
Home » 2019 (Page 7)
Yearly Archives: 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New patent, Opicapone, WO 2019123066, Unichem
New patent, Opicapone, WO 2019123066, Unichem
WO-2019123066
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019123066&tab=PCTDESCRIPTION
Process for the preparation of opicapone and its intermediates, useful for treating parkinson’s disease. Bial-Portela has developed and launched opicapone, for treating Parkinson’s disease.
Opicapone is a selective and reversible catechol-O-methyltransferase (COMT) inhibitor, use as adjunctive therapy for parkinson’s disease. Opicapone was approved by European Medicine Agency (EMA) on June 24, 2016 and it is developed and marketed as ONGENTYS® by Bial-Portela in Europe. Opicapone is chemically described as 2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-l-oxide and depicted below as compound of formula (I).
Opicapone and a process for preparation of it is disclosed in US 8,168,793. The process disclosescondensation of 3, 4-dibenzyloxy-5-nitrobenzoic acid with (Z)-2, 5-dichloro-N’-hydroxy-4, 6-dimethylnicotinimidamide in presence of N, N’-Carbonyl diimidazole in N, N’-dimethylformamide. The crude condensation intermediate was subjected to tetrabutylammonium fluoride (TBAF) mediated cyclization in tetrahydrofuran to give l,2,4-oxadiazole derivative, purifying it by precipitating in 1:1 mixture of dichloromethane: diethyl ether and recrystallized it in isopropyl alcohol. Oxidation of l,2,4-oxadiazole compound is carried out using 10 fold excess of urea hydrogen peroxide complex and trifluoroacetic anhydride in dichloromethane and was purified by column chromatography. Obtained N-oxide compound was converted into opicaponecompound of formula (I) by deprotection O-benzoyl groups by exposure it to boron tribromide (BBr3) in dichloromethane at -78°C to room temperature. Final product was purified in mixture of toluene and ethanol. Above synthetic stepsare outline in scheme 1.
c eme
This process has several drawbacks like cyclization reaction involve use of TBAF and THF. Use of expensive TBAF, leads to high cost in the production and therefore uneconomical for industrial production, whereas use of THF during this reaction has limitation due to peroxide contents. . Similarly diethyl ether is a potential fire hazard and can form peroxides rapidly and thus should be avoided in commercial scale production. Above cyclization is also carried out in presence of DMF and CDI at l20°C.
Similar approach was reported in W02008094053 which describes preparation of opicapone by one pot cyclization of 3,4-dibenzyloxy-5-nitrobenzoic acid with (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide using N,N’-Carbonyl diimidazole in N,N’-dimethylformamide followed by heating the reaction mixture at l35°C for 5 hours to obtain l,2,4-oxadiazole derivative. This oxadiazole derivative was purified by recrystallization from isopropyl alcohol. Further oxidation using urea hydrogen peroxide complex followed by o-debenzylation using boron tribromide (BBr3) was achieved to obtain Opicapone.
This process also suffers from drawback like use of elevated temperature (l35°C) and use of expensive BBr3.
US 9,126,988 also disclose process for the preparation of opicapone, whichinvolves several chemical steps: 1) nitrating vanillic acid in presence of nitric acid in acetic acidfollowed by recrystallization with acetic acid to get nitro compound with yield 40-46%; 2)which converted into acid chloride compoundby treating it with thionyl chloride in presence of catalytic amount of N, N-dimethylformamide in dichloromethane or l,4-dioxane; 3) condensing acid chloride compound with (Z)-2, 5-dichloro-N’-hydroxy-4, 6-dimethylnicotinimidamide in presence of excess amount of pyridine in N,N-dimethyl acetamide/ tetrahydrofuran/ dichloromethane or l,4-dioxane at 5-10 °C and then heating the reaction mixture at H0-l l5°C for 5-6 hours to get 1,2,4-oxadiazole compound; 4) which was oxidized using urea hydrogen peroxide complex and trifluoroacetic anhydride in dichloromethane to get N-oxide product which was purified by repeated recrystallization (2 or more times) using mixture of formic acid and toluene to get pure product with 59% yield; 5) O-methyl group was deprotected using aluminium chloride and pyridine in N-Methyl pyrrolidone at 60 C to obtain opicapone. After completion of reaction, the crude product was isolated by quenching the reaction mixture in mixture cone. HC1: water followed by filtration, washing with water: isopropyl alcohol and recrystallization from ethanol. Final purification was done in mixture of formic acid and isopropyl alcohol. Above synthetic steps are outline in scheme 2.
Scheme 2
As described above, cited literature processes suffers from some drawbacks like elevated reaction temperature and longer duration, use of excess amount of pyridine for cyclization reaction which is difficult to handle on large scale preparation. Another drawback of reported procedure is unsafe workup procedures for isolation of N-oxide as residual peroxides were not quenched by any peroxide quenching reagent. Also repeated crystallizations (more than two) are required for purification of N-oxide derivative to remove unreacted starting material which is tedious and time consuming process. Also for its purification mixture of solvent i.e. formic acid and toluene are used which hamper its recovery and is not cost effective process.
US 9,126,988 also disclosed process for the preparation of 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide compound of formula (IV), in which 2,5-dichloro-4,6-dimethylnicotinonitrile compound of formula (VIII) was reacted with hydroxyl amine solution in the presence of catalytic amount of 1,10-phenanthroline in methanokwater at 70-80°C for 6 hrs. After completion reaction mixture was cooled, filtered and dried to get 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (88%).
Bioorganic & Medicinal Chemistry 13 (2005) 5740-5749, Karl Bailey et.al. disclosed process for preparation of 3,4-dimethoxy-5-nitro benzoic acid compound of formula (Ilia). In which a solution of Cr03, concentrated H2S04 and water was added to solution of 3,4-dimethoxy-5-nitro benzaldehyde in acetone and water. The obtained solution was stirred for 24 hrs and then isopropanol was added to eliminate any unreacted Cr(VI) species to obtained crude green sludge, which was extracted into ethyl acetate and washed with 1M HC1 to remove remaining Cr(III) species. Obtained product is then recrystallized from water and ethanol to yield 69 % of 3,4-dimethoxy-5-nitro benzoic acid compound of formula (Ilia).
US 5,358, 948 also disclosed process for preparation of 3,4-dimethoxy-5-nitro benzoic acid compound of formula (Ilia). In which a solution of potassium permanganate was added to a solution of 3,4-dimethoxy-5-nitro benzaldehyde in acetone. The mixture was then stirred at 20°C for 18 hrs togives 3,4-dimethoxy-5-nitro benzoic acid compound of formula (Ilia) with 72% yield.
Disadvantage of the above cited literature (Karl Bailey et.al and US’ 948) processes are harsh, acidic condition and involve expensive reagents. The process is both uneconomical and time consuming, (18-24 hrs) hence not suitable for commercial production.
Oxidation of aldehydes to the corresponding carboxylic acids, on the other hand, are commonly carried out using KMn04 in acidic or basic media, or K2Cr207 in acidic medium or chromic acid. These heavy metal-based reagents are hazardous and the protocols produce metal wastes that require special handling owing to their toxicities.
It is therefore, desirable to provide efficient, robust, alternative simple process, cost effective process which is used on a large scale and allows product to be easily workup, purified and isolate without the disadvantages mentioned above.
Example 1: Preparation of 3, 4-dimethoxy-5-nitro benzoic acid (Ilia).
To a cooled solution of 3,4-dimethoxy-5-nitro benzaldehyde (lOOg, 0.474 mole) in DMF (500 ml) was added Oxone (294.1 g, 0.478 mole) lot wise at 5-10 °C. Reaction mixture was stirred for 30 minutes at same temperature, allowed to warm to room temperature and stirred for 2-3 hours. After completion, the reaction mixture was diluted with 1500 ml of water and filtered. The solid was washed with water until all peroxides removed and drying at 50°C under vacuum afforded 3,4-dimethoxy-5-nitro benzoic acid of formula (Ilia) (l02g, 95%).
Example 2: Preparation of 2 ,5-dichloro-N’ {[(3,4-dimethoxy-5-nitrophenyl) carbonyl]oxy}-4, 6-dimethylpyridine-3-carboximidamide (Va)
To a solution of 3,4-dimethoxy-5-nitro benzoic acid of formula (Ilia) (5 g, 0.022 mole)in 60 ml of acetonitrile was added N,N’-Carbonyldiimidazole (4.28g, 0.026 mole) in portions and the reaction mixture was stirred at room temperature for 1.5 hours. Then was added 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (5.4g, 0.023 mole) and stirring was continued for 3 hours. After completion, the reaction mixture was diluted with 240 ml of water and 300 ml of dichloromethane. Organic layer was separated and washed with water (200 ml x 3), concentrated under reduced pressure to obtain 2,5-dichloro-N'{ [(3,4-dimethoxy-5-nitrophenyl)carbonyl]oxy}-4,6-dimethylpyridine-3-carboximidamide of formula (Va) (8.67g, 88.9%).
Example 3: Preparation of 2, 5-dichloro-3-[5-(3, 4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine (Via)
To a solution of 2,5-dichloro-N'{ [(3,4-dimethoxy-5-nitrophenyl)carbonyl]oxy}- 4,6-dimethylpyridine-3-carboximidamide of formula (Va) (0.5g, 0.0011 mole)in 10 ml of dichloromethane was added isopropyl alcohol (1 ml) followed by KOH (0.075g, 0.001 lmole) dissolved in 0.1 ml of water. After stirring for 1 hour at room temperature the reaction mixture was diluted with 30 ml of dichloromethane and washed with water (lOml x 2). The reaction mixture was concentrated under reduced pressure to obtain 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine of formula (Via) (0.4 g, 83%).
Example 4: Preparation of 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine (Via) (One pot cyclization procedure)
To a stirred solution of 3,4-dimethoxy-5-nitro benzoic acid formula (Ilia) (lOOg, 0.44 mol)in 1000 ml of dichloromethane was added N,N’-Carbonyldiimidazole (86g, 0.53 mole) in portions and the reaction mixture was stirred at room temperature for 1.5 hours. Then was added 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (l08g, 0.46 mole) and stirring was continued for 3 hours. Isopropyl alcohol (200 ml) and KOH (30g, 0.53 mole) dissolved in 30 ml of water was then added to the reaction mixture. After stirring for 1 hour at room temperature the organic layer was washed with water (1000 ml x 2). Solvent was distilled out at atmospheric pressure, added 1000 ml of isopropyl alcohol and suspension was stirred at 55-60°C for 2 hours. The reaction mixture was allowed to cool to room temperature, stirred for 2 hours and filtered. The solid was washed with isopropyl alcohol (100 ml x 2) and dried at 50-60°C under vacuum to obtain 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine of formula (Via) (l60g, 85%).
Example 5: Preparation of 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine (Via) (cyclization procedure using thionyl chloride)
To a stirred solution of 3,4-dimethoxy-5-nitro benzoic acid of formula (Ilia) (lOOg, 0.44 mol) in 500 ml of dichloromethane was added 0.4 ml of N,N-dimethyl formamide followed bythionyl chloride (82g, 0.69 mole) drop wise at room temperature and the reaction mixture was heated at 40°C for 4 hours. After completion, dichloromethane and excess of thionyl chloride was distilled out under reduced pressure at 40°C. The obtained residue was dissolved in 500 ml of dichloromethane and was added to pre-cooled mixture of 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (l03g, 0.44 mole) and triethyl amine (73 ml, 0.53 mole) in 500 ml of dichloromethane at 5°C. After addition, the reaction mixture was allowed to warm to 25-30°C and stirred for 2 hours. Then was added isopropyl alcohol (200 ml) followed by KOH (62g, 1.1 mole) dissolved in 62 ml of water and stirring was continued for 2 hours at room temperature. The reaction mixture was washed with 1000 ml of water, 1N aqueous HC1 solution (500ml x 2) followed by 500 ml of 5% aqueous sodium bicarbonate solution. Solvent was distilled out at atmospheric pressure at 40°C. To the residue was added 1200 ml of methanol and the suspension was stirred at 55-60°C for 2 hours. The reaction mixture was allowed to cool to room temperature, maintained for 2 hours and filtered. The solid product was washed with methanol (100 ml x 2) and dried at 50°C under vacuum to obtain 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)- 1, 2, 4-oxadiazol-3-yl]-4, 6-dimethyl pyridine of formula (Via) (l65g, 88%).
Example 6: Preparation of 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine-l-oxide (Vila)
To a cooled solution of 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine of formula (Via) (25g, 0.0588 mole) in 300 ml of dichloromethane was added urea hydrogen peroxide complex (l8.26g, 0.194 mole) in portions followed by trifluoroacetic anhydride (37g, 0.176 mole) maintaining temperature below l0°C. After stirring at 5-l0°C for 1 hour, the reaction mixture was allowed to warm to room temperature and stirred for 5 hours. The reaction mixture was washed with water (300 ml x 2), 300ml of 5% aqueous sodium sulphite solution to quench residual peroxides and finally with 300 ml of water. Dichloromethane layer was distilled out at atmospheric pressure. The obtained solid was suspended in 250 ml of ethyl acetate and 12.5 ml of cone. HC1 was added at room temperature. The resulting suspension was then stirred at 65-70°C for 1 hour and allowed to cool to room temperature. After stirring for 2 hours, the reaction mixture was filtered, solid was washed with ethyl acetate (50 ml x 2) followed by water (50 x 3) and dried at 50°C under vacuum to obtain (5-(3,4-bis(methoxy)-5-nitrophenyl)-l,2,4-oxadiazol-3-yl)-2,5-dichloro-4,6-dimethylpyridine 1 -oxide of formula (Vila) (18g, 69%).
Example 7: Preparation of 5-[3-(2,5-Dichloro-4,6-dimethyl-l-oxido-3-pyridinyl)-l,2,4-oxadiazol-5-yl]-3-nitro-l,2-benzenediol (Opicapone, I)
To a cooled solution of 2,5-dichloro-3-[5-(3,4-dimethoxy-5-nitrophenyl)-l,2,4-oxadiazol-3-yl]-4,6-dimethylpyridine-l-oxide of formula (Vila) (25g, 0.056 mole) in 200 ml of N,N-Dimethylformamide was added AlCl3 (l l.34g, 0.085 mol) at 5-l0°C in portions. The reaction mixture was then heated at 85 °C for 6 hours. After completion, the reaction mixture was cooled to room temperature and poured onto cold mixture of cone. HC1 (200 ml) and water (400 ml). The reaction mixture was filtered, solid washed with water (100 ml X 3) followed by methanol (50 ml x2) and dried at 50°C under vacuum to obtain 5-[3-(2,5-Dichloro-4, 6-dimethyl- l-oxido-3-pyridinyl)- 1,2, 4-oxadiazol-5-yl]-3-nitro- 1,2-benzenediol of formula (I) (22 g, 94%).
Example 8: Preparation of 2, 5-dichloro-N’-hydroxy-4, 6-dimethyl nicotinimidamide of formula (IV)
To a suspension of 2,5-dichloro-4,6-dimethylnicotinonitrile of formula (VIII) (lOOg, 0.497 mole) in l,4-dioxane (400 ml) and water (900 ml) was added 50% aqueous solution of hydroxyl amine (l30g) and N-methyl morpholine (50.2g, 0.497) at room temperature. The reaction mixture was then stirred at 70-80°C for 10 hours. After completion, water (1100 ml) was added to the reaction mixture at 70-80°C and allowed to cool to room temperature. After stirring for 2 hours the reaction mixture was filtered, solid was washed with water (200ml x 3) and dried at 50°C under vacuum to obtain 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (68 g, 58%).
Example 9:Preparation of 2,5-dichloro-N’-hydroxy-4, 6-dimethylnicotinimidamide of formula (IV)
To a suspension of 2,5-dichloro-4,6-dimethylnicotinonitrile of formula (VIII) (lOOg, 0.497 mole) in methanol (600 ml) and water (800 ml) was added 50% aqueous solution of hydroxyl amine (l30g) and 2-methylpyrazine (7.02g, 0.0746) at room temperature. The reaction mixture was then stirred at 70-80°C for 6-8 hours. After completion, water (800 ml) was added to the reaction mixture at 70-80°C and allowed to cool to room temperature. After stirring for 2 hours the reaction mixture was filtered, solid was washed with water (200ml x 3) and dried at 50°C under vacuum to obtain 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (82 g, 70%).
Example 10: Preparation of 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV)
To a solution of hydroxylamine hydrochloride (86.4g, 1.243 mole) in 400 ml of water was added LiOH.H20 (52.7g, 1.25 mole) at room temperature and heated at 50°C for 30 minutes. To the reaction mixture was added 300 ml of methanol, 2-methylpyrazine (3.5 lg, 0.037 mole) and 2,5-dichloro-4,6-dimethylnicotinonitrile of formula (VIII) (50g, 0.248 mole) at 50°C. The reaction mixture was then stirred at 70-80°C for 6 hours. After completion, water (500 ml) was added to the reaction mixture at 70-80°C and allowed to cool to room temperature. After stirring for 2 hours the reaction mixture was filtered, solid was washed with water (lOOml x 3) and dried at 50°C under vacuum to obtain 2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide of formula (IV) (37.6 g, 64%).
Example 11: Purification of 5-[3-(2,5-Dichloro-4,6-dimethyl-l-oxido-3-pyridinyl)-l,2,4-oxadiazol-5-yl]-3-nitro-l,2-benzenediol (Opicapone, I)
The crude 5-[3-(2,5-Dichloro-4, 6-dimethyl- l-oxido-3-pyridinyl)- 1,2, 4-oxadiazol-5-yl]-3-nitro-l,2-benzenediol of formula (I)(25.0g) was suspended in 250 ml of N,N-dimethylformamide and reaction mixture was heated at 60-65°C to obtain clear solution. Then was added 500 ml of methanol and reaction mixture was cooled to room temperature. After stirring for 2-3 hours, the reaction mixture was filtered, solid was washed with methanol and dried at 50°C under vacuum to obtain 5-[3-(2, 5-Dichloro-4, 6-dimethyl- l-oxido-3-pyridinyl)- 1,2, 4-oxadiazol-5-yl]-3-nitro-l,2-benzenediol of formula (I) (22.0 g, 88%).
/////////////New patent, Opicapone, WO 2019123066, Unichem, WO2019123066
Quinupramine, キヌプラミン
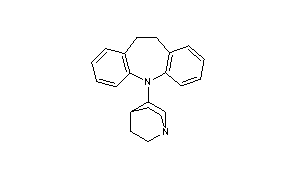
Quinupramine
キヌプラミン
- 5-(1-azabicyclo[2.2.2]oct-3-yl)-10,11-dihydro-5H-dibenz[b,f]azepine
- Formula:C21H24N2
- MW:304.44 g/mol
- CAS:31721-17-2
Quinupramine (brand names Kevopril, Kinupril, Adeprim, Quinuprine) is a tricyclic antidepressant (TCA) used in Europe for the treatment of depression.[1][2]
Pharmacologically, quinupramine acts in vitro as a strong muscarinic acetylcholine receptor antagonist (anticholinergic) and H1 receptorantagonist (antihistamine), moderate 5-HT2 receptor antagonist, and weak serotonin and norepinephrine reuptake inhibitor.[3] It has negligible affinity for the α1-adrenergic, α2-adrenergic, β-adrenergic, or D2 receptor.[3]
Clinically, quinupramine is reported to be stimulating similarly to imipramine, desipramine, and demexiptiline.[4] It can be inferred that its in vivo metabolites may have stronger effects on the reuptake of norepinephrine and/or serotonin than quinupramine itself
SYN
References
- ^ Swiss Pharmaceutical Society (2000). Index Nominum 2000: International Drug Directory (Book with CD-ROM). Boca Raton: Medpharm Scientific Publishers. p. 908. ISBN 3-88763-075-0.
- ^ José Miguel Vela; Helmut Buschmann; Jörg Holenz; Antonio Párraga; Antoni Torrens (2007). Antidepressants, Antipsychotics, Anxiolytics: From Chemistry and Pharmacology to Clinical Application. Weinheim: Wiley-VCH. p. 248. ISBN 978-3-527-31058-6.
- ^ Jump up to:a b Sakamoto H, Yokoyama N, Kohno S, Ohata K (December 1984). “Receptor binding profile of quinupramine, a new tricyclic antidepressant”. Japanese Journal of Pharmacology. 36 (4): 455–60. doi:10.1254/jjp.36.455. PMID 6098759.
- ^ Kent, Angela; M. Billiard (2003). Sleep: physiology, investigations, and medicine. New York: Kluwer Academic/Plenum. p. 233. ISBN 0-306-47406-9.
-
- DOS 2 030 492 (Sogeras; appl. 20.6.1970; GB-prior. 20.6.1969).
- GB 1 252 320 (Sogeras; valid from 29.5.1970; prior. 20.6.1969).
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 33 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.046.149 |
| Chemical and physical data | |
| Formula | C21H24N2 |
| Molar mass | 304.43 g/mol g·mol−1 |
//////////////Quinupramine, キヌプラミン
Quizartinib dihydrochloride, キザルチニブ塩酸塩
Quizartinib dihydrochloride
キザルチニブ塩酸塩
| Formula |
C29H32N6O4S. 2HCl
|
|---|---|
| CAS |
1132827-21-4
|
| Mol weight |
633.5891
|
JAPAN, PMDA APPROVED,. 2019/6/18, Vanflyta, To use as part of a treatment regimen for newly diagnosed acute myeloid leukemia that meets certain criteria
Drug Trials Snapshot
fda 2023, 7/20/2023
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor, originated Ambit Biosciences, and acquired by Daiichi Sankyo, that is currently under development for the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It reported good results in 2012 from a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
As of 2017 it has completed 5 clinical trials and another 7 are active.[3]
SYN

References
- ^ Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry. 52 (23): 7808–7816. doi:10.1021/jm9007533.
- ^ Drug Tames Refractory AML. ASH Dec 2012
- ^ Quizartinib studies
 |
|
| Names | |
|---|---|
| IUPAC name
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
|
|
| Other names
AC220
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C29H32N6O4S | |
| Molar mass | 560.67 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Quizartinib dihydrochloride, キザルチニブ塩酸塩, JAPAN 2019
OLD POST

N-(5-tert-butyl-isoxazol-3-yl)-N’-{ 4- [7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
| CAS | 950769-58-1 (free base) 1132827-21-4 (2HCl) |
| Formula | C29H32N6O4S |
| MW | 560.7 |
| Synonim | AC220, AC-010220 ASP-2689 |


Quizartinib
Ambit Biosciences
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor that is currently under development by Ambit Biosciencesfor the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]

AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
| Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML. | |
| Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ. |
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.

Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]

………………………..
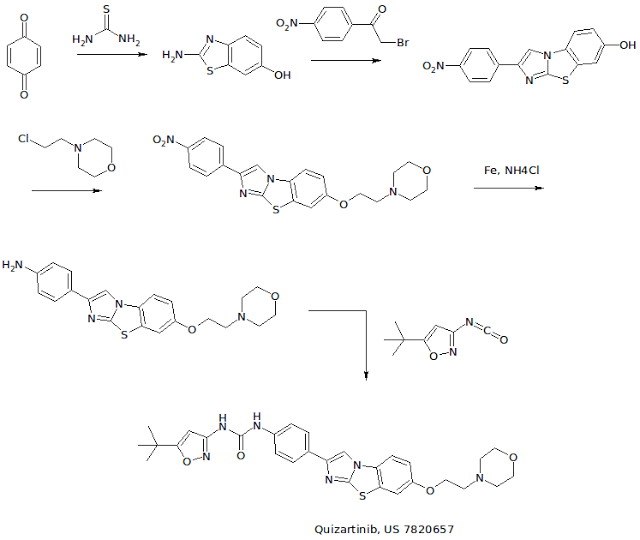
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo

N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;

(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);

(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);


(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am

(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;

(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-

(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :

A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches

[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method

2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
methanol/dichloromethane; Rf (product) = 0.35; Rf (starting material) = 0.40).
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol

1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches

* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method

2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening

TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole

1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs

4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole

[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs

4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.

Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
celite, washed celite filter cake celite, celite, celite,
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%

5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate

[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea

1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:

http://www.google.com/patents/WO2011056939A1?cl=en SE THIS FOR DELETED CLIPS


4. Example F-4
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride

1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
H. Analytical Data
1. N-(5-ieri-butyl-isoxazol-3-yl)-N’-{ 4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.


………………………
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
6][l,3]benzothiazole intermediate: 2-(4-nitrophenyl)imidazo[2,l-6][l,3]benzothiazol-7-ol,
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl2 1H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis

[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms

6.6.2.2 Cold Precipitation Experiments

NYl-4144519vl 102 6.6.2.3 Slurry Experiments
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl

NYl-4144519v l 104

NYM 144519vl 105

N Y l -4 1 4 4 5 1 9 v l 1 0 6

NYI-4I44519vi 107

N V I 4 1 4 4 5 1 9 1 0 8

“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl

NYI-4144519v l 109

Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor
J Med Chem 2009, 52(23): 7808
http://pubs.acs.org/doi/full/10.1021/jm9007533

General Procedure E for Preparation of Hydrochloride Salt
References
- Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry 52 (23): 7808–7816.
- Drug Tames Refractory AML. ASH Dec 2012
- NMR……….http://file.selleckchem.com/downloads/nmr/S152601-AC-220-HNMR-Selleck.pdf
- HPLC………http://file.selleckchem.com/downloads/hplc/S152601-AC-220-HPLC-Selleck.pdf


HS 10340

HS-10340
CAS 2156639-66-4

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma


PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
FDA approves first treatment Soliris (eculizumab) for neuromyelitis optica spectrum disorder, a rare autoimmune disease of the central nervous system
The U.S. Food and Drug Administration today approved Soliris (eculizumab) injection for intravenous use for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive. NMOSD is an autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord.
“Soliris provides the first FDA-approved treatment for neuromyelitis optica spectrum disorder, a debilitating disease that profoundly impacts patients’ lives,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval changes the landscape of therapy for patients with NMOSD. Having an approved therapy for this condition is the culmination of extensive work we have engaged in with drug companies to …
- June 27, 2019
The U.S. Food and Drug Administration today approved Soliris (eculizumab) injection for intravenous use for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive. NMOSD is an autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord.
“Soliris provides the first FDA-approved treatment for neuromyelitis optica spectrum disorder, a debilitating disease that profoundly impacts patients’ lives,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval changes the landscape of therapy for patients with NMOSD. Having an approved therapy for this condition is the culmination of extensive work we have engaged in with drug companies to expedite the development and approval of safe and effective treatments for patients with NMOSD, and we remain committed to these efforts for other rare diseases.”
In patients with NMOSD, the body’s immune system mistakenly attacks healthy cells and proteins in the body, most often in the optic nerves and spinal cord. Individuals with NMOSD typically have attacks of optic neuritis, which causes eye pain and vision loss. Individuals also can have attacks resulting in transverse myelitis, which often causes numbness, weakness, or paralysis of the arms and legs, along with loss of bladder and bowel control. Most attacks occur in clusters, days to months to years apart, followed by partial recovery during periods of remission. Approximately 50% of patients with NMOSD have permanent visual impairment and paralysis caused by NMOSD attacks. According to the National Institutes of Health, women are more often affected by NMOSD than men and African Americans are at greater risk of the disease than Caucasians. Estimates vary, but NMOSD is thought to impact approximately 4,000 to 8,000 patients in the United States.
NMOSD can be associated with antibodies that bind to a protein called aquaporin-4 (AQP4). Binding of the anti-AQP4 antibody appears to activate other components of the immune system, causing inflammation and damage to the central nervous system.
The effectiveness of Soliris for the treatment of NMOSD was demonstrated in a clinical study of 143 patients with NMOSD who had antibodies against AQP4 (anti-AQP4 positive) who were randomized to receive either Soliris treatment or placebo. Compared to treatment with placebo, the study showed that treatment with Soliris reduced the number of NMOSD relapses by 94 percent over the 48-week course of the trial. Soliris also reduced the need for hospitalizations and the need for treatment of acute attacks with corticosteroids and plasma exchange.
Soliris has a boxed warning to alert health care professionals and patients that life-threatening and fatal meningococcal infections have occurred in patients treated with Soliris, and that such infections may become rapidly life-threatening or fatal if not recognized and treated early. Patients should be monitored for early signs of meningococcal infections and evaluated immediately if infection is suspected. Use should be discontinued in patients who are being treated for serious meningococcal infections. Health care professionals should use caution when administering Soliris to patients with any other infection. In the NMOSD clinical trial, no cases of meningococcal infection were observed.
Soliris is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). Prescribers must enroll in the REMS program. Prescribers must counsel patients about the risk of meningococcal infection, provide the patients with the REMS educational materials and ensure patients are vaccinated with meningococcal vaccine(s). The drug must be dispensed with the FDA-approved patient Medication Guide that provides important information about the drug’s uses and risks.
The most frequently reported adverse reactions reported by patients in the NMOSD clinical trial were: upper respiratory infection, common cold (nasopharyngitis), diarrhea, back pain, dizziness, influenza, joint pain (arthralgia), sore throat (pharyngitis) and contusion.
The FDA granted the approval of Soliris to Alexion Pharmaceuticals.
Soliris was first approved by the FDA in 2007. The drug is approved to reduce destruction of red blood cells in adults with a rare blood disease called paroxysmal nocturnal hemoglobinuria, for the treatment of adults and children with a rare disease that causes abnormal blood clots to form in small blood vessels in the kidneys (atypical hemolytic uremic syndrome to inhibit complement-mediated thrombotic microangiopathy), and for the treatment of adults with Myasthenia Gravis who are anti-acetylcholine receptor antibody positive.
The FDA granted this application Priority Review. The use for NMOSD received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
///////////////fda 2019, Soliris, eculizumab, neuromyelitis optica spectrum disorder, Orphan Drug, Priority Review
CS 3001
CS-3001
BB 7, VX 033
- Molecular Weight, 478.37
C17 H18 Br F2 N3 O2 S2
CStone Pharmaceuticals Co Ltd, JUNE 2018 IND FILED CHINA
URAT1 inhibitor – useful for treating hyperuricemia and gout.
The compound was originally claimed in WO2017202291 , covering thiophene derivative URAT1 inhibitors, useful for treating hyperuricemia and gouty arthritis, assigned to Medshine Discovery Inc , but naming the inventors.and has been reported in some instances to be a URAT1 modulator. In June 2018, an IND application was filed in



WO-2019101058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019101058&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of URAT1 inhibitor (designated as Forms A and B) are claimed. The compounds are disclosed to be useful for treating hyperuricemia and gouty arthritis.
Novel crystalline forms of a URAT1 inhibitor, designated as Forms A and B, and their preparation.
For example, remove 2.0 mL of phosphoric acid into 2000 mL of water, sonicate for 10 minutes, mix, and let cool to room temperature as mobile phase A.
////////////CS-3001, BB 7, VX 033, CHINA, PRECLINICAL, CStone Pharmaceuticals, URAT1 inhibitor, hyperuricemia, gout
O=C(O)C(C)(C)Sc4nnc(Br)n4c2sc(c1CC(F)(F)CCc12)C3CC3
TL 487
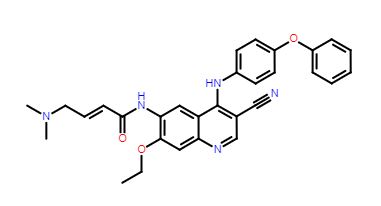
TL-487
- Molecular Weight, 507.58, MF C30 H29 N5 O3
Teligene Inc(2E)-N-[3-Cyano-7-ethoxy-4-[(4-phenoxyphenyl)amino]-6-quinolinyl]-4-(dimethylamino)-2-butenamide
(E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
PATENT
US 20150057312
PATENT
WO2013152135

PATENT
WO-2019096327
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019096327&redirectedID=true
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
///////////////TL-487, PRECLINICAL, CHINA, breast cancer, ovary cancer, olon cancer, BTK, HER-2 kinase inhibitor,
CN(C)C\C=C\C(=O)Nc3cc4c(Nc2ccc(Oc1ccccc1)cc2)c(cnc4cc3OCC)C#N
TENAPANOR


Tenapanor
Molecular FormulaC50H66Cl4N8O10S2
Average mass1145.049 Da
1234423-95-0 [RN]
1234423-95-0 (free base) 1234365-97-9 (2HCl)
9652
3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)-N-(26-((3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)phenyl)sulfonamido)-10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosyl)benzenesulfonamide
Benzenesulfonamide, N,N’-(10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosane-1,26-diyl)bis[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]-
12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-
1-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethyl]-3-[4-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethylcarbamoylamino]butyl]urea
17-[[[3-[(4S)-6,8-Dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-12,15-dioxa-2,7,9-triazaheptadecanamide
Tenapanor, also known as AZD-1722 and RDX 5791, is an inhibitor of the sodium-proton (Na(+)/H(+)) exchanger NHE3, which plays a prominent role in sodium handling in the gastrointestinal tract and kidney. Tenapanor possesses an excellent preclinical safety profile and, as of now, there are no serious concerns about its side effects.
Tenapanor is a drug developed by Ardelyx, which acts as an inhibitor of the sodium-proton exchanger NHE3. This antiporterprotein is found in the kidney and intestines, and normally acts to regulate the levels of sodium absorbed and secreted by the body. When administered orally, tenapanor selectively inhibits sodium uptake in the intestines, limiting the amount absorbed from food, and thereby reduces levels of sodium in the body.[1] This may make it useful in the treatment of chronic kidney disease and hypertension, both of which are exacerbated by excess sodium in the diet.[2]
Ardelyx and licensees Kyowa Hakko Kirin and Fosun Pharma are developing tenapanor, an NHE3 (Na+/H+ exchange-3) inhibitor that increases fluid content in the GI tract and which also reduces GI tract pain via an unknown TRPV-1-dependent pathway, for treating constipation-predominant irritable bowel syndrome (IBS-C) and hyperphosphatemia in patients with end stage renal disease (ESRD).
Syn

PATENT
WO2010078449
PATENT
WO-2019091503
A novel crystalline form of tenapanor free base, process for its preparation, composition comprising it and its use for the preparation of tenapanor with chemical purity >98.8% is claimed. Also claimed are salt forms of tenapanor, preferably tenapanor phosphate and their use for treating irritable bowel syndrome, constipation, hyperphosphatemia, final stage renal failure, chronic kidney disease and preventing excess sodium in patients with kidney and heart conditions. Further claimed are processes for the preparation of tenapanor comprising the steps of reaction of a diamine compound with 1,4-diisocyanatobutane, followed by deprotection and condensation to obtain tenapanor. Novel intermediates of tenapanor and their use for the preparation of tenapanor are claimed. Tenapanor is known to be a sodium hydrogen exchanger 3 inhibitor and analgesic.
enapanor, having the chemical name 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazaheptadecaneamide, is a selective inhibitor of the sodium protonic NHE3 antiporter. Orally administered tenapanor selectively inhibits the absorption of sodium in the intestine. This leads to an increase of water content in the digestive tract, improved bowel flow and normalization of the frequency of bowel movement and stool consistency. At the same time it exhibits antinociceptive activity and ability to lower serum phosphate levels. Because of these properties, it is clinically tested for the treatment of irritable bowel syndrome, especially when accompanied by constipation, treatment of hyperphosphatemia, especially in patients with dialysis with final stage renal failure, treatment of chronic kidney disease, and prevention of excess sodium in patients with kidney and heart conditions. The tenapanor molecule, which was first described in the international patent application WO 2010/078449, has the following structural formula:
In this document, tenapanor was prepared as bishydrochloride salt. The bishydrochloride salt was prepared only in the form of an amorphous foam, which, after solidification, required grinding for further processing. However, the thus obtained particles are of varying sizes, while a narrow particle size distribution is required for pharmaceutical use in order to ensure uniform behavior. The amorphous foam obtained in the said document is essentially a thickened reaction mixture or a slightly purified reaction mixture containing, in addition to tenapanor, various impurities. The possibilities to purify the reaction mixtures are limited. Moreover, amorphous foams tend to adsorb solvents, and it is usually difficult to remove (or dry out) the residual solvents from the amorphous foam. This is undesirable for pharmaceutical use. A typical feature of amorphous foams is a large specific surface, resulting in a greater interaction of the substance with the surrounding environment. This significantly increases the risk of decomposition of the substance, for example through air oxygen, moisture or light. The present invention aims at overcoming these problems.
It would be advantageous to provide tenapanor solid forms (tenapanor free base or tenapanor salts) which are precipitated in solid forms, thus allowing to filter off the liquid reaction mixture containing the impurities. This results in a significantly improved purity.
The process used in WO 2010/078449 for the preparation of bishydrochloride salt of tenapanor was based on the preparation of 3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzene-l-sulfonyl chloride of formula III from 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l, 2,3,4-te
Scheme 1
The said document also discloses resolution of the starting tetrahydroisoquinoline of formula II by L-or D-dibenzoylt
(II) (S-II) (R-II)
Scheme 2
WO 2010/078449 discloses further steps of preparation of tenapanor, as shown in Scheme 3.
(V) (I)
Scheme 3
Individual synthetic steps described in Scheme 3 result in low yields: 42% for the reaction of the chloride of formula III with 2-(2-(2-aminoethoxy)ethoxy)ethylamine of formula IV, and 59% for the subsequent reaction with 1,4-diisocyanatobutane of formula V. The products of both synthetic steps are isolated by preparative chromatography which is technologically an unsuitable isolation and purification technique. The low yields and the need to use preparative chromatography for the isolation are caused by an abundance of side products and impurities and by the inability of the intermediates as well as of the product to provide a crystalline form.
Thus present invention thus further aims at providing a method of preparation of tenapanor which would be economically effective, in particular in relation to the expensive starting compound 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline, and which would also enable industrial scale production, in particular by removing steps which cannot be scaled up effectively or which cannot be scaled up at all. Furthermore, the method of preparation of tenapanor should provide tenapanor in a form which is useful for use in pharmaceutical forms and does not have the disadvantages of an amorphous foam.
Tenapanor free base in the form of an amorphous solid foam was prepared by the procedure disclosed in patent application WO 2010/078449, Example 202. The chemical purity of the tenapanor prepared by this procedure was 96.5% (HPLC). The structure of tenapanor was verified by MS and H and 13C NMR spectra.
Step A
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline
Potassium carbonate (9.30 g) and anhydrous xylene (500 ml) were added to the reaction vessel. Benzyl mercaptane (25 g) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
(S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline 50 g in anhydrous xylene (500 ml), Pd2(dba)3 (3 g) and Xantphos (3 g). The resulting solution was stirred at 25 °C for 30 minutes and then added to a solution of benzyl mercaptane. The resulting reaction mixture was maintained at 140 °C for 16 h. The mixture was then concentrated and the residue was subjected to preparative chromatography on silica gel with the mobile phase ethyl acetate / petroleum ether (1: 100-1 :50). 20 g of product are obtained as a yellow oil (36% yield).
Ste B
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochloride
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (16 g) was dissolved in the reaction vessel in acetic acid/water (160 mL: 16 mL) mixture. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the well stirred mixture. After disappearance of the starting material, the reaction mixture was purged with nitrogen and concentrated in vacuo. A product (10 g, 66.6%) was obtained as a colorless substance.
Step C
Preparation of (S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l, 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
2-(2-(2-Aminoethoxy)ethoxy)ethylamine HC1 (30 g; 0.2 mol) and triethylamine (5.2 g; 52 mmol) were dissolved in dichloromethane (500 ml) and the mixture was chilled in an ice bath. (S)-3-(6,8-Dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (10 g; 26 mmol) was added in parts during 40 minutes to the chilled reaction mixture. The ice bath was removed and the reaction mixture was stirred at laboratory temperature for additional 30 minutes.
The dichloromethane solution was extracted three times by brine (2x 250 ml), dried over sodium sulphate, and concentrated in vacuo. The residue was purified using preparative chromatography on silica gel with dichloromethane-methanol mobile phase.
Yield 7.2 g. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Step D
Preparation of 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazah
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide (5g; 10 mmol) prepared in step A was dissolved in dichloromethane (50 ml). Triethylamine (1.5 g; 14.9 mmol) and 1 ,4-diisocyanatobutane (0.48 g; 3.4 mmol) were added to the solution. The reaction mixture was cooled using ice and stirred overnight. The resulting fine suspension was filtered off, the filtrate was concentrated and the obtained product was purified by preparative chromatography on on silica gel with dichloromethane-methanol mixture as a mobile phase
Yield: 2 g of tenapanor in the form of amorphous solid foam. HPLC purity 96.5 %.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. *H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H).
Ste E
Preparation of bishydrochloride salt of tenapanor
Tenapanor free base (1 g; 0.85 mmol) prepared in step B was dissolved in a mixture of methanol (10 ml) and 4M aqueous HCl (0.5 ml; 2 mmol) under mild reflux. The solution was concentrated on rotary vacuum evaporator, and the title product was obtained in the yield of 1 g of amorphous solid foam.
Example 1
Preparation of tenapanor, crystalline form I
Tenapanor free base (200 mg, 0.17 mmol), prepared as in step D of the comparative example, was dissolved in 0.4 ml acetonitrile under mild reflux. The clear solution was cooled at the rate of 1 °C/min with stirring to laboratory temperature (i.e., range from 22 °C to 26 °C) and then stirred for additional 2 hours at this temperature. The resulting crystals were isolated by filtration on sintered glass filter and dried for 6 hours in a vacuum oven at 40 °C. Crystallization yield was 170 mg of crystalline form I of tenapanor. HPLC showed a purity of 99.5%.
Examples 4 to 9 illustrate the inventive method of preparation of crystalline tenapanor.
Example 4
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinoline
DIPEA (9.6 mL) and anhydrous dioxane (100 mL) were added to a reaction vessel. Benzyl mercaptan (8.1 ml) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
In a second reaction vessel, (S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (21.2 g) in anhydrous dioxane (140 mL), Pd2(dba)3 (835 mg)and Xantphos (835 mg) were mixed. The resulting solution was stirred at 25 °C for 30 minutes and then added to the solution of benzyl mercaptan. The resulting reaction mixture was maintained at gentle reflux for 3 hours.
After cooling, the suspension obtained was filtered through a thin layer of celite. HC1 was added to the filtrate. The precipitated hydrochloride was isolated by filtration, washed well and dried. 21 g of pinkish product were obtained (81.6% yield).
Example 5
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochlorid
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline hydrochloride (11.1 g) was stirred in DCM/2M HC1 (70 mL:6 mL) mixture in a reaction vessel. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the vigorously stirred mixture. After disappearance of the starting material, the resulting suspension was bubbled through by nitrogen and the product was filtered off and washed with DCM. 9.2 g of white product was obtained (82.7% yield).
Example 6
In the reaction vessel, t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (21.8 g) was stirred in DCM. The mixture was cooled in an ice bath under an inert atmosphere. To the cooled solution was
added 1 ,4-diisocyanatobutane (6.14 g) and TEA (0.1 mL). The cooling bath was removed and the reaction mixture was further stirred for 2 h.
35% HCl was added to the reaction mixture and the mixture was stirred under gentle reflux overnight.
After cooling, the precipitated product was filtered off and washed with DCM.
The product was recrystallized from propan-2-ol. 22.3 g of white product was obtained (80% yield).
Example 7
Preparation of (5)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l , 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
(S)-3-(6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (11.7 g) prepared in Example 2 was stirred in dichloromethane (100 ml) and the suspension was cooled in an ice bath. To the cooled suspension was added a solution of t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (6.8 g) and DIPEA (14 ml) in DCM (50 ml). The resulting solution was stirred for 2 hours in an ice bath. The reaction mixture was extracted twice with water. Concentrated HCl (15 mL) was added to the dichloromethane solution and the mixture heated at gentle reflux for 2 h.
The precipitated product, after cooling, was extracted into water. The aqueous phase was separated and basified with Na2C03. The product as the free base was extracted into DCM and the dichloromethane solution was dried over sodium sulfate and concentrated in vacuo. 12.9 g of product were obtained.
Yield 93.4%. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Example 8
Preparation of 17-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-triazaheptadecanamide (tenapanor free base)
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4- tetrahydroisoquinolin- 4-yl)benzenesulfonamide (12.9 g) prepared in Example 4 was dissolved in dichloromethane (150 ml). To the solution was added triethylamine (0.3 ml) and 1,4-diisocyanatobutane (1.7 g). The reaction mixture was stirred at 25 °C for 2 h. The resulting reaction mixture was extracted with water and aqueous Na2C03. The dichloromethane solution of the product was dried over sodium sulfate and concentrated to a solid foam. Yield 13.9 g. The crude product was taken up in acetone (100 ml) and then recrystallized from methanol (80 ml). 7.3 g of white crystalline product was obtained. Yield 49.8%.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. !H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H)
Example 9
Preparation of 17-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-
(S)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (0.81 g) prepared in Example 2 and l,l’-(butane-l,4-diyl)bis(3-(2-(2-(2-aminoethoxy)ethoxy)ethyl)urea) dihydrochloride prepared according to Example 3 (0.48 g) were stirred in anhydrous ΝΜΡ (10 ml). To the suspension was added DIPEA (2 mL) and the resulting solution was stirred at 60 °C for 1.5 h. Water (10 mL) was added dropwise to the reaction mixture and the mixture was cooled to 5 °C. The precipitated product was isolated and stirred in acetone at 5 °C overnight. The beige product was filtered off (0.67 g) and recrystallized from methanol (12 ml).
0.53 g of a colorless crystalline product was obtained.
Yield 78.7 %. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S. DSC analysis showed the melting temperature of 130.5 °C.
Example 10
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 11
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and hydrobromic acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with hydrobromic acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 12
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 13
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and citric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with citric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Other pharmaceutically acceptable acids were tested by the procedures shown in Examples 10-13, but did not yield salts which would precipitate in amorphous stable solid form from the solution. The tested acids were: methanesulfonic acid, benzenesulfonic acid, oxalic acid, maleinic acid, tartaric acid, fumaric acid, trichloroacetic acid.
Example 14
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, a solution of phosphoric acid in THF (50 μ1/5 ml) is added dropwise during 10 minutes. The resulting suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with phosph (79 %) oric is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 430 mg of colourless salt of tenapanor with phosphoric acid. XRPD showed amorphousness of the product.
Example 15
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, hydrobromic acid (48%; 100 μΐ) is added dropwise during 10 minutes. A fine precipitate forms already during the dropwise addition of HBr, and the suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with HBr is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 397 mg (69 %) of colourless salt of tenapanor with HBr (1 :2). XRPD showed amorphousness of the product.
References
- ^ Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D (2014). “Intestinal inhibition of the na+/h+ exchanger 3 prevents cardiorenal damage in rats and inhibits na+ uptake in humans”. Sci Transl Med. 6 (227): 227ra36. doi:10.1126/scitranslmed.3007790. PMID 24622516.
- ^ Salt-buster drug cuts sodium absorbed from food. New Scientist, 14 March 2014
REFERENCES
1: Johansson SA, Knutsson M, Leonsson-Zachrisson M, Rosenbaum DP. Effect of Food Intake on the Pharmacodynamics of Tenapanor: A Phase 1 Study. Clin Pharmacol Drug Dev. 2017 Mar 24. doi: 10.1002/cpdd.341. [Epub ahead of print] PubMed PMID: 28339149.
2: Johansson S, Rosenbaum DP, Ahlqvist M, Rollison H, Knutsson M, Stefansson B, Elebring M. Effects of Tenapanor on Cytochrome P450-Mediated Drug-Drug Interactions. Clin Pharmacol Drug Dev. 2017 Mar 16. doi: 10.1002/cpdd.346. [Epub ahead of print] PubMed PMID: 28301096.
3: Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor Treatment of Patients With Constipation-Predominant Irritable Bowel Syndrome: A Phase 2, Randomized, Placebo-Controlled Efficacy and Safety Trial. Am J Gastroenterol. 2017 Feb 28. doi: 10.1038/ajg.2017.41. [Epub ahead of print] PubMed PMID: 28244495.
4: Carney EF. Dialysis: Efficacy of tenapanor in hyperphosphataemia. Nat Rev Nephrol. 2017 Apr;13(4):194. doi: 10.1038/nrneph.2017.27. PubMed PMID: 28239171.
5: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Åstrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J Am Soc Nephrol. 2017 Feb 3. pii: ASN.2016080855. doi: 10.1681/ASN.2016080855. [Epub ahead of print] PubMed PMID: 28159782.
6: Koliani-Pace J, Lacy BE. Update on the Management of Chronic Constipation. Curr Treat Options Gastroenterol. 2017 Mar;15(1):126-134. doi: 10.1007/s11938-017-0118-2. Review. PubMed PMID: 28116695.
7: Charoenphandhu N, Kraidith K, Lertsuwan K, Sripong C, Suntornsaratoon P, Svasti S, Krishnamra N, Wongdee K. Na(+)/H(+) exchanger 3 inhibitor diminishes hepcidin-enhanced duodenal calcium transport in hemizygous β-globin knockout thalassemic mice. Mol Cell Biochem. 2017 Mar;427(1-2):201-208. doi: 10.1007/s11010-016-2911-y. PubMed PMID: 27995414.
8: Thammayon N, Wongdee K, Lertsuwan K, Suntornsaratoon P, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Na(+)/H(+) exchanger 3 inhibitor diminishes the amino-acid-enhanced transepithelial calcium transport across the rat duodenum. Amino Acids. 2017 Apr;49(4):725-734. doi: 10.1007/s00726-016-2374-1. PubMed PMID: 27981415.
9: Afsar B, Vaziri ND, Aslan G, Tarim K, Kanbay M. Gut hormones and gut microbiota: implications for kidney function and hypertension. J Am Soc Hypertens. 2016 Dec;10(12):954-961. doi: 10.1016/j.jash.2016.10.007. Review. PubMed PMID: 27865823.
10: Johansson S, Leonsson-Zachrisson M, Knutsson M, Spencer AG, Labonté ED, Deshpande D, Kohler J, Kozuka K, Charmot D, Rosenbaum DP. Preclinical and Healthy Volunteer Studies of Potential Drug-Drug Interactions Between Tenapanor and Phosphate Binders. Clin Pharmacol Drug Dev. 2016 Sep 22. doi: 10.1002/cpdd.307. [Epub ahead of print] PubMed PMID: 27654985.
11: Ketteler M, Liangos O, Biggar PH. Treating hyperphosphatemia – current and advancing drugs. Expert Opin Pharmacother. 2016 Oct;17(14):1873-9. doi: 10.1080/14656566.2016.1220538. Review. PubMed PMID: 27643443.
12: Johansson S, Rosenbaum DP, Knutsson M, Leonsson-Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2016 Jul 1. [Epub ahead of print] PubMed PMID: 27368672.
13: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Stefansson BV, Rydén-Bergsten T, Greasley PJ, Johansson SA, Knutsson M, Carlsson BC. Effect of Tenapanor on Interdialytic Weight Gain in Patients on Hemodialysis. Clin J Am Soc Nephrol. 2016 Sep 7;11(9):1597-605. doi: 10.2215/CJN.09050815. PubMed PMID: 27340281; PubMed Central PMCID: PMC5012484.
14: Nusrat S, Miner PB Jr. New pharmacological treatment options for irritable bowel syndrome with constipation. Expert Opin Emerg Drugs. 2015;20(4):625-36. doi: 10.1517/14728214.2015.1105215. Review. PubMed PMID: 26548544.
15: Spencer AG, Greasley PJ. Pharmacologic inhibition of intestinal sodium uptake: a gut centric approach to sodium management. Curr Opin Nephrol Hypertens. 2015 Sep;24(5):410-6. doi: 10.1097/MNH.0000000000000154. Review. PubMed PMID: 26197202.
16: Zielińska M, Wasilewski A, Fichna J. Tenapanor hydrochloride for the treatment of constipation-predominant irritable bowel syndrome. Expert Opin Investig Drugs. 2015;24(8):1093-9. doi: 10.1517/13543784.2015.1054480. Review. PubMed PMID: 26065434.
17: Thomas RH, Luthin DR. Current and emerging treatments for irritable bowel syndrome with constipation and chronic idiopathic constipation: focus on prosecretory agents. Pharmacotherapy. 2015 Jun;35(6):613-30. doi: 10.1002/phar.1594. Review. PubMed PMID: 26016701.
18: Gerritsen KG, Boer WH, Joles JA. The importance of intake: a gut feeling. Ann Transl Med. 2015 Mar;3(4):49. doi: 10.3978/j.issn.2305-5839.2015.03.21. PubMed PMID: 25861604; PubMed Central PMCID: PMC4381464.
19: Labonté ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D. Gastrointestinal Inhibition of Sodium-Hydrogen Exchanger 3 Reduces Phosphorus Absorption and Protects against Vascular Calcification in CKD. J Am Soc Nephrol. 2015 May;26(5):1138-49. doi: 10.1681/ASN.2014030317. PubMed PMID: 25404658; PubMed Central PMCID: PMC4413764.
20: Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014 Mar 12;6(227):227ra36. doi: 10.1126/scitranslmed.3007790. PubMed PMID: 24622516.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.243.471 |
| Chemical and physical data | |
| Formula | C50H66Cl4N8O10S2 |
| Molar mass | 1145.046 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////Tenapanor, AZD 1722, RDX 5791, chronic kidney disease, hypertension
CN1CC(C2=CC(=CC(=C2C1)Cl)Cl)C3=CC(=CC=C3)S(=O)(=O)NCCOCCOCCNC(=O)NCCCCNC(=O)NCCOCCOCCNS(=O)(=O)C4=CC=CC(=C4)C5CN(CC6=C(C=C(C=C56)Cl)Cl)C
HM04 or H0900
![3-[(1R)-1-(2,3-Dichloro-4-pyrazin-2-ylphenyl)-2,2,2-trifluoroethyl]-1-methyl-1-(1-methylpiperidin-4-yl)urea.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91884613&t=l)
HM04 or H0900
Cas 1808913-24-7
(R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea
The compound was disclosed in WO2015134839 . Helsinn under license from Novo Nordisk , is investigating ghrelin antagonists for treating obesity, Prader-Willi syndrome and other metabolic disorders; in May 2015, the program was listed as being in preclinical development
Helps reducing ghrelin signaling activity and treating disorder associated with an increase in ghrelin level (eg food abuse, alcohol addiction, and Prader-Willi syndrome).
Ghrelin, a growth hormone-releasing peptide produced by ghrelinergic cells in the gastrointestinal tract, is understood to function as a neuropeptide that regulates energy metabolism by stimulating appetite. The modulation, for example inhibition, of ghrelin signaling, through the ghrelin/growth hormone secretagogue receptor (GHS-Rla), is an attractive target for pharmacological treatment of disorders associated with high ghrelin level. Potential disorders for treatment using ghrelin modulators include food abuse (such as binge eating, obesity, hyperphagia (or uncontrollable appetite), post-dieting body weight rebound (including post-dieting hyperphagia), alcohol addiction, and genetic diseases associated with increased ghrelin level (e.g., Prader-Willi syndrome (PWS)).
PATENT
US 20150252021

PATENT
WO2015134839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015134839
Example 1
nthesis of Intermediate lk
Intermediate k
Step 1:
To a solution of la (100 g, 0.62 mol) in DMF (1.2 L) was added N-bromosuccinimide (110 g, 0.62 mol) at 0 °C. The mixture was stirred at room temperature for 4 h, then water (800 mL) was added and the resulting mixture was extracted with EtOAc (3 x 500 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was triturated with petroleum ether to provide lb (133.7 g, 89% yield) as a brown solid. !H-NMR (CDC13, 300 MHz): δ= 7.30 (d, 1 H), 6.59 (d, 1 H), 4.22 (br, 2 H). LC-MS: 241 [M+l]+.
Step 2:
To a solution of lb (133.7 g, 0.55 mol) in dry CH2C12 (1.5 L) was added acetic anhydride (110 g, 0.62 mol) dropwise over a period of 20 minutes at room temperature. The mixture was stirred at room temperature overnight, then diluted with CH2C12 (300 mL) and washed with water (150 mL) and brine (200 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was triturated with petroleum ether (300 mL) to provide compound lc (143.0 g, 91% yield) as a white solid. ¾-NMR (CDC13, 400 MHz): δ= 8.26 (d, 1 H), 7.63 (br, 1 H), 7.54 (d, 1 H), 2.26 (s, 3 H). LC-MS: 280 [M-l]“.
Step 3:
A mixture of compound lc (50.0 g, 0.18 mol), butyl vinyl ether (Id, 89.0 g, 0.89 mol), bis(l,3-diphenylphosphino)propane (DPPP, 22.0 g, 0.053 mol), TEA (100 mL, 0.71 mol) and Pd(OAc)2 (6.4 g, 0.027 mol) in DMSO (1.2 L) was heated at 130 °C under N2 overnight. After the reaction was completed, the mixture was cooled to 0 °C and 2N HC1 (480 mL) was added dropwise over a period of 30 minutes. Then, the mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous a2S04 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide le (19.5 g, 45% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 8.46 (d, 1 H), 7.82 (br, 1 H), 7.51 (d, 1 H), 2.63 (s, 3 H), 2.29 (s, 3 H). LC-MS: 244 [M-l]“.
Step 4:
To a solution of le (21.9 g, 89.4 mmol) in MeOH (350 mL) was added 2N NaOH solution (350 mL) at room temperature. The mixture was heated at 50 °C overnight, then cooled and concentrated under reduced pressure. The resulting solid was triturated with water (100 mL) for 30 min and filtered to provide If (18.0 g, 98% yield) as a brown solid. ¾-NMR (CDC13, 400 MHz): 3= 7.48 (d, 1 H), 6.68 (d, 1 H), 4.56 (br, 2 H), 2.62 (s, 3 H). LC-MS: 202[M-1]\
Step 5:
To a mixture of compound If (18.0 g, 89.2 mmol) and ice (360 g) in cone. HC1 (180 mL) was added a solution of NaN02 (9.2 g, 133.7 mmol) in water (20 mL) dropwise over a period of 30 minutes, and the resulting mixture stirred in an ice bath for 30 min. A solution of KI (74.0 g, 446 mmol) in water (360 mL) was added dropwise over 45 min at 0 °C. The mixture was stirred for 30 min and then extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 :40) to provide lg (23.9 g, 86% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 7.6 (d, 1 H), 7.06 (d, 1 H), 2.62 (s, 3 H).
Step 6:
To a solution of lg (23.9 g, 76.1 mmol) in MeOH (100 mL)/THF (100 mL) was slowly added NaB¾ (2.9 g, 76.1 mmol) at 0 °C. The mixture was stirred at room temperature for 5 min, and then quenched with water (100 mL). The mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide lh (22.4 g, 93% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.26 (d, 1 H), 5.23 (q, 1 H), 2.17 (br, 1 H), 1.47 (d, 3 H).
Step 7:
To a mixture of lh (22.4 g, 70.9 mmol), phthalimide (12.5 g, 85.0 mmol) and PPh3 (22.3 g, 85.0 mmol) in dry THF (450 mL) was added DIAD (21.5 g, 106.3 mmol) at room temperature under N2 protection. The mixture was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 15) to provide li (18.5 g, 58% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.78-7.84 (m, 3 H), 7.70-7.73 (m, 2 H), 7.41-7.43 (d, 1 H), 5.76-5.81 (q, 1 H), 1.84 (d, 3 H).
Step 8:
A solution of li (7.2 g, 16.2 mmol) and hydrazine hydrate (98%, 4.0 g, 80.9 mmol) in MeOH (150 mL) was heated under reflux for 2 h, then cooled and concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with CH2C12 (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure to give lj (3.8 g, 75% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.25 (d, 1 H), 4.55 (q, 1 H), 1.36-1.38 (d, 3 H). LC-MS: 316 [M+l]+.
Step 9:
To a solution of lj (41. Og, 0.13 mol) in methyl tert-butyl ether (750 mL) was added slowly a solution of D-mandelic acid (7.8 g, 0.052 mol) in methyl tert-butyl ether (1 10 mL) at 45°C. The mixture was stirred at this temperature for 30 min then cooled and filtered. White solid obtained was partitioned between 5% NaOH solution (300 mL) and methyl tert-butyl ether (300 mL). The bi -phases were separated and the aqueous phase was extracted with methyl tert-butyl ether (300 mL). The combined organic layer was concentrated to provide Intermediate lk (12 g, 58.5% yield) as a white solid (ee%=98.0%, Chiralpak AD-H, 5 μπι, 4.6*250mm, mobile phase: Hex: EtOH : DEA=80 : 20 : 0.2), retention time = 6.408 min).
Example 2
Synthesis of Compoun
A suspension of N-methyl-4-piperidone 2a (13.3 g, 58.6 mmol), NH2Me (30% in MeOH, 100 mL) and Pd/C (0.66 g) in MeOH (200 mL) was heated at 60 °C under H2 atmosphere (50 psi) overnight, then cooled and filtered. The filtrate was concentrated under reduced pressure and the residue was dissolved in HC1 in dioxane (3N, 100 mL) and stirred for 30 min. The precipitate was filtered and washed with EtOAc (50 mL) to provide 2b (7.7g, 54% yield) as white powder. 1H-NMR (DMSO, 400 MHz): δ= 9.50 (br, 2 H), 3.48 (d, 2 H), 3.15-3.16 (m, 1 H), 2.96-3.01 (m, 2 H), 2.70 (s, 3 H), 2.51 (s, 3 H), 2.22-2.28 (m, 2 H), 1.94-2.02 (m, 2 H), LC-MS: 129 [M+l]+ .
Example 20
Synthesis of H0900
Step 1:
To a mixture of 16d (32 g, 120 mmol) in dry CH2CI2 (800 mL) was added Dess-Martin peroxide reagent (76 g, 180 mmol) portion- wise at 0 °C. The mixture was stirred at room temperature for 1 h, then diluted with DCM (800 mL), washed with aqueous NaHC03 solution (300 mL) and brine (300 mL). The organic phase was separated, dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford crude 18a (31.4 g) which was used directly in the next step without further purification.
Step 2:
To a solution of 18a (12 g, 40 mmol) and 3b (22.2 g, 60 mmol) in DME (560 mL) were added Pd(PPh3)4 (9.25 g, 8 mmol) and Cul (1.52 g, 8 mmol) at room temperature. The mixture was stirred at 90 °C overnight, then concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE = 1 :5) to provide 18b (8.0 g, 79.3%) as a white solid. LC-MS: 253 [M+l]+.
Step 3:
To a solution of 18b (7 g, 27.7 mmol) and (¾)-tert-butylsulfinamide (7.27 g, 30.56 mmol) in dry THF (200 mL) was added Ti(i-OPr)4 (15.7 g, 55.4 mmol) dropwise at room temperature. The mixture was stirred at 80 °C overnight, and then cooled. Ethyl acetate (40 mL) was added, the resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA:PE =1 :5) to provide 18c (6.8 g, 69%) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 9.10 (s, 1H), 8.97 (s, 1H), 8.72 (s, 1H), 8.64 (d, 1H),8.12 (d, 1H), 7.59 (d, 1H), 1.30 (s, 9H).LC-MS: 356 [M+l]+.
Step 4:
To a stirred solution of 18c (6.8 g, 19 mmol) and Tetrabutylammonium difluorotnphenylsilicate (15.8 g, 29 mmol) in dry THF (250 mL) was added a solution of TMSCF3 (11 g, 77 mmol) in anhydrous THF (50 mL) at -65 °C. The mixture was then stirred at -65 °C for 2 h, and at that point aqueous NH4CI solution (250 mL) was added. The mixture was diluted with ethyl acetate (250 mL), washed with brine (250 mL), dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE=1 :2) to provide 18d (4.3 g, 52%) as a yellow solid. LC-MS: 426 [M+l]+.
Step 5:
To a stirred solution of 18d (4.3 g, 10.1 mmol) in MeOH (40 mL) was added a solution of HCl/MeOH (4N, 40 mL) at room temperature. The mixture was stirred for 1 h, then concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 mL) to afford crude 18e (4.3g) which was directly in the next step without further purification. LC-MS: 322 [M+l]+.
Step 6:
To a solution of 18e (2.7 g, 7.1 mmol), 2b (3.4 g, 21.3 mmol) and TEA (80 mL) in DCM (220 mL) was added thiphosgene (3.15 g, 10.6 mmol) in DCM (40 mL) dropwise at 0 °C. The solution was warmed to ambient temperature and stirred for 1 h, then diluted with DCM ( 100 mL) and washed with aqueous Na2C03 solution (100 mL) and brine (100 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated. The residue was purified with silica gel column chromatography (silica, DCM : CH3OH=10 : 1) to provide crude H0900 (2.13 g, ee%=92.5%) which was further purified through chiral separation to afford H0900 (1.6 g, 49% yield) as a white solid. (ee%=98.5%, Chiralpak IC 5um, 4.6*250mm, Phase: Hex: EtOH:
DEA=90: 10:0.2), retention tine =12.829 min. 1H-NMR (CDC13, 400 MHz): δ= 8.86 (d, 1H), 8.63 (dd, 1H), 8.55 (d, 1H), 7.47 (d, 1H), 7.40 (d, 1H), 6.28 (m, 1H), 5.18 (d, 1H), 4.12 (m, 1H), 2.88 (t, 2H), 2.77 (s, 3H), 2.22 (s, 3H), 2.05 (m, 2H), 2.48 (m, 2H), 1.52 (m, 2H), 1.73-1.49 (m, 4H). LC-MS: 476 [M+l]+.
PATENT
WO-2019118298
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118298&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline fumarate salt forms of (R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea (also referred to as HM04 or H0900; designated as Forms 1-4), process for their preparation and compositions comprising them are claimed.
PWS occurs in approximately 1 in 10,000 births and is associated with deletion or lack of expression of region 15ql 1.2 of the paternal chromosome 15.
Characteristics of PWS include short stature, low muscle tone, and hyperphagia. Growth hormone replacement is frequently used to treat growth deficiencies and hypotonia. However, treatment for the insatiable appetite is lacking and PWS children can mature into adults suffering from obesity and type 2 diabetes. Levels of ghrelin are generally elevated in PWS; however, the relationship with ghrelin signaling and food intake in PWS remains unclear. See Purtell L., et ah, In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-l levels. Neuropeptides. 201 l;45(4):30l-7; Cummings D.E., et ah, Elevated plasma ghrelin levels in Prader Willi syndrome. Nature Medicine . 2002;8(7):643-4; DelParigi A., et ah, High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. The Journal of Clinical Endocrinology and Metabolism. 2002;87(l2):546l-4.
[005] Accordingly, it is desirable to find treatments that effectively inhibit GHSRla, that are tolerable to the patient, and that do not interfere with other functions of the growth hormones. GHSRla modulators, including inhibitors such as (R)-3-(l-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2, 2, 2-trifluoroethyl)-l -methyl- l-(l-m ethylpiperidin-4-yl) urea (HM04, H0900) depicted below, are reported in LT.S. Patent No. 9,546,157.
Step 1 : Synthesis of compound 2A
[00106] 2,2,6,6-tetramethylpiperidine (7.20 kg, 51.1 mol, 3.0 eq.,
KF=0.30%) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer and mixed at RT under nitrogen protection. THF (50 L) was added into the reactor and stirred. The vessel was purged with nitrogen three times and cooled to 0 °C. n-BuLi (20.4 L, 3.0 eq.; 2.5 M hexane solution) was added to the mixture dropwise while keeping the temperature at about 0 °C to about 5 °C for over one hour. The color of the solution turned yellow. The mixture was stirred at about 0 °C to about 5 °C for 30 minutes. The mixture was cooled to about -78 °C to about -70 °C to form Solution A.
[00107] Compound 1 (3.25 kg, 17.0 mol. 1.0 eq., KF=0.03%) was dissolved in 15 L of THF to form Solution B.
[00108] Solution B was added to solution A dropwise at a temperature of about -70 °C to about -78 °C over one hour and then stirred for 30 minutes to form solution C. Tri-isopropyl borate ((i-PrO)3B) (3.52 kg, 18.7 mol., 1.1 eq.) was added dropwise into solution C over 10 minutes. The reaction mixture was stirred at a
temperature of about -70 °C to about -78 °C for one hour. HC1 (40 L, 3M, 7.0 eq.) was added over 30 minutes to quench the reaction. A 10 degree rise in temperature was noted.
[00109] The resulting aqueous layer was separated and extracted with EtOAc (40 L). The aqueous layer was separated and extracted twice again with EtOAc (35 L, 30 L). The organic layers were combined resulting in about 160 L of liquid. The combined organic layer was washed twice with 50 L of a 1M aqueous HC1 solution saturated with NaCl. The organic layer was concentrated to about 5 L in a 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 8 hours.
[00110] The residual EtOAc was swapped with DME for 3 times (10 L x 3). The organic layer was concentrated in the 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 6 hours. Each time about 5 L of residual remained. DME (20 L) was added to the residual to obtain a deep brown solution of 14.2% compound 2A (3.55 kg in 25 kg of solution; 88.8% yield; 97.4% purity (AETC by HPLC, retention time = 1.6 minutes); 0.24% residual ethyl acetate). 1H-NMR (400 MHz, DMSO): 5=8.55 (s, 2H), 7.36 (d, 1H), 7.69 (d, 1H). A second batch of compound 2A was prepared by the same method to produce 3.29 kg (95.4% purity, 82.3% yield, 0.11% residual ethyl acetate).
[00111] Step 2: Synthesis of Compound 3A
C! , N
M
K2CO3 (I .O equiv)
2A OH
DME/H20 3:1 (20 vol), 50 e C 3A N
[00112] Compound 2 A (2.91 kg in 20.5 kg solution) was added into a 100
L reactor at room temperature under nitrogen. DME (45 mL), 2-chloropyrazin (1.42 kg,
12.4 mol., 1.0 eq.), and Pd(dppf)Cl2 (10% w/w, 291 g) were added sequentially, and each
mixed at room temperature under nitrogen. Nitrogen was bubbled into the mixture for 20
minutes and the resulting mixture was purged and filled with nitrogen (3 times). The
mixture was heated to 48-52°C over 60 minutes. K2CO3 (2.57 kg, 18.6 mol, 1.5 eq.) was
added to 22 L of water in another reactor at room temperature and then added dropwise to
the compound 2 A mixture over 10 minutes. The mixture was stirred at 48-52°C for 16
hours and then cooled to room temperature. This procedure was repeated twice and all
three batches were combined.
[00113] An aqueous solution of K2CO3 (1.0 kg) was dissolved in 22 L of
water and added to the combined mixture to adjust the pH to 9. TBME (50 L) was added
into the mixture and filtered (PET filter, 3-5 pm, 205g/m2) to remove about 50 g of
sticky, brown solid material (catalyst analog). The aqueous layer was twice separated and
extracted with TBME (40 L, 40L).
[00114] The aqueous layer was combined with the aqueous layer of a
fourth batch prepared according to the above method. The pH of the combined aqueous layers was adjusted to pH<3 with HC1 (2N, 48 L). The solid precipitated out slowly as
the mixture was stirred at room temperature for 1 hour. The mixture was filtered (PET
filter, 3-5 pm, 205g/m2) over 30 minutes to obtain 20 kg of wet product. ACN (40 L) was
added into a 100 L reactor equipped with an overhead stirrer at room temperature. The 20
kg of wet product was added into the reactor and the reaction mixture heated to reflux
and stirred at reflux for 4 hours. The reaction mixture was cooled to room temperature
over 3 hours (around 15 °C/hour) and filtered to obtain 8.5 kg of wet solid. The wet solid
was dried under vacuum (20-30 mmHg) at 50-55 °C for 15 hours to obtain compound 3 A
as a pale white solid (6.1 kg; 97.4% purity (AUC by HPLC, retention time = 3.7
minutes); 83.8% yield). 1H-NMR (400 MHz, DMSO): 5=7.67 (d, 1H), 7.82 (d, 1H), 8.75
(d, 1H), 8.82 (t, 1H), 8.98 (d, 1H), 13.89 (bs, 1H).
[00115] Step 3: Synthesis of compound 6A
3A 6A N
N
[00116] Compound 3 A (6.1 kg, 22.7 mol, 1.0 eq.) was added into a 100 L
reactor equipped with a temperature probe, overhead stirrer, and condenser. Methanol
(92 L) was added into the reactor at room temperature. The mixture was cooled to
0-10 °C and added with SOCk (5.4 kg, 45.3 mol, 2.0 eq.) dropwise at 0-10 °C over 30
minutes. The reaction mixture was heated to reflux (65 °C) and stirred at reflux for 15
hours. A suspension was formed. Most of the solvent and SOCk was removed under
vacuum distillation until about 30 L remained. The mixture was concentrated under
vacuum (30-40 mmHg) at 50-55 °C for about 6 hours. Water (10 L) was added to the residual at -5 to 15 °C. The pH was adjusted to 8-9 with an aqueous solution of K2CO3 (200 g, dissolved in 2L of water) at -5 to 15 °C. The resulting aqueous layer was extracted twice with isopropyl acetate (25 L, 25 L). The combination of organic layers (about 50 kg) was washed with 20 L of NaHCCb aqueous layer. The organic layer was separated and washed with 10 L of of an aqueous solution of NaHCCb. All the aqueous layers were combined (55.8 kg). The organic layer was filtered through a silica pad (30 cm) and the pad washed with extra isopropyl acetate until the compound 6 A was filtered from the silica gel (about 3 hours). The organic layer was concentrated to about 5 L. THF (10 L) was added to the residual and concentrated to about 5 L (3 times) under vacuum (30-40 mmHg) at 50-55 °C for about 3 hours. Another 10 L of THF was added to the residual concentrate, giving a concentrated solution of compound 6A (15.8 kg; 32.83%,
5.19 kg compound 6A in solution; 97.9% purity (AUC by HPLC, retention time = 8.5 min); 80.8% yield). 1H-NMR (400 MHz, DMSO): 5=3.98 (s, 3H), 7.54 (d, 1H), 7.78 (d, 1H), 8.63 (d, 1H), 8.72 (t, 1H), 8.94 (d, 1H).
[00117] Step 4: Synthesis of compound 6B
[00118] THF (26 L) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer under nitrogen. DIBAL-H (26 kg, 46 mol, 5.0 eq.) was added and the system purged and filled with nitrogen three times. The mixture was cooled to -78 to -70 °C to form solution A. A room temperature solution of compound 6A (2.6 kg, 9.2 mol, 1.0 eq.) in 52 L of THF was added dropwise at -78 to -70 °C over 30 minutes under nitrogen. The mixture was warmed to -30 °C over about 5-6 hours. The reaction mixture was stirred at -40 to -30 °C for 30 minutes. The mixture was slowly added to 42 L of 2N HCL over 1 hour reaching a maximum temperature of 35 °C. The mixture was extracted with 26 L of isopropyl acetate. The organic layer was separated and washed with 30 L of brine. This procedure was repeated and both batches of organic layer were combined and concentrated from about 100 L to about 5-10 L under vacuum.
A solid slowly formed during concentration. The mixture was cooled to 5-15 °C and stirred for 1 hour. The mixture was filtered (30-50 pm) over 30 minutes. The solid was dried under vacuum at 50 °C for 6 hours to obtain compound 6B as a brown solid (2.1 kg; 97.5% purity (AUC by HPLC, retention time = 8.6 min); 45.7% yield). 1H-NMR (400 MHz, DMSO): d = 4.65 (d, 2H), 5.68 (t, 1H), 7.62 (d, 1H), 7.68 (d, 1H), 8.72 (d, 1H),
8.80 (t, 1H), 8.94 (d, 1H).
[00119] Step 5: Synthesis of compound 7
[00120] DMSO (10 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 6B (2.05 kg, 8.04 mol, 1.0 eq.) was added under nitrogen at room temperature. Et3N (8 L) was added under nitrogen at RT and the mixture was then cooled to 15-20 °C.
SCb. pyridine (5.1 kg, 32.08 mol, 4.0 eq.) was dissolved into 10 L of DMSO at 5-15 °C in a separate flask and added to the mixture dropwise over 3.5 hours at about 20 °C. The reaction mixture was transferred to 70 L of ice-water. The suspension mixture was stirred at 0-10 °C for 1 hour and filtered (PET, 3-5 pm, 205 g/m2) by centrifuge over 1.5 hours to obtain compound 7 as a brown solid. The solid was dissolved in 35 L of DCM at room temperature. The resulting DCM layer was washed with 5 L of brine. The organic layer was separated and concentrated under vacuum at 40-45 °C to dryness to obtain compound 7 as a brown solid (2.33 kg; 96.3% purity (AEiC by HPLC, retention time = 9.2 minutes); 93.5% yield). 1H-NMR (400 MHz, DMSO): d = 7.67 (d, 1H), 7.99 (d, 1H), 8.67 (d, 1H), 8.75 (s, 1H), 8.99 (d, 1H), 10.56 (s, 1H).
[00121] Step 6: Synthesis of compound 8
[00122] THF (23 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 7 (2.3 kg, 9.1 mol, 1.0 eq.) and (S)-2-methylpropane-2-sulfmamide (1.21 kg, 10 mol, 1.1 eq.) were added sequentially to the flask under nitrogen. Ti(OEt)4 (6.22 kg, 27.3 mol, 3.0 eq.) was added dropwise to the flask over 1 hour at 30-35 °C under nitrogen. The system was purged with nitrogen three times and then the mixture was stirred at room temperature for 2 hours. Isopropyl acetate (40 L) was added to the reaction mixture. The entire reaction mixture was then charged to 20 L of brine while stirring slowly at RT. A lot of solid was formed and no heat release was observed. The solid (about 18 kg) was filtered using centrifuge, and then the solid was slurried with 20 L of isopropyl acetate again for 20 minutes, and filtered again, resulting is slightly less solid (17.3 kg). The filtrates were then combined and washed with 20 L of brine. The organic layer was separated and concentrated in a rotavapor under vacuum (30-40 mmHg) at 40-50 °C for about 4 hours to remove the solvents and obtain a brown oil (compound 8). The oil was dissolved in DMF to obtain a black solution (7.36 kg; 40.1%; 3.0 kg compound 8 in solution; 92.1% purity (AUC by HPLC, retention time = 9.7 minutes); >100% yield). 1H-NMR (400 MHz, CDCb): d = 1.30 (s, 9H), 7.59 (d, 1H), 8.11 (d, 1H), 8.64 (s, 1H), 8.73 (m, 1H), 8.97 (s, 1H), 9.10 (s, 1H).
[00123] Step 7: Synthesis of compound 11
O
S
10 s C
8
11 N
[00124] DMF (26 L, 10 v/w) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at 15 °C. Compound 8 (7.3 kg of
DMF solution, containing 2.9 kg, 8.1 mol, 1.0 eq.) and TBAA (2.44 kg, 8.1 mol, 1.0 eq.) were added sequentially to the flask under nitrogen. The mixture was cooled to 0-10 °C.
TMSCF3 (2.88 kg, 20.3 mol, 2.5 eq.) was then added to the flask over 60 min at 0-10 °C.
The reaction mixture was stirred at 0-5 °C under nitrogen protection for 3 hours.
Isopropyl acetate (60 L) was added to the mixture, followed by the addition of 45 L of
NaHCCb under stirring at 5-25 °C. The organic layer was separated, washed three times with NaHC03 (30 L x 3), and concentrated from 60 kg to 2.5 kg of brown oil. The oil product was dissolved in 20 L of TBME and filtered through a pad of silica gel (about 40 cm high, 30 cm diameter) over 2 hours to obtain 2.14 kg of compound 1 1 in TBME solution. The solution was concentrated at 45-50 °C to dryness to obtain compound 1 1 as a black oil (1.85 kg; 85.2% purity (AETC by HPLC, retention time = 9.1 minutes, 9.6 minutes for diastereoisomer); 53.6% yield). 1H-NMR (400 MHz, CDCh): d = 1.33 (s, 9H), 3.82-3.85 (d, 1H), 5.61-5.66 (m, 1H), 7.53-7.60 (m, 2H), 8.63-8.64 (d, 1H), 8.71-8.72 (m, 1H), 8.95 (s, 1H).
[00125] Step 8: Synthesis of compound 12 (free base)
[00126] Compound 1 1 (1.8 kg, 4.23 mol, 1.0 eq., crude) was added to a 50 L reactor equipped with a temperature probe and overhead stirrer under nitrogen at 25 °C. Anhydrous MeOH (18 L) was added to dissolve compound 1 1. Then MeOH/HCl (18 L, 1 N) was added dropwise at 25-30 °C over 10 minutes and the mixture was stirred at 25-30 °C for 1 hour. Water (15 L) was added to the reaction and the mixture concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 4 hours to remove the solvent. The pH of the mixture was adjusted to 10 with 5 L of K2CO3 solution. 20 L of EtOAc was then added to the mixture and the organic layer was separated and the aqueous layer extracted twice with EtOAc (15 L x 2). The organic layers were combined and washed with 10 L of brine. The combined organic layers contained 996 g of
compound 12 in 40 kg of EtOAc solution (84% purity (AUC by HPLC, retention time =
2.8 minutes). The organic layers were concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to a 7.5 kg volume of compound 12 in EtOAc solution (83% purity (AETC by HPLC, retention time = 2.7 minutes).
[00127] In a separate 50 L reactor equipped with a temperature probe and overhead stirrer, D-CSA was added (930 g, 4.0 mol, 1.0 eq. to 1.26 kg compound 12) and stirred at room temperature under nitrogen. EtOAc (10 L) and then the EtOAc solution of compound 12 (1.26 kg, 3.9 mol, 1.0 eq.) were each sequentially added to the reactor. The mixture was stirred at room temperature for 1 hour and slowly became a suspension. The mixture was filtered by centrifuge and washed with EtOAc to produce 2.3 kg of compound 12 as an off-white solid (96.0% purity).
[00128] The solid product, 20 L of EtOAc, and 10 L of 10% aqueous K2CO3 were added sequentially to a 50 L flask and stirred at room temperature until no solid remained (pH = 9-10). The organic layer was separated and the aqueous layer extracted twice with EtOAc (10 L x 2). The organic layers were combined (about 32 kg) and washed with 10 L of brine. The organic layer contained 716 g of compound 12 in
31.8 kg of solution.
[00129] The organic layer was concentrated under vacuum at 45-50 °C to about 8 L. Activated carbon (200 g) was added to the organic layer and the mixture stirred at 60-70 °C for 1 hour, cooled to room temperature, and filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to remove the activated carbon. The mixture was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to yield 710 g of compound 12 as a yellow solid (99.4% purity). [00130] D-CSA (410 g, 1.77 mol, 1.0 eq. to 680 g compound 12), 3.4 L iPrOH, and 68 mL of water were added sequentially to a 10 L reactor equipped with a temperature probe and overhead stirrer and stirred at room temperature under nitrogen. The mixture was heated to reflux (84 °C) to form solution A after 1 hour. Compound 12 (680 g) was dissolved in 3.4 L of iPrOH and added into solution A for one partition. A clear solution was formed and the temperature decreased to 65 °C. The mixture was stirred at 65 °C for about 15 minutes after which a solid appeared. The mixture was cooled to 10 °C over 2 hours, stirred at 10 °C for an additional 30 minutes, and filtered through a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to collect the 1.1 kg of white solid.
[00131] EtOAc (10 L), 1.1 kg of white solid product, and 5 L of 10% K2CO3 were added sequentially to a 20 L flask and mixed for 5 minutes. The solid dissolved (pH = 9-10). The EtOAc layer was separated and the aqueous layer extracted twice with EtOAc (5 L each). The organic layers were combined (about 20 L), washed with 5 L of brine, and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-55 °C for about 3 hours to remove most of the solution and until the residue weight reached 1 kg. Heptanes (1 L) was added to the mixture and stirred at room temperature for 30 minutes. The mixture was filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to obtain 419 g of compound 12 base as a white solid (99.7% purity). The filtrate was concentrated to 135 g of compound 12 as a yellow solid (98.7% purity). 1H-NMR (400 MHz, CDCh): d = 1.85 (bs, 2H), 5.17 (m, 1H), 7.56 (d, 1H), 7.68 (d, 1H), 8.62 (d, 1H), 8.70-8.71 (m, 1H), 8.93 (s, 1H). Combined, the products resulted in a 40.7% yield of compound 12.
[00132] Step 9: Synthesis of compound 10
10A 10
[00133] Pd/C (40 g, 5% w/w) was added into a 10 L autoclave reactor at room temperature under nitrogen. THF (2 L), 2 L of methylamine (27%-30% alcoholic solution, 2.1 eq.), and 800 g of compound 10A (7 mol, 1.0 eq.) were sequentially added into the reactor. The system was purged with hydrogen three times. The mixture was stirred at hydrogen pressure (50 psi) at 70-75 °C overnight and was then filtered using a Biichner funnel and filter paper (pore size: 30-50 pm) over 10 minutes to remove the Pd/C. The filtrate was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to obtain 933 g of yellow oil. The mixture was distilled without a column at atmospheric pressure and the 140-170 °C portion was collected to obtain 763 g of compound 10 as a colorless oil (98.6% purity (AUC by HPLC, retention time = 4.8 minutes); 84.2% yield; 8000 ppm residual ethanol). A portion of the oil (563 g) was distilled using a 3 cm column at atmospheric pressure and the 140-170 °C portion was collected to obtain 510 g of compound 10 (75.8% yield; 134 ppm residual ethanol). 1H-NMR (400 MHz, CDCb): d = 0.82 (bs, 1H), 1.10-1.12 (q, 2H), 1.66 (d, 2H), 1.73-1.81 (t, 2H), 2.05 (s, 3H), 2.08-2.19 (m, 1H), 2.22 (s, 3H), 2.60 (d, 2H).
[00134] Step 10: Synthesis of HM04 fumarate salt
[00135] DCM (1L), 200 g CDI (1.23 mol, 2.0 eq.), and 35 g DABCO (0.31 mol, 0.5 eq.) were sequentially added into a 3 L reactor equipped with a temperature probe and overhead stirrer, and stirred at room temperature under nitrogen. The mixture was cooled to -10 to -5 °C. Compound 12 (200 g) was dissolved in 1 L of DCM and added into the mixture dropwise over 1 hour, followed by stirring for 16 hours at -10 to -5 °C. Compound 10 (159 g, 1.24 mol, 2.0 eq.) was added at -10 to 0 °C over 10 minutes. The mixture was then warmed to 0 to 5 °C and held for 2 hours. The mixture was concentrated under vacuum at 40-45 °C to about 1 L. HC1 (1 L of 1 N) was added to the residual and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to remove the DCM. Another 3 L of 1N HC1 was added to the residual and extracted three times with TBME (4 L, 2 L, 2 L). The aqueous layer was slowly adjusted to pH = 9-10 with 20% aqueous K2CO3 (about 1.5 L) and extracted with DCM (2 L x 3). The organic layers were combined (about 4 L) and washed three times with 0.25 N KH2PO4 (1.2 L x 3). The organic layer was washed with 2 L of brine to bring the pH to neutral and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to 450 g (335 mL). MTBE (1.5 L) was added to the residual and distilled until 500 mL of liquid was collected. This step was repeated four times with the addition of 500 mL of TBME and collection of 500 mL of distillate, with the exception that 330 mL of liquid was collected at the final distillation. About 1 to 1.2 L of residual remained in the flask. The residual was slowly cooled to room temperature and stirred at room temperature overnight. The mixture was filtered, washed twice with TBME (400 mL x 2), and dried to obtain 192 g of HM04 free base a light yellow solid (99.3% purity (AUC by HPLC, retention time = 11.0 minutes). The product on the wall was dissolved in DCM and concentrated under vacuum to obtain 22 g of HM04 free base as a brown sticky oil (97.6% purity). The filtrate was concentrated under vacuum to obtain 22.5 g of yellow solid (94.0% purity).
[00136] HM04 free base (187 g, 0.39 mol, 1.0 eq., 99.3% purity) and 1.9 L of ACN were sequentially added to a 3 L flask equipped with a temperature probe and overhead stirrer and stirred at 15 °C under nitrogen to obtain a light-yellow suspension. Fumaric acid (45.6 g, 0.39 mol, 1.0 eq.) was added to the flask and generated a white suspension after 1 minute. The reaction suspension was stirred overnight at room temperature, filtered (15-20 pm, ash<0.l5), washed twice with ACN (50 mL x 2), and dried under vacuum at 50 °C for 6 hours to obtain 207 g of HM04 fumarate salt as a light yellow solid (99.4% purity (AUC by HPLC, retention time = 11.1 minutes); 57.8% yield; 3100 ppm residual ACN). The filtrate was concentrated under vacuum to obtain 20.1 g of HM04 fumarate salt as a light yellow solid (97.3% purity).
[00137] A portion of the product (117 g) was further dried in a vacuum oven (20-40 mmHg) to lower the residual acetonitrile content. After drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours, the residual acetonitrile content was measured as 3100 ppm, 2570 ppm, 1300 ppm, and 256 ppm, respectively. After the drying process, 98 g of HM04 fumarate salt was isolated (99.4% purity (AUC by HPLC, retention time = 11.0 minutes); 1H-NMR (400 MHz, DMSO): d = 1.49-1.58 (m, 2H),
1.81-1.92 (m, 2H), 2.44-2.53 (m, 5H), 2.78 (s, 3H), 3.12 (m, 2H), 4.06-4.13 (m, 1H), 6.36-6.41 (m, 1H), 6.55 (s, 2H), 7.47 (d, 1H), 7.73 (d, 1H), 8.11 (d, 1H), 8.75 (d, 1H),
8.81-8.82 (m, 1H), 8.99 (d, 1H). The yield of 98g of HM04 fumarate salt isolated after drying the partial batch was extrapolated over the whole batch to calculate an
approximate yield of 48% for step 10.
[00138] XRPD analysis of HM04 fumarate salt products obtained after drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours was performed (see Figures 6-9, respectively). The XRPD profile showed that the HM04 fumarate salt product was consistent with Form 1.
Example 6. Streamlined Synthesis of HM04 Fumarate Salt Form 1
[00139] The overall yield of HM04 fumarate salt produced using Step 10 of Example 5 was calculated as approximately 48%. In order to increase the overall yield, a streamlined synthesis was investigated that eliminated the step of isolating HM04 free base. In particular, step 10 of the method of Example 5 shown in Figure 5 was changed. An overview of the streamlined synthesis beginning after step 9 of Example 5 is shown in Figure 10.
[00140] Streamlined HM04 Fumarate Salt Trial 1 : PCM (121.4 g). CPI (20.0 g, 123 mmol, 2 eq.) and DABCO (3.5 g, 31 mmol) were sequentially added into an inertized 1 L reactor. The mixture was cooled to -10 °C. Separately, a solution of DCM (132.5 g) and compound 12 (20.0 g, 62.1 mmol) were charged into a vessel and stirred until a solution was obtained. This solution was dropped into the 1 L reactor over 33 minutes by keeping the internal temperature at -10 to -5 °C. At the end of the addition, the vessel was rinsed with DCM (7.0 g), which was then added to the reaction mixture.
After stirring overnight (19 hours) and positive IPC, compound 10 (15.9 g, 124 mmol, 2 eq.) was added over 15 minutes and the vessel rinsed with DCM (9.0 g). After heating at 0 °C, 1 hour of stirring, positive IPC, and a further 1.5 hours of stirring, the mixture was heated at room temperature and charged with water (200.1 g). The aqueous layer was separated and the organic layer extracted twice with 1 N HC1 (201, 200 g). The combined aqueous layers containing the product were washed with TBME (148 g). After removal of the organic layer, the aqueous layer was charged with DCM (265.0 g) and 50% K2CO3 solution (about 240 ml) until reaching pH 9.61.
[00141] Meanwhile, a solution of KH2PO4 (8.2 g) in water (240 g) was prepared. The organic layer containing the product was charged with the KH2PO4 solution until reaching pH 7.12 (142.2 g). After separation of the aqueous layer, the organic layer was washed with water (200 g). After separation of the aqueous layer, the organic layer was evaporated at 50 °C. ACN (314.4 g) was added and the solvent distilled again at 70-75 °C under vacuum. ACN (235.8 g) was added and the solvent distilled again under vacuum. ACN (141.5 g) was added, the resulting solution polish filtered and the filter washed with ACN (16 g). After heating at 60 °C, fumaric acid (7.2 g, 62 mmol) was added to the solution, causing a white precipitate. After cooling to 20 °C over 1 hour, the suspension was filtered and washed twice with TBME (2 x 30 g). After drying on the filter with nitrogen flow, 70.7 g of wet raw product was obtained. This was slurried with TBME (177.0 g) for 1 hour, filtered, and washed with TBME (70 g). After drying on the filter under nitrogen flow, 33.0 g of wet product was obtained. Heating at 50 °C under vacuum afforded the dry product as a white powder of HM04 fumarate salt (21.1 g;
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9926337 | SUBSTITUTED ASYMMETRIC UREAS AND MEDICAL USES THEREOF | 2016-12-02 | |
| US9546157 | p-Substituted Asymmetric Ureas and Medical Uses Thereof | 2015-03-06 | 2015-09-10 |
////////////HM04, H0900, Helsinn, Novo Nordisk, PRECLINICAL, obesity, Prader-Willi syndrome, ghrelin
CN(C1CCN(C)CC1)C(=O)N[C@H](c3ccc(c2cnccn2)c(Cl)c3Cl)C(F)(F)F
GFH 018
![(E)-3-[6-[2-(6-Methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=132246793&t=l)
GFH-018
CAS 2169299-67-4
GenFleet Therapeutics
Advanced solid tumor; Cancer
TGF-beta Receptor Type-1 (TGFBR1; ALK5; SKR4; TbetaR-I) Inhibitors
Signal Transduction Modulators
GFH-018 , a TGFBR1 inhibitor, being investigated by GenFleet as an oral tablet formulation, for the treatment of cancer, including advanced solid tumors and hepatocellular carcinoma, in March 2019, the company was developing GFH-018 as a class 1 chemical drug in China, with a clinical trial expected to begin in the second half of 2019.


PATENT
WO2017215506
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017215506
PATENT
WO-2019114792
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019114792&tab=FULLTEXT&maxRec=1000
Novel crystalline and salt (hydrochloride, sulfate and mesylate) forms of a TGF-βRI inhibitor, designated as Forms A and B, processes for their preparation and compositions comprising them are claimed for treating cancers. The compound was originally claimed in WO2017215506 , assigned to Medshine Discovery Inc alone.
Step C: 1-9 (4.50 g, 15.20 mmol), 1-6 (4.43 g, 18.24 mmol), sodium carbonate (4.83 g, 45.60 mmol), [1,1′-bis (diphenyl) Phosphine) ferrocene] palladium dichloride (556.07 mg, 759.96 μmol), 2-biscyclohexylphosphine-2′, 6′-dimethoxybiphenyl (311.98 mg, 759.96 μmol) and [2-( 2-Aminophenyl)phenyl]-chloro-palladium-cyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphine (547.64 mg, 759.96 μmol) was added to the dioxane (100 ml) and water (20 ml) in a mixed solvent. It was replaced with nitrogen three times and then heated to 90 to 100 ° C and stirred for 2 hours. The reaction mixture was poured into water (200 ml) and evaporated and evaporated. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified on a silica gel column (eluent: methylene chloride/methanol, v/v=30/1) to afford crude crude product in petroleum ether/ethyl acetate (v/v=5/1) After stirring for 12 hours, the solid was collected by filtration, and the solid was concentrated and dried under reduced pressure to give 1-10. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.49 (S, IH), 7.82-7.74 (m, 2H), 7.59-7.46 (m, 4H), 6.99 (dd, J = 2.6,6.1Hz, IH), 4.39 (d, J = 6.3 Hz, 2H), 2.90 – 2.70 (m, 4H), 2.20 (s, 3H).
192 mg of the compound of formula (I) was weighed into a glass bottle. 10 ml of a tetrahydrofuran:acetic acid (v/v=9/1) mixed solvent was added, and after ultrasonic assisted for 30 minutes, the sample was dissolved into a clear solution. Stir on a magnetic stirrer (40 ° C). After 1.05 equivalents of p-toluenesulfonic acid monohydrate was slowly added, the sample was stirred overnight. After naturally cooling to room temperature, the supernatant was discarded by centrifugation, stirred for 10 hours by adding 10 ml of tetrahydrofuran, and the supernatant was discarded by centrifugation, and the same procedure was repeated twice more. The obtained solid was dried in a vacuum oven at 40 ° C for 1 hour, and after milling, it was further dried in a vacuum oven at 30 ° C for 16 hours to obtain a crystal form B of the compound of the formula (II).
.///////////////////GFH-018, GFH 018, GenFleet Therapeutics, Advanced solid tumor, Cancer, PRECLINICAL
NC(=O)/C=C/c4n5ncnc5ccc4c2c3CCCn3nc2c1cccc(C)n1
