
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Uridine triacetate, ウリジントリアセタート FDA approves first emergency treatment for overdose of certain types of chemotherapy

December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
|
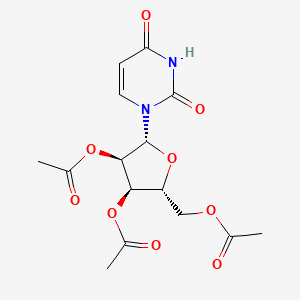
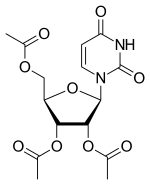
[(2R,3R,4R,5R)-3,4-bis(acetyloxy)-5-(2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-yl)oxolan-2-yl]methyl acetate
|
| Drug Name(s) | XURIDEN |
| FDA Application No. | (NDA) 208169 |
| Active Ingredient(s) | URIDINE TRIACETATE |
| Company | WELLSTAT THERAP |
| Original Approval or Tentative Approval Date | September 4, 2015 |
FDA APPROVAL SUMMARY
Chemotherapy induced poisoning, VISTOGARD, FDA 2015-12-11
Hereditary orotic aciduria, Xuriden, FIRST APPROVAL, 2015-09-04
|
|||||||
| External Identifiers |
|
|---|
Uridine triacetate is a drug used in the treatment of hereditary orotic aciduria[1] and to treat patients following an overdose ofchemotherapy drugs 5-fluorouracil or capecitabine, or in patients exhibiting early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of 5-fluorouracil or capecitabine administration.[2][3]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics and it is marketed in USA by BTG. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine triacetate is a prodrug of uridine.[4]
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTP incorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [3]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidine metabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
Marketed as the product Xuriden (FDA), uridine triacetate is indicated for the treatment of hereditary orotic aciduria. Marketed as the product Vistogard (FDA), uridine triacetate is indicated for the emergency treatment of adult and pediatric patients in the following situations: following a fluorouracil or capecitabine overdose regardless of the presence of symptoms; or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration.

Uridine Triacetate was approved by the U.S. Food and Drug Administration (FDA) on Sep 4, 2015. It was developed by Wellstat Therapeutics, then marketed as Xuriden® by Wellstat Therapeutics in US. Then it was also approved by FDA for overdose of certain types of chemotherapy on Dec 11, 2015 and marketed as Vistogard®.
Uridine Triacetate is a prodrug of the nucleoside uridine used to treat hereditary orotic aciduria. Hereditary orotic aciduria is inherited from a recessive gene. The disease is due to a defective or deficient enzyme, which results in the body being unable to normally synthesize uridine, a necessary component of ribonucleic acid (RNA). Signs and symptoms of the disease include blood abnormalities (anemia, decreased white blood cell count, decreased neutrophil count), urinary tract obstruction due to the formation of orotic acid crystals in the urinary tract, failure to thrive, and developmental delays.
Xuriden® is approved as oral granules that can be mixed with food or in milk or infant formula, and is administered once daily. The starting dosage is 60 mg/kg once daily; the dose may be increased to 120 mg/kg (not to exceed 8 grams) once daily for insufficient efficacy.
Mechanism Of Action
Uridine triacetate is an acetylated form of uridine. Following oral administration, uridine triacetate is deacetylated by nonspecific esterases present throughout the body, yielding uridine in the circulation (Figure 1).
Figure 1: Uridine Triacetate Conversion to Uridine

URIDEN provides uridine in the systemic circulation of patients with hereditary orotic aciduria who cannot synthesize adequate quantities of uridine due to a genetic defect in uridine nucleotide synthesis.
Uridine triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578] such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Uridine triacetate is also used for replacement therapy in the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase (UMPS) deficiency. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria.

Reference:1. J. Am. Chem. Soc. 1953, 75, 2017-2019.
2. Angew. Chem. internat. Edit. 1971, 10, 75.
3. US3116282.
PATENT
Production Example 1

5.6 g of uracil and 0.1 g of ammonium sulfate were dissolved in 22.4 ml of 1,1,1,3,3,3-hexamethyldisilazane and reacted at 120° C. for 2.5 hours. After the completion of the reaction, the reaction mixture was distilled to give 11.8 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine. 1H-NMR (400 MHz, in C2D6CO): δ=0.29 (s, 9H), 0.31 (s, 9H), 6.35 (d, J=5.6 Hz, 1H), 8.19 (d, J=5.5Hz, 1H)
Referential Example 11.21 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 1.15 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.8 ml of acetonitrile and cooled to 5° C. Next, 0.94 g of SnCl4 was added dropwise thereinto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted for 3 hours. The reaction mixture was analyzed by HPLC. Thus, β-uridine triacetate was obtained with a reaction yield of 83%.
Example 1

0.93 g of 2,4-bis(trimethylsilyloxy)-1,3-diazine obtained in PRODUCTION EXAMPLE 1 and 0.92 g of 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose were dissolved in 4.7 ml of acetonitrile and cooled to 4° C. Then 0.49 g of FeCl3 was added thereto at the same temperature. After stirring for 10 minutes at the same temperature, the mixture was heated to 50° C. and reacted. The reaction was monitored by HPLC. After the completion of the reaction, the reaction mixture was added dropwise at 4° C. into a cold aqueous solution of sodium hydrogencarbonate which had been preliminarily prepared. After filtering off the catalyst residue, the filtrate was separated and the aqueous layer was extracted with 20 ml portions of ethyl acetate thrice. The organic layers were combined, washed with a saturated aqueous solution of sodium chloride and dried over sodium sulfate. After distilling off the solvent, 1.2 g (purity 80%) of the target compound was obtained as a viscous white solid.
Namely, the target compound could be obtained at a yield comparable to REFERNTIAL EXAMPLE 1 wherein SnCl4 was employed as the catalyst. 1H-NMR (400 MHz, in CDCl3): δ=2.11 (s, 3H), 2.14 (s, 3H), 2.15 (s, 3H), 4.35 (m, 3H), 5.33 (m, 2H), 5.79 (d, J=8.2 Hz, 1H), 6.04 (d, J=4.9 Hz, 1H), 7.39 (d, J=8.2 Hz, 1H)

CLIP
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Vistogard (uridine triacetate) for the emergency treatment of adults and children who receive an overdose of the cancer treatment fluorouracil or capecitabine, or who develop certain severe or life-threatening toxicities within four days of receiving these cancer treatments.
“Treating cancer requires not only selecting which drug may be most effective and well tolerated, but ensuring the correct dose is given at proper intervals. While rare, unintentional overdose can occur,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is a first-of-its-kind therapy that can potentially save lives following overdose or life-threatening toxicity from these chemotherapy agents.”
Fluorouracil (taken by infusion) and capecitabine (taken orally) are similar types of chemotherapy that have been used for decades to treat several types of cancer, including breast and gastrointestinal cancers. An overdose of fluorouracil or capecitabine is rare, but when it occurs, the effects are serious and can be fatal.
Vistogard, taken orally, blocks cell damage and cell death caused by fluorouracil chemotherapy. Patients should take Vistogard as soon as possible after the overdose (whether or not they have symptoms) or early-onset (within four days) of severe or life-threatening toxicity. The patient’s health care provider will determine when he or she should return to the prescribed chemotherapy after treatment with Vistogard.
The efficacy and safety of Vistogard were studied in 135 adult and pediatric cancer patients who were treated in two separate trials and had either received an overdose of flourouracil or capecitabine, or had early-onset, unusually severe or life-threatening toxicities within 96 hours after receiving flourouracil (not due to an overdose). The studies’ primary measure was survival at 30 days or until chemotherapy could resume if prior to 30 days. Of those who were treated with Vistogard for overdose, 97 percent were still alive at 30 days. Of those treated with Vistogard for early-onset severe or life-threatening toxicity, 89 percent were alive at 30 days. In both studies, 33 percent of patients resumed chemotherapy in less than 30 days.
Vistogard is not recommended for treating non-emergency adverse reactions associated with flourouracil or capecitabine because Vistogard may lessen the efficacy of these drugs. The safety and efficacy of Vistogard initiated more than 96 hours following the end of treatment with flourouracil or capecitabine have not been established.
The most common side effects of treatment with Vistogard were diarrhea, vomiting and nausea.
The FDA granted Vistogard orphan drug designation, which provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Vistogard was also granted priority review and fast track designations, which are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions.
Vistogard is marketed by Wellstat Therapeutics Corporation based in Gaithersburg, Maryland.
CLIP
With support from Almac, Wellstat delivers for a rare disease.
Proximity of API and finished drug development helps uridine triacetate to market for two indications
By Rick Mullin
“The initial contact was a cold call by Almac in 2010 or 2011,” recalls Mike Bamat, senior vice president of R&D at Wellstat Therapeutics, a small drug company in Gaithersburg, Md. “There were probably a couple of calls. It was one of those things where timing is everything.”
Almac, a Craigavon, Northern Ireland-based pharmaceutical services company, was looking to get in on Wellstat’s development of uridine triacetate, a synthetic pyrimidine analog, as an antidote for fluorouracil and capecitabine toxicity and overdose in cancer patients receiving those chemotherapies. And the calls, which Almac records indicate followed some communication between the companies, happened to come just when Wellstat was looking to change service partners as it moved toward commercial development of the drug.

Uridine triacetate
Discovery: Wellstat Therapeutic’s research on the therapeutic potential of exogenous uridine leads to a determination that uridine triacetate is a safe means of delivering the agent
Applications: Treatment of hereditary orotic aciduria (HOA), an extremely rare disease in which the body does not produce uridine, causing overproduction of orotic acid; emergency treatment of toxic reaction to or overdose of the cancer treatments fluorouracil and capecitabine
Methods of action: Treating HOA, uridine triacetate restores intracellular nucleotide concentrations, normalizing orotic acid production; as a chemotherapy antidote, it increases intracellular levels of uridine to dilute fluorouracil and capecitabine
Years in development: Since 2008 for chemotherapy antidote, and 2013 for HOA
Approved: Xuriden for HOA, Sept. 4, 2015; Vistogard for chemotherapy antidote, Dec. 11, 2015
The job went to Almac, as did work that sprang up as the result of another phone call to Wellstat—this one from the U.S. Food & Drug Administration.
As Bamat explains, uridine triacetate caught FDA’s attention regarding another potential indication—an extremely rare and life-threatening disease called hereditary orotic aciduria, or HOA. A consequence of the body’s inability to produce uridine, a necessary component of ribonucleic acid, HOA can manifest in a range of symptoms including blood abnormalities, developmental delays, and urinary tract obstruction caused by overproduction of orotic acid. There have been 20 reported cases of HOA since the 1950s. Only four cases are currently known in the U.S., Bamat says, and likely fewer than 20 in the world.
Wellstat landed approvals for Xuriden, the HOA treatment, in September of last year and Vistogard, the chemotherapy antidote, in December.
The story of Xuriden centers on a raft of FDA incentives for super-rare diseases that enabled Wellstat to move forward on an expedited application for a drug that will never be made in any great volume. But bringing Xuriden and Vistogard to market may also be viewed as the story of a drug discovery firm becoming a commercial enterprise thanks to its partnership with a service provider.
As Wellstat began late-stage development of the chemotherapy antidote, its research partner at the time, QS Pharma, was acquired by the service firm WIL Research. The look and feel of the partnership changed, according to Bamat.
“We kind of lost the small, easy-to-work-with relationship we had with them,” he says. Wellstat also needed support on development and manufacturing of a finished drug product composed of granules delivered in packets or sachets. The drug is administered orally, usually sprinkled on food such as applesauce or yogurt.
Almac was deemed a good fit because of its experience with developing drugs in granule form for “sachet presentation,” a packaging method more common in Europe than in the U.S. The Northern Ireland firm’s ability to develop and manufacture the active pharmaceutical ingredient (API) and the drug product in one location—at its headquarters—would also prove to be a significant advantage.
The distance between Gaithersburg and Craigavon, however, was a concern, according to Bamat. “We debated it. Especially those of us who knew we would be going there,” he says. “We couldn’t just jump in a car and go. But we looked at a variety of things, including cost and value, and it was all very positive at Almac.”
According to David Downey, vice president of commercial operations at Almac, bringing Wellstat’s work on uridine triacetate to commercial production posed several challenges, the first being to secure supply of uridine starting material, which is extracted from sugar beets by Euticals, an Italian firm. Next was developing a method to control particle size in both the API and the finished product. Almac also had to validate process equipment as it scaled up production.
“Uridine triacetate is Wellstat’s first commercial product,” Downey says. “So we were provided with a process more fit for development than for commercial production.”
The basic formulation of a granule drug product is simple, according to Downey: The API and excipient are mixed in a dry blender. The challenge is developing an analytical regimen to assure the granules are blended uniformly. Meeting the challenge required a high level of coordination between API and drug product process development.
“Wellstat needed a partner that could support them from the API to the drug product,” Downey says. The physical proximity between the Almac facilities in Craigavon conducting API and drug product work was a key advantage, he claims.

“If you listen to our business development people, you’ll hear them use the term, ‘crossing car parks as opposed to crossing oceans,’ ” Downey says, explaining that many competitors who offer API and finished drug services run these operations thousands of kilometers apart from each other, sometimes on different continents.
Before it signed on with Almac, Wellstat had been working with uridine triacetate for about 10 years. Its focus on developing the antidote drug started in 2008. Branching into the HOA treatment, however, upped the stakes.
Clinical study development for an HOA therapy was expedited via a full house of regulatory incentives from FDA, according to Bamat. “We had orphan drug designation, rare pediatric designation, breakthrough therapy designation, and priority review,” he says. “So they really went all out in helping us develop this.”
Although Wellstat was interested in developing a life saving drug for children, it was concerned about paying for it, given the tiny market. “At that time, the rare pediatric disease priority review voucher program was just on the radar,” Bamat says. “FDA said, ‘Consider this new program. Maybe it’s a way that at some risk you could recoup some of your costs.’ We looked at it and were willing to take the risk.”
It paid off. Wellstat was able to sell its priority review voucher—which entitles a company that brings a rare pediatric drug to market to receive expedited review of a subsequent drug—to AstraZeneca last year for an undisclosed amount. Other vouchers sold in 2015 brought high sums, including $350 million for one that AbbVie bought from United Therapeutics in August.
Bamat says Wellstat is not likely to change focus after its success with uridine triacetate. It continues to investigate new indications for the compound and will likely work with Almac on anything going into commercial development.
He emphasizes the importance of maintaining an effective working relationship with an outsourcing partner. “My main consideration is that these are people we can really work with on a day-to-day, week-to-week basis,” Bamat says. “Will the communication be good? Will they be honest and transparent with us, and will we be the same for them? That was a key factor, and we felt it was a plus with Almac.”
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2-3 hours |
| Biological half-life | 2-2.5 hours |
| Excretion | Renal |
| Identifiers | |
| DrugBank | DB09144 |
| Chemical data | |
| Formula | C15H18Cl0N2O9S0 |
| Molar mass | 370.31 g·mol−1 |
References
- HIGHLIGHTS OF PRESCRIBING INFORMATION OF XURIDEN
- Jump up^ BTG Announces FDA Approval of VISTOGARD® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine
- Jump up^ “FDA Approved Drugs:Uridine Triacetate”. FDA. 2015-12-11. Retrieved 2016-04-29.
- “Uridine triacetate”. DrugBank.
| Patent ID | Date | Patent Title |
|---|---|---|
| US7807654 | 2010-10-05 | Compositions and methods for treatment of mitochondrial diseases |
| US2010222296 | 2010-09-02 | PYRIMIDINES, SUCH AS URIDINE, IN TREATMENTS FOR PATIENTS WITH BIPOLAR DISORDER |
| US7737128 | 2010-06-15 | Pyrimidines, such as uridine, in treatments for patients with bipolar disorder |
| US2010098678 | 2010-04-22 | Methods of Treatment of Mitochondrial Disorders |
| US2010041620 | 2010-02-18 | METHODS FOR IMPROVING FRONTAL BRAIN BIOENERGETIC METABOLISM |
| US2010041621 | 2010-02-18 | METHODS AND COMPOSITIONS FOR IMPROVING COGNITIVE PERFORMANCE |
| US7582619 | 2009-09-01 | Compositions and methods for treatment of mitochondrial diseases |
| US2008226684 | 2008-09-18 | METHOD AND PROCESS FOR THE PRODUCTION OF MULTI-COATED RECOGNITIVE AND RELEASING SYSTEMS |
| US7105498 | 2006-09-12 | Acylated uridine and cytidine and uses thereof |
| US6956028 | 2005-10-18 | Compositions and methods for treatment of mitochondrial diseases |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015307542 | 2015-10-29 | MODIFIED NUCLEIC ACID MOLECULES AND USES THEREOF |
| US2015167017 | 2015-06-18 | ALTERNATIVE NUCLEIC ACID MOLECULES AND USES THEREOF |
| US8821899 | 2014-09-02 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8771713 | 2014-07-08 | Method and process for the production of multi-coated recognitive and releasing systems |
| US8741316 | 2014-06-03 | Highly porous, recognitive polymer systems |
| US2012294869 | 2012-11-22 | Methods for Treating Fatty Liver Disease |
| US2012078529 | 2012-03-29 | DETERMINING THE SEVERITY OF 5-FLUOROURACIL OVERDOSE |
| US8067392 | 2011-11-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7915233 | 2011-03-29 | Compositions and methods for treatment of mitochondrial diseases |
| US7884202 | 2011-02-08 | Nucleobase Having Perfluoroalkyl Group and Process for Producing the Same |
Uridine triacetate
- Molecular FormulaC15H18N2O9
- Average mass370.311 Da
ウリジントリアセタート
[(2R,3R,4R,5R)-3,4-diacetyloxy-5-(2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl acetate
FDA APPROVED2015/9/4 . AS Xuriden
Uridine triacetate (INN),[1] formerly known as vistonuridine, is an orally active tri-acetylated prodrug of uridine[2] used:
- in the treatment of hereditary orotic aciduria (brand name Xuriden /ˈzʊərədɛn/ ZOOR-ə-den);[3]
- to treat patients following an overdose of chemotherapy drugs 5-fluorouracil (5-FU) or capecitabine regardless of the presence of symptoms, or who exhibit early-onset, severe or life-threatening toxicity affecting the cardiac or central nervous system, and/or early-onset, unusually severe adverse reactions (e.g., gastrointestinal toxicity and/or neutropenia) within 96 hours following the end of fluorouracil or capecitabine administration (brand name Vistogard).[4][5][6]
Uridine triacetate was developed, manufactured and distributed by Wellstat Therapeutics. Also, It was granted breakthrough therapy designation by FDA in 2015.
Uridine Triacetate is a synthetic uridine pro-drug that is converted to uridine in vivo. Uridine, a pyrimidine nucleotide, has been used in a variety of diseases including depressive disorders and inherited myopathies. (NCI04)
Uridine triacetate, formerly known as vistonuridine, is an orally active prodrug of the naturally occurring nucleoside uridine. It is used for the treatment of hereditary orotic aciduria (Xuriden), or for the emergency treatment of fluorouracil or capecitabine overdose or toxicity (Vistogard). It is provided in the prodrug form as uridine triacetate as this form delivers 4- to 6-fold more uridine into the systemic circulation compared to equimolar doses of uridine itself. When used for the treatment or prevention of toxicity associated with fluorouracil and other antimetabolites, uridine triacetate is utilized for its ability to compete with 5-fluorouracil (5-FU) metabolites for incorporation into the genetic material of non-cancerous cells. It reduces toxicity and cell-death associated with two cytotoxic intermediates: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). Normally, FdUMP inhibits thymidylate synthase required for thymidine synthesis and DNA replication and repair while FUTPincorporates into RNA resulting in defective strands. As a result, these metabolites are associated with various unpleasant side effects such as neutropenia, mucositis, diarrhea, and hand–foot syndrome. Like many other neoplastic agents, these side effects limit the doses of 5-FU that can be administered, which also affects the efficacy for treatment. By pre-administering with uridine (as the prodrug uridine triacetate), higher doses of 5-FU can be given allowing for improved efficacy and a reduction in toxic side effects [A18578]. It can also be used as a rescue therapy if severe side effects present within 96 hours after initiation of therapy. Uridine triacetate is also used for the treatment of hereditary orotic aciduria, also known as uridine monophosphate synthase deficiency. This rare congenital autosomal recessive disorder of pyrimidinemetabolism is caused by a defect in uridine monophosphate synthase (UMPS), a bifunctional enzyme that catalyzes the final two steps of the de novo pyrimidine biosynthetic pathway. As a result of UMPS deficiency, patients experience a systemic deficiency of pyrimidine nucleotides, accounting for most symptoms of the disease. Additionally, orotic acid from the de novo pyrimidine pathway that cannot be converted to UMP is excreted in the urine, accounting for the common name of the disorder, orotic aciduria. Furthermore, orotic acid crystals in the urine can cause episodes of obstructive uropathy. When administered as the prodrug uridine triacetate, uridine can be used by essentially all cells to make uridine nucleotides, which compensates for the genetic deficiency in synthesis in patients with hereditary orotic aciduria. When intracellular uridine nucleotides are restored into the normal range, overproduction of orotic acid is reduced by feedback inhibition, so that urinary excretion of orotic acid is also reduced.
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 65” (PDF). World Health Organization. p. 92. Retrieved 12 March 2017.
- ^ “Uridine triacetate — DrugBank Page”. 12 March 2017.
- ^ “Xuriden (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “Vistogard (uridine triacetate) Oral Granules. Full Prescribing Information” (PDF). Wellstat Therapeutics Corporation. Gaithersburg, MD 20878. Retrieved 12 March 2017.
- ^ “BTG Announces FDA Approval of Vistogard® (Uridine Triacetate) as Antidote to Overdose and Early Onset, Severe, or Life-Threatening Toxicities from Chemotherapy Drugs 5-Fluorouracil (5-FU) or Capecitabine”. BTG International Ltd. 11 December 2015. Retrieved 12 March 2017.
- ^ “Approved Drugs — Uridine Triacetate”. U.S. Food and Drug Administration. Retrieved 12 March 2017.
External links
Patents
- US7776838
- US5968914
- US6258795
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7776838 |
| Expiration | Aug 17, 2027 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
| FDA Orange Book Patents: 2 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6258795 |
| Expiration | Jul 10, 2019 |
| Applicant | WELLSTAT THERAP |
| Drug Application | N208159 (Prescription Drug: VISTOGARD. Ingredients: URIDINE TRIACETATE) |
 |
|
| Clinical data | |
|---|---|
| Trade names | Vistogard, Xuriden |
| Routes of administration |
Oral granules |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Pyrimidine catabolic pathway |
| Onset of action | Tmax = 2–3 hours |
| Elimination half-life | 2–2.5 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.021.710 |
| Chemical and physical data | |
| Formula | C15H18N2O9 |
| Molar mass | 370.31 g·mol−1 |
| 3D model (JSmol) | |
////////////Uridine triacetate, ウリジントリアセタート , FDA 2015, breakthrough therapy designation ,
|
CC(=O)OC[C@H]1O[C@H]([C@H](OC(C)=O)[C@@H]1OC(C)=O)N1C=CC(=O)NC1=O
|
FDA approves new oral therapy to treat ALK-positive lung cancer
December 11, 2015
Release
The U.S. Food and Drug Administration today approved Alecensa (alectinib) to treat people with advanced (metastatic) ALK-positive non-small cell lung cancer (NSCLC) whose disease has worsened after, or who could not tolerate treatment with, another therapy called Xalkori (crizotinib).
Lung cancer is the leading cause of cancer death in the United States, with an estimated 221,200 new diagnoses and 158,040 deaths in 2015, according to the National Cancer Institute. An ALK (anaplastic lymphoma kinase) gene mutation can occur in several different types of cancer cells, including lung cancer cells. ALK gene mutations are present in about 5 percent of patients with NSCLC. In metastatic cancer, the disease spreads to new parts of the body. In ALK-positive NSCLC metastatic patients, the brain is a common place for the disease to spread.
“Today’s approval provides a new therapy for a group of patients who would have few treatment options once their disease no longer responds to treatment with Xalkori,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to the primary effect on tumors in the lung, Alecensa clinical trials provide evidence of an effect on tumors that had spread to the brain, which is an important effect for clinicians to understand.”
Alecensa is an oral medication that blocks the activity of the ALK protein, which may prevent NSCLC cells from growing and spreading.
The safety and efficacy of Alecensa were studied in two single-arm clinical trials of patients with metastatic ALK-positive NSCLC whose disease was no longer controlled by treatment with Xalkori. Study participants received Alecensa twice daily to measure the drug’s effect on their lung cancer tumors. In the first study, 38 percent of participants experienced a partial shrinkage of their NSCLC tumors, an effect that lasted for an average of 7.5 months. In the second study, 44 percent of participants experienced a partial shrinkage of their NSCLC tumors, lasting for an average of 11.2 months. The trials also examined Alecensa’s effect on individuals’ brain metastases, a common occurrence in this population. Sixty-one percent of participants in the two trials who had measurable brain metastases experienced a complete or partial reduction in their brain tumors, lasting an average of 9.1 months.
The most common side effects of Alecensa are fatigue, constipation, swelling (edema) and muscle pain (myalgia). Alecensa may cause serious side effects, including liver problems, severe or life-threatening inflammation of the lungs, very slow heartbeats and severe muscle problems. Treatment with Alecensa may cause sunburn when patients are exposed to sunlight.
Alecensa was approved using the accelerated approval regulatory pathway, which allows the FDA to approve products for serious or life-threatening diseases based on evidence that the product has an effect on an outcome that is reasonably likely to predict clinical benefit. In the case of Alecensa, the tumor response to treatment, along with the duration of response, provided this evidence. Under the accelerated approval requirements, a confirmatory study is required to verify and describe the clinical benefit of Alecensa.
The FDA granted the Alecensa application breakthrough therapy designation and priority review status. These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions. Alecensa also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Alecensa is marketed by Genentech, based in San Francisco, California. Xalkori is marketed by Pfizer, based in New York, New York.
Synthesis
Read also
/////////////////
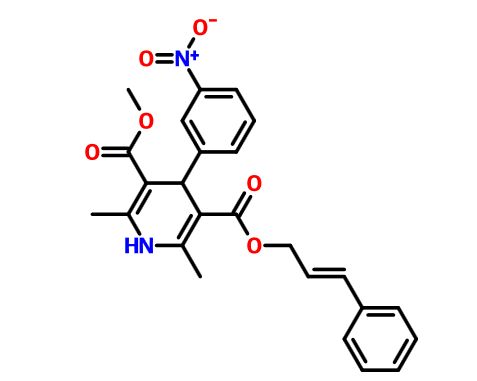
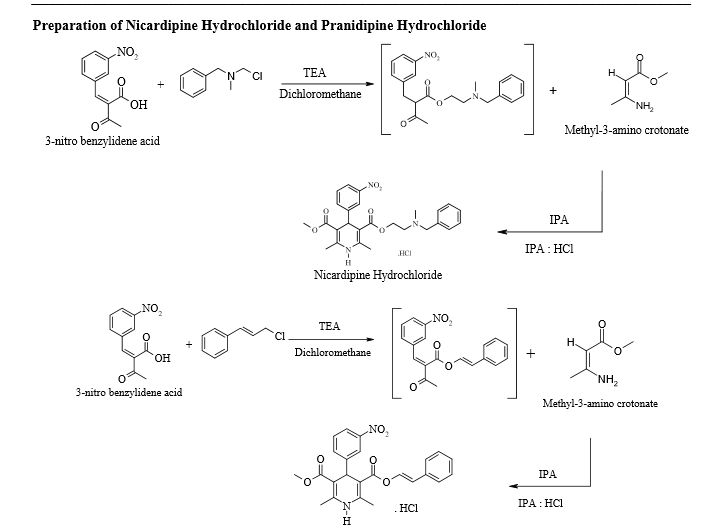
Pranidipine

Pranidipine , OPC-13340, FRC 8411
Acalas®
NDA Filing in Japan
A calcium channel blocker potentially for the treatment of angina pectoris and hypertension.
![]()
CAS No. 99522-79-9
- Molecular FormulaC25H24N2O6
- Average mass448.468
Pranidipine is a calcium channel blocker. It is a long acting calcium channel antagonist of the dihydropyridine group.[1]
![]()
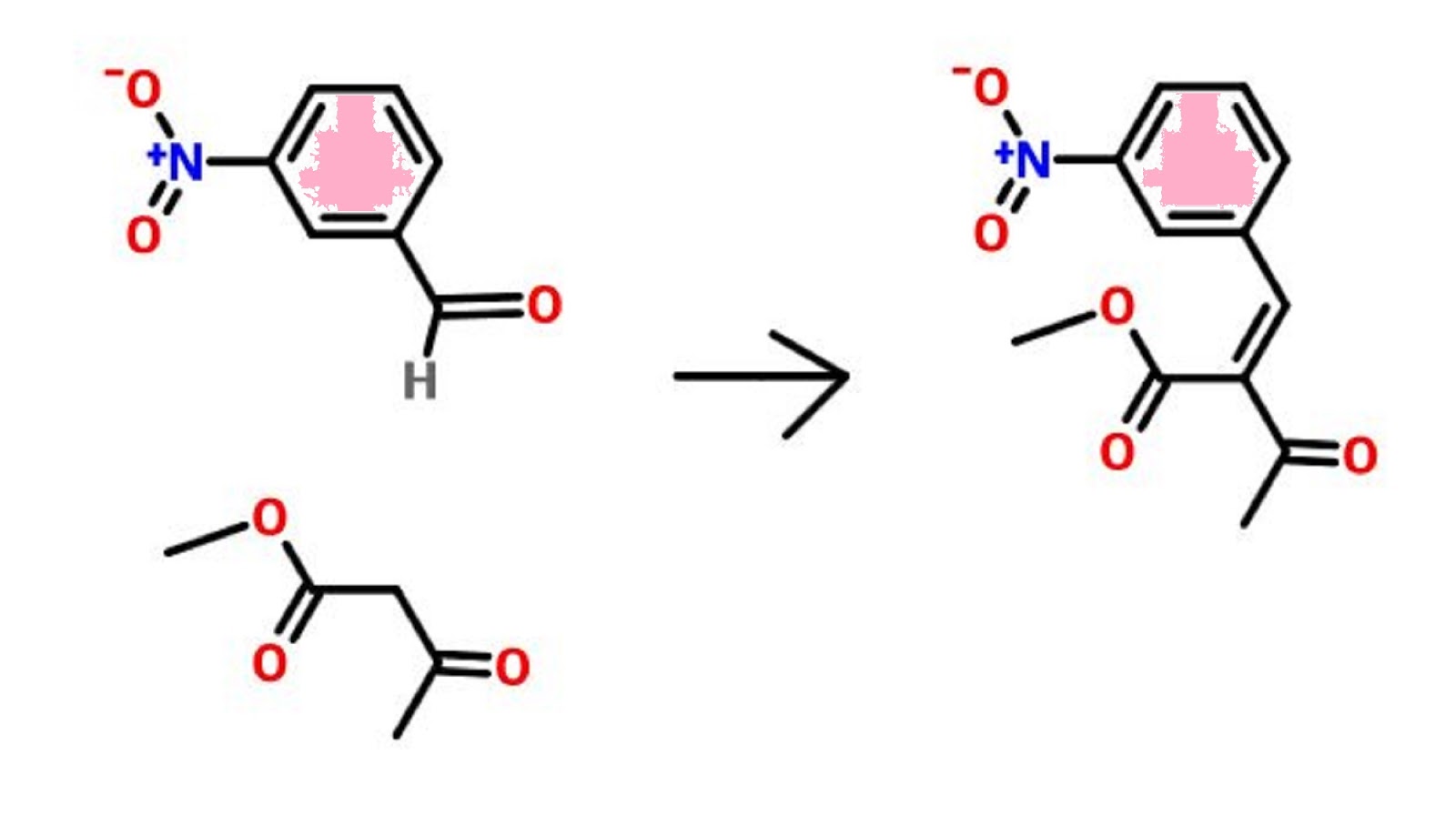
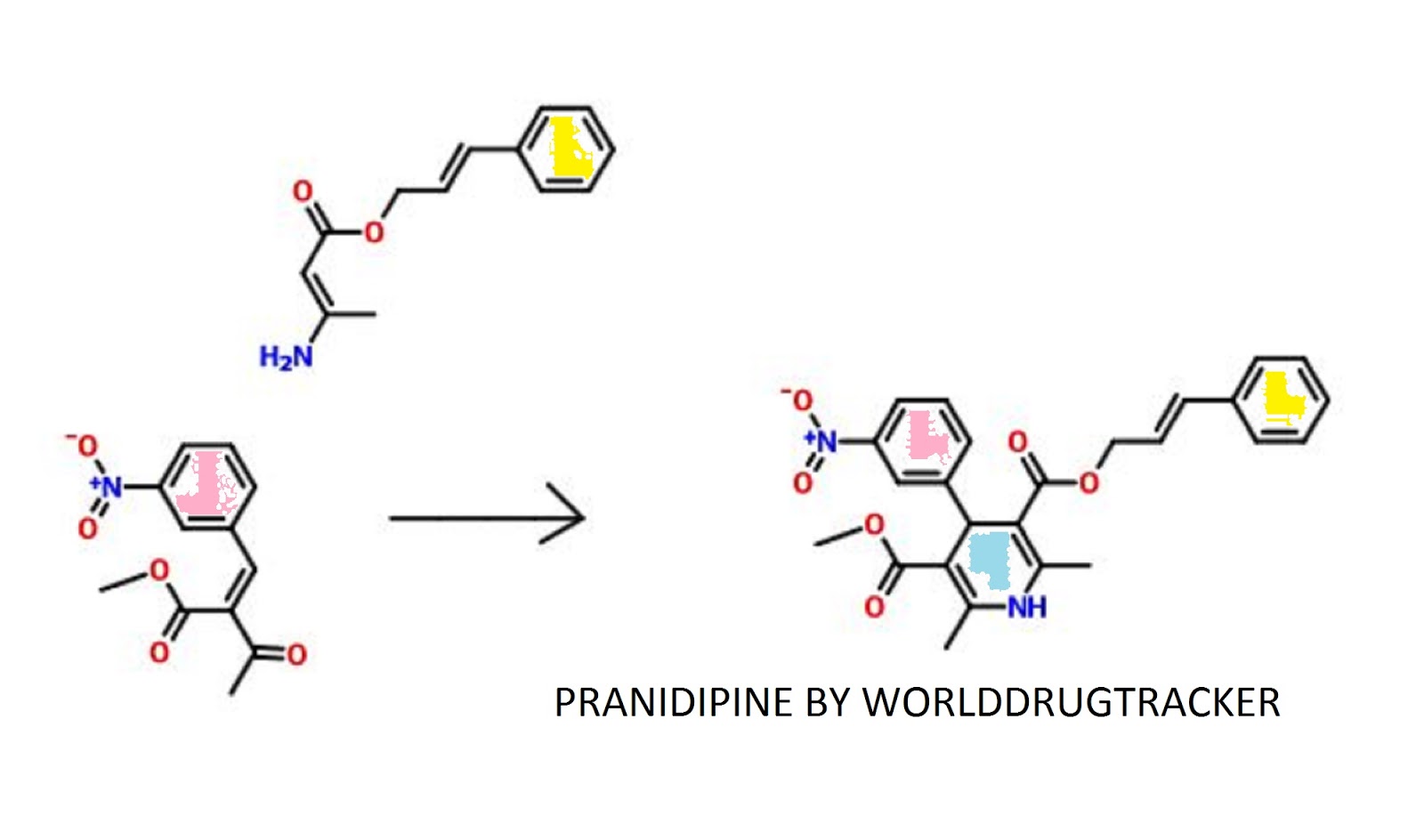
PATENT
EP 0173126
http://www.google.com/patents/EP0173126A1?cl=en
PAPER
Der Pharmacia Sinica, 2014, 5(1):11-17
pelagiaresearchlibrary.com/der-pharmacia-sinica/vol5-iss1/DPS-2014-5-1-11-17.pdf
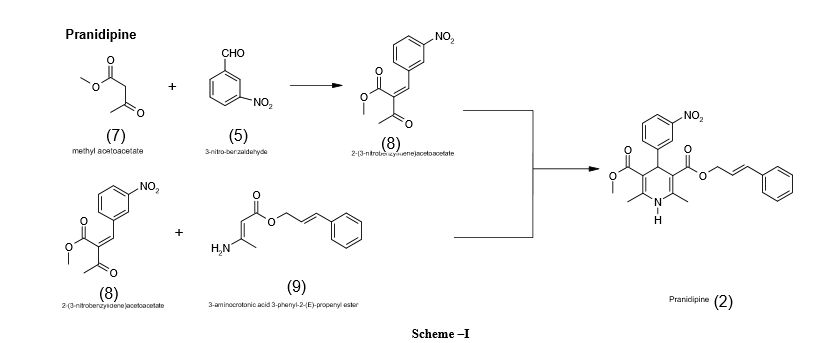
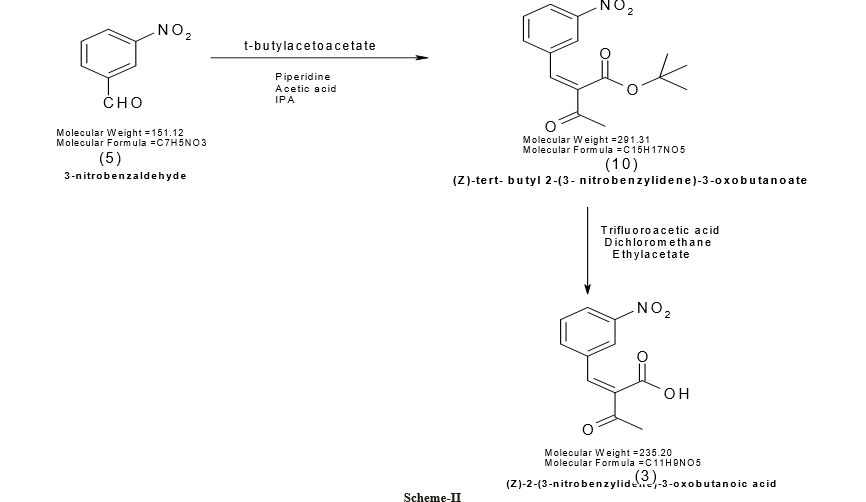
CLICK ON IMAGE FOR CLEAR VIEW
| Patent | Submitted | Granted |
|---|---|---|
| Process for the preparation of 1,4 – dihydropyridines and novel 1,4-dihydropyridines useful as therapeutic agents [US2003230478] | 2003-12-18 | |
| Advanced Formulations and Therapies for Treating Hard-to-Heal Wounds [US2014357645] | 2014-08-19 | 2014-12-04 |
| METHODS OF TREATING CARDIOVASCULAR AND METABOLIC DISEASES [US2014322199] | 2012-08-06 | 2014-10-30 |
| Protein Carrier-Linked Prodrugs [US2014323402] | 2012-08-10 | 2014-10-30 |
| sGC STIMULATORS [US2014323448] | 2014-04-29 | 2014-10-30 |
| TREATMENT OF ARTERIAL WALL BY COMBINATION OF RAAS INHIBITOR AND HMG-CoA REDUCTASE INHIBITOR [US2014323536] | 2012-12-07 | 2014-10-30 |
| Agonists of Guanylate Cyclase Useful For the Treatment of Gastrointestinal Disorders, Inflammation, Cancer and Other Disorders [US2014329738] | 2014-03-28 | 2014-11-06 |
| METHODS, COMPOSITIONS, AND KITS FOR THE TREATMENT OF CANCER [US2014335050] | 2012-05-25 | 2014-11-13 |
| ROR GAMMA MODULATORS [US2014343023] | 2012-09-18 | 2014-11-20 |
| High-Loading Water-Soluable Carrier-Linked Prodrugs [US2014296257] | 2012-08-10 | 2014-10-02
|

| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-20150342954-A1 |
2015-12-03 |
|
|||
|
2.
EP-2558474-B1 |
2015-11-25 |
EN
|
|
||
|
3.
US-20150307580-A1 |
2015-10-29 |
|
|||
|
4.
US-20150305974-A1 |
2015-10-29 |
|
|||
|
5.
WO-2015164658-A1 |
2015-10-29 |
EN
|
|
||
|
6.
EP-2527360-B1 |
2015-10-28 |
EN
|
|
||
|
7.
WO-2015157471-A1 |
2015-10-15 |
EN
|
|
||
|
8.
US-20150284411-A1 |
2015-10-08 |
|
|||
|
9.
US-20150283202-A1 |
2015-10-08 |
|
|||
|
10.
US-9150512-B2 |
2015-10-06 |
|
References
Jin Yang, Keisuke Fukuo, Shigeto Morimoto, Tadaaki Niinobu, Toshimitsu Suhara, Toshio Ogihara (2000). “Pranidipine Enhances the Action of Nitric Oxide Released From Endothelial Cells”. Hypertension 35: 82–85. doi:10.1161/01.hyp.35.1.82.
 |
|
| Names | |
|---|---|
| IUPAC name
methyl (2E)-phenylprop-2-en-1-yl 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate
|
|
| Other names
2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylic acid O5-methyl O3-[(E)-3-phenylprop-2-enyl] ester
|
|
| Identifiers | |
| 99522-79-9 |
|
| ChEMBL | ChEMBL1096842 |
| ChemSpider | 4940726 |
| Jmol interactive 3D | Image |
| MeSH | C048161 |
| PubChem | 6436048 |
| UNII | 9DES9QVH58 |
| Properties | |
| C25H24N2O6 | |
| Molar mass | 448.46786 |
//////////
CC1=C(C(C(=C(N1)C)C(=O)OCC=CC2=CC=CC=C2)C3=CC(=CC=C3)[N+](=O)[O-])C(=O)OC
see dipine series………..http://organicsynthesisinternational.blogspot.in/p/dipine-series.html

Nilvadipine – Wikipedia, the free encyclopedia

 manidipine
manidipine
Ataciguat

Ataciguat HMR-1766
Hoechst Marion Roussel De Gmbh
5-Chloro-2-[[(5-chloro-2-thienyl)sulfonyl]amino]-N-[4-(4-morpholinylsulfonyl)phenyl]benzamide
C21H19Cl2N3O6S3
UNII-QP166M390Q;
576.49306 g/mol
A guanylate cyclase activator potentially for the treatment of aortic valve stenosis.
![]()
CAS No. 254877-67-3
- Originator sanofi-aventis
- Developer Mayo Clinic; National Center for Advancing Translational Sciences; Sanofi; sanofi-aventis
- Class Anthranilic acids; Benzamides; Cardiovascular therapies; Chlorobenzenes; Morpholines; Small molecules; Sulfonamides; Thiophenes
- Mechanism of Action Guanylate cyclase stimulants
- 30 Jun 2015 Mayo Clinic plans a phase II trial for Aortic valve stenosis in USA (NCT02481258)
- 29 Jan 2014 Phase-I clinical trials in Aortic valve stenosis in USA (PO)
- 01 Jan 2010 Discontinued – Phase-II for Peripheral arterial occlusive disorders in Austria, Canada, France, Germany, Italy, Poland, Portugal, Russia, South Africa and USA (PO) prior to 2010
SYNTHESIS

The Intermediates hown above is used in next step shown below

Paper
Organic Letters (2013), 15(7), 1638-1641
http://pubs.acs.org/doi/abs/10.1021/ol400411v
http://pubs.acs.org/doi/suppl/10.1021/ol400411v/suppl_file/ol400411v_si_001.pdf

The Ru(II)-catalyzed intermolecular o-C–H amidation of arenes in N-benzoylated sulfoximine with sulfonyl azides is demonstrated. The reaction proceeds with broad substrate scope and tolerates various functional groups. Base hydrolysis of the amidation product provides the anthranilic acid derivatives and methylphenyl sulfoximine (MPS) directing group. This method is successfully employed for the synthesis of HMR 1766.
PATENT
WO 2009043495
http://www.google.com/patents/WO2009043495A1?cl=en
Patent
http://www.google.com/patents/WO2008124505A2?cl=en
HMR-1766 (ataciguat sodium, see patent publication WO2000002851)

PATENT
http://www.google.com/patents/WO2000002851A1?cl=en
| Patent | Submitted | Granted |
|---|---|---|
| TRA COMBINATION THERAPIES [US2007238674] | 2007-10-11 | |
| sGC STIMULATORS OR sGC ACTIVATORS ALONE AND IN COMBINATION WITH PDE5 INHBITORS FOR THE TREATMENT OF CYSTIC FIBROSIS [US2013035340] | 2011-02-03 | 2013-02-07 |
| SOLUBLE GUANYLATE CYCLASE (SGC) MODULATORS FOR TREATMENT OF LIPID RELATED DISORDERS [US2013123354] | 2013-01-08 | 2013-05-16 |
| Novel combination [US2005059660] | 2004-07-29 | 2005-03-17 |
| SGC STIMULATORS OF SGC ACTIVATORS IN COMBINATION WITH PDE5 INHBITORS FOR THE TREATMENT OF ERECTILE DYSFUNCTION [US2014288079] | 2014-03-18 | 2014-09-25 |
| Patent | Submitted | Granted |
|---|---|---|
| novel use of activators and stimulators of soluble guanylate cyclase for the prevention or treatment of renal disorders [US2010016305] | 2010-01-21 | |
| HETEROARYL-SUBSTITUTED PIPERIDINES [US8119663] | 2009-12-10 | 2012-02-21 |
| Use of soluble guanylate cyclase activators for the treatment of Raynaud’s Phenomenon [US2009215769] | 2009-08-27 | |
| Use of Activators of Soluble Guanylate Cyclase for Promoting Wound Healing [US2009221573] | 2009-09-03 | |
| Use of Suluble Guanylate Cyclase Acitvators for Treating Acute and Chronic Lung Diseases [US2009286781] | 2009-11-19 | |
| Use of Activators of Soluble Guanylate Cyclase for Treating Reperfusion Damage [US2009298822] | 2009-12-03 | |
| HETEROCYCLIC DERIVATIVE AND USE THEREOF [US2011028493] | 2011-02-03 | |
| SUBSTITUTED PIPERIDINES [US8202862] | 2010-12-02 | 2012-06-19 |
| METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS [US2009022729] | 2009-01-22 | |
| sGC STIMULATORS [US2014323448] | 2014-04-29 | 2014-10-30 |



/////////
C1COCCN1S(=O)(=O)C2=CC=C(C=C2)NC(=O)C3=C(C=CC(=C3)Cl)NS(=O)(=O)C4=CC=C(S4)Cl
MELOGLIPTIN

Melogliptin
Phase III
A DP-IV inhibitor potentially for treatment of type II diabetes.
![]()
EMD-675992; GRC-8200
CAS No. 868771-57-7
4(S)-Fluoro-1-[2-[(1R,3S)-3-(1H-1,2,4-triazol-1-ylmethyl)cyclopentylamino]acetyl]pyrrolidine-2(S)-carbonitrile


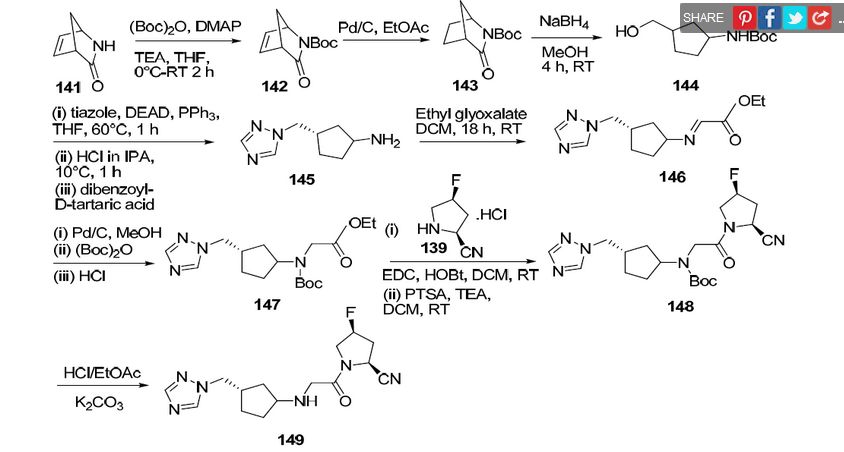
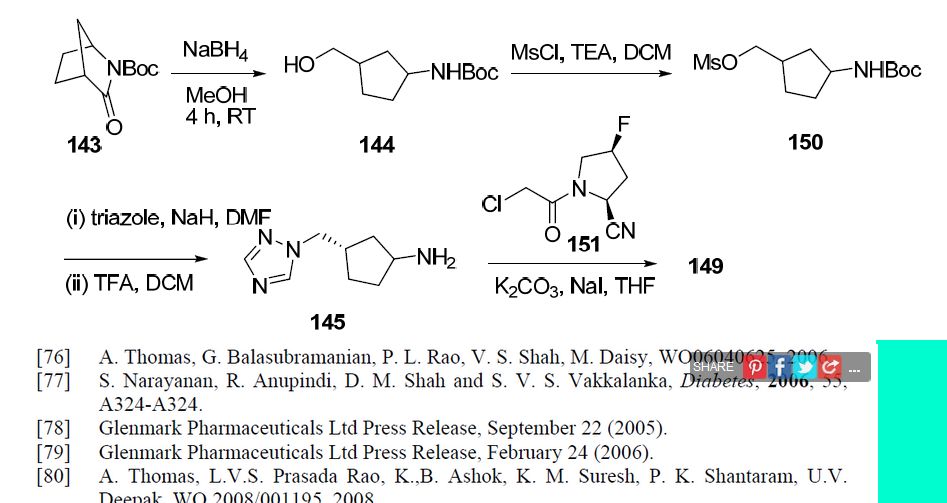
SEE..http://apisynthesisint.blogspot.in/2015/12/melogliptin.html
See more at: http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html

DISCLAIMER…….The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
/////////
Umbralisib, TGR-1202, a Phosphoinositide-3 kinase delta inhibitor, Rhizen Pharmaceuticals S.A./TG Therapeutics
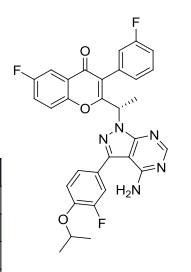

| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
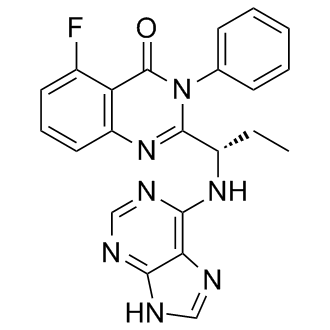
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)

Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.

REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
RP 6530, Tenalisib
 RP 6530
RP 6530(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one (Compound A1 is RP 6530).
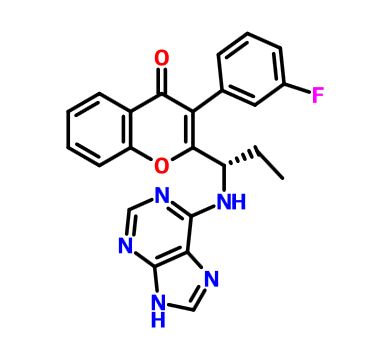
RP 6530
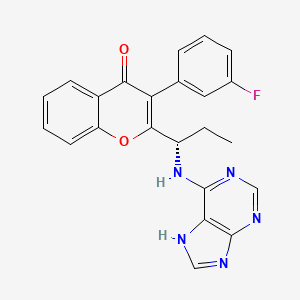
RP 6530, RP6530, RP-6530
Tenalisib
RP6530-1401, NCI-2015-01804, 124584, NCT02567656
(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
3-(3-fluorophenyl)-2-[(1S)-1-(7H-purin-6-ylamino)propyl]chromen-4-one
MW415.4, C23H18FN5O2
CAS 1639417-53-0, 1693773-94-2
RP6530 demonstrated high potency against PI3Kδ (IC50 =24.5 nM) and γ (IC50 = 33.2 nM) enzymes with selectivity over α (>300-fold) and β (>100-fold) isoforms. Cellular potency was confirmed in target-specific assays, namely anti-FcεR1-(EC50=37.8 nM) or fMLP (EC50 = 39.0 nM) induced CD63 expression in human whole blood basophils, LPS induced CD19+ cell proliferation in human whole blood (EC50=250 nM), and LPS induced CD45R+ cell proliferation in mouse whole blood (EC50=101 nM).
A PI3K inhibitor potentially for the treatment of hematologic malignancies.
An inhibitor of phosphoinositide-3 kinase (PI3K) δ/γ isoforms and anti-cellular proliferation agent for treatment of hematol. malignancies
Rhizen Pharmaceuticals is developing RP-6530, a PI3K delta and gamma dual inhibitor, for the potential oral treatment of cancer and inflammation In November 2013, a phase I trial in patients with hematologic malignancies was initiated in Italy ]\. In September 2015, a phase I/Ib study was initiated in the US, in patients with relapsed and refractory T-cell lymphoma. At that time, the study was expected to complete in December 2016
PATENTS……..WO 11/055215 , WO 12/151525.
Inventors
| Inventors | Meyyappan Muthuppalaniappan, Srikant Viswanadha, Govindarajulu Babu, Swaroop Kumar V.S. Vakkalanka, |
| Incozen Therapeutics Pvt. Ltd., Rhizen Pharmaceuticals Sa |
- Antineoplastics; Small molecules
- Mechanism of Action Phosphatidylinositol 3 kinase delta inhibitors; Phosphatidylinositol 3 kinase gamma inhibitors
- Phase I Haematological malignancies
- Preclinical Multiple myeloma
| Swaroop K. V. S. Vakkalanka, | |
| COMPANY | Rhizen Pharmaceuticals Sa |
https://clinicaltrials.gov/ct2/show/NCT02017613
PI3K delta/gamma inhibitor RP6530 An orally active, highly selective, small molecule inhibitor of the delta and gamma isoforms of phosphoinositide-3 kinase (PI3K) with potential immunomodulating and antineoplastic activities. Upon administration, PI3K delta/gamma inhibitor RP6530 inhibits the PI3K delta and gamma isoforms and prevents the activation of the PI3K/AKT-mediated signaling pathway. This may lead to a reduction in cellular proliferation in PI3K delta/gamma-expressing tumor cells. In addition, this agent modulates inflammatory responses through various mechanisms, including the inhibition of both the release of reactive oxygen species (ROS) from neutrophils and tumor necrosis factor (TNF)-alpha activity. Unlike other isoforms of PI3K, the delta and gamma isoforms are overexpressed primarily in hematologic malignancies and in inflammatory and autoimmune diseases. By selectively targeting these isoforms, PI3K signaling in normal, non-neoplastic cells is minimally impacted or not affected at all, which minimizes the side effect profile for this agent. Check for active clinical trials using this agent. (NCI Thesaurus)
| Company | Rhizen Pharmaceuticals S.A. |
| Description | Dual phosphoinositide 3-kinase (PI3K) delta and gamma inhibitor |
| Molecular Target | Phosphoinositide 3-kinase (PI3K) delta ; Phosphoinositide 3-kinase (PI3K) gamma |
| Mechanism of Action | Phosphoinositide 3-kinase (PI3K) delta inhibitor; Phosphoinositide 3-kinase (PI3K) gamma inhibitor |
| Therapeutic Modality | Small molecule |

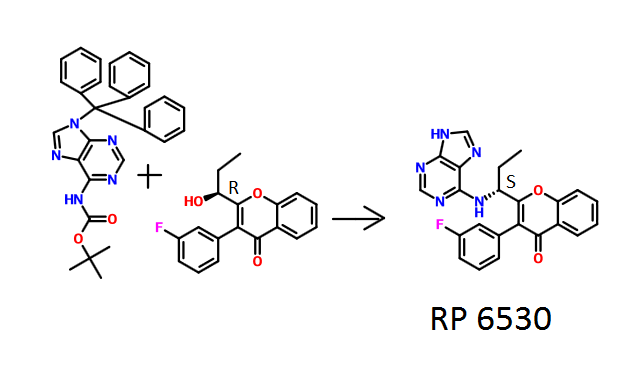
Dual PI3Kδ/γ Inhibition By RP6530 Induces Apoptosis and Cytotoxicity In B-Lymphoma Cells
RP6530 is a potent and selective dual PI3Kδ/γ inhibitor that inhibited growth of B-cell lymphoma cell lines with a concomitant reduction in the downstream biomarker, pAKT. Additionally, the compound showed cytotoxicity in a panel of lymphoma primary cells. Findings provide a rationale for future clinical trials in B-cell malignancies.

PI3K Dual Inhibitor (RP-6530)
| Therapeutic Area | Respiratory , Oncology – Liquid Tumors , Rheumatology | Molecule Type | Small Molecule |
|---|---|---|---|
| Indication | Peripheral T-cell lymphoma (PTCL) , Non-Hodgkins Lymphoma , Asthma , Chronic Obstructive Pulmonary Disease (COPD) , Rheumatoid Arthritis | ||
| Development Phase | Phase I | Rt. of Administration | Oral |
Description
Rhizen is developing dual PI3K gamma/delta inhibitors for liquid tumors and inflammatory conditions.
Mechanism of Action
While alpha and beta isoforms are ubiquitous in their distribution, expression of delta and gamma is restricted to circulating hematogenous cells and endothelial cells. Unlike PI3K-alpha or beta, mice lacking expression of gamma or delta do not show any adverse phenotype indicating that targeting of these specific isoforms would not result in overt toxicity. Dual delta/gamma inhibition is strongly implicated as an intervention strategy in allergic and non-allergic inflammation of the airways and other autoimmune diseases. Scientific evidence for PI3K-delta and gamma involvement in various cellular processes underlying asthma and COPD stems from inhibitor studies and gene-targeting approaches. Also, resistance to conventional therapies such as corticosteroids in several COPD patients has been attributed to an up-regulation of the PI3K delta/gamma pathway. Disruption of PI3K-delta/gamma signalling therefore provides a novel strategy aimed at counteracting the immuno-inflammatory response. Due to the pivotal role played by PI3K-delta and gamma in mediating inflammatory cell functionality such as leukocyte migration and activation, and mast cell degranulation, blocking these isoforms may also be an effective strategy for the treatment of rheumatoid arthritis as well.
Given the established criticality of these isoforms in immune surveillance, inhibitors specifically targeting the delta and gamma isoforms would be expected to attenuate the progression of immune response encountered in airway inflammation and rheumatoid arthritis.

Clinical Trials
Rhizen has identified an orally active Lead Molecule, RP-6530, that has an excellent pre-clinical profile. RP-6530 is currently in non-GLP Tox studies and is expected to enter Clinical Development in H2 2013.
In December 2013, Rhizen announced the start of a Phase I clinical trial. The study entitled A Phase-I, Dose Escalation Study to Evaluate Safety and Efficacy of RP6530, a dual PI3K delta /gamma inhibitor, in patients with Relapsed or Refractory Hematologic Malignancies is designed primarily to establish the safety and tolerability of RP6530. Secondary objectives include clinical efficacy assessment and biomarker response to allow dose determination and potential patient stratification in subsequent expansion studies.
Partners by Region
Rhizen’s pipeline consists of internally discovered (with 100% IP ownership) novel small molecule programs aimed at high value markets of Oncology, Immuno-inflammtion and Metabolic Disorders. Rhizen has been successful in securing critical IP space in these areas and efforts are on for further expansion in to several indications. Rhizen seeks partnerships to unlock the potential of these valuable assets for further development from global pharmaceutical partners. At present global rights on all programs are available and Rhizen is flexible to consider suitable business models for licensing/collaboration.
In 2012, Rhizen announced a joint venture collaboration with TG Therapeutics for global development and commercialization of Rhizen’s Novel Selective PI3K Kinase Inhibitors. The selected lead RP5264 (hereafter, to be developed as TGR-1202) is an orally available, small molecule, PI3K specific inhibitor currently being positioned for the treatment of haematological malignancies.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014195888Intermediates
Intermediate 1: 3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one: To a solution of 2-(l-bromopropyl)-3-(3-fluorophenyl)-4H-chromen-4-one1 (8.80 g, 24.36 mmol ) in DMSO (85 ml), n-butanol (5 ml) was added and heated to 120° C for 3h. The reaction mixture was cooled to room temperature (RT), quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (2.10 g, 29 %) which was used without further purification in next step.
Intermediate 2: 3-(3-fluorophenyl)-2-propionyl-4H-chromen-4-one: DMSO (1.90 ml, 26.82 mmol) was added to dichloromethane (70 ml) and cooled to -78°C. Oxalyl chloride (1.14 ml, 13.41 mmol) was then added. After 10 minutes, intermediate 1 (2.00 g, 6.70 mmol) in dichloromethane (20 ml) was added dropwise and stirred for 20 min. Triethylamine (7 ml) was added and stirred for lh. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow liquid (1.20 g, 60%) which was used as such in next step.
Intermediate 3: (+)/(-)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one :
To a solution of intermediate 2 (0.600 g, 2.02 mmol) in DMF (7.65 ml) under nitrogen purging, formic acid : trietylamine 5 : 2 azeotrope (1.80 ml) was added followed by [(S,S)tethTsDpenRuCl] (3.0 mg). The reaction mixture was heated at 80°C for 1.5 hours under continuous nitrogen purging. The reaction mixture was quenched with water, extected with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (0.450 g, 74%). Mass: 299.0 (M+).
Enantiomeric excess: 78%, enriched in the late eluting isomer (retention time: 9.72 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 4: (+)/(-)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one :
The title compound was obtained as yellow solid (0.500 g, 83%) by using a procedure similar to the one described for intermediate 3, using intermediate 2 (0.600 g, 2.02 mmol), DMF (7.65 ml), formic acid : trietylamine 5 : 2 azeotrope (1.80 ml) and [(R,R)tethTsDpenRuCl] (3.0 mg). Mass: 298.9 (M+). Enantiomeric excess: 74.8%, enriched in the fast eluting isomer (retention time: 8.52 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: (R)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one:
Step 1 : (R)-2-(l-(benzyloxy)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one: To 2-(3-fluorophenyl)-l-(2-hydroxyphenyl)ethanone (2.15 g, 9.36 mmol ), in dichloromethane ( 20 ml), HATU (4.27 g, 11.23 mmol), R-(+)2-benzyloxybutyric acid (2.00 g, 10.29 mmol) were added and stirred for lOmin, then triethylamine (14.0 ml, 101.1 mmol) was added dropwise and stirred at RT for 24h. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as yellow solid (1.65 g, 45%). JH-NMR (δ ppm, CDC13, 400 MHz): 8.24 (dd, / = 7.9,1.5 Hz, 1H), 7.74 (dt, / = 7.1,1.7 Hz, 1H), 7.58 (dd, / = 8.3,0.4 Hz, 1H), 7.44-7.06 (m, 10H), 4.51 (d, / = 7.8 Hz, 1H), 4.34 (d, / = 7.8 Hz, 1H), 4.25 (dd, / = 7.8,6.2 Hz, 1H), 2.17-1.90 (m, 2H), 0.95 (t, / = 7.5 Hz, 3H). Mass: 389.0 (M+).
Step 2: (R)-3-(3-fluorophenyl)-2-(l-hydroxypropyl)-4H-chromen-4-one : To (R)-2-(l-(benzyloxy)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one (1.50 g, 3.86 mmol) in dichloromethane (15 ml) cooled to 0°C and aluminium chloride (1.00 g, 7.72 mmol) was added portion wise and stirred at RT for 6h. The reaction mixture was quenched with 2N HC1 solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as yellow solid (0.552 g, 48%).‘ JH-NMR (δ ppm, CDC13, 400 MHz): ‘ 8.24 (dd, / = 8.0,1.6 Hz, 1H), 7.72 (m, , 1H), 7.52 (dd, / = 8.4,0.5 Hz, 1H), 7.44 (m, 2H), 7.12-7.01(m,3H), 4.49 (t, / = 7.0 Hz, 1H), 1.94 (m, 2H), 0.93 (t, / = 7.5 Hz, 3H). Mass: (299.0(M+). Purity: 96.93%.
25[a] D -14.73 (c = 1, CHCI3). Enantiomeric excess: 85.92%, enriched in the fast eluting isomer (retention time: 8.57 min.) as determined by HPLC on a chiralpak AS-3R column.
Compound A
(RS)- 2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 1 (2.50 g, 8.41 mmol) in THF (25 ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (4.81 g, 10.09 mmol) and triphenylphosphine (3.31 g, 12.62 mmol) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (2.5 ml, 12.62 mmol) was added and stirred at RT for 2h. The reaction mixture was concentrated and column chromatographed with ethyl acetate : petroleum ether to afford a yellow coloured intermediate. To the intermediate, dichloromethane (65 ml) and trifluoroacetic acid (7.9 ml) were added and the resulting mixture was stirred at RT for 12 h. The reaction mixture was then basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as pale-brown solid (1.05 g, 30 %). MP: 148-150°C. Mass: 415.6 (M+).
Compound Al
(S)-2-(l-(9H-purin-6-ylamino)propyl)-3-(3-fluorophenyl)-4H-chromen-4-one
Method A: To a solution of intermediate 3 (0.250 g, 0.838 mmol) in THF (5ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (0.479 g, 1.00 mmol) and triphenylphosphine (0.329 g, 1.25 mmol) were added and the resulting mixture was stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.25 mmol) was then added and stirred at RT for 12 h. The reaction mixture was concentrated and column chromatographed with ethyl acetate: pet.ether to afford the yellow coloured intermediate. To the intermediate in dichloromethane (6 ml), trifluoroacetic acid (1.2 ml) was added stirred at RT for 12 h. The reaction mixture was basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as an off-white solid (0.015 g, 4 %). MP: 137-140°C. JH-NMR (δ ppm, DMSO- , 400 MHz): 12.94 (s, 1H), 8.12-8.10 (m, 4H), 7.84-7.80 (m, 1H), 7.61 (d, / = 8.3 Hz, 1H), 7.50-7.41 (m, 2H), 7.28-7.18 (m, 3H), 5.20-5.06 (m, 1H), 2.10-1.90 (m, 2H), 0.84 (t, / = 3.7 Hz, 3H). Enantiomeric excess: 77.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 7.90 min.).
Method B : To a solution of intermediate 5 (2.60 g, 8.68 mmol) in THF (52 ml), tert-butyl 9-trityl-9H-purin-6-ylcarbamate (4.96 g, 10.42 mmol) and triphenylphosphine (2.76 g, 13.03 mmol) were added and the resulting mixture was stirred at RT for 5 min. Dusopropylazodicarboxylate (0.25 ml, 1.25 mmol) was then added and stirred at RT for 12 h. The reaction mixture was concentrated and column chromatographed with ethyl acetate: petroleum ether to afford the yellow coloured intermediate. To the intermediate in dichloromethane (55 ml), trifluoroacetic acid (14.2 ml) was added and stirred at RT for 12 h. The reaction mixture was basified with aqueous sodium bicarbonate solution, extracted with dichloromethane and dried over sodium sulphate. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as pale-yellow solid (1.00 g, 27 %). MP: 168-170°C. Mass: 416.5(M++1) Enantiomeric excess: 86.5% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 7.90 min.).
Method C : The title compound was separated by preparative SFC conditions from Compound A (1.090 g) on a CHIRALPAK AY-H column (250 x 30 mm; 5μπι) using methanol : C(¾ (35:65) as the mobile phase at a flow rate of 80 g / min. Off-white solid (0.378 g). e.e. 100%. Rt: 2.37 min. Mass: 416.1(M++1). MP: 149-152°C.
Scheme 1A

CAUTION ethyl compd below, NOT THE PRODUCT
Example 47
(S)-2-(l-(9H-purin-6-yIamino) ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one
[428] To a solution of intermediate 65 (2.0g, 8.68 mmoles) in dichloromethane (20ml), triethylamine (3.6ml, 26.06 mmoles) was added followed by N-Boc-Alanine (1.97g, 10.42 mmoles). To this mixture HATU (6.6g, 17.37 mmoles) was added and stirred at RT for 12h. The reaction mixture was quenched by the addition of water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the isoflavone intermediate (1.70g). To a solution of this intermediate (1.7g) in dichloromethane (20ml), trifluoroacetic acid (3 ml) was added and stirred at RT for 2h. The reaction mixture was concentrated, basified with sodium bicarbonate solution, extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the amine intermediate (0.641 g). To a solution of this amine intermediate (0.30g, 1.05 mmoles) in tert-butanol (6ml), N, N- diisopropylethylamine (0.36ml, 2.17 mmoles) and 6-bromopurine (0.168g, 0.847 mmoles) were added and refluxed for 24h. The reaction mixture was concentrated, diluted with water, extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: ethyl acetate to afford the title compound as off-white solid (0.041g, 10% yield). MP: 135-138 °C. Ή-NMR (δ ppm, DMSO-D6, 400 MHz): δ 12.95(s,lH), 8.15(t, / = 6.8Hz, 1H), 8.11(s, 1H), 8.08(s, 1H), 8.03(d, J = 7.8 Hz, 1H), 7.81(t ,J = 7.3Hz, 1H), 7.60 (d, J = 8.3Hz, 1H), 7.49 (t, J = 7.3Hz, 2H), 7.25(m,3H), 5.19(br m, 1H), 1.56(d, J = 6.9Hz,3H). Mass: 402.18(M+ +1).
Scheme 1
Base

This scheme provides a synthetic route for the preparation of compound of formula wherein all the variables are as described herein in above

15 14 10 12 12a
Abstract 2704: RP6530, a dual PI3K δ/γ inhibitor, potentiates ruxolitinib activity in the JAK2-V617F mutant erythroleukemia cell lines
– Author Affiliations
- 1Rhizen Pharmaceuticals SA, Fritz-Courvoisier 40, Switzerland;
- 2Incozen Therapeutics Pvt. Ltd., Hyderabad, India.
Abstract
Background: Myelofibrosis (MF) represents a life-threatening neoplasm that manifests particularly in the elderly population and is characterized by bone marrow fibrosis and extramedullary hematopoeisis. While ruxolitinib, a JAK1/2 inhibitor, has recently been approved by the USFDA for its disease modifying potential in MF patients, it is still not considered as a curative option. Targeting another kinase such as PI3K, downstream of JAK, could therefore be a more efficient way of treating myelofibrotic neoplasms. RP6530 is a novel, potent, and selective PI3K δ/γ inhibitor that demonstrated high potency against PI3Kδ (IC50 = 25 nM) and γ (IC50 = 33 nM) enzymes with selectivity over α (>300-fold) and β (>100-fold) isoforms. The objective of this study was to evaluate the effect of a combination of ruxolitinib and RP6530 in the JAK2-V617F mutant Human Erythroleukemia (HEL) cell line.
Methods: Passive resistance was conferred by incubating HEL cells with increasing concentrations of ruxolitinib over an 8-10-week period. Endogenous JAK2, PI3Kδ, PI3Kδ, and pAKT were estimated by Western Blotting. RP6530, ruxolitinib, and the combination of RP6530 + Ruxolitinib were tested for their effect on viability and apoptosis. Cell viability was assessed by a MTT assay. Induction of apoptosis was analyzed by Annexin V/PI staining.
Results: Resistance to ruxolitinib was confirmed by a right-ward shift in EC50 of ruxolitinib in a HEL cell proliferation assay (0.82 μM Vs. 12.2 μM). Endogeous pAKT expression was 3.7-fold higher in HEL-RR compared to HEL-RS cells indicating activation of the AKT signaling pathway. While single-agent activity of RP6530 was modest (33-46% inhibition @ 10 μM) in both HEL-RS and HEL-RR cells, addition of 10 μM RP6530 to ruxolitinib was synergistic resulting in a near-complete inhibition of proliferation (>90% for HEL-RS and >70% for HEL-RR). While the order of addition did not affect the potency of RP6530, addition of 5 μM RP6530, 4 h prior to the addition of ruxolitinib resulted in a significant reduction in EC50 of ruxolitinib (5.8 μM) in HEL-RR cells. On lines with cell proliferation data, incubation of 10 μM RP6530 with ruxolitinib for 72 h increased the percent of apoptotic cells (55% in HEL-RS and 37% in HEL-RR) compared to either agent alone (16-27% in HEL-RS and 17-21% in HEL-RR).
Conclusions: Ruxolitinib resistance in the V617F JAK-2 mutant HEL cells is accompanied by an increase in pAKT expression. Inhibition of pAKT via the addition of RP6530, a dual PI3K δ/γ inhibitor, resulted in a reversal of ruxolitinib resistance. Complementary activity was also observed in HEL-RS cells indicating that a combination of ruxolitinib and RP6530 could have a positive bearing on the clinical outcome in MF patients.
Citation Format: Swaroop Vakkalanka, Seeta Nyayapathy, Srikant Viswanadha. RP6530, a dual PI3K δ/γ inhibitor, potentiates ruxolitinib activity in the JAK2-V617F mutant erythroleukemia cell lines. [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 2704. doi:10.1158/1538-7445.AM2015-2704
- 01 Sep 2015 Phase-I clinical trials in Haematological malignancies (Second-line therapy or greater) in USA (PO) (NCT02567656)
- 18 Nov 2014 Preclinical trials in Multiple myeloma in Switzerland (PO) prior to November 2014
- 18 Nov 2014 Early research in Multiple myeloma in Switzerland (PO) prior to November 2014
| WO2011055215A2 | Nov 3, 2010 | May 12, 2011 | Incozen Therapeutics Pvt. Ltd. | Novel kinase modulators |
| WO2012151525A1 | May 4, 2012 | Nov 8, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO2013164801A1 | May 3, 2013 | Nov 7, 2013 | Rhizen Pharmaceuticals Sa | Process for preparation of optically pure and optionally substituted 2- (1 -hydroxy- alkyl) – chromen – 4 – one derivatives and their use in preparing pharmaceuticals |
| US20110118257 | May 19, 2011 | Rhizen Pharmaceuticals Sa | Novel kinase modulators | |
| US20120289496 | May 4, 2012 | Nov 15, 2012 | Rhizen Pharmaceuticals Sa | Novel compounds as modulators of protein kinases |
| WO 2011055215 |
WO2015175966
WO2015051252
-
The Distillery: Therapeutics 09/04/2014
BC Innovations, TherapeuticsIndication Target/marker/pathway Summary Licensing status Publication and contact information Cardiovascular disease Intimal hyperplasia Phosphoinositide 3-kinase-g (PI3Kg) Rodent studies suggest inhibiting … -
BC Innovations, Targets & MechanismsTargets & Mechanisms: PI3K inhibition: solid immunotherapy Table 1. A peek at PI3K inhibitors. According to a study in Nature by Ali et al., inhibition of phosphoinositide 3-kinase-d (PI3Kd) or the PI3K catalytic …
-
RP6530: Phase I started 12/23/2013
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland Product: RP6530 Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) delta; Phosphoinositide 3-kinase (PI3K) gamma Description: Dual … -
Rhizen preclinical data 07/01/2013
Week in Review, Preclinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland Product: RP6530 Business: Cancer Indication: Treat B cell lymphoma In vitro, 2-7 M RP6530 led to a >50% dose-dependent inhibition in growth of immortalized …
/////
c1cccc4c1C(/C(=C(/[C@H](CC)Nc3c2c(ncn2)ncn3)O4)c5cc(ccc5)F)=O
CCC(C1=C(C(=O)C2=CC=CC=C2O1)C3=CC(=CC=C3)F)NC4=NC=NC5=C4NC=N5
Alembic Pharma advances 1% on Rhizen-Novartis license agreement
Swiss subsidiary Rhizen Pharmaceuticals S.A. entered into an exclusive, worldwide license agreement with Novartis for the development and commercialization of Rhizen’s, inhaled dual Pl3K-delta gamma inhibitor and its closely related compounds for various indications.
read

Rhizen Pharmaceuticals Announces Exclusive Worldwide License Agreement for the Development and Commercialization of a Dual PI3K-delta gamma Inhibitor
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland , Dec. 09, 2015 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. announced today that they have entered into an exclusive, worldwide license agreement with Novartis for the development and commercialization of Rhizen’s, inhaled dual PI3K-delta gamma inhibitor and its closely related compounds for various indications.
Under the terms of the agreement, Rhizen will receive an upfront payment and is eligible to receive development, regulatory and sales milestones payments. In addition Rhizen is also eligible to receive tiered royalties on annual nets sales.
The lead compound is a novel, potent, and selective dual PI3K-delta gamma inhibitor with demonstrated anti-inflammatory and immuno-modulatory activity in pre-clinical systems and models representative of respiratory diseases. With a favorable ADME and PK profile and high therapeutic index in animals, the inhaled dual PI3K-delta gamma inhibitor holds promise in the treatment of human airway disorders.
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.
info@rhizen.com

RP 6503
//////
DR SRINIVASA REDDY gets NASI – Reliance Industries Platinum Jubilee Award (2015) for Application Oriented Innovations in Physical Sciences.

 DR SRINIVASA REDDY recieving NASI – Reliance Industries Platinum Jubilee Award (2015) for Application Oriented Innovations in Physical Sciences.
DR SRINIVASA REDDY recieving NASI – Reliance Industries Platinum Jubilee Award (2015) for Application Oriented Innovations in Physical Sciences.

Dr. D. Srinivasa Reddy, Senior Scientist, Division of Organic Chemistry, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune – 411 008
![]()

AWARD CLICK HERE….LINK

PUNE: Senior scientist of National Chemical Laboratory (NCL), D Srinivasa Reddy has been selected for this year’s NASI-Reliance Industries Platinum Jubilee award for the application oriented innovations in the area of physical sciences instituted by the National Academy of Sciences, India (NASI). Two awards each in physical sciences and two in biological sciences are given every year, a statement issued by NCL said.
Manindra Agrawal from Department of Computer Science and Engineering, Indian Institute of Kanpur is the other recipient of this award in physical sciences. Each award carries a plaque and Rs 2 lakh cash.
The award will be presented in the 85th annual session of National Academy of Sciences, India. Reddy’s research group interests are in the field of total synthesis and drug discovery by applying medicinal chemistry. He has also been involved in the synthesis of the agrochemicals like small molecules for crop protection, said the statement. The total synthesis of more than 20 natural products has been achieved in his lab including a sex pheromone that attracts the mealy bugs and has potential use in the crop protection.
On the medicinal chemistry front significant progress has been made by his group using a new concept called silicon-switch approach towards central nervous system drugs. Identification of new chemical entities for the potential treatment of diabetes and infectious diseases is being done in collaboration with industry partners.
Read about DR REDDY at
click
One Organic Chemist One Day: Dr. D. Srinivasa Reddy of …
Feb 23, 2015 – Dr. Reddy’s research group current interests are in the field of total synthesis and drug discovery by applying medicinal chemistry.
Dr. D. Srinivasa Reddy of NCL winner Shanti Swarup Bhatnagar Award 2015
Some details
The National Academy of Sciences, India (NASI) 5, Lajpatrai Road, Allahabad – 211002 NASI-RELIANCE INDUSTRIES PLATINUM JUBILEE AWARDS FOR THE YEAR 2015 FOR APPLICATION ORIENTED INNOVATIONS COVERING BOTH PHYSICAL AND BIOLOGICAL SCIENCES NASI invites nominations for the NASI-Reliance Industries Platinum Jubilee Awards for APPLICATION ORIENTED INNOVATIONS for the year 2015. The nominee should be an Indian citizen / Overseas Citizen of India working in India and below the age of 50 years as on April 15, 2015. Areas for the Awards – Physical Sciences, including – Chemistry, Engineering, Mathematics, Physics, Electronics, Nanotechnology, Information & Computer Sciences, Earth and Atmospheric Sciences; Biological Sciences, including – Agriculture, Animal and Plant Sciences, Environment, Biotechnology, Biochemistry, Bioprocess Engineering, Bioinformatics, all branches of medical sciences and Nanotechnology. Number and Value of Awards – two in Physical Sciences and two in Biological Sciences each year. Each award carries a Plaque and Rs. 2 lakhs in cash. Nominators and Last Date – All Science Secretaries, Secretary HRD, Director Generals – CSIR, ICMR & ICAR; Directors of IITs, IIITs, National Laboratories, Industries with well recognized R&D units including private research institutions; Vice-Chancellors of all Universities – Central & State; Directors of Indian Institute of Science, Bangalore, AIIMS, New Delhi, PGI’s of Medical Research, Chairman, UGC; Presidents of all Science and Engineering Academies (Engineering, Medical, Agriculture); All Fellows of the NASI. The nomination shall be made in the prescribed format. Twenty (20) copies of the nomination form along with only one soft copy (in CD), complete in all respect with the supporting documents, must reach the Academy latest by April 15, 2015. A candidate may only be nominated once. However, a nomination will remain valid for consideration for 3 years or the age eligibility which ever expires earlier. Selection of the Awardees – As per Regulations of NASI Presentation of the Awards – The presentation of the Awards will be made at the time of 85 th Annual Session of NASI. Details and Nomination form for the award are also available on NASI’s website http://www.nasi.org.in and http://www.nasi.nic.in …………….http://www.nasi.org.in/NASI-Reliance%20Industries%20Platinum%20Jubilee%20Awards%202015.pdf
SEE
http://www.nasi.org.in/NASI-Reliance%20Industries%20Platinum%20Jubilee%20Awardees%20(2015).htm


PUNE: Senior scientist at National Chemical Laboratory (NCL) D Srinivasa Reddy has bagged yet another award, the NASI-Reliance Industries Platinum Jubilee award for the year 2015.
This is the second award Reddy has won in a span of less than a fortnight. Earlier, he was selected for the bronze medal presented by the Chemical Research Society of India (CRSI). He has also been a recipient of excellence award from the National Drug Research Institute.
The platinum jubilee award is being conferred for his work in application-oriented innovations in physical sciences category.
Every year, this award recognises two scientists, each working in the fields of physical and biological sciences.
The other scientist who has been awarded in this category is Manindra Agarwal from IIT, Kanpur.
Reddy along with his team has been involved in several high-end research in drug discovery, mainly by applying medicinal chemistry. Other major works he undertook also involve synthesising of agrochemicals meant to be used for crop protection.
He also has vast experience working with various pharmaceutical companies prior to joining NCL in 2010.
The award consists of cash of Rs 2 lakh.
NASI
National Academy of Sciences, India (www.nasi.org.in) was founded in 1930 with the objective to provide a national forum for the scientists to help them publish their research work and discuss the issues for the solutions. NASI-Reliance together give two awards each in the areas of physical and biological sciences for the application oriented innovations started from the year 2006. Areas for the Awards – Physical Sciences, including Chemistry, Engineering, Mathematics, Physics, Electronics, Nanotechnology, Information & Computer Sciences, Earth and Atmospheric Sciences; Biological Sciences, including Agriculture, Animal and Plant Sciences, Environment, Biotechnology, Biochemistry, Bioprocess Engineering, Bioinformatics, all branches of medical sciences and Nanotechnology.
Read about DR REDDY at
One Organic Chemist One Day: Dr. D. Srinivasa Reddy of …
Feb 23, 2015 – Dr. Reddy’s research group current interests are in the field of total synthesis and drug discovery by applying medicinal chemistry.
Dr. D. Srinivasa Reddy of NCL winner Shanti Swarup Bhatnagar Award 2015
Some pictures of his group


NCL, PUNE
//////////DR SRINIVASA REDDY , NASI – Reliance Industries Platinum Jubilee Award, 2015, Application Oriented Innovations in Physical Sciences,
Identification of Medicinal Products Standards will apply in six Months
DRUG REGULATORY AFFAIRS INTERNATIONAL

The pharma sector must comply with IDMP standards in the EU region starting July 2016. This provides regulators with the means of easily comparing product data across regions and with different manufacturers.
Over the last couple of years the European Health Authorities in conjunction with the International Standards Organization (ISO) have been developing a set of global data standards referred to as Identification of Medicinal Products (IDMP).
The Identification of Medicinal Products (IDMP) standards were developed in response to a worldwide demand for internally harmonized specifications for medicinal products. The EU is the first to implement these standards, and the other ICH regions will follow. The pharma sector must comply with IDMP standards in the EU region starting July 2016. Following the EU, the other ICH (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use) countries will then begin their own adoption processes…
View original post 124 more words
