
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
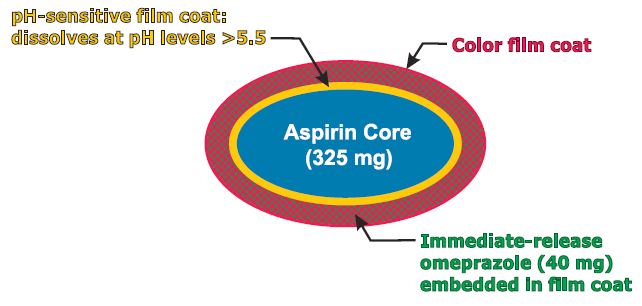
NDA PA32540, Pozen Submits NDA for Aspirin Alternatives
PA32540
PA-325/40 (EC-ASA 325 mg + IR omeprazole 40 mg)
enteric-coated aspirin (EC-ASA) 325 mg
MAR 27,2013
Pozen Inc. announced the submission of a new drug application to the U.S. Food and Drug Administration for the marketing approval of two potential cardiovascular drugs.
The move is a major step toward commercializing the drugs and means that the company thinks it has proven that the drugs are safe and effective.
Pozen’s PA32540 and PA8140 are both intended as alternatives to plain aspirin for the prevention of cardiovascular disease. Many people take aspirin to prevent heart problems, but long-term use of aspirin can cause ulcers.
Pozen’s drugs contain aspirin and the omeprazole, the active ingredient in heartburn drugs like Prilosec. The omeprazole is released as soon as the drug is taken and the aspirin is released over time.
Pozen is seeking approval for use in patients at risk of aspirin-induced ulcers.
Pozen CEO John Plachetka called the move “an important milestone for the drug” and said the company looks forward to completing a commercial deal with a partner “in the upcoming months.”
Pozen’s PA-325/40 is a combination pill containing enteric-coated aspirin 325 mg surrounded by a pH-sensitive film surrounded by an immediate-release omeprazole 40 mg.
SPOTLIGHT-Ganetespib Shows Potency Against Lung Cancer
Ganetespib
http://www.ama-assn.org/resources/doc/usan/ganetespib.pdf
A drug that indirectly impairs the function of several cancer-driving proteins, including anaplastic lymphoma kinase, may be an effective new treatment for patients with anaplastic lymphoma kinase-positive non-small cell lung cancer.
The drug, ganetespib, may also be effective for treating patients who have become resistant to the only FDA-approved targeted therapy for this disease, crizotinib, according to data published in Cancer Discovery, a journal of the American Association for Cancer Research.
“Lung cancer, a leading cause of death, is no longer thought of as a single disease, but rather as a group of diseases, each with a distinct genetic profile,” according to David Proia, Ph.D., associate director of cancer biology at Synta Pharmaceuticals Corporation, the company that funded the research. “This realization has paved the way for the design of new treatments tailored to the specific biological characteristics of a patient’s tumor.
“For example, patients with lung cancer caused by alterations in the anaplastic lymphoma kinase (ALK) protein typically respond well to crizotinib, which blocks that activity of the modified ALK and consequently kills off the cancer cells,” said Proia. “However, as is the case for many cancer drugs, most patients treated with crizotinib eventually become resistant to the drug.”
Proia and colleagues investigated ganetespib as an alternative treatment for ALK-positive non-small cell lung cancer (NSCLC). Ganetespib targets heat shock protein 90 (Hsp90), a chaperone for many different proteins, including ALK, to ensure proper functioning. When Hsp90 is blocked, ALK can no longer work properly and is destroyed by the cell. Once ALK is lost, the cancer cells die and the tumors shrink.
Ganetespib had 30 times greater potency than crizotinib against a cultured ALK-positive NSCLC cell line, resulting in the complete loss of ALK protein expression. In addition, the drug was active against ALK-positive lung cancer cell lines that had become resistant to the effects of crizotinib.
The researchers then compared ganetespib and crizotinib in mice xenografted with human ALK-positive NSCLC cancer cells. Ganetespib displayed greater antitumor activity and prolonged animal survival as compared to crizotinib. It was also shown that ganetespib had meaningful activity in a patient with ALK-driven NSCLC who had responded to, and then progressed, following crizotinib therapy.
“Ganetespib therapy represents a new option for treating ALK-dependent lung cancer in sequence with direct ALK inhibitors and/or for treating patients who relapse following direct ALK inhibitor therapy,” said Proia.
The U.S. Food and Drug Administration today approved Tecfidera (dimethyl fumarate) capsules to treat adults with relapsing forms of multiple sclerosis (MS).

Dimethyl fumarate
March 27, 2013
The U.S. Food and Drug Administration today approved Tecfidera (dimethyl fumarate) capsules to treat adults with relapsing forms of multiple sclerosis (MS).
MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function (relapses) are initially followed by recovery periods (remissions). Over time, recovery periods may be incomplete, leading to progressive decline in function and increased disability. MS patients often experience muscle weakness and difficulty with coordination and balance. Most people experience their first symptoms of MS between the ages of 20 and 40.
Dimethyl fumarate (DMF) is the methyl ester of fumaric acid. DMF was initially recognized as a very effective hypoxic cell radiosensitizer. Later, DMF combined with three other fumaric acid esters (FAE) was licensed in Germany as oral therapy for psoriasis (Fumaderm). Other diseases, such as necrobiosis lipoidica, granuloma annulare, and sarcoidosis were also found to respond to treatment with DMF in case reports or small patient series. Recently, phase III clinical trials found that DMF (BG-12) successfully reduced relapse rate and time to progression of disability in multiple sclerosis. DMF is thought to have immunomodulatory properties without significant immunosuppression.
In a non-medical use, DMF was applied as a biocide in furniture or shoes to prevent growths of mold during storage or transport in a humid climate. However, due to incidences of allergic reactions after skin contact the European Union banned DMF in consumer products since 1998, and since January 2009 the import of products containing DMF was also banned.
MHLW Japan approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome

Rufinamide
mar26, 2013
The agency approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome
Rufinamide is an anticonvulsant medication. It is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders. Rufinamide, a triazole derivative, was developed in 2004 by Novartis Pharma, AG, and is manufactured by Eisai.
Rufinamide was approved by the US Food and Drug Administration on November 14, 2008 as adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in children 4 years and older and adults. Its official FDA-approved labeling does not mention use in the treatment of partial seizures inasmuch as clinical trials submitted to the FDA were marginal. However, several recent clinical trials suggest that the drug has efficacy for partial seizures It is marketed under the brand name Banzel. It is also marketed in the European Union under the brand name Inovelon.
The mechanism of action of rufinamide is unknown. However, it is presumed to involve stabilization of the sodium channel inactive state, effectively keeping these ion channels closed. Although the direct mechanism of action may be different, several other antiepileptic agents also stabilize a sodium channel inactive state including phenytoin, carbamazepine, and lacosamide (stabilizes the slow inactive state).
MHLW Japan, GlaxoSmithKline and Genmab announced the approval of Arzerra (ofatumumab) by the MHLW for use in patients with relapsed/refractory CD20-positive chronic lymphocytic leukaemia.
mar 26, 2013
GlaxoSmithKline and Genmab announced the approval of Arzerra (ofatumumab) by the MHLW for use in patients with relapsed/refractory CD20-positive chronic lymphocytic leukaemia.
The thumbs-up triggers a milestone payment of 20 million Danish kroner to Genmab.
Ofatumumab(trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.
MHLW JAPAN has approved Nippon Shinyaku Co’s Regtect (acamprosate) for support maintenance of abstinence in patients with alcohol dependence

ACAMPROSATE
MAR 26 2013
MHLW JAPAN has approved Nippon Shinyaku Co’s Regtect (acamprosate) for support maintenance of abstinence in patients with alcohol dependence.
Acamprosate, also known as N-acetyl homotaurine and by the brand name Campral, is a drug used for treating alcohol dependence.
Acamprosate is thought to stabilize the chemical balance in the brain that would otherwise be disrupted by alcoholism, possibly by antagonizing glutamatergic N-methyl-D-aspartate receptors and agonizing gamma-aminobutyric acid (GABA) type A receptors. Reports indicate that acamprosate only works with a combination of attending support groups and abstinence from alcohol.Certain serious side effects include diarrhea, allergic reactions, irregular heartbeats, and low or high blood pressure, while less serious side effects include headaches, insomnia, and impotence. Acamprosate should not be taken by people with kidney problems or allergies to the drug.
Campral is manufactured and marketed in the United States by Forest Laboratories, while Merck KGaA markets it outside the US. It is sold as 333 mg white and odorless tablets of acamprosate calcium, which is the equivalent of 300 mg of acamprosate.
MAA, EU, Boehringer Ingelheim and Eli Lilly and Company announce acceptance of EMA application for investigational Type 2 Diabetes treatment empagliflozin

Empagliflozin
26 March 2013
Boehringer Ingelheim and Eli Lilly and Company today announced the European Medicines Agency (EMA) has accepted for review a marketing authorisation application (MAA) for the investigational sodium glucose cotransporter-2 (SGLT2) inhibitor, empagliflozin, for the treatment of Type 2 Diabetes (T2D) in adults. The acceptance of the MAA marks the beginning of the review process in the European Union for this potential oral diabetes treatment.
A New Drug Application (NDA) for empagliflozin was recently submitted to the Food and Drug Administration (FDA) in the United States for the treatment of Type 2 Diabetes mellitus (T2D) in adults.
Empagliflozin is part of a class of drugs being investigated for the reduction of blood glucose levels in adults with T2D. In clinical trials to date, SGLT2 inhibitors have been shown to reduce blood glucose by removing excess glucose independently of beta cell function and insulin resistance.
* Empagliflozin is an investigational compound. Its safety and efficacy have not yet been fully established
About Diabetes
An estimated 371 million people worldwide have Type 1 and Type 2 Diabetes. Type 2 Diabetes is the most common type, accounting for an estimated 90% of all diabetes cases.Diabetes is a chronic condition that occurs when the body either does not properly produce, or use, the hormone insulin.
Empagliflozin is drug which is being investigated in clinical trials for the oral treatment of type 2 diabetes by Boehringer Ingelheim and Eli Lilly and Company.[1][2] It is an inhibitor of the sodium glucose co-transporter-2 (SGLT-2), which is found almost exclusively in the proximal tubules of nephronic components in the kidneys. SGLT-2 accounts for about 90 percent of glucose reabsorption into the blood. Blocking SGLT-2 causes blood glucose to be eliminated through the urine via the urethra.[3][4]
SGLT-2 inhibitors such as empagliflozin reduce blood glucose by blocking glucose reabsorption in the kidney and thereby excreting glucose (i.e., blood sugar) via the urine.[5]
- Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P (January 2012). “Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors”. Diabetes Obes Metab 14 (1): 83–90. doi:10.1111/j.1463-1326.2011.01517.x. PMID 21985634.
- “Empagliflozin”. clinicaltrials.gov. U.S. National Institutes of Health. Retrieved 22 September 2012.
- Nair S, Wilding JP (January 2010). “Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus”. J. Clin. Endocrinol. Metab. 95 (1): 34–42. doi:10.1210/jc.2009-0473. PMID 19892839.
- Bays H (March 2009). “From victim to ally: the kidney as an emerging target for the treatment of diabetes mellitus”. Curr Med Res Opin 25 (3): 671–81. doi:10.1185/03007990802710422. PMID 19232040.
- Abdul-Ghani MA, DeFronzo RA (September 2008). “Inhibition of renal glucose reabsorption: a novel strategy for achieving glucose control in type 2 diabetes mellitus”. Endocr Pract 14 (6): 782–90. PMID 18996802.
Kyowa Hakko Kirin, has received approval for NOURIAST tablets 20 mg (istradefylline), a novel antiparkinsonian agent, has been approved for manufacturing and marketing in Japan

Istradefylline (KW-6002) is a selective antagonist at the A2A receptor. It has been found to be useful in the treatment of Parkinson’s disease.[1] Istradefylline reduces dyskinesia resulting from long-term treatment with classical antiparkinson drugs such as levodopa. Istradefylline is an analog of caffeine.
References
Peter A. LeWitt, MD, M. Guttman, James W. Tetrud, MD, Paul J. Tuite, MD, Akihisa Mori, PhD, Philip Chaikin, PharmD, MD, Neil M. Sussman, MD (2008). “Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces off time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005)”. Annals of Neurology 63 (3): 295–302. doi:10.1002/ana.21315. PMID 18306243.
TUE 26 MAR 2013
Japan, Kyorin, Pentasa, mesalazine suppositories launched
PENTASA (mesalamine) for oral administration is a controlled-release formulation of mesalamine, an amino-salicylate anti-inflammatory agent for gastrointestinal use. Chemically, mesalamine is 5-amino-2-hydroxybenzoic acid. It has a molecular weight of 153.14.
The structural formula is:

Cellular Biomedicine Group Marks the Launch of China Clinical Trial for TC-DC Therapy for Hepatocellular Carcinoma

March 21, 2013
Cellular Biomedicine Group announced that in the first week of March 2013, the company launched a clinical trial for TC-DC (Tumor Stem Cell Specific Dendritic Cell) therapy for hepatocellular carcinoma (HCC), the most common type of liver cancer. The clinical trial, which is already in progress, is the result of collaboration between CBMG, California Stem Cell (CSC) and Shanghai’s PLA 85 Hospital. It is the first immune cell clinical trial of its kind in China.
CBMG’s joint venture with CSC grants CBMG an exclusive license from CSC to develop and market CSC’s cancer (TC-DC) technology in Greater China. CBMG receives support from CSC’s California-based team of scientists and medical professionals, including CSC’s Dr. Hans Keirstead .
PLA 85 Hospital is a large general teaching hospital with 12 departments, located in Shanghai and has been granted Class A Hospital status at the Tertiary Level (the highest class). The hospital has over 600 beds with more than 200 professors and associate professors, including many well-known experts who are known for their pioneering work in the diagnosis and treatment of tumors. The principal Investigator of the trial and director of the Liver Disease Center of PLA 85 Hospital, ProfessorChengwei Chen , commented, “When I heard of the success this treatment had in clinical trials for other cancers in the U.S., I was very excited at the prospect of the hope it could bring to the millions of patients in China suffering from HCC. I am happy to lead this endeavor to help as many people as we can.” Said Dr. Steve Liu , Chairman of CBMG, “The launch of this trial is a major milestone for all of the physicians, scientists and other professionals at CBMG, CSC and PLA 85 Hospital who have contributed to this work.”
Multinational Contract Research Organization (CRO) CMIC-GCP has been contracted to manage the trial design and minimize delays.
Hepatocellular Carcinoma
Forty-five percent of the world’s HCC patients are in China, with over 300,000 new patients diagnosed every year. Currently the therapies commonly offered to most patients are surgery and local chemotherapy, with a 2-year recurrence rate of 51% and median survival time of 13 months.
CBMG’s research studies the effects of TC-DC (Tumor Stem Cell Specific Dendritic Cell) therapy. Dr. William Cao , President of CBMG said, “In simplified terms, TC-DC therapy takes a sample of the patient’s own dendritic, or immune cells and a sample of the patient’s tumor stem cells and places them together in the lab. The dendritic cell will learn the characteristics of the tumor stem cells, and is reintroduced to the patient’s body, where it can “train” the immune system to fight and destroy the tumor stem cells, which are the root cause of tumor recurrence and metastasis.”
About Cellular Biomedicine Group
Cellular Biomedicine Group, Inc. develops proprietary cell therapies for the treatment of certain degenerative diseases and cancers. Our developmental stem cell, progenitor cell, and immune cell projects are the result of research and development by scientists and doctors from China and the United States. Our flagship GMP facility, consisting of eight independent cell production lines, is designed, certified and managed according to U.S. standards. To learn more about CBMG, please visit:www.cellbiomedgroup.com
