
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
I (Anthony Crasto) am Editorial Board member for our Journal of Analytical & Pharmaceutical Research


I am on editorial board ……… Editorial Board member for our Journal of Analytical & Pharmaceutical Research………http://medcraveonline.com/JAPLR/editorial-board
This is possible with your cooperation and support
read…….http://medcraveonline.com/JAPLR/JAPLR-02-00010.pdf
http://medcraveonline.com/JAPLR/JAPLR-02-00011.pdf
Tackling the Challenges with Poorly Soluble Drugs
http://medcraveonline.com/JAPLR/JAPLR-01-00001.pdf
BTI-320 (formerly PAZ320), Soluble mannan polysaccharides from Boston Therapeutics for the treatment of type 2 diabetes in combination with oral agents or insulin
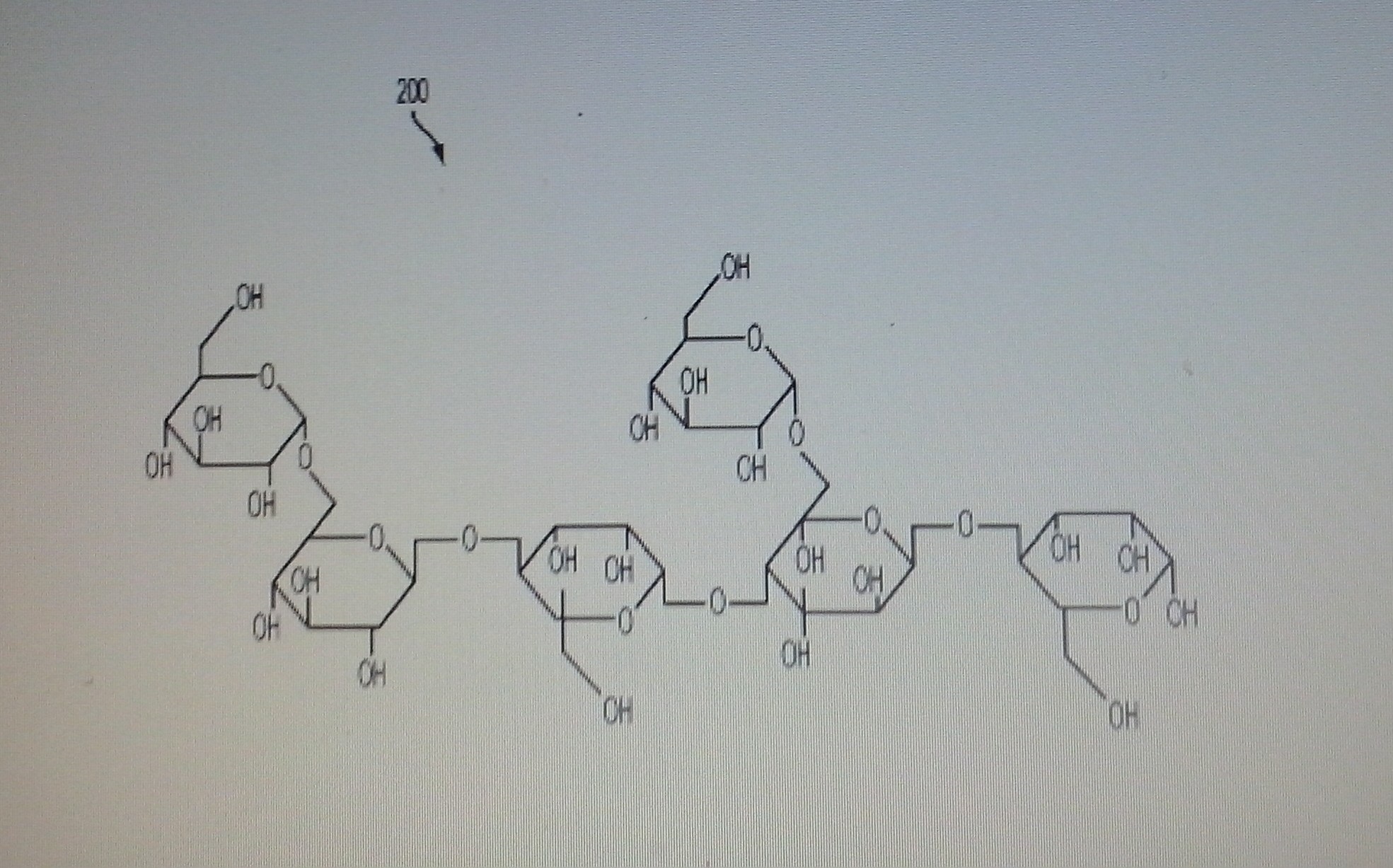
BTI-320 (formerly PAZ320)
PAZ 320
Non-insulin dependent diabetes
Alpha-glucosidase inhibitor; Hydrolase inhibitor; Sucrose alpha-glucosidase inhibitor
Composition of chemically purified (fractionation) soluble mannan polysaccharides from legume’s seeds
BTI-320 is in phase II clinical development at Boston Therapeutics for the treatment of type 2 diabetes in combination with oral agents or insulin, and also for the treatment of high-risk patients with pre-diabetes. A chewable tablet formulation is being developed. The product is already available as dietary supplement.
| Company | Boston Therapeutics Inc. |
| Description | Chewable polysaccharide that inhibits alpha glucosidase |
| Molecular Target | |
| Mechanism of Action | Alpha glucosidase inhibitor |
| Therapeutic Modality | Macromolecule: Polysaccharide |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |


PATENT
http://www.google.co.in/patents/WO2012061675A1?cl=en
A composition of chemically purified soluble mannans from legumes’ seeds (e.g. Ceratonia siliqua, Cæsalpinia spinosa Trigonelle foenum-graecum, and Cyamopsis tetragonolobus) and their use in the assembly of palatable dietary supplements is disclosed herein. The fractionation process provides high-quality physiologically soluble, chemically modified and purified homogeneous size polysaccharide fibers, devoid of natural impurities, for example proteins, alkaloids, glycoalkaloids, and/or environmental impurities including heavy metals, agricultural residues and microbial toxins. This process provides hypoallergenic dietary fibers devoid of any potential allergens, cytotoxins, and gastrointestinal toxins. A sequential process for assembly of the soluble fibers with plurality of molecular weights to create a time controlled dissolution of the functional high and low molecular weight fibers for improving solubility and palatability with improved dietary performance in the oral and gastro-intestinal system is also disclosed herein.
Fig. 1 illustrates a block flow diagram of an embodiment of a method for recovering purified mannan polysaccharides;
Fig. 2 illustrates a chemical structure of a mannan polysaccharide;
Fig. 3 illustrates a block flow diagram of an embodiment of a method for recovering high molecular weight (HMW) purified mannan polysaccharides;
Fig. 4 illustrates a block flow diagram of an embodiment of a method for recovering low molecular weight (LMW) purified mannan polysaccharides;
REFERENCES
https://clinicaltrials.gov/show/NCT02060916
https://clinicaltrials.gov/show/NCT02358668
BTI-320, a nonsystemic novel drug to control glucose uptake into the bloodstream, functions as a competitive inhibitor of sugar hydrolyzing enzymes
75th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 5-9, Boston) 2015, Abst 974-P
Boston Therapeutics’ Hong Kong Affiliate Advance Pharmaceutical’s BTI-320 Clinical Trial Reaches Mid-Point by Enrolling 30 Patients at the Chinese University of Hong Kong
Boston Therapeutics Press Release 2015, July 08
Insight into the molecular mechanism of action of BTI320, a non-systemic novel drug to control serum glucose levels in individuals with diabetes50th Annu Meet Eur Assoc Study Diabetes (EASD) (September 15-19, Vienna) 2014, Abst 545
////BTI-320, PAZ320, PHASE 2, BTI 320, PAZ 320, Macromolecule, Polysaccharide, Non-insulin dependent diabetes, Alpha-glucosidase inhibitor, Hydrolase inhibitor, Sucrose alpha-glucosidase inhibitor, phase II clinical development, Boston Therapeutics, Soluble mannan polysaccharides
Composition of chemically purified (fractionation) soluble mannan polysaccharides from legume’s seeds
POLYMER OF BELOW
CAS 9036-88-8, 51395-96-1
| refractive index : | 78.5 ° (C=1.4, H2O) |
Ailes;MANNAN;K-41K1;D-Mannan;NSC 174478;NSC 174479;NSC 174481;NSC 307194;NSC 174477;NSC 174473

| Chemical name: | 1,6-Anhydro-β-D-mannopyranose |
| Synonyms: | 1,6-Anhydro-D-mannose; 1,6-Anhydromannose; Mannosan; NSC 226600; |
| CAS Number: | 14168-65-1 |
| Possible CAS #: | NA |
| Molecular form.: | C₆H₁₀O₅ |
| Appearance: | White to Pale Beige Solid |
| Melting Point: | 182-184°C |
| Mol. Weight: | 162.14 |
Summary:
Mannans are major constitutents of hemicelluloses in plant tissue and are polymers composed of β(1→4)-linked mannose and glucose residues. Some contain galactopyranosyl side chains (see a galactomannan).
Slightly galactosylated mannans (4% galactose), considered as linear β(1→4)-D-mannans, have been isolated from the seed endosperm of vegetable ivory nut ( Phytelephas macrocarpa) and date ( Phoenix dactylifera) .
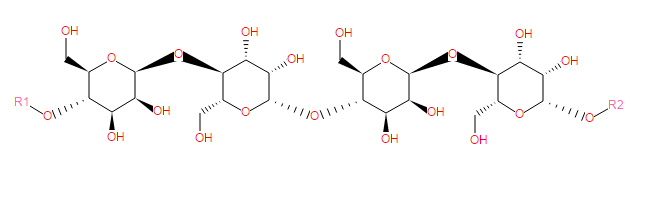
Glycan icon:

Child Classes: a 1,6-α-D-mannan backbone (0), a galactoglucomannan (0), a galactomannan (0), a glucomannan (0), a mannan oligosaccharide (1)
SMILES: C(O)C4(C(O[R1])C(O)C(O)C(OC3(C(O)C(O)C(OC2(C(O)C(O)C(OC1(C(O)C(O)C(O[R2])OC(CO)1))OC(CO)2))OC(CO)3))O4)
CAS:9036-88-8,
//////////
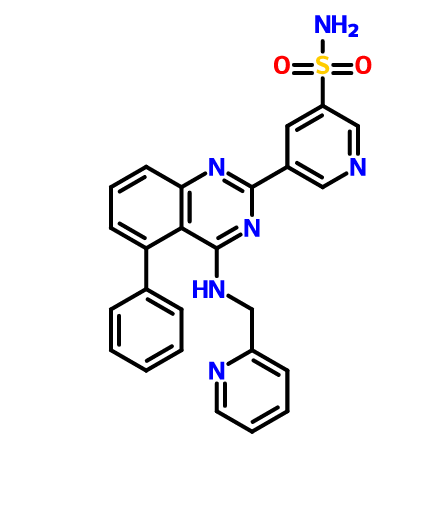
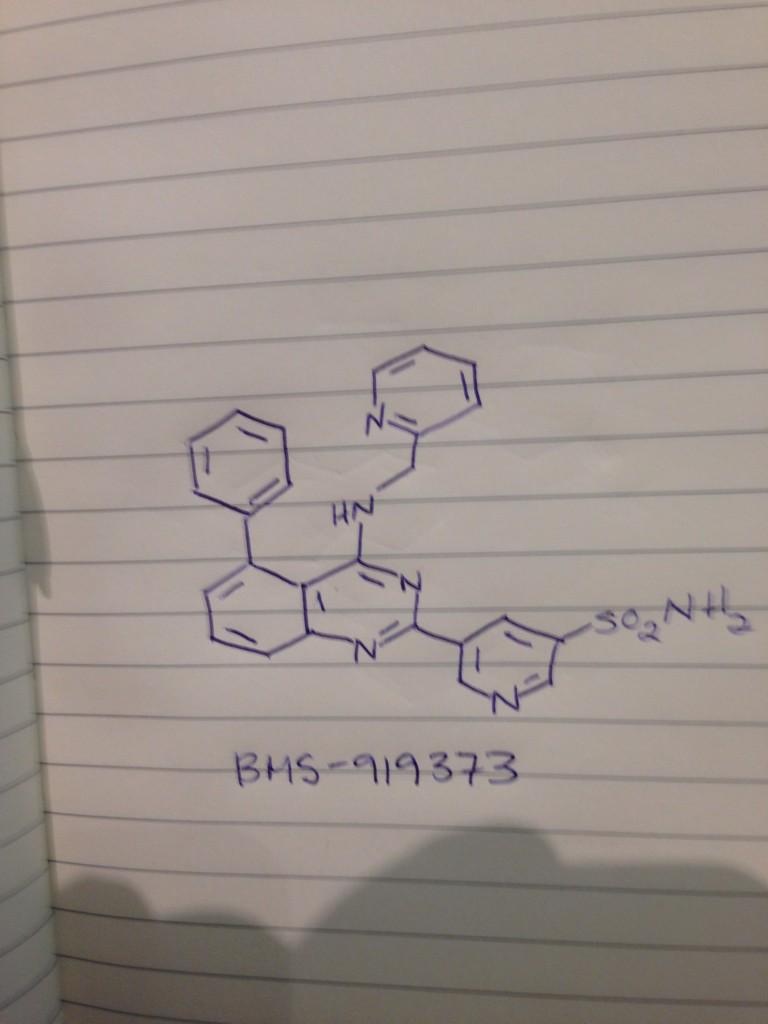
BMS 919373
 .
.
Bethany Halford on Twitter: “BMS-919373, from $BMS for …https://twitter.com/beth_halford/status/634105343719682048
Aug 19, 2015 – BMS–919373, from $BMS for atrial fibrillation #ACSBoston MEDI 1st disclosures @bmsnews pic.twitter.com/y3D4Yv2U7M.
BMS 919373
- Phase IIParoxysmal atrial fibrillation
- Phase IAcute coronary syndromes; Atrial fibrillation
-
- 01 Oct 2014Phase-I clinical trials in Atrial fibrillation in Canada (PO) (NCT02153437)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in Canada (PO) (NCT02156076)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in USA (PO)
- https://clinicaltrials.gov/ct2/show/NCT02153437
- https://clinicaltrials.gov/ct2/show/NCT02156076
- CAS HCL SALT 1272356-77-0
| Latest Stage of Development | Phase I |
| Standard Indication | Fibrillation |
| Indication Details | Treat atrial fibrillation |
Synthesis
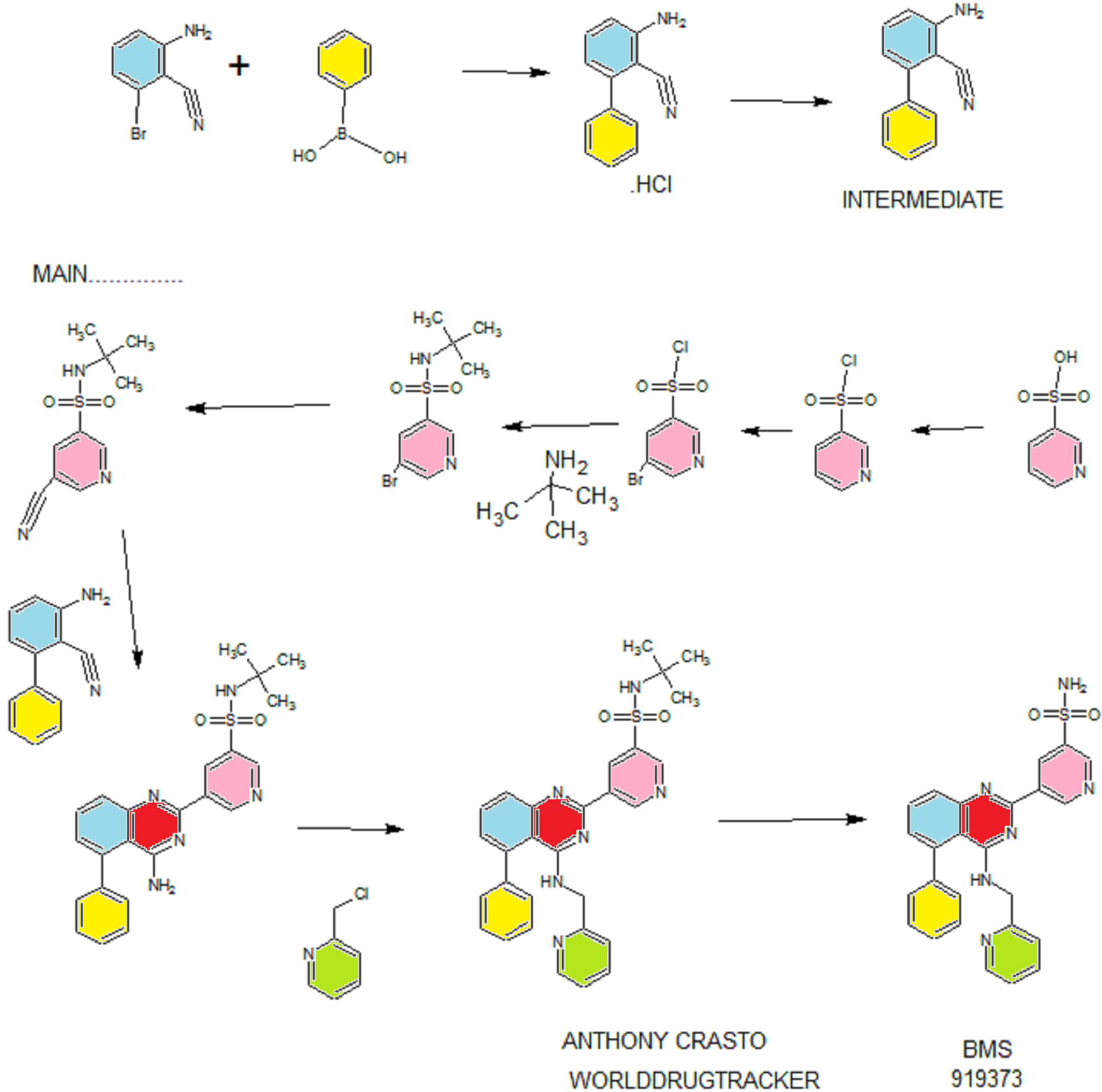
PATENT
WO 2011028741
http://www.google.co.in/patents/WO2011028741A1?cl=en
EXAMPLE 7
5-(5-Phenyl-4-(pyridin-2-ylmethylamino)quinazolin-2-yl)pyridine-3-sulfonamide

Step 1. Preparatio -Bromopyridine-3 -sulfonamide

See also U.S. Publication Nos. 2006/217387 and 2006/375834, and J. Org. Chem., 54:389 (1989). A mixture of pyridine-3 -sulfonic acid (10.3 g, 64.8 mmol), phosphorous pentachloride (20.82 g, 100 mmol) and phosphorous oxychloride (10 mL, 109 mmol) was heated to reflux where it stirred for 4h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was evaporated to dryness under reduced pressure to yield a residue. The residue was treated with bromine (6.00 mL, 1 16 mmol) and then heated to reflux where it stirred for 14h. After this time, the reaction mixture was cooled to 0 °C and then a saturated solution of NH4OH in ¾0 (40 mL) was slowly added. The resulting mixture was allowed to warm to room temperature where it stirred for 30 min. The reaction mixture was then filtered and the filter cake was washed with hexane to afford 5 -bromopyridine-3 -sulfonamide (6.0 g) as an off- white solid. The product was used without further purification. LCMS Method Q: retention time 0.75 min; [M+l] = 237.0.
Step 2. Preparation of pyridine-3-sulfonamide-5-ylboronic acid pinacol ester

See also WO2008/150827 Al and WO2008/144463. A mixture of 5- bromopyridine-3 -sulfonamide (1.5 g, 6.33 mmol), bis(pinacolato)diboron (2.41 g, 9.5 mmol) and potassium acetate (1.86 g, 19.0 mmol) in 1,4-dioxane (15 mL) was degassed with nitrogen for 15 min then (l, l’-bis(diphenylphosphino)- ferrocene)palladium (II) chloride dichloromethane complex (232 mg, 0.317 mmol) was added and the resulting mixture was degassed again with nitrogen for 10 min. At the conclusion of this period, the reaction mixture was heated in a microwave at 120 °C for 45 min. After this time, the reaction mixture was filtered through CELITE® and the filtrate was concentrated under reduced pressure to provide pyridine-3- sulfonamide-5-ylboronic acid pinacol ester (740 mg) as a brown solid. The product was used without further purification. XH NMR (400 MHz, DMSO-d6) δ (ppm): 8.83 (s, 1H), 8.80 (s, 1H), 8.26 (s, 1H), 7.56-7.74 (bs, 2H), 1.17 (s, 12H).
Step 3. Example 7

To a solution of 2-chloro-5-phenyl-N-(pyridin-2-ylmethyl)quinazolin-4- amine (150 mg, 0.43 mmol) in 1,4-dioxane (6 mL) and ¾0 (1 mL) under nitrogen was added pyridine-3-sulfonamide-5-ylboronic acid pinacol ester (185 mg, 0.65 mmol), and potassium carbonate (119 mg, 0.86 mmol). Upon completion of addition, the mixture was degassed with nitrogen for 15 minutes and then (1, 1′- bis(diphenylphosphino)ferrocene)palladium (II) chloride dichloromethane complex (31 mg, 0.043 mmol) was added. The resulting mixture was again degassed with nitrogen for 10 min. After this time, the mixture was heated to 90 °C where it stirred for 16h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was quenched by the addition of water and then transferred to a separation funnel. The aqueous layer was extracted with ethyl acetate. The combined organic portions were washed with water and saturated NaCl, dried over Na2S04, filtered and concentrated under reduced pressure. The resulting concentrate was purified by preparative TLC using 5% methanol in dichloromethane to afford Example 7 (50 mg) as a brown solid. ‘H NMR (400 MHz, DMSO-d6) δ (ppm): 9.81 (s, 1H), 9.17 (s, 1H), 9.09 (s, 1H), 8.24 (d, J= 4.4 Hz, 1H), 7.94 (d, J=7.2 Hz, 1H), 7.86 (t, J= 7.6 Hz, 1Η),7.75-7.72 (t, J= 7.6 Hz, 3H), 7.59-7.51 (m, 5H), 7.34 (d, J=7.2 Hz, 2H), 7.24 (t, J=6.4 Hz, 1H), 6.98 (t, J= 3.2 Hz, 1H), 4.77 (d, J= 4.0 Hz, 2H). LCMS Method Q: retention time 1.39 min; [M+l] = 469.0. HPLC Method B: purity 98.1%, retention time = 8.74 min. [00120] Alternatively, Example 7 can be synthesized as follows:
Step 1. Preparation of 5-Bromo-pyridine-3-sulfonyl chloride

PC15 (2.95 Kg, 14.16 moles) and POCl3 (2.45 Kg, 15.98 moles) were added into pyridine-3 -sulfonic acid (1.5 Kg, 9.42 mol) in 10 L RB flask equipped with mechanical stirrer under inert atmosphere. The reaction mass was heated to 120- 125°C where it stirred for 18 h. After this time, the reaction progress was monitored by HPLC, which indicated the reaction was complete. Excess POCI3 was removed under vacuum to give a residue. The residue was cooled to ambient temperature and bromine (1.2 Kg, 7.5 moles) was added. Upon completion of addition, the resulting mixture was heated to 120-125°C where it stirred for 5 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then poured into ice-water (10 L), and the resulting mixture was extracted with DCM (10.5 Lx2). The DCM extracts were combined and the solvent was removed under vacuum to yield crude product (1.8 Kg, 74.4% yield).
Step 2. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

Crude 5 -bromopyridine-3-sulfonyl chloride from step 1 above was dissolved in THF (14 L, 8 vol) and then transferred to a 20 L RB flask equipped with mechanical stirrer under inert atmosphere. The solution was cooled to 0-5°C and tert- butyl amine (1.95 Kg, 26.66 moles) was added at 0-5°C. Upon completion of addition, the reaction mixture was warmed to ambient temperature where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The solvent was evaporated under vacuum to give a thick residue. The residue was dissolved in ethyl acetate (18 L, 12 vol). The organic layer was separated, washed with water (9 L, 5 vol) and then concentrated under vacuum to yield a residue. Hexanes (9 L, 5 vol) were added to the residue and the product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.5 Kg, 54.28% overall yield). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 8.99 (d, J = 2Hz, 1H), 8.81 (d, J= 2 Hz, 1H), 8.29 (t, J= 2Hz, 1H). [M++l] = 293. Step 3. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

5 -Bromo-N-tert-butylpyridine-3 -sulfonamide (1.5 Kg, 5.11 moles) was dissolved in dimethylformamide (7.5 L, 5 vol) and the solution was added to a 20 L glass-lined reactor equipped with mechanical stirrer. The solution was degassed with nitrogen for 30 min. After this time, potassium ferrocyanide trihydrate (867 g, 2.05 moles), sodium carbonate (1.08 Kg, 10.189 moles), copper (I) iodide (73.2 g, 0.374 moles) and dichloro-bis (triphenylphosphine) palladium (II) (71.6 g, 0.102 moles) were added. Upon completion of addition, the reaction mixture was heated to 120- 125°C where it stirred for 4 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then filtered through a celite bed. Water (18 L, 12 vol) was added into the filtrate and the resulting mixture was extracted with ethyl acetate (7.5L*2). The organic layers were combined, washed with water and then concentrated to yield a thick residue. Hexanes (7.5 L, 5 vol) were added to the residue. The product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.0 Kg, 82.8% yield, 89% purity by HPLC). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 9.21 – 9.24 (d,d J= 7.2Hz, 3.2Hz, 2H), 8.70-8.71(m,lH), 7.98 (s, lH). [M++l] = 239.2.
Step 4. Preparation of 3-aminobiphenyl-2-carbonitrile

2-Amino-6-bromo-benzonitrile (1.0 Kg, 5.07 moles) and toluene (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer under inert atmosphere. Potassium acetate (996 g, 10.16 moles) and phenylboronic acid (866, 7.10 moles) were added into the solution and the solution was degassed with nitrogen for 30 min. After this time, dichloro-bis (triphenylphosphine) palladium (II) (17.8 g, 0.025 moles) was added to the reaction mixture at ambient temperature. The mixture was heated to 110°C, where it stirred for 17 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was completed. The reaction mixture was filtered through a celite bed. The filtrate was transferred back to the reactor and concentrated hydrochloric acid (-35%, 2 L, 2 vol) was charged to the reactor at ambient temperature. The HCl salt of the title compound precipitated out from the reaction and was collected by filtration. The HCl salt was transferred into the 20 L reactor and then made basic with 10% NaOH solution (pH 8-9). The resulting product was extracted with ethyl acetate (10 L, 10 vol). The ethyl acetate layer was washed with water (5 L, 5 vol) and then the solvent was evaporated under vacuum to give a residue. Hexanes (5 L, 5 vol) were added to the residue at 35-40°C, and the resulting slurry was cooled to ambient temperature. Once at the prescribed temperature, the product was collected by filtration to provide a pale yellow solid (802 g, 81.4%, 99% by HPLC). XH NMR (DMSO-D6, 400 MHz, δ ppm); 7.43-7.52 (m, 5H), 7.33-7.37 (m, 1H), 6.83 (d, J=8Hz, 1H), 6.62 (d, J=8Hz, 1H), 6.1 (s, 2H). ES-MS: [M++l] = 194.23.
Step 5. Preparation of 5-(4-amino-5-phenylquinazolin-2-yl)-N-tert-butylpyridine-3-

3-Aminobiphenyl-2-carbonitrile (1028 g, 5.30 moles), 5-bromo-N-tert- butylpyridine-3 -sulfonamide (1440 g, 5.55 moles) and 1,4-dioxane (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. Sodium tert-butoxide (1.275 Kg 12.870 moles) was added to the solution portion-wise at 20- 30°C. Upon completion of addition, the reaction mixture was heated to reflux where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 30-35°C and then poured into water (40 L, 40 vol). The resulting mixture was extracted with DCM (20 L*2). The DCM layers were combined, washed with water (10 L, 10 vol) and then dried over sodium sulfate. The solvent was evaporated under vacuum to give a residue. Isopropyl alcohol (1.2 L, 1.2 vol) was added to the residue at 40°C. The resulting precipitate slurry was cooled to 10-15°C and then stirred for 2 h. After this time, the precipitate was collected by filtration and dried at 50°C for 16 h to yield the product (1.9 Kg, 82.9% yield, 99% purity by HPLC). Ή NMR (DMSO-D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.11 (s, 2H), 7.83-7.94 (m, 4H), 7.49-7.60 (m, 5H), 7.31 (d,d /=6.8Hz,1.2Hz, 1H). ES-MS: [M++l] = 433.53.
Step 6. Preparation of N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide

2-(Chloromethyl) pyridine hydrochloride (564 g, 3.44 moles) and dimethyl acetamide (7L, 7 vol) were added to a 20 L RB flask- 1 equipped with mechanical stirrer under inert atmosphere. The resulting solution was cooled to 0- 5°C and triethylamine (346.3, 3.44 moles) was added at 0-5°C. 5-(4-Amino-5- phenylquinazolin-2-yl)-N-tert-butylpyridine-3-sulfonamide (1.0 Kg. 2.306 moles) and dimethylacetamide (4 L, 4 vol) were added to a separate 20 L RB flask-2 equipped with mechanical stirrer under inert atmosphere. This solution was cooled to 0-5°C and sodium tert-butoxide (884 g, 9.24 moles) was added at 0-5°C. The resulting solution was stirred to affect dissolution and then transferred to the RB flask- 1 at 0- 5°C. Upon completion of addition, the reaction mixture was stirred at 0-5°C for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The reaction mass was poured into water (60 L, 60 vol) with stirring. The crude product was collected by filtration and dried at 60°C for 12 h. After this time, the dried material was dissolved in THF (20 L, 20 vol). Upon dissolution, 6M HC1 in isopropyl alcohol (1 L, 1 vol) was added at 20-25°C. The crude HCL salt of the product was obtained a pale-yellow free flow solid (920 g, 71% yield, 93% purity by HPLC). The crude HC1 salt (1.345 Kg, 2.56moles), methanol (6.7 L, 5 vol) and dichloromethane (13.5 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The slurry was stirred for 20-30 min at 30°C. After this time, the solvent was distilled to 4 vol with respect to input under vacuum. The resulting slurry was cooled to 20-25°C, where stirred for 2 h. At the conclusion of this period, the slurry was filtered and dried at 50°C for 6 h to yield the product (1.1 Kg, 82% yield, 98% purity by HPLC). XH NMR (DMSO- D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.10-9.14 (m, 2H), 8.39 (s, 1H), 7.92-8.03 (m, 4H), 7.56-7.58 (m, 5H), 7.43-7.49 (m, 3H), 7.1 (bs, 1H), 4.88 (s, 2H), 1.17 (2, 9H).
Step 7. Example 7

N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide (1.0 Kg, 1.9 moles) and concentrated hydrochloric acid (7 L, 7 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The reaction mixture was heated to 90-100°C where it stirred for 1 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 5-10°C and the pH was adjusted to 1.7 to 2.0 using 12% aqueous sodium hydroxide solution. Once at the prescribed pH, the crude HC1 salt of the product was collected by filtration. The HC1 salt filter cake and ethanol (5 L, 5 vol) were added to 10 L glass-lined reactor equipped with a mechanical stirrer. The resulting mixture was made basic to pH 7-8 at 20-25°C using triethyl amine (2.25 Kg, 22.23 moles). Once at the prescribed pH, the basic mixture was stirred for 2 h. After this time, the free base of product was filtered and washed with water (10 L, 10 vol) followed by ethanol (2L, 2 vol). The resulting product was dried at 50-55°C for 8 h to yield Example 7 (644 g, 72% yield, 99.9% purity by HPLC).
XH NMR (DMSO-D6, 400 MHz, δ ppm); 9.81 (d, J=2.0Hz, 1H), 9.18 (t, J=2Hz, 1H), 9.1 1 (d, J=2Hz, 1H), 8.23 (d, J=4.4Hz, 1H), 7.92-7.94 (m, 1H), 7.83-7.87 (m, 1H), 7.78 (s, 2H), 7.70-7.72 (m, 1H), 7.50-7.59 (m, 5H), 7.31-7.34 (m, 2H), 7.22-7.25 (m, 1H), 6.95 (t, J=4Hz, 1H), 4.76 (d, J=4Hz, 2H). ES-MS: [M++l] = 469.
/////////atrial fibrillation, Potassium channel Kv1.5 (KCNA5) inhibitor, IKur antagonist, Bristol-Myers Squibb Co., BMS 919373, BMS-919373, PHASE 2
NS(=O)(=O)c1cc(cnc1)c4nc2cccc(c2c(NCc3ccccn3)n4)c5ccccc5
CRD 1152, CURADEV PHARMA PRIVATE LTD
Several candidates….one is…….CRD1152
ONE OF THEM IS CRD 1152
Kynurenine pathway regulators (solid tumors)

Compound 2
CAS1638121-21-7
N3-(3-Chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine

COMPD 190
CAS 1638118-99-6

COMPD248
7-Chloro-N3- (3-chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine, 166
DMSO-d6: δ 7.87 (d, J = 5.1 Hz, 1H), 7.25 (s, 2H), 7.16-7.10 (m, 2H), 6.88 (d, J = 5.1 Hz, 1H), 6.59 (dd, J′ = 6.2 Hz, J″ = 2.6 Hz, 1H), 6.48 (dt, J′ = 8.8 Hz, J″ = 6.7 Hz, J′′′ = 3.4 Hz, 1H) M + H] 312
OR
N3-(3,4- difluorophenyl)- 7-(pyridin-4- yl)furo[2,3- c]pyridine-2,3- diamine, 184
CD3CN: δ 8.72 (s, 2H), 8.26 (s, 3H), 7.07-7.03 (m, 2H), 6.47-6.40 (m, 2H), 5.74 (s, 1H), 5.55 (s, 2H) M + H] 339
OR

COMPD73
CAS 1638117-85-7
Several candidates………..CRD1152
 67
67
 66
66
| Company | Curadev Pharma Pvt. Ltd. |
| Description | Small molecule dual indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO1; IDO) inhibitor |
| Molecular Target | Indoleamine 2,3-dioxygenase (INDO) (IDO) ; Tryptophan 2,3-dioxygenase (TDO2) (TDO) |
| Mechanism of Action | Indoleamine 2,3-dioxygenase (INDO) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Roche |
Hoffmann-La Roche partners with Curadev Pharma Ltd. for IDO1 and TDO inhibitors (April 20, 2015)
Curadev Pharma Pvt Ltd., founded in 2010 and headquartered in New Delhi, announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors to treat cancer. The agreement covers the development of CRD1152, the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2,3-dioxygenase-1) and TDO (tryptophan-2,3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response. Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments, as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev is also eligible for milestones and royalties on any additional products resulting from the research collaboration.




Curadev Announces Research Collaboration and Licensing Agreement to Develop Cancer Immunotherapeutic
Curadev’s dual IDO and TDO immune tolerance inhibitor – a novel approach in cancer immunotherapy
Apr 20, 2015, 06:30 ET from Curadev
NEW DELHI, India, April 20, 2015 /PRNewswire/ —
Curadev Pharma Private Ltd. today announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors. The agreement covers the development of the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2, 3-dioxygenase-1) and TDO (tryptophan-2, 3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response.
Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan – to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
“We are very excited to be working with the global leader in oncology with their unrivalled expertise in clinical development,” said Arjun Surya, PhD, Chief Scientific Officer, Curadev. “The collaboration acknowledges our focused research efforts on patient-critical drug targets that have yielded a drug candidate that could make a significant difference in the development of novel treatments for patients suffering from cancer.”
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization to further extend Curadev’s findings, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments based on achievement of certain predetermined events and sales levels as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev would also be eligible for milestones and royalties on any additional products resulting from the research collaboration. Roche will fund future research, development, manufacturing and commercialization costs and will also provide additional research funding to Curadev for support of the research collaboration.
About Curadev
Headquartered in New Delhi, India, Curadev Pharma Private Limited was founded in 2010 by a team of professionals from the pharmaceutical and biotech sectors with the mission to improve human health and enhance the quality of human life by accelerating the discovery and delivery of new drugs. Curadev focuses on the creation and out-licensing of pre-IND assets and IND packages for drug development.
For further information:
Curadev Partnering
Manish Tandon – VP and Chief Financial Officer, manish@curadev.in
PATENT
US20160046596) INHIBITORS OF THE KYNURENINE PATHWAY
Monali Banerjee
Sandip Middya
Ritesh Shrivastava
Sushil Raina
Arjun Surya
Dharmendra B. Yadav
Veejendra K. Yadav
Kamal Kishore Kapoor
Aranapakam Venkatesan
Roger A. Smith
Scott K. Thompson
ONE ………….Example 2
Synthesis of N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine (Compound 2)
Step 1: 3-Methoxymethoxy-pyridine

Step 2: 3-Methoxymethoxy-pyridine-4-carbaldehyde

Step 3: 3-Hydroxy-pyridine-4-carbaldehyde

Step 4: 4-{[3-Chloro-4-fluoro-phenylimino]-methyl}-pyridin-3-ol

Step 5: N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine

Monali Banerjee – Director, R&D
Ms. Banerjee has more than 10 years of research experience, during which she has held positions of increasing responsibility. Her past organizations include TCG Lifesciences (Chembiotek) and Sphaera Pharma. Ms. Banerjee is a versatile scientist with a deep understanding of the fundamental issues that underlie various aspects of drug discovery. At Curadev, she has been responsible for target selection, patent analysis, pharmacophore design, assay development, ADME/PK and in vivo and in vitro pharmacology. Ms. Banerjee holds a Masters in Biochemistry and a Bachelors in Chemistry both from Kolkata University.
writeup
|
The essential amino acid Tryptophan (Trp) is catabolized through the kynurenine (KYN) pathway. The initial rate-limiting step in the kynurenine pathway is performed by heme-containing oxidoreductase enzymes, including tryptophan 2,3-dioxygenase (TDO), indoleamine 2,3-dioxygenase-1 (IDO1), and indoleamine 2,3-dioxygenase-2 (IDO2). IDO1 and IDO2 share very limited homology with TDO at the amino acid level and, despite having different molecular structures, each enzyme has the same biochemical activity in that they each catalyze tryptophan to form N-formylkynurenine. IDO1, IDO2, and/or TDO activity alter local tryptophan concentrations, and the build-up of kynurenine pathway metabolites due to the activity of these enzymes can lead to numerous conditions associated with immune suppression.
|
|
Kynurenine pathway dysregulation and IDO1 and/or TDO activity also correlate with cardiovascular risk factors, and kynurenines and IDO1 are markers for Atherosclerosis and other cardiovascular heart diseases such as coronary artery disease (Platten et al., Science, 2005, 310(5749):850-5, Wirlietner et al. Eur J Clin Invest. 2003 July; 33(7):550-4) in addition to kidney disease. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease (Pawlak et al., Atherosclerosis, 2009, (204)1:309-314). Studies show that kynurenine pathway metabolites are associated with endothelial dysfunction markers in the patients with chronic kidney disease (Pawlak et al., Advances in Medical Sciences, 2010, 55(2):196-203).
|
///////CRD1152, CRD-1152, CRD 1152, CURADEV PHARMA PRIVATE LTD, ROCHE, IDO1 and TDO inhibitors, COLLABORATION, CANCER, indoleamine-2,3-dioxygenase-1, Hoffmann-La Roche, kynurenine pathway regulators, solid tumors
GDC-0919; NLG-919; RG-6078

MF C18H22N2O
MW: 282.17321
GDC-0919; NLG-919; RG-6078, GDC0919; GDC-0919; GDC 0919; NLG919; NLG 919; NLG-919; RG6078; RG-6078; RG 6078.
1-cyclohexyl-2-(5H-imidazo[5,1-a]isoindol-5-yl)ethanol
CAS No.1402836-58-1
GDC-0919, also known as NLG919 and RG6078, is an orally available inhibitor of indoleamine 2,3-dioxygenase 1 (IDO1), with potential immunomodulating and antineoplastic activities. Upon administration, NLG919 targets and binds to IDO1, a cytosolic enzyme responsible for the oxidation of the essential amino acid tryptophan into kynurenine. By inhibiting IDO1 and decreasing kynurenine in tumor cells, this agent increases tryptophan levels, restores the proliferation and activation of various immune cells, including dendritic cells (DCs), natural killer (NK) cells, T-lymphocytes, and causes a reduction in tumor-associated regulatory T-cells (Tregs). Activation of the immune system, which is suppressed in many cancers, may induce a cytotoxic T-lymphocyte (CTL) response against the IDO1-expressing tumor cells

- Originator Lankenau Institute for Medical Research
- Developer Genentech; NewLink Genetics Corporation
- Class Antineoplastics; Small molecules
- Mechanism of Action Immunomodulators; Indoleamine-pyrrole 2,3-dioxygenase inhibitors
Phase I Solid tumours
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015210769 | 2015-07-30 | ANTIBODY MOLECULES TO PD-1 AND USES THEREOF |
| US2014066625 | 2014-03-06 | Fused Imidazole Derivatives Useful as IDO Inhibitors |
- 27 Sep 2015 Pharmacokinetics results from a phase-I clinical trial in Solid tumours presented at the European Cancer Congress 2015 (ECC-2015)
- 27 Sep 2015 Positive efficacy and safety results from a phase-I clinical trial in Solid tumours presented at the European Cancer Congress 2015 (ECC-2015)
- 31 Jul 2015 Phase-I clinical trials in Solid tumours (Combination therapy, Late-stage disease, Second-line therapy or greater) in USA (PO) (NCT02471846)
PATENT
http://www.google.com/patents/WO2012142237A1?cl=en

PATENT
Fused Imidazole Derivatives Useful as IDO Inhibitors
13041-cyclohexyl-2-(5H-imidazo[5,1- a]isoindol-5-yl)ethanol79 1H NMR (a mixture of diastereomers) 1.10-1.37 (m, 6H), 1.66-1.80 (m, 5H), 2.05 (m, 2H), 2.15 (m, 1H), 3.72 (m, 1H), 5.36 and 5.46 (two m, 1H), 7.16 (s, 1H), 7.25 (m, 1H), 7.34 (m, 1H), 7.43 (d, 1H, J = 7.6 Hz), 7.54 (d, 1H, J = 7.6 Hz), 7.80 (s, 1H)
| WO2011056652A1 * | Oct 27, 2010 | May 12, 2011 | Newlink Genetics | Imidazole derivatives as ido inhibitors |
| WO2012142237A1 * | Apr 12, 2012 | Oct 18, 2012 | Newlink Geneticks Corporation | Fused imidazole derivatives useful as ido inhibitors |
| WO2014159248A1 | Mar 10, 2014 | Oct 2, 2014 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| US8722720 | Oct 27, 2010 | May 13, 2014 | Newlink Genetics Corporation | Imidazole derivatives as IDO inhibitors |
| US9260434 | Oct 14, 2013 | Feb 16, 2016 | Newlink Genetics Corporation | Fused imidazole derivatives useful as IDO inhibitors |
| US20140066625 * | Oct 14, 2013 | Mar 6, 2014 | Newlink Genetics Corporation | Fused Imidazole Derivatives Useful as IDO Inhibitors |
| US20160002249 * | Jul 8, 2015 | Jan 7, 2016 | Newlink Genetics Corporation | Fused Imidazole Derivatives Useful as IDO Inhibitors |
REFERENCES
Nature Reviews Drug Discovery14,373(2015)doi:10.1038/nrd4658
http://www.ncbi.nlm.nih.gov/pubmed/21517759
http://www.roche.com/irp150128-annex.pdf
/////CRD1152, CRD 1152, CRD-1152, Curadev, Research Collaboration, Licensing Agreement, Develop, Cancer Immunotherapeutic, IDO1 and TDO inhibitors
OC(C1CCCCC1)CC(C2=C3C=CC=C2)N4C3=CN=C4
CFG 920, Novartis Scientists team up with Researchers at Aurigene, Bangalore, India,
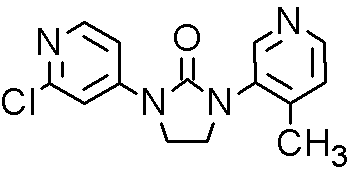
CFG920,
Inhibitor Of Prostate Cancer With Fewer Cardiac Side Effects
Cas 1260006-20-9
Novartis
Target: CYP17/CYP11B2
Disease: Castration-resistant prostate cancer
MF C14H13ClN4O
MW: 288.0778
Elemental Analysis: C, 58.24; H, 4.54; Cl, 12.28; N, 19.40; O, 5.54
Steroid 17-alpha-hydroxylase inhibitors
CFG920 is a CYP17 inhibitor, is also an orally available inhibitor of the steroid 17-alpha-hydroxylase/C17,20 lyase (CYP17A1 or CYP17), with potential antiandrogen and antineoplastic activities. Upon oral administration, CYP17 inhibitor CFG920 inhibits the enzymatic activity of CYP17A1 in both the testes and adrenal glands, thereby inhibiting androgen production. This may decrease androgen-dependent growth signaling and may inhibit cell proliferation of androgen-dependent tumor cells.
https://clinicaltrials.gov/ct2/show/NCT01647789
NCT01647789: A Study of Oral CFG920 in Patients With Castration Resistant Prostate Cancer2012
- 09 Nov 2015Adverse events, efficacy and pharmacokinetics data from the phase I part of a phase I/II trial in Prostate cancer (Metastatic disease) presented at the 27th AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics (AACR-NCI-EORTC-2015)
- 29 Jan 2013Phase-I clinical trials in Prostate cancer in Spain (PO)
- 10 Dec 2012Phase-I clinical trials in Prostate cancer in Canada (PO)
In August 2015, preclinical data were presented at the 250th ACS meeting in Boston, MA. In monkeys, treatment with CFG-920 (3 mg/kg, po) showed good bioavailability with F value of 93%, Tmax of 0.5 h, Cmax of 1382 nM.dn and AUC of 2364 nM.h, while CFG-920 (10 mg/kg, po) showed F value of 183%, Cmax of 1179 nM.dn and Tmax of 1.04 h
Bethany Halford on Twitter: “CFG920 – @Novartis CMOS for …

Novartis is developing CFG-920 (structure shown), an oral CYP17 inhibitor, for the potential treatment of metastatic castration-resistant prostate cancer. In March 2013, a phase I/II trial was initiated and at that time, the study was expected to complete in January 2015; in August 2015, clinical data were presented
2015 250th (August 19) Abs MEDI 341
Discovery of CFG920, a dual CYP17/CYP11B2 inhibitor, for the treatment of castration resistant prostate cancer
American Chemical Society National Meeting and Exposition
Christoph Gaul, Prakash Mistry, Henrik Moebitz, Mark Perrone, Bjoern Gruenenfelder, Nelson Guerreiro, Wolfgang Hackl, Peter Wessels, Estelle Berger, Mark Bock, Saumitra Sengupta, Venkateshwar Rao, Murali Ramachandra, Thomas Antony, Kishore Narayanan, Samiulla Dodheri, Aravind Basavaraju, Shekar Chelur


| Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, CSN Murthy is chief executive officer |

WO 2010149755

Prostate cancer is the most commonly occurring cancer in men. Doctors often treat the metastatic stage of the disease by depriving the patient of sex hormones via chemical or surgical castration. But if it progresses far enough, the cancer can survive this therapy, transforming into the castration-resistant form. “Once the cancer becomes castration-resistant, the prognosis is poor,” said Novartis’s Christoph Gaul.
In recent years, CYP17, a bifunctional 17α-hydroxylase/17,20-lyase cytochrome P450 enzyme, has emerged as a target for treating castration-resistant prostate cancer. The enzyme catalyzes the biosynthesis of sex hormones, including testosterone, and blocking it can starve prostate cancer of the androgens it needs to thrive.
Johnson & Johnson’s CYP17 inhibitor, abiraterone acetate (Zytiga), a steroid that binds irreversibly to CYP17, was approved by the Food & Drug Administration in 2011. But Novartis scientists thought they could make a better CYP17 inhibitor, Gaul told C&EN. They teamed up with researchers at Aurigene, in Bangalore, India, and came up with their clinical candidate, CFG920.
Unlike abiraterone, CFG920 isn’t a steroid, and it inhibits CYP17 reversibly. It also reversibly inhibits another cytochrome P450 enzyme, CYP11B2, which is involved in the synthesis of the mineralocorticoids, hormones that regulate cardiac function.
Treating prostate cancer patients by lowering their androgen levels turns out to have negative cardiac side effects: Patients’ lipid metabolism is thrown off and their mineralocorticoid levels jump, leading to increases in blood pressure. Those changes can be stressful for the heart. “If prostate cancer patients don’t die because of the cancer, a lot of times they die because of cardiac disease,” Gaul said.
Because CFG920 also keeps mineralocorticoid levels in check, Novartis is hoping the drug candidate will ameliorate some of the cardiac side effects of inhibiting CYP17. The compound is currently in Phase I clinical trials.
PATENT
WO 2010149755
Example 58
Prύpιn”ation ofI'(2’ChIoroψ}ri(ibi-^’\l)’3’f4’metMψ}τUin’3’yl)-imiJazoliJin’2’θne (5HA)-

Using the same reaction conditions as in Example 14. 1-(4-methyl-pyridin-3-yl)- itnida/olidin-2-onc ().-.!.4b: 600 mg. 3.3898 mmol) uas reacted with 2-chloro-4-iodo- py.idine (974 mg.4.067 mmol). 1 , 4-dioxane (60 mL). copper iodide (65 mg, 0.3398 mmol), /r<w.v-1.2-diamino cycK)hexane (0.12 ml,, 1.0169 mmol) and potassium phosphate (2.15 g, 10.1694 mmol) to afford 810 mg of the product (83% yield).
1H NMR (C1DCI3. 300 Mi l/): 6 8.5-8.4 (m. 211). 8.3 (d. IH), 7.6-7.5 (m, 2H). 7.2 (S. 111). 4.1-3.9 (ni. 4H), 2.35 <s. 3H)
LCVIS puιϊt>: 90.8%. nι-7 – 289.1 (M M)
HPl C: 97.14%
REFERENCES
1: Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015 Mar;95:80-7. doi: 10.1016/j.steroids.2014.12.021. Epub 2015 Jan 3. Review. PubMed PMID: 25560485; PubMed Central PMCID: PMC4323677.
2: Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. Int J Mol Sci. 2013 Jul 4;14(7):13958-78. doi: 10.3390/ijms140713958. Review. PubMed PMID: 23880851; PubMed Central PMCID: PMC3742227.
///////CFG-920, CYP17 inhibitor (prostate cancer), Novartis, CFG 920, Novartis scientists, team up , researchers , Aurigene, Bangalore, India,
Dr. Reddy’s Laboratories CEO G V Prasad has been recognized as one of India’s top 5 most valuable CEOs


CEO G V Prasad has been recognized as one of India’s top 5 most valuable CEOs Read more: bit.ly/CEOsRanking
http://businessworld.in/article/How-We-Ranked-The-CEOs/31-03-2016-92402/
////////Dr. Reddy’s Laboratories, CEO , G V Prasad, India’s top 5 most valuable CEOs
Novartis Molecule for functionally liver selective glucokinase activators for the treatment of type 2 diabetes


(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
(3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide)
cas 866772-52-3
NVP-LBX192
LBX-192

54 Discovery and Evaluation of NVP-LBX192, a Liver Targeted Glucokinase Activator
https://acs.confex.com/acs/nerm09/webprogram/Paper75087.html
| Molecular Formula: | C26H33N5O4S2 |
|---|---|
| Molecular Weight: | 543.70132 g/mol |
Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes
2009 52 (19) 6142 – 6152
Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes
Journal of Medicinal Chemistry
Bebernitz GR, Beaulieu V, Dale BA, Deacon R, Duttaroy A, Gao JP, Grondine MS, Gupta RC, Kakmak M, Kavana M, Kirman LC, Liang JS, Maniara WM, Munshi S, Nadkarni SS, Schuster HF, Stams T, Denny IS, Taslimi PM, Vash B, Caplan SL
2010 240th (August 22) Medi-198
Glucokinase activators with improved physicochemicalproperties and off target effects
American Chemical Society National Meeting and Exposition
Kirman LC, Schuster HF, Grondine MS et al
2010 240th (August 22) Medi-197
Investigation of functionally liver selective glucokinase activators
American Chemical Society National Meeting and Exposition
Schuster HF, Kirman LC, Bebernitz GC et al
PATENT
http://www.google.com/patents/US7750020
EXAMPLE 1 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

A. Phenylacetic Acid Ethyl Ester
A solution of phenylacetic acid (50 g, 0.36 mol) in ethanol (150 mL) is treated with catalytic amount of sulfuric acid (4 mL). The reaction mixture is refluxed for 4 h. The reaction is then concentrated in vacuo. The residue is dissolved in diethyl ether (300 mL) and washed with saturated aqueous sodium bicarbonate solution (2×50 mL) and water (1×100 mL). The organic layer dried over sodium sulfate filtered and concentrated in vacuo to give phenylacetic acid ethyl ester as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.2 (t, J=7.2, 3H), 3.6 (s, 2H), 4.1 (q, J=7.2, 2H), 7.3 (m, 5H); MS 165 [M+1]+.
B. (4-Chlorosulfonyl-phenyl)-acetic acid ethyl ester
To a cooled chlorosulfonic acid (83.83 g, 48 mL, 0.71 mol) under nitrogen is added the title A compound, phenylacetic acid ethyl ester (59 g, 0.35 mol) over a period of 1 h. Reaction temperature is brought to RT (28° C.), then heated to 70° C., maintaining it at this temperature for 1 h while stirring. Reaction is cooled to RT and poured over saturated aqueous sodium chloride solution (200 mL) followed by extraction with DCM (2×200 mL). The organic layer is washed with water (5×100 mL), followed by saturated aqueous sodium chloride solution (1×150 mL). The organic layer dried over sodium sulfate, filtered and concentrated in vacuo to give crude (4-chlorosulfonyl-phenyl)acetic acid ethyl ester. Further column chromatography over silica gel (60-120 mesh), using 100% hexane afforded pure (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester as a colorless oil.
C. [4-(4-Methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester
A solution of N-methylpiperazine (9.23 g, 10.21 ml, 0.092 mol), DIEA (13 g, 17.4 mL, 0.10 mol) and DCM 80 mL is cooled to 0° C., and to this is added a solution of the title B compound, (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester (22 g, 0.083 mol) in 50 mL of DCM within 30 min. Reaction mixture stirred at 0° C. for 2 h, and the reaction mixture is washed with water (100 mL), followed by 0.1 N aqueous hydrochloric acid solution (1×200 mL). The organic layer dried over sodium sulfate, filtered and concentrated under vacuo to give crude [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester. Column chromatography over silicagel (60-120 mesh), using ethyl acetate afforded pure [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester as white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 1.3 (t, J=7.4, 3H), 2.3 (s, 3H), 2.5 (m, 4H), 3.0 (br s, 4H), 3.7 (s, 2H), 4.2 (q, J=7.4, 2H), 7.4 (d, J=8.3, 2H), 7.7 (d, J=7.3, 2H); MS 327 [M+1]+.
D. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester
A solution of the title C compound, [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester (15 g, 0.046 mol) in a mixture of THF (60 mL) and DMTP (10 mL) is cooled to −78° C. under nitrogen. The resulting solution is stirred at −78° C. for 45 min and to this is added LDA (25.6 mL, 6.40 g, 0.059 mol, 25% solution in THF/Hexane). A solution of iodomethylcyclopentane (11.60 g, 0.055 mol) in a mixture of DMTP (12 mL) and THF (20 mL) is added over a period of 15 min at −78° C. and reaction mixture stirred at −78° C. for 3 h further, followed by stirring at 25° C. for 12 h. The reaction mixture is then quenched by the dropwise addition of saturated aqueous ammonium chloride solution (50 mL) and is concentrated in vacuo. The residue is diluted with water (50 mL) and extracted with ethyl acetate (3×100 mL). The organic solution is washed with a saturated aqueous sodium chloride (2×150 mL), dried over sodium sulfate, filtered and concentrated in vacuo. Column chromatography over silica gel (60-120 mesh), using 50% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 1.2 (t, J=7.1, 3H), 2.3 (s, 3H), 2.5 (br s, 4H), 3.0 (br s, 4H), 3.6 (m, 1H), 4.1 (q, J=7.1, 2H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H); MS 409 [M+1]+.
E. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid
A solution of the title D compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester (14 g, 0.034 mol) in methanol:water (30 mL:10 mL) and sodium hydroxide (4.11 g, 0.10 mol) is stirred at 60° C. for 8 h in an oil bath. The methanol is then removed in vacuo at 45-50° C. The residue is diluted with water (25 mL) and extracted with ether (1×40 mL). The aqueous layer is acidified to pH 5 with 3 N aqueous hydrochloric acid solution. The precipitated solid is collected by vacuum filtration, washed with water (20 mL), followed by isopropyl alcohol (20 mL). Finally, solid cake is washed with 100 mL of hexane and dried under vacuum at 40° C. for 6 h to give 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid as a white solid: 1H NMR (400 MHz, CDCl3) δ 1.1-2.0 (m, 11H), 2.4 (s, 3H), 2.7 (br s, 4H), 3.1 (br s, 4H), 3.6 (m, 1H), 7.5 (d, J=8.3, 2H), 7.6 (d, J=8.3, 2H); MS 381 [M+l]+.
F. 5-Methoxy-thiazolo[5,4-b]pyridin-2-ylamine
A solution of 6-methoxy-pyridin-3-ylamine (5.0 g, 0.0403 mol) in 10 mL of acetic acid is added slowly to a solution of potassium thiocyanate (20 g, 0.205 mol) in 100 mL of acetic acid at 0° C. followed by a solution of bromine (2.5 mL, 0.0488 mol) in 5 mL of acetic acid. The reaction is stirred for 2 h at 0° C. and then allowed to warm to RT. The resulting solid is collected by filtration and washed with acetic acid, then partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The insoluble material is removed by filtration and the organic layer is evaporated and dried to afford 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine as a tan solid.
G. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
A solution of the title E compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (5 g, 0.013 mol) in DCM (250 mL) is cooled to 0° C. and then charged HOBt hydrate (2.66 g, 0.019 mol), followed by EDCI hydrochloride (6 g, 0.031 mol). The reaction mixture is stirred at 0° C. for 5 h. After that the solution of the title F compound, 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine (2.36 g, 0.013 mol) and D1EA (8 mL, 0.046 mol) in a mixture of DCM (60 mL) and DMF (20 mL) is added dropwise over 30 min. Reaction temperature is maintained at 0° C. for 3 h, then at RT (28° C.) for 3 days. Reaction is diluted with (60 mL) of water and the organic layer is separated and washed with saturated sodium bicarbonate solution (2×50 mL) followed by water washing (2×50 mL) and saturated sodium chloride aqueous solution (1×150 mL). Finally the organic layer is dried over sodium sulfate, filtered, and evaporated under vacuo. The crude product is purified using column chromatography over silica gel (60-120 mesh), using 40% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 2.2 (s, 3H), 2.5 (br s, 4H), 3.1 (br s, 4H), 3.7 (m, 1H), 4.0 (s, 3H), 6.8 (d, J=8.8, 1H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H), 7.8 (d, J=8.8, 1H), 8.6 (s, 1H); MS 617 [M+1]+.
H. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride
The title G compound, 3-cyclopentyl-2-(4-methyl piperazinyl sulfonyl)phenyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)propionamide (2.8 g, 0.0051 mol) is added to a cooled solution of 10% hydrochloric acid in isopropanol (3.75 mL). The reaction mixture is stirred at 0° C. for 1 h and then at RT for 2 h. The solid is separated, triturated with 10 mL of isopropanol and collected by vacuum filtration and washed with 50 mL of hexane. The solid is dried at 70° C. for 48 h to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride as an off white solid.
EXAMPLE 2 (R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1,3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4° C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
- Column: Chiralcel OD-R (250×20 mm) Diacel make, Japan;
- Solvent A: water:methanol:acetonitrile (10:80:10 v/v/v);
- Solvent B: water:methanol:acetonitrile (05:90:05 v/v/v);
- Using gradient elution: gradient program (time, min/% B): 0/0, 20/0, 50/100, 55/0, 70/0;
- Flow rate: 6.0 mL/min; and
- Detection: by UV at 305 nm.
EXAMPLE 3 (S)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is prepared analogously to Example 2.
J MED CHEM 2009, 52, 6142-52
Investigation of Functionally Liver Selective Glucokinase Activators for the Treatment of Type 2 Diabetes
http://pubs.acs.org/doi/abs/10.1021/jm900839k

Type 2 diabetes is a polygenic disease which afflicts nearly 200 million people worldwide and is expected to increase to near epidemic levels over the next 10−15 years. Glucokinase (GK) activators are currently under investigation by a number of pharmaceutical companies with only a few reaching early clinical evaluation. A GK activator has the promise of potentially affecting both the β-cells of the pancreas, by improving glucose sensitive insulin secretion, as well as the liver, by reducing uncontrolled glucose output and restoring post-prandial glucose uptake and storage as glycogen. Herein, we report our efforts on a sulfonamide chemotype with the aim to generate liver selective GK activators which culminated in the discovery of 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide (17c). This compound activated the GK enzyme (αKa = 39 nM) in vitro at low nanomolar concentrations and significantly reduced glucose levels during an oral glucose tolerance test in normal mice.


PATENT
EP-1735322-B1
Example 2(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4°C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
- Column: Chiralcel OD-R (250 x 20 mm) Diacel make, Japan;
- Solvent A: water:methanol:acetonitrile (10:80:10 v/v/v);
- Solvent B: water:methanol:acetonitrile (05:90:05 v/v/v);
- Using gradient elution: gradient program (time, min / %B): 0/0, 20/0, 50/100, 55/0, 70/0;
- Flow rate: 6.0 mL/min; and
- Detection: by UV at 305 nm.
REFERENCES
US 7750020
WO-2005095418-A1
US-20080103167-A1
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015218151 | 2015-08-06 | NOVEL PHENYLACETAMIDE COMPOUND AND PHARMACEUTICAL CONTAINING SAME |
| US7750020 | 2010-07-06 | Sulfonamide-Thiazolpyridine Derivatives As Glucokinase Activators Useful The Treatment Of Type 2 Diabetes |
///NOVARTIS, DIABETES, Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes, 866772-52-3, Novartis Molecule, functionally liver selective glucokinase activators, treatment of type 2 diabetes , NVP-LBX192, LBX-192
c1(sc2nc(ccc2n1)OC)NC(C(c3ccc(cc3)S(=O)(=O)N4CCN(CC4)C)CC5CCCC5)=O
P7435 from Piramal Enterprises Mumbai, India
P7435
Piramal Enterprises Mumbai, India
P-7435; P7435-DGAT1, P7435, P 7435
- Molecular Weight, 454.47
GDAT1 inhibitor
- Phase IDiabetes mellitus; Lipid metabolism disorders
- ClassAntihyperglycaemics; Antihyperlipidaemics; Small molecules
- Mechanism of ActionDiacylglycerol O acyltransferase inhibitors

| Latest Stage of Development | Phase I |
| Standard Indication | Metabolic (unspecified) |
| Indication Details | Treat metabolic disorders |
https://clinicaltrials.gov/ct2/show/NCT01910571
https://clinicaltrials.gov/ct2/show/NCT01764425
- 24 Nov 2014Piramal Enterprises completes a phase I trial in healthy, overweight or obese subjects in USA (NCT01910571)
- 17 Jun 2014Adverse events and pharmacokinetics data from a phase I trial in healthy male volunteers presented at the 74th Annual Scientific Sessions of the American Diabetes Association (ADA-2014)
- 17 Jun 2014Pharmacodynamics data from preclinical studies in Dyslipidaemia and obesity presented at the 74th Annual Scientific Sessions of the American Diabetes Association (ADA-2014)

Chairman Ajay Piramal

Swati Piramal-The Vice Chairperson of Piramal Enterprises Ltd

Nandini Piramal, Executive Director, Piramal Enterprises
Piramal Enterprises gets US FDA approval for P7435 IND
http://www.pharmabiz.com/NewsDetails.aspx?aid=76992&sid=2
Our Bureau, Mumbai
Tuesday, August 06, 2013, 12:25 Hrs [IST]
Piramal Enterprises Ltd has received US Food and Drug Administration (FDA) approval for its Investigational New Drug (IND) P7435. This is a novel, potent and highly selective, oral diacylglycerolacyltransferase 1 (DGAT1) inhibitor.
P7435 has been developed by the NCE Research Division of PEL for the management of metabolic disorders such as lipid abnormalities and diabetes. It is well-established that increased lipid levels’ (including triglycerides) is one of the major risk factors for cardiovascular disease (CVD). It has been reported by the World Health Organisation, that CVD, is the number one cause of deaths globally, representing approximately 30 per cent of all deaths. Currently, there is a significant medical need for effective and safe drugs for the management of lipid abnormalities and metabolic disorders.
P7435 has demonstrated its lipid lowering potential in various preclinical studies by showing significant reduction in triglyceride levels, glucose and insulin levels,and decrease in food intake and body weight gain -factors which are associated with lipid abnormalities and metabolic disorders.
PEL has established the safety and tolerability of P7435 in a phase I trial recently completed in India. This extension trial in the US will further evaluate the safety and efficacy of P7435 in a larger population.
Dr Swati Piramal, vice chairperson, Piramal Enterprises, said, “The NCE Research division of PEL continues its ambitious diabetes/metabolic disorders programme to discover and develop NCEs to fight against diseases like diabetes and lipid disorders. With P7435 we are looking at addressing a serious need for effective and well-tolerated drugs that treat lipid disorders, which are commonly associated with diabetes and CVDs. Expansion of this trial will allow testing this NCE in a wider population,which is critical to the development of this drug and will provide therapeutic solutions not just to India but also to the rest of the world.”
The NCE Research division of Piramal Enterprises focuses on the discovery and development of innovative small molecule medicines to improve the lives of patients suffering from cancer, metabolic disorders and inflammatory conditions. The key elements of its strategy include capitalizing on Piramal’s strengths, in particular the India advantage, and leveraging external partnerships to achieve high levels of R&D productivity. Piramal’s state-of-the-art Research Centre in Mumbai has comprehensive capabilities spanning target identification all the way through clinical development. Its robust pipeline, including 8 compounds in clinical development, bears testimony to its innovative and rigorous drug discovery process.
PAPER
European Journal of Medicinal Chemistry (2012), 54, 324-342
http://www.sciencedirect.com/science/article/pii/S0223523412003133
PATENT
WO 2010023609
http://www.google.co.in/patents/WO2010023609A1?cl=en
/////////Piramal Enterprises, Mumbai, India, P-7435, P7435-DGAT1, P7435, P 7435, GDAT1 inhibitor
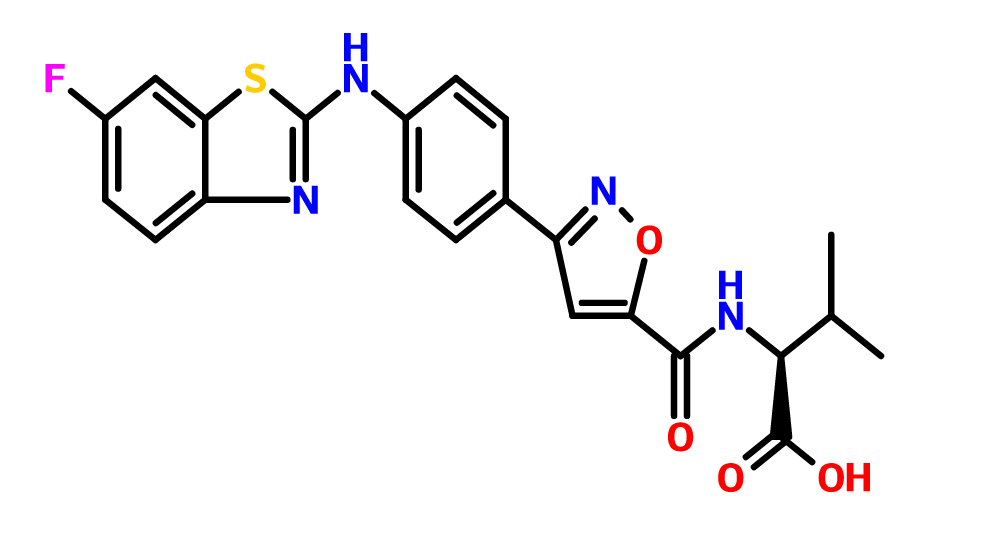
O=C(O)[C@@H](NC(=O)c1cc(no1)c2ccc(cc2)Nc3nc4ccc(F)cc4s3)C(C)C
AUNP-12 from Aurigene Discovery Technologies Limited
AUNP-12
AUR-012; Aurigene-012; NP-12, Aurigene; PD-1 inhibitor peptide (cancer), Aurigene; PD-1 inhibitor peptide (cancer), Aurigene/ Pierre Fabre; W-014A
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Laboratoires Pierre Fabre S.A. |
Aurigene Discovery Technologies Limited

-
Programmed Cell Death 1 or PD-1 (also referred to as PDCD1) is a 50 to 55 kD type I membrane glycoprotein (Shinohara T et al, Genomics, 1994, Vol. 23, No. 3, pp. 704-706). PD-1 is a receptor of the CD28 superfamily that negatively regulates T cell antigen receptor signalling by interacting with the specific ligands and is suggested to play a role in the maintenance of self tolerance.
-
PD-1 peptide relates to almost every aspect of immune responses including autoimmunity, tumour immunity, infectious immunity, transplantation immunity, allergy and immunological privilege.
-
The PD-1 protein’s structure comprise of—
-
- an extracellular IgV domain followed by
- a transmembrane region and
- an intracellular tail
-
-
The intracellular tail contains two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, which suggests that PD-1 negatively regulates TCR signals. Also, PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, (Y. Agata et al., Int Immunol 8, 765, May 1996) suggesting that compared to CTLA-4 ((Cytotoxic T-Lymphocyte Antigen 4, also known as CD152 (Cluster of differentiation 152) is a protein that also plays an important regulatory role in the immune system), PD-1 more broadly negatively regulates immune responses.
-
PD-1 has two ligands, PD-L1 (Programmed Death Ligand for PDCD1L1 or B7-H1) (Freeman G J et al, Journal of Experimental Medicine, 2000, Vol. 19, No. 7, pp. 1027-1034) and PD-L2 (Programmed Death Ligand 2 or PDCD1L2 or B7-DC) (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267), which are members of the B7 family. PD-L1 is known to be expressed not only in immune cells, but also in certain kinds of tumour cell lines (such as monocytic leukaemia-derived cell lines, mast cell tumour-derived cell lines, hematoma-derived cell lines, neuroblastoma-derived cell lines, and various mammary tumour-derived cell lines) and in cancer cells derived from diverse human cancer tissues (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267) and on almost all murine tumour cell lines, including PA1 myeloma, P815 mastocytoma, and B16 melanoma upon treatment with IFN-γ (Y. Iwai et al., Proc Natl Acad Sci USA 99, 12293, Sep. 17, 2002 and C. Blank et al., Cancer Res 64, 1140, February, 2004). Similarly PD-L2 expression is more restricted and is expressed mainly by dendritic cells and a few tumour cell lines. PD-L2 expression has been verified in Hodgkin’s lymphoma cell lines and others. There is a hypothesis that some of the cancer or tumour cells take advantage from interaction between PD-1 and PD-L1 or PD-L2, for suppressing or intercepting T-cell immune responses to their own (Iwai Y et al, Proceedings of the National Academy of Science of the United States of America, 2002, Vol. 99, No. 19, pp. 12293-12297).
-
Tumour cells and virus (including HCV and HIV) infected cells are known to express the ligand for PD-1 (to create Immunosuppression) in order to escape immune surveillance by host T cells. It has been reported that the PD-1 gene is one of genes responsible for autoimmune diseases like systemic lupus erythematosis (Prokunina et al, Nature Genetics, 2002, Vol. 32, No. 4, 666-669). It has also been indicated that PD-1 serves as a regulatory factor for the onset of autoimmune diseases, particularly for peripheral self-tolerance, on the ground that PD-1-deficient mice develop lupus autoimmune diseases, such as glomerulonephritis and arthritis (Nishimura H et al, International Immunology, 1998, Vol. 10, No. 10, pp. 1563-1572; Nishimura H et al, Immunity, 1999, Vol. 11, No. 2, pp. 141-151), and dilated cardiomyopathy-like disease (Nishimura H et al, Science, 2001, Vol. 291, No. 5502, pp. 319-332).
-
Hence, in one approach, blocking the interaction of PD-1 with its ligand (PD-L1, PD-L2 or both) may provide an effective way for specific tumour and viral immunotherapy.
-
Wood et al in U.S. Pat. No. 6,808,710 discloses method for down modulating an immune response comprising contacting an immune cell expressing PD-1 with an antibody that binds to PD-1, in multivalent form, such that a negative signal is transduced via PD-1 to thereby down modulate the immune response. Such an antibody may be a cross-linked antibody to PD-1 or an immobilized antibody to PD-1.
-
Freeman et al in U.S. Pat. No. 6,936,704 and its divisional patent U.S. Pat. No. 7,038,013 discloses isolated nucleic acids molecules, designated B7-4 nucleic acid molecules, which encode novel B7-4 polypeptides, isolated B7-4 proteins, fusion proteins, antigenic peptides and anti-B7-4 antibodies, which co-stimulates T cell proliferation in vitro when the polypeptide is present on a first surface and an antigen or a polyclonal activator that transmits an activating signal via the T-cell receptor is present on a second, different surface.
-
There are some reports regarding substances inhibiting immunosuppressive activity of PD-1, or interaction between PD-1 and PD-L1 or PD-L2, as well as the uses thereof. A PD-1 inhibitory antibody or the concept of a PD-1 inhibitory peptide is reported in WO 01/14557, WO 2004/004771, and WO 2004/056875. On the other hand, a PD-L1 inhibitory antibody or a PD-L1 inhibitory peptide is reported in WO 02/079499, WO 03/042402, WO 2002/086083, and WO 2001/039722. A PD-L2 inhibitory antibody or a PD-L2 inhibitory peptide is reported in WO 03/042402 and WO 02/00730.
-
WO2007005874 describes isolated human monoclonal antibodies that specifically bind to PD-L1 with high affinity. The disclosure provides methods for treating various diseases including cancer using anti-PD-L1 antibodies.
-
US2009/0305950 describes multimers, particularly tetramers of an extracellular domain of PD-1 or PD-L1. The application describes therapeutic peptides.
-
Further, the specification mentions that peptides can be used therapeutically to treat disease, e.g., by altering co-stimulation in a patient. An isolated B7-4 or PD-1 protein, or a portion or fragment thereof (or a nucleic acid molecule encoding such a polypeptide), can be used as an immunogen to generate antibodies that bind B7-4 or PD-1 using standard techniques for polyclonal and monoclonal antibody preparation. A full-length B7-4 or PD-1 protein can be used, or alternatively, the invention provides antigenic peptide fragments of B7-4 or PD-1 for use as immunogens. The antigenic peptide of B7-4 or PD-1 comprises at least 8 amino acid residues and encompasses an epitope of B7-4 or PD-1 such that an antibody raised against the peptide forms a specific immune complex with B7-4 or PD-1. Preferably, the antigenic peptide comprises at least 10 amino acid residues, more preferably at least 15 amino acid residues, even more preferably at least amino acid residues, and most preferably at least 30 amino acid residues.
-
Freeman et al in U.S. Pat. No. 7,432,059 appears to disclose and claim methods of identifying compounds that up modulate T cell activation in the presence of a PD-1-mediated signal. Diagnostic and treatment methods utilizing compositions of the invention are also provided in the patent.
-
Further, Freeman et al in U.S. Pat. No. 7,709,214 appears to cover methods for up regulating an immune response with agents that inhibit the interactions between PD-L2 and PD-1.
-
Despite existence of many disclosures as discussed above, however, a significant unmet medical need still exists due to the lack of effective peptides or modified peptides as therapeutic agents as alternatives in the therapeutic area. It is known that synthetic peptides offer certain advantages over antibodies such as ease of production with newer technologies, better purity and lack of contamination by cellular materials, low immunogenicity, improved potency and specificity. Peptides may be more stable and offer better storage properties than antibodies. Moreover, often peptides possess better tissue penetration in comparison with antibodies, which could result in better efficacy. Peptides can also offer definite advantages over small molecule therapeutics counterparts such as lesser degree of toxicity and lower probability of drug-drug interaction.
-
The present invention therefore may provide the solution for this unmet medical need by offering novel synthetic peptide and its derivatives which are based on the PD1 ectodomain.

Patent
http://www.google.com/patents/US20110318373
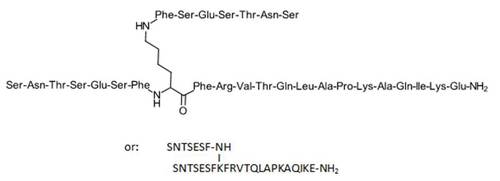
| 8. | SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2 (SEQ ID NO: 49) |
|

Example 2 Synthesis of

Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
-
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows
-
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 m L). The resin was washed with DMF (6×15 m L), DCM (6×15 m L) and DMF (6×15 m L). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 m mol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 m L; 5 equiv, 1 m mol) and HOBT (0.08 g; 5 equiv, 0.6 m mol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 m mol, 8 equivalent excess of HOBt (0.22 g, 1.6 m mol) and 10 equivalent excess of DIC (0.32 m L, 2 m mol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 m mol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 m mol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B: Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT-12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+); 816.2[M/4+H]+;
STRUCTURE , READER DISCRETION IS NEEDED
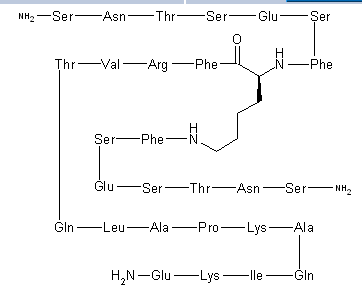
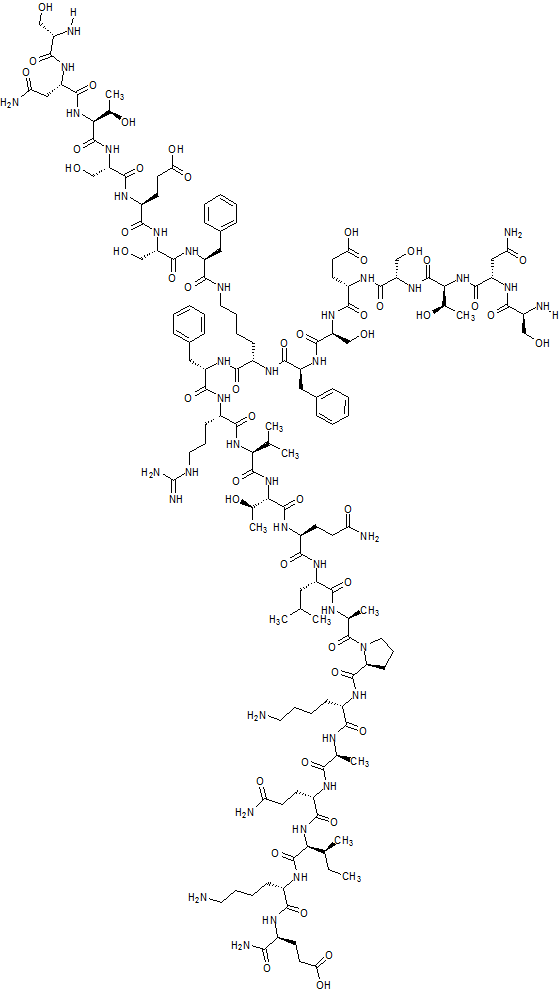
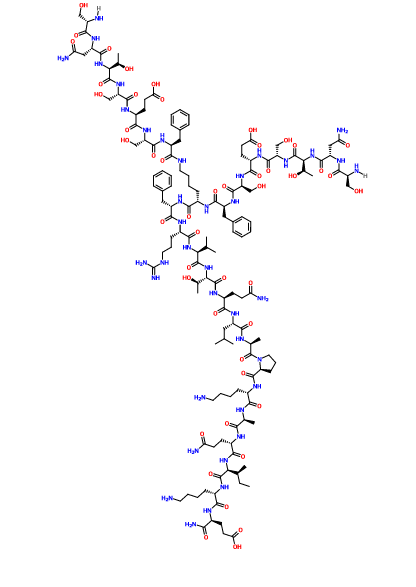
N2,N6-Bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-alpha-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-L-alpha-glutamine
C142 H226 N40 O48, 3261.553
SEE ALSO
US 2015087581
Compound 8 (SEQ ID NO: 49) SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2
Example 2Synthesis of Sequence Shown in SEQ ID NO: 49
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows.
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 mmol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 mL; 5 equiv, 1 mmol) and HOBT (0.08 g; 5 equiv, 0.6 mmol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 mmol, 8 equivalent excess of HOBt (0.22 g, 1.6 mmol) and 10 equivalent excess of DIC (0.32 mL, 2 mmol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 mmol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 mmol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B:Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT—12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+;); 816.2[M/4+H]+.
SMILES
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)N)[C@@H](C)O
NEXT………..
- Molecular Weight, 3261.55
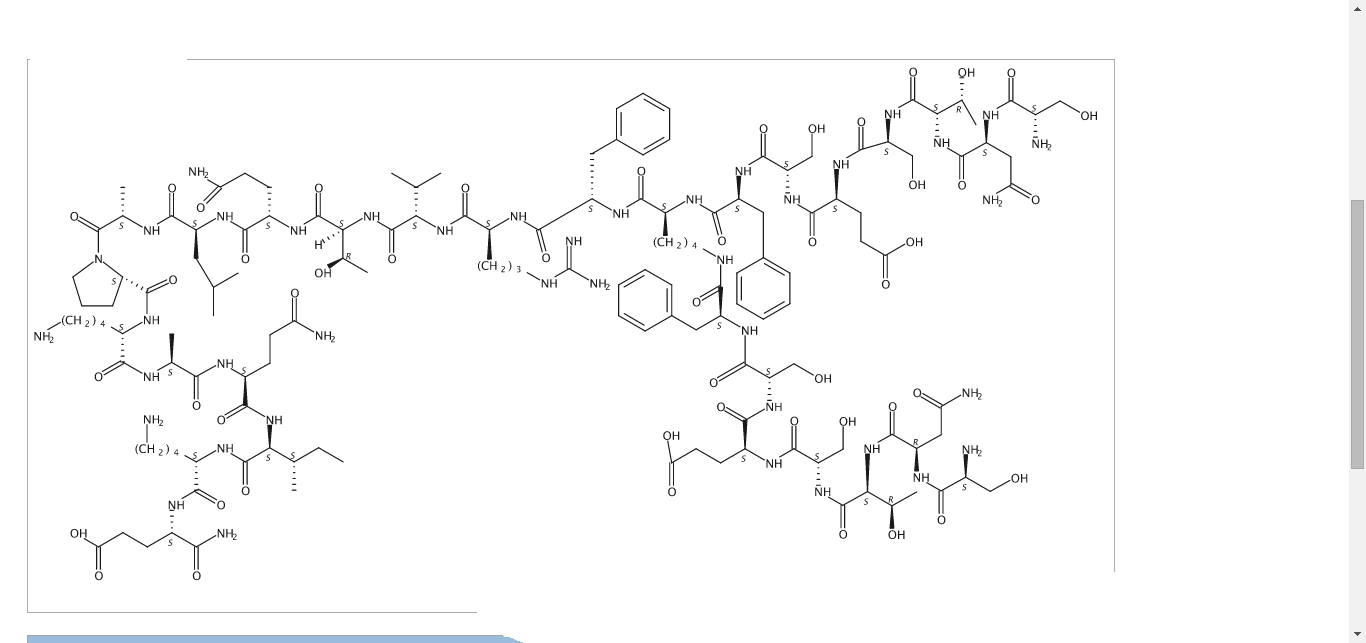
NEXT……….

Clips
Aurigene and Pierre Fabre Pharmaceuticals Announce a Licensing Agreement for a New Cancer Therapeutic in Immuno-oncology: AUNP12, an Immune Checkpoint Modulator Targeting the PD-1 Pathway
Pierre Fabre are thus reinforcing their oncology portfolio which already enjoys a combination of chemotherapies, monoclonal antibodies and immuno-conjugates assets at various development phases
Feb 13, 2014, 03:14 ET from Aurigene and Pierre Fabre Pharmaceuticals
CASTRES, France and BANGALORE, India, February 13, 2014 /PRNewswire/ —
Pierre Fabre, the third largest French pharmaceutical company, and Aurigene, a leading biotech company based in India, today announced that the two companies have entered into a collaborative license, development and commercialization agreement granting Pierre Fabre global Worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12.
AUNP-12 offers a breakthrough mechanism of action in the PD-1 pathway compared to other molecules currently in development in the highly promising immune therapy cancer space. AUNP-12 is the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities with emerging and established treatment regimens. AUNP-12 will be in development for numerous cancer indications.
Under the terms of this agreement, Aurigene will receive an upfront payment from Pierre Fabre. Aurigene will also receive additional milestone payments based upon the continued development, regulatory progresses and commercialization of AUNP-12.
“We are pleased that Pierre Fabre see the PD-1 program as a strategic asset in their portfolio. Overall, the deal structure, in line with the financial terms that have been seen in this space, demonstrate the importance that Pierre Fabre attach to the program,” said CSN Murthy, CEO, Aurigene.
“The plans that Pierre Fabre have detailed for the development of this differentiated asset highlight the long-term opportunities for this novel cancer therapeutic,” added Murali Ramachandra, Sr VP, Research, Aurigene.
“This agreement, in the field of oncology, is fully consistent with our vision to build Pierre Fabre’s future in prescription drugs, from a combination of cutting-edge internal R&D capabilities and license partnerships with innovative biotech companies like Aurigene,” stated Bertrand Parmentier, CEO, Pierre Fabre.
“With this deal, Pierre-Fabre Pharmaceuticals are reinforcing their portfolio of oncology assets and capitalizing on their proven capabilities in developing biological compounds such as monoclonal antibodies and immuno-conjugates. We have been impressed by the science at Aurigene and encouraged by the differentiated profile reported for AUNP-12,” added Frédéric Duchesne, President, Pierre Fabre Pharmaceuticals.
About immuno-oncology
Immuno-oncology is an emerging field in cancer therapy, where the body’s own immune system is harnessed to fight against cancer. This approach of targeting cancer through immune response has had a breakthrough when robust and sustained responses were obtained only upon blocking the immune checkpoint targets (such as PD-1 and CTLA4). Recent successes in clinical trials performed with such therapies suggest that immunotherapy should be considered alongside surgery, chemotherapy, radiotherapy and targeted therapy as the fifth cornerstone of cancer treatment.
PD-1 (Programmed cell Death 1) is a receptor that negatively regulates T-cell activation by interacting with specific ligands PD-L1 and PD-L2. Tumor cells express these ligands and thereby escape from the action of T-cells.
About AUNP-12
AUNP-12 is a branched 29-amino acid peptide sequence engineered from the PD-L1/ L2 binding domain of PD-1 It blocks the PD-1/PD-L1, PD-1/PD-L2 and PD-L1/CD80 pathways. AUNP-12 is highly effective in antagonizing PD-1 signaling, with desirable in vivo exposure upon subcutaneous dosing. It inhibits tumor growth and metastasis in preclinical models of cancer and is well tolerated with no overt toxicity at any of the tested doses.
About Aurigene
Aurigene is a biotech focused on development of innovative small molecule and peptide therapeutics for Oncology and Inflammation; key focus areas for Aurigene are Immuno-oncology, Epigenetics and the Th17 pathway. Aurigene’s PD-1 program is the first of several peptide-based immune checkpoint programs that are at different stages of Discovery.
Aurigene has partnered with several big pharma and mid-pharma companies in the US and Europe, and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies.
Aurigene’s pre-clinical pipeline includes (1) Selective and pan-BET Bromodomain inhibitors (2) RoR gamma reverse agonists (3) EZH2 inhibitors (4) NAMPT inhibitors and (5) Several immune check point peptide inhibitor programs.
For more information: http://aurigene.com/
About Pierre Fabre:
Pierre Fabre is a privately-owned health care company created in 1961 by Mr Pierre Fabre. It is the second largest French independent pharmaceutical group with 2013 sales amounting to about €2 billion (yet to be audited) across 140 countries. The company is structured around two divisions: Pharmaceuticals (Prescription drugs, OTC, Oral care) and Dermo-cosmetics. Prescription drugs are organized around four main franchises: oncology, dermatology, women’s health and neuropsychiatry. Pierre Fabre employs some 10 000 people worldwide, including 1 300 in R&D. The company allocates about 20% of its pharmaceuticals sales to R&D and relies on more than 25 years of experience in the discovery, development and global commercialization of innovative drugs in oncology. Pierre Fabre has a long commitment to oncology and immunology with major R&D centers in France: the Pierre Fabre immunology Centre (CIPF) in Saint Julien en Genevois and the Pierre Fabre Research Institute (IRPF) located on the Toulouse-Oncopole campus which has been officially recognized as a National Center of Excellence for cancer research since 2012.
REFERENCES
http://slideplayer.com/slide/5760496/
P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, “A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events,” Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), “Immunosuppression modulating compounds”, US Patent application US 2011/0318373, 29 Dec 2011.
P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, “Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway,” Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, “Therapeutic compounds for immunomodulation” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
P. G. N. Sasikumar and M. Ramachandra, “Immunomodulating cyclic compounds from the BC loop of human PD1” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, “Peptidomimetic compounds as immunomodulators” (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), “Modulators of immunoinhibitory receptor PD-1, and methods of use thereof”, PCT Patent Application WO/2011/082400, 7 Jul 2011.
M. Cordingley, “Battle of PD-1 blockade is on”, February 7, 2014 : http://discoveryview.ca/battle-of-pd-1-blockade-is-on/ [Accessed 25 February 2014].
Mr. CSN Murthy
Chief Executive Officer, Aurigene Discovery Technologies Ltd.

Mr. CSN Murthy began his career with ICICI Ventures, India’s first Venture Capital fund. He was subsequently a management consultant to the Pharma and Chemical sectors. Later, he worked in the Business Development and General Management functions in Pharmaceutical companies, including as the Chief Operating Officer of Gland Pharma Ltd. CSN holds a Bachelors degree in Chemical Engineering from the Indian Institute of Technology (IIT), Madras and an MBA from the Indian Institute of Management (IIM), Bangalore.
Dr.Thomas Antony
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr.Thomas Antony did his Ph.D in Biophysical Chemistry from University of Delhi and had his postdoctoral training at Jawaharlal Nehru University- Delhi, The University of Medicine and Dentistry of New Jersey- USA, and Max Planck Institute for Biophysical Chemistry- Germany. He is the recipient of many research fellowships, including Max Planck Fellowship and Humboldt Research Fellowship. He has more than 20 years of research experience. Dr.Thomas has published 24 research papers and he is the co-author of three international patents. His core area of expertise is in assay development and screening. At Aurigene, Dr.Thomas leads the Biochemistry and Structural Biology Divisions. He was the coordinator of Aurigene-University of Malaya collaboration programs.
Dr. Kavitha Nellore
Associate Research Director, Aurigene Discovery Technologies Ltd.

Dr. Kavitha Nellore obtained her PhD in Bioengineering from Pennsylvania State University, USA. During this time, she was a fellow of the Huck’s Institute of Life Sciences specializing in Biomolecular Transport Dynamics. She has been at Aurigene for more than a decade, and is currently leading a group of cell biologists at both Bangalore and Kuala Lumpur. At Aurigene, she leads multiple drug discovery programs in the therapeutic areas of inflammation, oncology and immuno-oncology. She plays a key role in target selection as well as validation efforts to add to Aurigene’s pipeline. Kavitha also played a key role in coordinating the Aurigene-University of Malaya collaboration.
To print: Click here or Select File and then Print from your browser's menu “Strategic partnerships will boost drug discovery activities in India”Wednesday, May 16, 2007 08:00 IST
What are the factors led to the scores of partnerships in research and drug discovery space? Biology demands expertise in areas such as DMPK, cell and assay biology and vivo expertise, which is not as common as chemistry expertise in India. There is also a paradigm shift in assignments for CROs, as they are gearing up to gain higher value from existing projects, including Intellectual Property (IP) generation. Probably, this is where Aurigene has a head start over other CROs. Although we are in research services, our success is in IP generated work. We work with world-class companies like Novo Nordisk, Rheoscience, Debiopharm, Forest Labs and Merck Serono on integrated discovery projects. What do you think is the uniqueness of Aurigene that sets it apart from other companies? It is understood that a major chunk of the business for CROs is from global customers. What is the reason for this? The whole concept of drug discovery outsourcing business has caught on in the last 18-month. Could you give us an overview of the market? In India, CROs operate differently. The innovation driven companies are taking on ‘collaborative drug discovery’ projects, leveraging the mutual strengths each has to offer the other (cost and expertise respectively). Therefore, the complexion of business is different from that of the Western countries. We would see large outsourcing companies positioning themselves as offshore partners for pharma-biotech majors. They could also go in for a BOT (Build Operate Transfer) model. Under this model, companies will set-up the facility and manage it to make it a ‘centre of excellence’, focusing on certain disease segments. Another option is that some companies can take up their own programmes to develop compounds and enter into licensing partnerships. This is similar to the US and Europe biotech style of working. Both of these practices are likely to happen in India. How much do you acknowledge science and strategy in Aurigene’s growth? What according to you is the future of the drug discovery sector? |
/////////AUNP-12, Aurigene, Pierre Fabre Pharmaceuticals, Licensing Agreement, New Cancer Therapeutic, Immuno-oncology, AUNP 12, Immune Checkpoint Modulator Targeting the PD-1 Pathway, PEPTIDES
FEW MORE COMPDS FROM PATENT
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54, Sequence Length: 29, 22, 7
multichain; modified (modifications unspecified)
SNTSESFK FRVTQ LAPKAQIKE, 1353564-66-5
SNTSESF
C142 H225 N39 O49
L-Glutamic acid, N2,N6-bis(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3262.54

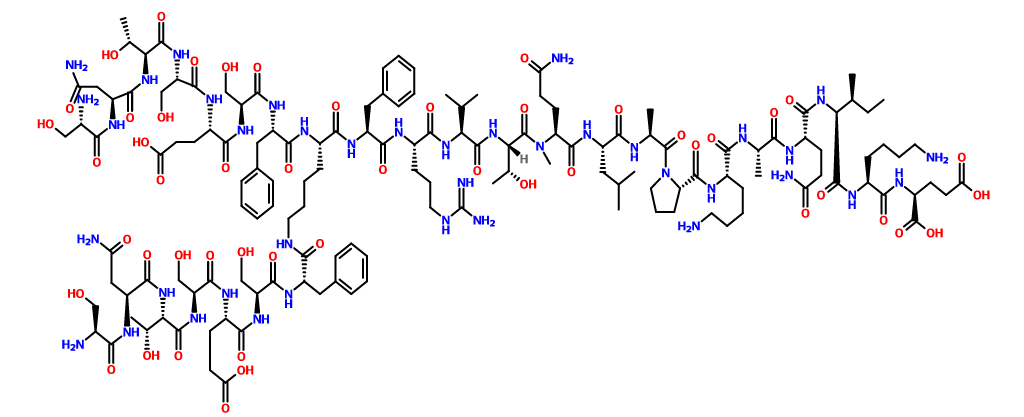
NEXT……………………
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF
CAS 1353564-64-3
C142 H226 N40 O48
L-α-Glutamine, L-seryl-D-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
MW 3261.55, Sequence Length: 29, 22, 7
multichain; modified
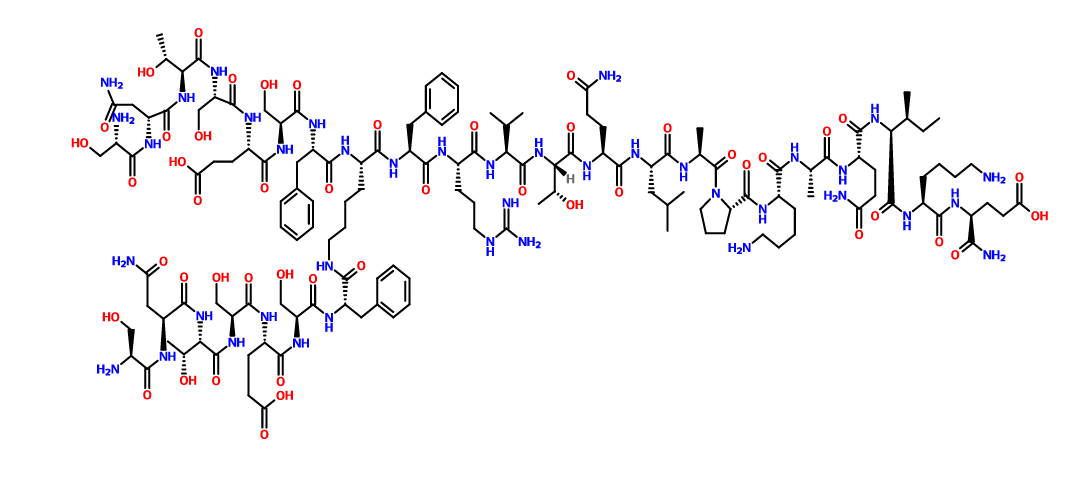

smiles
O=C(N[C@@H](CCCCNC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O)C(=O)N[C@@H](Cc2ccccc2)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N3CCC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(=O)O)C(N)=O)[C@H](Cc4ccccc4)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@@H](C)O
NEXT……………..
CAS 1353564-60-9
C142 H226 N40 O48
L-α-Glutamine, D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl-N6-(L-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFKFR VTQLAPKAQI KE
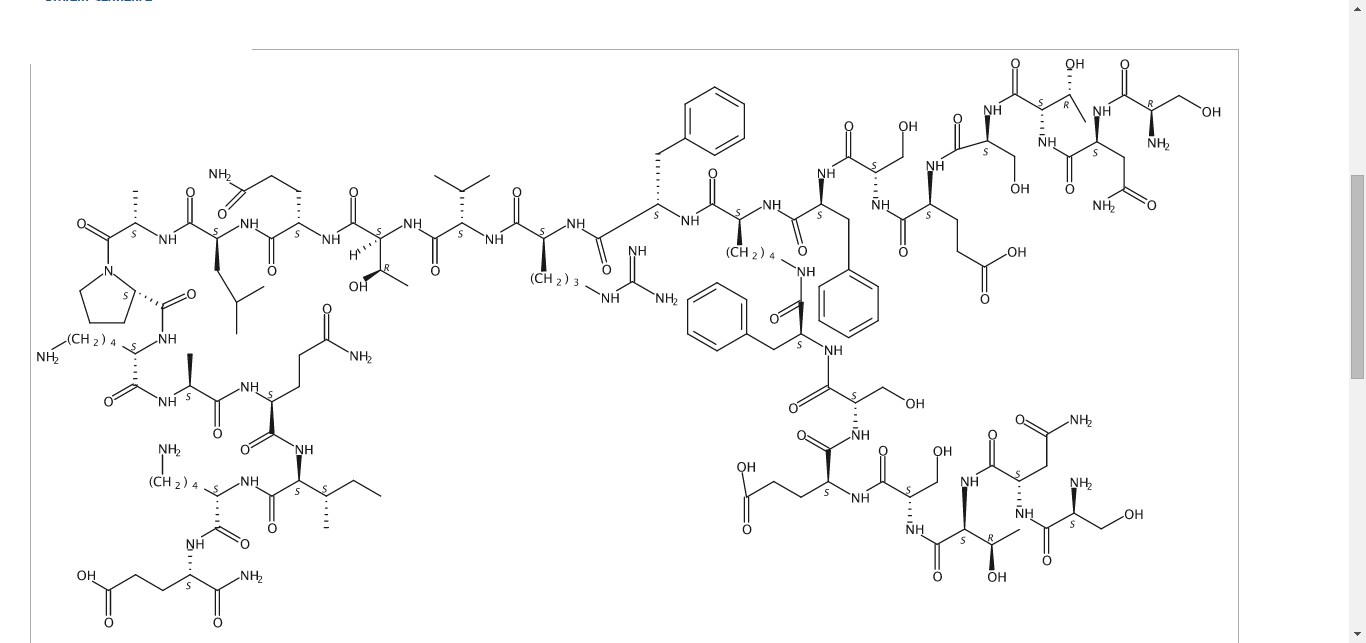
NRXT…………………….
. CAS 1353564-61-0
C142 H226 N40 O48
L-α-Glutamine, N2,N6-bis(D-seryl-L-asparaginyl-L-threonyl-L-seryl-L-α-glutamyl-L-seryl-L-phenylalanyl)-L-lysyl-L-phenylalanyl-L-arginyl-L-valyl-L-threonyl-L-glutaminyl-L-leucyl-L-alanyl-L-prolyl-L-lysyl-L-alanyl-L-glutaminyl-L-isoleucyl-L-lysyl-
3261.55
Sequence Length: 29, 22, 7multichain; modified
SNTSESFK FRVTQ LAPKAQI KE
SNTSESF

/////////////
