
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
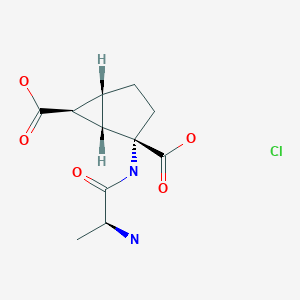
Talaglumetad hydrochloride
|
Talaglumetad hydrochloride
|
|
| Formula |
C11H16N2O5. HCl
|
|---|---|
| Exact mass |
292.0826
|
| Mol weight |
292.7161
|
CAS: 441765-97-5
441765-98-6 (free base)
IUPAC Name: (1R,4S,5S,6S)-4-[[(2S)-2-aminopropanoyl]amino]bicyclo[3.1.0]hexane-4,6-dicarboxylic acid hydrochloride
Synonyms: Talaglumetad HCl, Talaglumetad hydrochloride, LY 544344 hydrochloride,
UNII-X30300EU7I, D09008, 441765-97-5,
Bicyclo(310)hexane-2,6-dicarboxylic acid, 2-(((2S)-2-amino-1-oxopropyl)amino)-, monohydrochloride, (1S,2S,5R,6S)-
(1S,2S,5R,6S)-2-(L-Alanylamino)bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
(1S,2S,5R,6S)-2-[2(S)-Aminopropionamido]bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
(1S,2S,5R,6S)-2-[2(S)-Aminopropionamido]bicyclo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride
Treatment of anxiety and stress disorders [metabotropic glutamate [mGlu] agonist]
Talaglumetad hydrochloride, a prodrug of the type II metabotropic glutamate receptor agonist eglumetad, reached phase III clinical studies for the treatment of anxiety at Lilly.

-
In recent years, with the repeated cloning of glutamate receptor genes, it has become clear that there are surprisingly many subtypes of glutamate receptors. At present, glutamate receptors are broadly classified into two types: the “ionotropic type”, in which the receptor has an ion channel structure, and the “metabotropic type”, in which the receptor is coupled to G-proteins (Science, 258, 597-603, 1992). Ionotropic receptors are classified pharmacologically into three types: N-methyl-D-asparaginic acid (NMDA), α-amino-3-hydroxy-5-methyl isoxazole-4-propionate AMPA), and kynate (Science, 258, 597-603, 1992). Metabotropic receptors are classified into eight types, type 1 through type 8 (J. Neurosci., 13, 1372-1378, 1993; Neuropharmacol., 34, 1-26, 1995).
-
The metabotropic glutamate receptors are classified pharmacologically into three groups. Of these, group 2 (mGluR2/mGluR3) bind with adenylcyclase, and inhibit the accumulation of the Forskolin stimulation of cyclic adenosine monophosphate (cAMP) (Trends Pharmacol. Sci., 14, 13 (1993)), which suggests that compounds that act on group 2 metabotropic glutamate receptors should be useful for the treatment or prevention of acute and chronic psychiatric and neurological disorders. As a substance that acts on group 2 metabotropic glutamate receptors, (+)-(1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid has been disclosed in Japanese Unexamined Patent Publication, No. Hei 8-188561 [1996].
-
Fluorine atoms tend to be strongly electron-attractive and to confer high fat solubility, and compounds into which fluorine atoms are introduced greatly change their physical properties. Thus introducing fluorine atoms might greatly affect the absorbability, metabolic stability, and pharmacological effects of a compound. But it is by no means easy to introduce fluorine atoms. In fact, Japanese Unexamined Patent Publication No. Hei 8-188561 [1996] does not even discuss the introduction of fluorine atoms into (+)-(1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid.
………………………………………………………….
Process development of (1S,2S,5R,6S)-spiro[bicyclo[3.1.0]hexane-2′,5′-dioxo-2,4′-imidazolidine]-6-carboxylic acid, (R)-alpha-methylbenzenemethanamine salt (LSN344309)
Org Process Res Dev 2006, 10(1): 28
http://pubs.acs.org/doi/abs/10.1021/op049829e
LY544344 hydrochloride 6 is Talaglumetad

Process development and a pilot-plant process for the synthesis of 4 and its resolution to obtain (1S,2S,5R,6S)-spiro[bicyclo[3.1.0]hexane-2‘,5‘-dioxo-2,4‘-imidazolidine]-6-carboxylic acid, (R)-α-methylbenzenemethanamine salt (5) are described. Starting from the inexpensive raw 2-cyclopenten-1-one and sulfur ylide 1 the racemic bicyclo keto ester 2 was synthesized. Reaction of 2 with potassium cyanide and ammonium carbonate under Bücherer−Berg’s reaction conditions affords racemic 3 in 80% yield. Hydrolysis of 3 followed by the resolution with (R)-(+)-α-methylbenzylamine gave 4 in excellent yield and purity under optimized conditions. The improvement of the original discovery process to accommodate safety and environmental requirements for scale-up in manufacturing facilities is also discussed.
LY544344 hydrochloride 6 is a new chemical entity under investigation by Eli Lilly & Company as a potential treatment of neurological or psychiatric disorders related to the mammalian central nervous system (CNS)

Scheme 1. Original process for the synthesis of LSN344309 an intermediate of Talaglumetad
…………………………………………………….
Journal of Medicinal Chemistry (2005), 48(16), 5305-5320
http://pubs.acs.org/doi/full/10.1021/jm050235r

…………………………………………………….
WO 2002055485
OR;
http://www.google.im/patents/US20040138304?cl=un

………………………………………………………….
http://www.google.com/patents/EP1052246A1?cl=en

………………………………………………
REFERENCES
New approaches in the development of orally bioavailable selective group 2 metabotropic glutamate receptor agonists
Drugs Fut 2002, 27(Suppl. A): Abst C39
Utility of influx transporters to enhance oral bioavailability
241st ACS Natl Meet (March 27-30, Anaheim) 2011, Abst MEDI 163
The intestinal absorption of a prodrug of the mGlu2/3 receptor agonist LY354740 is mediated by PEPT1: In situ rat intestinal perfusion studies
J Pharm Sci 2010, 99(3): 1574
Dipeptides as effective prodrugs of the unnatural amino acid (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740), a selective group II metabotropic glutamate receptor agonist
J Med Chem 2005, 48(16): 5305
An efficient synthesis of LY544344.HCl: A prodrug of mGluR2 agonist LY354740
Tetrahedron Lett 2005, 46(43): 7299
Pharmacodynamics of a novel anxiolytic (LY544344)
24th CINP Congr (June 20-24, Paris) 2004, Abst P02.269
| WO2000004010A1 * | Jul 14, 1999 | Jan 27, 2000 | Stephen Richard Baker | Bicyclohexane derivatives |
| EP0696577A1 * | Aug 11, 1995 | Feb 14, 1996 | Eli Lilly And Company | Synthetic excitatory amino acids |
| EP1052246A1 * | Jan 27, 1999 | Nov 15, 2000 | Taisho Pharmaceutical Co. Ltd | Fluorine-containing amino acid derivatives |
Complaints and Recalls: new EU-GMP Chapter 8 published
DRUG REGULATORY AFFAIRS INTERNATIONAL
GMP News: Complaints and Recalls: new EU-GMP Chapter 8 published
|
View original post 450 more words
If a Facility stores Medicinal Products for more than 36 Hours GDP will apply
DRUG REGULATORY AFFAIRS INTERNATIONAL
GMP News: If a Facility stores Medicinal Products for more than 36 Hours GDP will apply
Since the EU Good Distribution Practice (GDP) Guide has been revised, a number of questions regarding its interpretation have been raised. One of these questions relates to storage facilities and so called distribution hubs. In the past, many facilities which have been involved in the supply chain were not managed under GDP and didn’t posses a licence for their activities.
The British Medicines Authority MHRA published a press release on 18 August 2014 to explain what they consider to be a facility which must be licensed and which needs to implement the GDP requirements. According to the MHRA: “The GDP Inspectorate is raising awareness of the impact of the new regulations to those parties that are either directly or indirectly affected and any freight consolidator or freight forwarder either in the air, sea or road transport sector…
View original post 59 more words
FDA publishes ICH Q4B – Annex 6 on Uniformity of Dosage Units
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
GMP News: FDA publishes ICH Q4B – Annex 6 on Uniformity of Dosage Units
On 16 June 2014, the FDA published the ICH harmonised Guideline entitled “Evaluation and Recommendation of Pharmacopoeial Texts for Use in the ICH Regions on Uniformity of Dosage Units General Chapter (Q4B Annex 6)”. This ICH Guideline thus came into force in the USA, too.
The objective of the ICH Q4B Working Group is to reach mutual recognition by regulatory authorities in the ICH regions for all testing methods listed in the ICH Q6A Guideline on Specifications. Through this, comparable testing laid down in the different pharmacopeias shouldn’t be performed separately when it has been assessed by the authorities that those are similar and interchangeable.
The Annex 6 states that the following official texts :
- Ph.Eur. 2.9.40 (Uniformity of Dosage Units
- JP 6.02 Uniformity of Dosage Units
- USP General Chapter <905> Uniformity of Dosage Units
View original post 91 more words
Glenmark’s TRPA1 antagonist ‘GRC 17536’ shows positive data in a proof of concept study

MUMBAI, India, Sep 17, 2014
- Glenmark's first in class TRPA1 antagonist, GRC 17536, has shown positive data in a Phase 2a proof of concept study in patients with painful diabetic neuropathy
Glenmark Pharmaceuticals today announced that its first in class Transient Receptor Potential Ankyrin 1 (TRPA1) antagonist, GRC 17536 has shown positive data in a Phase 2a double blind, placebo controlled, multi-centre, proof of concept study conducted on 138 patients in Europe and India.
A statistically significant and clinically relevant response was seen in a prospectively-identified, substantial sub-group of patients with moderate to severe pain who had relatively intact sensory responses as detected by a standardized testing methodology. GRC 17536 was well-tolerated with no evidence of CNS or other drug related side effects.
Patrick Keohane, Chief Medical Officer, Glenmark stated “Diabetic neuropathy remains a difficult to manage chronic clinical condition with limited therapeutic options. These initial efficacy and safety data with GRC 17536, a peripherally acting novel therapeutic, are encouraging, and Glenmark intends to be ready to file for a Phase 2b dose range finding study in patients with neuropathic pain before the end of this financial year. This announcement also reaffirms our position globally in the development of novel pain therapies”.
Commenting on this result, Dr. Michael Buschle, Chief Scientific Officer & President – Biologics, Glenmark Pharmaceuticals mentioned, “This is very promising and GRC 17536 may be useful for several indications which we will pursue”.
The Glenmark TRPA1 program includes indications in pain as well as respiratory. Inhaled doses of GRC 17536 are also being tested in a Phase 2A proof of concept study in patients with Chronic Cough.
, Managing Director and CEO, along with Dr. Michael Buschle, President Biologics, Glenmark Pharmaceuticals at a press conference in Mumbai on Monday. Photo: Paul Noronha")
Glenn Saldanha (left), Managing Director and CEO, along with Dr. Michael Buschle, President Biologics, Photo: Paul Noronha
Note on TRPA1
TRPA1 is an ion channel expressed on peripheral and spinal sensory neurons and it mediates pain signal transmission. It functions as a cellular sensor for detecting painful mechanical, biochemical and thermal stimuli that cause sensory nerve hyperactivity during chronic pathologies including chronic pain, inflammation, itch and cough. TRPA1 receptor is shown to induce pain hypersensitivity in animal models of diabetic neuropathic pain and its blockade attenuates pain hypersensitivity as well as later loss of the nerve fibers and their function. GRC 17536 is a potent, selective and first in class antagonist of TRPA1 receptor. Preclinical studies have demonstrated its effectiveness in animal models of neuropathic and inflammatory pain including the peripheral diabetic neuropathic pain, osteoarthritic pain, postoperative pain and chemotherapy induced pain which supports potential utility of TRPA1 blockade in therapeutic pain management.
About Glenmark Pharmaceuticals Ltd
Glenmark Pharmaceuticals Ltd. (GPL) is a research-driven, global, integrated pharmaceutical company and ranked among the top 80 Pharma & Biotech companies of the world in terms of revenues as per SCRIP 100 Rankings. Glenmark is a leading player in the discovery of new molecules both NCEs and NBEs. Glenmark has several molecules in various stages of clinical development and primarily focused in the areas of Inflammation, Pain and Oncology. The company has significant presence in branded formulations across emerging economies including India. Its subsidiary, Glenmark Generics Limited services the requirements of the US and Western Europe markets.

CARMEGLIPTIN………….a DPP-4 inhibitor

(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
(2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one
813452-14-1 (di-HCl)
916069-91-5 (mono-HCl)
Roche…….innovator

CARMEGLIPTIN
813452-18-5
(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one
(S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| 分子式: | C20H28FN3O3 |
| 分子量: | 377 |
813452-18-5, Carmegliptin, R-1579;carmegliptin, Carmegliptin (USAN/INN), SureCN419289, UNII-9Z723VGH7J, CHEMBL591118, CHEBI:699093, Ro-4876904, D08631, R-1579, B1Q
Type 2 diabetes is a chronic, progressive metabolic disease, affecting about 4% of the world population. The main goal of the management of type 2 diabetes is to achieve glycemic control as close to the nondiabetic range as practicable, in order to reduce the risk of late-stage complications.However, the therapeutic effect provided by existing medications is often not sustainable, since the multi-organ defects responsible for the disease are only insufficiently addressed.
Dipeptidyl peptidase-IV (DPP-IV) inhibitors have emerged as a new therapeutic option to treat type 2 diabetes.
Their rapid rise in popularity is due to the favourable safety profile (no hypoglycemia, no weight gain, no gastrointestinal problems—typical side effects associated with established anti-diabetic agents). DPP-IV is a ubiquitous serine protease, the inhibition of which prevents the degradation of glucagon-like peptide 1 (GLP-1). The resulting higher levels of GLP-1 have a beneficial impact on major players involved in the pathogenesis of type 2 diabetes: β-cells, liver, α-cells, gut, and brain.
Long-term studies with DPP-IV inhibitors in patients are underway in order to confirm the safety and sustainability of these effects, and, in particular, their ability to prevent the progressive loss of β-cell function.
SYNTHESIS

aReagents and conditions: a) HCO2Me, Δ; b) POCl3, MeCN; c) HO2CCH2CO2Et, neat, 120 °C; d) ethyl acrylate, neat; e) t-BuOK, neat (5 steps); f) NH4OAc, MeOH; g) NaBH4, TFA, THF; h) Boc2O, CH2Cl2; i) KOH, aq THF; j) DPPA, Et3N, TMSCH2CH2OH, PhMe, 80 °C; k) Et4NF, MeCN; l) chiral HPLC; m) Et3N, CH2Cl2; n) NaH, DMF; o) HCl, dioxane; p) HCl, 2-PrOH.

Scheme 2.
Reagents and conditions: (a) NH4OAc, MeOH, rt, 95%; (b) NaBH4, TFA, THF, 0 °C; (c) Boc2O, CH2Cl2, 83% over 2 steps; (d) KOH, aq THF, rt; (e) DPPA, Et3N, 2-(trimethylsilyl)ethanol, toluene, 80 °C; (f) Et4NF, CH3CN, 50 °C, 56% over 3 steps; (g) Et3N, CH2Cl2, (h) NaH, cat. NaI, DMF; (i) HCl, 1,4-dioxane.
Carmegliptin (2.70) is an anti-diabetes drug which is currently in late stage clinical trials. It represents a further structural advancement from the other existing marketed drugs in this class, sitagliptin (2.71, Januvia) and vildagliptin (2.72, Zomelis, Figure 7). These compounds are all members of the dipeptidyl peptidase 4 class (DPP-4), a transmembrane protein that is responsible for the degradation of incretins; hormones which up-regulate the concentration of insulin excreted in a cell. As DPP-4 specifically cleaves at proline residues, it is unsurprising that the members of this drug class exhibit an embedded pyrrolidine ring (or mimic) and additional decoration (a nitrile or fluorinated alkyl substituent is present in order to reach into a local lipophilic pocket). One specific structural liability of the 2-cyano-N-acylpyrrolidinyl motif (2.73) is its inherent susceptibility towards diketopiperazine formation (2.74, Scheme 29) [80], however, one way to inhibit this transformation is to position a bulky substituent on the secondary amine nucleophile as is the case in vildagliptine (2.72).
![[1860-5397-9-265-7]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-7.png)
Figure 7: Structures of DPP-4 inhibitors of the gliptin-type.
![[1860-5397-9-265-i29]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i29.png)
Scheme 29: Formation of inactive diketopiperazines from cis-rotameric precursors.
A single crystal X-ray structure of carmegliptin bound in the human DPP-4 active site has been published indicating how the fluoromethylpyrrolidone moiety extends into an adjacent lipophilic pocket [81]. Additional binding is provided by π–π interaction between the aromatic substructure and an adjacent phenylalanine residue as well as through several H-bonds facilitated by the adjacent polar substituents (Figure 8).
![[1860-5397-9-265-8]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-8.png)
Figure 8: Co-crystal structure of carmegliptin bound in the human DPP-4 active site (PDB 3kwf).
The reported synthesis of carmegliptin enlists a Bischler-Napieralski reaction utilising the primary amine 2.76 and methyl formate to yield the initial dihydroquinoline 2.77 as its HCl salt (Scheme 30) [82]. This compound was next treated with 3-oxoglutaric acid mono ethyl ester (2.78) in the presence of sodium acetate. Decarboxylation then yields the resulting aminoester 2.79 which was progressed through an intramolecular Mannich-type transformation using aqueous formaldehyde to allow isolation of enaminoester 2.80 after treatment of the intermediate with ammonium acetate in methanol.
The next step involves a very efficient crystallisation-induced dynamic resolution of the racemic material using the non-natural (S,S)-dibenzoyl-D-tartaric acid ((+)-DBTA). It is described that the desired (S)-enantiomer of compound 2.81 can be isolated in greater than 99% ee and 93% overall yield. This approach is certainly superior to the original separation of the two enantiomers (at the stage of the final product) by preparative chiral HPLC that was used in the discovery route (albeit it should be noted that both enantiomers were required for physiological profiling at the discovery stage).
Next, a 1,2-syndiastereoselective reduction of enaminoester 2.81 occurs with high diastereocontrol imposed by the convexed presentation of the substrate for the formal conjugate addition and subsequent protonation steps. This is followed by Boc-protection and interconversion of the ethyl ester to its amide derivative 2.82 in 80% overall yield for this telescoped process. The primary amide in 2.82 was then oxidised via a modern variant of the classical Hoffmann rearrangement using phenyliodine diacetate (PIDA).
Following extensive investigation it was found that slowly adding this reagent in a mixture of acetonitrile/water to a suspension of amide 2.82 and KOH gave clean conversion to the amine product in high yield. This new procedure was also readily scalable offering a cleaner, safer and more reliable transformation when compared to other related rearrangement reactions. During a further telescoped procedure amine 2.83 was treated with lactone 2.84 to regenerate the corresponding lactam after mesylate formation. Finally, removal of the Boc-group with aqueous hydrochloric acid furnished carmegliptin as its HCl salt.
![[1860-5397-9-265-i30]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i30.png)
Scheme 30: Improved route to carmegliptin.
- Peters, J.-U. Curr. Top. Med. Chem. 2007, 7, 579–595……………..80
- Mattei, P.; Boehringer, M.; Di Gorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U.Bioorg. Med. Chem. Lett. 2010, 20, 1109–1113. doi:10.1016/j.bmcl.2009.12.024………..81
- Albrecht, S.; Adam, J.-M.; Bromberger, U.; Diodone, R.; Fettes, A.; Fischer, R.; Goeckel, V.; Hildbrand, S.; Moine, G.; Weber, M. Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207……….82
………………………………………………………………………………………………………………..
Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207
http://pubs.acs.org/doi/full/10.1021/op2000207

A short and high-yielding synthesis of carmegliptin (1) suitable for large-scale production is reported. The tricyclic core was assembled efficiently by a decarboxylative Mannich addition−Mannich cyclization sequence. Subsequent crystallization-induced dynamic resolution of enamine 7 using (S,S)-dibenzoyltartaric acid was followed by diastereoselective enamine reduction to give the fully functionalized tricyclic core with its three stereogenic centers. The C-3 nitrogen was introduced by Hofmann rearrangement of amide 28, and the resulting amine 10was coupled with (S)-fluoromethyl lactone 31. Following cyclization to lactam 13 and amine deprotection, 1 was obtained in 27−31% overall yield with six isolated intermediates.
Preparation of (2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride (1) CARMEGLIPTIN
A suspension of carbamate 13 (136 kg, 285 mol) in a mixture of H2O (112 kg) and acetone (122 kg) was treated at 50 °C within 60 min with 37% aq HCl (98.0 kg). After 90 min at 47−52 °C the solution was polish filtered through a 5 μm filter. The first reactor and the transfer lines were washed with a hot (47−52 °C) mixture of H2O (13.0 kg) and acetone (116 kg). The filtrate was cooled to 25 °C and treated at this temperature within 80 min with acetone (1600 kg) whereupon the product crystallized out. The resulting suspension was stirred for 1 h at 25 °C and subsequently centrifuged. The crystals were washed in two portions with acetone (391 kg) and dried at 50 °C and <30 mbar until constant weight to afford 122.4 kg (95%) of the title compound as colorless crystals with an assay (HPLC) of 98.8% (w/w).
1H NMR (400 MHz, D2O) δ 2.11−2.22 (m, 1H); 2.45 (dd, J = 17.6 Hz, 6.7 Hz; 1H); 2.76 (dd, J = 17.6 Hz, 9.55 Hz, 1H); 2.90−3.05 (m, 1H); 3.08−3.19 (m, 2H); 3.24−3.36 (m, 1H); 3.43 (dd, J = 9.8 Hz, 5.75 Hz, 1H); 3.49−3.58 (m, 1H); 3.70−3.84 (m, 4H); 3.87 (s, 3H); 3.88 (s, 3H); 4.12 (td, J = 11.6 Hz, 4.5 Hz, 1H); 4.45−4.55 (m, 1H); 4.56−4.68 (m, 3H); 6.91 (s, 1H), 6.95 (s, 1H).
IR (cm−1): 3237, 2925, 1682, 496, 478.
MS (ESI): m/z 378.3 ([M + H]+ (free amine)).
Anal. Calcd for C20H30Cl2FN3O3: C, 53.34; H, 6.71; N, 9.33; Cl, 15.74; F 4.22; O, 10.66. Found: C, 53.04; H, 6.43; N, 9.45; Cl, 15.66; F, 4.29; O, 11.09.
REF FOR ABOVE
Mattei, P.; Böhringer, M.; Di Giorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Kocer, B.; Kuhn, B.; Löffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U. Bioorg. Med. Chem. Lett. 2010, 20, 1109
Böhringer, M.; Kuhn, B.; Lübbers, T.; Mattei, P.; Narquizian, R.; Wessel,H. P. (F. Hoffmann-La Roche AG). U.S. Pat. Appl. 2004/0259902, 2004.
…………………………………………………..
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
Bioorg Med Chem Lett 2010, 20(3): 1109
Bioorg Med Chem Lett 2010, 20(3): 1109
http://www.sciencedirect.com/science/article/pii/S0960894X09017296
-
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
- Pages 1109-1113
- Patrizio Mattei, Markus Boehringer, Patrick Di Giorgio, Holger Fischer, Michael Hennig, Joerg Huwyler, Buelent Koçer, Bernd Kuhn, Bernd M. Loeffler, Alexander MacDonald, Robert Narquizian, Etienne Rauber, Elena Sebokova, Urs Sprecher
-

 Scheme 3.
Scheme 3.Reagents and conditions: (a) preparative HPLC (Chiralpak® AD column), heptane/2-propanol 85:15, 37% (b) BH3.Me2S, THF, 0 °C; (c) (MeOCH2CH2)2NSF3, CH2Cl2, 67% (2 steps); (d), SOCl2, ZnCl2, 80 °C, 72 h, 61%; (e) Et3N, CH2Cl2; (f) NaH, DMF, 56% (2 steps); (g) HCl, 1,4-dioxane, 91%; (h) HCl, 2-propanol, 86%.
The synthesis of 8p is outlined ABOVE and required the enantiopure building blocks (S,S,S)-5 and 12. (S,S,S)-5 was obtained from the racemate by preparative chiral HPLC. Acid chloride 12 was prepared starting from (S)-paraconic acid (9). Reduction of 9 with borane–dimethyl sulfide complex afforded hydroxymethyl lactone 10. Since 10 is known to racemise rather readily, it was immediately treated with bis(2-methoxyethyl)aminosulfur trifluoride, thereby affording fluoromethyl lactone 11. This was converted to 12 by reaction with thionyl chloride in the presence of zinc chloride. The (S)-4-fluoromethyl-pyrrolidinone 8p was isolated as the dihydrochloride salt, a highly water soluble white crystalline solid, mp >275 °C.
…………………………………………………….
US 2013109859
The most preferred product is (2S,3S,11bS)-2-tert.-Butoxycarbonylamino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H pyrido[2,1-a]isoquinoline-3-carboxylic acid amide having the following structure:

It has been found that during the amidation of the ester epimerization takes place at position 3 and thus the 3R-epimer of the formula IVb is transformed to a larger extent in the 3S-epimer of formula V.

e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
A 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
…………………………………………………….
US 2008071087







(2S,3S,11bS)-(3-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester (8)
Example 8
Transformation of (2S,3S,11bS)-(3-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl) ]-carbamic acid tert-butyl ester into (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl) -4-fluoromethyl-pyrrolidin-2-one.a)
Preparation of 4-fluoromethyl-5H-furan-2-oneA 6 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 500 g (4.38 mmol) 4-hydroxymethyl-5H-furan-2-one and 2.0 L dichloromethane. The solution was cooled to −10° C. and 1.12 kg (4.82 mol) bis-(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) was added during 50 min, maintaining the temperature at −5 to −10° C. with a cooling bath. During the addition a yellowish emulsion formed, which dissolved to an orange-red solution after completed addition. This solution was stirred for 1.5 h at 15-20° C., then cooled to −10° C. A solution of 250 ml water in 1.00 L ethanol was added during 30 min, maintaining the temperature between −5 and −10° C., before the mixture was allowed to reach 15-20° C. It was then concentrated in a rotatory evaporator to a volume of ca. 1.6 L at 40° C./600-120 mbar. The residue was dissolved in 2.0 L dichloromethane and washed three times with 4.0 L 1N hydrochloric acid. The combined aqueous layers were extracted three times with 1.4 L dichloromethane. The combined organic layers were evaporated in a rotatory evaporator to give 681 g crude product as a dark brown liquid. This material was distilled over a Vigreux column at 0.1 mbar, the product fractions being collected between 71 and 75° C. (312 g). This material was re-distilled under the same conditions, the fractions being collected between 65 and 73° C., to give 299 g 4-fluoromethyl-5H-furan-2-one (58% yield; assay: 99%).MS: m/e 118 M+, 74,59,41.b) Preparation of (S)-4-fluoromethyl-dihydro-furan-2-oneA 2 L autoclave equipped with a mechanical stirrer was charged with a solution of 96.0 g 4-fluoromethyl-5H-furan-2-one (8.27×10−1 mol) in 284 mL methanol. The autoclave was sealed and pressurized several times with argon (7 bar) in order to remove any traces of oxygen. At ˜1 bar argon, a solution of 82.74 mg Ru(OAc)2((R)-3,5-tBu-MeOBlPHEP) (6.62×10−5 mol) (S/C 12500) in 100 mL methanol was added under stirring from a catalyst addition device previously charged in a glove box (O2 content <2 ppm) and pressurized with argon (7 bar). The argon atmosphere in the autoclave was replaced by hydrogen (5 bar). At this pressure, the reaction mixture was stirred (˜800 rpm) for 20 h at 30° C. and then removed from the autoclave and concentrated in vacuo. The residue was distilled to afford 91.8 g (94%) (S)-4-fluoromethyl-dihydro-furan-2-one. The chemical purity of the product was 99.7% by GC-area.c) Preparation of (2S,3S,11bS)-3-(3-Fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 50 g (128 mmol) (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester, 500 mL toluene and 2.51 g (25.6 mmol) 2-hydroxypyridine. To this slightly brownish suspension, 22.7 g (192 mmol) of (S)-4-fluoromethyl-dihydro-furan-2-one was added dropwise at RT. No exothermy was observed during the addition. The dropping funnel was rinsed portionwise with totally 100 mL toluene. The suspension was heated to reflux, whereas it turned into a dear solution starting from 60° C., after 40 min under reflux a suspension formed again. After totally 23 h under reflux, the thick suspension was cooled to RT, diluted with 100 mL dichloromethane and stirred for 30 min at RT. After filtration, the filter cake was washed portionwise with totally 200 mL toluene, then portionwise with totally 100 mL dichloromethane. The filter cake was dried at 50° C./10 mbar for 20 h, to give 60.0 g product (94% yield; assay: 100%).


 UNDESIRED
UNDESIRED

 DESIRED
DESIRED
 UNDESIRED
UNDESIRED


MS: m/e 496 (M+H)+, 437.
d) Preparation of (2S,3S,11bS)-3-((4S)-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel, a cooling bath and a nitrogen inlet was charged with 28 g (56.5 mmol) of (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino) -9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and 750 mL THF. The mixture was cooled to 0° C. and a solution of 6.17 mL (79 mmol) methanesulfonic acid in 42 mL THF was added during 10 min, maintaining the temperature at 0-5° C. At 0° C. a solution of 12.6 mL (90.2 mmol) triethylamine in 42 mL THF was added during 15 min. The resulting suspension was stirred for 80 min at 0-5° C., whereas it became gradually thicker. Then 141 mL (141 mmol) 1 M lithium-bis(trimethylsilyl)amide were added to the mixture during 15 min, whereas the suspension dissolved. The solution was allowed to reach RT during 60 min under stirring. 500 mL water was added without cooling, the mixture was extracted and the aqueous phase was subsequently extracted with 500 mL and 250 mL dichloromethane. The organic layers were each washed with 300 mL half saturated brine, combined and evaporated on a rotatory evaporator. The resulting foam was dissolved in 155 mL dichloromethane, filtered and again evaporated to give 30.5 g crude product as a slightly brownish foam. This material was dissolved in 122 mL methanol, resulting in a thick suspension, which dissolved on heating to reflux. After 20 min of reflux the solution was allowed to gradually cool to RT during 2 h, whereas crystallization started after 10 min. After 2 h the suspension was cooled to 0° C. for 1 h, followed by −25° C. for 1 h. The crystals were filtered off via a pre-cooled glass sinter funnel, washed portionwise with 78 mL TBME and dried for 18 h at 45° C./20 mbar, to give 21.0 g of the title product as white crystals (77% yield; assay: 99.5%).
MS: m/e 478 (M+H)+, 437, 422.
e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one dihydrochlorideA 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
These compounds are useful intermediates for the preparation of DPP-IV inhibitors as disclosed in PCT International Patent Appl. WO 2005/000848. More preferably, the invention relates to a process for the preparation of (2S,3S,11bS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester.
XXXXXXX
According to still another embodiment (Scheme 2, below) the (S)-4-fluoromethyl-dihydro-furan-2-one (VII) is directly coupled with the amino-pyrido[2,1-a]isoquinoline derivative (VI) to form the hydroxymethyl derivative of the pyrido[2,1-a]isoquinoline (VIII), which is then subsequently cyclized to the fluoromethyl-pyrrolidin-2-one derivative (IX). The latter can be deprotected to yield the desired pyrido[2,1-a]isoquinoline derivative (I).

In a further preferable embodiment, the process for the preparation of (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one or of a pharmaceutically acceptable salt thereof comprises the subsequent steps:
- e) coupling of the (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (amine of formula VI, wherein R2 and R3 are methoxy, R4 is hydrogen and Prot is Boc) with the (S)-4-fluoromethyl-dihydro-furan-2-one of formula

- f) cyclization of the obtained (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester in the presence of a base, and
- g) deprotecting the obtained (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester.

………………………………………………………….
PATENT
http://www.google.com.ar/patents/US7122555?cl=pt-PT
Example 23
RACEMIC
1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

a) 4-Fluoromethyl-dihydro-furan-2-one
A solution of 4-hydroxymethyl-dihydro-furan-2-one (Tetrahedron 1994, 50, 6839; 1.02 g, 8.78 mmol) and bis(2-methoxyethyl)aminosulfur trifluoride (3.88 g, 17.6 mmol) in chloroform (4.4 mL) was stirred at 40° C. for 1 h, then poured onto ice and partitioned between sat. aq. sodium hydrogencarbonate solution and dichloromethane. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, heptane-ethyl acetate gradient) afforded the title compound (576 mg, 56%). Colourless liquid, MS (EI) 118.9 (M+H)+.
b) 3-Chloromethyl-4-fluoro-butyryl chloride
A mixture of 4-fluoromethyl-dihydro-furan-2-one (871 mg, 7.37 mmol), thionyl chloride (4.39 g, 36.9 mmol), and zinc chloride (60 mg, 0.44 mmol) was stirred 72 h at 80° C., then excess thionyl chloride was removed by distillation. Kugelrohr distillation of the residue (85° C., 0.2 mbar) afforded the title compound (450 mg, 35%). Colourless liquid, 1H-NMR (300 MHz, CDCl3): 4.65–4.55 (m, 1H), 4.50–4.40 (m, 1H), 3.70–3.60 (m, 2H), 3.25–3.05 (m, 2H), 2.80–2.60 (m, 1H).
c) (RS,RS,RS)-[3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (RS,RS,RS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 5b) and 3-chloromethyl-4-fluoro-butyryl chloride. White solid, MS (ISP) 514.5 (M+H)+.
d) (RS,RS,RS)-[3-(4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5d from (RS,RS,RS)-[3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Off-white foam, MS (ISP) 478.5 (M+H)+.
e) 1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
The title compound was produced in accordance with the general method of Example 1e from (RS,RS,RS)-[3-(4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Light yellow oil, MS (ISP) 378.5 (M+H)+.
Examples 28 and 29
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
 UNDESIRED
UNDESIREDand
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

The title compounds were produced from 1-((RS,RS,RS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one (Example 23) by chromatographic separation (SiO2, CH2Cl2/MeOH/NH4OH 80:1:0.2, then 95:5:0.25).
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Yellow oil, Rf=0.45 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Light yellow solid, Rf=0.40 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
Example 30
(S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one Dihydrochloride
 DESIRED
DESIREDa) [(S,S,S)-3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (S,S,S)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 16b) and 3-chloromethyl-4-fluoro-butyryl chloride (Example 23b). Off-white solid.
b) [(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
Sodium hydride (55–65% dispersion in oil, 1.14 g, 28.5 mmol) was added to a suspension of [(S,S,S)-3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (6.72 g, 13.1 mmol) in N,N-dimethylformamide (95 mL) at r.t., then after 1 h the reaction mixture was poured onto ice and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, cyclohexane/2-propanol 4:1) afforded [(S,S,S)-3-((S)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 38%) and the epimer, [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.73 g, 44%).
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.6 (SiO2, cyclohexane/2-propanol 1:1).
[(S,S,S)-3-((R)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.4 (SiO2, cyclohexane/2-propanol 1:1).
-
- c) (S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 5.02 mmol) was converted to (S)-1-((S,S,S)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one in accordance with the general method of Example 1e. The product was dissolved in 2-propanol (10 mL) and treated with hydrogen chloride (5–6 M in 2-propanol, 37 mL). The suspension formed was stirred for 64 h at r.t., then the precipitate was collected by filtration and dried, to afford the title compound (2.04 g, 91%). White solid, m.p. >300° C.
Example 31(R)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
 UNDESIRED
UNDESIREDThe title compound was produced in accordance with the general method of Example 30c from [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (Example 30b). White solid, m.p. >300° C.

DR ANTHONY MELVIN CRASTO
MY BLOGS ON MED CHEM
New Drug Approvals ALL ABOUT DRUGS WORLD DRUG TRACKER MEDICINAL CHEMISTRY INTERNATIONALDRUG SYN INTERNATIONALSCALEUP OF DRUGS ALL FOR DRUGS ON WEBEUREKAMOMENTS
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS, BRAVESITES, GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO, YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN, FRIENDFEED, NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING
FDA approves AstraZeneca’s constipation drug Movantik
September 16, 2014
The US Food and Drug Administration has approved AstraZeneca’s Movantik for opioid-induced constipation in adults with chronic non-cancer pain.
Movantik (naloxegol), an oral once-a-day treatment licensed from Nektar Therapeutics, belongs to a class of drugs called peripherally-acting mu-opioid receptor antagonists, which are used to decrease the constipating effects of opioids. The drug’s safety and effectiveness were established in two trials of 1,352 participants who had taken opioids for at least four weeks for non-cancer related pain and had opioid-induced constipation.
Read more at: http://www.pharmatimes.com/Article/14-09-16/FDA_approves_AstraZeneca_s_constipation_drug_Movantik.aspx#ixzz3DdGiFse8
Acai counteracts oxidative stress, lengthens lifespan in fruit flies
24 AUG 2012
Bewildered by the array of antioxidant fruit juices on display in the supermarket and the promises they make? To sort out the antioxidant properties of fruits and berries, scientists at Emory University School of Medicine turned to fruit flies for help.
They found that a commercially available acai berry product can lengthen the lives of fruit flies, when the flies’ lives are made short through additional oxidative stress. Under certain conditions (a simple sugar diet) acai supplementation could triple flies’ lifespans, from eight to 24 days. Acai could also counteract the neurotoxic effects of the herbicide paraquat on the flies.
The results were recently published by the journal Experimental Gerontology, which awarded the paper its inaugural “Outstanding paper” prize. The lead author is Alysia Vrailas-Mortimer, a postdoctoral fellow in Emory University School of Medicine’s Department of Cell Biology.
Vrailas-Mortimer says she didn’t start out focusing on acai…
View original post 645 more words
AMIODARONE


Amiodarone
CAS : 1951-25-3
(2-Butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
2-butyl-3-benzofuranyl-4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl ketone; 2-butyl-3-[3,5-diiodo-4-(b-diethylaminoethoxy)benzoyl]benzofuran
Molecular Formula: C25H29I2NO3
Molecular Weight: 645.31
Percent Composition: C 46.53%, H 4.53%, I 39.33%, N 2.17%, O 7.44%
In December 1985, amiodarone was approved by the FDA for the treatment of arrhythmias.[6] This makes amiodarone one of the few drugs approved by the FDA without rigorous randomized clinical trials.
Amiodarone is an antiarrhythmic agent used for various types of cardiac dysrhythmias, both ventricular and atrial. It was discovered in 1961. Despite relatively common side-effects, it is used in arrhythmias that are otherwise difficult to treat with medication.

A more recent synthesis of amiodarone reports the cyclisation of α-phenoxyhexanal 389 under acidic conditions to yield the substituted benzofuran 390 (Scheme 76). A Friedel–Crafts acylation next introduces the aryl ring at the 3-position. Demethylation, iodination and a final alkylation with a diethylaminoethane fragment yields amiodarone [115-117].
- 115 Witczak, M.; Kwiecień, H. Synth. Commun. 2005, 35, 2223–2230. doi:10.1080/00397910500182747
Return to citation in text: [1] - Wang, Z. J. Synthetic Process for 2-Butyl-3-(hydroxy-3,5-diiodobenzoyl)-benzofuran. Chin. Patent 1,858,042, Nov 8, 2006……….116
Return to citation in text: [1] - Ha, H. R.; Stieger, B.; Grassi, G.; Altorfer, H. R.; Follath, F. Eur. J. Clin. Pharmacol. 2000, 55, 807–814.doi:10.1007/s002280050701….117
![[1860-5397-7-57-i76]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i76.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 76: Synthesis of amiodarone……….http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S76
Literature References:
Benzofuran derivative with multiple electrophysiological effects. Prepn: FR 1339389; R. Tondeur, F. Binon,US 3248401 (1963, 1966 to Soc. Belge l’Azote Prod. Chim. Marly).
Physicochemical properties: M. Bonati et al., J. Pharm. Sci. 73,829 (1984).
HPLC determn in plasma: M. De Smet, D. L. Massart, J. Pharm. Biomed. Anal. 6, 277 (1988).
Comprehensive description: T. A. Plomp, Anal. Profiles Drug Subs. 20, 1-120 (1991).
Review of pharmacology, clinical efficacy and safety: M. Chow, Ann. Pharmacother. 30, 637-643 (1996); B. N. Singh, Clin. Cardiol. 20, 608-618 (1997).
Clinical trial in cardiac resuscitation: P. J. Kudenchuk et al., N. Engl. J. Med. 341, 871 (1999); to prevent atrial fibrillation: D. Roy et al., ibid. 342, 913 (2000).
External links
- Siddoway LA (December 2003). “Amiodarone: guidelines for use and monitoring”. Am Fam Physician 68 (11): 2189–96. PMID 14677664.
- Amiodarone (MedicineNet.com)
- Amiodarone (FamilyPracticeNotebook.com)
- Amiodarone (The WorldWide Intensivist)
- U.S. National Library of Medicine: Drug Information Portal – Amiodarone
- Amiodarone (FDA MedWatch)
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2-{4-[(2-butyl-1-benzofuran-3-yl)carbonyl]-2,6-diiodophenoxy}ethyl)diethylamine | |
| Clinical data | |
| Trade names | Cordarone, Nexterone |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a687009 |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | oral or intravenous |
| Pharmacokinetic data | |
| Bioavailability | 20–55% |
| Metabolism | Liver |
| Half-life | 58 days (range 15-142 days) |
| Excretion | Primarily Hepatic and Biliary |
| Identifiers | |
| CAS number | 1951-25-3 |
| ATC code | C01BD01 |
| PubChem | CID 2157 |
| IUPHAR ligand | 2566 |
| DrugBank | DB01118 |
| ChemSpider | 2072 |
| UNII | N3RQ532IUT |
| KEGG | D02910 |
| ChEBI | CHEBI:2663 |
| ChEMBL | CHEMBL633 |
| Chemical data | |
| Formula | C25H29I2NO3 |
| Mol. mass | 645,31 g/mol |
Derivative Type: Hydrochloride
CAS Registry Number: 19774-82-4
Manufacturers’ Codes: L-3428
Trademarks: Amiodar (Sanofi Winthrop); Ancaron (Taisho); Cordarex (Sanofi Winthrop); Cordarone (Wyeth); Ortacrone (Sanofi Winthrop); Pacerone (Upsher-Smith); Tachydaron (AWD); Trangorex (Sanofi Winthrop)
Molecular Formula: C25H29I2NO3.HCl
Molecular Weight: 681.77
Percent Composition: C 44.04%, H 4.44%, I 37.23%, N 2.05%, O 7.04%, Cl 5.20%
Properties: Crystalline powder, mp 156°. Also reported as crystals from acetone, mp 159 ±2° (Bonati). Soly at 25° (g/100ml): chloroform 44.51; methylene chloride 19.20; methanol 9.98; ethanol 1.28; benzene 0.65; tetrahydrofuran 0.60; acetonitrile 0.32; 1-octanol 0.30; ether 0.17; 1-propanol 0.13; water 0.07; hexane 0.03 petroleum ether 0.001. Sparingly sol in isopropanol; slightly sol in acetone, dioxane, and carbon tetrachloride. pH (5% soln) 3.4-3.9. pKa (25°C) 6.56 ±0.06. uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10).
Melting point: mp 156°; mp 159 ±2° (Bonati)
pKa: pKa (25°C) 6.56 ±0.06
Absorption maximum: uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10)
Therap-Cat: Antiarrhythmic (class III).
Keywords: Antiarrhythmic.
AMIODARONE
|
|
Title: Amiodarone
CAS Registry Number: 1951-25-3
CAS Name: (2-Butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
Additional Names: 2-butyl-3-benzofuranyl-4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl ketone; 2-butyl-3-[3,5-diiodo-4-(b-diethylaminoethoxy)benzoyl]benzofuran
Molecular Formula: C25H29I2NO3
Molecular Weight: 645.31
Percent Composition: C 46.53%, H 4.53%, I 39.33%, N 2.17%, O 7.44%
Literature References: Benzofuran derivative with multiple electrophysiological effects. Prepn: FR 1339389; R. Tondeur, F. Binon, US 3248401 (1963, 1966 to Soc. Belge l’Azote Prod. Chim. Marly). Physicochemical properties: M. Bonati et al., J. Pharm. Sci. 73, 829 (1984). HPLC determn in plasma: M. De Smet, D. L. Massart, J. Pharm. Biomed. Anal. 6, 277 (1988). Comprehensive description: T. A. Plomp, Anal. Profiles Drug Subs. 20, 1-120 (1991). Review of pharmacology, clinical efficacy and safety: M. Chow, Ann. Pharmacother. 30, 637-643 (1996); B. N. Singh, Clin. Cardiol. 20, 608-618 (1997). Clinical trial in cardiac resuscitation: P. J. Kudenchuk et al., N. Engl. J. Med. 341, 871 (1999); to prevent atrial fibrillation: D. Roy et al., ibid. 342, 913 (2000).
Derivative Type: Hydrochloride
CAS Registry Number: 19774-82-4
Manufacturers’ Codes: L-3428
Trademarks: Amiodar (Sanofi Winthrop); Ancaron (Taisho); Cordarex (Sanofi Winthrop); Cordarone (Wyeth); Ortacrone (Sanofi Winthrop); Pacerone (Upsher-Smith); Tachydaron (AWD); Trangorex (Sanofi Winthrop)
Molecular Formula: C25H29I2NO3.HCl
Molecular Weight: 681.77
Percent Composition: C 44.04%, H 4.44%, I 37.23%, N 2.05%, O 7.04%, Cl 5.20%
Properties: Crystalline powder, mp 156°. Also reported as crystals from acetone, mp 159 ±2° (Bonati). Soly at 25° (g/100ml): chloroform 44.51; methylene chloride 19.20; methanol 9.98; ethanol 1.28; benzene 0.65; tetrahydrofuran 0.60; acetonitrile 0.32; 1-octanol 0.30; ether 0.17; 1-propanol 0.13; water 0.07; hexane 0.03 petroleum ether 0.001. Sparingly sol in isopropanol; slightly sol in acetone, dioxane, and carbon tetrachloride. pH (5% soln) 3.4-3.9. pKa (25°C) 6.56 ±0.06. uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10).
Melting point: mp 156°; mp 159 ±2° (Bonati)
pKa: pKa (25°C) 6.56 ±0.06
Absorption maximum: uv max (methanol): 208, 242 nm (E1%1cm 662 ±8, 623 ±10)
Therap-Cat: Antiarrhythmic (class III).
Keywords: Antiarrhythmic.
|
Amiodarone
-
- ATC:C01BD01
- Use:antiarrhythmic
- Chemical name:(2-butyl-3-benzofuranyl)[4-[2-(diethylamino)ethoxy]-3,5-diiodophenyl]methanone
- Formula:C25H29I2NO3
- MW:645.32 g/mol
- CAS-RN:1951-25-3
- InChI Key:IYIKLHRQXLHMJQ-UHFFFAOYSA-N
- InChI:InChI=1S/C25H29I2NO3/c1-4-7-11-22-23(18-10-8-9-12-21(18)31-22)24(29)17-15-19(26)25(20(27)16-17)30-14-13-28(5-2)6-3/h8-10,12,15-16H,4-7,11,13-14H2,1-3H3
- EINECS:217-772-1
- LD50:178 mg/kg (M, i.v.); >4 g/kg (M, p.o.)
Derivatives
hydrochloride
- Formula:C25H29I2NO3 • HCl
- MW:681.78 g/mol
- CAS-RN:19774-82-4
Substance Classes
Synthesis Path
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Cordarex | Sanofi-Aventis | |
| Cornaron | TAD Pharma | ||
| F | Corbionax Ge | Winthrop/Sanofi-Aventis | |
| Cordarone | Sanofi-Aventis | ||
| GB | Cordaron | Sanofi-Aventis | |
| I | Amiodar | Sigma-Tau | |
| Cordarone | Sanofi-Aventis | ||
| J | Ancaron | Sanofi-Aventis | |
| USA | Cordarone | Wyeth-Ayerst | as hydrochloride |
Formulations
- inj. sol. 150 mg/3ml; tabl. 100 mg, 200 mg
References
-
- FR 1 339 389 (Labaz; appl. 22.11.1962).
- US 3 248 401 (Labaz; 26.4.1966; D-prior. 24.11.1961).
-
2-butylbenzofuran:
- Buu-Hoï, N.P. et al.: J. Chem. Soc. (JCSOA9) 1964, 173.
PATENT
CN109053652-PREPARATION METHOD OF AMIODARONE HYDROCHLORIDE INTERMITTENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN235615504&_cid=P11-KL0AU0-06410-1
| Amiodarone hydrochloride is currently the most widely used antiarrhythmic drug. In addition, amiodarone hydrochloride has become the first choice for long-term medication for patients with arrhythmia due to its stable curative effect and minor side effects. In today’s society, people’s work rhythm is constantly increasing, the pressure they face is increasing, the number of patients with cardiovascular diseases is increasing, the demand for anti-arrhythmic drugs is greatly increasing, and the market share of amiodarone hydrochloride is bound to continue to increase. |
| 2-Butyl-3-(4-hydroxybenzoyl)benzofuran is an important intermediate of amiodarone hydrochloride. Patent CN104262304 uses compounds 1 and 2 as starting materials, and the resulting compound 3 is reacted with sodium methoxide as a base and toluene as a solvent to form compound 5; compound 5 is reacted with compound 7 under Lewis acid conditions to form compound 8; compound 8 Hydrolyzed under Lewis acid conditions, the reaction produces 2-butyl-3-(4-hydroxybenzoyl)benzofuran 9. |
| |
| This synthetic route has the following shortcomings: Compound 3 can be directly obtained without hydrolysis under the condition of sodium methoxide as base and toluene as solvent. However, the difficulty of demethylation is much greater than that of decarboxylation, resulting in the formation of compound 5. The conversion rate of the cyclization reaction is low, the by-products are many, and the separation is difficult; resulting in low total yield and increased cost. |
| The synthetic route reported in patent CN107382925, except that the route for preparing compound 5 from compound 3 is different, other reaction routes are the same, but the reaction conditions are different. In this reaction route, the aldehyde group of compound 3 is first protected with trimethyl orthoformate to produce compound 0, and then compound 0 is hydrolyzed to produce compound 4. Compound 4 is cyclized under the catalysis of p-toluenesulfonyl chloride to obtain compound 5. |
| |
| This route has the following disadvantages: when compound 3 is prepared to compound 5, trimethyl orthoformate is added to protect the aldehyde group, then the ester group is first hydrolyzed to form an acid, and then the protective group of the aldehyde group is removed; although side reactions of the aldehyde group can be reduced, but The reaction steps are added, and the trimethyl orthoformate is highly flammable and has potential safety hazards. The yield of compound 4 to compound 5 catalyzed by p-toluenesulfonyl chloride is low, and the cost is also increased. |
| Example 1: |
| Add 15.00kg (122.8mol, 1eq) compound 1 and 30.82kg (147.4mol, 1.2eq) compound 2 to 120.00kg (8w/w) ethyl acetate, add 24.00kg (73.7mol, 0.6eq) cesium carbonate, 1.00 kg (2.5 mol, 0.02 eq) methyl trioctyl ammonium chloride, stirred and heated to 75-85°C, reacted for 1 to 2 hours. After the reaction, the filtrate was filtered with suction, and the filtrate was washed with 60.00kg (4w/w) purified water, and the organic phase was concentrated to dryness under reduced pressure at 40°C to obtain 30.21kg (120.7mol) of compound 3 with a yield of about 98.3%. |
| 30.00kg (120.0mol, 1eq) of compound 3 was added to 14.40kg (360.0mol, 3eq) of 150.00kg (5w/w) aqueous solution of sodium hydroxide, and stirred at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w/w) ethyl acetate for extraction, add 30.00kg (1w/w) for the organic phase and wash once with purified water, and add 30.00kg (1w/w) for the organic phase Wash with saturated sodium chloride aqueous solution, add 1.50kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40° C. to obtain 27.30 kg (115.6 mol) of compound 4 with a yield of about 96.4%. |
| Add 27.00kg (114.3mol, 1eq) of compound 4 to 216.00kg (8.0w/w) of acetic anhydride, add 75.60kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 270.00kg (10w/w) purified water, stirred for 2 hours, and then extracted with 81.00kg (3w/w) ethyl acetate. The organic phase was washed twice with 27.00kg (1.0w/w) purified water, and the organic phase was washed with 27.00kg (1.0w/w) saturated aqueous sodium chloride solution, and dried with 2.70kg (0.1w/w) anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 14.26kg (81.8mol) of compound 5 was obtained, and the yield was about 71.6%. 1 HNMR(400MHz,d DMSO )δ:0.92~0.96(t,3H,-CH 3 ),1.37~1.42(m,2H,-CH 2 CH 3 ),1.68~1.72(m,2H,-CH 2 CH 2 CH 3 ),2.77~2.81(t,2H,Ar-CH 2 -CH 2 ), 6.59 (s, 1H, -ArH), 7.20 ~ 7.24 (m, 2H, ArH), 7.49 ~ 7.56 (m, 2 H, ArH). 13 CNMR (400 Hz, DMSO) δ: 159.76, 154.46, 129.07, 123.61, 122.95, 120.72, 111.03, 102.41, 29.71, 27.79, 22.12, 14.05 See attached drawings 1-2. |
| Add 17.00kg (111.7mol, 1eq) of compound 6 to 34.00kg (2w/w) toluene, add 33.31kg (280.0mol, 2.5eq) of thionyl chloride, heat to 75~85℃, keep warm for 2~4 hour. After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 18.99 kg (111.3 mol) of compound 7 as a colorless solution with a yield of about 97.4%. |
| Add 10.70kg (80.4mol, 1.0eq) of aluminum trichloride to 60.00kg of 1,2-dichloroethane at -20~-10℃, add 14.00kg (80.4mol, 1.0eq) of compound 5 under stirring, After stirring uniformly, 16.46kg (96.5mol, 1.2eq) of compound 7 is added at -20~-10°C, and the reaction is kept for 1~2 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 45.00kg purified water, the organic phase was washed with 45.00kg saturated sodium chloride aqueous solution, and 1.40kg was added. Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 55°C to obtain 20.11 kg (65.0 mol) of compound 8. The yield is about 81.2%. |
| Add 20.00kg (64.9mol, 1.0eq) of compound 8 to 60.00kg (3w/w) of toluene, add 9.52kg (71.4mol, 1.1eq) of aluminum trichloride, and raise it to 80~90℃ to react for 2~4 hours . After the reaction, transfer the reaction solution to 90.00kg (4.5w/w) purified water, adjust the pH to 2 with dilute hydrochloric acid, separate the organic phase and wash 2 times with 50.00kg (2.5w/w) purified water, and add 50.00 for the organic phase kg (2.5w/w) saturated sodium chloride aqueous solution wash, add 2.00kg (0.1w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. The wet product was filtered with suction and dried under vacuum at 80°C to obtain 16.60 kg (56.4 mol) of compound 9 as a white crystalline powder with a yield of about 86.9%. |
| 1 HNMR(400MHz,d DMSO )δ: 0.81~0.85(t,3H,-CH 3 ),1.24~1.29(m,2H,-CH 2 CH 3 ),1.68~1.70(m,2H,-CH 2 CH 2 CH 3 ),2.82~2.85(t,2H,Ar-CH 2 -CH 2 ), 6.91~7.72(m,8H,ArH), 10.46(m,1H,-OH). 13 CNMR(400 Hz,DMSO)δ:189.72,163.49,162.69,153.48,132.08,130.16,127.28,124.87,123.98 ,121.16,116.90,11 5.78,111.51,29.94,27.51,22.09,13.84. See attached drawings 3~4. |
| According to the operation of Example 1, the total yield was 46.6%. |
| Example 2: |
| Add 15.00kg (122.8mol, 1eq) of compound 1 and 30.82kg (147.4mol, 1.2eq) of compound 2 to 120.00kg (8w/w) ethyl acetate, add 10.19kg (73.7mol, 0.6eq) potassium carbonate, 1.00 kg (2.5 mol, 0.02 eq) methyl trioctyl ammonium chloride, stirred and heated to 75-85° C., reacted for 1 to 2 hours. After the reaction is completed, suction filtration, the filtrate is washed with 60.00kg (4w/w) purified water, and the organic phase is concentrated to dryness under reduced pressure at 40°C. Obtain 26.21kg (104.7mol) of compound 3, the yield is about 85.3%. |
| 26.00kg (103.9mol, 1eq) of compound 3 was added to 12.47kg (311.7mol, 3eq) of sodium hydroxide in 130.00kg (5w/w) methanol solution, and stirred at 20-30°C. After the reaction, it was concentrated to dryness under reduced pressure at 40°C. Add 130.00kg (5w/w) aqueous solution to dissolve, add 1N dilute hydrochloric acid to adjust the pH to 4, add 26.00kg (1w/w) ethyl acetate for extraction, add 26.00kg (1w/w) purified water for organic phase and wash once, organic phase Add 26.00kg (1w/w) saturated sodium chloride aqueous solution to wash, add 1.30kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40°C. 23.50 kg (99.5 mol) of compound 4 was obtained, and the yield was about 95.8%. |
| Add 23.00kg (97.4mol, 1eq) of compound 4 to 184.00kg (8.0w/w) of acetic anhydride, add 64.40kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 230.00kg (10w/w) purified water, stirred for 2 hours, and 69.00kg (3w/w) ethyl acetate was added for extraction. The organic phase was washed twice with 23.00kg (1.0w/w) purified water, the organic phase was washed with 23.00kg (1.0w/w) saturated sodium chloride aqueous solution, and 2.30kg (0.1w/w) was dried with anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 12.28kg (70.5mol) of compound 5 was obtained, and the yield was about 72.4%. |
| Add 15.00kg (98.6mol, 1eq) of compound 6 to 30.00kg (2w/w) of toluene, add 35.20kg (295.8mol, 3eq) of thionyl chloride, heat to 75~85℃, keep warm and react for 2~4 hours . After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 16.63 kg (97.5 mol) of compound 7 as a colorless solution, with a yield of about 98.9%. |
| Add 27.56kg (206.7mol, 3.0eq) of aluminum trichloride to 60.00kg of 1,2-dichloroethane at -20~-10℃, add 12.00kg (68.9mol, 1.0eq) of compound 5 with stirring, After stirring uniformly, 14.11kg (82.7mol, 1.2eq) of compound 7 is added at -20~-10℃, and the reaction is kept for 1~2 hours. After the reaction, the reaction solution was transferred to 80.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 40.00kg purified water, and the organic phase was washed with 40.00kg saturated sodium chloride aqueous solution, and 1.20kg was added. Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 55°C to obtain 17.17 kg (55.7 mol) of compound 8, with a yield of about 80.8%. |
| Add 17.00kg (55.1mol, 1.0eq) of compound 8 to 51.00kg (3w/w) of toluene, add 22.04kg (165.3mol, 3.0eq) of aluminum trichloride, raise to 80~90℃ and react for 2~4 hours . After the reaction, the reaction solution was transferred to 76.50kg (4.5w/w) purified water, adjusted to pH 2 with dilute hydrochloric acid, separated, the organic phase was washed twice with 42.50kg (2.5w/w) purified water, and the organic phase was added 42.50kg (2.5w/w) saturated sodium chloride aqueous solution was washed, and 1.70kg (0.1w/w) anhydrous sodium sulfate was added for drying. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. After suction filtration, the wet product was dried in vacuum at 80° C. to obtain 13.81 kg (46.9 mol) of compound 9 as a white crystalline powder with a yield of about 85.1%. |
| According to the operation of Example 2, the total yield was 40.2%. |
| Example 3: |
| Add 15.00kg (122.8mol, 1eq) compound 1 and 30.82kg (147.4mol, 1.2eq) compound 2 to 120.00kg (8w/w) toluene, add 24.00kg (73.7mol, 0.6eq) cesium carbonate, 1.00kg (2.5mol, 0.02eq) methyl trioctyl ammonium chloride, stir and raise the temperature to 75~85℃, and react for 1~2 hours. After the reaction is completed, suction filtration, the filtrate is washed with 60.00kg (4w/w) purified water, and the organic phase is concentrated to dryness under reduced pressure at 40°C. 30.36 kg (121.3 mol) of compound 3 was obtained, and the yield was about 98.8%. |
| 30.00kg (120.0mol, 1eq) of compound 3 was added to 20.20kg (360.0mol, 3eq) of 150.00kg (5w/w) aqueous solution of potassium hydroxide, and stirred at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w/w) ethyl acetate for extraction, add 30.00kg (1w/w) for the organic phase and wash once with purified water, and add 30.00kg (1w/w) for the organic phase Wash with saturated sodium chloride aqueous solution, add 1.50kg (0.05w/w) anhydrous sodium sulfate to dry. The organic phase was concentrated to dryness under reduced pressure at 40°C. 27.11 kg (114.7 mol) of compound 4 was obtained, and the yield was about 95.6%. |
| Add 27.00kg (114.3mol, 1eq) of compound 4 to 432.00kg (16.0w/w) of acetic anhydride, add 75.60kg (2.8w/w) of sodium acetate, raise the temperature to 90~100℃ for reaction, and react for 1~2 hours . After the reaction, the reaction solution was transferred to 540.00kg (20w/w) purified water, stirred for 5 hours, and 81.00kg (3w/w) ethyl acetate was added for extraction. The organic phase was washed twice with 27.00kg (1.0w/w) purified water, and the organic phase was washed with 27.00kg (1.0w/w) saturated aqueous sodium chloride solution, and dried with 2.70kg (0.1w/w) anhydrous sodium sulfate. The organic phase was concentrated to dryness under reduced pressure at 40°C. 14.57kg (83.6mol) of compound 5 was obtained, and the yield was about 73.1%. |
| Add 17.00kg (111.7mol, 1eq) of compound 6 to 34.00kg (2w/w) of toluene, add 35.20kg (295.8mol, 3eq) of thionyl chloride, heat to 75~85℃, keep the temperature and react for 2~4 hours . After the reaction, the solvent was distilled off under reduced pressure at 65°C to obtain 19.21 kg (112.6 mol) of compound 7 as a colorless solution, with a yield of about 100.0%. |
| Add 10.70kg (80.4mol, 1.0eq) of aluminum trichloride to 60.00kg of toluene at -20~-10℃, add 14.00kg (80.4mol, 1.0eq) of compound 5 with stirring, and after stirring, add at -20 16.46kg (96.5mol, 1.2eq) of compound 7 was added at ~-10°C, and the reaction was incubated for 1 to 2 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 45.00kg purified water, the organic phase was washed with 45.00kg saturated sodium chloride aqueous solution, and 1.40kg was added. Dry with water sodium sulfate. Add 10.72 kg (80.4 mol, 1.0 eq) of aluminum trichloride to the organic phase and raise it to 80-90° C. and react for 2 to 4 hours. After the reaction, the reaction solution was transferred to 90.00kg purified water, adjusted to pH 2 with dilute hydrochloric acid, separated into the organic phase and washed twice with 50.00kg purified water, the organic phase was washed with 50.00kg saturated sodium chloride aqueous solution, and 2.00kg Dry with water sodium sulfate. The organic phase was concentrated under reduced pressure at 65°C until a solid precipitated out, the temperature was lowered to 0°C, and the temperature was kept for 4 hours to crystallize. The wet product was filtered with suction and dried in vacuum at 80°C to obtain 14.47 kg (45.2 mol) of compound 9 as a white crystalline powder with a yield of 61.2%. |
| According to the operation of Example 3, the total yield was 42.3%. |
| Comparative Example 1 (CN104262304): |
| 700g (5.7mol, 1.0eq) of compound 1 was added to 2000g of ethyl acetate. After stirring, 1400g (6.7mol, 1.2eq) of compound 2 was slowly added. After the addition, 150g (0.46mol, 0.08eq) were added in sequence. Cesium carbonate and 50g (0.12mol, 0.02eq) methyl trioctyl ammonium chloride, and then slowly heated to 80 ℃, kept the reaction for 8 hours, at the end of the reaction, the ethyl acetate was evaporated under reduced pressure. After the evaporation, 2500g of toluene was added and controlled Warm to 20℃, continue to add 250g (4.6mol, 0.8eq) sodium methoxide, heat to reflux, keep the reaction for 5 hours; when the reaction is over, add dilute hydrochloric acid, adjust the pH to 7, stand still, separate the water phase; continue to add 600g Extract twice with water, discard the water phase, distill the organic phase under reduced pressure to remove the toluene, and collect the 135°C fraction. 383 g (2.2 mol) of compound 5 was obtained as a light yellow transparent liquid, and the yield was about 38.6%. |
| Add 360g (2.1mol, 1.0eq) of compound 5 to 2400g of toluene, heat to reflux, keep dehydration for 4 hours; after dehydration, cool to 15℃, add 420g (2.5mol, 1.2eq) of compound 7, 240g (1.8mol) , 0.86eq) zinc chloride, 60g (0.81mol, 0.4eq) N-nitrosodimethylamine, slowly increase the temperature to 80°C, keep the reaction for 10 hours; after the reaction is over, add dilute hydrochloric acid and adjust the pH to 1~2 , Continue to heat and stir for 2 hours, let stand, and discard the water phase; continue to add 450g of water to extract twice, discard the water phase, heat the organic phase to reflux, and keep dehydration for 4 hours; after dehydration, cool to 10°C, add 270g (2.1 mol, 1eq) Aluminum trichloride, slowly increase the temperature to 75°C, keep the reaction for 8 hours; after the reaction is over, add saturated sodium bicarbonate aqueous solution, adjust to pH 7, let stand, discard the water phase; add 360g saturated brine for extraction Three times, the water phase was discarded, the organic phase was distilled under reduced pressure, and it was steamed until a solid precipitated, and the temperature was reduced to 0°C, and the temperature was kept and stirred for 4 hours. After suction filtration, the wet product was vacuum dried at 80° C. for 8 hours to obtain 323 g (1.1 mol, 1 eq) of compound 9 as an off-white crystalline powder with a yield of about 52.4%. |
| According to the operation of Comparative Example 1, the total yield was 20.2%. |
| Comparative Example 2 (CN107382925): |
| 610g (5.0mol, 1.0eq) of compound 1 and 1728g (12.5mol, 2.5eq) of potassium carbonate were added to the mixture of 610kg (1w/w) DMF and 1830kg (3w/w) toluene, and heated to 60~ with stirring. Incubate at 70°C for half an hour, heat up to 80-100°C and add dropwise, 1098g (5.3mol, 1.05eq) of compound 2, react for 2 hours. After the reaction, 1830 g of water was added to wash twice, and the organic phase was concentrated to dryness under reduced pressure at 80°C. 1126 g (121.3 mol) of compound 3 was obtained, and the yield was about 90.1%. |
| 1100g (4.4mol, 1.0eq) of Compound 3 was added to 514g (4.8mol, 1.1eq) of trimethyl orthoformate and 7.6g (47mmol, 0.01eq) of p-toluenesulfonic acid in 11kg (10w/w) methanol solution, Stir at 20-30°C for 3 hours, add 2.4 g (44 mmol, 0.01 eq), and stir for 1 hour. Then it was concentrated to dryness under reduced pressure at 40°C. 1170 g (3.9 mol) of compound 0 was obtained, and the yield was about 89.7%. |
| Add 1100g (3.7mol, 1eq) of compound 0 to 2200g (2w/w) toluene, add 163g (4.1mol, 1.1eq) sodium hydroxide and 2200g (2w/w) water under stirring, 35~40 Stir and keep at ℃ for 4 hours. At the end of the reaction, 110g (0.1w/w) sodium chloride is added, and hydrochloric acid is added dropwise to adjust the pH to 1~2. The liquid was separated to obtain a toluene organic phase containing compound 4. Add 415g (4.1mol, 1.1eq) of triethylamine to the organic phase, raise it to 70~80℃ for reaction, add 782g (4.1mol, 1.1eq) of p-toluenesulfonyl chloride dropwise, and finish the reaction at 70~80℃ for 2 hours. A solution of 164g (4.1mol, 1.1eq) sodium hydroxide and 2200g (2w/w) water was added dropwise, and the reaction was kept at 70-80°C for 2 hours. Separate the liquids, wash the organic phase with 1100 g of 0.1M hydrochloric acid until neutral, and concentrate the organic phase to dryness at 80°C under reduced pressure. 453 g (2.6 mol) of compound 5 was obtained, and the yield was about 70.0%. |
| Add 409g (2.4mol, 1.0eq) compound 7, 336g (2.5mol, 1.05eq) aluminum trichloride to 60.00kg 1,2-dichloroethane at -20~-10℃, add 418g( 2.4 mol, 1.0 eq) Compound 5, incubated and reacted for 4 hours. Add 320g (2.4mol, 1.0eq) of aluminum trichloride to the organic phase, raise to reflux, and react under reflux for 6 hours. After the reaction, the reaction solution was transferred to 1000g purified water, the organic phase was separated and washed twice with 500g purified water, and the organic phase was purified by column chromatography to obtain 283g (1.0mol) of compound 9 as white crystalline powder with a yield of about 40.1%. . |
| According to the operation of Comparative Example 2, the total yield was 22.7%. |
| Description of the drawings |
| Figure 1 shows the proton nuclear magnetic resonance spectrum of compound 5; |
| Figure 2 shows the carbon nuclear magnetic resonance spectrum of compound 5; |
| Figure 3 shows the proton nuclear magnetic resonance spectrum of compound 9; |
| Figure 4 shows the carbon nuclear magnetic resonance spectrum of compound 9. |
PATENT
CN106946822
| Example 1: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione and 56.1g (0.5mol) of acrolein dimer in organic solvent tetrahydrofuran (2L). ), 126.9 g (0.5 mol) of elemental iodine and 3.4 g (25 mmol) of zinc chloride were added, and the above reaction solution was stirred and reacted in a reactor equipped with magnetic stirring at 40°C for 2 hours. After the reaction, the acidic reaction system was neutralized to neutrality with saturated sodium thiosulfate and saturated sodium bicarbonate respectively; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phase was combined and concentrated under reduced pressure; ethyl acetate and Diethyl ether was recrystallized and separated to obtain 98.6 g (0.32 mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 98.6g (0.32mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 2.1g (16mmol) of aluminum oxide was dissolved in 2L of acetonitrile; the mixture was stirred and reacted at 80°C for 5 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain 52.9 g (0.18 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 1 was 36%, wherein the yield of step (1) was 64%, and the yield of step (2) was 56%. |
| Example 2: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 58.5g (0.25mol) of 1-(4-methoxyphenyl)-1,3-heptanedione and 56.1g (0.5mol) of acrolein dimer in the organic solvent methylene chloride (2L), then add 165.8g (0.5mol) of carbon tetrabromide, BBr 3 6.26g (25mmol), the above reaction solution was stirred and reacted for 4 hours at 25°C in a reactor equipped with magnetic stirring. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with dichloromethane; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized to obtain 2-butane Benzyl-3-(4-methoxybenzoyl)benzofuran 49.3 g (0.16 mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 49.3g (0.16mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 1.1g (8mmol) of boron diethyl ether was dissolved in 2L of 1,2-dichloroethane; the mixture was stirred and reacted at 60℃ for 5 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution The aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain the 2-butyl-3-(4-hydroxybenzoyl) benzo Furan 32.37g (0.11mol). 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 2 was 44%, wherein the yield of step (1) was 64%, and the yield of step (2) was 68%. |
| Example 3: Preparation of 2-butyl-3-(4-methylbenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 56.1g (0.5mol) of acrolein dimer in organic solvent ethanol (2L) ), then add 66.8g (0.5mol) of N-chlorosuccinimide, ZrCl 4 5.8 g (25 mmol), the above reaction solution was stirred and reacted in a reactor equipped with magnetic stirring at 70°C for 8 hours. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane 104.7 g (0.34 mol) of phenyl-3-(4-methoxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 104.7g (0.34mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 6.5g (34mmol) of boron chloride was dissolved in 2L acetonitrile; the mixture was stirred and reacted at 0°C for 1 hour. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 79.4 g (0.27 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 3 was 54%, wherein the yield of step (1) was 68%, and the yield of step (2) was 79%. |
| Example 4: Preparation of 2-butyl-3-(4-hydroxybenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 112.0g (1.0mol) of acrolein dimer in organic solvent toluene (2L) ), then add 142.9g (0.5mol) of dibromoglycine, AlCl 3 5.8 g (50 mmol), the above reaction liquid was stirred and reacted in a reactor equipped with magnetic stirring at 100°C for 1 hour. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane Group-3-(4-methoxybenzoyl)benzofuran 110.9g (0.36mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 110.9g (0.36mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 10.2g (72mmol) of boron diethyl ether was dissolved in 2L acetonitrile; the mixture was stirred and reacted at 80°C for 2 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by liquid extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain the 2-butyl-3-(4-hydroxybenzoyl)benzofuran 82.3g (0.28mol) . 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 4 was 56%, wherein the yield of step (1) was 72%, and the yield of step (2) was 78%. |
| Example 5: Preparation of 2-butyl-3-(4-methylbenzoyl)benzofuran |
| Step (1): Take 117.1g (0.5mol) of 1-(4-methoxyphenyl)-1,3-heptanedione, and dissolve 28g (0.25mol) of acrolein dimer in the organic solvent dichloromethane ( 2L), then add 159.8g (0.5mol) of liquid bromine, ZnCl 2 6.8 g (50 mmol), the above reaction solution was stirred and reacted for 8 hours at 25°C in a reactor equipped with magnetic stirring. After the reaction is completed, the acidic reaction system is neutralized with saturated sodium bicarbonate; the aqueous phase obtained by liquid-liquid extraction with ethyl acetate; the organic phases are combined and concentrated under reduced pressure; ethyl acetate and ether are recrystallized and separated to obtain 2-butane Group-3-(4-methoxybenzoyl)benzofuran 58.5g (0.19mol). 1 H NMR(400MHz, CDCl 3 , TMS, 25℃) δ = 7.84 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.2 Hz, 1H), 7.36 (d, J = 7.3 Hz, 1H), 7.26 (dd, J = 9.4,5.9Hz,1H), 7.18(t,J=7.5Hz,1H), 6.96(d,J=8.8Hz,2H), 3.89(s,3H), 2.91(t,J=7.6Hz,2H) ,1.80–1.71(m,2H),1.36(dd,J=15.0,7.4Hz,2H), 0.89ppm(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=190.7,164.8,163.6,153.8,132.1,131.8,127.4,124.3,123.4,121.4,116.9,113.8,111.1,55.6,30.3,28.0,22.5,13.8ppm |
| |
| Step (2): Put 58.5g (0.19mol) of 2-butyl-3-(4-methoxybenzoyl)benzofuran obtained in step (1) into a reactor equipped with magnetic stirring. 7.2g (38mmol) of sulfonic acid was dissolved in 2L of toluene; the mixture was stirred and reacted at 100°C for 4 hours. After the reaction, the acidic reaction system was neutralized with saturated sodium bicarbonate solution to neutrality; The aqueous phase obtained by extraction; the organic phases were combined and concentrated under reduced pressure; ethyl acetate and ether were recrystallized and separated to obtain 41.2 g (0.14 mol) of the 2-butyl-3-(4-hydroxybenzoyl)benzofuran. 1 H NMR(400MHz, CDCl 3 ,TMS,25℃)δ=10.46(s,1H),7.68(d,J=8.6Hz,2H), 7.62(d,J=8.1Hz,1H), 7.33(dd,J=15.7,7.9Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.6 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 1.65 (dt, J = 15.0, 7.5 Hz, 2H),1.23(dd,J=14.7,7.3Hz,2H),0.80(t,J=7.4Hz,3H). 13 C NMR(100MHz,CDCl 3 ,25℃)δ=189.3,163.1,162.2,153.0,131.6,129.7,126.8,124.5,123.6,120.7,116.4,115.3,111.1,29.5,27.1,21.6,13.4ppm. |
| |
| The yield of Example 5 was 28%, wherein the yield of step (1) was 38%, and the yield of step (2) was 74% |
| In addition to the specific types of halogenated reagents, acid catalysts, and organic solvent raw materials used in the above embodiments, other halogenated reagents, acid catalysts, and organic solvents can also be used; wherein, the halogenated reagent in step (1) is preferably Liquid bromine, elemental iodine, N-iodosuccinimide, N-bromosuccinimide, N-chlorosuccinimide, 1,3-dichloro-5,5-dimethyl Any of hydantoin, dibromohydantoin, bromochlorohydantoin, carbon tetrabromide, and the acid catalyst is boron tribromide, boron trifluoride ether, aluminum chloride, hydrogen fluoride, zinc chloride, zirconium chloride At least one of the organic solvents is dichloromethane, 1,2-dichloroethane, acetonitrile, tetrahydrofuran, ethanol; the acid catalyst in step (2) is boron tribromide, trifluoride One of boron diethyl ether, aluminum chloride, hydrogen fluoride, sulfuric acid, and p-toluenesulfonic acid, and the organic solvent is any of methylene chloride, 1,2-dichloroethane, acetonitrile, tetrahydrofuran, ethanol, and toluene . The specific types of acid catalysts and organic solvents used in steps (1) and (2) of the present invention may be the same or different; in addition, the boiling point of the organic solvent used must be lower than the corresponding treatment temperature. |
| Unless otherwise specified, the various reaction raw materials in the present invention (eg, acrolein dimer, 1-(4-methoxyphenyl)-1,3-heptanedione, etc.) are commercially available The purity is preferably analytically pure. |
PATENT
CN109988131
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN248953969&_cid=P11-KL0BBI-08827-1
| Amiodarone (Amiodarone), also known as amiodarone, was first introduced as a coronary artery dilator, and Rosenbanm was the first to use it in the treatment of anti-arrhythmia in 1976. Amiodarone is extremely toxic. The lethal dose of intravenous injection is 10 times that of the therapeutic dose. The large oral lethal dose is negligible. Long-term larger doses are safe. Amiodarone hydrochloride (ADHC) is the hydrochloride of amiodarone, which was first marketed in Italy in 1984, and its structure is as follows: |
| |
| Amiodarone hydrochloride is a class III antiarrhythmic drug. It is mainly used clinically for supraventricular and ventricular tachyarrhythmias. It is also used for various organic heart diseases and acute coronary syndromes. It can be used as a symptomatic The first-line treatment of atrial fibrillation with left ventricular insufficiency or chronic heart failure. At present, it has become the drug of choice for the prevention of AMI with ventricular tachyarrhythmia, post-infarction ventricular arrhythmia, heart failure with arrhythmia, and sudden cardiac death. |
| In the existing synthetic methods, amiodarone hydrochloride is all obtained by etherification and salt formation with 2-butyl-(4-hydroxy-3,5-diiodobenzoyl)benzofuran as the key intermediate. The structure of this key intermediate is as follows: |
| |
| As a key intermediate of amiodarone hydrochloride, its synthesis is relatively mature. The traditional synthetic route has been applied to industrial production. The route is as follows: |
| |
| This process has the following problems: 1. Long route, complicated steps, and various operations; 2. Low yield and high cost; 3. Many wastes and high waste liquid treatment cost. Therefore, this route is no longer suitable for the current industrial and environmental protection requirements, and more efficient and green methods need to be developed. |
| Patent CN104262304 and CN1858042 improved the above synthesis method and developed a new synthesis method, the route is as follows: |
| |
| This method has been greatly improved compared with the earlier process, but the yield and purity of 2-butylbenzofuran prepared by the route method are low, which leads to a greatly reduced yield and purity of amiodarone hydrochloride. CN107382925 continues to improve the above process, and the purity and yield have increased, but column chromatography purification is required, which is not suitable for industrial large-scale production. |
| Example 1: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| |
| Put 5.5 g K 2 CO 3 , 076 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 60 mg of nickel catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 50°C and stirred for 18-22 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and twice with 40 ml of water. The filtrate was concentrated under reduced pressure to obtain 3.12 g of a dark yellow solid, namely 2-butylbenzofuran, with a yield of 89.7%. |
| 2. Synthesis of intermediate 2-butyl-(4-methoxybenzoyl)benzofuran: |
| |
| Add 5.50 g of aluminum trichloride and 24 ml of dichloromethane to a 100 ml reaction flask, stir and lower the temperature to 0°C, and add 7.0 g of p-methoxybenzoyl chloride dropwise to it within 5°C, and keep warm after dropping. Stir for 1 hour. Dissolve 6.00 g of 2-butylbenzofuran in 24 ml of dichloromethane and add dropwise to the above reaction solution within 5°C of temperature control. After dropping, slowly raise the temperature to 25°C, keep the temperature for 2 hours, and complete the reaction. After cooling to room temperature, it was poured into ice water for separation, the aqueous phase was extracted with dichloromethane, the organic layers were combined, washed twice with water, and the organic phase was concentrated under reduced pressure to obtain 10.60 g of oil. |
| 3. Synthesis of 2-butyl-(4-hydroxybenzoyl)benzofuran: |
| |
| Add 10.00 g of 2-butyl-(4-methoxybenzoyl) benzofuran, 4.50 g of aluminum trichloride and 40 ml of toluene into a 100 ml reaction flask. The temperature is raised to reflux, and the reaction is kept warm for 6 hours. The reaction is complete . Cool down to 0℃, pour into ice water, separate the liquids, extract the aqueous phase with toluene, combine the organic phases, add equal volumes of water, adjust the pH to above 12 with NaOH solution, separate the liquids, adjust the pH to less than 3 with hydrochloric acid for the aqueous phase , Filtered, and the filter cake was vacuum dried to obtain 8.16 g of light yellow solid. |
| 4. Synthesis of 2-butyl-(4-hydroxy-3,5-diiodobenzoyl)benzofuran: |
| |
| Add 8 g of 2-butyl-(4-hydroxybenzoyl) benzofuran, 15.18 g of iodine, 8.26 g of potassium carbonate and 48 ml of ethanol to a 100 ml reaction flask, and heat to reflux with stirring, and keep the reaction temperature for 2 hours. The reaction is over. The temperature was lowered to room temperature, filtered, the filtrate was added dropwise to the sodium metabisulfite aqueous solution, after dripping, the mixture was kept and stirred for 0.5 hours, filtered, the filter cake was washed twice with water, and the solid was vacuum dried to obtain 14.61 g of off-white solid. |
| 5. Synthesis of 2-butyl-[4-[2-(diethylamino)hydroxyethyl]-3,5-diiodobenzoyl]benzofuran: |
| |
| Add 12.00 g of 2-butyl-(4-hydroxy-3,5-diiodobenzoyl) benzofuran and 120 ml of toluene into a 250 ml reaction flask. The temperature is raised to 60°C. After the solid is dissolved, add to it 4.94 g of 2-diethylaminochloroethane hydrochloride, 5.65 g of potassium carbonate and 8.50 g of water were heated to reflux, stirred and reacted for 8 hours, and the reaction was completed. The reaction solution was washed 3 times with water, 0.60 g activated carbon was added to the organic phase, the temperature was raised to reflux and stirred for 1 hour, and then filtered with suction. The filtrate was concentrated under reduced pressure until solids began to precipitate, and the temperature was reduced to 0°C for crystallization. The cold toluene was washed twice, and the solid was vacuum-dried at 80°C to obtain 13.97 g of a finished white solid, which was the finished product of amiodarone hydrochloride. |
| Compared with the existing production process, the preparation method of amiodarone hydrochloride in this embodiment simplifies the operation and improves the convenience of operation and the stability of the product. Through the control of the catalyst and material ratio, the purity and yield of each intermediate are improved, and the product does not require column chromatography to purify, which saves costs and improves production efficiency, which provides convenience for industrial large-scale production. |
| Example 2: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 190 mg of ruthenium catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 50°C and stirred for 22-28 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.25 g of a dark yellow solid, which was 2-butylbenzofuran, with a yield of 93.4%. |
| The remaining steps are the same as in Example 1. |
| Example 3: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 89 mg of palladium catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 40°C under nitrogen and stirred for 26-30 hours. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.36 g of a dark yellow solid, which was 2-butylbenzofuran, with a yield of 96.6%. |
| The remaining steps are the same as in Example 1. |
| Example 4: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 100 mg of rhodium catalyst were added to it, and replaced with nitrogen 3 Next, the reaction was kept at 40°C under nitrogen protection and stirred for 20-24 hours. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 2.93 g of dark yellow solid, which is 2-butylbenzofuran, with a yield of 84.2%. |
| The remaining steps are the same as in Example 1. |
| Example 5: |
| 1. Preparation of intermediate 2-butylbenzofuran: |
| Put 5.5 g K 2 CO 3 , 0.76 g of CuI and 0.74 g of TBAI were added to a 100 ml reaction flask containing 30 ml of toluene, and 4.4 g of 2-iodophenol, 2.5 ml of 1-hexyne and 80 mg of gold catalyst were added to it, and replaced with nitrogen 3 Secondly, the reaction was kept at 40°C and stirred for 20-28 hours under nitrogen protection. The reaction solution was filtered, and the filtrate was washed with 40 ml of 5% NaOH aqueous solution and 40 ml of water twice. The filtrate was concentrated under reduced pressure to obtain 3.07 g of dark yellow solid, which is 2-butylbenzofuran, with a yield of 88.2%. |
| The remaining steps are the same as in Example 1. |
?///////////
Mitsubishi Tanabe And EnVivo in Phase III Trial Of Alzheimer’s Disease Treatment MT-4666

OR
Encenicline (EVP-6124, MT-4666)
EVP-6124 , MT-4666, α7-nAChR agonist, UNII-5FI5376A0X
Chemical Name: (R)-7-chloro-N-quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide
Therapy Type: Small Molecule
Target Type: Cholinergic System
CAS : 550999-75-2
- C16 H17 Cl N2 O S
- Benzo[b]thiophene-2-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-7-chloro-
- (R)-7-Chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide; EVP 6124
Condition(s): Alzheimer’s Disease, Schizophrenia
U.S. FDA Status: Alzheimer’s Disease (Phase 3), Schizophrenia (Phase 3)
Status in Select Countries: Investigational in Japan
Company: FORUM Pharmaceuticals Inc. (was EnVivo Pharmaceuticals), Mitsubishi Tanabe Pharma
Approved for: None AS ON SEPT 2014

CAS 550999-74-1
Benzo[b]thiophene-2-carboxamide, N-(3R)-1-azabicyclo[2.2.2]oct-3-yl-7-chloro-, monohydrochloride
(R)-7-Chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride
Mitsubishi Tanabe Pharma ..Encenicline-hydrochloride (EVP-6124) for Alzheimer’s disease by partner EnVivo Pharmaceuticals. Mitsubishi Tanabe has licensed EVP-6124 from EnVivo and is currently developing the drug under the code MT-4666.
The drug is a new alpha-7 potentiator intended to improve cognition in patients affected with Alzheimer’s disease. The drug is being tested in Phase III COGNITIV clinical trials in two categories: COGNITIV AD in patients with Alzheimer’s disease and COGNITIV CIAS in patients with cognitive impairment associated with schizophrenia.
Alzheimer’s disease affects five million people in the U.S. alone, or one in eight Americans over the age of 65. The disease is the sixth-leading cause of death in the country, with the number of affected patients expected to balloon to nearly triple by 2030. Alzheimer’s disease is a complex neurodegenerative disease that eventually leads to cellular loss and dysfunction in the brain resulting in decline of language skills and reasoning among others.
Phase III of COGNITIV AD clinical trial program involves about 1,600 patients with mild to moderate AD and who are presently receiving stable treatment with or have undergone previous acetylcholinesterase inhibitor treatment. The trials will be placebo-controlled, double-blind, and randomized. Patients in the trial will be randomized to receive either one of two doses of MT-4666 once daily against a placebo to assess safety and efficacy of the drug.
In the news release recently launched by EnVivo, CEO and president Deborah Dunsire said, “We are pleased to advance encenicline into Phase 3 clinical development in Alzheimer’s disease, a significant milestone for our company and promising step forward for patients who desperately need new therapies…Prior clinical studies of encenicline have demonstrated clinically significant improvements in cognitive function in patients with Alzheimer’s disease. For the millions of patients living with AD, we believe encenicline has the potential to make a meaningful difference.”
Encenicline hydrochloride is a partial, selective agonist of the α-7 nicotinic acetylcholine receptor (α7-nAChR). It is being developed for the treatment of cognitive deficits in schizophrenia and Alzheimer’s disease. Cholinergic function declines in Alzheimer’s, and currently approved acetylcholinesterase inhibitor therapies modestly improve cognitive deficits in patients with AD by way of boosting cholinergic transmission. The rationale of selective α7-nAChR agonists is that they will enhance cognition without causing side effects associated with overactivation of other nAChRs such as α4β2, or muscarinic AChRs. In rats, encenicline penetrates the blood-brain barrier and improves memory performance by potentiating the acetylcholine response. Encenicline has been reported to act as a co-agonist with acetylcholine. It sensitizes the α-7 nACh receptor to its natural ligand and renders sub-efficacious doses of AChEI drugs effective in restoring memory function in an object recognition task (Prickaerts et al., 2012).
This compound was originally developed at Bayer Healthcare and then licensed to Envivo Pharmaceuticals, which subsequently licensed development in Asia to Mitsubishi Tanabe Pharma Corporation. Envivo then changed its name to FORUM Pharmaceuticals Inc.
Encenicline is being tested in Alzheimer’s disease and schizophrenia. In Alzheimer’s, an ascending-dose Phase 1/2 study showed 0.1 to 1 mg/day of EVP-6124 to be safe and well-tolerated when given to 49 people with mild to moderate AD for 28 days. No serious side effects were reported. Secondary efficacy endpoints suggested that EVP-6124 given in addition to therapy with the acetyl cholinesterase inhibitors donepezil or rivastigmine appeared to improve attention, verbal fluency, and executive function as measured on tests in the CogState or NTB batteries (see conference news story). This study has posted results on clinicaltrials.gov.
A 24-week Phase 2 trial conducted in 409 people with mild to moderate Alzheimer’s disease in the United States and Eastern Europe compared 0.3, 1, and 2 mg of EVP-6124 per day to placebo, measuring cognition with ADAS-Cog as the primary outcome plus cognitive, functional, and psychiaric secondary outcomes. EVP 6124 was given as adjunct therapy to donepezil or rivastigmine. This trial was reported to have met its primary and most secondary endpoints, showing that people on the highest dose improved over baseline. EVP-6124 dose-dependently improved measures of attention, verbal and language fluency, and executive function. In this trial, all treatment groups initially improved, possibly due to a placebo effect, but by 12 weeks the groups separated and the placebo and low-dose groups declined (see conference news story). EVP-6124 was well-tolerated.
Mitsubishi Tanabe Pharma Corporation is conducting a Phase 2 trial for the treatment of Alzheimer’s disease in Japan.
In October 2013, two international Phase 3 trials began enrolling what are to be 790 patients in each trial with mild to moderate Alzheimer’s who are already taking an acetylcholinesterase inhibitor. The trials will compare two fixed, undisclosed add-on doses of EVP-6124 to placebo, all given as once-daily tablets for six months, for cognitive benefit as measured by the ADAS-Cog, clinical benefit as measured by the Clinical Dementia Rating Sum of Boxes (CDR-SB), as well as for safety and tolerability. Called COGNITIV AD, this Phase 3 program is is set to run through 2016.
For schizophrenia, a Phase 1 study comparing 0.3 and 1 mg/day of EVP-6124 to placebo in 28 people with the disease gave preliminary evidence for the compound’s safety, tolerability, and pharmacokinetics in this population. In addition, the compound yielded signals of bioactivity in the brain by way of EEG tests of evoked potentials, a measure of sensory gating affected in this disease. See study results on clinicaltrials.gov.
A subsequent 12-week Phase 2 trial compared 0.3 and 1 mg/day of EVP-6124 to placebo in 317 people with schizophrenia and measured safety and the compound’s efficacy on cognitive function. As presented at the American College of Neuropsychopharmacology meeting held in Hawaii December 2011, EVP 6124 met its primary endpoint of improvement on the CogState overall cognition index. The study also met secondary endpoints, showing improvement in clinical function as assessed by the Schizophrenia Cognition Rating Scale, and a decrease in negative symptoms (See company press release).
Two six-month, 700-patient Phase 3 studies, plus a six-month extention study, are ongoing. For all clinical trials of encenicline, see clincialtrials.gov.
http://www.google.com.ar/patents/WO2014051055A1?cl=pt-PT
Synthesis (hereinafter, the compound of Reference Example 25) carboxamide hydrochloride (Reference Example 25) (R) -7 – chloro-N-(quinuclidin-3 – – yl) benzo [b] thiophene-2:
[First Step]
Synthesis of carboxamide (R) -7 – chloro-N-(quinuclidin-3 – – yl) benzo [b] thiophene-2:
-N, N, N ‘, N’-tetra-7 – chloro-1 – benzothiophene -2 – – o-(yl benzotriazol-1) chloroform solution (210mg, 1.0mmol) of carboxylic acid in (10mL) was added (0.70mL, 4.0mmol) and (570mg, 1.5mmol), diisopropylethylamine methyl hexafluorophosphate, (R) – (200mg, 1.0mmol) amine hydrochloride – quinuclidine-3 was added, and the mixture was stirred at room temperature. 16 hours later, was added distilled water, 1.0N sodium hydroxide solution, and extracted with chloroform. Was washed with saturated brine and the organic layer was concentrated and then dried over anhydrous sodium sulfate. (Fuji Silysia Chemical amine silica gel DM1020, chloroform alone – chloroform / methanol = 90/10) on silica gel column chromatography of the crude product obtained was purified by the title compound; was obtained as a white solid (170mg 53%).
1 H-NMR (400MHz, DMSO-d 6)
δ :1.22-1 .38 (1H, m) ,1.53-1 .62 (2H, m) ,1.75-1 .82 (2H, m) ,2.63-2 .73 (4H , m) ,2.84-2 .94 (1H, m) ,3.07-3 .18 (1H, m) ,3.90-4 .00 (1H, m), 7.49 (1H, dd , J = 7.6,8.0 Hz), 7.59 (1H, d, J = 7.6Hz), 7.96 (1H, d, J = 8.0Hz), 8.31 (1H, s) ,8.62-8 .66 (1H, m).
MS (ESI): 321 [M + H] +
[Second Step]
Synthesis of the compound of Reference Example 25:

Ethyl acetate solution – solution of hydrogen chloride in ethyl acetate (170mg, 0.53mmol) of the (2.0mL) carboxamide – (R) -7 – chloro-N-(quinuclidin-3 – yl) benzo [b] thiophene-2 was added (4.0M, 0.20mL, 0.80mmol), and the mixture was stirred at room temperature. 10 minutes later, by which is filtered off and the resulting solid was washed with ethyl acetate and hexane, and dried, the compound of Reference Example 25; was obtained as a white solid (170mg 90%).
1 H-NMR (400MHz, DMSO-d 6)
δ :1.70-1 .78 (1H, m) ,1.86-1 .94 (2H, m) ,2.10-2 .19 (2H, m) ,3.18-3 .35 (5H , m) ,3.63-3 .72 (1H, m) ,4.27-4 .36 (1H, m), 7.50 (1H, d, J = 7.6,8.0 Hz), 7 .61 (1H, d, J = 7.6Hz), 7.98 (1H, d, J = 8.0Hz), 8.38 (1H, s) ,9.07-9 .10 (1H, m) ,9.80-9 .85 (1H, m).
MS (ESI): 321 [M + H] +
………………………………….
http://www.google.com/patents/EP1461335A1?cl=en
Example 69
N-[(3 R) – 1 – azabicyclo [2.2.2] oct-3-y 1]-7-chloro-1-benzothiophene-2-carboxamide hydrochloride DESIRED

x HCI
176.2 mg (0.83 mmol) of 7-chloro-l-benzothiophene-2-carboxylic acid, 150 mg (0.75 mmol)
R-3-Aminochinuklidin dihydrochloride, 343.7 mg (0.90 mmol) of HATU, 350.5 mg
(2.71 mmol) of N, N-diisopropylethylamine and 3.0 ml of DMF are reacted according to the general working procedure (variant B). The reaction mixture is purified by preparative HPLC. The product will be in a mixture of 4 M HCl solution in dioxane and methanol, and then concentrated. This gives 175.2 mg
(65.1% of theory) of the title compound.
1H NMR (200 MHz, DMSO-d 6): δ – 10.03 (s, IH, br), 9.17 (d, IH), 8.43 (s, IH), 7.98 (m, IH), 7.63 (m, IH ), 7.52 (dd, IH), 4.33 (m, IH), 3.77-3.10 (m, 6H), 2.28-
2.02 (m, 2H), 1.92 (m, 2H), 1.75 (m, IH) ppm.
HPLC: R t = 4.0 min (Method H)
MS (ESIpos): m / z = 321 (M + H) + (free base).
Example 70
N-[(3 S) – 1-azabicyclo [2.2.2] oct-3-yl]-7-chloro-1-benzothiophene-2-carboxamide hydrochloride UNDESIRED

x HCI
176.2 mg (0.83 mmol) of 7-chloro-l-benzothiophene-2-carboxylic acid, 150 mg (0.75 mmol) of S-3-Aminochinuklidin dihydrochloride, 343.7 mg (0.90 mmol) of HATU, 350.5 mg (2.71 mmol) of N, N- diisopropylethylamine and 3.0 ml of DMF are implemented according to the general procedure (Method B). The reaction mixture is purified by preparative HPLC. The product will be in a mixture of 4 M HCl solution in dioxane and methanol, and then concentrated. Obtained 231.9 mg (85.7% of theory) of the title compound. The analytical data are consistent with those of the enantiomeric compound from Example 69.
………………………………………
http://www.google.com.ar/patents/EP2727604A1?cl=en
(Reference Example 3)
Synthesis of (R)-7-Chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride (hereinafter referred to as the compound of Reference Example 3):
[First step]Synthesis of 7-Chloro-1-benzothiophene-2-carboxylic acid:

[Second step]Synthesis of (R)-7-Chloro-N-(quinuclidine-3-yl)benzo[b]thiophene-2-carboxamide:

To a solution (10 mL) of 7-chloro-1-benzothiophene-2-carboxylic acid (210 mg, 1.0 mmol) in chloroform, o-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (570 mg, 1.5 mmol) and diisopropylethylamine (0.70 mL, 4.0 mmol) were added. Thereafter, (R)-quinuclidine-3-amine hydrochloride (200 mg, 1.0 mmol) was added thereto, and the resulting mixture was stirred at room temperature. Sixteen hours later, distilled water and 1.0 N aqueous sodium hydroxide solution were added thereto, and the resultant was extracted with chloroform. The organic layer was washed with brine, then dried over anhydrous sodium sulfate and concentrated. The obtained crude product was purified by silica gel column chromatography (amine silica gel DM1020, Fuji Silysia Chemical Ltd., chloroform alone to chloroform/methanol = 90/10) to obtain the title compound (170 mg; 53%) as a white solid.
1H-NMR (400 MHz, DMSO-d6)
δ: 1.22-1.38 (1H, m), 1.53-1.62 (2H, m), 1.75-1.82 (2H, m), 2.63-2.73 (4H, m), 2.84-2.94 (1H, m), 3.07-3.18 (1H, m), 3.90-4.00 (1H, m), 7.49 (1H, dd, J=7.6, 8.0 Hz), 7.59 (1H, d, J=7.6 Hz), 7.96 (1H, d, J=8.0 Hz), 8.31 (1H, s), 8.62-8.66 (1H, m).
MS (ESI) [M+H]+ 321
1H-NMR (400 MHz, DMSO-d6)
δ: 1.22-1.38 (1H, m), 1.53-1.62 (2H, m), 1.75-1.82 (2H, m), 2.63-2.73 (4H, m), 2.84-2.94 (1H, m), 3.07-3.18 (1H, m), 3.90-4.00 (1H, m), 7.49 (1H, dd, J=7.6, 8.0 Hz), 7.59 (1H, d, J=7.6 Hz), 7.96 (1H, d, J=8.0 Hz), 8.31 (1H, s), 8.62-8.66 (1H, m).
MS (ESI) [M+H]+ 321
[Third step]Synthesis of Compound of Reference Example 3:
To a solution (2.0 mL) of (R)-7-chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide (170 mg, 0.53 mmol) in ethyl acetate, hydrogen chloride-ethyl acetate solution (4.0 M, 0.20 mL, 0.80 mmol) was added, and the resulting mixture was stirred at room temperature. Ten minutes later, the obtained solid was filtered off, washed with ethyl acetate and hexane, and dried to obtain the compound of Reference Example 3 (170 mg; 90%) as a white solid.
1H-NMR (400 MHz, DMSO-d6)
δ: 1.70-1.78 (1H, m), 1.86-1.94 (2H, m), 2.10-2.19 (2H, m), 3.18-3.35 (5H, m), 3.63-3.72 (1H, m), 4.27-4.36 (1H, m), 7.50 (1H, d, J=7.6, 8.0 Hz), 7.61 (1H, d, J=7.6 Hz), 7.98 (1H, d, J=8.0 Hz), 8.38 (1H, s), 9.07-9.10 (1H, m), 9.80-9.85 (1H, m).
MS (ESI) [M+H]+
321
1H-NMR (400 MHz, DMSO-d6)
δ: 1.70-1.78 (1H, m), 1.86-1.94 (2H, m), 2.10-2.19 (2H, m), 3.18-3.35 (5H, m), 3.63-3.72 (1H, m), 4.27-4.36 (1H, m), 7.50 (1H, d, J=7.6, 8.0 Hz), 7.61 (1H, d, J=7.6 Hz), 7.98 (1H, d, J=8.0 Hz), 8.38 (1H, s), 9.07-9.10 (1H, m), 9.80-9.85 (1H, m).
MS (ESI) [M+H]+
321
| WO1991012254A1 * | 15 Feb 1991 | 17 Aug 1991 | Novo Nordisk As | Substituted urea compounds and their preparation and use |
| WO2004069141A2 * | 5 Feb 2004 | 19 Aug 2004 | Strakan Ltd | Transdermal granisetron |
| WO2004076449A2 * | 20 Feb 2004 | 10 Sep 2004 | Jozef Klucik | 3-substituted-2(arylalkyl)-1-azabicycloalkanes and methods of use thereof |
| WO2008019372A2 * | 7 Aug 2007 | 14 Feb 2008 | Amr Technology Inc | 2-aminobenzoxazole carboxamides as 5ht3 modulators |
| WO2008096870A1 * | 8 Feb 2008 | 14 Aug 2008 | Astellas Pharma Inc | Aza-bridged-ring compound |
| JPH0881374A * | Title not available |
|
MW: 357.3032 |
||
| 2 |
MW: 320.8423 |
|
| 3 |
MW: 375.318 |
