

| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8969557 | ACC INHIBITORS AND USES THEREOF |
2012-11-09
|
2013-05-16
|
| US2017267690 | SOLID FORMS OF A THIENOPYRIMIDINEDIONE ACC INHIBITOR AND METHODS FOR PRODUCTION THEREOF |
2017-03-01
|
|
| US2016297834 | ACC INHIBITORS AND USES THEREOF |
2016-03-11
|
|
| US9453026 | ACC INHIBITORS AND USES THEREOF |
2015-01-23
|
2015-07-23
|
Home » 2016 (Page 26)
Yearly Archives: 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FIRSOCOSTAT, ND 630, GS-0976, NDI-010976
FIRSOCOSTAT, ND 630, NDI 010976, ND-630, NDI-010976
CAS: 1434635-54-7, UNII: XE10NJQ95M
PHASE 2, Non-alcoholic steatohepatitis, GILEAD
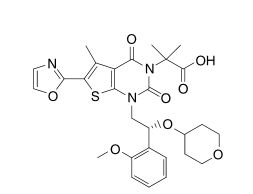
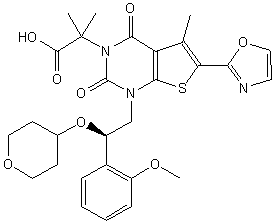
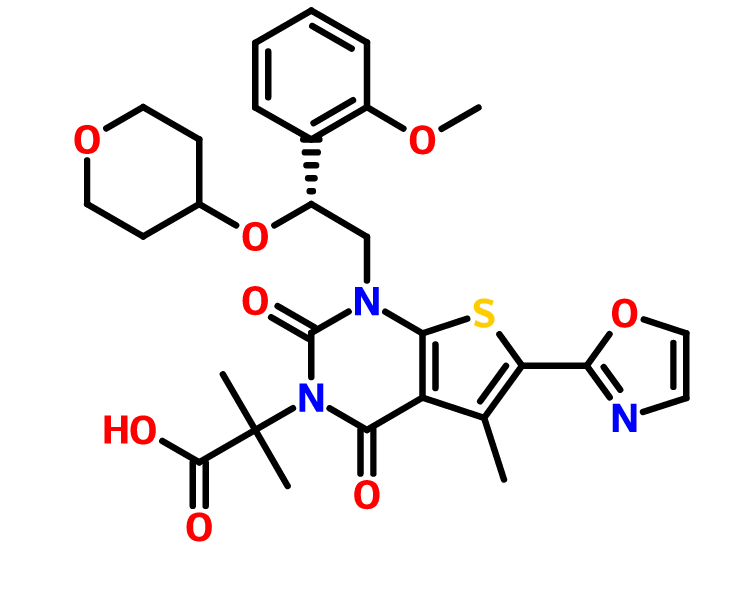
1,4-dihydro-1-[(2R)-2-(2-methoxyphenyl)-2-[(tetrahydro-2H-pyran-4-yl)oxy]ethyl]-α,α,5-trimethyl-6-(2-oxazolyl)-2,4-dioxo-thieno[2,3-d]pyrimidine-3(2H)-acetic acid
2-[l-[2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethyl]-5- methyl-6-(l,3-oxazol-2-yl)-2,4-dioxo-lH,2H,3H,4H-thieno[2,3-d]pyrimidin-3-yl]-2- methylpropanoic acid
2-[1-[(2R)-2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethyl]-5-methyl-6-(1,3-oxazol-2-yl)-2,4-dioxothieno[2,3-d]pyrimidin-3-yl]-2-methylpropanoic acid
CAS 1434635-54-7
Thieno[2,3-d]pyrimidine-3(2H)-acetic acid, 1,4-dihydro-1-[(2R)-2-(2-methoxyphenyl)-2-[(tetrahydro-2H-pyran-4-yl)oxy]ethyl]-α,α,5-trimethyl-6-(2-oxazolyl)-2,4-dioxo-
| Molecular Formula: | C28H31N3O8S |
|---|---|
| Molecular Weight: | 569.62604 g/mol |

| Company | Nimbus Therapeutics LLC |
| Description | Small molecule allosteric inhibitor of acetyl-coenzyme A carboxylase alpha (ACACA; ACC1) and acetyl-coenzyme A carboxylase beta (ACACB; ACC2) |
| Molecular Target | Acetyl-Coenzyme A carboxylase alpha (ACACA) (ACC1) ; Acetyl-Coenzyme A carboxylase beta (ACACB) (ACC2) |
| Mechanism of Action | Acetyl-coenzyme A carboxylase alpha (ACACA) (ACC1) inhibitor; Acetyl-coenzyme A carboxylase beta (ACACB) (ACC2) inhibitor |
| Therapeutic Modality | Small molecule |
Preclinical Diabetes mellitus; Hepatocellular carcinoma; Metabolic syndrome; Non-alcoholic steatohepatitis; Non-small cell lung cancer
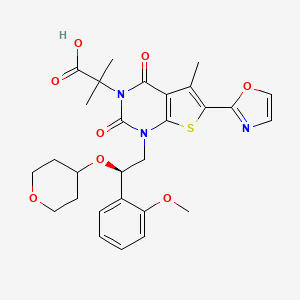
1,4-Dihydro-1-((2R)-2-(2-methoxyphenyl)-2-((tetrahydro-2H-pyran-4-yl)oxy)ethyl)-alpha,alpha,5-trimethyl-6-(2-oxazolyl)-2,4-dioxothieno(2,3-d)pyrimidine-3(2H)-acetic acid
In April 2016, Gilead Sciences and Nimbus Therapeutics, LLC announced that the companies have signed a definitive agreement under which Gilead will acquire Nimbus Apollo, Inc., a wholly-owned subsidiary of Nimbus Therapeutics, and its Acetyl-CoA Carboxylase (ACC) inhibitor program. Nimbus Therapeutics will receive an upfront payment of $400 million, with the potential to receive an additional $800 million in development-related milestones over time.
The Nimbus Apollo program includes the lead candidate NDI-010976, an ACC inhibitor, and other preclinical ACC inhibitors for the treatment of non-alcoholic steatohepatitis (NASH), and for the potential treatment of hepatocellular carcinoma (HCC) and other diseases.
In May 2016, Nimbus Therapeutics announced the recent closing of Gileads acquisition of Nimbus Apollo. The acquisitions completion triggered a $400 million upfront payment to Nimbus from Gilead.
In January 2016, fast track designation was assigned in the U.S. for this indication. In May 2016, Gilead Sciences acquired Nimbus Apollo from Nimbus Therapeutics, including its acetyl-CoA carboxylase (ACC) inhibitor program.
Gilead Sciences following the acquisition of Nimbus Apollo , is developing firsocostat , the lead from a program of acetyl-CoA carboxylase (ACC)-targeting compounds, for treating fatty liver disease including non-alcoholic steatohepatitis.
Acetyl CoA carboxylase 1/2 allosteric inhibitors – Nimbus
Therapeutics
The Liver Meeting 2015 – American Association for the Study of Liver Diseases (AASLD) – 2015 Annual Meeting, San Francisco, CA, USA
Nimbus compounds targeting liver disease in rat models
Data were presented by Geraldine Harriman, from Nimbus Therapeutics, from rat models using acetyl-CoA carboxylase (ACC) inhibitors NDI-010976 (ND-630) and N-654, which improved metabolic syndrome endpoints, decreased liver steatosis, decreased expression of inflammatory markers and improved fibrosis. The hepatotropic ACC inhibitor NDI-010976 had IC50 values of 2 and 7 nM for ACC1 and 2, respectively, EC50 values in HepG2 serum free and 10% serum of 9 and 66 nM, respectively, and 2-fold C2C12 fatty acid oxidation (FAOxn) stimulation at 200 nM. Rat FASyn (synthase), malonyl-CoA (liver) and malonyl-COA (muscle) respective ED50 values were 0.14 mg/kg po, 0.8 and 3 mg/kg. The rat respiratory quotient (RQ) MED was 3 mg/kg po. ADME data showed low multispecies intrinsic clearance (human, mouse, rat, dog, monkey). NDI-010976 was eliminated predominantly as the parent drug. Additionally, P450 inhibition was > 50 microM. In liver and muscle, NDI-010976 modulated key metabolic parameters including a dose-dependent reduction in the formation of the enzymatic product of acetyl coA carboxyloase malonyl coA; the ED50 value was lower in muscle. The drug also decreased FASyn dose dependently and increased fatty acid oxidation in the liver (EC50 = 0.14 mg/kg). In 28-day HS DIO rats, NDI-010976 favorably modulated key plasma and liver lipids, including decreasing liver free fatty acid, plasma triglycerides and plasma cholesterol; this effect was also seen in 37-day ZDF rats

PATENT
http://www.google.com/patents/WO2013071169A1?cl=en
Example 76: Synthesis of 2-[l-[2-(2-methoxyphenyl)-2-(oxan-4-yloxy)ethyl]-5- methyl-6-(l,3-oxazol-2-yl)-2,4-dioxo-lH,2H,3H,4H-thieno[2,3-d]pyrimidin-3-yl]-2- methylpropanoic acid (1-181).

Synthesis of compound 76.1. Into a 250-mL 3 -necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed oxan-4-ol (86 g, 842.05 mmol, 2.01 equiv) and FeCl3 (10 g). This was followed by the addition of 57.2 (63 g, 419.51 mmol, 1.00 equiv) dropwise with stirring at 0 °C. The resulting solution was stirred for 3 h at room temperature. The resulting solution was diluted with 500 mL of H20. The resulting solution was extracted with 3×1000 mL of ethyl acetate and the organic layers combined. The resulting solution was extracted with 3×300 mL of sodium chloride (sat.) and the organic layers combined and dried over anhydrous sodium sulfate. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10). This resulted in 22 g (21%) of 76.1 as a white solid.
Synthesis of compound 76.2. The enantiomers of 76.1 (22g) were resolved by chiral preparative HPLC under the following conditions (Gilson Gx 281): Column: Venusil Chiral OD-
H, 21.1 *25 cm, 5 μιη; mobile phase: hexanes (0.2% TEA) and ethanol (0.2% TEA) (hold at 10% ethanol (0.2%TEA) for 13 min); detector: UV 220/254 nm. 11.4 g (52%) of 76.2 were obtained as a white solid.
Synthesis of compound 76.3. Into a 500-mL 3-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 70.1 (12 g, 20.49 mmol, 1.00 equiv), tetrahydrofuran (200 mL), 76.2 (6.2 g, 24.57 mmol, 1.20 equiv) and DIAD (6.5 g, 32.18 mmol, 1.57 equiv). This was followed by the addition of a solution of triphenylphosphane (8.4 g, 32.03 mmol, 1.56 equiv) in tetrahydrofuran (100 mL) dropwise with stirring at 0 °C in 60 min. The resulting solution was stirred overnight at room temperature. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5). This resulted in 17 g (crude) of 76.3 as a white solid.
Synthesis of compound 76.4. Into a 500-mL 3-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 76.3 (17 g, crude), toluene (300 mL), Pd(PPh3)4 (1.7 g, 1.47 mmol, 0.07 equiv) and 2-(tributylstannyl)-l,3-oxazole (8.6 g, 24.02 mmol, 1.16 equiv). The resulting solution was stirred overnight at 110 °C. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10). Purification afforded 6 g of 76.4 as a white solid.
Synthesis of compound 1-181. Into a 250-mL 3-necked round-bottom flask, was placed 76.4 (6 g, 7.43 mmol, 1.00 equiv), tetrahydrofuran (100 mL), TBAF (2.3 g, 8.80 mmol,
I .18 equiv). The resulting solution was stirred for 1 h at room temperature. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (50: 1). This resulted in 3.4 g (80%) of Compound 1-181 as a white solid.
Purification: MS (ES): m/z 570 (M+H)+, 592 (M+Na)+.
1H NMR (300 MHz, DMSO- d6): δ 1.22-1.36 (m, 2H), 1.62 (m, 8H), 2.75 (s, 3H), 3.20-3.39 (m, 3H), 3.48-3.58 (m, 2H), 3.80 (s, 3H), 3.85-4.20 (m, 2H), 5.30 (m, 1H), 7.03 (m, 2H), 7.33-7.50 (m, 3H), 8.2 (s, 1H).

Preparation of ND-630.1,4-dihydro-1-[(2R)-2-(2-methoxyphenyl)-2-[(tetrahydro-2H-pyran-4-yl)oxy]ethyl]-α,α,5-trimethyl-6-(2-oxazolyl)-2,4-dioxo-thieno[2,3-d]pyrimidine-3(2H)-acetic acid, ND-630, was prepared as described (49)…….http://www.pnas.org/content/113/13/E1796.full.pdf
Harriman GC, Masse CE, Harwood HJ, Jr, Baht S, Greenwood JR (2013) Acetyl-CoA
carboxylase inhibitors and uses thereof. US patent publication US 2013/0123231.
CLIPS
Conference: 66th Annual Meeting of the American Association for the Study of Liver Diseases Conference Start Date: 13-Nov-2015
…candidates for minimizing IR injury in liver transplantation.Nimbus compounds targeting liver disease in rat modelsData were presented by Geraldine Harriman, from Nimbus Therapeutics, from rat models using acetyl-CoA carboxylase (ACC) inhibitors NDI-010976 (ND–630) and N-654, which improved metabolic syndrome endpoints, decreased liver steatosis, decreased expression of inflammatory markers and improved fibrosis. The hepatotropic ACC inhibitor NDI-010976 had IC50 values of 2 and 7 nM for ACC1 and 2, respectively…
REFERENCES
November 13-17 2015
The Liver Meeting 2015 – American Association for the Study of Liver Diseases (AASLD) – 2015 Annual Meeting San Francisco, CA, USA ,
The Liver Meeting 2015 – American Association for the Study of Liver Diseases (AASLD) – 2015 Annual Meeting San Francisco, CA, USA ,
| WO-2014182943 |
| WO-2014182945 |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015203510 | 2015-07-23 | ACC INHIBITORS AND USES THEREOF |
| US2013123231 | 2013-05-16 | ACC INHIBITORS AND USES THEREOF |
WO2017151816 ,
CN 107629069
CN 107629069
CN 107151251
WO 2013071169
WO 2016112305
PATENT
WO-2018161022
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018161022&tab=PCTDESCRIPTION&maxRec=1000
Solid forms, including a salts (such as choline, diethylamine, NN-dibenzylethylenediamine, ethanolamine) or co-crystal, of firsocostat and compositions comprising them are claimed, which exhibits Acetyl-CoA carboxylase inhibitory activity and useful for treating ACC mediated diseases such as metabolic disorders, neurological disorders, and infectious diseases. Also claimed are process for preparing firsocostat and intermediates useful for preparing them are claimed.
The present disclosure provides forms of Compound I or a compound of formula (I) having the formula:
Compound I may be referred to by formula (I):
(I)
or its chemical name of (R)-2-(l-(2-(2-methoxyphenyl)-2-((tetrahydro-2H-pyran-4-yl)oxy)ethyl)-5-methyl-6-(oxazol-2-yl)-2,4-dioxo-l,2-dihydrothieno[2,3-d]pyrimidin-3(4H)-yl)-2-methylpropanoic acid. U.S. Patent No. 8,969,557 discloses that Compound I exhibits ACC inhibitory activity. In the present disclosure, compounds may be presented in the form of chemical structures or names.
Scheme 1 represents an exemplary synthesis of a compound of formula (F) and may be carried out according to the embodiments described herein.
Scheme 1
(E) (F)
Scheme 2
(E-1 ) (I)
Scheme 3
Step (g)
Scheme 4
scheme 5
Example 1 : Synthesis of Compound B-2
B-2
[0401] Compound A-2 was combined with Compound G-1 (about 1 equivalents (“equiv”)) with K2CO3 (about 2.3 equiv) in dimethylacetamide. The mixture was stirred at room temperature. The resulting mixture was then diluted with ethyl acetate and washed with water and brine. The organic layer was separated and concentrated to dryness, and the resulting product was purified by column chromatography (eluent: 0 to about 28% ethyl acetate:
heptanes). The resulting product was Compound B-2. ¾ NMR (300 MHz, CDCh): δ 7.92 (d, J
= 8.4 Hz, 1H), 7.57 (m, 1H), 7.06 (m, 2H), 5.20 (s, 2H), 4.00 (s, 3H), 2.42 (s, 3H), 1.77 (s, 6H), 1.44 (s, 9H).
Example 2: Synthesis of a compound of formula (C)
(B) (C)
[0402] Compound of formula (B) or Compound B (which may be prepared as described in Example 1) and a (S,S)-Ruthenium catalyst, such as a Ruthenium catalyst as described herein, or a suitable antipode of the Ruthenium catalyst, are combined in the presence of potassium tert-butoxide (“KO^-Bu”) and isopropanol and refluxed to yield a compound of formula (C) or Compound C. Compound C is isolated and purified by methods described herein.
Example 3: Synthesis of Compound D-1
C-1 D-1
[0403] To Compound C-1 in dichloromethane is added 4-bromotetrahydro-2H-pyran. Upon addition of an organic base, the reaction mixture is stirred ovemight to yield a compound of formula D-1 or Compound D-1. Compound D-1 is isolated and purified by the methods described herein.
Example 4: S
D-1 E-2
[0404] Oxazole in THF is cooled to between about -80 °C and about -60 °C. Then, ft-butyllithium in hexanes is added while maintaining the temperature of the reaction below about -60 °C. The mixture is stirred at this temperature for 90 minutes. Zinc (II) chloride is added, maintaining the temperature of the mixture below about -60 °C, and the mixture is stirred at that temperature for about one hour before warming to about 10-20 °C. Compound D-1 is added to the reactor followed by tetrakis(triphenylphosphine)palladium(0) (“Pd(PPh3)4”), and the temperature is adjusted to between about 55-65 °C. The mixture is stirred at that temperature for about 12 hours to yield Compound E-2. Compound E-2 is isolated and purified by the methods described herein.
Example 5: Synthesis of Compound I
[0405] A sulfuric acid solution was prepared by addition of concentrated sulfuric acid (47 g,
4.7 w/w Compound E-2) to water (12 g, 1.2 v/w Compound E-2) followed by a water (15 g, 1.5 v/w Compound E-2) rinse forward. 2-Propanol (37 g, 4.7 v/w Compound E-2) was slowly charged to a reactor containing sulfuric acid solution at about 9 °C while maintaining the reaction contents at no more than about 40 °C, and the solution was cooled to about 5 °C .
Compound E-2 (10 g, 1.0 equiv) was charged to the solution, followed by a 2-propanol rinse forward (2 g, 0.25 v/w E-2). The contents were cooled to about 7 °C and stirred for a minimum of about 21 hours. The contents were slowly added into water, and the slurry was agitated for about 30 minutes. The slurry was filtered, and the filter cake was washed and dried under vacuum for about 4 hours. The crude wet cake was charged back to the reactor, followed by additions of ethyl acetate (40 g, 4.4 v/w Compound E-2) and water (100 g, 10 v/w Compound E-2). The slurry was adjusted to pH at about 8-9 with an about 20 wt% sodium hydroxide solution at about 22 °C, and then agitated for about 30 minutes at about 22 °C. The solution was allowed to settle. The top organic layer was collected and the bottom aqueous layer was washed with ethyl acetate (40 g, 4.4 v/w Compound E-2) at about 22 °C for about 30 minutes. The solution was allowed to settle, and the top organic layer was removed. 2-Methyltetrahydrofuran (86 g, 10 v/w Compound E-2) was then added, was adjusted to pH at about 4-5 with an about 4 N HCl solution at about 22 °C. The solution was agitated for about 30 minutes at about 22 °C and then allowed to settle. The bottom aqueous layer was extracted with 2-methyltetrahydrofuran (52 g, 6 v/w Compound E-2) at about 22 °C for about 30 minutes. After the solution was allowed to settle, the bottom aqueous layer was removed. The organic layers were combined and distilled under vacuum (jacket at about < 45 °C) to about 4V pot volume. Ethanol (55.4 g, 7 v/w
Compound E-2) was added and the reaction as distilled (repeated twice). Ethanol was again added (23.7 g,3 v/w Compound E-2), followed by water (30 g, 3 v/w Compound E-2). The reaction was heated to about 75 °C and then cooled over about 4 hours to about 50 °C, then to about 0 °C over about 5 hours. The reaction was then aged and filtered, and the solid was washed with a precooled mixture of ethanol (9.5 g, 1.2 v/w Compound E-2) and water (6 g, 0.6 v/w Compound E-2). The resulting product was washed to afford Compound of formula (I). ¾ NMR (400 MHz, CDCh): δ 7.70 (s, 1H), 7.57 (dd, J= 1.6 Hz, J= 7.6 Hz, 1H), 7.29 (td, J= 1.6 Hz, J = 8.0 Hz, 1H), 7.23 (d, J= 0.4 Hz, 1H), 7.02 (t, J= 7.6 Hz, 1H), 6.86 (d, J= 8.4 Hz, 1H), 5.39 (dd, J= 5.6 Hz, J= 8.0 Hz, 1H), 4.17-4.14 (m, 1H), 4.04 (br, 1H), 3.86 (s, 3H), 3.78-3.67 (m, 2H), 3.46-3.40 (m, 1H), 3.37-3.32 (m, 2H), 2.85 (s, 3H), 1.87 (s, 3H), 1.83 (s, 3H), 1.75-1.72 (m, 2H), 1.59-1.51 (m, 1H), 1.48-1.39 (m, 1H).
Example 6: Synthesis of Compound J-l
Step (a): Formation of Compound P-l
[0406] 2-Methoxyphenylmagnesium bromide (1 M in THF, 1.0 equiv.) was added to a solution of diethyl oxalate (1.1 equiv.) in THF (250 mL) at about -20 °C over approximately 20 min. After aging for about 45 min at about -20 °C, the resulting slurry was quenched with saturated NH4CI (250 mL) and was diluted with water (200 mL). This mixture was extracted with EtO Ac (400 mL), and the organic phase was washed with brine (200 mL). The organic phase was concentrated and the solvent was exchanged to THF. The resulting THF solution was used in the next step as is. ¾ NMR (400 MHz, CDCh): δ 7.90 (m, 1H), 7.61 (m, 1H), 7.10 (t, J = 7.6 Hz, 1H), 7.01 (d, J= 8.4 Hz 1H), 4.41 (q, J= 7.1 Hz, 2H), 3.88 (s, 3H), 1.41 (t, J= 7.1 Hz, 3H).
Alternate Preparation Compound P-l:
[0407] Anisole (1.0 equiv.) in THF (15 mL) was cooled to about -20 °C, and 2.5 M n-BuLi/hexane (1.1 equiv.) was added. The mixture was allowed to warm to about 0 °C and aged for about 2 hours, then warmed to room temperature overnight. The solution was then added to a solution of diethyl oxalate (4.0 equiv.) in THF (10 mL) at about -20 °C. The mixture was allowed to warm to about room temperature and aged for approximately 2 hours, then cooled to about 0 °C and quenched via addition of saturated NH4CI (30 mL). This mixture was extracted with EtOAc, and the organic phase was washed with brine and dried over MgSCk
Concentration afforded Compound P-1.
Alternate Preparation Compound P-1:
[0408] 2-Bromoanisole (1.0 equiv.) in THF (63 mL) was cooled to about -65 °C and 2.5M ft-BuLi/hexanes (1.0 equiv) was added. After aging for approximately 1 h, diethyl oxalate (4.0 equiv.) was charged, and the reaction mixture was allowed to warm to about room temperature. After approximately 1 h at about room temperature, the reaction mixture was cooled to about 0 °C, quenched by addition of saturated NH4CI (50 mL), and diluted with EtOAc. The aqueous phase was separated and was extracted with EtOAc. The combined organic phases were washed with brine and dried over MgS04. Concentration under high vacuum afforded a product that was passed through a plug of silica gel to afford Compound P-1.
Step (b): Hydrolysis of Compound P-1 and salt conversion to Compound O-l:
P-1 0-1
[0409] The resulting solution of ketoester, compound P-1, in THF (about 1.0 equiv.) was cooled over an ice bath and 2N NaOH (1.36 equiv.) was added. The reaction was agitated at about 0 °C and after reaction completion, the reaction was then acidified by addition of 6N HC1 (57 mL) to about pH<l and extracted with EtOAc (500 mL). The organic phase was washed with brine (200 mL). The organic phase was concentrated and then solvent exchanged to EtOAc. The resulting solution was cooled to about 0 °C and solid KOlBu (1.0 equiv.). The slurry was agitated for approximately 4 h and the solids were filtered, rinsed with EtOAc, and dried overnight at about 60 °C under vacuum to afford Compound O-l . ¾ NMR (400 MHz, DMSO-d6): 5 7.61 (d, J= 7.6 Hz, 1H), 7.49 – 7.41 (m, 1H), 7.04 (d, J= 8.4 Hz 1H), 6.96 (t, J = 7.4 Hz, 1H), 3.73 (s, 3H).
Step (c): Reduction of Compound O-l to Compound N-1:
0-1 N-1
[0410] To triethylamine (3.6 equiv.) precooled to about 0 °C, was added formic acid (9.0 equiv.) over about 30 min while maintaining a temperature less than about 30 °C. Solid RuCl (i?,i?)-Ts-DENEB catalyst (0.07 mol%) followed by ketoacid potassium salt (1.0 equiv.) were then charged to the mixture of triethylarnine/forrnic acid. The resulting slurry was warmed to about 50 °C and was stirred under nitrogen until the reaction was complete. The reaction was cooled over an ice bath and quenched by the addition of water (76 mL) followed by 10N NaOH (128 mL) to pH>13. Water (30 mL) and iPrAc (130 mL) were added and the organic layer was separated, and the aqueous phase was extracted with iPrAc (2 χ 130 mL). The aqueous phase was cooled and was acidified with concentrated HC1. This was extracted with iPrAc several times and the combined organic extract was concentrated and solvent exchanged to toluene, filtered hot, and then cooled to about 30 °C over approximately 2 h, aged for approximately 1 h, then filtered to afford solids that were then slurry-rinsed with toluene (50 mL) at room temperature and filtered. The wet cake was dried to afford Compound N-1. ¾ NMR (400 MHz, CDCh): δ 7.44 (d, J = 7.6 Hz, 1H), 7.40 – 7.36 (m, 1H), 7.06 (t, J = 7.6 Hz 1H), 6.98 (d, J = 8.4 Hz, 1H), 5.41 (s, 1H), 3.94 (s, 3H).
Step (d): Spiroketalization to afford Compound L-1:
N-1 L-1
[0411] Compound N-1 (1.0 equiv.), tetrahydropyran-4-one (compound M, 1.1 equiv.), and MTBE (30 mL) were sequentially charged and cooled to about 0 °C. Boron trifluoride THF complex (1.4 equiv.) was added over about 10 mins. After reaction completion, the reaction was slowly quenched with a pre-mixed solution of sodium bicarbonate (3.66 g) and water (40 mL). The solution was warmed to about 20 °C and diluted with toluene (40 mL) and stirred until dissolved. Agitation was stopped and the aqueous layer removed. The organic layer was washed with water (20 mL) and removed. The organic layer was collected and reactor rinsed forward with toluene (4 mL) to yield Compound L-1. ¾ NMR (400 MHz, CDCh): δ 7.42 – 7.38 (m, 1H), 7.32 (dd, J = 7.5, 1.5 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 5.52 (s, 1H), 3.97 – 3.79 (m, 7H), 2.18 – 1.97 (m, 4H).
Step (e): Reduction of Compound L-1 to Compound K-l :
L-1 K-1
[0412] A stock solution of spiroketal, compound L-1, in MeTHF/MTBE (1.0 equiv.) was charged to a reactor. The solution was then distilled to about 4 volumes. MeTHF (187 mL) was charged, and distilled down to about 5 volumes. The solution was cooled to about 20 °C. DCM (90 mL) was charged and the solution was cooled to about 10 °C and tert-butyl magnesium chloride (2 M in diethyl ether) (5.0 equiv.) was added over approximately 45 mins. Following addition, the contents were cooled to about 7 °C and aged overnight at about 10 °C, then to about 0 °C. A premixed solution of HC1 (45 mL) and water (126 mL) was then slowly added. The aqueous bottom layer was drained and the aqueous layer extracted with MeTHF (93 mL). The combined organic layers were washed with water (37 mL) and the remaining organic layer was distilled down to about 4 volumes. Isopropyl acetate (181 mL) was charged and the solution reduced to about 5 volumes. The reaction was cooled to about 72 °C and heptanes (58 mL) was charged and the solution was held for about 1 hour before cooling to about 0 °C over approximately 5 hours. The slurry was agitated at about 0 °C for >12 h and then filtered, rinsed with an isopropyl acetate (9 mL) and heptanes (18 mL) mixture, followed by water (54 mL). The solids were dried to yield compound K-l. ¾ NMR (400 MHz, CDCh): δ 8.49 (br. s, 1 H), 7.42 – 7.29 (m, 2H), 6.98 (t, J= 7.4 Hz, 1H), 6.92 (d, 8.3 Hz, 1H), 5.43 (s, 1H), 3.96 (dt, J = 11.5, 4.3 Hz, 1H), 3.89 (dt, J = 11.5, 4.3 Hz, 1H), 3.85 (s, 3H), 3.67 – 3.58 (m, 1H), 3.47 – 3.30 (m, 2H), 2.03 – 1.93 (m, 1H), 1.84 – 1.75 (m, 1H), 1.75 – 1.56 (m, 2H).
Step (f): Reduction of Com ound K-l to Compound J-1:
J-1
K-1
[0413] A solution of acid, compound K-l (1.0 equiv.), in THF (90 mL) was cooled to about 0 °C and NaBH4 (1.2 equiv.) was added followed by BF3 THF complex (1.5 equiv.). The solution was warmed to about 20 °C and agitated until the reaction was deemed complete. Upon completion, MeOH (24 mL) was added to the reaction mixture after adjusting the temperature to about 5 °C, and was stirred until the gas evolution ceased. EtOAc (102 mL) was charged followed by saturated NLUClaq solution (87 mL). The agitation was stopped and the aqueous layer was removed. The organic layer was distilled down to about 3 volumes under vacuum, and then heptane (46 mL) was charged. The resulting mixture was cooled to about 0 °C and agitated at this temperature for approximately 4 h before being filtered and rinsed with heptane (3 mL). The resulting solids were dried to yield compound J-1. ¾ NMR (400 MHz, CDCh): δ 7.42 (d, J = 7.2 Hz, 1H), 7.27 (m, 1H), 6.98 (m, 1H), 6.87 (d, J = 8.4 Hz, 1H), 5.06 (dd, J = 8.4, 2.8 Hz, 1H), 3.93 (m, 2H), 3.82 (s, 3H), 3.67 (m, 1H), 3.55 – 3.46 (m, 2H), 3.41 – 3.32 (m, 2H), 2.27 (d, J = 8.0 Hz, 1H), 2.01 (m, 1H), 1.80 – 1.70 (m, 1H), 1.65 (m, 2H).
Step (g): Alternate Direct Reduction of Compound L-1 to Compound J-1:
L-1 J-1
[0414] To a solution of ketal, compound L-1 (1 equiv.), in diglyme (0.7 mL) was added NaBH4 (3.6 equiv.) followed by BF3 THF complex (4.5 equiv.). Reaction mixture was agitated for about 18 hours and was quenched by dropwise addition of MeOH (1 mL) followed by saturated Ν¾(¾ solution (1 mL). EtOAc (2 mL) was added, shaken well and the aqueous layer was removed. Organic solvent was removed under reduced pressure to obtain the crude compound J-1.
Example 7: Alternate Synthesis to Compound N-1
Step (a): Addition of hydrogen cyanide to ortho-anisaldehyde, compound U-1, to form compound T-1
[0415] To an Eppendorf tube was added ort/ro-anisaldehyde, compound U-1 (1.0 equiv), followed by 0.4 M sodium acetate buffer pH 5 (0.25 mL) and fert-butyl methyl ether (0.75 mL). The mixture was shaken using a thermomixer at about 30 °C and about 1200 rpm to ensure
complete dissolution of the aldehyde. Once this was complete acetone cyanohydrin (1.15 equiv) is added to the reaction mixture followed by hydroxynitrilase enzyme (2 mg). The Eppendorf tube was shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzyme before being cooled to about 30 °C. The Eppendorf tube was then centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme from the organic layer. The organic layer was removed and concentrated to dryness to give crude compound T-l . ¾ NMR (400 MHz, CDCh): δ 7.45 – 7.39 (m, 2H), 7.04 – 6.96 (m, 2H), 5.63 (s 1H), 3.94 (s, 3H), 3.75 (br, 1H).
Step (b): Hydrolysis of c
T-1 N-1
[0416] Before starting the reaction the following stock solutions were prepared: A solution of the crude cyanohydrin (compound T-l) in DMSO (about 100 mg/mL); a solution of 50 mM potassium phosphate (pH 7) containing 2 mM dithiothreitol (DTT); and 1 mM ethylenediamine tetraacetic acid (EDTA). To an Eppendorf tube was added nitrilase enzyme (4 mg) followed by 1.1 mL of the reaction buffer solution and 0.05 mL of the solution containing the crude cyanohydrin (about 10 mg). The Eppendorf tube was shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzyme before being cooled to about 30 °C once more. The Eppendorf tube was centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme and then separate it from the supernatant. The supernatant was either sampled directly for reverse phase UPLC or extracted with DCM for normal phase HPLC. In the case of DCM extraction, after separating the layers the organic layer was concentrated to dryness before the appropriate diluent was added for normal phase HPLC. UPLC analysis showed a peak with retention time identical to a reference standard of compound N-1.
Example 8: Alternate S nthesis to Compound N-1
P-1 V-1 N-1
Step (a): Reduction of Compound P-1 to form 2 ‘-methoxy-ethyl mandelate, Compound V-1:
P-1 V-1
[0417] The following stock solutions were made prior to the start of the reaction: a solution of starting material in DMSO (about 100 mg/ mL), NADP+ or NAD+ in 0.1M phosphate buffer (as appropriate) (2 mg/mL), glucose dehydrogenase in 0.1 M phosphate buffer (4 mg/mL), and glucose in 0.1 M phosphate buffer (20 mg/mL). To an Eppendorf tube is charged the ketoreductase enzyme (2 mg) followed by 0.25 mL of buffer solution containing NAD(P)+, 0.25 mL of buffer solution containing glucose dehydrogenase (GDH) and 0.5 mL of buffer solution containing glucose. Finally, 0.05 mL of the stock solution containing the starting material, compound P-1 in DMSO is added. The Eppendorf tube was then shaken in a thermomixer at about 30 °C and about 1200 rpm overnight. The Eppendorf tube was then heated to about 60 °C at about 1400 rpm for about 15 mins in order to denature the enzymes before being cooled to about 30 °C. The Eppendorf tube was then centrifuged at about 13,400 rpm for about 15 mins in order to pellet the denatured enzyme and the supernatant removed. This was either sampled directly for reverse phase UPLC or extracted with DCM for normal phase HPLC. In the case of DCM extraction after separating the layers the organic layer was concentrated to dryness before the appropriate diluent was added for normal phase HPLC. UPLC analysis showed a peak with retention time identical to a reference standard of the product material.
Step (b) Hydrolysis of 2 ‘-methoxy-ethyl mandelate, compound V-1, to provide compound N-1:
V-1 N-1
[0418] A solution of 2′ -methoxy-ethyl mandelate (1.0 equiv.) in EtOH (30 mL) was cooled to about 0 °C and 1.25 M NaOH (30 mL) was slowly added. Upon reaction completion, the reaction was adjusted to about pH 1 with 1M HC1 (40 mL). The mixture was extracted three times with ethyl acetate (30 mL) and the combined organics were washed with a brine solution (25 mL). The combined organic layers were dried over sodium sulfate, filtered, and the solvent removed under vacuum to provide the product. NMR data reported as above.
CLIP
https://cen.acs.org/articles/94/i39/silent-liver-disease-epidemic.html

/////// ND 630, NDI 010976, ND-630, NDI-010976, NIMBUS, GILEAD, 1434635-54-7, PHASE 2
FIRSOCOSTAT, ND 630, GS-0976, NDI-010976, FAST TRACK, CS-6509
COc1ccccc1[C@H](CN2C(=O)N(C(=O)c3c(C)c(sc23)c4occn4)C(C)(C)C(=O)O)OC5CCOCC5
O=C(O)C(C)(C)N4C(=O)c1c(C)c(sc1N(C[C@H](OC2CCOCC2)c3ccccc3OC)C4=O)c5ncco5
WHO defines Requirements on Zones E and F
DRUG REGULATORY AFFAIRS INTERNATIONAL

In May, the WHO published a draft guideline which describes the recommendations for ventilation systems used in the manufacture of non-sterile dosage forms. It also contains for the first time a definition for microbial requirements with regard to the zones E and F. Read more about the ventilation sytems recommendations.

In May 2016, the WHO published a draft guideline which describes the recommendations for ventilation systems used in the manufacture of non-sterile dosage forms. From a technical point of view, the guideline is very interesting and includes a detail which may be overlooked: it contains – as first international GMP guideline – a proposal for the definition of microbiological requirements concerning the zones E and F. So far, the approach to extend the zoning via the zones A-D defined in Annex 1 to the zones E and F and thus define microbial limits had only been available in an Aide Memoire…
View original post 84 more words
Ponesimod

Ponesimod
Phase III
MW 460.97, C23 H25 Cl N2 O4 S
A sphingosine-1-phosphate receptor 1 (S1P1) agonist potentially for the treatment of multiple sclerosis.
(Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one
- (2Z,5Z)-5-[[3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]phenyl]methylene]-3-(2-methylphenyl)-2-(propylimino)-4-thiazolidinone
- 5-[3-Chloro-4-[((2R)-2,3-dihydroxypropyl)oxy]benz-(Z)-ylidene]-2-((Z)-propylimino)-3-(o-tolyl)thiazolidin-4-one
- ACT 128800
ACT-128800; RG-3477; R-3477
CAS No. 854107-55-4
update 18/3/21 FDA APPROVEDAS PONVORY
SYNTHESIS
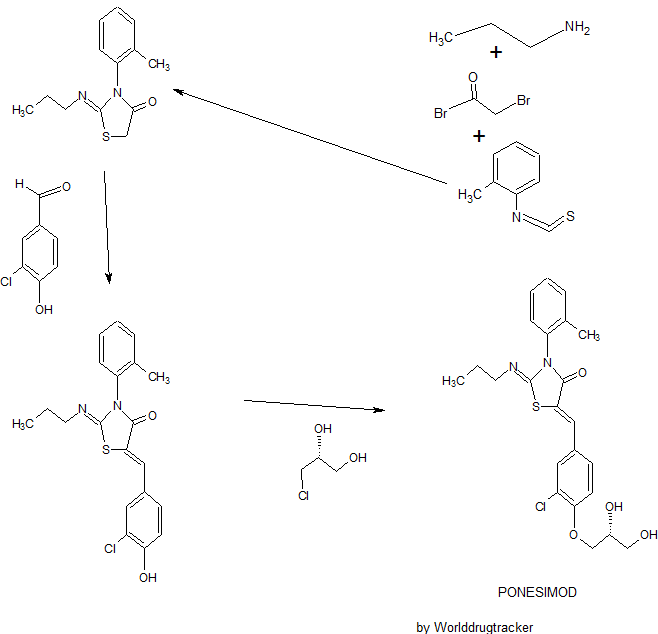
Ponesimod

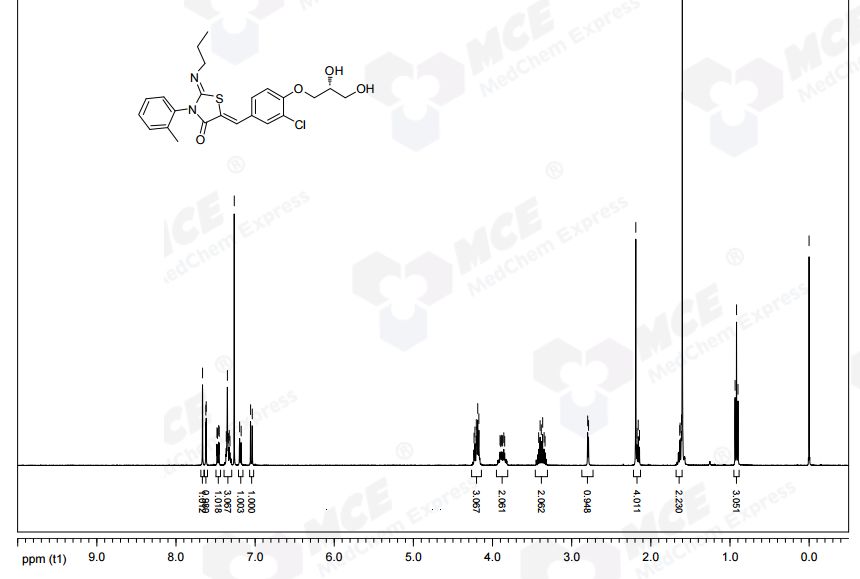
NMR CDCL3 FROM NET
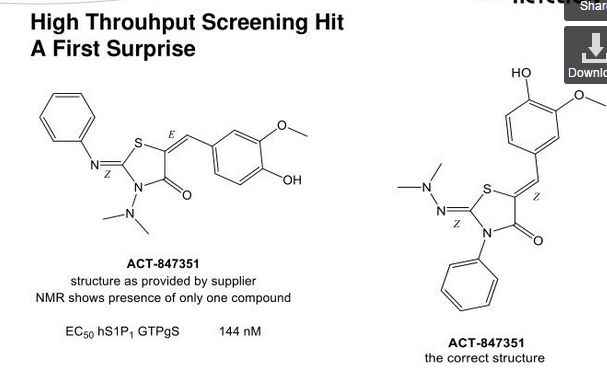
Ponesimod (INN, codenamed ACT-128800) is an experimental drug for the treatment of multiple sclerosis (MS) and psoriasis. It is being developed by Actelion.
The first oral treatment for relapsing multiple sclerosis, the nonselective sphingosine-1-phosphate receptor (S1PR) modulator fingolimod, led to identification of a pivotal role of sphingosine-1-phosphate and one of its five known receptors, S1P1R, in regulation of lymphocyte trafficking in multiple sclerosis. Modulation of S1P3R, initially thought to cause some of fingolimod’s side effects, prompted the search for novel compounds with high selectivity for S1P1R. Ponesimod is an orally active, selective S1P1R modulator that causes dose-dependent sequestration of lymphocytes in lymphoid organs. In contrast to the long half-life/slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation and its pharmacological effects are rapidly reversible. Clinical data in multiple sclerosis have shown a dose-dependent therapeutic effect of ponesimod and defined 20 mg as a daily dose with desired efficacy, and acceptable safety and tolerability. Phase II clinical data have also shown therapeutic efficacy of ponesimod in psoriasis. These findings have increased our understanding of psoriasis pathogenesis and suggest clinical utility of S1P1R modulation for treatment of various immune-mediated disorders. A gradual dose titration regimen was found to minimize the cardiac effects associated with initiation of ponesimod treatment. Selectivity for S1P1R, rapid onset and reversibility of pharmacological effects, and an optimized titration regimen differentiate ponesimod from fingolimod, and may lead to better safety and tolerability. Ponesimod is currently in phase III clinical development to assess efficacy and safety in relapsing multiple sclerosis. A phase II study is also ongoing to investigate the potential utility of ponesimod in chronic graft versus host disease.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Biology and pharmacology of sphingosine-1-phosphate receptor 1
The past decades have witnessed major advances in the treatment of autoimmune and chronic inflammatory diseases. A plethora of novel therapies targeting specific molecules involved in the inflammatory or immune system activation cascades have become available. These have significantly increased our understanding of disease pathogenesis and improved the management of immune-mediated disorders. However, most of the targeted therapies are biological drugs which need to be injected, are eliminated slowly (e.g. over several weeks) and can lose efficacy or tolerability due to their potential immunogenicity. In an attempt to overcome these hurdles, pharmaceutical research has made considerable efforts to develop novel oral targeted therapies for autoimmune and chronic inflammatory diseases.
Sphingosine-1-phosphate receptor 1 (S1P1R) is one of five known G protein-coupled receptors with nanomolar affinity for the lysophospholipid sphingosine-1-phosphate (S1P), which is generated through physiologic metabolism of the cell membrane constituent sphingomyelin by all cells [Brinkmann, 2007]. S1P receptors, including S1P1R, are widely expressed in many tissues [Chun et al. 2010]. S1P1R expression on lymphocytes controls their egress from thymus and secondary lymphoid organs [Cyster and Schwab, 2012]. Lymphocyte egress requires a gradient of S1P concentration, which is established by a high S1P concentration in blood and lymph compared with a low concentration in the interstitial fluid of lymphoid organs [Grigorova et al. 2009].
Synthetic S1P1 receptor modulators disrupt the interaction of the physiologic S1P ligand with S1P1R by promoting initial activation followed by sustained internalization and desensitization of S1P1R [Hla and Brinkmann, 2011; Pinschewer et al. 2011]. Experiments conducted in animal models of transplant rejection, multiple sclerosis, lupus erythematosus, arthritis and inflammatory bowel disease with the first-generation, nonselective S1P receptor modulator, fingolimod, have demonstrated the potential efficacy of this mode of action across several immune-mediated chronic inflammatory conditions [Brinkmann, 2007]. Fingolimod is a structural analog of sphingosine that is phosphorylated in the body by a sphingosine kinase to generate the bioactive form of the drug, fingolimod phosphate, which binds to multiple S1P receptors [Brinkmann, 2007]. Clinical trials in multiple sclerosis (MS) have confirmed the efficacy of fingolimod in relapsing MS, but not in primary progressive disease, and led to the approval of the first oral medication for the treatment of relapsing forms of MS in 2010 [Kappos et al. 2010].
The mechanism of action of fingolimod has increased our understanding of MS pathogenesis. T and B cells, but not natural killer (NK) cells, express functional S1P1R and are affected by fingolimod [Cyster and Schwab, 2012]. Furthermore, S1P1R is differentially expressed and regulated in functionally distinct subsets of lymphocytes and fingolimod has been shown to predominantly affect naïve T cells and central memory T cells (TCM) while sparing effector memory T cells (TEM), and terminally differentiated effector T cells (TE) in patients with relapsing MS [Mehling et al. 2008, 2011]. This has raised the possibility that, at least in MS, retention of TCM cells, which include pro-inflammatory T helper 17 (Th17) cells, by fingolimod may prevent their accumulation in the cerebrospinal fluid (CSF) and subsequent differentiation to TE cells in the central nervous system (CNS) [Hla and Brinkmann, 2011]. The effects of S1P1R modulation on B cells are less well defined. Recent data from patients with relapsing MS have shown predominant reduction of memory B cells and recently activated memory B cells (CD38int-high) in peripheral blood after treatment with fingolimod [Claes et al. 2014; Nakamura et al. 2014]. As memory B cells are implicated in the pathogenesis of MS and other autoimmune diseases, these observations suggest another potential mechanism underlying the therapeutic effects of S1P1R modulators.
Astrocytes, microglia, oligodendrocytes and neurons express various S1P receptors including S1P1R, S1P3R and S1P5R. Fingolimod has been shown to penetrate the CNS tissues and in vitro studies have shown activation of astrocytes and oligodendrocytes by fingolimod [Foster et al. 2007]. Conditional deletion of S1P1R on neural cells in mice reduced the severity of experimental autoimmune encephalomyelitis (EAE) and reductions in the clinical scores were paralleled by decreased demyelination, axonal loss and astrogliosis [Choi et al. 2011]. Unfortunately, there was no beneficial effect in a recently completed, large study of fingolimod in patients with primary progressive MS [Lublin et al. 2015], suggesting that the direct effect on CNS cells alone may not be sufficient. Taken together, these data suggest the possibility of a direct beneficial effect of S1P1R modulation in the brain of patients with relapsing MS [Dev et al. 2008]; however, its contribution to efficacy relative to the immunological effects remains unclear.
Initial studies in rodents suggested that modulation of S1P3R on cardiac myocytes by fingolimod was associated with a reduction of heart rate (HR) by activation of G-protein-coupled inwardly rectifying potassium channels (GIRK) that regulate pacemaker frequency, and the shape and duration of action potentials [Koyrakh et al. 2005; Camm et al. 2014]. Modulation of S1P2R and S1P3R on myofibroblasts by fingolimod was also shown to stimulate extracellular matrix synthesis [Sobel et al. 2013]. Modulation of these receptors on vascular smooth muscle cells appeared to be associated with vasoconstriction, leading to the slight increase in blood pressure observed with fingolimod treatment [Salomone et al. 2003; Watterson et al. 2005; Hu et al. 2006; Lorenz et al. 2007; Kappos et al. 2010]. These observations raised the possibility that some side effects associated with fingolimod treatment could be avoided by more selective S1P1R modulators, thus triggering the search for novel compounds.
Currently, there are several selective S1P1R modulators in clinical development [Gonzalez-Cabrera et al.2014; Subei and Cohen, 2015]. Here we review data and the development status of ponesimod, a selective S1P1R modulator developed by Actelion Pharmaceuticals Ltd.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Ponesimod, a selective, rapidly reversible, orally active, sphingosine-1-phosphate receptor modulator
Ponesimod (ACT-128800 (Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) is a selective, rapidly reversible, orally active, S1P1R modulator. Ponesimod emerged from the discovery of a novel class of S1P1R agonists based on the 2-imino-thiazolidin-4-one scaffold (Figure 1) [Bolli et al. 2010]. Ponesimod activates S1P1R with high potency [half maximal effective concentration (EC50) of 5.7 nM] and selectivity. Relative to the potency of S1P, the potency of ponesimod is 4.4 higher for S1P1R and 150-fold lower for S1P3R, resulting in an approximately 650-fold higher S1P1R selectivity compared with the natural ligand.

Chemical structure of ponesimod, C23H25N2O4CIS (molecular weight 460.98).http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Clinical trials
In a 2009–2011 Phase II clinical trial including 464 MS patients, ponesimod treatment resulted in fewer new active brain lesions thanplacebo, measured during the course of 24 weeks.[3][4]
In a 2010–2012 Phase II clinical trial including 326 patients with psoriasis, 46 or 48% of patients (depending on dosage) had a reduction of at least 75% Psoriasis Area and Severity Index (PASI) score compared to placebo in 16 weeks.[3][5]
SEE https://clinicaltrials.gov/ct2/show/NCT02425644
Adverse effects
Common adverse effects in studies were temporary bradycardia (slow heartbeat), usually at the beginning of the treatment,dyspnoea (breathing difficulties), and increased liver enzymes (without symptoms). No significant increase of infections was observed under ponesimod therapy.[3] QT prolongation is detectable but was considered to be too low to be of clinical importance in a study.[6]
Mechanism of action
Like fingolimod, which is already approved for the treatment of MS, ponesimod blocks the sphingosine-1-phosphate receptor. This mechanism prevents lymphocytes (a type of white blood cells) from leaving lymph nodes.[3] Ponesimod is selective for subtype 1 of this receptor, S1P1.[7]
PAPER
Bolli, Martin H.; Journal of Medicinal Chemistry 2010, V53(10), P4198-4211 CAPLUS
2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1Receptor Agonists
Drug Discovery Chemistry, Actelion Pharmaceuticals Ltd., Gewerbestrasse 16, CH-4123 Allschwil, Switzerland
J. Med. Chem., 2010, 53 (10), pp 4198–4211
DOI: 10.1021/jm100181s
Publication Date (Web): May 06, 2010
Copyright © 2010 American Chemical Society
*To whom correspondence should be addressed. Phone: + 41 61 565 65 70. Fax: + 41 61 565 65 00. E-mail:martin.bolli@actelion.com.

Sphingosine-1-phosphate (S1P) is a widespread lysophospholipid which displays a wealth of biological effects. Extracellular S1P conveys its activity through five specific G-protein coupled receptors numbered S1P1 through S1P5. Agonists of the S1P1 receptor block the egress of T-lymphocytes from thymus and lymphoid organs and hold promise for the oral treatment of autoimmune disorders. Here, we report on the discovery and detailed structure−activity relationships of a novel class of S1P1 receptor agonists based on the 2-imino-thiazolidin-4-one scaffold. Compound 8bo (ACT-128800) emerged from this series and is a potent, selective, and orally active S1P1 receptor agonist selected for clinical development. In the rat, maximal reduction of circulating lymphocytes was reached at a dose of 3 mg/kg. The duration of lymphocyte sequestration was dose dependent. At a dose of 100 mg/kg, the effect on lymphocyte counts was fully reversible within less than 36 h. Pharmacokinetic investigation of8bo in beagle dogs suggests that the compound is suitable for once daily dosing in humans.
(Z,Z)-5-[3-Chloro-4-((2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolyl-thiazolidin-4-one (8bo)
…………..DELETED…………… column chromatography on silica gel eluting with heptane:ethyl acetate 1:4 to give the title compound (1.34 g, 37%) as a pale-yellow foam.
1H NMR (CDCl3): δ 0.94 (t, J = 7.3 Hz, 3 H), 1.58−1.70 (m, 2 H), 2.21 (s, 3 H), 3.32−3.48 (m, 2 H), 3.82−3.95 (m, 3 H), 4.12−4.27 (m, 4 H), 7.07 (d, J = 8.8 Hz, 1 H), 7.21 (d, J = 7.0 Hz, 1 H), 7.31−7.39 (m, 3 H), 7.49 (dd, J = 8.5, 2.0 Hz, 1 H), 7.64 (d, J= 2.0 Hz, 1 H), 7.69 (s, 1 H).
13C NMR (CDCl3): δ 11.83, 17.68, 23.74, 55.42, 63.46, 69.85, 70.78, 133.48, 120.75, 123.71, 127.05, 128.25, 128.60, 129.43, 130.06, 131.13, 131.50, 134.42, 136.19, 146.98, 154.75, 166.12. LC-MS (ES+): tR 0.96 min. m/z: 461 (M + H).
HPLC (ChiralPak AD-H, 4.6 mm × 250 mm, 0.8 mL/min, 70% hexane in ethanol): tR 11.8 min. Anal. (C23H25N2O4SCl): C, H, N, O, S, Cl.
PATENT
WO 2014027330
https://www.google.com/patents/WO2014027330A1?cl=3Den
The present invention relates inter alia to a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (hereinafter also referred to as the “COMPOUND” or “compound (2)”), especially in crystalline form C which form is described in WO 2010/046835. The preparation of COMPOUND and its activity as immunosuppressive agent is described in WO 2005/054215. Furthermore, WO 2008/062376 describes a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one which can be used as an intermediate in the preparation of COMPOUND.
Example 1 a) below describes such a process of preparing (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one according to WO 2008/062376. According to WO 2008/062376 the obtained (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one can then be transformed into COMPOUND by using standard methods for the alkylation of phenols. Such an alkylation is described in Example 1 b) below. Unfortunately, this process leads to the impurity (2Z,5Z)-5-(3-chloro-4-((1 ,3-dihydroxypropan-2-yl)oxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one which is present in about 2% w/w in the crude product (see Table 1 ) and up to 6 recrystallisations are necessary in order to get this impurity below 0.4% w/w (see Tables 1 and 2) which is the specified limit based on its toxicological qualification.


the obtained (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one to form (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (2):

.
The reaction of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one can be performed under conditions which are typical for a Knoevenagel condensation. Such conditions are described in the literature for example in Jones, G., Knoevenagel Condensation in Organic Reaction, Wiley: New York, 1967, Vol. 15, p 204; or Prout, F. S., Abdel-Latif, A. A., Kamal, M. R., J. Chem. Eng. Data, 2012, 57, 1881-1886.
2-[(Z)-Propylimino]-3-o-tolyl-thiazolidin-4-one can be prepared as described in WO 2008/062376, preferably without the isolation and/or purification of intermediates such as the thiourea intermediate that occurs after reacting o-tolyl-iso-thiocyanate with n-propylamine. Preferably 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one obtained according to WO 2008/062376 is also not isolated and/or purified before performing the Knoevenagel condensation, i.e. before reacting 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one with (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ), i.e. in a preferred embodiment compound (2) is prepared in a one-pot procedure analogous to that described in WO 2008/062376.
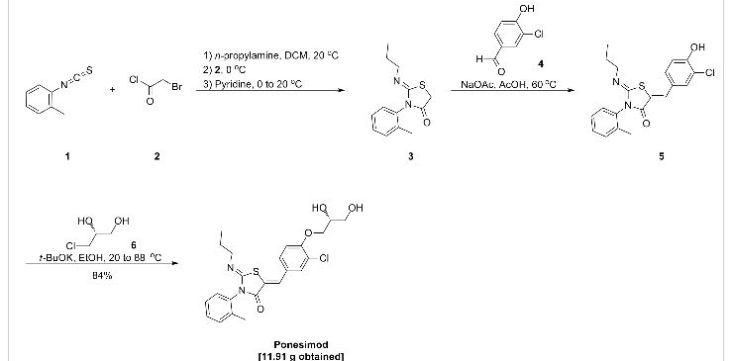
Example 1 : (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one

a) Preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one:

Acetic acid solution: To acetic acid (149.2 mL) are added sodium acetate (1 1 .1 1 g, 2.00 eq.) and 3-chloro-4-hydroxybenzaldehyde (10.60 g, 1.00 eq.) at 20 °C. The mixture is stirred at 20 °C until complete dissolution (2 to 3 h).
n-Propylamine (4.04 g, 1.00 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 40 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (13.53 g, 1.00 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). 70 mL of dichloromethane are distilled off under atmospheric pressure and jacket temperature of 60 °C. The temperature is adjusted to 42 °C and the acetic acid solution is added to the reaction mixture. The resulting solution is heated to 58 °C and stirred at this temperature for 15 h before IPC (conversion specification≥ 95 %). 25 mL of solvents are distilled off under vacuum 900 – 500 mbars and jacket temperature of 80 °C. The temperature is adjusted to 60 °C and water (80.1 mL) is added to the reaction mixture over 1 h. The resulting yellow suspension is stirred at 60 °C for 30 min. The suspension is cooled to 20 °C over 1 h and stirred at this temperature for 30 min.
The product is filtered and washed with a mixture of acetic acid (30 mL) and water (16 mL) and with water (50 mL) at 20 °C. The product is dried under vacuum at 50 °C for 40 h to afford a pale yellow solid; yield 25.93 g (78 %).
b) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
To a suspension of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one (10.00 g, 1.00 eq.) in ethanol (47.2 mL) is added (R)-3-chloro-1 ,2-
propanediol (3.37 g, 1.18 eq.) at 20 °C. Potassium tert-butoxide (3.39 g, 1.13 eq.) is added in portions at 20 °C. The resulting fine suspension is stirred at 20 °C for 25 min before being heated to reflux (88 °C). The reaction mixture is stirred at this temperature for 24 h before IPC (conversion specification≥ 96.0 %). After cooling down to 60 °C, acetonitrile (28.6 mL) and water (74.9 mL) are added. The resulting clear solution is cooled from 60 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.010 g, 0.001 eq.; crystalline form C can be prepared as described in WO 2010/046835) are added at 50 °C. The suspension is heated from 0 °C to 50 °C, cooled to 0 °C over 6 h and stirred at this temperature for 12 h.
The product is filtered and washed with a mixture of acetonitrile (23.4 mL) and water (23.4 mL) at 0 °C. The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield 1 1.91 g (84 %).
c) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
Recrystallisation I: The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in acetonitrile (30 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 12.8 mL).
Recrystallisation II: The wet product is dissolved in acetonitrile (27.0 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 1 1.3 mL).
Recrystallisation III: The wet product is dissolved in acetonitrile (24.3 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4- one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 10.1 mL).
Recrystallisation IV: The wet product is dissolved in acetonitrile (21.9 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 9.1 mL).
Recrystallisation V: The wet product is dissolved in acetonitrile (19.7 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 8.2 mL).
Recrystallisation VI: The wet product is dissolved in acetonitrile (23.9 mL) at 70 °C. Water (20 mL) is added at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h.
During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2- (propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed twice with a mixture of acetonitrile (4.5 mL) and water (4.5 mL) at -10 °C.
The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield: 7.0 g (70 %).
Example 2: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde

Potassium tert-butoxide (1 18 g, 1.20 eq.) is added to n-propanol (963 mL) followed by 3-chloro-4-hydroxybenzaldehyde (137 g, 1.00 eq.). To the mixture is added (R)-3-chloro-1 ,2-propanediol (126 g, 1.30 eq.). The suspension is heated to 90 °C and stirred at this temperature for 17 h. Solvent (500 mL) is distilled off at 120 °C external temperature and reduced pressure. Water is added (1.1 L) and solvent (500 mL) is removed by distillation. The turbid solution is cooled to 20 °C. After stirring for one hour a white suspension is obtained. Water (500 mL) is added and the suspension is cooled to 10 °C. The suspension is filtered and the resulting filter cake is washed with water (500 mL). The product is dried at 50 °C and reduced pressure to yield 149 g of a white solid (73%), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 3: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde
Potassium tert-butoxide (8.60 g, 1.20 eq.) is added to n-propanol (70 mL) below 15 °C, the temperature is allowed to rise. After the addition the temperature is corrected again to below 15 °C before addition of 3-chloro-4-hydroxybenzaldehyde (10 g, 1 .00 eq.). The suspension is heated to 40 °C and stirred for 30 min. (R)-3-Chloro-1 ,2-propanediol (9.18 g, 1.30 eq.) is added at 40 °C. The resulting suspension is heated to 60 °C and stirred at this temperature for 15 h then heated to 94 °C till meeting the IPC-specification (specification conversion≥ 90.0 %). The mixture is cooled to 30 °C and n-propanol is partially distilled off (-50 mL are distilled off) under reduced pressure and a maximum temperature of 50 °C, the jacket temperature is not allowed to raise above 60 °C.
Water (81 mL) is added and a second distillation is performed under the same conditions (24 mL are distilled off). The mixture is heated till homogeneous (maximum 54 °C) and then cooled to 24 °C. At 24 °C the mixture is seeded with crystalline (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of form A (0.013 g, 0.00085 eq.). How to obtain the crystalline seeds is described in Examples 2 and 5. The reaction mixture is cooled to 0 °C over 7.5 h.
The product is filtered and washed with water (2 x 35 mL) and once with methyl tert-butyl ether (20 mL) at 5 °C. The product is dried under vacuum at 40 °C for 20 h to afford an off-white solid; yield: 10.6 g (72 %), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 4: (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)- 3-(o-tolyl)thiazolidin-4-one
a) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
n-Propylamine (5.23 g, 1.32 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 15 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (14.88 g, 1.10 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). Dichloromethane is partially distilled off (66 mL are distilled off) under atmospheric pressure and jacket temperature of 60 °C. Ethanol (1 1 1.4 mL), sodium acetate (12.75 g, 2.30 eq.) and (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde from Example 3 (14.38 g, 0.93 eq.) are added. The remaining dichloromethane and a part of ethanol are distilled off (49.50 mL are distilled off) under atmospheric pressure and jacket temperature up to 85 °C. The reaction mixture (orange suspension) is stirred for 3 – 5 h under reflux (78 °C) before IPC (conversion specification≥ 97.0 %).
Water (88.83 mL) is added and the temperature adjusted to 40 °C before seeding with micronized (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.075 g, 0.0024 eq.). The reaction mixture is cooled to 0 °C over 5 h, heated up to 40 °C, cooled to 0 °C over 6 h and stirred at this temperature for 2 h.
The product is filtered and washed with a 1 :1 ethanohwater mixture (2 x 48 mL) at 0 °C. The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 24.71 g (86 %).
b) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in ethanol (40 mL) at 70 °C. The temperature is adjusted at 50 °C for seeding with micronised (2Z,5Z)-5-(3-chloro-4-((R)-2,3- dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.016 g, 0.0016 eq.). The reaction mixture is cooled from 50 °C to 0 °C over 4 h, heated up to 50 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h.
The product is filtered and washed with ethanol at 0 °C (2 x 12.8 mL). The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 9.2 g (92 %).
Example 5: Preparation of crystalline seeds of (R)-3-chloro-4-(2,3-dihydroxypropoxy)- benzaldehyde
10 mg of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of at least 99.5% purity by 1 H-NMR assay is dissolved in a 4 mL vial by adding 1 mL of pure ethanol (puriss p. a.). The solvent is allowed to evaporate through a small hole in the cap (approx. 2 mm of diameter) of the vial until complete dryness. The white solid residue is crystalline (R)-3-chloro-4-(2,3- dihydroxypropoxy)-benzaldehyde in crystalline form A. Alternatively, methanol or methylisobutylketone (both in puriss p. a. quality) is used. This procedure is repeated until sufficient seeds are made available.

PATENT
WO 2005054215
SEE https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005054215
| WO2005054215A1 | Nov 16, 2004 | Jun 16, 2005 | Actelion Pharmaceuticals Ltd | 5-(benz- (z) -ylidene) -thiazolidin-4-one derivatives as immunosuppressant agents |
| WO2008062376A2 | Nov 22, 2007 | May 29, 2008 | Actelion Pharmaceuticals Ltd | New process for the preparation of 2-imino-thiazolidin-4-one derivatives |
| WO2010046835A1 | Oct 19, 2009 | Apr 29, 2010 | Actelion Pharmaceuticals Ltd | Crystalline forms of (r) -5- [3-chloro-4- ( 2, 3-dihydroxy-propoxy) -benz [z] ylidene] -2- ( [z] -propylimino) -3-0-tolyl-thiazolidin-4-one |
| Reference | ||
|---|---|---|
| 1 | * | BOLLI, M.H. ET AL.: “2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1 Receptor Agonists“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 10, 2010, pages 4198-4211, XP55090073, ISSN: 0022-2623, DOI: 10.1021/jm100181s |
References
- “Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: Favorable impact of dose up-titration”. The Journal of Clinical Pharmacology 54: 179–88. Feb 2014. doi:10.1002/jcph.244. PMID 24408162.
- “Mass balance, pharmacokinetics and metabolism of the selective S1P1 receptor modulator ponesimod in humans”. Xenobiotica 45: 139–49. Feb 2015. doi:10.3109/00498254.2014.955832. PMID 25188442.
- H. Spreitzer (29 September 2014). “Neue Wirkstoffe – Ponesimod”. Österreichische Apothekerzeitung (in German) (20/2014): 42.
- “Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial”. Journal of Neurology, Neurosurgery 85: 1198–208. Nov 2014. doi:10.1136/jnnp-2013-307282. PMC 4215282. PMID 24659797.
- “Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial”. The Lancet 384: 2036–45. Dec 2014. doi:10.1016/S0140-6736(14)60803-5. PMID 25127208.
- “Effect of Ponesimod, a selective S1P1 Receptor Modulator, on the QT Interval in Healthy Subjects”. Basic 116: 429–37. May 2015.doi:10.1111/bcpt.12336. PMID 25287214.
- “Ponesimod”. Actelion. Retrieved 31 October 2014.

ABOUT PONESIMOD
Ponesimod is a potent orally active, selective sphingosine-1-phosphate receptor 1 (S1P1) immunomodulator.
Ponesimod prevents lymphocytes from leaving lymph nodes, thereby reducing circulating blood lymphocyte counts and preventing infiltration of lymphocytes into target tissues. The lymphocyte count reduction is rapid, dose-dependent, sustained upon continued dosing, and quickly reversible upon discontinuation. Initial data suggest that ponesimod does not cause lymphotoxicity by destroying/depleting lymphocytes or interfering with their cellular function. Other blood cells e.g. cells of the innate immune system are largely unaffected. Ponesimod is therefore considered a promising new oral agent for the treatment of a variety of autoimmune disorders.
CURRENT STATUS
OPTIMUM (Oral Ponesimod versus Teriflunomide In relapsing MUltiple sclerosis) is a Phase III multi-center, randomized, double-blind, parallel-group, active-controlled superiority study to compare the efficacy and safety of ponesimod to teriflunomide in patients with relapsing multiple sclerosis (RMS). The study aims to determine whether ponesimod is more efficacious than teriflunomide in reducing relapses. The study is expected to enroll approximately 1’100 patients, randomized in 2 groups in a 1:1 ratio to receive ponesimod 20 mg/day or teriflunomide 14 mg/day, and is expected to last a little over 3 years. An additional study to further characterize the utility and differentiation of ponesimod in multiple sclerosis is being discussed with Health Authorities.
Ponesimod is also evaluated in a Phase II open-label, single-arm, intra-subject dose-escalation study to investigate the biological activity, safety, tolerability, and pharmacokinetics of ponesimod in patients suffering from moderate or severe chronic graft versus host disease (GvHD)inadequately responding to first- or second-line therapy. The study will also investigate the clinical response to ponesimod treatment in these patients. Approximately 30 patients will be enrolled to receive ponesimod in escalating doses of 5, 10, and 20 mg/day over the course of 24 weeks. The study is being conducted at approximately 10 sites in the US and is expected to last approximately 18 months.
AVAILABLE CLINICAL DATA
The decision to move into Phase III development was based on the Phase IIb dose-finding study with ponesimod in patients with relapsing-remitting multiple sclerosis. A total of 464 patients were randomized into this study and the efficacy, safety and tolerability of three ponesimod doses (10, 20, and 40 mg/day) versus placebo, administered once daily for 24 weeks.
The primary endpoint of this study was defined as the cumulative number of new gadolinium-enhancing lesions on T1-weighted magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24 after study drug initiation. A key secondary endpoint of this study was the annualized relapse rate over 24 weeks of treatment. Patients who completed 24 weeks of treatment were offered the opportunity to enter into an extension study. This ongoing trial is investigating the long-term safety, tolerability, and efficacy of 10 and 20 mg/day of ponesimod in patients with relapsing-remitting multiple sclerosis, in a double-blind fashion. The study continues to provide extensive safety and efficacy information for ponesimod in this indication, with some patients treated for more than 6 years.
The safety database from all studies with ponesimod now comprises more than 1,300 patients and healthy volunteers.
MILESTONES
2015 – Phase III program in multiple sclerosis initiated
2011 – Phase IIb dose-finding study in multiple sclerosis successfully completed
2006 – Entry-into-man
2004 – Preclinical development initiated
KEY SCIENTIFIC LITERATURE
Olsson T et al. J Neurol Neurosurg Psychiatr. 2014 Nov;85(11):1198-208. doi: 10.1136/jnnp-2013-307282. Epub 2014 Mar 21
Freedman M.S, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 420 (P923).
Fernández Ó, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 417 (P919).
Piali L, Froidevaux S, Hess P, et al. J Pharmacol Exp Ther 337(2):547-56, 2011
Bolli MH, Abele S, Binkert C, et al. J Med Chem. 53(10):4198-211, 2010
Kappos L et al. N Engl J Med. 362(5):387-401, 2010
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2Z,5Z)-5-{3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]benzylidene}-3-(2-methylphenyl)-2-(propylimino)-1,3-thiazolidin-4-one
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | 2 main metabolites |
| Biological half-life | 31–34 hrs[1] |
| Excretion | Feces (57–80%, 26% unchanged), urine (10–18%)[2] |
| Identifiers | |
| CAS Number | 854107-55-4 |
| ATC code | none |
| PubChem | CID 11363176 |
| ChemSpider | 9538103 |
| ChEMBL | CHEMBL1096146 |
| Synonyms | ACT-128800 |
| Chemical data | |
| Formula | C23H25ClN2O4S |
| Molar mass | 460.974 g/mol |
////Ponesimod, Phase III , A sphingosine-1-phosphate receptor 1, S1P1 agonist, multiple sclerosis. ACT-128800; RG-3477; R-3477, autoimmune disease, lymphocyte migration, multiple sclerosis, psoriasis, transplantation
CCC/N=C\1/N(C(=O)/C(=C/C2=CC(=C(C=C2)OC[C@@H](CO)O)Cl)/S1)C3=CC=CC=C3C
ABT-530, Pibrentasvir

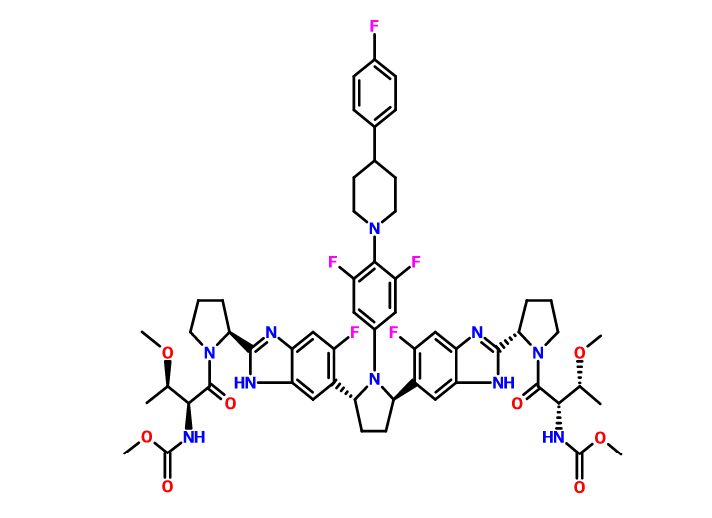
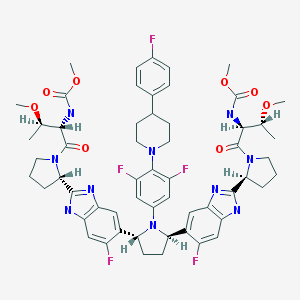
Pibrentasvir
ABT-530, Pibrentasvir, A 1325912.0
Dimethyl N,N’-([(2R,5R)-1-{3,5-difluoro-4-[4-(4-fluorophenyl)piperidin-1-yl]phenyl}pyrrolidine-2,5-diyl]bis{(6-fluoro-1H-benzimidazole-5,2-diyl)[(2S)-pyrrolidine-2,1-diyl][(2S,3R)-3-methoxy-1-oxobutane-1,2-diyl]})biscarbamate
Methyl {(2S,3R)-1-[(2S)-2-{5-[(2R,5R)-1-{3,5-difluoro-4-[4-(4-fluorophenyl)piperidin-1-yl]phenyl}-5-(6-fluoro-2-{(2S)-1-[N-(methoxycarbonyl)-O-methyl-L-threonyl]pyrrolidin-2-yl}-1H-benzimidazol-5-yl)pyrrolidin-2-yl]-6-fluoro-1H-benzimidazol-2-yl}pyrrolidin-1-yl]-3-methoxy-1-oxobutan-2-yl}carbamate
Dimethyl N,N’-(((2R,5R)-1-(3,5-difluoro-4-(4-(4-fluorophenyl)piperidin-1-yl)phenyl)pyrrolidine-2,5-diyl)bis((6-fluoro-1H-benzimidazole-5,2-diyl)((2S)-pyrrolidine-2,1-diyl)((2S,3R)-3-methoxy-1-oxobutane-1,2-diyl)))biscarbamate
Methyl ((2S,3R)-1-((2S)-2-(5-((2R,5R)-1-(3,5-difluoro-4-(4-(4-fluorophenyl)piperidin-1-yl)phenyl)-5-(6-fluoro-2-((2S)-1-(N-(methoxycarbonyl)-O-methyl-L-threonyl)pyrrolidin-2-yl)-1H-benzimidazol-5-yl)pyrrolidin-2-yl)-6-fluoro-1H-benzimidazol-2-yl)pyrrolidin-1-yl)-3-methoxy-1-oxobutan-2-yl)carbamate
Phase III
Abbott Laboratories INNOVATOR
A protease inhibitor potentially for the treatment of HCV infection.
Hepatitis C virus NS 5 protein inhibitors
![]()
CAS No. 1353900-92-1
| MF | C57H65F5N10O8 |
|---|
MW 1113.1925 MW
Pibrentasvir
- UNII-2WU922TK3L
- CLINICAL https://clinicaltrials.gov/search/intervention=A-1325912.0%20OR%20ABT-530%20OR%20Pibrentasvir

SYNTHESIS
PATENT
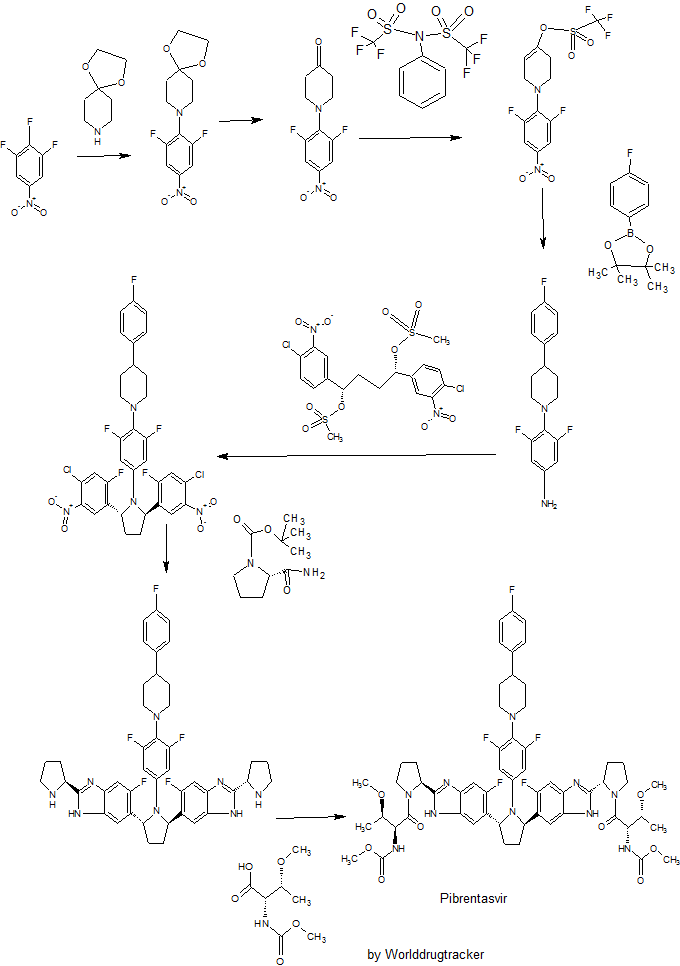
WO 2012051361
http://www.google.com/patents/WO2012051361A1?cl=en

Example 3.52 methyl {(2S,3R)-l-[(2S)-2-{5-[(2R,5R)-l-{3,5-difluoro-4-[4-(4- fluorophenyl)piperidin-l-yl]phenyl}-5-(6-fluoro-2-{(2.S)-l-[A^-(methoxycarbonyl)-0-methyl-L- threonyl]pyiTolidin-2-yl}-l f-benzimidazol-5-yl)pyiTolidin-2-yl]-6-fluoro-l f-benzimidaz yl}pyrrolidin-l-yl]-3-methoxy-l-oxobutan-2-yl}carbamatelH NMR (400 MHz, DMSO) δ 12.36 – 12.06 (m, 2H), 7.41 (dd, J = 11.2, 6.3, 1H), 7.34 (dd, J = 10.4, 4.8, 1H), 7.30 – 7.20 (m, 3H), 7.17 – 6.98 (m, 5H), 5.98 – 5.82 (m, 2H), 5.65 – 5.47 (m, 2H), 5.17 – 5.06 (m, 2H), 4.25 (dd, J = 15.6, 8.1, 2H), 3.88 – 3.74 (m, 3H), 3.53 (d, J = 1.3, 6H), 3.49 – 3.38 (m, 2H), 3.31 (d, 1H), 3.25 (d, J = 3.7, 1H), 3.13 (d, J = 1.3, 3H), 3.03 (d, J = 2.3, 3H), 3.00 – 2.84 (m, 3H), 2.60 – 2.53 (m, J = 2.5, 2H), 2.26 – 1.55 (m, 14H), 1.28 – 1.13 (m, 1H), 1.10 – 0.88 (m, 6H). MS (ESI; M+H) m/z = 1113.4.
PATENT
The present invention features crystalline polymorphs of methyl {(2S,3R)-1- [(2S)-2-{5-[(2R,5R)-l-{3,5-difluoro-4 4-(4-fluorophenyl)piperidin-l-yl]phenyl}-5-(6-fluoro-2-{(2S)- 1 -[N-(methoxycarbonyl)-0-methyl-L-threonyl]pyrrolidin-2-yl} – 1 H-benzimidazol-5-yl)pyrrolidin- -yl] -6-fluoro- 1 H-benzimidazol-2-yl} pyrrolidin- 1 -yl] -3 -methoxy- 1 -oxobutan-2-
yl} carbamate 
, herein “Compound I”). Compound I is a potent HCV NS5A inhibitor and is described in U.S. Patent Application Publication No. 2012/0004196, which is incorporated herein by reference in its entirety.
//////////1353900-92-1, PHASE 3, ABT-530, Pibrentasvir, ABT 530, A 1325912.0
C[C@H]([C@@H](C(=O)N1CCC[C@H]1c2[nH]c3cc(c(cc3n2)[C@H]4CC[C@@H](N4c5cc(c(c(c5)F)N6CCC(CC6)c7ccc(cc7)F)F)c8cc9c(cc8F)[nH]c(n9)[C@@H]1CCCN1C(=O)[C@H]([C@@H](C)OC)NC(=O)OC)F)NC(=O)OC)OC
C[C@H]([C@@H](C(=O)N1CCC[C@H]1c2[nH]c3cc(c(cc3n2)[C@H]4CC[C@@H](N4c5cc(c(c(c5)F)N6CCC(CC6)c7ccc(cc7)F)F)c8cc9c(cc8F)[nH]c(n9)[C@@H]1CCCN1C(=O)[C@H]([C@@H](C)OC)NC(=O)OC)F)NC(=O)OC)OC
Dr Anthony’s New Drug Approvals hits 13 lakh views in 212 countries
Dr Anthony’s New Drug Approvals hits 13 lakh views in 212 countries

An Indian helping millions

MAKING INDIANS FEEL PROUD
LINK
////////blog, Dr Anthony , New Drug Approvals, 13 lakh views, 212 countries, India
3,5-Dibromo-N-(4,6-difluorobenzo[d]thiazol-2-yl)thiophene-2-carboxamide having potent anti-norovirus activity

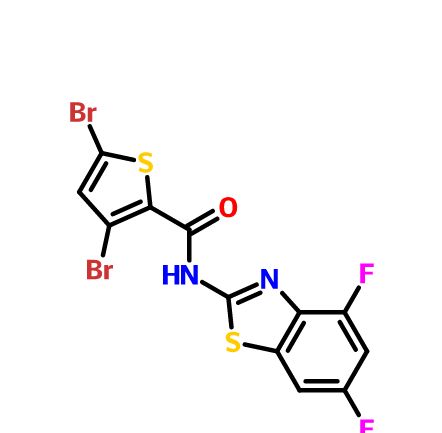
3,5-Dibromo-N-(4,6-difluorobenzo[d]thiazol-2-yl)thiophene-2-carboxamide
New and novel anti-norovirus agents
There is an urgent need for structurally novel anti-norovirus agents. In this study, we describe the synthesis, anti-norovirus activity, and structure–activity relationship (SAR) of a series of heterocyclic carboxamide derivatives. Heterocyclic carboxamide 1 (50% effective concentration (EC50)=37 µM) was identified by our screening campaign using the cytopathic effect reduction assay. Initial SAR studies suggested the importance of halogen substituents on the heterocyclic scaffold and identified 3,5-di-boromo-thiophene derivative 2j (EC50=24 µM) and 4,6-di-fluoro-benzothiazole derivative 3j (EC50=5.6 µM) as more potent inhibitors than 1. Moreover, their hybrid compound, 3,5-di-bromo-thiophen-4,6-di-fluoro-benzothiazole 4b, showed the most potent anti-norovirus activity with a EC50 value of 0.53 µM (70-fold more potent than 1). Further investigation suggested that 4b might inhibit intracellular viral replication or the late stage of viral infection.
3,5-Dibromo-N-(4,6-difluorobenzo[d]thiazol-2-yl)thiophene-2-carboxamide (4b)
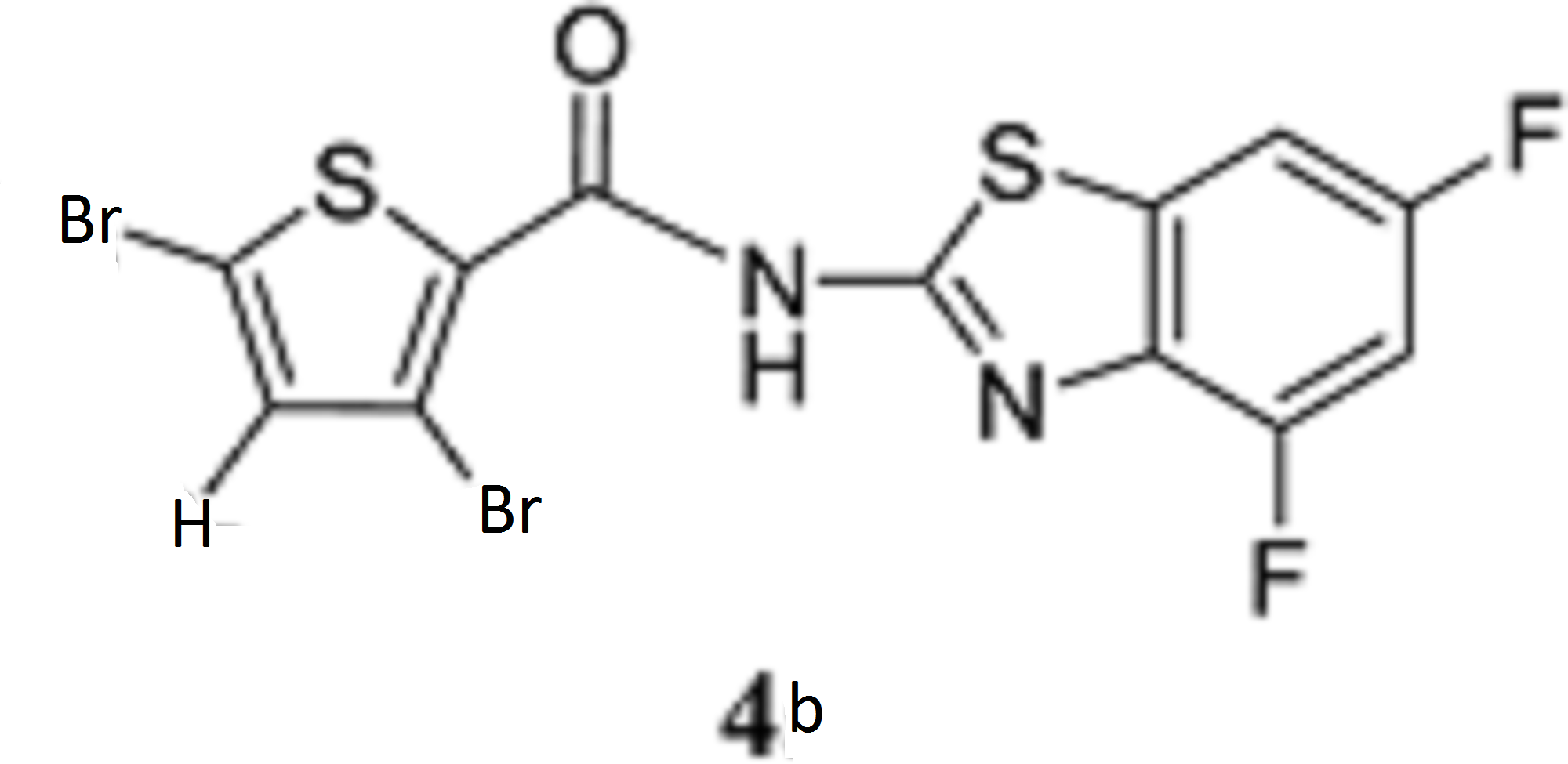
According to the same procedure used for 2f, starting from 3,5-dibromothiophene-2-carboxylic acid (286 mg, 1.00 mmol) and 4,6-difluorobenzo[d]thiazol-2-amine (204 mg, 1.10 mmol), 4b (270 mg, 60%) was obtained as white powder. mp: 245–246°C. 1H-NMR (DMSO-d6) δ: 7.43 (1H, dt, J=10.2, 2.0 Hz), 7.56 (1H, s), 7.83 (1H, dd, J=8.4, 2.0 Hz). 13C-NMR (DMSO-d6) δ: 102.2 (dd, J=28.0, 23.1 Hz), 104.7 (dd, J=26.4, 3.3 Hz), 114.3, 118.4, 131.4 (d, J=7.4 Hz), 134.3 (d, J=10.7 Hz), 134.9, 135.2, 152.7 (d, J=241.2, 20.7 Hz), 158.3 (dd, J=242.2, 10.7 Hz), 159.0, 159.7. HPLC purity: >99%, ESI-MS m/z 453 [M+H]+.
Antiviral Activity and Cytotoxicity of Tetra-halogenated Hybrid Compounds
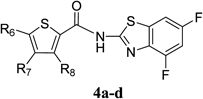 |
|||||
|---|---|---|---|---|---|
| Compound | R6 | R7 | R8 | EC50 (µM)a) | CC50 (µM)b) |
| 4a | Cl | H | H | 2.1 | >100 |
| 4b | Br | H | Br | 0.53 | >100 |
| 4c | Cl | H | Cl | 1.1 | >100 |
| 4d | Cl | Cl | H | 1.4 | 31 |
a) EC50 was evaluated by the CPE reduction assay. 280 TCID50/50 µL of MNV and a dilution series of each compound were incubated for 30 min. The mixture was exposed to RAW264.7 cells for 1 h (in duplicate). b) Cytotoxicity was evaluated by the WST-8 assay. RAW264.7 cells were treated with dilution series of each compound (in triplicate) for 72 h.
Discovery and Synthesis of Heterocyclic Carboxamide Derivatives as Potent Anti-norovirus Agents

How to Kill Norovirus

Three Methods:Killing Norovirus with Good HygieneKilling Norovirus in Your HomeTreating NorovirusCommunity Q&A
Norovirus is a contagious virus that affects many people each year. You can get norovirus through interaction with an infected person, by eating contaminated food, touching contaminated surfaces, or drinking contaminated water. However, there are ways to kill norovirus before it infects you. To do this, you will have to maintain personal hygiene and keep your home contamination-free.
Method1
Killing Norovirus with Good Hygiene
-
 1
1Wash your hands thoroughly. If you think you may have come into contact with the virus, you must wash your hands thoroughly to avoid the spread of infection. To wash your hands to avoid contamination, use soap and hot water. Alcohol hand sanitizer is generally considered ineffective against this particular kind of virus. You should wash your hands if[1]:
- You have come into contact with someone who has norovirus.
- Before and after you interact with someone with norovirus.
- If you visit a hospital, even if you don’t think you interacted with anyone with norovirus.
- After going to the bathroom.
- Before and after eating.
- If you are a nurse or doctor, wash your hands before and after coming into contact with an infected patient, even if you wear gloves.
-
 2
2Avoid cooking for others if you are sick. If you have been infected and are sick, do not handle any food or cook for others in your family. If you do, they are almost certain to get the infection too.
- If a family member is contaminated, do not let them cook for anyone else. Try to limit the amount of time healthy family members spend with the sick family member.
-
 3
3Wash your food before eating or cooking it. Wash all food items such as meats, fruits and vegetables thoroughly before consumption or for use in cooking. This is important as norovirus has the tendency to survive even at temperatures well above 140 degrees Fahrenheit (60 degrees Celsius).[2]
- Remember to carefully wash any vegetables or fruit, before consuming them, whether you prefer them fresh or cooked.
-
 4
4Cook your food thoroughly before eating it. Seafood should be cooked thoroughly before eating it. Quick steaming your food will generally not kill the virus, as it can survive the steaming process. Instead, bake or boil your food at temperatures higher than 140F (60C) if you are concerned about it’s origins.[3]
- If you suspect any kind of food of being contaminated, you should dispose of it immediately. For instance, if a contaminated family member handled the food, you should either throw the food out or isolate it and make sure that only the person who already has the virus eats it.




Method2
Killing Norovirus in Your Home
-
 1
1Use bleach to clean surfaces. Chlorine bleach is an effective cleaning agent that kills norovirus. Increase the concentration or buy a new bottle of chlorine bleach if the bleach you have has been open for more than a month. Bleach becomes less effective the longer it remains open. Before applying bleach to a visible surface, test it somewhere that is not easily seen to make sure that it won’t damage the surface. If the surface is damaged by bleach, you can also use phenolic solutions, such as Pine-Sol, to clean the surface. There are certain concentrations of chlorine bleach you can use for different surfaces.[4]
- For stainless steel surfaces and items used for food consumption: Dissolve one tablespoon of bleach in a gallon of water and clean the stainless steel.
- For non-porous surfaces like countertops, sinks, or tile floors: Dissolve one third of a cup of bleach in a gallon of water.
- For porous surfaces, like wooden floors: Dissolve one and two thirds of a cup of bleach in a gallon of water.
-
 2
2Rinse surfaces with clean water after using bleach. After cleaning the surfaces, leave the solution to work for 10 to 20 minutes. After the time period elapses, rinse the surface with clean water. After these two steps, close off the area, and leave it like that for one hour.
- Leave the windows open, if possible, as breathing in bleach can be hazardous to your health.
-
 3
3Clean areas exposed to feces or vomit. For areas exposed to feces or vomit contamination there are special cleaning procedures that you should try to follow. This is because the vomit or feces of a person contaminated with norovirus can easily cause you to become infected. To clean the vomit or feces:
- Put disposable gloves on. Consider wearing a facemask that covers your mouth and nose as well.
- Using paper towels, gently clean the vomit and feces. Be careful not to splash or drip while cleaning.
- Use disposable cloths to clean and disinfect the entire area with chlorine bleach.
- Use sealed plastic bags to dispose of all the waste materials.
-
 4
4Clean your carpets. If the feces or vomit gets on a carpeted area, there are other steps you can take to make sure that the area is clean and disinfected. To clean the carpeted area:
- Wear disposable gloves if you can while cleaning the carpets. You should also consider wearing a facemask that covers your mouth and nose.
- Use any absorbent material to clean all the visible feces or vomit. Place all contaminated materials in a plastic bag to prevent aerosols from forming. The bag should be sealed and put into the garbage can.
- The carpet should then be cleaned with steam at 170 degrees Fahrenheit (76 degrees Celsius) for about five minutes, or, if you want to save time, clean the carpet for one minute with 212 degrees Fahrenheit (100 degrees Celsius) steam.
-
 5
5Disinfect clothing. If any of your clothing or a family member’s clothing has become contaminated, or is suspected of having been contaminated, you should take care when washing the fabric. To clean clothing and linens:
- Remove any traces of vomit or feces by wiping it away with paper towels or a disposable absorbent material.
- Put the contaminated clothing into the washing machine in a pre-wash cycle. After this stage is complete, wash the clothes using a regular washing cycle and detergent. The clothes should be dried separately from the uncontaminated clothes. A drying temperature exceeding 170 degrees Fahrenheit is recommended.
- Do not wash contaminated clothing with uncontaminated cleaning.





Method3
Treating Norovirus
-
 1
1Recognize symptoms. If you think you may have been infected with norovirus, it is helpful to know what symptoms to look for. If you do have the virus, the following steps will help you to deal with the illness while it lasts. Symptoms include[5]:
- Fever. Just like in any other infection, the norovirus infection will cause fever. Fever is a way in which the body fights infection. The body temperature will rise, making the virus more vulnerable to the immune system. Your body temperature will most likely rise above 100.4 degrees Fahrenheit (38 degrees Celsius) when suffering from a Norovirus infection.
- Headaches. High body temperatures will cause blood vessels to dilate in your entire body, including your head. The high amount of blood inside your head will cause pressure to build up, and the protective membranes covering your brain will suffer inflammation and become painful.
- Stomach cramps. Norovirus infections usually settle in the stomach. Your stomach may become inflamed, causing pain.
- Diarrhea. Diarrhea is a common symptom of Norovirus contamination. It occurs as a defense mechanism, through which the body is trying to flush out the virus.
- Vomiting. Vomiting is another common symptom of an infection with Norovirus. Like in the case of diarrhea, the body is trying to eliminate the virus from the system by vomiting.
-
 2
2Understand that while there is no treatment, there are ways to manage symptoms. Unfortunately, there is no specific drug that acts against the virus. However, you can combat the symptoms that the norovirus causes. Remember that the virus is self-limiting, which means that it generally goes away on its own.
- The virus generally lasts for a few days to a week.
-
 3
3Drink lots of fluids. Consuming a lot of water and other fluids will help to keep you hydrated. This can help to keep your fever low and your headaches to a minimum. It is also important to drink water if you have been vomiting or have had diarrhea. When these too symptoms occur, it is very likely that you will become dehydrated.
- If you get bored with water, you can drink ginger tea, which may help to manage your stomach pains while also hydrating you.
-
 4
4Consider taking anti-vomiting drugs. Anti-emetic (vomit-preventing) drugs such as ondansetron and domperidone can be given to provide symptomatic relief if you are vomiting frequently.[6]
- However, keep in mind that these drugs can only be obtained with a prescription from your doctor.
-
 5Seek medical help if the infection is severe. As mentioned above, most infections subside after a few days. If the virus persists for longer than a week, you should consider seeking medical help. This is particularly important if the person who is sick is a child or elderly person, or a person with lowered immunity
5Seek medical help if the infection is severe. As mentioned above, most infections subside after a few days. If the virus persists for longer than a week, you should consider seeking medical help. This is particularly important if the person who is sick is a child or elderly person, or a person with lowered immunity





JNJ-54257099

JNJ-54257099,
1-((2R,4aR,6R,7R,7aR)-2-Isopropoxy-2-oxidodihydro-4H,6H-spiro[furo[3,2-d][1,3,2]dioxaphosphinine-7,2′-oxetan]-6-yl)pyrimidine-2,4(1H,3H)-dione
MW 374.28, C14 H19 N2 O8 P
CAS 1491140-67-0
2,4(1H,3H)-Pyrimidinedione, 1-[(2R,2′R,4aR,6R,7aR)-dihydro-2-(1-methylethoxy)-2-oxidospiro[4H-furo[3,2-d]-1,3,2-dioxaphosphorin-7(6H),2′-oxetan]-6-yl]-
1-((2R,4aR,6R,7R,7aR)-2-Isopropoxy-2-oxidodihydro-4H,6H-spiro[furo[3,2-d][1,3,2]dioxaphos-phinine-7,2′-oxetan]-6-yl)pyrimidine-2,4(1H,3H)-dione

Janssen R&D Ireland INNOVATOR
Ioannis Nicolaos Houpis, Tim Hugo Maria Jonckers, Pierre Jean-Marie Bernard Raboisson, Abdellah Tahri,

Tim Jonckers was born in Antwerp in 1974. He studied Chemistry at the University of Antwerp and obtained his Ph.D. in organic chemistry in 2002. His Ph.D. work covered the synthesis of new necryptolepine derivatives which have potential antimalarial activity. Currently he works as a Senior Scientist at Tibotec, a pharmaceutical research and development company based in Mechelen, Belgium, that focuses on viral diseases mainly AIDS and hepatitis. The company was acquired by Johnson & Johnson in April 2002 and recently gained FDA approval for its HIV-protease inhibitor PREZISTA™.

Principal Scientist at Janssen, Pharmaceutical Companies of Johnson and Johnson

DATA
Chiral SFC using the methods described(Method 1, Rt= 5.12 min, >99%; Method 2, Rt = 7.95 min, >99%).
1H NMR (400 MHz, chloroform-d) δ ppm 1.45 (dd, J = 7.53, 6.27 Hz, 6 H), 2.65–2.84 (m, 2 H), 3.98 (td, J = 10.29, 4.77 Hz, 1 H), 4.27 (t,J = 9.66 Hz, 1 H), 4.43 (ddd, J = 8.91, 5.77, 5.65 Hz, 1 H), 4.49–4.61 (m, 1 H), 4.65 (td, J = 7.78, 5.77 Hz, 1 H), 4.73 (d, J = 7.78 Hz, 1 H), 4.87 (dq, J = 12.74, 6.30 Hz, 1 H), 5.55 (br. s., 1 H), 5.82 (d, J = 8.03 Hz, 1 H), 7.20 (d, J = 8.03 Hz, 1 H), 8.78 (br. s., 1 H);
31P NMR (chloroform-d) δ ppm −7.13. LC-MS: 375 (M + H)+.
HCV is a single stranded, positive-sense R A virus belonging to the Flaviviridae family of viruses in the hepacivirus genus. The NS5B region of the RNA polygene encodes a RNA dependent RNA polymerase (RdRp), which is essential to viral replication. Following the initial acute infection, a majority of infected individuals develop chronic hepatitis because HCV replicates preferentially in hepatocytes but is not directly cytopathic. In particular, the lack of a vigorous T-lymphocyte response and the high propensity of the virus to mutate appear to promote a high rate of chronic infection. Chronic hepatitis can progress to liver fibrosis, leading to cirrhosis, end-stage liver disease, and HCC (hepatocellular carcinoma), making it the leading cause of liver transplantations. There are six major HCV genotypes and more than 50 subtypes, which are differently distributed geographically. HCV genotype 1 is the predominant genotype in Europe and in the US. The extensive genetic heterogeneity of HCV has important diagnostic and clinical implications, perhaps explaining difficulties in vaccine development and the lack of response to current therapy.
Transmission of HCV can occur through contact with contaminated blood or blood products, for example following blood transfusion or intravenous drug use. The introduction of diagnostic tests used in blood screening has led to a downward trend in post-transfusion HCV incidence. However, given the slow progression to the end-stage liver disease, the existing infections will continue to present a serious medical and economic burden for decades.
Therapy possibilities have extended towards the combination of a HCV protease inhibitor (e.g. Telaprevir or boceprevir) and (pegylated) interferon-alpha (IFN-a) / ribavirin. This combination therapy has significant side effects and is poorly tolerated in many patients. Major side effects include influenza-like symptoms, hematologic
abnormalities, and neuropsychiatric symptoms. Hence there is a need for more effective, convenient and better-tolerated treatments.
The NS5B RdRp is essential for replication of the single-stranded, positive sense, HCV RNA genome. This enzyme has elicited significant interest among medicinal chemists. Both nucleoside and non-nucleoside inhibitors of NS5B are known. Nucleoside inhibitors can act as a chain terminator or as a competitive inhibitor, or as both. In order to be active, nucleoside inhibitors have to be taken up by the cell and converted in vivo to a triphosphate. This conversion to the triphosphate is commonly mediated by cellular kinases, which imparts additional structural requirements on a potential nucleoside polymerase inhibitor. In addition this limits the direct evaluation of nucleosides as inhibitors of HCV replication to cell-based assays capable of in situ phosphorylation.
Several attempts have been made to develop nucleosides as inhibitors of HCV RdRp, but while a handful of compounds have progressed into clinical development, none have proceeded to registration. Amongst the problems which HCV-targeted
nucleosides have encountered to date are toxicity, mutagenicity, lack of selectivity, poor efficacy, poor bioavailability, sub-optimal dosage regimes and ensuing high pill burden and cost of goods.
Spirooxetane nucleosides, in particular l-(8-hydroxy-7-(hydroxy- methyl)- 1,6-dioxaspiro[3.4]octan-5-yl)pyrimidine-2,4-dione derivatives and their use as HCV inhibitors are known from WO2010/130726, and WO2012/062869, including
CAS-1375074-52-4.
There is a need for HCV inhibitors that may overcome at least one of the disadvantages of current HCV therapy such as side effects, limited efficacy, the emerging of resistance, and compliance failures, or improve the sustained viral response.
The present invention concerns HCV-inhibiting uracyl spirooxetane derivatives with useful properties regarding one or more of the following parameters: antiviral efficacy towards at least one of the following genotypes la, lb, 2a, 2b, 3,4 and 6, favorable
profile of resistance development, lack of toxicity and genotoxicity, favorable pharmacokinetics and pharmacodynamics and ease of formulation and administration.
Such an HCV-inhibiting uracyl spirooxetane derivative is a compound with formula I
including any pharmaceutically acceptable salt or solvate thereof.
PATENT
WO 2015077966
https://www.google.com/patents/WO2015077966A1?cl=en
Synthesis of compound (I)

(5) (6a)

Synthesis of compound (6a)
A solution of isopropyl alcohol (3.86 mL,0.05mol) and triethylamine (6.983 mL, 0.05mol) in dichloromethane (50 mL) was added to a stirred solution of POCI3 (5)
(5.0 mL, 0.055 lmol) in DCM (50 mL) dropwise over a period of 25 min at -5°C. After the mixture stirred for lh, the solvent was evaporated, and the residue was suspended in ether (100 mL). The triethylamine hydrochloride salt was filtered and washed with ether (20 mL). The filtrate was concentrated, and the residue was distilled to give the (6) as a colorless liquid (6.1g, 69 %yield).
Synthesis of compound (4):
CAS 1255860-33-3 is dissolved in pyridine and 1,3-dichloro-l, 1,3,3-tetraisopropyldisiloxane is added. The reaction is stirred at room temperature until complete. The solvent is removed and the product redissolved in CH2CI2 and washed with saturated NaHC03 solution. Drying on MgSC^ and removal of the solvent gives compound (2). Compound (3) is prepared by reacting compound (2) with p-methoxybenzylchloride in the presence of DBU as the base in CH3CN. Compound (4) is prepared by cleavage of the bis-silyl protecting group in compound (3) using TBAF as the fluoride source.
Synthesis of compound (7a)
To a stirred suspension of (4) (2.0 g, 5.13 mmol) in dichloromethane (50 mL) was added triethylamine (2.07 g, 20.46 mmol) at room temperature. The reaction mixture was cooled to -20°C, and then (6a) (1.2 g, 6.78mmol) was added dropwise over a period of lOmin. The mixture was stirred at this temperature for 15min and then NMI was added (0.84 g, 10.23 mmol), dropwise over a period of 15 min. The mixture was stirred at -15°C for lh and then slowly warmed to room temperature in 20 h. The solvent was evaporated, the mixture was concentrated and purified by column chromatography using petroleum ether/EtOAc (10: 1 to 5: 1 as a gradient) to give (7a) as white solid (0.8 g, 32 % yield).
Synthesis of compound (I)
To a solution of (7a) in CH3CN (30 mL) and H20 (7 mL) was add CAN portion wise below 20° C. The mixture was stirred at 15-20° C for 5h under N2. Na2S03 (370 mL) was added dropwise into the reaction mixture below 15°C, and then Na2C03 (370 mL) was added. The mixture was filtered and the filtrate was extracted with CH2C12
(100 mL*3). The organic layer was dried and concentrated to give the residue. The residue was purified by column chromatography to give the target compound (8a) as white solid. (Yield: 55%)
1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.45 (dd, J=7.53, 6.27 Hz, 6 H), 2.65 -2.84 (m, 2 H), 3.98 (td, J=10.29, 4.77 Hz, 1 H), 4.27 (t, J=9.66 Hz, 1 H), 4.43 (ddd, J=8.91, 5.77, 5.65 Hz, 1 H), 4.49 – 4.61 (m, 1 H), 4.65 (td, J=7.78, 5.77 Hz, 1 H), 4.73 (d, J=7.78 Hz, 1 H), 4.87 (dq, J=12.74, 6.30 Hz, 1 H), 5.55 (br. s., 1 H), 5.82 (d, J=8.03 Hz, 1 H), 7.20 (d, J=8.03 Hz, 1 H), 8.78 (br. s., 1 H); 31P NMR (CHLOROFORM-^) δ ppm -7.13; LC-MS: 375 (M+l)+
PATENT
https://www.google.co.in/patents/WO2013174962A1?cl=en
The starting material l-[(4R,5R,7R,8R)-8-hydroxy-7-(hydroxymethyl)-l,6-dioxa- spiro[3.4]octan-5-yl]pyrimidine-2,4(lH,3H)-dione (1) can be prepared as exemplified in WO2010/130726. Compound (1) is converted into compounds of the present invention via a p-methoxybenzyl protected derivative (4) as exemplified in the following Scheme 1. cheme 1

Examples
Scheme 2
Synthesis of compound (8a)

Synthesis of compound (2)
Compound (2) can be prepared by dissolving compound (1) in pyridine and adding l,3-dichloro-l,l,3,3-tetraisopropyldisiloxane. The reaction is stirred at room temperature until complete. The solvent is removed and the product redissolved in CH2CI2and washed with saturated NaHC03 solution. Drying on MgSC^ and removal of the solvent gives compound (2).
Synthesis of compound (3)
Compound (3) is prepared by reacting compound (2) with p-methoxybenzylchloride in the presence of DBU as the base in CH3CN.
Synthesis of compound (4)
Compound (4) is prepared by cleavage of the bis-silyl protecting group in compound (3) using TBAF as the fluoride source.
Synthesis of compound (6a)
A solution of isopropyl alcohol (3.86 mL,0.05mol) and triethylamine (6.983 mL, 0.05mol) in dichloromethane (50 mL) was added to a stirred solution of POCl3 (5) (5.0 mL, 0.055 lmol) in DCM (50 mL) dropwise over a period of 25 min at -5°C. After the mixture stirred for lh, the solvent was evaporated, and the residue was suspended in ether (100 mL). The triethylamine hydrochloride salt was filtered and washed with ether (20 mL). The filtrate was concentrated, and the residue was distilled to give the (6) as a colorless liquid (6.1g, 69 %yield).
Synthesis of compound (7a)
To a stirred suspension of (4) (2.0 g, 5.13 mmol) in dichloromethane (50 mL) was added triethylamine (2.07 g, 20.46 mmol) at room temperature. The reaction mixture was cooled to -20°C, and then (6a) (1.2 g, 6.78mmol) was added dropwise over a period of lOmin. The mixture was stirred at this temperature for 15min and then NMI was added (0.84 g, 10.23 mmol), dropwise over a period of 15 min. The mixture was stirred at -15°C for lh and then slowly warmed to room temperature in 20 h. The solvent was evaporated, the mixture was concentrated and purified by column chromatography using petroleum ether/EtOAc (10:1 to 5: 1 as a gradient) to give (7a) as white solid (0.8 g, 32 % yield).
Synthesis of compound (8a)
To a solution of (7a) in CH3CN (30 mL) and H20 (7 mL) was add CAN portion wise below 20°C. The mixture was stirred at 15-20°C for 5h under N2. Na2S03 (370 mL) was added dropwise into the reaction mixture below 15°C, and then Na2C03 (370 mL) was added. The mixture was filtered and the filtrate was extracted with CH2C12
(100 mL*3). The organic layer was dried and concentrated to give the residue. The residue was purified by column chromatography to give the target compound (8a) as white solid. (Yield: 55%)
1H NMR (400 MHz, CHLOROFORM- ) δ ppm 1.45 (dd, J=7.53, 6.27 Hz, 6 H), 2.65 – 2.84 (m, 2 H), 3.98 (td, J=10.29, 4.77 Hz, 1 H), 4.27 (t, J=9.66 Hz, 1 H), 4.43 (ddd, J=8.91, 5.77, 5.65 Hz, 1 H), 4.49 – 4.61 (m, 1 H), 4.65 (td, J=7.78, 5.77 Hz, 1 H), 4.73 (d, J=7.78 Hz, 1 H), 4.87 (dq, J=12.74, 6.30 Hz, 1 H), 5.55 (br. s., 1 H), 5.82 (d, J=8.03 Hz, 1 H), 7.20 (d, J=8.03 Hz, 1 H), 8.78 (br. s., 1 H); 31P NMR (CHLOROFORM-^) δ ppm -7.13; LC-MS: 375 (M+l)+ Scheme 3
Synthesis of compound (VI)

Step 1: Synthesis of compound (9)Compound (1), CAS 1255860-33-3 ( 1200 mg, 4.33 mmol ) and l,8-bis(dimethyl- amino)naphthalene (3707 mg, 17.3 mmol) were dissolved in 24.3 mL of
trimethylphosphate. The solution was cooled to 0°C. Compound (5) (1.21 mL, 12.98 mmol) was added, and the mixture was stirred well maintaining the temperature at 0°C for 5 hours. The reaction was quenched by addition of 120 mL of tetraethyl- ammonium bromide solution (1M) and extracted with CH2CI2 (2×80 mL). Purification was done by preparative HPLC (Stationary phase: RP XBridge Prep CI 8 ΟΒϋ-10μιη, 30x150mm, mobile phase: 0.25% NH4HCO3 solution in water, CH3CN) , yielding two fractions. The purest fraction was dissolved in water (15 mL) and passed through a manually packed Dowex (H+) column by elution with water. The end of the elution was determined by checking UV absorbance of eluting fractions. Combined fractions were frozen at -78°C and lyophilized. Compound (9) was obtained as a white fluffy solid (303 mg, (0.86 mmol, 20%> yield), which was used immediately in the following reaction. Step 2: Preparation of compound (VI)
Compound (9) (303 mg, 0.86 mmol) was dissolved in 8 mL water and to this solution was added N . N’- D ic y c ! he y !-4- mo rph line carboxamidine (253.8 mg, 0.86 mmol) dissolved in pyridine (8.4 mi.). The mixture was kept for 5 minutes and then
evaporated to dryness, dried overnight in vacuo overnight at 37°C. The residu was dissolved in pyridine (80 mL). This solution was added dropwise to vigorously stirred DCC (892.6 mg, 4.326 mmol) in pyridine (80 mL) at reflux temperature. The solution was kept refluxing for 1.5h during which some turbidity was observed in the solution. The reaction mixture was cooled and evaporated to dryness. Diethylether (50 mL) and water (50 mL) were added to the solid residu. N’N-dicyclohexylurea was filtered off, and the aqueous fraction was purified by preparative HPLC (Stationary phase: RP XBridge Prep C18 OBD-ΙΟμιη, 30x150mm, mobile phase: 0.25% NH4HCO3 solution in water, CH3CN) , yielding a white solid which was dried overnight in vacuo at 38°C. (185 mg, 0.56 mmol, 65% yield). LC-MS: (M+H)+: 333.
1H NMR (400 MHz, DMSO-d6) d ppm 2.44 – 2.59 (m, 2 H) signal falls under DMSO signal, 3.51 (td, J=9.90, 5.50 Hz, 1 H), 3.95 – 4.11 (m, 2 H), 4.16 (d, J=10.34 Hz, 1 H), 4.25 – 4.40 (m, 2 H), 5.65 (d, J=8.14 Hz, 1 H), 5.93 (br. s., 1 H), 7.46 (d, J=7.92 Hz, 1 H), 2H’s not observed
Paper
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00382,
Discovery of 1-((2R,4aR,6R,7R,7aR)-2-Isopropoxy-2-oxidodihydro-4H,6H-spiro[furo[3,2-d][1,3,2]dioxaphosphinine-7,2′-oxetan]-6-yl)pyrimidine-2,4(1H,3H)-dione (JNJ-54257099), a 3′-5′-Cyclic Phosphate Ester Prodrug of 2′-Deoxy-2′-Spirooxetane Uridine Triphosphate Useful for HCV Inhibition
Janssen Infectious Diseases − Diagnostics BVBA, Turnhoutseweg 30, 2340 Beerse, Belgium
J. Med. Chem., Article ASAP
DOI: 10.1021/acs.jmedchem.6b00382
Publication Date (Web): May 14, 2016
Copyright © 2016 American Chemical Society
*Phone: +32 014601168. E-mail: tjoncker@its.jnj.com.
JNJ-54257099 (9) is a novel cyclic phosphate ester derivative that belongs to the class of 2′-deoxy-2′-spirooxetane uridine nucleotide prodrugs which are known as inhibitors of the HCV NS5B RNA-dependent RNA polymerase (RdRp). In the Huh-7 HCV genotype (GT) 1b replicon-containing cell line 9 is devoid of any anti-HCV activity, an observation attributable to inefficient prodrug metabolism which was found to be CYP3A4-dependent. In contrast, in vitro incubation of 9 in primary human hepatocytes as well as pharmacokinetic evaluation thereof in different preclinical species reveals the formation of substantial levels of 2′-deoxy-2′-spirooxetane uridine triphosphate (8), a potent inhibitor of the HCV NS5B polymerase. Overall, it was found that 9 displays a superior profile compared to its phosphoramidate prodrug analogues (e.g., 4) described previously. Of particular interest is the in vivo dose dependent reduction of HCV RNA observed in HCV infected (GT1a and GT3a) human hepatocyte chimeric mice after 7 days of oral administration of 9
////////////JNJ-54257099, 1491140-67-0, JNJ54257099, JNJ 54257099
O=C(C=C1)NC(N1[C@H]2[C@]3(OCC3)[C@H](O4)[C@@H](CO[P@@]4(OC(C)C)=O)O2)=O
AM 2394
AM 2394
1-(6′-(2-hydroxy-2-methylpropoxy)-4-((5-methylpyridin-3-yl)oxy)-[3,3′-bipyridin]-6-yl)-3-methylurea
Urea, N-[6′-(2-hydroxy-2-methylpropoxy)-4-[(5-methyl-3-pyridinyl)oxy][3,3′-bipyridin]-6-yl]-N‘-methyl-
CAS 1442684-77-6
Chemical Formula: C22H25N5O4
Exact Mass: 423.1907
Array Biopharma Inc., Amgen Inc. INNOVATORS
AM-2394 is a potent and selective Glucokinase agonist (GKA), which catalyzes the phosphorylation of glucose to glucose-6-phosphate. AM-2394 activates GK with an EC50 of 60 nM, increases the affinity of GK for glucose by approximately 10-fold, exhibits moderate clearance and good oral bioavailability in multiple animal models, and lowers glucose excursion following an oral glucose tolerance test in an ob/ob mouse model of diabetes
Type 2 diabetes mellitus (T2DM) is a disease characterized by elevated plasma glucose in the presence of insulin resistance and inadequate insulin secretion. Glucokinase (GK), a member of the hexokinase enzyme family, catalyzes the phosphorylation of glucose to glucose-6-phosphate in the presence of ATP.


Glucokioase i exok ase IV or D> is a glycolytic enssyiris that plays, an importaat. role irt blood sugar regulation .related to glucose utifeattoti a»d metabolism in the liver and pancreatic beta •cells. Serving as a glucose sessor, gtoeokiuase controls lasma glucose, levels. Glucokinaae plays a doal rob in .reducing plasma glucose levels; glucose-mediated activation of the en¾ymc in hepatocytes facilitates hepatic giocose npiafcc aad glycogen synthesis, while that la pancreatic beta ceils ultimately induces ins lin seeretio«. Both of these effects in turn reduce plasma glucose levels.
Clinical evidence has shown that, glueokitiase variants with, decreased, and increased activities are associated with mature easel, diabetes of the y ung { O0Y2) and persistent: hyperinsul nemic hypoglycemia &( infancy (PHHI), respectively. lso, aoo n.sulin dependent diabetes rneilitos (NIDDM) patients have been reported to have inappropriately lo giueokaiase activity; Ftirtherrnare. overexpressioa of glucokiuase it* dietary or gesetie animal models of diabetes either prevents, aoKiiorafes, or reverses the progress of pathological. symptoms in the disease. For these reasons, compounds that activate gfecokiaase have been sought by the pitasaaceatjeai liidustry.
International patent application, Publication No. WO 2 7/OS3345, which was published on May 10, 200?, discloses as giocokinase act ators certain 2-an«.aopyridiiie derivatives bearing at the 3 -position a meihyieneoxy-dkrked aromatic group a d on. the ammo group a heteroaryl ring, such as dna/oly! or i A4-lmadiazoiyl
it has .now been found that pyridyl ureas are useful as glneokirtase activators. Cettain of these •compounds have been, found to have an outstanding combination of properties that especially adapts them, for oral use to control plasma glucose levels.
![]()

Novel Series of Potent Glucokinase Activators Leading to the Discovery of AM-2394
† Departments of Therapeutic Discovery, Metabolic Disorders, and Pharmacokinetics and Drug Metabolism, Amgen Inc., 1120 Veterans Boulevard, South San Francisco, California 94080, United States
‡ Departments of Metabolic Disorders, Comparative Biology and Safety Sciences and Pharmacokinetics and Drug Metabolism, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, United States
§ Array BioPharma Inc., 3200 Walnut Street, Boulder, Colorado 80301, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00140
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00140
*E-mail: pdransfi@amgen.com.

Glucokinase (GK) catalyzes the phosphorylation of glucose to glucose-6-phosphate. We present the structure–activity relationships leading to the discovery of AM-2394, a structurally distinct GKA. AM-2394 activates GK with an EC50 of 60 nM, increases the affinity of GK for glucose by approximately 10-fold, exhibits moderate clearance and good oral bioavailability in multiple animal models, and lowers glucose excursion following an oral glucose tolerance test in an ob/ob mouse model of diabetes.

PATENT
WO 2013086397
http://www.google.com/patents/WO2013086397A1?cl=en
COPYING ERROR
Example. 1734 t¾^Jtiyi¾rea

Step A: In 100 mL of DMA were corafeiaed 1 ^545miSO- -ll«omp ridinr2-yl)-3-i«e hir8a- (17.5 g, 70,5 ii!-!to!). 5-o:ieS:t}yI yiidlii~3- ). (9,24 g, S4.7 ΪΗΪΪΪΟ!}, sad CO · (10.1 g, 77.6 mmo!) mid heated to 90 *C for 5 days. After that time, the reaction was om lete a d to it was added water arid DCM and stirred vigorously for 3 hr. The resulting solid was isolated via vacuum .filtratiott nd the cake was wasted mill rater and DCM. The DCM in tli aqueous rime was dried vdth a stream of aidogeji aad vigorous sbrriug. Use resulting solid was then collected via vacuum filtration aad these solids were
Stirred vig rousl in f 0% MeOH irt EtOAc arid die res dtipg solid was colleeied. via vactiiars fiirfati m.
Trie two batches wen i coiiibiaed to yield I-(5-bmmo-4 5^»ie†fey pyiidin-3-yl xy)p Tidin-2- d 3~ metbySurea (I S J g, 5 3.7 om»)i, 76% yield).
S e .8: In 2 niL ofc ioxane

yI) iyridMJ-2-yios:y)pf¾ps3i-2-oI (0,098 g, 0.33 «ΜΠΟΪ), “ -i5-bs¾tao-4-{5-a3fidiy I py f idia-3 – ylosy)f5yridia-2-yl)-3-raethyl«rea (0.075 g, 0.22 tn ol.. t, and.2M poiass.ua» carbonate (0.33 ml, 0.67 m oi} artd tfets was s parged wi h At .for 10 mia before PdC§4dppl)*DCM (0.01 g g, 0.022 msttol) was added and dre reae!io a was sparged for aaotber 5 ma-, ir efore a was sealed and heated to 100 oversight The react! art was then loaded directly onto s ilica gel (50% acetone to PCM w4i. }%
MH40H) to afford i – (6′-(2diydioxy-2i-H5eth:ylpropCis:y) -4-{ 5″i:t re th y Ipy r i d i rt -3- io s y ) -3 ,3 : -bipyr id i rt -6- yl)-3-aie5¾ylt)rea φ.? 42 , 0.096 m ol, 43 % yield). !1 1 HMR (400 Mife, CDCij) 3 ppm 9.06 is,. !H),
S.33 is, 1H>, 8,27 (rs 2H), 8. Π (s, I H): K. (s, IHU 82 (dd, j-S.fi, 5.9 H HI), 1.21 (S !H), 6,«8
(d, Hz, i i i ). 6. ,4 (s:. m>, 4.25 (s, 2H), 2,87 (dj =4,3 Hz„ 3H) 2,37 (s, 3H>. 1 .33 is, <SH). Mass speetram (apci) tar/, : – 423.9 (M÷H).
REFERENCES
Novel Series of Potent Glucokinase Activators Leading to the Discovery of AM-2394
Paul J. Dransfield, Vatee Pattaropong, Sujen Lai, Zice Fu, Todd J. Kohn, Xiaohui Du, Alan Cheng, Yumei Xiong, Renee Komorowski, Lixia Jin, Marion Conn, Eric Tien, Walter E. DeWolf Jr., Ronald J. Hinklin, Thomas D. Aicher, Christopher F. Kraser, Steven A. Boyd, Walter C. Voegtli, Kevin R. Condroski, Murielle Veniant-Ellison, Julio C. Medina, Jonathan Houze, and Peter Coward
Publication Date (Web): May 23, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.6b00140
/////////Glucokinase activator, GKA, AM-2394, 1442684-77-6, AM 2394, Amgen
O=C(NC)NC1=CC(OC2=CC(C)=CN=C2)=C(C3=CC=C(OCC(C)(O)C)N=C3)C=N1
Obeticholic acid

Obeticholic acid
Obeticholic acid; 6-ECDCA; INT-747; 459789-99-2; 6-Ethylchenodeoxycholic acid; 6alpha-Ethyl-chenodeoxycholic acid;
(4R)-4-[(3R,5S,6R,7R,8S,9S,10S,13R,14S,17R)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]pentanoic acid
| Molecular Formula: | C26H44O4 |
|---|---|
| Molecular Weight: | 420.62516 g/mol |
NDA Filed
A farnesoid X receptor (FXR) agonist potentially for treatment of primary biliary cirrhosis and nonalcoholic steatohepatitis.
![]()
6-ECDCA; DSP-1747; INT-747
CAS No.459789-99-2
Obeticholic acid (abbreviated to OCA), is a semi-synthetic bile acid analogue which has the chemical structure 6α-ethyl-chenodeoxycholic acid. It has also been known as INT-747. It is undergoing development as a pharmaceutical agent for severalliver diseases and related disorders. Intercept Pharmaceuticals Inc. (NASDAQ symbol ICPT) hold the worldwide rights to develop OCA outside Japan and China, where it is licensed to Dainippon Sumitomo Pharma.[2]
REVIEW
INT-747(Obeticholic acid; 6-ECDCA) is a potent and selective FXR agonist(EC50=99 nM) endowed with anticholestatic activity. IC50 value: 99 nM(EC50) [1] Target: FXR agonist in vitro: The exposure of rat hepatocytes to 1 microM 6-ECDCA caused a 3- to 5-fold induction of small heterodimer partner (Shp) and bile salt export pump (bsep) mRNA and 70 to 80% reduction of cholesterol 7alpha-hydroxylase (cyp7a1), oxysterol 12beta-hydroxylase (cyp8b1), and Na(+)/taurocholate cotransporting peptide (ntcp) [2]. in vivo: In vivo administration of 6-ECDCA protects against cholestasis induced by E(2)17alpha [2]. high salt (HS) diet significantly increased systemic blood pressure. In addition, HS diet downregulated tissue DDAH expression while INT-747 protected the loss in DDAH expression and enhanced insulin sensitivity compared to vehicle controls [3]. Rats were gavaged with INT-747 or vehicle during 10 days after bile-duct ligation and then were assessed for changes in gut permeability, BTL, and tight-junction protein expression, immune cell recruitment, and cytokine expression in ileum, mesenteric lymph nodes, and spleen. After INT-747 treatment, natural killer cells and interferon-gamma expression markedly decreased, in association with normalized permeability selectively in ileum (up-regulated claudin-1 and occludin) and a significant reduction in BTL [4].
REFERENCES
| [1] | Verbeke L, et al. The FXR Agonist Obeticholic Acid Prevents Gut Barrier Dysfunction and Bacterial Translocation in Cholestatic Rats. Am J Pathol. 2015 Feb;185(2):409-19. |
| [2] | Ghebremariam YT, et al. FXR agonist INT-747 upregulates DDAH expression and enhances insulin sensitivity in high-salt fed Dahl rats. PLoS One. 2013 Apr 4;8(4):e60653. |
| [3] | Fiorucci S, et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J Pharmacol Exp Ther. 2005 May;313(2):604-12. |
| [4] | Pellicciari R, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002 Aug 15;45(17):3569-72. |
Invention and development
The natural bile acid, chenodeoxycholic acid, was identified in 1999 as the most active physiological ligand for the farnesoid X receptor (FXR), which is involved in many physiological and pathological processes. A series of alkylated bile acid analogues were designed, studied and patented by Roberto Pellicciari and colleagues at the University of Perugia, with 6α-ethyl-chenodeoxycholic acid emerging as the most highly potent FXR agonist.[3] FXR-dependent processes in liver and intestine were proposed as therapeutic targets in human diseases.[4] Obeticholic acid is the first FXR agonist to be used in human drug studies.

Clinical studies
OCA is undergoing development in phase 2 and 3 studies for specific liver and gastrointestinal disorders.[5]
Primary biliary cirrhosis
Primary biliary cirrhosis (PBC) is an auto-immune, inflammatory liver disease which produces bile duct injury, fibrosis, cholestasisand eventual cirrhosis. It is much more common in women than men and can cause jaundice, itching (pruritus) and fatigue.Ursodeoxycholic acid therapy is beneficial, but the disease often progresses and may require liver transplantation.[6] Animal studies suggested that treatment with FXR agonists should be beneficial in cholestatic diseases such as PBC.[7] OCA at doses between 10 mg and 50 mg was shown to provide significant biochemical benefit, but pruritus was more frequent with higher doses.[8][9] The results of a randomized, double-blind phase 3 study of OCA, 5 mg or 10 mg, compared to placebo (POISE) were presented in April 2014, and showed that the drug met the trial’s primary endpoint of a significant reduction in serum alkaline phosphatase, abiomarker predictive of disease progression, liver transplantation or death.[10]

Nonalcoholic steatohepatitis (NASH)
Non-alcoholic steatohepatitis is a common cause of abnormal liver function with histological features of fatty liver, inflammation andfibrosis. It may progress to cirrhosis and is becoming an increasing indication for liver transplantation. It is increasing in prevalence. OCA is proposed to treat NASH.[11] A phase 2 trial published in 2013 showed that administration of OCA at 25 mg or 50 mg daily for 6 weeks reduced markers of liver inflammation and fibrosis and increased insulin sensitivity.[12]
The Farnesoid X Receptor Ligand Obeticholic Acid in Nonalcoholic Steatohepatitis Treatment (FLINT) trial, sponsored by NIDDK, was halted early in January 2014, after about half of the 283 subjects had completed the study, when a planned interim analysis showed that a) the primary endpoint had been met and b) lipid abnormalities were detected and arose safety concerns. Treatment with OCA (25 mg/day for 72 weeks) resulted in a highly statistically significant improvement in the primary histological endpoint, defined as a decrease in the NAFLD Activity Score of at least two points, with no worsening of fibrosis. 45% (50 of 110) of the treated group had this improvement compared with 21% (23 of 109) of the placebo-treated controls.[13] However concerns about longterm safety issues such as increased cholesterol and adverse cardiovascular events may warrant the concomitant use of statins in OCA-treated patients.[14]
Portal hypertension
Animal studies suggest that OCA improves intrahepatic vascular resistance and so may be of therapeutic benefit in portal hypertension.[15] An open label phase 2a clinical study is under way.
Bile acid diarrhea
Bile acid diarrhea (also called bile acid malabsorption) can be secondary to Crohn’s disease or be a primary condition. Reduced median levels of FGF19, an ileal hormone that regulates increased hepatic bile acid synthesis, have been found in this condition.[16] FGF19 is potently stimulated by bile acids and especially by OCA.[17] A proof of concept study of OCA (25 mg/d) has shown clinical and biochemical benefit.[18]

SYNTHESIS
Take 10g of austempered cholic acid 89.6% purity crude (single hetero greater than 2%), 3 times its weight of acetone and added to their 20% by weight of triethylamine was added, was heated at reflux for 2h, cooled slowly to 10 ° C, the precipitated crystals were filtered to give Obey acid organic amine salt crystals.
Acidification [0020] The organic amine salts Obey acid crystals were dissolved with purified water after 10wt% by mass percentage to the PH value of 2.0 with dilute hydrochloric acid, filtered and dried to give purified Obey acid.
[0021] The purified Obey acid ethyl acetate dissolved by heating and then cooling to 20 ° C, the precipitated crystals were filtered and dried to obtain a purity of 98.7% recrystallization Obey acid (single hetero less than 0.1%), recovery was 84.5%.
PATENT
Obey acid (as shown in formula I) is a semi-synthetic chenodeoxycholic acid derivative, for the treatment of high blood pressure, the portal vein and liver diseases, including primary biliary cirrhosis, bile acid diarrhea, non-alcoholic steatohepatitis. Obey acid through activation of FXR receptors play a role, FXR is a nuclear receptor, is expressed mainly in the liver, intestine, kidney, and it can be adjusted with acids fat and carbohydrate metabolism related gene expression in bile, also regulate immune response. FXR activation can inhibit the synthesis of bile acids, bile acids prevent excessive accumulation of toxic reactions caused.
WO2002072598 debuted Obey acid preparation method (shown below), which in strong alkaline conditions to give compound VII by alkylation with ethyl iodide compound VI directly, through reducing compound VII prepared and carboxy deprotection Obey acid. However, due to direct alkylation with ethyl iodide poor selectivity and yield is too low, the synthesis process is difficult to achieve amplification synthesis.
Obey bile acid synthesis (WO2002072598)
WO2006122977 above synthesis process has been improved (see below), the process by the silicon compound IX into protected enol compound X, compound X and acetaldehyde after dehydration condensation to give compound Vb, after compound Vb in alkaline conditions under palladium on carbon hydrogenation to give compound XI, after a carbonyl compound XI reduction system Obey acid.
Obey bile acid synthesis (WO2006122977)
The synthetic process can be achieved, although the enlarged combined, however, the compound Vb produce large amounts of byproducts under strongly alkaline conditions palladium on carbon hydrogenation process for preparing high temperature and strong alkaline compound XI during this step leading to the separation of income a lower rate (about 60%), low yield of this step may be due to compound Vb and XI in unprotected hydroxy dehydration occurs under strongly basic (30% NaOH) and high temperature (95-105 ℃) conditions side effects caused.
synthesis of bile acids Obey,
Obey bile acid synthesis route is as follows:
PATENT
According to Obey acid 6 was prepared in the form of C Patent Document W02013192097A1 reaction of Example 1, as follows:
The 3 a – hydroxy -6 a – ethyl-7-keto -5 P – 24-oic acid (. 86g, 205 4mmol), water (688mL) and 50% (w / w) hydrogen sodium hydroxide solution (56. 4mL) and the mixture of sodium borohydride (7. 77g, 205. 4mmol) in a mixture of 50% (w / w) sodium hydroxide solution (1.5 mL of) and water (20 mL) in 90 ° in C to 105 ° C reaction. Was heated with stirring under reflux for at least 3 hours, the reaction was completed, the reaction solution was cooled to 80 ° C. Between 30 ° C at 50 ° C of citric acid (320. 2g, anhydrous), a mixture of n-butyl acetate (860 mL of) and water (491mL) to ensure an acidic pH of the aqueous phase was separated. Evaporation of the organic phase was distilled to give the residue was diluted with n-butyl acetate, slowly cooled to 15 ° C to 20 ° C, centrifugation. The crude product was crystallized from n-butyl acetate. After Obey acid isolated by n-butyl acetate (43mL, 4 times), dried samples were dried at 80 ° C under vacuum. To give 67. 34g (77. 9%) crystalline form C Obey acid.
References
- Gioiello, Antimo; Macchiarulo, Antonio; Carotti, Andrea; Filipponi, Paolo; Costantino, Gabriele; Rizzo, Giovanni; Adorini, Luciano; Pellicciari, Roberto (April 2011). “Extending SAR of bile acids as FXR ligands: Discovery of 23-N-(carbocinnamyloxy)-3α,7α-dihydroxy-6α-ethyl-24-nor-5β-cholan-23-amine”. Bioorganic & Medicinal Chemistry 19 (8): 2650–2658.doi:10.1016/j.bmc.2011.03.004.
- Wall Street Journal. “A $4 Billion Surprise for 45-Person Biotech”. Retrieved10 January 2014.
- Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM (August 2002). “6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity”. J. Med. Chem. 45(17): 3569–72. doi:10.1021/jm025529g. PMID 12166927.
- Rizzo G, Renga B, Mencarelli A, Pellicciari R, Fiorucci S (September 2005). “Role of FXR in regulating bile acid homeostasis and relevance for human diseases”. Curr. Drug Targets Immune Endocr. Metabol. Disord. 5 (3): 289–303. doi:10.2174/1568008054863781.PMID 16178789.
- “ClinicalTrials.gov”.
- Hirschfield GM, Gershwin ME (January 2013). “The immunobiology and pathophysiology of primary biliary cirrhosis”. Annu Rev Pathol 8: 303–30. doi:10.1146/annurev-pathol-020712-164014. PMID 23347352.
- Jump up^ Lindor, KD (May 2011). “Farnesoid X receptor agonists for primary biliary cirrhosis”.Current opinion in gastroenterology 27 (3): 285–8.doi:10.1097/MOG.0b013e32834452c8. PMID 21297469.
- Jump up^ Fiorucci S, Cipriani S, Mencarelli A, Baldelli F, Bifulco G, Zampella A (August 2011). “Farnesoid X receptor agonist for the treatment of liver and metabolic disorders: focus on 6-ethyl-CDCA”. Mini Rev Med Chem 11 (9): 753–62. doi:10.2174/138955711796355258.PMID 21707532.
- Jump up^ Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, Kowdley KV, Vincent C, Bodhenheimer HC, Parés A, Trauner M, Marschall HU, Adorini L, Sciacca C, Beecher-Jones T, Castelloe E, Böhm O, Shapiro D (2015). “Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid”.Gastroenterology 148 (4): 751–61.e8. doi:10.1053/j.gastro.2014.12.005.PMID 25500425.
- Jump up^ Intercept Pharma. “Press release: Intercept Announces Positive Pivotal Phase 3 POISE Trial Results”. Retrieved March 27, 2014.
- Jump up^ Adorini L, Pruzanski M, Shapiro D (September 2012). “Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis”. Drug Discov. Today 17 (17–18): 988–97.doi:10.1016/j.drudis.2012.05.012. PMID 22652341.
- Jump up^ Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D (September 2013). “Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease”. Gastroenterology 145 (3): 574–82.e1.doi:10.1053/j.gastro.2013.05.042. PMID 23727264.
- Jump up^ Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E (2015). “Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial”. Lancet 385 (9972): 956–65.doi:10.1016/S0140-6736(14)61933-4. PMID 25468160.
- Jump up^ http://www.thestreet.com/story/12714549/1/intercept-pharma-government-scientists-spar-over-negative-safety-of-liver-drug-emails-show.html?puc=yahoo&cm_ven=YAHOO
- Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, Vander Elst I, Windmolders P, Vanuytsel T, Nevens F, Laleman W (November 2013). “Obeticholic acid, a farnesoid-X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats”. Hepatology 59 (6): :2286–98. doi:10.1002/hep.26939. PMID 24259407.
- Walters JR, Tasleem AM, Omer OS, Brydon WG, Dew T, le Roux CW (November 2009). “A new mechanism for bile acid diarrhea: defective feedback inhibition of bile acid biosynthesis”. Clin. Gastroenterol. Hepatol. 7 (11): 1189–94.doi:10.1016/j.cgh.2009.04.024. PMID 19426836.
- Zhang JH, Nolan JD, Kennie SL, Johnston IM, Dew T, Dixon PH, Williamson C, Walters JR (May 2013). “Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids”. Am. J. Physiol. Gastrointest. Liver Physiol. 304 (10): G940–8.doi:10.1152/ajpgi.00398.2012. PMC 3652069. PMID 23518683.
- Walters JR, Johnston IM, Nolan JD, Vassie C, Pruzanski ME, Shapiro DA (January 2015). “The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid”. Aliment. Pharmacol. Ther. 41 (1): 54–64.doi:10.1111/apt.12999. PMID 25329562.
External links
- “Intercept pharmaceuticals”.
- NashBiotechs Several articles on drug candidates in NASH
| Patent ID | Date | Patent Title |
|---|---|---|
| US8546365 | 2013-10-01 | Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions |
| US8377916 | 2013-02-19 | Steroids as agonists for FXR |
| US8058267 | 2011-11-15 | STEROIDS AS AGONISTS FOR FXR |
| US7994352 | 2011-08-09 | Process for Preparing 3a(Beta)-7a(Beta)-Dihydroxy-6a(Beta)-Alkyl-5Beta-Cholanic Acid |
| US7932244 | 2011-04-26 | Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions |
| US7786102 | 2010-08-31 | Steroids as agonists for FXR |
| US2009062526 | 2009-03-05 | NOVEL METHOD OF SYNTHESIZING ALKYLATED BILE ACID DERIVATIVES |
| US7138390 | 2006-11-21 | Steroids as agonists for fxr |
| US2005107475 | 2005-05-19 | Methods of using farnesoid x receptor (frx) agonists |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016074419 | 2016-03-17 | Preparation and Uses of Obeticholic Acid |
| US2015359805 | 2015-12-17 | Bile Acid Derivatives as FXR Ligands for the Prevention or Treatment of FXR-Mediated Diseases or Conditions |
| US2015166598 | 2015-06-18 | Steroids as Agonists for FXR |
| US2014371190 | 2014-12-18 | Farnesoid X receptor modulators |
| US2014186438 | 2014-07-03 | COMPOSITIONS COMPRISING EPA AND OBETICHOLIC ACID AND METHODS OF USE THEREOF |
| US2014148428 | 2014-05-29 | Treatment of Pulmonary Disease |
| US2014057886 | 2014-02-27 | Bile Acid Derivatives as FXR Ligands for the Prevention or Treatment of FXR-Mediated Diseases or Conditions |
| US2014024631 | 2014-01-23 | Steroids as Agonists for FXR |
| US2013345188 | 2013-12-26 | Preparation and Uses of Obeticholic Acid |
| US8546365 | 2013-10-01 | Bile acid derivatives as FXR ligands for the prevention or treatment of FXR-mediated diseases or conditions |

 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(3α,5β,6α,7α)-6-Ethyl-3,7-dihydroxycholan-24-oic acidOR (4R)-4-[(3R,5S,6R,7R,8S,9S,10S,13R,14S,17R)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]pentanoic acid |
|
| Clinical data | |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 459789-99-2 |
| ATC code | A05AA04 (WHO) |
| PubChem | CID 447715 |
| IUPHAR/BPS | 3435 |
| ChemSpider | 394730 |
| UNII | 0462Z4S4OZ |
| KEGG | C15636 |
| ChEMBL | CHEMBL566315 |
| Synonyms | 6α-ethyl-chenodeoxycholic acid; INT-747 |
| Chemical data | |
| Formula | C26H44O4 |
| Molar mass | 420.62516 g/mol |
/////////6-ECDCA, DSP-1747, INT-747, 459789-99-2, Obeticholic acid
CC[C@@H]1[C@@H]2C[C@@H](CC[C@@]2([C@H]3CC[C@]4([C@H]([C@@H]3[C@@H]1O)CC[C@@H]4[C@H](C)CCC(=O)O)C)C)O
CCC1C2CC(CCC2(C3CCC4(C(C3C1O)CCC4C(C)CCC(=O)O)C)C)O
Flow Chemistry Symposium & Workshop 16-17 June at IICT, Hyderabad, India

MESSAGE FROM VIJAY KIRPALANI
A 2-day FLOW CHEMISTRY Symposium + Workshop has been organized on 16-17 June 2016 at
IICT Hyderabad, India by Flow Chemistry Society – India Chapter (in collaboration with IICT-Hyderabad & IIT-B)
with speakers from India, UK, Netherlands and Hungary.
Both days have intensive interactive sessions on the theory and industrial applications of Flow Chemistry followed by live demonstrations using 7 different Flow Reactor platforms — from microliters to 10,000 L/day industrial scale.
The Fees are Rs. 5,000 for Industry Delegates and Rs. 2,500 for Academic Delegates (+15% Service Tax) : contact : vk@pi-inc.co or msingh@cipla.com
I have attached a detailed program and look forward to meeting you at the event..


Best regards
Vijay Kirpalani
President
Flow Chemistry Society – India Chapter
email : vk@pi-inc.co
Tel: +91-9321342022 // +91-9821342022
ABOUT
IICT, Hyderabad, India



Dr. S. Chandrasekhar,
Director
CSIR-Indian Institute of Chemical Technology (IICT)
Hyderabad, India
SPEAKERS
Mr Vijay Kirpalani
President
Flow Chemistry Society – India Chapter, INDIA

Dr Charlotte Wiles , CHEMTRIX
UK &THE NETHERLANDS,UNIV OF HULL

Prof Anil Kumar( IIT-B), INDIA




Head of Research and Development at ThalesNano Inc, HUNGARY
GLIMPSE OF HYDERABAD INDIA






/////Flow Chemistry, Symposium, Workshop, 16-17 June, IICT, Hyderabad, India
