
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 38)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GEMCITABINE
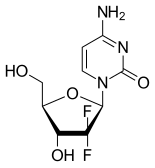

GEMCITABINE
95058-81-4
WeightAverage: 263.1981
Monoisotopic: 263.071762265
Chemical FormulaC9H11F2N3O4
4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Gemcitabine hydrochloride | U347PV74IL | 122111-03-9 | OKKDEIYWILRZIA-OSZBKLCCSA-N |
- LY-188011
- LY188011
Gemcitabine
CAS Registry Number: 95058-81-4
CAS Name: 2¢-Deoxy-2¢,2¢-difluorocytidine
Additional Names: 1-(2-oxo-4-amino-1,2-dihydropyrimidin-1-yl)-2-deoxy-2,2-difluororibose; dFdC; dFdCyd
Manufacturers’ Codes: LY-188011
Trademarks: Gemzar (Lilly)
Molecular Formula: C9H11F2N3O4
Molecular Weight: 263.20
Percent Composition: C 41.07%, H 4.21%, F 14.44%, N 15.97%, O 24.32%
Literature References: Prepn: L. W. Hertel, GB2136425; idem,US4808614 (1984, 1989 both to Lilly); L. W. Hertel et al.,J. Org. Chem.53, 2406 (1988); T. S. Chou et al.,Synthesis1992, 565. Antitumor activity: L. W. Hertel et al.,Cancer Res.50, 4417 (1990). Mode of action study: V. W. T. Ruiz et al.,Biochem. Pharmacol.46, 762 (1993). Clinical pharmacokinetics and toxicity: J. L. Abbruzzese et al.,J. Clin. Oncol.9, 491 (1991). Review of clinical studies: B. Lund et al.,Cancer Treat. Rev.19, 45-55 (1993).
Properties: Crystals from water, pH 8.5. [a]365 +425.36°; [a]D +71.51° (c = 0.96 in methanol). uv max (ethanol): 234, 268 (e 7810, 8560). LD10 i.v. in rats: 200 mg/m2 (Abbruzzese).
Optical Rotation: [a]365 +425.36°; [a]D +71.51°
Absorption maximum: uv max (ethanol): 234, 268 (e 7810, 8560)
Toxicity data: LD10 i.v. in rats: 200 mg/m2 (Abbruzzese)
Derivative Type: Hydrochloride
CAS Registry Number: 122111-03-9
Molecular Formula: C9H11F2N3O4.HCl
Molecular Weight: 299.66
Percent Composition: C 36.07%, H 4.04%, F 12.68%, N 14.02%, O 21.36%, Cl 11.83%
Properties: Crystals from water-acetone, mp 287-292° (dec). [a]D +48°; [a]365 +257.9° (c = 1.0 in deuterated water). uv max (water): 232, 268 nm (e 7960, 9360).
Melting point: mp 287-292° (dec)
Optical Rotation: [a]D +48°; [a]365 +257.9° (c = 1.0 in deuterated water)
Absorption maximum: uv max (water): 232, 268 nm (e 7960, 9360)
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Antimetabolites; Pyrimidine Analogs.
Gemcitabine is a nucleoside metabolic inhibitor used as adjunct therapy in the treatment of certain types of ovarian cancer, non-small cell lung carcinoma, metastatic breast cancer, and as a single agent for pancreatic cancer.
Gemcitabine hydrochloride was first approved in ZA on Jan 10, 1995, then approved by the U.S. Food and Drug Administration (FDA) on May 15, 1996, and approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on Aug 31, 2001. It was developed and marketed as Gemzar® by Eli Lilly.
Gemcitabine hydrochloride is a nucleoside metabolic inhibitor. It kills cells undergoing DNA synthesis and blocks the progression of cells through the G1/S-phase boundary. It is indicated for the treatment of advanced ovarian cancer that has relapsed at least 6 months after completion of platinum-based therapy, in combination with paclitaxel, for first-line treatment of metastatic breast cancer after failure of prior anthracycline-containing adjuvant chemotherapy, unless anthracyclines were clinically contraindicated, and it is also indicated in combination with cisplatin for the treatment of non-small cell lung cancer, and treated as a single agent for the treatment of pancreatic cancer.
Gemzar® is available as injection of lyophilized powder for intravenous use, containing 200 mg or 1000 mg of free Gemcitabine per vial. The recommended initial dosage is 1000 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for ovarian cancer, 1250 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for breast cancer, 1000 mg/m2 over 30 minutes on days 1, 8, and 15 of each 28 day cycle or 1250 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for non-small cell lung cancer, and 1000 mg/m2 over 30 minutes once weekly for the first 7 weeks, then one week rest, then once weekly for 3 weeks of each 28 day cycle for pancreatic cancer.
Approved Countries or AreaUpdate US, JP, CN, ZA
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-05-15 | First approval | Gemzar | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection, Lyophilized powder, For solution | Eq. 200 mg/1000 mg Gemcitabine/vial | Lilly | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-02-01 | New indication | Gemzar | Relapsed or refractory malignant lymphoma | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2011-02-23 | New indication | Gemzar | Advanced ovarian cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2010-02-05 | New indication | Gemzar | Advanced breast cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2008-11-25 | New indication | Gemzar | Urothelial cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2006-06-15 | New indication | Gemzar | Biliary cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2001-08-31 | First approval | Gemzar | Pancreatic cancer,Non small cell lung cancer (NSCLC) | Injection, Lyophilized powder, For suspension | 200 mg; 1 g | Lilly |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-04-15 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq. 1000 mg Gemcitabine per vial | 湖北一半天制药 | ||
| 2014-04-15 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq. 200 mg Gemcitabine per vial | 湖北一半天制药 | 6类 | |
| 2014-04-08 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq.1000 mg Gemcitabine per vial | 南京正大天晴制药 | 6类 | |
| 2011-12-02 | Marketing approval | 健择/Gemzar | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq. 200 mg/1000 mg Gemcitabine per vial | Lilly | |
| 2010-08-31 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | 1000 mg/200 mg | 北京协和药厂 | 6类 |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1995-01-10 | First approval | Gemzar | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection, Lyophilized powder, For solution | Eq. 200 mg/1000 mg Gemcitabine per vial | Lilly |
Gemcitabine, with brand names including Gemzar,[1] is a chemotherapy medication.[2] It treats cancers including testicular cancer,[3]breast cancer, ovarian cancer, non-small cell lung cancer, pancreatic cancer, and bladder cancer.[2][4] It is administered by intravenous infusion.[2] It acts against neoplastic growth, and it inhibits the replication of Orthohepevirus A, the causative agent of Hepatitis E, through upregulation of interferon signaling.[5]
Common side effects include bone marrow suppression, liver and kidney problems, nausea, fever, rash, shortness of breath, mouth sores, diarrhea, neuropathy, and hair loss.[2] Use during pregnancy will likely result in fetal harm.[2] Gemcitabine is in the nucleoside analog family of medication.[2] It works by blocking the creation of new DNA, which results in cell death.[2]
Gemcitabine was patented in 1983 and was approved for medical use in 1995.[6] Generic versions were introduced in Europe in 2009 and in the US in 2010.[7][8] It is on the WHO Model List of Essential Medicines.[9]
Medical uses
Gemcitabine treats various carcinomas. It is used as a first-line treatment alone for pancreatic cancer, and in combination with cisplatin for advanced or metastatic bladder cancer and advanced or metastatic non-small cell lung cancer. It is used as a second-line treatment in combination with carboplatin for ovarian cancer and in combination with paclitaxel for breast cancer that is metastatic or cannot be surgically removed.[10][11][12]
It is commonly used off-label to treat cholangiocarcinoma[13] and other biliary tract cancers.[14]
It is given by intravenous infusion at a chemotherapy clinic.[2]
Contraindications and interactions
Taking gemcitabine can also affect fertility in men and women, sex life, and menstruation. Women taking gemcitabine should not become pregnant, and pregnant and breastfeeding women should not take it.[15]
As of 2014, drug interactions had not been studied.[11][10]
SYN
. Hertel, L. W.; Kroin, J. S.; Misner, J. W.; Tustin, J. M. J. Org. Chem. 1988, 53, 2406– 2409.

NEXT
a) Noe, C. R.; Jasic, M.; Kollmann, H.; Saadat, K. WO009147, 2007.; b) Noe, C. R.; Jasic, M.; Kollmann, H.; Saadat, K. US0249119, 2008. Note: no stereochemistry was indica


NExT
15. Hanzawa, Y.; Inazawa, K.; Kon, A.; Aoki, H.; Kobayashi, Y. Tetrahedron Lett. 1987, 28, 659–662. 16. Wirth, D. D. EP0727432, 1996


Synthesis Reference
John A. Weigel, “Process for making gemcitabine hydrochloride.” U.S. Patent US6001994, issued May, 1995.US6001994Route 1
Reference:1. J. Org. Chem. 1988, 53, 2406-2409.
2. US4808614A.Route 2
Reference:1. CN102417533A.Route 3
Reference:1. Nucleosides, Nucleotides and Nucleic Acids 2010, 29, 113-122.Route 4
Reference:1. CN102617677A.Route 5
Reference:1. CN103012527A.
SYN
U.S. Patent No. 4,808,614 (the ‘614 patent) describes a process for synthetically producing gemcitabine, which process is generally illustrated in Scheme Scheme 1

5

SYN
U.S. Patent No. 4,965,374 (the ‘374 patent) describes a process for producing gemcitabine from an intermediate 3,5-dibenzoyl ribo protected lactone of the formula:

11 where the desired erythro isomer can be isolated in a crystalline form from a mixture of erythro and threo isomers. The process described in the ‘374 patent is generally outlined in Scheme 2.
Scheme 2

mixture of α and β anomers
SYN
U.S. Patent No. 5,521,294 (the ‘294 patent) describes l-alkylsulfonyl-2,2- difluoro-3 -carbamoyl ribose intermediates and intermediate nucleosides derived therefrom. The compounds are reportedly useful in the preparation of 2′-deoxy-2′,2’- difluoro-β-cytidine and other β-anomer nucleosides. The ‘294 patent teaches, inter alia, that the 3-hydroxy carbamoyl group on the difluororibose intermediate may enhance formation of the desired β-anomer nucleoside derivative. The ‘294 patent describes converting the lactone 4 to the dibenzoyl mesylate 13, followed by deprotection at the 3 position to obtain the 5-monobenzoyl mesylate intermediate 15, which is reacted with various isocyanates to obtain the compounds of formula 16. The next steps involve coupling and deprotection using methods similar to those described in previous patents. The process and the intermediates 15 and 16 are illustrated by scheme 3 below: Scheme 3

13 15
PhCOCK
PhNCO/TEA -o. -~- j*«0Ms
PhNHCOO -r F
16
1 coupling 2 deprotection


16 gemcitabine
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0008621514000500
PATENT
https://patents.google.com/patent/WO2008129530A1/en
Scheme 4

e3
13A deprotection isomer separation

deprotection


EXAMPLE 1
[0045] This example demonstrates the preparation of 2-deoxy-2,2-difluoro-D- ribofuranose-3,5-dicinnamate-l-p-toluenesulfonate.
[0046] Crude 2-deoxy-2,2-difluoro-D-riboufuranose-3,5-dicinnamate (2.5g, 6 mmol) was dissolved in dichloromethane (20 ml) in a round flask, and diethylamine (0.7g, 9.6 mmol) was added followed by p-toluenesulfonyl chloride (1.32 g, 6.92 mmol), which was added drop wise while cooling to 0-50C. The mixture was stirred for 1 hour, and washed with IN HCl (15 ml), concentrated solution OfNaHCO3 (15 ml), and dried over MgSO4. The solvent was distilled off under reduced pressure to obtain crude 2-deoxy-2,2-difluoro-D-ribofuranose-3,5-dicinnamate-l-p- toluenesulfonate as light oil. Yield: 3.22 g, (5.6 mmol), 93%.
EXAMPLE 2
[0047] This example demonstrates the preparation of 3′,5′-dicinnamoyl-2′-deoxy- 2′,2′-difluorocytidine.
[0048] Dry 1 ,2-dichloroethane (800 ml) was added to N,O-bis(trimethylsilyl)- cytosine (136 g, 487 mmol) under nitrogen blanket to produce a clear solution, followed by adding trimethylsilyl triflate (Me3SiOTf), (100 ml, 122.8 g, 520 mmol) and stirred for 30 minutes. A solution of 2-deoxy-2,2-difluoro-D-ribofuranose-3,5- dicinnamate-1-p-toluenesulfonate (128 g, 224 mmol) in 1 ,2-dichloroethane (400 ml) was added drop wise, and the mixture was refluxed overnight. After cooling, the solvent was distilled off to obtain crude 3,5-dicinnamoyl-N4-trimethylsilyl-2′-deoxy- 2′,2′-difluorocytidine as a light yellow solid. The residue was dissolved in ethyl acetate (1600 ml) and washed 3 times with water (3X400 ml). The ethyl acetate phase was mixed with concentrated solution OfNaHCO3 (800 ml) for about 5 minutes, and then the mixture was set aside for about 20 minutes without stirring. The thus formed solid, which was precipitated in the inter-phase of the two layers, was filtered off and washed with 60 ml of ethyl acetate. The solid was dried under reduced pressure to obtain 116.7 g (223 mmol, 99.5%) of the crude 3′,5′-dicinnamoyl- 2′-deoxy-2′,2′- difluorocytidine containing 73.3 % of the β-anomer and 11.8 % of the α-anomer.
EXAMPLE 3
[0049] This example demonstrates the preparation of 3′,5′-dicinnamoyl-2′-deoxy- 2′,2′-difluorocytidine.
[0050] Dry 1,2-dichloroethane (1.5 L) was added to bis(trimethylsilyl)cytosine (417 g, 1.49 mol) under nitrogen blanket to produce a clear solution followed by adding trimethylsilyl triflate (Me3SiOTf), (300 ml, 368.4 g, 1.56 mol) and stirred for 30 minutes. A solution of 2-deoxy-2,2-difluoro-D-ribofuranose-3,5-dicinnamate-l-p- toluenesulfonate (384 g, 673 mmol) in 1,2-dichloroethane (1.2 L) was added drop wise, and the mixture was refluxed overnight. After cooling, the solvent was distilled off to obtain crude 3,5-dicinnamoyl-N4-trimethylsilyl-2l-deoxy-2′,2′-difluorocytidine as a light yellow solid. The residue was dissolved in ethyl acetate (2.4 L) and washed 3 times with water (3X1.2 L). The ethyl acetate phase was mixed with concentrated solution OfNaHCO3 (1.34 L) for about 20 minutes. The thus formed solid, which was precipitated in the inter-phase of the two layers, was filtered off and washed with 180 ml of ethyl acetate. The solid was dried under reduced pressure to obtain 346.5 g (0.66 mol, 99.9% yield) of the crude 3l,5l-dicinnamoyl-2′-deoxy-2′,2′-difluorocytidine containing 43 % of the β-anomer and 52 % of the α-anomer.
EXAMPLE 4
[0051] This example demonstrates the preparation of gemcitabine hydrochloride. [0052] To a solution of ammonia-methanol (15.8 %, 4.57 L), the crude 3,5- dicirmamoyl-2′-deoxy-2′,2′-difluorocytidine of example 3 was added (346.5 g, 0.66 mol), and stirred at ambient temperature for 6 hours. The mixture was concentrated to afford a light yellow solid (306 g). Purified water (3 L) was added to the solid, followed by addition of ethyl acetate (1.8 L), and stirring was maintained for about 10 minutes. The aqueous layer was separated and the organic layer was extracted with water (1.05 L). The aqueous layers were combined and water was removed by evaporation under reduced pressure to obtain an oil (154.7 g). Water was added (660 ml) and the mixture was heated to 50-550C to dissolve the solid. The mixture was cooled to 5-1O0C during about one hour and mixed for about 16 hours at that temperature. The thus formed solid was filtered and dried to afford 46.75 g (0.177 mol), containing 98 % of the β-anomer and 1.3 % of the α-anomer. 0.5N HCl (936 ml) was added followed by addition of dichloromethane (300 ml) with stirring. The water phase was separated and the aqueous phase was washed with dichloromethane (300 ml). After filtration, the aqueous phase was concentrated to dryness under reduced pressure to obtain gemcitabine hydrochloride as a solid (46.9 g). The solid was dissolved in water (187 ml) at ambient temperature and the mixture was heated to 500C to afford a clear solution and cooled to ambient temperature. Acetone (1.4 L) was added and stirring was maintained for about one hour. Then, the precipitate was collected by filtration and washed twice with acetone (2X30 ml) and dried at 450C under vacuum to obtain 39.2 g of gemcitabine hydrochloride, containing 99.9% of the β-anomer
EXAMPLE 5
[0053] This example demonstrates the preparation of gemcitabine hydrochloride. [0054] To a solution of ammonia-methanol (about 15.8 %, 1.35 L), the crude 3′,5′- dicinnamoyl-2′-deoxy-2′,2′-difluorocytidine prepared as described in example 2 was added (96 g, 183.4 mmol), and stirred at ambient temperature for 4 hours. The mixture was concentrated to afford a light yellow solid (80.5 g). Purified water (1 L) was added to the solid, followed by addition of ethyl acetate (600 ml), and stirring was maintained for about 10 minutes. The aqueous layer was separated and the organic layer was extracted with water (350 ml). The aqueous layers were combined and water was removed by evaporation under reduced pressure to obtain an oil (46.4 g). Water was added (220 ml) and the mixture was heated to 50-550C to dissolve the solid. The mixture was cooled to 0-50C during about one hour and mixed for about 16 hours at that temperature. The thus formed solid was filtered and dried to afford 11.1 g of gemcitabine free base. 0.5N HCl (240 ml) was added followed by addition of dichloromethane (100 ml) with stirring. The water phase was separated and the aqueous phase was washed with dichloromethane (300 ml). After filtration, the aqueous phase was concentrated to dryness under reduced pressure to obtain gemcitabine hydrochloride as a solid (12.0 g). The solid was dissolved in water (48 ml) at ambient temperature and the mixture was heated to 5O0C to afford a clear solution and cooled to ambient temperature. Acetone (360 ml) was added and stirring was maintained for about one hour. Then, the precipitate was collected by filtration and washed twice with acetone (2X30 ml) and dried at 450C under vacuum to obtain 9.9 g of gemcitabine hydrochloride, containing 99.6% of the β-anomer.
EXAMPLE 6
[0055] This example demonstrates the slurrying procedure of the 3 ‘,5′- dicinnamoyl-2′-deoxy-2’,2l-difluorocytidine in different solvents. [0056] 1 g of the crude 3′,5′-dicinnamoyl-2′-deoxy-2l,2′-difluorocytidine, containing 73.7 % of the β-anomer and 17.5 % of the α-anomer, was placed in flask and 10 ml of a solvent was added and the mixture was mixed at ambient temperature for one hour. Then, the solid was obtained by filtration, washed with 5 ml of the solvent and dried. The liquid obtained after filtering the solid and the liquid obtained after washing the solid were combined (hereinafter the mother liquor). The ratio between the β-anomer and the α-anomer in the solid and in the mother liquor was determined by HPLC and the results are summarized in Table 1.
Table 1

/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Adverse effects
Gemcitabine is a chemotherapy drug that works by killing any cells that are dividing.[10] Cancer cells divide rapidly and so are targeted at higher rates by gemcitabine, but many essential cells also divide rapidly, including cells in skin, the scalp, the stomach lining, and bone marrow, resulting in adverse effects.[16]: 265
The gemcitabine label carries warnings that it can suppress bone marrow function and cause loss of white blood cells, loss of platelets, and loss of red blood cells, and that it should be used carefully in people with liver, kidney, or cardiovascular disorders. People taking it should not take live vaccines. The warning label also states it may cause posterior reversible encephalopathy syndrome, that it may cause capillary leak syndrome, that it may cause severe lung conditions like pulmonary edema, pneumonia, and adult respiratory distress syndrome, and that it may harm sperm.[10][17]
More than 10% of users develop adverse effects, including difficulty breathing, low white and red blood cells counts, low platelet counts, vomiting and nausea, elevated transaminases, rashes and itchy skin, hair loss, blood and protein in urine, flu-like symptoms, and edema.[10][15]
Common adverse effects (occurring in 1–10% of users) include fever, loss of appetite, headache, difficulty sleeping, tiredness, cough, runny nose, diarrhea, mouth and lip sores, sweating, back pain, and muscle pain.[10]
Thrombotic thrombocytopenic purpura (TTP) is a rare but serious side effect that been associated with particular chemotherapy medications including gemcitabine. TTP is a blood disorder and can lead to microangipathic hemolytic anemia (MAHA), neurologic abnormalities, fever, and renal disease.[18]
Pharmacology
Gemcitabine is hydrophilic and must be transported into cells via molecular transporters for nucleosides (the most common transporters for gemcitabine are SLC29A1 SLC28A1, and SLC28A3).[19][20] After entering the cell, gemcitabine is first modified by attaching a phosphate to it, and so it becomes gemcitabine monophosphate (dFdCMP).[19][20] This is the rate-determining step that is catalyzed by the enzyme deoxycytidine kinase (DCK).[19][20] Two more phosphates are added by other enzymes. After the attachment of the three phosphates gemcitabine is finally pharmacologically active as gemcitabine triphosphate (dFdCTP).[19] [21]
After being thrice phosphorylated, gemcitabine can masquerade as deoxycytidine triphosphate and is incorporated into new DNA strands being synthesized as the cell replicates.[2][19][20]
When gemcitabine is incorporated into DNA it allows a native, or normal, nucleoside base to be added next to it. This leads to “masked chain termination” because gemcitabine is a “faulty” base, but due to its neighboring native nucleoside it eludes the cell’s normal repair system (base-excision repair). Thus, incorporation of gemcitabine into the cell’s DNA creates an irreparable error that leads to inhibition of further DNA synthesis, and thereby leading to cell death.[2][19][20]
The form of gemcitabine with two phosphates attached (dFdCDP) also has activity; it inhibits the enzyme ribonucleotide reductase (RNR), which is needed to create new DNA nucleotides. The lack of nucleotides drives the cell to uptake more of the components it needs to make nucleotides from outside the cell, which also increases uptake of gemcitabine.[2][19][20][22]
Chemistry
Gemcitabine is a synthetic pyrimidine nucleoside prodrug—a nucleoside analog in which the hydrogen atoms on the 2′ carbon of deoxycytidine are replaced by fluorine atoms.[2][23][24]
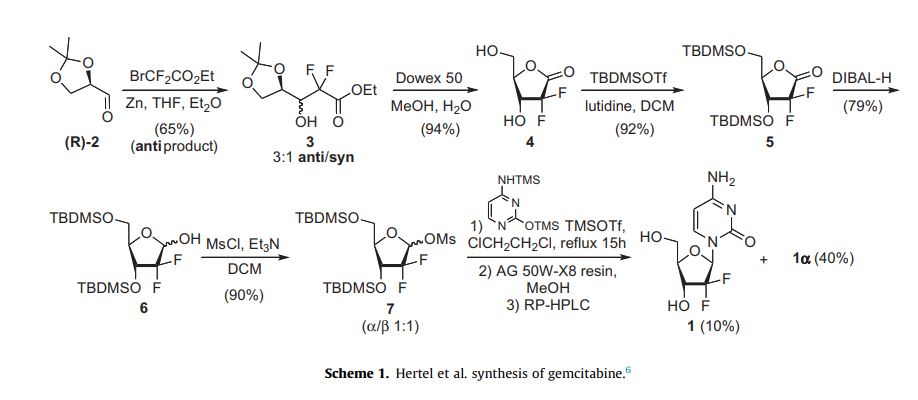
The synthesis described and pictured below is the original synthesis done in the Eli Lilly Company labs. Synthesis begins with enantiopure D-glyceraldehyde (R)-2 as the starting material which can made from D-mannitol in 2–7 steps. Then fluorine is introduced by a “building block” approach using ethyl bromodifluroacetate. Then, Reformatsky reaction under standard conditions will yield a 3:1 anti/syn diastereomeric mixture, with one major product. Separation of the diastereomers is carried out via HPLC, thus yielding the anti-3 gemcitabine in a 65% yield.[23][24] At least two other full synthesis methods have also been developed by different groups.[24]

Illustration of the original synthesis process used and published by Hertel et al. in 1988 of Lilly laboratories.
History[
Gemcitabine was first synthesized in Larry Hertel’s lab at Eli Lilly and Company during the early 1980s. It was intended as an antiviral drug, but preclinical testing showed that it killed leukemia cells in vitro.[25]
During the early 1990s gemcitabine was studied in clinical trials. The pancreatic cancer trials found that gemcitabine increased one-year survival time significantly, and it was approved in the UK in 1995[10] and approved by the FDA in 1996 for pancreatic cancers.[4] In 1998, gemcitabine received FDA approval for treating non-small cell lung cancer and in 2004, it was approved for metastatic breast cancer.[4]
European labels were harmonized by the EMA in 2008.[26]
By 2008, Lilly’s worldwide sales of gemcitabine were about $1.7 billion; at that time its US patents were set to expire in 2013 and its European patents in 2009.[27] The first generic launched in Europe in 2009,[7] and patent challenges were mounted in the US which led to invalidation of a key Lilly patent on its method to make the drug.[28][29] Generic companies started selling the drug in the US in 2010 when the patent on the chemical itself expired.[29][8] Patent litigation in China made headlines there and was resolved in 2010.[30]
Society and culture
As of 2017, gemcitabine was marketed under many brand names worldwide: Abine, Accogem, Acytabin, Antoril, axigem, Bendacitabin, Biogem, Boligem, Celzar, Citegin, Cytigem, Cytogem, Daplax, DBL, Demozar, Dercin, Emcitab, Enekamub, Eriogem, Fotinex, Gebina, Gemalata, Gembin, Gembine, Gembio, Gemcel, Gemcetin, Gemcibine, Gemcikal, Gemcipen, Gemcired, Gemcirena, Gemcit, Gemcitabin, Gemcitabina, Gemcitabine, Gemcitabinum, Gemcitan, Gemedac, Gemflor, Gemful, Gemita, Gemko, Gemliquid, Gemmis, Gemnil, Gempower, Gemsol, Gemstad, Gemstada, Gemtabine, Gemtavis, Gemtaz, Gemtero, Gemtra, Gemtro, Gemvic, Gemxit, Gemzar, Gentabim, Genuten, Genvir, Geroam, Gestredos, Getanosan, Getmisi, Gezt, Gitrabin, Gramagen, Haxanit, Jemta, Kalbezar, Medigem, Meditabine, Nabigem, Nallian, Oncogem, Oncoril, Pamigeno, Ribozar, Santabin, Sitagem, Symtabin, Yu Jie, Ze Fei, and Zefei.[1]
Research
Because it is clinically valuable and is only useful when delivered intravenously, methods to reformulate it so that it can be given by mouth have been a subject of research.[31][32][33]
Research into pharmacogenomics and pharmacogenetics has been ongoing. As of 2014, it was not clear whether or not genetic tests could be useful in guiding dosing and which people respond best to gemcitabine.[19] However, it appears that variation in the expression of proteins (SLC29A1, SLC29A2, SLC28A1, and SLC28A3) used for transport of gemcitabine into the cell lead to variations in its potency. Similarly, the genes that express proteins that lead to its inactivation (deoxycytidine deaminase, cytidine deaminase, and NT5C) and that express its other intracellular targets (RRM1, RRM2, and RRM2B) lead to variations in response to the drug.[19] Research has also been ongoing to understand how mutations in pancreatic cancers themselves determine response to gemcitabine.[34]
It has been studied as a treatment for Kaposi sarcoma, a common cancer in people with AIDS which is uncommon in the developed world but not uncommon in the developing world.[35]
References
- ^ Jump up to:a b c “Gemcitabine International Brands”. Drugs.com. Archived from the original on 25 May 2014. Retrieved 6 May 2017.
- ^ Jump up to:a b c d e f g h i j k l “Gemcitabine Hydrochloride”. The American Society of Health-System Pharmacists. Archived from the original on 2 February 2017. Retrieved 8 December 2016.
- ^ “Drug Formulary/Drugs/ gemcitabine – Provider Monograph”. Cancer Care Ontario. Retrieved 6 December 2020.
- ^ Jump up to:a b c National Cancer Institute (2006-10-05). “FDA Approval for Gemcitabine Hydrochloride”. National Cancer Institute. Archived from the original on 5 April 2017. Retrieved 22 April 2017.
- ^ Li Y, Li P, Li Y, Zhang R, Yu P, Ma Z, Kainov DE, de Man RA, Peppelenbosch MP, Pan Q (December 2020). “Drug screening identified gemcitabine inhibiting hepatitis E virus by inducing interferon-like response via activation of STAT1 phosphorylation”. Antiviral Research. 184: 104967. doi:10.1016/j.antiviral.2020.104967. PMID 33137361.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 511. ISBN 9783527607495.
- ^ Jump up to:a b Myers, Calisha (13 March 2009). “Gemcitabine from Actavis launched on patent expiry in EU markets”. FierceBiotech. Archived from the original on 11 September 2017.
- ^ Jump up to:a b “Press release: Hospira launches two-gram vial of gemcitabine hydrochloride for injection”. Hospira via News-Medical.Net. 16 November 2010. Archived from the original on 2 October 2015.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b c d e f g “UK label”. UK Electronic Medicines Compendium. 5 June 2014. Archived from the original on 10 July 2017. Retrieved 6 May 2017.
- ^ Jump up to:a b “US formLabel” (PDF). FDA. June 2014. Archived (PDF) from the original on 16 February 2017. Retrieved 6 May 2017. For label updates see FDA index page for NDA 020509 Archived 2017-04-29 at the Wayback Machine
- ^ Zhang XW, Ma YX, Sun Y, Cao YB, Li Q, Xu CA (June 2017). “Gemcitabine in Combination with a Second Cytotoxic Agent in the First-Line Treatment of Locally Advanced or Metastatic Pancreatic Cancer: a Systematic Review and Meta-Analysis”. Targeted Oncology. 12 (3): 309–321. doi:10.1007/s11523-017-0486-5. PMID 28353074. S2CID 3833614.
- ^ Plentz RR, Malek NP (December 2016). “Systemic Therapy of Cholangiocarcinoma”. Visceral Medicine. 32 (6): 427–430. doi:10.1159/000453084. PMC 5290432. PMID 28229078.
- ^ Jain A, Kwong LN, Javle M (November 2016). “Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice”. Current Treatment Options in Oncology. 17 (11): 58. doi:10.1007/s11864-016-0432-2. PMID 27658789. S2CID 25477593.
- ^ Jump up to:a b Macmillan Cancer Support. “Gemcitabine”. Macmillan Cancer Support. Archived from the original on 25 March 2017. Retrieved 6 May 2017.
- ^ Rachel Airley (2009). Cancer Chemotherapy. Wiley-Blackwell. ISBN 978-0-470-09254-5.
- ^ Siddall E, Khatri M, Radhakrishnan J (July 2017). “Capillary leak syndrome: etiologies, pathophysiology, and management”. Kidney International. 92 (1): 37–46. doi:10.1016/j.kint.2016.11.029. PMID 28318633.
- ^ Kasi PM (January 2011). “Thrombotic thrombocytopenic purpura and gemcitabine”. Case Reports in Oncology. 4 (1): 143–8. doi:10.1159/000326801. PMC 3114619. PMID 21691573.
- ^ Jump up to:a b c d e f g h i Alvarellos ML, Lamba J, Sangkuhl K, Thorn CF, Wang L, Klein DJ, Altman RB, Klein TE (November 2014). “PharmGKB summary: gemcitabine pathway”. Pharmacogenetics and Genomics. 24 (11): 564–74. doi:10.1097/fpc.0000000000000086. PMC 4189987. PMID 25162786.
- ^ Jump up to:a b c d e f Mini E, Nobili S, Caciagli B, Landini I, Mazzei T (May 2006). “Cellular pharmacology of gemcitabine”. Annals of Oncology. 17 Suppl 5: v7-12. doi:10.1093/annonc/mdj941. PMID 16807468.
- ^ Fatima, M., Iqbal Ahmed, M. M., Batool, F., Riaz, A., Ali, M., Munch-Petersen, B., & Mutahir, Z. (2019). Recombinant deoxyribonucleoside kinase from Drosophila melanogaster can improve gemcitabine based combined gene/chemotherapy for targeting cancer cells. Bosnian Journal of Basic Medical Sciences, 19(4), 342-349. https://doi.org/10.17305/bjbms.2019.4136
- ^ Cerqueira NM, Fernandes PA, Ramos MJ (2007). “Understanding ribonucleotide reductase inactivation by gemcitabine”. Chemistry. 13 (30): 8507–15. doi:10.1002/chem.200700260. PMID 17636467.
- ^ Jump up to:a b Brown K, Weymouth-Wilson A, Linclau B (April 2015). “A linear synthesis of gemcitabine”. Carbohydrate Research. 406: 71–5. doi:10.1016/j.carres.2015.01.001. PMID 25681996.
- ^ Jump up to:a b c Brown K, Dixey M, Weymouth-Wilson A, Linclau B (March 2014). “The synthesis of gemcitabine”. Carbohydrate Research. 387: 59–73. doi:10.1016/j.carres.2014.01.024. PMID 24636495.
- ^ Sneader, Walter (2005). Drug discovery: a history. New York: Wiley. p. 259. ISBN 978-0-471-89979-2.
- ^ “Gemzar”. European Medicines Agency. 24 September 2008. Archived from the original on 11 September 2017.
- ^ Myers, Calisha (18 August 2009). “Patent for Lilly’s cancer drug Gemzar invalidated”. FiercePharma. Archived from the original on 11 September 2017.
- ^ Holman, Christopher M. (Summer 2011). “Unpredictability in Patent Law and Its Effect on Pharmaceutical Innovation” (PDF). Missouri Law Review. 76 (3): 645–693. Archived from the original (PDF) on 2017-09-11. Retrieved 2017-05-06.
- ^ Jump up to:a b Ravicher, Daniel B. (28 July 2010). “On the Generic Gemzar Patent Fight”. Seeking Alpha. Archived from the original on 9 December 2012.
- ^ Wang M, Alexandre D (2015). “Analysis of Cases on Pharmaceutical Patent Infringement in Great China”. In Rader RR, et al. (eds.). Law, Politics and Revenue Extraction on Intellectual Property. Cambridge Scholars Publishing. p. 119. ISBN 9781443879262. Archived from the original on 2017-09-11.
- ^ Dyawanapelly S, Kumar A, Chourasia MK (2017). “Lessons Learned from Gemcitabine: Impact of Therapeutic Carrier Systems and Gemcitabine’s Drug Conjugates on Cancer Therapy”. Critical Reviews in Therapeutic Drug Carrier Systems. 34 (1): 63–96. doi:10.1615/CritRevTherDrugCarrierSyst.2017017912. PMID 28322141.
- ^ Birhanu G, Javar HA, Seyedjafari E, Zandi-Karimi A (April 2017). “Nanotechnology for delivery of gemcitabine to treat pancreatic cancer”. Biomedicine & Pharmacotherapy. 88: 635–643. doi:10.1016/j.biopha.2017.01.071. PMID 28142120.
- ^ Dubey RD, Saneja A, Gupta PK, Gupta PN (October 2016). “Recent advances in drug delivery strategies for improved therapeutic efficacy of gemcitabine”. European Journal of Pharmaceutical Sciences. 93: 147–62. doi:10.1016/j.ejps.2016.08.021. PMID 27531553.
- ^ Pishvaian MJ, Brody JR (March 2017). “Therapeutic Implications of Molecular Subtyping for Pancreatic Cancer”. Oncology. 31 (3): 159–66, 168. PMID 28299752. Archived from the original on 3 July 2017.
- ^ Krown SE (September 2011). “Treatment strategies for Kaposi sarcoma in sub-Saharan Africa: challenges and opportunities”. Current Opinion in Oncology. 23 (5): 463–8. doi:10.1097/cco.0b013e328349428d. PMC 3465839. PMID 21681092.
External links
- “Gemcitabine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | /dʒɛmˈsaɪtəbiːn/ |
| Trade names | Gemzar, others[1] |
| Other names | 2′, 2′-difluoro 2’deoxycytidine, dFdC |
| AHFS/Drugs.com | Monograph |
| Pregnancy category | AU: D |
| Routes of administration | Intravenous |
| ATC code | L01BC05 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | <10% |
| Elimination half-life | Short infusions: 32–94 minutes Long infusions: 245–638 minutes |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 95058-81-4 |
| PubChem CID | 60750 |
| IUPHAR/BPS | 4793 |
| DrugBank | DB00441 |
| ChemSpider | 54753 |
| UNII | B76N6SBZ8R |
| KEGG | D02368 |
| ChEBI | CHEBI:175901 |
| ChEMBL | ChEMBL888 |
| CompTox Dashboard (EPA) | DTXSID3040487 |
| ECHA InfoCard | 100.124.343 |
| Chemical and physical data | |
| Formula | C9H11F2N3O4 |
| Molar mass | 263.201 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////GEMCITABINE, LY 188011, LY188011, CANCER
NC1=NC(=O)N(C=C1)[C@@H]1O[C@H](CO)[C@@H](O)C1(F)F
Patent
Publication numberPriority datePublication dateAssigneeTitle
EP0577303A1 *1992-06-221994-01-05Eli Lilly And CompanyStereoselective glycosylation process
WO2006070985A1 *2004-12-302006-07-06Hanmi Pharm. Co., Ltd.METHOD FOR THE PREPARATION OF 2#-DEOXY-2#,2#-DIFLUOROCYTIDINE
WO2007027564A2 *2005-08-292007-03-08Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
WO2007069838A1 *2005-12-142007-06-21Dong-A Pharm.Co., Ltd.A manufacturing process of 2′,2′-difluoronucleoside and intermediate
Family To Family Citations
JPS541315B2 *1974-11-221979-01-23
US4526988A *1983-03-101985-07-02Eli Lilly And CompanyDifluoro antivirals and intermediate therefor
US4751221A *1985-10-181988-06-14Sloan-Kettering Institute For Cancer Research2-fluoro-arabinofuranosyl purine nucleosides
US5223608A *1987-08-281993-06-29Eli Lilly And CompanyProcess for and intermediates of 2′,2′-difluoronucleosides
US4965374A *1987-08-281990-10-23Eli Lilly And CompanyProcess for and intermediates of 2′,2′-difluoronucleosides
US5256798A *1992-06-221993-10-26Eli Lilly And CompanyProcess for preparing alpha-anomer enriched 2-deoxy-2,2-difluoro-D-ribofuranosyl sulfonates
US5371210A *1992-06-221994-12-06Eli Lilly And CompanyStereoselective fusion glycosylation process for preparing 2′-deoxy-2′,2′-difluoronucleosides and 2′-deoxy-2′-fluoronucleosides
US5256797A *1992-06-221993-10-26Eli Lilly And CompanyProcess for separating 2-deoxy-2,2-difluoro-D-ribofuranosyl alkylsulfonate anomers
US5480992A *1993-09-161996-01-02Eli Lilly And CompanyAnomeric fluororibosyl amines
US5521294A *1995-01-181996-05-28Eli Lilly And Company2,2-difluoro-3-carbamoyl ribose sulfonate compounds and process for the preparation of beta nucleosides
US5559222A *1995-02-031996-09-24Eli Lilly And CompanyPreparation of 1-(2′-deoxy-2′,2′-difluoro-D-ribo-pentofuranosyl)-cytosine from 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose
US5602262A *1995-02-031997-02-11Eli Lilly And CompanyProcess for the preparation of 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose
US5633367A *1995-03-241997-05-27Eli Lilly And CompanyProcess for the preparation of a 2-substituted 3,3-difluorofuran
GB9514268D0 *1995-07-131995-09-13Hoffmann La RochePyrimidine nucleoside
US5756775A *1995-12-131998-05-26Eli Lilly And CompanyProcess to make α,α-difluoro-β-hydroxyl thiol esters
CA2641719A1 *2006-02-072007-08-16Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
US20070249823A1 *2006-04-202007-10-25Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
Publication numberPriority datePublication dateAssigneeTitle
CN102617483A *2011-06-302012-08-01江苏豪森药业股份有限公司Process for recycling cytosine during preparing process of gemcitabine hydrochloride
WO2013164798A12012-05-042013-11-07Tpresso AgPackaging of dry leaves in sealed capsules
CN105566418A *2014-10-092016-05-11江苏笃诚医药科技股份有限公司2′,3′-di-O-acetyl-5′-deoxy-5-fluorocytidine synthesis method
EP3817732A4 *2018-08-032022-06-08Cellix Bio Private LimitedCompositions and methods for the treatment of cancer
Family To Family Citations
CA2641719A1 *2006-02-072007-08-16Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
US20070249823A1 *2006-04-202007-10-25Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
IT1393062B1 *2008-10-232012-04-11Prime Europ TherapeuticalsPROCEDURE FOR THE PREPARATION OF GEMCITABINE CHLORIDRATE
PublicationPublication DateTitle
CA2509687C2012-08-14Process for the production of 2'-branched nucleosides
WO2008129530A12008-10-30Gemcitabine production process
AU2005328519B22012-03-01Intermediate and process for preparing of beta- anomer enriched 21deoxy, 21 ,21-difluoro-D-ribofuranosyl nucleosides
EP1931693A22008-06-18Process for preparing gemcitabine and associated intermediates
EP2164856A12010-03-24Processes related to making capecitabine
US8193354B22012-06-05Process for preparation of Gemcitabine hydrochloride
EP3638685A12020-04-22Synthesis of 3'-deoxyadenosine-5'-0-[phenyl(benzyloxy-l-alaninyl)]phosphate (nuc-7738)
EP2456778A12012-05-30Process for producing flurocytidine derivatives
US20090221811A12009-09-03Process for preparing gemcitabine and associated intermediates
JP5114556B22013-01-09A novel highly stereoselective synthetic process and intermediate for gemcitabine
EP0350292B11994-05-04Process for preparing 2'-deoxy-beta-adenosine
KR100908363B12009-07-20Stereoselective preparation method of tri-O-acetyl-5-deoxy-β-D-ribofuranose and separation method thereof
US20070249823A12007-10-25Process for preparing gemcitabine and associated intermediates
WO2007070804A22007-06-21Process for preparing gemcitabine and associated intermediates
KR101259648B12013-05-09A manufacturing process of 2′,2′-difluoronucloside and intermediate
US8338586B22012-12-25Process of making cladribine
WO2010029574A22010-03-18An improved process for the preparation of gemcitabine and its intermediates using novel protecting groups and ion exchange resins
KR20080090950A2008-10-09Process for preparing gemcitabine and associated intermediates
WO2012115578A12012-08-30Synthesis of flg

NEW DRUG APPROVALS
ONE TIME
$9.00
OTERACIL POTTASIUM

OTERACIL
UNII4R7FFA00RX, CAS Number2207-75-2, WeightAverage: 195.175, Monoisotopic: 194.96823705, Chemical FormulaC4H2KN3O4
[K+].OC1=NC(=NC(=O)N1)C([O-])=O
1,3,5-Triazine-2-carboxylic acid, 1,4,5,6-tetrahydro-4,6-dioxo-, potassium salt (1:1)
218-627-5[EINECS]
2207-75-2[RN]
4,6-Dihydroxy-1,3,5-triazine-2-carboxylic acid potassium salt
- KOX
- NSC 28841
- Oxonate
- Oxonate, potassium
CDSCO APPROVED,01.02.2022

Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
Oteracil Potassium is the potassium salt of oxonate, an enzyme inhibitor that modulates 5- fluorouracil (5-FU) toxicity. Potassium oxonate inhibits orotate phosphoribosyltransferase, which catalyzes the conversion of 5-FU to its active or phosphorylated form, FUMP. Upon oral administration, Oxonate is selectively distributed to the intracellular sites of tissues lining the small intestines, producing localized inhibitory effects within the gastrointestinal tract. As a result, 5-FU associated gastrointestinal toxic effects are reduced and the incidence of diarrhea or mucositis is decreased in 5-FU related therapy.
Oteracil is an adjunct to antineoplastic therapy, used to reduce the toxic side effects associated with chemotherapy. Approved by the European Medicines Agency (EMA) in March 2011, Oteracil is available in combination with Gimeracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Oteracil’s main role within Teysuno is to reduce the activity of 5-FU within normal gastrointestinal mucosa, and therefore reduce’s gastrointestinal toxicity 1. It functions by blocking the enzyme orotate phosphoribosyltransferase (OPRT), which is involved in the production of 5-FU.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYNTHESIS
https://patents.google.com/patent/CN103435566A/zh


SYN
https://europepmc.org/article/pmc/pmc7717319
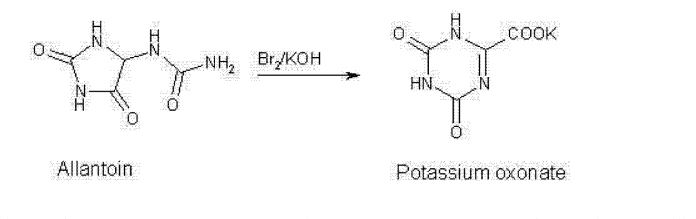
Poje et al. reported a two-step, gram-scale preparation of the TS-1 additive oteracil 21 (Scheme 16).226 Iodine-mediated-oxidation of uric acid 116 produced dehydroallantoin 117 as the major product, and subsequent treatment with potassium hydroxide resulted in the rearranged product oteracil 21.227

Synthesis of Oteracil 21a
aReagents and conditions: (a) LiOH, I2, H2O, 5 °C, 5 min, then AcOH, 75%; (b) aq KOH, 20 min, rt, 82%.
(226) Poje M; Sokolić-Maravić L The mechanism for the conversion of uric acid into allantoin and dehydro-allantoin: A new look at an old problem. Tetrahedron 1986, 42 (2), 747–751. [Google Scholar]
(227) Sugi M; Igi M EP Patent 0957096, 1999.
EP0957096A1 *1998-05-111999-11-17SUMIKA FINE CHEMICALS Co., Ltd.Method for producing potassium oxonate
CN101475539A *2009-02-112009-07-08鲁南制药集团股份有限公司Refining method for preparing high-purity oteracil potassium
CN102250025A *2011-05-182011-11-23深圳万乐药业有限公司Preparation method suitable for industrially producing oteracil potassium
CN102746244A *2012-07-272012-10-24南京正大天晴制药有限公司Refining method of oteracil potassium
//////////OTERACIL POTTASIUM, KOX, NSC 28841, Oxonate, Oxonate potassium, INDIA 2022, APPROVALS 2022, CANCER
[K+].OC1=NC(=NC(=O)N1)C([O-])=O

NEW DRUG APPROVALS
ONE TIME
$10.00
GIMERACIL
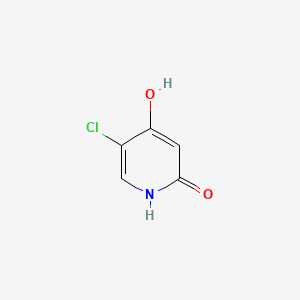

GIMERACIL
C5H4ClNO2, 145.54
5-chloro-4-hydroxy-1H-pyridin-2-one
5-Chloro-2,4-dihydroxypyridine
5-Chloro-4-hydroxy-2(1H)-pyridone
CDSCO APPROVED,01.02.2022
Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
| Combination of | |
|---|---|
| Tegafur | Antineoplastic drug |
| Gimeracil | Enzyme inhibitor |
| Oteracil | Enzyme inhibitor |
| Clinical data | |
| Trade names | Teysuno, TS-1 |
| Other names | S-1[1] |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by Tegafur |
| Pregnancy category | Contraindicated |
| Routes of administration | By mouth |
| ATC code | L01BC53 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) [2]EU: Rx-only [3]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 150863-82-4 |
| PubChem CID | 54715158 |
Tegafur/gimeracil/oteracil, sold under the brand names Teysuno and TS-1,[3][4] is a fixed-dose combination medication used for the treatment of advanced gastric cancer when used in combination with cisplatin,[3] and also for the treatment of head and neck cancer, colorectal cancer, non–small-cell lung, breast, pancreatic, and biliary tract cancers.[5]: 213
The most common severe side effects when used in combination with cisplatin include neutropenia (low levels of neutrophils, a type of white blood cell), anaemia (low red blood cell counts) and fatigue (tiredness).[3]
Tegafur/gimeracil/oteracil (Teysuno) was approved for medical use in the European Union in March 2011.[3] It has not been approved by the U.S. Food and Drug Administration (FDA).[5]: 213
Medical uses
In the European Union tegafur/gimeracil/oteracil is indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.[3]
Contraindications
In the European Union, tegafur/gimeracil/oteracil must not be used in the following groups:
- people receiving another fluoropyrimidine (a group of anticancer medicines that includes tegafur/gimeracil/oteracil) or who have had severe and unexpected reactions to fluoropyrimidine therapy;[3]
- people known to have no DPD enzyme activity, as well as people who, within the previous four weeks, have been treated with a medicine that blocks this enzyme;[3]
- pregnant or breastfeeding women;[3]
- people with severe leucopenia, neutropenia, or thrombocytopenia (low levels of white cells or platelets in the blood);[3]
- people with severe kidney problems requiring dialysis;[3]
- people who should not be receiving cisplatin.[3]
Mechanism of action
Tegafur is the actual chemotherapeutic agent. It is a prodrug of the active substance fluorouracil (5-FU).[3] Tegafur, is a cytotoxic medicine (a medicine that kills rapidly dividing cells, such as cancer cells) that belongs to the ‘anti-metabolites’ group. Tegafur is converted to the medicine fluorouracil in the body, but more is converted in tumor cells than in normal tissues.[3] Fluorouracil is very similar to pyrimidine.[3] Pyrimidine is part of the genetic material of cells (DNA and RNA).[3] In the body, fluorouracil takes the place of pyrimidine and interferes with the enzymes involved in making new DNA.[3] As a result, it prevents the growth of tumor cells and eventually kills them.[3]
Gimeracil inhibits the degradation of fluorouracil by reversibly blocking the dehydrogenase enzyme dihydropyrimidine dehydrogenase (DPD). This results in higher 5-FU levels and a prolonged half-life of the substance.[6]
Oteracil mainly stays in the gut because of its low permeability, where it reduces the production of 5-FU by blocking the enzyme orotate phosphoribosyltransferase. Lower 5-FU levels in the gut result in a lower gastrointestinal toxicity.[6]
Within the medication, the molar ratio of the three components (tegafur:gimeracil:oteracil) is 1:1:0.4.[7]
The maximum tolerated dose differed between Asian and Caucasian populations (80 mg/m2 and 25 mg/m2 respectively), perhaps due to differences in CYP2A6 genotype.[5]: 213
Research
It is being developed for the treatment of hepatocellular carcinoma.[8] and has activity in esophageal,(Perry Chapter 33) breast,[citation needed] cervical,[citation needed] and colorectal cancer.[9]
References
- ^ Liu TW, Chen LT (201). “S-1 with leucovorin for gastric cancer: how far can it go?”. Lancet Oncol. 17 (1): 12–4. doi:10.1016/S1470-2045(15)00478-7. PMID 26640038.
- ^ “Teysuno 20mg/5.8mg/15.8mg hard capsules – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 30 July 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Teysuno EPAR”. European Medicines Agency (EMA). Retrieved 30 July 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “ティーエスワン 患者さん・ご家族向け総合情報サイト | 大鵬薬品工業株式会社”.
- ^ Jump up to:a b c DeVita, DeVita; Lawrence, TS; Rosenberg, SA (2015). DeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology (10th ed.). LWW. ISBN 978-1451192940.
- ^ Jump up to:a b A. Klement (22 July 2013). “Dreier-Kombination gegen Magenkrebs: Teysuno”. Österreichische Apothekerzeitung (in German) (15/2013): 23.
- ^ Peters GJ, Noordhuis P, Van Kuilenburg AB et al. (2003). “Pharmacokinetics of S-1, an oral formulation of ftorafur, oxonic acid and 5-chloro-2,4-dihydroxypyridine (molar ratio 1:0.4:1) in patients with solid tumors”. Cancer Chemother. Pharmacol. 52 (1): 1–12. doi:10.1007/s00280-003-0617-9. PMID 12739060. S2CID 10858817.
- ^ “BCIQ”.
- ^ Miyamoto Y, Sakamoto Y, Yoshida N, Baba H (2014). “Efficacy of S-1 in colorectal cancer”. Expert Opin Pharmacother. 15 (12): 1761–70. doi:10.1517/14656566.2014.937706. PMID 25032886. S2CID 23637808.
External links
- “Tegafur”. Drug Information Portal. U.S. National Library of Medicine.
- “Gimeracil”. Drug Information Portal. U.S. National Library of Medicine.
- “Oteracil”. Drug Information Portal. U.S. National Library of Medicine.
Gimeracil is an adjunct to antineoplastic therapy, used to increase the concentration and effect of the main active componets within chemotherapy regimens. Approved by the European Medicines Agency (EMA) in March 2011, Gimeracil is available in combination with Oteracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Gimeracil’s main role within Teysuno is to prevent the breakdown of Fluorouracil (5-FU), which helps to maintin high enough concentrations for sustained effect against cancer cells 2. It functions by reversibly and selectively blocking the enzyme dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU 1. This allows higher concentrations of 5-FU to be achieved with a lower dose of tegafur, thereby also reducing toxic side effects.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////


SYNTHESIS

SYN
https://europepmc.org/article/pmc/pmc7717319
Synthesis of Gimeracil 20a
aReagents and conditions: (a) CH3C(OCH3)3, MeOH, then (CH3)2NHCH(OCH3)2, reflux, 92%; (b) aq AcOH, 130 °C, 2 h, 95%; (c) SO2Cl2, HOAc, 50 °C, 0.5 h, 91%; (d) 40% H2SO4, 130 °C, 4 h, 91%; (e) SO2Cl2, HOAc, 50 °C, 45 min, 86%; (f) 75% H2 SO4, 140 °C, 3 h, then NaOH, then pH 4–4.5, 89%


In 1953, Kolder and Hertog reported a synthesis of the TS-1 additive gimeracil 20, which was completed in seven steps using 4-nitropyridine N-oxide as starting material.222 Later, Yano et al. reported an alternative gram-scale synthesis (Scheme 15).223 The one-pot, three component condensation of malononitrile 111, 1,1,1-trimethoxyethane, and 1,1-dimethyoxytrimethylamine generated the dicyano intermediate 112, which was into 2(1H)-pyridinone 113.224 Selective chlorination of 113 was followed by acid-mediated demethylation, hydrolysis, and decarboxylation, to afford gimeracil 20. Interestingly, Xu et al. found that treatment of intermediate 113 with sulfuryl chloride resulted in dichloro 115 formation, which could still be converted to gimeracil 20 by treatment with sulfuric acid.225
(222) Kolder CR; den Hertog HJ Synthesis and reactivity of 5-chloro-2,4-dihydroxypyridine. Rec. Trav. Chim 1953, 72, 285–295. [Google Scholar]
(223) Yano S; Ohno T; Ogawa K Convenient and practical synthesis of 5-chloro-4-hydroxy-2(1H)-pyridinone. Heterocycles 1993, 36, 145–148. [Google Scholar]
(224) Mittelbach M; Kastner G; Junek H Synthesen mit Nitrilen, 71. Mitt. Zur Synthese von 4-Hydroxynicotinsaure aus Butadiendicarbonitrilen. Arch. Pharm 1985, 318 (6), 481–486. [Google Scholar]
(225) Xu Y; Mao D; Zhang F CN Patent 1915976, 2007.

NEW DRUG APPROVALS
THIS MAY NOT RUN WITHOUT SUBSCRIPTION HELP. AVOID CLOSURE OF THIS BLOG
$10.00
//////////GIMERACIL, APPROVALS 2022, INDIA 2022
OC1=CC(=O)NC=C1Cl
Danavorexton, TAK 925


Danavorexton, TAK 925
2114324-48-8
- Molecular FormulaC21H32N2O5S
- Average mass424.554 Da
1-Piperidinecarboxylic acid, 3-[(methylsulfonyl)amino]-2-[[(cis-4-phenylcyclohexyl)oxy]methyl]-, methyl ester, (2R,3S)-
Methyl (2R,3S)-3-[(methylsulfonyl)amino]-2-[[(cis-4-phenylcyclohexyl)oxy]methyl]-1-piperidinecarboxylate
- OriginatorTakeda
- ClassCyclohexanes; Esters; Ethers; Piperidines; Sleep disorder therapies; Small molecules; Sulfonamides
- Mechanism of ActionOrexin receptor type 2 agonists
- Orphan Drug StatusYes – Narcolepsy
- Phase IHypersomnia; Narcolepsy; Respiration disorders; Sleep apnoea syndrome
- 01 Jun 2022Takeda Pharmaceuticals completes a phase I clinical trials in Respiratory disorder (In adults) in Netherlands (IV) (ISRCTN63027076)
- 02 Apr 2022Efficacy and safety data from phase a Ib trial in Hypersomnia presented at the 74th Annual Meeting of the American Academy of Neurology 2022 (AAN-2022)
- 10 Mar 2022Phase-I clinical trials in Sleep apnoea syndrome in Australia (IV) (NCT05180890)
Danavorexton (developmental code name TAK-925) is a selective orexin 2 receptor agonist.[1] It is a small-molecule compound and is administered intravenously.[1][2] The compound was found to dose-dependently produce wakefulness to a similar degree as modafinil in a phase 1 clinical trial.[1][3] As of March 2021, danavorexton is under development for the treatment of narcolepsy, idiopathic hypersomnia, and sleep apnea.[2][1][4] It is related to another orexin receptor agonist known as TAK-994, the development of which was discontinued for safety reasons in October 2021.[1][5]
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00626
TAK-925, a potent, selective, and brain-penetrant orexin 2 receptor (OX2R) agonist, [methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4-phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate, 16], was identified through the optimization of compound 2, which was discovered by a high throughput screening (HTS) campaign. Subcutaneous administration of compound 16 produced wake-promoting effects in mice during the sleep phase. Compound 16 (TAK-925) is being developed for the treatment of narcolepsy and other related disorders.


aReagents and conditions: (a) chiral column separation; (b) RCOCl, Et3N, THF, rt (for 15 and 16); (c) ethyl chlorocarbonate, DIEA, THF, rt (for 17); (d) isocyanatoethane, Et3N, THF, 0 °C−rt (for 18).
Methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4- phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate (16) To a mixture of 14 (58 mg, 0.16 mmol) and Et3N (0.044 mL, 0.32 mmol) in THF (3 mL) was added methyl chlorocarbonate (0.024 mL, 0.32 mmol) at rt. The mixture was stirred at rt overnight. The mixture was quenched with water and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane/EtOAc, 1:1 to 0:100) to give 16 (64 mg, 0.15 mmol, 95%) as a colorless oil. Crystallization of 16 (1.8 g, 4.1 mmol) from EtOH-H2O gave 16 (1.7 g, 3.9 mmol, 95%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 1.40−1.55 (5H, m), 1.56−1.73 (5H, m), 1.87 (1H, brd, J = 13.2 Hz), 1.96 (1H, brd, J = 13.6 Hz), 2.44−2.57 (1H, m), 2.83 (1H, brs), 2.95 (3H, s), 3.40 (1H, brs), 3.53−3.62 (5H, m), 3.73 (1H, brt, J = 9.7 Hz), 3.84 (1H, brs), 4.47 (1H, brs), 7.15 (1H, brt, J = 7.2 Hz), 7.18 (1H, brs), 7.19 (2H, brd, J = 8.1 Hz), 7.27 (2H, brt, J = 7.4 Hz). 13C NMR (151 MHz, DMSO-d6, the minor rotamer’s signals are marked with an asterisk) δ24.05, 24.39*, 26.00, 26.17*, 27.60*, 27.79, 28.68, 30.15*, 37.54, 38.13*, 39.91, 42.99, 51.01, 52.07, 53.90*, 54.49, 61.48, 61.89*, 71.68, 125.68, 126.51, 128.14, 147.34, 155.27*, 156.08. MS (ESI/APCI) mass calculated for [M + H]+ (C21H33N2O5S) requires m/z 424.6, found m/z 425.2. mp 113 °C. Anal. Calcd for C21H32N2O5S: C, 59.41; H, 7.60; N, 6.60. Found: C, 59.45; H, 7.59; N, 6.55. [α] 20 D +16.3 (c 0.1, CHCl3
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | TAK-925 |
| Routes of administration | Intravenous[1][2] |
| Drug class | Orexin receptor agonist |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2114324-48-8 |
| PubChem CID | 130310079 |
| ChemSpider | 68011464 |
| UNII | 1QMD83K4YN |
| ChEMBL | ChEMBL4650341 |
| Chemical and physical data | |
| Formula | C21H32N2O5S |
| Molar mass | 424.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
References
- ^ Jump up to:a b c d e f Jacobson LH, Hoyer D, de Lecea L (January 2022). “Hypocretins (orexins): The ultimate translational neuropeptides”. J Intern Med. doi:10.1111/joim.13406. PMID 35043499.
- ^ Jump up to:a b c “Danavorexton – Takeda”. Adis Insight. Springer Nature Switzerland AG. Retrieved 7 March 2021.
- ^ Evans, R., Hazel, J., Faessel, H., Wu, J., Hang, Y., Alexander, R., … & Hartman, D. (2019). Results of a phase 1, 4-period crossover, placebo-controlled, randomized, single dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-925, a novel orexin 2 receptor agonist, in sleep-deprived healthy adults, utilizing modafinil as an active comparator. Sleep Medicine, 64, S106. https://scholar.google.com/scholar?cluster=10933819770107034612
- ^ Evans R, Tanaka S, Tanaka S, Touno S, Shimizu K, Sakui S, et al. (December 2019). “A Phase 1 single ascending dose study of a novel orexin 2 receptor agonist, TAK-925, in healthy volunteers (HV) and subjects with narcolepsy type 1 (NT1) to assess safety, tolerability, pharmacokinetics, and pharmacodynamic outcomes”. Sleep Medicine. 64: S105–S106. doi:10.1016/j.sleep.2019.11.290.
- ^ Tong A (6 October 2021). “Takeda flashes red light on ‘breakthrough’ narcolepsy drug after PhII trials turned up mysterious safety signal”. Endpoints News.
External links
///////////////Danavorexton, TAK 925, ORPHAN DRUG, PHASE 1

NEW DRUG APPROVALS
ONE TIME
$10.00
ATISOBAN


ATOSIBAN
cas 90779-69-4
WeightAverage: 994.19
Monoisotopic: 993.441208989
Chemical FormulaC43H67N11O12S2
(2S)-5-amino-2-{[(2S)-1-[(4R,7S,10S,13S,16R)-13-[(2S)-butan-2-yl]-7-(carbamoylmethyl)-16-[(4-ethoxyphenyl)methyl]-10-[(1R)-1-hydroxyethyl]-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentaazacycloicosane-4-carbonyl]pyrrolidin-2-yl]formamido}-N-(carbamoylmethyl)pentanamide
- Oxytocin, 1-(3-mercaptopropanoic acid)-2-(O-ethyl-D-tyrosine)-4-L-threonine-8-L-ornithine-
- 1,2-Dithia-5,8,11,14,17-pentaazacycloeicosane, cyclic peptide deriv.
- Antocile
- Antocin
- Antocin II
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Atosiban acetate | 0P5DNO7CEF | 914453-95-5 | SVDWBHHCPXTODI-QIWYXCRTSA-N |
- CAP-449
- CAP-476
- CAP-581
- F-314
- ORF 22164
- ORF-22164
- RW-22164
- RWJ 22164
- RWJ-22164
Atosiban, sold under the brand name Tractocile among others, is an inhibitor of the hormones oxytocin and vasopressin. It is used as an intravenous medication as a labour repressant (tocolytic) to halt premature labor. It was developed by Ferring Pharmaceuticals in Sweden and first reported in the literature in 1985.[5] Originally marketed by Ferring Pharmaceuticals, it is licensed in proprietary and generic forms for the delay of imminent preterm birth in pregnant adult women.
The most commonly reported side effect is nausea.[4]
Atosiban is an inhibitor of the hormones oxytocin and vasopressin. It is used intravenously to halt premature labor. Although initial studies suggested it could be used as a nasal spray and hence would not require hospital admission, it is not used in that form. Atobisan was developed by the Swedish company Ferring Pharmaceuticals. It was first reported in the literature in 1985. Atosiban is licensed in proprietary and generic forms for the delay of imminent pre-term birth in pregnant adult women.
Medical uses
Atosiban is used to delay birth in adult women who are 24 to 33 weeks pregnant, when they show signs that they may give birth pre-term (prematurely).[4] These signs include regular contractions lasting at least 30 seconds at a rate of at least four every 30 minutes,[4] and dilation of the cervix (the neck of the womb) of 1 to 3 cm and an effacement (a measure of the thinness of the cervix) of 50% or more.[4] In addition, the baby must have a normal heart rate.[4]
Pharmacology
Mechanism of action
Atosiban is a nonapeptide, desamino-oxytocin analogue, and a competitive vasopressin/oxytocin receptor antagonist (VOTra). Atosiban inhibits the oxytocin-mediated release of inositol trisphosphate from the myometrial cell membrane. As a result, reduced release of intracellular, stored calcium from the sarcoplasmic reticulum of myometrial cells and reduced influx of Ca2+ from the extracellular space through voltage-gated channels occur. In addition, atosiban suppresses oxytocin-mediated release of PGE and PGF from the decidua.[6]
In human preterm labour, atosiban, at the recommended dosage, antagonises uterine contractions and induces uterine quiescence. The onset of uterus relaxation following atosiban is rapid, uterine contractions being significantly reduced within 10 minutes to achieve stable uterine quiescence.
Other uses
Atosiban use after assisted reproduction
Atosiban is useful in improving the pregnancy outcome of in vitro fertilization-embryo transfer (IVF-ET) in patients with repeated implantation failure.[7] The pregnancy rate improved from zero to 43.7%.[8]
First- and second-trimester bleeding was more prevalent in ART than in spontaneous pregnancies. From 2004 to 2010, 33 first-trimester pregnancies with vaginal bleeding after ART with evident uterine contractions, when using atosiban and/or ritodrine, no preterm delivery occurred before 30 weeks.[9]
In a 2010 meta-analysis,[10] nifedipine is superior to β2 adrenergic receptor agonists and magnesium sulfate for tocolysis in women with preterm labor (20–36 weeks), but it has been assigned to pregnancy category C by the U.S. Food and Drug Administration, so is not recommended before 20 weeks, or in the first trimester.[9] A report from 2011 supports the use of atosiban, even at very early pregnancy, to decrease the frequency of uterine contractions to enhance success of pregnancy.[7]
Pharmacovigilance
Following the launch of atosiban in 2000, the calculated cumulative patient exposure to atosiban (January 2000 to December 2005) is estimated as 156,468 treatment cycles. To date, routine monitoring of drug safety has revealed no major safety issues.[11]
Regulatory affairs
Atosiban was approved in the European Union in January 2000 and launched in the European Union in April 2000.[12][4] As of June 2007, atosiban was approved in 67 countries, excluding the United States and Japan.[12] It was understood that Ferring did not expect to seek approval for atosiban in the US or Japan, focusing instead on development of new compounds for use in Spontaneous Preterm Labor (SPTL).[12] The fact that atosiban only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the US.[13]
Systematic reviews
In a systematic review of atosiban for tocolysis in preterm labour, six clinical studies — two compared atosiban to placebo and four atosiban to a β agonist — showed a significant increase in the proportion of women undelivered by 48 hours in women receiving atosiban compared to placebo. When compared with β agonists, atosiban increased the proportion of women undelivered by 48 hours and was safer compared to β agonists. Therefore, oxytocin antagonists appear to be effective and safe for tocolysis in preterm labour.[14]
A 2014 systematic review by the Cochrane Collaboration showed that while atosiban had fewer side effects than alternative drugs (such as ritodrine), other beta blockers, and calcium channel antagonists, it was no better than placebo in the major outcomes i.e. pregnancy prolongation or neonatal outcomes. The finding of an increase in infant deaths in one placebo-controlled trial warrants caution. Further research is recommended.[15]
PATENT
WO 2021207870
Atosiban (Atosiban) is an oxytocin and vasopressin V1A combined receptor antagonist, which can be used as a competitive antagonist of cyclic peptide oxytocin receptors in the uterus, decidua and fetal membrane. Atosiban is a disulfide-bonded cyclic polypeptide composed of 9 amino acids. It is a modified oxytocin molecule at positions 1, 2, 4 and 8. The N-terminal of the peptide is 3-mercaptopropionic acid (thiol and [ Cys] 6 thiol forms a disulfide bond), the C-terminal is in the form of an amide, and the second amino acid at the N-terminal is ethylated [D-Tyr(Et)] 2 . Atosiban is generally present in medicines in the form of acetate salt, commonly known as atosiban acetate. Its chemical formula is C 45 H 71 N 11 O 14 S 2 , its molecular weight is 994.19, and its structural formula is as follows:
[0003]
[0004]
In the prior art, atosiban is usually synthesized by a solid-phase peptide synthesis (SPPS) method, an amino resin is used as a starting carrier resin, and protected amino acids are sequentially connected, and the obtained atosiban is oxidized and then cleaved to obtain atosiban. However, the above-mentioned existing process has high cost, generates a large amount of solvent waste, and is not easy to monitor during the cyclization process. In addition, the above-mentioned prior art has deficiencies in the overall yield of crude peptides. Moreover, due to the existence of D-Tyr(Et) in the structure of atosiban, Fmoc-D-Tyr(Et) easily undergoes a racemization reaction during the peptide attachment process, resulting in [Tyr(Et) 2 ]-A The impurity of tosiban, which is similar in polarity to atosiban itself, is difficult to completely remove through purification, thus affecting the quality of atosiban.
[table 0001]
| Amino acid name | alphabetic symbols |
| Glycine | Gly |
| Ornithine | Orn |
| Proline | Pro |
| cysteine | Cys |
| Asparagine | Asn |
| Threonine | Thr |
| Isoleucine | Ile |
| D-tyrosine (oxyethyl) | D-Tyr(ET) |
Table 3 List of intermediates and Fmoc protected amino acids
[0043]
[table 0002]
| Fmoc-Orn(Boc)-OH |
| Fmoc-Pro-OH |
| Fmoc-Cys(Trt)-OH |
| Fmoc-Asn-OH |
| Fmoc-Thr(tBu)-OH |
| Fmoc-Ile-OH |
| Fmoc-D-Tyr(ET)-OH |
| Fmoc-Gly Rink Resin |
| Fmoc-Orn(Boc)-Gly Rink Resin |
| Fmoc-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |
| Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink Resin |



[0045]
According to the most preferred embodiment of the present invention, the method of the present invention comprises the following steps:
[0046]
The first step: Fmoc-Gly Rink resin can be directly purchased, which reduces the first step of synthesis and improves the synthesis efficiency;
[0047]
The second step: preparing a deprotection solution: the deprotection solution is a mixture of piperidine/N,N-dimethylformamide, preferably piperidine/N,N-dimethylformamide in a volume ratio of 1/4.
[0048]
The third step: preparation of Fmoc-Orn(Boc)-Gly Rink resin: deprotect the Fmoc-Gly Rink resin obtained in the first step, wash with DMF, add Fmoc-Orn(Boc)-OH in DMF solution, Condensation reaction is carried out under the condition of peptide coupling condensing agent to obtain Fmoc-Orn(Boc)-Gly Rink resin;
[0049]
The fourth step: preparation of Fmoc-Pro-Orn(Boc)-Gly Rink resin: the peptide resin obtained in the fourth step is deprotected and washed, and then reacted with Fmoc-Pro-OH under the condition of a peptide coupling agent to obtain Fmoc-Pro-Orn(Boc)-Gly Rink resin;
[0050]
The fifth step: preparation of Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the fifth step is deprotected and washed, and then reacted with Fmoc-Cys(Trt)-OH under the condition of peptide coupling agent to obtain Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0051]
The sixth step: preparation of Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the sixth step is deprotected and washed, and then reacted with Fmoc-Asn-OH under the condition of peptide coupling agent to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin ;
[0052]
The seventh step: preparation of Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the seventh step was deprotected and washed, and then reacted with Fmoc-Thr(tBu)-OH under the condition of a peptide coupling agent. Obtain Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0053]
The eighth step: preparation of Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the eighth step is deprotected and washed, and then reacted with Fmoc-Ile-OH under the condition of a peptide coupling agent to obtain Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn (Boc)-Gly Rink resin;
[0054]
The ninth step: preparation of Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the ninth step is deprotected and washed, and then reacted with Fmoc-D-Tyr(ET)-OH under the condition of a peptide coupling agent to obtain Fmoc-D-Tyr(RT)-Ile-Thr(tBu )-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0055]
The tenth step: preparation of Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the tenth step is deprotected and washed, and then reacted with Mpa(Trt) under the condition of a peptide coupling agent to obtain Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn -Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0056]
The eleventh step: Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin in TFA/TIS/EDT/H2O =90/54/10/5 TFA, cleaved for 3 hours, and filtered to obtain crude peptide solution;
[0057]
The twelfth step: sedimentation and washing of the crude peptide solution with methyl tert-butyl ether, centrifugation at 2000 rpm, and vacuum drying to obtain a pale yellow solid powder of atosiban linear crude peptide;
[0058]
The thirteenth step: prepare three solutions for atosiban cyclization: solution A-sodium acetate buffered aqueous solution, solution B-aqueous solution of linear peptide atosiban crude peptide acetic acid, solution C: 30%-60% hydrogen peroxide solution ;
[0059]
The fourteenth step: Mix the above three solutions of A, B, and C at 15-25 ° C, and stir for 1-3 hours after mixing, so that the Mpa at the 1st position and the Cys at the 6th position form a disulfide bond to obtain Cyclized atosiban crude peptide.
[0060]
Step fifteen: Purify crude atosiban by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes.
[0061]
The sixteenth step: freeze-dry the purified atosiban solution at -50 to -70° C. for 18-48 hours with a freeze dryer.
[0062]
The purity of atosiban obtained by the method of the invention is more than 99.5%, and the total product yield is 55%-65%.
[0063]
The advantage of the method for preparing atosiban of the present invention is:
[0064]
The traditional SPPS synthesis of atosiban usually produces a large amount of waste with high disposal costs. This process adopts high-temperature SPPS process and selects different condensing agent combinations, which is faster than the conventional SPPS process, the product purity can reach more than 99.9%, the purity is better than that of the conventional atosiban process, the impurity content is low, and the product quality is high. The total yield can reach 55%-65%.
Detailed ways
[0065]
The invention will now be described with reference to specific embodiments. It must be understood that these examples are merely illustrative of the invention and are not intended to limit the scope of the invention. Unless otherwise stated, percentages and parts are by weight. Unless otherwise specified, experimental materials and reagents used in the following examples were obtained from commercial sources.
[0066]
Example 1:
[0067]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.61 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-diisopropylcarbodiimide, 2-(7-benzotriazole)-N,N,N’,N ‘-Tetramethylurea hexafluorophosphate mixed in a volume ratio of 1:1, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:2 A solution-acetic acid-sodium acetate buffer aqueous solution (concentration is 30g/L), B solution-linear peptide atosiban crude peptide acetic acid aqueous solution and C solution: 60% hydrogen peroxide solution.
[0068]
The crude peptide yield was 85%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 56%.
[0069]
Example 2:
[0070]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, the other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-tert-dicyclohexylcarbodiimide, 1-hydroxybenzotriazole and Oxyma, which are mixed in a volume ratio of 1:1:1 , Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D- Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:2 solution A-formic acid-sodium formate buffer aqueous solution (concentration 25g/L), solution B-linear peptide atosiban crude peptide formic acid aqueous solution and solution C: 30% hydrogen peroxide solution, and oxygen was introduced.
[0071]
The crude peptide yield was 83%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity greater than 99.5%, and the total product yield is 57%.
[0072]
Example 3:
[0073]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence were connected in the following order, and the coupling reagents were N,N-diisopropylethylamine, 2-(7-benzotriazole)-N,N,N’,N’- Two kinds of tetramethylurea hexafluorophosphate mixed in a 1:1 volume ratio, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc- Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 75°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:3 solution A-sodium phosphate buffered aqueous solution (concentration 15g/L), solution B-linear peptide atosiban crude peptide phosphoric acid aqueous solution and solution C: DMSO aqueous solution (volume 1:1).
[0074]
The crude peptide yield was 80%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70 DEG C for 28 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 55%.
[0075]
Example 4:
[0076]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:3 by volume). Subsequently, the other amino acids in the sequence were connected in the following order, and the coupling reagents were selected from 2-oxime ethyl cyanoacetate, N,N-diisopropylcarbodiimide, and 1-hydroxybenzotriazole in a volume ratio of 1. :1:1 mix, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH , Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 80°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:4 solution of A-trifluoroacetic acid-aqueous ammonia solution (concentration of 45 g/L), solution B-aqueous solution of linear peptide atosiban crude peptide trifluoroacetic acid and solution C: saturated aqueous iodine solution.
[0077]
The crude peptide yield was 78%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 38 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 52%.
PATENT
WO/2022/141615
Atosiban Acetate Injection was first listed in Austria on March 23, 2000 under the trade name: Atosiban, a new type of anti-prematurity drug developed by Ferring GmbH, which is an oxytocin The analog is a competitive antagonist of oxytocin receptors in the uterus, decidua, and fetal membranes. It is a first-line drug recommended by the European Medical Association; it can inhibit the binding of oxytocin and oxytocin receptors, thereby directly inhibiting the effect of oxytocin. In the uterus, it can inhibit uterine contraction; it can also inhibit the hydrolysis of phosphatidylinositol.
Atosiban is a cyclic nonapeptide whose molecular formula is C 43 H 67 N 11 O 12 S 2 ; molecular weight is 994.19; CAS registration number is 90779-69-4; its peptide sequence is as follows:
Cyclo[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH 2
In the Chinese patents with announcement numbers CN101314613B and CN101696236B, the solid-phase synthesis of atosiban uses Rink Amide AM Resin resin solid-phase coupling stepwise to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr(tBu)- Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-Resin is directly oxidized in solid phase to generate disulfide bonds, and then cleaved to obtain atosiban. The Rink Amide AM Resin resin used in the prior art needs to be cracked under a strong acid environment, which is not conducive to product stability and has a greater operational risk; Mpr and Cys both have sulfhydryl groups, and the sulfhydryl groups have the ability to capture tBu to generate double tBu impurities, When the peptide resin after solid-phase oxidation is cleaved to remove the protective group and resin, due to the presence of tBu or tBu source Boc protective group, it requires high capture agent, which is not conducive to product quality control and reduces product yield.
The Chinese patent with publication number CN105408344B discloses a method for synthesizing atosiban starting from Fmoc-Orn-Gly-NH2, wherein Fmoc-Orn-Gly-NH2 is connected to trityl through the side chain of ornithine On the base resin, impurities can be effectively controlled. However, using dipeptide and trityl-type resin for coupling, the resin attached to the Orn side chain of the dipeptide increases the steric hindrance of the subsequent Pro coupling and prolongs the coupling time, which is easy to cause missing peptide impurities.
Example 1. Synthesis of Fmoc-Pro-Orn-Gly-NH 2 tripeptide
[0027]
Fmoc-Pro-OH (134.94 g, 400 mmol) and N-hydroxysuccinimide (46.00 g, 400 mmol) were weighed into 1600 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Orn(Boc)-NH 2 (92.92 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate to dryness under reduced pressure, add N-hydroxysuccinimide (46.00 g, 400 mmol) and 1600 ml of tetrahydrofuran to dissolve, and stir at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Gly-NH 2 (29.64 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution, and the reaction was continued at room temperature after dropping, and the monitoring of the raw materials was completed. The reaction was filtered, and the filtrate was concentrated under reduced pressure. Dry, add 1000 mL of 5% TFA/DCM solution to the reaction solution, continue to react for 1 h, and concentrate to dryness to obtain a yellow oil, which is recrystallized from isopropanol to obtain 171.56 g of white solid with a yield of 69%.
[0028]
Example 2. Synthesis of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.42 mmol/g
[0029]
Trityl resin (37.5 g, 30 mmol, substitution degree: 0.80 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 52.14 g of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.42 mmol/g.
[0030]
Example 3. Synthesis of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0031]
2-CTC Resin resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.80 g of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0032]
Example 4. Synthesis of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0033]
4-methyl-trityl resin (33.33 g, 30 mmol, substitution degree: 0.90 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.89 g of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0034]
Example 5. Synthesis of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0035]
4-Methoxy-trityl resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.69 g of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0036]
Example 6. Synthesis of Atosiban Linear Peptide Resin 1
[0037]
Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 (35.71 g) prepared in Example 2 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0038]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 4.86g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.68g DIC (pre-cooled to <0°C), Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 .
[0039]
Repeat the step of receiving the peptide and remove the Fmoc protective group. According to the amino acid sequence of atosiban, Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 was coupled to Fmoc- Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH give Mpa(Trt)-D-Tyr(Et)-Ile-Thr- Asn-Cys(Trt)-Pro-Orn (trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 48.72g after drying, and the resin weight gain was 89.0%.
[0040]
Example 7. Synthesis of atosiban linear peptide resin 2
[0041]
Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 (30.00 g) prepared in Example 3 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0042]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 13.65g HBTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0°C), put Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing solution was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 .
[0043]
Fmoc-D-Tyr(Et)-OH (86.30 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Ile-OH (26.24 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. The monitoring of the raw materials was completed. After filtration, the solution was concentrated under reduced pressure. , the concentrated solution was added to petroleum ether to separate out the solid, the solid was washed and then dried, recrystallized and dried with isopropanol to obtain 75.60 g of Fmoc-D-Tyr(Et)-Ile-OH with a yield of 75%.
[0044]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, sequentially couple Fmoc-Asn on Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 -OH, Fmoc-Thr-OH, Fmoc-D-Tyr(Et)-Ile-OH, Mpa(Trt)-OH to give Mpa(Trt)-D-Tyr(Et)-Ile-Thr-Asn-Cys(Trt )-Pro-Orn( 2 -CTC Resin)-Gly-NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.77g after drying, and the resin weight gain rate was 87.4%.
[0045]
Example 8. Synthesis of atosiban linear peptide resin 3
[0046]
Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 4 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod, and the ninhydrin test was positive, indicating that Fmoc had been removed.
[0047]
Weigh 17.57g Fmoc-Cys(Trt)-OH, 13.65g HBTU and 4.05g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0 ℃), activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35 ℃ for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF The resin was washed 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly-NH 2 .
[0048]
Mpa(Trt)-OH (69.69 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and HD-Tyr(Et)-OH (41.85 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry it, recrystallize and dry with isopropanol to obtain Mpa(Trt)-D-Tyr(Et)-OH 77.98g, yield 72%.
[0049]
Repeat the step of receiving the peptide and removing the Fmoc protective group, according to the amino acid sequence of atosiban, on Fmoc-Cys(Trt)-Pro-Orn (4-methyl-trityl resin)-Gly- NH 2 Fmoc-Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Mpa(Trt)-D-Tyr(Et)-OH were sequentially coupled to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr -Asn-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.91g after drying, and the resin weight gain rate was 88.3%.
[0050]
Example 9. Synthesis of atosiban linear peptide resin 4
[0051]
Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 5 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0052]
Fmoc-Asn-OH (70.87 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Cys(Trt)-OH (79.96 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry, recrystallize and dry with isopropanol to obtain Fmoc-Asn-Cys(Trt)-OH 102.17g, yield 73%.
[0053]
Weigh 20.99g Fmoc-Asn-Cys(Trt)-OH and 13.65g HCTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cool to <0°C) , activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35°C for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF to wash the resin , wash 6 times. After washing, the washing liquid was removed to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly-NH 2 .
[0054]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, in Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH were sequentially coupled on NH 2 to obtain Mpa(Trt)-D-Tyr(Et)-Ile- Thr-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.28g after drying, and the resin weight gain was 84.0%.
[0055]
Example 10. Synthesis of atosiban crude peptide 1
[0056]
Configure 487.2ml of TFA/DCM=2/98 (V/V) lysis solution, cool to 5-10°C, add 48.72g of peptide resin prepared in Example 6 into the lysis solution, at room temperature (20-35°C) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.77g of atosiban linear peptide, dissolve 14.30g of atosiban linear peptide in 0.75L of glacial acetic acid, add 6.75L of water to dilute, add 0.1M/L iodine ethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, That is, the crude atosiban peptide is obtained, and its HPLC spectrum is shown in Figure 1.
[0057]
Example 11. Synthesis of atosiban crude peptide 2
[0058]
Configure TFA/DCM=5/95 (V/V) lysate 448.6ml, cool to 5~10℃, add 42.77g of peptide resin prepared in Example 7 into the lysate, at room temperature (20~35℃) React for 3h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate, dry to obtain a solid, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.21g of atosiban linear peptide, dissolve 14.21g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 0.1M/L iodoethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, that is The crude atosiban peptide was obtained, and its HPLC chromatogram was similar to that in Figure 1.
[0059]
Example 12. Synthesis of atosiban crude peptide 3
[0060]
Configure 450.5ml of TFA/DCM=20/80(V/V) lysis solution, cool to 5~10℃, add 45.05g of peptide resin prepared in Example 8 into the lysis solution, at room temperature (20~35℃) React for 2h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.63g of atosiban linear peptide, dissolve 14.63g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 10% hydrogen peroxide solution, and react at room temperature for 1.0h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0061]
Example 13. Synthesis of atosiban crude peptide 4
[0062]
Configure TFA/DCM=1/99 (V/V) lysate 442.7ml, cool to 5~10℃, add 44.27g of peptide resin prepared in Example 9 into the lysate, at room temperature (20~35℃) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.13 g of atosiban linear peptide, dissolve 14.13 g of atosiban linear peptide in 1.5 L of glacial acetic acid, add 6 L of water to dilute, add 30% hydrogen peroxide solution, and react at room temperature for 1.0 h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0063]
Example 14. Purification of atosiban crude peptide 1
[0064]
The atosiban crude peptide prepared in Example 10 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.12g , the yield is 64%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is shown in Figure 2.
[0065]
Example 15. Purification of atosiban crude peptide 2
[0066]
The crude atosiban peptide obtained in Example 11 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 9.80 g , the yield is 62%, the purity is 99%, and the obtained atosiban peptide HPLC spectrum is similar to Figure 2.
[0067]
Example 16. Purification of atosiban crude peptide 3
[0068]
The crude atosiban peptide obtained in Example 12 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.28g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
[0069]
Example 17. Purification of atosiban crude peptide 4
[0070]
The crude atosiban peptide obtained in Example 13 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.27g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
PATENT
https://patents.google.com/patent/US9434767B2/es
Atosiban is a nonapeptide which contains three non-natural amino acids: D-Tyr(Et), Mpa and Orn, and a pair of disulfide bonds looped between Mpa and Cys, the structural formula is:
c[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH2.
By means of competing for oxytocin receptor with oxytocin, Atosiban can inhibit the combination between oxytocin and oxytocin receptor, and directly prevent the oxytocin from acting on uterus, and then inhibit the uterine contraction; as another hand, atosiban can also inhibit the hydrolysis of phosphatidylinositol and then block the generation of messenger and activity of Ca2+, with the decreasing of activity from oxytocin, the contraction of uterine is indirectly inhabited.
At present, there are many reports about synthesis process method in China and abroad A report in China shows that the inventor found a simple process by adopting solid phase oxidation, resulting in a low purity crude product, with low yield and low application value. The aforementioned reports about atosiban synthesis process reveal that most of them adopt the method using Boc solid phase synthetic and cleaving peptide with liquid ammonia, then oxidating with liquid phase oxidation, and purifying. Those respective processes result in “the three wastes” and are too complex for industrial production. See U.S. Pat. No. 4,504,469.
Example 1Preparing the Linear Atosiban Peptide Resin
(i) 6.25 g of Rink Amide resin (substitutability=0.8 mmol/g) is put into a reaction bottle, DMF is added into the bottle and washed twice, then swelled for 30 min with DMF. Fmoc protecting group of Rink Amide resin is removed with 30-40 ml of 20% DBLK, washed for 4 times with DMF, then washed twice with DCM after removal, the product is detected by ninhydrin detecting method, the resin is reddish-brown.
(ii) 4.46 g of Fmoc-Gly-OH and 2.43 g of HOBt dissolved in a suitable amount of DMF, which had been pre-activated with 3.05 ml DIC; the mixture is, added to the reaction bottle, and reacted for 2 h, the resin is negative by ninhydrin detecting method, after the reaction, the product is washed for 4 times with DMF, then washed twice with DCM, if the resin is positive, repeating the above condensation reaction until negative.
(iii) Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH and Mpa(Trt)-OH are coupled orderly.
Example 2Cleaving the Linear Atosiban Peptide Resin
5.15 g of linear atosiban is prepared by washing the linear atosiban peptide resin obtained from Example 1 for 3 times with 30 ml of methanol, adding the dry resin obtained to 150 ml of mixed solution with a volume ratio of TFA:H2O=95:5, reacting for 2 hours at 25° C. and filtering, washing the resin for 3 times with few trifluoroacetic acid, combining the filtrate and pouring into 1500 ml glacial ether, making rest for 2 hours, centrifugally separating the linear atosiban, washing for 3 times, and drying in a vacuum drier, MS: 995.3, HPLC: 91.5%, content: 65.5%, synthesis yield: 68%.
Example 3Oxidizing the Linear Atosiban
2.85 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 5% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.60 g of H2O2, reacting for 10 min at 25° C., monitoring with HPLC (HPLC: 75.6%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
Example 4Oxidizing the Linear Atosiban
3.01 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 30 min at 25° C., monitoring with HPLC (HPLC: 89.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.6%.
Example 5Oxidizing the Linear Atosiban
2.95 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 60 min at 25° C., monitoring with HPLC (HPLC: 83.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
The above is the further detailed description of the invention in conjunction with specific preferred examples, but it should not be considered that the specific examples of the invention are only limited to the these descriptions. For one of ordinary skill in the art, many deductions and replacements can be made without departing from the inventive concept. Such deductions and replacements should fall within the scope of protection of the invention.
Clips
https://www.mdpi.com/1420-3049/27/6/1920/htm
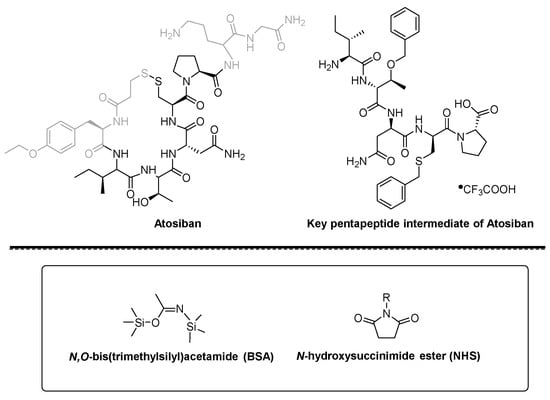
Figure 1. Structure of Atosiban, pentapeptide intermediate, BSA and NHS ester.
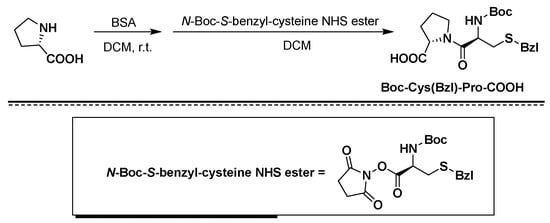
Figure 2. Synthesis of Boc-Cys(Bzl)-Pro-COOH using BSA/NHS as coupling agents.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clinical trials
Atosiban vs. nifedipine
A 2013 retrospective study comparing the efficacy and safety of atosiban and nifedipine in the suppression of preterm labour concluded that atosiban and nifedipine are effective in delaying delivery for seven days or more in women presenting with preterm labour.[16] A total of 68.3% of women in the atosiban group remained undelivered at seven days or more, compared with 64.7% in the nifedipine group.[16] They have the same efficacy and associated minor side effects.[16] However, flushing, palpitation, and hypotension were significantly higher in the nifedipine group.[16]
A 2012 clinical trial compared tocolytic efficacy and tolerability of atosiban with that of nifedipine.[17] Forty-eight (68.6%) women allocated to atosiban and 39 (52%) to nifedipine did not deliver and did not require an alternate agent at 48 hours, respectively (p=.03).[17] Atosiban has fewer failures within 48 hours.[17] Nifedipine may be associated with a longer postponement of delivery.[17]
A 2009 randomised controlled study demonstrated for the first time the direct effects of atosiban on fetal movement, heart rate, and blood flow.[18] Tocolysis with either atosiban or nifedipine combined with betamethasone administration had no direct fetal adverse effects.[18]
Atosiban vs. ritodrine
Multicentre, controlled trial of atosiban vs. ritodrine in 128 women shows a significantly better tocolytic efficacy after 7 days in the atosiban group than in the ritodrine group (60.3 versus 34.9%), but not at 48 hours (68.3 versus 58.7%). Maternal adverse events were reported less frequently in the atosiban group (7.9 vs 70.8%), resulting in fewer early drug terminations due to adverse events (0 versus 20%). Therefore, atosiban is superior to ritodrine in the treatment of preterm labour.[19]
Brand names
In India it is marketed under the brand name Tosiban by Zuventus healthcare ltd.
References
- ^ “Atosiban International Drug Names”. Drugs.com. 10 April 2020. Retrieved 29 April 2020.
- ^ “Tractocile 7.5 mg/ml Solution for Injection – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 29 April 2020.
- ^ “Tractocile 7.5 mg/ml Concentrate for Solution for Infusion – Summary of Product Characteristics (SmPC)”. (emc). 24 June 2013. Retrieved 29 April 2020.
- ^ Jump up to:a b c d e f g “Tractocile EPAR”. European Medicines Agency (EMA). Retrieved 29 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Akerlund M, Carlsson AM, Melin P, Trojnar J (1985). “The effect on the human uterus of two newly developed competitive inhibitors of oxytocin and vasopressin”. Acta Obstet Gynecol Scand. 64 (6): 499–504. doi:10.3109/00016348509156728. PMID 4061066. S2CID 25799128.
- ^ Sanu O, Lamont RF (2010). “Critical appraisal and clinical utility of atosiban in the management of preterm labor”. Ther Clin Risk Manag. 6: 191–199. doi:10.2147/tcrm.s9378. PMC 2861440. PMID 20463780.
- ^ Jump up to:a b Chou PY, Wu MH, Pan HA, Hung KH, Chang FM (June 2011). “Use of an oxytocin antagonist in in vitro fertilization-embryo transfer for women with repeated implantation failure: a retrospective study”. Taiwan J Obstet Gynecol. 50 (2): 136–40. doi:10.1016/j.tjog.2011.04.003. PMID 21791296.
- ^ Lan, VT; Khang, VN; Nhu, GH; Tuong, HM (September 2012). “Atosiban improves implantation and pregnancy rates in patients with repeated implantation failure”. Reprod Biomed Online. 25 (3): 254–60. doi:10.1016/j.rbmo.2012.05.014. PMID 22818095.
- ^ Jump up to:a b Wu, MY; Chen, SU; Yang, YS (December 2011). “Using atosiban in uterine contractions of early pregnancies after assisted reproduction”. J Formos Med Assoc. 110 (12): 800. doi:10.1016/j.jfma.2011.11.016. PMID 22248840.
- ^ Conde-Agudelo, A; Romero, R; Kusanovic, JP (2011). “Nifedipine in the management of preterm labor: a systematic review and metaanalysis”. Am J Obstet Gynecol. 204 (2): 134.e1–134.e20. doi:10.1016/j.ajog.2010.11.038. PMC 3437772. PMID 21284967.
- ^ Lamont, Ronald F; Kam, KY Ronald (March 2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163. ISSN 1747-4108.
- ^ Jump up to:a b c Lamont, Ronald F.; Kam, KY Ronald (2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163.
- ^ Lamont CD, Jørgensen JS, Lamont RF (September 2016). “The safety of tocolytics used for the inhibition of preterm labour”. Expert Opinion on Drug Safety. 15 (9): 1163–73. doi:10.1080/14740338.2016.1187128. PMID 27159501. S2CID 4937942.
It was for this reason and the fact that Tractocile (atosiban) only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the USA.
- ^ Coomarasamy, A; Knox, EM; Gee, H; Khan, KS (November 2002). “Oxytocin antagonists for tocolysis in preterm labour — a systematic review”. Med Sci Monit. 8 (11): RA268–73. PMID 12444392.
- ^ Flenady, Vicki; Reinebrant, Hanna E.; Liley, Helen G.; Tambimuttu, Eashan G.; Papatsonis, Dimitri N. M. (6 June 2014). “Oxytocin receptor antagonists for inhibiting preterm labour” (PDF). The Cochrane Database of Systematic Reviews (6): CD004452. doi:10.1002/14651858.CD004452.pub3. ISSN 1469-493X. PMID 24903678.
- ^ Jump up to:a b c d Saleh SS, Al-Ramahi MQ, Al Kazaleh FA (January 2013). “Atosiban and nifedipine in the suppression of preterm labour: a comparative study”. J Obstet Gynaecol. 33 (1): 43–5. doi:10.3109/01443615.2012.721822. PMID 23259877. S2CID 20753923.
- ^ Jump up to:a b c d Salim R, Garmi G, Nachum Z, Zafran N, Baram S, Shalev E (December 2012). “Nifedipine compared with atosiban for treating preterm labor: a randomized controlled trial”. Obstet Gynecol. 120 (6): 1323–31. doi:10.1097/aog.0b013e3182755dff. PMID 23168756. S2CID 22487349.
- ^ Jump up to:a b de Heus R, Mulder EJ, Derks JB, Visser GH (June 2009). “The effects of the tocolytics atosiban and nifedipine on fetal movements, heart rate and blood flow”. J Matern Fetal Neonatal Med. 22 (6): 485–90. doi:10.1080/14767050802702349. PMID 19479644. S2CID 35810758.
- ^ Shim JY, Park YW, YoonBH, Cho YK, Yang JH, Lee Y, Kim A. “Multicentre, parallelgroup, randomised, single-blind study of the safety and efficacy of atosibanversus ritodrine in the treatment of acute preterm labour in Korean women. BJOG 2006Nov;113(11):1228-34.
External links
- “Atosiban”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Tractocile, Antocin, others[1] |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous |
| ATC code | G02CX01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) [2][3]EU: Rx-only [4]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 90779-69-4 |
| PubChem CID | 5311010 |
| IUPHAR/BPS | 2213 |
| DrugBank | DB09059 |
| ChemSpider | 4470550 |
| UNII | 081D12SI0Z |
| KEGG | D03008 |
| ChEMBL | ChEMBL378642 |
| CompTox Dashboard (EPA) | DTXSID8048991 |
| ECHA InfoCard | 100.234.128 |
| Chemical and physical data | |
| Formula | C43H67N11O12S2 |
| Molar mass | 994.19 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Publication numberPriority datePublication dateAssigneeTitle
US4504469A *1982-12-211985-03-12Ferring AbVasotocin derivatives
WO2006119388A2 *2005-05-032006-11-09Novetide, Ltd.Methods for the production of peptide having a c-terminal amide
CN102127146A *2010-12-242011-07-20深圳翰宇药业股份有限公司Method for preparing atosiban acetate
DK0710243T3 *1993-06-292000-10-16Ferring BvSynthesis of cyclic peptides
CN101357937B *2007-07-312012-11-07上海苏豪逸明制药有限公司Method for synthesizing atosiban acetate from solid phase polypeptide
CN101314613B *2008-05-082012-04-25吉尔生化(上海)有限公司Solid phase synthesis method for atosiban
CN102127146B *2010-12-242013-04-24深圳翰宇药业股份有限公司Method for preparing atosiban acetate
CN102584953B *2012-02-092014-01-01深圳翰宇药业股份有限公司Purification method for atosiban
CN104098650B *2013-04-152019-04-09中国医学科学院药物研究所The synthesis and application of the intermediate of Atosiban
GB201310921D0 *2013-06-192013-07-31Chemical & Biopharmaceutical Lab Of Patras S APeptide-resin conjugate and use thereof
CN105949283A *2016-06-072016-09-21海南合瑞制药股份有限公司Atosiban acetate impurities and preparation and detection methods
CN106279367B *2016-08-152019-06-04海南合瑞制药股份有限公司A kind of atosiban acetate crystal and preparation method thereof
CN107312072A *2017-06-202017-11-03浙江湃肽生物有限公司A kind of method of purifies and separates Atosiban
ApplicationPriority dateFiling dateTitle
CN201010604790.62010-12-24
CN2010106047906A2010-12-242010-12-24Method for preparing atosiban acetate
CN2010106047902010-12-24
PCT/CN2011/0844142010-12-242011-12-22Method for preparing atosiban acetate
Similar Documents
PublicationPublication DateTitle
US9434767B22016-09-06Method for preparing atosiban acetate
CN103497245B2015-05-13Method for synthesizing thymalfasin
DK2421887T32015-07-27A process for the preparation of degarelix
CN104974237B2019-02-12A kind of method of segment method synthesis in solid state ziconotide
CN101747426B2013-01-16Method for synthesizing pramlintide
CN102702320B2013-08-21Method for preparing eptifibatide
CN106589069B2018-07-17A kind of preparation method of oxytocin
US9394341B22016-07-19Eptifibatide preparation method
CN104231051A2014-12-24Preparation method for linaclotide
CN104177490B2017-02-08Method for preparing salmon calcitonin acetate by fragment condensation
CN101372505B2011-03-30Method for preparing desmopressin acetate
CN105418736A2016-03-23Preparation method of terlipressin through combination of solid and liquid
CN106084015B2020-01-31method for synthesizing carbetocin
CN103467573B2017-12-15A kind of preparation method of carbetocin
CN103214568B2014-09-24Solid phase method of secretin
CN103992389A2014-08-20Method for solid cyclizing synthesis of desmopressin
CN109021087A2018-12-18A kind of method that solid liquid phase combination prepares ziconotide
CN104761619A2015-07-08Desmopressin acetate solid phase preparation technology
CN106854235B2020-09-18Solid phase fragment method for synthesizing carbetocin
CN106554391B2021-11-05Method for synthesizing marine biological peptide Xen2174
CN107216374A2017-09-29A kind of synthetic method of ziconotide
CN103992401B2017-05-17Method for preparing exenatide
CN105037496B2018-12-25A kind of preparation method of eptifibatide
WO2017097194A12017-06-15Completely-solid-phase preparation method for carbetocin
CN103980350A2014-08-13Solid-phase cyclization synthesis method of Atosiban
////////////// ATOSIBAN, CAP-449, CAP-476, CAP-581, F-314, ORF 22164, ORF-22164, RW-22164, RWJ 22164, RWJ-22164
[H][C@]1(NC(=O)[C@@]([H])(NC(=O)[C@@H](CC2=CC=C(OCC)C=C2)NC(=O)CCSSC[C@H](NC(=O)[C@H](CC(N)=O)NC1=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCN)C(=O)NCC(N)=O)[C@@H](C)CC)[C@@H](C)O

NEW DRUG APPROVALS
TO MAINTAIN SUBSCRIPTION OF THIS BLOG
$10.00
Darinaparsin
Darinaparsin
ダリナパルシン , Darvias
JAPAN 2022 APPROVED, PMDA 2022/6/20
(2S)-2-amino-5-[[(2R)-1-(carboxymethylamino)-3-dimethylarsanylsulfanyl-1-oxopropan-2-yl]amino]-5-oxopentanoic acid
Glycine, L-gamma-glutaMyl-S-(diMethylarsino)-L-cysteinyl-
| Formula | C12H22AsN3O6S |
|---|---|
| CAS | 69819-86-9 |
| Mol weight | 411.3062 |
| Efficacy | Antineoplastic |
|---|---|
| Comment | organic arsenical |
Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L
- OriginatorTexas A&M University; University of Texas M. D. Anderson Cancer Center
- DeveloperSolasia Pharma; ZIOPHARM Oncology
- ClassAmines; Antineoplastics; Arsenicals; Oligopeptides; Pentanoic acids; Small molecules; Sulfides
- Mechanism of ActionApoptosis stimulants; Cell cycle inhibitors; Reactive oxygen species stimulants
- Orphan Drug StatusYes – Peripheral T-cell lymphoma
- PreregistrationPeripheral T-cell lymphoma
- DiscontinuedLiver cancer; Lymphoma; Multiple myeloma; Non-Hodgkin’s lymphoma; Solid tumours
- 28 Mar 2022No recent reports of development identified for phase-I development in Peripheral-T-cell-lymphoma in China (IV, Injection)
- 26 Jan 2022ZIOPHARM Oncology is now called Alaunos Therapeutics
- 11 Dec 2021Safety and efficacy data from a phase II trial in Peripheral T-cell lymphoma presented at the 63rd American Society of Hematology Annual Meeting and Exposition (ASH-2021)
Darinaparsin is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects. Compared to inorganic arsenic compounds such as arsenic trioxide (As2O3), darinaparsin appears to exhibit a wide therapeutic window.
Darinaparsin, also know as ZIO-101 and SP-02, is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects.
Darinaparsin is an organic arsenical composed of dimethylated arsenic linked to glutathione, and is being investigated for antitumor properties in vitro and in vivo. While other arsenicals, including arsenic trioxide, have been used clinically, none have shown significant activity in malignancies outside of acute promyelocytic leukemia. Darinaparsin has significant activity in a broad spectrum of hematologic and solid tumors in preclinical models. Here, we review the literature describing the signaling pathways and mechanisms of action of darinaparsin and compare them to mechanisms of cell death induced by arsenic trioxide. Darinaparsin has overlapping, but distinct, signaling mechanisms. We also review the current results of clinical trials with darinaparsin (both intravenous and oral formulations) that demonstrate significant antitumor activity.
PAPER
Biochemical Pharmacology (Amsterdam, Netherlands), 126, 79-86; 2017



PATENT
WO 2015085208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015085208
Preparation of Darinaparsin
[0071] Sterile water (15.5 L) and ethyl alcohol (200 proof, 15.5 L) were charged in a reaction flask prior to the addition of L-glutathione (3.10 kg). While being stirred, the reaction mixture was cooled to 0-5 °C prior to the addition of triethylamine (1.71 L). Stirring was continued until most of the solids were dissolved and the solution was filtered. After filtration, the reaction mixture was cooled to 0-5 °C prior to the addition of chlorodimethylarsine (1.89 kg) over 115 minutes while maintaining the temperature at 0-5 °C. Stirring continued at 0-5 °C for 4 hours before acetone (30.6 L) was added over 54 minutes while maintaining the temperature at 0-5 °C. The suspension was stored at 0-5°C overnight prior to filtration. The solid was collected in a filter funnel, washed successively with ethyl alcohol (200 proof, 13.5 L) and acetone (13.5 L) and dried in suction for 23 minutes. A second similar run was performed and the collected solids from both runs were combined. Ethyl alcohol (200 proof, 124 L) and the combined solids (11.08 kg) were charged in a vessel. The slurry was stirred at ambient temperature for 2 hours before filtration, washing successively with ethyl alcohol (200 proof, 27 L) and acetone (27 L) and dried in suction for 60 minutes. The resulting solid was transferred to drying trays and dried in a vacuum oven at ambient temperature for 66 hours to provide darinaparsin as a solid with the differential scanning calorimetry (DSC) thermogram of Figure 1, with an extrapolated onset temperature at about 191.36° C and a peak temperature at about 195.65° C.
PATENT
WO 2010021928
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109°C was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
SGLU-1: Glutathione (14.0 g, 45.6 mmol) was stirred rapidly in glyme while dimethylchoroarsine (6.5 g, 45.6 mmol) was added dropwise. Pyridine (6.9 g, 91.2 mmol) was then added to the slurry and the mixture was subsequently heated to reflux. The heat was removed immediately and the mixture stirred at room temperature for 4 h. Isolation of the resultant insoluble solid and recrystallization from ethanol afforded 4 as the pyridine hydrochloride complex (75% yield). mp 115-118°C; NMR (D20) δ1.35 (s, 6H), 1.9-4.1 (m’s, 10H), 7.8-9.0 (m, 5H); mass spectrum (m/e) 140, 125, 110, 105, 79, 52, 45, 36.
PATENT
WO 2009075870
Step 1
Example 1. Preparation of Dimethylchloroarsine (DMCA). A 3-neck round-bottom flask (500 mL) equipped with mechanical stirrer, inlet for nitrogen, thermometer, and an ice bath was charged with cacodylic acid (33 g, 0.23 mol) and cone. hydrochloric acid (67 mL). In a separate flask, a solution of SnCl2·2H2O (54 g, 0.239 mol) in cone. hydrochloric acid (10 mL) was prepared. The SnCl2·2 H2O solution was added to the cacodylic acid in HCl solution under nitrogen while maintaining the temperature between 5 °C and 10 °C. After the addition was complete, the ice bath was removed and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was transferred to a separatory funnel and the upper layer (organic) collected. The bottom layer was extracted with dichloromethane (DCM) (2 × 25 mL). The combined organic extract was washed with 1 N HCl (2 × 10 mL) and water (2 × 20 mL). The organic extract was dried over MgSO4 and DCM was removed by rotary evaporation (bath temperature 80 °C, under nitrogen, atmospheric pressure). The residue was further distilled under nitrogen. Two tractions of DMCA were collected. The first fraction contained some DCM and the second fraction was of suitable quality (8.5 g, 26% yield). The GC analysis confirmed the identity and purity of the product.
Step 2
Example 3. Preparation of S-Dimethylarsinoglutathione (SGLU-1). In a 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel and thermometer under an inert atmosphere was prepared a suspension of glutathione (114.5 g, 0.37 mol) in a 1:1 (v/v) mixture of water/ethanol (1140 mL) and cooled to below 5 °C. The mixture was treated slowly (over 15 min) with triethylamine (63.6 mL, 0.46 mol) while maintaining the temperature below 20 °C. The mixture was cooled to 4 °C and stirred for 15 min and then the traces of undissolved material removed by filtration. The filtrate was transferred in a clean 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel, nitrogen inlet, and thermometer and DMCA (70 g, 0.49 mol) (lot # 543-07-01-44) was added slowly while maintaining the temperature at 3-4°C. The reaction mixture was stirred at 1-4°C for 4 h, and acetone (1.2 L) was added over a period of 1 h. The mixture was stirred for 90 min between 2 and 3°C and the resulting solid was isolated by filtration. The product was washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and the wet solids were suspended in ethanol 200 Proof (2000 mL). The product was isolated by filtration, washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and dried in vacuum for 2 days at RT to give 115 g (75%) of SGLU-1, HPLC purity > 99.5% (in process testing).
PATENT
WO 2007027344
Example 2 Preparation of S-Dimethylarsinoglutathione A 5 L, three necked round bottom flask was equipped with a mechanical stirrer assembly, thermometer, addition funnel, nitrogen inlet, and a drying tube was placed in a cooling bath. A polyethylene crock was charged with glutathione-reduced (200 g) and deionized water (2 L) and stirred under a nitrogen atmosphere to dissolve all solids. The mixture was filtered to remove any insoluble material and the filtrate was transferred to the 5 L flask. While stirring, ethanol, 200 proof (2 L) was added and the clear solution was cooled to 0-5° C. using an ice/methanol bath. Pyridine (120 g) was added followed by a dropwise addition of Me2AsCl (120 g) over a minimum of 1 hour. The reaction mixture was stirred at 0-5° C. for a minimum of 2 hours prior to removal of the cooling bath and allowing the mixture to warm to room temperature under a nitrogen atmosphere with stirring. The reaction mixture was stirred overnight (>15 hrs) at room temperature under a nitrogen atmosphere at which time a white solid may precipitate. The reaction mixture was concentrated to a slurry (liquid and solid) at 35-45° C. using oil pump vacuum to provide a white solid residue. As much water as possible is removed, followed by two coevaporations with ethanol to azeotrope the last traces of water. The white solid residue was slurried in ethanol, 200 pf. (5 L) under a nitrogen atmosphere at room temperature overnight. The white solid was filtered and washed with ethanol, 200 pf. (2×500 mL) followed by acetone, ACS (2×500 mL). The resulting solid was transferred to drying trays and vacuum oven dried overnight at 25-35° C. using oil pump vacuum to provide pyridinium hydrochloride-free S-dimethylarsinoglutathione as a white solid. melting point of 189-190° C.
PATENT
WO 20060128682
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109° C. was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
Pyridine Hydrochloride Free Synthesis of S-Dimethylarsinoglutathione (GLU) Dimethylarsinoglutathione is made using an adapted of Chen (Chen, G. C., et al. Carbohydrate Res. (1976) 50: 53-62) the contents of which are hereby incorporated by reference in their entirety. Briefly, dithiobis(dimethylarsinoglutamine) is dissolved in dichloromethane under nitrogen. Tetramethyldiarsine is added dropwise to the solution and the reaction is stirred overnight at room temperature under nitrogen and then exposed to air for 1 h. The mixture is then evaporated to dryness and the residue is washed with water and dried to give a crude solid that is recrystallized from methanol to give S-dimethylarsinoglutathione.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Solasia Announces Submission of New Drug Application for Anti-cancer Drug DARINAPARSIN for Peripheral T-Cell Lymphoma in Japan
Solasia Pharma K.K. (TSE: 4597, Headquarters: Tokyo, Japan, President & CEO: Yoshihiro Arai, hereinafter “Solasia”) today announced submission of a New Drug Application (NDA) for its new anti-cancer drug darinaparsin (generic name, development code: SP-02) as a treatment for relapsed or refractory peripheral T-cell lymphoma to the Ministry of Health, Labour and Welfare (MHLW). Based on positive results of R&D on darinaparsin, centered primarily on the results of the Asian Multinational Phase 2 Study (study results released in June 2020), Solasia filed an NDA for the drug with the regulatory authority in Japan ahead of anywhere else in the world.
Solasia expects to obtain regulatory approval in 2022 and to also launch in the same year. If approved and launched, darinaparsin would be the third drug Solasia successfully developed and brought to market since its founding and is expected to contribute to the treatment of PTCL.
Mr. Yoshihiro Arai, President and CEO of Solasia, commented as follows:
“No standard treatment has been established for relapsed or refractory PTCL as of yet. I firmly believe that darinaparsin, with its novel mechanism of action that differs from those of already approved drugs, will contribute to patients and healthcare providers at clinical sites as a new treatment option for relapsed or refractory PTCL. Since founding, Solasia has conducted R&D on five pipeline drugs. Of the five, we have successfully developed and brought to market two drugs, i.e., began providing them to patients, and today, we submitted an NDA for our first anti-cancer drug. Under our mission to provide patients with ‘Better Medicine for a Brighter Tomorrow’, we will continue aiming to contribute to patients’ treatment and enhanced quality of life. ”
About darinaparsin (SP-02)
Darinaparsin, an organoarsenic compound with anticancer activity, is a novel mitochondrial-targeted agent being developed for the treatment of various hematologic and solid tumors. The proposed mechanism of action of the drug involves the disruption of mitochondrial function, increased production of reactive oxygen species, and modulation of intracellular signal transduction pathways. Darinaparsin is believed to exert anticancer effect by inducing cell cycle arrest and apoptosis. Darinaparsin has been granted orphan drug designation in the US and EU.
For more information, please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Asian Multinational Phase 2 Study
The Asian Multinational Phase 2 Study was a multinational, multicenter, single-arm, open-label, non-randomized study to evaluate the efficacy and safety of darinaparsin monotherapy in patients with relapsed or refractory PTCL conducted in Japan, Korea, Taiwan, and Hong Kong. (CT.gov Identifier: NCT02653976).
Solasia plans to present the results of the study at an international academic conference to be held in the near future.
About peripheral T-cell lymphoma (PTCL)
Please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Solasia
Please visit at https://solasia.co.jp/en/
/////////////Darinaparsin, Darvias, JAPAN 2022, APPROVALS 2022, PMDA, ダリナパルシン , Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L, Orphan Drug
C[As](C)SCC(C(=O)NCC(=O)O)NC(=O)CCC(C(=O)O)N
Pimitespib
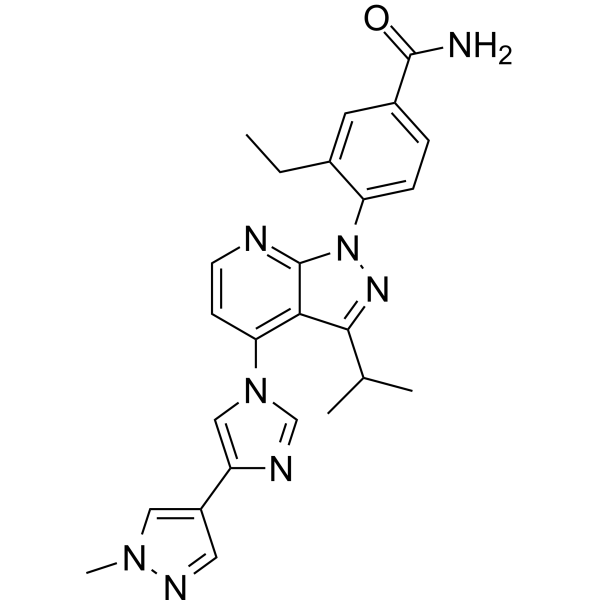
![Benzamide, 3-ethyl-4-[3-(1-methylethyl)-4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-1H-pyrazolo[3,4-b]pyridin-1-yl]-.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=67501411&t=l)
Pimitespib
TAS 116
CAS 1260533-36-5
Antineoplastic, Hsp 90 inhibitor
3-ethyl-4-[4-[4-(1-methylpyrazol-4-yl)imidazol-1-yl]-3-propan-2-ylpyrazolo[3,4-b]pyridin-1-yl]benzamide
Pimitespib (TAS-116) is an oral bioavailable, ATP-competitive, highly specific HSP90α/HSP90β inhibitor (Kis of 34.7 nM and 21.3 nM, respectively) without inhibiting other HSP90 family proteins such as GRP94. Pimitespib demonstrates less ocular toxicity.
| Formula | C25H26N8O |
|---|---|
| CAS | 1260533-36-5 |
| Mol weight | 454.5269 |
JAPAN APPROVED 2022/6/20, ピミテスピブ
| Jeselhy |
Taiho. originator
Pimitespib is a specific inhibitor of heat shock protein 90 (Hsp90) subtypes alpha and beta, with potential antineoplastic and chemo/radiosensitizing activities. Upon oral administration, pimitespib specifically binds to and inhibits the activity of Hsp90 alpha and beta; this results in the proteasomal degradation of oncogenic client proteins, which inhibits client protein dependent-signaling, induces apoptosis, and inhibits the proliferation of cells overexpressing HSP90alpha/beta. Hsp90, a family of molecular chaperone proteins that are upregulated in a variety of tumor cells, plays a key role in the conformational maturation, stability, and function of “client” proteins within the cell,; many of which are involved in signal transduction, cell cycle regulation and apoptosis, including kinases, cell-cycle regulators, transcription factors and hormone receptors. As TAS-116 selectively inhibits cytosolic HSP90alpha and beta only and does not inhibit HSP90 paralogs, such as endoplasmic reticulum GRP94 or mitochondrial TRAP1, this agent may have less off-target toxicity as compared to non-selective HSP90 inhibitors.
Patent
WO2011004610
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011004610
PATENT
CN108623496
| 3-Ethyl-4-fluorobenzonitrile is an important intermediate for the preparation of a variety of new drugs under development, such as TAS-116, a Phase II clinical drug of Taiho Pharmaceuticals for the treatment of gastrointestinal stromal tumors. |
| |
| Patent WO2005105760 discloses its preparation method. In the method, tetrakis(triphenylphosphine) palladium is used as a catalyst, and 3-bromo-4-fluorobenzonitrile is coupled with tetraethyl tin in a solvent hexamethylphosphoramide for a heating reaction for 15 hours to obtain 3 -Ethyl-4-fluorobenzonitrile. The method uses highly toxic tetraethyl tin, which brings great harm to operators and the environment, and is difficult to carry out industrial production. Meanwhile, the product 3-ethyl-4-fluorobenzonitrile obtained by the preparation method is an oily substance, which is purified by column chromatography with complicated operation, which is unfavorable for industrial production, and the specific purity of the product is not described. |
| |
| Therefore, looking for a new method for preparing 3-ethyl-4-fluorobenzonitrile with cheap and easy-to-obtain raw materials, safe and simple operation, high product purity and low cost suitable for industrial production, which will speed up the research process of related new drugs under development. , it is of great significance to reduce the production cost of related new drugs. |
| Example 1 3-Bromo-4-fluorobenzonitrile |
| |
| 3-Bromo-4-fluorobenzaldehyde (250g, 1.23mol) was dissolved in acetonitrile (1.5L), then hydroxylamine sulfonic acid (67g, 1.48mol) was added, and the reaction was refluxed for 4h. TLC showed that the conversion of the raw materials was complete, and the reaction solution was concentrated. To a small volume, add water (2L) and stir for 30min, cool to 5-10°C and continue stirring for 10min, filter, dissolve the filter cake with methyl tert-butyl ether (1.2L), wash twice with 500ml of water, saturated with 200ml Washed with sodium bicarbonate solution, dried over anhydrous sodium sulfate, filtered, the filtrate was adsorbed with activated carbon (10g), filtered, concentrated under reduced pressure to remove the solvent, added n-heptane (250ml), cooled and stirred in an ice-salt bath for 1h, filtered, reduced Press drying to give 3-bromo-4-fluorobenzonitrile (217 g, 88% yield). 1 H NMR (CDCl 3 ,400MHz):δ7.91(m,1H),7.63(m,1H),7.24(m,1H)。 |
| Example 2 3-Bromo-4-fluorobenzonitrile |
| |
| Add tetrahydrofuran (100ml) to a 250ml reaction flask, add 3-bromo-4-fluorobenzaldehyde (10g, 49.2mmol) and ammonia (40ml) under stirring, add elemental iodine (25g, 98.5mmol) in batches under cooling to 5°C ), then raised to ambient temperature and reacted for 2 to 3 hours, the reaction was completed, the reaction solution was poured into a 10% aqueous solution of sodium sulfite (200g), extracted twice with methyl tert-butyl ether (100ml), dried over anhydrous sodium sulfate , concentrated under reduced pressure to remove the solvent, added n-heptane (20 ml), cooled to 0-10 °C and stirred for 1 h, filtered, and dried under reduced pressure to obtain 3-bromo-4-fluorobenzonitrile (9.6 g, yield: 97.5 %). The NMR spectrum of this compound was determined and was identical to the product of Example 1. |
| Example 3 3-ethyl-4-fluorobenzonitrile |
| |
| 3-Bromo-4-fluorobenzonitrile (200 g, 1 mol) and [1,1-bis(diphenylphosphino)ferrocene]palladium(II) chloride dichloromethane complex (4.08 g, 5mmol) was dissolved in THF (1.2L), 1.0M/L diethylzinc n-hexane solution (600mL, 0.6mol) was added at 40-50°C, and the temperature was raised to 50-60°C for 4-5h. TLC showed The raw materials reacted completely. After the reaction solution was cooled to room temperature, it was added to 5% dilute hydrochloric acid (1 L), the layers were separated, the organic layer was washed twice with 500 ml of water, and then concentrated under reduced pressure to remove the solvent. Then n-hexane (600mL) and activated carbon (20g) were added, refluxed for 0.5h, cooled to room temperature, filtered, then added activated carbon (10g) to the filtrate, refluxed for 0.5h, cooled to room temperature, filtered, and cooled to -50°C to -60°C and filtered, and the filter cake was dried under reduced pressure at 10-20°C to obtain 3-ethyl-4-fluorobenzonitrile (112 g, yield: 75%) as an off-white solid, melting point 23.1-27.4°C. 1 H NMR (CDCl 3 , 400MHz): δ 7.50 (m, 2H), 7.09 (m, 1H), 2.69 (q, J=7.6Hz, 2H), 1.24 (t, 3H, J=7.6Hz), HPLC purity 99.6%. |
| HPLC assay conditions: |
| Chromatographic UV detector: DAD |
| Chromatography pump: 1100 quaternary pump |
| Chromatographic column: Agilent (USA) ZORBAX SB-C184.6×150mm, 5μm PN883975-902 Chromatographic conditions: |
| Mobile Phase A: Water |
| Mobile Phase B: Acetonitrile |
| |
| Injection volume: 5 μL, flow rate: 1.0 mL/min, column temperature: room temperature, detection wavelength: 210 nm. |


Acylation of 2-fluoro-4-iodopyridine with isobutyric anhydride in presence of BuLi and DIEA in THF at -78 °C gives 1-(2-fluoro-4-iodo-3-pyridinyl)-2-methylpropan-1-one ,
This upon cyclization using hydrazine hydrate at 65 °C gives 4-iodo-3-isopropylpyrazolo[3,4-b]pyridine.
N-Protection of intermediate with PMB-Cl in the presence of base NaH in solvent DMF at 0 °C affords 4-iodo-3-isopropyl-1-(4-methoxybenzyl)pyrazolo[3,4-b]pyridine,
This is coupled with 4-(4-imidazolyl)-1-methylpyrazole in the presence of Cu2O, 4,7-dimethoxy-1,10-phenanthroline, Cs2CO3 and PEG-diamine in solvent NMP or DMSO at 130 °C to furnish 4-[4-(4-pyrazolyl)-imidazol-1-yl]pyrazolo[3,4-b]pyridine derivative .
N-Deprotection of PMB-protected pyrazolo[3,4-b]pyridine derivative by using TFA and anisole gives free pyrazolo[3,4-b]pyridine ,
This on condensation with 3-ethyl-4-fluorobenzonitrile in the presence of Cs2CO3 in DMF at 95 °C yields 4-(pyrazolo[3,4-b]pyridin-1-yl)benzonitrile .
Finally, partial hydrolysis of nitrile by means of aqueous NaOH and H2O2 in DMSO/EtOH gives the Pimitespib TAS-116 .
CLIP
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b01085
J. Med. Chem.2019, 62, 2, 531–551
Publication Date:December 7, 2018
https://doi.org/10.1021/acs.jmedchem.8b0108

The molecular chaperone heat shock protein 90 (HSP90) is a promising target for cancer therapy, as it assists in the stabilization of cancer-related proteins, promoting cancer cell growth, and survival. A novel series of HSP90 inhibitors were discovered by structure–activity relationship (SAR)-based optimization of an initial hit compound 11a having a 4-(4-(quinolin-3-yl)-1H-indol-1-yl)benzamide structure. The pyrazolo[3,4-b]pyridine derivative, 16e (TAS-116), is a selective inhibitor of HSP90α and HSP90β among the HSP90 family proteins and exhibits oral availability in mice. The X-ray cocrystal structure of the 16e analogue 16d demonstrated a unique binding mode at the N-terminal ATP binding site. Oral administration of 16e demonstrated potent antitumor effects in an NCI-H1975 xenograft mouse model without significant body weight loss.
3-Ethyl-4-(3-Isopropyl-4-(4-(1-methyl-1H-Pyrazol-4-yl)-1H-Imidazol-1-yl)-1H-Pyrazolo[3,4-b]pyridin-1-yl)benzamide (16e). Yield 64% (2 steps), white powder. UPLC−MS (ESI) m/z: 454.8 [M + H]+ , tR = 1.19 min. UPLC purity 99.65%. 1 H NMR (400 MHz, CDCl3): δ 1.14 (t, J = 7.5 Hz, 3H), 1.25 (d, J = 7.0 Hz, 6H), 2.62 (q, J = 7.3 Hz, 2H), 3.18 (spt, J = 6.8 Hz, 1H), 3.98 (s, 3H), 5.88 (br s,1H), 6.22 (br s, 1H), 7.13 (d, J = 5.1 Hz, 1H), 7.39 (d, J = 1.1 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.78−7.81 (m, 3H), 7.86 (d, J = 1.5 Hz, 1H), 7.96 (d, J = 1.8 Hz, 1H), 8.59 (d, J = 4.7 Hz, 1H). HRMS: calcd for C25H26N8O, 455.2308 [M + H]+ ; found, 455.2311.
PAPER
Journal of Medicinal Chemistry (2021), 64(5), 2669-2677.
PATENT
WO 2016181990
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016181990
Compound 1 in the present invention is 3-ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazol-4-yl) -1H-imidazole-1-yl) -1H-. Pyrazolo [3,4-b] pyridin-1-yl} benzamide (formula below). Compound 1 is known to have HSP90 inhibitory activity and exhibit excellent antitumor activity. Compound 1 can be synthesized based on the production methods described in Patent Documents 1 and 2.
[0013]
[hua 1]
Patent Document 1: International Publication No. 2012/093708
Patent Document 2: International Publication No. 2011/004610
Comparative Example 1 3-Ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3, 4-b] Pyridine-1-yl} Synthesis of type I crystals of benzamide
3-Ethyl-4 obtained according to the production method described in International Publication No. 2012/093708 and International Publication No. 2011/004610. -{3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3,4-b] pyridin-1- A white solid (3.58 g) of yl} benzamide was added to ethanol (7.84 mL) and stirred at room temperature for 2 hours. After sampling, it was washed with ethanol (7.84 mL) and dried under reduced pressure at 70 to 80 ° C. for 20 hours to obtain type I crystals (yield: 2.40 g, yield: 61.2%, purity: 98.21%). rice field.
Further, as shown in FIG. 1, the type I crystal has a diffraction angle (2θ) of 8.1 °, 10.9 °, 12.1 °, 14.0 °, and 14.9 in the powder X-ray diffraction spectrum. °, 16.2 °, 17.7 °, 20.2 °, 21.0 °, 21.5 °, 22.6 °, 24.3 °, 25.4 ° 26.4 °, 27.0 ° , 28.3 °, 30.2 °, 30.9 °, 31.5 °, 32.7 °, 34.7 °, 35.4 ° and 36.6 ° showed characteristic peaks.
[0032]
1H-NMR (DMSO-d 6):δppm 9.35 (1H,d,J=4.88Hz), 8.93 (1H,d,J=1.22Hz), 8.84 (1H,brs), 8.72 (1H,d,J=1.95Hz), 8.70 (1H,s) ,8.63 (1H,d,J=1.22Hz), 8.60 (1H,dd,J=8.29,1.95Hz), 8.46 (1H,s) ,8.25 (1H,d,J=8.29Hz), 8.22 (1H,brs), 8.12 (1H,d,J=4.88Hz), 4.59 (3H,s) ,3.95 (1H,tt,J=6.83,6.83Hz), 3.21 (2H,q,J=7.56Hz), 1.83(6H,d,J=6.83Hz), 1.75 (3H,t,J=7.56Hz):LRMS(ESI)m/z 455[M+H]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015050235
Synthesis of Test Compound The
following synthesis example compounds (Synthesis Examples 1 to 3) were synthesized according to the method described in WO2011 / 004610.
[0361]
Synthesis Example 1: 4- {3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazol-1-yl) -1H-pyrazolo [3,4-b] pyridine -1-yl} -3-methylbenzamide
[0362]
[Changing 22]
PATENT
WO 2011004610
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011004610
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Pimitespib
3-Ethyl-4-{4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-3-(propan-2-yl)-1H-pyrazolo[3,4-b]pyridin-1-yl}benzamide
C25H26N8O : 454.53
[1260533-36-5]
//////////Pimitespib, ピミテスピブ, JAPAN 2022, APPROVALS 2022, TAS 116, Jeselhy
O=C(N)C1=CC=C(N2N=C(C(C)C)C3=C(N4C=C(C5=CN(C)N=C5)N=C4)C=CN=C32)C(CC)=C1
VP1-001
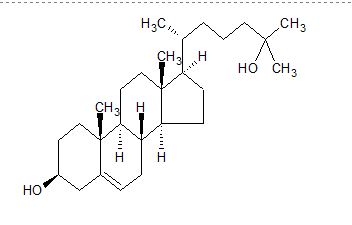


VP1-001
CAS 2140-46-7
Cholest-5-ene-3,25-diol, (3β)-Molecular Weight, 402.65, C27 H46 O2
- Cholest-5-ene-3β,25-diol (8CI)
- (3β)-Cholest-5-ene-3,25-diol
- 25-Hydroxy-5-cholestene-3β-ol
- 25-Hydroxycholesterol
- 5-Cholesten-3β,25-diol
Compound 29 – ViewPoint Therapeutics
- Originator University of California at San Francisco; University of Michigan; University of Washington
- Developer University of California at San Francisco; University of Michigan; University of Washington; ViewPoint Therapeutics
- Class Small molecules; Sterols
- Mechanism of Action Amyloid inhibitors; Protein aggregation inhibitors; Protein folding inhibitors
- 28 Jun 2020 No recent reports of development identified for research development in Presbyopia in USA (Ophthalmic, Drops)
- 28 Dec 2019 No recent reports of development identified for preclinical development in Cataracts in USA (Ophthalmic, Drops)
- 01 Apr 2016 ViewPoint Therapeutics receives SBIR grant from National Eye Institute for VP1 001 development in Cataracts (ViewPoint Therapeutics website)
CLIP
https://europepmc.org/article/PMC/6676924
We previously identified an oxysterol, VP1-001 (also known as compound 29), that partially restores the transparency of lenses with cataracts. To understand the mechanism of VP1-001, we tested the ability of its enantiomer, ent-VP1-001, to bind and stabilize αB-crystallin (cryAB) in vitro and to produce a similar therapeutic effect in cryAB(R120G) mutant and aged wild-type mice with cataracts. VP1-001 and ent-VP1-001 have identical physicochemical properties. These experiments are designed to critically evaluate whether stereoselective binding to cryAB is required for activity.

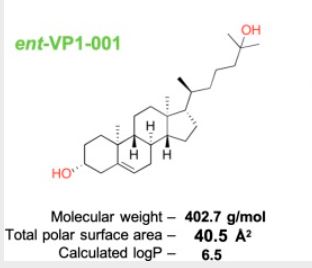
SYN https://europepmc.org/articles/PMC6676924/bin/iovs-60-07-63_s01.pdf
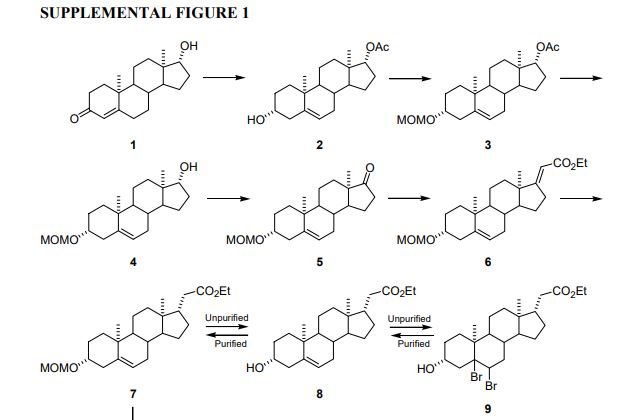


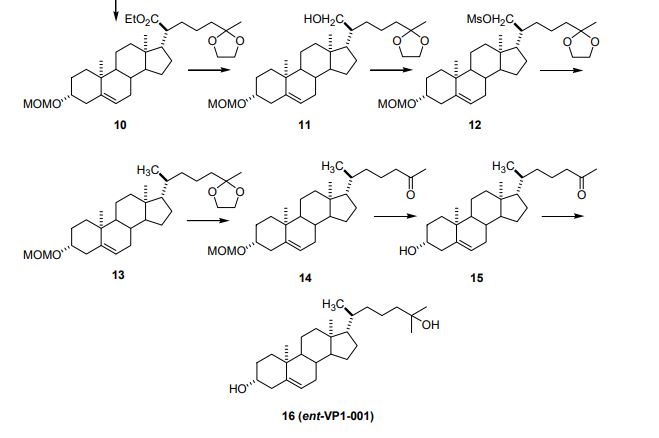
ref
- (2) Ogawa, Shoujiro; Steroids 2009, Vol74(1), Pg81-87
- (7) Beckwith, A. L. J.; Journal of the Chemical Society 1961, Pg3162-4
- (8) Beckwith, A. L. J.; Proceedings of the Chemical Society, London 1958, Pg194-5
- (9) Dauben, William G.; Journal of the American Chemical Society 1950, Vol72, Pg4248-50
- (10) Fieser, Louis F.; Journal of Organic Chemistry 1957, Vol22, Pg1380-4
- Ogawa, Shoujiro; Steroids 2009, Vol74(1), Pg81-87
https://www.sciencedirect.com/science/article/abs/pii/S0039128X08002432
A solution of the 3-acetate 2a (200mg, 0.45mmol) in 5%
methanolic KOH (20mL) was refluxed for 1 h. After evaporation of the solvent, the residue was dissolved in CH2Cl2, and
the solution was washed with water, dried with Drierite® and
evaporated in vacuo. Recrystallization of the oily residue from
aq. methanol gave the 3,25-dihydroxy-5-ene 2b as colorless
needles: yield, 163mg (90%). m.p. 178–180 ◦C. (lit. 178–180 ◦C
[32]). IR (KBr), max cm−1: 3301 (OH). 1H NMR (CDCl3) ı: 0.68
(3H, s, 18-CH3), 0.93 (3H, d, J 5.4, 21-CH3), 1.01 (3H, s, 19-CH3),
1.21 (6H, s, 26- and 27-CH3), 3.50 (1H, br. m, 3-H), 5.34 (1H, br.
s, 6-H). 13C NMR (CDCl3) ı: 11.9 (C-18), 18.7 (C-21), 19.4 (C-19),
20.7 (C-23), 21.1 (C-11), 24.3 (C-15), 28.2 (C-16), 29.2 and 29.3 (C26 and C-27), 31.6 (C-2), 31.9 (C-7), 31.9 (C-8), 35.7 (C-20), 36.4
(C-22), 36.5 (C-10), 37.2 (C-1), 39.8 (C-12), 42.3 (C-4 and C-13),
44.4 (C-24), 50.1 (C-9), 56.0 (C-17), 56.7 (C-14), 71.1 (C-25), 71.8
(C-3), 121.7 (C-6), 140.7 (C-5). LR-MS (EI), m/z: 402 (M+, 65%), 384
(M−H2O, 100%), 369 (M−H2O–CH3, 53%), 366 (M−2H2O, 21%),
351 (M−2H2O–CH3, 34%), 317 (M−H2O–ring A–part of ring B,
13%), 299 (M−2H2O–ring A–part of ring B, 29%), 273 (M−S.C.,
75%), 255 (M−H2O–S.C., 31%), 245 (24%), 231 (M−S.C.–ring D,
22%), 213 (M−H2O-CH3–S.C.–part of ring D, 43%). HR-MS (EI),
calculated for C27H46O2 [M+], 402.3498; found m/z: 402.3498.


//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
TO MAINTAIN SUBSCRIPTION OF THIS BLOG
$10.00
CLIP
A Noninvasive Alternative to Cataract Surgery?
Investigators are exploring chemical compounds to restore transparency to the crystalline lens.
Surgery is an effective but costly means of managing cataracts, and, like all surgical interventions, it carries risks. Moreover, a substantial number of people worldwide, particularly in developing countries, lack access to cataract surgery. The World Health Organization estimates that 65.2 million people globally are blind or visually impaired from cataracts.1 The development of an eye drop that restores transparency and flexibility to the crystalline lens would therefore be a game-changer as a less expensive, noninvasive option for treating a leading cause of blindness.
RESEARCH RESULTS
The crystalline lens is composed of epithelial and fiber cells. One of the major lens proteins in the fiber cells is alpha-crystallin, a chaperone protein thought to maintain homeostasis of the crystalline lens, thereby preserving its transparency and flexibility. As a person ages, alpha-crystallin proteins become prone to misfolding, causing them to clump together and form insoluble high-molecular-weight protein aggregates, which can lead to cataract formation.
Compound 29 (full name, 5-cholesten-3beta,25-diol), also known as VP1-001 (ViewPoint Therapeutics) and as 25-hydroxy-cholesterol, is an oxysterol, a derivative of cholesterol. Usha Andley, PhD, FARVO, is an investigator conducting research on this chemical compound’s use as a treatment for cataracts.2,3 Another oxysterol being investigated for this purpose is lanosterol. In a recent study, neither oxysterol was effective at reducing opacities of in vitro cultured lenses treated with various reagents to induce opacification in vitro.4 In an interview with ME, however, Dr. Andley stated that VP1-001 appears to be more effective than lanosterol at reducing lens opacity. VP1-001 differs from lanosterol in terms of solubility, she said, allowing VP1-001 to penetrate the eye better.
The goal of the research being conducted by Dr. Andley and her colleagues, she said, is twofold:
No. 1: To ensure that the compound is not toxic to the cornea; and
No. 2: To show that the compound is capable of reducing the tendency of alpha-crystallins to aggregate and perhaps reverse their aggregation.
In proof-of-concept studies, VP1-001 was incorporated into an eye drop formulation of 8% cyclodextrin. Dr. Andley and colleagues administered the drops three times per week in a mouse model for 2 to 4 weeks. According to Dr. Andley, the compound seemed to increase the stability of the alpha-crystallin protein so that it increased the soluble fraction of proteins from mouse and human lens cataracts. The compound also increased the solubility of two other lens crystallins, beta- and gamma-crystallin, in the mouse lens, and it seemed to reduce the abundance of high-molecular-weight aggregates in the lens.2,3
ViewPoint Therapeutics is currently developing this technology for use in humans. According to Dr. Andley, who is working with the company, ViewPoint Therapeutics is using different model systems for in vitro and protein-binding studies in an attempt to improve on earlier results with the chemical compound. Their research has identified new compounds, nonsterol ligands for alpha-crystallin, that exhibited in vitro activity and efficacy similar to or better than those of VP1-001 in mouse models of cataracts.5 The discovery of these nonsterols supports the hypothesis that pharmacologic chaperones targeting alpha-crystallin can prevent or reverse cataracts, Dr. Andley said.
CHALLENGES AND FUTURE DIRECTIONS
In studies to date, Dr. Andley and her colleagues have treated mice in one eye with the VP1-001 formulation, and the contralateral untreated eyes have served as controls. They then compared the two eyes after the conclusion of treatment (Figure). She is looking forward to conducting masked and randomized studies that include baseline measurements of lens opacity. According to Dr. Andley, this research will begin this year. Animal testing, however, can advance understanding only so far. Human testing of safety and efficacy will be a major step forward in the research on VP1-001 and newer variants.
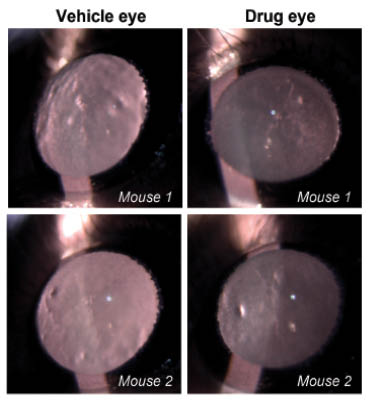
Figure | Representative slit-lamp images from aged wild-type mouse lenses show the extent of opacity treated with vehicle (left) or drug (right). Mice were treated topically with the drug in one eye and vehicle in the contralateral eye three times per week for 2 weeks. Slit-lamp examinations were performed on conscious, live mice. Mouse 1 and 2 were treated with VP1-001.
A major challenge in the development of a pharmacologic treatment for cataract is how to determine if a chemical compound can be maintained in the lens at a sufficient concentration and for an adequate duration to achieve the desired outcome. A second challenge relates to detecting change. Dr. Andley and her colleagues are seeking a more quantitative method by which to assess the extent of lenticular opacity before and after treatment. They have a modified Lens Opacities Classification System, she said, but it is less objective than methods such as Scheimpflug photography. A goal, then, is to develop a more standardized, objective way of measuring results.
In addition to age-related cataract, Dr. Andley suggested, a topical agent could be advantageous for the treatment of congenital cataracts that form because of a mutation in alpha A crystallin or alpha B crystallin. Retaining the crystalline lens instead of extracting it would allow pediatric eyes to develop normally, she noted.
The idea of an eye drop formulation to treat cataracts may seem like science fiction, but Dr. Andley expects it to become a more realistic possibility within the next few years. The utility of such a chemical compound could extend beyond cataracts to the treatment of presbyopia. The hypothesis, she said, is that softening the crystalline lens would improve accommodative amplitude.
1. World Health Organization. Blindness and vision impairment. https://www.who.int/news-room/fact-sheets/detail/blindness-and-visual-impairment. Published October 8, 2019. Accessed January 22, 2020.
2. Makley LN, McMenimen KA, DeVree BT, et al. Pharmacological chaperone for α-crystallin partially restores transparency in cataract models. Science. 2015;350(6261):674-677.
3. Molnar KS, Dunyak BM, Su B, et al. Mechanism of action of VP1-001 in cryAB(R120G)-associated and age-related cataracts. Invest Ophthalmol Vis Sci. 2019;60(10):3320-3331.
4. Daszynski DM, Santhoshkumar P, Phadte AS, et al. Failure of oxysterols such as lanosterol to restore lens clarity from cataracts. Sci Rep. 2019;9(1):8459.
5. Dunyak B, Su B, Molnar K, et al. Discovery of non-sterol aB-crystallin ligands as potential cataract therapeutics. Invest Ophthalmol Vis Sci. 2019;60(9):5691.
///////////VP1-001 , occular
Vutrisiran sodium, ALN 65492, Votrisiran
RNA, (Um-sp-(2′-deoxy-2′-fluoro)C-sp-Um-Um-Gm-(2′-deoxy-2′-fluoro)G-Um-Um-(2′-deoxy-2′-fluoro)A-Cm-Am-Um-Gm-(2′-deoxy-2′-fluoro)A-Am-(2′-deoxy-2′-fluoro)A-Um-Cm-Cm-Cm-Am-sp-Um-sp-Cm), complex with RNA (Um-sp-Gm-sp-Gm-Gm-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)A-(2′-deoxy-2′-fluoro)U-Gm-Um-Am-Am-Cm-Cm-Am-Am-Gm-Am) 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate] (1:1)
Vutrisiran Sodium
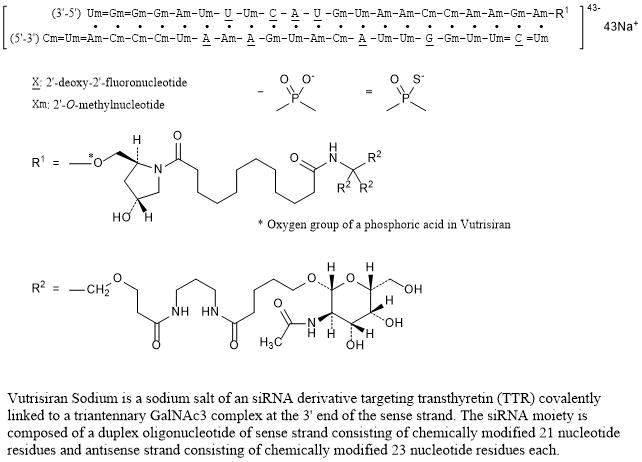
Nucleic Acid Sequence
Sequence Length: 44, 23, 2113 a 9 c 8 g 14 umultistranded (2); modified
Vutrisiran sodium
- ALN 65492
- Votrisiran
C530H672F9N171Na43O323P43S6 : 17289.77
[1867157-35-4 , Vutrisiran]
| Formula | C530H672F9N171O323P43S6.43Na ORC530H672F9N171Na43O323P43S6 |
|---|---|
| CAS | 1867157-35-4 , VURISIRAN |
| Mol weight | 17289.7661 |
FDA APPROVED, AMVUTTRA, 2022/6/13
| ブトリシランナトリウム |
| Efficacy | Gene expression regulator |
|---|---|
| Disease | Polyneuropathy of hereditary transthyretin-mediated amyloidosis [D |
| Comment | RNA interference (RNAi) drug Treatment of transthyretin (TTR)-mediated amyloidosis (ATTR amyloidosis) |
UNII28O0WP6Z1P UNII
Vutrisiran
Vutrisiran Sodium is a sodium salt of an siRNA derivative targeting transthyretin (TTR) covalently linked to a triantennary GalNAc3 complex at the 3’ end of the sense strand. The siRNA moiety is composed of a duplex oligonucleotide of sense strand consisting of chemically modified 21 nucleotide residues and antisense strand consisting of chemically modified 23 nucleotide residues each.
Vutrisiran is a double-stranded small interfering ribonucleic acid (siRNA) that targets wild-type and mutant transthyretin (TTR) messenger RNA (mRNA).7 This siRNA therapeutic is indicated for the treatment of neuropathies associated with hereditary transthyretin-mediated amyloidosis (ATTR), a condition caused by mutations in the TTR gene.2 More than 130 TTR mutations have been identified so far,3 but the most common one is the replacement of valine with methionine at position 30 (Val30Met).2 The Val30Met variant is the most prevalent among hereditary ATTR patients with polyneuropathy, especially in Portugal, France, Sweden, and Japan.2
TTR mutations lead to the formation of misfolded TTR proteins, which form amyloid fibrils that deposit in different types of tissues. By targeting TTR mRNA, vutrisiran reduces the serum levels of TTR.6,7 Vutrisiran is commercially available as a conjugate of N-acetylgalactosamine (GalNAc), a residue that enables the delivery of siRNA to hepatocytes.5,7 This delivery platform gives vutrisiran high potency and metabolic stability, and allows for subcutaneous injections to take place once every three months.8 Another siRNA indicated for the treatment of polyneuropathy associated with hereditary ATTR is patisiran.2 Vutrisiran was approved by the FDA in June 2022.
CLIP
https://www.nature.com/articles/s41392-020-0207-x
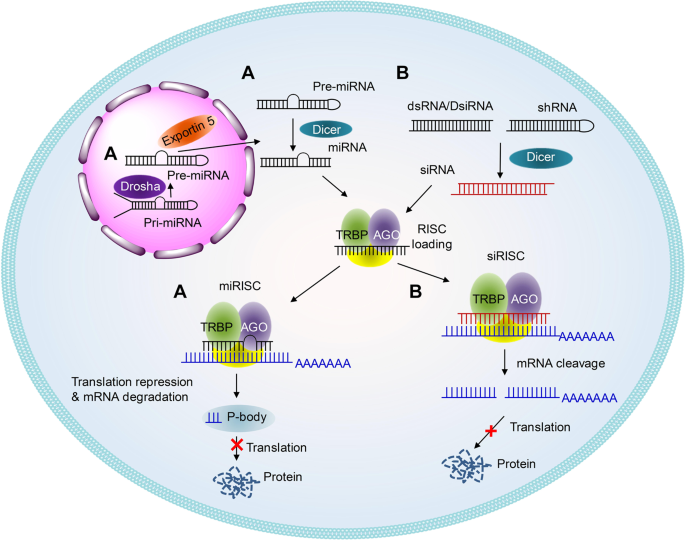
Schematic illustrations of the working mechanisms of miRNA (a) and siRNA (b)
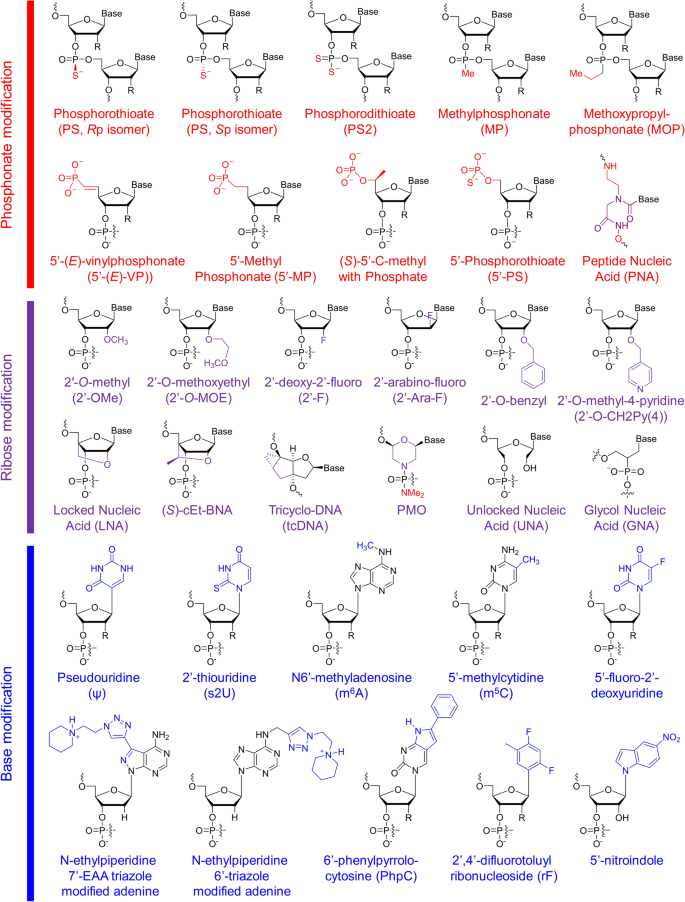
Structures of chemical modifications and analogs used for siRNA and ASO decoration. According to the modification site in the nucleotide acid, these structures can be divided into three classes: phosphonate modification, ribose modification and base modification, which are marked in red, purple and blue, respectively. R = H or OH, for RNA or DNA, respectively. (S)-cEt-BNA (S)-constrained ethyl bicyclic nucleic acid, PMO phosphorodiamidate morpholino oligomer
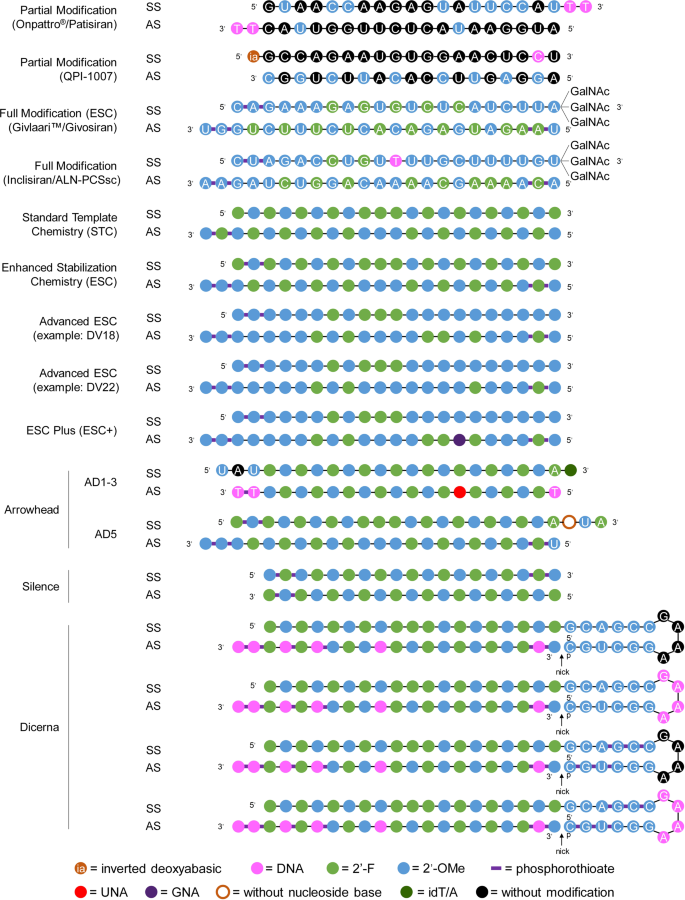
Representative designs for the chemical modification of siRNA. The sequences and modification details for ONPATTRO®, QPI-1007, GIVLAARI™ and inclisiran are included. The representative siRNA modification patterns developed by Alnylam (STC, ESC, advanced ESC and ESC+) and arrowhead (AD1-3 and AD5) are shown. Dicerna developed four GalNAc moieties that can be positioned at the unpaired G–A–A–A nucleotides of the DsiRNA structure. 2′-OMe 2′-methoxy, 2′-F 2′-fluoro, GNA glycol nucleic acid, UNA unlocked nucleic acid, SS sense strand, AS antisense strand
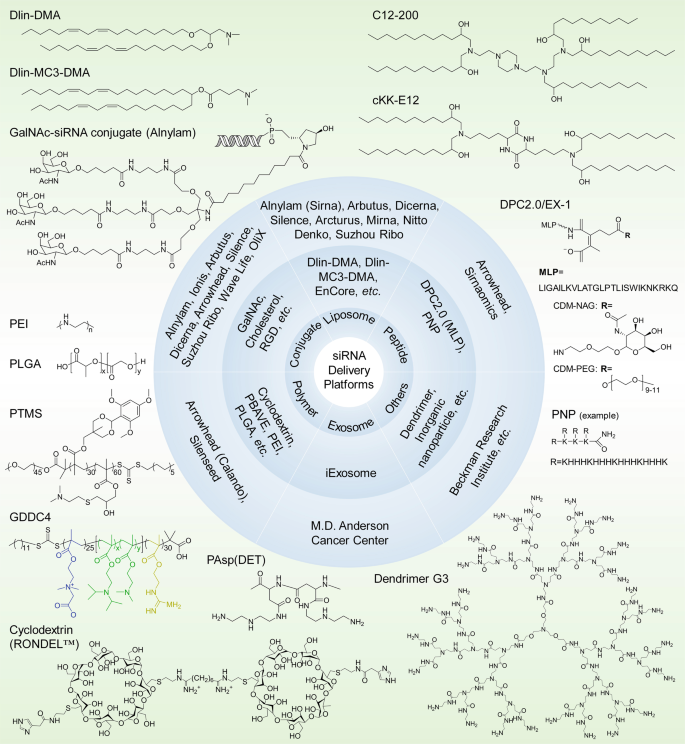
siRNA delivery platforms that have been evaluated preclinically and clinically. Varieties of lipids or lipidoids, siRNA conjugates, peptides, polymers, exosomes, dendrimers, etc. have been explored and employed for siRNA therapeutic development by biotech companies or institutes. The chemical structures of the key component(s) of the discussed delivery platforms, including Dlin-DMA, Dlin-MC3-DMA, C12-200, cKK-E12, GalNAc–siRNA conjugates, MLP-based DPC2.0 (EX-1), PNP, PEI, PLGA-based LODER, PTMS, GDDC4, PAsp(DET), cyclodextrin-based RONDEL™ and dendrimer generation 3 are shown. DLin-DMA (1,2-dilinoleyloxy-3-dimethylaminopropane), DLin-MC3-DMA (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino) butanoate, DPC Dynamic PolyConjugates, MLP membrane-lytic peptide, CDM carboxylated dimethyl maleic acid, PEG polyethylene glycol, NAG N-acetylgalactosamine, PNP polypeptide nanoparticle, PEI poly(ethyleneimine), LODER LOcal Drug EluteR, PLGA poly(lactic-co-glycolic) acid, PTMS PEG-PTTMA-P(GMA-S-DMA) poly(ethylene glycol)-co-poly[(2,4,6-trimethoxybenzylidene-1,1,1-tris(hydroxymethyl))] ethane methacrylate-co-poly(dimethylamino glycidyl methacrylate), GDDC4 PG-P(DPAx-co-DMAEMAy)-PCB, where PG is guanidinated poly(aminoethyl methacrylate) PCB is poly(carboxybetaine) and P(DPAx-co-DMAEMAy) is poly(dimethylaminoethyl methacrylate-co-diisopropylethyl methacrylate), PEG-PAsp(DET) polyethylene glycol-b-poly(N′-(N-(2-aminoethyl)-2-aminoethyl) aspartamide), PBAVE polymer composed of butyl and amino vinyl ether, RONDEL™ RNAi/oligonucleotide nanoparticle delivery
REF
Nucleic Acids Research (2019), 47(7), 3306-3320.
Drug Metabolism & Disposition (2019), 47(10), 1183-1201.
PATENT
WO 2020128816
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020128816
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and an additional therapeutic agent for the treatment of transthyretin amyloidosis. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent for the treatment of transthyretin amyloidosis.
The present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof a combination of a benzoxazole derivative transthyretin stabilizer or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. Particularly, the present invention relates to pharmaceutical compositions and methods of treatment comprising administering to a patient in need thereof 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof and one or more additional therapeutic agent. The compositions and methods of the invention are useful in stabilizing transthyretin, inhibiting transthyretin misfolding, proteolysis, and treating amyloid diseases associated thereto.
Transthyretin (TTR) is a 55 kDa homotetrameric protein present in serum and cerebral spinal fluid and which functions as a transporter of L-thyroxine (T4) and holo-retinol binding protein (RBP). TTR has been found to be an amyloidogenic protein that, under certain conditions, can be transformed into fibrils and other aggregates which can lead to disease pathology such as polyneuropathy or cardiomyopathy in humans.
US Patent Nos. 7,214,695; 7,214,696; 7,560,488; 8, 168.683; and 8,653,119 each of which is incorporated herein by reference, discloses benzoxazole derivatives which act as transthyretin stabilizers and are of the formula
or a pharmaceutically acceptable salt thereof; wherein Ar is 3,5-difluorophenyl, 2,6-difluorophenyl, 3,5-dichlorophenyl, 2,6-dichlorophenyl, 2-(trifluoromethyl)phenyl or 3-(trifluoromethyl)phenyl. Particularly, 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis) of the formula
is disclosed therein. Tafamidis is an orally active transthyretin stabilizer that inhibits tetramer dissociation and proteolysis that has been approved in certain jurisdictions for the treatment of transthyretin polyneuropathy (TTR-PN) and is currently in development for the treatment of transthyretin cardiomyopathy (TTR-CM). US Patent No. 9,249, 112, also incorporated herein by reference, discloses polymorphic forms of the meglumine salt of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis meglumine). US Patent No. 9,770,441 discloses polymorphic forms of the free acid of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid (tafamidis), and is also incorporated by reference herein.
Summary of the Invention
The present invention provides pharmaceutical compositions and methods comprising the compound 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent. Particular embodiments of this invention are pharmaceutical compositions and methods comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents selected from the group consisting of agents that lower plasma levels of TTR such as an antisense therapy, TTR gene editing therapy, transcriptional modulators, translational modulators, TTR protein degraders and antibodies that bind and reduce TTR levels; amyloid reduction therapies such as anti amyloid antibodies (either TTR selective or general), stimulators of amyloid clearance, fibril disruptors and therapies that inhibit amyloid nucleation; other TTR stabilizers; and TTR modulators such as therapeutics which inhibit TTR cleavage. Particularly, the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and the present invention provides pharmaceutical compositions and methods comprising tafamidis or tafamidis meglumine salt with one or more additional therapeutic agents. More particularly, the present invention provides pharmaceutical compositions and
methods comprising a polymorphic form of tafamidis free acid or a polymorphic form of tafamidis meglumine salt with one or more additional therapeutic agents.
The present invention also provides a method of treating or preventing transthyretin amyloidosis in a patient, the method comprising administering to a patient in need thereof a therapeutically or prophylactically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole- 6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agents.
A particular embodiment of the present method of treatment is the method comprising a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered orally. Additional embodiments of this invention are methods of treatment as described above wherein the 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof, and one or more additional therapeutic agent are administered parenterally (intravenously or subcutaneously). Further embodiments of this invention are methods of treatment wherein the 2-(3,5-dichlorophenyl)-1, 3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally and the one or more additional therapeutic agent is administered either orally or parenterally. Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR Another embodiment of the present invention is wherein a pharmaceutical composition comprising 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agent is administered parenterally and then 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR 5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof is administered orally. A particular method of treatment is a method of treating TTR amyloidosis such as TTR polyneuropathy or TTR
cardiomyopathy, the method comprising administering to a patient in need thereof a therapeutically effective amount of 2-(3,5-dichlorophenyl)-1,3-benzoxazole-6-carboxylic acid or a pharmaceutically acceptable salt or prodrug thereof in combination with one or more additional therapeutic agents.
Brief Description of the Drawings
REF
Biochemical Pharmacology (Amsterdam, Netherlands) (2021), 189, 114432.
PATENT
WO 2021041884
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021041884
Exemplary RNAi agents that reduce the expression of TTR include patisiran and vutrisiran.
The ter s “antisense polynucleotide agent”, “antisense oligonucleotide”, “antisense compound”, and “antisense agent” as used interchangeably herein, refer to an agent comprising a single-stranded oligonucleotide that specifically binds to the target nucleic acid molecules via hydrogen bonding (e.g., Watson-Crick, Hoogsteen, or reversed Hoogsteen hydrogen bonding) and inhibits the expression of the targeted nucleic acid by an antisense mechanism of action, e.g., by RNase H. In some embodiments, an antisense agent is a nucleic acid therapeutic that acts by reducing the expression of a target gene, thereby reducing the expression of the polypeptide encoded by the target gene. Exemplary antisense agents that reduce the expression of TTR include inotersen and Ionis 682884/ ION-TTR-LRx (see, e.g., WO2014179627 which is incorporated by reference in its entirety). Further antisense agents that reduce the expression of TTR are provided, for example in WO2011139917 and WO2014179627, each of which is incorporated by reference in its entirety.
REF
Clinical Pharmacology & Therapeutics (Hoboken, NJ, United States) (2021), 109(2), 372-382
Annals of Plastic Surgery (2021), 86(2S_Suppl_1), S23-S29.
Journal of Cardiovascular Pharmacology (2021), 77(5), 544-548.
Annals of Pharmacotherapy (2021), 55(12), 1502-1514.
Kidney International (2022), 101(2), 208-211
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
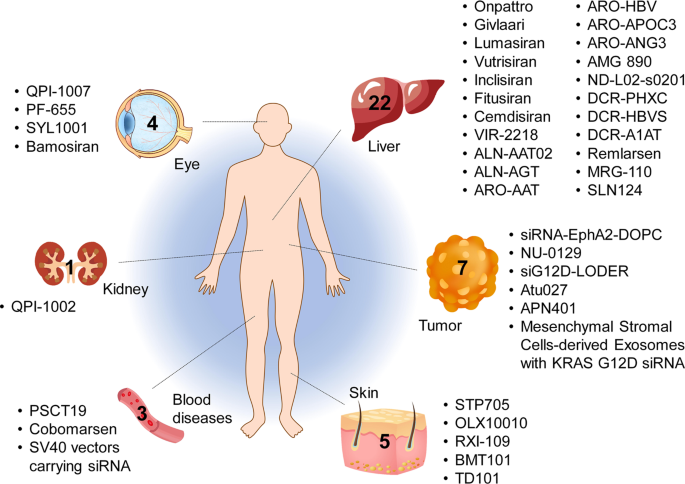
Tissues targeted by siRNA and miRNA therapeutics currently being investigated at the clinical stage. The corresponding therapeutic names are shown beside the tissues
CLIP
Vutrisiran An Investigational RNAi Therapeutic for ATTR Amyloidosis Vutrisiran has not been approved by the U.S. Food and Drug Administration, European Medicines Agency, or any other regulatory authority and no conclusions can or should be drawn regarding the safety or effectiveness of this investigational therapeutic. Overview • Vutrisiran is an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, which encompasses both hereditary ATTR (hATTR) amyloidosis and wild-type ATTR (wtATTR) amyloidosis.1, 2 • Vutrisiran inhibits the production of disease-causing transthyretin (TTR) protein by the liver, leading to a reduction in the level of TTR in the blood.1, 2 • Vutrisiran is administered subcutaneously (under the skin) and utilizes one of Alnylam’s delivery platforms known as the Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate delivery platform.1, 2 • Vutrisiran is administered every three months.2 • Vutrisiran is under review by the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Brazilian Health Regulatory Agency (ANVISA). Vutrisiran has been granted Orphan Drug Designation in the U.S. and the European Union (EU) for the treatment of ATTR amyloidosis. Vutrisiran has also been granted a Fast Track designation in the U.S. for the treatment of the polyneuropathy of hATTR amyloidosis in adults. In the U.S. vutrisiran has received an action date under the Prescription Drug User Fee Act (PDUFA) of April 14, 2022. The Company received orphan drug designation in Japan. Alnylam has global commercial rights to vutrisiran, assuming regulatory approvals. Clinical Development • A Phase 1 clinical study of vutrisiran was conducted in 80 healthy volunteers (60 received vutrisiran and 20 received placebo). Vutrisiran demonstrated an acceptable safety profile and a single dose reduced serum TTR for a period of at least 90 days.2 • The safety and efficacy of vutrisiran are being evaluated in the HELIOS Phase 3 clinical program, currently consisting of two clinical trials: HELIOS-A and HELIOS-B. • HELIOS-A is a randomized, open-label, global multi-center Phase 3 study of 164 adult patients with hATTR amyloidosis with polyneuropathy.1 • The primary endpoint of HELIOS-A is change from baseline in the modified Neuropathy Impairment Score +7 (mNIS+7) at 9 months. • Secondary endpoints at 9 months include the Norfolk Quality of Life-Diabetic Neuropathy (Norfolk QoL-DN) Total Score and the 10-Meter Walk Test (10-MWT). • The 9-month endpoints will be analyzed at 18 months with the addition of other secondary endpoints. • HELIOS-B is a randomized, double-blind, placebo-controlled Phase 3 study of 655 adult patients with ATTR amyloidosis with cardiomyopathy (including both hATTR and wtATTR amyloidosis).3 • The primary endpoint will evaluate the efficacy of vutrisiran versus placebo for the composite outcome of all-cause mortality and recurrent cardiovascular (CV) events (CV hospitalizations and urgent heart failure (HF) visits) at 30-36 months. • Secondary endpoints include the change from baseline in the 6-minute walk test (6-MWT), health status measured using the Kansas City Cardiomyopathy Questionnaire Overall Summary (KCCQ-OS), echocardiographic assessments of mean left ventricular wall thickness and global longitudinal strain, the N-terminal prohormone B-type natriuretic peptide (NT-proBNP) as a cardiac biomarker, and all-cause mortality, rate of recurrent CV events, and composite of all-cause mortality and recurrent all-cause hospitalizations and urgent HF visits at month 30 or 30-36 months. Page 2 © 2021 Alnylam Pharmaceuticals, Inc. All rights reserved. TTRsc02-USA-00012 v4 About ATTR Amyloidosis • ATTR amyloidosis is a rare, underdiagnosed, rapidly progressive, debilitating, and fatal disease caused by misfolded TTR that accumulates as amyloid fibrils in multiple tissues including the nerves, heart, and GI tract. There are two types of ATTR amyloidosis: hATTR amyloidosis and wtATTR amyloidosis.4,5,6 • hATTR amyloidosis is an inherited condition that is caused by variants (i.e., mutations) in the transthyretin (TTR) gene.5,7,8 TTR protein is produced primarily in the liver and is normally a carrier of vitamin A.9 The variant results in misfolded TTR proteins that accumulate as amyloid deposits in multiple tissues, including the nerves, heart and gastrointestinal (GI) tract.5, 6, 7 It is a multisystem disease that can include sensory and motor, autonomic, and cardiac symptoms. The condition can have a debilitating impact on a patient’s life and may lead to premature death with a median survival of 4.7 years following diagnosis.8,10 It is estimated that there are approximately 50,000 patients with hATTR amyloidosis worldwide.11 • wtATTR amyloidosis is a non-hereditary condition that occurs when misfolded wild-type TTR accumulates as amyloid deposits in multiple organs. It predominantly manifests as cardiac symptoms, but other systems are also involved, and commonly leads to heart failure and mortality within 2.5 to 5.5 years.12,13,14,15,16,17,18,19 wtATTR amyloidosis affects an estimated 200,000-300,000 people worldwide.20 • Alnylam is committed to developing multiple treatment options for people who are living with ATTR amyloidosis to help manage the debilitating and progressive nature of the disease. For more information about vutrisiran, please contact media@alnylam.com. For more information on HELIOS-A (NCT03759379) and HELIOS-B (NCT04153149) please visit http://www.clinicaltrials.gov or contact media@alnylam.com. Current information as of November 2021
CLIP
Alnylam announces extension of review period for new drug vutrisiran to treat ATTR amyloidosis
Alnylam announces 3-month extension of review period for new drug application for vutrisiran to treat ATTR amyloidosis.
Alnylam Pharmaceuticals, Inc., a RNAi therapeutics company, announced that the FDA has extended the review timeline of the New Drug Application (NDA) for vutrisiran, an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis, to allow for the review of newly added information related to the new secondary packaging and labelling facility.
Alnylam recently learned that the original third-party secondary packaging and labelling facility the Company planned to use for the vutrisiran launch was recently inspected and the inspection requires classification for the FDA to take action on the vutrisiran NDA. The inspection observations were not directly related to vutrisiran. In order to minimize delays to approval, Alnylam has identified a new facility to pack and label vutrisiran and submitted an amendment to the NDA for review by the FDA. The updated Prescription Drug User Fee Act (PDUFA) goal date to allow for this review is July 14, 2022. No additional clinical data have been requested by the FDA.
////////////Vutrisiran sodium, APPROVALS 2022, FDA 2022, FDA APPROVED, AMVUTTRA, 2022/6/13, ブトリシランナトリウム , ALN 65492, Votrisiran, siRNA

NEW DRUG APPROVALS
ONE TIME TO SUSTAIN AND MAINTAIN THIS BLOG
$10.00
DIFLUPREDNATE

(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluor-11-hydroxy-3,20-dioxopregna-1,4-dien-17-ylbutyrat[German][ACD/IUPAC Name]
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate[ACD/IUPAC Name]
23674-86-4[RN]
245-815-4[EINECS]
2652
6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
DIFLUPREDNATE
CAS# 23674-86-4
- Molecular FormulaC27H34F2O7
- Average mass508.552 Da
- W 6309
- W-6309
- DFBA
- Difluoroprednisolone butyrate acetate
S8A06QG2QE
TU3831500
дифлупреднат[Russian][INN]
ديفلوبريدنات[Arabic][INN]
二氟泼尼酯[Chinese][INN]
(1R,3aS,3bS,5S,9aS,9bR,10S,11aS)-1-[2-(acetyloxy)acetyl]-5,9b-difluoro-10-hydroxy-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl butanoate
(6a,11b)-21-(Acetyloxy)-6,9-difluoro-11-hydroxy-17-(1-oxobutoxy)pregna-1,4-diene-3,20-dione
(6α,11β)-21-(acetyloxy)-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butanoate
(6α,11β)-21-Acetoxy-6,9-difluoro-11-hydroxy-3,20-dioxopregna-1,4-dien-17-yl butyrate
23674-86-4[RN], 245-815-4[EINECS], 2652, 6a,9a-Difluoroprednisolone-21-acetate-17-butyrate
Difluprednate is a topical corticosteroid used for the symptomatic treatment of inflammation and pain associated with ocular surgery.
Difluprednate is a corticosteroid, It is chemically a butyrate ester of 6(alpha),9(alpha)-difluoro prednisolone acetate. Accordingly, difluprednate is sometimes abbreviated DFBA, for difluoroprednisolone butyrate acetate.
Difluprednate is a topical corticosteroid indicated for the treatment of infammation and pain associated with ocular surgery. It is a butyrate ester of 6(α), 9(α)-difluoro prednisolone acetate. Difluprednate is abbreviated DFBA, or difluoroprednisolone butyrate acetate. It is indicated for treatment of endogenous anterior uveiti.
Approval
On June 24, 2008, the US Food and Drug Administration (FDA) approved difluprednate for the treatment of post-operative ocular inflammation and pain.[1] It is marketed by Alcon under the tradename Durezol.
Depositor-Supplied Patent Identifiers
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020325543-A1 | Diagnostic method | 2017-11-20 | |
| WO-2012088044-A2 | Compositions and methods for improving ocular surface health, corneal clarity, optical function and maintaining visual acuity | 2010-12-20 | |
| US-7790905-B2 | Pharmaceutical propylene glycol solvate compositions | 2002-02-15 | 2010-09-07 |
| US-7927613-B2 | Pharmaceutical co-crystal compositions | 2002-02-15 | 2011-04-19 |
PATENT
WO/2022/118271DIFLUPREDNATE FOR REDUCING THE ADVERSE EFFECTS OF OCULAR INFLAMMATION
SYN 1
Synthetic Reference
Process for preparation of Difluprednate from sterol fermentation product; Ding, Kai; Xu, Feifei; Assignee Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Peop. Rep. China; East China University of Science and Technology; 2014; Patent Information; Aug 06, 2014; CN; 103965277; A

SYN 2
Synthetic Reference
Preparation method of Difluprednate; Tian, Yuan; Zhou, Shengan; Guo, Bin; Xu, Zhiguo; Assignee Guangzhou Renheng Pharmaceutical Technology Co., Ltd., Peop. Rep. China 2017; Patent Information; May 10, 2017; CN; 106632561; A

SYN3
Synthetic Reference
Shailesh, Singh; Bharat, Suthar; Jain, Ashish; Gaikwad, Vinod; Kulkarni, Kuldip. Process for preparing difluprednate. Assignee Ajanta Pharma Ltd., India. IN 2013MU02535. (2015).

SYN4
Synthetic Reference
Sun, Hongbin; Chen, Bo. Method for preparation of Difluprednate. Assignee China Pharmaceutical University, Peop. Rep. China. CN 103509075. (2014).

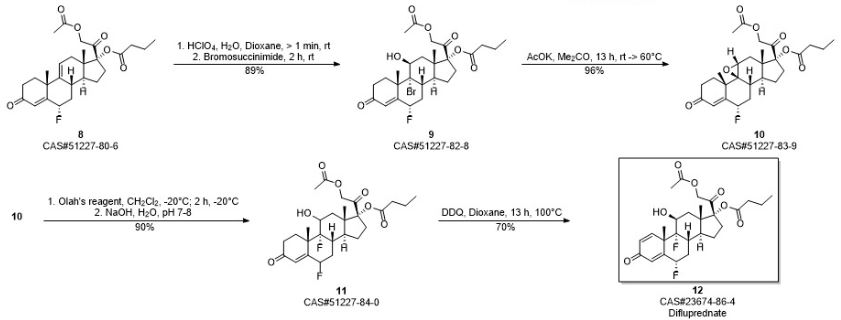
PATENT
https://patents.google.com/patent/CN103509075A/en

Embodiment 1:4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound)
10g hydrocortisone-21 acetic ester (formula II compound) is joined in 250mL eggplant type bottle, add 50mL N, dinethylformamide and 8.8mL pyridine, slowly heat up and make material dissolution complete, slowly cooling afterwards, slowly be added dropwise to 4.4mL methylsulfonyl chloride, add rear solution to be yellow completely.Be warming up to 85 ℃ of stirrings, the reaction solution thick one-tenth that can slowly become sticky is faint yellow, adds slightly some DMFs and makes reaction solution dilution, can normally stir, and keeps this thermotonus one hour, and reaction solution slowly becomes grey black during this period.TLC follows the tracks of (sherwood oil: ethyl acetate=1: 1) show that reaction finishes.Stop heating, treat that the backward reaction solution of slow cooling adds 200mL methyl alcohol, stir 1min, reaction flask is placed in to crystallization under ice-water bath.Suction filtration after 1h, makes water and methanol wash filter cake, crude product productive rate 100%.With methyl alcohol-methylene dichloride mixed solvent system recrystallization, obtain sterling, M.P.231-235 ℃, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.55(1H,s,11-H),5.07(1H,d,J=5Hz,21-H),4.84(1H,d,J=5Hz,21-H),2.15(3H,s,H-21-OAc),1.31(3H,s,19-CH 3),0.65(3H,s,18-CH 3),0.66-2.90(m,17H,backbone).
Embodiment 2:4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound)
By 9.4g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester (formula III compound) and 10g4-Dimethylamino pyridine add in 1000mL eggplant-shape bottle, add again 50mL diethylene glycol dimethyl ether and 260mL methylene dichloride, heated and stirred makes dissolution of solid, slowly adds 32mL butyryl oxide slightly after cooling, is warming up to 80 ℃ of return stirrings.After 23h, TLC follows the tracks of, and raw material primitive reaction is complete, stops heating and stirs.Vacuum concentration is removed methylene dichloride.After being down to room temperature, add frozen water in reaction flask, white solid standing to be separated out.Suction filtration, saturated sodium bicarbonate aqueous solution washing leaching cake, dries under infrared lamp, obtain 4,9 (11)-diene-17 α, 21-dihydroxyl-3,20-ketone-21-acetic ester 17 iophenoxic acid esters (formula IV compound) sterling 10.65g, M.P220-224 ℃, productive rate 95.9%. 1H-NMR(500MHz,CDCl 3):δ(ppm)5.75(1H,s,4-H),5.54(1H,m,11-H),4.87(1H,d,J=4.8Hz,O=C-CH 2-O,21-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.75(2H,m,2-H),0.70(3H,s,18-CH 3),0.95(3H,t,J=4.4Hz),1.34(3H,s,18-CH 3),1.66(2H,m,-CH 2CH 3),2.17(3H,s,O=C-CH 3),2.32(2H,t,J=4.3Hz,O=C-CH 2),? 13C-NMR(75MHz,CDCl 3):δ(ppm)199.1,198.9,173.4,170.4,169.1,144.1,124.1,118.5,94.5,66.9,48.2,46.3,40.9,37.5,36.4,34.2,33.8,32.7,32.2,32.1,30.6,26.2,24.5,20.5,18.3,13.7,13.6;ESI-MS?m/z:457.2[M+H +],479.2[M+Na +];HRMS?for?C 27H 36O 6+Na +?calcd?479.2410,found479.2402.
Embodiment 3:3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters (formula V)
10g4, pregnant steroid-17 α of 9 (11)-diene, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters add in 250mL eggplant type bottle, then add 80mL methylvinyl acetate, slowly drip while stirring the 1mL vitriol oil.Be warming up to 80 ℃ of stirring reactions, solution is thin out yellow clarification slowly.(sherwood oil: ethyl acetate=3: 1), raw material reaction is complete produces new point to TLC after 30min.Stop heating, wait to be cooled to 50 ℃, add 1mL triethylamine, be stirred to and be down to room temperature.Add water in reaction solution, ethyl acetate aqueous layer extracted three times, saturated common salt water washing organic phase twice, anhydrous sodium sulfate drying.After 30min, steam organic solvent and obtain brown color oily matter.Column chromatography is purified and is obtained 3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.74(1H,s,4-H),5.53(1H,s,11-H),5.45(1H,s,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.17(3H,s,19-CH 3),0.96(3H,t,J=7.5Hz),0.70(3H,s,18-CH 3).
Embodiment 4:4, fluoro-17 α of 9 (11)-diene-6-, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters
10g3,5,9 (11) pregnant steroid-3 of triolefin, 17 α, 21 trihydroxy–3,20-diketone-3,21-diacetate esters 17 iophenoxic acid esters are dissolved in 60mL acetonitrile, and under nitrogen protection ,-4 ℃ are stirred half an hour.Slowly drip the acetonitrile suspension 40mL of Selecfluor in reaction flask, under nitrogen protection, react 2 hours, TLC (sherwood oil: ethyl acetate=3: 1) monitoring reaction, raw material reaction is complete.Stopped reaction, adds water in reaction flask, ethyl acetate extraction three times, saturated common salt water washing twice, anhydrous sodium sulfate drying.Vacuum concentration is removed organic solvent, obtain faint yellow solid 4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI), productive rate 85%. 1H-NMR(500MHz,CDC1 3):δ(ppm)5.90(1H,d,J=4.5Hz,4-H),5.59(1H,s,11-H),5.07(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.46(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),0.73(3H,s,19-CH 3).
Embodiment 5:4,9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3,20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII)
14g4, 9 (11)-diene-6 α-fluoro-17 α, 21-dihydroxyl-3, 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VII) and 9 (11)-diene-6 β-fluoro-17 α, 21-dihydroxyl-3, the mixture of 20-diketone-21-acetic ester 17 iophenoxic acid esters (formula VI) adds in dry three-necked bottle, add while stirring 400mL acetum, under room temperature, slowly pass into anhydrous hydrogen chloride gas (98% vitriol oil is added dropwise in 37% concentrated hydrochloric acid solution and makes) until saturated, be stirred to raw material and be dissolved into yellow solution completely, continue to stir 2h, TLC monitoring reacts completely, stop stirring, in reaction solution, add the aqueous solution, after separating out solid, suction filtration, saturated sodium bicarbonate aqueous solution washing, dry, be weighed as 13g, productive rate is 93%. 1H?NMR(300MHz,CDCl 3):δ(ppm)6.10(s,1H),5.61(s,1H),5.41-5.16(m,1H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.82(dd,J=28.3,15.7Hz,3H),2.50(s,2H),2.32(t,J=7.4Hz,2H),2.17(s,3H),1.96(s,5H),1.66(d,J=7.4Hz,2H),1.46(s,2H),1.33(s,3H),0.96(s,3H),0.71(s,3H).
Embodiment 6:6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula VIII)
13g 6 α-fluoro-4; 9; (11)-diene-pregnant steroid-3,20-22 ketone-17-butyric ester-20-acetic ester is dissolved in and fills 300mL1, in the eggplant type bottle of 4 dioxane; add while stirring 40mL 0.46mol/L high chloro acid solution; under room temperature, stir after several minutes, add 14g N-succinimide in reaction system, under nitrogen protection, stir; raw material dissolves gradually, and it is faint yellow that reaction solution is.(the sherwood oil: ethyl acetate=12: 5) monitoring, raw material primitive reaction is complete, adds 10%Na of TLC after 2h 2sO 3unnecessary N-succinimide is fallen in aqueous solution cancellation, and checks (it is blue that test paper no longer becomes) with starch-kalium iodide test paper.Add water in reaction flask, ethyl acetate extraction three times, twice of saturated common salt water washing organic phase, anhydrous sodium sulfate drying organic phase, after 30min, be spin-dried for organic phase, obtain faint yellow oily matter, column chromatography purification (sherwood oil: ethyl acetate=12: 1) obtain white solid 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3, the about 14g of 20-diketone-17-butyric ester-20-acetic ester, productive rate is 89%. 1H-NMR(300MHz,CDCl 3):δ(ppm)5.93(1H,d,J=4.5,4-H),5.06(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),1.84(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.02(3H,s,19-CH 3),4.72(1H,s,11-H);ESI-MS?m/z:593.3,595.3[M+Na +].
Embodiment 7:6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula IX)
14g 6 α-fluoro-9 α-bromo-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in 500mL eggplant type bottle, adds 200mL acetone, stirs raw material is fully dissolved, and adds afterwards 3g Potassium ethanoate, is warming up to 60 ℃ of return stirring 13h.TLC (sherwood oil: ethyl acetate=2: 1) monitoring finds that new product occurs.Stop heating, in reaction solution, add water, ethyl acetate extraction, anhydrous sodium sulfate drying organic phase, after standing 30min, steams except organic solvent, obtains yellow oil, productive rate 96%.Column chromatography is purified, and obtains white solid powder, and nuclear-magnetism confirmation structure is 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester. 1H-NMR(300MHz,CDC1 3):δ(ppm)6.11(1H,d,J=4.5Hz,4-H),5.31(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),0.94(3H,s,18-CH 3),0.97(3H,t,J=7.5Hz),1.55(3H,s,19-CH 3),3.52(1H,s,11-H);ESI-MS?m/z:491.2[M+H +],513.2[M+Na +].
Embodiment 8:6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (formula X)
100mg 6 α-fluoro-9 β, 11 beta epoxides-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester drops in the Plastic Bottle of tetrafluoroethylene, adds 2mL methylene dichloride to dissolve, and stirs at-20 ℃.1mL Olah reagent with under 1mL methylene dichloride low temperature, mix after, be slowly added dropwise in reaction system, maintain low temperature and stir 2 hours, TLC monitoring reaction finishes.Reaction flask shifts out low-temp reaction groove, is slowly added dropwise to the 1mol/L NaOH aqueous solution by excessive HF cancellation, is adjusted to pH7~8.Add chloroform in reaction system, extraction, organic layer is used respectively aqueous hydrochloric acid and the saturated common salt water washing of 3mol/L, anhydrous sodium sulfate drying, after standing 30min, steams except organic solvent, column chromatography is further purified and is obtained white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, productive rate 90%. 1H-NMR(300MHz,CDCl 3):δ(ppm)?6.11(1H,d,J=4.5Hz,4-H),5.27(1H,m,6-H),4.64-4.91(2H,ABq,J=16.6Hz,21-H),2.17(3H,s,-COCH 3),4.40(1H,d,J=4.5Hz,11-H),1.02(3H,s,18-CH 3),0.96(3H,t,J=7.5Hz),1.52(3H,s,19-CH 3);ESI-MS?m/z:533.3[M+Na +]
Embodiment 9:6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester (difluprednate) (formula I)
40mg 6 α, 9 α-fluoro-11 beta-hydroxies-4-alkene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester is dissolved in 3mL dioxane, adds 28mgDDQ, and 100 ℃ of return stirrings heat up.TLC monitoring reaction (sherwood oil: ethyl acetate=12: 8) after 13h, generate the larger product of polarity, steam except organic solvent dioxane, obtain brown color oily matter, add a small amount of methylene dichloride lysate, suction filtration, elimination solid residue, filtrate is washed with sodium bicarbonate aqueous solution after adding a small amount of methylene dichloride again, steams except organic phase rear pillar Chromatographic purification, obtain white solid powder 6 α, 9 α-fluoro-11 beta-hydroxies-Isosorbide-5-Nitrae-diene-pregnant steroid-3,20-diketone-17-butyric ester-20-acetic ester, be title molecule difluprednate, productive rate 70%. 1h-NMR (300MHz, CDCl 3): δ (ppm) 7.20 (1H, d, J=4.5Hz, 1-H), 6.43 (1H, s, 4-H), 6.38 (1H, d, J=6Hz, 2-H), 5.36 (1H, m, 6-H), 4.64-4.91 (2H, ABq, J=16.6Hz, 21-H), 4.43 (1H, d, J=4.5Hz, 11-H), 2.27 (2H, m ,-CH 2-CH 3), 2.17 (3H, s, O=C-CH 3), 1.55 (3H, s, 19-CH 3), 1.02 (3H, s, 18-CH 3), 0.93 (3H, t, J=4.5Hz, 0=C-CH 2cH 2cH 3); ESI-MS m/z:509.3[M+H +]; HRMS for C 27h 35o 7f 2+ H +calcd 509.2351, found 509.2356.M.P.188-190 ℃ (literature value M.P.190-194 ℃); [α] d22=+30.1 ° of (literature values [α] d22=+31.7 °).
Claims (6)
Hide Dependent
1. a method of preparing difluprednate, as following reaction formula:
Specifically comprise the following steps:
(1) by hydrocortisone-21-acetic ester (formula II compound):
Carry out dehydration reaction, generate formula III compound:
(2) formula III compound is carried out to butyric acid esterification, obtains formula IV compound:
(3) formula IV compound is carried out to the reaction of enolization esterifying reagent, obtains formula V compound:
(4) formula V compound is reacted with fluoro reagent and obtains formula VI and formula VII compound:
(5) by formula VI compound, through configuration reversal, reaction obtains formula VII compound;
(6) formula VII compound is reacted with N-bromo-succinimide and water, obtains formula VIII compound:
(7) formula VIII compound epoxidation under alkaline condition is obtained to formula IX compound:
(8) formula IX compound is reacted with fluorination reagent and obtains formula X compound:
(9) dehydrogenation of formula X compound oxidation is obtained to formula I compound (difluprednate).
2. method as claimed in claim 1, is characterized in that, in step (2), formula III compound is obtained to formula IV compound through fourth esterification, and the fourth esterifying reagent adopting is butyryl oxide or butyryl chloride; The alkaline catalysts adopting is pyridine, triethylamine or DMAP; The solvent adopting is methylene dichloride, diethylene glycol dimethyl ether, 1, the mixture of the optional solvents in 2-ethylene dichloride, dioxane, trichloromethane, DMF, methyl-sulphoxide, N,N-dimethylacetamide or above-mentioned solvent.
3. method as claimed in claim 1, is characterized in that, in step (3), formula IV compound is obtained to formula V compound through enolization esterification, and the enolization esterifying reagent adopting is diacetyl oxide, Acetyl Chloride 98Min., methylvinyl acetate or vinyl-acetic ester; The catalyzer adopting is the vitriol oil or tosic acid; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, methylvinyl acetate, vinyl-acetic ester or above-mentioned solvent.
4. method as claimed in claim 1, is characterized in that, in step (4), formula V compound is obtained to formula VI compound and formula VII compound through fluoridizing, and the fluoro reagent adopting is Selectfluor or Accufluor; The solvent adopting is the mixture of the optional solvents in methylene dichloride, chloroform, toluene, acetonitrile or above-mentioned solvent.
5. method as claimed in claim 1, it is characterized in that, in step (8), formula IX compound is obtained to formula X compound through fluoridizing open loop, the fluorination reagent adopting is aqueous hydrogen fluoride solution, hydrogen fluoride pyridine solution (Olah reagent) or hydrogen fluoride triethylamine solution; The solvent adopting is methylene dichloride, chloroform, 1, the mixture of the optional solvents in 2-ethylene dichloride, tetrahydrofuran (THF), toluene or above-mentioned solvent; Range of reaction temperature is-50~50 ℃.
6. a key intermediate compound for synthetic difluprednate, shown in IV compound:

Patent
Publication numberPriority datePublication dateAssigneeTitle
US3780177A *1967-06-161973-12-18Warner Lambert Co17-butyrate,21-ester derivatives of 6alpha,9alpha-difluoroprednisolone,compositions and use
US4525303A *1982-06-211985-06-25Dainippon Ink And Chemicals Inc.Process for preparation of steroids
CN101397321A *2007-09-292009-04-01天津药业研究院有限公司Preparation of hydrocortisone and derivatives thereof
CN102076344A *2008-05-282011-05-25瓦利杜斯生物医药有限公司Non-hormonal steroid modulators of nf-kb for treatment of disease
CN102134266A *2010-12-302011-07-27北京市科益丰生物技术发展有限公司Preparation method of melengestrol acetate
Publication numberPriority datePublication dateAssigneeTitle
CN102964412A *2012-11-272013-03-13山东省医药工业研究所Novel crystal form and preparation method of difluprednate
CN103965277A *2014-05-192014-08-06中国科学院上海有机化学研究所Method for synthesizing difluprednate from sterol fermentation product
CN106632561A *2016-12-162017-05-10广州仁恒医药科技股份有限公司Method for preparing difluprednate
CN106749464A *2016-12-292017-05-31奥锐特药业有限公司Steroidal epoxide carries out open loop, the method for fluorination reaction and its device
CN107915766A *2016-10-112018-04-17江苏福锌雨医药科技有限公司A kind of preparation method of fludrocortison acetate
CN108503679A *2018-04-032018-09-07广州仁恒医药科技股份有限公司A kind of purification process of Difluprednate intermediate
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Clinical trials
Difluprednate ophthalmic emulsion 0.05% is also being studied in other ocular inflammatory diseases, including a phase 3 study evaluating difluprednate for the treatment of anterior uveitis[2][3]

NEW DRUG APPROVALS
TO MAINTAIN THIS BLOG SUBSCRIPTIONS
$10.00
References
- ^ “Sirion Therapeutics Announces FDA Approval of Durezol for Treatment of Postoperative Ocular Inflammation and Pain” (Press release). Sirion Therapeutics, Inc. 2008-06-24. Retrieved 2008-06-30.
- ^ Clinical trial number NCT00501579 for “Study of Difluprednate in the Treatment of Uveitis” at ClinicalTrials.gov
- ^ Sheppard JD, Toyos MM, Kempen JH, Kaur P, Foster CS (May 2014). “Difluprednate 0.05% versus prednisolone acetate 1% for endogenous anterior uveitis: a phase III, multicenter, randomized study”. Investigative Ophthalmology & Visual Science. 55 (5): 2993–3002. doi:10.1167/iovs.13-12660. PMC 4581692. PMID 24677110.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609025 |
| License data | US FDA: Difluprednate |
| Routes of administration | eye drops |
| ATC code | D07AC19 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 23674-86-4 |
| PubChem CID | 32037 |
| DrugBank | DB06781 |
| ChemSpider | 391990 |
| UNII | S8A06QG2QE |
| KEGG | D01266 |
| ChEBI | CHEBI:31485 |
| ChEMBL | ChEMBL1201749 |
| CompTox Dashboard (EPA) | DTXSID0046773 |
| ECHA InfoCard | 100.041.636 |
| Chemical and physical data | |
| Formula | C27H34F2O7 |
| Molar mass | 508.559 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////////DIFLUPREDNATE, W 6309, W-6309, DFBA, Difluoroprednisolone butyrate acetate, S8A06QG2QE, TU3831500, дифлупреднат , ديفلوبريدنات , 二氟泼尼酯 , OCCULAR, PAIN
CCCC(=O)OC1(CCC2C1(CC(C3(C2CC(C4=CC(=O)C=CC43C)F)F)O)C)C(=O)COC(=O)C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
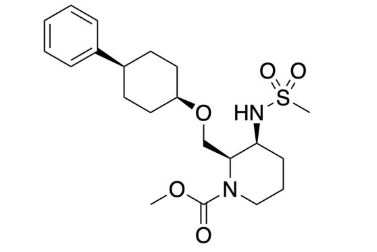{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}