and 10 °C. After the addition was complete, the ice bath was removed and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was transferred to a separatory funnel and the upper layer (organic) collected. The bottom layer was extracted with dichloromethane (DCM) (2 × 25 mL). The combined organic extract was washed with 1 N HCl (2 × 10 mL) and water (2 × 20 mL). The organic extract was dried over MgSO4 and DCM was removed by rotary evaporation (bath temperature 80 °C, under nitrogen, atmospheric pressure). The residue was further distilled under nitrogen. Two tractions of DMCA were collected. The first fraction contained some DCM and the second fraction was of suitable quality (8.5 g, 26% yield). The GC analysis confirmed the identity and purity of the product.
Step 2
Example 3. Preparation of S-Dimethylarsinoglutathione (SGLU-1). In a 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel and thermometer under an inert atmosphere was prepared a suspension of glutathione (114.5 g, 0.37 mol) in a 1:1 (v/v) mixture of water/ethanol (1140 mL) and cooled to below 5 °C. The mixture was treated slowly (over 15 min) with triethylamine (63.6 mL, 0.46 mol) while maintaining the temperature below 20 °C. The mixture was cooled to 4 °C and stirred for 15 min and then the traces of undissolved material removed by filtration. The filtrate was transferred in a clean 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel, nitrogen inlet, and thermometer and DMCA (70 g, 0.49 mol) (lot # 543-07-01-44) was added slowly while maintaining the temperature at 3-4°C. The reaction mixture was stirred at 1-4°C for 4 h, and acetone (1.2 L) was added over a period of 1 h. The mixture was stirred for 90 min between 2 and 3°C and the resulting solid was isolated by filtration. The product was washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and the wet solids were suspended in ethanol 200 Proof (2000 mL). The product was isolated by filtration, washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and dried in vacuum for 2 days at RT to give 115 g (75%) of SGLU-1, HPLC purity > 99.5% (in process testing).
PATENT
WO 2007027344
Example 2 Preparation of S-Dimethylarsinoglutathione A 5 L, three necked round bottom flask was equipped with a mechanical stirrer assembly, thermometer, addition funnel, nitrogen inlet, and a drying tube was placed in a cooling bath. A polyethylene crock was charged with glutathione-reduced (200 g) and deionized water (2 L) and stirred under a nitrogen atmosphere to dissolve all solids. The mixture was filtered to remove any insoluble material and the filtrate was transferred to the 5 L flask. While stirring, ethanol, 200 proof (2 L) was added and the clear solution was cooled to 0-5° C. using an ice/methanol bath. Pyridine (120 g) was added followed by a dropwise addition of Me2AsCl (120 g) over a minimum of 1 hour. The reaction mixture was stirred at 0-5° C. for a minimum of 2 hours prior to removal of the cooling bath and allowing the mixture to warm to room temperature under a nitrogen atmosphere with stirring. The reaction mixture was stirred overnight (>15 hrs) at room temperature under a nitrogen atmosphere at which time a white solid may precipitate. The reaction mixture was concentrated to a slurry (liquid and solid) at 35-45° C. using oil pump vacuum to provide a white solid residue. As much water as possible is removed, followed by two coevaporations with ethanol to azeotrope the last traces of water. The white solid residue was slurried in ethanol, 200 pf. (5 L) under a nitrogen atmosphere at room temperature overnight. The white solid was filtered and washed with ethanol, 200 pf. (2×500 mL) followed by acetone, ACS (2×500 mL). The resulting solid was transferred to drying trays and vacuum oven dried overnight at 25-35° C. using oil pump vacuum to provide pyridinium hydrochloride-free S-dimethylarsinoglutathione as a white solid. melting point of 189-190° C.
PATENT
WO 20060128682
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109° C. was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
Pyridine Hydrochloride Free Synthesis of S-Dimethylarsinoglutathione (GLU) Dimethylarsinoglutathione is made using an adapted of Chen (Chen, G. C., et al. Carbohydrate Res. (1976) 50: 53-62) the contents of which are hereby incorporated by reference in their entirety. Briefly, dithiobis(dimethylarsinoglutamine) is dissolved in dichloromethane under nitrogen. Tetramethyldiarsine is added dropwise to the solution and the reaction is stirred overnight at room temperature under nitrogen and then exposed to air for 1 h. The mixture is then evaporated to dryness and the residue is washed with water and dried to give a crude solid that is recrystallized from methanol to give S-dimethylarsinoglutathione.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Solasia Announces Submission of New Drug Application for Anti-cancer Drug DARINAPARSIN for Peripheral T-Cell Lymphoma in Japan
Solasia Pharma K.K. (TSE: 4597, Headquarters: Tokyo, Japan, President & CEO: Yoshihiro Arai, hereinafter “Solasia”) today announced submission of a New Drug Application (NDA) for its new anti-cancer drug darinaparsin (generic name, development code: SP-02) as a treatment for relapsed or refractory peripheral T-cell lymphoma to the Ministry of Health, Labour and Welfare (MHLW). Based on positive results of R&D on darinaparsin, centered primarily on the results of the Asian Multinational Phase 2 Study (study results released in June 2020), Solasia filed an NDA for the drug with the regulatory authority in Japan ahead of anywhere else in the world.
Solasia expects to obtain regulatory approval in 2022 and to also launch in the same year. If approved and launched, darinaparsin would be the third drug Solasia successfully developed and brought to market since its founding and is expected to contribute to the treatment of PTCL.
Mr. Yoshihiro Arai, President and CEO of Solasia, commented as follows:
“No standard treatment has been established for relapsed or refractory PTCL as of yet. I firmly believe that darinaparsin, with its novel mechanism of action that differs from those of already approved drugs, will contribute to patients and healthcare providers at clinical sites as a new treatment option for relapsed or refractory PTCL. Since founding, Solasia has conducted R&D on five pipeline drugs. Of the five, we have successfully developed and brought to market two drugs, i.e., began providing them to patients, and today, we submitted an NDA for our first anti-cancer drug. Under our mission to provide patients with ‘Better Medicine for a Brighter Tomorrow’, we will continue aiming to contribute to patients’ treatment and enhanced quality of life. ”
About darinaparsin (SP-02)
Darinaparsin, an organoarsenic compound with anticancer activity, is a novel mitochondrial-targeted agent being developed for the treatment of various hematologic and solid tumors. The proposed mechanism of action of the drug involves the disruption of mitochondrial function, increased production of reactive oxygen species, and modulation of intracellular signal transduction pathways. Darinaparsin is believed to exert anticancer effect by inducing cell cycle arrest and apoptosis. Darinaparsin has been granted orphan drug designation in the US and EU.
For more information, please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Asian Multinational Phase 2 Study
The Asian Multinational Phase 2 Study was a multinational, multicenter, single-arm, open-label, non-randomized study to evaluate the efficacy and safety of darinaparsin monotherapy in patients with relapsed or refractory PTCL conducted in Japan, Korea, Taiwan, and Hong Kong. (CT.gov Identifier: NCT02653976).
Solasia plans to present the results of the study at an international academic conference to be held in the near future.
About peripheral T-cell lymphoma (PTCL)
Please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Solasia
Please visit at https://solasia.co.jp/en/
/////////////Darinaparsin, Darvias, JAPAN 2022, APPROVALS 2022, PMDA, ダリナパルシン , Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L, Orphan Drug
C[As](C)SCC(C(=O)NCC(=O)O)NC(=O)CCC(C(=O)O)N
Pimitespib
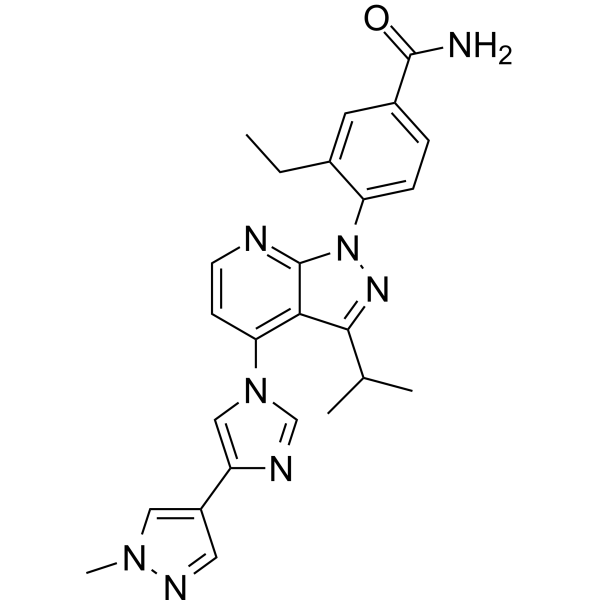
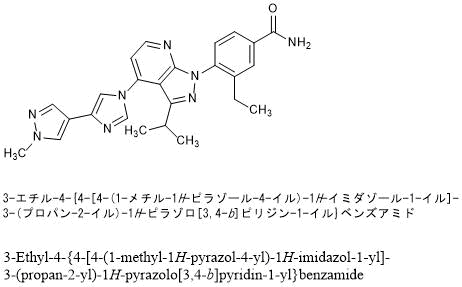
![Benzamide, 3-ethyl-4-[3-(1-methylethyl)-4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-1H-pyrazolo[3,4-b]pyridin-1-yl]-.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=67501411&t=l)
Pimitespib
TAS 116
CAS 1260533-36-5
Antineoplastic, Hsp 90 inhibitor
3-ethyl-4-[4-[4-(1-methylpyrazol-4-yl)imidazol-1-yl]-3-propan-2-ylpyrazolo[3,4-b]pyridin-1-yl]benzamide
Pimitespib (TAS-116) is an oral bioavailable, ATP-competitive, highly specific HSP90α/HSP90β inhibitor (Kis of 34.7 nM and 21.3 nM, respectively) without inhibiting other HSP90 family proteins such as GRP94. Pimitespib demonstrates less ocular toxicity.
| Formula | C25H26N8O |
|---|---|
| CAS | 1260533-36-5 |
| Mol weight | 454.5269 |
JAPAN APPROVED 2022/6/20, ピミテスピブ
| Jeselhy |
Taiho. originator
Pimitespib is a specific inhibitor of heat shock protein 90 (Hsp90) subtypes alpha and beta, with potential antineoplastic and chemo/radiosensitizing activities. Upon oral administration, pimitespib specifically binds to and inhibits the activity of Hsp90 alpha and beta; this results in the proteasomal degradation of oncogenic client proteins, which inhibits client protein dependent-signaling, induces apoptosis, and inhibits the proliferation of cells overexpressing HSP90alpha/beta. Hsp90, a family of molecular chaperone proteins that are upregulated in a variety of tumor cells, plays a key role in the conformational maturation, stability, and function of “client” proteins within the cell,; many of which are involved in signal transduction, cell cycle regulation and apoptosis, including kinases, cell-cycle regulators, transcription factors and hormone receptors. As TAS-116 selectively inhibits cytosolic HSP90alpha and beta only and does not inhibit HSP90 paralogs, such as endoplasmic reticulum GRP94 or mitochondrial TRAP1, this agent may have less off-target toxicity as compared to non-selective HSP90 inhibitors.
Patent
WO2011004610
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011004610
PATENT
CN108623496
| 3-Ethyl-4-fluorobenzonitrile is an important intermediate for the preparation of a variety of new drugs under development, such as TAS-116, a Phase II clinical drug of Taiho Pharmaceuticals for the treatment of gastrointestinal stromal tumors. |
| |
| Patent WO2005105760 discloses its preparation method. In the method, tetrakis(triphenylphosphine) palladium is used as a catalyst, and 3-bromo-4-fluorobenzonitrile is coupled with tetraethyl tin in a solvent hexamethylphosphoramide for a heating reaction for 15 hours to obtain 3 -Ethyl-4-fluorobenzonitrile. The method uses highly toxic tetraethyl tin, which brings great harm to operators and the environment, and is difficult to carry out industrial production. Meanwhile, the product 3-ethyl-4-fluorobenzonitrile obtained by the preparation method is an oily substance, which is purified by column chromatography with complicated operation, which is unfavorable for industrial production, and the specific purity of the product is not described. |
| |
| Therefore, looking for a new method for preparing 3-ethyl-4-fluorobenzonitrile with cheap and easy-to-obtain raw materials, safe and simple operation, high product purity and low cost suitable for industrial production, which will speed up the research process of related new drugs under development. , it is of great significance to reduce the production cost of related new drugs. |
| Example 1 3-Bromo-4-fluorobenzonitrile |
| |
| 3-Bromo-4-fluorobenzaldehyde (250g, 1.23mol) was dissolved in acetonitrile (1.5L), then hydroxylamine sulfonic acid (67g, 1.48mol) was added, and the reaction was refluxed for 4h. TLC showed that the conversion of the raw materials was complete, and the reaction solution was concentrated. To a small volume, add water (2L) and stir for 30min, cool to 5-10°C and continue stirring for 10min, filter, dissolve the filter cake with methyl tert-butyl ether (1.2L), wash twice with 500ml of water, saturated with 200ml Washed with sodium bicarbonate solution, dried over anhydrous sodium sulfate, filtered, the filtrate was adsorbed with activated carbon (10g), filtered, concentrated under reduced pressure to remove the solvent, added n-heptane (250ml), cooled and stirred in an ice-salt bath for 1h, filtered, reduced Press drying to give 3-bromo-4-fluorobenzonitrile (217 g, 88% yield). 1 H NMR (CDCl 3 ,400MHz):δ7.91(m,1H),7.63(m,1H),7.24(m,1H)。 |
| Example 2 3-Bromo-4-fluorobenzonitrile |
| |
| Add tetrahydrofuran (100ml) to a 250ml reaction flask, add 3-bromo-4-fluorobenzaldehyde (10g, 49.2mmol) and ammonia (40ml) under stirring, add elemental iodine (25g, 98.5mmol) in batches under cooling to 5°C ), then raised to ambient temperature and reacted for 2 to 3 hours, the reaction was completed, the reaction solution was poured into a 10% aqueous solution of sodium sulfite (200g), extracted twice with methyl tert-butyl ether (100ml), dried over anhydrous sodium sulfate , concentrated under reduced pressure to remove the solvent, added n-heptane (20 ml), cooled to 0-10 °C and stirred for 1 h, filtered, and dried under reduced pressure to obtain 3-bromo-4-fluorobenzonitrile (9.6 g, yield: 97.5 %). The NMR spectrum of this compound was determined and was identical to the product of Example 1. |
| Example 3 3-ethyl-4-fluorobenzonitrile |
| |
| 3-Bromo-4-fluorobenzonitrile (200 g, 1 mol) and [1,1-bis(diphenylphosphino)ferrocene]palladium(II) chloride dichloromethane complex (4.08 g, 5mmol) was dissolved in THF (1.2L), 1.0M/L diethylzinc n-hexane solution (600mL, 0.6mol) was added at 40-50°C, and the temperature was raised to 50-60°C for 4-5h. TLC showed The raw materials reacted completely. After the reaction solution was cooled to room temperature, it was added to 5% dilute hydrochloric acid (1 L), the layers were separated, the organic layer was washed twice with 500 ml of water, and then concentrated under reduced pressure to remove the solvent. Then n-hexane (600mL) and activated carbon (20g) were added, refluxed for 0.5h, cooled to room temperature, filtered, then added activated carbon (10g) to the filtrate, refluxed for 0.5h, cooled to room temperature, filtered, and cooled to -50°C to -60°C and filtered, and the filter cake was dried under reduced pressure at 10-20°C to obtain 3-ethyl-4-fluorobenzonitrile (112 g, yield: 75%) as an off-white solid, melting point 23.1-27.4°C. 1 H NMR (CDCl 3 , 400MHz): δ 7.50 (m, 2H), 7.09 (m, 1H), 2.69 (q, J=7.6Hz, 2H), 1.24 (t, 3H, J=7.6Hz), HPLC purity 99.6%. |
| HPLC assay conditions: |
| Chromatographic UV detector: DAD |
| Chromatography pump: 1100 quaternary pump |
| Chromatographic column: Agilent (USA) ZORBAX SB-C184.6×150mm, 5μm PN883975-902 Chromatographic conditions: |
| Mobile Phase A: Water |
| Mobile Phase B: Acetonitrile |
| |
| Injection volume: 5 μL, flow rate: 1.0 mL/min, column temperature: room temperature, detection wavelength: 210 nm. |


Acylation of 2-fluoro-4-iodopyridine with isobutyric anhydride in presence of BuLi and DIEA in THF at -78 °C gives 1-(2-fluoro-4-iodo-3-pyridinyl)-2-methylpropan-1-one ,
This upon cyclization using hydrazine hydrate at 65 °C gives 4-iodo-3-isopropylpyrazolo[3,4-b]pyridine.
N-Protection of intermediate with PMB-Cl in the presence of base NaH in solvent DMF at 0 °C affords 4-iodo-3-isopropyl-1-(4-methoxybenzyl)pyrazolo[3,4-b]pyridine,
This is coupled with 4-(4-imidazolyl)-1-methylpyrazole in the presence of Cu2O, 4,7-dimethoxy-1,10-phenanthroline, Cs2CO3 and PEG-diamine in solvent NMP or DMSO at 130 °C to furnish 4-[4-(4-pyrazolyl)-imidazol-1-yl]pyrazolo[3,4-b]pyridine derivative .
N-Deprotection of PMB-protected pyrazolo[3,4-b]pyridine derivative by using TFA and anisole gives free pyrazolo[3,4-b]pyridine ,
This on condensation with 3-ethyl-4-fluorobenzonitrile in the presence of Cs2CO3 in DMF at 95 °C yields 4-(pyrazolo[3,4-b]pyridin-1-yl)benzonitrile .
Finally, partial hydrolysis of nitrile by means of aqueous NaOH and H2O2 in DMSO/EtOH gives the Pimitespib TAS-116 .
CLIP
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b01085
J. Med. Chem.2019, 62, 2, 531–551
Publication Date:December 7, 2018
https://doi.org/10.1021/acs.jmedchem.8b0108

The molecular chaperone heat shock protein 90 (HSP90) is a promising target for cancer therapy, as it assists in the stabilization of cancer-related proteins, promoting cancer cell growth, and survival. A novel series of HSP90 inhibitors were discovered by structure–activity relationship (SAR)-based optimization of an initial hit compound 11a having a 4-(4-(quinolin-3-yl)-1H-indol-1-yl)benzamide structure. The pyrazolo[3,4-b]pyridine derivative, 16e (TAS-116), is a selective inhibitor of HSP90α and HSP90β among the HSP90 family proteins and exhibits oral availability in mice. The X-ray cocrystal structure of the 16e analogue 16d demonstrated a unique binding mode at the N-terminal ATP binding site. Oral administration of 16e demonstrated potent antitumor effects in an NCI-H1975 xenograft mouse model without significant body weight loss.
3-Ethyl-4-(3-Isopropyl-4-(4-(1-methyl-1H-Pyrazol-4-yl)-1H-Imidazol-1-yl)-1H-Pyrazolo[3,4-b]pyridin-1-yl)benzamide (16e). Yield 64% (2 steps), white powder. UPLC−MS (ESI) m/z: 454.8 [M + H]+ , tR = 1.19 min. UPLC purity 99.65%. 1 H NMR (400 MHz, CDCl3): δ 1.14 (t, J = 7.5 Hz, 3H), 1.25 (d, J = 7.0 Hz, 6H), 2.62 (q, J = 7.3 Hz, 2H), 3.18 (spt, J = 6.8 Hz, 1H), 3.98 (s, 3H), 5.88 (br s,1H), 6.22 (br s, 1H), 7.13 (d, J = 5.1 Hz, 1H), 7.39 (d, J = 1.1 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.78−7.81 (m, 3H), 7.86 (d, J = 1.5 Hz, 1H), 7.96 (d, J = 1.8 Hz, 1H), 8.59 (d, J = 4.7 Hz, 1H). HRMS: calcd for C25H26N8O, 455.2308 [M + H]+ ; found, 455.2311.
PAPER
Journal of Medicinal Chemistry (2021), 64(5), 2669-2677.
PATENT
WO 2016181990
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016181990
Compound 1 in the present invention is 3-ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazol-4-yl) -1H-imidazole-1-yl) -1H-. Pyrazolo [3,4-b] pyridin-1-yl} benzamide (formula below). Compound 1 is known to have HSP90 inhibitory activity and exhibit excellent antitumor activity. Compound 1 can be synthesized based on the production methods described in Patent Documents 1 and 2.
[0013]
[hua 1]
Patent Document 1: International Publication No. 2012/093708
Patent Document 2: International Publication No. 2011/004610
Comparative Example 1 3-Ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3, 4-b] Pyridine-1-yl} Synthesis of type I crystals of benzamide
3-Ethyl-4 obtained according to the production method described in International Publication No. 2012/093708 and International Publication No. 2011/004610. -{3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3,4-b] pyridin-1- A white solid (3.58 g) of yl} benzamide was added to ethanol (7.84 mL) and stirred at room temperature for 2 hours. After sampling, it was washed with ethanol (7.84 mL) and dried under reduced pressure at 70 to 80 ° C. for 20 hours to obtain type I crystals (yield: 2.40 g, yield: 61.2%, purity: 98.21%). rice field.
Further, as shown in FIG. 1, the type I crystal has a diffraction angle (2θ) of 8.1 °, 10.9 °, 12.1 °, 14.0 °, and 14.9 in the powder X-ray diffraction spectrum. °, 16.2 °, 17.7 °, 20.2 °, 21.0 °, 21.5 °, 22.6 °, 24.3 °, 25.4 ° 26.4 °, 27.0 ° , 28.3 °, 30.2 °, 30.9 °, 31.5 °, 32.7 °, 34.7 °, 35.4 ° and 36.6 ° showed characteristic peaks.
[0032]
1H-NMR (DMSO-d 6):δppm 9.35 (1H,d,J=4.88Hz), 8.93 (1H,d,J=1.22Hz), 8.84 (1H,brs), 8.72 (1H,d,J=1.95Hz), 8.70 (1H,s) ,8.63 (1H,d,J=1.22Hz), 8.60 (1H,dd,J=8.29,1.95Hz), 8.46 (1H,s) ,8.25 (1H,d,J=8.29Hz), 8.22 (1H,brs), 8.12 (1H,d,J=4.88Hz), 4.59 (3H,s) ,3.95 (1H,tt,J=6.83,6.83Hz), 3.21 (2H,q,J=7.56Hz), 1.83(6H,d,J=6.83Hz), 1.75 (3H,t,J=7.56Hz):LRMS(ESI)m/z 455[M+H]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015050235
Synthesis of Test Compound The
following synthesis example compounds (Synthesis Examples 1 to 3) were synthesized according to the method described in WO2011 / 004610.
[0361]
Synthesis Example 1: 4- {3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazol-1-yl) -1H-pyrazolo [3,4-b] pyridine -1-yl} -3-methylbenzamide
[0362]
[Changing 22]
PATENT
WO 2011004610
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011004610
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Pimitespib
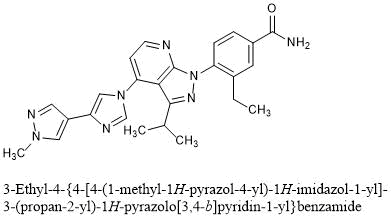
3-Ethyl-4-{4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-3-(propan-2-yl)-1H-pyrazolo[3,4-b]pyridin-1-yl}benzamide
C25H26N8O : 454.53
[1260533-36-5]
//////////Pimitespib, ピミテスピブ, JAPAN 2022, APPROVALS 2022, TAS 116, Jeselhy
O=C(N)C1=CC=C(N2N=C(C(C)C)C3=C(N4C=C(C5=CN(C)N=C5)N=C4)C=CN=C32)C(CC)=C1
Vutrisiran sodium, ALN 65492, Votrisiran
RNA, (Um-sp-(2′-deoxy-2′-fluoro)C-sp-Um-Um-Gm-(2′-deoxy-2′-fluoro)G-Um-Um-(2′-deoxy-2′-fluoro)A-Cm-Am-Um-Gm-(2′-deoxy-2′-fluoro)A-Am-(2′-deoxy-2′-fluoro)A-Um-Cm-Cm-Cm-Am-sp-Um-sp-Cm), complex with RNA (Um-sp-Gm-sp-Gm-Gm-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)A-(2′-deoxy-2′-fluoro)U-Gm-Um-Am-Am-Cm-Cm-Am-Am-Gm-Am) 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate] (1:1)
Vutrisiran Sodium
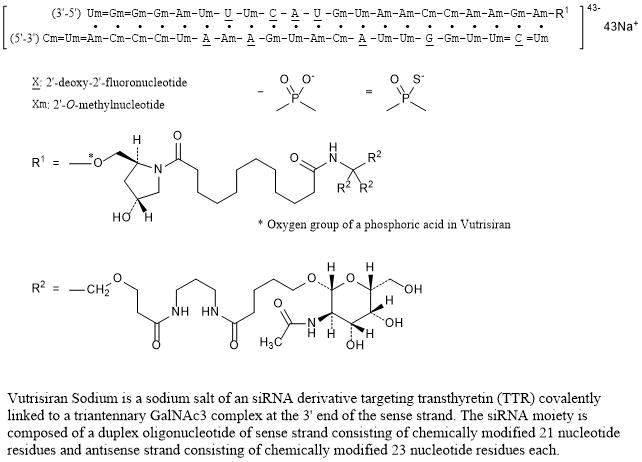
Nucleic Acid Sequence
Sequence Length: 44, 23, 2113 a 9 c 8 g 14 umultistranded (2); modified
Vutrisiran sodium
- ALN 65492
- Votrisiran
C530H672F9N171Na43O323P43S6 : 17289.77
[1867157-35-4 , Vutrisiran]
| Formula | C530H672F9N171O323P43S6.43Na ORC530H672F9N171Na43O323P43S6 |
|---|---|
| CAS | 1867157-35-4 , VURISIRAN |
| Mol weight | 17289.7661 |
FDA APPROVED, AMVUTTRA, 2022/6/13
| ブトリシランナトリウム |
| Efficacy | Gene expression regulator |
|---|---|
| Disease | Polyneuropathy of hereditary transthyretin-mediated amyloidosis [D |
| Comment | RNA interference (RNAi) drug Treatment of transthyretin (TTR)-mediated amyloidosis (ATTR amyloidosis) |
UNII28O0WP6Z1P UNII
Vutrisiran
Vutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each.
Vutrisiran is a double-stranded small interfering ribonucleic acid (siRNA) that targets wild-type and mutant transthyretin (TTR) messenger RNA (mRNA).7 This siRNA therapeutic is indicated for the treatment of neuropathies associated with hereditary transthyretin-mediated amyloidosis (ATTR), a condition caused by mutations in the TTR gene.2 More than 130 TTR mutations have been identified so far,3 but the most common one is the replacement of valine with methionine at position 30 (Val30Met).2 The Val30Met variant is the most prevalent among hereditary ATTR patients with polyneuropathy, especially in Portugal, France, Sweden, and Japan.2
TTR mutations lead to the formation of misfolded TTR proteins, which form amyloid fibrils that deposit in different types of tissues. By targeting TTR mRNA, vutrisiran reduces the serum levels of TTR.6,7 Vutrisiran is commercially available as a conjugate of N-acetylgalactosamine (GalNAc), a residue that enables the delivery of siRNA to hepatocytes.5,7 This delivery platform gives vutrisiran high potency and metabolic stability, and allows for subcutaneous injections to take place once every three months.8 Another siRNA indicated for the treatment of polyneuropathy associated with hereditary ATTR is patisiran.2 Vutrisiran was approved by the FDA in June 2022.
CLIP
https://www.nature.com/articles/s41392-020-0207-x
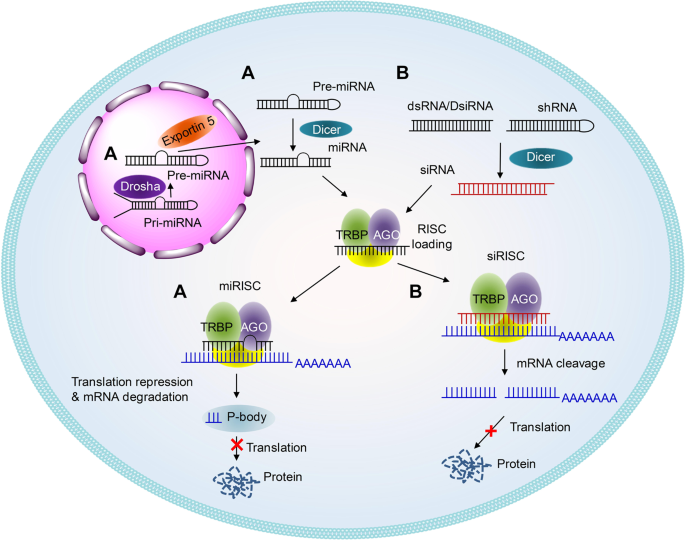
Schematic illustrations of the working mechanisms of miRNA (a) and siRNA (b)
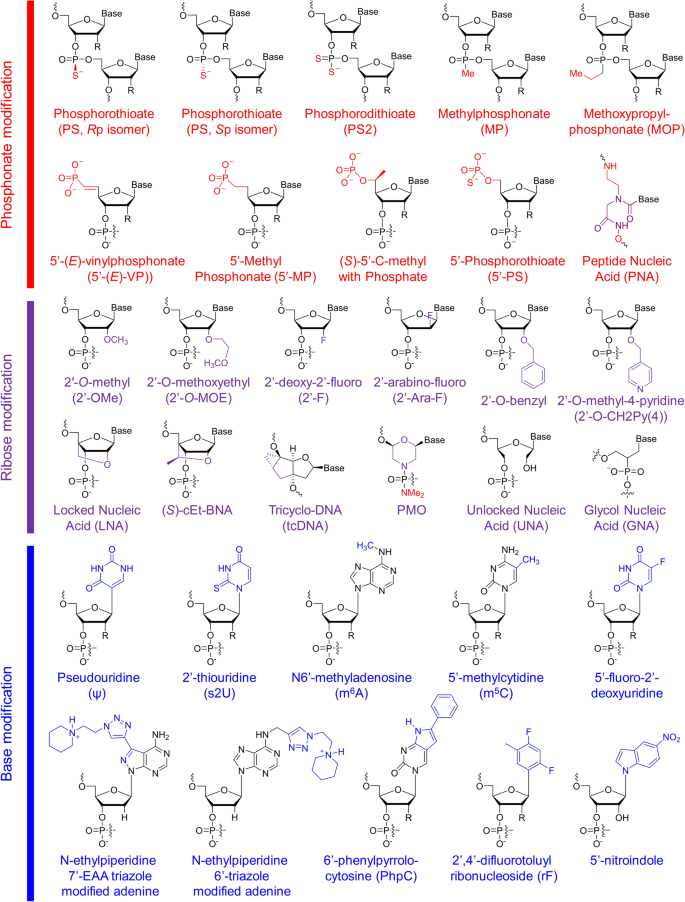
Structures of chemical modifications and analogs used for siRNA and ASO decoration. According to the modification site in the nucleotide acid, these structures can be divided into three classes: phosphonate modification, ribose modification and base modification, which are marked in red, purple and blue, respectively. R = H or OH, for RNA or DNA, respectively. (S)-cEt-BNA (S)-constrained ethyl bicyclic nucleic acid, PMO phosphorodiamidate morpholino oligomer
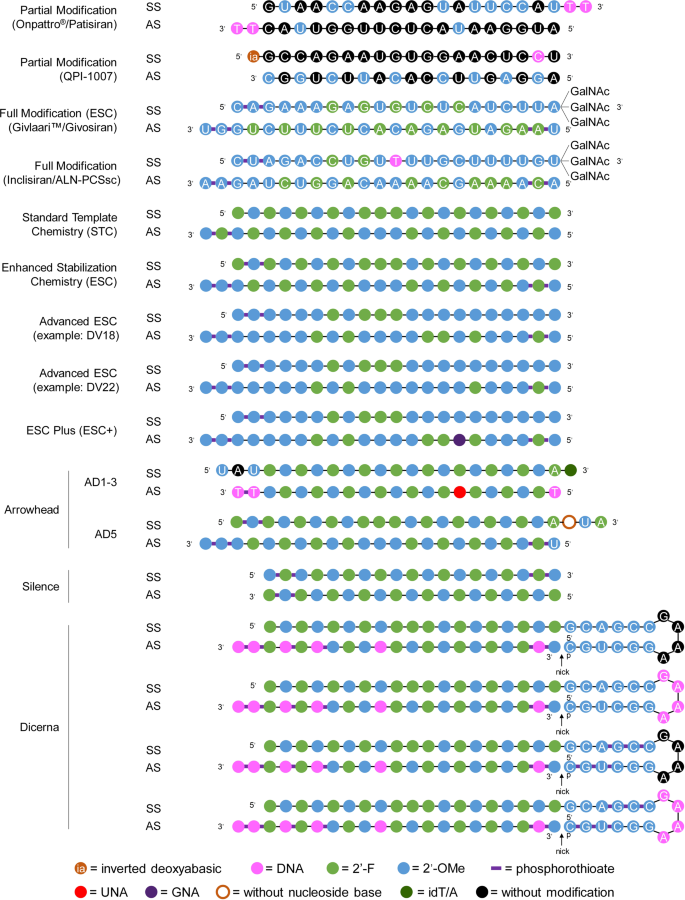
Representative designs for the chemical modification of siRNA. The sequences and modification details for ONPATTRO®, QPI-1007, GIVLAARI™ and inclisiran are included. The representative siRNA modification patterns developed by Alnylam (STC, ESC, advanced ESC and ESC+) and arrowhead (AD1-3 and AD5) are shown. Dicerna developed four GalNAc moieties that can be positioned at the unpaired G–A–A–A nucleotides of the DsiRNA structure. 2′-OMe 2′-methoxy, 2′-F 2′-fluoro, GNA glycol nucleic acid, UNA unlocked nucleic acid, SS sense strand, AS antisense strand
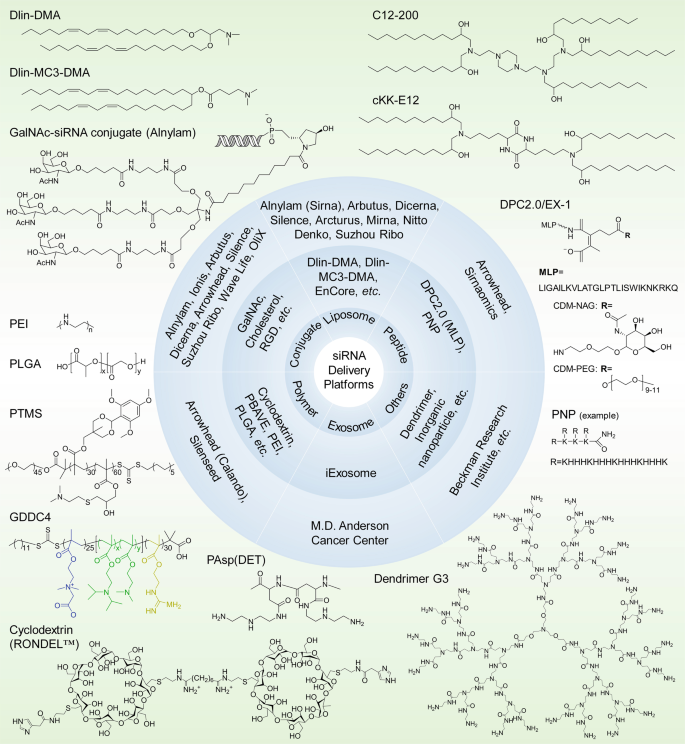
siRNA delivery platforms that have been evaluated preclinically and clinically. Varieties of lipids or lipidoids, siRNA conjugates, peptides, polymers, exosomes, dendrimers, etc. have been explored and employed for siRNA therapeutic development by biotech companies or institutes. The chemical structures of the key component(s) of the discussed delivery platforms, including Dlin-DMA, Dlin-MC3-DMA, C12-200, cKK-E12, GalNAc–siRNA conjugates, MLP-based DPC2.0 (EX-1), PNP, PEI, PLGA-based LODER, PTMS, GDDC4, PAsp(DET), cyclodextrin-based RONDEL™ and dendrimer generation 3 are shown. DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate, DPC Dynamic PolyConjugates, MLP membrane-lytic peptide, CDM carboxylated dimethyl maleic acid, PEG polyethylene glycol, NAG N-acetylgalactosamine, PNP polypeptide nanoparticle, PEI poly(ethyleneimine), LODER LOcal Drug EluteR, PLGA poly(lactic-co-glycolic) acid, PTMS PEG-PTTMA-P(GMA-S-DMA) poly(ethylene glycol)-co-poly[(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl))] ethane methacrylate-co-poly(dimethylamino glycidyl methacrylate), GDDC4 PG-P(DPAx-co-DMAEMAy)-PCB, where PG is guanidinated poly(aminoethyl methacrylate) PCB is poly(carboxybetaine) and P(DPAx-co-DMAEMAy) is poly(dimethylaminoethyl methacrylate-co-diisopropylethyl methacrylate), PEG-PAsp(DET) polyethylene glycol-b-poly(N′-(N-(2-aminoethyl)-2-aminoethyl) aspartamide), PBAVE polymer composed of butyl and amino vinyl ether, RONDEL™ RNAi/oligonucleotide nanoparticle delivery
REF
Nucleic Acids Research (2019), 47(7), 3306-3320.
Drug Metabolism & Disposition (2019), 47(10), 1183-1201.
PATENT
WO 2020128816
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020128816
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and an additional therapeutic agent for the treatment of transthyretin amyloidosis. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent for the treatment of transthyretin amyloidosis.
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. The compositions and methods of the invention are useful in stabilizing transthyretin, inhibiting transthyretin misfolding, proteolysis, and treating amyloid diseases associated thereto.
Transthyretin (TTR) is a 55 kDa homotetrameric protein present in serum and cerebral spinal fluid and which functions as a transporter of L-thyroxine (T4) and holo-retinol binding protein (RBP). TTR has been found to be an amyloidogenic protein that, under certain conditions, can be transformed into fibrils and other aggregates which can lead to disease pathology such as polyneuropathy or cardiomyopathy in humans.
US Patent Nos. 7,214,695; 7,214,696; 7,560,488; 8, 168.683; and 8,653,119 each of which is incorporated herein by reference, discloses benzoxazole derivatives which act as transthyretin stabilizers and are of the formula
or a pharmaceutically acceptable salt thereof; wherein Ar is 3,5-difluorophenyl, 2,6-difluorophenyl, 3,5-dichlorophenyl, 2,6-dichlorophenyl, 2-(trifluoromethyl)phenyl or 3-(trifluoromethyl)phenyl. Particularly, 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis) of the formula
is disclosed therein. Tafamidis is an orally active transthyretin stabilizer that inhibits tetramer dissociation and proteolysis that has been approved in certain jurisdictions for the treatment of transthyretin polyneuropathy (TTR-PN) and is currently in development for the treatment of transthyretin cardiomyopathy (TTR-CM). US Patent No. 9,249, 112, also incorporated herein by reference, discloses polymorphic forms of the meglumine salt of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis meglumine). US Patent No. 9,770,441 discloses polymorphic forms of the free acid of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis), and is also incorporated by reference herein.
Summary of the Invention
The present invention provides pharmaceutical compositions and methods comprising the compound 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent. Particular embodiments of this invention are pharmaceutical compositions and methods comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents selected from the group consisting of agents that lower plasma levels of TTR such as an antisense therapy, TTR gene editing therapy, transcriptional modulators, translational modulators, TTR protein degraders and antibodies that bind and reduce TTR levels; amyloid reduction therapies such as anti amyloid antibodies (either TTR selective or general), stimulators of amyloid clearance, fibril disruptors and therapies that inhibit amyloid nucleation; other TTR stabilizers; and TTR modulators such as therapeutics which inhibit TTR cleavage. Particularly, the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and
methods comprising a polymorphic form of tafamidis free acid or a polymorphic form of tafamidis meglumine salt with one or more additional therapeutic agents.
The present invention also provides a method of treating or preventing transthyretin amyloidosis in a patient, the method comprising administering to a patient in need thereof a therapeutically or prophylactically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole- 6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents.
A particular embodiment of the present method of treatment is the method comprising a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered orally. Additional embodiments of this invention are methods of treatment as described above wherein the 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered parenterally (intravenously or subcutaneously). Further embodiments of this invention are methods of treatment wherein the 2-(3,5-dichlorophenyl)-1, 3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally and the one or more additional therapeutic agent is administered either orally or parenterally. Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR
cardiomyopathy, the method comprising administering to a patient in need thereof a therapeutically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agents.
Brief Description of the Drawings
REF
Biochemical Pharmacology (Amsterdam, Netherlands) (2021), 189, 114432.
PATENT
WO 2021041884
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021041884
Exemplary RNAi agents that reduce the expression of TTR include patisiran and vutrisiran.
The ter s “antisense polynucleotide agent”, “antisense oligonucleotide”, “antisense compound”, and “antisense agent” as used interchangeably herein, refer to an agent comprising a single-stranded oligonucleotide that specifically binds to the target nucleic acid molecules via hydrogen bonding (e.g., Watson-Crick, Hoogsteen, or reversed Hoogsteen hydrogen bonding) and inhibits the expression of the targeted nucleic acid by an antisense mechanism of action, e.g., by RNase H. In some embodiments, an antisense agent is a nucleic acid therapeutic that acts by reducing the expression of a target gene, thereby reducing the expression of the polypeptide encoded by the target gene. Exemplary antisense agents that reduce the expression of TTR include inotersen and Ionis 682884/ ION-TTR-LRx (see, e.g., WO2014179627 which is incorporated by reference in its entirety). Further antisense agents that reduce the expression of TTR are provided, for example in WO2011139917 and WO2014179627, each of which is incorporated by reference in its entirety.
REF
Clinical Pharmacology & Therapeutics (Hoboken, NJ, United States) (2021), 109(2), 372-382
Annals of Plastic Surgery (2021), 86(2S_Suppl_1), S23-S29.
Journal of Cardiovascular Pharmacology (2021), 77(5), 544-548.
Annals of Pharmacotherapy (2021), 55(12), 1502-1514.
Kidney International (2022), 101(2), 208-211
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
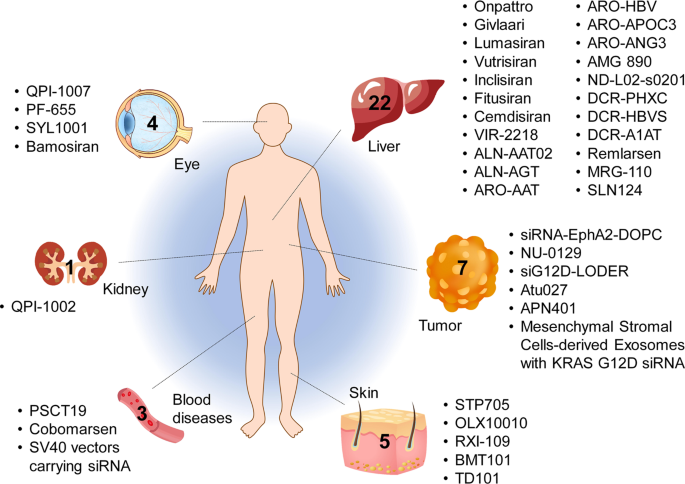
Tissues targeted by siRNA and miRNA therapeutics currently being investigated at the clinical stage. The corresponding therapeutic names are shown beside the tissues
CLIP
Vutrisiran An Investigational RNAi Therapeutic for ATTR Amyloidosis Vutrisiran has not been approved by the U.S. Food and Drug Administration, European Medicines Agency, or any other regulatory authority and no conclusions can or should be drawn regarding the safety or effectiveness of this investigational therapeutic. Overview • Vutrisiran is an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, which encompasses both hereditary ATTR (hATTR) amyloidosis and wild-type ATTR (wtATTR) amyloidosis.1, 2 • Vutrisiran inhibits the production of disease-causing transthyretin (TTR) protein by the liver, leading to a reduction in the level of TTR in the blood.1, 2 • Vutrisiran is administered subcutaneously (under the skin) and utilizes one of Alnylam’s delivery platforms known as the Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate delivery platform.1, 2 • Vutrisiran is administered every three months.2 • Vutrisiran is under review by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Brazilian Health Regulatory Agency (ANVISA). Vutrisiran has been granted Orphan Drug Designation in the U.S. and the European Union (EU) for the treatment of ATTR amyloidosis. Vutrisiran has also been granted a Fast Track designation in the U.S. for the treatment of the polyneuropathy of hATTR amyloidosis in adults. In the U.S. vutrisiran has received an action date under the Prescription Drug User Fee Act (PDUFA) of April 14, 2022. The Company received orphan drug designation in Japan. Alnylam has global commercial rights to vutrisiran, assuming regulatory approvals. Clinical Development • A Phase 1 clinical study of vutrisiran was conducted in 80 healthy volunteers (60 received vutrisiran and 20 received placebo). Vutrisiran demonstrated an acceptable safety profile and a single dose reduced serum TTR for a period of at least 90 days.2 • The safety and efficacy of vutrisiran are being evaluated in the HELIOS Phase 3 clinical program, currently consisting of two clinical trials: HELIOS-A and HELIOS-B. • HELIOS-A is a randomized, open-label, global multi-center Phase 3 study of 164 adult patients with hATTR amyloidosis with polyneuropathy.1 • The primary endpoint of HELIOS-A is change from baseline in the modified Neuropathy Impairment Score +7 (mNIS+7) at 9 months. • Secondary endpoints at 9 months include the Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QoL-DN) Total Score and the 10-Meter Walk Test (10-MWT). • The 9-month endpoints will be analyzed at 18 months with the addition of other secondary endpoints. • HELIOS-B is a randomized, double-blind, placebo-controlled Phase 3 study of 655 adult patients with ATTR amyloidosis with cardiomyopathy (including both hATTR and wtATTR amyloidosis).3 • The primary endpoint will evaluate the efficacy of vutrisiran versus placebo for the composite outcome of all-cause mortality and recurrent cardiovascular (CV) events (CV hospitalizations and urgent heart failure (HF) visits) at 30-36 months. • Secondary endpoints include the change from baseline in the 6-minute walk test (6-MWT), health status measured using the Kansas City Cardiomyopathy Questionnaire Overall Summary (KCCQ-OS), echocardiographic assessments of mean left ventricular wall thickness and global longitudinal strain, the N-terminal prohormone B-type natriuretic peptide (NT-proBNP) as a cardiac biomarker, and all-cause mortality, rate of recurrent CV events, and composite of all-cause mortality and recurrent all-cause hospitalizations and urgent HF visits at month 30 or 30-36 months. Page 2 © 2021 Alnylam Pharmaceuticals, Inc. All rights reserved. TTRsc02-USA-00012 v4 About ATTR Amyloidosis • ATTR amyloidosis is a rare, underdiagnosed, rapidly progressive, debilitating, and fatal disease caused by misfolded TTR that accumulates as amyloid fibrils in multiple tissues including the nerves, heart, and GI tract. There are two types of ATTR amyloidosis: hATTR amyloidosis and wtATTR amyloidosis.4,5,6 • hATTR amyloidosis is an inherited condition that is caused by variants (i.e., mutations) in the transthyretin (TTR) gene.5,7,8 TTR protein is produced primarily in the liver and is normally a carrier of vitamin A.9 The variant results in misfolded TTR proteins that accumulate as amyloid deposits in multiple tissues, including the nerves, heart and gastrointestinal (GI) tract.5, 6, 7 It is a multisystem disease that can include sensory and motor, autonomic, and cardiac symptoms. The condition can have a debilitating impact on a patient’s life and may lead to premature death with a median survival of 4.7 years following diagnosis.8,10 It is estimated that there are approximately 50,000 patients with hATTR amyloidosis worldwide.11 • wtATTR amyloidosis is a non-hereditary condition that occurs when misfolded wild-type TTR accumulates as amyloid deposits in multiple organs. It predominantly manifests as cardiac symptoms, but other systems are also involved, and commonly leads to heart failure and mortality within 2.5 to 5.5 years.12,13,14,15,16,17,18,19 wtATTR amyloidosis affects an estimated 200,000-300,000 people worldwide.20 • Alnylam is committed to developing multiple treatment options for people who are living with ATTR amyloidosis to help manage the debilitating and progressive nature of the disease. For more information about vutrisiran, please contact media@alnylam.com. For more information on HELIOS-A (NCT03759379) and HELIOS-B (NCT04153149) please visit http://www.clinicaltrials.gov or contact media@alnylam.com. Current information as of November 2021
CLIP
Alnylam announces extension of review period for new drug vutrisiran to treat ATTR amyloidosis
Alnylam announces 3-month extension of review period for new drug application for vutrisiran to treat ATTR amyloidosis.
Alnylam Pharmaceuticals, Inc., a RNAi therapeutics company, announced that the FDA has extended the review timeline of the New Drug Application (NDA) for vutrisiran, an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, to allow for the review of newly added information related to the new secondary packaging and labelling facility.
Alnylam recently learned that the original third-party secondary packaging and labelling facility the Company planned to use for the vutrisiran launch was recently inspected and the inspection requires classification for the FDA to take action on the vutrisiran NDA. The inspection observations were not directly related to vutrisiran. In order to minimize delays to approval, Alnylam has identified a new facility to pack and label vutrisiran and submitted an amendment to the NDA for review by the FDA. The updated Prescription Drug User Fee Act (PDUFA) goal date to allow for this review is July 14, 2022. No additional clinical data have been requested by the FDA.
////////////Vutrisiran sodium, APPROVALS 2022, FDA 2022, FDA APPROVED, AMVUTTRA, 2022/6/13, ブトリシランナトリウム , ALN 65492, Votrisiran, siRNA

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN AND MAINTAIN THIS BLOG
$10.00
Tirzepatide
YXEGTFTSDY SIXLDKIAQK AFVQWLIAGG PSSGAPPPS

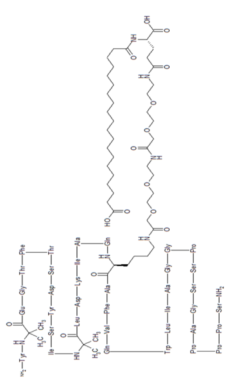
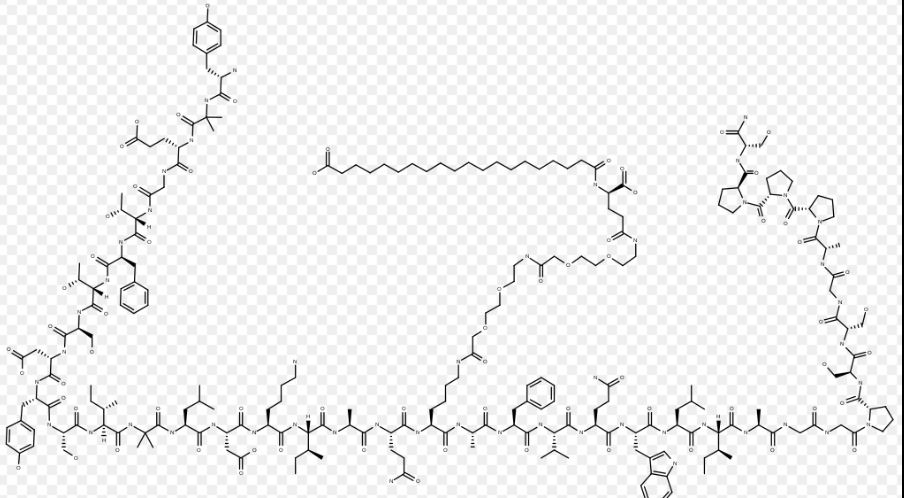


Tirzepatide
チルゼパチド
LY3298176,
| Formula | C225H348N48O68 |
|---|---|
| CAS | 2023788-19-2 |
| Mol weight | 4813.4514 |
FDA APPROVED 2022/5/13, Mounjaro
| Class | Antidiabetic agent GLP-1 receptor agonist |
|---|---|
| Efficacy | Antidiabetic, Gastric inhibitory polypeptide receptor agonist, Glucagon-like peptide 1 (GLP-1) receptor agonist |
| Disease | Type 2 diabetes mellitus |

Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
C225H348N48O68 : 4813.45
[2023788-19-2]
L-Serinamide, L-tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-
Other Names
- L-Tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-L-serinamide
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are hormones involved in blood sugar control.[2] Tirzepatide is a first-in-class medication that activates both the GLP-1 and GIP receptors, which leads to improved blood sugar control.[2] Tirzepatide was approved for medical use in the United States in May 2022.[2]
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00108

The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
PATENT
- WO2016111971
- US2020023040
- WO2019245893
- US2020155487
- US2020155650
- WO2020159949CN112592387
- WO2021066600CN112661815
- WO2021154593
- US2021338769

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Tirzepatide in indicated to improve blood sugar control in adults with type 2 diabetes, as an addition to diet and exercise.[2]
Contraindications
Tirzepatide should not be used in people with a personal or family history of medullary thyroid cancer or in people with multiple endocrine neoplasia syndrome type 2.[2]
Adverse effects
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Pharmacology
Tirzepatide is an analogue of gastric inhibitory polypeptide (GIP), a human hormone which stimulates the release of insulin from the pancreas. Tirzepatide is a linear polypeptide of 39 amino acids which has been chemically modified by lipidation to improve its uptake into cells and its stability to metabolism.[9] The compound is administered as a weekly subcutaneous injection.[10] It completed phase III trials globally in 2021.[11][12]
Mechanism of action
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3] Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonyl protecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
References
- ^ Jump up to:a b “Highlights of prescribing information” (PDF). accessdata.fda.gov. FDA. May 2022. Retrieved 14 May 2022.
- ^ Jump up to:a b c d e f g h i “FDA Approves Novel, Dual-Targeted Treatment for Type 2 Diabetes”. U.S. Food and Drug Administration (FDA) (Press release). 13 May 2022. Retrieved 13 May 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, et al. (January 2021). “Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes”. The Journal of Clinical Endocrinology and Metabolism. 106 (2): 388–396. doi:10.1210/clinem/dgaa863. PMC 7823251. PMID 33236115.
- ^ Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. (December 2018). “LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept”. Molecular Metabolism. 18: 3–14. doi:10.1016/j.molmet.2018.09.009. PMC 6308032. PMID 30473097.
- ^ Jump up to:a b Min T, Bain SC (January 2021). “The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials”. Diabetes Therapy. 12 (1): 143–157. doi:10.1007/s13300-020-00981-0. PMC 7843845. PMID 33325008.
- ^ Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. (November 2018). “Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial”. The Lancet. 392 (10160): 2180–2193. doi:10.1016/S0140-6736(18)32260-8. PMID 30293770.
- ^ Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. (June 2020). “Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens”. Diabetes, Obesity & Metabolism. 22 (6): 938–946. doi:10.1111/dom.13979. PMC 7318331. PMID 31984598.
- ^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID 35133415.
- ^ Jump up to:a b Ahangarpour M, Kavianinia I, Harris PW, Brimble MA (January 2021). “Photo-induced radical thiol-ene chemistry: a versatile toolbox for peptide-based drug design”. Chemical Society Reviews. Royal Society of Chemistry. 50 (2): 898–944. doi:10.1039/d0cs00354a. PMID 33404559. S2CID 230783854.
- ^ Bastin M, Andreelli F (2019). “Dual GIP-GLP1-Receptor Agonists In The Treatment Of Type 2 Diabetes: A Short Review On Emerging Data And Therapeutic Potential”. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 12: 1973–1985. doi:10.2147/DMSO.S191438. PMC 6777434. PMID 31686879.
- ^ “Tirzepatide significantly reduced A1C and body weight in people with type 2 diabetes in two phase 3 trials from Lilly’s SURPASS program” (Press release). Eli Lilly and Company. 17 February 2021. Retrieved 28 October 2021 – via PR Newswire.
- ^ “Lilly : Phase 3 Tirzepatide Results Show Superior A1C And Body Weight Reductions In Type 2 Diabetes”. Business Insider. RTTNews. 19 October 2021. Retrieved 28 October 2021.
- ^ Jump up to:a b Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. (September 2020). “Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist”. JCI Insight. 5 (17). doi:10.1172/jci.insight.140532. PMC 7526454. PMID 32730231.
- ^ Østergaard S, Paulsson JF, Kofoed J, Zosel F, Olsen J, Jeppesen CB, et al. (October 2021). “The effect of fatty diacid acylation of human PYY3-36 on Y2 receptor potency and half-life in minipigs”. Scientific Reports. 11 (1): 21179. Bibcode:2021NatSR..1121179O. doi:10.1038/s41598-021-00654-3. PMC 8551270. PMID 34707178.
- ^ Jump up to:a b US patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
- ^ Frederick MO, Boyse RA, Braden TM, Calvin JR, Campbell BM, Changi SM, et al. (2021). “Kilogram-Scale GMP Manufacture of Tirzepatide Using a Hybrid SPPS/LPPS Approach with Continuous Manufacturing”. Organic Process Research & Development. 25 (7): 1628–1636. doi:10.1021/acs.oprd.1c00108. S2CID 237690232.
- ^ Sagonowsky, Eric (26 October 2021). “As Lilly gears up for key 2022 launches, Trulicity, Taltz and more drive solid growth”. Fierce Pharma. Retrieved 9 April 2022.
- ^ Kellaher, Colin (28 April 2022). “Eli Lilly’s Tirzepatide Meets Main Endpoints in Phase 3 Obesity Study >LLY”. Dow Jones Newswires. Retrieved 29 April 2022 – via MarketWatch.
- ^ Kahan, Scott; Garvey, W. Timothy (28 April 2022). “SURMOUNT-1: Adults achieve weight loss of 16% or more at 72 weeks with tirzepatide”. healio.com. Retrieved 29 April 2022.
- ^ Taylor, Nick Paul (28 April 2022). “SURMOUNT-able: Lilly’s tirzepatide clears high bar set by Novo’s Wegovy in obesity”. FierceBiotech. Retrieved 29 April 2022.
- ^ Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. (August 2021). “Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes”. The New England Journal of Medicine. 385 (6): 503–515. doi:10.1056/NEJMoa2107519. PMID 34170647. S2CID 235635529.
- ^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Further reading
- Bhagavathula AS, Vidyasagar K, Tesfaye W (September 2021). “Efficacy and Safety of Tirzepatide in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized Phase II/III Trials”. Pharmaceuticals (Basel). 14 (10). doi:10.3390/ph14100991. PMC 8537322. PMID 34681215.
- Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID 33030356.
- Ryan DH (September 2021). “Next Generation Antiobesity Medications: Setmelanotide, Semaglutide, Tirzepatide and Bimagrumab: What do They Mean for Clinical Practice?”. J Obes Metab Syndr. 30 (3): 196–208. doi:10.7570/jomes21033. PMC 8526285. PMID 34518444.
External links
- “Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
- Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
- Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
- Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
- Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
CLIP
FDA approves Lilly’s Mounjaro™ (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
May 13, 2022
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro™ (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program
The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
- SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
- SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
- SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
- SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
- SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity
ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro™ (tirzepatide) injection1
Mounjaro™ (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS
Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
- Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
- It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings
Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
- Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
- Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects
The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
- Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
- Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
- Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
- Do you take other diabetes medicines, such as insulin or sulfonylureas?
- Do you have a history of diabetic retinopathy?
- Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
- Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
- Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
- Read the Instructions for Use that come with Mounjaro.
- Use Mounjaro exactly as your healthcare provider says.
- Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
- Use Mounjaro 1 time each week, at any time of the day.
- Do not mix insulin and Mounjaro together in the same injection.
- If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more
For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
Please click to access full Prescribing Information and Medication Guide.
TR CON CBS MAY2022
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Lilly Cautionary Statement Regarding Forward-Looking Statements
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro™ (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
- Mounjaro. Prescribing Information. Lilly USA, LLC.
- Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
- Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
- Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
- Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
- Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
CLIP
Lilly’s tirzepatide delivered up to 22.5% weight loss in adults with obesity or overweight in SURMOUNT-1
April 28, 2022
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
- Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
- Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
- Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
CLIP
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide
Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program
SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
| Clinical data | |
|---|---|
| Trade names | Mounjaro |
| Other names | LY3298176, GIP/GLP-1 RA |
| License data | US DailyMed: Tirzepatide |
| Routes of administration | subcutaneous |
| Drug class | Antidiabetic, GLP-1 receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2023788-19-2 |
| PubChem CID | 156588324 |
| IUPHAR/BPS | 11429 |
| DrugBank | DB15171 |
| ChemSpider | 76714503 |
| UNII | OYN3CCI6QE |
| KEGG | D11360 |
| ChEMBL | ChEMBL4297839 |
| Chemical and physical data | |
| Formula | C225H348N48O68 |
| Molar mass | 4813.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Tirzepatide, FDA 2022, APPROVALS 2022, Mounjaro, PEPTIDE, チルゼパチド , LY3298176,
UNIIOYN3CCI6QE
chart 1 Structure of GLP-1 & TZP & Exenatide & Somalutide
Olipudase alfa
| HPLSPQGHPA RLHRIVPRLR DVFGWGNLTC PICKGLFTAI NLGLKKEPNV ARVGSVAIKL CNLLKIAPPA VCQSIVHLFE DDMVEVWRRS VLSPSEACGL LLGSTCGHWD IFSSWNISLP TVPKPPPKPP SPPAPGAPVS RILFLTDLHW DHDYLEGTDP DCADPLCCRR GSGLPPASRP GAGYWGEYSK CDLPLRTLES LLSGLGPAGP FDMVYWTGDI PAHDVWHQTR QDQLRALTTV TALVRKFLGP VPVYPAVGNH ESTPVNSFPP PFIEGNHSSR WLYEAMAKAW EPWLPAEALR TLRIGGFYAL SPYPGLRLIS LNMNFCSREN FWLLINSTDP AGQLQWLVGE LQAAEDRGDK VHIIGHIPPG HCLKSWSWNY YRIVARYENT LAAQFFGHTH VDEFEVFYDE ETLSRPLAVA FLAPSATTYI GLNPGYRVYQ IDGNYSGSSH VVLDHETYIL NLTQANIPGA IPHWQLLYRA RETYGLPNTL PTAWHNLVYR MRGDMQLFQT FWFLYHKGHP PSEPCGTPCR LATLCAQLSA RADSPALCRH LMPDGSLPEA QSLWPRPLFC (Disulfide bridge: 43-119, 46-111, 74-85, 175-180, 181-204, 339-385, 538-542, 548-561) |
Olipudase alfa
Xenpozyme, Japan 2022, APPROVALS 2022, 2022/3/28
PEPTIDE, オリプダーゼアルファ (遺伝子組換え)
Alternative Names: Acid sphingomyelinase Niemann Pick disease type B – Sanofi; Acid-sphingomyelinase – Sanofi; GZ-402665; Recombinant human acid sphingomyelinase – Sanofi; rhASM – Sanofi; Sphingomyelinase-C (synthetic human) – Sanofi; Synthetic human sphingomyelinase-C – Sanofi; Xenpozyme
| Formula | C2900H4373N783O791S24 |
|---|---|
| CAS | 927883-84-9 |
| Mol weight | 63631.0831 |
| Efficacy | Lysosomal storage disease treatment, Enzyme replacement (acid sphingomyelinase) |
|---|---|
| Comment | Enzyme replacement therapy product Treatment of Niemann-Pick disease type A/B |
- OriginatorGenzyme Corporation
- DeveloperSanofi
- ClassRecombinant proteins; Sphingomyelin phosphodiesterases
- Mechanism of ActionSphingomyelin-phosphodiesterase replacements
- Orphan Drug StatusYes – Niemann-Pick diseases
- RegisteredNiemann-Pick diseases
- 28 Mar 2022Registered for Niemann-Pick diseases (In adolescents, In children, In adults) in Japan (IV) – First global approval
- 09 Feb 2022FDA assigns PDUFA action date of (03/07/2022) for Olipudase alfa (In children, In adults) for Niemann-Pick diseases
- 09 Feb 2022Adverse e
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase alfa was associated with significant improvements in clinically relevant disease end points among patients with chronic visceral acid sphingomyelinase (ASM) deficiency (ASMD), according to results from the phase 2/3 ASCEND trial presented at the 17th Annual WORLDSymposium.
ASMD is a rare, debilitating lysosomal storage disease characterized by a deficiency of the enzyme acid sphingomyelinase, which results in the accumulation of sphingomyelin in various tissues of the body. Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM.
The multicenter, randomized, double-blind, placebo-controlled ASCEND trial evaluated the efficacy and safety of olipudase alfa in 36 adults with chronic visceral ASMD. Patients were randomly assigned 1:1 to receive olipudase alfa 3mg/kg intravenously every 2 weeks or placebo for 52 weeks. The coprimary end points were the percent change in spleen volume and percent-predicted diffusing capacity of the lung for carbon monoxide (DLCO).
At week 52, treatment with olipudase alfa resulted in a 39.45% reduction in spleen volume, compared with a 0.5% increase for placebo (P <.0001). A decrease in spleen volume of at least 30% was observed in 17 patients (94%) treated with olipudase afla compared with no patients treated with placebo. Additionally, olipudase alfa significantly improved lung function by 22% from baseline compared with 3% for patients receiving placebo (P =.0004), as measured by percent predicted DLCO.
Olipudase alfa also met key secondary end points including a 31.7% reduction in liver volume (vs a 1.4% reduction for placebo; P <.0001) and a 16.8% improvement in mean platelet counts (vs 2.5% with placebo; P =.019) at week 52. Significant improvements in HDL, LDL, AST, ALT, chitotriosidase (54% vs 12% with placebo; P =.0003), and lyso-sphingomyelin (78% vs 6% with placebo) were also observed in the olipudase alfa group at week 52.
With regard to Splenomegaly Related Score, a patient-reported outcome measurement that evaluates patient symptoms associated with an enlarged spleen, findings showed no meaningful difference between olipudase alfa and placebo (-8 point vs -9.3 points, respectively).
As for safety, olipudase alfa was well tolerated with most adverse events being mild to moderate in severity. There were no treatment-related serious adverse events and no adverse event-related discontinuations.
Disclosure: Some authors have declared affiliations with or received funding from the pharmaceutical industry. Please refer to the original study for a full list of disclosures.
Reference
Wasserstein M, Arash-Kaps L, Barbato A, et al. Adults with chronic acid sphingomyelinase deficiency show significant visceral, pulmonary, and hematologic improvements after enzyme replacement therapy with olipudase-alfa: 1-year results of the ASCEND placebo-controlled trial. Presented at: 17th Annual WORLDSymposium; February 8-12, 2021. Abstract 265.
CLIP
https://www.sanofi.com/en/media-room/press-releases/2021/2021-12-06-14-00-00-2346501
EMA accepts regulatory submission for olipudase alfa, the first potential therapy for ASMD
- Olipudase alfa has been granted PRIority MEdicines (PRIME) designation in Europe, Breakthrough Therapy designation in the United States, and SAKIGAKE designation in Japan
- European regulatory decision anticipated second half of 2022
DECEMBER 6, 2021
The European Medicines Agency (EMA) has accepted for review under an accelerated assessment procedure the Marketing Authorization Application (MAA) for olipudase alfa, Sanofi’s investigational enzyme replacement therapy which is being evaluated for the treatment of acid sphingomyelinase deficiency (ASMD). Historically referred to as Niemann-Pick disease (NPD) type A and type B, ASMD is a rare, progressive, and potentially life-threatening disease for which no treatments are currently approved. The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan. If approved, olipudase alfa will become the first and only therapy for the treatment of ASMD.
“Today’s milestone has been decades in the making and our gratitude goes to the ASMD community who has stood by us with endless patience while olipudase alfa advanced through clinical development,” said Alaa Hamed, MD, MPH, MBA, Global Head of Medical Affairs, Rare Diseases, Sanofi. “Olipudase alfa represents the kind of potentially life-changing innovation that is possible when industry, medical professionals and the patient community work together toward a common goal.”
The MAA is based on positive results from two separate clinical trials (ASCEND and ASCEND-Peds) evaluating olipudase alfa in adult and pediatric patients with non-central nervous system (CNS) manifestations of ASMD type A/B and ASMD type B.
Olipudase alfa has received special designations from regulatory agencies worldwide, recognizing the innovation potential of the investigational therapy.
“Scientific innovation is the greatest source of hope for people living with diseases like ASMD where there are no approved treatments and is a critical component for ensuring a viable healthcare ecosystem,” said Bill Sibold, Executive Vice President of Sanofi Genzyme. “At Sanofi, we have a long history of pioneering scientific innovation, and we remain committed to finding solutions to address unmet medical needs, including those of the rare disease community.”
The EMA awarded olipudase alfa the PRIority MEdicines designation, also known as PRIME, intended to aid and expedite the regulatory process for investigational medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options.
The U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation to olipudase alfa. This designation is intended to expedite the development and review of drugs intended to treat serious or life-threatening diseases and conditions. The criteria for granting Breakthrough Therapy designation include preliminary clinical evidence indicating that the molecule may demonstrate substantial improvement on a clinically significant endpoint over available therapies.
In Japan, olipudase alfa was awarded the SAKIGAKE designation, which is intended to promote research and development in Japan for innovative new medical products that satisfy certain criteria, such as the severity of the intended indication. In September, Sanofi filed the J-NDA submission for olipudase alfa.
About ASMD
ASMD results from a deficient activity of the enzyme acid sphingomyelinase (ASM), which is found in special compartments within cells called lysosomes and is required to breakdown lipids called sphingomyelin. If ASM is absent or not functioning as it should, sphingomyelin cannot be metabolized properly and accumulates within cells, eventually causing cell death and the malfunction of major organ systems. The deficiency of the lysosomal enzyme ASM is due to disease-causing variants in the sphingomyelin phosphodiesterase 1 gene (SMPD1). The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan.
ASMD represents a spectrum of disease caused by the same enzymatic deficiency, with two types that may represent opposite ends of a continuum sometimes referred to as ASMD type A and ASMD type B. ASMD type A is a rapidly progressive neurological form of the disease resulting in death in early childhood due to central nervous system complications. ASMD type B is a serious and potentially life-threatening disease that predominantly impacts the lungs, liver, and spleen, as well as other organs. ASMD type A/B represents an intermediate form that includes varying degrees of neurologic involvement. Patients with ASMD type A/B or ASMD type B were studied in the ASCEND trial program. Another type of NPD is NPD type C, which is unrelated to ASMD.
About olipudase alfa
Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM, allowing for the breakdown of sphingomyelin. Olipudase alfa is currently being investigated to treat non-CNS manifestations of ASMD. Olipudase alfa has not been studied in ASMD type A patients. Olipudase alfa is an investigational agent and the safety and efficacy have not been evaluated by the FDA, EMA, or any other regulatory authority worldwide.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
///////Olipudase alfa, japan 2022, APPROVALS 2022, Xenpozyme, PEPTIDE, オリプダーゼアルファ (遺伝子組換え) , ORPHAN DRUG, GZ-402665 , GZ 402665

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Andexanet alfa
(heavy chain)
IVGGQECKDG ECPWQALLIN EENEGFCGGT ILSEFYILTA AHCLYQAKRF KVRVGDRNTE
QEEGGEAVHE VEVVIKHNRF TKETYDFDIA VLRLKTPITF RMNVAPACLP ERDWAESTLM
TQKTGIVSGF GRTHEKGRQS TRLKMLEVPY VDRNSCKLSS SFIITQNMFC AGYDTKQEDA
CQGDAGGPHV TRFKDTYFVT GIVSWGEGCA RKGKYGIYTK VTAFLKWIDR SMKTRGLPKA
KSHAPEVITS SPLK
(light chan)
ANSFLFWNKY KDGDQCETSP CQNQGKCKDG LGEYTCTCLE GFEGKNCELF TRKLCSLDNG
DCDQFCHEEQ NSVVCSCARG YTLADNGKAC IPTGPYPCGK QTLER
(Disulfide bridge: H7-H12, H27-H43, H108-L98, H156-H170, H181-H209, L16-L27, L21-L36, L38-L47, L55-L66, L62-L75, L77-L90)
Andexanet alfa
JAPAN 2022, PEPTIDE
| Ondexxya |
| 2022/3/28 |
| Anticoagulant reversal (factor Xa inhibitors) |
CAS: 1262449-58-0
アンデキサネットアルファ (遺伝子組換え)
- Andexanet alfa
- r-Antidote
- rfXa Inhibitor Antidote
- PRT-4445
- PRT064445
Andexanet alfa, sold under the trade name Andexxa among others, is an antidote for the medications rivaroxaban and apixaban, when reversal of anticoagulation is needed due to uncontrolled bleeding.[1] It has not been found to be useful for other factor Xa inhibitors.[2] It is given by injection into a vein.[2]
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots, heart attacks, strokes, or cardiac arrest.[2] It works by binding to rivaroxaban and apixaban.[2]
It was approved for medical use in the United States in May 2018.[1] It was developed by Portola Pharmaceuticals.[3]
ndexanet alfa is a recombinant human coagulation Factor Xa that promotes blood coagulation. It was developed by Portola Pharmaceuticals and was approved in in May 2018. It is marketed as Andexxa for intravenous injection or infusion and is indicated for the reversal of anticoagulation in combination with rivaroxaban and apixaban in cases of life-threatening or uncontrolled bleeding. Rivaroxaban and apixaban are Factor Xa inhibitors that promote anticoagulation in situations where blood clotting is unfavourable, such as in deep vein thrombosis and pulmonary embolism. However, the use of these agents is associated with a risk for uncontrollable bleeding episodes that can lead to can cause serious or fatal bleeding. Andexanet alfa is currently under regulatory review by the European Union and is undergoing clinical development in Japan 1.
Andexanet alfa works by binding to Factor Xa inhibitors and prevent them from interacting with endogenous Factor Xa. It displayed high affinity (0.53–1.53 nmol/L) to apixaban, betrixaban, edoxaban and rivaroxaban 1. However, the effectiveness of andexanet alfa on treating bleeding related to any FXa inhibitors other than apixaban and rivaroxaban was not demonstrated, thus such use is limited 7. Its pharmacokinetic properties are not reported to be affected by factor Xa inhibitors 1. Andexanet alfa retains the structural similarity to that of endogenous human factor Xa, but exists in its mature functional form without the need for activation via the intrinsic or extrinsic coagulation pathways 5 and remains catalytically inactive due to structural modification 1. The procoagulation potential of andexanet alfa is eliminated through the removal of a 34-residue fragment containing Gla: via this truncation, andexanet alfa is unable to bind to membrane surfaces and assemble the prothrombinase complex 5. It also prevents andexanet alfa from taking up space on phospholipid surface membranes, so that native FXa may bind and assemble the prothrominase complex 5. The amino acid residue modification from serine to alanine in the binding site of the catalytic domain allows more effective binding to FXa inhibitors and deters the andexanet alfa from converting prothrombin to thrombin 5.
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

Structure of andexant alfa. Andexanet alfa is a modified activated human factor Xa (FXa) that binds FXa with high affinity and a 1:1 stoichiometric ratio but does not have intrinsic catalytic activity (the amino acid serine at position 419 is replaced by alanine) and lacks the membrane-binding-carboxyglutamic acid domain (Gla domain) of native FX. The Gla domains are responsible for the binding of FXa to phospholipids
Medical uses
Andexanet alfa is used to stop life threatening or uncontrollable bleeding in people who are taking rivaroxaban or apixaban.[1]
There are no randomised clinical trials as of 2019. Studies in healthy volunteers show that the molecule binds factor Xa inhibitors and counters their anti-Xa-activity.[4] The only published clinical trial is a prospective, open label, single group study.[5] This study reports results on 352 people and demonstrates a reduction of anti-Xa-activity while also showing an excellent or good hemostatic efficacy in 82%. While people who were expected to die in 30 days were excluded from the study, 14% of participants died. There was no relationship between hemostatic efficacy and reduced anti-Xa-activity.[6] The FDA has demanded a randomised clinical trial: the first results are not expected before 2023.[7]
Adverse effects
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots or cardiac arrest.[2]
Andexanet alfa has a boxed warning that it is associated with arterial and venous blood clots, ischemic events, cardiac arrest, and sudden deaths.[1]
Pharmacology
Mechanism of action
Andexanet alfa is a biologic agent, a recombinant modified version of human activated factor X (FXa).[8] Andexanet alfa differs from native FXa due to the removal of a 34 residue fragment that contains the Gla domain. This modification reduces andexanet alfa’s procoagulant potential. Additionally, a serine to alanine (S419A) mutation in the active site eliminates its activity as a prothrombin to thrombin catalyst, but still allows the molecule to bind to FXa inhibitors.[9] FXa inhibitors bind to andexanet alfa with the same affinity as to natural FXa. As a consequence in the presence of andexanet alfa natural FXa is partially freed, which can lead to effective hemostasis.[3][10] In other words, it acts as a decoy receptor. Andexanet alfa reverses effect of all anticoagulants that act directly through FXa or by binding antithrombin III. The drug is not effective against factor IIa inhibitor dabigatran.[11]
History[edit]
It was approved in the United States in 2018 based on data from two phase III studies on reversing the anticoagulant activity of FXa inhibitors rivaroxaban and apixaban in healthy volunteers.[4] As a condition of its accelerated approval there is a study being conducted comparing it to other currently used reversal agents (“usual care”).[5][12]
Society and culture
Economics
Initial pricing (AWP) is $58,000 per reversal (800 mg bolus + 960 mg infusion, $3,300 per 100 mg vial) which is higher than reversal agents for other DOAC agents (idarucizumab for use in dabigatran reversal is $4,200 per reversal).[13]
References
- ^ Jump up to:a b c d e “Andexxa- andexanet alfa injection, powder, lyophilized, for solution”. DailyMed. 21 September 2020. Retrieved 12 November 2020.
- ^ Jump up to:a b c d e f g “Andexxa Monograph for Professionals”. Drugs.com. Retrieved 19 December 2018.
- ^ Jump up to:a b Dolgin E (March 2013). “Antidotes edge closer to reversing effects of new blood thinners”. Nature Medicine. 19 (3): 251. doi:10.1038/nm0313-251. PMID 23467222. S2CID 13340319.
- ^ Jump up to:a b Siegal DM, Curnutte JT, Connolly SJ, Lu G, Conley PB, Wiens BL, Mathur VS, Castillo J, Bronson MD, Leeds JM, Mar FA, Gold A, Crowther MA (December 2015). “Andexanet Alfa for the Reversal of Factor Xa Inhibitor Activity”. New England Journal of Medicine. 373 (25): 2413–24. doi:10.1056/NEJMoa1510991. PMID 26559317.
- ^ Jump up to:a b Connolly SJ, Crowther M, Eikelboom JW, Gibson CM, Curnutte JT, Lawrence JH, et al. (April 2019). “Full Study Report of Andexanet Alfa for Bleeding Associated with Factor Xa Inhibitors”. New England Journal of Medicine. 380 (14): 1326–1335. doi:10.1056/NEJMoa1814051. PMC 6699827. PMID 30730782.
- ^ Justin Morgenstern, “Andexanet Alfa: More garbage science in the New England Journal of Medicine”, First10EM blog, February 11, 2019. Available at: https://first10em.com/andexanet-alfa/.
- ^ “A Randomized Clinical Trial of Andexanet Alfa in Acute Intracranial Hemorrhage in Patients Receiving an Oral Factor Xa Inhibitor”. 11 January 2022.
- ^ Lu, Genmin; DeGuzman, Francis R.; Lakhotia, Sanjay; Hollenbach, Stanley J.; Phillips, David R.; Sinha, Uma (2008-11-16). “Recombinant Antidote for Reversal of Anticoagulation by Factor Xa Inhibitors”. Blood. 112 (11): 983. doi:10.1182/blood.V112.11.983.983. ISSN 0006-4971.
- ^ Kaatz, Scott; Bhansali, Hardik; Gibbs, Joseph; Lavender, Robert; Mahan, Charles E.; Paje, David G. (2017-09-13). “Reversing factor Xa inhibitors – clinical utility of andexanet alfa”. Journal of Blood Medicine. 8: 141–149. doi:10.2147/JBM.S121550. PMC 5602457. PMID 28979172.
- ^ Lu G, Deguzman FR, Hollenbach SJ, et al. (March 2013). “A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa”. Nature Medicine. 19 (4): 446–51. doi:10.1038/nm.3102. PMID 23455714. S2CID 11235887.
- ^ H. Spreitzer (23 December 2013). “Neue Wirkstoffe – Andexanet Alfa”. Österreichische Apothekerzeitung (in German) (26/2013): 40.
- ^ “Trial of Andexanet in ICH Patients Receiving an Oral FXa Inhibitor”. ClinicalTrials.gov. 11 January 2022.
- ^ “Lexi Comp Drug Information Online”. 24 May 2018.
Further reading
- Connolly SJ, Milling TJ, Eikelboom JW, Gibson CM, Curnutte JT, Gold A, et al. (September 2016). “Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors”. New England Journal of Medicine. 375 (12): 1131–41. doi:10.1056/NEJMoa1607887. PMC 5568772. PMID 27573206.
External links
- “Andexanet alfa”. Drug Information Portal. U.S. National Library of Medicine.
- “Ondexxya (andexanet alfa): Avoid use of andexanet prior to heparinization”. European
| Clinical data | |
|---|---|
| Trade names | Andexxa, Ondexxya, others |
| Other names | Coagulation factor Xa (recombinant), inactivated-zhzo, PRT06445, r-Antidote, PRT4445 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Andexanet_alfa |
| Routes of administration | Intravenous injection |
| ATC code | V03AB38 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-only [1]EU: Rx-only |
| Pharmacokinetic data | |
| Metabolism | Not studied |
| Elimination half-life | 5 h to 7 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1262449-58-0 |
| IUPHAR/BPS | 7576 |
| DrugBank | DB14562 |
| ChemSpider | none |
| UNII | BI009E452R |
| KEGG | D11029 |
| ChEMBL | ChEMBL3301583 |
//////////Andexanet alfa, JAPAN 2022, APPROVALS 2022, アンデキサネットアルファ (遺伝子組換え) , Ondexxya , PRT-4445, PRT064445

NEW DRUG APPROVALS
$10.00
Carotegrast methyl
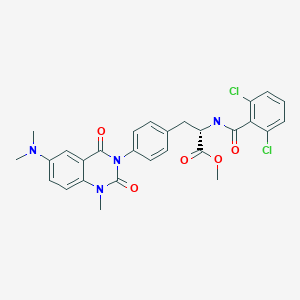
Carotegrast methyl
| Formula | C28H26Cl2N4O5 |
|---|---|
| CAS | 401905-67-7 |
| Mol weight | 569.4358 |
PMDA APROVED, CAROGRA, カロテグラストメチル
| ON 2022/3/28 |
Antiasthmatic, Integrin alpha 4 inhibitor
- An alpha4 integrin antagonist.
401905-67-7[RN]
L-Phenylalanine, N-(2,6-dichlorobenzoyl)-4-[6-(dimethylamino)-1,4-dihydro-1-methyl-2,4-dioxo-3(2H)-quinazolinyl]-, methyl ester
methyl (2S)-2-[(2,6-dichlorophenyl)formamido]-3-{4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl]phenyl}propanoate
Methyl N-(2,6-dichlorobenzoyl)-4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,4-dihydro-3(2H)-quinazolinyl]-L-phenylalaninate
Carotegrast Methyl
Methyl (2S)-2-(2,6-dichlorobenzamido)-3-{4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]phenyl}propanoate
C28H26Cl2N4O5 : 569.44
[401905-67-7]
PATENT
WO 2008062859
https://patents.google.com/patent/WO2008062859A1/en



Step 1
(Method 2): The title compound was prepared starting from 2-amino-5-dimethylamino- benzoic acid methyl ester dihydrochloride through the hydrolysis under basic condition To 5.0 g of 2-amino-5-dimethylamino-benzoic acid methyl ester di-hydrochloride, there were added 15 mL of water and 15.6 mL of a 6M aqueous solution of sodium hydroxide and the resulting mixture was heated to 40°C for 2 hours. After the confirmation of the progress of the reaction according to HPLC, the reaction system was cooled to room temperature, a 6M hydrochloric acid aqueous solution was dropwise added to the reaction system to thus neutralize the same and to separate out crystals (pH 4.9) and then the reaction system was stirred at 10°C for 2 hours. The solid thus obtained was isolated through the filtration under reduced pressure, washed with 30 mL of water and then dried under reduced pressure at 60°C for 14 hours. Title compound 3.14 g was obtained as gray-colored solid. The physical properties determined were almost identical to those observed for the same compound prepared in the above-mentioned synthesis example. H-NMR (400MHz, DMSO-d6): δ 8.21 (bs, 3H), 7.10 (d, 1H, J=2.8Hz), 6.97 (dd, 1H, J=9.1, 2.8Hz), 6.70 (d, 1H, J=9.1 Hz), 2.72 (s, 6H); 13C-NMR (100MHz, DMSO-d6): δ168.89, 144.55, 141.61, 123.29, 117.90, 114.78, 110.11,41.95; MS (ESI+): m/z 181.3 (MH+), (ESI-): m/z 179.2 (M-H–).
Step 2
Step 1: Synthesis of Nα-(2,6-dichlorobenzoyl) -4-{2-ethoxycarbonylamino-5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester To 1.96 g of 2-amino-5-dimethylaminobenzoic acid, there were added 12 mL of acetonitrile and 5.29 mL of pyridine to form a suspension and then the resulting suspension was cooled to 4°C. To this suspension there was dropwise added 4.17 mL of ethyl chloroformate over 5 minutes and then the mixture was stirred at 25°C for one hour. After confirming the disappearance of the starting material by HPLC, 0.7 mL of ethanol was added to the mixture to thus decompose the excess ethyl chloroformate and the mixture was further stirred for additional one hour. To this reaction solution there were added 4.0 g of 4-amino-Nα-(2,6-dichlorobenzoyl)-L-phenylalanine methyl ester and 12 mL of N,Ndimethylformamide, and the resulting mixture was stirred overnight. Subsequently, 48 mL of methanol was drop-wise added, the resulting mixture was stirred at 10°C overnight and then the solid separated from the mixture was isolated through filtration under reduced pressure. The solid was then washed with 8 mL of methanol and dried at 70°C for 5 hours under reduced pressure. Title compound 5.50 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 10.29 (s, 1H), 9.42 (bs, 1H), 9.24 (d, 1H, J=7.9Hz), 7.73 (bs, 1H), 7.62 (d, 2H, J=8.4Hz), 7.48-7.44 (m, 2H), 7.41 (dd, 1H, J=9.5, 6.2Hz), 7.27 (d, 2H,J=8.4Hz), 7.01 (d, 1H, J=2.7Hz), 6.93 (dd, 1H, J=9.1, 2.9Hz), 4.71 (ddd, 1H, J=9.2, 8.1, 5.7Hz), 4.05 (q, 2H, J=7.0Hz), 3.66 (s, 3H), 3.10 (dd, 1H, J=14.0, 5.6Hz), 2.96 (dd, 1H, J=14.0, 9.2Hz), 2.93 (s, 6H), 1.18 (t, 3H, J=7.2Hz); MS (ESI+): m/z 601.2 (MH+) and 623.2 (M+Na), (ESI–): m/z 599.1 (M-H–).
Step 3
Step2: Synthesis of Na-(2,6-dichlorobenzoyl)-4-{6-dimethylamino-1-methylquinazoline-2,4[1H,3H]-dion-3-yl}-L-phenylalanine methyl ester To 2.0 g of Na-(2,6-dichlorobenzoyl)-4-{2-ethoxycarbonylamino -5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester prepared in above-mentioned step 1, were added 16 mL of N,N-dimethylfbrmamide, 0.8 mL of methanol and 0.91 g of potassium carbonate, followed by the stirring of the resulting mixture at 25°C overnight. To this reaction solution, there was added 0.75 mL of methyl p-toluenesulfonate for subjecting the methyl ester to alkylation at 25~40°C. After confirming the disappearance of the starting material by HPLC, 0.75 mL of acetic acid was added to quench the reaction, 16 mL of water was dropped and the solid was separated. Further, 8 mLof N,N-dimethylformamide/water = 1/1 mixed liquid was added to the resulting mixture, followed by the stirring of the mixture at 25°C. Then the solid thus separated was isolated through filtration under reduced pressure and then washed with 8 mL of water. Thereafter, the isolated solid was dried at 70°C for 4 hours under reduced pressure. Desired compound 1.77 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 9.28 (d, 1H, J=8.1 Hz), 7.48-7.36 (m, 6H), 7.31 (dd, 1H, J=3.0, 9.0Hz), 7.24 (d, 1H, J=3.0Hz), 7.20-7.15 (m, 2H), 4.18 (ddd, 1H, J=10.2, 8.1,4.8Hz), 3.69 (s, 3H), 3.49 (s, 3H), 3.22 (dd, 1H, J=14.1, 4.8Hz), 3.02 (dd, 1H, J=14.2, 10.5Hz), 2.94 (s, 6H); MS (ESI+): m/z 569.2 (MH+) and 591.1 (M+Na), (ESI-): m/z 567.2 (M-H–).
PATENT
https://patents.google.com/patent/WO2004074264A1/en
PATENT’ WO 2003070709
https://patents.google.com/patent/WO2003070709A1/en
PATENT
WO 2002016329
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Carotegrast methyl, CAROGRA, カロテグラストメチル , JAPAN 2022, APPROVALS 2022,
COC(=O)[C@H](Cc1ccc(cc1)N2C(=O)N(C)c3ccc(cc3C2=O)N(C)C)NC(=O)c4c(Cl)cccc4Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
RISPERIDONE

Risperidone
EU APPROVED 2022/2/14, Okedi
- R-64,766
- R-64766
- RCN-3028
- RCN3028
Risperidone, R-64766, Risperdal M-Tab, Risperdal Consta, Rispolept, Belivon, Risperdal
| Formula | C23H27FN4O2 |
|---|---|
| CAS | 106266-06-2 |
| Mol weight | 410.4845 |
3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-2-methyl-4H,6H,7H,8H,9H-pyrido[1,2-a]pyrimidin-4-one
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Risperidone tartrate | 0S6B72E3LK | 666179-92-6 | KSWIOGDSXUFKOC-LREBCSMRSA-N |
Risperidone
CAS Registry Number: 106266-06-2
CAS Name: 3-[2-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one
Manufacturers’ Codes: R-64766
Trademarks: Belivon (Organon); Risperdal (J & J)
Molecular Formula: C23H27FN4O2, Molecular Weight: 410.48
Percent Composition: C 67.30%, H 6.63%, F 4.63%, N 13.65%, O 7.80%
Literature References: Combined serotonin (5-HT2) and dopamine (D2) receptor antagonist. Prepn: L. E. J. Kennis, J. Vandenberk, EP196132; eidem,US4804663 (1986, 1989 both to Janssen). Pharmacology: P. A. J. Janssen et al.,J. Pharmacol. Exp. Ther.244, 685 (1988). Receptor binding studies: J. E. Leysen et al.,ibid.247, 661 (1988). HPLC determn in plasma: A. Avenoso et al.,J. Chromatogr. B746, 173 (2000). Clinical study in psychoses: Y. G. Gelders et al.,Pharmacopsychiatry23, 206 (1990); in autism: L. Scahill et al., N. Engl. J. Med.347, 314 (2002). Brief review: M. G. Livingston, Lancet343, 457-460 (1994). Review of pharmacology and therapeutic potential: S. Grant, A. Fitton, Drugs48, 253-273 (1994); B. Green, Curr. Med. Res. Opin.16, 57-65 (2000); of clinical experience in schizophrenia: H.-J. Möller, Expert Opin. Pharmacother.6, 803-818 (2005),
Properties: Crystals from DMF + 2-propanol, mp 170.0°. LD50 in male, female mice, rats, dogs (mg/kg): 29.7, 26.9, 34.3, 35.4, 14.1, 18.3 i.v.; 82.1, 63.1, 113, 56.6, 18.3, 18.3 orally (Janssen, 1988).
Melting point: mp 170.0°
Toxicity data: LD50 in male, female mice, rats, dogs (mg/kg): 29.7, 26.9, 34.3, 35.4, 14.1, 18.3 i.v.; 82.1, 63.1, 113, 56.6, 18.3, 18.3 orally (Janssen, 1988)
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Benzisoxazoles; Serotonin-Dopamine Antagonist.
Risperidone, sold under the brand name Risperdal among others, is an atypical antipsychotic[2] used to treat schizophrenia and bipolar disorder.[2] It is taken either by mouth or by injection (subcutaneous or intramuscular).[2] The injectable versions are long-acting and last for 2-4 weeks.[6]
Common side effects include movement problems, sleepiness, dizziness, trouble seeing, constipation, and increased weight.[2][7] Serious side effects may include the potentially permanent movement disorder tardive dyskinesia, as well as neuroleptic malignant syndrome, an increased risk of suicide, and high blood sugar levels.[2][6] In older people with psychosis as a result of dementia, it may increase the risk of death.[2] It is unknown if it is safe for use in pregnancy.[2] Its mechanism of action is not entirely clear, but is believed to be related to its action as a dopamine and serotonin antagonist.[2]
Study of risperidone began in the late 1980s and it was approved for sale in the United States in 1993.[2][8][4] It is on the World Health Organization’s List of Essential Medicines.[9] It is available as a generic medication.[6] In 2019, it was the 149th most commonly prescribed medication in the United States, with more than 4 million prescriptions.[10][11]
Synthesis ReferenceUS4804663
SYN
| EP 0196132; ES 8705881; JP 1986221186; US 4804663 |
The Friedel-Crafts condensation of 1,3-difluorobenzene (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
SYN
ES 2050069
The intermediate 3-(2-chloroethyl)-2-methyl-6, 7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (V) has been obtained as follows: The cyclization of 2-aminopyridine (I) with 3-acetyltetrahydrofuran-2-one (II) by means of polyphosphoric acid (PPA) at 160 C gives 3-(2-hydroxyethyl)-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (III), which is hydrogenated with H2 over Pd/C in ethanol/water to yield the tetrahydro derivative (IV). Finally, the OH group of (IV) is treated with SOCl2 in dichloromethane to afford the target 2-chloroethyl intermediate (V).
SYN
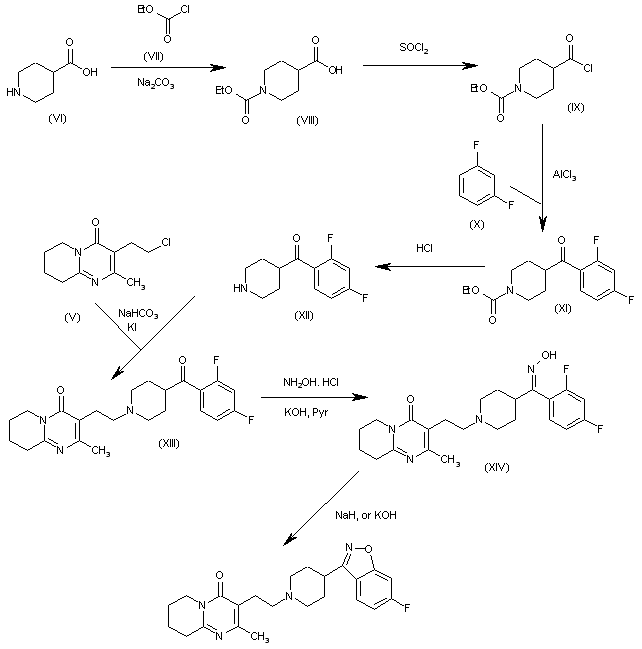
The condensation of piperidine-4-carboxylic acid (VI) with ethyl chloroformate (VII) by means of Na2CO3 in toluene/water gives 1-(ethoxycarbonyl)piperidine-4-carboxylic acid (VIII), which is treated with SOCl2 to yield the corresponding acyl chloride (IX). The Friedel-Crafts condensation of (IX) with refluxing 1,3-difluorobenzene (X) by means of AlCl3 gives 4-(2,4-difluorobenzoyl)piperidine-1-carboxylic acid ethyl ester (XI), which is treated with concentrated HCl at 100 C to yield 4-(2,4-difluorobenzoyl)piperidine (XII). The condensation of piperidine (XII) with the 2-chloroethyl intermediate (V) by means of KI and NaHCO3 in refluxing acetonitrile affords the adduct (XIII), which is treated with hydroxylamine hydrochloride and KOH in refluxing pyridine/ethanol to provide the corresponding oxime (XIV). Finally, this compound is cyclized by means of KOH in refluxing water or with NaH in refluxing THF to afford in both cases the target 1,2-benzisoxazole.
SYN
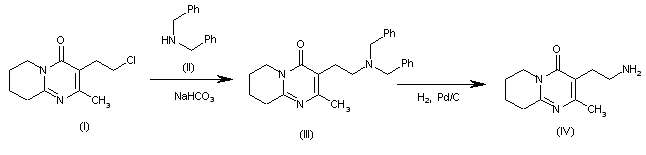
The intermediate 3-(2-aminoethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (IV) has been obtained as follows: The condensation of 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (I) with dibenzylamine (II) by means of NaHCO3 in refluxing acetonitrile gives the tertiary amine (III), which is debenzylated by hydrogenation with H2 over Pd/C in warm ethanol to afford the target intermediate (IV).
SYN
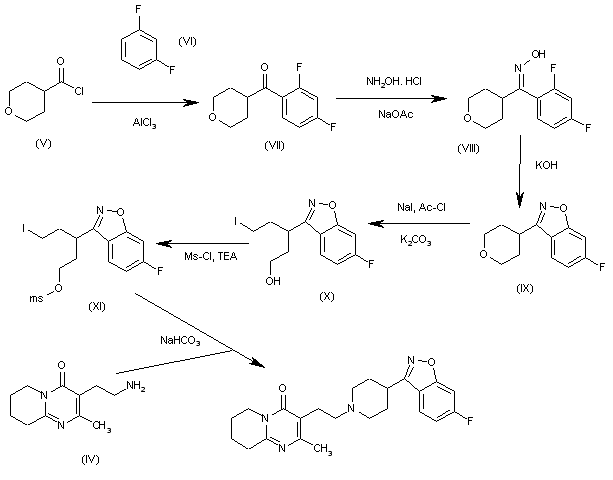
The condensation of tetrahydropyran-4-carbonyl chloride (V) with refluxing 1,3-difluorobenzene (VI) by means of AlCl3 gives 1-(2,4-difluorophenyl)-1-(tetrahydropyran-4-yl)methanone (VII), which is treated with hydroxylamine hydrochloride and sodium acetate in refluxing ethanol/water to yield the corresponding oxime (VIII). The cyclization of (VIII) by means of KOH in refluxing methanol affords 6-fluoro-3-(tetrahydropyran-4-yl)-1,2-benzisoxazole (IX), which is treated with NaI and Ac-Cl and then with K2CO3 in refluxing acetonitrile to provide the 5-iodopentanol derivative (X). The reaction of the OH group of (X) with Ms-Cl and TEA in dichloromethane gives the corresponding mesylate (XI), which is finally cyclized with the intermediate amine (IV) by means of NaHCO3 in refluxing acetonitrile to yield the target piperidine.
SYN

SYN
Eur. Pat. Appl. 196132

SYN
- Production Route of Risperidone
- (CAS NO.: ), with other name of 4H-Pyrido(1,2-a)pyrimidin-4-one, 6,7,8,9-tetrahydro-3-(2-(4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl)ethyl)-2-methyl-, could be produced through many synthetic methods.Following is one of the synthesis routes:
The Friedel-Crafts condensation of 1,3-di (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
- SYN
Piperidine-Based Nonfused Biheterocycles With C–N and C–C Coupling
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Risperidone (15970)
Risperidone (7.2.1) (Risperdal) is the first second-generation antipsychotic that was specifically designed as a combined D2 and serotonin 5-HT(2A) receptor antagonist, thus following the pharmacological mechanism thought to be responsible for the antipsychotic effects. After its advent in the 1990s as the first novel second-generation antipsychotic, risperidone has achieved worldwide acceptance. It was initially approved for use in schizophrenia, mania of bipolar disorder, and irritability and aggression of autism. But it is also effectively used in other instances of psychosis, including schizoaffective disorder, depression with psychotic features, and psychosis secondary to general medical conditions. Risperidone may be effective in other conditions such as major depression, various anxiety disorders, delirium, dementia, for Alzheimer’s dementia, which occurs in 6–8% of persons older than 65 and increases to 30% among those 85 years or older, and substance abuse disorders [84–113].
Risperidone is proposed for inclusion in the WHO Model List of Essential Medications for treatment of schizophrenia, mania, and autism.
Risperidone (7.2.1) was synthesized starting from 1-acetyl-4-piperidine-carbonyl chloride (7.2.4), which was used to acylate 1,3-difluorobenzene (7.2.5) in dichloromethane using aluminum chloride as Lewis acid. The reaction gave 1-(4-(2,4-difluorobenzoyl)piperidin-1-yl)ethan-1-one (7.2.6). The protecting acetyl group of the last was removed off by hydrolysis in 6 N hydrochloric acid on reflux, which gave (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7). The obtained product was converted further to corresponding oxime (7.2.8) on reaction with hydroxylamine hydrochloride in ethanol in the presence of N,N-diethylenethanamine. Synthesized oxime (7.2.8) was cyclized to 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole (7.2.9) on reflux with 50% potassium hydroxide solution in water. At the final stage the obtained product (7.2.9) was alkylated with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (7.2.10) on heating at 85–90°C in dimethylformamide in the presence of sodium carbonate and potassium iodide, which gave the desired product, risperidone (7.2.1) [114,115]. Later, another method of (7.2.7) → (7.2.1) transformation was proposed, which involved the reductive alkylation of (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7) with aldehyde (7.2.11) and sodium cyanoborohydride, which gave compound (7.2.12), coherently converted to oxime (7.2.13) and further to the desired compound, risperidone (7.2.1) [116] (Scheme 7.7).

///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Risperidone is mainly used for the treatment of schizophrenia, bipolar disorder, and irritability associated with autism.[12]
Schizophrenia
Risperidone is effective in treating psychogenic polydipsia and the acute exacerbations of schizophrenia.[13][14]
Studies evaluating the utility of risperidone by mouth for maintenance therapy have reached varying conclusions. A 2012 systematic review concluded that evidence is strong that risperidone is more effective than all first-generation antipsychotics other than haloperidol, but that evidence directly supporting its superiority to placebo is equivocal.[15] A 2011 review concluded that risperidone is more effective in relapse prevention than other first- and second-generation antipsychotics with the exception of olanzapine and clozapine.[16] A 2016 Cochrane review suggests that risperidone reduces the overall symptoms of schizophrenia, but firm conclusions are difficult to make due to very low-quality evidence. Data and information are scarce, poorly reported, and probably biased in favour of risperidone, with about half of the included trials developed by drug companies. The article raises concerns regarding the serious side effects of risperidone, such as parkinsonism.[17] A 2011 Cochrane review compared risperidone with other atypical antipsychotics such as olanzapine for schizophrenia:[18]
| Summary |
|---|
| Risperidone seems to produce somewhat more extrapyramidal side effects and clearly more prolactin increase than most other atypical antipsychotics. It may also differ from other compounds in the occurrence of other adverse effects such as weight gain, metabolic problems, cardiac effects, sedation, and seizures. Nevertheless, the large proportion of participants leaving studies early and incomplete reporting of outcomes makes drawing firm conclusions difficult.[18] |
| showOutcomeFindings in wordsFindings in numbersQuality of evidence |
Long-acting injectable formulations of antipsychotic drugs provide improved compliance with therapy and reduce relapse rates relative to oral formulations.[19][20] The efficacy of risperidone long-acting injection appears to be similar to that of long acting injectable forms of first generation antipsychotics.[21]
Bipolar disorder
Second-generation antipsychotics, including risperidone, are effective in the treatment of manic symptoms in acute manic or mixed exacerbations of bipolar disorder.[22][23][24] In children and adolescents, risperidone may be more effective than lithium or divalproex, but has more metabolic side effects.[25] As maintenance therapy, long-acting injectable risperidone is effective for the prevention of manic episodes but not depressive episodes.[26] The long-acting injectable form of risperidone may be advantageous over long acting first generation antipsychotics, as it is better tolerated (fewer extrapyramidal effects) and because long acting injectable formulations of first generation antipsychotics may increase the risk of depression.[27]
Autism
Compared to placebo, risperidone treatment reduces certain problematic behaviors in autistic children, including aggression toward others, self-injury, challenging behaviour, and rapid mood changes.[28] The evidence for its efficacy appears to be greater than that for alternative pharmacological treatments.[29] Weight gain is an important adverse effect.[4][30] Some authors recommend limiting the use of risperidone and aripiprazole to those with the most challenging behavioral disturbances in order to minimize the risk of drug-induced adverse effects.[31] Evidence for the efficacy of risperidone in autistic adolescents and young adults is less persuasive.[32]
Other uses
Risperidone has shown promise in treating therapy-resistant obsessive–compulsive disorder, when serotonin reuptake inhibitors alone are not sufficient.[33]
Risperidone has not demonstrated a benefit in the treatment of eating disorders or personality disorders, except for limited evidence in schizotypal personality disorder.[34]
While antipsychotic medications such as risperidone have a slight benefit in people with dementia, they have been linked to higher incidence of death and stroke.[34] Because of this increased risk of death, treatment of dementia-related psychosis with risperidone is not FDA approved and carries a black box warning.[4]
Forms
Available forms of risperidone include tablet, oral dissolving tablet, oral solution, and powder and solvent for suspension for injection.[35]
Adverse effects
See also: List of adverse effects of risperidone
Common side effects include movement problems, sleepiness, dizziness, trouble seeing, constipation, and increased weight.[2][7] About 9 to 20% of people gained more than 7% of the baseline weight depending on the dose.[2] Serious side effects may include the potentially permanent movement disorder tardive dyskinesia, as well as neuroleptic malignant syndrome, an increased risk of suicide, and high blood sugar levels.[2][6] In older people with psychosis as a result of dementia, it may increase the risk of death.[2]
While atypical antipsychotics appear to have a lower rate of movement problems as compared to typical antipsychotics, risperidone has a high risk of movement problems among the atypicals.[36][37] Atypical antipsychotics however are associated with a greater amount of weight gain.[37]
Drug interactions
- Carbamazepine and other enzyme inducers may reduce plasma levels of risperidone.[4] If a person is taking both carbamazepine and risperidone, the dose of risperidone will likely need to be increased. The new dose should not be more than twice the patient’s original dose.[4]
- CYP2D6 inhibitors, such as SSRI medications, may increase plasma levels of risperidone and those medications.[4]
- Since risperidone can cause hypotension, its use should be monitored closely when a patient is also taking antihypertensive medicines to avoid severe low blood pressure.[4]
- Risperidone and its metabolite paliperidone are reduced in efficacy by P-glycoprotein inducers such as St John’s wort[38][39]
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotic treatment to avoid acute withdrawal syndrome or rapid relapse.[40] Some have argued the additional somatic and psychiatric symptoms associated with dopaminergic super-sensitivity, including dyskinesia and acute psychosis, are common features of withdrawal in individuals treated with neuroleptics.[41][42][43][44] This has led some to suggest the withdrawal process might itself be schizomimetic, producing schizophrenia-like symptoms even in previously healthy patients, indicating a possible pharmacological origin of mental illness in a yet unknown percentage of patients currently and previously treated with antipsychotics. This question is unresolved, and remains a highly controversial issue among professionals in the medical and mental health communities, as well as the public.[45]
Dementia
Older people with dementia-related psychosis are at a higher risk of death if they take risperidone compared to those who do not. Most deaths are related to heart problems or infections.[4]
Pharmacology
Pharmacodynamics
See also: Atypical antipsychotic § Pharmacodynamics, and Antipsychotic § Comparison of medications
| Site | Ki (nM) | Action |
|---|---|---|
| 5-HT1A | 423 | Antagonist |
| 5-HT1B | 14.9 | Antagonist |
| 5-HT1D | 84.6 | Antagonist |
| 5-HT2A | 0.17 | Inverse agonist |
| 5-HT2B | 61.9 | Inverse agonist |
| 5-HT2C | 12.0 | Inverse agonist |
| 5-HT5A | 206 | Antagonist |
| 5-HT6 | 2,060 | Antagonist |
| 5-HT7 | 6.60 | Irreversible antagonist[47] |
| α1A | 5.0 | Antagonist |
| α1B | 9.0 | Antagonist |
| α2A | 16.5 | Antagonist |
| α2B | 108 | Antagonist |
| α2C | 1.30 | Antagonist |
| D1 | 244 | Antagonist |
| D2 | 3.57 | Antagonist |
| D2S | 4.73 | Antagonist |
| D2L | 4.16 | Antagonist |
| D3 | 3.6 | Inverse agonist |
| D4 | 4.66 | Antagonist |
| D5 | 290 | Antagonist |
| H1 | 20.1 | Inverse agonist |
| H2 | 120 | Inverse agonist |
| mACh | >10,000 | Negligible |
Risperidone pharmacodynamics excluding D-amino acid oxidase inhibition
Risperidone has been classified as a “qualitatively atypical” antipsychotic agent with a relatively low incidence of extrapyramidal side effects (when given at low doses) that has more pronounced serotonin antagonism than dopamine antagonism. Risperidone contains the functional groups of benzisoxazole and piperidine as part of its molecular structure. Although not a butyrophenone, it was developed with the structures of benperidol and ketanserin as a basis. It has actions at several 5-HT (serotonin) receptor subtypes. These are 5-HT2C, linked to weight gain, 5-HT2A, linked to its antipsychotic action and relief of some of the extrapyramidal side effects experienced with the typical neuroleptics.[48]
It has been found that D-amino acid oxidase, the enzyme that catalyses the breakdown of D-amino acids (e.g. D-alanine and D-serine — the neurotransmitters) is inhibited by risperidone.[49]
Risperidone acts on the following receptors:
Dopamine receptors: This drug is an antagonist of the D1 (D1, and D5) as well as the D2 family (D2, D3 and D4) receptors, with 70-fold selectivity for the D2 family. This drug has “tight binding” properties, which means it has a long half-life and like other antipsychotics, risperidone blocks the mesolimbic pathway, the prefrontal cortex limbic pathway, and the tuberoinfundibular pathway in the central nervous system. Risperidone may induce extrapyramidal side effects, akathisia and tremors, associated with diminished dopaminergic activity in the striatum. It can also cause sexual side effects, galactorrhoea, infertility, gynecomastia and, with chronic use reduced bone mineral density leading to breaks, all of which are associated with increased prolactin secretion.[48]
Serotonin receptors: Its action at these receptors may be responsible for its lower extrapyramidal side effect liability (via the 5-HT2A/2C receptors) and improved negative symptom control compared to typical antipsychotics such as haloperidol for instance. Its antagonistic actions at the 5-HT2C receptor may account, in part, for its weight gain liability.[medical citation needed]
Alpha α1 adrenergic receptors: This action accounts for its orthostatic hypotensive effects and perhaps some of the sedating effects of risperidone.[48]
Alpha α2 adrenergic receptors: Perhaps greater positive, negative, affective and cognitive symptom control.[50]
Histamine H1 receptors: effects on these receptors account for its sedation and reduction in vigilance. This may also lead to drowsiness and weight gain.[48]
Voltage-gated sodium channels: Because it accumulates in synaptic vesicles, Risperidone inhibits voltage-gated sodium channels at clinically used concentrations.[51]
Though this medication possesses similar effects to other typical and atypical antipsychotics, it does not possess an affinity for the muscarinic acetylcholine receptors. In many respects, this medication can be useful as an “acetylcholine release-promoter” similar to gastrointestinal drugs such as metoclopramide and cisapride.[medical citation needed]
Pharmacokinetics
Risperidone undergoes hepatic metabolism and renal excretion. Lower doses are recommended for patients with severe liver and kidney disease.[4] The active metabolite of risperidone, paliperidone, is also used as an antipsychotic.[52]
Society and culture

Risperdal (risperidone) 4 mg tablets (UK)
Legal status
Risperidone was approved by the United States Food and Drug Administration (FDA) in 1993 for the treatment of schizophrenia.[63] In 2003, the FDA approved risperidone for the short-term treatment of the mixed and manic states associated with bipolar disorder. In 2006, the FDA approved risperidone for the treatment of irritability in autistic children and adolescents.[64] The FDA’s decision was based in part on a study of autistic people with severe and enduring problems of violent meltdowns, aggression, and self-injury; risperidone is not recommended for autistic people with mild aggression and explosive behavior without an enduring pattern.[65] On 22 August 2007, risperidone was approved as the only drug agent available for treatment of schizophrenia in youths, ages 13–17; it was also approved that same day for treatment of bipolar disorder in youths and children, ages 10–17, joining lithium.
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Okedi, intended for the treatment of schizophrenia in adults for whom tolerability and effectiveness has been established with oral risperidone.[66] The applicant for this medicinal product is Laboratorios Farmacéuticos Rovi, S.A.[66]
Availability
Janssen’s patent on risperidone expired on 29 December 2003, opening the market for cheaper generic versions from other companies, and Janssen’s exclusive marketing rights expired on 29 June 2004 (the result of a pediatric extension). It is available under many brand names worldwide.[1]
Risperidone is available as a tablet, an oral solution, and an ampule, which is a depot injection.[1]
Lawsuits
On 11 April 2012, Johnson & Johnson (J&J) and its subsidiary Janssen Pharmaceuticals Inc. were fined $1.2 billion by Judge Timothy Davis Fox of the Sixth Division of the Sixth Judicial Circuit of the U.S. state of Arkansas.[67] The jury found the companies had downplayed multiple risks associated with risperidone (Risperdal). The verdict was later reversed by the Arkansas State Supreme court.[68]
In August 2012, Johnson & Johnson agreed to pay $181 million to 36 U.S. states in order to settle claims that it had promoted risperidone and paliperidone for off-label uses including for dementia, anger management, and anxiety.[69]
In November 2013, J&J was fined $2.2 billion for illegally marketing risperidone for use in people with dementia.[70]
In 2015, Steven Brill posted a 15-part investigative journalism piece on J&J in The Huffington Post, called “America’s most admired lawbreaker”, which was focused on J&J’s marketing of risperidone.[71][72]
J&J has faced numerous civil lawsuits on behalf of children who were prescribed risperidone who grew breasts (a condition called gynecomastia); as of July 2016 there were about 1,500 cases in Pennsylvania state court in Philadelphia, and there had been a February 2015 verdict against J&J with $2.5 million awarded to a man from Alabama, a $1.75M verdict against J&J that November, and in 2016 a $70 million verdict against J&J.[73] In October 2019, a jury awarded a Pennsylvania man $8 billion in a verdict against J&J.[74]
Names
Brand names include Risperdal, Risperdal Consta, Risperdal M-Tab, Risperdal Quicklets, Risperlet, Okedi, and Perseris.[75]
References
- ^ Jump up to:a b c Drugs.com International trade names for risperidone Archived 18 March 2016 at the Wayback Machine Page accessed 15 March 2016
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Risperidone”. The American Society of Health-System Pharmacists. Archived from the original on 2 December 2015. Retrieved 1 December 2015.
- ^ “Risperdal Consta 25 mg powder and solvent for prolonged-release suspension for injection – Summary of Product Characteristics (SmPC)”. (emc). 6 December 2018. Retrieved 29 January 2022.
- ^ Jump up to:a b c d e f g h i j “Risperdal- risperidone tablet Risperdal M-Tab- risperidone tablet, orally disintegrating Risperdal- risperidone solution”. DailyMed. Retrieved 31 December 2019.
- ^ “Okedi EPAR”. European Medicines Agency (EMA). 15 December 2021. Retrieved 2 March 2022.
- ^ Jump up to:a b c d Hamilton R (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. pp. 434–435. ISBN 9781284057560.
- ^ Jump up to:a b Hasnain M, Vieweg WV, Hollett B (July 2012). “Weight gain and glucose dysregulation with second-generation antipsychotics and antidepressants: a review for primary care physicians”. Postgraduate Medicine. 124 (4): 154–67. doi:10.3810/pgm.2012.07.2577. PMID 22913904. S2CID 39697130.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Washington, D.C.: American Psychiatric Pub. p. 627. ISBN 9781585623099.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Risperidone – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ “Respiridone”. The American Society of Health-System Pharmacists. Archived from the original on 13 April 2011. Retrieved 3 April 2011.
- ^ Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”. Lancet. 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019. S2CID 32085212.
- ^ Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harvard Review of Psychiatry. 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID 23656760. S2CID 22523977.
- ^ Barry SJ, Gaughan TM, Hunter R (June 2012). “Schizophrenia”. BMJ Clinical Evidence. 2012. PMC 3385413. PMID 23870705.
- ^ Glick ID, Correll CU, Altamura AC, Marder SR, Csernansky JG, Weiden PJ, et al. (December 2011). “Mid-term and long-term efficacy and effectiveness of antipsychotic medications for schizophrenia: a data-driven, personalized clinical approach”. The Journal of Clinical Psychiatry. 72 (12): 1616–27. doi:10.4088/JCP.11r06927. PMID 22244023.
- ^ Rattehalli RD, Zhao S, Li BG, Jayaram MB, Xia J, Sampson S (December 2016). “Risperidone versus placebo for schizophrenia” (PDF). The Cochrane Database of Systematic Reviews. 2016 (12): CD006918. doi:10.1002/14651858.CD006918.pub3. PMC 6463908. PMID 27977041.
- ^ Jump up to:a b Komossa K, Rummel-Kluge C, Schwarz S, Schmid F, Hunger H, Kissling W, Leucht S (January 2011). “Risperidone versus other atypical antipsychotics for schizophrenia”. The Cochrane Database of Systematic Reviews (1): CD006626. doi:10.1002/14651858.CD006626.pub2. PMC 4167865. PMID 21249678.
- ^ Leucht C, Heres S, Kane JM, Kissling W, Davis JM, Leucht S (April 2011). “Oral versus depot antipsychotic drugs for schizophrenia–a critical systematic review and meta-analysis of randomised long-term trials”. Schizophrenia Research. 127 (13): 83–92. doi:10.1016/j.schres.2010.11.020. PMID 21257294. S2CID 2386150.
- ^ Lafeuille MH, Dean J, Carter V, Duh MS, Fastenau J, Dirani R, Lefebvre P (August 2014). “Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia”. Current Medical Research and Opinion. 30 (8): 1643–55. doi:10.1185/03007995.2014.915211. PMID 24730586. S2CID 24814527.
- ^ Nielsen J, Jensen SO, Friis RB, Valentin JB, Correll CU (May 2015). “Comparative effectiveness of risperidone long-acting injectable vs first-generation antipsychotic long-acting injectables in schizophrenia: results from a nationwide, retrospective inception cohort study”. Schizophrenia Bulletin. 41 (3): 627–36. doi:10.1093/schbul/sbu128. PMC 4393684. PMID 25180312.
- ^ Muralidharan K, Ali M, Silveira LE, Bond DJ, Fountoulakis KN, Lam RW, Yatham LN (September 2013). “Efficacy of second generation antipsychotics in treating acute mixed episodes in bipolar disorder: a meta-analysis of placebo-controlled trials”. Journal of Affective Disorders. 150 (2): 408–14. doi:10.1016/j.jad.2013.04.032. PMID 23735211.
- ^ Nivoli AM, Murru A, Goikolea JM, Crespo JM, Montes JM, González-Pinto A, et al. (October 2012). “New treatment guidelines for acute bipolar mania: a critical review”. Journal of Affective Disorders. 140 (2): 125–41. doi:10.1016/j.jad.2011.10.015. PMID 22100133.
- ^ Yildiz A, Vieta E, Leucht S, Baldessarini RJ (January 2011). “Efficacy of antimanic treatments: meta-analysis of randomized, controlled trials”. Neuropsychopharmacology. 36 (2): 375–89. doi:10.1038/npp.2010.192. PMC 3055677. PMID 20980991.
- ^ Peruzzolo TL, Tramontina S, Rohde LA, Zeni CP (2013). “Pharmacotherapy of bipolar disorder in children and adolescents: an update”. Revista Brasileira de Psiquiatria. 35 (4): 393–405. doi:10.1590/1516-4446-2012-0999. PMID 24402215.
- ^ Gitlin M, Frye MA (May 2012). “Maintenance therapies in bipolar disorders”. Bipolar Disorders. 14 Suppl 2: 51–65. doi:10.1111/j.1399-5618.2012.00992.x. PMID 22510036. S2CID 21101054.
- ^ Gigante AD, Lafer B, Yatham LN (May 2012). “Long-acting injectable antipsychotics for the maintenance treatment of bipolar disorder”. CNS Drugs. 26 (5): 403–20. doi:10.2165/11631310-000000000-00000. PMID 22494448. S2CID 2786921.
- ^ Jesner OS, Aref-Adib M, Coren E (January 2007). “Risperidone for autism spectrum disorder”. The Cochrane Database of Systematic Reviews (1): CD005040. doi:10.1002/14651858.CD005040.pub2. PMID 17253538.
- ^ Kirino E (2014). “Efficacy and tolerability of pharmacotherapy options for the treatment of irritability in autistic children”. Clinical Medicine Insights. Pediatrics. 8: 17–30. doi:10.4137/CMPed.S8304. PMC 4051788. PMID 24932108.
- ^ Sharma A, Shaw SR (2012). “Efficacy of risperidone in managing maladaptive behaviors for children with autistic spectrum disorder: a meta-analysis”. Journal of Pediatric Health Care. 26 (4): 291–9. doi:10.1016/j.pedhc.2011.02.008. PMID 22726714.
- ^ McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN, Veenstra-Vanderweele J (May 2011). “A systematic review of medical treatments for children with autism spectrum disorders”. Pediatrics. 127 (5): e1312–21. doi:10.1542/peds.2011-0427. PMID 21464191. S2CID 2903864.
- ^ Dove D, Warren Z, McPheeters ML, Taylor JL, Sathe NA, Veenstra-VanderWeele J (October 2012). “Medications for adolescents and young adults with autism spectrum disorders: a systematic review”. Pediatrics. 130 (4): 717–26. doi:10.1542/peds.2012-0683. PMC 4074627. PMID 23008452.
- ^ Dold M, Aigner M, Lanzenberger R, Kasper S (April 2013). “Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials”. The International Journal of Neuropsychopharmacology. 16 (3): 557–74. doi:10.1017/S1461145712000740. PMID 22932229.
- ^ Jump up to:a b Maher AR, Theodore G (June 2012). “Summary of the comparative effectiveness review on off-label use of atypical antipsychotics”. Journal of Managed Care Pharmacy. 18 (5 Suppl B): S1–20. doi:10.18553/jmcp.2012.18.s5-b.1. PMID 22784311.
- ^ Joint Formulary Committee. British National Formulary (online) London: BMJ Group and Pharmaceutical Press http://www.medicinescomplete.com [Accessed on 2 February 2020]
- ^ Divac N, Prostran M, Jakovcevski I, Cerovac N (2014). “Second-generation antipsychotics and extrapyramidal adverse effects”. BioMed Research International. 2014: 656370. doi:10.1155/2014/656370. PMC 4065707. PMID 24995318.
- ^ Jump up to:a b Pillay J, Boylan K, Carrey N, Newton A, Vandermeer B, Nuspl M, MacGregor T, Jafri SH, Featherstone R, Hartling L (March 2017). “First- and Second-Generation Antipsychotics in Children and Young Adults: Systematic Review Update”. Comparative Effectiveness Reviews (184): ES–24. PMID 28749632. Report 17-EHC001-EF. Bookshelf ID: NBK442352.
Compared with FGAs, SGAs may decrease the risk for experiencing any extrapyramidal symptom (EPS). FGAs probably cause lower gains in weight and BMI.
- ^ Wang, J. S.; Ruan, Y.; Taylor, R. M.; Donovan, J. L.; Markowitz, J. S.; Devane, C. L. (2004). “The Brain Entry of Risperidone and 9-hydroxyrisperidone Is Greatly Limited by P-glycoprotein”. The International Journal of Neuropsychopharmacology. 7 (4): 415–9. doi:10.1017/S1461145704004390. PMID 15683552.
- ^ Gurley BJ, Swain A, Williams DK, Barone G, Battu SK (July 2008). “Gauging the clinical significance of P-glycoprotein-mediated herb-drug interactions: comparative effects of St. John’s wort, Echinacea, clarithromycin, and rifampin on digoxin pharmacokinetics”. Molecular Nutrition & Food Research. 52 (7): 772–9. doi:10.1002/mnfr.200700081. PMC 2562898. PMID 18214850.
- ^ BMJ Group, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISSN 0260-535X.
Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
- ^ Chouinard G, Jones BD (January 1980). “Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics”. The American Journal of Psychiatry. 137 (1): 16–21. doi:10.1176/ajp.137.1.16. PMID 6101522.
- ^ Miller R, Chouinard G (November 1993). “Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias, neuroleptic-induced supersensitivity psychosis and refractory schizophrenia”. Biological Psychiatry. 34 (10): 713–38. doi:10.1016/0006-3223(93)90044-E. PMID 7904833. S2CID 2405709.
- ^ Chouinard G, Jones BD, Annable L (November 1978). “Neuroleptic-induced supersensitivity psychosis”. The American Journal of Psychiatry. 135 (11): 1409–10. doi:10.1176/ajp.135.11.1409. PMID 30291.
- ^ Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, et al. (March 2005). “Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis”. Proceedings of the National Academy of Sciences of the United States of America. 102 (9): 3513–8. Bibcode:2005PNAS..102.3513S. doi:10.1073/pnas.0409766102. PMC 548961. PMID 15716360.
- ^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655. S2CID 6267180.
- ^ National Institute of Mental Health. PDSD Ki Database (Internet) [cited 2013 Aug 10]. ChapelHill (NC): University of North Carolina. 1998-2013. Available from: “Archived copy”. Archived from the original on 8 November 2013. Retrieved 16 May 2016.
- ^ Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M (October 2006). “Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor”. Molecular Pharmacology. 70 (4): 1264–70. doi:10.1124/mol.106.024612. PMID 16870886. S2CID 1678887.
- ^ Jump up to:a b c d Brunton L, Chabner B, Knollman B. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, Twelfth Edition. McGraw Hill Professional; 2010.
- ^ Abou El-Magd RM, Park HK, Kawazoe T, Iwana S, Ono K, Chung SP, et al. (July 2010). “The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia”. Journal of Psychopharmacology. 24 (7): 1055–67. doi:10.1177/0269881109102644. PMID 19329549. S2CID 39050369.
- ^ Hecht EM, Landy DC (February 2012). “Alpha-2 receptor antagonist add-on therapy in the treatment of schizophrenia; a meta-analysis”. Schizophrenia Research. 134 (2–3): 202–6. doi:10.1016/j.schres.2011.11.030. PMID 22169246. S2CID 36119981.
- ^ Brauner, Jan M.; Hessler, Sabine; Groemer, Teja W.; Alzheimer, Christian; Huth, Tobias (2014). “Risperidone inhibits voltage-gated sodium channels”. European Journal of Pharmacology. 728: 100–106. doi:10.1016/j.ejphar.2014.01.062. PMID 24508524.
- ^ “The DrugBank database”. Archived from the original on 17 November 2011.
- ^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
- ^ Jump up to:a b Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID 6931472.
- ^ Jump up to:a b Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
- ^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID 6143748.
- ^ Jump up to:a b Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA (April 1979). “Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man”. British Journal of Clinical Pharmacology. 7 (4): 325–31. doi:10.1111/j.1365-2125.1979.tb00941.x. PMC 1429660. PMID 444352.
- ^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID 4992598.
- ^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID 3545764.
- ^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID 7185768.
- ^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
- ^ “Electronic Orange Book”. Food and Drug Administration. April 2007. Archived from the original on 19 August 2007. Retrieved 24 May 2007.
- ^ “FDA approves the first drug to treat irritability associated with autism, Risperdal” (Press release). FDA. 6 October 2006. Archived from the original on 28 August 2009. Retrieved 14 August 2009.
- ^ Scahill L (July 2008). “How do I decide whether or not to use medication for my child with autism? Should I try behavior therapy first?”. Journal of Autism and Developmental Disorders. 38 (6): 1197–8. doi:10.1007/s10803-008-0573-7. PMID 18463973. S2CID 20767044.
- ^ Jump up to:a b “Okedi: Pending EC decision”. European Medicines Agency. 15 December 2021. Retrieved 18 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Companies belittled risks of Risperdal, slapped with huge fine” Archived 12 April 2012 at the Wayback Machine, Los Angeles Times, 11 April 2012.
- ^ Thomas K (20 March 2014). “Arkansas Court Reverses $1.2 Billion Judgment Against Johnson & Johnson”. The New York Times. Archived from the original on 5 November 2015.
- ^ “NY AG: Janssen pays $181M over drug marketing”. The Seattle Times. 30 August 2012. Archived from the original on 7 April 2016.
- ^ “Johnson & Johnson to Pay More Than $2.2 Billion to Resolve Criminal and Civil Investigations”. Department of Justice, Office of Public Affairs. 4 November 2013. Archived from the original on 5 March 2015. Retrieved 23 December 2020.
- ^ Ashbrook T (22 September 2015). “Johnson & Johnson And The Big Lies Of Big Pharma”. On Point. Archived from the original on 22 November 2016.
- ^ Brill S (September 2015). “America’s Most Admired Lawbreaker”. The Huffington Post. Archived from the original on 2 October 2015.
- ^ Feeley J (1 July 2016). “J&J Hit With $70 Million Risperdal Verdict Over Male Breasts”. Bloomberg News. Archived from the original on 7 May 2017.
- ^ “Jury says J&J must pay $8 billion in case over male breast growth linked to Risperdal”. Reuters. 9 October 2019. Retrieved 9 October 2019.
- ^ “Risperidone: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 28 September 2020.
Further reading
- Dean L (2017). “Risperidone Therapy and CYP2D6 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 28520384. Bookshelf ID: NBK425795.
“Risperidone”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Risperdal, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a694015 |
| License data | US DailyMed: Risperidone |
| Pregnancy category | AU: C |
| Routes of administration | By mouth, intramuscular, subcutaneous |
| Drug class | Atypical antipsychotic[2] |
| ATC code | N05AX08 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only) [3]US: ℞-only [4]EU: Rx-only [5] |
| Pharmacokinetic data | |
| Bioavailability | 70% (by mouth)[2] |
| Metabolism | Liver (CYP2D6 mediated to 9-hydroxyrisperidone)[2] |
| Elimination half-life | 20 hours (by mouth), 3–6 days (IM)[2] |
| Excretion | Urinary (70%) feces (14%)[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 106266-06-2 |
| PubChem CID | 5073 |
| PubChem SID | 475100 |
| IUPHAR/BPS | 96 |
| DrugBank | DB00734 |
| ChemSpider | 4895 |
| UNII | L6UH7ZF8HC |
| KEGG | D00426 |
| ChEBI | CHEBI:8871 |
| ChEMBL | ChEMBL85 |
| PDB ligand | 8NU (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID8045193 |
| ECHA InfoCard | 100.114.705 |
| Chemical and physical data | |
| Formula | C23H27FN4O2 |
| Molar mass | 410.493 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////////Risperidone, R-64,766, R-64766, RCN-3028, RCN3028

NEW DRUG APPROVALS
ONE TIME
$10.00
Lutetium Lu 177 vipivotide tetraxetan
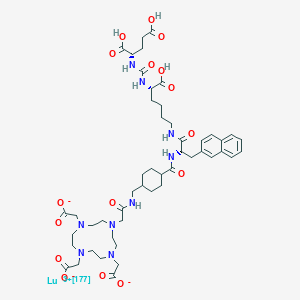


Lutetium Lu 177 vipivotide tetraxetan
FDA APPROVED 2022/3/23, Pluvicto
To treat prostate-specific membrane antigen-positive metastatic castration-resistant prostate cancer following other therapies
| Formula | C49H65N9O16. Lu. 3H |
|---|---|
| CAS | 1703749-62-5 |
| Mol weight | 1214.0819 |
| Antineoplastic, Radioactive agent | |
| Disease | Prostate cancer (PSMA positive) |
|---|
ルテチウム(177Lu)ビピボチドテトラキセタン;
UNII-G6UF363ECX, WHO 11429
G6UF363ECX
177Lu-Psma-617
Vipivotide tetraxetan Lu-177
177Lu-Labeled PSMA-617
2-[4-[2-[[4-[[(2S)-1-[[(5S)-5-carboxy-5-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]pentyl]amino]-3-naphthalen-2-yl-1-oxopropan-2-yl]carbamoyl]cyclohexyl]methylamino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lutetium-177(3+)
(177Lu)Lutetium 2,2′,2”-[10-(2-{[(trans-4-{[(2S)-1-{[(5S)-5-carboxy-5-({[(1S)-1,3-dicarboxypropyl]carbamoyl}amino)pentyl]amino}-3-(2-naphthyl)-1-oxo-2-propanyl]carbamoyl}cyclohexyl)methyl]amino}-2- oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (non-preferred name)
1983157-55-6[RN]
PSMA-617 LU-177
Lutetium Lu 177 Vipivotide Tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells. Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells. Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation. PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.
Lutetium (177Lu) vipivotide tetraxetan, sold under the brand name Pluvicto, is a radiopharmaceutical medication used for the treatment of prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] Lutetium (177Lu) vipivotide tetraxetan is a targeted radioligand therapy.[2][3]
The most common adverse reactions include fatigue, dry mouth, nausea, anemia, decreased appetite, and constipation.[2]
Lutetium (177Lu) vipivotide tetraxetan is a radioconjugate composed of PSMA-617, a human prostate-specific membrane antigen (PSMA)-targeting ligand, conjugated to the beta-emitting radioisotope lutetium Lu 177 (177Lu), with potential antineoplastic activity against PSMA-expressing tumor cells.[4] Upon intravenous administration of lutetium Lu 177 vipivotide tetraxetan, vipivotide tetraxetan targets and binds to PSMA-expressing tumor cells.[4] Upon binding, PSMA-expressing tumor cells are destroyed by 177Lu through the specific delivery of beta particle radiation.[4] PSMA, a tumor-associated antigen and type II transmembrane protein, is expressed on the membrane of prostatic epithelial cells and overexpressed on prostate tumor cells.[4]
Lutetium (177Lu) vipivotide tetraxetan was approved for medical use in the United States in March 2022.[2][5]
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
History[edit]
Efficacy was evaluated in VISION (NCT03511664), a randomized (2:1), multicenter, open-label trial that evaluated lutetium (177Lu) vipivotide tetraxetan plus best standard of care (BSoC) (n=551) or BSoC alone (n=280) in men with progressive, prostate-specific membrane antigen (PSMA)-positive metastatic castration-resistant prostate cancer (mCRPC).[2] All participants received a GnRH analog or had prior bilateral orchiectomy.[2] Participants were required to have received at least one androgen receptor pathway inhibitor, and 1 or 2 prior taxane-based chemotherapy regimens.[2] Participants received lutetium (177Lu) vipivotide tetraxetan 7.4 GBq (200 mCi) every 6 weeks for up to a total of 6 doses plus BSoC or BSoC alone.[2]
The U.S. Food and Drug Administration granted the application for lutetium (177lu) vipivotide tetraxetan priority review and breakthrough therapy designations.[2]
References
- ^ “Highlights of prescribing information: PLUVICTOTM (lutetium Lu 177 vipivotide tetraxetan) injection, for intravenous use” (PDF). Advanced Accelerator Applications USA, Inc. Novartis. March 2022.
- ^ Jump up to:a b c d e f g h i j “FDA approves Pluvicto for metastatic castration-resistant prostate can”. U.S. Food and Drug Administration. 23 March 2022. Retrieved 23 March 2022. This article incorporates text from this source, which is in the public domain.
- ^ Neels OC, Kopka K, Liolios C, Afshar-Oromieh A (December 2021). “Radiolabeled PSMA Inhibitors”. Cancers. 13 (24): 6255. doi:10.3390/cancers13246255. PMC 8699044. PMID 34944875.
- ^ Jump up to:a b c d “Lutetium Lu 177 Vipivotide Tetraxetan (Code C148145)”. NCI Thesaurus. 28 February 2022. Retrieved 23 March 2022. This article incorporates text from this source, which is in the public domain.
- ^ “Novartis Pluvicto approved by FDA as first targeted radioligand therapy for treatment of progressive, PSMA positive metastatic castration-resistant prostate cancer” (Press release). Novartis. 23 March 2022. Retrieved 23 March 2022.
External links
- “Lutetium lu 177 vipivotide tetraxetan”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Pluvicto |
| Other names | 177Lu-PSMA-617, Lutetium Lu 177 vipivotide tetraxetan (USAN US) |
| License data | US DailyMed: Pluvicto |
| Routes of administration | Intravenous |
| Drug class | Radiopharmaceutical |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1703749-62-5 |
| PubChem CID | 122706785 |
| ChemSpider | 58828499 |
| UNII | G6UF363ECX |
| KEGG | D12335 |
| Chemical and physical data | |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
////////////Lutetium Lu 177 vipivotide tetraxetan, ルテチウム(177Lu)ビピボチドテトラキセタン, FDA 2022, APPROVALS 2022, PROSTRATE CANCER, WHO 11429
C1CC(CCC1CNC(=O)CN2CCN(CCN(CCN(CC2)CC(=O)[O-])CC(=O)[O-])CC(=O)[O-])C(=O)NC(CC3=CC4=CC=CC=C4C=C3)C(=O)NCCCCC(C(=O)O)NC(=O)NC(CCC(=O)O)C(=O)O.[Lu+3]
Vipivotide tetraxetan (Synonyms: PSMA-617)
CAS No. : 1702967-37-0
Vipivotide tetraxetan (PSMA-617) is a high potent prostate-specific membrane antigen (PSMA) inhibitor, with a Ki of 0.37 nM.

NEW DRUG APPROVALS
one time
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}