
Home » 2021 (Page 19)
Yearly Archives: 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fosdenopterin hydrobromide
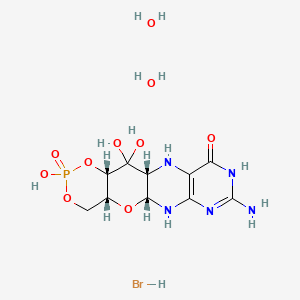

Fosdenopterin hydrobromide
FDA APPR 2021/2/26, NULIBRY
BBP-870/ORGN001
a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.
| ホスデノプテリン臭化水素酸塩水和物; |
| Formula | C10H14N5O8P. 2H2O. HBr |
|---|---|
| CAS | 2301083-34-9DIHYDRATE |
| Mol weight | 480.1631 |
2301083-34-9
(1R,10R,12S,17R)-5-amino-11,11,14-trihydroxy-14-oxo-13,15,18-trioxa-2,4,6,9-tetraza-14λ5-phosphatetracyclo[8.8.0.03,8.012,17]octadeca-3(8),4-dien-7-one;dihydrate;hydrobromide
1,3,2-DIOXAPHOSPHORINO(4′,5′:5,6)PYRANO(3,2-G)PTERIDIN-10(4H)-ONE, 8-AMINO-4A,5A,6,9,11,11A,12,12A-OCTAHYDRO-2,12,12-TRIHYDROXY-, 2-OXIDE, HYDROBROMIDE, HYDRATE (1:1:2), (4AR,5AR,11AR,12AS)-
| CYCLIC PYRANOPTERIN MONOPHOSPHATE MONOHYDROBROMIDE DIHYDRATE |
(4aR,5aR,11aR,12aS)-8-Amino-2,12,12-trihydroxy-4a,5a,6,7,11,11a,12,12aoctahydro-2H-2lambda5-(1,3,2)dioxaphosphinino(4′,5′:5,6)pyrano(3,2-g)pteridine-2,10(4H)-dione, hydrobromide (1:1:2)
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide, hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780



C10H14N5O8P, Average: 363.223
150829-29-1
- ALXN-1101
- WHO 11150
- Synthesis ReferenceClinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA: Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. J Med Chem. 2013 Feb 28;56(4):1730-8. doi: 10.1021/jm301855r. Epub 2013 Feb 19.
Fosdenopterin (or cyclic pyranopterin monophosphate, cPMP), sold under the brand name Nulibry, is a medication used to reduce the risk of death due to a rare genetic disease known as molybdenum cofactor deficiency type A (MoCD-A).[1]
Adverse effects
The most common side effects include complications related to the intravenous line, fever, respiratory infections, vomiting, gastroenteritis, and diarrhea.[1]
Mechanism of action
People with MoCD-A cannot produce cyclic pyranopterin monophosphate (cPMP) in their body.[1] Fosdenopterin is an intravenous medication that replaces the missing cPMP.[1][2] cPMP is a precursor to molybdopterin, which is required for the enzyme activity of sulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
History
Fosdenopterin was developed by José Santamaría-Araujo and Guenter Schwarz at the German universities TU Braunschweig and the University of Cologne.[4][5]
The effectiveness of fosdenopterin for the treatment of MoCD-A was demonstrated in thirteen treated participants compared to eighteen matched, untreated participants.[1][6] The participants treated with fosdenopterin had a survival rate of 84% at three years, compared to 55% for the untreated participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for fosdenopterin priority review, breakthrough therapy, and orphan drug designations along with a rare pediatric disease priority review voucher.[1] The FDA granted the approval of Nulibry to Origin Biosciences, Inc., in February 2021.[1] It is the first medication approved for the treatment of MoCD-A.[1]
References
- ^ Jump up to:a b c d e f g h i j “FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A”. U.S. Food and Drug Administration (FDA) (Press release). 26 February 2021. Retrieved 26 February 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ DrugBank DB16628 . Accessed 2021-03-05.
- ^ Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G (April 2004). “The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor”. The Journal of Biological Chemistry. 279 (16): 15994–9. doi:10.1074/jbc.M311815200. PMID 14761975.
- ^ Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Gröne HJ, et al. (June 2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli”. Human Molecular Genetics. 13 (12): 1249–55. doi:10.1093/hmg/ddh136. PMID 15115759.
- ^ Tedmanson S (5 November 2009). “Doctors risk untried drug to stop baby’s brain dissolving”. TimesOnline.
- ^ Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, et al. (November 2015). “Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study”. Lancet. 386 (10007): 1955–63. doi:10.1016/S0140-6736(15)00124-5. PMID 26343839. S2CID 21954888.
External links
- “Fosdenopterin”. Drug Information Portal. U.S. National Library of Medicine.
Molybdenum cofactor deficiency (MoCD) is an exceptionally rare autosomal recessive disorder resulting in a deficiency of three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde oxidase.1 Signs and symptoms begin shortly after birth and are caused by a build-up of toxic sulfites resulting from a lack of SOX activity.1,5 Patients with MoCD may present with metabolic acidosis, intracranial hemorrhage, feeding difficulties, and significant neurological symptoms such as muscle hyper- and hypotonia, intractable seizures, spastic paraplegia, myoclonus, and opisthotonus. In addition, patients with MoCD are often born with morphologic evidence of the disorder such as microcephaly, cerebral atrophy/hypodensity, dilated ventricles, and ocular abnormalities.1 MoCD is incurable and median survival in untreated patients is approximately 36 months1 – treatment, then, is focused on improving survival and maintaining neurological function.
The most common subtype of MoCD, type A, involves mutations in MOCS1 wherein the first step of molybdenum cofactor synthesis – the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP) – is interrupted.1,3 In the past, management strategies for this disorder involved symptomatic and supportive treatment,5 though efforts were made to develop a suitable exogenous replacement for the missing cPMP. In 2009 a recombinant, E. coli-produced cPMP was granted orphan drug designation by the FDA, becoming the first therapeutic option for patients with MoCD type A.1
Fosdenopterin was approved by the FDA on Februrary 26, 2021, for the reduction of mortality in patients with MoCD type A,5 becoming the first and only therapy approved for the treatment of MoCD. By improving the three-year survival rate from 55% to 84%,7 and considering the lack of alternative therapies available, fosdenopterin appears poised to become a standard of therapy in the management of this debilitating disorder.
Fosdenopterin replaces an intermediate substrate in the synthesis of molybdenum cofactor, a compound necessary for the activation of several molybdenum-dependent enzymes including sulfite oxidase (SOX).1 Given that SOX is responsible for detoxifying sulfur-containing acids and sulfites such as S-sulfocysteine (SSC), urinary levels of SSC can be used as a surrogate marker of efficacy for fosdenopterin.7 Long-term therapy with fosdenopterin has been shown to result in a sustained reduction in urinary SSC normalized to creatinine.7
Animal studies have identified a potential risk of phototoxicity in patients receiving fosdenopterin – these patients should avoid or minimize exposure to sunlight and/or artificial UV light.7 If sun exposure is necessary, use protective clothing, hats, and sunglasses,7 in addition to seeking shade whenever practical. Consider the use of a broad-spectrum sunscreen in patients 6 months of age or older.8
Molybdenum cofactor deficiency (MoCD) is a rare autosomal-recessive disorder in which patients are deficient in three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde dehydrogenase.1 The loss of SOX activity appears to be the main driver of MoCD morbidity and mortality, as the build-up of neurotoxic sulfites typically processed by SOX results in rapid and progressive neurological damage. In MoCD type A, the disorder results from a mutation in the MOCS1 gene leading to deficient production of MOCS1A/B,7 a protein that is responsible for the first step in the synthesis of molybdenum cofactor: the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP).1,4
Fosdenopterin is an exogenous form of cPMP, replacing endogenous production and allowing for the synthesis of molybdenum cofactor to proceed.7
- Mechler K, Mountford WK, Hoffmann GF, Ries M: Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015 Dec;17(12):965-70. doi: 10.1038/gim.2015.12. Epub 2015 Mar 12. [PubMed:25764214]
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, Hennermann JB, Jameson E, Konig K, McGregor TL, Font-Montgomery E, Santamaria-Araujo JA, Santra S, Vaidya M, Vierzig A, Wassmer E, Weis I, Wong FY, Veldman A, Schwarz G: Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015 Nov 14;386(10007):1955-63. doi: 10.1016/S0140-6736(15)00124-5. Epub 2015 Sep 3. [PubMed:26343839]
- Iobbi-Nivol C, Leimkuhler S: Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim Biophys Acta. 2013 Aug-Sep;1827(8-9):1086-101. doi: 10.1016/j.bbabio.2012.11.007. Epub 2012 Nov 29. [PubMed:23201473]
- Mendel RR: The molybdenum cofactor. J Biol Chem. 2013 May 10;288(19):13165-72. doi: 10.1074/jbc.R113.455311. Epub 2013 Mar 28. [PubMed:23539623]
- FDA News Release: FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A [Link]
- OMIM: MOLYBDENUM COFACTOR DEFICIENCY, COMPLEMENTATION GROUP A (# 252150) [Link]
- FDA Approved Drug Products: Nulibry (fosdenopterin) for intravenous injection [Link]
- Health Canada: Sun safety tips for parents [Link]
SYN
Journal of Biological Chemistry (1995), 270(3), 1082-7.
https://linkinghub.elsevier.com/retrieve/pii/S0021925818829696
PATENT
WO 2005073387
PATENT
WO 2012112922
PAPER

Journal of Medicinal Chemistry (2013), 56(4), 1730-1738
https://pubs.acs.org/doi/10.1021/jm301855r

Cyclic pyranopterin monophosphate (1), isolated from bacterial culture, has previously been shown to be effective in restoring normal function of molybdenum enzymes in molybdenum cofactor (MoCo)-deficient mice and human patients. Described here is a synthesis of 1 hydrobromide (1·HBr) employing in the key step a Viscontini reaction between 2,5,6-triamino-3,4-dihydropyrimidin-4-one dihydrochloride and d-galactose phenylhydrazone to give the pyranopterin (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one (10) and establishing all four stereocenters found in 1. Compound 10, characterized spectroscopically and by X-ray crystallography, was transformed through a selectively protected tri-tert-butoxycarbonylamino intermediate into a highly crystalline tetracyclic phosphate ester (15). The latter underwent a Swern oxidation and then deprotection to give 1·HBr. Synthesized 1·HBr had in vitro efficacy comparable to that of 1 of bacterial origin as demonstrated by its enzymatic conversion into mature MoCo and subsequent reconstitution of MoCo-free human sulfite oxidase–molybdenum domain yielding a fully active enzyme. The described synthesis has the potential for scale up.





PAPER
European Journal of Organic Chemistry (2014), 2014(11), 2231-2241.
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/ejoc.201301784
Abstract
The first synthesis of an oxygen‐stable analogue of the natural product cyclic pyranopterin monophosphate (cPMP) is reported. In this approach, the hydropyranone ring is annelated to pyrazine by a sequence comprising ortho‐lithiation/acylation of a 2‐halopyrazine, followed by nucleophilic aromatic substitution. The tetrose substructure is introduced from the chiral pool, from D‐galactose or D‐arabitol.

Abstract
Molybdenum cofactor (Moco) deficiency is a lethal hereditary metabolic disease. A recently developed therapy requires continuous intravenous supplementation of the biosynthetic Moco precursor cyclic pyranopterin monophosphate (cPMP). The limited stability of the latter natural product, mostly due to oxidative degradation, is problematic for oral administration. Therefore, the synthesis of more stable cPMP analogues is of great interest. In this context and for the first time, the synthesis of a cPMP analogue, in which the oxidation‐labile reduced pterin unit is replaced by a pyrazine moiety, was achieved starting from the chiral pool materials D‐galactose or D‐arabitol. Our synthesis, 13 steps in total, includes the following key transformations: i) pyrazine lithiation, followed by acylation; ii) closure of the pyrane ring by nucleophilic aromatic substitution; and iii) introduction of phosphate.
Patent
https://patents.google.com/patent/US9260462B2/en
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclic pyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, untreatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis (see U.S. Pat. No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
Scheme 3.

Scheme 4.

(I).
Scheme 6.

(I).

Scheme 8.

(I).

Scheme 10.

EXAMPLESExample 1Preparation of Precursor Z (cPMP)


Experimental
Air sensitive reactions were performed under argon. Organic solutions were dried over anhydrous MgSO4 and the solvents were evaporated under reduced pressure. Anhydrous and chromatography solvents were obtained commercially (anhydrous grade solvent from Sigma-Aldrich Fine Chemicals) and used without any further purification. Thin layer chromatography (t.l.c.) was performed on glass or aluminum sheets coated with 60 F254 silica gel. Organic compounds were visualized under UV light or with use of a dip of ammonium molybdate (5 wt %) and cerium(IV) sulfate 4H2O (0.2 wt %) in aq. H2SO4 (2M), one of I2 (0.2%) and KI (7%) in H2SO4 (1M), or 0.1% ninhydrin in EtOH. Chromatography (flash column) was performed on silica gel (40-63 μm) or on an automated system with continuous gradient facility. Optical rotations were recorded at a path length of 1 dm and are in units of 10−1 deg cm2 g−1; concentrations are in g/100 mL. 1H NMR spectra were measured in CDCl3, CD3OD (internal Me4Si, δ 0 ppm) or D2O(HOD, δ 4.79 ppm), and 13C NMR spectra in CDCl3 (center line, δ 77.0 ppm), CD3OD (center line, δ 49.0 ppm) or DMSO d6 (center line δ 39.7 ppm), D2O (no internal reference or internal CH3CN, δ 1.47 ppm where stated). Assignments of 1H and 13C resonances were based on 2D (1H—1H DQF-COSY, 1H—13C HSQC, HMBC) and DEPT experiments. 31P NMR were run at 202.3 MHz and are reported without reference. High resolution electrospray mass spectra (ESI-HRMS) were recorded on a Q-TOF Tandem Mass
Spectrometer. Microanalyses were performed by the Campbell Microanalytical Department, University of Otago, Dunedin, New Zealand.
A. Preparation of (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one mono hydrate (1)
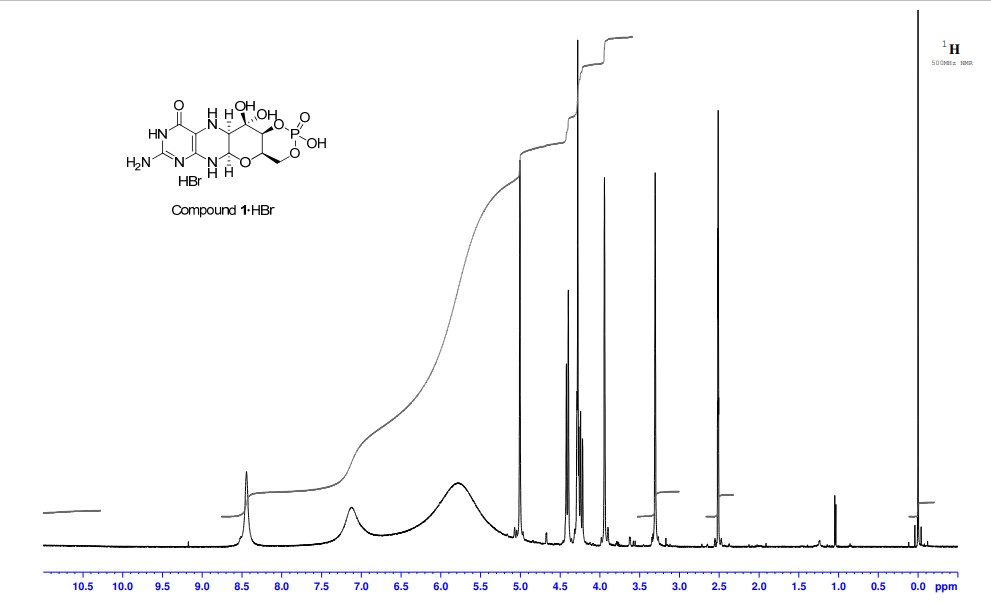
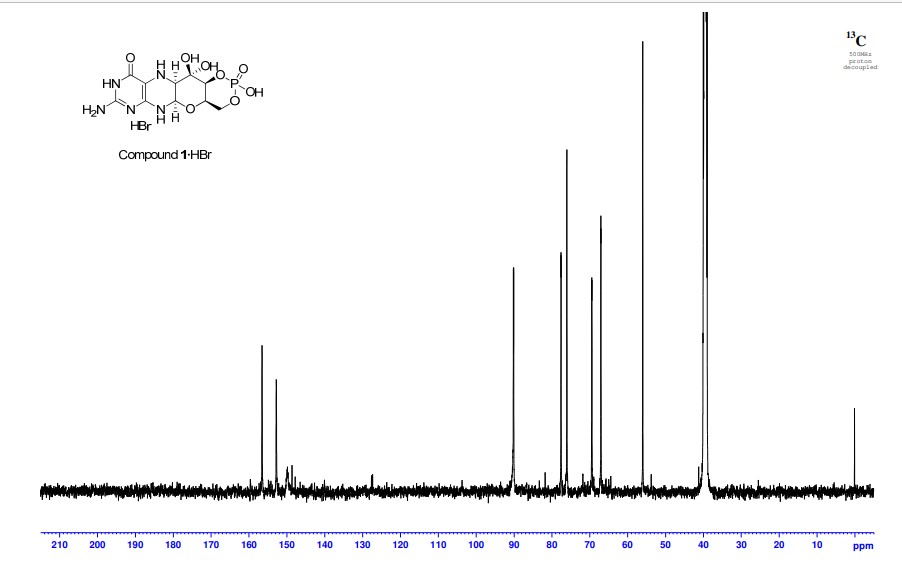
2,5,6-Triamino-3,4-dihydropyrimidin-4-one dihydrochloride (Pfleiderer, W.; Chem. Ber. 1957, 90, 2272; Org. Synth. 1952, 32, 45; Org. Synth. 1963, Coll. Vol. 4, 245, 10.0 g, 46.7 mmol), D-galactose phenylhydrazone (Goswami, S.; Adak, A. K. Tetrahedron Lett. 2005, 46, 221-224, 15.78 g, 58.4 mmol) and 2-mercaptoethanol (1 mL) were stirred and heated to reflux (bath temp 110° C.) in a 1:1 mixture of MeOH—H2O (400 mL) for 2 h. After cooling to ambient temperature, diethyl ether (500 mL) was added, the flask was shaken and the diethyl ether layer decanted off and discarded. The process was repeated with two further portions of diethyl ether (500 mL) and then the remaining volatiles were evaporated. Methanol (40 mL), H2O (40 mL) and triethylamine (39.4 mL, 280 mmol) were successively added and the mixture seeded with a few milligrams of 1. After 5 min a yellow solid was filtered off, washed with a little MeOH and dried to give 1 as a monohydrate (5.05 g, 36%) of suitable purity for further use. An analytical portion was recrystallized from DMSO-EtOH or boiling H2O. MPt 226 dec. [α]D 20 +135.6 (c1.13, DMSO). 1H NMR (DMSO d6): δ 10.19 (bs, exchanged D2O, 1H), 7.29 (d, J=5.0 Hz, slowly exchanged D2O, 1H), 5.90 (s, exchanged D2O, 2H), 5.33 (d, J=5.4 Hz, exchanged D2O, 1H), 4.66 (ddd, J˜5.0, ˜1.3, ˜1.3 Hz, 1H), 4.59 (t, J=5.6 Hz, exchanged D2O, 1H), 4.39 (d, J=10.3 Hz, exchanged D2O, 1H), 3.80 (bt, J˜1.8 Hz, exchanged D2O, 1H), 3.70 (m, 1H), 3.58 (dd, J=10.3, 3.0 Hz, 1H), 3.53 (dt, J=10.7, 6.4 Hz, 1H), 3.43 (ddd, J=11.2, 5.9, 5.9 Hz, 1H), 3.35 (t, J=6.4 Hz, 1H), 3.04 (br m, 1H). 13C NMR (DMSO d6 center line 6 39.7): δ 156.3 (C), 150.4 (C), 148.4 (C), 99.0 (C), 79.4 (CH), 76.5 (CH), 68.9 (CH), 68.6 (CH), 60.6 (CH2), 53.9 (CH). Anal. calcd. for C10H15N5O5H2O 39.60; C, 5.65; H, 23.09; N. found 39.64; C, 5.71; H, 22.83; N.
B. Preparation of Compounds 2 (a or b) and 3 (a, b or c)
Di-tert-butyl dicarbonate (10.33 g, 47.3 mmol) and DMAP (0.321 g, 2.63 mmol) were added to a stirred suspension of 1 (1.5 g, 5.26 mmol) in anhydrous THF (90 mL) at 50° C. under Ar. After 20 h a clear solution resulted. The solvent was evaporated and the residue chromatographed on silica gel (gradient of 0 to 40% EtOAc in hexanes) to give two product fractions. The first product to elute was a yellow foam (1.46 g). The product was observed to be a mixture of two compounds by 1H NMR containing mainly a product with seven Boc groups (2a or 2b). A sample was crystallized from EtOAc-hexanes to give 2a or 2b as a fine crystalline solid. MPt 189-191° C. [α]D 20 −43.6 (c 0.99, MeOH). 1H NMR (500 MHz, CDCl3): δ 5.71 (t, J=1.7 Hz, 1H), 5.15 (dt, J=3.5, ˜1.0, 1H), 4.97 (t, J=3.8, 1H), 4.35 (br t, J=˜1.7, 1H), 4.09-3.97 (m, 3H), 3.91 (m, 1H), 1.55, 1.52, 1.51, 1.50, 1.45 (5s, 45H), 1.40 (s, 18H). 13C NMR (125.7 MHz, CDCl3): δ 152.84 (C), 152.78 (C), 151.5 (C), 150.9 (C), 150.7 (2×C), 150.3 (C), 149.1 (C), 144.8 (C), 144.7 (C), 118.0 (C), 84.6 (C), 83.6 (C), 83.5 (C), 82.7 (3×C), 82.6 (C), 76.3 (CH), 73.0 (CH), 71.4 (CH), 67.2 (CH), 64.0 (CH2), 51.4 (CH), 28.1 (CH3), 27.8 (2×CH3), 27.7 (CH3), 27.6 (3×CH3). MS-ESI+ for C45H72N5O19 +, (M+H)+, Calcd. 986.4817. found 986.4818. Anal. calcd. for C45H71N5O19H2O 54.39; C, 7.39; H, 6.34; N. found 54.66; C, 7.17; H, 7.05; N. A second fraction was obtained as a yellow foam (2.68 g) which by 1H NMR was a product with six Boc groups present (3a, 3b or 3c). A small amount was crystallized from EtOAc-hexanes to give colorless crystals. [α]D 2O −47.6 (c, 1.17, CHCl3). 1H NMR (500 MHz, CDCl3): δ 11.10 (br s, exchanged D2O, 1H), 5.58 (t, J=1.8 Hz, 1H), 5.17 (d, J=3.4 Hz, 1H), 4.97 (t, J=3.9 Hz, 1H), 4.62 (s, exchanged D2O, 1H), 4.16 (dd, J=11.3, 5.9 Hz, 1H), 4.12 (dd, J=11.3, 6.4 Hz, 1H), 3.95 (dt, J=6.1, 1.1 Hz, 1H), 3.76 (m, 1H), 1.51, 1.50, 1.49, 1.48, 1.46 (5s, 54H). 13C NMR (125.7 MHz, CDCl3): δ 156.6 (C), 153.0 (C), 152.9 (C), 151.9 (C), 150.6 (C), 149.4 (2×C), 136.2 (C), 131.8 (C), 116.9 (C), 85.0 (2×C), 83.3 (C), 82.8 (C), 82.49 (C), 82.46 (C), 73.3 (CH), 71.5 (CH), 67.2 (CH), 64.5 (CH2), 51.3 (CH), 28.0, 27.72, 27.68, 27.6 (4×CH3). MS-ESI+ for C40H64N5O17 +, (M+H)+calcd. 886.4287. found 886.4289.
C. Preparation of Compound 4a, 4b or 4c
Step 1—The first fraction from B above containing mainly compounds 2a or 2b (1.46 g, 1.481 mmol) was dissolved in MeOH (29 mL) and sodium methoxide in MeOH (1M, 8.14 mL, 8.14 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
Step 2—The second fraction from B above containing mainly 3a, 3b or 3c (2.68 g, 3.02 mmol) was dissolved in MeOH (54 mL) and sodium methoxide in MeOH (1M, 12.10 mL, 12.10 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
The products from step 1 and step 2 above were combined and chromatographed on silica gel (gradient of 0 to 15% MeOH in CHCl3) to give 4a, 4b or 4c as a cream colored solid (1.97 g). 1H NMR (500 MHz, DMSO d6): δ 12.67 (br s, exchanged D2O, 1H), 5.48 (d, J=5.2 Hz, exchanged D2O, 1H), 5.43 (t, J=˜1.9 Hz, after D2O exchange became a d, J=1.9 Hz, 1H), 5.00 (br s, exchanged D2O, 1H), 4.62 (d, J=5.7 Hz, exchanged D2O, 1H), 4.27 (d, J=6.0 Hz, exchanged D2O, 1H), 3.89 (dt, J=5.2, 3.8 Hz, after D2O became a t, J=3.9 Hz, 1H), 3.62 (dd, J=6.0, 3.7 Hz, after D2O exchange became a d, J=3.7 Hz, 1H), 3.52-3.39 (m, 4H), 1.42 (s, 9H), 1.41 (s, 18H). 13C NMR (125.7 MHz, DMSO d6): δ 157.9 (C), 151.1, (C), 149.8 (2×C), 134.6 (C), 131.4 (C), 118.8 (C), 83.5 (2×C), 81.3 (C), 78.2 (CH), 76.5 (CH), 68.1 (CH), 66.8 (CH), 60.6 (CH2), 54.4 (CH), 27.9 (CH3), 27.6 (2×CH3). MS-ESI+ for C25H40N5O11 +, (M+H)+ calcd. 586.2719. found 586.2717.
D. Preparation of Compound 5a, 5b or 5c
Compound 4a, 4b or 4c (992 mg, 1.69 mmol) was dissolved in anhydrous pyridine and concentrated. The residue was dissolved in anhydrous CH2Cl2 (10 mL) and pyridine (5 mL) under a nitrogen atmosphere and the solution was cooled to −42° C. in an acetonitrile/dry ice bath. Methyl dichlorophosphate (187 μL, 1.86 mmol) was added dropwise and the mixture was stirred for 2 h 20 min. Water (10 mL) was added to the cold solution which was then removed from the cold bath and diluted with ethyl acetate (50 mL) and saturated NaCl solution (30 mL). The organic portion was separated and washed with saturated NaCl solution. The combined aqueous portions were extracted twice further with ethyl acetate and the combined organic portions were dried over MgSO4 and concentrated. Purification by silica gel flash column chromatography (eluting with 2-20% methanol in ethyl acetate) gave the cyclic methyl phosphate 5a, 5b or 5c (731 mg, 65%). 1H NMR (500 MHz, CDCl3,): δ 11.72 (bs, exchanged D2O, 1H), 5.63 (t, J=1.8 Hz, 1H), 5.41 (s, exchanged D2O, 1H), 4.95 (d, J=3.2 Hz, 1H), 4.70 (dt, J=12.4, 1.8 Hz, 1H), 4.42 (dd, J=22.1, 12.1 Hz, 1H). 4.15 (q, J=3.7 Hz, 1H), 3.82 (s, 1H), 3.75 (s, 1H), 3.58 (d, J=11.7 Hz, 3H), 2.10 (bs, exchanged D20, 1H+H2O), 1.50 (s, 9H), 1.46 (s, 18H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0): δ 157.5 (C), 151.2 (C), 149.6 (2×C), 134.5 (C), 132.3 (C), 117.6 (C), 84.7 (2×C), 82.8 (C), 77.3 (CH), 74.8 (d, J=4.1 Hz, CH), 69.7 (CH2), 68.8 (d, J=4.1 Hz, CH), 68.6 (d, J=5.9 Hz, CH), 56.0 (d, J=7.4 Hz, CH3), 51.8 (CH), 28.1 (CH3), 27.8 (CH3). MS-ESI+ for C26H40N5NaO13P+ (M+Na)+, calcd. 684.2252. found 684.2251.
E. Preparation of Compound 6a, 6b or 6c
Compound 5a, 5b or 5c (223 mg, 0.34 mmol) was dissolved in anhydrous CH2Cl2 (7 mL) under a nitrogen atmosphere. Anhydrous DMSO (104 μL, 1.46 mmol) was added and the solution was cooled to −78° C. Trifluoroacetic anhydride (104 μL, 0.74 mmol) was added dropwise and the mixture was stirred for 40 min. N,N-diisopropylethylamine (513 μL, 2.94 mmol) was added and the stirring was continued for 50 min at −78° C. Saturated NaCl solution (20 mL) was added and the mixture removed from the cold bath and diluted with CH2Cl2 (30 mL). Glacial acetic acid (170 μL, 8.75 mmol) was added and the mixture was stirred for 10 min. The layers were separated and the aqueous phase was washed with CH2Cl2 (10 mL). The combined organic phases were washed with 5% aqueous HCl, 3:1 saturated NaCl solution:10% NaHCO3 solution and saturated NaCl solution successively, dried over MgSO4, and concentrated to give compound 6a, 6b or 6c (228 mg, quant.) of suitable purity for further use. 1H NMR (500 MHz, CDCl3): δ 5.86 (m, 1 H), 5.07 (m, 1 H), 4.70-4.64 (m, 2 H), 4.49-4.40 (m, 1 H), 4.27 (m, 1 H), 3.56, m, 4 H), 1.49 (s, 9 H), 1.46 (s, 18 H) ppm. 13C NMR (500 MHz, CDCl3): δ 157.5 (C), 151.1 (C), 150.6 (2 C), 134.6 (C), 132.7 (C), 116.6 (C), 92.0 (C), 84.6 (2 C), 83.6 (C), 78.0 (CH), 76.0 (CH), 70.4 (CH2), 67.9 (CH), 56.2 (CH3) δ6.0 (CH), 28.2 (3CH3), 26.8 (6 CH3) ppm. 31P NMR (500 MHz, CDCl3): δ−6.3 ppm.
F. Preparation of compound 7: (4aR,5aR,11aR,12aS)-1,3,2-Dioxaphosphorino[4′,5′:5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4-a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-2-oxide
Compound 6a, 6b or 6c (10 mg, 14.8 μmol was dissolved in dry acetonitrile (0.2 mL) and cooled to 0° C. Bromotrimethylsilane (19.2 μL, 148 μmol) was added dropwise and the mixture was allowed to warm to ambient temperature and stirred for 5 h during which time a precipitate formed. HCl(aq) (10 μl, 37%) was added and the mixture was stirred for a further 15 min. The mixture was centrifuged for 15 min (3000 g) and the resulting precipitate collected. Acetonitrile (0.5 mL) was added and the mixture was centrifuged for a further 15 min. The acetonitrile wash and centrifugation was repeated a further two times and the resulting solid was dried under high vacuum to give compound 7 (4 mg, 75%). 1H NMR (500 MHz, D2O): δ 5.22 (d, J=1.6 Hz, 1H), 4.34 (dt, J=13, 1.6 Hz, 1H), 4.29-4.27 (m, 1H), 4.24-4.18 (m, 1H), 3.94 (br m, 1H), 3.44 (t, J=1.4 Hz, 1H). 31P NMR (500 MHz, D2O): δ −4.8 MS-ESI+ for C10H15N5O8P+, (M+H)+calcd. 364.0653. found 364.0652.
Example 2Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. Coli in the In vitro Synthesis of Moco
In vitro synthesis of Moco was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. Moco synthesis also involved the use of the purified components E. coli MPT synthase, gephyrin, molybdate, ATP, and apo-sulfite oxidase. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. The assay is based on the conversion of cPMP into MPT, the subsequent molybdate insertion using recombinant gephyrin and ATP, and finally the reconstitution of human apo-sulfite oxidase.
As shown in FIG. 1, Moco synthesis from synthetic cPMP was confirmed, and no differences in Moco conversion were found in comparison to E. coli purified cPMP.
Example 3Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. coli in the In vitro Synthesis of MPT
In vitro synthesis of MPT was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. MPT synthesis also involved the use of in vitro assembled MPT synthase from E. coli. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. Three repetitions of each experiment were performed and are shown in FIGS. 2 and 3.
As shown in FIGS. 2 and 3, MPT synthesis from synthetic cPMP confirmed, and no apparent differences in MPT conversion were found when compared to E. coli purified cPMP. A linear conversion of cPMP into MPT is seen in all samples confirming the identity of synthetic cPMP (see FIG. 2). Slight differences between the repetitions are believed to be due to an inaccurate concentration determination of synthetic cPMP given the presence of interfering chromophores.
Example 4Preparation of Precursor Z (cPMP)
A. Preparation of Starting Materials

B. Introduction of the protected Phosphate

The formation of the cyclic phosphate using intermediate [10] (630 mg) gave the desired product [11] as a 1:1 mixture of diastereoisomers (494 mg, 69%).

C. Oxidation and Overall Deprotection of the Molecule
Oxidation of the secondary alcohol to the gem-diol did prove successful on intermediate [12], but the oxidized product [13] did show significant instability and could not be purified. For this reason, deprotection of the phosphate was attempted before the oxidation. However, the reaction of intermediate [11] with TMSBr led to complete deprotection of the molecule giving intermediate [14]. An attempt to oxidize the alcohol to the gem-diol using Dess-Martin periodinane gave the aromatized pteridine [15].
Oxidation of intermediate [11] with Dess-Martin periodinane gave a mixture of starting material, oxidized product and several by-products. Finally, intermediate [11] was oxidized using the method described Example 1. Upon treatment, only partial oxidation was observed, leaving a 2:1 mixture of [11]/[16]. The crude mixture was submitted to the final deprotection. An off white solid was obtained and analyzed by 1H-NMR and HPLC-MS. These analyses suggest that cPMP has been produced along with the deprotected precursor [11].
Because the analytical HPLC conditions gave a good separation of cPMP from the major impurities, this method will be repeated on a prep-HPLC in order to isolate the final material.
CLIP
BridgeBio Pharma And Affiliate Origin Biosciences Announces FDA Acceptance Of Its New Drug Application For Fosdenopterin For The Treatment Of MoCD Type A
Application accepted under Priority Review designation with Breakthrough Therapy Designation and Rare Pediatric Disease Designation previously grantedThere are currently no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury for infants and children.This is BridgeBio’s first NDA acceptanceSAN FRANCISCO, September 29, 2020 – BridgeBio Pharma, Inc. (Nasdaq: BBIO) and affiliate Origin Biosciences today announced the US Food and Drug Administration (FDA) has accepted its New Drug Application (NDA) for fosdenopterin (previously BBP-870/ORGN001), a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.The NDA has been granted Priority Review designation. Fosdenopterin has previously been granted Breakthrough Therapy Designation and Rare Pediatric Disease Designation in the US and may be eligible for a priority review voucher if approved. It received Orphan Drug Designation in the US and Europe. This is BridgeBio’s first NDA acceptance.“We want to thank the patients, families, scientists, physicians and all others involved who helped us reach this critical milestone,” said BridgeBio CEO and founder Neil Kumar, Ph.D. “MoCD Type A is a devastating disease with a median survival of less than four years and we are eager for our investigational therapy to be available to patients, who currently have no approved treatment options. BridgeBio exists to help as many patients as possible afflicted with genetic diseases, no matter how rare. We are grateful that the FDA has accepted our first NDA for priority review and we look forward to submitting our second NDA later this year for infigratinib for second line treatment of cholangiocarcinoma.”About Fosdenopterin
Fosdenopterin is being developed for the treatment of patients with MoCD Type A. Currently, there are no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury with a median survival between 3 to 4 years. Fosdenopterin is a first-in-class cPMP hydrobromide dihydrate and is designed to treat MoCD Type A by replacing cPMP and permitting the two remaining MoCo synthesis steps to proceed, with activation of MoCo-dependent enzymes and elimination of sulfites.About Molybdenum Cofactor Deficiency (MoCD) Type A
MoCD Type A is an ultra-rare, autosomal recessive, inborn error of metabolism caused by disruption in molybdenum cofactor (MoCo) synthesis which is vital to prevent buildup of s-sulfocysteine, a neurotoxic metabolite of sulfite. Patients are often infants with severe encephalopathy and intractable seizures. Disease progression is rapid with a high infant mortality rate.Those who survive beyond the first few month’s experience profuse developmental delays and suffer the effects of irreversible neurological damage, including brain atrophy with white matter necrosis, dysmorphic facial features, and spastic paraplegia. Clinical presentation that can be similar to hypoxic-ischemic encephalopathy (HIE) or other neonatal seizure disorders may lead to misdiagnosis and underdiagnosis. Immediate testing for elevated sulfite levels and S-sulfocysteine in the urine and very low serum uric acid may help with suspicion of MoCD.About Origin Biosciences
Origin Biosciences, an affiliate of BridgeBio Pharma, is a biotechnology company focused on developing and commercializing a treatment for Molybdenum Cofactor Deficiency (MoCD) Type A. Origin is led by a team of veteran biotechnology executives. Together with patients and physicians, the company aims to bring a safe, effective treatment for MoCD Type A to market as quickly as possible. For more information on Origin Biosciences, please visit the company’s website at www.origintx.com.
About BridgeBio Pharma
BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized genetic diseases at their source. BridgeBio was founded in 2015 to identify and advance transformative medicines to treat patients who suffer from Mendelian diseases, which are diseases that arise from defects in a single gene, and cancers with clear genetic drivers. BridgeBio’s pipeline of over 20 development programs includes product candidates ranging from early discovery to late-stage development. For more information visit bridgebio.com.
| Clinical data | |
|---|---|
| Trade names | Nulibry |
| Other names | Precursor Z, ALXN1101 |
| License data | US DailyMed: Fosdenopterin |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 150829-29-1 |
| PubChem CID | 135894389 |
| DrugBank | DB16628 |
| ChemSpider | 17221217 |
| UNII | 4X7K2681Y7 |
| KEGG | D11779 |
| ChEMBL | ChEMBL2338675 |
| CompTox Dashboard (EPA) | DTXSID90934067 |
| Chemical and physical data | |
| Formula | C10H14N5O8P |
| Molar mass | 363.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESNC1=NC(=O)C2=C(N[C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]4C(O)(O)[C@@H]3N2)N1 | |
| hideInChIInChI=1S/C10H14N5O8P/c11-9-14-6-3(7(16)15-9)12-4-8(13-6)22-2-1-21-24(19,20)23-5(2)10(4,17)18/h2,4-5,8,12,17-18H,1H2,(H,19,20)(H4,11,13,14,15,16)/t2-,4-,5+,8-/m1/s1Key:CZAKJJUNKNPTTO-AJFJRRQVSA-N |
//////////Fosdenopterin hydrobromide, ホスデノプテリン臭化水素酸塩水和物 , ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780, BBP-870/ORGN001, Priority Review designation, Breakthrough Therapy Designation, Rare Pediatric Disease Designation, Orphan Drug Designation, molybdenum cofactor deficiency, ALXN-1101, WHO 11150, FDA 2021, APPROVALS 2021
#Fosdenopterin hydrobromide, #ホスデノプテリン臭化水素酸塩水和物 , #ALXN1101 HBr, #UNII-X41B5W735T, X41B5W735T, #D11780, #BBP-870/ORGN001, #Priority Review designation, #Breakthrough Therapy Designation, #Rare Pediatric Disease Designation, #Orphan Drug Designation, #molybdenum cofactor deficiency, #ALXN-1101, #WHO 11150, #FDA 2021, #APPROVALS 2021
C1C2C(C(C3C(O2)NC4=C(N3)C(=O)NC(=N4)N)(O)O)OP(=O)(O1)O.O.O.Br
Melphalan flufenamide hydrochloride
 .HCl
.HCl
Melphalan flufenamide hydrochloride
メルファランフルフェナミド塩酸塩;
L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester, hydrochloride
| Formula | C24H30Cl2FN3O3. HCl |
|---|---|
| CAS | 380449-54-7 |
| Mol weight | 534.8786 |
FDA APPROVED PEPAXTO, 2021/2/26
| Efficacy | Antineoplastic, Alkylating agent |
|---|---|
| Disease | Multiple myeloma |
- Ethyl (2S)-2-[(2S)-2-amino-3-{4-[bis(2-chloroethyl)amino]phenyl}propanamido]-3-(4-fluorophenyl)propanoate
- J 1
- J 1 (prodrug)
- L-Melphalanyl-L-p-fluorophenylalanine ethyl ester
- Melflufen
- Melphalan flufenamide
- Pepaxto
- Prodrug J 1
Melflufen
- Molecular FormulaC24H30Cl2FN3O3
- Average mass498.418 Da
- SP ROT +33.0 ° Conc: 1.3 g/100mL; chloroform ; 589.3 nm, Oncology Research 2003, V14(3), P113-132
мелфалана флуфенамид [Russian] [INN]ميلفالان فلوفيناميد [Arabic] [INN]氟美法仑 [Chinese] [INN]380449-51-4[RN]
9493Ethyl 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-L-phenylalaninate
F70C5K4786L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester
Melphalan flufenamide, sold under the brand name Pepaxto, is an anticancer medication used to treat multiple myeloma.[3][4]
The most common adverse reactions include fatigue, nausea, diarrhea, pyrexia and respiratory tract infection.[3]
Melphalan flufenamide is a peptidase enhanced cytotoxic (PEnC) that exerts a targeted delivery of melphalan in cells with high expression of aminopeptidases, such as aminopeptidase N, which has been described as over-expressed in human malignancies.Aminopeptidase N plays a functional role in malignant angiogenesis.
Melphalan flufenamide was approved for medical use in the United States in February 2021.[4][5]
Medical uses
Melphalan flufenamide is indicated in combination with dexamethasone for the treatment of adults with relapsed or refractory multiple myeloma, with relapsed or refractory multiple myeloma who have received at least four prior lines of therapy and whose disease is refractory to at least one proteasome inhibitor, one immunomodulatory agent, and one CD-38 directed monoclonal antibody.[3][4]
Metabolism
Melphalan flufenamide is metabolized by aminopeptidase hydrolysis and by spontaneous hydrolysis on N-mustard.[6] Its biological half-life is 10 minutes in vitro.
Origin and development
Melphalan flufenamide is a peptidase enhanced cytotoxic (PEnC) with a targeted delivery within tumor cells of melphalan, a widely used classical chemotherapeutic belonging to a group of alkylating agents developed more than 50 years ago. Substantial clinical experience has been accumulated about melphalan since then. Numerous derivatives of melphalan, designed to increase the activity or selectivity, have been developed and investigated in vitro or in animal models.[7] Melphalan flufenamide was synthesized, partly due to previous experience of an alkylating peptide cocktail named Peptichemio[8] and its anti-tumor activity is being investigated.
Pharmacology
Compared to melphalan, melphalan flufenamide exhibits significantly higher in vitro and in vivo activity in several models of human cancer.[9][10][11][12][13][14][15][16] A preclinical study, performed at Dana–Farber Cancer Institute, demonstrated that melphalan flufenamide induced apoptosis in multiple myeloma cell lines, even those resistant to conventional treatment (including melphalan).[17] In vivo effects in xenografted animals were also observed, and the results confirmed by M Chesi and co-workers – in a unique genetically engineered mouse model of multiple myeloma – are believed to be predictive of clinical efficacy.[18]
Structure
Chemically, the drug is best described as the ethyl ester of a dipeptide consisting of melphalan and the amino acid derivative para-fluoro-L-phenylalanine.
Pharmacokinetics
Pharmacokinetic analysis of plasma samples showed a rapid formation of melphalan; concentrations generally exceeded those of melphalan flufenamide during ongoing infusion. Melphalan flufenamide rapidly disappeared from plasma after infusion, while melphalan typically peaked a few minutes after the end of infusion. This suggests that melphalan flufenamide is rapidly and widely distributed to extravasal tissues, in which melphalan is formed and thereafter redistributed to plasma.[19]
This rapid disappearance from plasma is likely due to hydrolytic enzymes.[20] The Zn(2+) dependent ectopeptidase (also known as alanine aminopeptidase), degrades proteins and peptides with a N-terminal neutral amino acid. Aminopeptidase N is frequently overexpressed in tumors and has been associated with the growth of different human cancers suggesting it as a suitable target for anti-cancerous therapy.[21]
Adverse effects
In a human Phase 1 trial, no dose-limiting toxicities (DLTs) were observed at lower doses. At doses above 50 mg, reversible neutropenias and thrombocytopenias were observed, and particularly evident in heavily pretreated patients.[22] These side-effects are shared by most chemotherapies, including alkylating agents in general.
Drug interactions
No drug interaction studies have been reported. Several in vitro studies indicate that melphalan flufenamide may be successfully combined with standard chemotherapy or targeted agents.[23][24]
Therapeutic efficacy
In a Phase 1/2 trial, in solid tumor patients refractory to standard therapy, response evaluation showed disease stabilization in a majority of patients.[25][26] In relapsed and refractory multiple-myeloma (RRMM) patients, promising activity was seen in heavily pre-treated RRMM patients where conventional therapies had failed; the median Progression-Free Survival was 9.4 months and the Duration of Response was 9.6 months.[27] An overall response rate of 41% and a clinical benefit rate of 56% were also shown, with similar results seen across patient populations regardless of their refractory status. Hematologic toxicity was common, but manageable with cycle prolongations, dose modifications and supportive therapy, and non-hematologic treatment-related adverse events were infrequent.
History
Efficacy was evaluated in HORIZON (NCT02963493), a multicenter, single-arm trial.[3] Eligible patients were required to have relapsed refractory multiple myeloma.[3] Patients received melphalan flufenamide 40 mg intravenously on day 1 and dexamethasone 40 mg orally (20 mg for patients ≥75 years of age) on day 1, 8, 15 and 22 of each 28-day cycle until disease progression or unacceptable toxicity.[3] Efficacy was evaluated in a subpopulation of 97 patients who received four or more prior lines of therapy and were refractory to at least one proteasome inhibitor, one immunomodulatory agent, and a CD38-directed antibody.[3]
The application for melphalan flufenamide was granted priority review and orphan drug designations.[3]
Society and culture
Names
Melphalan flufenamide is the International nonproprietary name (INN).[28]
PAPER
Organic Process Research & Development (2019), 23(6), 1191-1196.
https://pubs.acs.org/doi/pdf/10.1021/bk-2020-1369.ch005
Ethyl (2S)-2-[(2S)-2-amino-3-[bis-(2-chloroethyl)amino]phenyl]propaneamido]-3-(4-fluorophenyl)propanoate hydrochloride, (melphalan flufenamide or Melflufen), is an alkylating agent intended for the treatment of multiple myeloma. Initially only milligram quantities were synthesized, following a route starting from pharmaceutical-grade melphalan. Along with the pharmaceutical development, adjustments were made to the original medicinal chemistry route. This resulted in material for early clinical trials, but it became obvious that further development was necessary. Development resulted in a route in which two phenyl alanine derivatives were coupled to give a dipeptide. This intermediate was further manipulated to give an aniline which could be converted into the desired compound melflufen. The aniline derivative was converted to the corresponding N,N–bis-chloroethylaniline using chloroacetic acid and borane. Deprotection and conversion to the hydrochloride gave melflufen in good yield and excellent purity. Production was performed without chromatography at multi-kilogram scale to supply the API for Phase III studies and commercial validation batches.
PAPER
Antineoplastics
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Melphalan
Melphalan, l-3-[p-[bis-(2-chloroethyl)amino]phenyl]alanine (30.2.1.13), is a structural analog of chlorambucil in which the butyric acid fragment is replaced with an aminoacid fragment, alanine. This drug is synthesized from l-phenylalanine, the nitration of which with nitric acid gives 4-nitro-l-phenylalanine (30.2.1.8). Reacting this with an ethanol in the presence of hydrogen chloride gives the hydrochloride of 4-nitro-l-phenylalanine ethyl ester (30.2.1.9), the amino group of which is protected by changing it to phthalamide by a reaction with succinic anhydride to give 30.2.1.10. The nitro group in this molecule is reduced to an amino group using palladium on calcium carbonate as a catalyst. The resulting aromatic amine (30.2.1.11) is then reacted with ethylene oxide, which forms a bis-(2-hydroxyethyl)-amino derivative (30.2.1.12). The hydroxy groups in this molecule are replaced with chlorine atoms upon reaction with thionyl chloride, after which treatment with hydrochloric acid removes the phthalamide protection, giving melphalan (30.2.13) [47–50].

Melaphalan is used intravenously and orally to treat multiple myeloma and cancers of the breast, neck, and ovaries. A synonym of this drug is alkeran.
The racemic form of this drug, d,l-3-[p-[bis-(2-chloroethyl)amino]phenyl]alanine, is also widely used under the name sarcolysine or racemelfalan.
PATENT WO 2001096367PAPEROncology Research (2003), 14(3), 113-132PATENTWO 2016180740https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016180740
Alkylating agents, such as drugs derived from nitrogen mustard, that is bis(2-chloroethyl)amine derivatives, are used as chemotherapeutic drugs in the treatment of a wide variety of cancers. Melphalan, or p-bis-(2-chloroethyl)-amino-L-phenylalanine (compound (Id), CAS No. 148-82-3), is an alkylating agent which is a conjugate of nitrogen mustard and the amino acid phenylalanine (US 3,032,584). Melphalan is used clinically in the treatment of metastatic melanomas, but has limited efficacy, dose-limiting toxicities and resistance can develop.
Melphalan flufenamide ethyl ester (L-melphalanyl-L-p-fluorophenylalanine ethyl ester, melflufen, compound (Ib)) is a derivative of melphalan conjugated to the amino acid phenylalanine, creating a dipeptide (WO 01/96367):
The monohydrochloride salt of melflufen (L-melphalanyl-L-p-fluorophenylalanine ethyl ester monohydrochloride; hydrochloride salt of (Ib); CAS No. 380449-54-7) is referred to as melflufen hydrochloride.
When studied in cultures of human tumor cells representing approximately 20 different diagnoses of human cancers, including myeloma, melflufen showed 50- to 100-fold higher potency compared with that of melphalan (http://www.oncopeptides.se/products/melflufen/ accessed 26 March 2015). Data disclosed in Arghya, et al, abstract 2086 “A Novel Alkylating Agent Melphalan Flufenamide Ethyl Ester Induces an Irreversible DNA Damage in Multiple Myeloma Cells” (2014) 5th ASH Annual Meeting and Exposition, suggest that melflufen triggers a rapid, robust and irreversible DNA damage, which may account for its ability to overcome melphalan-resistance in multiple myeloma cells. Melflufen is currently undergoing phase I/IIa clinical trials in multiple myeloma.
A process for preparing melflufen in hydrochloride salt form is described in WO 01/96367, and is illustrated in Scheme 1, below. In that process N-tert-butoxycarbonyl-L-melphalan is reacted with p-fluorophenylalanine ethyl ester to give N-tert-butoxycarbonyl-L-melphalanyl-L-p-fluorophenylalanine ethyl ester. After purification by gradient column chromatography the yield of that step is 43%.
Scheme 1. Current route to melflufen (in hydrochloride salt form)
As shown in Scheme 1, the known process for preparing melflufen (in hydrochloride salt form) uses the cytotoxic agent melphalan as a starting material, and melflufen is synthesised in a multistep sequence. Melphalan is highly toxic, thus the staring materials and all of the intermediates, and also the waste stream generated, are extremely toxic. That is a major disadvantage in terms of safety, environmental impact and cost when using the process on a large scale. Therefore, an improved and safer method is highly desired, especially for production of melflufen on a large scale. Further, the purity of commercially available melphalan is poor due to its poor stability, the yield in each step of the process is poor, and purity of the final product made by the known process is not high.
A process for preparing melphalan is described in WO 2014/141294. In WO 2014/141294 the step to introduce the bis(2-chloroethyl) group into the molecule comprises conversion of a primary phenyl amine to a tertiary phenyl amine diol, by reaction with ethylene oxide gas. This gives a 52.6% yield. The amine diol is then converted to a bis(2-chloroethyl) phenylamine by reaction with phosphoryl chloride. Using ethylene oxide, or chloroethanol, to convert an aromatic amine to the corresponding bis-(2-hydroxy ethyl) amine, followed by
chlorination of that intermediate, is a common technique for producing aromatic bis-(2-chloroethyl) amines. It is also known to start from a chloroarene and let it undergo a SNAr-reaction with diethanolamine. The present inventors have applied those methods to produce melflufen (in its salt form), shown in Scheme 2 below.
Scheme 2. Alternative pathways to melflufen
The inventors have found that using ethylene oxide in THF (route (a) of Scheme 2), no alkylation occurs at 55 °C; increasing the temperature to 60 °C lead to the dialkylated intermediate being formed, but the reaction was very slow. To increase yield and reaction rate the reaction would require high temperatures, but this would cause increased pressure so that the reaction would need be performed in a pressure reactor. Such conditions are likely lead to formation of side products. Similar reaction conditions but using a 50:50 mixture of ethylene oxide and acetic acid (route (b) of Scheme 2) lead to faster reaction times but formation of side products. Using potassium carbonate and chloroethanol (route (c) of Scheme 2) also lead to formation of side product, possibly due to the chloroethanol undergoing partial trans-esterification with the ethyl ester.
The inventors also attempted chlorination of the di-alkylated compound. Chlorination of the bis-(2-hydroxyethyl) compound (4) of Scheme 2 using thionyl chloride in dichloromethane led to significant de-protected side product formation. Chlorination of the bis-(2-hydroxyethyl) compound (4) of Scheme 2 using POCl3 required high temperature and long
reaction times. In addition, both thionyl chloride and POCl3 are challenging to handle at large scale due to safety concerns. The inventors also converted the bis-(2-hydroxyethyl) compound (4) of Scheme 2 to the corresponding dimesylate by treatment with methanesulfonyl chloride and triethylamine. The dimesylate was treated then with sodium chloride in DMF at 120 °C. However, the crude product of this reaction contained significant side products making this route unsuitable to be used economically at scale.
In summary, none of these routes were found to be suitable for large scale production of high purity melflufen. They do not work well for the synthesis of melflufen, resulting in poor yields and are inefficient. Further, the routes shown in Scheme 2 require multiple steps to form the N, N-bis-chloroethyl amine and use toxic reagents.
Example 1 – Synthesis of compound (VIc)
To a reactor with overhead stirring, equipped with nitrogen inlet and reflux condenser, was charged Boc-nitrophenylalanine (compound (IVc)) (35.0 g, 112.8 mmol, 1 eq.), followed by acetone (420 mL), N-methylmorpholine (43.4 mL, 394.8 mmol, 3.5 eq.), fluoro-L-phenylalanine ethyl ester hydrochloride (compound (V)) (28.5 g, 115 mmol, 1.02 eq.), EDC (23.8 g, 124.1 mmol, 1.1 eq.) and HOBt·H2O (1.7 g, 11.3 mmol, 0.1 eq.). The slurry was stirred at room temperature for 18.5 h which led to full consumption of compound (IVc) according to HPLC. Water (180 mL) and 2-MeTHF (965 mL) were charged. Approximately 640 g solvent was then removed by evaporation (TJ: 35 °C) from the clear two phase orange mixture. 360 mL 2-MeTHF was then added and evaporated off twice. The water phase was acidified to pH 3 via addition of 58 mL 2 M sulfuric acid. The organic layer was heated to 35-40 °C and was then sequentially washed with water (90 mL), twice with saturated aqueous NaHCO3 solution (90 mL) and then brine (90 mL) and finally water (90 mL). To the 2-MeTHF dissolved product was added heptane (270 mL) drop wise at 35-40 °C before the mixture was allowed to reach room temperature overnight with stirring. Another 135 mL heptane was added drop wise before the beige slurry was cooled to 10 °C. The product was isolated and was rinsed with 100 mL cold 2-MeTHF/heptane 6/4. Product compound (VIc) was stored moist (82.5 g). A small sample of the product was analyzed by limit of detection (LOD) which revealed the solid to contain 43.8% solvent residues. Based on this, the purified product was obtained in a yield of 82 %. The purity was determined by HPLC to be: 99.4 area%.
1 H-NMR (300 MHz, DMSO-D6) δ 8.48 (broad d, 1H, J=7.5 Hz), 8.16 (2H, d, J=8.7 Hz), 7.55 (2H, d, J=9 Hz), 7.28 (2H, dd, J=8,7, 8.1 Hz), 7.12-7.02 (3H, m), 4.49 (1H, dd, J=14.4, 7.2 Hz), 4.32-4.24 (1 H, m), 4.04 (2H, dd, J=14.4, 7.2 Hz), 3.08-2.95 (3H,m), 2.84 (1H, dd, J=13.2, 10.8 Hz), 1.27 (s, 9H), 1.11 (3H, t, J=7.2Hz)
13C-NMR (75 MHz, DMSO-D6) δ 171.4 (C=O), 171.2 (C=O), 161.2 (C-F, d, J=242.3 Hz), 155.2 (C=O), 146.6 (C), 146.2 (C), 133.1 (C), 131.1 (2 carbon, CH, d, J=8.3 Hz), 130.6 (2 carbon, CH), 123.1 (C), 114.9 (2 carbon, CH, J=20.4 Hz), 78.1 (C), 60.6 (CH2), 55.1 (CH), 53.6 (CH), 37.3 (CH2), 35.9 (CH2), 28.0 (3 carbons, CH3), 14.0 (CH3)
Example 2 – Synthesis of compound (IIc)
To a hydrogenation autoclave was added wet solid product compound (VIc) (approximately 4.9 g dry weight, 9.7 mmol, 1 eq.), 2-MeTHF (75 mL) and 3 w/w% of a 5% Pd/C-catalyst (147 mg, 50% moist). The reaction mixture was degased with nitrogen and then 1 barg hydrogen gas was charged. Stirring was set to 600 rpm and TJ to 36 °C. The reaction was completed in four hours, The hydrogenation autoclave was rinsed with 10 mL 2-MeTHF and the rinsing portion was added to the reaction solution in the E-flask. Charcoal (250 mg, 5 wt%) was then added and the resulting mixture was stirred for 15 minutes at room temperature before it was filtered. The filter was rinsed with 10 mL 2-MeTHF and the rinsing portion was added to the filter. The light yellow/pink filtrate contained white precipitated product. The slurry was heated to approximately 40 °C to dissolve the solid before heptane (42 mL) was added drop wise during one hour. The heating was turned off and the mixture was allowed to reach room temperature with overnight stirring. Additional 21 mL heptane was the added before the mixture was cooled to approximately 7 °C (ice/water bath). The solid was isolated and was washed through with 10 mL cold 2-MeTHF/heptane 6/4. The moist solid (5.7 g) was vacuum dried at 35 °C overnight which gave a dry weight of
compound (IIc) of 4.2 g which corresponds to a yield of 91 %. The purity was determined by HPLC to be 99.1 area%.
1H-NMR (300 MHz, DMSO-D6) δ 8.26 (1H, d, J=7.5Hz), 7.26 (dd, 2H, J=8.1, 5.7 Hz), 7.09 (2H, t, J=8.7 Hz), 6.86 (2H, d, J=8.1 Hz), 6.71 (1H, d, J=8.7 Hz), 6.45 (1H, d, J=8.1 Hz), 4.87 (2H, s), 4.45 (1H, dd, J=14.4, 7.5 Hz), 4.07-4.00 (3H, m), 3.06-2.91 (2H, m), 2.71 (1H, dd, J=13.8, 3.9 Hz), 2.54-2.46 (1H, m), 1.31 (s, 9H), 1.11 (3H, t, J=6.9 Hz).
13C-NMR (75 MHz, DMSO-D6) δ 171.4 (C=O), 171.2 (C=O), 161.2 (C-F, d, J=242.3 Hz), 155.1 (C=O), 146.9 (C), 133.2 (C, d, J=3.0 Hz), 131.1 (2 carbon, CH, d, J=8.3 Hz), 129.5 (2 carbon, CH), 124.8 (C), 114.8 (2 carbon, CH, J=21.1 Hz), 113.6 (2 carbon, CH), 77.9 (C), 60.5 (CH2), 56.0 (CH), 53.5 (CH), 36.7 (CH2), 35.9 (CH2), 28.1 (3 carbons, CH3), 13.9 (CH3)
The present inventors have repeated Example 2 several times using crude compound (VIc) or recrystallised compound (VIc) (purity: 99.1 area%) as starting material and varying various reaction conditions, e.g. pressure of H2, w/w% of Pd/C, solvent and temperature. The crude purity (97.2 area%) was a slightly higher when recrystallized compound (VIc) was used as starting material than when using crude compound (VIc), in which case the crude purity is generally 95-96 area%. Final yield and purity is also slightly higher than when starting from crude compound (VIc) (98-98.5 area%).
The present inventors have also repeated Example 2 several times varying the Pd/C w/w%, temperature, pressure of H2 and concentration using 2-MeTHF as the solvent. A high conversion of Compound (VIc) (>99.5 area%) was achieved for Pd/C w/w% from 3 to 6 bar; temperature ranges from 30 to 40 °C, H2 pressure from 1 to 6 barg, and for varying reaction concentrations. The resulting crude purity was similar in all attempts (95.3-96.2 area%), as was the purity of the isolated product after crystallization from 2-MeTHF/heptane (98.0-98.5 area%).
Example 3 – Preparation of compound (IIIc)
(i) carried out using BH3SMe2 in the presence of chloroacetic acid salt
In a 0.5 L dried reactor with overhead stirrer, compound (IIc) (6.99 g, 14.76 mmol) was added, followed by anhydrous tetrahydrofuran (46 mL), chloroacetic acid (36.3 g, 383.8 mmol), chloroacetic acid sodium salt (17.2 g, 147.6 mmol) at TI=5-13°C. A solution of
BH3SMe2 (14.6 g, 191.9 mmol, 18.2 mL) was then added over 45 minutes. After the addition, the reaction temperature was adjusted to TI=25-30°C and kept for 2 hr after reaching this temperature. The reaction was slowly quenched with ethanol (17.7 g, 383.8 mmol, 22.4 mL) and was stirred overnight at TJ=5°C and then slowly diluted with distilled water (138 mL) to precipitate the product, compound (IIIc). The temperature was adjusted to TI=15°C and the stirring rate was increased before addition of a solution of aqueous K2CO3 (8.0 M, 27 mL) to pH = 7.0-7.5. The reaction slurry was collected on a filter and reaction vessel and filter-cake were washed with water (2×40 mL). The filter-cake was re-slurred in water (200 mL) for 1 hr at TJ=20°C and then filtered again. Washing with water (50 mL), followed by drying at TJ=35°C under high vacuum, produced the crude white product, compound (IIIc), in 7.85 g (88.8%) uncorrected yield. HPLC purity 97.5 area %.
Crude compound (IIIc) (7.5 gram) prepared according to the described procedure was charged to a reactor and washed down with 2-MeTHF (80 mL). Heating at TJ=50°C dissolved the substance. Heptane (80 mL) was added with stirring at TI=45-50°C and then stirred before adjusting the temperature to TJ=10°C. The precipitated solid was collected by filtration and dried at TJ=35°C under high vacuum which produced white product, compound (IIIc), in 6.86 g (91.5%). HPLC purity 99.1 area %.
1H-NMR (300 MHz, DMSO-D6) δ 8.30 (1H, d, J=7.8 Hz), 7.26 (2H, dd, J=8.1, 6 Hz), 7.09-7.05 (3H, m), 6.79 (1H, d, J=8.9 Hz), 6.63 (2H, d, J=8.4 Hz), 4.49-4.42 (1H, dd, J=14.7, 7.5 Hz), 4.07-3.99 (3H, m), 3.68 (8H, s), 3.06-2.91 (2H, m), 2.76 (1H, dd, J=13.8, 4.2 Hz), 2.56 (1H, m), 1.29 (9H, s), 1.1 (3H, t, J=6.6 Hz)
13C-NMR (75 MHz, DMSO-D6) δ 172.1 (C=O), 171.3 (C=O), 161.2 (C-F, d, J=242.3 Hz), 155.2 (C=O), 144.7 (C), 133.2 (C, d, J=3.0 Hz), 131.1 (2 carbon, CH, d, J=7.5 Hz), 130.2 (2 carbon, CH), 126.1 (C), 114.9 (2 carbon, CH, J=21.1 Hz), 111.6 (2 carbon, CH), 78.0 (C), 60.6 (CH2), 55.9 (CH), 53.5 (CH), 52.2 (CH2), 41.2 (CH2), 36.4 (CH2), 35.9 (CH2), 28.1 (3 carbons, CH3), 14.0 (CH3)
(ii) Carried out using BH3SMe2 in the presence of chloroacetic acid salt
In a 0.5 L dried reactor with overhead stirrer, compound (IIe) (7.5 g, 15.84 mmol) was added, followed by 2-MeTHF (150 mL). The mixture was heated to 45 °C to form a clear solution. The solution was cooled to 4 °C and chloroacetic acid (38.9 g, 411.8 mmol), followed by chloroacetic acid sodium salt (18.4 g, 158.4 mmol) was added at TI=5-13°C. A solution of BH3SMe2 (15.6 g, 205.9 mmol, 19.5 mL) was then added over 90 minutes. After the addition, the reaction temperature was adjusted to TI=20-25°C and kept for 5 hr after reaching this temperature. The reaction was slowly quenched with water at TI=15-25 °C (150 g, 8333 mmol, 150 mL), pH=3.5 in water phase, and left overnight without stirring at TI=6 °C.
Product, compound (IIIc), had precipitated out in the organic phase and the temperature was adjusted to TI=35 °C while stirring, and two clear phases formed. The phases were allowed to separate and the water phase was removed. The organic phase was washed three times with 20% NaCl(aq). pH in the three water phases were: 1.7, 1.1, and 1.1. After the removal of the third water phase, the organic phase was transferred to a round bottom flask and concentrated to half its volume on an evaporator. Product, compound (IIIc), started to precipitate out and the product slurry was allowed to mature at 6 °C for 19 hr. The slurry was collected on a filter and round bottom flask and filter-cake were washed with 2-MeTHF:n-heptane (2×40 mL), followed by drying at TJ=35 °C under high vacuum, to produce the crude white product, compound (IIIc), in 8.3 g (87.6%) uncorrected yield. HPLC purity 99.4 area % .
(iii) Carried out using borane-tetrahydrofuran in the presence of chloroacetic acid salt
In a 100 mL dried round bottom flask with magnet stirrer bar, compound (IIc) (0.75 g, 1.58 mmol) was added under a slow nitrogen flow followed by anhydrous tetrahydrofuran (6 mL), chloroacetic acid (3.89 g, 41.2 mmol), and chloroacetic acid sodium salt (1.84 g, 15.8 mmol). At TI=5-13°C °C a 1 M solution of BH3THF (20.6 mmol, 20.6 mL) was added over 30
minutes. After the addition the reaction temperature was adjusted between TI=23-28 °C and kept for 2 hr after reaching this temperature. In process control sample (HPLC) indicated in-complete reaction and the jacket temperature was set to TJ=40°C and when the internal temperature reached TI=40°C the reaction was kept at this temperature for 2 hr when in-process sample (HPLC) showed 6.7 area% starting material, 7.1% acylation adduct
(impurity) and 84.1% compound (IIIc). The reaction was progressed at TI=23°C and left for 4 days before slowly quenched with ethanol (2.4 g, 3 mL). Water (100 mL) was added and the pH adjusted with 1 M aqueous K2CO3 to pH 7. The reaction slurry was collected on a filter and reaction vessel and filter-cake were washed with water (2×20 mL) followed by drying at TJ=35°C under high vacuum produced the crude colorless product in 0.85 g (89.6%) uncorrected yield. HPLC purity was 94.3 area %, with one major impurity attributed to a chloroacylation adduct of the starting material in 3.8 area %.
(iv) Carried out using BH3SMe2 without addition of chloroacetic acid salt
In a 100 mL dried round bottom flask with magnet stirrer bar, compound (IIc) (0.75 gram, 1.58 mmol) was added under a slow nitrogen flow followed by anhydrous tetrahydrofuran (6 mL) and chloroacetic acid (3.89 g, 41.2 mmol). At TI=5-16°C a solution of BH3SMe2 (1.56 g, 20.6 mmol, 2.0 mL) was added over 30. After the addition the reaction temperature was adjusted between TI=25°C and kept for 2.5 h after reaching this temperature. A process control sample (HPLC) indicated melflufen (Compound (Ib)), the Boc-deprotected form of Compound (IIIc), in 66 area %. The reaction was slowly quenched with ethanol (2.9 g, 3.7 mL). The pH of the reaction was adjusted with 1 M aqueous K2CO3 solution to pH=8, followed by addition of EtOAc (40 mL). Layers were separated and the aqueous layer re-extracted with EtOAc (50 mL). The organic layers were combined and reduced at <30 mbar / 35°C to an oil. The oil was re-distilled from EtOAc (30 mL) twice and the residue was dried at TJ=23°C / 5 mbar to leave 1.6 g brownish oil. HPLC purity of Compound (Ib) was 66.1 area %.
Example 4 – Preparation of compound (Ib) as hydrochloride salt
Boc-melflufen (compound (IIIc)) (5.0 g, 8.3 mmol) was charged to a round bottomed flask, equipped with magnet stirrer bar, and nitrogen inlet. 1.3 M HCl (anhydrous) in ethanol (64 mL, 83.5 mmol, 10 eq.) was added. After 19 h the conversion was 99.4%. The solvents were partially distilled at TJ=33°C on a rotary evaporator, followed by the addition of ethanol (18 mL). This was repeated twice. Seed crystals were added and after 30 minutes product had precipitated. The slurry was stirred for 21 h and was then concentrated. Methyl tert-butyl ether (MTBE) (108 mL) was added at room temperature with an even rate over 30 minutes. After 100 minutes of stirring at room temperature the precipitate was collected by vacuum filtration and washed with 2×25 mL ethanol: MTBE (1:6). Drying was performed overnight at TJ=35°C / 5 mbar in vacuum oven. Yield of compound (Ib) in the form of its hydrochloride salt, 4.0 g (90%). HPLC-purity 98.7 area%.
1H-NMR (300 MHz, MeOH-D4) δ 7.26 (2H, dd, J=8.4, 8.1 Hz), 7.17 (2H, d, J=8.4 Hz), 7.02 (2H, dd, J=9, 8.4 Hz), 6.74 (2H, d, J=8.4 Hz), 4.69 (1H, dd, J=7.8, 6.3 Hz), 4.15 (2H, dd, J=14.1, 7.2 Hz), 4.04 (1H, dd, J=8.4, 5.4 Hz), 3.76 (4H, dd, J=6.3, 6 Hz), 3.67 (4H, dd, 6.6, 5.7 Hz), 3.17 (2H, dd, J=14.4, 6 Hz), 3.06-2.88 (2H, m), 1.22 (3H, t, J=7.2 Hz)
13C-NMR (75 MHz, MeOH-D4) δ 172.2 (C=O), 169.8 (C=O), 163.4 (C-F, d, J=244.5 Hz), 147.4 (C), 133.9 (C, d, J=3 Hz), 132.1 (2 carbon, CH, d, J=7.5 Hz), 131.8 (2 carbon, CH), 123.4 (C), 116.2 (2 carbon, CH, d, J=21.9 Hz), 113.7 (2 carbon, CH), 62.6 (CH2), 55.6 (CH), 55.5 (CH), 54.3 (CH2), 41.6 (CH2), 37.6 (CH2), 37.6 (CH2), 14.5 (CH3)
Example 4 was repeated successfully in the presence ethyl acetate and with varying concentrations of HCl from 1.3 M to 2.5 M and at varying temperatures from 6 °C to room temperature.PAPERhttps://pubs.acs.org/doi/10.1021/acs.oprd.9b00116 Organic Process Research & Development (2019), 23(6), 1191-1196.Melflufen is a novel cytostatic currently in phase III clinical trials for treatment of multiple myeloma. Development of a process suitable for production is described. The two key features of the novel method are late introduction of the alkylating pharmacophore and an improved method for formation of the bis-chloroethyl group.


1H NMR spectrum of L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester, hydrochloride (1) (in D4–MeOH).

13C NMR spectrum of L-Phenylalanine, 4-[bis(2-chloroethyl)amino]-L-phenylalanyl-4-fluoro-, ethyl ester, hydrochloride (1) (in D4–MeOH).

References
- ^ Berglund, Åke; Ullén, Anders; Lisyanskaya, Alla; Orlov, Sergey; Hagberg, Hans; Tholander, Bengt; Lewensohn, Rolf; Nygren, Peter; Spira, Jack; Harmenberg, Johan; Jerling, Markus; Alvfors, Carina; Ringbom, Magnus; Nordström, Eva; Söderlind, Karin; Gullbo, Joachim (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Strese, Sara; Wickström, Malin; Fuchs, Peder Fredlund; Fryknäs, Mårten; Gerwins, Pär; Dale, Tim; Larsson, Rolf; Gullbo, Joachim (2013). “The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo”. Biochemical Pharmacology. 86(7): 888–95. doi:10.1016/j.bcp.2013.07.026. PMID 23933387.
- ^ Jump up to:a b c d e f g h i “FDA grants accelerated approval to melphalan flufenamide for relapsed”. U.S. Food and Drug Administration(FDA). 26 February 2021. Retrieved 1 March 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “FDA Approves Oncopeptides’ Pepaxto (melphalan flufenamide) for Patients with Triple-Class Refractory Multiple Myeloma” (Press release). Oncopeptides AB. 1 March 2021. Retrieved 1 March 2021 – via PR Newswire.
- ^ “Pepaxto: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 1 March 2021.
- ^ Gullbo, J; Tullberg, M; Våbenø, J; Ehrsson, H; Lewensohn, R; Nygren, P; Larsson, R; Luthman, K (2003). “Structure-activity relationship for alkylating dipeptide nitrogen mustard derivatives”. Oncology Research. 14 (3): 113–32. doi:10.3727/000000003771013071. PMID 14760861.
- ^ Wickstrom, M.; Lovborg, H.; Gullbo, J. (2006). “Future Prospects for Old Chemotherapeutic Drugs in the Target-Specific Era; Pharmaceutics, Combinations, Co-Drugs and Prodrugs with Melphalan as an Example”. Letters in Drug Design & Discovery. 3(10): 695. doi:10.2174/157018006778631893.
- ^ Gullbo, J; Dhar, S; Luthman, K; Ehrsson, H; Lewensohn, R; Nygren, P; Larsson, R (2003). “Antitumor activity of the alkylating oligopeptides J1 (L-melphalanyl-p-L-fluorophenylalanine ethyl ester) and P2 (L-prolyl-m-L-sarcolysyl-p-L-fluorophenylalanine ethyl ester): Comparison with melphalan”. Anti-Cancer Drugs. 14 (8): 617–24. doi:10.1097/00001813-200309000-00006. PMID 14501383. S2CID 10282399.
- ^ Berglund, Åke; Ullén, Anders; Lisyanskaya, Alla; Orlov, Sergey; Hagberg, Hans; Tholander, Bengt; Lewensohn, Rolf; Nygren, Peter; Spira, Jack; Harmenberg, Johan; Jerling, Markus; Alvfors, Carina; Ringbom, Magnus; Nordström, Eva; Söderlind, Karin; Gullbo, Joachim (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Strese, Sara; Wickström, Malin; Fuchs, Peder Fredlund; Fryknäs, Mårten; Gerwins, Pär; Dale, Tim; Larsson, Rolf; Gullbo, Joachim (2013). “The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo”. Biochemical Pharmacology. 86(7): 888–95. doi:10.1016/j.bcp.2013.07.026. PMID 23933387.
- ^ Wickström, M; Johnsen, J. I.; Ponthan, F; Segerström, L; Sveinbjörnsson, B; Lindskog, M; Lövborg, H; Viktorsson, K; Lewensohn, R; Kogner, P; Larsson, R; Gullbo, J (2007). “The novel melphalan prodrug J1 inhibits neuroblastoma growth in vitro and in vivo”. Molecular Cancer Therapeutics. 6 (9): 2409–17. doi:10.1158/1535-7163.MCT-07-0156. PMID 17876040.
- ^ Gullbo, J; Lindhagen, E; Bashir-Hassan, S; Tullberg, M; Ehrsson, H; Lewensohn, R; Nygren, P; de la Torre, M; Luthman, K; Larsson, R (2004). “Antitumor efficacy and acute toxicity of the novel dipeptide melphalanyl-p-L-fluorophenylalanine ethyl ester (J1) in vivo”. Investigational New Drugs. 22 (4): 411–20. doi:10.1023/B:DRUG.0000036683.10945.bb. PMID 15292711. S2CID 31613292.
- ^ Gullbo, J; Wickström, M; Tullberg, M; Ehrsson, H; Lewensohn, R; Nygren, P; Luthman, K; Larsson, R (2003). “Activity of hydrolytic enzymes in tumour cells is a determinant for anti-tumour efficacy of the melphalan containing prodrug J1”. Journal of Drug Targeting. 11(6): 355–63. doi:10.1080/10611860310001647140. PMID 14668056. S2CID 25203458.
- ^ Gullbo, J; Dhar, S; Luthman, K; Ehrsson, H; Lewensohn, R; Nygren, P; Larsson, R (2003). “Antitumor activity of the alkylating oligopeptides J1 (L-melphalanyl-p-L-fluorophenylalanine ethyl ester) and P2 (L-prolyl-m-L-sarcolysyl-p-L-fluorophenylalanine ethyl ester): Comparison with melphalan”. Anti-Cancer Drugs. 14 (8): 617–24. doi:10.1097/00001813-200309000-00006. PMID 14501383. S2CID 10282399.
- ^ Chauhan, D.; Ray, A.; Viktorsson, K.; Spira, J.; Paba-Prada, C.; Munshi, N.; Richardson, P.; Lewensohn, R.; Anderson, K. C. (2013). “In Vitro and in Vivo Antitumor Activity of a Novel Alkylating Agent, Melphalan-Flufenamide, against Multiple Myeloma Cells”. Clinical Cancer Research. 19 (11): 3019–31. doi:10.1158/1078-0432.CCR-12-3752. PMC 4098702. PMID 23584492.
- ^ Viktorsson, K; Shah, C. H.; Juntti, T; Hååg, P; Zielinska-Chomej, K; Sierakowiak, A; Holmsten, K; Tu, J; Spira, J; Kanter, L; Lewensohn, R; Ullén, A (2016). “Melphalan-flufenamide is cytotoxic and potentiates treatment with chemotherapy and the Src inhibitor dasatinib in urothelial carcinoma”. Molecular Oncology. 10 (5): 719–34. doi:10.1016/j.molonc.2015.12.013. PMC 5423156. PMID 26827254.
- ^ Chauhan, D; Ray, A; Viktorsson, K; Spira, J; Paba-Prada, C; Munshi, N; Richardson, P; Lewensohn, R; Anderson, K. C. (2013). “In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells”. Clinical Cancer Research. 19 (11): 3019–31. doi:10.1158/1078-0432.CCR-12-3752. PMC 4098702. PMID 23584492.
- ^ Chesi, M; Matthews, G. M.; Garbitt, V. M.; Palmer, S. E.; Shortt, J; Lefebure, M; Stewart, A. K.; Johnstone, R. W.; Bergsagel, P. L. (2012). “Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy”. Blood. 120 (2): 376–85. doi:10.1182/blood-2012-02-412783. PMC 3398763. PMID 22451422.
- ^ Berglund, Åke; Ullén, A; Lisyanskaya, A; Orlov, S; Hagberg, H; Tholander, B; Lewensohn, R; Nygren, P; Spira, J; Harmenberg, J; Jerling, M; Alvfors, C; Ringbom, M; Nordström, E; Söderlind, K; Gullbo, J (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Wickström, M; Viktorsson, K; Lundholm, L; Aesoy, R; Nygren, H; Sooman, L; Fryknäs, M; Vogel, L. K.; Lewensohn, R; Larsson, R; Gullbo, J (2010). “The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan”. Biochemical Pharmacology. 79 (9): 1281–90. doi:10.1016/j.bcp.2009.12.022. PMID 20067771.
- ^ Wickström, M; Larsson, R; Nygren, P; Gullbo, J (2011). “Aminopeptidase N (CD13) as a target for cancer chemotherapy”. Cancer Science. 102 (3): 501–8. doi:10.1111/j.1349-7006.2010.01826.x. PMC 7188354. PMID 21205077.
- ^ Berglund, Åke; Ullén, A; Lisyanskaya, A; Orlov, S; Hagberg, H; Tholander, B; Lewensohn, R; Nygren, P; Spira, J; Harmenberg, J; Jerling, M; Alvfors, C; Ringbom, M; Nordström, E; Söderlind, K; Gullbo, J (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Wickström, M; Haglund, C; Lindman, H; Nygren, P; Larsson, R; Gullbo, J (2008). “The novel alkylating prodrug J1: Diagnosis directed activity profile ex vivo and combination analyses in vitro”. Investigational New Drugs. 26 (3): 195–204. doi:10.1007/s10637-007-9092-1. PMID 17922077. S2CID 19915448.
- ^ Chauhan, D; Ray, A; Viktorsson, K; Spira, J; Paba-Prada, C; Munshi, N; Richardson, P; Lewensohn, R; Anderson, K. C. (2013). “In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells”. Clinical Cancer Research. 19 (11): 3019–31. doi:10.1158/1078-0432.CCR-12-3752. PMC 4098702. PMID 23584492.
- ^ Berglund, Åke; Ullén, A; Lisyanskaya, A; Orlov, S; Hagberg, H; Tholander, B; Lewensohn, R; Nygren, P; Spira, J; Harmenberg, J; Jerling, M; Alvfors, C; Ringbom, M; Nordström, E; Söderlind, K; Gullbo, J (2015). “First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies”. Investigational New Drugs. 33 (6): 1232–41. doi:10.1007/s10637-015-0299-2. PMID 26553306. S2CID 8207569.
- ^ Viktorsson, K; Shah, C. H.; Juntti, T; Hååg, P; Zielinska-Chomej, K; Sierakowiak, A; Holmsten, K; Tu, J; Spira, J; Kanter, L; Lewensohn, R; Ullén, A (2016). “Melphalan-flufenamide is cytotoxic and potentiates treatment with chemotherapy and the Src inhibitor dasatinib in urothelial carcinoma”. Molecular Oncology. 10 (5): 719–34. doi:10.1016/j.molonc.2015.12.013. PMC 5423156. PMID 26827254.
- ^ https://ash.confex.com/ash/2015/webprogram/Paper85666.html
- ^ World Health Organization (2012). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 67”. WHO Drug Information. 26 (1): 72. hdl:10665/109416.
External links
- “Melphalan flufenamide”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02963493 for “A Study of Melphalan Flufenamide (Melflufen) in Combination With Dexamethasone in Relapsed Refractory Multiple Myeloma Patients (HORIZON)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pepaxto |
| Other names | Melflufen, 4-[Bis-(2-chloroethyl)amino]-L-phenylalanine-4-fluoro-L-phenylalanine ethyl ester, J1[1][2] |
| License data | US DailyMed: Melphalan_flufenamide |
| Legal status | |
| Legal status | US: ℞-only [3] |
| Pharmacokinetic data | |
| Metabolism | Aminopeptidase hydrolysis, Spontaneous hydrolyisis on N-mustard |
| Elimination half-life | 10 min in vitro[medical citation needed] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 380449-51-4 |
| PubChem CID | 9935639 |
| DrugBank | DB16627 |
| ChemSpider | 8111267 |
| UNII | F70C5K4786 |
| ChEMBL | ChEMBL4303060 |
| Chemical and physical data | |
| Formula | C24H30Cl2FN3O3 |
| Molar mass | 498.42 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCCOC(=O)[C@H](CC1=CC=C(C=C1)F)NC(=O)[C@H](CC2=CC=C(C=C2)N(CCCl)CCCl)N | |
| hideInChIInChI=1S/C24H30Cl2FN3O3/c1-2-33-24(32)22(16-18-3-7-19(27)8-4-18)29-23(31)21(28)15-17-5-9-20(10-6-17)30(13-11-25)14-12-26/h3-10,21-22H,2,11-16,28H2,1H3,(H,29,31)/t21-,22-/m0/s1Key:YQZNKYXGZSVEHI-VXKWHMMOSA-N |
//////////Melphalan flufenamide hydrochloride, Melphalan flufenamide, FDA 2021, APPROVALS 2021, PEPAXTO, メルファランフルフェナミド塩酸塩 , J 1
#Melphalan flufenamide hydrochloride, #Melphalan flufenamide, #FDA 2021, #APPROVALS 2021, #PEPAXTO, メルファランフルフェナミド塩酸塩 , #J 1
AZD1222 (ChAdOx1), Oxford–AstraZeneca COVID-19 vaccine, COVISHIELD


AZD1222 (ChAdOx1)
| Identifiers | |
|---|---|
| CAS Number | 2420395-83-9 |
ChAdOx1 nCoV- 19 Corona Virus Vaccine (Recombinant) COVISHIELD™
- DNA (recombinant simian adenovirus Ox1 ΔE1E3 vector human cytomegalovirus promoter plus human tissue plasminogen activator signal peptide fusion protein with severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1 spike glycoprotein codon optimized-specifying)
The University of Oxford, AstraZeneca vaccine is a vaccine that aims to protect against COVID-19.
Manufacturer/developer: AstraZeneca, University of OxfordResearch name: AZD1222 (ChAdOx1)Vaccine type: Non-Replicating Viral VectorAdministration method: Intramuscular injection
Biological Components:
Covishield is a viral vector vaccine. It uses a weakened, non-replicating strain of Chimpanzee cold virus (adenovirus) to carry genetic material of the spike protein of SARS-CoV-2 into human cells
_2021_C.jpg)
Vial of the Oxford–AstraZeneca vaccine manufactured by the Serum Institute of India (marketed as Covishield in India and in a few other countries).[5]
COVISHIELD INGREDIENTS
L-Histidine Ethanol
L-Histidine Hydrochloride Monohydrate,Magnesium Chloride
Hexahydrate Polysorbate 80*, Sucrose, Sodium Chloride
Disodium Edetate Dihydrate (EDTA) , Water for injection
Polysorbate 80 which is an ingredient of Covishield is known to cause anaphylactic reactions in patients as can be read here whereas Covaxin has no such component.
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Astrazeneca Covid-19 Vaccine | Injection, suspension | 50000000000 {VP}/0.5mL | Intramuscular | AstraZeneca Pharmaceuticals LP | 2020-12-22 | Not applicable |  |
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Injection, suspension | Intramuscular | 50000000000 {VP}/0.5mL |
Storage Conditions: can be stored at 2 to 8 degrees Celsius making them convenient to store and transport.
Mechanism of Immunization: Covishield – This vaccine produces antibodies against only a specific region of the virus. It contains a portion of the DNA that codes for the spike protein (S-protein). Once inside the cells, the DNA part first needs to enter the nucleus to create its mirror image (complementary RNA). Then this RNA comes out in the cytoplasm as a messenger and starts making S-protein through a machine available for this purpose called ribosome. Since it is S-protein that provokes immunity it may not be as close to natural immunity as created by Covaxin. If there are any long-term side effects of the DNA material remaining inside the nucleus (e.g. integration in human DNA) is not yet known. So far, DNA vaccines were only being tried out for treating cancer patients and never used for preventing infections in normal subjects.
Clinical Development: Covishield has been developed by AstraZeneca with Oxford university in the UK and is being manufactured by the Serum Institute India (SII) in Pune. Covishield has completed phase 3 trials in S. Africa, Brazil and UK. 90% of the subjects in these studies were under the age of 55 making the efficacy and safety data applicable to this age group. The company has presented bridging study results in Indian population to the regulatory authorities based on which the approval was granted by DCGI. This data is not yet available in the public domain
Dosage Regimen: Covishield has been recommended to be taken in 2 doses. Observation of data from the UK shows improved protection with a gap of 12 weeks between 2 doses; though currently the expert committee set up by the Drug Controller General of India (DCGI) has recommended a gap of 4 weeks. Covaxin has been recommended to be taken in 2 doses 4 weeks apart.
Efficacy: Covishield has an average efficacy of 70% when 2 doses are administered 4 weeks apart. This data is from a meta-analysis (pooled analysis of multiple studies) of 4 Covishield trials in 11,636 patients out of which 3 trials were single blind and one double blind in 3 different countries. The efficacy of Covishield was published in The Lancet (link to the article). Observation of data has shown that the efficacy improves as the gap between the 2 doses is increased reaching a reported efficacy of 82.4% with a 12-week gap. Since, the phase-3 trials were conducted with a 4-week interval, it has become the standard.
Protection against Mutations: Preliminary research shows both vaccines are effective against the variant of the novel coronavirus first detected in the UK but there is no data on their efficacy against the mutants found in South Africa and Brazil. Data against these 2 variants is yet to be generated for both these vaccines.

. Consent: Covishield does not require any consent form as it has completed the phase-3 clinical trials
Who should not take Covishield?
Serum Institute of India’s factsheet said one should not get the Covishield vaccine if the person had a severe allergic reaction after a previous dose of this vaccine. Like Bharat Biotech, the SII factsheet also says that if a person is pregnant or plans to become pregnant or is breastfeeding she should tell the healthcare provider before taking the jab. People who have taken another anti-Covid vaccine should not take Covishield.
The ingredients of the Covishield vaccine are “L-Histidine, L-Histidine hydrochloride monohydrate, Magnesium chloride hexahydrate, Polysorbate 80, Ethanol, Sucrose, Sodium chloride, Disodium edetate dihydrate (EDTA), Water for injection,” it pointed out.
Side-effects of Covishield
Some of the very common side effects of the vaccines are tenderness, pain, warmth, redness, itching, swelling or bruising where the injection is given, generally feeling unwell, chills or feeling feverish, headache or joint aches.
Covishield is made by Serum Institute of India (SII) and Covaxin is manufactured by Bharat Biotech.
Over 50 lakh people have registered themselves on the Co-WIN portal since the window opened on Monday morning, the Centre said. Nearly 5 lakh beneficiaries above 60 or those aged 45-60 with comorbidities have received the first jab of Covid-19 vaccine till Tuesday evening.
Meanwhile, the govt has permitted all private hospitals to give Covid-19 vaccine if they adhere to the laid down norms and also asked the states and union territories to utilise the optimum capacity of private medical facilities empanelled under three categories. The states and Union Territories were also urged not to store, reserve, conserve or create a buffer stock of the COVID-19 vaccines, the Union Health Ministry said in a statement.
Sources: https://www.bbc.com/news/world-asia-india-55748124
The Oxford–AstraZeneca COVID-19 vaccine, codenamed AZD1222,[7] is a COVID-19 vaccine developed by Oxford University and AstraZeneca given by intramuscular injection, using as a vector the modified chimpanzee adenovirus ChAdOx1.[18][19][20][21] One dosing regimen showed 90% efficacy when a half-dose was followed by a full-dose after at least one month, based on mixed trials with no participants over 55 years old.[6] Another dosing regimen showed 62% efficacy when given as two full doses separated by at least one month.[6]
The research is being done by the Oxford University’s Jenner Institute and Oxford Vaccine Group with the collaboration of the Italian manufacturer Advent Srl located in Pomezia, which produced the first batch of the COVID-19 vaccine for clinical testing.[22] The team is led by Sarah Gilbert, Adrian Hill, Andrew Pollard, Teresa Lambe, Sandy Douglas and Catherine Green.[23][22]
On 30 December 2020, the vaccine was first approved for use[11][24] in the UK’s vaccination programme,[25] and the first vaccination outside of a trial was administered on 4 January 2021.[26] The vaccine has since been approved by several medicine agencies worldwide, such as the European Medicines Agency,[12][14] and the Australian Therapeutic Goods Administration (TGA),[9] and has been approved for an Emergency Use Listing (EUL) by the World Health Organization.[27]
Vaccine platform
The AZD1222 vaccine is a replication-deficient simian adenovirus vector, containing the full‐length codon‐optimised coding sequence of SARS-CoV-2 spike protein along with a tissue plasminogen activator (tPA) leader sequence.[28][29].
The adenovirus is said replication-deficient because some of its essential genes were deleted and replaced by a gene coding for the spike. Following vaccination, the adenovirus vector enters the cells, releases its genes, those are transported to the cell nucleus, thereafter the cell’s machinery does the transcription in mRNA and the translation in proteins.
The one of interest is the spike protein, an external protein that enables the SARS-type coronavirus to enter cells through the enzymatic domain of ACE2.[30] Producing it following vaccination will prompt the immune system to attack the coronavirus through antibodies and T-cells if it later infects the body.[6]
History
2020 development
In February 2020, the Jenner Institute agreed a collaboration with the Italian company Advent Srl for the production of the first batch of a vaccine candidate for clinical trials.[31]
In March 2020,[32][33] after the Gates Foundation urged the University of Oxford to find a large company partner to get its COVID-19 vaccine to market, the university backed off from its earlier pledge to donate the rights to any drugmaker.[34] Also, the UK government encouraged the University of Oxford to work with AstraZeneca instead of Merck & Co., a US based company over fears of vaccine hoarding under the Trump administration.[35]
In June 2020, the US National Institute of Allergy and Infectious Diseases (NIAID) confirmed that the third phase of testing for potential vaccines developed by Oxford University and AstraZeneca would begin in July 2020.[36]
Clinical trials
In July 2020, AstraZeneca partnered with IQVIA to speed up US clinical trials.[37]
On 31 August 2020, AstraZeneca announced that it had begun enrolling adults for a US-funded, 30,000-subject late-stage study.[38]
On 8 September 2020, AstraZeneca announced a global halt to the vaccine trial while a possible adverse reaction in a participant in the United Kingdom was investigated.[39][40][41] On 13 September, AstraZeneca and the University of Oxford resumed clinical trials in the United Kingdom after regulators concluded it was safe to do so.[42] AstraZeneca was criticised for vaccine safety after concerns from experts noting the company’s refusal to provide details about serious neurological illnesses in two participants who received the experimental vaccine in Britain.[43] While the trial resumed in the UK, Brazil, South Africa, Japan[44] and India, it remained on pause in the US till 23 October 2020[45] while the Food and Drug Administration (FDA) investigated a patient illness that triggered the clinical hold, according to the United States Department of Health and Human Services (HHS) Secretary Alex Azar.[46]
On 15 October 2020, Dr João Pedro R. Feitosa, a 28-year-old doctor from Rio de Janeiro, Brazil, who received a placebo instead of the test vaccine in a clinical trial of AZD1222, died from COVID-19 complications.[47][48][49] The Brazilian health authority Anvisa announced that the trial would continue in Brazil.[50]
Results of Phase III trial
On 23 November 2020, Oxford University and AstraZeneca announced interim results from the vaccine’s ongoing Phase III trials.[6][51] There was some criticism of the methods used in the report, which combined results of 62% and 90% from different groups of test subjects given different dosages to arrive at a 70% figure.[52][53][54] AstraZeneca said it would carry out a further multi-country trial using the lower dose which had led to a 90% claim.[55]
The full publication of the interim results from four ongoing Phase III trials on 8 December 2020 clarified these reports.[56] In the group who received the first dose of active vaccine more than 21 days earlier, there were no hospitalisations or severe disease, unlike those receiving the placebo. Serious adverse events were balanced across the active and control arms in the studies, i.e. the active vaccine did not have safety concerns. A case of transverse myelitis was reported 14 days after booster vaccination as being possibly related to vaccination, with an independent neurological committee considering the most likely diagnosis to be of an idiopathic, short segment, spinal cord demyelination. The other two cases of transverse myelitis, one in the vaccine group and the other in the control group, were considered to be unrelated to vaccination.[56]
A subsequent analysis, published on 19 February, has shown an efficacy of 76% 22 days after the first dose and increase to 81.3% when the second dose is given 12 weeks or more after the first.[57]
2021 development
In February 2021, Oxford–AstraZeneca indicated developments to adapt the vaccine to target new variants of the coronavirus,[58] with expectation of a modified vaccine being available “in a few months” as a “booster jab”.[59] A key area of concern is whether the E484K mutation could impact the immune response and, possibly, current vaccine effectiveness.[60] The E484K mutation is present in the South African (B.1.351) and Brazilian (B.1.1.28) variants, with a small number of cases of the mutation also detected in infections by the original SARS-CoV-2 virus and the UK/Kent (B.1.1.7) variant.[60]
Scottish Study
A study was carried out by universities across Scotland of the effectiveness of first dose of Pfizer–BioNTech and Oxford–AstraZeneca COVID-19 vaccines against hospital admissions in Scotland, based on a national prospective cohort study of 5.4 million people. Between 8 December 2020 to 15 February 2021, 1,137,775 patients were vaccinated in the study, 490,000 of which were with the Oxford–AstraZeneca vaccine. The first dose of the Oxford–AstraZeneca vaccine was associated with a vaccine effect of 94% for COVID-19 related hospitalisation at 28–34 days post-vaccination. Results for both vaccines combined showed a vaccine effect for prevention of COVID-19 related hospitalisation which was comparable when restricting the analysis to those aged ≥80 years (81%). The majority of the patients over the age of 65 were given the Oxford–AstraZeneca vaccine. As of 22 February 2021, the study had not been peer-reviewed.[61][62]
Approvals
On 27 November 2020, the UK government asked the Medicines and Healthcare products Regulatory Agency to assess the AZD1222 vaccine for temporary supply,[63] and it was approved for use on 30 December 2020, as their second vaccine to enter the national rollout.[64]
On 4 January 2021, Brian Pinker, 82, became the first person to receive the Oxford–AstraZeneca COVID-19 vaccine outside of clinical trials.[26]
The European Medicines Agency (EMA) received an application for a conditional marketing authorisation (CMA) for the vaccine on 12 January 2021. A press release stated that a recommendation on this could be issued by the agency by 29 January, with the European Commission then making a decision on the CMA within days.[3] The Hungarian regulator unilaterally approved the vaccine instead of waiting for EMA approval.[65]
On 29 January 2021, the EMA recommended granting a conditional marketing authorisation for AZD1222 for people 18 years of age and older,[12][13] and the recommendation was accepted by the European Commission the same day.[14][66]
On 30 January 2021, the Vietnamese Ministry of Health approved the AstraZeneca vaccine for domestic inoculation, the first to be approved in Vietnam.[67]
The vaccine has also been approved by Argentina,[68] Bangladesh,[69] Brazil,[70] the Dominican Republic,[71] El Salvador,[72] India,[73][74] Malaysia,[75] Mexico,[76] Nepal,[77] Pakistan,[78] the Philippines,[79] Sri Lanka,[80] and Taiwan[81] regulatory authorities for emergency usage in their respective countries.
On 7 February 2021, the vaccine roll out in South Africa was suspended. Researchers from the University of the Witwatersrand said in a prior-to-peer analysis that the AstraZeneca vaccine provided minimal protection against mild or moderate disease infection among young people.[82][83] The BBC reported on 8 February 2021 that Katherine O’Brien, director of immunisation at the World Health Organization, indicated she felt it was “really plausible” the AstraZeneca vaccine could have a “meaningful impact” on the South African variant particularly in preventing serious illness and death.[84] The same report also indicated the Deputy Chief Medical Officer for England Jonathan Van-Tam said the (Witwatersrand) study did not change his opinion that the AstraZeneca vaccine was “rather likely” to have an effect on severe disease from the South African variant.[84]
On 10 February 2021, South Korea granted its first approval of a COVID-19 vaccine to AstraZeneca, allowing the two-shot regimen to be administered to all adults, including the elderly. The approval came with a warning, however, that consideration is needed when administering the vaccine to individuals over 65 years of age due to limited data from that demographic in clinical trials.[85][86]
On 10 February 2021, the World Health Organization (WHO) issued interim guidance and recommended the AstraZeneca vaccine for all adults, its Strategic Advisory Group of Experts also having considered use where variants were present and concluded there was no need not to recommend it.[87]
On 16 February 2021, the Australian Therapeutic Goods Administration (TGA) granted provisional approval for COVID-19 Vaccine AstraZeneca.[9][1]
On 26 February 2021, the vaccine was authorized with terms and conditions by Health Canada.[88]
Production and supply
The vaccine is stable at refrigerator temperatures and costs around US$3 to US$4 per dose.[89] On 17 December, a tweet by the Belgian Budget State Secretary revealed the European Union (EU) would pay €1.78 (US$2.16) per dose.[90]
According to AstraZeneca’s vice-president for operations and IT, Pam Cheng, the company would have around 200 million doses ready worldwide by the end of 2020, and capacity to produce 100 million to 200 million doses per month once production is ramped up.[52]
In June 2020, further to making 100 million doses available to the UK’s NHS for their vaccination programme,[91] AstraZeneca and Emergent BioSolutions signed a US$87 million deal to manufacture doses of the vaccine specifically for the US market. The deal was part of the Trump administration’s Operation Warp Speed initiative to develop and rapidly scale production of targeted vaccines before the end of 2020.[92] Catalent will be responsible for the finishing and packaging process.[93] The majority of manufacturing work will be done in the UK.[citation needed]
On 4 June 2020, the World Health Organization‘s (WHO) COVAX facility made initial purchases of 300 million doses from the company for low- to middle-income countries.[94] Also, AstraZeneca and Serum Institute of India reached a licensing agreement to supply 1 billion doses of the Oxford University vaccine to middle- and low-income countries, including India.[95][96]
On 29 September 2020, a grant from the Bill and Melinda Gates Foundation allowed COVAX to secure an additional 100 million COVID-19 vaccine doses either from AstraZeneca or from Novavax at US$3 per dose.[97]
On 13 June 2020, AstraZeneca signed a contract with the Inclusive Vaccines Alliance, a group formed by France, Germany, Italy, and the Netherlands, to supply up to 400 million doses to all European Union member states.[98][99][100] However, the European Commission intervened to stop the deal being formalised. It took over negotiations on behalf of the whole EU, signing a deal at the end of August.[101]
In August 2020, AstraZeneca agreed to provide 300 million doses to the USA for US$1.2 billion, implying a cost of US$4 per dose. An AstraZeneca spokesman said the funding also covers development and clinical testing.[102] It also reached technology transfer agreement with Mexican and Argentinean governments and agreed to produce at least 400 million doses to be distributed throughout Latin America. The active ingredients would be produced in Argentina and sent to Mexico to be completed for distribution.[103]
In September 2020, AstraZeneca agreed to provide 20 million doses to Canada.[104][105]
In October 2020, Switzerland signed an agreement with AstraZeneca to pre-order up to 5.3 million doses.[106][107]
On 5 November 2020, a tripartite agreement was signed between the government of Bangladesh, Serum Institute of India and Beximco Pharma of Bangladesh. Under the agreement Bangladesh ordered 30 million doses of Oxford–AstraZeneca vaccine from Serum through Beximco for $4 per shot.[108]
In November 2020, Thailand ordered 26 million doses of vaccine from AstraZeneca.[109] It would cover 13 million people,[110] approximately 20% of the population, with the first lot expected to be delivered at the end of May.[111][112][113] The public health minister indicated the price paid was $5 per dose;[114] AstraZeneca (Thailand) explained in January 2021 after a controversy that the price each country paid depended on production cost and differences in supply chain, including manufacturing capacity, labour and raw material costs.[115] In January 2021, the Thai cabinet approved further talks on ordering another 35 million doses[116] and the Thai FDA approved the vaccine for emergency use for 1 year.[117][118] Siam Bioscience, a company owned by Vajiralongkorn, will received technological transfer,[119] and has the capacity to manufacture up to 200 million doses a year for export to ASEAN.[120]
Also in November, the Philippines agreed to buy 2.6 million doses,[121] reportedly worth around ₱700 million (approximately $5.6/dose).[122]
In December 2020, South Korea signed a contract with AstraZeneca to secure 20 million doses of its vaccine, reportedly worth equivalently to those signed by Thailand and the Philippines,[123] with the first shipment expected as early as January 2021. As of January 2021, the vaccine remains under review by the South Korea Disease Control and Prevention Agency.[124][125] AstraZeneca signed a deal with South Korea’s SK Bioscience to manufacture its vaccine products. The collaboration calls for the SK affiliate to manufacture AZD1222 for local and global markets.[126]
On 7 January 2021, the South African government announced that they had secured an initial 1 million doses from the Serum Institute of India, to be followed by another 500,000 doses in February.[127]
Myanmar signed a contract with Serum Institute of India to secure 30 million doses of its vaccine in December 2020. Myanmar will get doses for 15 million people from February 2021.[128]
On 22 January 2021, AstraZeneca announced that in the event the European Union approved the COVID-19 Vaccine AstraZeneca, initial supplies would be lower than expected due to production issues at Novasep in Belgium. Only 31 million of the previously predicted 80 million doses would be delivered to the European Union by March 2021.[129] In an interview with Italian newspaper La Repubblica, AstraZeneca’s CEO Pascal Soriot said the delivery schedule for the doses in the European Union was two months behind schedule. He mentioned low yield from cell cultures in one large-scale European site.[130] Analysis published in The Guardian also identified an apparently low yield from bioreactors in the Belgium plant and noted the difficulties in setting up this form of process, with variable yields often occurring.[131] As a result, the European Union imposed export controls on vaccine doses; controversy erupted as to whether doses were being diverted to the UK, and whether or not deliveries to Northern Ireland would be disrupted.[132]
On 24 February 2021, Ghana became the first country in Africa to receive the Covid-19 vaccine through the COVAX initiative, where the facility sent six hundred thousand doses of AstraZeneca/Oxford jabs to Accra.[133]
Summary
Background
A safe and efficacious vaccine against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), if deployed with high coverage, could contribute to the control of the COVID-19 pandemic. We evaluated the safety and efficacy of the ChAdOx1 nCoV-19 vaccine in a pooled interim analysis of four trials.
Methods
This analysis includes data from four ongoing blinded, randomised, controlled trials done across the UK, Brazil, and South Africa. Participants aged 18 years and older were randomly assigned (1:1) to ChAdOx1 nCoV-19 vaccine or control (meningococcal group A, C, W, and Y conjugate vaccine or saline). Participants in the ChAdOx1 nCoV-19 group received two doses containing 5 × 1010 viral particles (standard dose; SD/SD cohort); a subset in the UK trial received a half dose as their first dose (low dose) and a standard dose as their second dose (LD/SD cohort). The primary efficacy analysis included symptomatic COVID-19 in seronegative participants with a nucleic acid amplification test-positive swab more than 14 days after a second dose of vaccine. Participants were analysed according to treatment received, with data cutoff on Nov 4, 2020. Vaccine efficacy was calculated as 1 - relative risk derived from a robust Poisson regression model adjusted for age. Studies are registered at ISRCTN89951424 and ClinicalTrials.gov, NCT04324606, NCT04400838, and NCT04444674.
Findings
Between April 23 and Nov 4, 2020, 23 848 participants were enrolled and 11 636 participants (7548 in the UK, 4088 in Brazil) were included in the interim primary efficacy analysis. In participants who received two standard doses, vaccine efficacy was 62·1% (95% CI 41·0–75·7; 27 [0·6%] of 4440 in the ChAdOx1 nCoV-19 group vs71 [1·6%] of 4455 in the control group) and in participants who received a low dose followed by a standard dose, efficacy was 90·0% (67·4–97·0; three [0·2%] of 1367 vs 30 [2·2%] of 1374; pinteraction=0·010). Overall vaccine efficacy across both groups was 70·4% (95·8% CI 54·8–80·6; 30 [0·5%] of 5807 vs 101 [1·7%] of 5829). From 21 days after the first dose, there were ten cases hospitalised for COVID-19, all in the control arm; two were classified as severe COVID-19, including one death. There were 74 341 person-months of safety follow-up (median 3·4 months, IQR 1·3–4·8): 175 severe adverse events occurred in 168 participants, 84 events in the ChAdOx1 nCoV-19 group and 91 in the control group. Three events were classified as possibly related to a vaccine: one in the ChAdOx1 nCoV-19 group, one in the control group, and one in a participant who remains masked to group allocation.
Interpretation
ChAdOx1 nCoV-19 has an acceptable safety profile and has been found to be efficacious against symptomatic COVID-19 in this interim analysis of ongoing clinical trials.
Funding
UK Research and Innovation, National Institutes for Health Research (NIHR), Coalition for Epidemic Preparedness Innovations, Bill & Melinda Gates Foundation, Lemann Foundation, Rede D’Or, Brava and Telles Foundation, NIHR Oxford Biomedical Research Centre, Thames Valley and South Midland’s NIHR Clinical Research Network, and AstraZeneca.
References
- ^ Jump up to:a b c “COVID-19 Vaccine AstraZeneca”. Therapeutic Goods Administration (TGA). 16 February 2021. Retrieved 16 February2021.
- ^ Jump up to:a b “Information for Healthcare Professionals on COVID-19 Vaccine AstraZeneca”. Medicines and Healthcare products Regulatory Agency (MHRA). 30 December 2020. Retrieved 4 January 2021.
- ^ Jump up to:a b “EMA receives application for conditional marketing authorisation of COVID-19 Vaccine AstraZeneca”. European Medicines Agency (EMA). 12 January 2021. Retrieved 12 January2021.
- ^ Jump up to:a b “Regulatory Decision Summary – AstraZeneca COVID-19 Vaccine”. Health Canada. 26 February 2021. Retrieved 26 February 2021.
- ^ Jump up to:a b “Already produced 40–50 million dosages of Covishield vaccine, says Serum Institute”. The Hindu. 28 December 2020.
- ^ Jump up to:a b c d e “AZD1222 vaccine met primary efficacy endpoint in preventing COVID-19”. Press Release (Press release). AstraZeneca. 23 November 2020. Retrieved 5 January 2021.
- ^ Jump up to:a b “AstraZeneca COVID-19 Vaccine (AZD1222)” (PDF). AstraZeneca. 27 January 2021.
- ^ “AstraZeneca and Oxford University announce landmark agreement for COVID-19 vaccine”. AstraZeneca (Press release). 30 April 2020. Retrieved 13 January 2021.
- ^ Jump up to:a b c d e “COVID-19 Vaccine AstraZeneca PI”. Therapeutic Goods Administration (TGA).
- ^ “AstraZeneca COVID-19 Vaccine monograph” (PDF). AstraZeneca. 26 February 2021.
- ^ Jump up to:a b “Conditions of Authorisation for COVID-19 Vaccine AstraZeneca”. Medicines and Healthcare products Regulatory Agency (MHRA). 30 December 2020. Retrieved 4 January 2021.
- ^ Jump up to:a b c “COVID-19 Vaccine AstraZeneca EPAR”. European Medicines Agency (EMA).
- ^ Jump up to:a b “EMA recommends COVID-19 Vaccine AstraZeneca for authorisation in the EU”. European Medicines Agency (EMA)(Press release). 29 January 2021. Retrieved 29 January 2021.
- ^ Jump up to:a b c “European Commission authorises third safe and effective vaccine against COVID-19”. European Commission (Press release). Retrieved 29 January 2021.
- ^ “아스트라제네카社 코로나19 백신 품목허가”. 식품의약품안전처(in Korean). 식품의약품안전처. 10 February 2021. Retrieved 10 February 2021.
- ^ “BPOM Terbitkan Izin Penggunaan Darurat Vaksin Covid-19 AstraZeneca”. Kompas.com. 10 March 2021. Retrieved 10 March2021.
- ^ “150,000 doses of AstraZeneca vaccine arrive in Serbia”. Government of Serbia. 21 February 2021. Retrieved 2 March 2021.
- ^ Walsh N, Shelley J, Duwe E, Bonnett W (27 July 2020). “The world’s hopes for a coronavirus vaccine may run in these health care workers’ veins”. São Paulo: CNN. Archived from the original on 3 August 2020. Retrieved 3 August 2020.
- ^ “Investigating a Vaccine Against COVID-19”. ClinicalTrials.gov(Registry). United States National Library of Medicine. 26 May 2020. NCT04400838. Archived from the original on 11 October 2020. Retrieved 14 July 2020.
- ^ “A Phase 2/3 study to determine the efficacy, safety and immunogenicity of the candidate Coronavirus Disease (COVID-19) vaccine ChAdOx1 nCoV-19”. EU Clinical Trials Register(Registry). European Union. 21 April 2020. EudraCT 2020-001228-32. Archived from the original on 5 October 2020. Retrieved 3 August 2020.
- ^ O’Reilly P (26 May 2020). “A Phase III study to investigate a vaccine against COVID-19”. ISRCTN (Registry). doi:10.1186/ISRCTN89951424. ISRCTN89951424.
- ^ Jump up to:a b “Oxford team to begin novel coronavirus vaccine research”. University of Oxford. 7 February 2020. Retrieved 28 November2020.
- ^ “COVID-19 Oxford Vaccine Trial”. COVID-19 Oxford Vaccine Trial. Retrieved 11 April 2020.
- ^ “Covid-19: Oxford-AstraZeneca coronavirus vaccine approved for use in UK”. BBC News Online. 30 December 2020. Retrieved 30 December 2020.
- ^ “Second COVID-19 vaccine authorised by medicines regulator”. GOV.UK (Press release). 30 December 2020. Retrieved 6 March2021.
- ^ Jump up to:a b “Covid: Brian Pinker, 82, first to get Oxford-AstraZeneca vaccine”. BBC News Online. 4 January 2021. Retrieved 4 January 2021.
- ^ “Coronavirus disease (COVID-19): Vaccines”. World Health Organization (WHO). Retrieved 6 March 2021.
- ^ Arashkia A, Jalilvand S, Mohajel N, Afchangi A, Azadmanesh K, Salehi-Vaziri M, et al. (2020). “Severe acute respiratory syndrome-coronavirus-2 spike (S) protein based vaccine candidates: State of the art and future prospects”. Reviews in Medical Virology. n/a(n/a): e2183. doi:10.1002/rmv.2183. PMC 7646037. PMID 33594794.
- ^ Watanabe, Y.; Mendonça, L.; Allen, E. R.; Howe, A.; Lee, M.; Allen, J. D.; Chawla, H.; Pulido, D.; Donnellan, F.; Davies, H.; Ulaszewska, M.; Belij-Rammerstorfer, S.; Morris, S.; Krebs, A. S.; Dejnirattisai, W.; Mongkolsapaya, J.; Supasa, P.; Screaton, G. R.; Green, C. M.; Lambe, T.; Zhang, P.; Gilbert, S. C.; Crispin, M. (2021), “Native-like SARS-CoV-2 spike glycoprotein expressed by ChAdOx1 nCoV-19/AZD1222 vaccine”, bioRxiv : The Preprint Server for Biology: 2021.01.15.426463, doi:10.1101/2021.01.15.426463, PMC 7836103, PMID 33501433
- ^ Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, Jiang C (February 2008). “SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway”. Cell Research. 18 (2): 290–301. doi:10.1038/cr.2008.15. PMC 7091891. PMID 18227861.
- ^ “Oxford team to begin novel coronavirus vaccine research”. University of Oxford. Retrieved 2 January 2021.
- ^ “Covid Vaccine Front-Runner Is Months Ahead of Her Competition”. Bloomberg Businessweek. 15 July 2020.
- ^ “Bill Gates, the Virus and the Quest to Vaccinate the World”. The New York Times. 23 November 2020.
- ^ “They Pledged to Donate Rights to Their COVID Vaccine, Then Sold Them to Pharma”. Kaiser Health News. Retrieved 28 January 2021.
- ^ Strasburg J, Woo S (21 October 2020). “Oxford Developed Covid Vaccine, Then Scholars Clashed Over Money”. The Wall Street Journal.
- ^ Coleman J (10 June 2020). “Final testing stage for potential coronavirus vaccine set to begin in July”. The Hill. Retrieved 11 June 2020.
- ^ “AZN, IQV Team Up To Accelerate COVID-19 Vaccine Work, RIGL’s ITP Drug Repurposed, IMV On Watch”. RTTNews. Retrieved 15 July 2020.
- ^ “Phase 3 Clinical Testing in the US of AstraZeneca COVID-19 Vaccine Candidate Begins”. National Institutes of Health (NIH). 30 August 2020. Retrieved 1 September 2020.
- ^ “AstraZeneca Covid-19 vaccine study is put on hold”. Stat. 8 September 2020. Retrieved 10 September 2020.
- ^ “AstraZeneca Covid-19 vaccine study is put on hold”. 8 September 2020.
- ^ Wu KJ, Thomas K (8 September 2020). “AstraZeneca Pauses Vaccine Trial for Safety Review”. The New York Times. ISSN 0362-4331. Retrieved 10 September 2020.
- ^ Loftus P (13 September 2020). “AstraZeneca Covid-19 Vaccine Trials Resume in U.K.”. The Wall Street Journal. Retrieved 13 September 2020.
- ^ Grady D, Wu KJ, LaFraniere S (19 September 2020). “AstraZeneca, Under Fire for Vaccine Safety, Releases Trial Blueprints”. The New York Times. ISSN 0362-4331. Retrieved 22 September 2020.
- ^ “AstraZeneca resumes vaccine trial in talks with US”. Japan Today. 3 October 2020.
- ^ “FDA authorises restart of the COVID-19 AZD1222 vaccine US Phase III trial”. AstraZeneca (Press release). Retrieved 1 December 2020.
- ^ “U.S. health secretary says AstraZeneca trial in United States remains on hold: CNBC”. Reuters. 23 September 2020. Retrieved 24 September 2020.
- ^ “‘What’s the deal?’ Researchers in paused vaccine trial search for answers”. NBC News.
- ^ “Volunteer in AstraZeneca Covid-19 vaccine trial dies in Brazil”. NBC News.
- ^ Voluntário brasileiro que participava dos testes da vacina de Oxford e morreu com a Covid era médico e ex-aluno da UFRJ, Globo
- ^ Simões E, Burger L (22 October 2020). “AstraZeneca COVID-19 vaccine trial Brazil volunteer dies, trial to continue”. Reuters. Retrieved 22 October 2020.
- ^ “Oxford University breakthrough on global COVID-19 vaccine”(Press release). University of Oxford. 23 November 2020. Retrieved 12 January 2021.
- ^ Jump up to:a b Callaway E (23 November 2020). “Why Oxford’s positive COVID vaccine results are puzzling scientists”. Nature. 588(7836): 16–18. Bibcode:2020Natur.588…16C. doi:10.1038/d41586-020-03326-w. PMID 33230278. S2CID 227156970.
- ^ “Oxford/AstraZeneca Covid vaccine ‘dose error’ explained”. BBC News. 27 November 2020. Retrieved 27 November 2020.
- ^ Robbins R, Mueller B (25 November 2020). “After Admitting Mistake, AstraZeneca Faces Difficult Questions About Its Vaccine”. The New York Times. ISSN 0362-4331. Retrieved 27 November 2020.
- ^ Boseley S (26 November 2020). “Oxford/AstraZeneca vaccine to undergo new global trial”. The Guardian. Retrieved 27 November2020.
- ^ Jump up to:a b Voysey M, Clemens SA, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. (January 2021). “Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK”. Lancet. 397 (10269): 99–111. doi:10.1016/S0140-6736(20)32661-1. PMC 7723445. PMID 33306989.
- ^ Voysey M, Costa Clemens SA, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. (February 2021). “Single-dose administration and the influence of the timing of the booster dose on immunogenicity and efficacy of ChAdOx1 nCoV-19 (AZD1222) vaccine: a pooled analysis of four randomised trials”. Lancet. 397(10277): 881–891. doi:10.1016/S0140-6736(21)00432-3. PMC 7894131. PMID 33617777.
- ^ Ellyatt H (8 February 2021). “AstraZeneca races to adapt Covid vaccine as South Africa suspends rollout”. CNBC. Retrieved 8 February 2021.
- ^ Triggle N (8 February 2021). “Covid: Are fears over Oxford-AstraZeneca jab justified?”. BBC. Retrieved 9 February 2021.
- ^ Jump up to:a b Wise J (February 2021). “Covid-19: The E484K mutation and the risks it poses”. BMJ. 372: n359. doi:10.1136/bmj.n359. PMID 33547053.
- ^ “Covid-19: First doses of vaccines in Scotland led to a substantial fall in hospital admissions”. the BMJ. Retrieved 25 February2021.
- ^ “scotland first vaccine data preprint” (PDF). Retrieved 25 February 2021.
- ^ “Government asks regulator to approve supply of Oxford/AstraZeneca vaccine”. Government of the United Kingdom. 27 October 2020. Retrieved 28 November 2020.
- ^ “Oxford University/AstraZeneca vaccine authorised by UK medicines regulator”. Government of the United Kingdom. 30 December 2020. Retrieved 30 December 2020.
- ^ “Everything You Need to Know About the Oxford-AstraZeneca Vaccine”. 23 January 2021.
- ^ “COVID-19 Vaccine AstraZeneca”. Union Register of medicinal products. Retrieved 18 February 2021.
- ^ Nikkei staff writers. “Coronavirus: Week of Jan. 24 to Jan. 30, Vietnam approves AstraZeneca vaccine”. Nikkei Asia.
- ^ Laing A (30 December 2020). “Argentine regulator approves AstraZeneca/Oxford COVID-19 vaccine -AstraZeneca”. Reuters.
- ^ “Oxford University-Astrazeneca vaccine: Bangladesh okays it for emergency use”. The Daily Star. 4 January 2021. Retrieved 7 January 2021.
- ^ Sabóia G, Mazieiro G, de Andrade H, Adorno L (17 January 2021). “Anvisa aprova uso emergencial das vacinas CoronaVac e AstraZeneca no Brasil” [Anvisa approves emergency use of the CoronaVac and AstraZeneca vaccines in Brazil]. UOL (in Portuguese). Retrieved 17 January 2021.
- ^ “La República Dominicana aprueba la vacuna de AstraZeneca contra la covid-19”. Agencia EFE (in Spanish). 31 December 2020.
- ^ “El Salvador greenlights AstraZeneca, Oxford University COVID-19 vaccine”. Reuters. 30 December 2020.
- ^ Gaurav K (1 January 2021). “Govt’s expert panel approves AstraZeneca/Oxford Covid-19 vaccine for emergency use”. Hindustan Times.
- ^ Prusty N, Jamkhandikar S (1 January 2021). “India drug regulator approves AstraZeneca COVID vaccine, country’s first – sources”. Reuters.
- ^ “Malaysia’s NPRA Approves AstraZeneca, Sinovac Covid-19 Vaccines”. CodeBlue. 2 March 2021. Retrieved 2 March 2021.
- ^ Comisión Federal para la Protección contra Riesgos Sanitarios. “AUTORIZACIÓN PARA USO DE EMERGENCIA A VACUNA ASTRAZENECA COVID-19”. gob.mx (in Spanish). Retrieved 7 January 2021.
- ^ “Nepal approves AstraZeneca COVID vaccine for emergency use – government statement”. Reuters. 15 January 2021.
- ^ Shahzad A (16 January 2021). “Pakistan approves AstraZeneca COVID-19 vaccine for emergency use”. Reuters. Retrieved 16 January 2021.
- ^ “Philippine regulator approves emergency use of AstraZeneca vaccine”. Reuters. 28 January 2021. Retrieved 28 January 2021.
- ^ “Sri Lanka approves vaccine amid warnings of virus spread”. AP NEWS. 22 January 2021. Retrieved 22 January 2021.
- ^ “Taiwan grants emergency authorisation for AstraZeneca COVID-19 vaccine”. MSN. Retrieved 22 February 2021.
- ^ “Latest – Oxford Covid-19 vaccine trial results – Wits University”. wits.ac.za. Retrieved 8 February 2021.
- ^ “South Africa halts AstraZeneca vaccinations after data shows little protection against mutation”. CNBC. 7 February 2021. Retrieved 8 February 2021.
- ^ Jump up to:a b “Covid: Boris Johnson ‘very confident’ in vaccines being used in UK”. BBC News. 8 February 2021. Retrieved 9 February 2021.
- ^ Kim HJ (10 February 2021). “S. Korea approves AstraZeneca’s COVID-19 vaccine for all adults”. Yonhap News Agency. Retrieved 10 February 2021.
- ^ Maresca T (10 February 2021). “South Korea approves AstraZeneca COVID-19 vaccine”. United Press International. Retrieved 10 February 2021.
- ^ “AstraZeneca-Oxford vaccine can be used for people aged over 65 – WHO”. RTÉ News and Current Affairs. 10 February 2021. Retrieved 12 February 2021.
- ^ Canada, Health. “COVID-19 vaccines and treatments portal: AstraZeneca COVID-19 Vaccine (ChAdOx1-S [recombinant])”. Health Canada. Retrieved 26 February 2021.
- ^ Belluz J (23 November 2020). “Why the AstraZeneca-Oxford Covid-19 vaccine is different”. Vox. Retrieved 26 November 2020.
- ^ Stevis-Gridneff M, Sanger-Katz M, Weiland N (18 December 2020). “A European Official Reveals a Secret: The U.S. Is Paying More for Coronavirus Vaccines”. The New York Times. Retrieved 19 December 2020.
- ^ “AstraZeneca to begin making vaccine“. BBC. 5 June 2020. Retrieved 1 July 2020.
- ^ “AstraZeneca, Emergent BioSolutions sign $87M deal to produce U.S. supply of COVID-19 vaccine”. FiercePharma. Retrieved 12 June 2020.
- ^ “AstraZeneca taps Catalent for COVID-19 vaccine finishing, packaging at Italian plant”. FiercePharma. Retrieved 16 June2020.
- ^ So AD, Woo J (December 2020). “Reserving coronavirus disease 2019 vaccines for global access: cross sectional analysis”. BMJ. 371: m4750. doi:10.1136/bmj.m4750. PMC 7735431. PMID 33323376. cited “Agreements with CEPI and Gavi and the Serum Institute of India will bring vaccine to low and middle-income countries and beyond” (Press release). AstraZeneca. 4 June 2020.
- ^ Rajagopal D (4 June 2020). “AstraZeneca & Serum Institute of India sign licensing deal for 1 billion doses of Oxford vaccine”. The Economic Times.
- ^ Kumar M (7 August 2020). “Covid-19 vaccine: Serum Institute signs up for 100 million doses of vaccines for India, low and middle-income countries”. The Financial Express.
- ^ So & Woo (2020), p. 3 cited “New collaboration makes further 100 million doses of COVID-19 vaccine available to low- and middle- income countries” (Press release). Gavi, the Vaccine Alliance. 29 September 2020.[permanent dead link]
- ^ “Covid-19: France, Italy, Germany and Netherlands sign vaccine deal for Europe”. France 24. 13 June 2020. Retrieved 15 June2020.
- ^ “AstraZeneca agrees to supply Europe with 400 mil doses of COVID-19 vaccine”. Japan Today. Retrieved 15 June 2020.
- ^ Calatayud A. “AstraZeneca to supply Europe with Covid-19 vaccine”. MarketWatch. Retrieved 15 June 2020.
- ^ Peston R (26 January 2021). “What is the dispute between the EU and AstraZeneca over Covid jabs?”. ITV News. Retrieved 27 January 2021.
- ^ Roland D (21 May 2020). “U.S. to Invest $1.2 Billion to Secure Potential Coronavirus Vaccine From AstraZeneca, Oxford University”. The Wall Street Journal. Retrieved 6 August 2020.
- ^ “AstraZeneca set to start making 400 million COVID-19 vaccines for Latam early in 2021”. Reuters. Retrieved 17 January 2021.
- ^ “With no successful vaccine candidates yet, Canada signs deal to secure 20M more COVID-19 vaccine doses”. CBC News. 25 September 2020.
- ^ Health Canada (2 October 2020). “Health Canada begins first authorization review of a COVID-19 vaccine submission”. gcnws. Retrieved 30 December 2020.
- ^ “Swiss sign next vaccine agreement with AstraZeneca”. SWI swissinfo.ch. Retrieved 16 October 2020.
- ^ “COVID-19 vaccine: Swiss federal government signs agreement with AstraZeneca”. admin.ch. Retrieved 16 October 2020.
- ^ “Dhaka to have 330 vaccination points”. The Daily Star. Retrieved 25 January 2021.
- ^ “เรื่องน่ารู้ของวัคซีนโควิด-19 ที่ไทยสั่งซื้อ”. BBC ไทย (in Thai). Retrieved 5 January 2021.
- ^ “ทำความรู้จัก ออกซ์ฟอร์ด-แอสทราเซเนกา วัคซีนที่ไทยเลือก”. มติชนออนไลน์ (in Thai). 2 January 2021. Retrieved 5 January 2021.
- ^ “ครม.ไฟเขียวงบซื้อวัคซีนโควิดเพิ่ม35ล้านโดส ฉีดให้คนไทย66ล้าน”. โพสต์ทูเดย์ (in Thai). Retrieved 5 January 2021.
- ^ “ข่าวดี ไทยเริ่มผลิตวัคซีน ‘โควิด-19’ ในประเทศ รอบที่ 2 แล้ว”. ไทยรัฐออนไลน์. 3 January 2021.
- ^ “สธ. แจง AstraZeneca เป็นผู้คัดเลือก Siam Bioscience ผลิตวัคซีนราคาทุน ขายถูกสุดในตลาด โต้ธนาธร ไม่ได้แทงม้าตัวเดียว”. THE STANDARD. 19 January 2021.
- ^ “ข่าวดี! ไทยจองซื้อวัคซีนโควิด-19 แอสตราเซเนกา “ราคาต้นทุน”” (in Thai). hfocus.org. 23 November 2020. Archived from the original on 23 November 2020.
- ^ “วัคซีนโควิด: แอสตร้าเซเนก้าชี้แจงเหตุผลเลือกสยามไบโอไซเอนซ์เป็นผู้ผลิต”. BBC News ไทย. 26 January 2021.
- ^ “โควิด-19: ทำไมรัฐบาลเลือก สยามไบโอไซเอนซ์ ผลิตวัคซีนเพื่อคนไทยและเพื่อนบ้าน”. BBC News ไทย. 15 January 2021.
- ^ “AstraZeneca vaccine approved, 50,000 doses due in February”. Bangkok Post. 21 January 2021.
- ^ “FDA approves AstraZeneca”. Bangkok Post. 22 January 2021.
- ^ “นายกฯ สำนึกในพระมหากรุณาธิคุณ ร.10 ทรงให้ “สยามไบโอไซเอนซ์” รองรับวัคซีนโควิด-19″. BBC ไทย (in Thai). 27 November 2020. Retrieved 5 January 2021.
- ^ “35m more shots to be bought in 2021”. Bangkok Post. 5 January 2021.
- ^ “Philippines, AstraZeneca Sign Deal for 2.6 Million Doses”. Bloomberg. 27 November 2020.
- ^ “Over 200 firms to ink deal for more COVID vaccines with gov’t, AstraZeneca”. Philippine Daily Inquirer. 11 January 2021.
- ^ “Korea signs agreement with AstraZeneca for COVID vaccine”. The Korea Times. 30 November 2020.
- ^ Shin H (3 December 2020). “South Korea reaches deal to buy AstraZeneca’s COVID-19 vaccine candidate: media”. Reuters. Retrieved 5 January 2021.
- ^ Cha S (4 January 2021). “S.Korea reviews AstraZeneca COVID-19 vaccine, expands ban on gatherings”. Reuters. Retrieved 5 January 2021.
- ^ Kim YC (30 November 2020). “Korea signs agreement with AstraZeneca for COVID vaccine”. The Korea Times. Retrieved 30 January 2021.
- ^ Felix J (7 January 2021). “SA will get 1 million doses of Covid-19 vaccine from India this month”. News24.com. Retrieved 7 January 2021.
- ^ “Myanmar will get doses for 15 million people this February”. 7day.news. Retrieved 8 January 2021.
- ^ Agencies (22 January 2021). “Covid: Oxford/AstraZeneca vaccine delivery to EU to be cut by 60%”. The Guardian. Retrieved 23 January 2021.
- ^ “Pascal Soriot: “There are a lot of emotions on vaccines in EU. But it’s complicated””. la Repubblica (in Italian). 26 January 2021. Retrieved 27 January 2021.
- ^ Boseley S (26 January 2021). “Why has AstraZeneca reduced promised vaccine supply to EU and is UK affected?”. The Guardian. Retrieved 27 January 2021.
- ^ “EU tightens vaccine export rules, creates post-Brexit outcry”. 30 January 2021.
- ^ “Ghana receives first historic shipment of COVID-19 vaccinations from international COVAX facility”. UN News. Retrieved 24 February 2021.
External links
| Scholia has a profile for AZD1222 (Q95042269). |
- “Medical Information site for COVID-19 Vaccine AstraZeneca”. AstraZeneca.
- “Vaccines: contract between European Commission and AstraZeneca now published”. European Commission.
- “How the Oxford-AstraZeneca Covid-19 Vaccine Works”. The New York Times.
- Background document on the AZD1222 vaccine against COVID-19 developed by Oxford University and AstraZeneca. World Health Organization (WHO) (Report).
- Australian Public Assessment Report for ChAdOx1-S (PDF) (Report).
| Box containing 100 AstraZeneca COVID-19 vaccine doses | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Clinical data | |
| Trade names | COVID-19 Vaccine AstraZeneca,[1][2][3] AstraZeneca COVID-19 Vaccine,[4] Covishield[5] |
| Other names | AZD1222,[6][7] ChAdOx1 nCoV-19,[8] ChAdOx1-S,[9] |
| License data | EU EMA: by INN |
| Pregnancy category | AU: B2[9][1] |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [9]CA: Schedule D; Authorized by interim order [4][10]UK: Conditional and temporary authorisation to supply [2][11]EU: Conditional marketing authorisation [12][13][14]KR – Approved[15]IND, INA[16], BD, AG, SV, DOM, MEX, NE, BR, SL, SRB[17]: Emergency Authorization only |
| Identifiers | |
| CAS Number | 2420395-83-9 |
| DrugBank | DB15656 |
| UNII | B5S3K2V0G8 |
////////AZD1222, ChAdOx1, Oxford–AstraZeneca, COVID 19 vaccine, COVISHIELD, CORONA, COVID 19, CORONA VIRUS
#AZD1222, #ChAdOx1, #Oxford–AstraZeneca, #COVID 19 vaccine, #COVISHIELD, #CORONA, #COVID 19, #CORONA VIRUS
Pyridostigmine
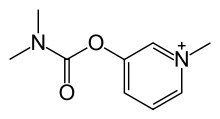
Pyridostigmine
- Molecular FormulaC9H13N2O2
- Average mass181.211 Da
155-97-5[RN]3-[(Dimethylcarbamoyl)oxy]-1-methylpyridinium
3-Dimethylcarbamoyloxy-1-methyl-pyridinium5-21-02-00078 (Beilstein Handbook Reference)[Beilstein]
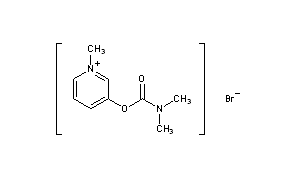
Pyridostigmine BromideCAS Registry Number: 101-26-8CAS Name: 3-[[(Dimethylamino)carbonyl]oxy]-1-methylpyridinium bromideAdditional Names: 3-hydroxy-1-methylpyridinium bromide dimethylcarbamate; 1-methyl-3-hydroxypyridinium bromide dimethylcarbamate; 3-(dimethylcarbamyloxy)-1-methylpyridinium bromideManufacturers’ Codes: Ro-1-5130Trademarks: Kalymin (Temmler); Mestinon (Roche); Regonol (Organon)Molecular Formula: C9H13BrN2O2Molecular Weight: 261.12Percent Composition: C 41.40%, H 5.02%, Br 30.60%, N 10.73%, O 12.25%Literature References: Reversible inhibitor of acetylcholinesterase.
Prepn: Urban, US2572579 (1951 to Hoffmann-La Roche). Mechanism of protective effect in soman poisoning: X. Deyi et al.,Fundam. Appl. Toxicol.1, 217 (1981). Evaluation of effect on neuromuscular function: M. Glikson et al.,ibid.16, 288 (1991). Evaluation of side effects profile under desert conditions: J. E. Cook et al.,Mil. Med.157, 250 (1992). Review of prophylactic effect in nerve agent poisoning: R. M. Dawson, J. Appl. Toxicol.14, 317 (1994).Properties: Shiny, hygroscopic crystals from abs ethanol, mp 152-154°. Freely sol in water, alcohol. Practically insol in ether, acetone, benzene. Aq solns may be sterilized by autoclaving with steam.Melting point: mp 152-154°Therap-Cat: Cholinergic; in treatment of myasthenia gravis. Pre-exposure antidote to chemical warfare agents.Keywords: Cholinergic.
Pyridostigmine is a medication used to treat myasthenia gravis.[1] It is also used together with atropine to end the effects of neuromuscular blocking medication of the non-depolarizing type.[2] It is typically given by mouth but can also be used by injection.[2] The effects generally begin within 45 minutes and last up to 6 hours.[2]
Common side effects include nausea, diarrhea, frequent urination, and abdominal pain.[2] More severe side effects include low blood pressure, weakness, and allergic reactions.[2] It is unclear if use in pregnancy is safe for the fetus.[2] Pyridostigmine is an acetylcholinesterase inhibitor in the cholinergic family of medications.[2] It works by blocking the action of acetylcholinesterase and therefore increases the levels of acetylcholine.[2]
Pyridostigmine was patented in 1945 and came into medical use in 1955.[3] It is on the World Health Organization’s List of Essential Medicines.[4] Pyridostigmine is available as a generic medication.[2]
Medical uses
Pyridostigmine is used to treat muscle weakness in people with myasthenia gravis or forms of congenital myasthenic syndrome and to combat the effects of curariform drug toxicity. Pyridostigmine bromide has been FDA approved for military use during combat situations as an agent to be given prior to exposure to the nerve agent Soman in order to increase survival. Used in particular during the first Gulf War, pyridostigmine bromide has been implicated as a causal factor in Gulf War syndrome.[5]
Pyridostigmine sometimes is used to treat orthostatic hypotension.[6] It may also be of benefit in chronic axonal polyneuropathy.[7]
It is also being prescribed ‘off-label’ for the postural tachycardia syndrome as well as complications resulting from Ehlers–Danlos syndrome.[7][8]
Contraindications
Pyridostigmine bromide is contraindicated in cases of mechanical intestinal or urinary obstruction and should be used with caution in patients with bronchial asthma.[9][10]
Side effects
Common side effects include:[9]
- Sweating
- Diarrhea
- Nausea
- Vomiting
- Abdominal cramps
- Increased salivation
- Tearing
- Increased bronchial secretions
- Constricted pupils
- Facial flushing due to vasodilation
- Erectile dysfunction
Additional side effects include:[9]
- Muscle twitching
- Muscle cramps and weakness
Mechanism of action
Pyridostigmine inhibits acetylcholinesterase in the synaptic cleft, thus slowing down the hydrolysis of acetylcholine. It is a quaternary carbamate inhibitor of cholinesterase that does not cross the blood–brain barrier which carbamylates about 30% of peripheral cholinesterase enzyme. The carbamylated enzyme eventually regenerates by natural hydrolysis and excess ACh levels revert to normal.
The ACh diffuses across the synaptic cleft and binds to receptors on the post synaptic membrane, causing an influx of Na+, resulting in depolarization. If large enough, this depolarization results in an action potential. To prevent constant stimulation once the ACh is released, an enzyme called acetylcholinesterase is present in the endplate membrane close to the receptors on the post synaptic membrane, and quickly hydrolyses ACh.
Names
Pyridostigmine bromide is available under the trade names Mestinon (Valeant Pharmaceuticals), Regonol and Gravitor (SUN Pharma).
Chemistry
Pyridostigmine, 3-[(dimethylaminocarbonyl)oxy]-1-methyl pyridinium bromide, is synthesized from 3-hydroxypyridine, which is reacted with dimethylaminocarbamoyl chloride, which gives 3-(dimethylaminocarbamoyl)pyridine. The last is reacted with methylbromide, giving pyridostigmine.
- R. Urban, U.S. Patent 2,572,579 (1951).
Syn
youtube
SYN
Method of synthesis
i. 3-hydroxypiridine is reacted with dimethylaminocarbamoyl chloride to give 3-(dimethylaminocarbamoyl)pyridine.
ii. The above formed compound is reacted with methylbromide to produce pyridostigmine. [2]


CLIP

Paper
Journal of Biological Chemistry (1961), 236, 1498-500.
Zeitschrift fuer Klinische Medizin (1985) (1986), 41(7), 495-8
Zhonghua Yaoxue Zazhi (1993), 45(6), 601-14.
Trends in Organic Chemistry (2011), 15, 25-31.
PATENT
WO 9822458
PATENT
WO 2008074816
https://patents.google.com/patent/WO2008074816A1/en
References
- ^ World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 429. hdl:10665/44053. ISBN 9789241547659.
- ^ Jump up to:a b c d e f g h i “Neostigmine Bromide”. The American Society of Health-System Pharmacists. Archived from the original on 21 December 2016. Retrieved 8 December 2016.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 540. ISBN 9783527607495. Archived from the original on 2016-12-20.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Golomb BA (March 2008). “Acetylcholinesterase inhibitors and Gulf War illnesses”. Proceedings of the National Academy of Sciences of the United States of America. 105 (11): 4295–300. Bibcode:2008PNAS..105.4295G. doi:10.1073/pnas.0711986105. JSTOR 25461411. PMC 2393741. PMID 18332428. Lay summary – Reuters (March 10, 2008).
- ^ Gales BJ, Gales MA (2007). “Pyridostigmine in the treatment of orthostatic intolerance”. Annals of Pharmacotherapy. 41 (2): 314–8. doi:10.1345/aph.1H458. PMID 17284509. S2CID 22855759.
- ^ Jump up to:a b Gales BJ, Gales MA (February 2007). “Pyridostigmine in the treatment of orthostatic intolerance”. The Annals of Pharmacotherapy. 41 (2): 314–8. doi:10.1345/aph.1H458. PMID 17284509. S2CID 22855759.
- ^ Kanjwal K, Karabin B, Sheikh M, et al. (June 2011). “Pyridostigmine in the treatment of postural orthostatic tachycardia: a single-center experience”. Pacing and Clinical Electrophysiology. 34 (6): 750–5. doi:10.1111/j.1540-8159.2011.03047.x. PMID 21410722. S2CID 20405336.
- ^ Jump up to:a b c Mestinon | Home Archived 2008-05-13 at the Wayback Machine
- ^ Mestinon Official FDA information, side effects and uses Archived 2008-05-24 at the Wayback Machine
External links[
- “Pyridostigmine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Mestinon, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682229 |
| Pregnancy category | AU: C |
| Routes of administration | by mouth, intravenous |
| ATC code | N07AA02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 7.6 +/- 2.4% |
| Elimination half-life | 1.78 +/- 0.24hrs |
| Excretion | kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 155-97-5 |
| PubChem CID | 4991 |
| DrugBank | DB00545 |
| ChemSpider | 4817 |
| UNII | 19QM69HH21 |
| KEGG | D00487 |
| ChEMBL | ChEMBL1115 |
| CompTox Dashboard (EPA) | DTXSID20165786 |
| Chemical and physical data | |
| Formula | C9H13N2O2 |
| Molar mass | 181.215 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESO=C(Oc1ccc[n+](c1)C)N(C)C | |
| hideInChIInChI=1S/C9H13N2O2/c1-10(2)9(12)13-8-5-4-6-11(3)7-8/h4-7H,1-3H3/q+1 Key:RVOLLAQWKVFTGE-UHFFFAOYSA-N |
/////////////Pyridostigmine,
Buspirone
Buspirone
- Molecular FormulaC21H31N5O2
- Average mass385.503 Da
- буспиронبوسبيرون丁螺酮
251-489-4[EINECS]253-072-2[EINECS]36505-84-7[RN]8-[4-(4-Pyrimidin-2-yl-piperazin-1-yl)-butyl]-8-aza-spiro[4.5]decane-7,9-dione8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione
- 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione
- Buspin
- Buspirone
- Spitomin
BuspironeCAS Registry Number: 36505-84-7CAS Name: 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dioneMolecular Formula: C21H31N5O2Molecular Weight: 385.50Percent Composition: C 65.43%, H 8.11%, N 18.17%, O 8.30%Literature References: Non-benzodiazepine anxiolytic; 5-hydroxytryptamine (5-HT1) receptor agonist. Prepn: Y. H. Wu et al.,J. Med. Chem.15, 477 (1972); Y. H. Wu, J. W. Rayburn, DE2057845 (1971 to Bristol-Myers); eidem,US3717634 (1973 to Mead-Johnson). Pharmacology: L. E. Allen et al.,Arzneim.-Forsch.24, 917 (1974). Comparison with diazepam in treatment of anxiety: H. L. Goldberg, R. J. Finnerty, Am. J. Psychiatry136, 1184 (1979); A. F. Jacobson et al.,Pharmacotherapy5, 290 (1985). Nonsynergistic effect with alcohol: T. Seppala et al.,Clin. Pharmacol. Ther.32, 201 (1982). Disposition and metabolism: S. Caccia et al.,Xenobiotica13, 147 (1983). Series of articles on chemistry, pharmacology, addictive potential, and clinical trials: J. Clin. Psychiatry43, pp 1-116 (1982); on pharmacology, safety and clinical comparison with clorazepate: Am. J. Med.80, Suppl. 3B, 1-51 (1986). Review of pharmacology and therapeutic efficacy: K. L. Goa, A. Ward, Drugs32, 114-129 (1986). Review: M. W. Jann, Pharmacotherapy8, 100-116 (1988); D. P. Taylor, FASEB J.2, 2445-2452 (1988).
Derivative Type: HydrochlorideCAS Registry Number: 33386-08-2Trademarks: Ansial (Vita); Ansiced (Abello); Axoren (Glaxo Wellcome); Bespar (BMS); Buspar (BMS); Buspimen (Menarini); Buspinol (Zdravlje); Buspisal (Lesvi); Narol (Almirall)Molecular Formula: C21H31N5O2.HClMolecular Weight: 421.96Percent Composition: C 59.77%, H 7.64%, N 16.60%, O 7.58%, Cl 8.40%Properties: Crystals from abs ethanol, mp 201.5-202.5°. LD50 i.p. in rats: 136 mg/kg (Allen).Melting point: mp 201.5-202.5°Toxicity data: LD50 i.p. in rats: 136 mg/kg (Allen)
Therap-Cat: Anxiolytic.Keywords: Anxiolytic; Arylpiperazines; Serotonin Receptor Agonist.
Buspirone, sold under the brand name Buspar, among others, is a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder.[9][10] Benefits support its short term use.[11] It has not been found to be effective in treating psychosis.[9] It is taken by mouth, and it may take up to four weeks to have an effect.[9][10]
Common side effects of buspirone include nausea, headaches, dizziness, and difficulty concentrating.[9][11] Serious side effects may include hallucinations, serotonin syndrome, and seizures.[11] Its use in pregnancy appears to be safe but has not been well studied, while use during breastfeeding is not recommended.[11][12] It is a serotonin 5-HT1A receptor agonist.[2]
Buspirone was first made in 1968 and approved for medical use in the United States in 1986.[9][10] It is available as a generic medication.[11] In 2018, it was the 92nd most-commonly prescribed medication in the United States, with more than 8 million prescriptions.[13][14]
Medical uses
Anxiety
Buspirone is used for the short-term treatment of anxiety disorders or symptoms of anxiety.[15][16][17][18][19] It is generally less preferred than selective serotonin reuptake inhibitors (SSRIs).[10]
Buspirone has no immediate anxiolytic effects, and hence has a delayed onset of action; its full clinical effectiveness may require 2–4 weeks to manifest itself.[20] The drug has been shown to be similarly effective in the treatment of generalized anxiety disorder (GAD) to benzodiazepines including diazepam, alprazolam, lorazepam, and clorazepate.[2] Buspirone is not known to be effective in the treatment of other anxiety disorders besides GAD,[21] although there is some limited evidence that it may be useful in the treatment of social phobia as an adjunct to selective serotonin reuptake inhibitors (SSRIs).[2][22]
Other uses
Sexual dysfunction
There is some evidence that buspirone on its own may be useful in the treatment of hypoactive sexual desire disorder (HSDD) in women.[23]
Miscellaneous
Buspirone is not effective as a treatment for benzodiazepine withdrawal, barbiturate withdrawal, or alcohol withdrawal/delirium tremens.[24]
SSRI and SNRI antidepressants such as paroxetine and venlafaxine may cause jaw pain/jaw spasm reversible syndrome (although it is not common), and buspirone appears to be successful in treating bruxism on SSRI/SNRI-induced jaw clenching.[25][26]
Contraindications
Buspirone has these contraindications:[27][28]
- Hypersensitivity to buspirone
- Metabolic acidosis, as in diabetes
- Should not be used with MAO inhibitors
- Severely compromised liver and/or kidney function
Side effects
Main article: List of side effects of buspirone
Known side effects associated with buspirone include dizziness, headaches, nausea, nervousness, and paresthesia.[2] Buspirone is relatively well tolerated, and is not associated with sedation, cognitive and psychomotor impairment, muscle relaxation, physical dependence, or anticonvulsant effects.[2] In addition, buspirone does not produce euphoria[20] and is not a drug of abuse.[16]
It is unclear if there is a risk of tardive dyskinesia or other movement disorders with buspirone.[9]
Overdose
Buspirone appears to be relatively benign in cases of single-drug overdose, although no definitive data on this subject appear to be available.[29] In one clinical trial, buspirone was administered to healthy male volunteers at a dosage of 375 mg/day, and produced side effects including nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress.[15][16][18] In early clinical trials, buspirone was given at dosages even as high as 2,400 mg/day, with akathisia, tremor, and muscle rigidity observed.[30] Deliberate overdoses with 250 mg and up to 300 mg buspirone have resulted in drowsiness in about 50% of individuals.[30] One death has been reported in association with 450 mg buspirone together with alprazolam, diltiazem, alcohol, cocaine.[30]
Interactions
Buspirone has been shown in vitro to be metabolized by the enzyme CYP3A4.[8] This finding is consistent with the in vivo interactions observed between buspirone and these inhibitors or inducers of cytochrome P450 3A4 (CYP3A4), among others:[27]
- Itraconazole: Increased plasma level of buspirone
- Rifampicin: Decreased plasma levels of buspirone
- Nefazodone: Increased plasma levels of buspirone
- Haloperidol: Increased plasma levels of haloperidol
- Carbamazepine: Decreased plasma levels of buspirone
- Grapefruit: Significantly increases the plasma levels of buspirone.[31] See grapefruit–drug interactions.
- Fluvoxamine: Moderately increase plasma levels of buspirone.[32]
Elevated blood pressure has been reported when buspirone has been administered to patients taking monoamine oxidase inhibitors (MAOIs).[27]
Pharmacology
Pharmacodynamics
| Site | Ki (nM) | Species | Ref |
|---|---|---|---|
| 5-HT1A | 3.98–214 21 (median) | Human | [33][34] |
| 5-HT1B | >100,000 | Rat | [35] |
| 5-HT1D | 22,000–42,700 | Human | [36][37] |
| 5-HT2A | 138 759–1,300 | Human Rat | [38] [35][38] |
| 5-HT2B | 214 | Human | [38] |
| 5-HT2C | 490 1,100–6,026 | Human Rat/pig | [38] [35][38] |
| 5-HT3 | >10,000 | Rat | [39][40] |
| 5-HT4 | >10,000 | Rat | [40] |
| 5-HT6 | 398 | Mouse | [41] |
| 5-HT7 | 375–381 | Rat | [42][43] |
| α1 | 1,000 | Rat | [35] |
| α2 | 6,000 | Rat | [44] |
| α2A | 7.3 (1-PP) | Human | [35] |
| β | 8,800 | Rat | [35] |
| D1 | 33,000 | Rat | [35] |
| D2 | 484 240 | Human Rat | [45] [35] |
| D3 | 98 | Human | [45] |
| D4 | 29 | Human | [45] |
| mACh | 38,000 | Rat | [35] |
| GABAA (BDZ) | >100,000 | Rat | [35] |
| Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. |
Buspirone acts as an agonist of the serotonin 5-HT1A receptor with high affinity.[2][35] It is a partial agonist of both presynaptic 5-HT1A receptors, which are inhibitory autoreceptors, and postsynaptic 5-HT1A receptors.[2] It is thought that the main effects of buspirone are mediated via its interaction with the presynaptic 5-HT1A receptor, thus reducing the firing of serotonin-producing neurons.[2] Buspirone also has lower affinities for the serotonin 5-HT2A, 5-HT2B, 5-HT2C, 5-HT6, and 5-HT7 receptors.[33]
In addition to binding to serotonin receptors, buspirone is an antagonist of the dopamine D2 receptor with weak affinity.[2][35] It preferentially blocks inhibitory presynaptic D2 autoreceptors, and antagonizes postsynaptic D2 receptors only at higher doses.[2] In accordance, buspirone has been found to increase dopaminergic neurotransmission in the nigrostriatal pathway at low doses, whereas at higher doses, postsynaptic D2 receptors are blocked and antidopaminergic effects such as hypoactivity and reduced stereotypy, though notably not catalepsy, are observed in animals.[2] Buspirone has also been found to bind with much higher affinity to the dopamine D3 and D4 receptors, where it is similarly an antagonist.[45]
A major metabolite of buspirone, 1-(2-pyrimidinyl)piperazine (1-PP), occurs at higher circulating levels than buspirone itself and is known to act as a potent α2-adrenergic receptor antagonist.[44][46][47] This metabolite may be responsible for the increased noradrenergic and dopaminergic activity observed with buspirone in animals.[46][48] In addition, 1-PP may play an important role in the antidepressant effects of buspirone.[48] Buspirone also has very weak and probably clinically unimportant affinity for the α1-adrenergic receptor.[35][49] However, buspirone has been reported to have shown “significant and selective intrinsic efficacy” at the α1-adrenergic receptor expressed in a “tissue- and species-dependent manner”.[49]
Unlike benzodiazepines, buspirone does not interact with the GABAA receptor complex.[2][50]
Pharmacokinetics
Buspirone has a low oral bioavailability of 3.9% relative to intravenous injection due to extensive first-pass metabolism.[2] The time to peak plasma levels following ingestion is 0.9 to 1.5 hours.[2] It is reported to have an elimination half-life of 2.8 hours,[2] although a review of 14 studies found that the mean terminal half-life ranged between 2 and 11 hours, and one study even reported a terminal half-life of 33 hours.[4] Buspirone is metabolized primarily by CYP3A4, and prominent drug interactions with inhibitors and inducers of this enzyme have been observed.[7][8] Major metabolites of buspirone include 5-hydroxybuspirone, 6-hydroxybuspirone, 8-hydroxybuspirone, and 1-PP.[4][5][6] 6-Hydroxybuspirone has been identified as the predominant hepatic metabolite of buspirone, with plasma levels that are 40-fold greater than those of buspirone after oral administration of buspirone to humans.[5] The metabolite is a high-affinity partial agonist of the 5-HT1A receptor (Ki = 25 nM) similarly to buspirone, and has demonstrated occupancy of the 5-HT1A receptor in vivo.[5] As such, it is likely to play an important role in the therapeutic effects of buspirone.[5] 1-PP has also been found to circulate at higher levels than those of buspirone itself and may similarly play a significant role in the clinical effects of buspirone.[46][48]

Phase I Metabolism of buspirone in humans[51][52][8]
History
Buspirone was first synthesized, by a team at Mead Johnson, in 1968,[21] but was not patented until 1975.[54][55] It was initially developed as an antipsychotic drug acting on the D2 receptor, but was found to be ineffective in the treatment of psychosis; it was then used as an anxiolytic instead.[2] In 1986, Bristol-Myers Squibb gained FDA approval for buspirone in the treatment of GAD.[21][56] The patent placed on buspirone expired in 2001 and it is now available as a generic drug.
Society and culture

Buspar (buspirone) 10-mg tablets
Generic names
Buspirone is the INN, BAN, DCF, and DCIT of buspirone, while buspirone hydrochloride is its USAN, BANM, and JAN.[1][57][58][59]
Brand name
Buspirone was primarily sold under the brand name Buspar.[57][59] Buspar is currently listed as discontinued by the US Federal Drug Administration.[60] In 2010, in response to a citizen petition, the US FDA determined that Buspar was not withdrawn for sale because of reasons of safety or effectiveness.[61]
2019 shortage
Due to interrupted production at a Mylan Pharmaceuticals plant in Morgantown, West Virginia, the United States experienced a shortage of buspirone in 2019.[62]
Research
Some tentative research supports other uses such as the treatment of depression and behavioral problems following brain damage.[2]
Chemistry
Buspirone is a member of the azapirone chemical class, and consists of azaspirodecanedione and pyrimidinylpiperazine components linked together by a butyl chain.
Analogues
Structural analogues of buspirone include other azapirones like gepirone, ipsapirone, perospirone, and tandospirone.[53]
Synthesis

Buspirone synthesis:[54] DE 2057845 U.S. Patent 3,717,634 U.S. Patent 3,907,801 U.S. Patent 3,976,776
Alkylation of 1-(2-pyrimidyl)piperazine (1) with 3-chloro-1-cyanopropane (2, 4-chlorobutyronitrile) gives 3, which is reduced either by hydrogenation over Raney nickel catalyst, or with LAH. The resulting 1° amine (4) from the previous step is then reacted with 3,3-tetramethyleneglutaric anhydride (5, 8-Oxaspiro[4.5]decane-7,9-dione) in order to yield buspirone (6).
PAPERS
- Koziol, Anna E.; Acta Crystallographica, Section E: Structure Reports Online 2006, V62(12), Po5616-o5618
- Mou, Jie; Organic Preparations and Procedures International 2008, V40(4), P391-394
- Kairisalo, Pekka Juhani; FI 72975 B 1987
- Journal of medicinal chemistry (1983), 26(2), 194-203
- Journal of medicinal chemistry (1986), 29(8), 1476-82.
- Medicinal research reviews (1990), 10(3), 283-326.
- Heterocycles (1993), 36(7), 1463-9
- Journal of medicinal chemistry (1996), 39(5), 1125-9.
- Journal of medicinal chemistry (1996), 39(16), 3195-202.
- Nature Catalysis, 3(10), 843-850; 2020
PAPER
https://pubs.rsc.org/en/content/articlelanding/2019/GC/C8GC03328E#!divAbstract
- Green Chemistry, 21(1), 59-63; 2019
Abstract
A continuous flow method for the direct conversion of alcohols to amines via a hydrogen borrowing approach is reported. The method utilises a low loading (0.5%) of a commercial catalyst system ([Ru(p-cymene)Cl2]2 and DPEPhos), reagent grade solvent and is selective for primary alcohols. Successful methylation of amines using methanol and the direct dimethylamination of alcohols using commercial dimethylamine solution are reported. The synthesis of two pharmaceutical agents Piribedil (5) and Buspirone (25) were accomplished in good yields employing these new methods.

http://www.rsc.org/suppdata/c8/gc/c8gc03328e/c8gc03328e2.pdf
8-(4-hydroxybutyl)-8-azaspiro[4.5]decane-7,9-dione (23): A solution of 3,3-tetramethyleneglutaric anhydride (0.25 mol/L in THF) was combined in a tee piece with a solution of 4-amino-1-butanol (0.25 mol/L in THF) and reacted in a 20 mL reactor coil (stainless steel, 20 min residence time) heated at 250 °C. The output was concentrated in vacuo and the residue purified by column chromatography on silica gel to afford the product in 84% yield (Rf = 0.31, 63% DCM/AcOEt). 1H NMR (400 MHz, CDCl3) δ = 3.78 (t, J = 7.2 Hz, 2H), 3.65 (t, J = 6.0 Hz, 2H), 2.58 (s, 4H), 1.77 – 1.64 (m, 4H), 1.64 – 1.53 (m, 4H), 1.53 – 1.43 (m, 4H). 13C NMR (100 MHz, CDCl3) δ = 172.33, 62.28, 44.87, 39.47, 39.14, 37.54, 29.81, 24.35, 24.17. HRMS for [C13H22NO3] + calculated 240.1594 found 240.1605.

8-(4-(4-(pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione (Buspirone, 25): The flow system was flushed with THF, the back-pressure regulator was set to 50 bar, and the coil reactor heated to 250 °C. Then a solution (10 mL overall volume) containing 1-(2-pyrimidyl)piperazine (2 mmol), 8-(4-hydroxybutyl)- 8-azaspiro[4.5]decane-7,9-dione (23) (2 mmol), dichloro(p-cymene)ruthenium(II) dimer (0.08 mmol) and bis[(2- diphenylphosphino)phenyl] ether (DPEPhos, 0.17 mmol) was pumped at 0.8 ml/min through a heated coil (8 mL, Phoenix reactor). The output solution obtained in steady state (monitored using the FlowUV) was concentrated in vacuo and purified by column chromatography on silica gel to afford the desired product in 76% yield (Rf = 0.29, 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 4.7 Hz, 2H), 6.48 (t, J = 4.7 Hz, 1H), 3.84 (t, J = 5.1 Hz, 4H), 3.79 (t, J = 6.8 Hz, 2H), 2.60 (s, 4H), 2.50 (t, J = 5.1 Hz, 4H), 2.40 (t, J = 6.8 Hz, 2H), 1.79 – 1.65 (m, 4H), 1.65 – 1.42 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 172.19, 161.63, 157.68, 109.77, 58.31, 53.06, 44.92, 43.60, 39.48, 39.35, 37.56, 26.04, 24.19, 24.19. HRMS for [C21H32N5O2] + calculated 386.2551 found 386.2570.


PAPER
Organic Preparations and Procedures International, 40(4), 391-394; 2008
https://www.tandfonline.com/doi/abs/10.1080/00304940809458099



PATENTS
US 3907801
ES 526304
EP 395192
EP 565274
EP 634411
EP 680961
US 5521313
Indian Pat. Appl., 2011MU01860,
PATENTS
WO 2014152737
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152737
Syn
J Med Chem 1972,15(5),477-479
DE 2057845; FR 2073406; GB 1332194; US 3717634
The condensation of 1-(2-pyrimidinyl)piperazine (I) with 3-chloro-1-cyanopropane (II) by means of Na2CO3 in n-butanol gives 4-(2-pyrimidinyl)-1-(3-cyanopropyl)piperazine (III). This product is reduced with LiAlH4 or with H2 and Raney-Ni yielding 4-(2-pyrimidinyl)-1-(4-aminobutyl)piperazine (IV), which is finally condensed with 8-oxaspiro[4.5]decane-7,9-dione-(3,3-tetramethylene-glutaric anhydride) (V) in pyridine.
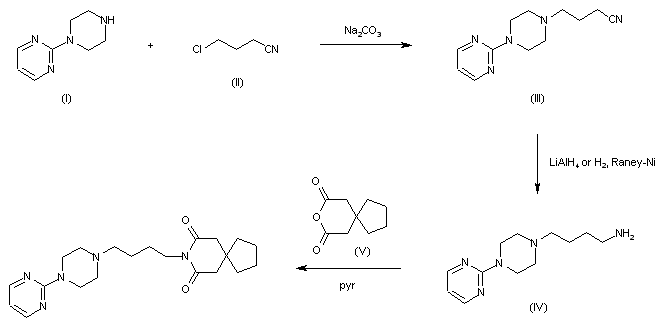
CLIP
Anxiolytics (Tranquilizers)
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Buspirone
Buspirone, 8-[4-[4-(2-pyrimidyl)-1-piperazinyl]butyl]-8-azaspiro [4,5] decan-7,9-dione (5.2.6), is synthesized by the reaction of 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) with 8-oxaspiro[4,5]decan-7,9-dione (5.2.5). In turn, 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) is synthesized by the reaction of 1-(2-pyrimidyl)piperazine with 4-chlorobutyronitrile, giving 4-(2-pyrimidyl)-1-(3-cyanopropyl)piperazine (5.2.3), which is hydrogenated with Raney nickel into buspirone (5.2.4) [51–55].

Buspirone is an extremely specific drug that could possibly represent a new chemical class of anxiolytics—azaspirones. As an anxiolytic, its activity is equal to that of benzodiazepines; however, it is devoid of anticonvulsant and muscle relaxant properties, which are characteristic of benzodiazepines. It does not cause dependence or addiction. The mechanism of its action is not conclusively known. It does not act on the GABA receptors, which occurs in benzodiazepine use; however, it has a high affinity for seratonin (5-HT) receptors and a moderate affinity for dopamine (D2) receptors. Buspirone is effective as an anxiolytic. A few side effects of buspirone include dizziness, drowsiness, headaches, nervousness, fatigue, and weakness. This drug is intended for treatment of conditions of anxiety in which stress, muscle pain, rapid heart rate, dizziness, fear, etc. are observed; in other words, conditions of anxiety not associated with somewhat common, usual, and everyday stress. Synonyms for buspirone are anizal, axoren, buspar, buspimen, buspinol, narol, travin, and others.
CLIP
Applications of Biocatalysis for Pharmaceuticals and Chemicals
Ramesh N. Patel, in Organic Synthesis Using Biocatalysis, 2016
5.2 Enzymatic Preparation of 6-Hydroxybuspirone
Buspirone (Buspar®, 59, Figure 11.17) is a drug used for the treatment of anxiety and depression, thought to produce its effects by binding to the serotonin 5HT1A receptor [114–116]. Mainly as a result of hydroxylation reactions, it is extensively converted to various metabolites and blood concentrations return to low levels a few hours after dosing [117]. A major metabolite, 6-hydroxybuspirone, produced by the action of liver cytochrome P450 CYP3A4, was present at much higher concentrations in human blood than buspirone itself. For development of 6-hydroxybuspirone as a potential antianxiety drug, preparation and testing of the two enantiomers as well as the racemate was of interest. An enantioselective microbial reduction process was developed for the reduction of 6-oxobuspirone 60 to (R)-6-hydroxybuspirone 61a or (S)-6-hydroxybuspitone 61b. About 150 microbial cultures were screened for the enantioselective reduction of 60. Rhizopus stolonifer SC 13898, Neurospora crassa SC 13816, Mucor racemosus SC 16198, and Pseudomonas putida SC 13817 gave >50% reaction yields and >95% ee of (S)-6-hydroxybuspirone 61a. The yeast strains Hansenula polymorpha SC 13845 and Candida maltosa SC 16112 gave (R)-6-hydroxybuspirone in >60% reaction yield and >97% ee [118]. The NADPH-dependent (R)-reductase (RHBR) from H. polymorpha SC 13845 was purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADPH required for reduction, glucose-6-phosphate dehydrogenase gene from Saccharomyces cerevisiae was cloned and coexpressed in the same E. coli strain. Recombinant cultures coexpressing (R)-reductase (RHBR) and glucose 6-phosphate dehydrogenase catalyzed the reduction of 6-ketobuspirone to (R)-6-hydroxybuspirone 61a in 99% yield and 99.9% ee at 50 g/L substrate input [119].

The NADH-dependent (S)-reductase (SHBR) from P. putida SC 16269 was also purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADH required for reduction, the NAD+ dependent formate dehydrogenase gene from Pichia pastoris was also cloned and co-expressed in the same E. coli strain. Recombinant E. coli coexpressing (S)-reductase and formate dehydrogenase was used to catalyze the reduction of 6-ketobuspirone to (S)-6-hydroxybuspirone 61b, in >98% yield and >99.8% ee at 50 g/L substrate input [119].
PATENT
https://patents.google.com/patent/US6686361
The present invention relates to methods of treating anxiety and depression using R-6-hydroxy-buspirone and pharmaceutical compositions containing R-6-hydroxy-buspirone.
Buspirone, chemically: 8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione, is approved for the treatment of anxiety disorders and depression by the United States Food and Drug Administration. It is available under the trade name BUSPAR® from Bristol-Myers Squibb Company.
Studies have shown that buspirone is extensively metabolized in the body. (See, for example, Mayol, et al., Clin. Pharmacol. Ther., 37, p. 210, 1985). One of the metabolites is 6-hydroxy-8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione having Formula I. This metabolite is also known as BMS 28674, BMS 442608, or

as 6-hydroxy-buspirone. This compound is believed to be the active metabolite of buspirone and its use in treating anxiety disorders and depression is disclosed in U.S. Pat. No. 6,150,365. The specific stereochemistry of 6-hydroxy-buspirone has not been described previously. Neither racemic 6-hydroxy-buspirone nor its enantiomers are commercially available at the present time.
Preclinical studies demonstrate that 6-hydroxy-buspirone, like buspirone, demonstrates a strong affinity for the human 5-HT1A receptor. In functional testing, 6-hydroxy-buspirone produced a dose-dependent anxiolytic response in the rat pup ultrasonic vocalization test, a sensitive method for assessment of anxiolytic and anxiogenic effects (Winslow and Insel, 1991, Psychopharmacology, 105:513-520).
Clinical studies in volunteers orally dosed with buspirone demonstrate that 6-hydroxy-buspirone blood plasma levels were not only 30 to 40 times higher but were sustained compared to buspirone blood plasma levels. The time course of 6-hydroxy-buspirone blood plasma levels, unlike buspirone blood plasma levels, correlate more closely with the sustained anxiolytic effect seen following once or twice a day oral dosing with buspirone.
Although buspirone is an effective treatment for anxiety disorders and depression symptomatology in a significant number of patients treated, about a third of patients get little to no relief from their anxiety and responders often require a week or more of buspirone treatment before experiencing relief from their anxiety symptomatology. Further, certain adverse effects are reported across the patient population. The most commonly observed adverse effects associated with the use of buspirone include dizziness, nausea, headache, nervousness, lightheadedness, and excitement. Also, since buspirone can bind to central dopamine receptors, concern has been raised about its potential to cause unwanted changes in dopamine-mediated neurological functions and a syndrome of restlessness, appearing shortly after initiation of oral buspirone treatment, has been reported in small numbers of patients. While buspirone lacks the prominent sedative effects seen in more typical anxiolytics such as the benzodiazepines, patients are nonetheless advised against operating potentially dangerous machinery until they experience how they are affected by buspirone.
It can be seen that it is desirable to find a medicament with buspirone’s advantages but which demonstrates more robust anxiolytic potency with a lack of the above described adverse effects.
Formation of 6-hydroxy-buspirone occurs in the liver by action of enzymes of the P450 system, specifically CYP3A4. Many substances such as grapefruit juice and certain other drugs; e.g. erythromycin, ketoconazole, cimetidine, etc., are inhibitors of the CYP3A4 isozyme and may interfere with the formation of this active metabolite from buspirone. For this reason it would be desirable to find a compound with the advantages of buspirone but without the drug—drug interactions when coadministered with agents affecting the activity level of the CYP3A4 isozyme.
EXAMPLE 3One-Step Synthesis of 6-Hydroxy-buspirone (I)
Buspirone (19.3 g, 50 mmole) was dissolved in dry THF (400 mL) and the resulting solution was cooled to −78° C. A solution of KN(SiMe3)2 in toluene (100 mL, 1 M) was added slowly. After the reaction mixture was stirred at −78° C. for 1 h, a solution of 2-(phenylsulfonyl)-3-phenyloxaziridine (Davis reagent, prepared according to literature method: F. A. Davis, et al., Org. Synth., 1988, 66, 203) (17.0 g, 65 mmole) in dry THF (150 mL, precooled to −78° C.) was added quickly via a cannular. After stirred for 30 mins at −78° C., the reaction was quenched with 1 N HCl solution (500 mL). It was extracted with EtOAc (3×500 mL). The aqueous layer was separated, neutralized with saturated sodium bicarbonate solution, and extracted with EtOAc (3×500 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a white solid residue which was subjected to column chromatography using CH2Cl2/MeOH/NH4OH (200:10:1) as the eluent to give pure 6-hydroxy-buspirone (I, 7.2 g) and a mixture of buspirone and 6-hydroxy-buspirone (I). The mixture was purified by above column chromatography to afford another 3.3 g of pure 6-hydroxy-buspirone (I).
1H NMR (CDCl3) δ8.30 (d, J=4.7 Hz, 2H), 6.48 (t, J=4.7 Hz, 1H), 4.20 (s, 1H), 3.83-3.72 (m, 5H), 3.55 (s, 1H), 2.80 (d, J=17.5 Hz, 1H), 2.55-2.40 (m, 7H), 2.09-2.03 (m, 1H), 1.76-1.54 (m, 10 H), 1.41-1.36 (m, 1H), 1.23-1.20 (m, 1H).
References
- ^ Jump up to:a b Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 192–. ISBN 978-1-4757-2085-3.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r Loane C, Politis M (June 2012). “Buspirone: what is it all about?”. Brain Research. 1461: 111–8. doi:10.1016/j.brainres.2012.04.032. PMID 22608068. S2CID 11734819.
- ^ Jump up to:a b c “buspirone (Rx) – BuSpar, Buspirex, more.” Medscape Reference. WebMD. Retrieved 14 November 2013.
- ^ Jump up to:a b c Gammans RE, Mayol RF, LaBudde JA (March 1986). “Metabolism and disposition of buspirone”. The American Journal of Medicine. 80 (3B): 41–51. doi:10.1016/0002-9343(86)90331-1. PMID 3515929.
- ^ Jump up to:a b c d e Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 490–. ISBN 978-1-58562-309-9.
- ^ Jump up to:a b Wong H, Dockens RC, Pajor L, Yeola S, Grace JE, Stark AD, et al. (August 2007). “6-Hydroxybuspirone is a major active metabolite of buspirone: assessment of pharmacokinetics and 5-hydroxytryptamine1A receptor occupancy in rats”. Drug Metabolism and Disposition. 35 (8): 1387–92. doi:10.1124/dmd.107.015768. PMID 17494642. S2CID 25558546.
- ^ Jump up to:a b c Mahmood I, Sahajwalla C (April 1999). “Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug”. Clinical Pharmacokinetics. 36 (4): 277–87. doi:10.2165/00003088-199936040-00003. PMID 10320950. S2CID 1102318.
- ^ Jump up to:a b c d Zhu M, Zhao W, Jimenez H, Zhang D, Yeola S, Dai R, et al. (April 2005). “Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes”. Drug Metabolism and Disposition. 33 (4): 500–7. doi:10.1124/dmd.104.000836. PMID 15640381. S2CID 10142905.
- ^ Jump up to:a b c d e f “Buspirone Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b c d Wilson, T. K.; Tripp, J. (January 2018). “Buspirone”. StatPearls. PMID 30285372.
- ^ Jump up to:a b c d e British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 338. ISBN 9780857113382.
- ^ “Buspirone Pregnancy and Breastfeeding Warnings”. Drugs.com. Retrieved 3 March 2019.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Buspirone Hydrochloride – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b “BUSPIRONE HCL (buspirone hydrochloride) tablet [Watson Laboratories, Inc.]”. DailyMed. Watson Laboratories, Inc. July 2013. Retrieved 14 November 2013.
- ^ Jump up to:a b c “BUSPAR® (buspirone hydrochloride) Tablets 5 mg & 10 mg PRODUCT INFORMATION” (PDF). TGA eBusiness Services. Aspen Pharma Pty Ltd. January 2010. Retrieved 14 November2013.
- ^ Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Jump up to:a b “Buspirone 10mg Tablets”. electronic Medicines Compendium. Actavis UK Ltd. 10 September 2012. Retrieved 14 November 2013.
- ^ Joint Formulary Committee. British National Formulary (BNF). Pharmaceutical Press. p. 224.
- ^ Jump up to:a b Sadock BJ, Sadock VA, Ruiz P (22 September 2014). Kaplan and Sadock’s Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry. Wolters Kluwer Health. pp. 3211–. ISBN 978-1-4698-8375-5.
- ^ Jump up to:a b c Howland RH (November 2015). “Buspirone: Back to the Future”. Journal of Psychosocial Nursing and Mental Health Services. 53 (11): 21–4. doi:10.3928/02793695-20151022-01. PMID 26535760.
- ^ Masdrakis VG, Turic D, Baldwin DS (2013). “Pharmacological treatment of social anxiety disorder”. Anxiety Disorders. Modern Trends in Pharmacopsychiatry. 29. pp. 144–53. doi:10.1159/000351960. ISBN 978-3-318-02463-0. PMID 25225024.
- ^ Goldstein I, Kim NN, Clayton AH, DeRogatis LR, Giraldi A, Parish SJ, et al. (January 2017). “Hypoactive Sexual Desire Disorder: International Society for the Study of Women’s Sexual Health (ISSWSH) Expert Consensus Panel Review”. Mayo Clinic Proceedings. 92 (1): 114–128. doi:10.1016/j.mayocp.2016.09.018. PMID 27916394.
- ^ Sontheimer DL, Ables AZ (March 2001). “Is imipramine or buspirone treatment effective in patients wishing to discontinue long-term benzodiazepine use?”. The Journal of Family Practice. 50(3): 203. PMID 11252203.
- ^ Garrett AR, Hawley JS (April 2018). “SSRI-associated bruxism: A systematic review of published case reports”. Neurology. Clinical Practice. 8 (2): 135–141. doi:10.1212/CPJ.0000000000000433. PMC 5914744. PMID 29708207.
- ^ Prisco V, Iannaccone T, Di Grezia G (2017-04-01). “Use of buspirone in selective serotonin reuptake inhibitor-induced sleep bruxism”. European Psychiatry. Abstract of the 25th European Congress of Psychiatry. 41: S855. doi:10.1016/j.eurpsy.2017.01.1701.
- ^ Jump up to:a b c “Buspirone monograph”. Drugs.com. Retrieved 2011-08-27.
- ^ Geddes J, Gelder MG, Mayou R (2005). Psychiatry. Oxford [Oxfordshire]: Oxford University Press. p. 237. ISBN 978-0-19-852863-0.
- ^ Fulton B, Brogden RN (1997). “Buspirone”. CNS Drugs. 7 (1): 68–88. doi:10.2165/00023210-199707010-00007. ISSN 1172-7047.
- ^ Jump up to:a b c Dart RC (2004). Medical Toxicology. Lippincott Williams & Wilkins. pp. 886–. ISBN 978-0-7817-2845-4.
- ^ Lilja JJ, Kivistö KT, Backman JT, Lamberg TS, Neuvonen PJ (December 1998). “Grapefruit juice substantially increases plasma concentrations of buspirone”. Clinical Pharmacology and Therapeutics. 64 (6): 655–60. doi:10.1016/S0009-9236(98)90056-X. PMID 9871430. S2CID 22009095.
- ^ Lamberg TS, Kivistö KT, Laitila J, Mårtensson K, Neuvonen PJ (1998). “The effect of fluvoxamine on the pharmacokinetics and pharmacodynamics of buspirone”. European Journal of Clinical Pharmacology. 54 (9–10): 761–6. doi:10.1007/s002280050548. PMID 9923581. S2CID 21939719.
- ^ Jump up to:a b c Roth BL, Driscol J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- ^ Boess FG, Martin IL (1994). “Molecular biology of 5-HT receptors”. Neuropharmacology. 33 (3–4): 275–317. doi:10.1016/0028-3908(94)90059-0. PMID 7984267. S2CID 35553281.
- ^ Jump up to:a b c d e f g h i j k l m Hamik A, Oksenberg D, Fischette C, Peroutka SJ (July 1990). “Analysis of tandospirone (SM-3997) interactions with neurotransmitter receptor binding sites”. Biological Psychiatry. 28 (2): 99–109. doi:10.1016/0006-3223(90)90627-e. PMID 1974152. S2CID 25608914.
- ^ Peroutka SJ, Switzer JA, Hamik A (1989). “Identification of 5-hydroxytryptamine1D binding sites in human brain membranes”. Synapse. 3 (1): 61–6. doi:10.1002/syn.890030109. PMID 2521959.
- ^ Waeber C, Schoeffter P, Palacios JM, Hoyer D (June 1988). “Molecular pharmacology of 5-HT1D recognition sites: radioligand binding studies in human, pig and calf brain membranes”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 337 (6): 595–601. doi:10.1007/bf00175783. PMID 2975354. S2CID 21344978.
- ^ Jump up to:a b c d e Bonhaus DW, Weinhardt KK, Taylor M, DeSouza A, McNeeley PM, Szczepanski K, et al. (1997). “RS-102221: a novel high affinity and selective, 5-HT2C receptor antagonist”. Neuropharmacology. 36 (4–5): 621–9. doi:10.1016/s0028-3908(97)00049-x. PMID 9225287. S2CID 24930608.
- ^ Nelson DR, Thomas DR (May 1989). “[3H]-BRL 43694 (Granisetron), a specific ligand for 5-HT3 binding sites in rat brain cortical membranes”. Biochemical Pharmacology. 38 (10): 1693–5. doi:10.1016/0006-2952(89)90319-5. PMID 2543418.
- ^ Jump up to:a b Borsini F, Giraldo E, Monferini E, Antonini G, Parenti M, Bietti G, Donetti A (September 1995). “BIMT 17, a 5-HT2A receptor antagonist and 5-HT1A receptor full agonist in rat cerebral cortex”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 352 (3): 276–82. doi:10.1007/bf00168557. PMID 8584042. S2CID 19340842.
- ^ Plassat JL, Amlaiky N, Hen R (August 1993). “Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase”. Molecular Pharmacology. 44 (2): 229–36. PMID 8394987.
- ^ Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, et al. (September 1993). “A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms”. Neuron. 11 (3): 449–58. doi:10.1016/0896-6273(93)90149-l. PMID 8398139. S2CID 28729004.
- ^ Ruat M, Traiffort E, Leurs R, Tardivel-Lacombe J, Diaz J, Arrang JM, Schwartz JC (September 1993). “Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation”. Proceedings of the National Academy of Sciences of the United States of America. 90 (18): 8547–51. Bibcode:1993PNAS…90.8547R. doi:10.1073/pnas.90.18.8547. PMC 47394. PMID 8397408.
- ^ Jump up to:a b Blier P, Curet O, Chaput Y, de Montigny C (July 1991). “Tandospirone and its metabolite, 1-(2-pyrimidinyl)-piperazine–II. Effects of acute administration of 1-PP and long-term administration of tandospirone on noradrenergic neurotransmission”. Neuropharmacology. 30 (7): 691–701. doi:10.1016/0028-3908(91)90176-c. PMID 1681447. S2CID 44297577.
- ^ Jump up to:a b c d Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P (March 2013). “Modification of cocaine self-administration by buspirone (buspar®): potential involvement of D3 and D4 dopamine receptors”. The International Journal of Neuropsychopharmacology. 16 (2): 445–58. doi:10.1017/S1461145712000661. PMC 5100812. PMID 22827916.
- ^ Jump up to:a b c Tunnicliff G (September 1991). “Molecular basis of buspirone’s anxiolytic action”. Pharmacology & Toxicology. 69 (3): 149–56. doi:10.1111/j.1600-0773.1991.tb01289.x. PMID 1796057.
- ^ Zuideveld KP, Rusiç-Pavletiç J, Maas HJ, Peletier LA, Van der Graaf PH, Danhof M (December 2002). “Pharmacokinetic-pharmacodynamic modeling of buspirone and its metabolite 1-(2-pyrimidinyl)-piperazine in rats”. The Journal of Pharmacology and Experimental Therapeutics. 303 (3): 1130–7. doi:10.1124/jpet.102.036798. PMID 12438536. S2CID 14139919.
- ^ Jump up to:a b c Fava M (2007). “The combination of buspirone and bupropion in the treatment of depression”. Psychotherapy and Psychosomatics. 76 (5): 311–2. doi:10.1159/000104708. PMID 17700052. S2CID 46284917.
- ^ Jump up to:a b Stern TA, Fava M, Wilens TE, Rosenbaum JF (27 April 2015). Massachusetts General Hospital Psychopharmacology and Neurotherapeutics E-Book. Elsevier Health Sciences. pp. 29–. ISBN 978-0-323-41323-7.
- ^ Nutt DJ, Ballenger JC (15 April 2008). Anxiety Disorders. John Wiley & Sons. pp. 395–. ISBN 978-0-470-98683-7.
- ^ Dockens RC, Salazar DE, Fulmor IE, Wehling M, Arnold ME, Croop R (November 2006). “Pharmacokinetics of a newly identified active metabolite of buspirone after administration of buspirone over its therapeutic dose range”. Journal of Clinical Pharmacology. 46(11): 1308–12. doi:10.1177/0091270006292250. PMID 17050795.
- ^ Jajoo HK, Mayol RF, LaBudde JA, Blair IA (1989). “Metabolism of the antianxiety drug buspirone in human subjects”. Drug Metabolism and Disposition. 17 (6): 634–40. PMID 2575499.
- ^ Taylor DP, Moon SL (July 1991). “Buspirone and related compounds as alternative anxiolytics”. Neuropeptides. 19 Suppl: 15–9. doi:10.1016/0143-4179(91)90078-w. PMID 1679210. S2CID 13730683.
- ^ Jump up to:a b Allen LE, Ferguson HC, Kissel JW (May 1972). “Psychosedative agents. 2. 8-(4-Substituted 1-piperazinylalkyl)-8-azaspiro(4.5)decane-7,9-diones”. Journal of Medicinal Chemistry. 15 (5): 477–9. doi:10.1021/jm00275a009. PMID 5035267.
- ^ US Patent 3907801 N-(8 (4-pyridyl-piperazino)-alkyl(9 -azaspiroalkanediones
- ^ United States Federal Drug Administration (September 9, 1986). Approval Type-1 New Molecular Entry.https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/018731Orig1s000rev.pdf
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 149–. ISBN 978-3-88763-075-1.
- ^ Morton IK, Hall JM (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 57–. ISBN 978-94-011-4439-1.
- ^ Jump up to:a b “Buspirone”.
- ^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 2019-09-20.
- ^ “Determination That BUSPAR (Buspirone Hydrochloride) Tablets, 10 Milligrams, 15 Milligrams, and 30 Milligrams, Were Not Withdrawn From Sale for Reasons of Safety or Effectiveness”. Federal Register. 2010-10-19. Retrieved 2019-09-20.
- ^ Rabin RC (2019-02-01). “Shortage of Anxiety Drug Leaves Patients Scrambling”. The New York Times. ISSN 0362-4331. Retrieved 2019-09-20.
External links
- Media related to Buspirone at Wikimedia Commons
- “Buspirone”. Drug Information Portal. U.S. National Library of Medicine.
////////////Buspirone, буспирон , بوسبيرون , 丁螺酮 , Anxiolytic,Arylpiperazines, Serotonin Receptor Agonist, Ansial, Vita, Ansiced, Abello, Axoren, Glaxo Wellcome, Bespar, BMS, Buspar, Buspimen, Menarini, Buspinol, Zdravlje, Buspisal, Lesvi, Narol, Almirall,
#Buspirone, #буспирон , #بوسبيرون , #丁螺酮 , #Anxiolytic, #Arylpiperazines, #Serotonin Receptor Agonist, #Ansial, #Vita, #Ansiced, #Abello, #Axoren, #Glaxo Wellcome, #Bespar, #BMS, #Buspar, #Buspimen, Menarini, Buspinol, Zdravlje, Buspisal, Lesvi, Narol, Almirall,
Azelnidipine
Azelnidipine
C33H34N4O6, 582.6 g/mol
CAS 123524-52-7
3-(1-Benzhydrylazetidin-3-yl) 5-isopropyl 2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate
CS-905, RS-9054
Approved India cdsco 2020
SYN REF https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4245158/
MP 95-98 °C AND NMR WO 2004058745 . EP 266922
Azelnidipine is a dihydropyridine calcium channel blocker. It is marketed by Daiichi-Sankyo pharmaceuticals, Inc. in Japan. It has a gradual onset of action and produces a long-lasting decrease in blood pressure, with only a small increase in heart rate, unlike some other calcium channel blockers. It is currently being studied for post-ischemic stroke management.
Azelnidipine (INN; marketed under the brand name CalBlock — カルブロック) is a dihydropyridine calcium channel blocker. Azelnidipine is L and T calcium channel blocker. It is sold in Japan by Daiichi-Sankyo pharmaceuticals, Inc. Unlike nicardipine, it has a gradual onset and has a long-lasting hypotensive effect, with little increase in heart rate. Drug Controller General Of India (DCGI) has approved the use of azelnipine in India. It is launched under the brand name Azusa (ajanta pharma ltd.)[1] In 2020.
Chemical Synthesis
A solution of benzhydrylamine (46) and epichlorohydrin (47) was mixed without adding solvent to give azetidinol 48 in 57% yield. DCC coupling between cyanoacetic acid (49) and azetidinol 48 in hot THF gave ester 50 in 93% yield. Cyanoester 50 was treated with ethanol and HCl gas in chloroform to give imidate HCl salt 51, which was treated with ammonia gas in chloroform and ammonium acetate in acetonitrile to give the corresponding amidinoacetate 52. A modified Hantzsch reaction was employed to construct the 2-amino-1,4- dihydropyridine core structure. Compound 52 was condensed with 2-(3-nitrobenzylidene)acetic acid isopropyl ester (55) in the presence of NaOMe in refluxing isopropanol to give the cyclized product, azelnidipine (V) in 74% yield. Benzylideneacetoacetate 55 was obtained through the Knoevenagel reaction employing 3-nitrobenzaldehyde (53) and isopropyl acetoacetate (54) in isopropanol containing a catalytic amount of piperidinium acetate at 45-55oC in 65% yield.
PATENT
EP 266922
IN 201621044802
CN 106279109
CN 107188885
CN 105461691
CN 103509003
CN 103183663
CN 102382104
JP 2012020970 A
PAPER
Bioanalysis (2019), 11(4), 251-266.
PAPER
Asian Journal of Chemistry (2014), 26(15), 4675-4678.
PAPER
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=26_16_30

Azelnidipine is designated chemically as 3-(1-benzhydrylazetidin-3-yl)-5-isopropyl-2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate. Its literature synthesis (Scheme-I) involves 3-nitrobenzaldehyde 5 with isopropyl acetoacetate 6. The product of (Z)-isopropyl 2-(3- nitrobenzylidene)-3-oxobutanoate (7a, b, c), on treatment with piperidine and acetic acid, coupling of (7) and 1-benzhydrylazetidin-3-yl 3-amino-3-iminopropanoate acetate (8) gave azelnidipine (1).
PAPER
International Research Journal of Pharmacy (2012), 3(8), 191-192.
Chemical & Pharmaceutical Bulletin (1995), 43(5), 797-817.
PATENT
https://patents.google.com/patent/WO2014139410A1/en
The invention belongs to the technical field of medicine and provides an important intermediate of dihydropyridine calcium antagonist adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidine The synthesis process of ester acetate. Background technique
Azelnidipine is a new type of dihydropyridine calcium channel blocker developed by Sankyo and Ube Industries of Japan. It was approved for sale in Japan in late May 2003 under the trade name Calblock. Adipine has a selective blockade of calcium channels in arterial smooth muscle cells, it can dilate blood vessels, reduce peripheral vascular resistance and arterial pressure, and is widely used clinically for mild or moderate essential hypertension, renal disorders with hypertension And treatment of severe hypertension. Compared with nicardipine and nifedipine dihydropyridine calcium channel blockers, adipine is superior in selectivity, long-lasting and long-lasting, and has little effect on the heart.

阿折地平的结构式
A flat floor structure
At present, references to the preparation of agdipine include: European patents EP0266922; Chinese patent CN201010516967.7; Chinese Journal of Medicinal Chemistry, 2010, 20 (3): 192-194; Chinese Journal of Pharmaceutical Industry, 2008, 39 (3): 163-165; Chemical Industry and Engineering, 2009, 26 ( 1 ): 15-18; Qilu Pharmacy, 2005, 24 (6): 365-366. The preparation method of adipine in these literatures is based on the reaction of epichlorohydrin and diphenylamine with N-alkylation, cyclization, esterification, Pinner synthesis, neutralization, and oxime reaction. The intermediate 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3-azetidinyl acetate is prepared first, followed by 2-(3-nitrobenzylidene)acetyl Acepinedipine was obtained by the Hantzsch condensation of isopropyl acetate.
The control of the solvent and reaction conditions in the esterification, Pinner synthesis and neutralization three-step reaction in this route is critical. Using the preparation methods provided by these documents, we found that the operation was cumbersome and the yield and purity were not satisfactory.
In the esterification reaction, according to the method specifically reported in the above literature, the highest yield of the obtained product is only 85%, and the purity is poor, it is difficult to purify, and it is difficult to obtain a solid product.

副产物 (7 )和(8 )结构式 发明内容 We have found that 3-amino-3-iminopropionic acid-1- (3) is prepared by a three-step reaction from cyanoacetate-1-diphenylhydrazin-3-azetidinyl ester (3) according to the method specifically reported in the above literature. Diphenylhydrazino)-3-azetidinyl acetate (6), the reaction operation is cumbersome, and it is easy to produce by-products of hydrolysis of ester bonds and hydrolysis of imid bonds (7) and (8), three-step reaction. The total yield is only 20~30%, and the purification of the product is difficult, which seriously affects the quality of the final product and greatly increases the production cost.
Byproducts (7) and (8) structural formula Summary of the invention
It is an object of the present invention to provide a process for the preparation of the key intermediate of adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate. The adipine intermediate of the present invention 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate acetate has the following structural formula:

The preparation method of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate of the present invention comprises the following steps: 1) Esterification: 1-diphenylhydrazin-3-azetidinol (2), cyanoacetic acid (1) and N,N-dicyclohexylcarbodiimide (DCC) in organic solvent at 0~ Reacting at 80 ° C, to obtain 7-diphenylindolyl-3-azetidinyl cyanoacetate (3);
2) Pinner reaction: Add intermediate (3), absolute ethanol to dichlorosilane, stir and cool
To -20~25 °C, dry hydrogen chloride gas is passed, and then the reaction solution is kept sealed at -20~25 °C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl) -3-azetidinyl ester hydrochloride (4);
3) Neutralization reaction: The intermediate (4) is dissolved in dichloromethane, and the base is added at -5 to 25 ° C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylhydrazine). Benzyl-3-azetidinyl ester (5);
4) Formation reaction: The intermediate (5) is dissolved in acetonitrile, ammonium acetate is added, and the temperature is raised to 40 to 60 ° C to obtain 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3. – azetidinium acetate compound (6). detailed description
Example
1. Preparation of cyanoacetic acid-1-diphenylhydrazine-3-azetidine (esterification)

Method 1: Add 1-diphenylhydrazin-3-azetidinol (2, 235 g, 0.983 mol) and cyanoacetic acid (1, 100 g, 1.18 mol) to 1.5 mL of dichloromethane, and stir until fully dissolved. Ν, Ν-dicyclohexylcarbodiimide (DCC, 243 g, 1.18 mol) was added at 0-10 ° C and allowed to react at room temperature for 3 h. After the completion of the reaction, the reaction mixture was cooled to 0 to 5 ° C, and filtered, filtered, washed with a small portion of dichloromethane. The organic solvent was evaporated to dryness under reduced pressure and dried to give 275 g of white solid.
Method 2: chloroform was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 5 hours, the HPLC purity was 98.7%, and the product yield was 95.3%.
Method 3: Ethyl acetate was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 2 h, the HPLC purity was 98.9%, and the product yield was 96.1%.

Method 4: Using hydrazine as the reaction solvent, the operation was the same as above, and the reaction was carried out at 55 ° C for 7 h, the HPLC purity was 98.5%, and the product yield was 94.7%. 2. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester hydrochloride (Pinner reaction)
Intermediate 3 (270 g, 0.882 mol), absolute ethanol (61.8 mL, 1.06 mol) was added to 1.5 L of dry dichloromethane, cooled to -5 to 0 ° C in a water salt bath, and dried. HC1 gas for 2.5 h, after the completion of the aeration, the reaction solution was kept under stirring at 0 ° C for 6 h.
Allow to stand overnight at 0-4 °C. After completion of the reaction, the solvent was evaporated under reduced pressure to give an oily viscous intermediate 4 .
3. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester

Method 1: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add dry diethylamine (182 mL, 1.76 mol) to the solution, adjust pH 7-8, continue to stir after the dropwise addition. 2h. The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo.
Method 2: Diamine is used for neutralization, and the operation is the same as above.
Method 3: Triethylamine is used for neutralization, and the operation is the same as above.
Method 4: Ethylenediamine is used for neutralization, and the operation is the same as above.
Method 5: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add potassium carbonate (242.88 g, 1.76 mol) to the solution in portions, adjust pH 7-8, continue stirring for 2 h. . The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo. Method 6: Neutralize with sodium carbonate, and operate as above.
Method 7: Neutralize with sodium hydroxide, and operate as above.

4. Preparation of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate (formed into 脒)
To the intermediate 5, 1.2 L of acetonitrile was added, and after dissolution, ammonium acetate (68.0 g, 0.882 mol) was added, and the mixture was heated to 55 ° C for 6 h. After the reaction, it was naturally cooled, crystallization, suction filtration, acetonitrile washing cake, and dried to give 236 g of a white solid. The total yield of the three-step reaction was 69.9 73.1%.
PAPER
https://pubs.rsc.org/en/content/articlelanding/2015/cc/c4cc09337b#!divAbstract
Abstract
A protocol for the coupling of 3-iodoazetidines with Grignard reagents in the presence of an iron catalyst has been developed. A variety of aryl, heteroaryl, vinyl and alkyl Grignards were shown to participate in the coupling process to give the products in good to excellent yields. Furthermore, a short formal synthesis towards a pharmacologically active molecule was shown.

http://www.rsc.org/suppdata/cc/c4/c4cc09337b/c4cc09337b1.pdfPATENThttps://patents.google.com/patent/CN103509003A/zhAzelnidipine, whose chemical name is 3-(1-diphenylmethylazetidin-3-yl) 5-isopropyl 2-amino-1,4-dihydro-6-methyl 4-(3-nitrophenyl)-3,5-pyridinedicarboxylate, developed by Japan Sankyo Co., Ltd. and approved to be marketed in Japan in late May 2003. The existing synthesis method of azedipine is cumbersome, and the preparation of intermediate (VI) adopts column chromatography method, and the purification of product (I) also uses column chromatography method, which is not suitable for industrial production.
A method for preparing azeldipine, which is characterized in that it is prepared by the following steps.
[0006]

Description of the drawings:
Figure 1 is a flow chart of the synthesis process of azeldipine.
[0025] Example 12-Preparation of (3-nitrobenzylidene) isopropyl acetoacetate (III)
[0026] Add 2.1kg of 3-nitrobenzaldehyde and 5L of isopropanol to the reaction kettle, start stirring, add 3kg of isopropyl acetoacetate, and stir. Add 43ml of anhydrous piperidine and 12ml of glacial acetic acid, and continue to stir until the solid is completely dissolved. Heat the temperature to 45°C and keep the reaction for 6h, then lower the temperature, stir and crystallize for 16h. Filter and collect the resulting filter cake. Put the obtained filter cake and 16L ethanol (industrial) into the reaction kettle, start stirring, beating, filtering, and collecting the filter cake. Put the filter cake in the baking tray, put it in the oven, and dry at 70-80°C. Collect the product 2-(3-nitrobenzylidene) isopropyl acetoacetate (III), about 2.7 kg.
[0027] Example 21-Preparation of benzhydryl-3-hydroxyazetidine (Intermediate V)
[0028] 9.6L of methanol, 5.4kg of benzhydrylamine (IV) and 3.33kg of epichlorohydrin were added to the reaction kettle, stirred at room temperature for 48 hours, the reaction was completed, the temperature was raised to 68°C, and the reaction was refluxed for 72h. Cool to room temperature. Concentrate under reduced pressure to remove methanol, and collect the filter cake by filtration. The filter cake was put into the reaction kettle, 19.2L of ether and 13.75L of 3mol/L NaOH solution were added, stirred, and the water layer was released after standing still. The ether layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was collected. The ether was recovered under reduced pressure to dryness to obtain about 3.05 kg of 1-benzyl-3-hydroxyazetidine (Intermediate V).
[0029] Example 3 Preparation of cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI)
[0030] Put about 3.05g of intermediate (V), 27L of tetrahydrofuran and 1.7kg of cyanoacetic acid into the reactor, start stirring, turn on the chilled water of the reactor to cool down, and slowly add 3.1kgN, N’-dicyclohexyl to the reactor Diimine, control the temperature at IO0C -15°C, after the addition, close the chilled water in the reactor. Turn on the heating system, slowly increase the temperature to 55-60°C, and react for 10 hours. The material liquid was cooled to room temperature, filtered, and the filtrate was concentrated to dryness. Put 16.8L of ethyl acetate into the reaction kettle, stir to dissolve, then wash with water, dry with anhydrous sodium sulfate, filter, and collect the filtrate. Ethyl acetate was recovered under reduced pressure, petroleum ether was added to the solid residue, stirred, and filtered to obtain cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI), about 3.19 kg.
[0031] Example 4 Preparation of amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (VII)
[0032] Put 25L of dichloromethane, about 3.19kg of intermediate (VI), and 430g of ethanol into the reactor, start stirring, cool to below 0°C, and pass in hydrogen chloride gas until the temperature stabilizes below 0°C, at 0°C Let stand for 14 hours at °C. Concentrate under reduced pressure to remove most of the hydrogen chloride gas and recover the solvent dichloromethane. Add 25L of dichloromethane to the residue of the reaction kettle, stir, cool to below 0°C, and pass in ammonia until the temperature stabilizes below 0°C, and filter . The filtrate was poured into the reactor, concentrated under reduced pressure to recover the solvent to obtain a colorless liquid, added 22.8L of acetonitrile and 905g of amine acetate, heated to 55-60°C for 1.5 hours, stopped the reaction, filtered while hot, and recovered the filtrate under reduced pressure Solvent to dryness, add 3L of ether to the residue to crystallize, filter, and dry to obtain amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (Intermediate VII) about 3.2kg .
[0033] Example 5. Add about 3.2kg of Intermediate (VII), about 2.7kg of Intermediate (III), 21L of isopropanol and 585g of sodium methoxide to the reaction kettle, start stirring, heat to reflux and react for 4 hours, and cool to Below 10°C, filter, the filtrate is decompressed to recover the solvent to dryness, add 35L ethyl acetate to the residue to dissolve, wash with 6.5LX3 water, release the water layer, add anhydrous sodium sulfate to the ethyl acetate layer to dry, filter , Collect the filtrate, recover ethyl acetate under reduced pressure, add 4.2L of toluene to the residue,
3.4L of n-hexane was heated to dissolve, filtered, the filtrate was stirred to room temperature to crystallize, filtered and collected and dried, and the product was placed in an oven at 45-55°C to dry to obtain the crude azedipine (I), about 2.3kg.
[0034] Example 6, Refining
[0035] Put 8.8L ethyl acetate and 8.8L n-hexane into the reaction kettle, turn on the stirring, put about 2.3kg of the crude azeldipine into the reaction kettle, slowly heat up until the material is dissolved, add 180g of activated carbon and stir for 0.5h, while it is hot Filter, hydraulically filter the material to the crystallization dad, wash the filter cake with 5.5L ethyl acetate and 4.5L n-hexane solution, combine with the filtrate, cool to 0~5°C to crystallize, filter, collect the product, and place it in a hot air circulating oven After drying at 45-55°C, 2.2 g of azeldipine is obtained. The purity is 99.6% as measured by high performance liquid chromatography. The refined yield is 96.0%.
[0036] Example 7 Azedipin Refining
[0037] The mixed solvent was prepared according to the volume ratio of ethyl acetate and n-hexane of 2:1, 22L of the mixed solvent was put into the reactor, about 2.3kg of azedipine crude product was put into the reactor, and the temperature was slowly heated until the material was dissolved, Add 180g of activated carbon and stir for 0.5h, filter while hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid with the filtrate, cool to 0~5°C for crystallization, filter, collect the product, and circulate the hot air Dry in an oven at a temperature of 45-55°C to obtain 2.2 g of azeldipine fine product, with a purity of 99.7% measured by high performance liquid chromatography.
[0038] Example 8 prepared a mixed solvent at a volume ratio of ethyl acetate and n-hexane of 1.5:1, put 22L of the mixed solvent into the reactor, put about 2.3kg of crude azeldipine into the reactor, and slowly heated to Dissolve the material, add 180g of activated carbon and stir for 0.5h, filter while it is hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid and the filtrate, cool to 0~5°C to crystallize, filter, and collect the product. Dry in a hot air circulating oven at a temperature of 45-55°C to obtain
2.2g azeldipine is a fine product with a purity of 99.6% measured by high performance liquid chromatography.
PATENT
https://patents.google.com/patent/CN103183663B/zh
Azelnidipine (Azelnidipine) is a new type of dihydropyridine calcium channel blocker jointly developed by Sankyo Co., Ltd. and Ube Industries Co., Ltd., which inhibits the entry of calcium ions into excitable tissues and causes peripheral blood vessels And coronary artery vasodilation plays a role in lowering blood pressure. Clinically, it is widely used in patients with mild or moderate symptoms of primary hypertension, hypertension with renal dysfunction, and severe hypertension. Compared with similar antihypertensive drugs, azeldipine has a slow and long-lasting antihypertensive effect.
[0004] The chemical structure of azeldipine is similar to that of nifedipine:

[0006] The Chinese patent CN87107150.9 reported the compound earlier and gave a detailed introduction to its synthesis; afterwards, most of the synthesis of azeldipine adopts this route:

[0008] The reaction takes o-nitrobenzaldehyde and isopropyl acetoacetate as raw materials to prepare intermediate compound 5; takes benzhydrylamine and epichlorohydrin as raw materials to prepare compound 2, compound 2 and cyanoacetic acid act in DCC Compound 3 is prepared by the next reaction. Compound 3 is added with ethanol under the action of hydrogen chloride gas, ammonia gas ammonolysis, and acetate anion exchange to obtain compound 4. Compound 4 and compound 5 are under the action of sodium methoxide to obtain compound 1, namely azeldipine.
[0009] Wherein: Compound 3 can be purchased as an industrial product, or can be prepared according to the traditional method reported in the literature; Compound 5 is prepared according to the traditional method reported in the literature.
[0010] In the process of preparing amidine 4 in the traditional reaction route, hydrogen chloride gas and ammonia gas need to be passed in successively. Therefore, the reaction requires anhydrous reagents. According to literature reports, the reaction yield is about 70%. From the perspective of industrial synthesis, The application of anhydrous reagents will undoubtedly increase the cost, while the use of gas will increase the difficulty of operation and require the use of high-pressure equipment. At the same time, post-reaction processing is difficult and industrial production is difficult. Therefore, this step of the reaction requires further improvement.
With acetonitrile as a solvent, the crude product of reaction 2) was stirred until dissolved, ammonium acetate was added, and acetate anion exchange was performed to obtain the amidine compound 4;

[0018] The second step: use toluene as a solvent, compound 4 and compound 5 in the use of sodium amide to obtain compound 1, namely azedipine

[0020] The preferred technical solution of the present invention is characterized in that the temperature of reaction 1) is controlled below _5°C
Example 1: Preparation of azeldipine
[0030] Add 50 g of compound 3, 1500 mL of dichloromethane, and 16.64 mL of absolute ethanol to a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5 °C to saturation, and after saturation, keep the reaction at -5 °C for 24 hours. Protect from light and nitrogen, slowly add the above reaction system to 1665ml of ammonia water with a concentration of 2.5-3.0% under the control of 0-5°C. After the addition, stir for 0.5h, stand for 0.5h, and separate the liquids. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C -60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 57.55 g of amidine 4, the yield was 91.2%, the HPLC purity was 99.63%, and the melting point was 130-132.3°C.
[0031] 50g amidine 4, 43.5g compound 5, 1000mL toluene, and 7.7g sodium amide were added into a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was purified once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 66.87g of α-crystal form of Azedipine, yield 88.2%, melting point: 121-123°C.
[0032] Example 2; Preparation of Azeldipine
[0033] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -6°C to -8°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to ammonia water with a concentration of 2.5-3.0%, adjust the pH to 7.8-8.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.0 lg of amidine 4 with a yield of 93.5%, an HPLC purity of 99.52%, and a melting point of 130.1-132.0°C.
[0034] 50g amidine 4, 43.5g compound 5, 1000mL toluene and 7.7g sodium amide were added to a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was refined once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 68.31 g of α-crystal azedipine, yield 90.01%, melting point: 121 -123 °C.
[0035] Example 3: Preparation of Amidine 4
[0036] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -7°C to -9°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to the ammonia water with a concentration of 2.5-3.0%, adjust the pH to 8.5-9.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.5 g of amidine 4, HPLC purity 99.78%, melting point: 130.7-132·2°C.

Step 2: Using toluene as a solvent, compound 4 and compound 5 under the action of sodium amide to obtain compound 1, namely azeldipine

PATENThttps://patents.google.com/patent/CN102453023A/zh
detailed description
[0007] In the synthesis workshop, benzhydrylamine is used as a raw material to be synthesized by addition, cyclization, esterification, acidification, ammoniation, condensation and other reactions. The crude azeodipine is refined, dried, mixed and packaged in a clean area. Fold the ground. The specific response is as follows:
[0008] 1. Addition and cyclization reaction
[0009] Methanol, benzhydrylamine, and epichlorohydrin were added to the reaction kettle, stirred at room temperature for 24hr, the reaction was completed, the reaction was heated to reflux for 24hr, cooled, filtered to collect the precipitated solid, and then the mother liquor was concentrated to recover the raw materials, and the heating was continued to reflux 18 After hours, collect the product, add dichloromethane and H2O to the obtained solid, adjust the pH to 10-11 with 40% NaOH while stirring in an ice bath, stand still, separate the organic layer, dry with anhydrous magnesium sulfate, and recover the dichloromethane under reduced pressure To dryness, a colorless solid compound III (1-benzyl-3-hydroxyazetidine) is obtained. After improvement, the raw materials are fully reacted, and the reaction yield of this step is improved. The mass yield is 75%. % Mentioned 85%.
[0010]

[0011] 2. Esterification reaction
[0012] Add THF, compound (III), and cyanoacetic acid to the reaction kettle, stir evenly, add DCC in batches under ice bath stirring, control the temperature at 10°C~15°C, after the addition, remove the ice water bath, and slowly heat up React at 55°C~60°C for 18h. After the reaction is complete, cool, filter to remove insoluble materials, concentrate the filtrate to dryness, add ethyl acetate to the residue to dissolve, wash with water, dry with anhydrous magnesium sulfate, and recover ethyl acetate under reduced pressure. The residue was added with petroleum ether and stirred for crystallization, and the solid was collected by filtration to obtain compound IV (1-diphenylmethyl-3-azetidinyl cyanoacetate).
[0013]

[0014] 3. Acidification and amination reaction
[0015] Dichloromethane, ethanol and intermediate (IV) were added to the reaction kettle respectively, mixed and stirred, cooled to about _5 ° C in an ice salt bath, and dried hydrogen chloride gas was introduced until saturation (about 1.5 hours) after . Let stand overnight at about -5°C, recover the solvent under reduced pressure at room temperature, add dichloromethane to the residue and stir, cool to about _5°C in an ice-salt bath, pass in the dried ammonia gas until saturation (about 3 hours) , Filtration to remove the insoluble matter, and the filtrate was decompressed to recover solvent at room temperature. Acetonitrile and ammonium acetate were added to the residue respectively, and the temperature was raised to 55~60°C for 2 hours with stirring. After the reaction was completed, it was cooled and filtered. 3-Azacyclobutanylamidinoacetate acetate), the reaction in this step is controlled at about _5°C, and the transesterification
The side reaction is reduced, and the reaction yield is improved.
[0016]

[0017] 4. Condensation reaction
[0018] Add isopropanol, intermediate (III’), sodium methoxide and compound V to the reaction kettle, mix and stir, heat to reflux and react for 5 hours. After the reaction is complete, cool and filter, and the filtrate is decompressed to recover the solvent to dryness, leaving residue Add ethyl acetate to dissolve, wash with water, dry with anhydrous magnesium sulfate, recover ethyl acetate under reduced pressure to 1/4 of the total volume, add n-hexane, and stir at 50°C for 30 min. After cooling and crystallization, the solid was collected by filtration, and air-dried at 45°C to obtain the crude azedipine (I). After the crude product was dissolved in ethyl acetate-n-hexane mixed solvent, activated carbon was added for decolorization and impurity removal to achieve the purpose of purification.

[0020] The refined product is dissolved in dioxane, refluxed with n-hexane, cooled and crystallized, and dried to obtain a solid that is boiled in cyclohexane, cooled and filtered, and dried to obtain α-crystalline form Azedipine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN102453023A *2010-10-212012-05-16大丰市天生药业有限公司Process for producing azelnidipineCN103130700A *2013-03-142013-06-05沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN103509003A *2012-06-272014-01-15威海威太医药技术开发有限公司Preparation method of azelnidipine
JP3491506B2 *1997-10-142004-01-26宇部興産株式会社Method for producing dihydropyridine derivativeCN101475521B *2008-11-132010-11-10青岛黄海制药有限责任公司Method for synthesizing acetate of 1-benzhydryl-3-azetidine amidino acetic ester
TitleLIU, JIAN-FENG ET AL.: “Improved Synthesis of Azelnidipine”, CHINESE JOURNAL OF MEDICINAL CHEMISTRY, vol. 20, no. 3, 30 June 2010 (2010-06-30), pages 192 – 194 *ZHANG, KAI ET AL.: “Synthesis of Azelnidipine”, CHINESE JOURNAL OF PHARMACEUTICALS, vol. 39, no. 3, 31 March 2008 (2008-03-31), pages 163 – 165, XP025959789, DOI: doi:10.1016/j.ejphar.2008.12.041 *
CN103130700B *2013-03-142015-04-29沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN104860855B *2014-12-082017-06-16宁夏紫光天化蛋氨酸有限责任公司A kind of preparation method of the methylmercapto butyric acid ester of 2 hydroxyl of the D of high-purity, L 4CN105949102A *2016-06-202016-09-21许昌豪丰化学科技有限公司Production method of azelnidipine intermediatePublication numberPriority datePublication dateAssigneeTitleWO2014139410A1 *2013-03-142014-09-18Shenyang Zhonghai Pharmaceutical Co., Ltd.A kind of preparation method of azeldipine intermediateCN105461691A *2015-12-312016-04-06Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106279109A *2016-08-182017-01-04Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106543061A *2016-10-202017-03-29Weihai Disu Pharmaceutical Co., Ltd.Preparation method of N-diphenylmethylcyclobutane-3-alcohol
References
- ^ Oizumi K, Nishino H, Koike H, Sada T, Miyamoto M, Kimura T (September 1989). “Antihypertensive effects of CS-905, a novel dihydropyridine Ca++ channel blocker”. Jpn. J. Pharmacol. 51 (1): 57–64. doi:10.1254/jjp.51.57. PMID 2810942.
| Clinical data | |
|---|---|
| Trade names | CalBlock,AZUSA,Azovas |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | none |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 123524-52-7 |
| PubChem CID | 65948 |
| ChemSpider | 59352 |
| UNII | PV23P19YUG |
| KEGG | D01145 |
| ChEMBL | ChEMBL1275868 |
| CompTox Dashboard (EPA) | DTXSID3020120 |
| ECHA InfoCard | 100.162.151 |
| Chemical and physical data | |
| Formula | C33H34N4O6 |
| Molar mass | 582.657 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILES[O-][N+](=O)c1cccc(c1)C5C(/C(=O)OC(C)C)=C(\NC(\N)=C5\C(=O)OC4CN(C(c2ccccc2)c3ccccc3)C4)C | |
| hideInChIInChI=1S/C33H34N4O6/c1-20(2)42-32(38)27-21(3)35-31(34)29(28(27)24-15-10-16-25(17-24)37(40)41)33(39)43-26-18-36(19-26)30(22-11-6-4-7-12-22)23-13-8-5-9-14-23/h4-17,20,26,28,30,35H,18-19,34H2,1-3H3 Key:ZKFQEACEUNWPMT-UHFFFAOYSA-N |
/////////Azelnidipine, CS-905, RS-9054, INDIA 2020, APPROVALS 2020
#Azelnidipine, #CS-905, #RS-9054, #INDIA 2020, #APPROVALS 2020
CC1=C(C(C(=C(N1)N)C(=O)OC2CN(C2)C(C3=CC=CC=C3)C4=CC=CC=C4)C5=CC(=CC=C5)[N+](=O)[O-])C(=O)OC(C)C
EVEROLIMUS
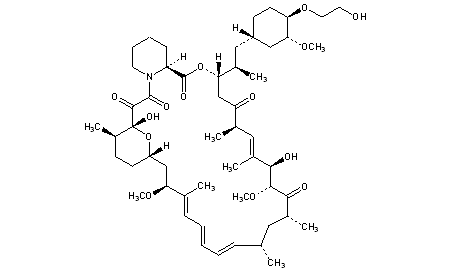
Everolimus
159351-69-6[RN]
23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydr oxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,26R,27R,34aS)-
23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-
42-O-(2-Hydroxyethyl)rapamycin
- Synonyms:RAD-001, SDZ-RAD, Afinitor
- ATC:L04AA18
Use:immunosuppressantChemical name:42-O-(2-hydroxyethyl)rapamycinFormula:C53H83NO14
- MW:958.24 g/mol
- CAS-RN:159351-69-6
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015);
$1,902 Million (Y2014);
$1,558 Million (Y2013);
$1,007 Million (Y2012);
$630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-08-29 | New dosage form | Afinitor Disperz | Renal cell carcinoma , Advanced breast cancer, Pancreatic cancer, Renal angiomyolipoma, Tuberous sclerosis complex (TSC) | Tablet, For suspension | 2 mg/3 mg/5 mg | Novartis | Priority |
| 2010-04-20 | New strength | Zortress | Advanced renal cell carcinoma (RCC) | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2009-03-30 | Marketing approval | Afinitor | Advanced renal cell carcinoma (RCC) | Tablet | 2.5 mg/5 mg/7.5 mg/10 mg | Novartis | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2016-06-02 | New indication | Afinitor | neuroendocrine tumors (NET), Advanced gastrointestinal tumors | Tablet | Novartis | ||
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis | Orphan; Conditional Approval |
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet, Orally disintegrating | 2 mg/3 mg/5 mg | Novartis | Orphan; Conditional Approval |
| 2009-08-03 | Marketing approval | Afinitor | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-12-22 | New indication | Certican | Rejection of suppression renal transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2007-01-26 | Marketing approval | Certican | Rejection in heart transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-02-13 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 2.5 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 10 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 5 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2003-07-18 | Marketing approval | Certican | Rejection of organ transplantation, Renal cell carcinoma | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.

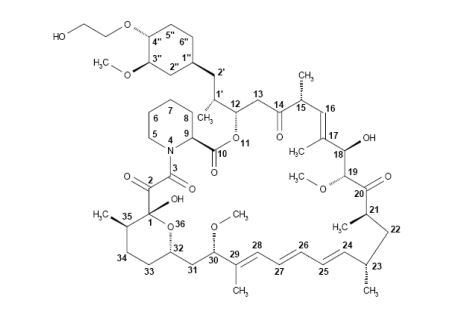
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex.
Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
Everolimus is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors as targeted therapy for use in a number of cancers.[medical citation needed]
It is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
- Advanced kidney cancer (US FDA approved in March 2009)[3]
- Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
- Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) in patients who are not suitable for surgical intervention (US FDA October 2010)[5]
- Progressive or metastatic pancreatic neuroendocrine tumors not surgically removable (May 2011)[6]
- Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
- Prevention of organ rejection after liver transplant(Feb 2013)
- Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
- Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Clinical trials
As of October 2010, Phase III trials are under way in gastric cancer, hepatocellular carcinoma, and lymphoma.[12] The experimental use of everolimus in refractory chronic graft-versus-host disease was reported in 2012.[13]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[19]
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future 1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN

Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2

Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3

Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4

Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5

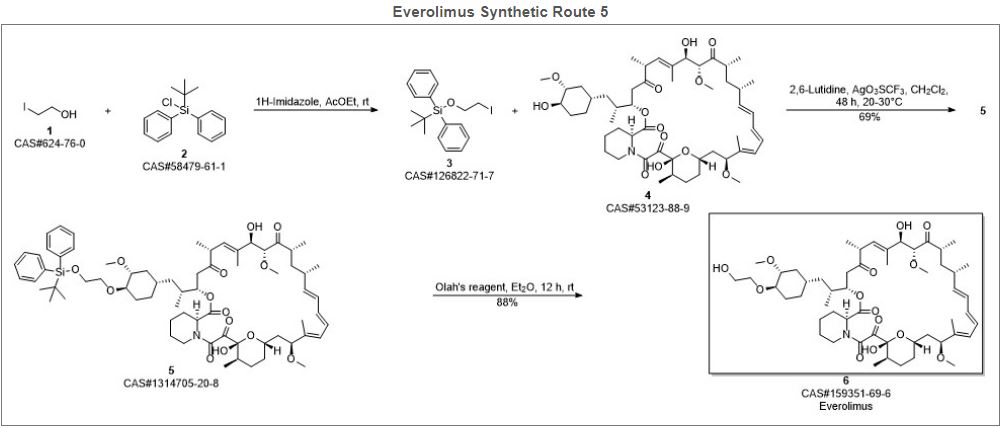
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6

Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
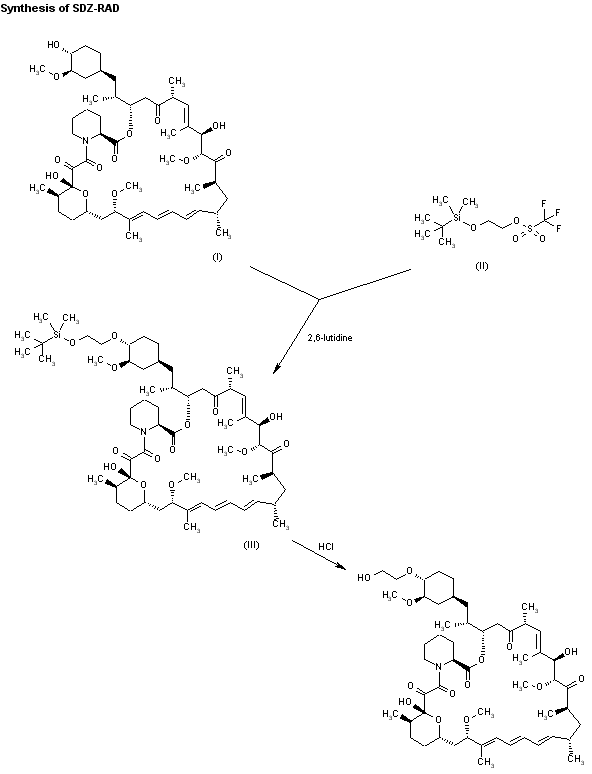
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
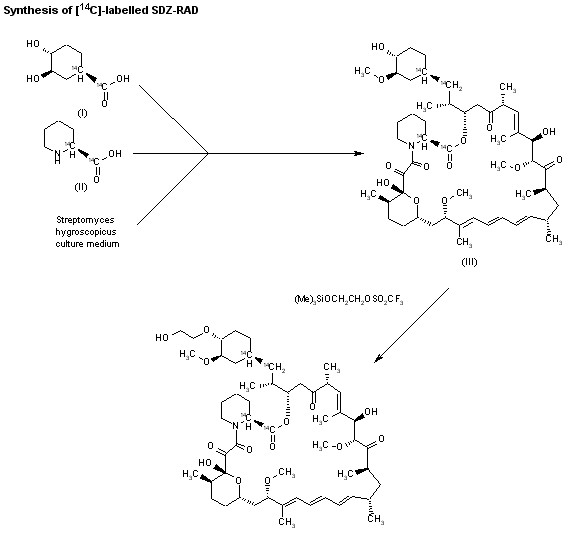
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
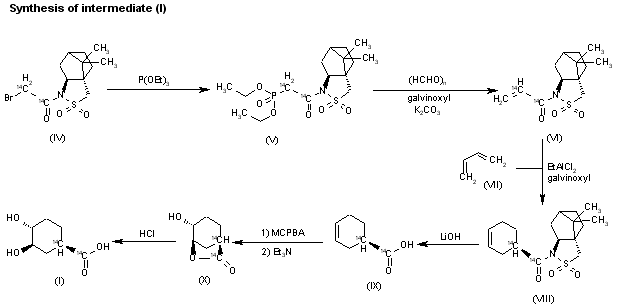
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
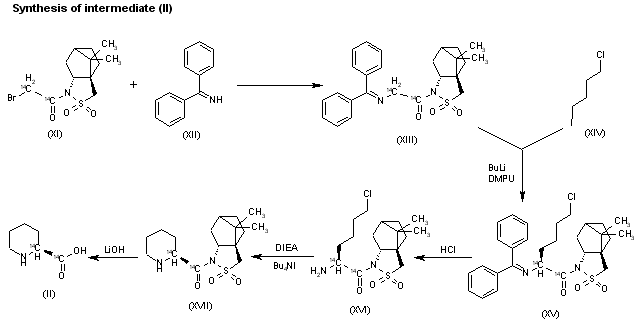
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:

See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.

(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis

Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):

According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.

(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).

Only 8% yield of the corresponding compound (4B)

and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
PATENT
https://patents.google.com/patent/WO2016020664A1/en
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),

It is a derivative of sirolimus of formula III),

and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),

wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),

was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche

Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),

as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),

wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Clip


References
- a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
- b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
- US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
- US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
- fermentation of rapamycin (sirolimus):
- Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
- The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
- US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
- WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
- EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
- US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
clip
https://doi.org/10.1039/C7MD00474EIssue 1, 2018
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]

Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
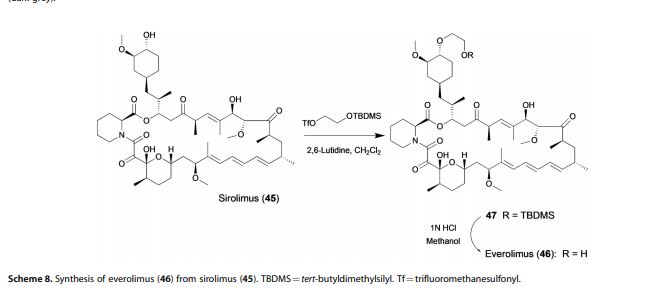
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
by Stu Borman
JUNE 27, 2018 | APPEARED IN VOLUME 96, ISSUE 27
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.

“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.

When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
References

- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Formica RN, Lorber KM, Friedman AL, Bia MJ, Lakkis F, Smith JD, Lorber MI (March 2004). “The evolving experience using everolimus in clinical transplantation”. Transplantation Proceedings. 36 (2 Suppl): 495S–499S. doi:10.1016/j.transproceed.2004.01.015. PMID 15041395.
- ^ “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 30 March 2009. Retrieved 6 April 2009.
- ^ “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 22 April 2010. Archived from the original on 25 April 2010. Retrieved 26 April 2010.
- ^ “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 November 2010.
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- ^ “US FDA approves Novartis drug Afinitor for breast cancer”. Reuters. 20 July 2012.
- ^ Jump up to:a b Everolimus (Afinitor). Feb 2016
- ^ Everolimus (Afinitor). April 2018
- ^ Lintern, Shaun (14 April 2015). “Policy delays risk ‘preventable deaths’, doctors warn NHS England”. Health Service Journal. Retrieved 20 April 2015.
- ^ “Couple forced to sell home after NHS refuse to fund daughter’s treatment for rare illness”. Daily Express. 11 May 2015. Retrieved 12 May 2015.
- ^ http://www.genengnews.com/gen-news-highlights/novartis-afinitor-cleared-by-fda-for-treating-sega-tumors-in-tuberous-sclerosis/81244159/
- ^ Lutz M, Kapp M, Grigoleit GU, Stuhler G, Einsele H, Mielke S (April 2012). “Salvage therapy with everolimus improves quality of life in patients with refractory chronic graft-versus-host disease” (PDF). Bone Marrow Transplant. 47 (S1): S410–S411.
- ^ “Positive Trial Data Leads Novartis to Plan Breast Cancer Filing for Afinitor by Year End”. 2011.
- ^ Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB (October 2012). “Genome sequencing identifies a basis for everolimus sensitivity”. Science. 338 (6104): 221. Bibcode:2012Sci…338..221I. doi:10.1126/science.1226344. PMC 3633467. PMID 22923433.
- ^ [1]
- ^ Jump up to:a b Zhavoronkov A (2020). “Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections”. Aging. 12 (8): 6492–6510. doi:10.18632/aging.102988. PMC 7202545. PMID 32229705.
- ^ Jump up to:a b c Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (February 2016). “Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system”. Aging Cell. 15 (1): 28–38. doi:10.1111/acel.12405. PMC 4717280. PMID 26463117.
- ^ Wang S, Raybuck A, Shiuan E, Jin J (2020). “Selective inhibition of mTORC1 in tumor vessels increases antitumor immunity”. JCI Insight. 5 (15): e139237. doi:10.1172/jci.insight.139237. PMC 7455083. PMID 32759497.
- ^ Jump up to:a b “Archived copy”. Archived from the original on 8 March 2014. Retrieved 26 February 2014.
- ^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID 12944570.
- ^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID 24767339.
- ^ Jeng L, Thorat A, Yang H, Yeh C-C, Chen T-H, Hsu S-C. Impact of Everolimus On the Hepatocellular Carcinoma Recurrence After Living Donor Liver Transplantation When Used in Early Stage: A Single Center Prospective Study [abstract]. Am J Transplant. 2015; 15 (suppl 3). http://www.atcmeetingabstracts.com/abstract/impact-of-everolimus-on-the-hepatocellular-carcinoma-recurrence-after-living-donor-liver-transplantation-when-used-in-early-stage-a-single-center-prospective-study/. Accessed 1 September 2015.
- ^ Thorat A, Jeng LB, Yang HR, Yeh CC, Hsu SC, Chen TH, Poon KS (November 2017). “Assessing the role of everolimus in reducing hepatocellular carcinoma recurrence after living donor liver transplantation for patients within the UCSF criteria: re-inventing the role of mammalian target of rapamycin inhibitors”. Annals of Hepato-Biliary-Pancreatic Surgery. 21 (4): 205–211. doi:10.14701/ahbps.2017.21.4.205. PMC 5736740. PMID 29264583.
- ^ Jeng LB, Lee SG, Soin AS, Lee WC, Suh KS, Joo DJ, Uemoto S, Joh J, Yoshizumi T, Yang HR, Song GW, Lopez P, Kochuparampil J, Sips C, Kaneko S, Levy G (December 2017). “Efficacy and safety of everolimus with reduced tacrolimus in living-donor liver transplant recipients: 12-month results of a randomized multicenter study”. American Journal of Transplantation. 18 (6): 1435–1446. doi:10.1111/ajt.14623. PMID 29237235.
- ^ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (July 2009). “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice”. Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- ^ Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (December 2014). “mTOR inhibition improves immune function in the elderly”. Science Translational Medicine. 6 (268): 268ra179. doi:10.1126/scitranslmed.3009892. PMID 25540326. S2CID 206685475.
Further reading
- Sedrani R, Cottens S, Kallen J, Schuler W (August 1998). “Chemical modification of rapamycin: the discovery of SDZ RAD”. Transplantation Proceedings. 30 (5): 2192–4. doi:10.1016/S0041-1345(98)00587-9. PMID 9723437.
External links
- “Everolimus”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | Everolimus /ˌɛvəˈroʊləməs/ |
| Trade names | Afinitor, Zortress |
| Other names | 42-O-(2-hydroxyethyl)rapamycin, RAD001 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609032 |
| License data | EU EMA: by INNUS DailyMed: EverolimusUS FDA: Everolimus |
| Pregnancy category | AU: C[1] |
| Routes of administration | By mouth |
| ATC code | L01EG02 (WHO) L04AA18 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Elimination half-life | ~30 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 159351-69-6 |
| PubChem CID | 6442177 |
| DrugBank | DB01590 |
| ChemSpider | 21106307 |
| UNII | 9HW64Q8G6G |
| KEGG | D02714 |
| ChEMBL | ChEMBL1908360 |
| CompTox Dashboard (EPA) | DTXSID0040599 |
| ECHA InfoCard | 100.149.896 |
| Chemical and physical data | |
| Formula | C53H83NO14 |
| Molar mass | 958.240 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCO[C@@H]1CC[C@H](C[C@H]1OC)C[C@@H](C)[C@@H]4CC(=O)[C@H](C)/C=C(\C)[C@@H](O)[C@@H](OC)C(=O)[C@H](C)C[C@H](C)\C=C\C=C\C=C(/C)[C@@H](OC)C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(=O)N3CCCC[C@H]3C(=O)O4 | |
| hideInChIInChI=1S/C53H83NO14/c1-32-16-12-11-13-17-33(2)44(63-8)30-40-21-19-38(7)53(62,68-40)50(59)51(60)54-23-15-14-18-41(54)52(61)67-45(35(4)28-39-20-22-43(66-25-24-55)46(29-39)64-9)31-42(56)34(3)27-37(6)48(58)49(65-10)47(57)36(5)26-32/h11-13,16-17,27,32,34-36,38-41,43-46,48-49,55,58,62H,14-15,18-26,28-31H2,1-10H3/b13-11+,16-12+,33-17+,37-27+/t32-,34-,35-,36-,38-,39+,40+,41+,43-,44+,45+,46-,48-,49+,53-/m1/s1 Key:HKVAMNSJSFKALM-GKUWKFKPSA-N |
//////////////// RAD-001, SDZ RAD, Certican, Novartis, Immunosuppressant, Everolimus, Afinitor, эверолимус , إيفيروليموس , 依维莫司 ,

Everolimus
159351-69-6[RN]
23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydr oxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,26R,27R,34aS)-
23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone, 9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-, (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-
42-O-(2-Hydroxyethyl)rapamycin
- Synonyms:RAD-001, SDZ-RAD, Afinitor
- ATC:L04AA18
Use:immunosuppressantChemical name:42-O-(2-hydroxyethyl)rapamycinFormula:C53H83NO14
- MW:958.24 g/mol
- CAS-RN:159351-69-6
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015);
$1,902 Million (Y2014);
$1,558 Million (Y2013);
$1,007 Million (Y2012);
$630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-08-29 | New dosage form | Afinitor Disperz | Renal cell carcinoma , Advanced breast cancer, Pancreatic cancer, Renal angiomyolipoma, Tuberous sclerosis complex (TSC) | Tablet, For suspension | 2 mg/3 mg/5 mg | Novartis | Priority |
| 2010-04-20 | New strength | Zortress | Advanced renal cell carcinoma (RCC) | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2009-03-30 | Marketing approval | Afinitor | Advanced renal cell carcinoma (RCC) | Tablet | 2.5 mg/5 mg/7.5 mg/10 mg | Novartis | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2016-06-02 | New indication | Afinitor | neuroendocrine tumors (NET), Advanced gastrointestinal tumors | Tablet | Novartis | ||
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis | Orphan; Conditional Approval |
| 2011-09-02 | Marketing approval | Votubia | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet, Orally disintegrating | 2 mg/3 mg/5 mg | Novartis | Orphan; Conditional Approval |
| 2009-08-03 | Marketing approval | Afinitor | Advanced breast cancer, Renal cell carcinoma , Pancreatic cancer | Tablet | 2.5 mg/5 mg/10 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-12-22 | New indication | Certican | Rejection of suppression renal transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis | |
| 2007-01-26 | Marketing approval | Certican | Rejection in heart transplantation | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-02-13 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 2.5 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 10 mg | Novartis | |
| 2013-01-22 | Marketing approval | 飞尼妥/Afinitor | Advanced renal cell carcinoma (RCC), Subependymal giant cell astrocytoma | Tablet | 5 mg | Novartis |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2003-07-18 | Marketing approval | Certican | Rejection of organ transplantation, Renal cell carcinoma | Tablet | 0.25 mg/0.5 mg/0.75 mg | Novartis |
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex.
Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
Everolimus is a medication used as an immunosuppressant to prevent rejection of organ transplants and in the treatment of renal cell cancer and other tumours. Much research has also been conducted on everolimus and other mTOR inhibitors as targeted therapy for use in a number of cancers.[medical citation needed]
It is the 40-O-(2-hydroxyethyl) derivative of sirolimus and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR).
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
- Advanced kidney cancer (US FDA approved in March 2009)[3]
- Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
- Subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis (TS) in patients who are not suitable for surgical intervention (US FDA October 2010)[5]
- Progressive or metastatic pancreatic neuroendocrine tumors not surgically removable (May 2011)[6]
- Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
- Prevention of organ rejection after liver transplant(Feb 2013)
- Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
- Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Clinical trials
As of October 2010, Phase III trials are under way in gastric cancer, hepatocellular carcinoma, and lymphoma.[12] The experimental use of everolimus in refractory chronic graft-versus-host disease was reported in 2012.[13]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
mTORC1 inhibition by everolimus has been shown to normalize tumor blood vessels, to increase tumor-infiltrating lymphocytes, and to improve adoptive cell transfer therapy.[19]
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future 1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
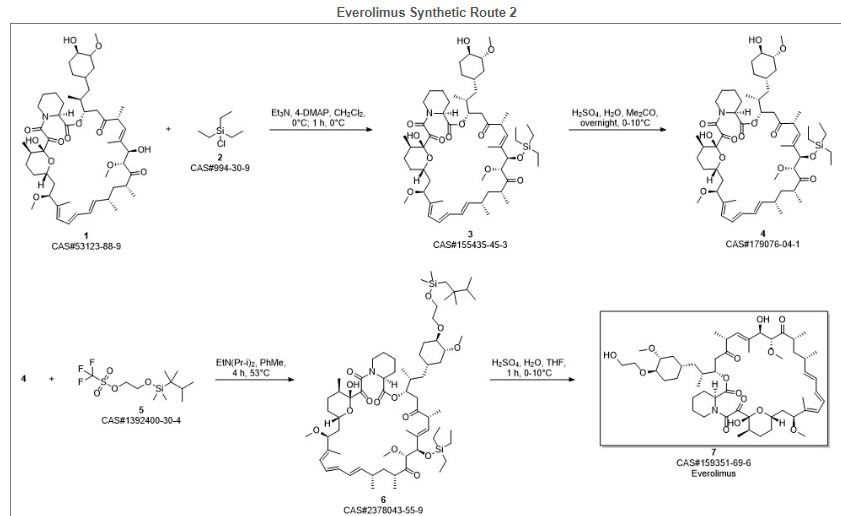
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
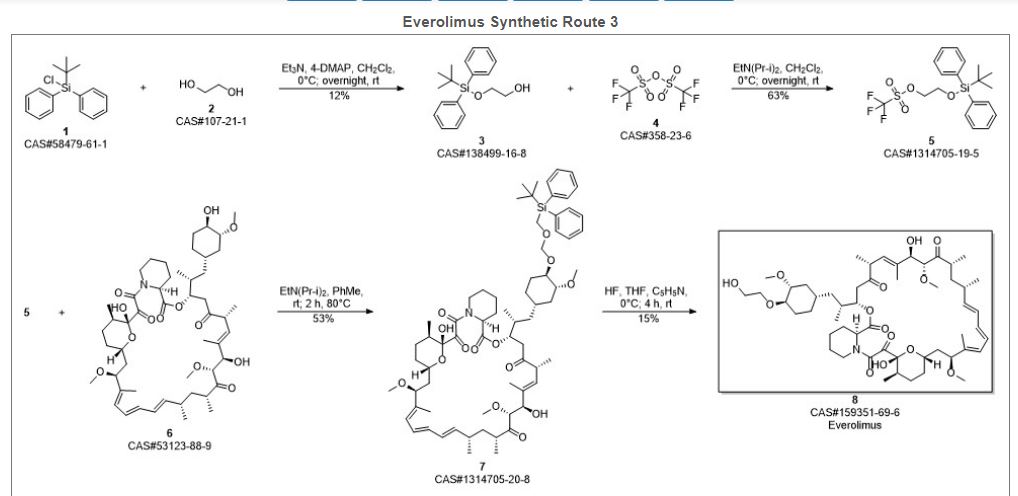
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
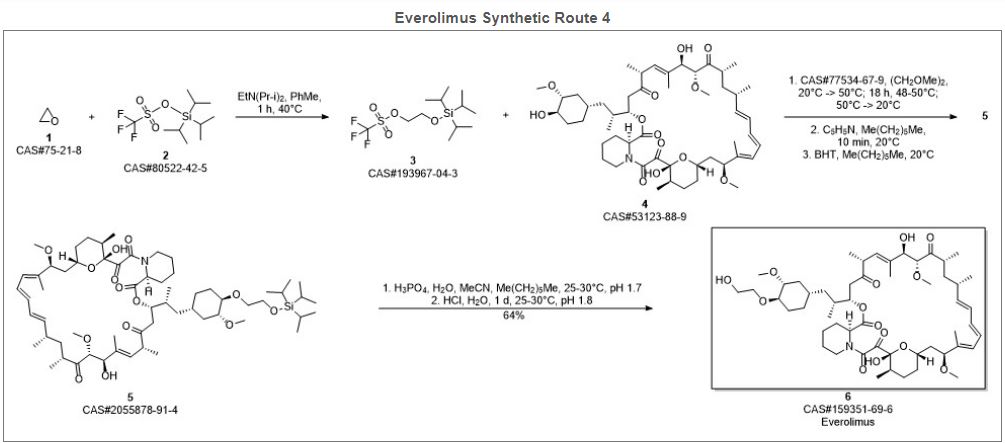
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
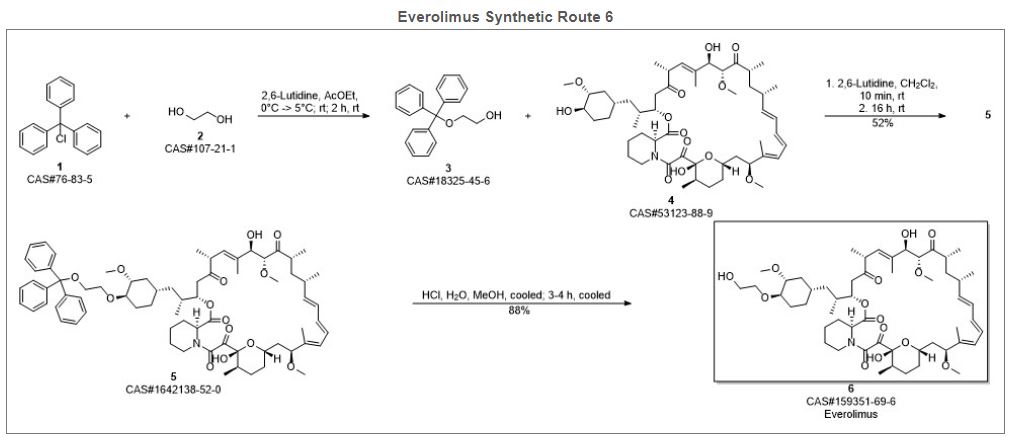
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
…………..
SYNTHESIS
https://www.google.com/patents/WO2012103960A1
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
RAP-OH + L-CH2-CH2-0-P — –> RAP-O-CH2-CH2-O-P — – > RAP-O-CH2-CH2-OH
(2) (4) (1)
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
xample 6: 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
……………………….
SYNTHESIS
https://www.google.co.in/patents/WO1994009010A1
Example 8: 40-O-(2-Hydroxy)ethyl-rapamycin
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
PATENT
https://patents.google.com/patent/WO2016020664A1/en
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Enzymes
Synthesis Path
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Certican | Novartis ,2004 | |
| F | Certican | Novartis | |
| I | Certican | Novartis | |
| J | Certican | Novartis |
Formulations
- tabl. 0.25 mg, 0.5 mg, 0.75 mg
References
- a WO 9 409 010 (Sandoz-Erfindungen; 28.4.1994; GB-prior. 9.10.1992).
- b US 6 277 983 (American Home Products; 21.8.2001; USA-prior. 27.9.2000).
- US 6 384 046 (Novartis; 7.5.2002; GB-prior. 27.3.1996).
- US 20 040 115 (Univ. of Pennsylvania; 15.1.2004; USA-prior. 9.7.2002).
- fermentation of rapamycin (sirolimus):
- Chen, Y. et al.: Process Biochemistry (Oxford, U. K.) (PBCHE5) 34, 4, 383 (1999).
- The Merck Index, 14th Ed., 666 (3907) (Rahway 2006).
- US 3 929 992 (Ayerst McKenna & Harrison Ltd.; 30.12.1975; USA-prior. 29.9.1972).
- WO 9 418 207 (Sandoz-Erfindungen; 18.8.1994; GB-prior. 2.2.1993).
- EP 638 125 (Pfizer; 17.4.1996; J-prior. 27.4.1992).
- US 6 313 264 (American Home Products; 6.11.2001; USA-prior. 8.3.1994).
clip
https://doi.org/10.1039/C7MD00474EIssue 1, 2018
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.

[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
by Stu Borman
JUNE 27, 2018 | APPEARED IN VOLUME 96, ISSUE 27
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
References
- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Formica RN, Lorber KM, Friedman AL, Bia MJ, Lakkis F, Smith JD, Lorber MI (March 2004). “The evolving experience using everolimus in clinical transplantation”. Transplantation Proceedings. 36 (2 Suppl): 495S–499S. doi:10.1016/j.transproceed.2004.01.015. PMID 15041395.
- ^ “Afinitor approved in US as first treatment for patients with advanced kidney cancer after failure of either sunitinib or sorafenib” (Press release). Novartis. 30 March 2009. Retrieved 6 April 2009.
- ^ “Novartis receives US FDA approval for Zortress (everolimus) to prevent organ rejection in adult kidney transplant recipients” (Press release). Novartis. 22 April 2010. Archived from the original on 25 April 2010. Retrieved 26 April 2010.
- ^ “Novartis’ Afinitor Cleared by FDA for Treating SEGA Tumors in Tuberous Sclerosis”. 1 November 2010.
- ^ https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm254350.htm
- ^ “US FDA approves Novartis drug Afinitor for breast cancer”. Reuters. 20 July 2012.
- ^ Jump up to:a b Everolimus (Afinitor). Feb 2016
- ^ Everolimus (Afinitor). April 2018
- ^ Lintern, Shaun (14 April 2015). “Policy delays risk ‘preventable deaths’, doctors warn NHS England”. Health Service Journal. Retrieved 20 April 2015.
- ^ “Couple forced to sell home after NHS refuse to fund daughter’s treatment for rare illness”. Daily Express. 11 May 2015. Retrieved 12 May 2015.
- ^ http://www.genengnews.com/gen-news-highlights/novartis-afinitor-cleared-by-fda-for-treating-sega-tumors-in-tuberous-sclerosis/81244159/
- ^ Lutz M, Kapp M, Grigoleit GU, Stuhler G, Einsele H, Mielke S (April 2012). “Salvage therapy with everolimus improves quality of life in patients with refractory chronic graft-versus-host disease” (PDF). Bone Marrow Transplant. 47 (S1): S410–S411.
- ^ “Positive Trial Data Leads Novartis to Plan Breast Cancer Filing for Afinitor by Year End”. 2011.
- ^ Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, Viale A, Heguy A, Peng L, Chan TA, Bochner B, Bajorin DF, Berger MF, Taylor BS, Solit DB (October 2012). “Genome sequencing identifies a basis for everolimus sensitivity”. Science. 338 (6104): 221. Bibcode:2012Sci…338..221I. doi:10.1126/science.1226344. PMC 3633467. PMID 22923433.
- ^ [1]
- ^ Jump up to:a b Zhavoronkov A (2020). “Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections”. Aging. 12 (8): 6492–6510. doi:10.18632/aging.102988. PMC 7202545. PMID 32229705.
- ^ Jump up to:a b c Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (February 2016). “Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system”. Aging Cell. 15 (1): 28–38. doi:10.1111/acel.12405. PMC 4717280. PMID 26463117.
- ^ Wang S, Raybuck A, Shiuan E, Jin J (2020). “Selective inhibition of mTORC1 in tumor vessels increases antitumor immunity”. JCI Insight. 5 (15): e139237. doi:10.1172/jci.insight.139237. PMC 7455083. PMID 32759497.
- ^ Jump up to:a b “Archived copy”. Archived from the original on 8 March 2014. Retrieved 26 February 2014.
- ^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID 12944570.
- ^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID 24767339.
- ^ Jeng L, Thorat A, Yang H, Yeh C-C, Chen T-H, Hsu S-C. Impact of Everolimus On the Hepatocellular Carcinoma Recurrence After Living Donor Liver Transplantation When Used in Early Stage: A Single Center Prospective Study [abstract]. Am J Transplant. 2015; 15 (suppl 3). http://www.atcmeetingabstracts.com/abstract/impact-of-everolimus-on-the-hepatocellular-carcinoma-recurrence-after-living-donor-liver-transplantation-when-used-in-early-stage-a-single-center-prospective-study/. Accessed 1 September 2015.
- ^ Thorat A, Jeng LB, Yang HR, Yeh CC, Hsu SC, Chen TH, Poon KS (November 2017). “Assessing the role of everolimus in reducing hepatocellular carcinoma recurrence after living donor liver transplantation for patients within the UCSF criteria: re-inventing the role of mammalian target of rapamycin inhibitors”. Annals of Hepato-Biliary-Pancreatic Surgery. 21 (4): 205–211. doi:10.14701/ahbps.2017.21.4.205. PMC 5736740. PMID 29264583.
- ^ Jeng LB, Lee SG, Soin AS, Lee WC, Suh KS, Joo DJ, Uemoto S, Joh J, Yoshizumi T, Yang HR, Song GW, Lopez P, Kochuparampil J, Sips C, Kaneko S, Levy G (December 2017). “Efficacy and safety of everolimus with reduced tacrolimus in living-donor liver transplant recipients: 12-month results of a randomized multicenter study”. American Journal of Transplantation. 18 (6): 1435–1446. doi:10.1111/ajt.14623. PMID 29237235.
- ^ Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (July 2009). “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice”. Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- ^ Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (December 2014). “mTOR inhibition improves immune function in the elderly”. Science Translational Medicine. 6 (268): 268ra179. doi:10.1126/scitranslmed.3009892. PMID 25540326. S2CID 206685475.
Further reading
- Sedrani R, Cottens S, Kallen J, Schuler W (August 1998). “Chemical modification of rapamycin: the discovery of SDZ RAD”. Transplantation Proceedings. 30 (5): 2192–4. doi:10.1016/S0041-1345(98)00587-9. PMID 9723437.
External links
- “Everolimus”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | Everolimus /ˌɛvəˈroʊləməs/ |
| Trade names | Afinitor, Zortress |
| Other names | 42-O-(2-hydroxyethyl)rapamycin, RAD001 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609032 |
| License data | EU EMA: by INNUS DailyMed: EverolimusUS FDA: Everolimus |
| Pregnancy category | AU: C[1] |
| Routes of administration | By mouth |
| ATC code | L01EG02 (WHO) L04AA18 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyEU: Rx-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Elimination half-life | ~30 hours[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 159351-69-6 |
| PubChem CID | 6442177 |
| DrugBank | DB01590 |
| ChemSpider | 21106307 |
| UNII | 9HW64Q8G6G |
| KEGG | D02714 |
| ChEMBL | ChEMBL1908360 |
| CompTox Dashboard (EPA) | DTXSID0040599 |
| ECHA InfoCard | 100.149.896 |
| Chemical and physical data | |
| Formula | C53H83NO14 |
| Molar mass | 958.240 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESOCCO[C@@H]1CC[C@H](C[C@H]1OC)C[C@@H](C)[C@@H]4CC(=O)[C@H](C)/C=C(\C)[C@@H](O)[C@@H](OC)C(=O)[C@H](C)C[C@H](C)\C=C\C=C\C=C(/C)[C@@H](OC)C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(=O)N3CCCC[C@H]3C(=O)O4 | |
| hideInChIInChI=1S/C53H83NO14/c1-32-16-12-11-13-17-33(2)44(63-8)30-40-21-19-38(7)53(62,68-40)50(59)51(60)54-23-15-14-18-41(54)52(61)67-45(35(4)28-39-20-22-43(66-25-24-55)46(29-39)64-9)31-42(56)34(3)27-37(6)48(58)49(65-10)47(57)36(5)26-32/h11-13,16-17,27,32,34-36,38-41,43-46,48-49,55,58,62H,14-15,18-26,28-31H2,1-10H3/b13-11+,16-12+,33-17+,37-27+/t32-,34-,35-,36-,38-,39+,40+,41+,43-,44+,45+,46-,48-,49+,53-/m1/s1 Key:HKVAMNSJSFKALM-GKUWKFKPSA-N |
//////////////// RAD-001, SDZ RAD, Certican, Novartis, Immunosuppressant, Everolimus, Afinitor, эверолимус , إيفيروليموس , 依维莫司 ,
#RAD-001, #SDZ RAD, #Certican, #Novartis, #Immunosuppressant, #Everolimus, #Afinitor, #эверолимус , #إيفيروليموس , #依维莫司 ,
DETOMIDINE

DETOMIDINE1H-Imidazole, 4-[(2,3-dimethylphenyl)methyl]-
4-(2,3-Dimethylbenzyl)-1H-imidazole
507876631-46-4[RN]7N8K34P2XH
- Molecular FormulaC12H14N2
- Average mass186.253 Da
UNII-7N8K34P2XHдетомидинديتوميدين地托咪定
Formal Name5-[(2,3-dimethylphenyl)methyl]-1H-imidazole, monohydrochlorideCAS Number90038-01-0Synonyms
- Domosedan
- MPV 253AII
Molecular FormulaC12H14N2 • HClFormula Weight222.7DetomidineCAS Registry Number: 76631-46-4CAS Name: 4-[(2,3-Dimethylphenyl)methyl]-1H-imidazoleAdditional Names: 4-(2¢,3¢-dimethylbenzyl)imidazoleMolecular Formula: C12H14N2Molecular Weight: 186.25Percent Composition: C 77.38%, H 7.58%, N 15.04%Literature References: a2-Adrenoceptor agonist with sedative and analgesic activity. Prepn: A. J. Karjalayne, K. O. A. Kurkela, EP24829; eidem,US4443466 (1981, 1984 both to Farmos). Physical studies: E. Laine et al.,Acta Pharm. Suec.20, 451 (1983). Crystal structure: L. H. J. Lajunen et al.,ibid.21, 163 (1984). Pharmacology: R. Virtanen, L. Nyman, Eur. J. Pharmacol.108, 163 (1985); R. Virtanen, E. MacDonald, ibid.115, 277 (1985). Mechanism of action: eidem,J. Vet. Pharmacol. Ther.8, 30 (1985).Properties: Crystals from acetone, mp 114-116°. LD50 i.v. in mice: 35 mg/kg (Karjalayne, Kurkela).Melting point: mp 114-116°Toxicity data: LD50 i.v. in mice: 35 mg/kg (Karjalayne, Kurkela) Derivative Type: HydrochlorideTrademarks: Domosedan (Farmos)Molecular Formula: C12H14N2.HClMolecular Weight: 222.71Percent Composition: C 64.72%, H 6.79%, N 12.58%, Cl 15.92%Properties: Crystals, mp 160°. Converts reversibly to monohydrate at room temp, 80% humidity.Melting point: mp 160° Therap-Cat-Vet: Sedative.
Detomidine is an imidazole derivative and α2-adrenergic agonist,used as a large animal sedative, primarily used in horses. It is usually available as the salt detomidine hydrochloride. It is a prescription medication available to veterinarians sold under the trade name Dormosedan.
Currently, detomidine is only licensed for use in horses in the US but it is also licensed for use in cattle in Europe and Australasia.[1]
Properties
Detomidine is a sedative with analgesic properties.[2] α2-adrenergic agonists produce dose-dependent sedative and analgesic effects, mediated by activation of α2 catecholamine receptors, thus inducing a negative feedback response, reducing production of excitatory neurotransmitters. Due to inhibition of the sympathetic nervous system, detomidine also has cardiac and respiratory effects and an antidiuretic action.[3]
Effects
UsesA profound lethargy and characteristic lowering of the head with reduced sensitivity to environmental stimuli (sound, pain, etc.) are seen with detomidine. A short period of reduced coordination is characteristically followed by immobility and a firm stance with front legs spread. Following administration there is an initial increase in blood pressure, followed by bradycardia and second degree atrioventricular block (this is not pathologic in horses). The horse commonly sweats to excess, especially on the flanks and neck. Other side effects reported include pilo erection (hair standing erect), ataxia, salivation, slight muscle tremors, and (rarely) penile prolapse.
Sedation and anaesthetic premedication in horses and other large animals, commonly combined with butorphanol for increased analgesia and depth of sedation. In conjunction with ketamine it may also be used for intravenous anaesthesia of short duration.
The drug is normally administered by the intravenous route, and is fastest and most efficient when given intravenously . However, in recalcitrant animals, detomidine may be administered by the intramuscular or sublingual routes. The dose range advised by the manufacturers is 20–40 µg/kg intravenous for moderate sedation, but this dose may need to be higher if given intramuscularly.
When given intravenously, detomidine usually takes effect in 2–5 minutes, and recovery is full within 30–60 minutes. However, this is highly dependent upon the dosage, environment, and the individual animal; some horses are highly resistant to sedation.
Detomidine is a poor premedication when using ketamine as an anesthetic in horses.As detomidine is an arrhythmogenic agent, extreme care should be exercised in horses with cardiac disease, and in the concurrent administration of other arrhythmogenics. The concurrent use of potentiated sulfonamide antibiotics is considered particularly dangerous.
Anesthetic recoveries in horses that have received ketamine following a detomidine premedication are often violent with the horse having multiple failures to stand resulting in trauma to itself. Xylazine is a superior premedication with ketamine resulting in safer recoveries.
PATENT
EP-03782989
Novel crystalline forms of detomidine hydrochloride monohydrate, processes for their preparation and compositions comprising them are claimed. Also claimed is their use as alpha2-adrenoreceptor agonists.
Detomidine hydrochloride (1H imidazole,4-[(2,3-dimethylphenyl)methyl]-hydrochloride (CAS Number: 90038-01-0) is a synthetic alpha 2-adrenoreceptor agonist with sedative and analgesic properties widely used for sedation of large animals like horses and cattle. This substance displays various other pharmacologic effects related to the cardiovascular and respiratory system as well as on muscles. Detomidine hydrochloride is available as a parenteral solution with 10 mg/ml as active ingredient which is indicated for use as a sedative and analgesic to facilitate minor surgical and diagnostic procedures in mature horses and yearlings (e.g. DORMOSEDAN®). Furthermore, detomidine hydrochloride is supplied as an oromucosal (i.e. sublingual) gel (e.g. DORMOSEDAN GEL®) with 7.6 mg/ml as active ingredient which is indicated for sedation and restraint in horses.
Further details regarding the clinical pharmacology and side effects as well as contraindications for this drug substance (i.e. active pharmaceutical ingredient) can be found in: Veterinary Psychopharmacology; Sharon L. et al., 2nd edition (2019), Wiley & Sons (pages 161 – 162). According to these authors detomidine has not been used in humans to date.
Detomidini hydrochloridum ad usum veterinarium is included in the EUROPEAN PHARMACOPOEIA (Ph. Eur. 9.0) but currently not included in the United States Pharmacopoeia (USP). It has to be noted that in the absence of a statement regarding a specific hydrate form, like a degree of hydration or mono-, di-, etc., in the title of the monograph – as is the case for detomidine hydrochloride – the anhydrous form is indicated for this substance.
According to a prior version of the respective monograph, namely Ph. Eur. 8.0, the substance exists as a white or almost white, hygroscopic, crystalline powder. The substance is soluble in water, freely soluble in ethanol (96 %), very slightly soluble in methylene chloride and practically insoluble in acetone. The molecular weight (M r) amounts to 222.7. The melting point (mp) is specified at about 160 °C. In the current monograph (Ph. Eur. 9.0) the content of detomidine hydrochloride is specified at 99.0 % to 101.0 percent (anhydrous substance).
[0003] In the current monograph (Ph. Eur. 9.0) the content of detomidine hydrochloride is specified at 99.0 % to 101.0 % (anhydrous substance).
The current monograph includes the three following known impurities:
Impurity A: (RS)-(2,3-dimethylphenyl) (1H-imidazol-4-yl)-methanol
Impurity B: (RS)-(1-benzyl-1H-imidazol-5-yl)(2,3-dimethylphenyl)-methanol
Impurity C: 4-[(2,3-dimethylcylohexyl)methyl]-1H-imidazole
The related substances are specified at ≤ 0.20 % for any unspecified impurities and ≤ 0.5 % for total impurities with a reporting threshold of 0.10 %.
The water content of detomidine hydrochloride as determined by Karl Fischer (KF) titration is limited to ≤ 2.0 % for release as well as shelf-life testing. As detomidine hydrochloride is hygroscopic, the compound has to be stored in airtight containers.
[0004] A synthesis method for detomidine was disclosed in US 4,584,383.
Specific details on the last two steps of a synthesis method for detomidine hydrochloride (including a reaction scheme) were published in Drugs Future 10, 17 (1985).
[0005] Detomidine hydrochloride is known to exist in two crystalline forms, namely the anhydrous form, as described above, and the monohydrate form B (M r: 240.7, CAS Number: 90038-00-9) which can easily interconvert, depending on ambient temperature and air humidity ( Veldre, K. et al., Eur. Journ. Pharm. 44, 273-280 (2011)). At 80 % air humidity and room temperature the monohydrate is reversibly formed. The theoretical water content of detomidine hydrochloride monohydrate amounts to 7.48 %.
[0006] To date, all commercially available (i.e. veterinary) drug products (i.e. parenteral solutions and oromucosal gels) only contain the anhydrous form. In general, hygroscopic substances like detomidine hydrochloride tend to absorb moisture so that they have to be protected from a humid environment during production and storage of the drug substance and corresponding drug product to avoid an inacceptable uptake of water. It has to be noted that such uptake during storage will reduce the content of the drug substance so that this would have to be taken into consideration during production of the corresponding drug product, like pharmaceutical preparation.
[0007] The problem to be solved is to provide a pure and stable active pharmaceutical ingredient (API), namely detomidine hydrochloride monohydrate, that can advantageously be used for the production of pharmaceutical compositions comprising the active pharmaceutical ingredient detomidine hydrochloride.
Example 1
Preparation of detomidine hydrochloride monohydrate (DHM)
[0053] Detomidine hydrochloride was synthesized starting from 1-benzyl-imidazole-4-carboxyaldehyde and 2,3-dimethylphenlymagnesiumbromide according to the two-step synthesis described in Drugs Future 10, 17 (1985).
[0054] For the second step of this synthesis (RS)-(3-Benzyl-3 H-imidazol-4-yl)-(2,3-dimethyl-phenyl)-methanol (HCl) was suspended in a mixture of water and hydrochloric acid. The catalyst (i. e. palladium on activated carbon) suspended in demineralized water was added. Hydrogenation (i.e. removal of the benzyl group and reduction of the hydroxyl group with hydrogen (H 2/Pd-C in HCl)) was performed at elevated temperature (50 – 80 °C) and the obtained suspension was filtered after the hydrogenation was finished. Subsequently ethyl acetate and a solution of ammonium hydroxide were added under continuous stirring. After discontinuation of stirring, phase separation occured after which the aqueous phase was repeatedly extracted with ethyl acetate. The combined organic phases were washed with demineralized water and filtered.
[0055] After addition of 5 – 6 N hydrogen chloride in 2-propanol and cooling precipitation of detomidine hydrochloride occured. After filtration the filtercake (i.e. raw product) was washed with ethyl acetate and dried.
[0056] A fraction of the resulting raw product (i.e. 5 g batch RSO E-190604 RP) was recrystallized from 5 g demineralized water by heating (until complete dissolution was obtained) and subsequent cooling on an ice bath. The resulting crystals were separated by filtration and the resulting filter cake washed with 2-propanol. Subsequently, the washed product was dried under vacuum (10 mbar) at 23 °C. The obtained yield for the white crystalline substance amounted to 66.0 % of the theory.
[0057] The resulting drug substance showed a water content (KF) of 7.49 %. The corresponding DSC curve was in line with the expectation (see for example Figure 1) and showed the two typical peaks routinely observed for DHM. Other than 2-propanol used for final washing none of the other solvents employed during the overall synthesis of this compound were found above the respective LOQ by GC-FID.
Example 2
Impurities after preparation of detomidine hydrochloride monohydrate (DHM)
[0058] A larger batch of detomidine hydrochloride (i.e. 50 g NK E-190709-I A K1) was synthesized in line with Example 1. However, the final crystals obtained after recrystallization from 50 ml demineralized water were washed with 25 ml demineralized water instead of 2-propanol. Drying was performed at 21 °C and 10 mbar until constant weight. The obtained yield for the white crystalline substance amounted to 87.2 % of the theory which was markedly higher than the yield obtained in Example 1. The water content of this substance was determined at 7.54 % (KF) and the corresponding DSC curve showed two peaks with an onset at 95.7 °C and 159.3 °C.
[0059] As shown below, recrystallization of the initial raw product from water (incl. washing) resulted in significant removal/reduction of impurities eluting before the detomidine peak (i.e. more polar compounds, e.g. Impurity A) as well as impurities eluting behind the detomidine peak (i.e. less polar compounds, e.g. Impurity C).
| Sample | Relevant compounds as detected by HPLC [area%]* | ||||
| Impurity A | Impurity RRT 0.84 | Detomidine | Impurity RRT 1.75 | Impurity C | |
| Raw product | 0.11 | 0.33 | 99.40 | 0.04 | 0.04 |
| Final crystallizate (K1) | 0.06 | 0.06 | 99.80 | 0.01 | 0.02 |
| *Table includes all compounds found at or above 0.04 area% in the initial raw product in the order in which they eluted from the HPLC column |
[0060] The final substance showed a very high HPLC purity of 99.80 area% (Ph. Eur. test method) and only a limited number of unknown impurities in addition to those
PATENT
https://patents.google.com/patent/WO2006108910A1/en
Example 1. Preparation of 4-[(2,3-dimethylbenzyl)]imidazole hydrochloride
(detomidine HCl)
l-Benzyl-5-(2,3-dimethylphenylhydroxymethyl)imidazole (20 kg), water (225 1), 30 % HCl (20 1), ethanol (5 1) and palladium on charcoal 10 % (4.4 kg) are charged. The mixture is stirred under 2.2 bar overpressure of hydrogen at 75 ± 5 °C for 24 hours. The reaction mixture is filtered at 45 ± 3 0C and the filter cake is washed with water (30 1). 170 1 of water is distilled off under reduced pressure and 30 % HCl (8 1) is added. The solution is cooled to 3 ± 3 0C during 2 h. The solution is seeded with crystals of detomidine HCl at 40 ± 5 °C, 30 ± 5 0C, 20 ± 5 °C and at 10 ± 5 0C, until the crystallization starts. The mixture is agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The crude product and water (250 1) are charged. The solution is heated to about 50 °C and stirred for 1 hour. The solution is cooled to 10 °C during 1.5 hour. The solution is filtered and 180 1 of water is distilled off under vacuum. 30 % HCl (20 1) is added and the solution is warmed to 60 0C, and then cooled to 3 ± 3 °C during 2 hours. The solution is seeded as above until the crystallization starts and agitated for two hours. The crystalline compound is collected by centrifogation and washed with toluene. The product is dried under vacuum at 39 ± 5 °C for 20 hours, at 61 ± 5 °C for 6 hours and at 85 ± 5 °C for 16 hours. The yield is 10.5 kg (78 %).
PATENT
https://patents.google.com/patent/US20080287685A1/en
- Detomidine which is 4-[(2,3-dimethylbenzyl)]imidazole of formula I
- is a well known pharmaceutical agent currently used as its hydrochloride salt in animal sedation.
- [0003]The synthesis of detomidine is described in U.S. Pat. Nos. 4,443,466 and 4,584,383. The preparation of detomidine hydrochloride salt is described in U.S. Pat. No. 4,584,383, wherein detomidine obtained from the hydrogenation step is first recovered from alkaline solution as a free base after which the crystalline product is converted into its hydrochloride salt by treatment with HCl-isopropanol in ethyl acetate.
- [0020]1-Benzyl-5-(2,3-dimethylphenylhydroxymethyl)imidazole (20 kg), water (225 l), 30% HCl (20 l), ethanol (5 l) and palladium on charcoal 10% (4.4 kg) are charged. The mixture is stirred under 2.2 bar overpressure of hydrogen at 75±5° C. for 24 hours. The reaction mixture is filtered at 45±3° C. and the filter cake is washed with water (30 l). 170 l of water is distilled off under reduced pressure and 30% HCl (8 l) is added. The solution is cooled to 3±3° C. during 2 h. The solution is seeded with crystals of detomidine HCl at 40±5° C., 30±5° C., 20±5° C. and at 10±5° C., until the crystallization starts. The mixture is agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The crude product and water (250 l) are charged. The solution is heated to about 50° C. and stirred for 1 hour. The solution is cooled to 10° C. during 1.5 hour. The solution is filtered and 180 l of water is distilled off under vacuum. 30% HCl (20 l) is added and the solution is warmed to 60° C., and then cooled to 3±3° C. during 2 hours. The solution is seeded as above until the crystallization starts and agitated for two hours. The crystalline compound is collected by centrifugation and washed with toluene. The product is dried under vacuum at 39±5° C. for 20 hours, at 61±5° C. for 6 hours and at 85±5° C. for 16 hours. The yield is 10.5 kg (78%).
PATENT
https://patents.google.com/patent/WO2020016827A1/en
Detomidine
Detomidine, 4-[(2,3-dimethylphenyl)methyl]-lH-Imidazole, is an a-2-andregenic agonist available under the brand name Equimidine® and Dormosedan® for use as a veterinary sedative. Detomidine is not currently approved for human use.
Detomidine and related compounds, including its 3,4 dimethyl isomer, iso-detomidine (4-(3,4- Dimethylbenzyl)-lH-imidazole) were first described in US4,443,466. Both the‘466 patent and the later US4, 584,383 describe the synthetic method of manufacturing detomidine as being based on coupling of an imidazole moiety with l-Bromo-2, 3-dimethyl benzene using a Grignard reaction. RU2448095 describes an alternative route of synthesis of the molecule based on coupling in presence of a Titanium catalyst. According to both the‘383 and‘095 patents, detomidine is purified by crystallization of its hydrochloride salt from water. The chemical structures of detomidine HC1 and iso-detomidine are shown below:

Detomidine HC1 Iso-detomidine
Two solid state forms of detomidine HC1 are known, the anhydrous and monohydrate forms.
Synthesis of the anhydrous form by crystallization of the monohydrate and further decomposition at elevated temperatures is described in US7,728,l47. Synthesis of the anhydrous form via decomposition of the monohydrate in reduced pressure is described in Laine et al (1983). According to Veldre et al (2011), the anhydrous and monohydrate forms of detomidine HC1 can easily interconvert depending on temperature and humidity.
The European Pharmacopeia 9.0 monograph (January 2014) describes detomidine HC1 for veterinary use. The monograph lists the established HPLC method for identification of detomidine and its impurities as using a Symmetry C8, 5 pm, 4.6 x 150 mm column, with a mobile phase of Ammonium phosphate buffer pH 7.9 – 65% and Acetonitrile – 35% at a flow rate of 1.0 mL/min and UV detection at 220 nm. That procedure is listed as recording three distinct impurities of detomidine:
Impurity A: (RS)-(2, 3 -dimethylphenyl)(l/f-imidazol-4-yl)m ethanol
– l/f-imidazol-5-yl)(2,3-dimethylphenyl)m ethanol

Impurity C: 4-| (2.3 -dimcthy ley clohcxyl)m ethyl |- 1 /7-im ida/olc

PCT/US18/012579 discloses topical formulations of detomidine and their uses in treating pain.
Purified detomidine for use in human pharmaceutical formulations is not known in the art.
EXAMPLE 5: Purification of organic impurities from detomidine HC1 monohvdrate
Two potential procedures for purification of organic impurities from sourced monohydrate were compared. The first attempted procedure was by direct re-crystallization of detomidine HC1 from 2.88 volumes of water, while the second included carbon treatment and precipitation of detomidine free base followed by the free base being reacted with HC1 and crystallized as monohydrate. Both procedures used the same non-GMP, off white anhydrous detomidine HC1 starting material which had previously been shown in Table 7 to contain 0.21% of iso-detomidine and 0.07% of Impurity A. All the re-crystallized materials were found to have practically the same purity level. The direct re-crystallization procedure was found to provide a product with a high yield and purity and at the same time provides a practical and scalable crystallization process which could be controlled by process parameters such as seeding and cooling rate.
Example 5 a: Direct recrvstallization
Anhydrous detomidine HC1 (4.5g) was introduced to a round-bottom flask with a magnetic stirrer and thermometer. Deionized water (l3ml) was then added and the mixture stirred and heated in a water bath. At 39°C, the complete dissolution of solids was observed, providing a clear yellow solution with a pH = 4.
The batch was gradually cooled by stirring. At 3 l°C, intensive crystallization was observed. The resulting slurry was cooled in an ice-water bath for 20 min and filtered. Flask and cake were then washed with 2 ml of cold deionized water and 3.97g of a white to cream colored solid was collected. 2.03g of the material was dried in a vacuum desiccator at ambient temperature and 20 mbar to a constant weight over 23 hrs producing a dry monohydrate – l .96g off-white crystalline solid (sample 1).
An additional l .9 lg of the material was dried in a vacuum oven at 90°C under house vacuum to a constant weight over about 24.5 hrs producing a dry anhydrate , l .68g off-white solid (sample 2)
The two samples were subjected to physical characterization and purity analysis by HPLC. The XRPD spectra and DSC and TGA thermograms of sample 1 are presented in Figures 8 -10 and of sample 2 are presented in Figures 11-13, respectively.
As shown in Table 11, direct re-crystallization resulted in the effective purification from all organic impurities, but was not effective for color. The content of iso-detomidine and of Impurity A was reduced to a level below the QL, but the off white color remained after re-crystallization.
Table 11 : properties following direct recrystallization (sample 1)

1 – below the QL
2 – system peak
Example 5b(i): Carbon treatment and detomidine free base isolation
Anhydrous detomidine HC1 (70.3g) and deionized water (220ml) were introduced to a 0.5 liter jacketed glass reactor equipped with a mechanical stirrer, thermocoupler and a circulating oil bath for heating and cooling.
The mixture was heated while stirring. At 40°C, complete dissolution was observed. Active carbon (CXV type, 5.2g) was added to the clear yellow solution and the batch stirred at 45°C for 50 minutes. Following this, the batch was filtered on through paper filter on Buchner funnel, reactor and filter washed with deionized water (20ml).
The slightly yellowish clear filtrate was reintroduced to the 0.5 liter reactor, stirred and 40% NaOH solution was added at 40°C. After 10ml NaOH solution was added, a pH of 7 was reached and precipitation began. An additional 13ml of NaOH was added over 1 hour at 42 – 52°C and intensive stirring (400 – 450 rpm) performed. The mixture at the end of the addition of NaOH had a pH of 13.
The batch was stirred at 33 – 35°C overnight then cooled to l6°C over 4 hours and stirred at this temperature for an additional hour. The resultant solid was filtered on Buchner filter, reactor and cake washed with two portions of deionized water (2><200ml). The wet solid (86g) was dried in a vacuum oven at 45°C to constant weight to produce a dry product (53.2g, Yield 90.7%) – white powder, m.p.=l 18.6 – 119.2
The dry detomidine base was analyzed for purity by HPLC, the results presented in Table 12. Table 12: Properties of detomidine base (intermediate in sample 2)

1 – system peak
Example 5b(nT Monohvdrate crystallization from detomidine base
The dry detomidine free base (53.0g) from Example 5b(i) was introduced together with 37% HC1 (29.7g) and deionized water (159g) into a 0.5 liter jacketed glass reactor equipped with a mechanical stirrer, a thermocoupler and a circulating oil bath for heating and cooling. The batch was stirred and heated to 45°C, at 37°C complete dissolution of solid was observed. The clear solution had a pH of 1. The solution was cooled gradually to 37°C and seeded with detomidine HC1 monohydrate and cooled gradually to 3°C over 4 hours, and then the batch was stirred for 45 minutes at this temperature. The solid was filtered on Buchner filter, reactor and cake washed with cold deionized water (80ml). The wet solid (61.9g) was dried in vacuum oven for 16 hours at 45°C to produce a dry product (57.8g, Yield 84.3%) – white crystalline powder (sample 2)
The dry detomidine HC1 monohydrate was analyzed for water by CKF (¾0 = 7.46%) and for purity by HPLC with the results presented in Table 13. Microscopic observation for particle morphology (regular prisms) was performed and the microscopic photograph is shown in Figure
14.
Table 13 : Properties of detomidine HC1 (sample 2)

1 – system peak
Example 5c: Re-crvstallization of detomidine HC1 to monohvdrate. bench scale experiment Anhydrous detomidine HC1 (754.6g) 37% HC1 (116. Og) and deionized water (2008g) were introduced to a 3 liter glass jacketed reactor equipped with a mechanical stirrer, two baffles, a thermocoupler and a circulating oil bath for heating and cooling. The batch was stirred and heated to 52°C, at 47°C complete dissolution was observed and the clear solution was found to have a pH of 0-0.5.
The solution was cooled gradually and at 45°C seeded with detomidine HC1 monohydrate (0.5g). Crystallization initiation was observed at 43°C and the batch was then cooled to 1.5°C during 5 hours and stirred for 12 hours at this temperature. The solid was filtered on Buchner filter and conditioned on the filter with vacuum for 40 minutes. The wet product (817g) was dried in vacuum oven to constant weight. For the first 13 hours, the material was dried at 30°C and 35-27 mbar, then for an additional 7 hours at 40°C and 30-18 mbar to produce a dry product (771.2g, Yield 94.6%) – white crystalline powder (Batch“90” in Tables 8-9; sample 3)
Dry detomidine HC1 monohydrate was analyzed for water by CKF (FhO = 7.37%) and for purity by HPLC, the results presented in Table 14. The physical characterization results are shown in Table 10 above.
The material was subjected to physical characterization and microscopic observation for particle morphology (regular prisms) microscopic photograph presented in Figure 7.
Table 14: Properties of detomidine HC1 (sample 3)
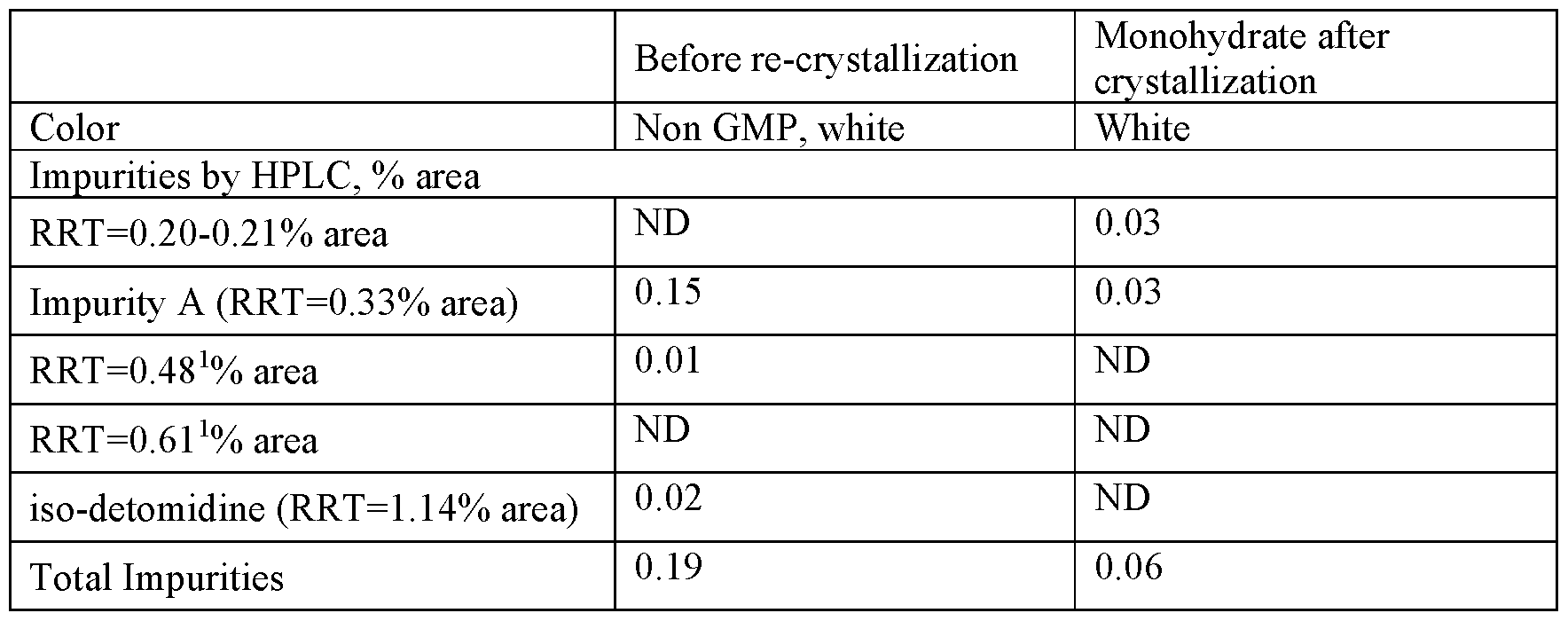
1 – system peak
EXAMPLE 6: Synthesis of iso -detomidine
Scheme 1 outlines a process for the synthesis of iso-detomidine was developed to produce a solid iso-detomidine HC1 in high yield and substantially free of impurities.

Scheme 1 : Route of synthesis of iso-detomidine
Example 6a: Sandmever Reaction
3,4 dimethyl aniline (150g, 1.24M) was mixed with acetonitrile (0.6 liter) in a 5 liter flask, chilled to lO°C and water (1.2 liter) added dropwise over 5 minutes. The mixture was cooled to 5°C with ice-ethanol bath and concentrated H2SO4 (98% wt, 363g 3.71M) was added dropwise over 30 min at 5-l0°C. Sodium nitrite (NaNC ) aqueous solution (89.7g in 300 ml water, 1.30M) was then added dropwise over 30 min at 0-5°C to give a brown solution. The resulting solution of diazonium salt was stirred at 0-5°C for an additional 30 min.
In another 5 liter flask KI (225g, 1.36M) was dissolved in water (0.8 liter) during stirring and cooled. The diazonium salt solution was added dropwise to the KI solution at 7-l3°C during 35 min, the batch stirred at 7-l3°C for 1.25 hr to give a black solution. MTBE (2.0 liter) was then added to the reaction mixture and Na2SC>4 (23.4g) was introduced in small portions during 5 min.
The mixture was settled and the organic phase separated and washed with two portions of brine (2 500ml). The organic solution was concentrated under vacuum to a volume of about 250ml.
The product was purified by vacuum distillation at ca. 40Pa, BP = 52 – 60°C to give 246g of intermediate 1 as a brown oil with a product yield of 86%.
Example 6b: TRT protection reaction
lH-Imidazole-4-carbaldehyde (45.2g, 0.47M) and acetonitrile (0.8 liter) are introduced into a 2 liter flack and cooled to 8°C, then TRT-C1 (131. Og, 0.47M) was added at 8°C and TEA (57. lg, 0.56M) was added dropwise during 20 min. The reaction mixture was stirred at 8 to l8°C for 2 hrs.
The reaction mixture was poured into a stirring mixture of water (0.72 liter) and MTBE (0.72 liter) and stirred for 10 minutes. The resulting solid was isolated by filtration on Buchner funnel and dissolved with THF (3 liter). The solution was dried over Na2SC>4 and concentrated to remove most of the solvent.
MTBE (400 ml) and PE (200ml) was added to the residue, the mixture stirred at 8°C for 16 hrs. The precipitated solid was isolated by filtration on Buchner filter and dried in air for 16 hrs at room temperature. Then the filter cake is dried by azeotropic drying with 2-Me-THF (2×500 ml) to give l29g of intermediate 2 as white solid with a yield of 66.5%.
Example 6c: Grignard reaction
A 2M solution of i-PrMgCl in THF (0.275 liter, 0.55M) and THF (1.0 liter) was introduced to a 2 liter flask at l2°C. Intermediate 1 (121.8g, 0.525M) was added dropwise during 20 min. The mixture was stirred at l2-l5°C for 3 hrs.
Intermediate 2 (84.6g, 0.25M) was added in small portions without cooling during 30 min, with a temperature rise to 25°C, to give a light brown solution. The solution was stirred for 2.5hrs at l5°C and added to aqueous solution of NH4CI (117g in 0.7 liter water) during 10 min at 5°C. PE (1.6 liter) was added during 5 min and the mixture stirred for extra 25 min.
Precipitated solid filtered on Buchner funnel and then re-slurred with mixture of MTBE (400 ml), water (600 ml) and PE (200 ml). Then the solid was filtered on Buchner funnel and re-slurred with MeOH (700 ml) at 60°C for 10 min, cooled to 20°C with cold water bath and filtered again on Buchner funnel. The solid product was dried in an air oven at 45 °C for 2 hrs to give 112 g of intermediate 3 as a white solid with a yield of 89.9%.
Example 6d: Reductive dehvdroxylation and de-protection
Intermediate 3 (l07g, 0.240M) and DCM (1.10 liter) were introduced to a 2 liter flask at 1 l°C, TFA (214 ml) was added dropwise over 5 mins with a temperature rise to l4°C.
The mixture was stirred for about 5 mins and EhSiH (94.4g, 0.794M) added dropwise during 5 mins. After stirring at 25-30°C for 16 hrs the mixture was concentrated by rotary evaporation at 40°C to a residue.
The residue of evaporation was dissolved in DCM (600 ml) and washed with 1.5M aq. HC1 (0.241iter). Organic phase was separated and washed with aq. NaOH (11.5g in 200ml water), pH of aqueous phase 13. Two phases were separated and the organic phase washed with brine (200 ml) dried over Na2S04 and filtered. The resulting solution was concentrated by rotary evaporation.
The evaporation residue was dissolved in mixture of EtOAc (500 ml) and EtOH (30 ml) and then 4M HC1 solution in dioxane (40 ml) was added dropwise in 5 minutes, pH = 1 – 2 adjusted and a white solid precipitated out.
The solid product was filtered on Buchner funnel, the cake dried in air for 16 hrs to give 36g of white solid.
The solid product was re-crystallized from iPrOH / Acetone. The dry cake (36g) and iPrOH were introduced into a 1 liter flask and heated to dissolution. Acetone (360 ml) was added to the resulting colorless solution at reflux during 10 mins. The mixture was cooled to 8°C and stirred at this temperature for additional 4.5 hrs. The solid product was filtered on Buchner funnel and dried in air for 36 hrs. 29.2g of iso-detomidine as a white solid was obtained with a yield of 54.4%. The 1H-NMR spectra of iso-detomidine is shown in Figure 15. EXAMPLE 7 : Re-crvstallization of detomidine HC1 spiked with 2% iso-detomidine
Detomidine HC1 monohydrate (26. Og), iso-detomidine HC1 (0.52g) and deionized water (68.7g) were introduced to a 100 ml glass jacketed reactor equipped with a mechanical stirrer, a thermocouple and a circulating oil bath for heating and cooling. The batch was stirred and heated to 51°C, at 47°C complete dissolution was observed.
The solution was cooled gradually and at 42°C seeded with detomidine HC1 monohydrate. Crystallization initiation was observed at 39°C and then the batch was cooled to 3°C for 5 hours, filtered on Buchner filter and conditioned on the filter with vacuum. The wet product (20.7 g) was dried in vacuum oven to constant weight to produce a dry product (20.13g, Yield 75.9%) – white crystalline powder
Dry detomidine HC1 monohydrate was analyzed for PSD and morphology, the results are presented in Table 8 (Sample. No. 91). The purity of re-crystallized material was analyzed using the optimized HPLC process disclosed herein, and the results are presented in Table 15.
Table 15 : Properties of detomidine HC1 following recrystallization from iso-detomidine spiked material

a area %
b Spiked amount, calculated
References
- ^ Clarke, Kathy W.; Hall, Leslie W.; Trim, Cynthia M., eds. (2014). “Principles of sedation, anticholinergic agents, and principles of premedication”. Veterinary Anaesthesia. pp. 79–100. doi:10.1016/B978-0-7020-2793-2.00004-9. ISBN 978-0-7020-2793-2.
- ^ England GC, Clarke KW (November 1996). “Alpha 2 adrenoceptor agonists in the horse–a review”. The British Veterinary Journal. 152 (6): 641–57. doi:10.1016/S0007-1935(96)80118-7. PMID 8979422.
- ^ Fornai F, Blandizzi C, del Tacca M (1990). “Central alpha-2 adrenoceptors regulate central and peripheral functions”. Pharmacological Research. 22 (5): 541–54. doi:10.1016/S1043-6618(05)80046-5. PMID 2177556.
External links
- “Medication Protocols for Horses”. The Ontario Association of Equine Practitioners. 2005. Archived from the original on 2007-04-17.
- Compendium of data sheets for animal medicines. National Office of Animal Health. 2005. ISBN 978-0-9548037-0-4.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATCvet code | QN05CM90 (WHO) |
| Legal status | |
| Legal status | Veterinary use only |
| Pharmacokinetic data | |
| Elimination half-life | 30 min |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 76631-46-4 |
| PubChem CID | 56032 |
| ChemSpider | 50586 |
| UNII | 7N8K34P2XH |
| KEGG | D07795 |
| ChEMBL | ChEMBL2110829 |
| CompTox Dashboard (EPA) | DTXSID00227457 |
| Chemical and physical data | |
| Formula | C12H14N2 |
| Molar mass | 186.258 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCc2cccc(Cc1cnc[nH]1)c2C | |
| hideInChIInChI=1S/C12H14N2/c1-9-4-3-5-11(10(9)2)6-12-7-13-8-14-12/h3-5,7-8H,6H2,1-2H3,(H,13,14) Key:RHDJRPPFURBGLQ-UHFFFAOYSA-N |
////////////// DETOMIDINE, UNII-7N8K34P2XH , детомидин ,ديتوميدين, 地托咪定 , Domosedan, Farmos, SEDATIVE
#DETOMIDINE, #UNII-7N8K34P2XH , #детомидин ,#ديتوميدين, #地托咪定 , #Domosedan, #Farmos, #SEDATIVE
https://patents.google.com/patent/WO2020016827A1/en
EXAMPLES
EXAMPLE 1 : Elemental analysis of impurities found in commercially available anhydrous detomidine HC1
Example la: Anhydrous detomidine HC1 was sourced from two commercial API suppliers. Properties of the commercial batches, GMP1, GMP2 and GMP3, are presented below.
Elemental impurity analysis was performed by inductively coupled plasma mass spectrometry (ICP-MS) on four different batches of sourced anhydrate. The results of the analysis are found in Table 1.
Table 1 : Elemental impurities in anhydrous detomidine HC1
11 Elements having levels L.T. 0.5 mg/kg (Ti, As, Hg, Pb, Mo, Pt, etc) are not presented in the table
The screening of elemental impurities shows that the GMP products contained significant levels of Pd (0.9 – 5.3 mg/kg). Pd is understood to be a catalyst used in the synthesis of detomidine (e.g., in reduction/hydrogenation methods).
Example lb: Characterization of commercially sourced material
Samples of the anhydrous detomidine products described in Table 1 were analyzed for water content and characterized by microscope, XRPD and thermal analyses. The results are summarized in Table 2.
Table 2: Characterization of commercial anhydrous detomidine HC1

a Anhydrous + mono hydrate The values presented in T able 2 demonstrate that the commercial samples of detomidine HC1 labeled as anhydrous contain some amount of monohydrate and this amount varied depending on storage conditions and packaging.
EXAMPLE 2: Stability assessment of anhvdrate and monohvdrate forms of detomidine base and detomidine HC1
Pure forms of crystalline free base, and HC1 salt (both monohydrate and anhydrate) were prepared from commercially sourced anhydrous detomidine HC1 as outlined in Table 3, and characterized using XRPD and thermal analysis. The solids were crystallized from aqueous solutions and then dried under different conditions. The crystallization and drying conditions are summarized in Table 3.
Table 3: Preparation of detomidine HC1 crystalline forms

The properties of the solids crystallized according to Table 3 are described in Table 4.
Table 4: Properties of Detomidine HC1 crystalline forms


These results demonstrate that crystallization from 2.8 – 2.9 volumes of water is effective for isolation and purification of the detomidine HC1 monohydrate drug substance. Drying of the monohydrate under mild conditions (20-40 mbar and temperatures from at least ambient to about 45 °C) provided pure monohydrate without traces of the anhydrous form.
The same monohydrate dried at elevated temperature (30-40 mbar 90°C) converted completely into the anhydrous form. The vacuum dried, hermetically closed anhydrate did not absorb water from the atmosphere and did not convert into the monohydrate. After exposure to atmospheric air, however, the anhydrate absorbed water and converted to a mixture of anhydrate and
monohydrate.
Melting points (m.p.) of the intermediate detomidine free base and hydrochloride of Sample 5 measured in open capillary corresponded with the published literature and the DSC data and are presented in Table 5. In order to evaluate effect of humidity on different forms of detomidine, a hydration study was performed. Samples of detomidine free base and hydrochloride salt were subjected to DVS analysis. These observations are in accordance with the DVS results shown in Figures 5 and 6, for detomidine free base and detomidine HC1, respectively.
Table 5: Composition and properties of known solid forms of detomidine



a -literature data
The free base was found to be crystalline and insoluble in water but it reacted readily with aqueous HC1 giving soluble detomidine hydrochloride.
Crystallization from water provided effective purification of the detomidine HC1 and formation of large regular crystals. Anhydrous detomidine hydrochloride appeared as small irregular particles whereas the possibility to control particle size distribution by crystallization parameters existed for the monohydrate.
The detomidine free base was found to be non-hygroscopic, but also able to absorb more than 1% of water at relative humidity (RH) >50%. An increase of humidity from RH 70% to RH >90% did not lead to absorption of additional water to monohydrate. During the dehydration cycle, the monohydrate began to lose water at RH -10% and converted into the anhydrate at RH =0%. Anhydrate did not absorb water at RH <30% and transformed completely to into the monohydrate at RH between 30% and 50%.
Four cycles of hydration-dehydration demonstrated good reproducibility of anhydrate- monohydrate interconversion.
An anhydrous detomidine HC1 of Sample 2 was shown to absorb water to a level of cKF 7.7% which corresponds well to the theoretical amount of water in the monohydrate form (Table 5).
The hydration profile of detomidine hydrochloride showed that the monohydrate is stable in a wide range of humidity between 10% and >90% RH. At the same time, the anhydrous form is not stable in atmospheric air and absorbs water at RH = 30 – 50%.
This data demonstrates that the anhydrous form is challenging in the aspects of water content and solid form stability and that detomidine HC1 monohydrate is more suitable for pharmaceutical development.
Example 3 : Impurity analysis of commercially sourced detomidine HC1
Using the established Pharmacopeia HPLC protocol (Symmetry C8, 5 pm, 4.6 x 150 mm column, with a mobile phase of 65% Ammonium phosphate buffer pH 7.9 and 35% Acetonitrile at a flow rate of 1.0 mL/min and UV detection at 220 nm), sourced samples of detomidine HC1 were assayed for impurities. As shown in Figure 1, a previously unreported peak was identified, which partially overlapped with that of detomidine. By LC-MS/MS analysis, this impurity was shown to have the same molecular weight as detomidine.
The established Pharmacopeia HPLC protocol did not separate the detomidine from the impurity. Therefore, for further identification of the elusive impurity, new HPLC protocols for assaying detomidine HC1 were developed. One protocol (“HPLC Protocol A”) comprised using a SunFire C8 column, IOqA, 3.5 pm, 4.6 x l50mm column with an initial mobile phase of 70% Ammonium Phosphate buffer solution, pH 7.9 and 30% Acetonitrile, at a flow rate of 1.0 mL/min and UV detection at 220 nm. To remove late eluting peaks, the flush gradient shown in Table 6 was applied after each run. This HPLC protocol allowed for a resolution factor of 3.9 between detomidine and the unidentified impurity. The quantitation level (QL) for impurities and degradation products is 0.025%. The detection level (DL) for impurities and degradation products is 0.01%.
Table 6: Flush gradient for HPLC protocol

Given its molecular weight, it was hypothesized that the impurity was iso-detomidine.
A solution of 100 pg/ml detomidine HC1 and about 1 pg/mL (about 1% of the working concentration) of detomidine impurity A and iso-detomidine were prepared and assayed using the new HPLC protocol (HPLC Protocol A), disclosed hereinabove. Figure 2 is a chromatogram showing that the previously unreported peak is confirmed as being iso-detomidine.
The analysis of commercially sourced detomidine HC1 revealed a significant additional impurity. Table 7 provides levels of the various detomidine impurities in different commercial batches. In all batches, total impurities were observed at levels of > 0.1% area.
Table 7: Impurity levels (% area) in commercial batches of detomidine.


provided by commercial supplier after undergoing the reciystallization process of Example 5, provided by inventors.
Further analysis of the peak at RRT=0.38 showed that it actually consisted of 2 separate, overlapping peaks. As shown in Figure 3, LC-MS/MS analysis confirmed one of these peaks as iso-impurity A. Further analysis, as shown in Figure 4, identified the second peak as (2,3- dimcthylphcnylX 1 //-imidazol-4-yl) methanone.
EXAMPLE 4: Optimization of the crystallization method of detomidine HC1 monohvdrate from commercial batches of anhydrous detomidine HC1
Crystallization experiments on 25, 65, and 770 gram scale were performed in 100 ml, 500 ml and 3 liter jacketed glass reactors, respectively, equipped with CBT (curved blade turbine) mechanical stirrers, circulating oil bath, thermocouples, and condensers. Stirrer speed in all experiments was between 300 – 600 rpm. Variable process parameters were: amounts of HC1, solvent ratio, cooling time/rate, seeding and cake wash. The parameters and the variation ranges were chosen according to production conditions. The crystallization parameters are summarized in Table 8.
Table 8: Crystallization parameters

a Seeding with detomidine HC1 monohydrate
b Time 24 hrs
c Seeding with anhydrous detomidine HC1
d 5.5 hrs cooling and overnight stirring at 1-3° C
e Spiked with 2% iso -detomidine
The drying parameters and solid properties of batches shown in Table 8 are described in Table 9. Table 9: Drying parameters and solid properties of detomidine monohydrate crystals

microscopic observation: Rods – aspect ratio > 2; prisms – aspect ratio < 2
u)M = mono hydrate
The data presented in Tables 8 and 9 demonstrate that crystallization from water and drying under technical vacuum gives pure detomidine HC1 monohydrate without traces of the detomidine HC1 anhydrous form. Variations of HC1 excess from 0 to 0.5 mole/mole base, cooling time from 1.5 to 24 hours and drying time from 15 to 33 hours appear to have no effect on the obtained properties of the solid form. All crystallization products appeared as pure detomidine HC1 monohydrate.
The crystallization initiation method also had no effect on crystalline form. The batches seeded with anhydrous material gave the same monohydrate as batches seeded with monohydrate and batches which crystallized spontaneously.
Contact with water for 24 hrs completely converted the anhydrous form into the monohydrate, even without complete dissolution (re-slurry).
Crystallization of the monohydrate from water gave large clear crystalline particles with a mean crystal size 0.3 – 0.7 mm, with some crystals larger than 2 mm in size. The shape of the crystals was rod-like or prism-like, if the aspect ratio of the crystals was < 2 the crystals were reported in Table 8 as prisms. A ratio of HC1 to base within the range 1.0 – 1.5 mole : mole and water to solid ratio within the range 2.1 – 2.8 V/wt were found to have no significant effect on the particle size distribution (PSD). However, a ratio of HC1 to base of about 1.5 were found to increase yields of highly pure detomidine HC1 monohydrate from under 90% (60.8%-86.4%) to over 90% (9l .4%-95.9%). Seeding also appeared to have no significant effect on PSD.
The cooling rate was found to have a weak effect on PSD. There was no effect observed for cooling over a time range between 1.5 and 5.5 hrs (mean cooling rate 0.10-0.3 l°C/min).
Slurry -to-slurry recrystallization of anhydrous material resulted in a strong reduction in particle size with the d(0.5) decreasing from 300-500m to 87m. These crystals were found irregular with no signs of prism-like or rod-like habit. In contrast, the re-slurry procedure applied to a mixture of anhydrate and monohydrate (15:85) gave a mixture of rod and prism-like crystals with d(0.5)=4l5p.
Batch size was found to have no significant effect on crystal size and shape. After scaling up from a 26g batch in 100 ml reactor to 770g in a 3 liter reactor, the PSD was very similar to that of small scale batches.
Prolonged cooling resulted in a “rounded” form of crystals. This effect was observed in two experiments, as seen in the microscopic photograph in Figure 7. In the first experiment the crystallizing suspension was cooled for 8 hrs, and in the second one it was stirred at low temperature for 12 hrs (batches 83 and 90 in Tables 8 and 9).
Under the conditions described, cooling had a strong effect on the process yield. Two re-slurry experiments were performed at the same water volume ratio as most of experiments (2.80 V/wt) but these two batches were not cooled and filtered at 24°C. In these experiments the yield dropped from 86% to 60-65% (batches 84, 85 in Tables 8 and 9).
Acceptable yields were obtained in cooled batches within the solvent volume ratio range 2.1 – 2.8 V/wt with the cooling temperature between about l.5°C – 4°C
An increase of HC1 to base molar ratio from 1 to 1.5 was found to raise the yield from 86% to 95%. Cake wash reduced the yield by 2 – 3%. Re-crystallization in presence of 2% iso- detomidine reduced the yield from 84 – 85% to 76%. The purity of the samples prepared according to methods disclosed in Tables 8 and 9, determined using the optimized HPLC method, are presented in Table 10. Table 10

E
Fluvoxamine

Fluvoxamine
- Molecular FormulaC15H21F3N2O2
- Average mass318.335 Da
- 54739-18-3
(E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]-1-pentanone O-(2-Aminoethyl)oxime1-Pentanone, 5-methoxy-1-[4-(trifluoromethyl)phenyl]-, O-(2-aminoethyl)oxime, (1E)-2-[({(1E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino)oxy]ethanamine
2-{[(E)-{5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino]oxy}ethanamine1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)-
387954739-18-3[RN]5583954[Beilstein]5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oximeA selective serotonin reuptake inhibitor that is used in the treatment of DEPRESSION and a variety of ANXIETY DISORDERS.
Fluvoxamine, sold under the brand name Luvox among others, is an antidepressant of the selective serotonin reuptake inhibitor (SSRI) class[5] which is used primarily for the treatment of obsessive–compulsive disorder (OCD).[6] It is also used to treat depression and anxiety disorders, such as panic disorder, social anxiety disorder, and post-traumatic stress disorder.[7][8]
FLUVOXAMINE MALEATE
C19H25F3N2O6, 434.4 g/mol
1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)-, (Z)-2-butenedioate (1:1)
(Z)-but-2-enedioic acid;2-[(E)-[5-methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene]amino]oxyethanamine
Luvox
61718-82-9
CAS 54739-20-7
Fevarin, Luvox CR
Synonyms
- 5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oxime, maleate (1:1)
- 5-Methoxy-4′-trifluoromethylvalerophenone (E)-O-2-aminoethyloxime monomaleate
- DU23000
- Fevarin
- Fluvoxamine maleate
- Luvox
- Luvox CR
- SME 3110
- UNII-5LGN83G74V
| Originator Company | Solvay SA |
| Active Companies | AbbVie Inc; Abbott Laboratories; Meiji Seika Pharma Co Ltd; Solvay SA |
| Launched (Obsessive compulsive disorder – EU – Dec-1983) |
In the EU, the product is indicated for the treatment of obsessive compulsive disorder (OCD) and for the treatment of major depressive disorder (MDD)
In Japan, Luvox is indicated for the treatment of adult or pediatric OCD, social anxiety disorder (SAD) and MDD
USFDA The drug was approved for the treatment of OCD and SAD in April 2008
CHINA
In 2000, the drug was launched in China for the treatment of OCD and MDD
Patents and Generics
FDA exclusivity expired in the US in June 2000. Generic versions have been on the market since that time. Generic fluvoxamine was still available in the US by May 2007, despite the fact the Solvay/Jazz product had not been relaunched . By October 2004, the drug was also off patent in most European countries .
Medical uses
Fluvoxamine is approved in the United States for OCD,[9][6] and social anxiety disorder.[10] In other countries (e.g., Australia,[11][12] the UK,[13] and Russia[14]) it also has indications for major depressive disorder. In Japan it is currently[when?] approved to treat OCD, SAD and MDD.[15][16] Fluvoxamine is indicated for children and adolescents with OCD.[17] The drug works long-term, and retains its therapeutic efficacy for at least one year.[18] It has also been found to possess some analgesic properties in line with other SSRIs and tricyclic antidepressants.[19][20][21]
There is tentative evidence that fluvoxamine is effective for social phobia in adults.[22] Fluvoxamine is also effective for GAD, SAD, panic disorder and separation anxiety disorder in children and adolescents.[23] There is tentative evidence that fluvoxamine may help some people with negative symptoms of chronic schizophrenia.[24][25]
A double-blind controlled study found that fluvoxamine may prevent clinical deterioration in outpatients with symptomatic COVID-19. The study had important limitations: it was run fully remotely; it had a small sample size (150) and short follow-up duration (15 days).[26] The accompanying editorial noted that, although this study is important enough to choose out of more than 10,000 other COVID-19 related submissions, it “presents only preliminary information” and “the findings should be interpreted as only hypothesis generating; they should not be used as the basis for current treatment decisions.”[27] Similarly, the study authors themselves cautioned that “the trial’s results should not be treated as a measure of fluvoxamine’s effectiveness against COVID-19 but as an encouraging indicator that the drug warrants further testing.”[28] A prospective open-labelled cohort study showed similar results.[29]
Adverse effects
Gastrointestinal side effects are more common in those receiving fluvoxamine than with other SSRIs.[30] Otherwise, fluvoxamine’s side-effect profile is very similar to other SSRIs.[2][9][11][13][31][32]Common (1–10% incidence) adverse effects
- Nausea
- Vomiting
- Weight loss
- Yawning
- Loss of appetite
- Agitation
- Nervousness
- Anxiety
- Insomnia
- Somnolence (drowsiness)
- Tremor
- Restlessness
- Headache
- Dizziness
- Palpitations
- Tachycardia (high heart rate)
- Abdominal pain
- Dyspepsia (indigestion)
- Diarrhea
- Constipation
- Hyperhidrosis (excess sweating)
- Asthenia (weakness)
- Malaise
- Sexual dysfunction (including delayed ejaculation, erectile dysfunction, decreased libido, etc.)
- Xerostomia (dry mouth)
Uncommon (0.1–1% incidence) adverse effects
- Arthralgia
- Hallucination
- Confusional state
- Extrapyramidal side effects (e.g. dystonia, parkinsonism, tremor, etc.)
- Orthostatic hypotension
- Cutaneous hypersensitivity reactions (e.g. oedema [buildup of fluid in the tissues], rash, pruritus)
Rare (0.01–0.1% incidence) adverse effects
- Mania
- Seizures
- Abnormal hepatic (liver) function
- Photosensitivity (being abnormally sensitive to light)
- Galactorrhoea (expulsion of breast milk unrelated to pregnancy or breastfeeding)
Unknown frequency adverse effects
- Hyperprolactinaemia (elevated plasma prolactin levels leading to galactorrhoea, amenorrhoea [cessation of menstrual cycles], etc.)
- Bone fractures
- Glaucoma
- Mydriasis
- Urinary incontinence
- Urinary retention
- Bed-wetting
- Serotonin syndrome — a potentially fatal condition characterised by abrupt onset muscle rigidity, hyperthermia (elevated body temperature), rhabdomyolysis, mental status changes (e.g. coma, hallucinations, agitation), etc.
- Neuroleptic malignant syndrome — practically identical presentation to serotonin syndrome except with a more prolonged onset
- Akathisia — a sense of inner restlessness that presents itself with the inability to stay still
- Paraesthesia
- Dysgeusia
- Haemorrhage
- Withdrawal symptoms
- Weight changes
- Suicidal ideation and behaviour
- Violence towards others[33]
- Hyponatraemia
- Syndrome of inappropriate antidiuretic hormone secretion
- Ecchymoses
Interactions[edit]

Luvox (fluvoxamine) 100 mg film-coated scored tablets
Fluvoxamine inhibits the following cytochrome P450 enzymes:[34][35][36][37][38][39][40][41][42]
- CYP1A2 (strongly) which metabolizes agomelatine, amitriptyline, caffeine, clomipramine, clozapine, duloxetine, haloperidol, imipramine, phenacetin, tacrine, tamoxifen, theophylline, olanzapine, etc.
- CYP3A4 (moderately) which metabolizes alprazolam, aripiprazole, clozapine, haloperidol, quetiapine, pimozide, ziprasidone, etc.[43]
- CYP2D6 (weakly) which metabolizes aripiprazole, chlorpromazine, clozapine, codeine, fluoxetine, haloperidol, olanzapine, oxycodone, paroxetine, perphenazine, pethidine, risperidone, sertraline, thioridazine, zuclopenthixol, etc.[44]
- CYP2C9 (moderately) which metabolizes nonsteroidal anti-inflammatory drugs, phenytoin, sulfonylureas, etc.
- CYP2C19 (strongly) which metabolizes clonazepam, diazepam, phenytoin, etc.
- CYP2B6 (weakly) which metabolizes bupropion, cyclophosphamide, sertraline, tamoxifen, valproate, etc.
By so doing, fluvoxamine can increase serum concentration of the substrates of these enzymes.[34]
The plasma levels of oxidatively metabolized benzodiazepines (e.g., triazolam, midazolam, alprazolam and diazepam) are likely to be increased when co-administered with fluvoxamine. However the clearance of benzodiazepines metabolized by glucuronidation (e.g., lorazepam, oxazepam, temazepam)[45][46] is unlikely to be affected by fluvoxamine.[47] It appears that benzodiazepines metabolized by nitro-reduction (clonazepam, nitrazepam) are unlikely to be affected by fluvoxamine.[48] Using fluvoxamine and alprazolam together can increase alprazolam plasma concentrations.[49] If alprazolam is coadministered with fluvoxamine, the initial alprazolam dose should be reduced to the lowest effective dose.[50][51]
Fluvoxamine and ramelteon coadministration is not indicated.[52][53]
Fluvoxamine has been observed to increase serum concentrations of mirtazapine, which is mainly metabolized by CYP1A2, CYP2D6, and CYP3A4, by 3- to 4-fold in humans.[54] Caution and adjustment of dosage as necessary are warranted when combining fluvoxamine and mirtazapine.[54]
Fluvoxamine seriously affects the pharmacokinetics of tizanidine and increases the intensity and duration of its effects. Because of the potentially hazardous consequences, the concomitant use of tizanidine with fluvoxamine, or other potent inhibitors of CYP1A2, should be avoided.[55]
Fluvoxamine’s interaction with St John’s wort can lead to increased serotonin levels and potentially lead to serotonin syndrome.[citation needed]
Pharmacology
| Site | Ki (nM) |
|---|---|
| SERT | 2.5 |
| NET | 1,427 |
| 5-HT2C | 5,786 |
| α1-adrenergic | 1,288 |
| σ1 | 36 |
Fluvoxamine is a potent selective serotonin reuptake inhibitor with around 100-fold affinity for the serotonin transporter over the norepinephrine transporter.[35] It has negligible affinity for the dopamine transporter or any other site, with the sole exception of the σ1 receptor.[59][60] It behaves as a potent agonist at this receptor and has the highest affinity (36 nM) of any SSRI for doing so.[59] This may contribute to its antidepressant and anxiolytic effects and may also afford it some efficacy in treating the cognitive symptoms of depression.[61] Unlike fluoxetine, fluvoxamine’s metabolites are inactive, without a significant effect on serotonin or norepinephrine uptake.[62]
History
Fluvoxamine was developed by Kali-Duphar,[63] part of Solvay Pharmaceuticals, Belgium, now Abbott Laboratories, and introduced as Floxyfral in Switzerland in 1983.[63] It was approved by the U.S. Food and Drug Administration (FDA) in 1994, and introduced as Luvox in the US.[64] In India, it is available, among several other brands, as Uvox by Abbott.[65] It was one of the first SSRI antidepressants to be launched, and is prescribed in many countries to patients with major depression.[66] It was the first SSRI, a non-TCA drug, approved by the U.S. FDA specifically for the treatment of OCD.[67] At the end of 1995, more than ten million patients worldwide had been treated with fluvoxamine.[68][failed verification] Fluvoxamine was the first SSRI to be registered for the treatment of obsessive compulsive disorder in children by the FDA in 1997.[69] In Japan, fluvoxamine was the first SSRI to be approved for the treatment of depression in 1999[70][71] and was later in 2005 the first drug to be approved for the treatment of social anxiety disorder.[72] Fluvoxamine was the first SSRI approved for clinical use in the United Kingdom.[73]
Society and culture
Manufacturers include BayPharma, Synthon, and Teva, among others.[74]
SYN


SYN
J. Zhejiang Univ. (Medical Sci.) (2003), 32 (5), 441-442

PATENT
WO 2014178064
The present invention relates to an improved and industrially applicable process for the preparation of fluvoxamine maleate of formula I,
Fluvoxamine or (E)-5-methoxy-1 -[4-(trifluoromethyl)phenyl]pentan- 1 -one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in US patent 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25°C.
Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanbl and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of
various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in US patent 4,085,225, the oxime of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at -5 to 0°C, then aminated with ammonia in methanol at 100°C using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.
The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages involve like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as wel l as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
US patent 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trtfluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C to obtain fluvoxamine base in
toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35°C may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
Thus, present invention fulfills the need of the art and provides an improved and industrially applicable process for preparation of fluvoxamine maleate, which provides fluvoxamine maleate in high purity and overall good yield.
EXAMPLES:
Stage – 1 : Preparation of (1E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyI pheny 1) pentan-1-imine formula III
To a stirred solution of 5-methoxy- 1 -(4-trifluoromethylphenyl) pentan-1 -one ( 150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30°C. The reaction mass was heated 45-50°C for 10- 15 minutes followed by maintaining the reaction mass at temperature 45-50°C for 8-9 hours under stirring. The reaction mass was cooled to 25-30°C and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50°C. The obtained slurry was cooled to 25-30°C and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5- 10 minutes at temperature 25-30°C followed by cooling the mass at temperature -5°C to – 10°C, stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at -5 to -10°C and further in vacuum at 25-30°C for 2-3 hours to give 138 – 142 gm of title compound. HPLC purity: >98.5%
Stage – 2: Preparation of crude fluvoxamine maleate formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes ( 1 14.64 gm) and water (69 ml), stage-1 (1 15 gm) was added at temperature 40-45°C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (86.36 gm) drop wise into the reaction mixture at temperature 40-45°C and maintained for 1 -2 hour. Water (1 150 ml) was added in to the reaction mixture at temperature 25-30°C and stirred for 20-25 minutes. Then toluene (575 ml x 2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water ( 1 1 50 x 5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30°C for 2-3 hours. The reaction mixture was cooled to 0-5°C and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and suck dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30°C, filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage – 3: Preparation of pure fluvoxamine maleate formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45°C. Stage -2 ( 1 50 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5- 10 minutes, filtered and cooled to 25°C. Toluene (68 ml) was added into the reaction mixture at temperature 25°C and stirred for 30 minutes. Filtered the solid, washed with 10-15°C chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one formula II
To a solution of cone. HCl (600 ml) and water ( 160 ml), organic residue (250 gm) of ( 1 £)+( 1 Z) of 1 -N-hydroxy-5-methoxy- 1 -[4-(trifluoromethyl) phenyl]pentan-1 -imine and traces of 5-methoxy- 1 -[4-(trifluoromethyl)phenyl]pentan- 1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30°C under stirring. The reaction mixture was heated to 67-75°C and maintained for 13-14 hours followed by cool ing the reaction mixture at temperature 25-30°C. Then after hexane (500 x 2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30°C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30°C. Further the layers were separated and hexane layer was added activated charcoal ( 12.5 gm) and stirred for 20-30 minutes at temperature 30-35°C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30°C followed by cooling at 0 to -5°C and stirred for 30-40 minutes at 0 to -5°C. The reaction mixture was filtered and dried to get 150 to l 75 gm of title compound. HPLC purity: >99%.
PATENT
US 20140243544
IN 2013MU01290/WO 2014178064
WO 2014035107
PATENT
https://patents.google.com/patent/US9783492B2/en
Fluvoxamine or (E)-5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in U.S. Pat. No. 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25° C.

Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanol and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in U.S. Pat. No. 4,085,225, the mine of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at −5 to 0° C., then aminated with ammonia in methanol at 100° C. using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.

The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages in like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as well as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
U.S. Pat. No. 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trifluoromethylvalerophenone oxime compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C. to obtain fluvoxamine base in toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35° C. may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
EXAMPLES
Stage-1: Preparation of (1 E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyl phenyl)pentan-1-imine Formula III
To a stirred solution of 5-methoxy-1-(4-trifluoromethylphenyl)pentan-1one (150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30° C. The reaction mass was heated 45-50° C. for 10-15 minutes followed by maintaining the reaction mass at temperature 45-50° C. for 8-9 hours under stirring. The reaction mass was cooled to 25-30° C. and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50° C. The obtained slurry was cooled to 25-30° C. and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5-10 minutes at temperature 25-30° C. followed by cooling the mass at temperature −5° C. to −10° C., stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at −5 to −10° C. and further in vacuum at 25-30° C. for 2-3 hours to give 138-142 gm of title compound. HPLC purity: >98.5%
Stage-2: Preparation of Crude Fluvoxamine Maleate Formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes (114.64 gm) and water (69 ml), stage-1 (115 gm) was added at temperature 40-45° C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (8636 gm) drop wise into the reaction mixture at temperature 40-45° C. and maintained for 1-2 hour. Water (1150 ml) was added in to the reaction mixture at temperature 25-30° C. and stirred for 20-25 minutes. Then toluene (575 ml×2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water (1150×5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30° C. for 2-3 hours. The reaction mixture was cooled to 0-5° C. and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and such dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30° C., filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage-3: Preparation of Pure Fluvoxamine Maleate Formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45° C. Stage-2 (150 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5-10 minutes, filtered and cooled to 25° C. Toluene (68 ml) was added into the reaction mixture at temperature 25° C. and stirred for 30 minutes. Filtered the solid, washed with 10-15° C. chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one Formula II
To a solution of conc. HCl (600 ml) and water (160 organic residue (250 gm) of (1 E)+(1 Z) of 1-N-hydroxy-5-methoxy-1-[4trifluoromethyl)phenyl]pentan-1-imine and traces of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30° C. under stirring. The reaction mixture was heated to 67-75° C. and maintained for 13-14 hours followed by cooling the reaction mixture at temperature 25-30° C. Then after hexane (500×2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30° C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250 ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30° C. Further the layers were separated and hexane layer was added activated charcoal (12.5 gm) and stirred for 20-30 minutes at temperature 30-35° C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30° C. followed by cooling at 0 to −5° C. and stirred for 30-40 minutes at 0 to −5° C. The reaction mixture was filtered and dried to get 150 to 175 gm of title compound. HPLC purity: >99%.
Claims (5)Hide Dependent
We claim:1. An improved process for the preparation of fluvoxamine maleate of formula I,

wherein the improvements comprises the steps of:a). condensing the compound of formula II,

with hydroxylamine hydrochloride in the presence of sodium carbonate granules at temperature 45-50° C. in suitable solvent to form a compound of formula III, wherein the compound of formula III comprises a mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine, and wherein the mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine comprises 98% of E-isomer and 2% of Z-isomer;

b). isolating compound of formula III;c). treating compound of formula III with 2-chloroethylamine hydrochloride in the presence of base in suitable solvent at 40-45° C. to form compound of formula IV;

d). extracting compound of formula IV with suitable solvent to form an organic layer;e). treating organic layer of step d) with maleic acid;f). isolating crude fluvoxamine maleate of formula I; andg). optionally purifying fluvoxamine maleate of formula I.
2. The process according to claim 1, wherein in step a), said suitable solvent is selected from the group consisting of alcohol, ketone, nitrile, and hydrocarbons in any suitable proportion or mixtures thereof;in step c), said base is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, lithium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, triethylamine and diisopropylethyamine;in step c), said solvent is selected from the group consisting of dimethylformamide (DMF), dimethylsulphoxide (DMSO) and hexamethylphosphoramide (HMPA) in any suitable proportion or mixtures thereof; andin step d) said suitable solvent is selected from the group consisting of toluene and xylene.3. A process for the isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one of formula II from mixture of (1E)+(1Z) of 1-N -hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III by treating compound of formula III with aqueous hydrochloric acid, wherein the mixture of (1E)+(1Z) of 1-N-hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III comprises 98% of E-isomer and 2% of Z-isomer.4. The process according to claim 3, wherein the reaction is performed at temperature 65-75°C.5. The process according to claim 1, wherein in step a), said suitable solvent is methanol.
Publication numberPriority datePublication dateAssigneeTitleUS4081551A *1975-03-201978-03-28U.S. Philips CorporationOxime ethers having anti-depressive activityUS4085225A1975-03-201978-04-18U.S. Philips CorporationOxime ethers having anti-depressive activityCN1079733A *1993-04-081993-12-22中国科学院成都有机化学研究所The synthetic method of a-benzoin oximeUS6433225B11999-11-122002-08-13Sun Pharamaceutical Industries, Ltd.Process for the preparation of fluvoxazmine maleateCN101654419A *2009-09-122010-02-24西北师范大学Preparation method of fluvoxamine maleate
Syn
US 6433225 SUN
https://patents.google.com/patent/US6433225B1/en
EXAMPLE 1
To a stirred mixture of toluene (1.20 lit.), PEG-400 (0.4 lit) and powdered potassium hydroxide (86.0 g on 100% basis, 1.53 mol.) at ambient temperature is added 5-methoxy-4′-trifluoromethylvalerophenone oxime (100 g, 0.363 mol.), followed by 2-chloroethyl amine hydrochloride (50.56 g, 0.435 mol.). The mixture is stirred at 30-35° C. for 2 hours. Water (1.2 lit.) is then added, stirred for 30 mins. and the aqueous layer is separated out. The organic layer is washed with water (˜3×500 ml) until the washings are neutral. To the washed organic layer is added a solution of maleic acid (14.14 g, 0.363 mol.) in water (65 ml) and the mixture is stirred at 25-30° C. temperature for 2 hours, then cooled to 5-10° C. when the maleate salt crystallizes out. The crystallized fluvoxamine maleate is filtered, washed with toluene (200 ml) and sucked to dryness. The crude fluvoxamine maleate thus obtained is dissolved in water (300 ml) at 50-55° C. to get a clear solution, then gradually cooled to 5-8° C. and then further stirred at this temperature for 2 hours. The recrystallised fluvoxamine maleate is filtered, washed with chilled water (5° C., 100 ml) and sucked dry. The product is finally dried at 50-55° C. to constant weight. The fluvoxamine maleate obtained complies with the specifications of British Pharmacopoeia, 1999.EXAMPLE 2
This process when scaled up in pilot plant on 4.0 kg scale input of 5-methoxy-4′-trifluoromethylvalerophenone oxime gave 4.5 kg (71.2%) of fluvoxamine maleate, complying to the specifications of British Pharmacopoeia, 1999.

SYN
US 4085225
https://patents.google.com/patent/US4085225A/en
EXAMPLE 15-Methoxy-4′-trifluoromethylvalerophenone O-(2-aminoethyl) oxime maleate (1:1).
20.4 Mmol (5.3 g) of 5-methoxy-4′-trifluoromethylvalerophenone (melting point 43°-44° C), 20.5 mmol (3.1 g) of 2-aminooxyethylaminedihydrochloride and 10 ml of pyridine were refluxed for 15 hours in 20 ml of absolute ethanol. After evaporating the pyridine and the ethanol in vacuo, the residue was dissolved in water. This solution was washed with petroleum ether and 10 ml of 50% sodium hydroxide solution were then added. Then three extractions with 40 ml of ether were carried out. The ether extract was washed successively with 20 ml of 5% sodium bicarbonate solution and 20 ml of water. After drying on sodium sulphate, the ether layer was evaporated in vacuo. Toluene was then evaporated another three times (to remove the pyridine) and the oil thus obtained was dissolved in 15 ml of absolute ethanol. An equimolar quantity of maleic acid was added to said solution and the solution was then heated until a clear solution was obtained. The ethanol was then removed in vacuo and the residue was crystallized from 10 ml of acetonitrile at +5° C. After sucking off and washing with cold acetonitrile, it was dried in air. The melting point of the resulting title compound was 120°-121.5° C.

SYN
GB 1535226

References
- ^ Jump up to:a b Use During Pregnancy and Breastfeeding
- ^ Jump up to:a b c d e f “Product Information Luvox”. TGA eBusiness Services. Abbott Australasia Pty Ltd. 15 January 2013. Retrieved 21 October 2013.
- ^ van Harten J (March 1993). “Clinical pharmacokinetics of selective serotonin reuptake inhibitors”. Clinical Pharmacokinetics. 24 (3): 203–20. doi:10.2165/00003088-199324030-00003. PMID 8384945. S2CID 84636672.
- ^ “Luvox”. ChemSpider. Royal Society of Chemistry. Archived from the original on 15 November 2013. Retrieved 21 October 2013.
- ^ “Fluvoxamine Maleate Information”. U.S. Food and Drug Administration(FDA). 15 July 2015. Archived from the original on 29 November 2019. Retrieved 28 November 2019.
- ^ Jump up to:a b McCain JA (July 2009). “Antidepressants and suicide in adolescents and adults: a public health experiment with unintended consequences?”. P T. 34(7): 355–78. PMC 2799109. PMID 20140100.
- ^ Figgitt DP, McClellan KJ (October 2000). “Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders”. Drugs. 60 (4): 925–54. doi:10.2165/00003495-200060040-00006. PMID 11085201.
- ^ Irons J (December 2005). “Fluvoxamine in the treatment of anxiety disorders”. Neuropsychiatric Disease and Treatment. 1 (4): 289–99. PMC 2424117. PMID 18568110.
- ^ Jump up to:a b “Fluvoxamine Maleate tablet, coated prescribing information”. DailyMed. 14 December 2018. Retrieved 28 November 2019.
- ^ “Luvox CR approved for OCD and SAD”. MPR. 29 February 2008. Retrieved 2 March 2019.
- ^ Jump up to:a b Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ “Luvox Tablets”. NPS MedicineWise. Retrieved 22 October 2018.
- ^ Jump up to:a b Joint Formulary Committee (2013). British National Formulary (BNF)(65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ “Summary of Full Prescribing Information: Fluvoxamine”. Drug Registry of Russia (RLS) Drug Compendium (in Russian). Retrieved 21 March 2015.
- ^ “2005 News Releases”. Astellas Pharma. Retrieved 16 September 2018.
- ^ “International Approvals: Ebixa, Depromel/Luvox, M-Vax”. http://www.medscape.com. Retrieved 16 September 2018.
- ^ “US-FDA Fluvoxamine Product Insert”. March 2005.
- ^ Wilde MI, Plosker GL, Benfield P (November 1993). “Fluvoxamine. An updated review of its pharmacology, and therapeutic use in depressive illness”. Drugs. 46(5): 895–924. doi:10.2165/00003495-199346050-00008. PMID 7507038.
- ^ Kwasucki J, Stepień A, Maksymiuk G, Olbrych-Karpińska B (2002). “[Evaluation of analgesic action of fluvoxamine compared with efficacy of imipramine and tramadol for treatment of sciatica–open trial]”. Wiadomosci Lekarskie. 55 (1–2): 42–50. PMID 12043315.
- ^ Schreiber S, Pick CG (August 2006). “From selective to highly selective SSRIs: a comparison of the antinociceptive properties of fluoxetine, fluvoxamine, citalopram and escitalopram”. European Neuropsychopharmacology. 16 (6): 464–8. doi:10.1016/j.euroneuro.2005.11.013. PMID 16413173. S2CID 39278756.
- ^ Coquoz D, Porchet HC, Dayer P (September 1993). “Central analgesic effects of desipramine, fluvoxamine, and moclobemide after single oral dosing: a study in healthy volunteers”. Clinical Pharmacology and Therapeutics. 54 (3): 339–44. doi:10.1038/clpt.1993.156. PMID 8375130. S2CID 8229797.
- ^ Williams T, Hattingh CJ, Kariuki CM, Tromp SA, van Balkom AJ, Ipser JC, Stein DJ (October 2017). “Pharmacotherapy for social anxiety disorder (SAnD)”. The Cochrane Database of Systematic Reviews. 10 (10): CD001206. doi:10.1002/14651858.CD001206.pub3. PMC 6360927. PMID 29048739.
- ^ Cheer SM, Figgitt DP (2002). “Spotlight on fluvoxamine in anxiety disorders in children and adolescents”. CNS Drugs. 16 (2): 139–44. doi:10.2165/00023210-200216020-00006. PMID 11825104. S2CID 26774895.
- ^ Silver H (2001). “Fluvoxamine as an adjunctive agent in schizophrenia”. CNS Drug Reviews. 7 (3): 283–304. doi:10.1111/j.1527-3458.2001.tb00200.x. PMC 6741705. PMID 11607044.
- ^ Polcwiartek C, Nielsen J (March 2016). “The clinical potentials of adjunctive fluvoxamine to clozapine treatment: a systematic review”. Psychopharmacology. 233 (5): 741–50. doi:10.1007/s00213-015-4161-1. PMID 26626327. S2CID 12168939.
- ^ Lenze EJ, Mattar C, Zorumski CF, Stevens A, Schweiger J, Nicol GE, Miller JP, Yang L, Yingling M, Avidan MS, Reiersen AM (December 2020). “Fluvoxamine vs Placebo and Clinical Deterioration in Outpatients With Symptomatic COVID-19: A Randomized Clinical Trial”. JAMA. 324 (22): 2292–2300. doi:10.1001/jama.2020.22760. PMID 33180097.
- ^ Seymour CW, Bauchner H, Golub RM (December 2020). “COVID-19 Infection-Preventing Clinical Deterioration”. JAMA. 324 (22): 2300. doi:10.1001/jama.2020.21720. PMID 33180115.
- ^ [+https://scitechdaily.com/antidepressant-fluvoxamine-may-prevent-covid-19-infections-from-worsening/ “Antidepressant Fluvoxamine May Prevent COVID-19 Infections From Worsening”] Check
|url=value (help). - ^ https://academic.oup.com/ofid/advance-article/doi/10.1093/ofid/ofab050/6124100
- ^ Brayfield A, ed. (13 August 2013). Fluoxetine Hydrochloride. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 November 2013.
- ^ Taylor D, Paton C, Shitij K (2012). The Maudsley prescribing guidelines in psychiatry. West Sussex: Wiley-Blackwell. ISBN 978-0-470-97948-8.
- ^ “Faverin 100 mg film-coated tablets – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Abbott Healthcare Products Limited. 14 May 2013. Retrieved 21 October 2013.
- ^ “Top Ten Legal Drugs Linked to Violence”. Time. 7 January 2011. Retrieved 10 September 2014.
- ^ Jump up to:a b Ciraulo DA, Shader RI (2011). Ciraulo DA, Shader RI (eds.). Pharmacotherapy of Depression (2nd ed.). Springer. p. 49. doi:10.1007/978-1-60327-435-7. ISBN 978-1-60327-435-7.
- ^ Jump up to:a b Brunton L, Chabner B, Knollman B (2010). Goodman and Gilman’s The Pharmacological Basis of Therapeutics (12th ed.). New York: McGraw-Hill Professional. ISBN 978-0-07-162442-8.
- ^ Baumann P (December 1996). “Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors”. Clinical Pharmacokinetics. 31 (6): 444–69. doi:10.2165/00003088-199631060-00004. PMID 8968657. S2CID 31923953.
- ^ DeVane CL, Gill HS (1997). “Clinical pharmacokinetics of fluvoxamine: applications to dosage regimen design”. The Journal of Clinical Psychiatry. 58 Suppl 5 (Suppl 5): 7–14. PMID 9184622.
- ^ DeVane CL (1998). “Translational pharmacokinetics: current issues with newer antidepressants”. Depression and Anxiety. 8 Suppl 1 (Suppl 1): 64–70. doi:10.1002/(SICI)1520-6394(1998)8:1+<64::AID-DA10>3.0.CO;2-S. PMID 9809216.
- ^ Bondy B, Spellmann I (March 2007). “Pharmacogenetics of antipsychotics: useful for the clinician?”. Current Opinion in Psychiatry. 20 (2): 126–30. doi:10.1097/YCO.0b013e328017f69f. PMID 17278909. S2CID 23859992.
- ^ Kroon LA (September 2007). “Drug interactions with smoking”. American Journal of Health-System Pharmacy. 64 (18): 1917–21. doi:10.2146/ajhp060414. PMID 17823102.
- ^ Waknine Y (13 April 2007). “Prescribers Warned of Tizanidine Drug Interactions”. Medscape News. Medscape. Retrieved 1 February 2008.
- ^ “Fluvoxamine (Oral Route) Precautions”. Mayo Clinic. Retrieved 2 November2018.
- ^ Hemeryck A, Belpaire FM (February 2002). “Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update”. Current Drug Metabolism. 3 (1): 13–37. doi:10.2174/1389200023338017. PMID 11876575.
- ^ “Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers”.
- ^ Raouf M (2016). Fudin J (ed.). “Benzodiazepine Metabolism and Pharmacokinetics” (PDF).
- ^ Peppers MP (1996). “Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease”. Pharmacotherapy. 16 (1): 49–57. doi:10.1002/j.1875-9114.1996.tb02915.x. PMID 8700792. S2CID 1389910.
- ^ “fluvoxamine maleate: PRODUCT MONOGRAPH” (PDF). 2016.
- ^ “Luvox Data Sheet” (PDF). Medsafe, New Zealand. 2017.
- ^ Suzuki Y, Shioiri T, Muratake T, Kawashima Y, Sato S, Hagiwara M, Inoue Y, Shimoda K, Someya T (April 2003). “Effects of concomitant fluvoxamine on the metabolism of alprazolam in Japanese psychiatric patients: interaction with CYP2C19 mutated alleles”. European Journal of Clinical Pharmacology. 58 (12): 829–33. doi:10.1007/s00228-003-0563-9. PMID 12698310. S2CID 32559753.
- ^ Gerlach M, Warnke A, Greenhill L (2014). Psychiatric Drugs in Children and Adolescents: Basic Pharmacology and Practical Applications. Springer-Verlag Wien. p. 131. ISBN 978-3-7091-1500-8.
- ^ Fleishaker JC, Hulst LK (1994). “A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine”. European Journal of Clinical Pharmacology. 46 (1): 35–9. doi:10.1007/bf00195913. PMID 8005185. S2CID 2161450.
- ^ Obach RS, Ryder TF (August 2010). “Metabolism of ramelteon in human liver microsomes and correlation with the effect of fluvoxamine on ramelteon pharmacokinetics”. Drug Metabolism and Disposition. 38 (8): 1381–91. doi:10.1124/dmd.110.034009. PMID 20478852. S2CID 8421997.
- ^ Pandi-Perumal SR, Spence DW, Verster JC, Srinivasan V, Brown GM, Cardinali DP, Hardeland R (12 April 2011). “Pharmacotherapy of insomnia with ramelteon: safety, efficacy and clinical applications”. Journal of Central Nervous System Disease. 3: 51–65. doi:10.4137/JCNSD.S1611. PMC 3663615. PMID 23861638.
- ^ Jump up to:a b Anttila AK, Rasanen L, Leinonen EV (October 2001). “Fluvoxamine augmentation increases serum mirtazapine concentrations three- to fourfold”. The Annals of Pharmacotherapy. 35 (10): 1221–3. doi:10.1345/aph.1A014. PMID 11675851. S2CID 44807359.
- ^ Granfors MT, Backman JT, Neuvonen M, Ahonen J, Neuvonen PJ (April 2004). “Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction”. Clinical Pharmacology and Therapeutics. 75(4): 331–41. doi:10.1016/j.clpt.2003.12.005. PMID 15060511. S2CID 25781307.
- ^ Ishikawa M, Ishiwata K, Ishii K, Kimura Y, Sakata M, Naganawa M, et al. (October 2007). “High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503”. Biological Psychiatry. 62 (8): 878–83. doi:10.1016/j.biopsych.2007.04.001. PMID 17662961. S2CID 728565.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Arlington, VA: American Psychiatric Pub. p. 354. ISBN 978-1-585-62386-0. OCLC 320111564.
- ^ Yahata M, Chiba K, Watanabe T, Sugiyama Y (September 2017). “Possibility of Predicting Serotonin Transporter Occupancy From the In Vitro Inhibition Constant for Serotonin Transporter, the Clinically Relevant Plasma Concentration of Unbound Drugs, and Their Profiles for Substrates of Transporters”. Journal of Pharmaceutical Sciences. 106 (9): 2345–2356. doi:10.1016/j.xphs.2017.05.007. PMID 28501470.
- ^ Jump up to:a b Hashimoto K (September 2009). “Sigma-1 receptors and selective serotonin reuptake inhibitors: clinical implications of their relationship”. Central Nervous System Agents in Medicinal Chemistry. 9 (3): 197–204. doi:10.2174/1871524910909030197. PMID 20021354.
- ^ Westenberg HG, Sandner C (April 2006). “Tolerability and safety of fluvoxamine and other antidepressants”. International Journal of Clinical Practice. 60 (4): 482–91. doi:10.1111/j.1368-5031.2006.00865.x. PMC 1448696. PMID 16620364.
- ^ Hindmarch I, Hashimoto K (April 2010). “Cognition and depression: the effects of fluvoxamine, a sigma-1 receptor agonist, reconsidered”. Human Psychopharmacology. 25 (3): 193–200. doi:10.1002/hup.1106. PMID 20373470. S2CID 26491662.
- ^ Hrdina PD (July 1991). “Pharmacology of serotonin uptake inhibitors: focus on fluvoxamine”. Journal of Psychiatry & Neuroscience. 16 (2 Suppl 1): 10–8. PMC 1188307. PMID 1931931.
- ^ Jump up to:a b Sittig’s Pharmaceutical Manufacturing Encyclopedia (PDF) (3rd ed.). William Andrew. 2008. p. 1699. ISBN 978-0-8155-1526-5. Retrieved 17 October2013.
- ^ Leslie LK, Newman TB, Chesney PJ, Perrin JM (July 2005). “The Food and Drug Administration’s deliberations on antidepressant use in pediatric patients”. Pediatrics. 116 (1): 195–204. doi:10.1542/peds.2005-0074. PMC 1550709. PMID 15995053.
- ^ “Brand Index―Fluvoxamine India”. Archived from the original on 19 October 2013. Retrieved 18 October 2013.
- ^ Omori IM, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire H, Churchill R, Furukawa TA (March 2010). “Fluvoxamine versus other anti-depressive agents for depression”. The Cochrane Database of Systematic Reviews (3): CD006114. doi:10.1002/14651858.CD006114.pub2. PMC 4171125. PMID 20238342.
- ^ “OCD Medication”. Archived from the original on 14 October 2013. Retrieved 17 October 2013.
- ^ “Fluvoxamine Product Monograph” (PDF). 1999.
- ^ “Luvox Approved For Obsessive Compulsive Disorder in Children and Teens”. Archived from the original on 16 January 2009. Retrieved 8 February 2014.
- ^ Higuchi T, Briley M (February 2007). “Japanese experience with milnacipran, the first serotonin and norepinephrine reuptake inhibitor in Japan”. Neuropsychiatric Disease and Treatment. 3 (1): 41–58. doi:10.2147/nedt.2007.3.1.41. PMC 2654524. PMID 19300537.
- ^ “Human Metabolome Database: Showing metabocard for Fluvoxamine (HMDB0014322)”. http://www.hmdb.ca. Retrieved 15 September 2018.
- ^ “Solvay’s Fluvoxamine maleate is first drug approved for the treatment of social anxiety disorder in Japan”.
- ^ Walker R, Whittlesea C, eds. (2007) [1994]. Clinical Pharmacy and Therapeutics (4th ed.). Edinburgh: Churchill Livingstone Elsevier. ISBN 978-0-7020-4293-5.
- ^ “Fluvoxamine”. http://www.drugbank.ca. Retrieved 22 October 2019.
External links
- “Fluvoxamine”. Drug Information Portal. U.S. National Library of Medicine.
/////////DU23000, Fevarin, Fluvoxamine maleate, Luvox, Luvox CR, SME 3110, UNII-5LGN83G74V, Fluvoxamine, sme 3110, DU 23000
#DU23000, #Fevarin, #Fluvoxamine maleate, #Luvox, #Luvox CR, #SME 3110, #UNII-5LGN83G74V, #Fluvoxamine, #sme 3110, #DU 23000
Evinacumab
(Heavy chain)
EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA ISGDGGSTYY
ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL RNTIFGVVIP DAFDIWGQGT
MVTVSSASTK GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF
LGGPSVFLFP PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE
QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS
QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK
SRWQEGNVFS CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK ASSLESGVPS
RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H140-L214, H153-H209, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’140-L’214, H’153-H’209, H’267-H’327, H’373-H’431, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Evinacumab
エビナクマブ (遺伝子組換え)
Immunoglobulin G4, anti-(human protein ANGPTL3 (angiopoietin-like 3)) (human monoclonal REGN1500 heavy chain), disulfide with human monoclonal REGN1500 light chain, dimer
| Formula | C6480H9992N1716O2042S46 |
|---|---|
| CAS | 1446419-85-7 |
| Mol weight | 146081.9345 |
Protein Sequence
Sequence Length: 1334, 453, 453, 214, 214multichain; modified (modifications unspecified)
FDA APPROVED, 2021/2/11, EVKEEZA
Antihyperlipidemic, Anti-angiopietin like 3
Monoclonal antibody
Treatment of dyslipidemia
- REGN 1500
- REGN-1500
- REGN1500
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1EVQLVESGGG VIQPGGSLRL SCAASGFTFD DYAMNWVRQG PGKGLEWVSA51ISGDGGSTYY ADSVKGRFTI SRDNSKNSLY LQMNSLRAED TAFFYCAKDL101RNTIFGVVIP DAFDIWGQGT MVTVSSASTK GPSVFPLAPC SRSTSESTAA151LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS LSSVVTVPSS201SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP251PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE301QFNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR351EPQVYTLPPS QEEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT401PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS CSVMHEALHN HYTQKSLSLS451LGK
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence:
1DIQMTQSPST LSASVGDRVT ITCRASQSIR SWLAWYQQKP GKAPKLLIYK51ASSLESGVPS RFSGSGSGTE FTLTISSLQP DDFATYYCQQ YNSYSYTFGQ101GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV151DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG201LSSPVTKSFN RGEC
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| bridge | Cys-22 – Cys-96 | disulfide bridge |
| bridge | Cys-140 – Cys-214” | disulfide bridge |
| bridge | Cys-153 – Cys-209 | disulfide bridge |
| bridge | Cys-232 – Cys-232′ | disulfide bridge |
| bridge | Cys-235 – Cys-235′ | disulfide bridge |
| bridge | Cys-267 – Cys-327 | disulfide bridge |
| bridge | Cys-373 – Cys-431 | disulfide bridge |
| bridge | Cys-22′ – Cys-96′ | disulfide bridge |
| bridge | Cys-140′ – Cys-214”’ | disulfide bridge |
| bridge | Cys-153′ – Cys-209′ | disulfide bridge |
| bridge | Cys-267′ – Cys-327′ | disulfide bridge |
| bridge | Cys-373′ – Cys-431′ | disulfide bridge |
| bridge | Cys-23” – Cys-88” | disulfide bridge |
| bridge | Cys-134” – Cys-194” | disulfide bridge |
| bridge | Cys-23”’ – Cys-88”’ | disulfide bridge |
| bridge | Cys-134”’ – Cys-194”’ | disulfide bridge |
PATENTS
WO 2017024062
US 20170305999
Evinacumab, sold under the brand name Evkeeza, is a monoclonal antibody medication for the treatment of homozygous familial hypercholesterolemia (HoFH).[1][2]
Evinacumab is a recombinant human IgG4 monoclonal antibody targeted against angiopoietin-like protein 3 (ANGPTL3) and the first drug of its kind. The ANGPTL family of proteins serve a number of physiologic functions – including involvement in the regulation of lipid metabolism – which have made them desirable therapeutic targets in recent years.2 Loss-of-function mutations in ANGPTL3 have been noted to result in hypolipidemia and subsequent reductions in cardiovascular risk, whereas increases in function appear to be associated with cardiovascular risk, and it was these observations that provided a rationale for the development of a therapy targeted against ANGPTL3.3
In February 2021, evinacumab became the first-and-only inhibitor of ANGPTL3 to receive FDA approval after it was granted approval for the adjunctive treatment of homozygous familial hypercholesterolemia (HoFH) under the brand name “Evkeeza”.8 Evinacumab is novel in its mechanism of action compared with other lipid-lowering therapies and therefore provides a unique and synergistic therapeutic option in the treatment of HoFH.
Common side effects include nasopharyngitis (cold), influenza-like illness, dizziness, rhinorrhea (runny nose), and nausea. Serious hypersensitivity (allergic) reactions have occurred in the Evkeeza clinical trials.[2]
Evinacumab binds to the angiopoietin-like protein 3 (ANGPTL3).[2] ANGPTL3 slows the function of certain enzymes that break down fats in the body.[2] Evinacumab blocks ANGPTL3, allowing faster break down of fats that lead to high cholesterol.[2] Evinacumab was approved for medical use in the United States in February 2021.[2][3]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable | | |
| Evkeeza | Injection, solution, concentrate | 150 mg/1mL | Intravenous | Regeneron Pharmaceuticals, Inc. | 2021-02-11 | Not applicable | |
History
The effectiveness and safety of evinacumab were evaluated in a double-blind, randomized, placebo-controlled, 24-week trial enrolling 65 participants with homozygous familial hypercholesterolemia (HoFH).[2] In the trial, 43 participants received 15 mg/kg of evinacumab every four weeks and 22 participants received the placebo.[2] Participants were taking other lipid-lowering therapies as well.[2]
The primary measure of effectiveness was the percent change in low-density lipoprotein (LDL-C) from the beginning of treatment to week 24.[2] At week 24, participants receiving evinacumab had an average 47% decrease in LDL-C while participants on the placebo had an average 2% increase.[2]
The U.S. Food and Drug Administration (FDA) granted the application for evinacumab orphan drug, breakthrough therapy, and priority review designations.[2] The FDA granted approval of Evkeeza to Regeneron Pharmaceuticals, Inc.[2]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761181s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves add-on therapy for patients with genetic form of severely”. U.S. Food and Drug Administration (FDA). 11 February 2021. Retrieved 12 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA Approves First-in-class Evkeeza (evinacumab-dgnb) for Patients with Ultra-rare Inherited Form of High Cholesterol” (Press release). Regeneron Pharmaceuticals. 11 February 2021. Retrieved 12 February 2021 – via PR Newswire.
Further reading
- Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. (July 2017). “Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease”. N Engl J Med. 377 (3): 211–221. doi:10.1056/NEJMoa1612790. PMC 5800308. PMID 28538136.
External links
- “Evinacumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Angiopoietin-like 3 (ANGPTL3) |
| Clinical data | |
| Trade names | Evkeeza |
| Other names | REGN1500, evinacumab-dgnb |
| License data | US DailyMed: Evinacumab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 1446419-85-7 |
| DrugBank | DB15354 |
| ChemSpider | none |
| UNII | T8B2ORP1DW |
| KEGG | D11753 |
| Chemical and physical data | |
| Formula | C6480H9992N1716O2042S46 |
| Molar mass | 146083.95 g·mol−1 |
//////////////
#Evinacumab, #Peptide, #APPROVALS 2021, #FDA 2021, #Monoclonal antibody, #dyslipidemia, #エビナクマブ (遺伝子組換え) , #REGN 1500, #REGN-1500, #REGN1500, #Anthony melvin crasto, #world drug tracker. # new drug approvals, #pharma

{kind=link}
{kind=link}
_I_(cropped).jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
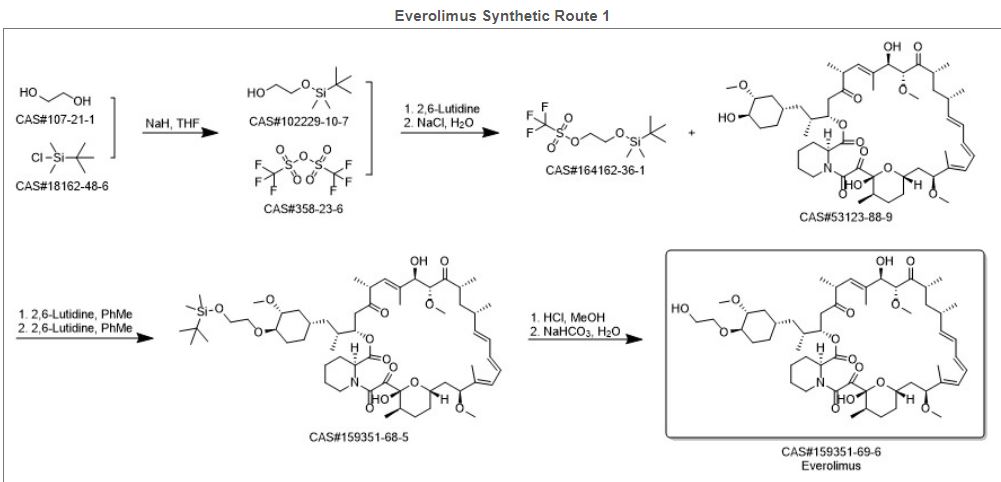{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}