
Saracatinib
NCGC00241099, cas 379231-04-6
893428-71-2 (trihydrate)
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methyl-1-piperazinyl)ethoxy]-5-[(tetrahydro-2H-pyran-4-yl)oxy]-4-quinazolinamine
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydropyran-4-yloxy)quinazolin-4-amine
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline
AZD0530
C27H32ClN5O5
542.03
AstraZeneca Pharmaceuticals LP
Saracatinib (AZD0530) is a highly selective, orally available, dual-specific Src/Abl kinase inhibitor with IC50 of 2.7 and 30 nM for c-Src and Abl kinase, respectively.Saracatinib (AZD0530) demonstrated potent antimigratory and antiinvasive effects in vitro, and inhibited metastasis in a murine model of bladder cancer. Antiproliferative activity of AZD0530 in vitro varied between cell lines (IC50=0.2 ~10 mM).
c-Src, Bcr–Abl, Yes1, Lck.target
AZD0530 is orally available 5-, 7-substituted anilinoquinazoline with anti-invasive and anti-tumor activities. AZD0530 is a dual-specific inhibitor of Src and Abl, protein tyrosine kinases that are overexpressed in chronic myeloid leukemia cells. This agent binds to and inhibits these tyrosine kinases and their effects on cell motility, cell migration, adhesion, invasion, proliferation, differentiation, and survival. Specifically, AZD0530 inhibits Src kinase-mediated osteoclast bone resorption.
AZD-0530 is a highly selective, dual-specific small molecule Src/Abl kinase inhibitor currently in phase II/III clinical trials at AstraZeneca for the treatment of ovarian cancer. Phase II clinical trials are also under way at the company for the treatment of solid tumors and hematological neoplasms. The Mayo Clinic is developing AZD-0530 in phase II clinical studies for the treatment of metastatic pancreas cancer.
Additional phase II trials are under way at the National Cancer Institute (NCI) for the treatment of colorectal cancer, prostate cancer, breast cancer, lung cancer, stomach cancer, soft tissue sarcoma, stage II or IV melanoma and thymic malignancies. A phase II trial for pancreatic cancer has been suspended. Src and Abl kinase are highly expressed in various human tumor types. No recent development has been reported for research into the treatment of head and neck cancer.
Phase II study of Saracatinib (AZD0530) for for the treatment of patients with hormone receptor-negative metastatic breast cancer : Nine patients were treated on study. After a median of 2 cycles (range 1-3), no patient had achieved CR, PR, or SD >6 months. The median time to treatment failure was 82 days (12-109 days).The majority (89%) of patients discontinued saracatinib because of disease progression. One patient acquired potentially treatment-related grade 4 hypoxia with interstitial infiltrates and was removed from the study. Common adverse events included fatigue, elevated liver enzymes, nausea, hyponatremia, dyspnea, cough, and adrenal insufficiency. CONCLUSIONS: These efficacy results were not sufficiently promising to justify continued accrual to this study. Based on this series, saracatinib does not appear to have significant single-agent activity for the treatment of patients with ER(-)/PR(-) MBC. (source: Clin Breast Cancer. 2011 Oct;11(5):306-11.)
Phase II study of Saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: Saracatinib has insufficient activity as a single agent in patients with advanced gastric adenocarcinoma to warrant further investigation. Further development in gastric cancer would require rational drug combinations or identification of a tumor phenotype sensitive to Src inhibition. (source: Invest New Drugs. 2011 Mar 12. [Epub ahead of print]).
Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Nine patients were enrolled. All patients had received prior radiotherapy and six patients had received prior chemotherapy for recurrent or metastatic disease. The most common adverse event was fatigue. Eight patients had progression of disease by response evaluation criteria in solid tumors (RECIST) within the first eight-week cycle and one patient was removed from the study after 11 days due to clinical decline with stable disease according to the RECIST criteria. Median overall survival was six months. The study was closed early due to lack of efficacy according to the early stopping rule. CONCLUSION: Single-agent saracatinib does not merit further study in recurrent or metastatic HNSCC. (source: Anticancer Res. 2011 Jan;31(1):249-53.)

893428-72-3 Saracatinib difumarate
893428-73-4 also
Saracatinib (AZD0530) is a Src inhibitor for c-Src with IC50 of 2.7 nM.
…………………………….

WO 2001094341
http://www.google.com/patents/WO2001094341A9?cl=en

………………….
WO 2006064217
http://www.google.fm/patents/EP1871769A2?cl=en
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline which compound is disclosed as Compound No. 73 within the Table in Example 14 of International Patent Application WO 01/94341. That compound is described herein by way of the Formula I
and as AZD0530, the code number by which the compound is known.
AZD0530 is an inhibitor of the Src family of non-receptor tyrosine kinase enzymes and, thereby, is a selective inhibitor of the motility of tumour cells and a selective inhibitor of the dissemination and invasiveness of mammalian cancer cells leading to inhibition of metastatic tumour growth. In particular, the compound AZD0530 is an inhibitor of c-Src non-receptor tyrosine kinase and should be of value as an anti-invasive agent for use in the containment and/or treatment of solid tumour disease in the human or animal body. The route for preparing the compound of the Formula I that is disclosed in International Patent Application WO 01/94341 involves the reaction of the compound 4-(6-chloro-2,3-methylenedioxyanilino)-7-hydroxy-5-tetrahydropyran-4-yloxyquinazoline with an alkylating agent to form the 2-(4-methylpiperazin-l-yl)ethoxy side-chain at the 7-position. The product of the reaction is disclosed in WO 01/94341 in the form of a dihydrochloride salt and in the form of a free base.
Example 14 4-(6-chloro-2,3-methylenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline (route 4)
Under an atmosphere of nitrogen gas, l-(2-hydroxyethyl)-4-methylpiperazine (13.93 g) was added to a stirred mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-fluoro- 5-tetrahydropyran-4-yloxyquinazoline (12.9 g), sodium te/t-pentoxide (9.87 g) and 1 ,2-diethoxyethane (37.5 ml). Water (1.34 g) and 1,2-diethoxyethane (25 ml) were added and the resultant reaction mixture was stirred and heated to 86°C for 18 hours. The reaction mixture was cooled to 5O0C and, under vacuum distillation at approximately 60 millibar pressure, approximately 50 ml of reaction solvent was distilled off. The reaction mixture was neutralised to pH 7.0 to 7.6 by the addition of a mixture of concentrated aqueous hydrochloric acid (36%, 10 ml) and water (84 ml) at a rate that kept the temperature of the reaction mixture at a maximum of 6O0C. With the temperature of the reaction mixture being kept at 6O0C, the reaction mixture was extracted with ethyl acetate (225 ml). The organic solution was washed with water (50 ml). Water (25 ml) was added and, with the temperature being kept at 6O0C, the mixture was stirred for 10 minutes, then allowed to stand for 30 minutes and the aqueous layer was separated. The organic layer was concentrated to a volume of about 100 ml by distillation of solvent at about 9O0C under atmospheric pressure. The residual mixture was cooled during 1 hour to 450C and held at that temperature for 2 hours to allow crystallisation of product. The mixture was warmed briefly to 550C and then cooled during 4 hours to 180C and held at that temperature for 1 hour. The crystalline precipitate was isolated by filtration and washed in turn with water (17 ml) and with tø’t-butyl methyl ether (17 ml). There was thus obtained 4-(6-chloro-2,3-πiethylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a trihydrate (11 g; 88% purity by HPLC using Method B, retention time 7.3 minutes); NMR Spectrum: (CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,lH), 6.05 (s, 2H), 6.55 (s, IH), 6.75 (d, IH), 6.85 (s, IH), 7.0 (d, IH), 8.55 (s, IH), 9.25 (s, IH).
A portion (10 g) of the material so obtained was placed on a filter and dried at ambient temperature in a stream of dry nitrogen gas. The resultant material was dissolved at 6O0C in dry isopropanol (140 ml) whilst maintaining a dry nitrogen atmosphere. The solution was allowed to cool to ambient temperature and to stand under a dry nitrogen atmosphere for 2 days. The resultant crystalline solid was isolated by filtration under a dry nitrogen atmosphere. The material (8 g) so obtained was a crystalline anhydrous form of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l -yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline, m.p. 142 to 1440C.
Example 15
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (200 ml) and water (10 ml) was heated to 75°C. A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (40 ml) was heated to 😯0C. A portion (80 ml) of the warmed solution of the quinazoline compound was added to the fumaric acid solution whilst the temperature was maintained at 750C. The resultant mixture was stirred at 750C for 75 minutes. The remainder of the quinazoline compound solution was added during 1 hour whilst the temperature was maintained at 750C. Isopropanol (50 ml) was added and the resultant mixture was stirred at 750C for 7 hours. The mixture was cooled slowly over at least 25 minutes to 5O0C and was stirred at that temperature for 6 hours. The mixture was cooled slowly over at least 20 minutes to 2O0C and was stirred at that temperature for 18.5 hours. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro- 2,3-methylenedioxyanilino)-7-[2-(4-methylρiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (37.0 g); m.p. 233-2370C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H)5 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H)3 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)3 6.6 (s, 4H)5 6.83 (d5 IH)3 6.84 (d, IH)5 6.91 (d3 IH)5 7.05 (d, IH)3 8.33 (s, IH)3 9.18 (s, IH).
Example 16
4-(6-chloro-2,3-methyIenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yIoxyquinazolme difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (210 ml) and water (30 ml) was heated to 4O0C and the mixture was filtered. The filter was washed with isopropanol (20 ml) and the washings were added to the warm filtrate. The resultant solution was warmed to 75°C.
A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (20 ml) was heated to 700C and the resultant mixture was filtered. A portion (110 ml) of the fumaric acid solution was added to the warmed solution of 4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin- 1 -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline whilst the temperature was maintained at 75°C. Seed crystals of 4-(6-chloro-
253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (0.02 g) were added and the resultant mixture was stirred at 750C for 1 hour. The remainder of the fumaric acid solution was added during 1 hour whilst the temperature was maintained at 750C and the resultant mixture was stirred at 750C for 14 hours.
The mixture was cooled slowly over at least 2 hours to 200C and was stirred at that temperature for 1 hour. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difumarate salt (35.8 g); m.p. 234-237°C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H)5 2.1-2.17 (m5 2H)5 2.33 (s5 3H)5 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)5 6.6 (s, 4H), 6.83 (d, IH)5 6.84 (d, IH)5 6.91 (d, IH)5 7.05 (d, IH)5 8.33 (s5 IH)5 9.18 (s5 IH).
Example 17 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline sesquifumarate salt
A mixture of 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difurnarate (0.15 g) and water (20 ml) was warmed using a heat gun to obtain a solution. The sample was allowed to evaporate slowly at ambient temperature to a volume of about 3 ml under a flow of air for 24 hours whereupon a precipitate had started to form. The mixture was placed in a refridgerator at 4°C for 2 days. The resultant precipitate was isolated by filtration and washed with water. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a sesquifumarate tetrahydrate salt (0.084 g) which was characterised using XRPD5 DSC5 TGA5 FTIR and solution NMR techniques.
………………..
A simplified process for the manufacture of AZD0530, a potent SRC kinase inhibitor
Org Process Res Dev 2011, 15(3): 688
http://pubs.acs.org/doi/abs/10.1021/op100161y

Process research and development of a synthetic route towards a novel SRC kinase inhibitor is described. The Medicinal Chemistry route was very long and suffered from extensive use of chlorinated solvents and chromatography. A number of steps in the Medicinal Chemistry route were also unattractive for large-scale use for a variety of reasons. The route was modified to produce a shorter synthetic scheme that started from more readily available materials. By using the modified route, the title compound was manufactured on kilogram scale without recourse to chromatography and in significantly fewer steps. The scaled synthesis required two Mitsunobu couplings, which were developed and scaled successfully. An interesting hydrazine impurity was identified in the second Mitsunobu coupling; a mechanism for its formation is proposed, and a method for its control is described. The formation and control of some other interesting impurities are also described.
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
To a slurry of 30 (2.139 kg at 92% w/w, 4.73 mol) and DTAD (2.771 kg, 12.03 mol) in THF (31 L) at ambient temperature in a 100 L vessel was added a solution of triphenylphosphine (3.057 kg, 11.66 mol) in THF (8 L) over 15 min. A THF (2 L) line wash was applied, and the mixture was stirred for 10 min. The reaction mixture was cooled to 15 °C, and a filtered (to remove undissolved particulates) solution of 31 (1.050 L, 1.049 kg, 7.27 mol) in THF …DELETED…………………………….The mixture was filtered and the cake washed with IPA (7 L as a slurry wash and 7 L as a displacement wash) before drying to constant weight under reduced pressure at 50 °C to give AZD0530 difumarate (3.546 kg at 89% w/w, 4.08 mol, 86% yield).
Final Purification of N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
AZD0530 difumarate (4.234 kg at 89% w/w, 4.87 mol) was refluxed in a mixture of IPA (10.L) and water (10.L, Fresenius). A solution was not obtained, so further IPA (450 mL) and water (450 mL, Fresenius) were added, and the mixture was refluxed. The resulting solution was cooled to 68 °C and screened over 3.5 min through a 20 μm in-line filter into a vessel preheated to 65 °C. IPA(20.4 L) at 65 °C was added via the first vessel and in-line filter, and the resulting solution was stirred at 65 °C for 2 h. Crystallisation was evident after 20 min. The mixture was allowed to self-cool to ambient temperature overnight before filtering and washing the cake with a mixture (prescreened through a 20 μm membrane) of water (640 mL) and IPA (5.76 L). The cake was washed with IPA (6.4 L, prescreened) and MTBE (6.4 L, prescreened) and dried to constant weight under reduced pressure at 50 °C to give AZD0530 difumarate (2.865 kg, at 95.2% w/w, 3.52 mol, 72% yield). Spectroscopic analysis was in agreement with the reported data…………Ford, J. G.; McCabe, J. F.; O’Kearney-McMullan, A.; O’Keefe, P.; Pointon, S. M.; Powell, L.; Purdie, M.; Withnall, J. WO/2006/064217, 2006.
……………………….
SEE…………N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor
J Med Chem 2006, 49(22): 6465
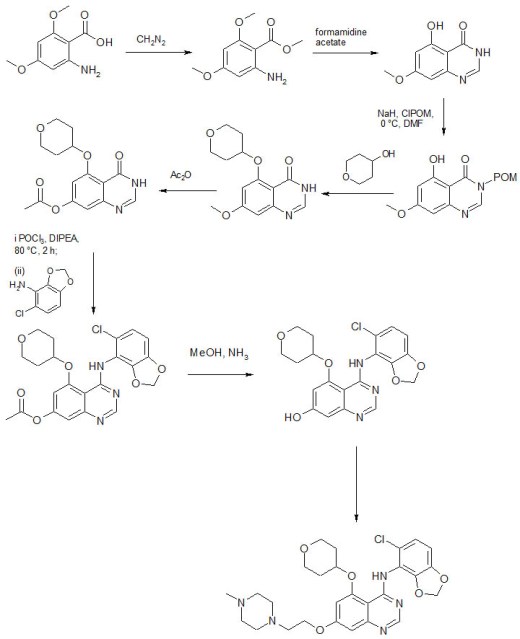
…………..
1: Hannon RA, Finkelman RD, Clack G, Iacona RB, Rimmer M, Gossiel F, Baselga J, Eastell R. Effects of Src kinase inhibition by saracatinib (AZD0530) on bone turnover in advanced malignancy in a Phase I study. Bone. 2012 Jan 8. [Epub ahead of print] PubMed PMID: 22245630.
2: Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina TA. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer. 2011 Oct;11(5):306-11. Epub 2011 May 3. PubMed PMID: 21729667; PubMed Central PMCID: PMC3222913.
3: Mackay HJ, Au HJ, McWhirter E, Alcindor T, Jarvi A, Macalpine K, Wang L, Wright JJ, Oza AM. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Invest New Drugs. 2011 Mar 12. [Epub ahead of print] PubMed PMID: 21400081.
4: Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, Stambuk H, Haque S, Pfister DG. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011 Jan;31(1):249-53. PubMed PMID: 21273606.
5: Renouf DJ, Moore MJ, Hedley D, Gill S, Jonker D, Chen E, Walde D, Goel R, Southwood B, Gauthier I, Walsh W, McIntosh L, Seymour L. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2010 Dec 18. [Epub ahead of print] PubMed PMID: 21170669.
6: Dalton RN, Chetty R, Stuart M, Iacona RB, Swaisland A. Effects of the Src inhibitor saracatinib (AZD0530) on renal function in healthy subjects. Anticancer Res. 2010 Jul;30(7):2935-42. PubMed PMID: 20683035.
7: Arcaroli JJ, Touban BM, Tan AC, Varella-Garcia M, Powell RW, Eckhardt SG, Elvin P, Gao D, Messersmith WA. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010 Aug 15;16(16):4165-77. Epub 2010 Aug 3. PubMed PMID: 20682712.
8: Morrow CJ, Ghattas M, Smith C, Bönisch H, Bryce RA, Hickinson DM, Green TP, Dive C. Src family kinase inhibitor Saracatinib (AZD0530) impairs oxaliplatin uptake in colorectal cancer cells and blocks organic cation transporters. Cancer Res. 2010 Jul 15;70(14):5931-41. Epub 2010 Jun 15. PubMed PMID: 20551056; PubMed Central PMCID: PMC2906706.
9: Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton JA, Finkelman RD, Eastell R. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J Bone Miner Res. 2010 Mar;25(3):463-71. PubMed PMID: 19775203.
10: Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, Warren MV, Walker J, Green TP, Jimeno A, Messersmith WA, Hidalgo M. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009 Jun 15;15(12):4138-46. Epub 2009 Jun 9. PubMed PMID: 19509160.
11: Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, Tan CK, Slingerland JM. Combined Src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clin Cancer Res. 2009 May 15;15(10):3396-405. PubMed PMID: 19451593.
12: Lara PN Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, Posadas E, Stadler W, Gandara DR. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009 Mar;20(3):179-84. PubMed PMID: 19396016; PubMed Central PMCID: PMC3225398.
13: Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, Elvin P, Boyer B, Carragher N, Plé PA, Bermingham A, Holdgate GA, Ward WH, Hennequin LF, Davies BR, Costello GF. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009 Jun;3(3):248-61. Epub 2009 Feb 7. PubMed PMID: 19393585.
14: de Vries TJ, Mullender MG, van Duin MA, Semeins CM, James N, Green TP, Everts V, Klein-Nulend J. The Src inhibitor AZD0530 reversibly inhibits the formation and activity of human osteoclasts. Mol Cancer Res. 2009 Apr;7(4):476-88. PubMed PMID: 19372577.
15: Schweppe RE, Kerege AA, French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009 Jun;94(6):2199-203. Epub 2009 Mar 17. PubMed PMID: 19293266; PubMed Central PMCID: PMC2690419.
16: Purnell PR, Mack PC, Tepper CG, Evans CP, Green TP, Gumerlock PH, Lara PN, Gandara DR, Kung HJ, Gautschi O. The Src inhibitor AZD0530 blocks invasion and may act as a radiosensitizer in lung cancer cells. J Thorac Oncol. 2009 Apr;4(4):448-54. PubMed PMID: 19240653; PubMed Central PMCID: PMC2716757.
17: Gwanmesia PM, Romanski A, Schwarz K, Bacic B, Ruthardt M, Ottmann OG. The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia positive leukaemia cell lines. BMC Cancer. 2009 Feb 13;9:53. PubMed PMID: 19216789; PubMed Central PMCID: PMC2654659.
18: Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008 Oct 23;27(49):6365-75. Epub 2008 Aug 4. PubMed PMID: 18679417.
Src inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo.
Simpkins et al. Clin Cancer Res. 2012 Aug 15. PMID: 22896656.
Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo.
Liu et al. Int J Cancer. 2012 May 24. PMID: 22623106.
Common PIK3CA mutants and a novel 3′ UTR mutation are associated with increased sensitivity to saracatinib.
Arcaroli et al. Clin Cancer Res. 2012 May 1;18(9):2704-14. PMID: 22553375.
Phase I study of saracatinib (AZD0530) in combination with paclitaxel and/or carboplatin in patients with solid tumours.
Kaye et al. Br J Cancer. 2012 May 22;106(11):1728-34. PMID: 22531637.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....































