
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
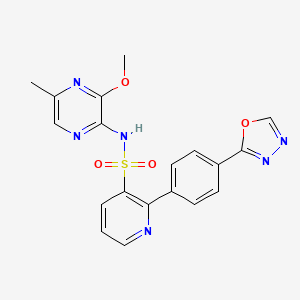
Zibotentan

Zibotentan (INN) (earlier code name ZD4054) is an anti-cancer candidate.[1] It is an endothelin receptor antagonist.[2]
It failed a phase III clinical trial for prostate cancer[3] but other trials are planned.[4] Tolerability of zibotentan plus docetaxel has been evaluated.[5]
SYN

https://www.google.com/patents/WO1996040681A1?cl=en
Bromination of 2-amino-5-methylpyrazine (I) with Br2 in CHCl3 affords the bromopyrazine (II). Subsequent bromide displacement in (II) by means of sodium methoxide gives rise to the methoxypyrazine (III). The amino group of (III) is then protected by acylation with isobutyl chloroformate, to produce carbamate (IV). Diazotization of 3-amino-2-chloropyridine (V), followed by treatment with sulfur dioxide in the presence of CuCl furnishes sulfonyl chloride (VI). Carbamate (IV) is then acylated by means of NaH and sulfonyl chloride (VI) in DMF to furnish the N-sulfonyl carbamate (VII). Esterification of 4-carboxyphenylboronic acid (VIII) with H2SO4 in MeOH gives 4-(methoxycarbonyl)phenylboronic acid (IX). Mitsunobu coupling between boronic acid (IX) and chloropyridine (VII) furnishes adduct (X). Methyl ester (X) is converted into hydrazide (XI) by treatment with hydrazine hydrate in refluxing methanol. Then, cyclization of the acyl hydrazide (XI) with boiling triethyl orthoformate gives rise to the target oxadiazole derivative.
https://www.google.com/patents/WO1996040681A1?cl=en
Example 36
Hydrazine hydrate (1.2 ml) was added to a solution of N-(isobutoxycarbonyl)-2- (4-memoxycarbonylphenyl)-N-(3-metJ oxy-5-methylpyrazin-2-yl)pyridine-3-sulphonamide (1.54 g) in methanol (15 ml) and the mixture was heated and stiπed under reflux for 24 hours then cooled. The solid was collected and dried under reduced pressure to give the free sulphonamido-acylhydrazide (0.857 g); 1H NMR (cVDMSO): 2.2 (s, 3H), 3.7 (s, 3H), 6.7 (br s, 2H), 7.3 (s, IH), 7.5 (m, 3H), 7.8 (d, 2H), 8.4 (d, IH), 8.75 (dd, IH), 9.8 (br s, IH). A solution of this acylhydrazide (207 mg) in triethylorthoformate (5 ml) was heated under reflux for 17 hours then cooled. The resultant solid was collected and purified by chromatography on a silica gel Mega Bond Elut column, eluting with 0-10% methanol/dichloromethane to give N-(3-methoxy-5-mef ylpyrazin-2-yl)-2-(4-[l,3,4-oxadiazol-2-yl]phenyl)pyridine-3- sulphonamide (39 mg) as a solid; 1H NMR (DMSO-do): 2.2 (br s, 3H), 3.8 (s, 3H), 7.4 (br s, IH), 7.6-7.8 (m, 3H), 8.0 (m, 2H), 8.5 (dd, IH), 8.9 (dd, IH), 9.4 (s, IH); mass spectrum (+ve ESP): 425 (M+H)+.
………………………….
http://www.google.im/patents/EP1904490A1?cl=en
N-(3-methoxy-5-methylpyrazin-2-yl)-2- (4-[l,3,4-oxadiazol-2-yl]phenyl)pyridine-3-sulphonamide (hereafter “Compound (I)). More specifically the invention relates to the ethanolamine salt of Compound (I) (hereafter “Compound (I) ethanolamine salt), and to pharmaceutical compositions containing it. The invention further relates to the use of Compound (I) ethanolamine salt in the manufacture of medicament for use in treating cancer and to methods of treating cancer in a warm blooded animal such as man using this salt. The invention further relates to the use of Compound (I) ethanolamine salt in producing Compound (I) during manufacture.
Compound (I) is an endothelin antagonist. The endothelins are a family of endogenous 21 amino acid peptides comprising three isoforms, endothelin-1 (ET-I), endothelin-2 and endothelin-3. The endothelins are formed by cleavage of the Trp2I-Val22 bond of their corresponding proendothelins by an endothelin converting enzyme. The endothelins are among the most potent vasoconstrictors known and have a characteristic long duration of action. They exhibit a wide range of other activities including cell proliferation and mitogenesis, extravasation and chemotaxis, and also interact with a number of other vasoactive agents.
The endothelins are released from a range of tissue and cell sources including vascular endothelium, vascular smooth muscle, kidney, liver, uterus, airways, intestine and leukocytes. Release can be stimulated by hypoxia, shear stress, physical injury and a wide range of hormones and cytokines. Elevated endothelin levels have been found in a number of disease states in man including cancers.
Recently, endothelin A receptor antagonists have been identified as potentially of value in the treatment of cancer (Cancer Research, 56, 663-668, February 15th, 1996 and Nature Medicine, Volume 1, Number 9, September 1999, 944-949).
Cancer affects an estimated 10 million people worldwide. This figure includes incidence, prevalence and mortality. More than 4.4 million cancer cases are reported from Asia, including 2.5 million cases from Eastern Asia, which has the highest rate of incidence in the world. By comparison, Europe has 2.8 million cases, North America 1.4 million cases, and Africa 627,000 cases. In the UK and US, for example, more than one in three people will develop cancer at some point in their life, Cancer mortality in the U.S. is estimated to account for about 600,000 a year, about one in every four deaths, second only to heart disease in percent of all deaths, and second to accidents as a cause of death of children 1-14 years of age. The estimated cancer incidence in the U.S. is now about 1,380,000 new cases annually, exclusive of about 900,000 cases of non-melanotic (basal and squamous cell) skin cancer.
Cancer is also a major cause of morbidity in the UK with nearly 260,000 new cases (excluding non-melanoma skin cancer) registered in 1997. Cancer is a disease that affects mainly older people, with 65% of cases occurring in those over 65. Since the average life expectancy in the UK has almost doubled since the mid nineteenth century, the population at risk of cancer has grown. Death rates from other causes of death, such as heart disease, have fallen in recent years while deaths from cancer have remained relatively stable. The result is that 1 in 3 people will be diagnosed with cancer during their lifetime and 1 in 4 people will die from cancer. In people under the age of 75, deaths from cancer outnumber deaths from diseases of the circulatory system, including ischaemic heart disease and stroke. In 2000, there were 151,200 deaths from cancer. Over one fifth (22 per cent) of these were from lung cancer, and a quarter (26 per cent) from cancers of the large bowel, breast and prostate.
Worldwide, the incidence and mortality rates of certain types of cancer (of stomach, breast, prostate, skin, and so on) have wide geographical differences which are attributed to racial, cultural, and especially environmental influences. There are over 200 different types of cancer but the four major types, lung, breast, prostate and colorectal, account for over half of all cases diagnosed in the UK and US. Prostate cancer is the fourth most common malignancy among men worldwide, with an estimated 400,000 new cases diagnosed annually, accounting for 3.9 percent of all new cancer cases. Current options for treating cancers include surgical resection, external beam radiation therapy and / or systemic chemotherapy. These are partially successful in some forms of cancer, but are not successful in others. There is a clear need for new therapeutic treatments. Compound (I) is exemplified and described in WO96/40681 as Example 36. WO96/40681 claims the endothelin receptors described therein for the treatment of cardiovascular diseases. The use of Compound (I) in the treatment of cancers and pain is described in WO04/018044. Compound (I) has the following structure:

Compound (I)
In WO04/018044 an endothelin human receptor binding assay is described. The pICjo (negative log of the concentration of compound required to displace 50% of the ligand) for Compound (I) at the ETA receptor was 8.27 [8.23 – 8.32] (n=4). Compound (I) is thus an excellent endothelin antagonist.
WO96/40681 and WO04/018044 disclose, in general terms, certain pharmaceutically acceptable salts of the compounds disclosed therein. Specifically it is stated that suitable pharmaceutically-acceptable salts include, for example, salts with alkali metal (such as sodium, potassium or lithium), alkaline earth metals (such as calcium or magnesium), ammonium salts, and salts with organic bases affording physiologically acceptable cations, such as salts with methylamine, dimethylamine, trimethylamine, piperidine and morpholine. In addition, it was stated that suitable pharmaceutically-acceptable salts include, pharmaceutically-acceptable acid- addition salts with hydrogen halides, sulphuric acid, phosphoric acid and with organic acids such as citric acid, maleic acid, methanesulphonic acid and p-toluenesulphonic acid.
Example 2 Formation of Compound (I) using ethanolamine
The above organic layer from Example 1 was adjusted to 42°C and isopropyl alcohol (114 ml), water (170ml) and ethanolamine (28.2 ml) were added and stirred at 42°C for 90 mins. The reaction mixture was allowed to cool to 2O0C and the lower aqueous phase separated and filtered through a 1 μm filter. The aqueous phase was then charged over 40min to a stirred solution of acetic acid (141 g) and water (33.5 g) at 500C and then cooled to 2O0C over 60 mins. The product was isolated by filtration and washed with a mixture of isopropyl alcohol (48.5 ml) and water (48.5 ml) and then isopropyl alcohol (48.5 ml). The product was dried overnight in a vacuum oven at 55°C. Weight 43.08g, Strength = 100%, 86.7%yield. 1H NMR (400 MHz5 DMSOd6) 9.87 (IH, s), 9.14 (IH, s), 8.81 (lH,d), 8.52 (IH, d), 7.98 (2H, d), 7.65 (2H, d), 7.62 (IH, dd), 7.41 (IH, bs), 3.80 (3H, s), 2.23 (3H, s). Mass Spectra MH+ 425.1036 (Ci9Hi7N6O4S calculated 425.1032).
| Patent | Submitted | Granted |
|---|---|---|
| Substituted pyrazin-2-yl-sulphonamide-(3-pyridyl) compounds and uses thereof [US6060475] | 2000-05-09 | |
| COMPOSITION 064 [US8168221] | 2009-04-16 | 2012-05-01 |
| THERAPEUTIC TREATMENT-014 [US2009062246] | 2009-03-05 | |
| Ethanolamine Salt of N- (3-Methoxy-5-Methylpyrazin-2Yl) -2- (4-[1, 3, 4-Oxadiazole-2-Yl] Phenyl) Pyridine-3-Sulphonamide [US2008221124] | 2008-09-11 | |
| N-HETEROARYL-PYRIDINESULFONAMIDE DERIVATIVES AND THEIR USE AS ENDOTHELIN ANTAGONISTS [WO9640681] | 1996-12-19 |
| Zibotentan | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 186497-07-4 |
| PubChem | 9910224 |
| ChemSpider | 8085875 |
| UNII | 8054MM4902 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C19H16N6O4S |
| Molar mass | 424.43 g mol−1 |
References
- James and Growcott (2009). “Drugs of the Future”.
- Jump up^ Tomkinson H, Kemp J, Oliver S, Swaisland H, Taboada M, Morris T (2011). “Pharmacokinetics and tolerability of zibotentan (ZD4054) in subjects with hepatic or renal impairment: two open-label comparative studies”. BMC Clin Pharmacol 11: 3. doi:10.1186/1472-6904-11-3.PMC 3070638. PMID 21414193.
- http://www.fiercebiotech.com/story/azs-zibotentan-flunks-late-stage-prostate-cancer-trial/2010-09-27
- http://www.genengnews.com/gen-news-highlights/pfizer-astrazeneca-and-actelion-separately-report-phase-iii-trial-failures/81243985/
- Jump up^ Trump DL, Payne H, Miller K, et al. (September 2011). “Preliminary study of the specific endothelin a receptor antagonist zibotentan in combination with docetaxel in patients with metastatic castration-resistant prostate cancer”. Prostate 71 (12): 1264–75.doi:10.1002/pros.21342. PMID 21271613.
External links
Ayurveda………..Medicinal Benefits of Liquorice (Mulethi) (मुलेठी, 甘草, شیرین بیان)

Licorice or Mulethi is a medicinal herb which is used in various Ayurvedic medicines. Its underground stems and roots are used for medicinal purpose. It has antioxidant, antimicrobial, anti-inflammatory and hepatoprotective properties.
Mulethi is useful in cough, sore throat, bronchitis, sexual weakness, skin problems, jaundice, hoarseness, vata dosha, ulcers etc. It has demulcent and expectorant properties.
read…………MY OLD ARTICLE

Liquorice, or licorice, (/ˈlɪk(ə)rɪʃ/ lik-(ə-)rish or /ˈlɪk(ə)rɪs/ lik-(ə-)ris)[2] is the root of Glycyrrhiza glabra from which a sweet flavour can be extracted. The liquorice plant is a legume native to southern Europe, India, and parts of Asia. It is not botanically related to anise, star anise, or fennel, which are sources of similar flavouring compounds. The word liquorice / licorice is derived (via the Old French licoresse) from the Greek γλυκύρριζα (glukurrhiza), meaning “sweet root”,[3] from γλυκύς (glukus), “sweet”[4] + ῥίζα (rhiza), “root”,[5][6] the name provided by Dioscorides.[7] It has been traditionally known and used as medicine in Ayurveda for rejuvenation.[8] It is called asadhimadhuram (அதிமதுரம்) in Tamil, irattimadhuram in Malayalam, yastimadhu (यस्टिमधु) in Sanskrit, mulethi (मुलेठी) in Hindi, andjethimadh (જેઠીમધ) in Gujarati language.[9]
Licorice (Glycyrrhiza glabra), locally known as mulethi, has been revered for centuries as a medicinal herb in Ayurveda. Besides possessing numerous medicinal properties, it is also a popular flavoring herb as it is 50 times sweeter than sugar, due to the presence of a compound called glycyrrhizin.
Through research, the anti-oxidant, anti-inflammatory, anti-microbial, analgesic (pain-relieving) and expectorant properties of this is sweet, moist herb have been established worldwide. It is also diuretic, rejuvenating and mildly laxative in nature. These properties have helped Licorice find a place in both Eastern and Western medicine for treating an array of ailments, ranging from cold and cough to arthritis, respiratory, digestive and liver problems.

The Sanskrit name for licorice is Yashtimadhu, which literally means “sweet root”. It is sweet, cool and heavy to digest. The Rasa (taste) of this herb is madhura (sweet), which makes it beneficial for vata and pitta doshas, while it’s Virya (action) is sheetal (cooling), which generally increases kapha when consumed in large doses over long term.
The medicinal property of mulethi is mainly because of the presence of powerful phytochemicals namely flavonoids, chalcones, saponins and xenoestrogens. Glycyrrhizin (salts of glycyrrhizic acid) is a popular saponin found in roots of mulethi that is responsible for the characteristic sweet taste (50 times more sweet than sugar) flavor. Liquiritin, licoflavonol, liquiritigenin, etc are the common chalcones that provide the distinct yellowish color to mulethi; while, the aroma of its root is mainly because of anethole. Here are the ten health benefits of mulethi:

Information
Latin name: Glycyrrhiza glabra
Sanskrit: Madhuyashti
Hindi: Mulhatti, Jethimadh, Mithilakdi
English: Sweetwood, Liquorice, Licorice
Bengali: Jashtimadhu
Gujrati: Jethi Madh
Marathi: Jeshtamadhu
Kannada: Jeshthamadhu
Malayalam: Itarttimadhuram, Erattimadhuram
Tamil: Atimadhuram
Telugu: Atimadhuramu

Anti-microbial activity – Roots of mulethi are very effective in protecting against virus, bacteria and fungi due to the presence of Glycyrrhizin that blocks the microbial growth. The root extract possesses the power to control malaria (as per preliminary research), influenza and also helps in the treatment of herpes resulting in virus suppression and severity of sores.
Anti-inflammatory activity – Liquorice has powerful anti-inflammatory and anti-allergic activity and can be used to treat chronic inflammation like rheumatic problems & arthritis, skin diseases and autoimmune diseases. It is also used for preventing any inflammatory conditions related to eye and also to treat conjunctivitis with the help of glycyrrhizin activity that counteracts negative effects caused by cortisol.
Improves immunity – Root extracts of mulethi aids in increasing the production of lymphocytes and macrophage thereby improving your defense mechanism & preventing microbial attack. It also helps in minimizing immune related allergic reactions and autoimmune complications.

Memory improvement – Roots of licorice exert supportive effect on the adrenal gland and thus indirectly aid in stimulating the brain. It not only decreases the effects of amnesia & improves learning but its antioxidant property (mulethi contains flavonoids) renders a shielding effect on the brain cells.
Anti-ulcer activity – The potent antioxidant and anti-inflamatory properties of licorice makes it the best natural medicinal aid to treat ulcers of stomach, intestine and mouth. The compound carbenoxolone synthesized from glycyrrhizin plays key role in healing mouth and gastric ulcers along with reducing gastric secretions and promoting development of intestinal mucus lining.
Liver protection – Licorice is one of the most common traditional remedy used to treat jaundice. Its antioxidant property is the key for preventing your liver from the action of free radicals and toxic materials. This herb is also reported to exhibit protection against diclofenac induced toxicity and also, in inhibiting damage of liver.

Digestive aid – Roots of licorice are also used to deal with stomach and digestion problems with the help of glycyrrhizin and its compound, carbenoxolone. It is one of the ancient home remedies for relieving constipation, acidity, heartburn, stomach discomfort, inflammation of digestive system and gastro esophageal acid reflux. As a mild laxative, it plays an effective role in bowel movements and also for treatment of allergic cough in addition to maintaining normal pH levels.
Hormonal regulation – The phytoestrogenic compounds present in mulethi roots exert valuable action against women hormonal imbalance problems, menopause symptoms like hot flashes & exhaustion, mood swings, etc. It is also found to help in cortisol production and relieving premenstrual issues like nausea and menstrual cramps. Licorice powder acts as the traditional medicine for nursing mothers to regulate body hormones and aid in milk secretion.
Heart healthy effects – Research studies have proved that licorice roots help in controlling cholesterol levels by increasing the body’s flow of bile and also reducing high blood cholesterol levels. The anti-oxidant property of licorice acts in increasing the blood capillary health, reducing inflammation, prevents blood vessel damage and block development of arterial plaque.
Other effects – Licorice roots work wonders in treatment of depression, diabetes and respiratory tract infection like sore throat (hoarseness of voice), cold and cough, etc in addition to rendering effective skin benefits, oral hygiene and weight loss. It is found to act as a cancer cure remedy, a potent aphrodisiac and a powerful analgesic agent.
Description
It is a herbaceous perennial, growing to 1 m in height, with pinnate leaves about 7–15 cm (3–6 in) long, with 9–17 leaflets. The flowers are 0.8–1.2 cm (1/3 to 1/2 in) long, purple to pale whitish blue, produced in a loose inflorescence. The fruit is an oblong pod, 2–3 cm (1 in) long, containing several seeds.[10] The roots are stoloniferous.[11]
Chemistry
The scent of liquorice root comes from a complex and variable combination of compounds, of which anethole is up to 3% of total volatiles. Much of the sweetness in liquorice comes from glycyrrhizin, which has a sweet taste, 30–50 times the sweetness of sugar. The sweetness is very different from sugar, being less instant, tart, and lasting longer.
The isoflavene glabrene and the isoflavane glabridin, found in the roots of liquorice, are phytoestrogens.[12][13]
Cultivation and uses
Liquorice, which grows best in well-drained soils in deep valleys with full sun, is harvested in the autumn two to three years after planting.[10] Countries producing liquorice include Iran, Afghanistan, the People’s Republic of China, Pakistan, Iraq, Azerbaijan, Uzbekistan, Turkmenistan, and Turkey.[14]
The world’s leading manufacturer of liquorice products is M&F Worldwide, which manufactures more than 70% of the worldwide liquorice flavours sold to end users.[15]
Safe dosage
Licorice is available in various forms – root, powder and extracts. Licorice root can be chewed directly while licorice tea (prepared by boiling licorice root in water) is also extremely beneficial as a home remedy.
Daily intake of 5-6 grams of licorice powder is considered safe while 250-500 mg of concentrated extracts can be taken thrice a day. Unsupervised use in high doses is not recommended for long term. People with hypertension or heart disease, pregnant women and breastfeeding mothers should avoid using licorice without prior consulation with an Ayurveda doctor.
 plant
plant

Medicine
The compound glycyrrhizin (or glycyrrhizic acid), found in liquorice, has been proposed as being useful for liver protection in tuberculosis therapy, but evidence does not support this use, which may in fact be harmful.[24] Glycyrrhizin has also demonstrated antiviral, antimicrobial, anti-inflammatory, hepatoprotective, and blood pressure-increasing effects in vitro and in vivo, as is supported by the finding that intravenous glycyrrhizin (as if it is given orally very little of the original drug makes it into circulation) slows the progression of viral and autoimmune hepatitis.[25][26] Liquorice has also demonstrated promising activity in one clinical trial, when applied topically, against atopic dermatitis.[27] Additionally, liquorice has also proven itself effective in treating hyperlipidaemia (a high amount of fats in the blood).[28] Liquorice has also demonstrated efficacy in treating inflammation-induced skin hyperpigmentation.[29][30] Liquorice may also be useful in preventing neurodegenerative disorders and dental caries.[31][32][33]
The antiulcer, laxative, antidiabetic, anti-inflammatory, immunomodulatory, antitumour and expectorant properties of liquorice have been investigated.[34]
Folk medicine
In traditional Chinese medicine, liquorice (मुलेठी, 甘草, شیرین بیان) is believed to “harmonize” the ingredients in a formula and to carry the formula to the 12 “regular meridians”.[35]
References
- “Glycyrrhiza glabra information from NPGS/GRIN”. http://www.ars-grin.gov. Retrieved 6 March 2008.
- licorice. Merriam-Webster’s Medical Dictionary, © 2007 Merriam-Webster, Inc.
- γλυκύρριζα, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus
- γλυκύς, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus
- Jump up^ ῥίζα, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus<
- Jump up^ liquorice, on Oxford Dictionaries
- Jump up^ google books Maud Grieve, Manya Marshall – A modern herbal: the medicinal, culinary, cosmetic and economic properties, cultivation and folk-lore of herbs, grasses, fungi, shrubs, & trees with all their modern scientific uses, Volume 2 Dover Publications, 1982 & Pharmacist’s Guide to Medicinal Herbs Arthur M. Presser Smart Publications, 1 Apr 2001 2012-05-19
- Jump up^ Balakrishna, Acharya (2006). Ayurveda: Its Principles & Philosophies. New Delhi, India: Divya prakashan. p. 206. ISBN 8189235567.
- Jump up^ “Top 10 health benefits of Mulethi or Liquorice”.
- ^ Jump up to:a b Huxley, A., ed. (1992). New RHS Dictionary of Gardening. ISBN 0-333-47494-5
- Jump up^ Brown, D., ed. (1995). “The RHS encyclopedia of herbs and their uses”. ISBN 1-4053-0059-0
- Jump up^ Somjen, D.; Katzburg, S.; Vaya, J.; Kaye, A. M.; Hendel, D.; Posner, G. H.; Tamir, S. (2004). “Estrogenic activity of glabridin and glabrene from licorice roots on human osteoblasts and prepubertal rat skeletal tissues”. The Journal of Steroid Biochemistry and Molecular Biology 91 (4–5): 241–246. doi:10.1016/j.jsbmb.2004.04.008. PMID 15336701.
- Jump up^ Tamir, S.; Eizenberg, M.; Somjen, D.; Izrael, S.; Vaya, J. (2001). “Estrogen-like activity of glabrene and other constituents isolated from licorice root”. The Journal of steroid biochemistry and molecular biology 78 (3): 291–298. doi:10.1016/S0960-0760(01)00093-0. PMID 11595510.
- ^ Jump up to:a b c M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2010.
- Jump up^ M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2001.
- Jump up^ Erik Assadourian, Cigarette Production Drops, Vital Signs 2005, at 70.
- Jump up^ M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2005.
- ^ Jump up to:a b c Marvin K. Cook, The Use of Licorice and Other Flavoring Material in Tobacco (Apr. 10, 1975).
- Jump up^ Boeken v. Phillip Morris Inc., 127 Cal. App. 4th 1640, 1673, 26 Cal. Rptr. 3d 638, 664 (2005).
- Jump up^ [1] the online Dutch food composition database]
- Jump up^ “Right good food from the Ridings”. AboutFood.com. 25 October 2007.
- Jump up^ “Where Liquorice Roots Go Deep”. Northern Echo. Retrieved 9 December 2008.
- Jump up^ http://science.howstuffworks.com/life/botany/licorice-info.htm
- Jump up^ Liu Q, Garner P, Wang Y, Huang B, Smith H (2008). “Drugs and herbs given to prevent hepatotoxicity of tuberculosis therapy: systematic review of ingredients and evaluation studies”.BMC Public Health (Systematic review) 8: 365. doi:10.1186/1471-2458-8-365. PMC 2576232. PMID 18939987.
- Jump up^ Chien, CF; Wu, YT; Tsai, TH (January 2011). “Biological analysis of herbal medicines used for the treatment of liver diseases.”. Biomedical Chromatography 25 (1-2): 21–38.doi:10.1002/bmc.1568. PMID 21204110.
- Jump up^ Yasui, S; Fujiwara, K; Tawada, A; Fukuda, Y; Nakano, M; Yokosuka, O (December 2011). “Efficacy of intravenous glycyrrhizin in the early stage of acute onset autoimmune hepatitis.”.Digestive Diseases and Sciences 56 (12): 3638–47. doi:10.1007/s10620-011-1789-5. PMID 21681505.
- Jump up^ Reuter, J; Merfort, I; Schempp, CM (2010). “Botanicals in dermatology: an evidence-based review.”. American Journal of Clinical Dermatology 11 (4): 247–67. doi:10.2165/11533220-000000000-00000. PMID 20509719.
- Jump up^ Hasani-Ranjbar, S; Nayebi, N; Moradi, L; Mehri, A; Larijani, B; Abdollahi, M (2010). “The efficacy and safety of herbal medicines used in the treatment of hyperlipidemia; a systematic review.”. Current pharmaceutical design 16 (26): 2935–47. doi:10.2174/138161210793176464. PMID 20858178.
- Jump up^ Callender, VD; St Surin-Lord, S; Davis, EC; Maclin, M (April 2011). “Postinflammatory hyperpigmentation: etiologic and therapeutic considerations.”. American Journal of Clinical Dermatology12 (2): 87–99. doi:10.2165/11536930-000000000-00000. PMID 21348540.
- Jump up^ Leyden, JJ; Shergill, B; Micali, G; Downie, J; Wallo, W (October 2011). “Natural options for the management of hyperpigmentation.”. Journal of the European Academy of Dermatology and Venereology 25 (10): 1140–5. doi:10.1111/j.1468-3083.2011.04130.x. PMID 21623927.
- Jump up^ Kannappan, R; Gupta, SC; Kim, JH; Reuter, S; Aggarwal, BB (October 2011). “Neuroprotection by spice-derived nutraceuticals: you are what you eat!” (PDF). Molecular Neurobiology 44(2): 142–59. doi:10.1007/s12035-011-8168-2. PMC 3183139. PMID 21360003.
- Jump up^ Gazzani, G; Daglia, M; Papetti, A (April 2012). “Food components with anticaries activity.”. Current Opinion in Biotechnology 23 (2): 153–9. doi:10.1016/j.copbio.2011.09.003.PMID 22030309.
- Jump up^ Messier, C; Epifano, F; Genovese, S; Grenier, D (January 2012). “Licorice and its potential beneficial effects in common oro-dental diseases.”. Oral Diseases 18 (1): 32–9.doi:10.1111/j.1601-0825.2011.01842.x. PMID 21851508.
- Jump up^ Shibata, S (October 2000). “A drug over the millennia: pharmacognosy, chemistry, and pharmacology of licorice.”. Yakugaku Zasshi 120 (10): 849–62. PMID 11082698.
- Jump up^ Bensky, Dan; et al. (2004). Chinese Herbal Medicine: Materia Medica, Third Edition. Eastland Press. ISBN 0-939616-42-4.
- Jump up^ Olukoga, A; Donaldson, D (June 2000). “Liquorice and its health implications.”. The Journal of the Royal Society for the Promotion of Health 120 (2): 83–9.doi:10.1177/146642400012000203. PMID 10944880.
- Jump up^ Armanini, D; Fiore, C; Mattarello, MJ; Bielenberg, J; Palermo, M (September 2002). “History of the endocrine effects of licorice.”. Experimental and Clinical Endocrinology & diabetes 110 (6): 257–61. doi:10.1055/s-2002-34587. PMID 12373628.
- Jump up^ Omar, Hesham R; Komarova,, Irina; El-Ghonemi,, Mohamed; Ahmed, Fathy; Rashad, Rania; Abdelmalak, Hany D; Yerramadha, Muralidhar Reddy; Ali, Yaseen; Camporesi, Enrico M. “How much is too much? in Licorice abuse: time to send a warning message from Therapeutic Advances in Endocrinology and Metabolism”. http://www.ncbi.nlm.nih.gov. SAGE Publications. Retrieved 13 January 2015.
38 Toxicology Center[2]
External links
Labeling under flow conditions: Understanding added applications
Stepping outside traditional synthetic labs into specialty applications is not always something we are looking for in the literature, but it is an excellent way to see different techniques which might be utilized in your own labs. Neil Vasdev’s group at the Harvard Medical School specializes in labeling compounds for more advanced analysis – imaging techniques as tracers for the study of advanced disease states. His group has been using flow chemistry and flow hydrogenation for some time so I thought it be interesting for everyone to see the work.

Two recent publications illustrate their research. In the first publication Chem Commun 2013, 49, 8755 the group uses three examples where they incorporate a label for study into an advanced intermediate C11 or F18 through a microfluidic reaction, followed by a strategic deprotection of a benzyl group or CBz under flow hydrogenation. Without going into significant detail, the group absolutely needed an…
View original post 201 more words
Logistics of process R&D: transforming laboratory methods to manufacturing scale

The manufacture of a | omeprazole (racemic product; top), and esomeprazole (the (S)-enantiomer; bottom), including b | a flow chart of the process for the …
Nature Reviews Drug Discovery 2, 654-664 (August 2003) | doi:10.1038/nrd1154
Logistics of process R&D: transforming laboratory methods to manufacturing scale
Abstract
In the past, process R&D — which is responsible for producing candidate drugs in the required quantity and of the requisite quality — has had a low profile, and many people outside the field remain unaware of the challenges involved. However, in recent years, the increasing pressure to achieve shorter times to market, the demand for considerable quantities of candidate drugs early in development, and the higher structural complexity — and therefore greater cost — of the target compounds, have increased awareness of the importance of process R&D. Here, I discuss the role of process R&D, using a range of real-life examples, with the aim of facilitating integration with other parts of the drug discovery pipeline.
Novartis obtains European approval for Cosentyx to treat psoriasis

Novartis obtains European approval for Cosentyx to treat psoriasis
Swiss drug-maker Novartis has received approval from the European Commission (EC) for its Cosentyx (secukinumab, formerly known as AIN457) to treat moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy.SEE
 PSORIAIS
PSORIAIS
secukinumab

Secukinumab is a human monoclonal antibody designed for the treatments of uveitis, rheumatoid arthritis, ankylosing spondylitis, and psoriasis. It targets member A from the cytokine family of interleukin 17.[1][2] At present, Novartis Pharma AG, the drug’s developer, plans to market it under the trade name “Cosentyx.” [3] It is highly specific to the human immunoglobulin G1k (IgG1k) subclass.[2]
In July 2014 secukinumab established superiority to placebo and to etanercept for the treatment of chronic plaque psoriasis in Phase III clinical trials.[4] In October 2014, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee unanimously voted to recommend the drug for FDA approval, although this vote in and of itself does not constitute an approval. However, the FDA typically follows recommendations from these committees.[5] In October 2014, Novartis announced that the drug had achieved a primary clinical endpoint in two phase III clinical trials for ankylosing spondylitis.[6] As of 28 October, the relevant FDA committee had not yet responded to these results. In early November 2014, Novartis also released the results of a Phase 3 study on Psoriatic Arthritis that yielded very promising results.[7]

Although the drug was originally intended to treat rheumatoid arthritis, phase II clinical trials for this condition yielded disappointing results.[8] Similarly, while patients in a phase II clinical trial for [psoriatic arthritis] did show improvement over placebo, the improvement did not meet adequate endpoints and Novartis is considering whether to do more research for this condition.[9] Novartis has said that it is targeting approval and release in early 2015 for plaque psoriasis and ankyloding spondylitis indications.
It is also in a phase II clinical trial for Multiple Sclerosis [10] as it has exhibited efficacy in treating experimental autoimmune encephalomyelitis (EAE), an animal model of MS.
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833.
- http://www.medscape.com/viewarticle/835331
- Langley RG, Elewski BE, Mark Lebwohl M, et al., for the ERASURE and FIXTURE Study Groups (July 24, 2014). “Secukinumab in Plaque Psoriasis — Results of Two Phase 3 Trials”. N Engl J Med 371: 326–338. doi:10.1056/NEJMoa1314258.
- committees.http://www.familypracticenews.com/index.php?id=2934&type=98&tx_ttnews=306073[dead link]
- http://inpublic.globenewswire.com/2014/10/23/Novartis+AIN457+secukinumab+meets+primary+endpoint+in+two+Phase+III+studies+in+ankylosing+spondylitis+a+debilitating+joint+condition+of+the+spine+HUG1864939.html
- http://www.medpagetoday.com/MeetingCoverage/ACR/48743
- http://www.medscape.com/viewarticle/806510_6
- http://www.ncbi.nlm.nih.gov/pubmed/23361084
- http://clinicaltrials.gov/show/NCT01874340
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL17A |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | |
| ATC code | L04AC10 |
| DrugBank | DB09029 |
| Synonyms | AIN457 |
| Chemical data | |
| Formula | C6584H10134N1754O2042S44 |
| Molecular mass | 147.94 kDa |
Rupatadine

Rupatadine
Platelet activating factor receptor antagonist; Histamine H1 receptor antagonist
Allergic rhinitis; Urticaria
| J. Uriach & Cia. S.A. |

Uriach developed and launched rupatadine for treating of allergic rhinitis and urticaria. Family members of the product case EP577957, have SPC protection in the EU until 2016.
As of January 2015, Newport Premium™ reports that Cadila Pharmaceuticals is producing or capable of producing commercial quantities of rupatadine fumarate and holds an active USDMF since September 2012.
Rupatadine is a second generation antihistamine and PAF antagonist used to treat allergies. It was discovered and developed by J. Uriach y Cia, S. A.[1] and is marketed under several trade names such as Rupafin, Alergoliber, Rinialer, Pafinur, Rupax and Ralif.
Therapeutic indications approved
Rupatadine fumarate has been approved for the treatment of allergic rhinitis and chronic urticaria in adults and children over 12 years. The defined daily dose (DDD) is 10 mg orally.

Available form
Rupatadine is available as round, light salmon coloured tablets containing 10 mg of rupatadine (as fumarate) to be administered orally, once a day.
Side effects
Rupatadine is a non-sedating antihistamine. However, as in other non sedating second-generation antihistamines, the most common side effects in controlled clinical studies were somnolence, headaches and fatigue.
Mechanism of action
Rupatadine is a second generation, non-sedating, long-acting histamine antagonist with selective peripheral H1 receptor antagonist activity. It further blocks the receptors of the platelet-activating factor (PAF) according to in vitro and in vivo studies.[2]
Rupatadine possesses anti-allergic properties such as the inhibition of the degranulation of mast cells induced by immunological and non-immunological stimuli, and inhibition of the release of cytokines, particularly of the TNF in human mast cells and monocytes.[3]

Pharmacokinetics
Rupatadine has several active metabolites such as desloratadine, 3-hydroxydesloratadine, 6-hydroxydesloratadine and 5-hydroxydesloratadine.
History
Rupatadine discovery, pre-clinical and clinical development was performed by J. Uriach y Cia, S. A., a Spanish pharmaceutical company. It was launched in 2003 in Spain by J. Uriach y Cia, S. A., with the brand name of Rupafin. The registration of the product is approved in 23 countries from the EU, 8 Central American countries, Brazil, Argentina, Chile, Turkey and 14 African countries.
Efficacy in humans
The efficacy of rupatadine as treatment for allergic rhinitis (AR) and chronic idiopathic urticaria (CIU) has been investigated in adults and adolescents (aged over 12 years) in several controlled studies, showing a rapid onset of action and a good safety profile even in prolonged treatment periods of a year.[3][4][5]
-
Rupatadine (I) is an authorized antihistaminic agent and has IUPAC name 8-Chloro-6,11-dihydro-11-[1-[(5-methyl-3-pyridinyl)methyl]-4-piperidinylidene]-5H-benzo[5,6]cyclohepta[1,2-b]pyridine, CAS number 158876-82-5 for the free base and the following chemical formula:

-
Rupatadine is currently marketed in 10 mg (rupatadine) tablets as rupatadine fumarate (CAS 182349-12-8 for the fumarate salt) for the treatment of allergic rhinitis and urticaria in adults and teenagers.
-
Rupatadine free base was first disclosed in EP0577957 .
-
Spanish patent application ES2087818 discloses the monofumarate salt of rupatadine (i.e. rupatadine fumarate) and aqueous liquid pharmaceutical compositions of rupatadine fumarate. In particular, this document discloses a syrup containing rupatadine fumarate at 4 g/L, sucrose, a flavouring agent, a sweetening agent and water; and a solution for injection which contains rupatadine fumarate at 20 g/L, benzyl alcohol, propyleneglycol and water.
-
Despite the aqueous liquid pharmaceutical compositions disclosed in EP0577957 and ES2087818 , the inventors have found that the solubility in water of rupatadine fumarate is 2.9 g/L (see Reference example 1) and therefore these prior art formulations may have stability problems due to supersaturation of rupatadine free base or rupatadine fumarate and would not be suitable for use as a medicament.
-
CN101669901 and CN101669926 disclose liquid formulations of rupatadine free base using cyclodextrins to dissolve rupatadine.
-
CN101669901 is directed to liquid formulations of rupatadine free base for ophthalmic delivery comprising rupatadine, a solvent and a cyclodextrin.
-
CN10169926 is directed to liquid formulations of rupatadine free base for nasal delivery comprising rupatadine, a solvent and a cyclodextrin. It is stated that rupatadine has low solubility in water (1.39 mg/mL to 0.82 mg/mL at pH 3.0 to 7.0, table 9 in CN10169926 ) and the problem of its low solubility is solved using cyclodextrins (tables 10-12 of CN10169926 ) in order to obtain liquid formulations.



The reaction of 2-cyano-3-methylpyridine (I) with H2SO4 in t-BuOH gives the N-tert-butylamide (II), which is treated with two equivalents of BuLi and the corresponding dianion alkylated with 3-chlorobenzyl chloride to afford amide (III). The treatment of (III) with POCl3 gives nitrile (IV) which is cyclized to ketone (V) by subsequent treatment with CF3SO3H and aqueous HCl. Reaction of ketone (V) with the Grignard derivative prepared from chloride (VI) affords alcohol (VII) which is finally dehydrated by H2SO4 to give UR-12592 (1), as shown in Scheme 20491401a. The key intermediate (VI) is synthesized through the condensation of 5-methylnicotinic acid (VIII) with 4-hydroxypiperidine by means of DCC in DMF to give amide (IX), followed by reduction with POCl3 and NaBH4 to give the amino alcohol (X) which is treated with SOCl2. Scheme 20491402a. Description White crystals, m.p. 196-8 癈. References 1. Carceller, E., Jim閚ez, P.J., Salas, J. (J. Uriach & Cia SA). Process for the preparation of 8-chloro-6,11-dihydro-11-[1-[(5-methyl-3-pyridinyl)methyl]-4 -piperidinylidene]-5H-benzo[5,6]cyclohepta[1,2-b]pyridine. ES 9602107.

8-chloro-11-(1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
-
To a solution of 1.7 mL (15 mmol) of 3,5-lutidine in 100 mL of CCl₄ was added 2.6 g (15 mmol) of NBS and the mixture was stirred at reflux under an argon atmosphere for 2 h. Then, the mixture was allowed to cool, the solid obtained was filtered off and to the filtrate was added 2.4 g (7.5 mmol) of the compound obtained in preparation 1 and 20 mg of 4-(dimethylamino)pyridine. The resulting mixture was stirred at room temperature for 18 h and 1.68 mL of triethylamine was added. It was diluted with 100 mL of dichloromethane and washed with 0.5N NaHCO₃ solution and with water. The organic phase was dried over sodium sulfate and the solvent was removed, to give 5.7 g of a residue that was chromatographed on silica gel (chloroform : methanol : ammonia, 60 : 2 : 0.2). 1.3 g of the title compound of the example was obtained as a white solid (yield: 40%).
mp: 58-61°C;
IR (KBr) ν: 3419, 3014, 1635, 1576, 1472 cm⁻¹;
¹H RMN (80 MHz, CDCl₃) δ (TMS): 8.39 (m, 3H, ar), 7.48 (m, 1H, ar), 7.37 (m, 1H, ar), 7.12 (m, 4H, ar), 3.45 (s, 2H, CH₂N), 3.36 (m, 2H), 3.1-2.1 (m, 13H). ¹³C RMN (20.15 MHz, CDCl₃) δ (TMS): 157.20 (C), 148.93 (CH), 147.46 (CH), 146.48 (CH), 139.50 (C), 138.56 (C), 137.06 (CH), 133.3 (C), 132.54 (C), 130.67 (CH), 128.80 (CH), 125.85 (CH), 121.92 (CH), 59.84 (CH₂), 54.63 (CH₂), 31.70 (CH₂), 31.32 (CH₂), 30.80 (CH₂), 30.56 (CH₂), 18.14 (CH₃).
WO2006114676
http://www.google.com/patents/WO2006114676A2?cl=en
Scheme-1

Example 1
Preparation of3-bromomethyl-5-methylpyridine hydrochloride: A mixture of carbon tetrachloride (4000ml), azobisisobutyronitrile (45.96gm, 0.279mol), 3,5-lutidine (150gm, 1.399mol) and N-bromosuccinamide (299.4gm, 1.682mol) is refluxed for 2 hours. The reaction mixture is cooled to room temperature and solid is filtered. HCl gas is passed to the filtrate and solid obtained is separated and filtered. Yield is 196gm Yield is 67.66%. Example 2
Preparation of Rupatadine :
A mixture of desloratadine (5.0gm, 0.016mol), methylene chloride (15ml), tetrabutylammonium bromide (0.575gm, 0.0018mol) and sodium hydroxide solution (2.5gm, 0.064mol in 8ml water) is cooled to 0 to 50C. 3-bromomethyl-5- methylpyridine hydrochloride (7.18gm, 0.032mol) in methylene chloride (35ml) is added to above mixture. The reaction mixture is stirred at 0 to 50C for 1 hour and at room temperature for 12 hours. Layers are separated and organic layer is washed with dilute HCl solution and water. Methylene chloride is distilled. Yield = 9.5g %Yield =
67.66%.
Example 3
Preparation of “Rupatadine fumarate:
A solution of fumaric acid (3.3gm) in methanol (46ml) is added to solution of
Rupatadine (4.5gm) in ethyl acetate (30ml) at room temperature. The reaction mass is cooled to -5 to O0C for 4 hours. Rupatadine fumarate is separated filtered and Washed with ethylacetate. Yield = 5.5 gm.
…………………………..
NEW PATENT
EP-02824103…An improved process for the preparation of rupatadine fumarate, Cadila Pharmaceuticals Ltd
Process for the preparing rupatadine intermediate (particularly 5-methylpyridine-3-methanol) comprises reduction of 5-methyl nicotinic acid alkyl ester using alkali metal borohydride is claimed. For a prior filing see WO2006114676, claiming the process for preparation of rupatadine fumarate.
……………………………………
http://pubs.acs.org/doi/abs/10.1021/jm00043a009
References
- Patents: EP 577957, US 5407941, US 5476856
- Merlos, M.; Giral, M.; Balsa, D.; Ferrando, R.; Queralt, M.; Puigdemont, A.; García-Rafanell, J.; Forn, J. (1997). “Rupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF)”. The Journal of Pharmacology and Experimental Therapeutics 280 (1): 114–121. PMID 8996188.
- Picado, C. S. (2006). “Rupatadine: Pharmacological profile and its use in the treatment of allergic disorders”. Expert Opinion on Pharmacotherapy 7 (14): 1989–2001. doi:10.1517/14656566.7.14.1989. PMID 17020424.
- Keam, S. J.; Plosker, G. L. (2007). “Rupatadine: A review of its use in the management of allergic disorders”. Drugs 67 (3): 457–474. doi:10.2165/00003495-200767030-00008. PMID 17335300.
- Mullol, J.; Bousquet, J.; Bachert, C.; Canonica, W. G.; Gimenez-Arnau, A.; Kowalski, M. L.; Martí-Guadaño, E.; Maurer, M.; Picado, C.; Scadding, G.; Van Cauwenberge, P. (2008). “Rupatadine in allergic rhinitis and chronic urticaria”. Allergy 63: 5–28. doi:10.1111/j.1398-9995.2008.01640.x. PMID 18339040.
|
1 to 8 of 8
|
||
|---|---|---|
| Patent | Submitted | Granted |
| 8-chloro-11-[1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden]-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine [US5407941] | 1995-04-18 | |
| Treatment of PAF and histamine-mediated diseases with 8-chloro-11-[1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden]-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine [US5476856] | 1995-12-19 | |
| Process for the synthesis of n-(5-methylnicotinoyl)-4 hydroxypiperidine, a key intermediate of rupatadine [US6803468] | 2004-03-04 | 2004-10-12 |
| $g(b)2-ADRENERGIC RECEPTOR AGONISTS [EP1003540] | 2000-05-31 | |
| $g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS [EP1019360] | 2000-07-19 | |
| 8-Chloro-11-[1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden]-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine. [EP0577957] | 1994-01-12 | 1995-07-12 |
| NOVEL CRYSTALLINE FORM OF RUPATADINE FREE BASE [US2009197907] | 2009-08-06 | |
| METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS [WO9966944] | 1999-12-29 | |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 8-Chloro-6,11-dihydro-11-[1-[(5-methyl-3-pyridinyl)methyl]-4-piperidinylidene]-5H-benzo[5,6]cyclohepta[1,2-b]pyridine fumarate | |
| Clinical data | |
| Trade names | Rupafin, Alergoliber, Rinialer, Pafinur, Rupax, Ralif |
| AHFS/Drugs.com | International Drug Names |
| Legal status |
|
| Routes | Oral |
| Pharmacokinetic data | |
| Protein binding | 98–99% |
| Metabolism | Hepatic, CYP-mediated |
| Half-life | 5.9 hours |
| Excretion | 34.6% urine, 60.9% faeces |
| Identifiers | |
| CAS number | 158876-82-5 182349-12-8 (fumarate) |
| ATC code | R06AX28 |
| PubChem | CID 133017 |
| ChemSpider | 117388 |
| UNII | 2AE8M83G3E |
| ChEMBL | CHEMBL91397 |
| Chemical data | |
| Formula | C26H26ClN3 |
| Molecular mass | 415.958 g/mol |
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide

N-[2-(7-Benzyl-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl)ethyl]acetamide
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide
Acetamide, N-[2-[7-(cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl]-
339.47, C22 H29 N O2
cas 1287785-08-3
Melatonin MT2 Agonists
Takeda……..innovator
Treatment of Sleep Disorders,
-
Melatonin (N-acetyl-5-methoxytryptamine), which is a hormone synthesized and secreted principally in the pineal gland, increases in dark circumstances and decreases in light circumstances. Melatonin exerts suppressively on pigment cells and the female gonads, and acts as a synchronous factor of biological clock while taking part in transmittance of photoperiodic code. Therefore, melatonin is expected to be used for the therapy of diseases related with melatonin activity, such as reproduction and endocrinic disorders, sleep-awake rhythm disorders, jet-lag syndrome and various disorders related to aging, etc.
-
Recently, it has been reported that the production of melatonin melatonin could reset the body’s aging clock (see Ann. N. Y. Acad. Sci., Vol. 719, pp. 456-460 (1994)). As previously reported, however, melatonin is easily metabolized by metabolic enzymes in vivo (see Clinical Examinations, Vol. 38, No. 11, pp. 282-284 (1994)). Therefore, it cannot be said that melatonin is suitable as a pharmaceutical substance.
-
Various melatonin agonists and antagonists such as those mentioned below are known.
- (1) EP-A-578620 discloses compounds of:
-
-

- (2) EP-A-420064 discloses a compound of:

- (3) EP-A-447285 discloses a compound of:

- (4) EP-A-662471 discloses a compound of:

- (5) EP-A-591057 discloses a compound of:

- (6) EP-A-527687 discloses compounds of:

X=S, 0, Y=CH
X=0, NH, Y=N - (7) EP-A-506539 discloses compounds of:

-
-
Tricyclic or more poly-cyclic compounds with a cyclic ether moiety, such as those mentioned below, are known.
- (1) Compounds of:

are disclosed in Tetrahedron Lett., Vol. 36, p. 7019 (1995).
- (2) Compounds of:


are disclosed in J. Med. Chem., Vol. 35, p. 3625 (1992).
- (3) Compounds of:

are disclosed in Tetrahedron, Vol. 48, p. 1039 (1992).
- (4) Compounds of:

are disclosed in Tetrahedron Lett., Vol. 32, p. 3345 (1991).
- (5) A compound of:

is disclosed in Bioorg. Chem., Vol. 18, p. 291 (1990).
- (6) A compound of:

is disclosed in J. Electroanal. Chem. Interfacial Electrochem., Vol. 278, p. 249 (1990).
see
- (1) Compounds of:














http://www.google.co.in/patents/EP0885210B1?cl=en
Highly Potent MT2-Selective Agonists
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide (15)
LY-156735 (TIK-301, PD-6735)….for the treatment of sleep latency in patients with primary insomnia

N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide
LY-156735 (TIK-301, PD-6735) is a melatonin MT1 and MT2 agonist which is under development for the treatment of insomnia and other sleep disorders.[1]
Beta-methyl-6-chloromelatonin (PD-6735) is a melatonin MT1 and MT2 agonist which had been in phase II trials at Phase 2 Discovery for the treatment of sleep latency in patients with primary insomnia, however, no recent development has been reported.
The melatonin agonist exhibits high selectivity and provides a novel mode of action different from that of benzodiazepine receptor ligands currently on the market.
Furthermore, the drug candidate is believed to be non-addicting, therefore, offering an advantage over marketed sleep medications. Originally discovered by Lilly, PD-6735 was licensed to Phase 2 Discovery in 2002 for further development.
Orphan drug designation has been assigned in the U.S. for the treatment of circadian rhythm sleep disorders in blind people with no light perception and for the treatment of neuroleptic-induced tardive dyskinesia in schizophrenia patients.
In 2007, the product candidate was licensed to Tikvah Therapeutics by Phase 2 Discovery for worldwide development and commercialization for the treatment of sleep disorder, depression and circadian rhythm disorder.
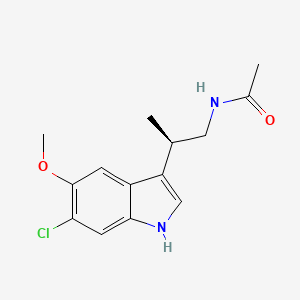
beta -alkylmelatonins as ovulation inhibitors [US4997845]1991-03-05
BETA-ALKYLMELATONINS [EP0281242]1988-09-07 GRANT1992-08-12

The condensation of 6-chloro-5-methoxy-1H-indole (I) with Meldrum’s acid (II) and acetaldehyde (III) catalyzed by L-proline in acetonitrile gives the adduct (IV), which is treated with Cu and ethanol in refluxing pyridine to yield 3-(6-chloro-5-methoxy-1H-indol-3-yl)butyric acid ethyl ester (V). The reaction of (V) with hydrazine at 140 C affords the hydrazide (VI), which is treated with NaNO2 and Ac-OH to provide the corresponding azide that, without isolation, is thermolyzed and rearranged in toluene at 80?C to give 7-chloro-6-methoxy-4-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (VII). The cleavage of the lactam ring of (VII) with KOH in refluxing ethanol/water yields 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (VIII). The decarboxylation of (VIII) by means of refluxing aq. 3M HCl affords 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole (IX), which is finally acylated with acetic anhydride and pyridine in toluene to provide the target 6-chloromelatonin as a racemic compound.
EP 0281242;……….http://www.google.com/patents/EP0281242B1?cl=en
Example 3
- Preparation of β-Methyl-6-chloromelatonin
-
Following the procedure of Example 1, a solution of 10.0 g (0.055 mole) of 5-methoxy-6-chloroindole, 3.1 ml (2.44 g, 0.055 mole) of acetaldehyde, and 7.94 g (0.055 mole) of Meldrum’s acid in 90 ml of acetonitrile was stirred for 48 hours. The solvent was removed under vacuum, and the adduct thus prepared was recrystallized by dissolving in warm toluene and immediately cooling. The adduct was obtained as slightly pink crystals; m.p. = 145°C; yield = 16.5 g (85%). The elemental analysis of the product showed a slightly elevated percentage of carbon. However, the NMR spectrum indicated that the product was pure and had the indicated structure.
Analysis calc. for C₁₇H₁₈NO₅Cl- Theory:
- C, 58.04; H, 5.16; N, 3.98; Cl, 10.08
- Found :
- C, 59.34; H, 5.15; N, 3.84; Cl, 9.69
-
The solvolysis and decarboxylation of the adduct (11.0 g; 31.3 mmoles) using ethanol, pyridine, and copper dust was carried out by the procedure of Example 1. The yield of 3-(5-methoxy-6-chloro-1H-indol-3-yl)pentanoic acid ethyl ester, a pale yellow oil, after chromatography over silica gel using 10% EtOAc/90% toluene was 8.68 g (94%).
Analysis calc. for C₁₅H₁₈NO₃Cl- Theory:
- C, 60.91; H, 6.13; N, 4.74; Cl, 11.99
- Found :
- C, 60.67; H, 5.86; N, 4.93; Cl, 11.73
-
A mixture of 8.68 g (29.3 mmoles) of the above ethyl ester and 6 ml of hydrazine hydrate was heated at 140°C under nitrogen in a flask fitted with an air cooled condensor. After 6½ hours, the excess hydrazine hydrate was removed under vacuum. The 2-methyl-2-(5-methoxy-6-chloro-3-indolyl)-propionhydrazide thus prepared was recrystallized from ethyl acetate; Yield = 7.13 g (86%); m.p. = 154-155°C.
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.14; H, 5.51; N, 14.49; Cl, 12.78
-
The above hydrazide (7.13 g, 25 mmoles) was converted to the corresponding acyl azide, the azide thermolyzed and rearranged at 80° in toluene, and the rearranged product cyclized with HCl according to the procedure of Example 1. The yield of crude, light tan, lactam, 1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole, product, (m.p. = 249-252°C) was 4.77 g (72%).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58
- Found :
- C, 59.45; H, 4.77; N, 10.72
-
The crude lactam (4.77 g, 18 mmoles) was hydrolyzed with aqueous ethanolic KOH as described in Example 1. The yield of crude amino acid, 2-carboxy-3-(1-amino-2-propyl)-5-methoxy-6-chloroindole, was 3.98 g (78%). The crude product (3.0 g; 10.6 mmoles) was decarboxylated, using the procedure of Example 1, by refluxing in 100 ml of 3M HCl overnight. The acidic solution was decolorized with activated carbon and was then basified with 5M NaOH. The amine was extracted into diethyl ether. After drying the ether extract over Na₂SO₄, the diethyl ether was removed in vacuo leaving as a residue the crystallized tryptamine, 3-(1-amino-2-propyl)-5-methoxy-6-chloroindole; m.p. 133-4°C. The yield, after recrystallization from toluene/hexane, was 1.62 g (64%).
Analysis calc. for C₁₂H₁₅N₂OCl- Theory:
- C, 60.38; H, 6.33; N, 11.74; Cl, 14.85
- Found :
- C, 60.11; H, 6.05; N, 11.93; Cl, 15.06
-
A solution of 1.51 g (6.3 mmoles) of the above tryptamine in 10 ml of toluene and 2.5 ml of pyridine was treated with 1.5 ml of acetic anhydride. After allowing the reaction mixture to stand for three hours at room temperature, the volatile materials were removed under vacuum. The residue was dissolved in ethyl acetate, and washed with aqueous NaHCO₃, and brine. The ethyl acetate solution was dried over Na₂SO₄, and the solvent removed by evaporation. The residual oil was crystallized from toluene/hexane yielding 6-chloro-β-methylmelatonin, (m.p. = 133-5°C; 1.09 g, 61%).
Analysis calc. for C₁₄H₁₇N₂O₂Cl- Theory:
- C, 59.89; H, 6.10; N, 9.98; Cl, 12.63
- Found :
- C, 60.03; H, 6.22; N, 9.75; Cl, 12.92
…………………………………………….
PATENT
http://www.google.com/patents/EP0281242B1?cl=en

The intermediate diazonium salt (XIII) has been obtained as follows: the hydrogenation of 3-chloro-4-methoxynitrobenzene (XI) with H2 over Pt/Al2O3 in toluene gives the corresponding aniline (XII), which is diazotized with NaNO2/HCl and treated with sodium tetrafluoroborate to yield the target diazonium salt intermediate (XIII). The reduction of pulegone (I) with H2 over Pd/C gives the menthol (II), which is oxidized with CrO3/H2SO4 to yield 3(R),7-dimethyl-6-oxooctanoic acid (IV), which can also be obtained by direct oxidation of (l)-menthol (III) under the same conditions.
The oxidation of (IV) with trifluoroperacetic acid (trifluoroacetic anhydride/H2O2) in dichloromethane yields the 3(R)-methylhexanedioic acid isopropyl monoester (V), which is treated with NaOEt in ethanol to obtain the corresponding ethyl monoester (VI). The reaction of (VI) with diethyl carbonate, EtONa, and “Adogen 464” (a phase transfer catalyst) in ethanol affords 5,5-bis(ethoxycarbonyl)-3(S)-methylpentanoic acid (VII), which is treated with oxalyl chloride to provide the expected acyl chloride (VIII). The reaction of (VIII) with sodium azide and benzyl alcohol gives the intermediate azide that rearranges to the benzyl carbamate (IX).
The reductive cyclization of (IX) with H2 over Pd/C in ethanol yields 5(R)-methyl-2-oxopiperidine-3-carboxylic acid ethyl ester (X), which is condensed with the intermediate diazonium salt (XIII) to afford the hydrazono derivative (XIV). The cyclization of (XIV) in hot formic acid provides 7-chloro-6-methoxy-4(R)-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (XV), which is treated with KOH In refluxing ethanol/water to cleave the lactam ring, yielding 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (XVI). The decarboxylation of (XVI) by means of refluxing 3M HCl affords 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole (XVII), which is finally acylated with Ac2O and pyridine in toluene to provide the target 6-chloromelatonin as a pure enantiomer.
Example 7
- Preparation of S-(-)-β-methyl-6-chloromelatonin and R-(+)-β-methyl-6-chloromelatonin
-
A solution of 4.0 g (21 mmoles) of 3-chloro-4-methoxynitrobenzene in 200 ml of toluene was hydrogenated over 0.4 g of 5% platinum on alumina. The catalyst was removed by filtration and the solvent evaporated from the filtrate. The crude 3-chloroanisidine prepared was placed in solution in diethyl ether and treated with ethereal HCl to produce the hydrochloride salt, which was collected and dried; weight = 2.48 g (61% yield).
-
A mixture of 2.40 g (12.4 mmoles) of 3-chloroanisidine hydrochloride in 7 ml of 4M HCl was treated, at 0°C, with 0.86 g (12.5 mmoles) of sodium nitrite in 5 ml of water. After stirring at 0°C for an hour the solution was filtered and the filtrate added slowly to an ice cold solution of 2.6 g (24 mmoles) of sodium fluoroborate in 8 ml of water. After stirring at 0°C for an hour the salt was collected and washed successively with, cold 5% sodium fluoroborate solution, cold methanol, and ether. The dried 3-chloro-4-methoxybenzene diazonium fluoroborate thus prepared weighed 2.2 g (69% yield).
-
A mixture of 2.03 g (11.0 mmole) of (R)-(-)-3-ethoxycarbonyl-5-methyl-2-piperidone and 30 ml of 0.75M NaOH was stirred at room temperature (24°C) overnight. The solution was cooled to 0°C and the pH lowered to 3.5 with 3M hydrochloric acid. The diazonium salt (2.8 g, 10.9 mmoles) was added in small portions and the reaction mixture cooled to about 0°C overnight. The product, R-(-)-3-(3-chloro-4-methoxy)phenylhydrazono-5-methyl-2-piperidone, was collected, washed with water, and dried; weight = 2.30 g (75% yield); m.p. = 205°C. A small sample was further purified by chromatography over a short silica gel column using ethyl acetate as the eluant. [α]²⁵ = -58° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.79; H, 5.78: N, 14.72; Cl, 12.69
-
A mixture of 2.20 g (7.8 moles) of the R-(-) hydrazone and 20 ml of 90% formic acid was heated at 85° for three hours then slowly diluted with an equal volume of water. The mixture was allowed to cool and then chilled overnight. The dark precipitate was collected, washed with water, then recrystallized from acetone/water, yielding 1.20 g (60% yield) of S-(-)-1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole; m.p. = 248°C. [α]²⁵ = -12.2° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58; Cl, 13.39
- Found :
- C, 59.16; H, 4.88; N, 10.80; Cl, 13.15
-
The conversion of (S)-(-)-lactam to (S)-(-)-6-chloro-β-methylmelatonin was carried out as described previously in Example 3. The product, S-(-)-β-methyl-6-chloromelatonin, was spectroscopically identical to the racemate, but gave an optical rotation of [α]²⁵ = -13.2° (c = 10, MeOH).
-
(R)-(+)-6-chloro-β-methylmelatonin was synthesized from (S)-(+)-3-ethoxycarbonyl-5-methyl-2-piperidone in the same manner as described above. The stereoisomer was identical to the (S)-(-) material except for the sign of rotation.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-[(2R)-(6-Chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide | |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 118702-11-7 |
| ATC code | ? |
| PubChem | CID 219018 |
| ChemSpider | 189853 |
| Chemical data | |
| Formula | C14H17ClN2O2 |
| Molecular mass | 280.757 |
References
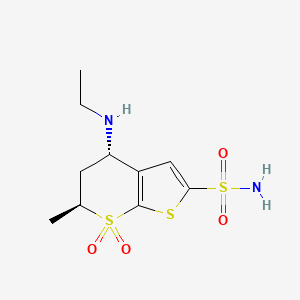
Dorzolamide Hydrochloride


| HS Code: | 2935009090 |
|---|
130693-82-2..HCL
Dorzolamide (trade name Trusopt) is a carbonic anhydrase inhibitor. It is an anti-glaucoma agent, and acts by decreasing the production of aqueous humour.[1] It is optically applied in the form of a 2% eye drops.[2]
History
This drug, developed by Merck, was the first drug in human therapy (market introduction 1995) which resulted from structure-baseddrug design. It was developed to circumvent the systemic side effects of acetazolamide which has to be taken orally.[2]
Uses
Dorzolamide hydrochloride is used to lower increased intraocular pressure in open-angle glaucoma and ocular hypertension.
Pharmacodynamics
It lowers IOP by about 20%.[2]
Side effects
Ocular stinging, burning, itching and bitter taste.[2] it causes shallowing of the anterior chamber and leads to transient Myopia.

Dorzolamide Hydrochloride and its derivatives is known. U.S. Pat. No. 5,688,968 describes preparation of Dorzolamide HCl starting from chiral 5,6-dihydro-4-(S)-hydroxy-6-(S)-methyl-4H-thiopyran-7,7-dioxide, as depicted in scheme 1:

The process described in BP 0 296 879 (equivalent of U.S. Pat. No. 4,797,413) is of particular relevance. EP 0 296 879 describes the synthesis of Dorzolamide Hydrochloride starting from thiophene-2-thiol as depicted in scheme 2 and 3



The process described in EP 0,296,879 (scheme 2) has the following disadvantages: (a) The starting material Thiophene-2-thiol is unstable and undergoes oxidation to form disulfide, leading to lower yield of viii; (b) the yield of sulfonamide (xii) from sulphonic acid (x) is very poor (35%) and requires use of 18-crown-6 ether, which is expensive; (c) oxidation of alcohol (xiii) to sulfone is carried out using oxone which is expensive and hazardous; and separation of cis/trans isomer is done by column chromatography which is industrially inconvenient.
| Systematic (IUPAC) name | |
|---|---|
| (4S,6S)-2-ethylamino-4-methyl-5,5-dioxo- 5λ6,7-dithiabicyclo[4.3.0]nona-8,10-diene-8-sulfonamide |
|
| Clinical data | |
| Trade names | Trusopt |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602022 |
|
|
| Legal status | |
| Routes | Topical (eye drops) |
| Pharmacokinetic data | |
| Protein binding | ~33% |
| Half-life | 4 months |
| Identifiers | |
| CAS number | 130693-82-2 |
| ATC code | S01EC03 |
| PubChem | CID 5284549 |
| DrugBank | DB00869 |
| ChemSpider | 4447604 |
| UNII | 9JDX055TW1 |
| KEGG | D07871 |
| ChEBI | CHEBI:4702 |
| ChEMBL | CHEMBL218490 |
| Chemical data | |
| Formula | C10H16N2O4S3 |
| Molecular mass | 324.443 g/mol |
TRUSOPT® (dorzolamide hydrochloride ophthalmic solution) is a carbonic anhydrase inhibitor formulated for topical ophthalmic use.
Dorzolamide hydrochloride is described chemically as: (4S-trans)-4-(ethylamino)-5,6-dihydro-6methyl-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide monohydrochloride. Dorzolamide hydrochloride is optically active. The specific rotation is
Its empirical formula is C10H16N2O4S3•HCl and its structural formula is:
 |
Dorzolamide hydrochloride has a molecular weight of 360.9 and a melting point of about 264°C. It is a white to off-white, crystalline powder, which is soluble in water and slightly soluble in methanol and ethanol.
TRUSOPT Sterile Ophthalmic Solution is supplied as a sterile, isotonic, buffered, slightly viscous, aqueous solution of dorzolamide hydrochloride. The pH of the solution is approximately 5.6, and the osmolarity is 260-330 mOsM. Each mL of TRUSOPT 2% contains 20 mg dorzolamide (22.3 mg of dorzolamide hydrochloride). Inactive ingredients are hydroxyethyl cellulose, mannitol, sodium citrate dihydrate, sodium hydroxide (to adjust pH) and water for injection. Benzalkonium chloride 0.0075% is added as a preservative.
The dorzolamide hydrochloride product is prepared from the aminated intermediate of Formula IV by the following scheme.


[00056] Preparation of dorzolamide hydrochloride product from the animated intermediate of Formula IV
[00057] Fuming sulfuric acid (20%, 5 1) is cooled to -7°±2°C and the aminated intermediate of Formula IV (2.5 Kg) is added to it in portions during stirring. The temperature of the reaction mixture is increased to 20°+5°C during addition of the aminated intermediate of Formula IV. The reaction mixture is stirred for 22 hours at 20°±5°C. Thionyl chloride (20 1) is added to the stirred reaction mixture at 20±5°C. The reaction mixture is heated to 60°-65°C and stirred for 24 hours at this temperature. The mixture is cooled back to 40°±2°C and the excess amount of thionyl chloride is evaporated at this temperature under vacuum. (The volume of the residue: ~9 1.) The residue is cooled to -5°+2°C.
[00058] Ethyl acetate (75 1) is cooled to -10°±5°C and the residue is added to it at this temperature. The temperature of the diluted solution: 10°-25°C. Aqueous ammonia (25%, 75 1) is cooled to -10°±5°C and the residue is added to it at this temperature during effective stirring, while maintaining the temperature below 300C. The final pH: ~11. The slurry is cooled to 0°+2°C and stirred for 14 hours at this temperature. The formed ammonium sulfate is filtered and the cake is washed with ethyl acetate (2x 20 1 and 10 1). Ethyl acetate is evaporated from the filtrate at 38°±2°C under vacuum. The residue is heated to 38°±2°C, washed with toluene (3×37.5 1) at this temperature. Water (25 1) is added to the aqueous phase, cooled to 20°-25°C and extracted with ethyl acetate (3x 75 1, 37.5 1, and 37.5 1). The collected ethyl acetate phase is concentrated to ~ 100 1 at 38°±2°C under vacuum. The residue is cooled to 20°-25°C and hydrogen chloride in ethanol (5%, 10.8 1) is added to it during stirring. The formed slurry is stirred for 1 hour at 20°-25°C then cooled to 0°-4°C and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×20 1) and dried at 55°-60°C under vacuum for 4-8 hours to give Dorzolamide hydrochloride salt (~2 Kg).
[00059] Crude Dorzolamide hydrochloride salt (9 Kg) is solved in water (225 1) at 20°-25°C and the pH is set to 8.0-8.5 by addition of 25% of aqueous ammonia (2 1). The formed slurry is extracted with ethyl acetate (5×72 1). The collected ethyl acetate phase is concentrated to 180 1 by vacuum distillation. The residue is cooled to 20°-25°C, ethyl acetate (45 1) and hydrogen chloride in ethanol (5%, 22.5 1) are added to it during stirring (pH:~1.0). The formed slurry is stirred for 1 hour at 20°-25°C then cooled to 0°-4°C and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×30 1), and dried at 55°-60°C under vacuum for 4-8 hours to give purified Dorzolamide hydrochloride salt (~8.2Kg).
[00060] Purified Dorzolamide hydrochloride salt (8 Kg) dissolved in water
(24 1) at 95°-105°C and treated with active carbon (80 g). After filtration, the water solution is cooled gradually to 0°-4°C and stirred for 3-5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with cooled water (2×5 1) and dried at 55°-60°C under vacuum for 4-8 hours to give crystallized DRZ HCl salt (~6.6 Kg).
http://www.google.com/patents/US20060142595
The invention provides a process for preparing 5,6-dihydro-4-(S)-(ethylamino)-6-(S)methyl-4H-thieno[2,3b]thiopyran-2-sulphonamide-7,7-dioxide hydrochloride of formula (I), comprising of nine steps, as depicted in scheme 4 below:


Example 9Preparation of 5,6 dihydro-4H-4-(S)-ethylamino-6-(S)-methylthieno[2,3-b]thiopyran-2-sulfonamide-7,7 dioxide Hydrochloride (I)
A mixture of compound from example 8 (15 gm0.0462 mole) and di-p-toluyl-D-tartaric acid monohydrate (4.55 gm, 0.01125 mole) in n-propanol (1600 ml) was heated to boiling and hot solution filtered through a filter-aid pad with a layer of charcoal. The filtrate was concentrated by boiling to a volume of about (400 ml) and then allowed to crystallize. After standing overnight the crystals were filtered off and material recrystallized twice more from n-propanol (400 ml) to yield a 2:1 salt of free base to acid. Combined mother liquors from this recrystallization were saved for stage B. The salt was then treated with a saturated solution of sodium bicarbonate and mid extracted with ethyl acetate. The organic extract were dried, filtered and concentrated to dryness to yield (3.2 gm) of freebase. The hydrochloride salt was prepared from 5,6 N HCl ethanol and crystallized from methanol-isopropanol to yield (2.83 gm) of (+) isomer, SOR 8.23 (C 0.9 methanol) M.P. 283-285° C. The combine mother liquor was treated with saturated solution of sodium bicarbonate and mixture extracted with ethyl acetate. The organic exacts were dried, filtered and concentrated to dryness. The residue was treated with di-p-toluyl-L-tartaric acid monohydrate (4.55 gm, 0.01125 mole) in n-propanol (1600 ml) and the isomer separated by the process described previously to give title compound (I) (3.75 gm, 22.48%) SOR=−8.34 (C 1, Methaol) M.P. 283 to 285° C.,
………………………………………..
http://www.google.com/patents/WO2008135770A2?cl=en
Dorzolamide is chemically termed as (4S,6S)-4-(ethylamino)-5,6-dihydro-6-methyl-4H- thieno[2,3-/?]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride. Dorzolamide hydrochloride is represented by following structural Formula I:
HN ‘CH,

Formula I
Dorzolamide hydrochloride is known to be a carbonic anhydrase inhibitor useful in the treatment of ocular hypertension.
A process for the preparation of dorzolamide and its derivatives was first described in EP 0296879. The process of particular relevance is depicted in scheme 1. Scheme 1

(viϋ) (ix) Trans and Cis (x)

Trans (xi) Trans(+) (xii) ( I )
The process disclosed in scheme 1 has following disadvantages.
(a) The reduction of the ketone of sulfonamide (vi) using absolute ethanol is carried out at reflux and then stirred at room temperature for several hours to complete the reaction. This longer duration of reaction produces many impurities.
(b) Oxidation of alcohol (vii) to sulfone (viii) is carried out using oxone. The oxone has many disadvantages such as it is irritating to the eyes, skin, nose and throat. It should be used with adequate ventilation and exposure to its dust should be minimized. Traces of heavy metal salts catalyze the decomposition of oxone. It is practically insoluble in all organic solvents hence a phase transfer catalyst is required.
(c) Activation of the 4-hydroxy group of the sulfoaminated hydroxysulfone (viii) and nucleophilic substitution by desired ethylamine, results in all diastereomeric products (x) i.e. trans and cis isomers, which must be separated by column chromatography and resolved, further using resolving agent. As a result, product loss is greater when the desired product is the more active enantiomer.
An alternate route for the preparation of dorzolamide hydrochloride by the Ritter reaction is disclosed in EP0296879 and consists of the treatment of a aliphatic hydroxyl with a nitrile and a strong acid to form an amide. The process disclosed is as depicted in Scheme 2.
Scheme 2

(viii) (ix-a ) Trans and Cis (x)

Trans(+) (xii)
Trans (+/-) (xi) ( I )
The reaction involves conversion of hydroxysulfones (viii) to the corresponding acetoamidosulfones (ix-a) with retention of configuration followed by reduction of the amido group, chromatographic separation and resolution to obtain the desired trans isomer (I).
The prior art teaches the use of an excess quantity of sulfuric acid to carry out the Ritter reaction and hence a large quantity of ice is required for quenching the reaction mass. When the reaction mass in concentrated sulfuric acid comes into contact with ice, a large amount of localized heat is generated causing decomposition of material. Since a huge amount of water is required for quenching the reaction mass, the amount of ethyl acetate required for extraction is also substantially large. The work-up using water is not advisable nor applicable industrially.
United States Patent 5688968 describes an alternative route of preparation of dorzolamide hydrochloride starting from chiral 5,6-dihydro-4-(S)-hydroxy-6-(S)-methyl-4H-thiopyran-7,7- dioxide, as depicted in Scheme 3:
Scheme 3

(xiv) (XV)
(xiii)

(xvi) (xvii ) Trans:Cis:: 95: 5 (xviii)
HN CH,


(xix) ( I )
The process described in Scheme 3 has the following disadvantages: (a) Use of expensive chiral hydroxysulfone starting material. The process for the preparation of the chiral hydroxysulfone starting material is disclosed in U.S. Patents Nos. 5,157,129, 5,474,919 and 5,760,249. In these processes, the chiral hydroxysulfone is obtained by the asymmetric enzymatic reduction of the corresponding ketosulfone, or by cyclization of the chiral thienyl thiobutyric acid, obtained, in turn, from a chiral hydroxyester or lactone, and the subsequent stereospecific reduction of the resulting ketone, (b) The process according to this patent uses maleic acid to separate the undesired cis- isomer from dorzolamide. However this maleate salt formation to remove the cis isomer is only suitable when the ratio of trans/cis is greater than 95:5. That means, the maleate salt formation of dorzolamide does not the remove cis isomer exclusively when the cis isomer content is more than 5%. It sometimes requires repeated purification to achieve the desired chiral purity.
Another alternate route for the preparation of dorzolamide hydrochloride is disclosed in United States patent no.7109353 which involves the use of sodium perborate as an oxidant, as depicted in Scheme 4.
Scheme 4
chlorinating agent, cyclinization



Vl IV

VIl VlIl IX
The process disclosed in Scheme 4 has following disadvantages (a) Conversion of (i) to (ii) requires the mixture to be refluxed for 18-20 hrs which is time consuming and may cause impurity in the product.
(b) As the process uses the Ritter reaction to convert (vi) to (vii), a large amount of water is required to quench the hot mass of reaction which is not practical in an industrial set-up. (c) Sodium perborate is used as an oxidizing agent to convert (v) to (vi), which has got bleaching properties, and the handling of it may be injurious when done so for a prolonged period.
Yet another process for the preparation of dorzolamide is disclosed in United States publication no. 20060155132 which involves protecting the chiral 5,6-dihydro-4-(R)- hydroxy-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran-7,7-dioxide as depicted in Scheme 5.
Scheme 5
protected amination benzyl sulphonyl chloride



The process disclosed in Scheme 5 has the following disadvantages, (a) The conversion process of compound (II) to (III) requires a very low temperature which ranges from -30° to 00C. (b) The amination process requires 16- 20 hrs, which is time consuming and may cause impurity in the product. All these disadvantages of the prior art are overcome by the process in accordance with the present invention.
Scheme 8

Example 4
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide
A suspension of 5,6-dihydro-4H-4-acetylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide (83.25 gms, 0.24 moles) in THF (832 ml) was cooled to 00C and sodium borohydride (49.11 gms, 1.29 moles) was added in lots maintaining temperature below 5°C. Reaction mass was stirred for 15 minutes at 5°C and boron trifluoride diethyl- etherate (249.75 ml, 287.2 gms, 2.02 moles) was added below 5°C. The reaction mass was stirred for 5 hours at 0°C to 5°C. Temperature of the reaction mass was raised to 25°C to 300C and stirred for 18 hours. The reaction mass was quenched in 1M sulphuric acid solution (1082 ml) below 5°C, temperature raised to 25°C to 30°C and stirred for 1 hour. The solvent was distilled under reduced pressure at 800C. The reaction mass was cooled to 100C and p H adjusted to 7 – 8 using 50% sodium hydroxide solution. Material was extracted in 1665 ml ethyl acetate once and 832 ml twice. The combined organic layers were washed with saturated sodium chloride solution, dried over sodium sulphate, charcoalised, filtered on hyflo, distilled to get title compound (77.42 gms). HPLC: 80:20::Trans:Cis
Example 7
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide hydrochloride
(a) Dorzolamide di-p-toluyl-L-tartrate salt as prepared in example 6 (44.26 gms, 0.085 moles) was taken in ethyl acetate (557.0 ml), basified with saturated sodium bicarbonate solution. Reaction mass was stirred for 15 minutes at 25°C to 3O0C and aqueous layer was extracted with ethyl acetate (278 ml X 2). The organic layers were combined, washed with brine solution, dried over sodium sulphate, and charcoalized. To the clear solution, IPA + HCL (16.35 ml, 0.089 moles) was added, stirred for 30 minutes and ethyl acetate was removed by distillation at atmospheric pressure at 85°C to about 280 ml volume, cooled to 25-3O0C, stirred for 12 hours at same temperature and filtered to get 26.0 gms of dorzolamide hydrochloride. Trans (-) dorzolamide hydrochloride > 99.5% Trans (+) dorzolamide hydrochloride < 0.5% Cis Isomer <0.1%
(b) Dorzolamide hydrochloride was obtained in a similar manner in quantitative yield from the salt of example 6(b).
(c) Dorzolamide hydrochloride was obtained in a similar manner in quantitative yield from the salt of example 6(c).
Example 8
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide -7,7-dioxide hydrochloride without isolation of base
Dorzolamide di-p-toluyl-L-tartrate (50 gms, 0.096 moles) prepared as per example 6, was charged in a round bottom flask along with isopropanol (1000 ml). The reaction mass was heated to 800C and charged with IPA-HCI (20 ml) dropwise to pH 3 to 4. The reaction mass was heated to reflux for 5-10 minutes. The clear solution obtained was concentrated to 100 ml. The reaction mass was charged with 300 ml ethyl acetate, cooled to 25°C, stirred for 12 to 14 hours at same temperature. The resulting dorzolamide hydrochloride was isolated by filtration and washed with ethyl acetate (50 ml), dried under vacuum at 60- 65 0C for 5-6 hours. Yield- 30 gms.
Trans (-) dorzolamide hydrochloride > 99.5% Trans (+) dorzolamide hydrochloride < 0.5% Cis Isomer <0.1%
…………………………………………………………..
http://www.google.com/patents/EP0453288A1

………………………………………
http://www.google.com/patents/US20060155132
Dorzolamide hydrochloride, known chemically as 5,6-dihydro-4-(S)-ethylamino-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran-2-sulfonamide-7,7-dioxyde hydrochloride, is a topically effective carbonic anhydrase inhibitor useful in the treatment of ocular hypertension.
Dorzolamide hydrochloride has the structure of Formula I:

U.S. Pat. Nos. 4,677,155 and 4,797,413 disclose Dorzolamide. In the prior art synthesis of dorzolamide, a chiral hydroxysulfone is used as a starting material. The chiral hydroxysulfone starting material can be obtained using the processes disclosed in U.S. Pat. Nos. 5,157,129, 5,474,919, and 5,760,249. In the disclosed processes, the chiral hydroxysulfone is obtained by the asymmetric enzymatic reduction of the corresponding ketosulfone, or by cyclization of the chiral thienyl thiobutyric acid, obtained, in turn, from a chiral hydroxyester or lactone, and the subsequent stereospecific reduction of the resulting ketone.
Processes for the preparation of dorzolamide hydrochloride are described in U.S. Pat. Nos. 4,797,413, 5,157,129, and 5,688,968 and in U.S. patent application Publication Ser. No. 2003/0220509. The disclosed processes involve conversion of a hydroxysulfone to the corresponding acetamidosulfone by a Ritter reaction with retention of configuration, followed by introduction of a sulfonamido group, and the subsequent reduction of the amido group to an amine, providing the desired product.
The process disclosed in U.S. Pat. No. 4,797,413 includes activation of the 4-hydoxy group of the sulfonaminated hydroxysulfone with tosyl chloride and the introduction of the desired alkylamino group by nucleophilic substitution, resulting in all diastereomeric products, which must be separated and resolved. As a result, at least 75 percent of the product is lost when the desired product is the more active enantiomer.

EXAMPLE 2
Preparation of 5,6-dihydro-4-(S)-ethylamino-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran 7,7-dioxide hydrochloride salt (Formula IV)
Tetrahydrofuran (50 l) and triethyl amine (4.8 l) are added to 4-(R)-hydroxy-5,6-dihydro-6-(S)-methyl-4H-thieno[2,3b]thiopyran-7,7-dioxide (5 Kg) and stirred under a nitrogen atmosphere at room temperature. The solution is cooled to −10° C. Benzylsulfonyl chloride (5.4 Kg) solved in THF (15 l) is added to the DRZ-19 THF solution in portions while maintaining the temperature below 0° C. The feeding funnel is washed with THF (2 l). The reaction mixture is stirred at 0° C. for 2-4 hours. The formed TEA HCl is filtered and the cake is washed with THF (2×10 l) Ethylamine in THF (30%, 63.7 l) is added to the filtrate and the reaction mixture is stirred at 20°-25° C. for 16 hours. Ethylamine gas prepared by heating of 70% EtNH2water solution (50 l) is absorbed in cooled THF (30 l). Water (20 l) is added to the reaction mixture and THF is evaporated from the filtrate at 40°±5° C. under vacuum. The residue is cooled to 20°-25° C., ethyl acetate (60 l) is added to it and stirred vigorously. After phase separation, the organic phase is washed with water (20 l). The ethyl acetate phase is heated to 40°±2° C. and hydrochloric acid (4M, ˜8-10 l) is added to it during stirring to set pH 2.0-2.5. The formed slurry is cooled to −8°±2° C. and stirred for 3 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (30 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give the desired salt (˜5 Kg).
Preparation of dorzolamide hydrochloride product from the aminated intermediate of Formula IV
Fuming sulfuric acid (20%, 5 l) is cooled to −7°±2° C. and the aminated intermediate of Formula IV (2.5 Kg) is added to it in portions during stirring. The temperature of the reaction mixture is increased to 20°±5° C. during addition of the aminated intermediate of Formula IV. The reaction mixture is stirred for 22 hours at 20°±5° C. Thionyl chloride (20 l) is added to the stirred reaction mixture at 20°±5° C. The reaction mixture is heated to 60°-65° C. and stirred for 24 hours at this temperature. The mixture is cooled back to 40°±2° C. and the excess amount of thionyl chloride is evaporated at this temperature under vacuum. (The volume of the residue: ˜9 l.) The residue is cooled to −5°±2° C.
Ethyl acetate (75 l) is cooled to −10°±5° C. and the residue is added to it at this temperature. The temperature of the diluted solution: 10°-25° C. Aqueous ammonia (25%, 75 l) is cooled to −10°±5° C. and the residue is added to it at this temperature during effective stirring, while maintaining the temperature below 30° C. The final pH: ˜11. The slurry is cooled to 0°±2° C. and stirred for 14 hours at this temperature. The formed ammonium sulfate is filtered and the cake is washed with ethyl acetate (2×20 l and 10 l ). Ethyl acetate is evaporated from the filtrate at 38°±2° C. under vacuum. The residue is heated to 38°±2° C., washed with toluene (3×37.5 l) at this temperature. Water (25 l) is added to the aqueous phase, cooled to 20°-25° C. and extracted with ethyl acetate (3×75 l, 37.5 l, and 37.5 l). The collected ethyl acetate phase is concentrated to ˜100 l at 38°±2° C. under vacuum. The residue is cooled to 20°-25° C. and hydrogen chloride in ethanol (5%, 10.8 l) is added to it during stirring. The formed slurry is stirred for 1 hour at 20°-25° C. then cooled to 0°-4° C. and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×20 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give Dorzolamide hydrochloride salt (˜2 Kg).
Crude Dorzolamide hydrochloride salt (9 Kg) is solved in water (225 l) at 20°-25° C. and the pH is set to 8.0-8.5 by addition of 25% of aqueous ammonia (2 l). The formed slurry is extracted with ethyl acetate (5×72 l). The collected ethyl acetate phase is concentrated to 180 l by vacuum distillation. The residue is cooled to 20°-25° C., ethyl acetate (45 l) and hydrogen chloride in ethanol (5%, 22.5 l) are added to it during stirring (pH:˜1.0). The formed slurry is stirred for 1 hour at 20°-25° C. then cooled to 0°-4° C. and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×30 l), and dried at 55°-60° C. under vacuum for 4-8 hours to give purified Dorzolamide hydrochloride salt (˜8.2 Kg).
Purified Dorzolamide hydrochloride salt (8 Kg) dissolved in water (24 l) at 95°-105° C. and treated with active carbon (80 g). After filtration, the water solution is cooled gradually to 0°-4° C. and stirred for 3-5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with cooled water (2×5 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give crystallized DRZ HCl salt (˜6.6 Kg).
…………………………………………………………

Reaction of (I) with acetic anhydride-sulfuric acid in methylene chloride provided the sulfonic acid in 98% yield. Conversion to the sulfonyl chloride with phosphorous pentachloride in methylene chloride followed by treatment with aqueous ammonia gave the sulfonamide (II). Reduction of the carbonyl function with sodium borohydride and oxidation of the thiopyran sulfur with Oxone(R) yielded (IV). The 4-hydroxy substituent was converted to the acetylamino functionality under Ritter conditions. Reduction of (V) with borane-dimethylsulfide complex yielded (VI) as a mixture of diasteriomers. Chromatography on silica gel gave the trans-racemate, which was resolved into its individual enantiomers through the di-p-toluoyl-L-tartaric acid salt. The absolute configuration of the S,S-enantiomer, MK-507, was established by single crystal X-ray analysis.
……………………………………………………….
http://bonanzasite.com/synthesis-of-dorzolamide-hydrochloride

………………………..

//////////A new synthesis of MK-0507 has been described: The condensation of 3(R)-(tosyloxy)butyric acid methyl ester (I) with lithium 2-thienylmercaptide (II) in formamide-THF gives 3(S)-(2-thienylthio)butyric acid methyl ester (III), which is hydrolyzed with aqueous HCl to the corresponding free acid (IV). The intramolecular Friedel-Crafts’cyclization of (IV) with trifluoroacetic anhydride yields 6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-4-one (V), which is reduced with LiAlH4 in toluene to afford 4(R)-hydroxy-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran (VI). Epimerization of (VI) with sulfuric acid gives the alcohol (VII) in a cis:trans ratio of 24:76%. Oxidation of (VII) with H2O2 and sodium tungstate yields the 7,7-dioxide (VIII; cis-trans mixture), which is acetylated with acetic anhydride to the acetate (IX). The reaction of (IX) with acetonitrile and sulfuric acid affords N-[6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-4-yl]acetamide 7,7-dioxide (X; cis-trans mixture), which is sulfonated with chlorosulfonic acid and then treated with SOCl2 to give 4-acetamide-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonyl chloride 7,7-dioxide (XI; cis-trans mixture). The reaction of (XI) with concentrated aqueous NH4OH in THF yields the corresponding sulfonamide (XII), which by reduction with BH3-dimethylsulfide in THF affords 4-(ethylamino)-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide (XIII; cis-trans mixture). Finally, this mixture is treated with maleic acid in acetone and the resulting maleates are submitted to fractionated crystallization, giving the maleate of the (4S,6S)-isomer, which is treated first with NaHCO3 and then with HCl to give MK-0507; [alpha](25)589 -17.1 C (c 1, H2O).
 CAS NO. 130693-82-2, DORZOLAMIDE HCL H-NMR spectral analysis |
 CAS NO. 130693-82-2, DORZOLAMIDE HCL C-NMR spectral analysis |
References
- Dorzolamide at Drugs.com. Revised: 12/2011
- KD Tripari MD. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
Further reading
- Kubinyi H (1999). “Chance favors the prepared mind–from serendipity to rational drug design”. J Recept Signal Transduct Res19 (1–4): 15–39. doi:10.3109/10799899909036635. PMID 10071748.
- Plummer C, MacKay E, Gelatt K (2006). “Comparison of the effects of topical administration of a fixed combination of dorzolamide-timolol to monotherapy with timolol or dorzolamide on IOP, pupil size, and heart rate in glaucomatous dogs”. Vet Ophthalmol 9 (4): 245–9. doi:10.1111/j.1463-5224.2006.00469.x. PMID 16771760.
- Grover S, Apushkin M, Fishman G (2006). “Topical dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa”. Am J Ophthalmol 141 (5): 850–8. doi:10.1016/j.ajo.2005.12.030. PMID 16546110.
- Almeida G, Faria e Souza S (2006). “Effect of topical dorzolamide on rabbit central corneal thickness”. Braz J Med Biol Res 39(2): 277–81. doi:10.1590/S0100-879X2006000200015. PMID 16470316.
Reference:
CIPLA LIMITED; CURTIS, Philip, Anthony Patent: WO2008/135770 A2, 2008 ; Location in patent: Page/Page column 21-22 ;
RAGACTIVES, S.L. Patent: US2003/220509 A1, 2003 ; Location in patent: Page/Page column 12 ;
WO2011/101704 A1, ;
Literature References:
Carbonic anhydrase inhibitor. Prepn: J. J. Baldwin et al., EP 296879; eidem, US 4797413 (1988, 1989 both to Merck & Co.). Mechanism of action study: R.-F. Wang et al., Arch. Ophthalmol. 109, 1297 (1991).
HPLC determn in plasma and urine: B. K. Matuszewski, M. L. Constanzer, Chirality 4, 515 (1992).
Clinical evaluations in glaucoma and ocular hypertension: E. A. Lippa et al., Ophthalmology 98, 308 (1991); E. A. Lippa et al., Arch. Ophthalmol. 110, 495 (1992).
| Reference | ||
|---|---|---|
| 1 | * | KAMEI K. ET AL.: ‘Chemical structure, physico-chemical properties and stability of dorzolamide hydrochloride‘ IYAKUHIN KENKYU vol. 25, no. 6, 1994, pages 438 – 452, XP008040715 |
| 2 | * | QUINT M.-P. ET AL.: ‘Dorsolamide hydrochloride‘ ANALYTICAL PROFILES OF DRUG SUBSTANCES AND EXCIPIENTS vol. 26, 1999, pages 283 – 316, XP008040718 |
| EP2128161A1 * | May 30, 2008 | Dec 2, 2009 | Ragactives, S.L. | Process for obtaining 4-hydroxy-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-7,7-dioxide and its enantiomers, and applications thereof |
| WO2008135770A2 * | May 7, 2008 | Nov 13, 2008 | Cipla Ltd | Process for preparing dorzolam ide |
| WO2009144263A2 * | May 28, 2009 | Dec 3, 2009 | Ragactives, S.L.U. | PROCESS FOR OBTAINING 4-HYDROXY-6-METHYL-5, 6-DIHYDRO-4H-THIENO [2,3-b] THIOPYRAN-7, 7-DIOXIDE AND ITS ENANTIOMERS, AND APPLICATIONS THEREOF |
| WO2014005943A1 * | Jun 28, 2013 | Jan 9, 2014 | Zach System S.P.A. | Process for preparing enantiomerically enriched oxamides |
| US8263787 | May 7, 2008 | Sep 11, 2012 | Cipla Limited | Process for preparing dorzolamide |
| WO1994021645A1 * | Mar 16, 1994 | Sep 29, 1994 | Thomas J Blacklock | ENANTIOSELECTIVE SYNTHESIS OF 5,6-DIHYDRO-(S)-4-(ETHYLAMINO)-(S)-6-METHYL-4H-THIENO[2,3-b]THIOPYRAN-2-SULFONAMIDE 7,7-DIOXIDE AND RELATED COMPOUNDS |
| EP0296879A1 * | Jun 23, 1988 | Dec 28, 1988 | Merck & Co., Inc. | Substituted aromatic sulfonamides as antiglaucoma agents |
| US5474919 * | Sep 13, 1994 | Dec 12, 1995 | Merck & Co., Inc. | Bioconversion process for the synthesis of transhydroxy sulfone by Rhodotorula rubra or Rhodotorula piliminae |
| US5760249 * | Aug 28, 1996 | Jun 2, 1998 | Merck & Co., Inc. | Synthesis of hydroxysulfone and related compounds |
| US20060142595 * | Dec 28, 2004 | Jun 29, 2006 | Council Of Scientific & Industrial Research | Starting by reacting a 2-halothiophene with a Grignard reagent in a solvent in situ with sulfur, triethylamine hydrochloride, crotonic acid and a base; product is chlorinated, cyclized, chlorosulfonated and aminated, reduced, oxidized, amidated, hydrogenated, neutralized, recrystallized and resolved |
| US5157129 | Apr 18, 1990 | Oct 20, 1992 | Merck & Co., Inc. | Enantiospecific synthesis of s-(+)-5,6-dihydro-4-(r-amino)-4h-thieno(2,3-b)thiopyran-2-sulfonamide-7,7-dioxide |
| US5474919 | Sep 13, 1994 | Dec 12, 1995 | Merck & Co., Inc. | Bioconversion process for the synthesis of transhydroxy sulfone by Rhodotorula rubra or Rhodotorula piliminae |
| US5688968 | Jan 6, 1995 | Nov 18, 1997 | Merck & Co., Inc. | Enantioselective synthesis of 5,6-dihydro-(S)-4-(ethylamino)-(S)-6-methyl-4H-thieno 2,3-B!thiopyran-2-sulfonamide 7,7-dioxide |
| US5760249 | Aug 28, 1996 | Jun 2, 1998 | Merck & Co., Inc. | Synthesis of hydroxysulfone and related compounds |
| US7109353 | Dec 28, 2004 | Sep 19, 2006 | Council Of Scientific And Industrial Research | Process for preparing 5,6-dihydro-4-(S)-(ethylamino)-6-(S) methyl-4H-thieno[2,3b]thiopyran-2-sulphonamide-7,7-dioxide HCl |
| US20060155132 | Jan 6, 2006 | Jul 13, 2006 | Kovacs Laszlo Z | Method of making dorzolamide hydrochloride |
| EP0296879A1 | Jun 23, 1988 | Dec 28, 1988 | Merck & Co., Inc. | Substituted aromatic sulfonamides as antiglaucoma agents |
| WO1994021645A1 | Mar 16, 1994 | Sep 29, 1994 | Thomas J Blacklock | ENANTIOSELECTIVE SYNTHESIS OF 5,6-DIHYDRO-(S)-4-(ETHYLAMINO)-(S)-6-METHYL-4H-THIENO[2,3-b]THIOPYRAN-2-SULFONAMIDE 7,7-DIOXIDE AND RELATED COMPOUNDS |
| WO2008135770A2 | May 7, 2008 | Nov 13, 2008 | Cipla Ltd | Process for preparing dorzolam ide |

Sandoz’s Zarzio (filgrastim) would be the first ‘biosimilar’ drug available in the US

A key advisory committee of the US Food and Drug Administration (FDA) has voted in favour of licencing a copycat version of a biological drug. If approved, Sandoz’s Zarxio (filgrastim) would be the first ‘biosimilar’ drug available in the US.
read at……..http://www.rsc.org/chemistryworld/2015/01/us-poised-approve-first-biosimilar-drug
On 7 January, the FDA’s Oncological Drugs Advisory Committee unanimously cleared Sandoz’ version of filgrastim – marketed as Neupogen by Amgen – for all five indications approved for the Amgen drug. The medication is used to prevent infection and low white blood cell counts caused by chemotherapy.


| Systematic (IUPAC) name | |
|---|---|
| Human granulocyte colony stimulating factor | |
| Clinical data | |
| Trade names | Neupogen |
| AHFS/Drugs.com | monograph |
| Legal status |
?
|
| Identifiers | |
| CAS number | 143011-72-7 |
| ATC code | L03AA02 |
| DrugBank | DB00099 |
| UNII | PVI5M0M1GW |
| ChEMBL | CHEMBL1201567 |
| Chemical data | |
| Formula | C845H1343N223O243S9 |
| Molecular mass | 18802.8 g/mol |
Filgrastim is a granulocyte colony-stimulating factor (G-CSF) analog used to stimulate the proliferation and differentiation ofgranulocytes;[1] it is a pharmaceutical analog of naturally occurring G-CSF. It is produced by recombinant DNA technology. The gene for human granulocyte colony-stimulating factor is inserted into the genetic material of Escherichia coli. The G-CSF then produced byE. coli is different from G-CSF naturally made in humans.

Commercialization
Filgrastim is marketed under several brand names, including:
| Company | Brand |
|---|---|
| Cadila Pharmaceuticals | Filcad |
| Abbott Laboratories | Imumax |
| Dr. Reddy’s Laboratories | Grafeel |
| Intas Biopharmaceuticals | Neukine |
| Amgen | Neupogen[2] |
| Emcure Pharmaceuticals | Emgrast |
| Reliance Life Sciences | Religrast |
| Sandoz | Zarzio |
| Biocon | Nufil |

Apricus Biosciences is currently developing and testing a product under the brand name Nupen which can deliver filgrastim through the skin to improve post-chemotherapy recovery of neutrophil counts.

Therapeutic uses
Filgrastim is used to treat neutropenia,[3] stimulating the bone marrow to increase production of neutrophils. Causes of neutropenia include chemotherapy and bone marrow transplantation.
Filgrastim is also used to increase the number of hematopoietic stem cells in the blood before collection by leukapheresis for use in hematopoietic stem cell transplantation.
Mechanism of Action: Filgrastim is a human granulocyte colony stimulating factor (G-CSF) produced by recombinant DNA technology. G-CSF regulates the production of neutrophils within the bone marrow; endogenous G-CSF is a glycoprotein produced by monocytes, fibroblasts, and endothelial cells.
G-CSF is a colony stimulating factor which has been shown to have minimal direct in vivo or in vitro effects on the production of other haematopoietic cell types.NEUPOGEN (filgrastim) is the name for recombinant methionyl human granulocyte colony stimulating factor (r-metHuG-CSF). ref: [1]
Contraindications
Filgrastim should not be used in patients with known hypersensitivity to E. coli-derived proteins.
Adverse effects
The most commonly observed adverse effect is mild-to-moderate bone pain after repeated administration and local skin reactions at the site of injection.[4] Other observed adverse effects include serious allergic reactions (including a rash over the whole body, shortness of breath, wheezing, dizziness, swelling around the mouth or eyes, fast pulse, and sweating), ruptured spleen (sometimes resulting in death), alveolar hemorrhage, acute respiratory distress syndrome, and hemoptysis.[4] Severe sickle cell crises, in some cases resulting in death, have been associated with the use of filgrastim in patients with sickle cell disorders.[5]
Interactions
Drug interactions between filgrastim and other drugs have not been fully evaluated. Drugs which may potentiate the release of neutrophils‚ such as lithium‚ should be used with caution.
Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes; this should be considered when interpreting bone-imaging results.[6]
Filgrastim has not been studied in pregnant women and its effects on the foetus is unknown. If taking filgrastim while pregnant, it is possible that traces of the drug could be found in the baby’s blood. It is not known if the drug can get into human breast milk.
References
- Beveridge, R. A.; Miller, J. A.; Kales, A. N.; Binder, R. A.; Robert, N. J.; Harvey, J. H.; Windsor, K.; Gore, I.; Cantrell, J.; Thompson, K. A.; Taylor, W. R.; Barnes, H. M.; Schiff, S. A.; Shields, J. A.; Cambareri, R. J.; Butler, T. P.; Meister, R. J.; Feigert, J. M.; Norgard, M. J.; Moraes, M. A.; Helvie, W. W.; Patton, G. A.; Mundy, L. J.; Henry, D.; Sheridan, B.; Staddon, A.; Ford, P.; Katcher, D.; Houck, W.; Major, W. B. (1998). “A Comparison of Efficacy of Sargramostim (Yeast-Derived RhuGM-CSF) and Filgrastim (Bacteria-Derived RhuG-CSF) in the Therapeutic Setting of Chemotherapy-Induced Myelosuppression”. Cancer Investigation 16 (6): 366–373. doi:10.3109/07357909809115775. PMID 9679526.
- “FDA Reviews What Could Be First Biosimilar”. Discov. Dev. Mag. (Rockaway, New Jersey, United States). Associated Press. 25 July 2014.
- Crawford, J.; Glaspy, J. A.; Stoller, R. G.; Tomita, D. K.; Vincent, M. E.; McGuire, B. W.; Ozer, H. (2005). “Final Results of a Placebo-Controlled Study of Filgrastim in Small-Cell Lung Cancer: Exploration of Risk Factors for Febrile Neutropenia”. Supportive Cancer Therapy 3 (1): 36–46. doi:10.3816/SCT.2005.n.023. PMID 18632435.
- Neupogen “Neupogen: Patient Information Leaflet”. Amgen. Retrieved 24 June 2013.
- “NEUPOGEN® Patient Guide”. Amgen. Retrieved 24 June 2013.
- “Neupogen”. RxList. 4 June 2012. Retrieved 23 June 2013.
Further reading
- Budiono Santoso; Chris J. van Boxtel; Boxtel, Christoffel Jos van (2001). Drug benefits and risks: international textbook of clinical pharmacology. New York: Wiley. ISBN 0-471-89927-5.
- “Neupogen information”. Retrieved 20 October 2005.

{kind=link}