
Home » 0rphan drug status (Page 21)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
The US FDA has issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate)高三尖杉酯碱 for chronic myeloid leukaemia (CML).


Omacetaxine mepesuccinate 高三尖杉酯碱
Alkaloid from Cephalotaxus harringtonia; FDA approved orphan drug status for Ceflatonin in the treatment of chronic myeloid leukemia due to being an inducer of apoptosis in myeloid cells and inhibitor of angiogenesis.
26833-87-4 CAS NO
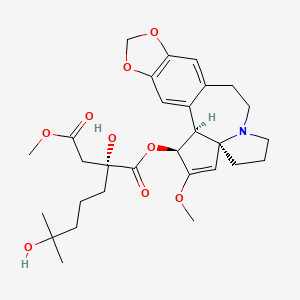
1-((1S,3aR,14bS)-2-Methoxy-1,5,6,8,9,14b-hexahydro-4H-cyclopenta(a)(1,3)dioxolo(4,5-h)pyrrolo(2,1-b)(3)benzazepin-1-yl) 4-methyl (2R)-2-hydroxy-2-(4-hydroxy-4-methylpentyl)butanedioate
1-((11bS,12S,14aR)-13-methoxy-2,3,5,6,11b,12-hexahydro-1H-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-d]cyclopenta[b]pyrrolo[1,2-a]azepin-12-yl) 4-methyl 2-hydroxy-2-(4-hydroxy-4-methylpentyl)succinate
Also known as: NSC-141633,
- BRN 5687925
- Ceflatonin
- CGX-635
- Homoharringtonine
- Myelostat
- NSC 141633
- Omacetaxine mepesuccinate
- Omapro
- Synribo
- UNII-6FG8041S5B
- 高三尖杉酯碱
CGX-635-14 (formulation), CGX-635, HHT, ZJ-C, Myelostat, Ceflatonin
USFDA on 26th October 2012 APPROVED
| Formula | C29H39NO9 |
|---|---|
| Mol. mass | 545.62 g/mol |
The US Food and Drug Administration has now issued full approval for Israeli drugmaker Teva’s Synribo (omacetaxine mepesuccinate) for chronic myeloid leukaemia (CML).
Synribo is indicated for adult patients with chronic phase (CP) or accelerated phase (AP) CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).
Read more at: http://www.pharmatimes.com/Article/14-02-17/US_green_light_for_Teva_s_CML_drug_Synribo.aspx#ixzz2tdkbGFcw
Homoharringtonine is an angiogenesis-inhibiting and apoptosis-inducing alkaloid which was approved in October 2012 by the FDA for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKI). In November 2012, the product was commercialized as Synribo(R) on the U.S. market by Teva.
The original developer, ChemGenex, selected homoharringtonine for the combination trials due to its complementary mechanism of action that can reduce Bcr-Abl protein expression associated with resistance to imatinib mesylate.
In 2004, the compound received orphan drug designation from the EMEA for the treatment of AML and CML. Orphan drug designation was granted by the FDA for the treatment of CML in 2006 and for the treatment of myelodysplasia in 2009. Fast track designation was assigned to homoharringtonine for CML in 2006. In 2009, the product was licensed to Hospira by ChemGenex Pharmaceuticals for development and marketing in Europe, the Middle East and parts of Africa.
Homoharringtonine, AKA HHT or omacetaxine mepesuccinate, is a cephalotaxine ester and protein synthesis inhibitor with established clinical activity as a single agent in hematological malignancies. Homoharringtonine is synthesized from cephalotaxine, which is an extract from the leaves of the plant, Cephalotaxus species. In October 2005, homoharringtonine received Orphan Drug designation from the EMEA for the treatment of chronic myeloid leukemia (CML). Then in March 2006, homoharringtonine received Orphan Drug status from the FDA for the treatment of CML. In November 2006, homoharringtonine, for the treatment of CML, was granted Fast Track designation by the FDA. Most recently, in October 2012, homoharringtonine was marketed under the brand name Synribo” and FDA approved for patients who are intolerant and/or resistant to two or more tyrosine kinase inhibitors used to treat accelerated or chronic phase CML
Omacetaxine mepesuccinate is administered subcutaneously and acts differently from TKIs. It may have a therapeutic advantage for patients who have failed TKIs. Omacetaxine is currently in global phase 2/3 clinical trials for CML and has been granted Orphan Drug designations by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMEA) as well as Fast Track status by the FDA. In vitro and animal model trails are promising and recent results showed that omacetaxine has potential to treat resistant leukemia mainly CML and ALL.
| PATENT | ||
|---|---|---|
|
3-25-2011
|
CEPHALOTAXUS ESTERS, METHODS OF SYNTHESIS, AND USES THEREOF
|
Tetrahedron Letters,Vo1.23,No.34,pp 3431-3434 … – Brock University
Omacetaxine mepesuccinate (INN, trade name Synribo) is a semi-synthetic analogue of an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of chronic myelogenous leukemia (CML). It was approved by the US FDA in October 2012 for the treatment of adult patients with CML with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs).[1]
Omacetaxine mepesuccinate is a semisynthetic derivative of the cytotoxic plant alkaloid homoharringtonine isolated from the evergreen tree Cephalotaxus with potential antineoplastic activity. Omacetaxine mepesuccinate binds to the 80S ribosome in eukaryotic cells and inhibits protein synthesis by interfering with chain elongation. This agent also induces differentiation and apoptosis in some cancer cell types. Omacetaxine mepesuccinate (INN, or homoharringtonine, trade name Synribo) is an alkaloid from Cephalotaxus harringtonia that is indicated for treatment of Chronic Myelogenous Leukemia. It was approved by the USFDA on 26th October 2012 for the treatment of adult patients with chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs)
Omacetaxine is indicated for use as a treatment for patients with chronic myeloid leukaemia who are intolerant of tyrosine kinase inhibitors.[2][3]
In June 2009, results of a long-term open label Phase II study were published, which investigated the use of omacetaxine infusions in CML patients. After twelve months of treatment, about one third of patients showed a cytogenetic response.[4] A study in patients who had failed imatinib and who had the drug resistant T315I mutation achieved cytogenetic response in 28% of patients and haematological response in 80% of patients, according to preliminary data.[5]
Phase I studies including a small number of patients have shown benefit in treating myelodysplastic syndrome (MDS, 25 patients)[6] and acute myelogenous leukaemia (AML, 76 patients).[7] Patients with solid tumors did not benefit from omacetaxine.[8]
Omacetaxine is a protein translation inhibitor. It inhibits protein translation by preventing the initial elongation step of protein synthesis. It interacts with the ribosomal A-site and prevents the correct positioning of amino acid side chains of incoming aminoacyl-tRNAs. Omacetaxine acts only on the initial step of protein translation and does not inhibit protein synthesis from mRNAs that have already commenced translation.[9]
Omacetaxine mepesuccinate
SYNRIBO contains the active ingredient omacetaxine mepesuccinate, a cephalotaxine ester. It is a protein synthesis inhibitor. Omacetaxine mepesuccinate is prepared by a semi-synthetic process from cephalotaxine, an extract from the leaves of Cephalotaxus sp. The chemical name of omacetaxine mepesuccinate is cephalotaxine, 4-methyl (2R)-hydroxyl-2-(4-hydroxyl-4-methylpentyl) butanedioate (ester).
Omacetaxine mepesuccinate has the following chemical structure:
 |
The molecular formula is C29H39NO9 with a molecular weight of 545.6 g/mol. SYNRIBO for injection is a sterile, preservative-free, white to off-white, lyophilized powder in a single-use vial. Each vial contains 3.5 mg omacetaxine mepesuccinate and mannitol.
SYNRIBO is intended for subcutaneous administration after reconstitution with 1.0 mL of 0.9% Sodium Chloride Injection, USP. The pH of the reconstituted solution is between 5.5 and 7.0.
…………………………………..
INTRODUCTION
Harringtonines 3 are particular cephalotaxanes formed by attachement of a branched hydroxyacyloxy side-chain at the 3-position of various cephalotaxines moieties. Harringtoriines are natural esters of cephalotaxines exhibiting generally a strong cytotoxic activity. However the lost only one atom of this minimal structure lead to a dramatic lost of activity (see below). Some example of harringtonines are harringtonine
3a, homoharringtonine 3b, drupangtonine 3c, anhydroharringtonine 3d and neoharringtonine 3e.
SCHEME 1 DEFINITION NOMENCLATURE AND NUMBERING OF CEPHALOTAXANES


Examples of harringtonines

Examples of cephalotaxines

Harringtonine 3a (n = 2) Anhydroharringtonine 3d Homoharringtonine 3b (n = 3)

(-)-Cephalotaxine 2a


Drupacine 2b Drupangtonine 3c Neoharringtonine 3e (n = 2)
…………………………………
The term “cephalotaxanes” refers to compounds or salts thereof which have a basic skeleton of formula

where p is equal to 1 or 2 (it being possible for the two units to be identical or different and linked via a single bond or an oxygen atom), which can contain various oxygenated substituents (aliphatic or aromatic ethers, free or esterified alcohols, substituted or free enols and/or phenols, bridged ethers, and more generally any substituent usually encountered in the natural state on compounds of this type).
Harringtonines are alkaloids which are of high interest in anticancer chemotherapy, in particular on certain haematosarcomas which are multi-resistant to the existing therapies. The selectivity of harringtonines, which is based on a novel mechanism of action relating to protein synthesis, is such that this series is favoured with a great future in anticancer therapy.
Several literature compilations give a seemingly exhaustive review of all of the knowledge relating to cephalotaxanes, these compilations being, chronologically: [C. R. Smith, Jr, R. G. Powell and K. L. Mikolajczack, Cancer Treat. Rep., Vol. 60, 1157 (1976); C. R. Smith, Jr, L. Kenneth, K. L. Mikolajczack and R. G. Powell in “Anticancer Agent Based on Natural Product Model”, 391 (1980); Liang Huang and Zhi Xue in “The Alkaloids”, Vol. XXIII (A. Brossi Ed.), 157 (1984); M. Suffness and G. A. Cordell in “The Alkaloids, Chemistry and Pharmacology” (A. Brossi Ed.), Vol. 25, 57-69, 295-298 (1’987); P. J. O’Dwyer, S. A. King, D. F. Hoth, M. Suffness and B. Leyland-Jones, Journal of Clinical Oncology, 1563 (1986); T. Hudlicky, L. D. Kwart and J. W. Reed, in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, 199 (1998)].
Antiparasitic activities, in particular on the haematozoon of malaria, have also been recognized [J. M. Whaun and N. D. Brown, Ann Trop. Med. Par., Vol. 84, 229 (1990)].
Homo-harringtonine (HHT), the most active member of the series, is active at and above daily doses of 2.5 mg/m2 of body area per 24 hours, i.e., as a guide, at doses twenty times lower than that for Taxol. HHT has already undergone fourteen phase I and II clinical trials and it is the only known product capable of a 70% reinduction of full haematological remissions in patients suffering from chronic myeloid leukaemias that have become resistant to alpha-interferon [S. O’Brien, H. Kantarjian, M. Keating, M. Beran, C. Koler, L. E. Robertson, J. Hester, M. Rios, M. Andreeff and M. Talpaz, Blood, 332 (1995); Leukemia Insights, Vol. 3, No. 1 (1998)].
Harringtonines were extracted over 35 years ago from an exclusively Asiatic cephalotaxacea known as Cephalotaxus harringtonia, following the programme of research into novel anticancer agents in the plant kingdom developed by the National Cancer Institute. In fact, the Cephalotaxus alkaloids consist essentially (at least 50%) of cephalotaxine, a biosynthetic precursor of the harringtonines, the latter individually representing only a few percent of the total alkaloids.
Besides their low concentration in the natural state in plant starting material, harringtonines are mixed with many congeners which have very similar chemical structures. Thus, in a high resolution high performance liquid chromatography (HPLC) chromatogram of a semi-purified alkaloid extract, no less than several tens of cephalotaxine esters are counted.
Numerous antileukemia drugs have been investigated but so far, there is no single drug that is effective and safe. As discussed in U.S. 3,497,593, an alkaloid from Tylophora plant is said to have antitumor activity against mouse leukemia (L-1210). U.S. 3,928,584 discloses an organic composition derived from tree sap and is said to have activity against mouse leukemia P-388. Also U.S. 4,431,639 discloses that an extract of Rhisoma Stractylis promotes the production of lymphocytes in the circulating blood, consequently eliminating cancer growth
-
Harringtonine or Homoharringtonine, hereinafter referred to as HH, has been known to be effective against acute chronic granulocytic and monocytic leukemia (Journal of Chinese Internal Medicine 3:162-164, 1978). However, it is highly toxic and causes damage to heart and hematopoietic organs. The results of experiments in animals, such as mice, rabbits and dogs, indicate that most of them die from cardiotoxicity after receiving the drug. Therefore, there is a need to improve the HH drug for safe use against leukemia. This drug is of special importance in that all known antileukemia drugs are effective against lymphatic leukemia and there are no effective drugs for treating nonlymphatic leukemia
All the literature from 1972 to the present date [Mikolajczack et al., Tetrahedron, 1995 (1972); T. Hudlicky, L. D. Kwart and J. W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S. W. Pelletier Ed.), Vol. 5, 639 (1987); M. A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxane 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3 totally preformed to give a harringtonine 4b, i.e. the conversion 2a+3e(4b as described in the example featured in the scheme below

- ……………………………………………………..
SYNTHESIS
Tetrahedron Lett 1982,23(34),3431, J Org Chem 1983,48(26),5321

The oxidation of 2-methyl-1-cyclopentene-1-carbaldehyde (I) with O3 and Ag2O gives 2,6-dioxoheptanoic acid (II), which is esterified with cephalotaxine (III) by means of (COCl)2, yielding the ester (IV). Reformatsky reaction of (IV) with methyl bromoacetate (V) and Zn affords the adduct (VI), which is treated with an excess of methylmagnesium iodide to provide the target homoharringtonine (as a single diastereomer), along with some starting cephalotaxine that is separated by chromatography.
………………………………
SYNTHESIS
EP 1064285; FR 2776292; WO 9948894, Tetrahedron Lett 1999,402931

The intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII) has been obtained by several related methods: 1. The Grignard condensation of 4-methyl-3-pentenyl bromide (I) with diethyl oxalate (II) in HF gives the 2-oxoheptenoate (III), which is condensed with methyl acetate (IV) by means of LiHMDS in THF to yield 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V).
The cyclization of (V) by means of Ts-OH in hot toluene or by means of hot aqueous formic acid affords 2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid ethyl ester (VI), which is hydrolyzed with KOH in boiling water to provide the corresponding dicarboxylic acid (VII). Finally, this compound is regioselectively monoesterified by means of BF3/MeOH in methanol to furnish the intermediate (racemic)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (VIII). 2.
The reaction of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with HCl in hot methanol gives 3-(ethoxycarbonyl)-3,7-dihydroxy-7-methyloctanoic acid methyl ester (IX), which is then cyclized by means of ZnCl2 in hot dichloroethane to yield the previously described intermediate (VIII). 3. The hydrolysis of 3-(ethoxycarbonyl)-3-hydroxy-7-methyl-6-octenoic acid methyl ester (V) with KOH in refluxing methanol/water gives the corresponding diacid (X), which is regioselectively monoesterified by means of BF3/MeOH in methanol to yield 3-carboxy-3-hydroxy-7-methyl-6-octenoic acid methyl ester (XI).
Finally, this compound is cyclized by means of Ts-OH in hot toluene to afford the previously described carboxylic intermediate (VIII). The racemic acid (VIII) is submitted to optical resolution by esterification with quinine (XII) by means of 2,4,6-trichlorobenzoyl chloride and TEA or DCC to give a diastereomeric mixture of esters (XIII) that is separated by preparative HPLC to obtain the desired diastereomer (XIV).
The hydrolysis of (XIV) with KOH in refluxing ethanol/water gives the corresponding chiral dicarboxylic acid (XV), which is regioselectively monoesterified with BF3/MeOH in methanol to yield the chiral (R)-2-(methoxycarbonylmethyl)-6,6-dimethyltetrahydropyran-2-carboxylic acid (XVI).
The esterification of (XVI) with cephalotaxine (XVII) by means of 2,4,6-trichlorobenzoyl chloride and TEA in toluene affords the corresponding ester (XVIII), which is treated with HBr in dichloromethane/HOAc, providing the bromoester (XIX). Finally, this compound is treated with NaHCO3, CaCO3 or BaCO3 in acetone/water to give the target hydroxyester.
………………………………………….
EXTRACTION
-
Throughout the specification, the concentration of the solvent is the same as first given unless stated otherwise. Redeuced pressure means about 2,27 kPa (17 mm Hg. abs), l is liter, kg is kilogram. ml is milliliter. Yield in weight %.
Example 1. HH is extracted from the skins, stems, leaves and seeds of Cephalotaxus fortunel Hook and other related species, such as Cephalotaxus sinensis Li, C. hainanensis, and C. wilsoniana, including C.oliveri mast and C.harringtonia. -
1 kg of finely ground Cephalotaxus fortunel Hook is extracted with 8 l of 90% ethanol at room temperature for 24 hrs. The solution is filtered to yield a filtrate A and filtercake. The filtercake is percolated with ethanol and filtered again to yield filtrate B. A and B are combined and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, 2% HCl is added to adjust the pH to 2.5. The solids are separated from the solution by filtration to yield a filtrate C. The solids are washed once with 2% HCl and filtered to yield a filtrate D. C and D are combined and the pH adjusted to 9.5 by adding saturated sodium carbonate solution. The alkaline filtrate is extracted with chloroform and the chloroform layer separated from the aqueous layer. This extration process is repeated five times. All the chloroform extracts are combined and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue respectively.
-
The solid alkaloid is then dissolved in 20 ml. of 6% citric acid in water. The solution is divided into three equal portions. These are adjusted to pH 7,8 and 9 by adding saturated sodium carbonate solution.
-
The portions having pH 8 and 9 are combined and extracted with chloroform. The chloroform extracts are distilled under reduced pressure, whereby chloroform is removed and recovered and a solid residue of crude Harringtonine is obtained.
-
The crude Harringtonine is dissolved in pure ethanol i.e. alkaloid : anhydrous ethanol 1:10 , and crystallized. The crystals are refined by recrystalliation in diethyl ether. Overall yield of Harringtonine is about 0.1% including yield from mixed HH from the subsequent process.
Harringtonine has the following chemical structure:
wherein R is

- melting point:
- 135° – 137°C
- crystal:
- colorless
- infrared spectrum:
- 3750, 1660, 1505, 1490, 1050, and 945 cm⁻¹.

-
The portion having a pH of 7 and the mother liquors from the foregoing crystallization of Harringtonine are combined and passed through a liquid chromatographic column of diameter to height ratio 1:50 packed with alumina. The column is finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids are mixture of HH. The mixed HH is then separated from each other by countercurrent distribution employing chloroform and pH 5 buffer. The first fraction of the countercurrent distribution is Homoharringtonine and the last fraction of the countercurrent distribution is Harringtonine. Homoharringtonine is purified by crystallization in methyl alcohol.
Homoharringtonine has the following chemical structure:
wherein R is

- yield:
- 0.02%
- melting point:
- 144° – 146°C
- infrared spectrum:
- 3500∼3400, 1750, 1665, 1030 and 940 cm⁻¹.







…………………………………………………………………………..
EXTRACTION
All the literature from 1972 to the present date [Mikolajczack et al.,Tetrahedron, 1995 (1972); T. Hudlicky, L.D. Kwart and J.W. Reed in “Alkaloid: Chemical and Biological Perspectives” (S.W. Pelletier Ed.), Vol. 5, 639 (1987); M.A. Miah, T. Hudlicky and J. Reed in “The Alkaloids”, Vol. 51, p. 236 (1998)] mention the impossibility hitherto of esterifying the highly sterically hindered secondary hydroxyl of cephalotaxine 2a with the tertiary carboxyl of the alkanoyl chain of harringtonic acid 3e totally preformed to give a harringtonine 4b , i.e. the conversion 2a + 3e ( 4b as described in the example featured in the scheme below

Example 46
Preparation of purified (-) cephalotaxine from total alkaloidic extract of Cephalotaxus sp
-
[0319]

-
Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica,; 23; 7; 835 (1980)]
-
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1 éq) showed the following results:
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).
Presence of 11 ± 5 % de (+)-cephalotaxine.
[α]22 = -134,0° (c = 0,214; CHCl3) : calculated rate 25 ± 5 % - Batch B: slightly racemized (1%)
[α]19 = -173,3° (c = 0,208; CHCl3)
- Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine) ; 5.99 (1H, OCH2O (-)-cephalotaxine) and 5.76 (1H, OCH2O (-)-cephalotaxine).

Enantiomeric enrichment of the natural cephalotaxine:
-
Crude chromatographied cephalotaxine (20g) was dissolved at 55°C in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity.
[α]20 D =-130° (C1, CHD3) corresponding to 10 % of racemization. The crystallized product thus obtained (20g) was dissolved again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-)-cephalotaxine [α]20 D= -185°(C1,CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20 = 0,5° (C1 ; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (-)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (-)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical evaluation of the enantiomeric purity of natural cephalotaxine:
-
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19.
The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11% ± 3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
Example 47Preparation of homoharringtonine, from anhydro-homoharringtonine:

1)° Method A
-
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at -10°C. After stirring at -10°C for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3 × 230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
-
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (214 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at -10°C a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at -10°C for 3 hours, was added water (13 ml) and then the temperature was raised to 20°C. After stirring at 20°C for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3 × 20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.




……………………
SEMISYNTHESIS
EXAMPLE 27 Preparation of homoharringtonine as a pharmaceutical use from crude semi-synthetic homoharringtonine resulting from example 25 by preparative high-performance liquid chromatography

1°) Method A
Crude homoharringtonine (35 g) is dissolved in buffer (triethylamine (1.55/1000) in deionised water and orthophosphoric acid to adjust pH to 3. The solution was filtered then injected on a preparative high-performance liquid chromatograph equipped with axial compression and high pressure pump (stationary phase: n-octadecylsilane, 15 μm, porosity 100, 1 kg; mobile phase; buffer/tetrahydrofurane 85/15). Elution was performed at a flow rate of 0.2 l/min. Fractions contain was monitored by U.V. detector and TLC. Retained fraction were finally checked by HPLC then combined, alkalinised with 2.5% aqueous ammonia and extracted with dichloromethane (4×400 ml). After concentration under reduced pressure homoharringtonine is obtained as a pale yellow resin which on trituration in a 8/2 water-methanol mixture gave pure homoharringtonine as a white crystalline solid (mp=127° C.), HPLC purity was higher than 99.8%.
2°) Method B
Same procedure of purification as method A was performed but mobile phase buffer/methanol (68/32) was used instead buffer/tetrahydrofurane.
3°) Method C
Same procedure of purification as method A was performed but mobile phase buffer/acetonitrile (85/15) was used instead buffer/tetrahydrofurane.
EXAMPLE 28 Preparation of homoharringtonine as a pharmaceutical use from semi-purified natural cephalotaxine
Crude homoharringtonine, prepared according to Example 25 from a partially racemized natural cephalotaxine and purified by chromatography and crystallisation according to the method A of Example 27, gave an homoharringtonine showing a non natural enantiomeric epi-homoharringtonine content less than 0.05%.
EXAMPLE 46 Preparation of purified (−) cephalotaxine from total alkaloidic extract of cephatotaxus sp

Partially racemized cephalotaxine [H. Wenkui; L. Yulin; P. Xinfu, Scientia Sinica; 23; 7; 835 (1980)]
1H NMR of two batches of cephalotaxine (extracted in the same conditions as above) with the optically active NMR shift reagent europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate (1éq) showed the following results:
Batch A: 1H NMR 400 MHz (CDCl3)(δ ppm): 6.06 (1H, OCH2O (+)-cephalotaxine) and 5.82 (1H, OCH2O (+)-cephalotaxine); 5.99 (1H, OCH2O (−)-cephalotaxine) and 5.76 (1H, OCH2O (−)-cephalotaxine). Presence of 11±5% de (+)-cephalotaxine. [α]22=−134,0°(c=0,214; CHCl3): calculated rate 25±5%
Batch B: slightly racemized (1%) [α]19=−173,3°(c=0,208; CHCl3)
Enantiomeric Enrichment of the Natural Cephalotaxine:
Crude chromatographied cephalotaxine (20 g) was dissolved at 55° C. in dry methanol (100 ml). Crystallization occurs by cooling with rotary evaporator and after filtration the product thus obtained showed 99.9% of HPLC purity, [α]20 D=−130°(C1, CHD3) corresponding to 10% of racemization. The crystallized product thus obtained (20 g) was dissolyed again in hot methanol (100 ml).
Slowly cooling the solution allows translucent prisms composed of pure enantiomeric (-−)-cephalotaxine [α]20 D=−185°(C1, CHCl3).
After filtration, the mother liquors was allowed to slowly evaporate at room temperature and crystals in the form of macled needles exclusively composed of racemic cephalotaxine [α]D 20=0,5°(C1; CHCl3) were obtained.
After filtration, the second mother liquors allowed prisms composed of (−)-cephalotaxine identical to this obtained at the first crystallization.
After filtration, the third mother liquors still allowed macled needles (urchins) composed of (±)-cephalotaxine.
The cycle is repeated three times. The combined prismatic crystals was recrystallized once to give enantiomerically pure (−)-cephalotaxine, while the combined macled needles treated in the same way gives 100% racemic cephalotaxine.
Chemical Evaluation of the Enantiomeric Purity of Natural Cephalotaxine:
A sample of partially racemized natural cephalotaxine was inserted in the process, which sequence is described in the Examples 1,2,3,4,5,6,15,19 and 21, by using a pure (2R)-homoharrintonic acid resulting from Example 19. The HPLC analysis of the diastereomeric mixture of anhydro-homoharrintonine thus obtained showed a significant enantio-epi-homoharringtonine rate (11%±3%) corresponding to the (+)-cephalotaxine content in the racemic mixture of origin (it has been demonstrated that the two antipodes of the homoharringtonic acid react in a stoechiometric way comparable to the pure enantiomeric cephalotaxine).
EXAMPLE 47
Preparation of homoharringtonine, from anhydro-homoharringtonine

1°) Method A
A commercial solution of hydrobromic acid in acetic acid (17.4 ml, 86.6 mmol, HBr 30% w/w) was added to a stirred solution of anhydrohomoharringtonine resulting from Example 21 (50.8 g, 9.63 mmol) in anhydrous dichloromethane (25.6 ml) at −10° C. After stirring at −10° C. for 3 hours was added water (240 ml) and the reaction mixture was become viscous. The temperature was allowed to rise to room temperature and after stirring for 2.5 hours was added sodium carbonate 0.76M (406 ml) to pH 8. The resulting aqueous layer was saturated with sodium chloride, then was extracted with dichloromethane (3×230 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness to afford a foam. After phase reverse chromatography below-mentioned were obtained 4.03 g of homoharringtonine (77%). The product thus obtained showed identical characteristics to this resulting from Example 25.
2°) Method B
To a stirred solution of anhydrohomoharringtonine resulting from Example 21 (21.4 mg, 0.406 mmol) in anhydrous dichloromethane (1.1 ml) was added at −10° C. a commercial solution of hydrobromic acid in acetic acid (0.728 ml, 3.6 mmol, HBr 30% w/w). After stirring at −10° C. for 3 hours, was added water (13 ml) and then the temperature was raised to 20° C. After stirring at 20° C. for 3 hours, was added a sodium carbonate solution (0.76M; 31.5 ml) up to pH 8. The resulting aqueous layer, after saturation with sodium chloride, was extracted with dichloromethane (3×20 ml) and the combined organic layers were dried over magnesium sulfate and evaporated to dryness. The resulting crude product was purified by phase reverse chromatography below-mentioned to provide homoharringtonine (166 mg, 75%). The product thus obtained showed identical characteristics to this resulting from Example 25.
…………………………………
EXTRACTION
The remarkable clinical efficacy of Homoharringtonine (HHT) resulting in lot of observations of complete remission of leukemia and other solid cancer in human being since 1988. Recently, research articles reported that the HHT efficacy in glaucoma, inhibition of Hepatities B virus replication and using in bone marrow transplantation. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” [Susan O’Brien, at al.; Sequential homoharringtonine and interferon-α in the treatment of early chronic phase chronic myelogenous leukemia; Blood, Vol 93, No 12 (June 15), 1999: pp 4149-4153]. Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.” (Susan O’Brien, at al.; Simultaneous homoharringtonine and interferon-α in the treatment of patients with chronic-phase chronic myelogenous leukemia; American Cancer Society; Apr. 1, 2002, Vol 94, No. 7).
On Nov. 8, 1988, U.S. Pat. No. 4,783,454 titled Process for producing harringtonine and homoharringtonine disclosed the technique of isolation of a purified HHT from bark of Cephalotaxus. However, the natural source ofCephalotaxus is very limited. Trees of Cephalotaxus grow slowly. Bark ofCephalotaxus has very low content of HHT. Extracting HHT from bark ofCephalotaxus the yield was about 0.02% only. More important to harvest bark ofCephalotaxus will kill and destroy trees. Supply of HHT is very short now. Therefore, it is necessary to find a new manufacturing method.
DETAILED DESCRIPTION
Great progress has been made in research on Homoharringtonine (HHT) production and on future generation HHT drug since 1988. For example, the University of Texas M.D. Anderson Cancer Center and National Cancer Institute reported that “Ninety-two percent of patients achieved CHR with HHT.” Another article reported that “the median number of days on HHT per month was 2 days with a median follow-up of 26 months; the estimated 2-year survival rate was 90%.”
The good clinical results of HHT in treating cancer brought to the major problem, which is the supply of HHT both short term and long term. It is apparent that a huge amount of bark of Cephalotaxus is needed for collection, extraction and purification of HHT. It is clear that due to the slow growth of the trees ofCephalotaxus, which is a nature source of HHT, and the killing of trees by harvesting bark is not a sustainable resource for HHT production.
Present invention disclosed new methods for producing HHT. The new methods of producing HHT are shown as follows.
1. Tissue Culture (Plant Cell Culture):
Culture manipulation to promote secretion of HHT is a new way for an extracellular product HHT. The biosynthetic methods can yield more HHT through precursor of HHT feeding. The production of HHT increased significantly after the addition of the precursors and special biochemical agents. Content of precursor of HHT abounds in tree and it is very cheap. The present methods include several significant developments in technique of culture plant tissues that are
-
- (a) yields of HHT selected from rapid growth, resistance to infections organisms; and
- (b) HHT can excrete into media.
Traditional method of plant culture is very difficult to overcome the problem of high cost. Therefore, traditional method appears too long to have commercial value. HHT is secondary metabolite of Cephalotaxus. Secondary compound acts in defense against the harmful effects of toxins, carcinogens or mutagens found in the plant. In fact, traditional method is very difficult to increase HHT contenting in plant tissues. The present new method uses a special biochemical agent for increasing content of HHT and more easily to purify HHT from other metabolites.
More important is that the key of the present new technique for producing high content of HHT in plant cell culture is to increase production of HHT by directed fermentation through precursor of HHT feeding. The present new methods are used special metabolite of Cephalotaxus for markedly enhance production of HHT. Therefore, the present invention disclosed a new source for the long term of producing HHT.
2. Using Precursor of HHT:
Recent research’s results have established that direct production of HHT from its precursor and advances in biosynthetic understanding for HHT metabolism. Biosynthesis or semisynthesis of HHT from major nonactivity ingredients is well established through great advances in special biochemistry reactions. Using precursor of HHT for semisynthesis and increase of production in plant cell culture are new developing methods for producing HHT.
3. Using Leaves:
Our new method use leaves of tree of Cephalotaxus not use the bark. So far, the extraction of HHT is used bark. The leaves are harvested from the trees ofCephalotaxus, which grow in mountains of South China. The natural source of leaves is very abundance. The new methods do not use bark. Therefore, it can avoid destroy trees. The natural source of Cephalotaxus tree is very limited and slow growing. In fact, bark of Cephalotaxus has very low yield of HHT. The yield of HHT from Cephalotaxus bark is about 50-100 mg/kg of dried bark. The present new method, therefore, has a great economic and environmental value.
4. Semisynthesis:
HHT has received important chemical studies particularly in regard to structure and anticancer activity relationship and semisynthesis.
A great progress in biochemistry allows semisynthesis to use precursor of HHT from leaves of Cephalotaxus and to produce HHT. The total chemical synthesis of HHT appears too long to have commercial value too. Semisynthesis method can yield a high efficient conversion of precursor to HHT. It is other better biological source for manufacturing HHT. This new method uses closing chemical analogues to convert to HHT. This analogue is produced from leaves or other organ of Cephalotaxus. The present invention disclosed that new methods and techniques of manufacturing HHT could avoid chopping down Cephalotaxus trees which governmental environmentalists are trying to have declared a threatened species.
5. Using Taxol Residual
The anticancer drug Taxol is the most promising new chemotherapeutic agents that developed for cancer treatment in the past twenty years. Taxol has a unique mechanism of action. It has been shown to promote tubulin polymerization and stabilize microtubules against depolymerization. The FDA approved the clinical use of Taxol for several types of cancer. So far, annual sales of Taxol are more than $2 billion in market. Taxol is extracted from bark or leaves of an evergreen tree named Taxus species including Taxus brevifolia (or called Pacific yew). After Taxol has been extracted from bark or leaves, all residual materials of Taxus brecifolia named Taxus residual, which are waste.
Both taxol and HHT can be extracted from yew tree. The content of taxol is less than 0.01% in yew tree. The content of HHT in yew tree is about 0.01% -0.22%. The content of HHT is much higher than content of Taxol. Taxol extracted from bark of yew is difficult and expensive. One reason is that the presences of closely related congeners are similar to Taxol. A major congener is Cephalomannine (CPM), which is a waster of process in manufacturing of Taxol.
The chemical and physical characters are very close between Taxol and Cephalomannine (CPM).
CPM characterized by the same ring structure as Taxol and distinguishes from them only in C-13 ester structure. The present invention disclosed that CPM and related derivative are used to produce HHT.
The following specific examples will provide detailed illustrations of methods of producing relative drugs, according to the present invention and pharmaceutical dosage units containing demonstrates its effectiveness in treatment of cancer cells. These examples are not intended, however, to limit or restrict the scope of the invention in any way, and should not be construed as providing conditions, parameters, reagents, or
EXAMPLE 1
Production of HHT by Culture Cells
So far, HHT is extracted from bark and skins of Cephalotaxus species. However, growth of Cephalotaxus species is very slow and concentration of HHT in plant is extremely low. Furthermore, it is difficult to harvest the plants because of their low propagation rate and the danger of drastic reduced in plant availability. Also, cost of total chemical synthesis of HHT is very expensive and is not available for commerce now. For the reasons given above it is more difficult to obtainCephalotaxus on a large scale for long time. Therefore, Cephalotaxus cell cultures are one of best methods for obtaining HHT. In this present invention, special elicitation is disclosed and it will significantly increase production of HHT.
The methods of cell and tissue culture are disclosed as below.
Parts of bark, stems, leaves, or roots of Cephalotaxus species were surface disinfected by treatment in 70% ethanol for 10 minutes and followed by 0.1 HgCl2for 3 minutes. Plant materials were washed five times for 10 minutes each by sterilized water. Parts of plant were cut into small pieces (0.5-1 mm) and put pieces to Murashige and Skoog’s (MS) medium and supplemented with derivative of new active ingredient of phylum mycota (IPM), precursor of HHT which is a derivative of Cephalotaxus (CEP), tyrosine (TYR) naphthaleneacetic acid (NAA), Kinetin (3 mg/L), and 3% sucrose (w/v). PH of medium was adjusted to 5.7˜5.8. Agar (10 g/L) added to medium. Callus tissues are collected from agar media and suspension cultured cells were harvested by filtration and cultured in MS medium.
The cultures were kept in a culture room at 26° C.±1° C. Friable callus tissues were obtained. The callu was inoculated into 4 L of MS liquid medium containing sucrose, derivative of CEP, PHE, TYR, NAA and Kinetin. Then callus tissues were cultivated 26° C. for 35 days on rotary shaker operated at 120 rpm in the dark. Cells were subcultured into fresh medium of same composition every 2 weeks and maintained at 120 rpm at 26°±1° C. Packed cell volume (PCV), fresh weight (FW), dry weight (DW), concentration of HHT and concentration of sugar were determined every 5th day. The cells were harvested and dried.
In general, callus and suspension cultures of cephalotaxus species grow very slow and no production of free or esterified HHT. However, according to the present invention, addition of IPM to cultures cause a drastic increasing in HHT after 30 days of incubation. For example, in control group (no IPM), HHT in cultured cells is 0.020 mg/g dry weight, but in treatment group (addition of IPM) HHT is about 0.050 mg/g dry weight. Therefore, IPM can increase 250% of content of HHT. It has resulted in plant cell culture systems that producing HHT at concentration higher than those produced by the mother plant. The production of HHT increases significantly after the addition of precursors (CEP). Addition of CEP can increase HHT. Obviously, the present invention provided a new commercial and economic method for producing HHT. The IPM and precursors (CEP) play key role in cultured cells.
EXAMPLE 2
Semi-Synthesis of HHT
HHT shows a significant inhibitory activity against leukemia and other cancer. Concentration of HHT, however, has only 0.01% in natural sources. Cephalotazine (CEP) is major alkaloids present in plant extracts and the concentration ofCephalotaxus has about 1%. Therefore, concentration of CEP is about 100 times higher then HHT in nature plant sources. But CEP is inactive. For the reason given above, semisynthesis of HHT from CEP will increase huge natural sources of HHT.
-
- (1) Extraction of CEP
10 kg of dried stems or leaves or roots of Cephalotaxus species were milled, placed in a percolator, along 80 L of 95% of ethanol, and allowed to stand 24 hours. The ethanol was recovered under reduced pressure (below 40° C.). 20 L of 5% tartaric acid was added to concentrated ethanol solution. The ammonia water was added to the acidic solution and adjusted pH to 9. The solution of pH 9 was filtered and yielded a filtrate. The filtrate was extracted with CHCl3. CHCl3 was recovered under reduced pressure and residue was obtained. The residue was chromatographed packed with alumna and eluted by CHCl3-MeOH (9:1). Eluate was concentrated under reduced pressure. Residue was dried under vacuum. The product is CEP.
-
- (2) Semisynthesized HHT from CEP
Materials and Methods
Melting points were determined on a Fisher-Johns apparatus. Infrared spectra were obtained on a Perkin-Elmer 567 infrared spectrophotometer or on a Beckman 4230 IR spectrophotometer. Peak positions were given in cm−1. The IR spectra of solid samples were measured as potassium bromide dispersions, and the spectra of liquids were determined in chloroform or carbon tetrachloride solutions. NMR spectra were measured on a Varian A-60, Perkin-Elmer R-32, Varian EM-390, or Brüker WH-90 NMR spectrometer. Chemical-shift values were given in parts per million downfield from Me4Si as an internal standard. Mass spectra were run on an AE1 MS-12 Finnigan 3300, or CEC21-110B mass spectrometer.
Preparative thin-layer chromatography was accomplished using 750-μm layers of aluminum oxide HF-254 (type E), aluminum oxide 60 PF-254 (type E), silica gel HF-254 (type 60 PF-254), or silica gel GF-254. Visualization was by short-wave ultraviolet light. Grace silica gel, Grade 923, and Woelm neutral aluminum oxide, activity III, were used for column chromatography. Analytical thin-layer chromatography was run on plastic sheets precoated with aluminum oxide F-254 neutral (type T), 200-μm thick, and on Polygram Sil G/UV254 (silica gel), 250 μm on plastic sheets. Visualization was usually by short-wave ultraviolet light, phosphomolybdic acid, or iodoplatinate.
Preparation of α-Ketoester-Harringtonine
1 g of Benzene-α-acetone Na was put into 10 L of benzene. Mixture was stirred at room temperature then was dissolved in 10 L of pyridine and stirred at 0° C. Oxalic chloride was added from a dropping funnel to solution of pyridine. Stirring was continued while the solution warmed to room temperature and stand overnight. Excess reagent was removed. This solution was dissolved in CH2Cl2and cooled to near 0° C. in an ice water bath. 5 g of CEP, 2.5 L of CH2Cl2 and 2.5 L of pyridine were added to cold CH2Cl2 solution. Manipulations were done in a dry N2 atmosphere and all glassware heat-dried just before use. The suspension was stirred at room temperature and overnight. The mixture was washed with 10% Na2CO3 and saturated aqueous NaCl, then dried with auhydrous magenesium sulfate, and filtered and the solvents were removed in vacuo. Evaporation provided as an amorphous solid α-ketoester-harringtonine (mp 143˜145° C.).
Semi-Synthesis of HHT
10 L of CH3CHBrCOOEt and activated zin dust and THF were added to the α-ketoester-harringtonine (at −78° C.) for 6 hours followed by slow warming to room temperature with stirred. The reaction mixture was diluted with 10 L CHCl3 and 10 L H2O and solid Na2CO3 was added. CHCl3 was evaporated under reduced pressure and residue was obtained.
The residue was purified by chromatography on alumina. The column was flushed with chloroform and followed by chloroform-methanol (9:1). The solvents were recovered under reduced pressure to provide as a solid. Solid was dissolved in pure ethanol and crystallized. The crystals were refined by recrystalization in diethyl ether. The crystals dried under vacuum. The product is HHT, which has the following characters:
[α]D −119° (C=0.96),
MSm/e (%): 689 (M+, 3), 314 (3), 299 (20), 298 (100), 282 (3), 266 (4), 20 (3), 150 (8), 131 (12), 73 (18)
EXAMPLE 3
HHT Extracted from Plant Tissue
Extraction of HHT has several major methods which including extraction by organic solvent, chromatograph and adjust pH.
HHT was extracted from plant tissue culture, plant cells or leaves of Cephalotaxusspecies.
1 kg of ground Cephalotaxus fortunei Hook was extracted with 10 liters of water at room temperature for 24 hrs. To filtered the solution to yield a filtrate. Ten liters of 90% ethanol added to filtrate. The mixture was Centrifugalized to yield a sediment. Percolated the sediment with ethanol and filter again to yield filtrate, combined filtrates, and distilled under reduced pressure to recover ethanol and an aqueous residue. To this residue, added 10% of HCl to adjust the pH to 2.5. To separated the solids from the solution by filtration to yield a filtrate (1). Washed the solids once with 2% HCl and filtered to yield a filtrate (2). Combined (1) and (2) and adjusted the pH to 9.5 by adding saturated sodium carbonate solution. Extracted the alkaline filtrate with chloroform and separated the chloroform layer from the aqueous layer. To repeated this extraction process five times. Combined all the chloroform extracts and distilled at reduced pressure to recover chloroform and alkaloid as a solid residue obtained. The solid alkaloid was then dissolved in 6% citric acid in water. The solution was divided into three equal portions. These were adjusted to pH 7, 8 and 9 by adding saturated sodium carbonate solution. The portions having pH 8 and 9 were combined and extracted with chloroform. The chloroform extracts were distilled under reduced pressure, whereby chloroform was removed and recovered and crude HHT was obtained. The crude HHT was dissolved in pure ethanol and crystallized. The crystals were refined by recrystallization in diethyl ether. The crude HHT obtained.
The portion having a pH of 7 passed through a liquid chromatographic column packed with alumina of diameter to height 1:50. The column was finally flushed with chloroform and followed by chloroform-methanol of 9:1 mixture. The resulting alkaloids were mixture crude of HHT. Combined crude HHT and then separated from each other by countercurrent distribution employing chloroform and pH 5 buffers. The first fraction of the countercurrent distribution was HHT. HHT was purified by crystallization in methyl alcohol. The crystallization was purified by recrystallization in methyl alcohol and dried under vacuum.
…………………….
Example 1 : Preparation of harringtonine drug substance by purification of commercial natural harringtonine
A. Analytical profile of starting product
By combination of HPLC analysis with UV detection (see Figure 6) and mass spectrometry detection (see figure 7 and 8) a total of 6.5% of related compound (identified as b,c: position isomer of harringtonine = 3.4%; d: homoharringtonine = 3%; e: 4′-demethyl harringtonine = 0.01%; f: drupacine derivative: 0.05%) are found in the starting product.
B. Chromatography of natural harringtonine
Natural harringtonine (5 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 80 mm; length: 1000 mm) containing 1000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi). Unwanted fractions are discarded based upon in-line UV spectrophotometric detection. Kept fractions are collected in 16 separate containers which each are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase). During the development phase, a dual in-line UV-MS detection is used. After discarding of the fractions representing more than 0.5 % of the total content of harringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure. Then crude concentrated solution of harringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 1.5 %. C. Crystallization of raw harringtonine
Under a laminar flow hood, the above raw harringtonine (4.1 grams) is dissolved in methanol (5ml), at 30°C. The resulting alcoholic solution was filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized rotary flask. Then, desionized water (50mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of harringtonine is kept under vacuum and rotation is continued during appearance of white crystals of pure harringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (10 mL x 2). The white translucent crystals are then dried using high vacuum at 40°C for 24 hours. Overall yield is 76%. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 400 therapeutic units dosed at 10mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 7 and 9 compare HPLC chromatogram before and after purification in using this process. Table II shows the comparison of the corresponding related compound content.

For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC) thermogravimetry, 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 5 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline harringtonine obtained by this process. A series of sharp absorption bands are noted at 615, 654, 674, 689, 709, 722, 750, 761 805, 850, 928, 989, 1022, 1033, 1062, 1083, 1112, 1162, 1205, 1224, 1262, 1277, 1308, 1340, 1364, 1382, 1438 1486, 1508, 1625, 1656, 1725, 1745, 2883, 2936, 2972, 3079, 3353, 3552 and 3647 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG) Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 12 mg of harringtonine drug substance were accurately weighed (12.4471 mg) into a DSC pan. The sample was heated from 25°C to 200°C at a rate of 10°C/min. The DSC data were obtained following a standard method in the art. The DSC curve of crystalline harringtonine drug substance ((Figure 4), exhibits a melting endotherm at 79.5 °C . No subsequent decomposition occurred under the upper tested temperature 200°C. Simultaneous TG measurement, indicated a loss on drying of 1.3 % which did not correspond to a lost of structural molecule of solvent or water.
Example 2: Preparation of homoharringtonine drug substance by purification of raw semi- synthetic (hemi-synthetic) homoharringtonine
A. Analytical profile of starting product
Crude reaction mixture of raw homoharringtonine contains a potential of 250 grams of homoharringtonine DS together with process impurities such as catalyst, unchanged starting product (anhydro-homo-harringtonine), and some related side product. HPLC analysis with UV detection (see left-side chromatogram on Figure 10) indicated a total of 9 % of related impurities. B. Chromatography of semi-synthetic homoharringtonine
Raw semi-synthetic homoharringtonine (550 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 450 mm; length: 1000 mm) containing 48,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 540 L/hour). Unwanted fractions are discarded based upon by- passed in-line UV spectrophotometric detector. Kept fractions are collected in 30 separate stainless steel containers (20 or 50 L each) which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 % (see rigth-side chromatogram on Figure 10)
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above raw homoharringtonine DS (210 grams) is dissolved in methanol (240 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (2400mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs. Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water (450 mL x 2). The white cryitals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 88% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current Good Manufacturing Practices were applied. This clinical batch corresponds to 40,000 therapeutic units dosed at 5mg.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 11 shows HPLC chromatogram before and after crystallization. Total of related impurities of homoharringtonine DS is 0.03%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry.
Infrared Spectrometry:
Identical IR spectra were obtained by either the KBr pellet and/or mineral oil mull preparation technique. Figure 3 shows typical infrared spectrum (KBr) for unambiguous identification at the solid state of the crystalline homoharringtonine obtained by this process. A series of sharp absorption bands are noted at 612, 703, 771 , 804, 826, 855, 879, 932, 1029, 1082, 1119,
1135, 1161 , 1191 , 1229, 1274, 1344, 1367, 1436, 1457, 1488, 1505, 1653, 1743, 2814, 2911 ,
2958, 3420, and 3552 cm“1
Differential Scanning Calorimetry (DSC) And Thermogravimetry (TG)
Measurement of DSC and TG were obtained on a Mettler Toledo STAR System. Approximately 11 mg of homoharringtonine drug substance were accurately weighed (10.6251 mg) into a DSC pan. The sample was heated from 25°C to 250°C at a rate of 5°C/min. The
DSC data were obtained following a standard method in the art. The DSC curve of crystalline homoharringtonine drug substance (Figure 1), exhibits a melting endotherm at 145.6 °C.
Melting range performed by the capillary method (Bucchi Apparatus) gave 143-145°C. Literature indicated 144-146°C [Anonymous, Acta Bot. Sin. 22, 156 (1980) cited by L. Huang and Z. Xue, Cephalotaxus Alkaloids, in “The Alkaloids”, vol. XXIII, pp157, (1988).
Crystallization medium was not published. This is the only literature reference regarding melting point of a crystalline form of HHT] X-Ray Powder Diffraction
X-ray powder diffraction pattern was collected on a INEL microdiffractomer, model
DIFFRACTINEL. Powdered homoharringtonine DS was packed in a glass capillary tube and was analyzed according to a standard method in the art. The X-ray generator was opered at 45 kV and 40 mA, using the copper Kalpha line as the radiation source. The sample was rotated along the chi axis and data was collected between 0 and 120 deg 2-theta. A collection time of 1200 sec was used. As showed on Figure 2, the x-ray powder diffraction for this crystalline form of homoharringtonine shows a typical pattern including major reflection peaks at approximately 7.9, 9.2, 10.9, 14.9 16.0, 17.7, 19.5, 19.7, 21.78, 23.1 , 25.3, 25.4 and 25.7 deg 2-theta.
Example 3: Preparation of homoharringtonine drug substance by purification of a commercial sample of impure homoharringtonine from Chinese source
A. Analytical profile of starting product
Analytical HPLC chromatogram of natural homoharringtonine (China National Pharmaceutical) is displayed on Figure 12 (bottom left).
B. Chromatography of Natural Homoharringtonine
Natural homoharringtonine (25 grams) is injected on a preparative high-pressure liquid chromatography (HPLC) system (Prochrom stainless steel; permanent axial compression; diameter: 200 mm; length: 1000 mm) containing 12,000 grams of reverse phase octadecylsilane specially dedicated for basic compounds as stationary phase. Then elution is performed in using a gradient of pH 3 buffered methanol-water solution as mobile phase (pressure 1200 psi, flow-rate 120 IJhour). Unwanted fractions are discarded based upon bypassed in-line UV spectrophotometric detector. Kept fractions are collected in 22 separate stainless steel containers which are individually checked in using an analytical HPLC system exhibiting a different selectivity pattern (octadecylsilane as stationary phase and buffered acetonitrile-water system as mobile phase) and equipped with a diode array detector. After discarding of the fractions representing more than 0.5 % of the total content of homoharringtonine, fractions which complied with pre-established specification were gathered, neutralized then evaporated under reduce pressure in using a mechanically stirred thin film evaporator. Then crude concentrated solution of homoharringtonine are alkalinized at pH 8.5 with aqueous ammonia and partitioned with dichloromethane. Resulting organic solution is concentrated under high vacuum. In-process HPLC analysis indicated a total of related compound lower than 0.5 %.
C. Crystallization of homoharringtonine DS
In a controlled clean room, under a laminar flow hood, the above chromatographied homoharringtonine DS (18 grams) is dissolved in methanol (35 mL), at 30°C. The resulting alcoholic solution is filtered on a 0.25 μ sterile Millipore filter to remove microparticules and germs and collected in a sterilized pilot rotary flask. Then, desionized water (300 mL) is added and methanol is completely removed under vacuum at 30°C in using a decontaminated pilot rotary evaporator. After removing methanol, heating is stopped and the aqueous solution of homoharringtonine DS is kept under vacuum and rotation is continued during appearance of white crystals of pure homoharringtonine. The stirring is continued until no more crystal occurs.
Under a laminar flow hood, the suspension of is poured on a sintered glass filter with house vacuum. The resulting crystalline solid cake is washed two times with cold desionized water
(50 mL x 2). The white crystals are then dried using high vacuum at 60°C for 48 hours. Overall yield is 84% from potential content of homoharringtonine in raw semi-synthetic homoharringtonine. All operations were documented prior to start the process and full current
Good Manufacturing Practices were applied.
D. Analysis
Routine analytical procedure includes solvent residues, loss on drying, water determination, melting point, IR and NMR spectrum, related compound and assay by HPLC. Figure 12 (bottom right) shows HPLC chromatogram after crystallization. Total of related impurities of homoharringtonine DS is 0.05%.
For the aim of further characterization, more advanced studies were performed including differential scanning calorimetry (DSC), thermogravimetry (TD), 2D NMR, solid NMR and X-ray powder diffractometry. Infrared Spectra, Differential Scanning Calorimetry (DSC) and X-Ray Powder Diffraction gave patterns strictly superimposable to the one of example 2 obtained from semi-synthetic homoharringtonine (Figure 3, 1 , and 2, respectively).
………………………………….
KOREAN PAPER.. LINK
Title: 한국산 개비자(Cephalotaxus koreans)에서의 Harringtonine과 Homoharringtonine의 확인 및 함량 분석
Author: 박호일 ; 이연 (한국생물공학회)
Source: 한국생물공학회지 = Korean journal of biotechnology and bioengineering; ISSN:1225-7117 @ 1225-7117 @ ; VOL.11; NO.6; PAGE.689-695; (1996)
Pub.Country: Korea
Language: Korean
Abstract: Harringtonine and homoharringtonine known as anti-cancer agents were isolated from Korean native plumyew(Cephalotaxus koreana) using column chromatography(CHCl3:MeOH=19:1, Rf=0.28). The structure of the mixture of two compounds was characterized by 1H-NMR. Comparison of our spectra of harringtonine and homoharringtonine with previously reported ones indicated that the two are identical. The contents of harringtonine and homoharringtonine in the needles, stems, and roots of Korean native plumyew were determined by high performance liquid chromatography(HPLC). The contents of both compounds varied with the site of location and the part of plant. The content of harringtonine was higher in needles and roots than in stems, whereas the content of homoharringtonlne was lower than harringtonine. Homoharringtonine contents in needles at Mt. Palgong, Mt. Dukyu, Mt. Baekyang, Mt. Jiri, and Namhae were higher than in stems and roots. But homoharringtonine contents in needles al Mt. Jokye and Jindo were lower than in stems and roots.
http://img.kisti.re.kr/originalView/originalView.jsp
……………………………………………………………………………….
SYNTHESIS OF HOMOHARRINGTONINE AND SEPARATION OF ITS STEREOMERS
-
[PDF]
Chapter 1 Drug Discovery from Plants – Springer
LC-NMR-MS and LC-SPE-NMR to accelerate their future discovery. Keywords …..Ceflatonine (34), a synthetic version of homoharringtonine produced by.
…………………………………………………………………………….
References
- “Synribo (omacetaxine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 18 February 2014.
- “SYNRIBO (omacetaxine mepesuccinate) injection, powder, lyophilized, for solution [Cephalon, Inc.]”. DailyMed. Cephalon, Inc. October 2012. Retrieved 18 February 2014.
- Sweetman, S, ed. (14 November 2012). Omacetaxine Mepesuccinate. “Martindale: The Complete Drug Reference”. Medicines Complete(Pharmaceutical Press).
- Li, Y. F.; Deng, Z. K.; Xuan, H. B.; Zhu, J. B.; Ding, B. H.; Liu, X. N.; Chen, B. A. (2009). “Prolonged chronic phase in chronic myelogenous leukemia after homoharringtonine therapy”. Chinese medical journal122 (12): 1413–1417. PMID 19567163. edit
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. (2009). “Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009”. Cancer 115 (23): 5382–5393.doi:10.1002/cncr.24601. PMID 19739234. edit
- Wu, L.; Li, X.; Su, J.; Chang, C.; He, Q.; Zhang, X.; Xu, L.; Song, L.; Pu, Q. (2009). “Effect of low-dose cytarabine, homoharringtonine and granulocyte colony-stimulating factor priming regimen on patients with advanced myelodysplastic syndrome or acute myeloid leukemia transformed from myelodysplastic syndrome”. Leukemia & Lymphoma50 (9): 1461. doi:10.1080/10428190903096719. edit
- Gu, L. F.; Zhang, W. G.; Wang, F. X.; Cao, X. M.; Chen, Y. X.; He, A. L.; Liu, J.; Ma, X. R. (2010). “Low dose of homoharringtonine and cytarabine combined with granulocyte colony-stimulating factor priming on the outcome of relapsed or refractory acute myeloid leukemia”.Journal of Cancer Research and Clinical Oncology 137 (6): 997–1003.doi:10.1007/s00432-010-0947-z. PMID 21152934. edit
- Kantarjian, H. M.; Talpaz, M.; Santini, V.; Murgo, A.; Cheson, B.; O’Brien, S. M. (2001). “Homoharringtonine”. Cancer 92 (6): 1591–1605.doi:10.1002/1097-0142(20010915)92:6<1591::AID-CNCR1485>3.0.CO;2-U. PMID 11745238. edit
- Wetzler M, Segal D. Omacetaxine as an Anticancer Therapeutic: What is Old is New Again. Current Pharmaceutical Design 2011;17:59-64
- Concise total synthesis of (±)-cephalotaxine via a transannulation strategy: Development of a facile reductive oxy-nazarov cyclization
Org Lett 2011, 13(13): 3538 - The first semi-synthesis of enantiopure homoharringtonine via anhydrohomoharringtonine from a preformed chiral acyl moiety
Tetrahedron Lett 1999, 40: 2931 - Synthesis of homoharringtonine and its derivative by partial esterification of cephalotaxine
Tetrahedron Lett 1982, 23(34): 3431 - Construction of chiral tertiary alcohol stereocenters via the (2,3)-Meisenheimer rearrangement: Enantioselective synthesis of the side-chain acids of homoharringtonine and harringtonine
J Org Chem 2013, 78(2): 339 - Studies in Cephalotaxus alkaloids. Stereospecific total synthesis of homoharringtonine
J Org Chem 1983, 48(26): 5321 - Chemistry – A European Journal, 2008 , vol. 14, 14 pg. 4293 – 4306
| WO2000040269A2 * | Jan 5, 2000 | Jul 13, 2000 | Clarence C Lee | Pharmaceutical compositions for treatment of diseased tissues |
| WO2002032904A1 * | Oct 17, 2000 | Apr 25, 2002 | Oncopharm Corp | New cephalotaxanes, their method of preparation and their use in treatment of cancers, leukemias, parasites including thus resistant to usual chemotherapeutic agents and as reversal agents |
| EP0393575A1 * | Apr 17, 1990 | Oct 24, 1990 | G.D. Searle & Co. | Neoplasia treatment compositions containing antineoplastic agent and side-effect reducing protective agent |
| USH271 * | Dec 18, 1985 | May 5, 1987 | The United States Of America As Represented By The Secretary Of The Army | Treatment of malaria with esters of cephalotaxine |
| US7169774 | Jun 25, 2004 | Jan 30, 2007 | Stragen Pharma S.A. | Cephalotaxane derivatives and their processes of preparation and purification |
| US7842687 | May 25, 2006 | Nov 30, 2010 | Chemgenex Pharmaceuticals, Inc. | Cephalotaxane derivatives and their processes of preparation and purification |
| US8466142 | Mar 3, 2009 | Jun 18, 2013 | Sloan-Kettering Institute For Cancer Research | Cephalotaxus esters, methods of synthesis, and uses thereof |
| Reference | ||
|---|---|---|
| 1 | * | KANTARJIAN H.M. ET AL: “Chronic myelogenous leukemia – Progress at the M. D. Anderson Cancer Center over the past two decades and future directions: First Emil J Freireich Award Lecture.” CLINICAL CANCER RESEARCH, (1997) 3/12 II (2723-2733). , XP001095529 |
| 2 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 1. Pharmacokinetic study in dogs and HHT determination in blood in using LC-MS method.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 179B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095449 |
| 3 | * | LEVY, VINCENT (1) ET AL: “Subcutaneous homoharringtonine (SQ HHT ): 2. Tolerance in humans and case report of a refractory patient with AML treated by very small dose of SQ HHT.” BLOOD, (NOVEMBER 16, 2001) VOL. 98, NO. 11 PART 2, PP. 202B. HTTP://WWW.BLOODJOURNAL.ORG/. PRINT. MEETING INFO.: 43RD ANNUAL MEETING OF THE AMERICAN SOCIETY OF HEMATOLOGY, PART 2 ORLANDO, FLORIDA, USA DECEMBER 07-11, 2001 , XP001095450 |
| 4 | * | WHAUN J M ET AL: “TREATMENT OF CHLOROQUINE -RESISTANT MALARIA WITH ESTERS OF CEPHALOTAXINE HOMOHARRINGTONINE.” ANN TROP MED PARASITOL(1990) 84(3), 229-237, XP008006193 |
1H NMR

13 CNMR

HPLC

TASIMELTION…FDA Approves Hetlioz: First Treatment for Non-24 Hour Sleep-Wake Disorder in Blind Individuals

TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
VEC-162, BMS-214778, 609799-22-6, Hetlioz, UNII-SHS4PU80D9,

January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
Tasimelteon
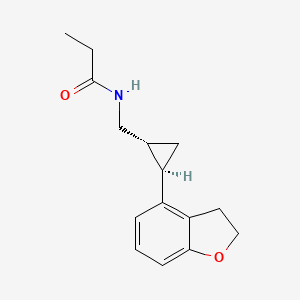
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
References
- ‘Time-bending drug’ for jet lag. BBC News. 2 December 2008
- Vachharajani, Nimish N., Yeleswaram, Krishnaswamy, Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Shantha MW Rajaratnam, Mihael H Polymeropoulos, Dennis M Fisher, Thomas Roth, Christin Scott, Gunther Birznieks, Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet 373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Audio interview with Joseph Hull of Harvard, spring 2011
- Vanda Pharmaceuticals seeks FDA approval
- Recent progress in the development of agonists and antagonists for melatonin receptors.Zlotos DP.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
 TASIMELTION
TASIMELTION
PATENTS
|
10-15-2010
|
PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE
|
|
|
8-21-2009
|
TREATMENT FOR DEPRESSIVE DISORDERS
|
|
|
5-10-2000
|
Benzopyran derivatives as melatonergic agents
|
|
|
11-10-1999
|
Benzodioxa alkylene ethers as melatonergic agents
|
|
|
6-19-1998
|
BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS
|
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc. 1938, 60, 2794.
- Oppolzer, W.; Chapuis, C.; Bernardinelli, G. Helv. Chim. Acta 1984, 67, 1397.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, G.; Carroll,, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Oppolzer, W.; Mills, R. J.; Pachinger, W.; Stevenson, T. Helv. Chim. Acta 1986, 69, 1542; Oppolzer, W.; Schneider, P. Helv. Chim. Acta 1986, 69, 1817; Oppolzer, W.; Mills, R. J.; Réglier, M. Tetrahedron Lett. 1986, 27, 183; Oppolzer, W.; Poli. G.Tetrahedron Lett. 1986, 27, 4717; Oppolzer, W.; Poli, G.; Starkemann, C.; Bernardinelli, G. Tetrahedron Lett. 1988, 29, 3559.
- Oppolzer, W.; Barras, J-P. Helv. Chim. Acta 1987, 70, 1666.
- Curran, D. P.; Kim, B. H.; Daugherty, J.; Heffner, T. A. Tetrahedron Lett. 1988, 29, 3555.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
Org. Synth. 1990, 69, 158 |
- Org. Syn. Coll. Vol. 8, 110
- Org. Syn. Coll. Vol. 9, 212
-
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Reychler, M. A. Bull. Soc. Chim. III 1889, 19, 120.
- Armstrong, H. E.; Lowry, T. M. J. Chem. Soc., Trans. 1902, 81, 1441.
- Dauphin, G.; Kergomard, A.; Scarset, A. Bull. Soc. Chim. Fr. 1976, 862.
- Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc. 1982, 104, 5412.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc. 1988, 110, 8477.
- Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
- Oppolzer, W. Tetrahedron 1987, 43, 1969.
- Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I 1988, 1753.
- Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087.
- For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
- Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
- Davis, F. A.; Sheppard, A. C. Tetrahedron 1989, 45, 5703.
- Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc. 1987, 109, 3370.
- Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron 1985, 41, 4747.
- Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett. 1986, 27, 5079.
- Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc. 1983, 105, 3123.
- Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett. 1987, 28, 5115.
- Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett. 1988, 29, 4269.
- Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem. 1986, 51, 2402.
- Davis, F. A.; Haque, M. S. J. Org. Chem. 1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem. 1989, 54, 2021.
- Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem. 1987, 52, 5288.
- Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett. 1989, 30, 779.
- Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc. 1990, 112, 6679.

dedicated to lionel my son



my daughter Aishal

THEY KEEP ME GOING
PANOBINOSTAT

Panobinostat
HDAC inhibitors, orphan drug
cas 404950-80-7
2E)-N-hydroxy-3-[4-({[2-(2-methyl-1H-indol-3-yl)ethyl]amino}methyl)phenyl]acrylamide
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide)
Molecular Formula: C21H23N3O2 Molecular Weight: 349.42622
- Faridak
- LBH 589
- LBH589
- Panobinostat
- UNII-9647FM7Y3Z
A hydroxamic acid analog histone deacetylase inhibitor from Novartis.
NOVARTIS, innovator
Histone deacetylase inhibitors
Is currently being examined in cutaneous T-cell lymphoma, CML and breast cancer.
clinical trials click here phase 3
DRUG SUBSTANCE–LACTATE AS IN http://www.google.com/patents/US7989639 SEE EG 31

Panobinostat (LBH-589) is an experimental drug developed by Novartis for the treatment of various cancers. It is a hydroxamic acid[1] and acts as a non-selective histone deacetylase inhibitor (HDAC inhibitor).[2]
panobinostat
Panobinostat is a cinnamic hydroxamic acid analogue with potential antineoplastic activity. Panobinostat selectively inhibits histone deacetylase (HDAC), inducing hyperacetylation of core histone proteins, which may result in modulation of cell cycle protein expression, cell cycle arrest in the G2/M phase and apoptosis. In addition, this agent appears to modulate the expression of angiogenesis-related genes, such as hypoxia-inducible factor-1alpha (HIF-1a) and vascular endothelial growth factor (VEGF), thus impairing endothelial cell chemotaxis and invasion. HDAC is an enzyme that deacetylates chromatin histone proteins. Check for
As of August 2012, it is being tested against Hodgkin’s Lymphoma, cutaneous T cell lymphoma (CTCL)[3] and other types of malignant disease in Phase III clinical trials, against myelodysplastic syndromes, breast cancer and prostate cancer in Phase II trials, and against chronic myelomonocytic leukemia (CMML) in a Phase I trial.[4][5]
Panobinostat is a histone deacetylase (HDAC) inhibitor which was filed for approval in the U.S. in 2010 for the oral treatment of relapsed/refractory classical Hodgkin’s lymphoma in adult patients. The company is conducting phase II/III clinical trials for the oral treatment of multiple myeloma, chronic myeloid leukemia and myelodysplasia. Phase II trials are also in progress for the treatment of primary myelofibrosis, post-polycythemia Vera, post-essential thrombocytopenia, Waldenstrom’s macroglobulinemia, recurrent glioblastoma (GBM) and for the treatment of pancreatic cancer progressing on gemcitabine therapy. Additional trials are under way for the treatment of hematological neoplasms, prostate cancer, colorectal cancer, renal cell carcinoma, non-small cell lung cancer (NSCLC), malignant mesothelioma, acute lymphoblastic leukemia, acute myeloid leukemia, head and neck cancer and gastrointestinal neuroendocrine tumors. Early clinical studies are also ongoing for the treatment of HER2 positive metastatic breast cancer. Additionally, phase II clinical trials are ongoing at Novartis as well as Neurological Surgery for the treatment of recurrent malignant gliomas as are phase I/II initiated for the treatment of acute graft versus host disease. The National Cancer Institute had been conducting early clinical trials for the treatment of metastatic hepatocellular carcinoma; however, these trials were terminated due to observed dose-limiting toxicity. In 2009, Novartis terminated its program to develop panobinostat for the treatment of cutaneous T-cell lymphoma. A program for the treatment of small cell lung cancer was terminated in 2012. Phase I clinical trials are ongoing for the treatment of metastatic and/or malignant melanoma and for the treatment of sickle cell anemia. The University of Virginia is conducting phase I clinical trials for the treatment of newly diagnosed and recurrent chordoma in combination with imatinib. Novartis is evaluating panobinostat for its potential to re-activate HIV transcription in latently infected CD4+ T-cells among HIV-infected patients on stable antiretroviral therapy.
Mechanistic evaluations revealed that panobinostat-mediated tumor suppression involved blocking cell-cycle progression and gene transcription induced by the interleukin IL-2 promoter, accompanied by an upregulation of p21, p53 and p57, and subsequent cell death resulted from the stimulation of caspase-dependent and -independent apoptotic pathways and an increase in the mitochondrial outer membrane permeability. In 2007, the compound received orphan drug designation in the U.S. for the treatment of cutaneous T-cell lymphoma and in 2009 and 2010, orphan drug designation was received in the U.S. and the E.U., respectively, for the treatment of Hodgkin’s lymphoma. This designation was also assigned in 2012 in the U.S. and the E.U. for the treatment of multiple myeloma.
Cardiovascular disease is the leading cause of morbidity and mortality in the western world and during the last decades it has also become a rapidly increasing problem in developing countries. An estimated 80 million American adults (one in three) have one or more expressions of cardiovascular disease (CVD) such as hypertension, coronary heart disease, heart failure, or stroke. Mortality data show that CVD was the underlying cause of death in 35% of all deaths in 2005 in the United States, with the majority related to myocardial infarction, stroke, or complications thereof. The vast majority of patients suffering acute cardiovascular events have prior exposure to at least one major risk factor such as cigarette smoking, abnormal blood lipid levels, hypertension, diabetes, abdominal obesity, and low-grade inflammation.
Pathophysiologically, the major events of myocardial infarction and ischemic stroke are caused by a sudden arrest of nutritive blood supply due to a blood clot formation within the lumen of the arterial blood vessel. In most cases, formation of the thrombus is precipitated by rupture of a vulnerable atherosclerotic plaque, which exposes chemical agents that activate platelets and the plasma coagulation system. The activated platelets form a platelet plug that is armed by coagulation-generated fibrin to form a biood clot that expands within the vessel lumen until it obstructs or blocks blood flow, which results in hypoxic tissue damage (so-called infarction). Thus, thrombotic cardiovascular events occur as a result of two distinct processes, i.e. a slowly progressing long-term vascular atherosclerosis of the vessel wall, on the one hand, and a sudden acute clot formation that rapidly causes flow arrest, on the other. This invention solely relates to the latter process.
Recently, inflammation has been recognized as an important risk factor for thrombotic events. Vascular inflammation is a characteristic feature of the atherosclerotic vessel wall, and inflammatory activity is a strong determinant of the susceptibility of the atherosclerotic plaque to rupture and initiate intravascular clotting. Also, autoimmune conditions with systemic inflammation, such as rheumatoid arthritis, systemic lupus erythematosus and different forms of vasculitides, markedly increase the risk of myocardial infarction and stroke.
Traditional approaches to prevent and treat cardiovascular events are either targeted 1) to slow down the progression of the underlying atherosclerotic process, 2) to prevent clot formation in case of a plaque rupture, or 3) to direct removal of an acute thrombotic flow obstruction. In brief, antiatherosclerotic treatment aims at modulating the impact of general risk factors and includes dietary recommendations, weight loss, physical exercise, smoking cessation, cholesterol- and blood pressure treatment etc. Prevention of clot formation mainly relies on the use of antiplatelet drugs that inhibit platelet activation and/or aggregation, but also in some cases includes thromboembolic prevention with oral anticoagulants such as warfarin. Post-hoc treatment of acute atherothrombotic events requires either direct pharmacological lysis of the clot by thrombolytic agents such as recombinant tissue-type plasminogen activator or percutaneous mechanical dilation of the obstructed vessel.
Despite the fact that multiple-target antiatherosclerotic therapy and clot prevention by antiplatelet agents have lowered the incidence of myocardial infarction and ischemic stroke, such events still remain a major population health problem. This shows that in patients with cardiovascular risk factors these prophylactic measures are insufficient to completely prevent the occurrence of atherothrombotic events.
Likewise, thrombotic conditions on the venous side of the circulation, as well as embolic complications thereof such as pulmonary embolism, still cause substantial morbidity and mortality. Venous thrombosis has a different clinical presentation and the relative importance of platelet activation versus plasma coagulation are somewhat different with an preponderance for the latter in venous thrombosis, However, despite these differences, the major underlying mechanisms that cause thrombotic vessel occlusions are similar to those operating on the arterial circulation. Although unrelated to atherosclerosis as such, the risk of venous thrombosis is related to general cardiovascular risk factors such as inflammation and metabolic aberrations.

Panobinostat can be synthesized as follows: Reduction of 2-methylindole-3-glyoxylamide (I) with LiAlH4 affords 2-methyltryptamine (II). 4-Formylcinnamic acid (III) is esterified with methanolic HCl, and the resulting aldehyde ester (IV) is reductively aminated with 2-methyltryptamine (II) in the presence of NaBH3CN (1) or NaBH4 (2) to give (V). The title hydroxamic acid is then obtained by treatment of ester (V) with aqueous hydroxylamine under basic conditions.
Panobinostat is currently being used in a Phase I/II clinical trial that aims at curing AIDS in patients on highly active antiretroviral therapy (HAART). In this technique panobinostat is used to drive the HI virus’s DNA out of the patient’s DNA, in the expectation that the patient’s immune system in combination with HAART will destroy it.[6][7]
 panobinostat
panobinostat
Panobinostat has been found to synergistically act with sirolimus to kill pancreatic cancer cells in the laboratory in a Mayo Clinic study. In the study, investigators found that this combination destroyed up to 65 percent of cultured pancreatic tumor cells. The finding is significant because the three cell lines studied were all resistant to the effects of chemotherapy – as are many pancreatic tumors.[8]
Panobinostat has also been found to significantly increase in vitro the survival of motor neuron (SMN) protein levels in cells of patients suffering fromspinal muscular atrophy.[9]
Panobinostat was able to selectively target triple negative breast cancer (TNBC) cells by inducing hyperacetylation and cell cycle arrest at the G2-M DNA damage checkpoint; partially reversing the morphological changes characteristic of breast cancer cells.[10]
Panobinostat, along with other HDAC inhibitors, is also being studied for potential to induce virus HIV-1 expression in latently infected cells and disrupt latency. These resting cells are not recognized by the immune system as harboring the virus and do not respond to antiretroviral drugs.[11]
Panobinostat inhibits multiple histone deacetylase enzymes, a mechanism leading to apoptosis of malignant cells via multiple pathways.[1]
The compound N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide) has the formula

as described in WO 02/22577. Valuable pharmacological properties are attributed to this compound; thus, it can be used, for example, as a histone deacetylase inhibitor useful in therapy for diseases which respond to inhibition of histone deacetylase activity. WO 02/22577 does not disclose any specific salts or salt hydrates or solvates of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide.
The compounds described above are often used in the form of a pharmaceutically acceptable salt. Pharmaceutically acceptable salts include, when appropriate, pharmaceutically acceptable base addition salts and acid addition salts, for example, metal salts, such as alkali and alkaline earth metal salts, ammonium salts, organic amine addition salts, and amino acid addition salts, and sulfonate salts. Acid addition salts include inorganic acid addition salts such as hydrochloride, sulfate and phosphate, and organic acid addition salts such as alkyl sulfonate, arylsulfonate, acetate, maleate, fumarate, tartrate, citrate and lactate. Examples of metal salts are alkali metal salts, such as lithium salt, sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt, and zinc salt. Examples of ammonium salts are ammonium salt and tetramethylammonium salt. Examples of organic amine addition salts are salts with morpholine and piperidine. Examples of amino acid addition salts are salts with glycine, phenylalanine, glutamic acid and lysine. Sulfonate salts include mesylate, tosylate and benzene sulfonic acid salts.
……………………………..
GENERAL METHOD OF SYNTHESIS
ADD YOUR METHYL AT RIGHT PLACE
As is evident to those skilled in the art, the many of the deacetylase inhibitor compounds of the present invention contain asymmetric carbon atoms. It should be understood, therefore, that the individual stereoisomers are contemplated as being included within the scope of this invention.
The hydroxamate compounds of the present invention can be produced by known organic synthesis methods. For example, the hydroxamate compounds can be produced by reacting methyl 4-formyl cinnamate with tryptamine and then converting the reactant to the hydroxamate compounds. As an example, methyl 4-formyl cinnamate 2, is prepared by acid catalyzed esterification of 4-formylcinnamic acid 3 (Bull. Chem. Soc. Jpn. 1995; 68:2355-2362). An alternate preparation of methyl 4-formyl cinnamate 2 is by a Pd- catalyzed coupling of methyl acrylate 4 with 4-bromobenzaldehyde 5.
CHO

Additional starting materials can be prepared from 4-carboxybenzaldehyde 6, and an exemplary method is illustrated for the preparation of aldehyde 9, shown below. The carboxylic acid in 4-carboxybenzaldehyde 6 can be protected as a silyl ester (e.g., the t- butyldimethylsilyl ester) by treatment with a silyl chloride (e.g., f-butyldimethylsilyl chloride) and a base (e.g. triethylamine) in an appropriate solvent (e.g., dichloromethane). The resulting silyl ester 7 can undergo an olefination reaction (e.g., a Horner-Emmons olefination) with a phosphonate ester (e.g., triethyl 2-phosphonopropionate) in the presence of a base (e.g., sodium hydride) in an appropriate solvent (e.g., tetrahydrofuran (THF)). Treatment of the resulting diester with acid (e.g., aqueous hydrochloric acid) results in the hydrolysis of the silyl ester providing acid 8. Selective reduction of the carboxylic acid of 8 using, for example, borane-dimethylsuflide complex in a solvent (e.g., THF) provides an intermediate alcohol. This intermediate alcohol could be oxidized to aldehyde 9 by a number of known methods, including, but not limited to, Swern oxidation, Dess-Martin periodinane oxidation, Moffatt oxidation and the like.

The aldehyde starting materials 2 or 9 can be reductively aminated to provide secondary or tertiary amines. This is illustrated by the reaction of methyl 4-formyl cinnamate 2 with tryptamine 10 using sodium triacetoxyborohydride (NaBH(OAc)3) as the reducing agent in dichloroethane (DCE) as solvent to provide amine 11. Other reducing agents can be used, e.g., sodium borohydride (NaBH ) and sodium cyanoborohydride (NaBH3CN), in other solvents or solvent mixtures in the presence or absence of acid catalysts (e.g., acetic acid and trifluoroacetic acid). Amine 11 can be converted directly to hydroxamic acid 12 by treatment with 50% aqueous hydroxylamine in a suitable solvent (e.g., THF in the presence of a base, e.g., NaOH). Other methods of hydroxamate formation are known and include reaction of an ester with hydroxylamine hydrochloride and a base (e.g., sodium hydroxide or sodium methoxide) in a suitable solvent or solvent mixture (e.g., methanol, ethanol or methanol/THF).

NOTE ….METHYL SUBSTITUENT ON 10 WILL GIVE YOU PANOBINOSTAT
……………………………….
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide
lactate
(34, panobinostat, LBH589)
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for str see above link
α-methyl-β-(β-bromoethyl)indole (29) was made according to method reported by Grandberg et al.(2. Grandberg, I. I.; Kost, A. N.; Terent’ev, A. P. Reactions of hydrazine derivatives. XVII. New synthesis of α-methyltryptophol. Zhurnal Obshchei Khimii 1957, 27, 3342–3345. )
The bromide 29 was converted to amine 30 by using similar method used by Sletzinger et al.(3. Sletzinger, M.; Ruyle, W. V.; Waiter, A. G. (Merck & Co., Inc.). Preparation of tryptamine
derivatives. U.S. Patent US 2,995,566, Aug 8, 1961.)
To a 500 mL flask, crude 2-methyltryptamine 30 (HPLC purity 75%, 1.74 g, 7.29 mmol) and 3-(4-
formyl-phenyl)-acrylic acid methyl ester 31 (HPLC purity 84%, 1.65 g, 7.28 mmol) were added,
followed by DCM (100 mL) and MeOH (30 mL). The clear solution was stirred at room temp for 30
min, then NaBH3CN (0.439 g, 6.99 mmol) was added in small portions. The reaction mixture was
stirred at room temp overnight. After removal of the solvents, the residue was diluted with DCM and
added saturated NaHCO3 aqueous solution, extracted with DCM twice. The DCM layer was dried
and concentrated, and the resulting residue was purified by flash chromatography (silica, 0–10%
MeOH in DCM) to afford 33 as orange solid (1.52 g, 60%). LC–MS m/z 349.2 ([M + H]+). 33 was
converted to hydroxamic acid 34 according to procedure D (Experimental Section), and the freebase
34 was treated with 1 equiv of lactic acid in MeOH–water (7:3) to form lactic acid salt which was
further recrystallized in MeOH–EtOAc to afford the lactic acid salt of 34as pale yellow solid. LC–MS m/z 350.2 ([M + H − lactate]+).
= DELTA
1H NMR (DMSO-d6) 10.72 (s, 1H, NH), 7.54 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 16 Hz, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 6.97 (td, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8Hz, 2H), 7.01 (t, J = 7.4, 0.9 Hz, 1H), 6.91 (td, J = 7.4, 0.9 Hz, 1H), 6.47 (d, J = 15.2 Hz, 1H), 3.94(q, J = 6.8 Hz, 1H, lactate CH), 3.92 (s, 2H), 2.88 and 2.81 (m, each, 4H, AB system, CH2CH2),2.31 (s, 3H), 1.21 (d, J = 6.8 Hz, 3H).;
13C NMR (DMSO-d6) 176.7 (lactate C=O), 162.7, 139.0,
137.9, 135.2, 134.0, 132.1, 129.1, 128.1, 127.4, 119.9, 119.0, 118.1, 117.2, 110.4, 107.0, 66.0, 51.3,
48.5, 22.9, 20.7, 11.2.
…………………………………………..
PANOBINOSTAT DRUG SUBSTANCE SYNTHESIS AND DATA
http://www.google.com/patents/US7989639

A flow diagram for the synthesis of LBH589 lactate is provided in FIG. A. A nomenclature reference index of the intermediates is provided below in the Nomenclature Reference Index:
| Nomenclature reference index | |
| Compound | Chemical name |
| 1 | 4-Bromo-benzaldehyde |
| 2 | Methyl acrylate |
| 3 | (2E)-3-(formylphenyl)-2-propenoic acid, methyl ester |
| 4 | 3-[4-[[[2-(2-Methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2- | |
| propenoic acid, methyl ester, monohydrochloride | |
| 5 | (2E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| 6 | 2-hydroxypropanoic acid, compd. with 2(E)-N- |
| hydroxy-3-[4-[[[2-(2-methyl-1H- | |
| indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| Z3a | 2-Methyl-1H-indole-3-ethanamine |
| Z3b | 5-Chloro-2-pentanone |
| Z3c | Phenylhydrazine |
The manufacture of LBH589 lactate (6) drug substance is via a convergent synthesis; the point of convergence is the condensation of indole-amine Z3a with aldehyde 3.
The synthesis of indole-amine Z3a involves reaction of 5-chloro-2 pentanone (Z3b) with phenylhydrazine (Z3c) in ethanol at reflux (variation of Fischer indole synthesis).
Product isolation is by an extractive work-up followed by crystallization. Preparation of aldehyde 3 is by palladium catalyzed vinylation (Heck-type reaction; Pd(OAc)2/P(o-Tol)3/Bu3N in refluxing CH3CN) of 4-bromo-benzyladehyde (1) with methyl acrylate (2) with product isolation via precipitation from dilute HCl solution. Intermediates Z3a and 3 are then condensed to an imine intermediate, which is reduced using sodium borohydride in methanol below 0° C. (reductive amination). The product indole-ester 4, isolated by precipitation from dilute HCl, is recrystallized from methanol/water, if necessary. The indole ester 4 is converted to crude LBH589 free base 5 via reaction with hydroxylamine and sodium hydroxide in water/methanol below 0° C. The crude LBH589 free base 5 is then purified by recrystallization from hot ethanol/water, if necessary. LBH589 free base 5 is treated with 85% aqueous racemic lactic acid and water at ambient temperature. After seeding, the mixture is heated to approximately 65° C., stirred at this temperature and slowly cooled to 45-50° C. The resulting slurry is filtered and washed with water and dried to afford LBH589 lactate (6).
If necessary the LBH589 lactate 6 may be recrystallised once again from water in the presence of 30 mol % racemic lactic acid. Finally the LBH589 lactate is delumped to give the drug substance. If a rework of the LBH589 lactate drug substance 6 is required, the LBH589 lactate salt is treated with sodium hydroxide in ethanol/water to liberate the LBH589 free base 5 followed by lactate salt formation and delumping as described above.
All starting materials, reagents and solvents used in the synthesis of LBH589 lactate are tested according to internal specifications or are purchased from established suppliers against a certificate of analysis.
EXAMPLE 7 Formation of Monohydrate Lactate Salt
About 40 to 50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base was suspended in 1 ml of a solvent as listed in Table 7. A stoichiometric amount of lactic acid was subsequently added to the suspension. The mixture was stirred at ambient temperature and when a clear solution formed, stirring continued at 4° C. Solids were collected by filtration and analyzed by XRPD, TGA and 1H-NMR.
| TABLE 7 | |||||
| LOD, % | |||||
| Physical | Crystallinity | (Tdesolvation) | |||
| Solvent | T, ° C. | Appear. | and Form | Tdecomposit. | 1H-NMR |
| IPA | 4 | FFP | excellent | 4.3 (79.3) | — |
| HA | 156.3 | ||||
| Acetone | 4 | FFP | excellent | 4.5 (77.8) | 4.18 (Hbz) |
| HA | 149.5 | ||||
The salt forming reaction in isopropyl alcohol and acetone at 4° C. produced a stoichiometric (1:1) lactate salt, a monohydrate. The salt is crystalline, begins to dehydrate above 77° C., and decomposes above 150° C.
EXAMPLE 18 Formation of Anhydrous Lactate Salt
DL-lactic acid (4.0 g, 85% solution in water, corresponding to 3.4 g pure DL-lactic acid) is diluted with water (27.2 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (10.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (110.5 g) is added, and the suspension is heated to 65° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 min at 65° C. During the addition of the lactate salt solution, the suspension converted into a solution. The addition funnel is rinsed with demineralized water (9.1 g), and the solution is stirred at 65° C. for an additional 30 minutes. The solution is cooled down to 45° C. (inner temperature) and seed crystals (10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate monohydrate) are added at this temperature. The suspension is cooled down to 33° C. and is stirred for additional 20 hours at this temperature. The suspension is re-heated to 65° C., stirred for 1 hour at this temperature and is cooled to 33° C. within 1 hour. After additional stirring for 3 hours at 33° C., the product is isolated by filtration, and the filter cake is washed with demineralized water (2×20 g). The wet filter-cake is dried in vacuo at 50° C. to obtain the anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt as a crystalline product. The product is identical to the monohydrate salt (form HA) in HPLC and in 1H-NMR, with the exception of the integrals of water signals in the 1H-NMR spectra.
In additional salt formation experiments carried out according to the procedure described above, the product solution was filtered at 65° C. before cooling to 45° C., seeding and crystallization. In all cases, form A (anhydrate form) was obtained as product.
EXAMPLE 19 Formation of Anhydrous Lactate Salt
DL-lactic acid (2.0 g, 85% solution in water, corresponding to 1.7 g pure DL-lactic acid) is diluted with water (13.6 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (5.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (54.85 g) is added, and the suspension is heated to 48° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 minutes at 48° C. A solution is formed. Seed crystals are added (as a suspension of 5 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form A, in 0.25 g of water) and stirring is continued for 2 additional hours at 48° C. The temperature is raised to 65° C. (inner temperature) within 30 minutes, and the suspension is stirred for additional 2.5 hours at this temperature. Then the temperature is cooled down to 48° C. within 2 hours, and stirring is continued at this temperature for additional 22 hours. The product is isolated by filtration and the filter cake is washed with demineralized water (2×10 g). The wet filter-cake is dried in vacuo at 50° C. to obtain anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A) as a crystalline product.
EXAMPLE 20 Conversion of Monohydrate Lactate Salt to Anhydrous Lactate Salt
DL-lactic acid (0.59 g, 85% solution in water, corresponding to 0.5 g pure DL-lactic acid) is diluted with water (4.1 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
10 g of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt monohydrate is placed in a 4-necked reaction flask. Water (110.9 g) is added, followed by the addition of the lactic acid solution. The addition funnel of the lactic acid is rinsed with water (15.65 g). The suspension is heated to 82° C. (inner temperature) to obtain a solution. The solution is stirred for 15 minutes at 82° C. and is hot filtered into another reaction flask to obtain a clear solution. The temperature is cooled down to 50° C., and seed crystals are added (as a suspension of 10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form, in 0.5 g of water). The temperature is cooled down to 33° C. and stirring is continued for additional 19 hours at this temperature. The formed suspension is heated again to 65° C. (inner temperature) within 45 minutes, stirred at 65° C. for 1 hour and cooled down to 33° C. within 1 hour. After stirring at 33° C. for additional 3 hours, the product is isolated by filtration and the wet filter cake is washed with water (50 g). The product is dried in vacuo at 50° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 21 Formation of Anhydrous Lactate Salt
DL-lactic acid (8.0 g, 85% solution in water, corresponding to 6.8 g pure DL-lactic acid) was diluted with water (54.4 g), and the solution was heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and was used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (20 g) is placed in a 1 L glass reactor, and ethanol/water (209.4 g of a 1:1 w/w mixture) is added. The light yellow suspension is heated to 60° C. (inner temperature) within 30 minutes, and the lactic acid solution is added during 30 minutes at this temperature. The addition funnel is rinsed with water (10 g). The solution is cooled to 38° C. within 2 hours, and seed crystals (20 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form) are added at 38° C. After stirring at 38° C. for additional 2 hours, the mixture is cooled down to 25° C. within 6 hours. Cooling is continued from 25° C. to 10° C. within 5 hours, from 10° C. to 5° C. within 4 hours and from 5° C. to 2° C. within 1 hour. The suspension is stirred for additional 2 hours at 2° C., and the product is isolated by filtration. The wet filter cake is washed with water (2×30 g), and the product is dried in vacuo at 45° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 28 Formation of Lactate Monohydrate Salt
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 75 ml of acetone were charged in a 250 ml 3-neck flask equipped with a magnetic stirrer and an addition funnel. To the stirred suspension were added dropwise 10 ml of 1 M lactic acid in water (10 mmol) dissolved in 20 ml acetone, affording a clear solution. Stirring continued at ambient and a white solid precipitated out after approximately 1 hour. The mixture was cooled in an ice bath and stirred for an additional hour. The white solid was recovered by filtration and washed once with cold acetone (15 ml). It was subsequently dried under vacuum to yield 3.94 g of the lactate monohydrate salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (86.2%).
References
- Revill, P; Mealy, N; Serradell, N; Bolos, J; Rosa, E (2007). “Panobinostat”. Drugs of the Future 32 (4): 315. doi:10.1358/dof.2007.032.04.1094476. ISSN 0377-8282.
- Table 3: Select epigenetic inhibitors in various stages of development from Mack, G. S. (2010). “To selectivity and beyond”. Nature Biotechnology 28 (12): 1259–1266.doi:10.1038/nbt.1724. PMID 21139608. edit
- ClinicalTrials.gov NCT00425555 Study of Oral LBH589 in Adult Patients With Refractory Cutaneous T-Cell Lymphoma
- ClinicalTrials.gov: LBH-589
- Prince, HM; M Bishton (2009). “Panobinostat (LBH589): a novel pan-deacetylase inhibitor with activity in T cell lymphoma”. Hematology Meeting Reports (Parkville, Australia: Peter MacCallum Cancer Centre and University of Melbourne) 3 (1): 33–38.
- Simons, J (27 April 2013). “Scientists on brink of HIV cure”. The Telegraph.
- ClinicalTrials.gov NCT01680094 Safety and Effect of The HDAC Inhibitor Panobinostat on HIV-1 Expression in Patients on Suppressive HAART (CLEAR)
- Mayo Clinic Researchers Formulate Treatment Combination Lethal To Pancreatic Cancer Cells
- Garbes, L; Riessland, M; Hölker, I; Heller, R; Hauke, J; Tränkle, Ch; Coras, R; Blümcke, I; Hahnen, E; Wirth, B (2009). “LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate”. Human Molecular Genetics 18 (19): 3645–3658. doi:10.1093/hmg/ddp313.PMID 19584083.
- Tate, CR; Rhodes, LV; Segar, HC; Driver, JL; Pounder, FN; Burow, ME; and Collins-Burow, BM (2012). “Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat”. Breast Cancer Research 14 (3).
- TA Rasmussen, et al. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Human Vaccines & Immunotherapeutics 9:5, 1-9, May 2013.
- Drugs of the Future 32(4): 315-322 (2007)
- WO 2002022577…
- WO 2007146718
- WO 2013110280
- WO 2010009285
- WO 2010009280
- WO 2005013958
- WO 2004103358
- WO 2003048774…
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
-
23009203 11-26-2012 Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. Journal of medicinal chemistry -
21634430 7-14-2011 Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. Journal of medicinal chemistry -
21417419 4-28-2011 Discovery, synthesis, and pharmacological evaluation of spiropiperidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. Journal of medicinal chemistry -
19317450 4-23-2009 Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. Journal of medicinal chemistry -
15650931 1-1-2005 The American Society of Hematology–46th Annual Meeting and Exposition. HDAC, Flt and farnesyl transferase inhibitors. IDrugs : the investigational drugs journal -
US7989639 8-3-2011 PROCESS FOR MAKING SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010286409 11-12-2010 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010179208 7-16-2010 Use of HDAC Inhibitors for the Treatment of Bone Destruction US2010160257 6-25-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF MYELOMA US2010137398 6-4-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF GASTROINTESTINAL CANCERS US2009306405 12-11-2009 PROCESS FOR MAKING N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE AND STARTING MATERIALS THEREFOR US2009281159 11-13-2009 USE OF HDAC INHIBITORS FOR THE TREATMENT OF LYMPHOMAS US2009264439 10-23-2009 Combination of a) N–4-(3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia US2009197936 8-7-2009 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2009012066 1-9-2009 Method of Use of Deacetylase Inhibitors
| US2008319045 | 12-26-2008 | Combination of Histone Deacetylase Inhibitors and Radiation |
| US2008221126 | 9-12-2008 | Use of Hdac Inhibitors for the Treatment of Myeloma |
| US2008176849 | 7-25-2008 | DEACETYLASE INHIBITORS |
| US2006189674 | 8-25-2006 | Deacetylase inhibitors |
| US7067551 | 6-28-2006 | Deacetylase inhibitors |
| US2006100140 | 5-12-2006 | Combination of a) n-{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]2-methylphenyl}-4- (3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia |
| US6833384 | 12-22-2004 | Deacetylase inhibitors |
| US6552065 | 4-23-2003 | Deacetylase inhibitors |
| GB776693A | Title not available | |||
| GB891413A | Title not available | |||
| GB2185020A | Title not available | |||
| WO2002022577A2 | Aug 30, 2001 | Mar 21, 2002 | Kenneth Walter Bair | Hydroxamate derivatives useful as deacetylase inhibitors |
| WO2003016307A1 | Aug 6, 2002 | Aug 19, 1993 | Jolie Anne Bastian | β3 ADRENERGIC AGONISTS |
| WO2003039599A1 | Nov 5, 2002 | May 15, 2003 | Ying-Nan Pan Chen | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| WO2005105740A2 | Apr 26, 2005 | Nov 10, 2005 | Serguei Fine | Preparation of tegaserod and tegaserod maleate |
| WO2006021397A1 | Aug 22, 2005 | Mar 2, 2006 | Recordati Ireland Ltd | Lercanidipine salts |
…………………………………..
extras
5. Mocetinostat (MGCD0103), including pharmaceutically acceptable salts thereof. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Mocetinostat, has the following chemical structure and name:

Vorinostat, including pharmaceutically acceptable salts thereof. Marks et al., Nature Biotechnology 25, 84 to 90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Vorinostat has the following chemical structure and name:

Belinostat (PXD-101 , PX-105684)
(2E)-3-[3-(anilinosulfonyl)phenyl]-N-hydroxyacrylamide

……………………………………………….
Dacinostat (LAQ-824, NVP-LAQ824,)
((E)-N-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1 H-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide

Entinostat (MS-275, SNDX-275, MS-27-275)
4-(2-aminophenylcarbamoyl)benzylcarbamate

(a) The HDAC inhibitor Vorinostat™ or a salt, hydrate, or solvate thereof.

Vorinostat………………..
(b) The HDAC inhibitor Givinostat or a salt, hydrate, or solvate thereof.

Givinostat or a salt, hydrate, or solvate thereof.
BELINOSTAT, FAST TRACK, ORPHAN DRUG, A hydroxamate-type inhibitor of histone deacetylase.

Belinostat (PXD101)
PHASE 2, FAST TRACK FDA , ORPHAN STATUS
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..

MIDOSTAURIN …with potential antiangiogenic and antineoplastic activities …

MIDOSTAURIN
READ …COMPLETE SYNTHESIS AT
Idelalisib ….US FDA Accepts NDA for Gilead’s Idelalisib for the Treatment of Refractory Indolent Non-Hodgkin’s Lymphoma

An antineoplastic agent and p110delta inhibitor
(S)-2-(1-(9H-purin-6-ylamino)propyl)-5-fluoro-3-phenylquinazolin-4(3H)-one
Icos (Originator)
- CAL-101
- GS-1101
- Idelalisib
- UNII-YG57I8T5M0
M.Wt: 415.43
Formula: C22H18FN7O
CAS No.: 870281-82-6
CAL-101 Solubility: DMSO ≥80mg/mL Water <1.2mg/mL Ethanol ≥33mg/mL
5-Fluoro-3-phenyl-2-[(1S)-1-(7H-purin-6-ylamino)propyl]-4(3H)-quinazolinone
idelalisib
Idelalisib (codenamed GS-1101 or CAL-101) is a drug under investigation for the treatment of chronic lymphocytic leukaemia. It is in Phase III clinical trials testing drug combinations with rituximab and/or bendamustine as of 2013. The substance acts as aphosphoinositide 3-kinase inhibitor; more specifically, it blocks P110δ, the delta isoform of the enzyme phosphoinositide 3-kinase.[1][2]
GDC-0032 is a potent, next-generation beta isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with IC 50 of 0.29 nM/0.12 nM/0.97nM,> 10 fold over Selective PI3K [beta].
GS-1101 is a novel, orally available small molecule inhibitor of phosphatidylinositol 3-kinase delta (PI3Kdelta) develop by Gilead and is waiting for registration in U.S. for the treatment of patients with indolent non-Hodgkin’s lymphoma that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy and for the treatment of chronic lymphocytic leukemia. The compound is also in phase III clinical evaluation for the treatment of elderly patients with previously untreated small lymphocytic lymphoma (SLL) and acute myeloid leukemia. Clinical trials had been under way for the treatment of inflammation and allergic rhinitis; however, no recent development has been reported. Preclinical studies have shown that GS-1101 has desirable pharmaceutical properties. The compound was originally developed by Calistoga Pharmaceuticals, acquired by Gilead on April 1, 2011.
clinical trials, click link
http://clinicaltrials.gov/search/intervention=CAL-101%20OR%20GS-1101%20OR%20Idelalisib
FOSTER CITY, Calif.–(BUSINESS WIRE)–Jan. 13, 2014– Gilead Sciences, Inc. (Nasdaq: GILD) announced today that the U.S. Food and Drug Administration (FDA) has accepted for review the company’s New Drug Application (NDA) for idelalisib, a targeted, oral inhibitor of PI3K delta, for the treatment of refractory indolent non-Hodgkin’s lymphoma (iNHL). FDA has granted a standard review for the iNHL NDA and has set a target review date under the Prescription Drug User Fee Act (PDUFA) of September 11, 2014.
The NDA for iNHL, submitted on September 11, 2013, was supported by a single arm Phase 2 study (Study 101-09) evaluating idelalisib in patients with iNHL that is refractory (non-responsive) to rituximab and to alkylating-agent-containing chemotherapy. Following Gilead’s NDA submission for iNHL, FDA granted idelalisib a Breakthrough Therapy designation for relapsed chronic lymphocytic leukemia (CLL). The FDA grants Breakthrough Therapy designation to drug candidates that may offer major advances in treatment over existing options. Gilead submitted an NDA for idelalisib for the treatment of CLL on December 6, 2013.
About Idelalisib
Idelalisib is an investigational, highly selective oral inhibitor of phosphoinositide 3-kinase (PI3K) delta. PI3K delta signaling is critical for the activation, proliferation, survival and trafficking of B lymphocytes and is hyperactive in many B-cell malignancies. Idelalisib is being developed both as a single agent and in combination with approved and investigational therapies.
Gilead’s clinical development program for idelalisib in iNHL includes Study 101-09 in highly refractory patients and two Phase 3 studies of idelalisib in previously treated patients. The development program in CLL includes three Phase 3 studies of idelalisib in previously treated patients. Combination therapy with idelalisib and GS-9973, Gilead’s novel spleen tyrosine kinase (Syk) inhibitor, also is being evaluated in a Phase 2 trial of patients with relapsed or refractory CLL, iNHL and other lymphoid malignancies.
Additional information about clinical studies of idelalisib and Gilead’s other investigational cancer agents can be found at http://www.clinicaltrials.gov. Idelalisib and GS-9973 are investigational products and their safety and efficacy have not been established.
About Indolent Non-Hodgkin’s Lymphoma
Indolent non-Hodgkin’s lymphoma refers to a group of largely incurable slow-growing lymphomas that run a relapsing course after therapy and can lead ultimately to life-threatening complications such as serious infections and marrow failure. Most iNHL patients are diagnosed at an advanced stage of disease, and median survival from time of initial diagnosis for patients with the most common form of iNHL, follicular lymphoma, is 8 to 10 years. The outlook for refractory iNHL patients is significantly poorer.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North and South America, Europe and Asia Pacific.
The delta form of PI3K is expressed primarily in blood-cell lineages, including cells that cause or mediate hematologic malignancies, inflammation, autoimmune diseases and allergies. By specifically inhibiting only PI3K delta, a therapeutic effect is exerted without inhibiting PI3K signalling that is critical to the normal function of healthy cells. Extensive studies have shown that inhibition of other PI3K forms can cause significant toxicities, particularly with respect to glucose metabolism, which is essential for normal cell activity.
In 2011, orphan drug designation was assigned to GS-1101 in the U.S. for the treatment of CLL. In 2013, several orphan drug designations were assigned to the compound in the E.U. and U.S.: for the treatment of follicular lymphoma, for the treatment of mucosa-associated lymphoid tissue lymphoma (MALT), for the treatment of nodal marginal zone lymphoma, for the treatment of splenic marginal zone lymphoma, and for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma. Orphan drug designation was also assigned in the U.S. for the treatment of lymphoplasmacytic lymphoma with or without Walenstom’s macroglobulinemia and, in the E.U., for the treatment of Waldenstrom’s macroglobulinemia (lymphoplasmacytic lymphoma).
Later in 2013, some of these orphan drug designations were withdrawn in the E.U.; for the treatment of chronic lymphocytic leukemia / small lymphocytic lymphoma, for the treatment of extranodal marginal-zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), for the treatment of of nodal marginal-zone lymphoma and for the treatment of splenic marginal-zone lymphoma. In 2013, the FDA granted a breakthrough therapy designation for the treatment of chronic lymphocytic leukemia.
- H. Spreitzer (13 May 2013). “Neue Wirkstoffe – Ibrutinib und Idelalisib”. Österreichische Apothekerzeitung (in German) (10/2013): 34.
- Wu, M.; Akinleye, A.; Zhu, X. (2013). “Novel agents for chronic lymphocytic leukemia”.Journal of Hematology & Oncology 6: 36. doi:10.1186/1756-8722-6-36.PMC 3659027. PMID 23680477.
CAL-101 is an Oral Delta Isoform-Selective PI3 Kinase Inhibitor.
| US8207153 | 6-27-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2012015964 | 1-20-2012 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2011306622 | 12-16-2011 | METHODS OF TREATING HEMATOLOGICAL DISORDERS WITH QUINAZOLINONE COMPOUNDS IN SELECTED SUBJECTS |
| US7932260 | 4-27-2011 | Quinazolinones as Inhibitors of Human Phosphatidylinositol 3-Kinase Delta |
| US2011044942 | 2-25-2011 | METHODS OF TREATMENT FOR SOLID TUMORS |
| US2010256167 | 10-8-2010 | QUINAZOLINONES AS INHIBITORS OF HUMAN PHOSPHATIDYLINOSITOL 3-KINASE DELTA |
| US2010202963 | 8-13-2010 | THERAPIES FOR HEMATOLOGIC MALIGNANCIES |
| WO2005113556A1 * | 12 May 2005 | 1 Dec 2005 | Icos Corp | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005117889A1 * | 12 Nov 2004 | 15 Dec 2005 | Didier Bouscary | Methods for treating and/or preventing aberrant proliferation of hematopoietic |
| WO2005120511A1 * | 4 Jun 2005 | 22 Dec 2005 | Joel S Hayflick | Methods for treating mast cell disorders |
| WO2006089106A2 * | 16 Feb 2006 | 24 Aug 2006 | Icos Corp | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| US20060106038 * | 25 May 2005 | 18 May 2006 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
The synthesis of a compound in accordance with formula I is first exemplified using steps A-E below, which provide a synthetic procedure for compound 107, the structure of which is shown below.

(107) is idelalisib
……………….
Synthesis of 2-fluoro-6-nitro-N-phenyl-benzamide (108)
Step A: A solution of 2-fluoro-6- nitrobenzoic acid (100 g, 0.54 mol) and dimethylformamide (5 mL) in dichloromethane (600 mL) was treated dropwise with oxalyl chloride (2 M in dichloromethane, 410 mL, 0.8 mol, 1.5 eq) over 30 min. After stirring 2 h at room temperature, the reaction was concentrated to an orange syrup with some solids present. The syrup was dissolved in dry dioxane (80 mL) and slowly added to a suspension of aniline (49 mL, 0.54 mol, 1 eq) and sodium bicarbonate (90 g, 1.08 mol, 2 eq) in a mixture of dioxane (250 mL) and water (250 mL) at 6 0C. The temperature reached 27°C at the end of the addition. After 30 min, the reaction mixture was treated with water (1.2 L). The precipitate was collected by vacuum filtration, washed with water (300 mL) , air dried in the funnel, and dried in vacuo at 50°C for 24 h to afford an off-white solid product (139 g, 99%). 1H NMR (300 MHz, DMSO-d6) δ 10.82 (s, IH), 8.12 (d, J = 7.7 Hz, IH), 7.91-7.77 (m, 2H), 7.64 (d, J = 7.7 Hz, 2H), 7.38 (t, J = 7.9 Hz, 2H), 7.15 > (t, J = 7.4 Hz, IH), ESI-MS m/z 261 (MH+). The reaction described above and compound 108 are shown below.

………………………..
Synthesis of(S) – [1- (2-fluoro-6-nitro-benzoyl) -phenyl-aminocarbonyl] – propyl-carbamic acid tert-butyl ester (109)
Step B: A suspension of compound 108 (0.5 mol) and dimethylformamide (5 mL) in thionyl chloride (256 mL, 2.5 mol, 5 eq) was stirred at 85°C for 5 hours. The reaction mixture was concentrated in vacuo to a brown syrup. The syrup was dissolved in dichloromethane (200 mL) and was slowly added to a solution of N-BOC-L-2-aminobutyric acid (112 g, 0.55 mol, 1.1 eq) and triethylamine (77 mL, 0.55 mol, 1.1 eq) in dichloromethane (600 mL) at 10 0C. After stirring at room temperature for 3 h, salts were removed by filtration, and the solution was washed with 100 mL of water, saturated sodium bicarbonate, water, 5% citric acid, and saturated sodium chloride. The organic phase was dried with magnesium sulfate and concentrated to a red syrup. The syrup was dissolved in dichloromethane (450 mL) and purified by flash chromatography on a silica gel plug (15 x 22 cm, 4 L dry silica) eluted with hexanes/ethyl acetate (10%, 8 L; 15%, 8 L; 20%, 8 L; 25%, 4 L) to yield the compound 109 as an off-white solid (147 g, 66%). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.0 Hz, IH), 7.84 (t, J = 8.6 Hz, IH), 7.78- 7.67 (m, IH), 7.65-7.49 (m, 3H), 7.40-7.28 ( m, 2H), 7.19 (d, J = 7.5 Hz, IH), 4.05 (broad s, IH), 1.75- 1.30 (m, 2H), 1.34 (s, 9H), 0.93 (broad s, 3H). ESI- MS m/z 446.3 (MH+) . The reaction described above and compound 109 are shown below.

Synthesis of(S) – [1- (5-fluoro-4-oxo-3-phenyl-3 , 4-dihydro-quinazolin-2- yl) -propyl] -carbamic acid tert-butyl ester (110)
Step C: A solution of compound 109 (125 mmol, 1 eq) in acetic acid (500 mL) was treated with zinc dust (48.4 g, 740 mmol, 6 eq) added in 3 portions, and the reaction mixture was allowed to cool to below 35°C between additions. After stirring for 2 h at ambient temperature, solids were filtered off by vacuum filtration and washed with acetic acid (50 mL) . The filtrate was concentrated in vacuo, dissolved in EtOAc (400 mL) , washed with water (300 mL) , and the water layer was extracted with EtOAc (300 mL) . The combined organic layers were washed with water (200 mL) , sat’d sodium bicarbonate (2 x 200 mL) , sat’d NaCl (100 mL) , dried with MgSO4, and concentrated to a syrup. The syrup was dissolved in toluene (200 mL) and purified by flash chromatography on a silica gel plug (13 x 15 cm, 2 L dry silica) eluted with hexanes/ethyl acetate (10%, 4 L; 15%, 4 L; 17.5%, 8 L; 25%, 4 L) to yield compound 110 as an off-white foamy solid (33.6 g, 69%). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (td, J = 8.2, 5.7 Hz, IH), 7.64-7.48 (m, 5H), 7.39 (broad d, J = 7.6 Hz, IH), 7.30 (dd, J = 8.3 Hz, IH), 7.23 (d, J = 7.6 Hz, IH), 4.02-3.90 (m, IH), 1.76-1.66 (m, IH), 1.62-1.46 (m, IH), 1.33 (s, 9H), 0.63 (t, J= 7.3 Hz, 3H). ESI-MS m/z 398.3 (MH+). The reaction described above and compound 110 are shown below.

…………..
Syn of (S) -2- (1-amino-propyl) -5-fluoro-3-phenyl-3H-quinazolin-4- one (111)
Step D: A solution of compound 110 (85 mmol) in dichloromethane (60 mL) was treated with trifluoroacetic acid (60 mL) . The reaction mixture was stirred for 1 h, concentrated in vacuo, and partitioned between dichloromethane (150 mL) and 10% K2CO3 (sufficient amount to keep the pH greated than 10) . The aqueous layer was extracted with additional dichloromethane (100 raL) , and the combined organic layers were washed with water (50 mli) and brine (50 mL) . After drying with Mg SO4, the solution was concentrated to provide compound 111 as an off-white solid (22 g, 88%) . 1H NMR (300 MHz,
CDCl3) δ 7.73-7.65 (m, IH), 7.62-7.49 (m, 4H), 7.32- 7.22 (m, 2H), 7.13-7.06 (m, IH), 3.42 (dd, J= 7.5, 5.2 Hz, IH), 1.87-1.70 (m, IH), 1.58-1.43 (m, IH), 0.80 (t, J = 7.4 Hz, 3H) . ESI-MS m/z 298.2 (MH+) . The reaction described above and compound 111 are shown below.

………………
syn of (S) -5-fluoro-3-phenyl-2- [1- (9H-purin-6-ylamino) -propyl] – 3H-quinazolin-4-one (107)
Step E: A suspension of compound 111(65.6 mmol, 1 eq) , 6-bromopurine (14.6 g, 73.4 mmol, 1.1 eq) , and DIEA (24.3 mL, 140 mmol, 2 eq) in tert- butanol (40 mL) was stirred for 24 h at 800C. The reaction mixture was concentrated in vacuo and treated with water to yield a solid crude product that was collected by vacuum filtration, washed with water, and air dried. Half of the obtained solid crude product was dissolved in MeOH (600 mL) , concentrated onto silica gel (300 mL dry) , and purified by flash chromatography (7.5 x 36 cm, eluted with 10 L of 4% MeOH/CH2Cl2) to yield a solid product. The solid product was then dissolved in EtOH (250 mL) and concentrated in vacuo to compound 107 idelalisib as a light yellow solid (7.2 g, 50%).
1H NMR (300 MHz, 80 0C, DMSO-d5) δ 12.66 (broad s, IH), 8.11 (s, IH), 8.02 (broad s, IH), 7.81-7.73 (m, IH),7.60-7.42 (m, 6H), 7.25-7.15 (m, 2H), 4.97 (broad s, IH), 2.02-1.73 (m, 2H), 0.79 (t, J= 7.3 Hz, 3H).
ESI-MS m/z 416.2 (MH+).
C, H, N elemental analysis (C22Hi8N7OF-EtOH- 0.4 H2O).
Chiral purity 99.8:0.2 (S:R) using chiral HPLC (4.6 x 250 mm Chiralpak ODH column, 20 °C, 85:15 hexanes : EtOH, 1 rnL/min, sample loaded at a concentration of 1 mg/mL in EtOH) . The reaction described above and compound 107 idelalisib are shown below.

| WO2001030768A1 * | 26 Oct 2000 | 3 May 2001 | Gustave Bergnes | Methods and compositions utilizing quinazolinones |
| WO2001081346A2 * | 24 Apr 2001 | 1 Nov 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2003035075A1 * | 27 Aug 2002 | 1 May 2003 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| WO2005016348A1 * | 13 Aug 2004 | 24 Feb 2005 | Jason Douangpanya | Method of inhibiting immune responses stimulated by an endogenous factor |
| WO2005016349A1 * | 13 Aug 2004 | 24 Feb 2005 | Thomas G Diacovo | Methods of inhibiting leukocyte accumulation |
| WO2005067901A2 * | 7 Jan 2005 | 28 Jul 2005 | Carrie A Northcott | Methods for treating and preventing hypertension and hypertension-related disorders |
APREMILAST, … ORALLY ACTIVE PDE4 INHIBITOR

APREMILAST
PDE4 inhibitor
N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl]-4-acetylaminoisoindolin-l,3-dione,
(S)—N-{2-[1-(3-ethoxy-4-methoxy-phenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
(S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
Molecular Formula: C22H24N2O7S Molecular Weight: 460.50016
608141-41-9 CAS NO
Celgene (Originator)
CC-10004 (apremilast) is an oral compound that is being studied in multiple Phase III clinical trials for the treatment of psoriasis, psoriatic arthritis and other chronic inflammatory diseases. We successfully completed our early stage studies, demonstrating clinical activity and tolerability and meeting safety endpoints in a placebo controlled proof-of mechanism trial in moderate-to-severe psoriasis and psoriatic arthritis. With the initiation of six multi-center international clinical trials, we are advancing the clinical development of CC-10004.
CC-10004, , Apremilast (USAN), SureCN302992, Apremilast (CC-10004), QCR-202,
- Apremilast
- CC 10004
- CC-10004
- CC10004
- UNII-UP7QBP99PN
- CLINICAL TRIALS….http://clinicaltrials.gov/search/intervention=Apremilast+OR+CC-10004
Apremilast is an orally available small molecule inhibitor of PDE4 being developed byCelgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
 APREMILAST
APREMILAST
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.
“Apremilast is Celgene’s lead oral phosphodiesterase IV inhibitor and anti-TNF alpha agent in phase III clinical studies at Celgene for the oral treatment of moderate to severe plaque-type psoriasis and for the oral treatment of psoriatic arthritis.
Early clinical development is also ongoing for the treatment of acne, Behcet’s disease, cutaneous sarcoidosis, prurigo nodularis, ankylosing spondylitis, atopic or contact dermatitis and rheumatoid arthritis. No recent development has been reported for research for the treatment of skin inflammation associated with cutaneous lupus erythematosus.
In 2011, Celgene discontinued development of the compound for the management of vision-threatening uveitis refractory to other modes of systemic immunosuppression due to lack of efficacy.
Celgene had been evaluating the potential of the drug for the treatment of asthma; however, no recent development has been reported for this research. The drug candidate is also in phase II clinical development at the William Beaumont Hospital Research Institute for the treatment of chronic prostatitis or chronic pelvic pain syndrome and for the treatment of vulvodynia (vulvar pain).
In 2013, orphan drug designations were assigned to the product in the U.S. and the E.U. for the treatment of Behcet’s disease.
Celgene Corp has been boosted by more impressive late-stage data on apremilast, an oral drug for psoriatic arthritis, this time in previously-untreated patients.
The company is presenting data from the 52-week PALACE 4 Phase III study of apremilast tested in PsA patients who have not taken systemic or biologic disease modifying antirheumatic drugs (DMARDs) at the American College of Rheumatology meeting in San Diego. The results from the 527-patient trial show that at week 16, patients on 20mg of the first-in-class oral inhibitor of phosphodiesterase 4 (PDE4) achieved an ACR20 (ie a 20% improvement in the condition) response of 29.2% and 32.3% for 30mg aapremilast, compared with 16.9% for those on placebo.
After 52 weeks, 53.4% on the lower dose and 58.7% on 30mg achieved an ACR20 response. ACR50 and 70 was reached by 31.9% and 18.1% of patients, respectively, for apremilast 30mg. The compound was generally well-tolerated and discontinuation rates for diarrhoea and nausea were less than 2% over 52 weeks.
Commenting on the data, Alvin Wells, of the Rheumatology and Immunotherapy Center in Franklin, Wisconsin, noted that apremilast demonstrated long-term safety and tolerability and significant clinical benefit in treatment-naive patients. He added that “these encouraging results suggest that apremilast may have the potential to be used alone and as a first-line therapy”. Celgene is also presenting various pooled data from the first three trials in the PALACE programme which, among other things, shows that apremilast significantly improves swollen and tender joints.
Treatment for PSA, which affects about 30% of the 125 million people worldwide who have psoriasis, currently involves injectable tumour necrosis factor (TNF) inhibitors, notably AbbVie’s Humira (adalimumab) and Pfizer/Amgen’s Enbrel (etanercept), once patients have not responded to DMARDs (at least in the UK). While the biologics are effective, the side effect profile can be a concern, due to the risk of infection and tuberculosis and many observers believe that apremilast will prove popular with patients and doctors due to the fact that it is oral, not injectable.
Apremilast was filed for PsA with the US Food and Drug Administration in the first quarter and will be submitted on both sides of the Atlantic for psoriasis before year-end. The European filing will also be for PsA.
Apremilast impresses for Behcet’s disease
Celgene has also presented promising Phase II data on apremilast as a treatment for the rare inflammatory disorder Behcet’s disease. 71% of patients achieved complete response at week 12 in clearing oral ulcers
 APREMILAST
APREMILAST
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06.
- Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor.
Man HW, Schafer P, Wong LM, Patterson RT, Corral LG, Raymon H, Blease K, Leisten J, Shirley MA, Tang Y, Babusis DM, Chen R, Stirling D, Muller GW.
J Med Chem. 2009 Mar 26;52(6):1522-4. doi: 10.1021/jm900210d.
- Therapeutics: Silencing psoriasis.Crow JM.Nature. 2012 Dec 20;492(7429):S58-9. doi: 10.1038/492S58a. No abstract available.
- NMR…http://file.selleckchem.com/downloads/nmr/S803401-Apremilast-HNMR-Selleck.pdf
- WO 2003080049
- WO 2013126495
- WO 2013126360
- WO 2003080049
- WO 2006065814
- US2003/187052 A1 …..MP 144 DEG CENT
- US2007/155791
-
J. Med. Chem., 2008, 51 (18), pp 5471–5489DOI: 10.1021/jm800582j
-
J. Med. Chem., 2011, 54 (9), pp 3331–3347DOI: 10.1021/jm200070e

…………………………………………
INTRODUCTION
2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione is a PDE4 inhibitor that is currently under investigation as an anti-inflammatory for the treatment of a variety of conditions, including asthma, chronic obstructive pulmonary disease, psoriasis and other allergic, autoimmune and rheumatologic conditions. S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.

Existing methods for synthesizing (S)-aminosulfone 1 involve resolution of the corresponding racemic aminosulfone by techniques known in the art. Examples include the formation and crystallization of chiral salts, and the use of chiral high performance liquid chromatography. See, e.g., Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972). In one example, as depicted in Scheme 1 below, (5)-aminosulfone 1 is prepared by resolution of racemic aminosulfone 3 with N-Ac-L-Leu. Racemic aminosulfone 3 is prepared by converting 3-ethoxy-4-methoxybenzonitrile 4 to enamine intermediate 5 followed by enamine reduction and borate hydrolysis. This process has been reported in U.S. Patent
Application Publication No. 2010/0168475.

CH2CI2, NaOH

Scheme 1
The procedure for preparing an enantiomerically enriched or enantiomerically pure aminosulfone, such as compound 1, may be inefficient because it involves the resolution of racemic aminosulfone 3. Thus, a need exists as to asymmetric synthetic processes for the preparation of an enantiomerically enriched or enantiomerically pure aminosulfone, particularly for manufacturing scale production. Direct catalytic asymmetric hydrogenation of a suitable enamine or ketone intermediate is of particular interest because it eliminates the need for either classic resolution or the use of stoichiometric amount of chiral auxiliary, and thus, may be synthetically efficient and economical.
……………………………………….
SYNTHESIS OF KEY INTERMEDIATE
Example 1
Synthesis of 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine

[00232] A slurry of dimethylsulfone (85 g, 903 mmol) in THF (480 ml) was treated with a
1.6M solution of n-butyllithium in hexane (505 ml, 808 mmol) at 0 – 5 °C. The resulting mixture was agitated for 1 hour then a solution of 3-ethoxy-4-methoxybenzonitrile (80 g, 451 mmol) in THF (240 ml) was added at 0 – 5 °C. The mixture was agitated at 0 – 5 °C for 0.5 hour, warmed to 25 – 30 °C over 0.5 hour and then agitated for 1 hour. Water (1.4 L) was added at 25 – 30 °C and the reaction mass was agitated overnight at room temperature (20 – 30 °C). The solid was filtered and subsequently washed with a 2: 1 mixture of water :THF (200 ml), water (200 ml) and heptane (2 x 200 ml). The solid was dried under reduced pressure at 40 – 45 °C to provide the product as a white solid (102 g, 83% yield); 1H NMR (DMSO-d6) δ 1.34 (t, J=7.0 Hz, 3H), 2.99 (s, 3H), 3.80 (s, 3H), 4.08 (q, J=7.0 Hz, 2H), 5.03 (s, 1H), 6.82 (s, 2H), 7.01 (d, J=8.5 Hz, 1H), 7.09 – 7.22 (m, 2H).
Example 2
Synthesis of (R)- 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine

[00233] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (36 mg, 0.074 mmol) and (i?)-l-[(5)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (40 mg, 0.074 mmol) in 25 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. The mixture was evaporated and the residue was purified by chromatography on a CI 8 reverse phase column using a water-acetonitrile gradient. The appropriate fractions were pooled and evaporated to -150 mL. To this solution was added brine (20 mL), and the resulting solution was extracted with EtOAc (3 x 50 mL). The combined organic layers were dried (MgS04) and evaporated to provide the product as a white crystalline solid (1.4 g, 70% yield); achiral HPLC (Hypersil BDS C8, 5.0 μπι, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 9.11 (99.6%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 7.32 (97.5%), 8.26 (2.47%); 1H NMR (DMSO-de) δ 1.32 (t, J= 7.0 Hz, 3H), 2.08 (s, 2H), 2.96 (s, 3H), 3.23 (dd, J= 3.6, 14.4 Hz, 1H), 3.41 (dd, J= 9.4, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 7.0 Hz, 2H), 4.26 (dd, J= 3.7, 9.3 Hz, 1H), 6.89 (s, 2H), 7.02 (s, 1H); 13C NMR (DMSO-d6) δ 14.77, 41.98, 50.89, 55.54, 62.03, 63.68, 111.48, 111.77, 118.36, 137.30, 147.93, 148.09. Example 3
Synthesis of (6 -l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine N-Ac-L-Leu salt

[00234] A solution of bis(l,5-cyclooctadiene)rhodium(I) trifluoromethanesulfonate (17 mg, 0.037 mmol) and (5)-l-[(i?)-2-(diphenylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (20 mg, 0.037 mmol) in 10 mL of 2,2,2-trifluoroethanol was prepared under nitrogen. To this solution was then charged l-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethenamine (2.0 g, 7.4 mmol). The resulting mixture was heated to 50 °C and hydrogenated under 90 psig hydrogen pressure. After 18 h, the mixture was cooled to ambient temperature and removed from the hydrogenator. Ecosorb C-941 (200 mg) was added and the mixture was stirred at ambient temperature for 3 h. The mixture was filtered through Celite, and the filter was washed with additional trifluoroethanol (2 mL). Then, the mixture was heated to 55 °C, and a solution of N- acetyl-L-leucine (1.3 g, 7.5 mmol) was added dropwise over the course of 1 h. Stirring proceeded at the same temperature for 1 h following completion of the addition, and then the mixture was cooled to 22 °C over 2 h and stirred at this temperature for 16 h. The crystalline product was filtered, rinsed with methanol (2 x 5 mL), and dried under vacuum at 45 °C to provide the product as a white solid (2.6 g, 80% yield); achiral HPLC (Hypersil BDS Cg, 5.0 μιη, 250 x 4.6 mm, 1.5 mL/min, 278nm, 90/10 gradient to 80/20 0.1% aqueous TFA/MeOH over 10 min then gradient to 10/90 0.1% aqueous TFA/MeOH over the next 15 min): 8.57 (99.8%); chiral HPLC (Chiralpak AD-H 5.0 μιη Daicel, 250 x 4.6 mm, 1.0 mL/min, 280 nm, 70:30:0.1 heptane-z-PrOH-diethylamine): 8.35 (99.6%); 1H NMR (DMSO-<¾) δ 0.84 (d, 3H), 0.89 (d, J= 6.6 Hz, 3H), 1.33 (t, J= 7.0 Hz, 3H), 1.41 – 1.52 (m, 2H), 1.62 (dt, J= 6.7, 13.5 Hz, 1H), 1.83 (s, 3H), 2.94 (s, 3H), 3.28 (dd, J= 4.0, 14.4 Hz, 1H), 3.44 (dd, J= 9.1, 14.4 Hz, 1H), 3.73 (s, 3H), 4.02 (q, J= 6.9 Hz, 2H), 4.18 (q, J= 7.7 Hz, 1H), 4.29 (dd, J= 4.0, 9.1 Hz, 1H), 5.46 (br, 3H), 6.90 (s, 2H), 7.04 (s, 1H), 8.04 (d, J= 7.9 Hz, 1H); Anal. (C20H34N2O7S) C, H, N. Calcd C, 53.79; H, 7.67; N 6.27. Found C, 53.78; H, 7.57; N 6.18.
SUBSEQUENT CONVERSION
S-enantiomer form of 2-[l-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4- acetylaminoisoindoline-l ,3-dione can be prepared by reacting (5)-aminosulfone 1 with intermediate 2.
……………………………………
 APREMILAST
APREMILAST
GENERAL SYNTHESIS AND SYNTHESIS OF APREMILAST




(apremilast)
[0145] Preparation of 3-Ethoxy-4-methoxybenzonitrile (Compound 2). 3-Ethoxy-
4-methoxybenzaldehyde (Compound 1, 10.0 gm, 54.9 mmol, Aldrich) and hydroxylamine hydrochloride (4.67 gm, 65.9 mmol, Aldrich) were charged to a 250 mL three-necked flask at room temperature, followed by the addition of anhydrous acetonitrile (50 mL). The reaction mixture was stirred at room temperature for thirty minutes and then heated to reflux (oil bath at 85 °C). After two hours of reflux, the reaction mixture was cooled to room temperature, and added 50 mL of deionized water. The mixture was concentrated under reduced pressure to remove acetonitrile and then transferred to a separatory funnel with an additional 80 mL of deionized water and 80 mL dichloromethane. The aqueous layer was extracted with dichloromethane (3 x 50 mL). The combined organic layers were washed successively with water (80 mL) and saturated sodium chloride (80 mL). The organic layer was dried over anhydrous sodium sulfate (approximately 20 gm). The organic layer was filtered and concentrated under reduced pressure to give a yellow oil. Purification by silica gel chromatography (0 to 1 % MeOH/DCM ) afforded 3-Ethoxy-4-methoxybenzonitrile
(Compound 2) as a white solid (7.69 gm, 79 % yield). MS (ESI positive ion) m/z 178.1 (M + 1). HPLC indicated >99% purity by peak area. 1H-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 3.83 (s, 3H), 4.05 (q, 2H), 7.10 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.40 (dd, J = 2.0 Hz, 1H).
[0146] Preparation of l-(3-Ethoxy-4-methoxyphenyi)-2-
(niethylsulfonyl)ethanamine (Compound 3). Dimethyl sulfone (2.60 gm, 27.1 mmol, Aldrich) and tetrahydrofuran (10 mL, Aldrich) were charged to a 250 mL three-necked flask at room temperature. The mixture was cooled to 0 – 5 °C, and the solution gradually turned white. n-Butyllithium (10.8 mL, 27.1 mmol, 2.5 M solution in hexanes, Aldrich) was added to the flask at a rate such that the reaction mixture was maintained at 5 – 10 °C. The mixture was stirred at 0 – 5 °C for one hour, turning light-yellow. 3-Ethoxy-4-methoxybenzonitrile (Compound 2, 4.01 gm, 22.5 mmol) in tetrahydrofuran (8 mL) was then charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for another 15 minutes. After warming to room temperature, the reaction mixture was stirred for another 1.5 hours and then transferred to a second 250 mL three-necked flask containing a suspension of sodium borohydride (1.13 gm, 29.3 mmol, Aldrich) in
tetrahydrofuran (1 1 mL), maintained at – 5 – 0 °C for 30 minutes. Trifluoroacetic acid (“TFA,” 5.26 mL, 68.3 mmol, Aldrich) was charged to the flask at a rate such that the reaction mixture was maintained at 0 – 5 °C. The mixture was stirred at 0 – 5 °C for 40 minutes and an additional 17 hours at room temperature. The reaction mixture was then charged with 2.7 mL of deionized water over five minutes at room temperature. The mxiture was stirred at room temperature for 15 hours. Aqueous NaOH (10 N, 4.9 mL) was charged to the flask over 15 minutes at 45 °C. The mixture was stirred at 45 °C for two hours, at 60 °C for 1.5 hours, and at room temperature overnight. After approximately 17 hours at room temperature the mixture was cooled to 0 °C for thirty minutes and then concentrated under reduced pressure. The residual material was charged with deionized water (3 mL) and absolute ethanol (3 mL) and stirred at 0 – 5 °C for 2 hours. The mixture was filtered under vacuum, and the filtered solid was washed with cold absolute ethanol (3 x 5 mL), followed by deionized water until the pH of the wash was about 8. The solid was air dried overnight, and then in a vacuum oven at 60 °C for 17 hours to afford Compound 3 as a white solid (4.75 gm, 77 %). MS (ESI positive ion) m/z 274.1 (M + 1). Ή-NMR (500 MHz, DMSO-c¾): δ ppm 1.32 (t, J = 7.0 Hz, 3H), 2.08 (bs, 2H), 2.95 (s, 3H), 3.23 (dd, J = 4.0 Hz, 1H), 3.40 (dd, J = 9.5 Hz, 1H), 3.72 (s, 3H), 4.01 (q, J = 7.0 Hz, 2H), 4.25 (dd, J = 3.5 Hz, 1H), 6.88 (s, 2H), 7.02 (s, 1H).
[0147] Preparation of 4-Nitroisobenzofuran-l,3-dione (Compound 5). Into a 250 mL round bottom flask, fitted with a reflux condenser, was placed 3-nitrophthalic acid (21.0 gm, 99 mmol, Aldrich) and acetic anhydride (18.8 mL, 199 mmol, Aldrich). The solid mixture was heated to 85 °C, under nitrogen, with gradual melting of the solids. The yellow mixture was heated at 85 °C for 15 minutes, and there was noticeable thickening of the mixture. After 15 minutes at 85 °C, the hot mixture was poured into a weighing dish, and allowed to cool. The yellow solid was grinded to a powder and then placed on a cintered funnel, under vacuum. The solid was washed with diethyl ether (3 x 15 mL), under vacuum and allowed to air dry overnight, to afford 4-nitroisobenzofuran-l ,3-dione, Compound 5, as a light-yellow solid (15.8 gm, 82 %). MS (ESI positive ion) m/z 194.0 (M + 1). TLC: Rf = 0.37 (10% MeOH/DCM with 2 drops Acetic acid) Ή-NMR (500 MHz, DMSO-i¾: δ ppm 8.21 (dd, J = 7.5 Hz, 1H), 8.39 (dd, J = 7.5 Hz, 1H), 8.50 (dd, J = 7.5 Hz, 1 H).
[0148] Preparation of 2-(l-(3-Ethoxy-4-methoxyphenyI)-2-
(methylsulfonyl)ethyl)-4-nitroisoindoline-l,3-dione (Compound 6). Into a 2 – 5 mL microwave vial was added 4-nitroisobenzofuran-l ,3-dione (Compound 5, 0.35 gm, 1.82 mmol), the amino-sulfone intermediate (Compound 3, 0.50 gm, 1.82 mmol) and 4.0 mL of glacial acetic acid. The mixture was placed in a microwave at 125 °C for 30 minutes. After 30 minutes the acetic acid was removed under reduced pressure. The yellow oil was taken up in ethyl acetate and applied to a 10 gm snap Biotage samplet. Purification by silica gel chromatography (0 to 20 % Ethyl Acetate/Hexanes) afforded Compound 6 as a light-yellow solid (0.67 gm, 82 %). MS (ESI positive ion) m/z 449.0 (M + 1). TLC: Rf = 0.19
(EtOAc:Hexanes, 1 : 1). HPLC indicated 99% purity by peak area. Ή-NMR (500 MHz, DMSO-c¾: δ ppm 1.32 (t, 3H), 2.99 (s, 3H), 3.73 (s, 3H), 4.02 (m, 2H), 4.21 (dd, J = 5.0 Hz, 1H), 4.29 (dd, J = 10.0 Hz, 1H), 5.81 (dd, J = 5.0 Hz, 1H), 6.93 (d, J – 8.5 Hz, 1H), 7.00 (dd, J = 2.0 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 8.07 (t, J = 15.5 Hz, 1H), 8.19 (dd, J = 8.5 Hz, 1H), 8.30 (dd, J = 9.0 Hz, 1H).
[0149] Preparation of 4-Amino-2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsulfonyl)ethyl)isoindoline-l,3-dione (Compound 7). Compound 6 (0.54 gm, 1.20 mmol) was taken up in ethyl acetate / acetone (1 : 1 , 24 mL) and flowed through the H-cube™ hydrogen reactor using a 10 % Pd/C CatCart™ catalyst cartridge system (ThalesNano, Budapest Hungary). After eluting, the yellow solvent was concentrated under reduced pressure to give Compound 7 as a yellow foam solid (0.48 gm, 95 %). MS (ESI positive ion) m/z 419.1 (M + 1). 1H-NMR (500 MHz, DMSO-<¾): δ ppm 1.31 (t, J = 7.0 Hz, 3H), 2.99 (s, 3H), 3.72 (s, 3H), 4.04 (q, J = 7.0 Hz, 2H), 4.09 (m, 1H), 4.34 (m, 1H), 5.71 (dd, J = 5.5 Hz, 1H), 6.52 (bs, 2H), 6.92-6.98 (m, 3H), 7.06 (bs, 1 H), 7.42 (dd, J = 7.0 Hz, 1H).
[0150] Preparation of N-(2-(l-(3-ethoxy-4-methoxyphenyl)-2-
(methylsuIfonyl)ethyl)-l,3-dioxoisoindolin-4-yl)acetamide (Apremilast, Compound 8).
Into a 2-5 mL microwave vial was placed Compound 7 (0.18 gm, 0.43 mmol), acetic anhydride (0.052 mL, 0.53 mmol) and acetic acid (4 mL). The microwave vial was placed into a Biotage microwave and heated to 125 °C for 30 minutes. The solvents were removed under reduced pressure and the residue was purified by silica gel chromatography (0 to 5% MeOH/DCM) to afford apremilast (Compound 8) as a yellow oil (0.14 gm, 71%). HPLC indicated 94.6% purity by peak area.
1H-NMR (500 MHz, DMSO-c 6): δ ppm 1.31 (t, 3H), 2.18 (s, 3H), 3.01 (s, 3H), 3.73 (s, 3H), 4.01 (t, J = 7.0 Hz, 2H), 4,14 (dd, J = 4.0 Hz, 1H), 4.33 (m, 1H), 5.76 (dd, J = 3.0 Hz, 1H), 6.95 (m, 2H), 7.06 (d, J = 1.5 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 8.43 (d, J = 8.5 Hz, 1H), 9.72 (bs, 1H).
……………………..
SYNTHESIS
5. EXAMPLES
Certain embodiments provided herein are illustrated by the following non-limiting examples.
5.1 PREPARATION OF (+)-2-[l-(3-ETHOXY-4-METHOXYPHENYL)-2- METHANESULFONYLETHYLJ-4- ACETYL AMINOISOINDOLIN-1,3- DIONE (APREMILAST)

5.1.1 Preparation of 3-aminopthalic acid
10% Pd/C (2.5 g), 3-nitrophthalic acid (75.0 g, 355 mmol) and ethanol (1.5 L) were charged to a 2.5 L Parr hydrogenator under a nitrogen atmosphere. Hydrogen was charged to the reaction vessel for up to 55 psi. The mixture was shaken for 13 hours, maintaining hydrogen pressure between 50 and 55 psi. Hydrogen was released and the mixture was purged with nitrogen 3 times. The suspension was filtered through a celite bed and rinsed with methanol. The filtrate was concentrated in vacuo. The resulting solid was reslurried in ether and isolated by vacuum filtration. The solid was dried in vacua to a constant weight, affording 54 g (84%> yield) of 3-aminopthalic acid as a yellow product. 1H-NMR (DMSO-d6) δ: 3.17 (s, 2H), 6.67 (d, 1H), 6.82 (d, 1H), 7.17 (t, 1H), 8-10 (brs, 2H). 13C-NMR(DMSO-d6) δ: 112.00, 115.32, 118.20, 131.28, 135.86, 148.82, 169.15, 170.09.
5.1.2 Preparation of 3-acetamidopthalic anhydride
A I L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 3-aminophthalic acid (108 g, 596 mmol) and acetic anhydride (550 mL). The reaction mixture was heated to reflux for 3 hours and cooled to ambient temperature and further to 0-5. degree. C. for another 1 hour. The crystalline solid was collected by vacuum filtration and washed with ether. The solid product was dried in vacua at ambient temperature to a constant weight, giving 75 g (61% yield) of 3-acetamidopthalic anhydride as a white product. 1H-NMR (CDCI3) δ: 2.21 (s, 3H), 7.76 (d, 1H), 7.94 (t, 1H), 8.42 (d, 1H), 9.84 (s, 1H).
5.1.3 Resolution of 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)- ethyl-2-amine
A 3 L 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser and charged with 2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine (137.0 g, 500 mmol), N-acetyl-L-leucine (52 g, 300 mmol), and methanol (1.0 L). The stirred slurry was heated to reflux for 1 hour. The stirred mixture was allowed to cool to ambient temperature and stirring was continued for another 3 hours at ambient temperature. The slurry was filtered and washed with methanol (250 mL). The solid was air-dried and then dried in vacuo at ambient temperature to a constant weight, giving 109.5 g (98% yield) of the crude product (85.8% ee). The crude solid (55.0 g) and methanol (440 mL) were brought to reflux for 1 hour, cooled to room temperature and stirred for an additional 3 hours at ambient temperature. The slurry was filtered and the filter cake was washed with methanol (200 mL). The solid was air-dried and then dried in vacuo at 30°C. to a constant weight, yielding 49.6 g (90%> recovery) of (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-ylamine-N-acety 1-L-leucine salt (98.4% ee). Chiral HPLC (1/99 EtOH/20 mM KH2P04 @pH 7.0, Ultron Chiral ES-OVS from Agilent Technologies, 150 mm.times.4.6 mm, 0.5 mL/min., @240 nm): 18.4 min (S-isomer, 99.2%), 25.5 min (R-isomer, 0.8%)
5.1.4 Preparation of (+)-2-[l-(3-ethoxy-4-methoxyphenyl)-2- methanesulfonylethyl] -4-acetylaminoisoindolin- 1 ,3-dione
A 500 mL 3 -necked round bottom flask was equipped with a mechanical stirrer,
thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), 3-acetamidophthalic anhydride (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50°C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x
2), saturated aqeous NaHC03 (250 mL.times.2), brine (250 mL.times.2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL.times.2). The product was dried in vacuo at 60°C. to a constant weight, affording 19.4 g (75% yield) of Compound 3 APREMILAST with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 @pH 3.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., @240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%).
1H-NMR (CDC13) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H).
13C-NMR(DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………………………………..
NMR
1H-NMR (CDCl3) δ: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). 13C-NMR (DMSO-d6) δ: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48.
…………….

aReagents and conditions: (a) LiN(SiMe3)2, then Me2SO2/n-BuLi/BF3Et2O, −78 °C; (b) N-Ac-l-leucine, MeOH; (c) HOAc, reflux.
……………………
SARCOIDOSIS
Sarcoidosis is a disease of unknown cause. Sarcoidosis is characterized by the presence of granulomas in one or more organ systems. The most common sites of involvement are the lungs and the lymph nodes in the mediastinum and hilar regions. However, sarcoidosis is a systemic disease and a variety of organ systems or tissues may be the source of primary or concomitant clinical manifestations and morbidity. The clinical course of sarcoidosis is extremely variable, and ranges from a mild or even asymptomatic disease with spontaneous resolution to a chronic progressive disease leading to organ system failure and, in 1-5% of cases, death. See Cecil
Textbook of Medicine, 21st ed. (Goldman, L., Bennett, J. C. eds), W. B. Saunders Company, Philadelphia, 2000, p. 433-436.
While the cause of sarcoidosis is unknown, a substantial body of information suggests that immune mechanisms are important in disease pathogenesis. For example, sarcoidosis is
characterized by enhanced lymphocyte and macrophage activity. See Thomas, P.D. and
Hunninghake, G.W., Am. Rev. Respir. Dis., 1987, 135: 747-760. As sarcoidosis progresses, skin rashes, erythema nodosum and granulomas may form. Granulomas or fibrosis caused by sarcoidosis can occur throughout the body, and may affect the function of vital organs such as the lungs, heart, nervous system, liver or kidneys. In these cases, the sarcoidosis can be fatal. See
http://www.nlm.nih.gov/medlineplus/sarcoidosis.html (accessed November 12, 2009).
Moreover, a variety of exogenous agents, both infectious and non-infectious, have been hypothesized as a possible cause of sarcoidosis. See Vokurka et ah, Am. J. Respir. Crit. Care Med., 1997, 156: 1000-1003; Popper et al, Hum. Pathol, 1997, 28: 796-800; Almenoff et al, Thorax, 1996, 51 : 530-533; Baughman et al., Lancet, 2003, 361 : 1111-1118. These agents include mycobaceria, fungi, spirochetes, and the agent associated with Whipple’s disease. Id.
Sarcoidosis may be acute or chronic. Specific types of sarcoidosis include, but are not limited to, cardiac sarcoidosis, cutaneous sarcoidosis, hepatic sarcoidosis, oral sarcoidosis, pulmonary sarcoidosis, neurosarcoidosis, sinonasal sarcoidosis, Lofgren’s syndrome, lupus pernio, uveitis or chronic cutaneous sarcoidosis.
As the lung is constantly confronted with airborne substances, including pathogens, many researchers have directed their attention to identification of potential causative transmissible agents and their contribution to the mechanism of pulmonary granuloma formation associated with sarcoidosis. See Conron, M. and Du Bois, R.M., Clin. Exp. Allergy, 2001, 31 : 543-554; Agostini et al, Curr. Opin. Pulm. Med. , 2002, 8: 435-440.
Corticosteroid drugs are the primary treatment for the inflammation and granuloma formation associated with sarcoidosis. Rizatto et al. , Respiratory Medicine, 1997, 91 : 449-460. Prednisone is most often prescribed drug for the treatment of sarcoidosis. Additional drugs used to treat sarcoidosis include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, minocycline, doxycycline and chloroquin. TNF-a blockers such as thalidomide and infliximab have been reported to be effective in treating patients with sarcoidosis. Baughman et al, Chest, 2002, 122: 227-232; Doty et al, Chest, 2005, 127: 1064-1071. Antibiotics have also been studied for the treatment of sarcoidosis, such as penicillin antibiotics, cephalosporin antibiotics, macrolide antibiotics, lincomycin antibiotics, and tetracycline antibiotics. Specific examples include minocycline hydrochloride, clindamycin, ampicillin, or clarithromycin. See, e.g., U.S. Patent Publication No. 2007/0111956.
There currently lacks a Food and Drug Administration-approved therapeutic agent for the treatment of sarcoidosis, and many patients are unable to tolerate the side effects of the standard corticosteroid therapy. See Doty et al, Chest, 2005, 127: 1064-1071. Furthermore, many cases of sarcoidosis are refractory to standard therapy. Id. Therefore, a demand exists for new methods and compositions that can be used to treat patients with sarcoidosis.
……………..
PATENTS
|
8-15-2012
|
PROCESSES FOR THE PREPARATION OF AMINOSULFONE COMPOUNDS
|
|
|
11-4-2011
|
HETEROCYCLIC COMPOUNDS AS PHOSPHODIESTERASE INHIBITORS
|
|
|
5-27-2011
|
Nanosuspension of a Poorly Soluble Drug via Microfluidization Process
|
|
|
5-28-2010
|
METHODS AND COMPOSITIONS USING PDE4 INHIBITORS FOR THE TREATMENT AND MANAGEMENT OF CANCERS
|

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
TOSEDOSTAT ….An aminopeptidase inhibitor with antineoplastic activity.

TOSEDOSTAT
An aminopeptidase inhibitor with antineoplastic activity.
- CHR 2797
- CHR-2797
- Tosedostat
- UNII-KZK563J2UW
- BB-76163Vernalis (Originator)
| CAS No. | 238750-77-1 |
| Chemical Name: | Tosedostat |
| Synonyms: | BB-76163;Chr-2797;tosedostat;CHR2797 (Tosedostat);Tosedostat (CHR2797);α-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-benzeneaceticacidcyclopentlyester;alpha-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]benzeneacetic acid cyclopentyl ester;Benzeneacetic acid, alpha-(((2R)-2-((1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl)-4-methyl-1-oxopentyl)amino)-, cyclopentyl ester, (alphas)- |
| Molecular Formula: | C21H30N2O6 |
| Formula Weight: | 406.47 |
CHR-2797 is an oral, once-daily experimental cancer therapy in phase II clinical development at Chroma Therapeutics for the oral treatment of refractory acute myeloid leukemia in elderly patients. It is also in early clinical development for the treatment of refractory solid tumors alone or in combination with chemotherapy.
No recent development has been reported for phase I/II studies evaluating CHR-2797 as monotherapy in hematologic/blood cancer. A phase I/II clinical trial of the compound in combination with erlotinib for non-small cell lung cancer was terminated in 2010 due to very poor recruitment of patients to the study.
Cell Therapeutics is also conducting phase II clinical trials of the compound for the treatment of myelodysplasia and acute myeloid leukemia.
CHR- 2797 is an inhibitor of aminopeptidases and has demonstrated strong preclinical efficacy as monotherapy in addition to demonstrating strong synergy with a number of leading cancer therapies in a range of cancer cells. It was originally licensed from Vernalis, where it was being evaluated for its potential in treating multiple sclerosis; however development in this indication has been discontinued.
In 2008, orphan drug designation was assigned to CHR-2797 in the U.S. for the treatment of acute myeloid leukemia. In 2011, the compound was licensed to Cell Therapeutics by Chroma Therapeutics in Central America, North America and South America for exclusive marketing and codevelopment for the oral treatment of blood-related cancers and other cancers.
In corporate news, biopharmaceutical company Cell Therapeutics, Inc. (CTIC) was up more than 6% and near 52 week highs after saying Thursday that the U.S. FDA has removed the partial clinical hold on tosedostat and all studies underway have been allowed to continue. Tosedostat is under development for the treatment of blood-related cancers. It is currently being studied in Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndromes.

Tosedostat is a proprietary orally bioavailable inhibitor of the M1 family of aminopeptidases with potential antineoplastic activity.
Tosedostat is converted intracellularly into a poorly membrane-permeable active metabolite (CHR-79888) which inhibits the M1 family of aminopeptidases, particularly puromycin-sensitive aminopeptidase (PuSA), and leukotriene A4 (LTA4) hydrolase; inhibition of these aminopeptidases in tumor cells may result in amino acid deprivation, inhibition of protein synthesis due to a decrease in the intracellular free amino acid pool, an increase in the level of the proapoptotic protein Noxa, and cell death.
Noxa is a member of the BH3 (Bcl-2 homology 3)-only subgroup of the proapoptotic Bcl-2 (B-cell CLL/lymphoma 2) protein family
Cell Therapeutics announced that it has received notification from the U.S. Food and Drug Administration (FDA) that the partial clinical hold on tosedostat (IND 075503) has been removed and all studies underway may continue. Tosedostat is a first-in-class selective inhibitor of aminopeptidases, which are required by tumor cells to provide amino acids necessary for growth and tumor cell survival, and is under development for the treatment of blood-related cancers.
Tosedostat is currently being studied in the United States and European Union in investigator-sponsored and cooperative group-sponsored Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDS).
“We are pleased that the FDA has responded favorably to the tosedostat clinical trial data provided and removed the partial clinical hold to allow further development of tosedostat in ongoing and future studies,” said John Pagel, MD, PhD, Associate Member, Clinical Research Division, Fred Hutchinson Cancer Research Center; Associate Professor, Medical Oncology Division, University of Washington School of Medicine; and Principal Investigator in the tosedostat first-line AML/MDS trial.
Recently, WO 93/20047 disclosed a class of hydroxamic acid based MMP inhibitors which also are active in inhibiting TNF production.
As mentioned above, MMP inhibitors have been proposed with hydroxamic acid or carboxylic acid zinc binding groups. The following patent publications disclose hydroxamic acid-based MMP inhibitors:
US 4599361 (Searle) EP-A-0236872 (Roche) EP-A-0274453 (Bellon) WO 90/05716 (British Bio-technology) WO 90/05719 (British Bio-technology) WO 91/02716 (British Bio-technology) EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) EP-A-0497192 (Roche) WO 92/13831 (British Bio-technology) WO 92/17460 (SmithKline Beecham) WO 92/22523 – (Research Corporation Technologies) WO 93/09090 (Yamanouchi) WO 93/09097 (Sankyo) WO 93/20047 (British Bio-technology) WO 93/24449 (Celltech) WO 93/24475 (Celltech) EP-A-0574758 (Roche) The following patent publications disclose carboxylic acid-based MMP inhibitors:
EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) WO 93/24449 (Celltech) WO 93/24475 (Celltech)
|
|
|
TOSEDOSTAT
| WO1996033166A1 * | 17 Apr 1996 | 24 Oct 1996 | Du Pont Merck Pharma | Hydroxamic and carboxylic acids as metalloprotease inhibitors |
| WO1998011063A1 * | 8 Sep 1997 | 19 Mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| GB2268934A * | Title not available |
| US5652262 * | 14 mar 1994 | 29 lug 1997 | British Biotech Pharmaceutical, Ltd. | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| US5821262 * | 4 ott 1994 | 13 ott 1998 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as inhibitors of cytokine production |
| US5861436 * | 29 apr 1997 | 19 gen 1999 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| EP0423943A2 | 19 set 1990 | 24 apr 1991 | Beecham Group p.l.c. | Use of collagenase inhibitors in the treatment of demyelinating diseases, in particular multiple sclerosis |
| JPH03157372A | Titolo non disponibile | |||
| WO1997049674A1 | 20 giu 1997 | 31 dic 1997 | Francesca Abrate | Matrix metalloproteinase inhibitors |
| WO1998011063A1 | 8 set 1997 | 19 mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| WO1999040910A1 | 27 gen 1999 | 19 ago 1999 | Andrew Paul Ayscough | Anti-inflammatory agents |
| WO1999044602A1 | 5 mar 1999 | 10 set 1999 | British Biotech Pharm | Inflammatory cell inhibitors |
| WO1999046241A1 | 12 mar 1998 | 16 set 1999 | British Biotech Pharm | Cytostatic agents |
| WO2000044373A1 * | Jan 27, 2000 | Aug 3, 2000 | Raymond Paul Beckett | Antibacterial hydroxamic acid derivatives |
| US6545051 | Jan 27, 2000 | Apr 8, 2003 | British Biotech Pharmaceuticals, Ltd. | Antibacterial hydroxamic acid derivatives |
Drugs Fut 2009, 34(2): 115
PLoS One (2013), 8(2), e57641.
WO 1999046241
WO 1995019956
WO 1998011063
US 6462023
US 20100260674
WO 2000044373
WO 9940910
NMR
http://file.selleckchem.com/downloads/nmr/S152202-CHR-2797-NMR-Selleck.pdf
Anti-Metastatic and Anti-Invasive Agents Compounds which have the property of inhibiting the action of the metalioproteinase enzymes involved in connective tissue breakdown and remodelling, such as fibroblast collagenase (Type 1 ), PMN-collagenase, 72 kDa-gelatinase, 92 kDa- gelatinase, stromelysin, stromelysin-2 and PUMP-1 (known as “matrix metalloproteinases”, and herein referred to as MMPs) have been proposed and are being tested in the clinic for the treatment of solid tumours. Cancer cells are particularly adept at utilising the MMPs to achieve rapid remodelling of the extracellular matrix, thereby providing space for tumour expansion and permitting metastasis. MMP inhibitors should minimise these processes and thus slow or prevent cancer progression.
In view of the rapid emergence of multidrug-resistant bacteria, the development of antibacterial agents with novel modes of action that are effective against the growing number of resistant bacteria, particularly the vancomycin resistant enterococci and β-lactam antibiotic-resistant bacteria, such as methicillin-resistant Staphylocccus aureus, is of utmost importance.
The natural antibiotic actinonin (see for example J. C. S Perkin I, 1975, 819) is a hydroxamic acid derivative of Structure (A):

In ddition to actinonin, various structural analogues of actinonin have also been shown to have antibacterial activity (see for example Broughton et al. (Devlin et al. Journal of the Chemical Society. Perkin Transactions 1 (9):830-841, 1975; Broughton et al. Journal of the Chemical Society. Perkin Transactions 1 (9):857-860, 1975).
The matlystatin group of compounds, share a number of structural similarities with actinonin. Both are peptidic molecules with functional hydroxamic acid metal binding groups (Ogita et al., J. Antibiotics. 45(11):1723-1732; Tanzawa et al., J. Antibiotics. 45(11):1733-1737; Haruyama et al., J. Antibiotics. 47(12):1473-1480; Tamaki et al., J. Antibiotics. 47(12):1481-1492).
………………………………………………………….
EXAMPLE 44 2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl-pentanoylamine]-2-phenyl-ethanoic acid cyclopentyl ester

The above compound was prepared using procedures similar to those described in example 8 using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1H, s), 5.2-5.14 (1H, m), 4.02 (1H, d, J=6.9 Hz), 2.94-2.85 (1H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1H, m) and 0.86 (6H, dd, J=6.5, 11 5 Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1, 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; 8 (MeOD), 7.33-7.19 (5H, m), 5.3 (1H, s), 5.11-5.06 (1H, m), 3.81 (1H, d, J=7.3 Hz), 2.83-2.74 (lH, m), 1.83-1.45 (10H, bm), 1.12-1.03 (lH, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3 Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2
Example 1
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

HO Ξ CONHOH
Prepared using procedures similar to those described in Preparative Example A using phenylglycine cyclopentyl ester.
Diastereoisomer A
Η-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d,
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, 14 dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2,
58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
Η-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d, J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.

tosedostat
http://www.google.it/patents/US6545051

42
| WO98/11063 | WO99/46241 ex 1b | WO 98/11063 analogy ex 8 |

43
| WO98/11063 | WO99/46241 ex 1a | WO 98/11063 analogy ex 8 |
……………………………………………………………………
entry 65 in http://www.google.com/patents/WO2000044373A1
……………………………………………………………………………………………………….
http://www.google.com/patents/WO1999044602A1
Example 43
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester
TC

HO Ξ CONHOH
Prepared using procedures similar to those described in example 8 of WO 98/11063, using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d, 34
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d,
J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and
0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3,
129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
……………………………..
3R-isobutyl-4S-methoxy-dihydrofuran-2,5-dione (WO 97/02239)
…………………………………………………………………………..
2(S)-Amino(phenyl)ethanoic acid cyclopentyl ester

…………………………………………………………………..
2(R)-[2,2-Dimethyl-5-oxo-1,3-dioxolan-4(S)-yl]-4-methylpentanoic acid pentafluorophenyl ester

…………………………………………………………..
intermediates
238750-91-9
α-amino-, cyclopentyl ester Benzeneacetic acid,
……………….
cas 240489-34-3
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

…………………..
will be updated very soon… keep watching

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

MARIZOMIB, Salinosporamide A
MARIZOMIB
http://www.ama-assn.org/resources/doc/usan/marizomib.pdf
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. 6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione, 4-(2-chloroethyl)-1-[(S)-(1S)-2-
cyclohexen-1-ylhydroxymethyl]-5-methyl-, (1R,4R,5S)-
2. (1R,4R,5S)-4-(2-chloroethyl)-1-{(S)-[(1S)-cyclohex-2-en-1-yl]hydroxymethyl}-5-methyl-
6-oxa-2-azabicyclo[3.2.0]heptane-3,7-dione
MOLECULAR FORMULA C15H20ClNO4
MOLECULAR WEIGHT 313.8
MANUFACTURER Nereus Pharmaceuticals, Inc.
NOTE….Nereus Pharmaceuticals was acquired by Triphase Research and Development in 2012.
CODE DESIGNATION NPI-0052
CAS REGISTRY NUMBER 437742-34-2
Scripps Institution of Oceanography (Originator)
mp, 168–170° C. (authentic sample: 168–170° C., 169–171° C. in Angew. Chem. Int. Ed., 2003, 42, 355–357); mixture mp, 168–170C.
[α]23 D −73.2 (c 0.49, MeOH), −72.9 (c 0.55, MeOH, in Angew. Chem. Int. Ed., 2003, 42, 355–357);
FTIR (film) νmax: 3406, 2955, 2920, 2844, 1823, 1701, 1257, 1076, 1012, 785, 691 cm−1;
1H NMR (CDCl3, 500 MHz): δ 10.62 (1H, br), 6.42 (1H, d, J=10.5 Hz), 5.88 (1H, m), 4.25 (1H, d, J=9.0 Hz), 4.14 (1H, m), 4.01 (1H, m), 3.17 (1H, t, J=7.0 Hz), 2.85 (1H, m), 2.48 (1H, m), 2.32 (2H, m), 2.07 (3H, s), 1.91 (2H, m), 1.66 (2H, m), 1.38 (1H, m);
13C NMR (CDCl3, 125 MHz): δ 176.92, 169.43, 129.08, 128.69, 86.32, 80.35, 70.98, 46.18, 43.28, 39.31, 29.01, 26.47, 25.35, 21.73, 20.00;
HRMS (ESI) calcd. for (M−H)− C15H19ClNO4 312.1003, found 312.1003.
Marizomib, a highly potent proteasome inhibitor, is in early clinical development at Triphase Research and Development I Corp for the treatment of relapsed or relapsed/refractory multiple myeloma. Phase I clinical trials have also been carried out for the treatment of solid tumors and lymphoma; however, no recent developments have been reported for these studies.
HDAC inhibitors halt tumor cell differentiation and growth, and when combined with marizomib in preclinical in vitro and in vivo studies, show additive and synergistic antitumor activities.
The compound was discovered from a new marine-obligate gram-positive actinomycete (Salinispora tropica). Preclinical studies suggest that this next-generation compound may be superior to other proteasome inhibitors, with broader target inhibition, faster onset and longer duration of action, higher potency, and oral and intravenous availability. By inhibiting proteasomes, marizomib prevents the breakdown of proteins involved in signal transduction, which blocks growth and induces apoptosis in cancer cells.
In 2013, orphan drug designation was assigned in the U.S. for the treatment of multiple myeloma.
The compound was originally developed by Nereus Pharmaceuticals, which was acquired by Triphase Research and Development in 2012.
marizomib is a naturally-occurring salinosporamide, isolated from the marine actinomycete Salinospora tropica, with potential antineoplastic activity. Marizomib irreversibly binds to and inhibits the 20S catalytic core subunit of the proteasome by covalently modifying its active site threonine residues; inhibition of ubiquitin-proteasome mediated proteolysis results in an accumulation of poly-ubiquitinated proteins, which may result in the disruption of cellular processes, cell cycle arrest, the induction of apoptosis, and the inhibition of tumor growth and angiogenesis. This agent more may more potent and selective than the proteasome inhibitor bortezomib
Marizomib (NPI-0052) is an oral, irreversible ββ-lactone derivative that binds selectively to the active proteasomal sites. In vivo studies with marizomib demonstrate reduced tumor growth without significant toxicity in myeloma xenograft models. A phase I trial in refractory and relapsed MM is under way.
Salinosporamide A is a potent proteasome inhibitor used as an anticancer agent that recently entered phase I human clinical trials for the treatment of multiple myeloma only three years after its discovery.[1][2] This novel marine natural product is produced by the recently described obligate marine bacteria Salinispora tropica and Salinispora arenicola, which are found in ocean sediment. Salinosporamide A belongs to a family of compounds, known collectively as salinosporamides, which possess a densely functionalized γ-lactam-β-lactone bicyclic core.
Salinosporamide A was discovered by William Fenical and Paul Jensen from Scripps Institution of Oceanography in La Jolla, CA. In preliminary screening, a high percentage of the organic extracts of cultured Salinospora strains possessed antibiotic and anticancer activities, which suggests that these bacteria are an excellent resource for drug discovery.Salinospora strain CNB-392 was isolated from a heat-treated marine sediment sample and cytotoxicity-guided fractionation of the crude extract led to the isolation of salinosporamide A. Although salinosporamide A shares an identical bicyclic ring structure with omuralide, it is uniquely functionalized. Salinosporamide A displayed potent in vitro cytotoxicity against HCT-116 human colon carcinoma with an IC50 value of 11 ng mL-1. This compound also displayed potent and highly selective activity in the NCI’s 60-cell-line panel with a mean GI50 value (the concentration required to achieve 50% growth inhibition) of less than 10 nM and a greater than 4 log LC50 differential between resistant and susceptible cell lines. The greatest potency was observed against NCI-H226 non-small cell lung cancer, SF-539 CNS cancer, SK-MEL-28 melanoma, and MDA-MB-435 breast cancer (all with LC50 values less than 10 nM). Salinosporamide A was tested for its effects on proteasome function because of its structural relationship to omuralide. When tested against purified 20S proteasome, salinosporamide A inhibited proteasomal chymotrypsin-like proteolytic activity with an IC50 value of 1.3 nM.[3] This compound is approximately 35 times more potent than omuralide which was tested as a positive control in the same assay. Thus, the unique functionalization of the core bicyclic ring structure of salinosporamide A appears to have resulted in a molecule that is a significantly more potent proteasome inhibitor than omuralide.[1]
Salinosporamide A inhibits proteasome activity by covalently modifying the active site threonine residues of the 20S proteasome.
Biosynthesis

Salinosporamide A and B building blocks

Proposed biosynthesis of the nonproteinogenic amino-acid beta-hydroxycyclohex-2′-enylanine (3) (R = H or S~PCP) via a shunt in the phenylalanine biosynthetic pathway

Biosynthesis
It was originally hypothesized that salinosporamide B was a biosynthetic precursor to salinosporamide A due to their structural similarities.
It was thought that the halogenation of the unactivated methyl group was catalyzed by a non-heme iron halogenase.[4][5]Recent work using 13C-labeled feeding experiments reveal distinct biosynthetic origins of salinosporamide A and B.[4][6]
While they share the biosynthetic precursors acetate and presumed β-hydroxycyclohex-2′-enylalanine (3), they differ in the origin of the four-carbon building block that gives rise to their structural differences involving the halogen atom. A hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) pathway is most likely the biosynthetic mechanism in which acetyl-CoA and butyrate-derived ethylmalonyl-CoA condense to yield the β-ketothioester (4), which then reacts with (3) to generate the linear precursor (5).
The first stereoselective synthesis was reported by Rajender Reddy Leleti and E. J.Corey.[7] Later several routes to the total synthesis of salinosporamide A have been reported.[7][8][9][10]
In vitro studies using purified 20S proteasomes showed that salinosporamide A has lower EC50 for trypsin-like (T-L) activity than does Bortezomib. In vivo animal model studies show marked inhibition of T-L activity in response to salinosporamide A, whereas bortezomib enhances T-L proteasome activity.
Initial results from early-stage clinical trials of salinosporamide A in relapsed/refractory multiple myeloma patients were presented at the 2011 American Society of Hematology annual meeting.[11] Further early-stage trials of the drug in a number of different cancers are ongoing.[12]
- Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W (2003). “Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora”. Angew. Chem. Int. Ed. Engl. 42 (3): 355–7.doi:10.1002/anie.200390115. PMID 12548698.
- Chauhan D, Catley L, Li G et al. (2005). “A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib”. Cancer Cell 8 (5): 407–19.doi:10.1016/j.ccr.2005.10.013. PMID 16286248.
- K. Lloyd, S. Glaser, B. Miller, Nereus Pharmaceuticals Inc.
- Beer LL, Moore BS (2007). “Biosynthetic convergence of salinosporamides A and B in the marine actinomycete Salinispora tropica”. Org. Lett. 9 (5): 845–8.doi:10.1021/ol063102o. PMID 17274624.
- Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT (2006). “Nature’s inventory of halogenation catalysts: oxidative strategies predominate”. Chem. Rev.106 (8): 3364–78. doi:10.1021/cr050313i.PMID 16895332.
- Tsueng G, McArthur KA, Potts BC, Lam KS (2007). “Unique butyric acid incorporation patterns for salinosporamides A and B reveal distinct biosynthetic origins”. Applied Microbiology and Biotechnology 75 (5): 999–1005. doi:10.1007/s00253-007-0899-7.PMID 17340108.
- Reddy LR, Saravanan P, Corey EJ (2004). “A simple stereocontrolled synthesis of salinosporamide A”. J. Am. Chem. Soc. 126 (20): 6230–1. doi:10.1021/ja048613p.PMID 15149210.
- Ling T, Macherla VR, Manam RR, McArthur KA, Potts BC (2007). “Enantioselective Total Synthesis of (-)-Salinosporamide A (NPI-0052)”.Org. Lett. 9 (12): 2289–92. doi:10.1021/ol0706051. PMID 17497868.
- Ma G, Nguyen H, Romo D (2007). “Concise Total Synthesis of (±)-Salinosporamide A, (±)-Cinnabaramide A, and Derivatives via a Bis-Cyclization Process: Implications for a Biosynthetic Pathway?”. Org. Lett. 9 (11): 2143–6. doi:10.1021/ol070616u. PMC 2518687.PMID 17477539.
- Endo A, Danishefsky SJ (2005). “Total synthesis of salinosporamide A”. J. Am. Chem. Soc. 127 (23): 8298–9.doi:10.1021/ja0522783. PMID 15941259.
- “Marizomib May Be Effective In Relapsed/Refractory Multiple Myeloma (ASH 2011)”. The Myeloma Beacon. 2012-01-23. Retrieved 2012-06-10.
- ClinicalTrials.gov: Marizomib
……………………………………………………
IMPORTANT PAPERS
Total synthesis of salinosporamide A
Org Lett 2008, 10(19): 4239
Entry to heterocycles based on indium-catalyzed conia-ene reactions: Asymmetric synthesis of (-)-salinosporamide A
Angew Chem Int Ed 2008, 47(33): 6244
A concise and straightforward total synthesis of (+/-)-salinosporamide A, based on a biosynthesis model
Org Biomol Chem 2008, 6(15): 2782
Formal synthesis of salinosporamide A starting from D-glucose
Synthesis (Stuttgart) 2009, 2009(17): 2983
Stereoselective functionalization of pyrrolidinone moiety towards the synthesis of salinosporamide A
Tetrahedron 2012, 68(32): 6504
………………
Salinosporamide A(1) was recently discovered by Fenical et al. as a bioactive product of a marine microorganism that is widely distributed in ocean sediments. Feeling, R. H.; Buchanan, G. O.; Mincer, T. J.; Kauffman, C. A.; Jensen, P. R.; Fenical, W., Angew. Chem. Int. Ed., 2003, 42, 355–357.

Structurally Salinosporamide A closely resembles the terrestrial microbial product omuralide (2a) that was synthesized by Corey et al. several years ago and demonstrated to be a potent inhibitor of proteasome function. See, (a) Corey, E. J.; Li, W. D., Z. Chem. Pharm. Bull., 1999, 47, 1–10; (b) Corey, E. J., Reichard, G. A.; Kania, R., Tetrahedron Lett., 1993, 34, 6977–6980; (c) Corey, E. J.; Reichard, G. A., J. Am. Chem. Soc., 1992, 114, 10677–10678; (d) Fenteany, G.; Standaert, R. F.; Reichard, G. A.; Corey, E. J.; Schreiber, S. L., Proc. Natl. Acad. Sci. USA, 1994, 91, 3358–3362.
Omuralide is generated by β-lactonization of the N-acetylcysteine thiolester lactacystin (2b) that was first isolated by the Omura group as a result of microbial screening for nerve growth factor-like activity. See, Omura, S., Fujimoto, T., Otoguro, K., Matsuzaki, K., Moriguchi, R., Tanaka, H., Sasaki, Y., Antibiot., 1991, 44, 113–116; Omura, S., Matsuzaki, K., Fujimoto, T., Kosuge, K., Furuya, T., Fujita, S., Nakagawa, A., J. Antibiot., 1991, 44, 117–118.
Salinosporamide A, the first compound Fenical’s group isolated from Salinospora, not only had a never-before-seen chemical structure 1, but is also a highly selective and potent inhibitor of cancer-cell growth. The compound is an even more effective proteasome inhibitor than omuralide and, in addition, it displays surprisingly high in vitro cytotoxic activity against many tumor cell lines (IC50values of 10 nM or less). Fenical et al. first found the microbe, which they’ve dubbed Salinospora, off the coasts of the Bahamas and in the Red Sea. See,Appl. Environ. Microbiol., 68, 5005 (2002).
Fenical et al. have shown that Salinospora species requires a salt environment to live. Salinospora thrives in hostile ocean-bottom conditions: no light, low temperature, and high pressure. The Fenical group has now identified Salinosporain five oceans, and with 10,000 organisms per cm3 of sediment and several distinct strains in each sample; and according to press reports, they’ve been able to isolate 5,000 strains. See, Chemical & Engineering News, 81, 37 (2003).
A great percentage of the cultures Fenical et al. have tested are said to have shown both anticancer and antibiotic activity. Like omuralide 2a, salinosporamide A inhibits the proteasome, an intracellular enzyme complex that destroys proteins the cell no longer needs. Without the proteasome, proteins would build up and clog cellular machinery. Fast-growing cancer cells make especially heavy use of the proteasome, so thwarting its action is a compelling drug strategy. See, Fenical et al., U.S. Patent Publication No. 2003-0157695A1
PATENTS
WO 2005113558
http://www.google.com/patents/US7183417
Part I. Synthesis of the Salinosporamide A(1)
EXAMPLE 1

(4S, 5R) Methyl 4,5-dihydro-2 (4-methoxyphenyl)-5-methyloxazole-4-carboxylate (4)
A mixture of (2S, 3R)-methyl 2-(4-methoxybenzamido)-3-hydroxybutanoate (3) (35.0 g, 131 mmol) and p-TsOH.H2O (2.5 g, 13.1 mmol) in toluene (400 mL) was heated at reflux for 12 h. The reaction mixture was diluted with water (200 mL) and extracted with EtOAc (3×200 mL). The combined organic layers were washed with water, brine and dried over Na2SO4. The solvent was removed in vacuo to give crude oxazoline as yellow oil. Flash column chromatography on silica gel (eluent 15% EtOAc-Hexanes) afforded the pure oxazoline (26.1 g, 80%) as solid.
Rf=0.51 (50% ethyl acetate in hexanes), mp, 86–87° C.; [α]23 D+69.4 (c 2.0, CHCl3); FTIR (film) νmax: 2955, 1750, 1545, 1355, 1187, 1011, 810 cm−1; 1HNMR(CDCl3, 400 MHz): δ 7.87 (2H, d, J=9.2 Hz), 6.84 (2H, d, J=8.8 Hz), 4.90 (1H, m), 4.40 (1H, d, J=7.6 Hz), 3.79 (3H,s), 3.71 (3H, s), 1.49 (3H, d, J=6.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 171.93, 165.54, 162.64, 130.52, 119.80, 113.85, 78.91, 75.16, 55.51, 52.73, 21.14; HRMS (ESI) calcd for C13H16NO4 (M+H)+.250.1079, found 250.1084.
EXAMPLE 2

(4R, 5R)-Methyl 4-{(benzyloxy) methyl)}-4,5-dihydro-2-(4-methoxyphenyl)-5-methyloxazole-4-carboxylate (5)
To a solution of LDA (50 mmol, 1.0 M stock solution in THF) was added HMPA (24 mL, 215 mmol) at −78° C. and then oxazoline 4 (12.45 g, 50 mmol, in 20 mL THF) was added dropwise with stirring at −78° C. for 1 h to allow complete enolate formation. Benzyloxy chloromethyl ether (8.35 mL, 60 mmol) was added at this temperature and after stirring the mixture at −78° C. for 4 h, it was quenched with water (50 mL) and warmed to 23° C. for 30 min. Then the mixture was extracted with ethyl acetate (3×50 mL) and the combined organic phases were dried (MgSO4) and concentrated in vacuo. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:4 then 1:3) to give the benzyl ether 5 (12.7 g, 69%).
Rf=0.59 (50% ethyl acetate in hexanes). [α]23 D−6.3 (c 1.0, CHCl3); FTIR (film) (νmax; 3050, 2975, 1724, 1642, 1607, 1252, 1027, 745, 697 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.96 (2H, d, J=9.2 Hz), 7.26 (5H, m), 6.90 (2H, J=8.8 Hz), 4.80 (1H, m), 4.61 (2H, s), 3.87 (3H, m), 3.81 (3H, s), 3.73 (3H, s), 1.34 (3H, d, J=6.8 Hz); 13C NMR (CDCl3, 100 MHZ): 6171.23, 165.47, 162.63, 138.25, 130.64, 128.52, 127.87, 127.77, 120.15, 113.87, 81.40, 79.92, 73.91, 73.43, 55.58, 52.45, 16.92; HRMS (ESI) calcd for C21H24O5 (M+H)+370.1654, found 370.1644.
EXAMPLE 3

(2R,3R)-Methyl 2-(4-methoxybenzylamino)-2-((benzyloxy)methyl)-3hydroxybutanoate (6)
To a solution of oxazoline 5 (18.45 g, 50 mmol) in AcOH (25 mL) at 23° C. was added in portions NaCNBH3 (9.3 g, 150 mmol). The reaction mixture was then stirred at 40° C. for 12 h to allow complete consumption of the starting material. The reaction mixture was diluted with water (100 mL), neutralized with solid Na2CO3 and the aqueous layer was extracted with ethyl acetate (3×100 mL). The combined organic phases were dried over NaSO4 and concentrated in vacuo. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5) to give the N-PMB amino alcohol 6 (16.78 g, 90%).
Rf=0.50 (50% ethyl acetate in hexanes). [α]23 D−9.1(c 1.0, CHCl3); FTIR (film) νmax; 3354, 2949, 1731, 1511, 1242, 1070, 1030, 820, 736, 697 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.32 (7H, m), 6.87 (2H, d, J=8.8 Hz), 4.55 (2H, m), 4.10 (1H, q, J=6.4 Hz), 3.85 (2H, dd, J=17.2, 10.0 Hz), 3.81 (3H, s,), 3.77 (3H, s), 3. 69 (2H, dd, J=22.8, 11.6 Hz), 3.22 (2H, bs), 1.16 (3H, d, J=6.0 Hz); 13C NMR (CDCl3, 100 MHz): δ 173.34, 159.03, 137.92, 132.51, 129.78, 128.67, 128.07, 127.98, 114.07, 73.80, 70.55, 69.82, 69.65, 55.51, 55.29, 47.68, 18.15; HRMS (ESI) calcd. for C21H28NO5 (M+H)+ 374.1967, found 374.1974.
EXAMPLE 4

(2R,3R)-Methyl-2-(N-(4-methoxybenzyl)acrylamido)-2-(benzyloxy)methyl)-3-hydroxybutanoate (7)
A solution of amino alcohol 6 (26.2 g, 68.5 mmol) in Et2O (200 mL) was treated with Et3N (14.2 mL, 102.8 mmol) and trimethylchlorosilane (10.4 mL, 82.2 mmol) at 23° C. and stirred for 12 h. After completion, the reaction mixture was diluted with ether (200 mL) and then resulting suspension was filtered through celite. The solvent was removed to furnish the crude product (31.2 g, 99%) in quantitative yield as viscous oil. A solution of this crude trimethylsilyl ether (31.1 g) in CH2Cl2 (200 mL) was charged with diisopropylethylamine (14.2 mL, 81.6 mmol) and then cooled to 0° C. Acryloyl chloride (6.64 mL, 82.2 mmol) was added dropwise with vigorous stirring and the reaction temperature was maintained at 0° C. until completion (1 h). The reaction mixture was then diluted with CH2Cl2 (100 mL) and the organic layer was washed with water and brine. The organic layer was separated and dried over Na2SO4. The solvent was removed to afford the crude acrylamide 7 as a viscous oil. The crude product was then dissolved in Et2O (200 mL) and stirred with 6N HCl (40 mL) at 23° C. for 1 h. The reaction mixture was diluted with water (100 mL) and concentrated to provide crude product. The residue was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5 to 1:1) to give pure amide 7 (28.3 g, 96%) as colorless solid, mp 88–89° C.
Rf=0.40 (50% ethyl acetate in hexanes), [α]23 D−31.1 (c 0.45, CHCl3), FTIR (film) νmax; 3435, 2990, 1725, 1649, 1610, 1512, 1415, 1287, 1242, 1175, 1087, 1029, 732, 698 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.25 (5H, m), 7.15 (2H, d, J=6.0 Hz), 6.85 (2H, d, J=7.5 Hz), 6.38 (2H, d, J=6.0 Hz), 5.55 (1H, t, J=6.0 Hz), 4.81 (2H, s), 4.71 (1H, q, J=6.5 Hz), 4.35 (2H, s), 4.00 (1H, d, J=10.0 Hz), 3.80 (1H, d, J=10.0 Hz), 3.76 (3H, s), 3.75 (3H, s), 3.28 (1H, bs), 1.22 (3H, d, J=6.0 Hz); 13C NMR (CDCl3, 125 MHz): δ 171.87, 168.74, 158.81, 137.73, 131.04, 129.68, 128.58, 128.51, 127.94, 127.72, 127.20, 127.14, 114.21, 73.71, 70.42, 69.76, 67.65, 55.45, 52.52, 49.09, 18.88; HRMS (ESI) calcd. for C24H30NO6 (M+H)+428.2073, found 428.2073.
EXAMPLE 5

(R)-Methyl-2-(N-(4-methoxybenzyl)acrylamido)-2-(benzyloxy)methyl)-3-oxybutanoate (8)
To a solution of amide 7 (10.67 g, 25.0 mmol) in CH2Cl2 (100 mL) was added Dess-Martin periodinane reagent (12.75 g, 30.0 mmol, Aldrich Co.) at 23° C. After stirring for 1 h, the reaction mixture was quenched with aq NaHCO3—Na2S2O3 (1:1, 50 mL) and extracted with ethyl acetate (3×50 mL). The organic phase was dried and concentrated in vacuo to afford the crude ketone. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes) to give pure keto amide 8 (10.2 g, 96%).
Rf=0.80 (50% ethyl acetate in hexanes), mp 85 to 86° C.; [α]23 D−12.8 (c 1.45, CHCl3); FTIR (film) νmax: 3030, 2995, 1733, 1717, 1510, 1256, 1178, 1088, 1027, 733, 697 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.30 (2H, d, J=8.0), 7.25 (3H, m), 7.11 (2H, m), 6.88 (2H, d, J=9.0 Hz), 6.38 (2H, m), 5.63 (1H, dd, J=8.5, 3.5 Hz), 4.93 (1H, d, J=18.5 Hz), 4.78 (1H, d, J=18.5, Hz), 4.27 (2H, m), 3.78 (3H, s), 3.76 (3H, s), 2.42 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 198.12, 169.23, 168.62, 158.01, 136.95, 130.64, 130.38, 128.63, 128.13, 127.77, 127.32, 114.33, 77.49, 73.97, 70.66, 55.49, 53.09, 49.03, 28.24; HRMS (ESI) calcd. for C24H28NO6 (M+H)+ 426.1916, found 426.1909.
EXAMPLE 6

(2R,3S)-Methyl-1-(4-methoxybenzyl)-2-((benzyloxy)methyl)-3-hydroxy-3-methyl-4-methylene-5-oxopyrrolidine-2-carboxylate (9+10)
A mixture of keto amide 8 (8.5 g, 20.0 mmol) and quinuclidine (2.22 g, 20.0 mmol) in DME (10 mL) was stirred for 5 h at 23° C. After completion, the reaction mixture was diluted with ethyl acetate (50 mL) washed with 2N HCl, followed by water and dried over Na2SO4. The solvent was removed in vacuo to give the crude adduct (8.03 g, 94.5%, 3:1 ratio of 9 to 10 dr) as a viscous oil. The diastereomeric mixture was separated at the next step, although small amounts of 9 and 10 were purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:10 to 1:2) for analytical purposes.
Major Diastereomer (9).
[α]23 D−37.8 (c 0.51, CHCl3); FTIR (film) vmax: 3450, 3055, 2990, 1733, 1683, 1507, 1107, 1028, 808,734 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.29 (5H, m), 7.15 (2H, d, J=7.5 Hz), 6.74 (2H, d, J=8.5 Hz), 6.13 (1H, s), 5.57 (1H, s), 4.81 (1H, d, J=14.5 Hz), 4.45(1H, d, J=15.0 Hz), 4.20 (1H, d, J=12.0 Hz), 4.10 (1H, d, J=12.0 Hz) 3.75 (3H, s), 3.70 (1H, d, J=10.5 Hz), 3.64 (3H, s), 3.54 (1H, d, J=10.5 Hz), 2.55 (1H, bs, OH), 1.50 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 169.67, 168.42, 158.97, 145.96, 137.57, 130.19, 130.12, 128.53, 127.83, 127.44, 116.79, 113.71, 76.32, 76.00, 73.16, 68.29, 55.45, 52.63, 45.36, 22.64; HRMS (ESI) calcd. for C24H28NO6 (M+H)+ 426.1916, found 426.1915.
Minor Diastereomer (10).
[α]23 D−.50.1 (c 0.40, CHCl3); FTIR (film) νmax: 3450, 3055, 2990, 1733, 1683, 1507, 1107, 1028, 808, 734 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.29 (5H, m), 7.12 (2H, d, J=7.5 Hz), 6.73 (2H, d, J=8.5 Hz), 6.12 (1H, s), 5.57 (1H, s), 4.88 (1H, d, J=15.5 Hz), 4.31 (1H, d, J=15.0 Hz), 4.08 (3H, m), 3.99 (1H, d, J=12.0 Hz) 3.73 (3H, s), 3.62 (3H, s), 3.47 (1H, bs, OH), 3.43 (1H, d, J=10.0 Hz), 1.31 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 169.65, 167.89, 159.13, 147.19, 136.95, 130.29, 129.76, 128.74, 128.19, 127.55, 116.80, 113.82, 76.21, 75.66, 73.27, 68.02, 55.45, 52.52, 45.24, 25.25; HRMS (ESI) calcd. for (M+H)+ C24H28NO6 426.1916, found 426.1915.
EXAMPLE 7
Silylation of 9 and 10 and Purification of 11.
To a solution of lactams 9 and 10 (7.67 g, 18 mmol) in CH2Cl2 (25 ml) was added Et3N (7.54 ml, 54 mmol), and DMAP (2.2 g, 18 mmol) at 0° C., and then bromomethyl-dimethylchlorosilane (5.05 g, 27 mmol) (added dropwise). After stirring the mixture for 30 min at 0° C., it was quenched with aq NaHCO3 and the resulting mixture was extracted with ethyl acetate (3×50 mL). The combined organic layers were washed with water, brine and dried over Na2SO4. The solvent was removed in vacuo to give a mixture of the silated derivatives of 9 and 10 (9.83 g, 95%). The diastereomers were purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5 to 1:4) to give pure diastereomer 11 (7.4 g, 72%) and its diastereomer (2.4 g, 22%).
Silyl Ether (11).
Rf=0.80 (30% ethyl acetate in hexanes). [α]23 D−58.9 (c 0.55, CHCl3); FTIR (film) νmax; 3050, 2995, 1738, 1697, 1512, 1405, 1243, 1108, 1003, 809, 732 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.27 (5H, m), 7.05 (2H, d, J=7.0 Hz), 6.71 (2H, d, J=8.5 Hz), 6.18 (1H, s), 5.53 (1H, s), 4.95 (1H, d, J=15.5 Hz), 4.45 (1H, d, J=15.0 Hz), 4.02 (1H, J=12.0 Hz), 3.86 (1H, d, J=11.5 Hz) 3.72 (3H, s), 3.68 (3H, s), 3.65 (1H, d, J=10.5 Hz), 3.30 (1H, d, J=10.0 Hz), 2.34 (2H, d, J=2.0 Hz), 1.58 (3H, s), 0.19 (3H, s), 0.18 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 168.62, 168.12, 158.93, 145.24, 137.53, 130.32, 130.30, 128.49, 127.76,127.22, 117.26, 113.60, 78.55, 78.03, 72.89, 68.45, 55.43, 52.37, 45.74, 21.87, 17.32, −0.72, −0.80; HRMS (ESI) Calcd. for C27H35BrNO6Si (M+H)+ 576.1417, found 576.1407.
EXAMPLE 8

Conversion of (11) to (12).
To a solution of compound 11 (5.67 g 10 mmol) in benzene (250 mL) at 80° C. under nitrogen was added a mixture of tributyltin hydride (4.03 ml, 15 mmol) and AIBN (164 mg, 1 mmol) in 50 ml benzene by syringe pump over 4 h. After the addition was complete, the reaction mixture was stirred for an additional 4 h at 80° C. and the solvent was removed in vacuo. The residue was dissolved in hexanes (20 mL) and washed with saturated NaHCO3 (3×25 mL), water and dried over Na2SO4. The solvent was removed in vacuo to give crude product. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5) to afford the pure 12 (4.42 g, 89%).
Rf=0.80 (30% ethyl acetate in hexanes). [α]23 D−38.8 (c 0.25, CHCl3); FTIR (film) νmax; 3025, 2985, 1756, 1692, 1513, 1247, 1177, 1059, 667 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.28 (5H, m), 7.09 (2H, d, J=7.0 Hz), 6.73 (2H, d, J=9.0 Hz), 4.96(1H, d, J=15.0 Hz), 4.35 (1H, d, J=15.5 Hz), 3.97 (1H, d, J=12.5 Hz), 3.86 (1H, d, J=12.0 Hz), 3.80 (1H, d, J=10.0 Hz), 3.72 (3H, s), 3.65 (3H, s), 3.27 (1H, d, J=10.5 Hz), 2.67 (1H, t, J=4.0 Hz), 2.41 (1H, m), 1.79 (1H, m), 1.46 (3H, s), 0.77 (1H, m), 0.46 (1H, m), 0.10 (3H, s), 0.19 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 175.48, 169.46, 158.76, 137.59, 131.04, 129.90, 128.58, 127.88, 127.52, 113.59, 113.60, 81.05, 78.88, 73.12, 69.03, 55.45, 51.94, 48.81, 45.50, 22.79, 17.06, 7.76, 0.54; HRMS (ESI) calcd. for (M+H)+ C27H36NO6Si 498.2312, found 498.2309.
EXAMPLE 9

Debenzylation of (12).
A solution of 12 (3.98 g, 8 mmol) in EtOH (50 ml) at 23° C. was treated with 10% Pd—C (˜1 g) under an argon atmosphere. The reaction mixture was evacuated and flushed with H2 gas (four times) and then stirred vigorously under an atmosphere of H2 (1 atm, H2 balloon) at 23° C. After 12 h, the reaction mixture was filtered through Celite and concentrated in vacuo to give the crude debenzylation product (3.08 g, 95%) which was used for the next step. A small amount crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:3) for analytical purposes. Rf=0.41 (50% ethyl acetate in hexanes).
mp, 45–47° C.; [α]23 D−30.9 (c 0.55, CHCl3); FTIR (film) νmax: 3432, 3020, 2926, 1735, 1692, 1512, 1244, 1174, 1094, 1024, 870, 795 cm−1; 1H NMR (CDCl3, 400 MHz): δ 7.36 (2H, d, J=8.5 Hz), 6.83 (2H, d, J=8.5 Hz), 5.16 (1H, d, J=15.0 Hz), 4.29 (1H, d, J=15.0 Hz), 3.92 (1H, m), 3.78 (3H, s), 3.68 (3H, s), 3.45 (1H, m), 2.53 (1H, t, J=4.0 Hz), 2.42 (1H, m), 1.82 (1H, m), 1.50 (3H, s), 1.28 (1H, m), 0.75 (1H, m), 0.47 (1H, m), 0.11 (3H, s), 0.02 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 175.82, 169.51, 159.32, 131.00, 129.72, 114.52, 80.79, 80.13, 61.85, 55.48, 51.99, 49.29, 45.06, 23.11, 17.03, 7.44, 0.54; HRMS (ESI) calcd. for C20H30NO6Si (M+H)+ 408.1842, found 408.1846.
EXAMPLE 10
Oxidation to Form Aldehyde (13).
To a solution of the above alcohol from debenzylation of 12 (2.84 g, 7 mmol) in CH2Cl2 (30 mL) was added Dess-Martin reagent (3.57 g, 8.4 mmol) at 23° C. After stirring for 1 h at 23° C., the reaction mixture was quenched with aq NaHCO3—Na2S2O3 (1:1, 50 mL) and extracted with ethyl acetate (3×50 mL). The organic phase was dried and concentrated in vacuo to afford the crude aldehyde. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:5) to give pure aldehyde 13 (2.68 g, 95%). Rf=0.56 (50% ethyl acetate in hexanes).
mp, 54–56° C.; [α]23 D−16.5 (c 0.60, CHCl3); FTIR (film) νmax: 3015, 2925, 1702 1297, 1247, 1170, 1096, 987, 794 cm−1; 1H NMR (CDCl3, 500 MHz): δ 9.62 (1H, s), 7.07 (2H, d, J=8.0 Hz), 6.73 (2H, d, J=8.5 Hz), 4.49 (1H, quart, J=8.5 Hz), 3.70 (3H, s), 3.67 (3H, s), 2.36 (2H, m), 1.75 (1H, m), 1.37 (3H, s), 0.73 (1H, m), 0.48 (1H, m), 0.07 (3H, s), 0.004 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 197.26, 174.70, 167.36, 158.07, 130.49, 128.96, 113.81, 83.97, 82.36, 55.34, 52.43, 47.74, 46.32, 23.83, 16.90, 7.52, 0.56, 0.45; HRMS (ESD calcd. for C20H28NO6Si (M+H)+ 406.1686, found 406.1692.
EXAMPLE 11

Conversion of (13) to (14).
To a solution of freshly prepared cyclohexenyl zinc chloride (10 mL, 0.5 M solution in THF, 5 mmol) (see Example 15 below) at −78° C. under nitrogen was added a −78° C. solution of aldehyde 13 (1.01 g, in 3 ml of THF, 2.5 mmol). After stirring for 5 h at −78° C. reaction mixture was quenched with water (10 mL) then extracted with ethyl acetate (3×10 mL). The combined organic layers were dried over Na2SO4 and solvent was removed in vacuo to give crude product (20:1 dr). The diastereomers were purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:10 to 1:2 affords the pure major diastereomer 14 (1.0 g, 83%) and a minor diastereomer (50 mg 5%). For 14: Rf=0.56 (50% ethyl acetate in hexanes).
mp, 79–81° C.; [a]23 D−28.5 (c 1.45, CHCl3); FTIR (film) νmax: 3267, 2927, 2894, 2829, 1742, 1667, 1509, 1248, 1164, 1024, 795 cm−1; 1H NMR (CDCl3, 500 MHz): δ 7.34 (2H, d, J=8.5 Hz), 6.81 (2H, d, J=9.0 Hz), 5.84 (1H, m), 5.73 (1H, m), 4.88 (1H, d, J=15.5 Hz), 4.39 (1H, d, J=14.5 Hz), 4.11 (1H, t, J=6.5 Hz), 3.77 (3H, s), 3.58 (3H, s), 3.00 (1H, m), 2.95 (1H, d, J=9.0 Hz), 2.83 (1H, t, J=3.5 Hz), 3.36 (1H, m), 2.27 (1H, m), 1.98 (2H, m), 1.74 (3H, m), 1.62 (3H, s), 1.14 (2H, m), 0.59 (1H, m), 0.39 (11H, m), 0.13 (3H, s), 0.03 (3H, s); 13C NMR (CDCl3, 125 MHz): δ 176.80, 170.03, 158.27, 131.86, 131.34, 128.50, 126.15, 113.40, 83.96, 82.45, 77.17, 55.45, 51.46, 48.34, 48.29, 39.08, 28.34, 25.29, 22.45, 21.09, 17.30, 7.75, 0.39, 0.28; HRMS (ESI) calcd. for C26H38NO6Si (M+H)+ 488.2468, found 488.2477.
EXAMPLE 12

Tamao-Fleming Oxidation of (14) to (15).
To a solution of 14 (0.974 g, 2 mmol) in THF (5 mL) and MeOH (5 mL) at 23° C. was added KHCO3 (0.8 g, 8 mmol) and KF (0.348 g, 6 mmol). Hydrogen peroxide (30% in water, 5 mL) was then introduced to this mixture. The reaction mixture was vigorously stirred at 23° C. and additional hydrogen peroxide (2 ml) was added after 12 h. After 18 h, the reaction mixture was quenched carefully with NaHSO3 solution (15 mL). The mixture was extracted with ethyl acetate (3×25 mL) and the combined organic layers were washed with water and dried over Na2SO4. The solvent was removed in vacuo to give the crude product. The crude product was purified by column chromatography (silica gel, ethyl acetate) to give the pure triol 15 (0.82 g, 92%).
Rf=0.15 (in ethyl acetate). mp, 83–84° C.; [α]23 D: +5.2 (c 0.60, CHCl3); FTIR (film) νmax; 3317, 2920, 2827, 1741, 1654, 1502, 1246, 1170, 1018, 802 cm−1; 1HNMR(CDCl3, 500 MHz): δ 7.77 (2H, d, J=8.0 Hz), 6.28 (2H, d, J=8.0 Hz), 5. 76 (1H, m), 5.63 (1H, d, J=10.0 Hz), 4.74 (1H, d, J=15.5 Hz), 4.54 (1H, d, J=15.0 Hz), 4.12 (1H, d, J=2.5 Hz), 3.80 (1H, m), 3.76 (3H, s), 3.72 (1H, m), 3.68 (3H, s), 3.00 (1H, m), 2.60 (1H, br), 2.20 (1H, m), 1.98 (2H, s), 1.87 (1H, m), 1.80 (1H, m), 1.71 (2H, m), 1.61 (3H, s), 1.14 (2H, m); 13C NMR (CDCl3, 125 MHz): δ 178.99, 170.12, 158.27, 131.30, 130.55, 128.13, 126.39, 113.74, 81.93, 80.75, 76.87, 61.61, 55.45, 51.97, 51.32, 48.07, 39.17, 27.71, 27.13, 25.22, 21.35, 21.22; HRMS (ESI) calcd. for C24H34NO7 (M+H)+ 448.2335, found 448.2334.
EXAMPLE 13

Deprotection of (15) to (16).
To a solution of 15 (0.670 g, 1.5 mmol) in acetonitrile (8 mL) at 0° C. was added a pre-cooled solution of ceric ammonium nitrate (CAN) (2.46 g 4.5 mmol in 2 mL H2O). After stirring for 1 h at 0° C. the reaction mixture was diluted with ethyl acetate (50 mL), washed with saturated NaCl solution (5 mL) and organic layers was dried over Na2SO4. The solvent was removed in vacuo to give the crude product which was purified by column chromatography (silica gel, ethyl acetate) to give the pure 16 (0.4 g, 83%).
Rf=0.10 (5% MeOH in ethyl acetate). mp, 138 to 140° C.; [α]23 D+14.5 (c 1.05, CHCl3); FTIR (film) νmax 3301, 2949, 2911, 2850, 1723, 1673, 1437, 1371, 1239, 1156, 1008, 689 cm−1; 1H NMR (CDCl3, 600 MHz): δ 8.48 (1H, br), 6.08 (1H, m), 5. 75 (1H, d, J=9.6 Hz), 5.29 (1H, br), 4.13 (1H, d, J=6.6 Hz), 3.83 (3H, m), 3.79 (1H, m), 3.72 (1H, m), 2.84 (1H, d, J=10.2 Hz), 2.20 (1H, m), 2.16 (1H, br), 1.98 (3H, m), 1.77 (3H, m), 1.59 (1H, m), 1.54 (3H, s), 1.25 (1H, m). 13C NMR (CDCl3, 125 MHz): δ 180.84, 172.95, 135.27, 123.75, 82.00, 80.11, 75.56, 62.39, 53.14, 51.78, 38.95, 28.79, 26.48, 25.04, 20.66, 19.99; HRMS (ESI) calcd. (M+H)+ for C16H26NO6 328.1760, found 328.1752.
EXAMPLE 14

Conversion of (16) to Salinosporamide A(1).
A solution of triol ester 16 (0.164 g, 0.5 mmol) in 3 N aq LiOH (3 mL) and THF (1 mL) was stirred at 5° C. for 4 days until hydrolysis was complete. The acid reaction mixture was acidified with phosphoric acid (to pH 3.5). The solvent was removed in vacuo and the residue was extracted with EtOAc, separated, and concentrated in vacuo to give the crude trihydroxy carboxylic acid 16a (not shown). The crude acid was suspended in dry CH2Cl2 (2 mL), treated with pyridine (0.5 mL) and stirred vigorously at 23° C. for 5 min. To this solution was added BOPCl (152 mg, 0.6 mmol) at 23° C. under argon, and stirring was continued for 1 h. The solvent was removed under high vacuum and the residue was suspended in dry CH3CN (1 mL) and treated with pyridine (1 mL). To this solution was added PPh3Cl2 (333 mg, 1.0 mmol) at 23° C. under argon with stirring. After 1 h the solvent was removed in vacuo. The crude product was purified by column chromatography (silica gel, ethyl acetate-CH2Cl2, 1:5) to give the pure β-lactone 1 (100 mg, 64%) as a colorless solid.
Rf=0.55 (50% ethyl acetate in hexane). mp, 168–170° C. (authentic sample: 168–170° C., 169–171° C. in Angew. Chem. Int. Ed., 2003, 42, 355–357); mixture mp, 168–170C. [α]23 D −73.2 (c 0.49, MeOH), −72.9 (c 0.55, MeOH, in Angew. Chem. Int. Ed., 2003, 42, 355–357); FTIR (film) νmax: 3406, 2955, 2920, 2844, 1823, 1701, 1257, 1076, 1012, 785, 691 cm−1; 1H NMR (CDCl3, 500 MHz): δ 10.62 (1H, br), 6.42 (1H, d, J=10.5 Hz), 5.88 (1H, m), 4.25 (1H, d, J=9.0 Hz), 4.14 (1H, m), 4.01 (1H, m), 3.17 (1H, t, J=7.0 Hz), 2.85 (1H, m), 2.48 (1H, m), 2.32 (2H, m), 2.07 (3H, s), 1.91 (2H, m), 1.66 (2H, m), 1.38 (1H, m);13C NMR (CDCl3, 125 MHz): δ 176.92, 169.43, 129.08, 128.69, 86.32, 80.35, 70.98, 46.18, 43.28, 39.31, 29.01, 26.47, 25.35, 21.73, 20.00; HRMS (ESI) calcd. for (M−H)− C15H19ClNO4 312.1003, found 312.1003.
| 544814 | Oct 1, 2007 | Jun 9, 2009 | Nereus Pharmaceuticals, Inc. | [3.2.0] Heterocyclic compounds and methods of using the same |
| US7579371 | Jun 15, 2006 | Aug 25, 2009 | Nereus Pharmaceuticals, Inc. | Methods of using [3.2.0] heterocyclic compounds and analogs thereof |
| US7824698 | Feb 4, 2008 | Nov 2, 2010 | Nereus Pharmaceuticals, Inc. | Lyophilized formulations of Salinosporamide A |
| US7842814 | Apr 6, 2007 | Nov 30, 2010 | Nereus Pharmaceuticals, Inc. | Total synthesis of salinosporamide A and analogs thereof |
| US7910616 | May 12, 2009 | Mar 22, 2011 | Nereus Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8003802 | Mar 6, 2009 | Aug 23, 2011 | Nereus Pharmaceuticals, Inc. | Total synthesis of Salinosporamide A and analogs thereof |
| US8067616 | Oct 27, 2010 | Nov 29, 2011 | Nereus Pharmaceuticals, Inc. | Total synthesis of salinosporamide A and analogs thereof |
| US8168803 | Jun 10, 2008 | May 1, 2012 | Nereus Pharmaceuticals, Inc. | Methods of using [3.2.0] heterocyclic compounds and analogs thereof |
| US8217072 | Jun 18, 2004 | Jul 10, 2012 | The Regents Of The University Of California | Salinosporamides and methods for use thereof |
| US8222289 | Dec 15, 2009 | Jul 17, 2012 | The Regents Of The University Of California | Salinosporamides and methods for use thereof |
| US8227503 | Mar 21, 2011 | Jul 24, 2012 | Nereus Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8314251 | Jul 15, 2011 | Nov 20, 2012 | Nereus Pharmaceuticals, Inc. | Total synthesis of salinosporamide A and analogs thereof |
| US8389564 | May 14, 2012 | Mar 5, 2013 | Venkat Rami Reddy Macherla | Proteasome inhibitors |
| US8394816 | Dec 5, 2008 | Mar 12, 2013 | Irene Ghobrial | Methods of using [3.2.0] heterocyclic compounds and analogs thereof in treating Waldenstrom’s Macroglobulinemia |
| Name: | Marizomib | |
| Synonyms: | 6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione, 4-(2-chloroethyl)-1-[(S)-(1S)-2-cyclohexen-1-ylhydroxymethyl]-5-methyl-, (1R,4R,5S)-; Other Names: (-)-Salinosporamide A; ML 858; Marizomib; NPI 0052; Salinosporamide A | |
| CAS Registry Number: | 437742-34-2 | |
| Molecular Formula: | C15H20ClNO4 | |
| Molecular Weight: | 313.1 | |
| Molecular Structure: |  |
Teva Gets Orphan Drug Designation for Treanda
Teva Announces Additional Regulatory Exclusivity for TREANDA® (Bendamustine HCI) for Injection
Orphan Designation combined with pediatric extension provides regulatory exclusivity through April 2016 for indolent B-cell non-Hodgkin lymphoma indication
JERUSALEM, November 27, 2013 –(BUSINESS WIRE)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) today announced that the U.S. Food and Drug Administration (FDA) has granted orphan drug exclusivity for TREANDA through October 2015 for indolent B-cell non-Hodgkin lymphoma (iNHL) that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen.http://www.pharmalive.com/teva-announces-additional-regulatory-exclusivity-for-treanda
read my old post, contains synthesis
https://newdrugapprovals.wordpress.com/2013/09/19/fda-oks-tevas-injectable-treanda/
