
Home » Posts tagged 'TYPE 2 DIABETES' (Page 2)
Tag Archives: TYPE 2 DIABETES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lobeglitazone Sulfate
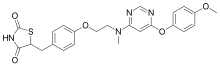
Lobeglitazone Sulfate, CKD-501
(Duvie®) Approved
Chong Kun Dang (Originator)
A dual PPARα and PPARγ agonist used to treat type 2 diabetes.
![]()
Trade Name:Duvie®MOA:Dual PPARα and PPARγ agonistIndication:Type 2 diabetes
CAS No. 607723-33-1(FREE)
763108-62-9(Lobeglitazone Sulfate)
2,4-Thiazolidinedione, 5-((4-(2-((6-(4-methoxyphenoxy)-4- pyrimidinyl)methylamino)ethoxy)phenyl)methyl)-, sulfate (1:1);
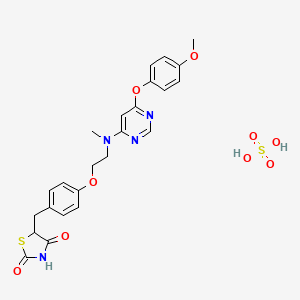
Lobeglitazone (trade name Duvie, Chong Kun Dang) is an antidiabetic drug in the thiazolidinedione class of drugs. As an agonistfor both PPARα and PPARγ, it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin.[3]
Lobeglitazone sulfate was approved by the Ministry of Food and Drug Safety (Korea) on July 4, 2013. It was developed and marketed as Duvie® by Chong Kun Dang Corporation.
Lobeglitazone is an agonist for both PPARα and PPARγ, and it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes.
Duvie® is available as tablet for oral use, containing 0.5 mg of free Lobeglitazone. The recommended dose is 0.5 mg once daily.
Lobeglitazone which was reported in our previous works belongs to the class of potent PPARα/γ dual agonists (PPARα EC50: 0.02 μM, PPARγ EC50: 0.018 μM, rosiglitazone; PPARα EC50: >10 μM, PPARγ EC50: 0.02 μM, pioglitazone PPARα EC50: >10 μM, PPARγ EC50: 0.30 μM). Lobeglitazone has excellent pharmacokinetic properties and was shown to have more efficacious in vivo effects in KKAy mice than rosiglitazone and pioglitazone.17 Due to its outstanding pharmacokinetic profile, lobeglitazone was chosen as a promising antidiabetes drug candidate.
Medical uses
Lobeglitazone is used to assist regulation of blood glucose level of diabetes mellitus type 2 patients. It can be used alone or in combination with metformin.[4]
Lobeglitazone was approved by the Ministry of Food and Drug Safety (Korea) in 2013, and the postmarketing surveillance is on progress until 2019.[4][5]
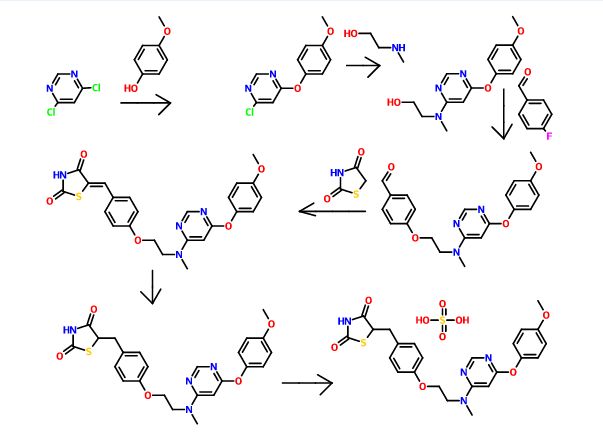
SYNTHESIS
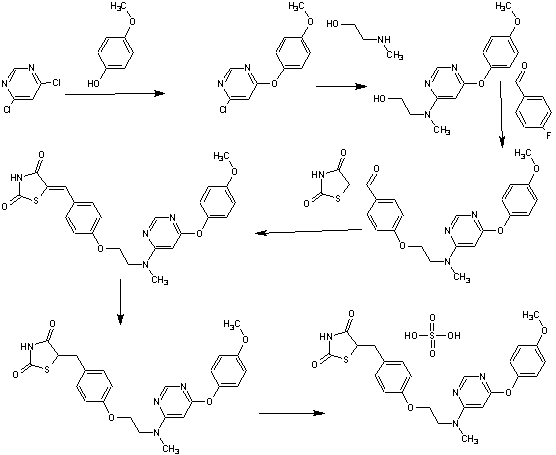

PAPER
Org. Process Res. Dev. 2007, 11, 190-199.
Process Development and Scale-Up of PPAR α/γ Dual Agonist Lobeglitazone Sulfate (CKD-501)
http://pubs.acs.org/doi/abs/10.1021/op060087u

A scaleable synthetic route to the potent PPARα/γ dual agonistic agent, lobeglitazone (1), used for the treatment of type-2 diabetes was developed. The synthetic pathway comprises an effective five-step synthesis. This process involves a consecutive synthesis of the intermediate, pyrimidinyl aminoalcohol (6), from the commercially available 4,6-dichloropyrimidine (3) without the isolation of pyrimidinyl phenoxy ether (4). Significant improvements were also made in the regioselective 1,4-reduction of the intermediate, benzylidene-2,4-thiazolidinedione (10), using Hantzsch dihydropyridine ester (HEH) with silica gel as an acid catalyst. The sulfate salt form of lobeglitazone was selected as a candidate compound for further preclinical and clinical study. More than 2 kg of lobeglitazone sulfate (CKD-501, 2) was prepared in 98.5% purity after the GMP batch. Overall yield of 2 was improved to 52% from 17% of the original medicinal chemistry route.
Silica gel TLC Rf = 0.35 (detection: iodine char chamber, ninhydrin solution, developing solvents: CH2Cl2/MeOH, 20:1); mp 111.4 °C; IR (KBr) ν 3437, 3037, 2937, 2775, 1751, 1698, 1648, 1610, 1503, 1439, 1301, 1246, 1215, 1183 cm-1; 1H NMR (400 MHz, CDCl3) δ 3.09 (m, 4H), 3.29 (m, 1H), 3.76 (s, 3H), 3.97 (m, 2H), 4.14 (m, 2H), 4.86 (m, 1H), 6.06 (bs, 1H), 6.86 (m, 2H), 7.00 (m, 2H), 7.13 (m, 4H), 8.30 (s, 1H), 11.99 (s, NH); 13C NMR (100 MHz, CDCl3) δ 37.1, 38.2, 53.7, 53.8, 56.3, 62.2, 65.8, 86.0, 115.1, 116.0, 123.0, 129.8, 131.2, 145.7, 153.4, 157.9, 158.1, 161.1, 166.5, 172.4, 172.5, 176.3, 176.5; MS (ESI)m/z (M + 1) 481.5; Anal. Calcd for C24H26N4O9S2: C, 49.82; H, 4.53; N, 9.68; S, 11.08. Found: C, 49.85; H, 4.57; N, 9.75; S, 11.15.
PATENT
References
- Lee JH, Noh CK, Yim CS, Jeong YS, Ahn SH, Lee W, Kim DD, Chung SJ. (2015). “Kinetics of the Absorption, Distribution, Metabolism, and Excretion of Lobeglitazone, a Novel Activator of Peroxisome Proliferator-Activated Receptor Gamma in Rats.”.Journal of Pharmaceutical sciences 104 (9): 3049–3059.doi:10.1002/jps.24378. PMID 25648999.
- Kim JW, Kim JR, Yi S, Shin KH, Shin HS, Yoon SH, Cho JY, Kim DH, Shin SG, Jang IJ, Yu KS. (2011). “Tolerability and pharmacokinetics of lobeglitazone (CKD-501), a peroxisome proliferator-activated receptor-γ agonist: a single- and multiple-dose, double-blind, randomized control study in healthy male Korean subjects.”. Clinical therapeutics 33 (11): 1819–1830.doi:10.1016/j.clinthera.2011.09.023. PMID 22047812.
- Lee JH, Woo YA, Hwang IC, Kim CY, Kim DD, Shim CK, Chung SJ. (2009). “Quantification of CKD-501, lobeglitazone, in rat plasma using a liquid-chromatography/tandem mass spectrometry method and its applications to pharmacokinetic studies.”. Journal of Pharmaceutical and Biomedical Analysis 50 (5): 872–877.doi:10.1016/j.jpba.2009.06.003. PMID 19577404.
- “MFDS permission information of Duvie Tablet 0.5mg”(Release of Information). Ministry of Food and Drug Safety. Retrieved2014-10-23.
- “국내개발 20번째 신약‘듀비에정’허가(20th new drug developed in Korea ‘Duvie Tablet’ was approved)”. Chong Kun Dang press release. 2013-07-04. Retrieved 2014-10-23.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-[(4-[2-([6-(4-Methoxyphenoxy)pyrimidin-4-yl]-methylamino)ethoxy]phenyl)methyl]-1,3-thiazolidine-2,4-dione
|
|
| Clinical data | |
| Trade names | Duvie |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | >99%[1] |
| Metabolism | liver (CYP2C9, 2C19, and 1A2)[1] |
| Biological half-life | 7.8–9.8 hours[2] |
| Identifiers | |
| CAS Number | 607723-33-1 |
| PubChem | CID 9826451 |
| DrugBank | DB09198 |
| ChemSpider | 8002194 |
| Synonyms | CKD-501 |
| Chemical data | |
| Formula | C24H24N4O5S |
| Molar mass | 480.53616 g/mol |
///Lobeglitazone Sulfate, CKD-501, Duvie®, Approved KOREA, Chong Kun Dang, A dual PPARα and PPARγ agonist , type 2 diabetes.
CN(CCOC1=CC=C(C=C1)CC2C(=O)NC(=O)S2)C3=CC(=NC=N3)OC4=CC=C(C=C4)OC.OS(=O)(=O)O
Henagliflozin

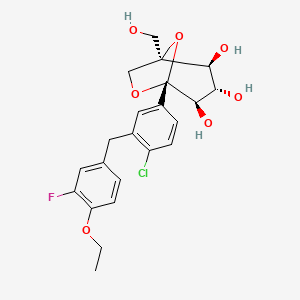

Henagliflozin, SHR-3824 ,
CAS 1623804-44-3
C22-H24-Cl-F-O7, 454.8756
PHASE 2 for the treatment of type 2 diabetes
China 20222, approvals 2022
HengRui (Originator)
| Jiangsu Hengrui Medicine Co Ltd |
UNII-21P2M98388; 21P2M98388; Henagliflozin; SHR3824; SHR-3824;

- HENAGLIFLOZIN PROLINE
- 4IO819SW6M
- 570.0 g/mol
- C27H33ClFNO9
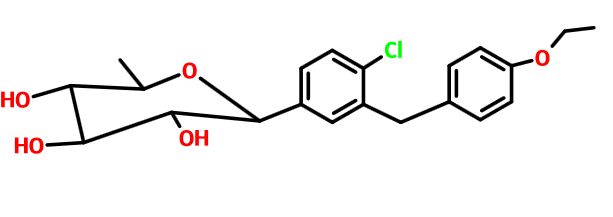
- (1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol;(2R)-pyrrolidine-2-carboxylic acid
In April 2016, Jiangsu Hengrui Medicine is developing henagliflozin (phase 2 clinical trial), a sodium-glucose cotransporter-2 (SGLT-2) inhibitor, for treating type 2 diabetes.
SGLT1 and SGLT2 inhibitors, useful for treating eg diabetes.
Henagliflozin proline is in phase II clinical trials by Jiangsu Hengrui (江苏恒瑞) for the treatment of type 2 diabetes.
1,6-dehydrated-1-C{4-chloro-3-[(3-fluoro-4-ethoxyphenyl)methyl]phenyl}-5-C-(hydroxymethyl)-β-L-idopyranose L-proline
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide
(1R,2S,3S,4R,5R)-5-[4-chloro-3-[(4-ethoxy-3-fluorophenyl)methyl]phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
Henagliflozin is a pharmaceutical drug for the treatment of type 2 diabetes.[1] In China, it is approved for adult patients with type 2 diabetes to improve the glycemic control.[2][3]
Henagliflozin, like other drugs of the gliflozin class, inhibits the transporter protein sodium/glucose cotransporter 2 (SGLT2) which leads to a reduction in blood glucose levels.[4]
Shanghai Hengrui Pharmaceutical Co., Ltd., 上海恒瑞医药有限公司, Jiangsu Hengrui Medicine Co., Ltd., 江苏恒瑞医药股份有限公司, Less «
- 01 May 2015 Jiangsu HengRui Medicine Co. initiates enrolment in a phase I drug interaction trial in volunteers in China (NCT02500485)
- 12 Feb 2015 Jiangsu HengRui Medicine plans a phase I trial for Type-2 diabetes mellitus in China (NCT02366377)
- 01 Feb 2015 Jiangsu HengRui Medicine initiates enrolment in a phase I trial for Type-2 diabetes mellitus in China (NCT02366351)
Henagliflozin is a novel sodium-glucose transporter 2 inhibitor and presents a complementary therapy to metformin for patients with T2DM due to its insulin-independent mechanism of action. This study evaluated the potential pharmacokinetic drug-drug interaction between henagliflozin and metformin in healthy Chinese male subjects. 2. In open-label, single-center, single-arm, two-period, three-treatment self-control study, 12 subjects received 25 mg henagliflozin, 1000 mg metformin or the combination. Lack of PK interaction was defined as the ratio of geometric means and 90% confidence interval (CI) for combination: monotherapy being within the range of 0.80-1.25. 3. Co-administration of henagliflozin with metformin had no effect on henagliflozin area under the plasma concentration-time curve (AUC0-24) (GRM: 1.08; CI: 1.05, 1.10) and peak plasma concentration (Cmax) (GRM: 0.99; CI: 0.92, 1.07). Reciprocally, co-administration of metformin with henagliflozin had no clinically significant on metformin AUC0-24 (GRM: 1.09, CI: 1.02, 1.16) although there was an 11% increase in metformin Cmax (GRM 1.12; CI 1.02, 1.23). All monotherapies and combination therapy were well tolerated. 4. Henagliflozin can be co-administered with metformin without dose adjustment of either drug.
PATENT
PATENT
WO2012019496
https://www.google.com/patents/WO2012019496A1?cl=en
Example 4
(1 ^ 2345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) 6,8 – alcohol dioxide


first step
1-ethoxy-2-fluoro – benzene
A mixture of 2-fluoro-phenol 4a (6.7 g, 60 mmol) was dissolved in 66 mL of acetone, was added iodoethane (6.3 mL,
78 mmol) and potassium carbonate (12.4 g, 90 mmol), at reflux in an oil bath for 5 hours. The reaction solution was concentrated under reduced pressure, was added 100 mL of ethyl acetate and 60 mL of water, separated, the aqueous phase was extracted with ethyl acetate (30 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, to give the title product 1-ethoxy-2-fluoro – benzene 4b (6.9 g, red oil). yield: 82.1%.
MS m / z (ESI): 280.2 [2M + 1]
The second step
(5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone A mixture of 5-bromo-2-chloro – benzoyl chloride 2a (12.4 g, 48.8 mmol) was dissolved a 100 mL of dichloromethane was added 1-ethoxy-2-fluoro – benzene 4b (6.84 g, 48.8 mmol), cooled to 0 ° C, was added portionwise aluminum (5.86 g, 44 mmol) chloride, 16 h. Was added dropwise under ice-cooling to the reaction mixture 20 mL of 2 M HCl solution, separated, the aqueous phase was extracted with 30 mL of dichloromethane, and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title The product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanone 4c (12.7 g, yellow solid), yield: 72.6%.
MS m / z (ESI): 358.9 [M + l] Step
(5 – bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol (5-Bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) -methanone 4c (12.7 g, 35.5 mmol) was dissolved in methanol and a 100 mL of tetrahydrofuran (ν: ν = 1: 1) mixed solvent, under an ice bath was added portionwise sodium borohydride (2.68 g, 70 mmol), and reacted at room temperature for 30 minutes. Add 15 mL of acetone, the reaction solution was concentrated under reduced pressure, 150 mL of ethyl acetate was added to dissolve the residue, washed with saturated sodium chloride solution (50 mLx2). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure The filtrate, to give the title product (5-bromo-2-chloro – phenyl) – (4-ethoxy-3-fluoro-phenyl) – methanol 4d (12.7 g, orange oil), was used directly without isolation next reaction.
the fourth step
4 – [(5-bromo-2-chloro-phenyl) – methyl] Small-ethoxy-2-fluoro – benzene (5-bromo-2-chloro – phenyl) – (4-ethoxy -3 – fluoro – phenyl) methanol 4d (12.7 g, 35.3 mmol) was dissolved in a 100 mL of dichloromethane was added triethylsilane (16.9 mL, 106 mmol), was added dropwise boron trifluoride etherate (8.95 mL, 70.6 mmol ), for 3 hours. Was added 50 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with ethyl acetate (100 mLx2), the organic phases combined, dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography to elute B surfactant system resulting residue was purified to give the title product 4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (10 g, as a pale yellow oil ) yield: 82.4%.
1H NMR (400 MHz, CDC1 3 ): δ 7.33-7.27 (m, 3H), 6.95-6.90 (m, 3H), 4.14 (q, 2H), 4.01 (s, 2H), 1.49 (t, 3H)
the fifth step
(2 3R, 4S, 5 ^ 6R) -2- [4- chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) – 2-methoxy – tetrahydro-pyran-3,4,5-triol
4 – [(5-bromo-2-chloro – phenyl) methyl] -1-ethoxy-2-fluoro – benzene 4e (7.36 g, 21.4 mmol) was dissolved in 30 mL of tetrahydrofuran, cooled to -78 ° C, was added dropwise a solution of n-butyllithium in hexane (10.27 mL, 25.7 mmol), at -78 ° C to react 1 hour, a solution of 20 mL (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy) -6- (trimethylsilyloxy) tetrahydropyran-2-one 2f (llg, 23.6 mmol) in tetrahydrofuran at -78 ° C under reaction 2 h, 2.8 mL of methanesulfonic acid and 71 mL of methanol, the reaction at room temperature for 16 hours. Was added 100 mL of saturated sodium carbonate solution, the reaction solution was concentrated under reduced pressure, to the residue was added 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (100 mLx3), organic phases were combined, dried over anhydrous magnesium sulfate, filtered, The filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (2 3R, 4S, 5 6R) -2- [4- chloro-3 – [(4-ethoxyphenyl 3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – tetrahydro-pyran-3,4,5-triol 4f (5.7 g, white solid ) yield: 58.3%.
1H NMR (400 MHz, CD 3 OD): δ 7.56 (s, 1H), 7.48 (dd, 1H), 7.37 (dd, 1H), 6.95-6.87 (m, 3H), 4.08-4.07 (m, 4H) , 3.91 (m, 1H), 3.93-3.73 (m, 2H), 3.56-3.53 (m, 1H), 3.45-3.43 (m, 1H), 3.30 (s, 2H), 3.08 (s, 3H), 1.35 (t, 3H)
The sixth step
(2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro – phenyl) methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol the (2 3R, 4S, 5 6R) -2- [4- chloro-3- [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6- (hydroxymethyl) -2-methoxy – 4f tetrahydropyran-3,4,5-triol (5.7 g, 12.5 mmol) was dissolved in 50 mL of pyridine, followed by adding tert-butyldimethylsilyl chloride (2.26 g, 15 mmol) and 4-dimethylaminopyridine (305 mg, 2.5 mmol), for 16 hours. The reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate, washed with a saturated copper sulfate solution (50 mLx3). The combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 3R, 4S, 5 6R) -6- [(tert-butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, colorless oil), without isolation directly used for the next reaction.
Seventh Step
[[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethyl-silane (2 3R, 4S, 5 6R) -6- [(tert butyl (dimethyl) silyl) oxymethyl] -2- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2-methoxy yl – tetrahydro-pyran-3,4,5-triol 4g (7.14 g, 12.5 mmol) was dissolved in 100 mL N, N- dimethylformamide was added 60% sodium hydride under ice-cooling (2.5 g , 62.5 mmol), and reacted at room temperature for 40 minutes completed the opening force, was added benzyl bromide (7.5 mL, 62.5 mmol), reaction of 16 hours. 20 mL of methanol, the reaction solution was concentrated under reduced pressure, was added 200 mL of ethyl acetate and 50 mL of water to dissolve the residue, separated, the aqueous phase was extracted with ethyl acetate (50 mL), the organic phase was washed with water (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate , filtered, and the filtrate was concentrated under reduced pressure to give the title product [[(2R, 3R, 4S, 5R, 6 ^ -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4- ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.5 g , yellow oil) yield: 99.8%.
Step Eight
[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol
The [[(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl yl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methoxy] – tert-butyl – dimethylsilane 4h (10.52 g, 12.5 mmol) was dissolved in 50 mL of methanol dropwise add acetyl chloride CO.13 mL, 1.9 mmol), for 1 hour. The reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy–6 – [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g , yellow oil yield: 83.6%.
Step Nine
(2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde
Oxalyl chloride (1.17 mL, 13.6 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 ° C, were added dropwise 20 mL of dimethyl sulfoxide (1.56 mL, 21.9 mmol) in methylene chloride and 50 mL [(2R, 3R, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-yl] methanol 4i (7.6 g, 10.45 mmol) in methylene chloride, and reacted at -78 ° C for 30 min, triethylamine (7.25 mL, 52.3 mmol), 2 hours at room temperature was added 50 mL 1 M HCl solution, separated, the organic phase was washed with saturated sodium chloride solution (50 mL x 2), the aqueous phase was extracted with dichloromethane (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (2 ^ 3456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4 – ethoxy-3-fluoro-phenyl) – methyl] phenyl] -6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.58 g, colorless oil), was used directly without isolation next reaction.
The tenth step
(2S, 3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl ] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 6-methoxy – tetrahydropyran-2-carbaldehyde 4j (7.6 g, 10.45 mmol) was dissolved in 80 mL 1,4- dioxane, followed by adding 15.8 mL 37% aqueous formaldehyde and sodium hydroxide solution (31.35 mL, 31.35 mmol), reacted at 70 ° C for 16 h. Add 50 mL of saturated sodium chloride solution, extracted with ethyl acetate (50 mLx4), the organic phase was washed with saturated sodium bicarbonate solution (50 mL), washed with saturated sodium chloride solution (50 mL), the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the title product (23,456 benzyloxy-3,4,5-tris – 6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran – 2- formaldehyde 4k (7.9g, as a colorless oil), without isolation directly used for the next reaction.
Step Eleven
[(3 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol
The (23456 3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] – 2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-carbaldehyde 4k (7.9 g, 10.45 mmol) was dissolved in 50 mL of tetrahydrofuran and methanol (v: v = 2: 3) mixed solvent , was added sodium borohydride (794 mg, 20.9 mmol), for 30 minutes. Add a small amount of acetone, the reaction solution was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product, 5R, 6 -3,4,5-tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -2- (hydroxymethyl ) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, colorless oil). yield: 14.1%.
Step Twelve
[(12345 ^ -2,3,4-tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol
The [(3S, 4S, 5R, 6 -3,4,5- tris-benzyloxy-6- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -2- (hydroxymethyl) -6-methoxy – tetrahydropyran-2-yl] methanol 4m (l.ll g, 1.46 mmol) was dissolved in 20 mL of dichloromethane, cooled to -10 ° C, was added trifluoroacetic acid (0.23 mL, 3 mmol), and reacted at room temperature for 2 hours. 20 mL of saturated sodium bicarbonate solution, separated, the aqueous phase was extracted with dichloromethane (20 mL> <2), and the combined organic phase was dried over anhydrous magnesium sulfate, filtered, and the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems B resultant residue was purified to give the title product [(1 2 3 4R, 5 -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] 6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4nC830 mg, colorless oil). yield: 78.3%.
MS m / z (ESI): 742.3 [M + 18]
Thirteenth Step
(12345-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8 dioxa-bicyclo [3.2.1] octane-2,3,4-triol
The [(1 2 3 4R, 5S) -2,3,4- tris-benzyloxy-5- [4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] benzene yl] -6,8-dioxa-bicyclo [3.2.1] octane-1-yl] methanol 4n (830 mg, 1.14 mmol) was dissolved in 20 mL of tetrahydrofuran and methanol (v: v = l: l) the a mixed solvent of o-dichlorobenzene was added (1.3 mL, 1 1.4 mmol) and Pd / C (500 mg, 10%), purged with hydrogen three times, the reaction for 3 hours. The reaction solution was filtered, rinsed with a small amount of ethyl acetate, the filtrate was concentrated under reduced pressure, purified by silica gel column chromatography with eluent systems resulting A residue was purified to give the title product (1S, 2 3S, 4R, 5 -5- [ 4-chloro-3 – [(4-ethoxy-3-fluoro-phenyl) – methyl] phenyl] -1- (hydroxymethyl) -6,8-dioxa-bicyclo [3.2.1] octane-2,3,4-triol 4 (420 mg, white solid), yield: 81.0% MS m / z (ESI):. 472.2 [m + 18]
1H NMR (400 MHz, CD 3 OD): δ 7.47 (s, 1H), 7.42-7.35 (m, 2H), 6.95-6.87 (m, 3H), 4.16-4.14 (m, 1H), 4.06-4.02 ( m, 4H), 3.85-3.70 (m, 2H), 3.67-3.54 (m, 4H), 1.37 (t, 3H)
References
- Weng J, Zeng L, Zhang Y, Qu S, Wang X, Li P, et al. (August 2021). “Henagliflozin as add-on therapy to metformin in patients with type 2 diabetes inadequately controlled with metformin: A multicentre, randomized, double-blind, placebo-controlled, phase 3 trial”. Diabetes, Obesity & Metabolism. 23 (8): 1754–1764. doi:10.1111/dom.14389. PMID 33769656.
- Wang G (17 February 2022). “Monthly Report: New Drug Approvals in China, January 2022”. BaiPharm.
Henagliflozin Proline Tablets
- “Henagliflozin – Jiangsu HengRui Medicine”. AdisInsight. Springer Nature Switzerland AG.
- He X, Liu G, Chen X, Wang Y, Liu R, Wang C, et al. (July 2023). “Pharmacokinetic and Pharmacodynamic Interactions Between Henagliflozin, a Novel Selective SGLT-2 Inhibitor, and Warfarin in Healthy Chinese Subjects”. Clinical Therapeutics. 45 (7): 655–661. doi:10.1016/j.clinthera.2023.06.002. PMID 37451912.
 |
|
| Clinical data | |
|---|---|
| Trade names | Rui Qin; 瑞沁 |
| Other names | SHR3824; SHR-3824 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H24ClFO7 |
| Molar mass | |
////////Henagliflozin, SHR-3824 , PHASE 2, type 2 diabetes, UNII-21P2M98388, 21P2M98388, SHR 3824, SHR3824, approvals 2022, china 2022, Henagliflozin proline
CCOc1ccc(cc1F)Cc2cc(ccc2Cl)[C@]34[C@@H]([C@H]([C@@H]([C@](O3)(CO4)CO)O)O)O
SYN
Synthesis 2024, 56, 906–943
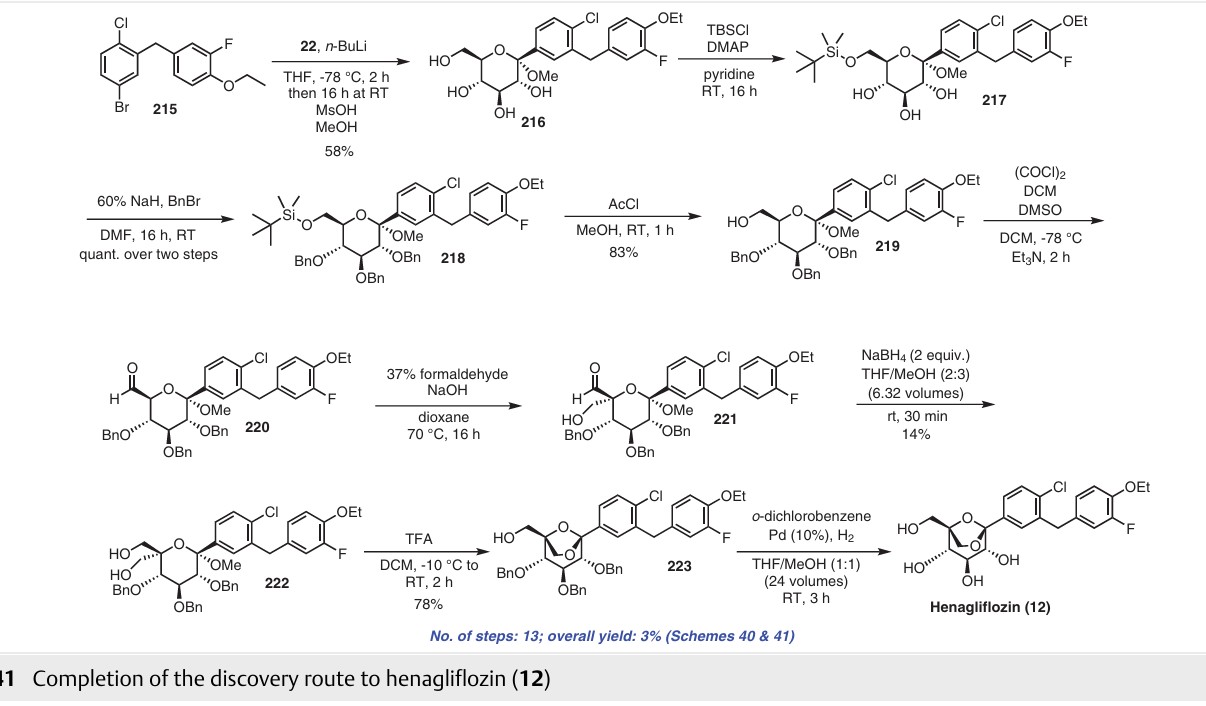
Henagliflozin (12) (also known as SHR3824), developed by Lexicon Pharmaceuticals (Princeton, NJ, USA), is a potent and selective SGLT inhibitor administered orally. In 2013, the first synthetic route for the preparation of henagliflozin (12) was described and claimed by two pharmaceutical companies: Shanghai Hengrui Pharmaceutical Co., Ltd., and Jiangsu Hengrui Medicine Co., Ltd. Several other C-aryl-glucoside-type derivatives were prepared and registered in the United States under patent application number US8609622B2.67 Among these derivatives, the synthesis of henagliflozin (12) was carried out using a thirteen-step process, resulting in an overall yield of 3% (Schemes 40 and 41). The process consisted of the formation of the key intermediate 215 starting from commercially available 2-fluorophenol (211). In the first step, phenolic compound 211 was converted into 212 in 82% yield using ethyl bromide and po
tassium carbonate in acetone. The Friedel–Crafts reaction of acid chloride 26c′ using AlCl3 in DCM afforded intermediate 213 in 72% yield, which was further reduced to 214 using NaBH4 in a mixture of THF/MeOH. Without further isolation, the reduction of 214 was carried out using Et3SiH and BF3·Et2O in DCM to give 215 (Scheme 40). The intermediate 215 was taken forward for lithium halogen exchange using n-BuLi followed by addition of the lithiated compound to O-silyl-protected compound 22 at
low temperature to afford a lactol intermediate. The obtained lactol intermediate was protected using
MsOH/MeOH to give the desired product 216 in 58% yield. Under the above conditions, deprotection of the O-silylgroups of the C-glucoside 22 was also observed. Further, under basic conditions, the secondary hydroxy group of 216 was silyl protected using tert-butyldimethylsilyl chloride (TBSCl) and DMAP to afford compound 217, which was treated with NaH and BnBr to give benzylated compound
218 in excellent yield. In methanol solution, deprotection of the silyl protecting group of compound 218 using acetylchloride afforded 219. Swern oxidation of the hydroxy compound 219 in the presence of oxalyl chloride and DMSO gave intermediate 220, which was used for the next step without isolation. The crude compound 220 was treated with NaOH and 37% formaldehyde solution to afford 221.
Dihydroxy intermediate 222 was then obtained in low yield via reduction of the aldehyde group of compound 221 with sodium borohydride in THF/MeOH mixture. Next, treatment of 222 with trifluoroacetic acid gave compound 223. Debenzylation of compound 223 was carried out by Pd/C
catalytic hydrogenation to afford the final product henaglifozin (12) (Scheme 41).
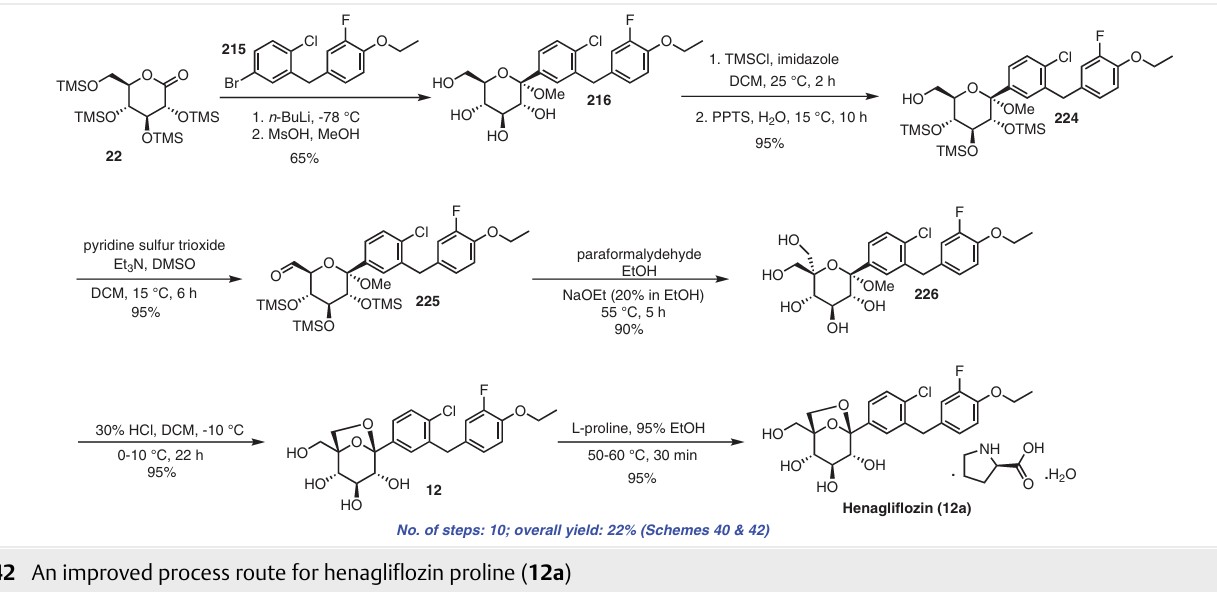
The highlight of the synthesis is the design of the route with minimal isolation stages and intermediates possessing unstable functional groups were subjected to subsequent transformations in situ. The drawbacks of the above synthetic process are the use of a protection and deprotection
strategy that led to low throughput and the final compound being obtained in low yield. Reduction of the aldehyde in 221 mediated by sodium borohydride resulted in a poor yield of product 222, and this procedure is not recommend ed for scale-up due to safety concerns. Additionally, the use
of palladium in the last step of the synthesis involves the risk of this toxic metal leaching into the final product. To address the issue with the discovery route, Yongjun and co-workers reported an alternative approach to obtain compound 12 (Scheme 42).68 The authors published the synthesis of henagliflozin proline (12a) starting from TMS protected D-glucolactone 22 and aglycone intermediate The diol 226 was obtained after carrying out a disproportionation reaction on the aldehyde using paraformaldehyde under strong alkaline conditions. Intramolecular etherification of diol 226 using 30% HCl gave henagliflozin
(12) in 95% yield, which was further treated with L-proline to give henagliflozin proline monohydrate 12a. The authors reported several advantages such as easy steps, cost-effective procedures, simple product purification and an overall method that was amenable for commercialization. This Addition of the aglycone intermediate 215 was carried out with 22 followed by mesylation of the OH group to provide 216 in 65% yield. Further, all the secondary hydroxy groups of intermediate 216 were selectively protected us ing TMSCl, imidazole and PPTS to give 224 in 95% yield. The free primary hydroxy group of 224 was oxidized using pyridine sulfur trioxide in triethylamine and DMSO to afford process involves 10 steps and gave an overall yield of 22% of henagliflozin proline (12a) (Schemes 40 and 42)
REF 67, 68
(67) Yang, F.; Tang, P. C.; Dong, Q.; Tu, W.; Fan, J.; Guan, D.; Shen, G.;Wang, Y.; Yuan, J.; Zhang, L. US8609622B2, 2013.
(68) Chun, K.; Peng, Z.; Qichao, L.; Bo, Z.; Zhen, W.; Guorong, Z.;Yongjun, T. CN 112375087A, 2020.
.
Tianagliflozin IND filed by Tianjin Institute of Pharmaceutical research


Tianagliflozin,
taigeliejing, 6-deoxydapagliflozin
| Molecular Formula: | C21H25ClO5 |
|---|---|
| Molecular Weight: | 392.8732 g/mol |
IND Filing…Tianjin Institute of Pharmaceutical research
Tianjin Institute Of Pharmaceutical Research,
(3R,4S,5S,6R)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methyloxane-3,4,5-triol
1–[4–Chloro–3–(4–ethoxybenzyl)phenyl]–1,6–dideoxy–β–d–glucopyranose
![]()
CAS N. 1461750-27-5
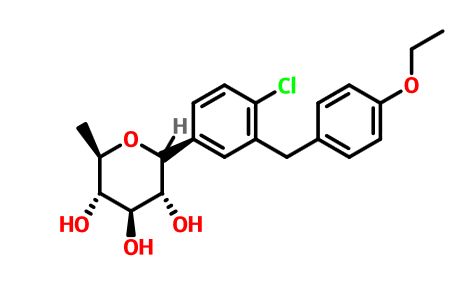

The structures of dapagliflozin and 6-deoxydapagliflozin (1)
,deletion of the 6-OH in the sugar moiety of dapagliflozin led to the discovery of a more potent SGLT2 inhibitor, 6-deoxydapagliflozin (1, ). In an in vitro assay, 1 was a more active SGLT2 inhibitor, with IC 50 = 0.67 nM against human SGLT2 (hSGLT2), as compared with 1.1 nM for dapagliflozin, leading to the identification of 1 as the most active SGLT2 inhibitor discovered so far in this field. Also in an in vivo assay, 1 also introduced more urinary glucose in a rat urinary glucose excretion test (UGE) and exhibited more potent blood glucose inhibitory activity in a rat oral glucose tolerance test (OGTT) than dapagliflozin.
Tianjin Institute Of Pharmaceutical Research,天津药物研究院

SPECTRAL DATA of Tianagliflozin
1 as a white solid (3.65 g, 93 %). R f = 0.35 (EtOAc);
m.p.: 148–149 °C;
1H NMR (400 MHz, DMSO-d 6): δ = 7.35 (d, 1H, J = 8.4 Hz), 7.25 (s, 1H), 7.18 (d, 1H, J = 8.0 Hz), 7.08 (d, 2H, J = 8.4 Hz), 6.81 (d, 2H, J = 8.4 Hz), 4.95 (d, 1H, J = 5.2 Hz, OH), 4.90 (d, 1H, J = 4.4 Hz, OH), 4.79 (d, 1H, J = 5.6 Hz, OH), 3.92–4.01 (m, 5H), 3.24–3.29 (m, 1H), 3.18–3.22 (m, 1H), 3.09–3.15 (m, 1H), 2.89–2.95 (m, 1H), 1.29 (t, 3H, J = 7.0 Hz, CH2 CH 3 ), 1.15 (d, 3H, J = 6.0 Hz, CHCH 3 ) ppm;
13C NMR (100 MHz, DMSO-d 6): δ = 156.85, 139.65, 137.82, 131.83, 131.16, 130.58, 129.52, 128.65, 127.14, 114.26, 80.71, 77.98, 75.77, 75.51, 74.81, 62.84, 37.55, 18.19, 14.62 ppm;
IR (KBr): v¯¯¯ = 3,564 (w), 3,385 (s), 2,981 (s), 2,899 (s), 2,861 (s), 1,613 (m), 1,512 (s), 1,477 (m), 1,247 (s), 1,102 (s), 1,045 (s), 1,012 (s) cm−1;
HR–MS: calcd for C21H29ClNO5 ([M + NH4]+) 410.1729, found 410.1724.
PATENT
CN 103864737
http://www.google.com/patents/CN103864737A?cl=en
PATENT
WO 2014094544
http://www.google.com/patents/WO2014094544A1?cl=en


-27-


1 D1 -6 Optionally, the step (7 ‘) is the step (7’) in place:
LS l- [4 – D (I- Dl- 6)

A.

(DMSO-d 6, 400 MHz), δ 7.35 (d, 1H, J = 8.0 Hz), 7.28 (d, 1H, J ‘. 2.0 Hz), 7.17 (dd, IH, / = 2.0 Hz and 8.4 Hz), 7.05 (d, 2H, J: 8.8 Hz), 6.79 (d, 2H, 8.8 Hz): 4.924,95 (m, 2H), 4,81 (d, IH, 6,0 Hz), 3.93- 3.99 (m, 5H), 3,85 (d, 1H, J = 10,4 Hz), 3,66 (dd, IH, 5,2 Hz and 11,6 Hz), 3.17-3,28 (m, 3H), 3.02-3.08 (m: IH), 1.28 (t, 3H, J = 7,0 Hz), 0,80 (s, 9H), -0.05 (s, 3H), -0.09 (s, 3H) .
PATENT
[0066] The added 100mL dried over anhydrous methanol 0. 5g of sodium metal, nitrogen at room temperature with stirring, until the sodium metal disappeared. Followed by addition of 5. 2g (10mmol) of compound 6, stirring was continued at room temperature for 3 hours. To the reaction system was added 5g strong acid cation exchange resin, stirred at room temperature overnight, the reaction mixture until pH = 7. The resin was removed by suction, and the filtrate evaporated to dryness on a rotary evaporator, the residue was further dried on a vacuum pump to give the product I-D1-6, as a white foamy solid.
PATENT
WO 2014139447
PATENT related
http://www.google.com/patents/WO2013044608A1?cl=en
http://link.springer.com/article/10.1007%2Fs40242-014-4043-9#/page-1
Design of SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: A History Driven by Biology to Chemistry.
Abstract
A brief history of the design of sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors is reviewed. The design of O-glucoside SGLT2 inhibitors by structural modification of phlorizin, a naturally occurring O-glucoside, in the early stage was a process mainly driven by biology with anticipation of improving SGLT2/SGLT1 selectivity and increasing metabolic stability. Discovery of dapagliflozin, a pioneering C-glucoside SGLT2 inhibitor developed by Bristol-Myers Squibb, represents an important milestone in this history. In the second stage, the design of C-glycoside SGLT2 inhibitors by modifications of the aglycone and glucose moiety of dapagliflozin, an original structural template for almost all C-glycoside SGLT2 inhibitors, was mainly driven by synthetic organic chemistry due to the challenge of designing dapagliflozin derivatives that are patentable, biologically active and synthetically accessible. Structure-activity relationships (SAR) of the SGLT2 inhibitors are also discussed.
http://www.ncbi.nlm.nih.gov/pubmed/25557661
Paper

Discovery of 6-Deoxydapagliflozin as a Highly Potent Sodium-dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes
http://www.ingentaconnect.com/content/ben/mc/2014/00000010/00000003/art00009?crawler=true
CLIP

A facile synthesis of 6-deoxydapagliflozin


The synthetic route to the target compound 1 is shown in Scheme 3. The starting material methyl 2,3,4-tri-O-benzyl-6-deoxy-6-iodo-α–d-glucopyranoside (3) was prepared from commercially available methyl α–d-glucopyranoside (2) according to a known method [5, 6].
Iodide 3 was reductively deiodinated to give 4 in 91 % yield under hydrogenolytic conditions using 10 % Pd/C as catalyst in the presence of Et3N as base in THF/MeOH at room temperature.
when the iodide 3 was treated with Barton–McCombie reagent (n-Bu3SnH/AIBN) [7] in toluene at room temperature no reaction occurred; however, when the reaction was carried out at elevated temperatures, such as reflux, a complex mixture formed with only a trace amount (3 %, entry 1) of the desired product 4.
When the iodide 3 was treated with LiAlH4 in THF at 0 °C to room temperature, another complex mixture was produced with only a trace amount (2 %, entry 2) of 4.
When Pd(OH)2 was used as the hydrogenolysis catalyst instead of 10 % Pd/C, the desired 4 was indeed formed (14 %, entry 4), but most of the starting material was converted to a few more polar byproducts, which were believed to result from the cleavage of at least one of the benzyl groups.
pdf available
Monatshefte für Chemie – Chemical Monthly
December 2013, Volume 144, Issue 12, pp 1903-1910
////////IND Filing, SGLT-2 inhibitor, type 2 diabetes, Tianagliflozin, taigeliejing, 6-deoxydapagliflozin, 1461750-27-5
Clc1c(cc(cc1)C2[C@@H]([C@H]([C@@H]([C@H](O2)C)O)O)O)Cc3ccc(cc3)OCC
RO-28-1675 for Type 2 Diabetes

RO-28-1675
- (2R)-3-Cyclopentyl-2-[4-(methanesulfonyl)phenyl]-N-(thiazol-2-yl)propionamide
- Ro 028-1675
- Ro 0281675
- Ro 28-1675
3-Cyclopentyl-2(R)-[4-(methylsulfonyl)phenyl]-N-(2-thiazolyl)propionamide
| MW | 378.51 | .-70.4 °
Conc 0.027 g/100mL; chloroform, 589 nm; 23 °C
|
|
|---|---|---|---|
| Formula | C18H22N2O3S2 | ||
| CAS No | 300353-13-3 |
Glucokinase Activators
Ro 28-1675 (Ro 0281675) is a potent allosteric GK activator with a SC1.5 value of 0.24± 0.0019 uM.
Roche (Innovator)
PHASE 1 Type 2 DIABETES,
IC50 value: 0.24± 0.0019 uM (SC1.5) [1]
Target: Glucokinase activator
The R stereoisomer Ro 28-1675 activated GK with a SC1.5 of 0.24 uM, while the S isomer did not activated GK up to 10 uM. Oral administration of Ro 28-1675 (50 mg/Kg) to male C57B1/6J mice caused a statistically significant reduction in fasting glucose levels and improvement in glucose tolerance relative to the vehicle treated animals [1].
Comparison of rat PK parameters indicated that Ro 28-1675 displayed lower clearance and higher oral bioavailability compared to 9a.
Following a single oral dose, Ro 28-1675 reduced fasting and postprandial glucose levels following an OGTT, was well tolerated, and displayed no adverse effects related to drug administration other than hypoglycemia at the maximum dose (400 mg).
 .
.
RO-28-1675 as glucokinase activator.
Joseph Grimsby et al., of Roche have recently discovered activators of glucokinase that increase kcat and decrease the S0.5 for glucose, and these may offer a treatment for type II diabetes. Glucokinase (GK) plays a key role in whole-body glucose homeostasis by catalyzing the phosphorylation of glucose in cells that express this enzyme, such as pancreatic β cells and hepatocytes.
By screening of a library of 120,000 structurally diverse synthetic compounds, they found one small molecule that increased the enzymatic activity of GK. Chemical optimization of this initial molecule led to the synthesis of RO-28-0450 as a lead GK activator which is a class of antidiabetic agents that act as nonessential, mixed-type GK activators (GKAs) that increase the glucose affinity and maximum velocity (Vmax) of GK. RO-28-0450 is a racemic compound.
Activation of GK was exquisitely sensitive to the chirality of the molecule: The R enantiomer, RO-28-1675, was found to be a potent GKA, whereas the S enantiomer, RO-28-1674, was inactive. RO-28-1675 also reversed the inhibitory action of the human glucokinase regulatory protein (GKRP). The activators binding in a glucokinase regulatory site originally was discovered in patients with persistent hyperinsulinemic hypoglycemi.
The result of RO-28-1675 as a potent small molecule GKA may shed light to the chemical biologists to devise strategy for developing activators. Thus for a success to this end we must focus on highly regulated enzymes, or cooperative enzymes such as glucokinase, where nature has provided binding sites that are designed to modulate catalysis.
.SYNTHESIS


Paper

Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl-propionamide (2.10 g, 74%) as a white foam.
[α] 23 589 = –70.4° (c=0.027, chloroform).
EI-HRMS m/e calcd for C18H22N2O3S2 (M+ ) 378.1072, found 378.1081.
1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 10.48 (br. s., 1 H), 7.88 (d, J=8.6 Hz, 2 H), 7.53 (d, J=8.6 Hz, 2 H), 7.50 (d, J=3.5 Hz, 1 H), 7.06 (d, J=3.5 Hz, 1 H), 3.76 (t, J=7.7 Hz, 1 H), 3.03 (s, 3 H), 2.28 (dt, J=13.6, 7.7 Hz, 1 H), 1.88 – 1.98 (m, 1 H), 1.42 – 1.84 (m, 7 H), 1.07 – 1.19 (m, 2 H).
Anal. Calcd for C18H22N2O3S2: C, 56.94; H, 5.59; N, 7.28. Found: C, 57.12; H, 5.86; N, 7.40.
PATENT
WO 2000058293
http://www.google.com/patents/WO2000058293A2?cl=en
Example 3 (A) 3-CyclopentyI-2-(4-methanesulfonyl-phenyI)-N-thiazol-2-yI-propionamide

A solution of dπsopropylamine (3.3 mL, 23.5 mmol) in dry tetrahydrofuran (50 mL) and 1.3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdιnone (10 mL) was cooled to -78°C under nitrogen and then treated with a 10M solution of n-butyllithium m hexanes (2.35 mL, 23 5 mmol) The yellow reaction mixture was stiπed at -78°C for 30 mm and then treated dropwise with a solution of 4-methylsulfonylphenylacetιc acid (2.40 g, 11.2 mmol) in a small amount of dry tetrahydrofuran. After approximately one-half of the 4- methylsulfonylphenylacetic acid m dry tetrahydrofuran was added, a precipitate formed Upon further addition of the remaining 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture became thick in nature After complete addition of the 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture was very thick and became difficult to stir An additional amount of dry tetrahydrofuran (20 mL) was added to the thick reaction mixture, and the reaction mixture was stirred at –
78 C for 45 mm, at which time, a solution of lodomethylcyclopentane (2.35 g, 11.2 mmol) in a small amount of dry tetrahydrofuran was added dropwise The reaction mixture was allowed to warm to 25°C where it was stiπed for 15 h. The reaction mixture was quenched with water (100 mL), and the resulting yellow reaction mixture was concentrated in vacuo to remove tetrahydrofuran. The aqueous residue was acidified to pH = 2 using concentrated hydrochloπc acid The aqueous layer was extracted with ethyl acetate The organic phase was dπed over magnesium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 230-400 mesh, 1/3 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (1.80 g, 52%) as a white solid: mp 152-154°C; EI-HRMS m/e calcd for C15H20O4S (Nf) 296.1082, found 296.1080
A solution of 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (4.91 g, 16.56 mmol) and tnphenylphosphine (6.52 g, 24.85 mmol) m methylene chloπde (41 mL) was cooled to 0°C and then treated with N-bromosuccinimide (5.01 g, 28.16 mmol) m small portions The reaction mixture color changed from light yellow to a darker yellow then to brown After the complete addition of N-bromosuccinimide, the reaction mixture was allowed to warm to 25°C over 30 min. The brown reaction mixture was then treated with 2-aminothiazole (4.98 g, 49.69 mmol). The resulting reaction mixture was stiπed at 25°C for 19 h. The reaction mixture was then concentrated in vacuo to remove methylene chloride. The remaining black residue was diluted with a 10% aqueous hydrochloric acid solution (400 mL) and then extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate then 1/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)-N-thiazol-2- yl-propionamide (4.49 g, 72%) as a white solid: mp 216-217°C; EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1071.
Example 13
(2R)-3-Cyclopentyl-2-(4-methanesuIfonylphenyl)-N-thiazol-2-yl-propionamide

A solution of ^-( ethanesulfonyl)phenyl acetic acid (43 63 g, 0.204 mol) in methanol (509 mL) was treated slowly with concentrated sulfunc acid (2 mL) The resulting reaction mixture was heated under reflux for 19 h The reaction mixture was allowed to cool to 25°C and then concentrated in vacuo to remove methanol The residue was diluted with ethyl acetate (800 mL) The organic phase was washed with a saturated aqueous sodium bicarbonate solution (1 x 200 mL), washed with a saturated aqueous sodium chlonde solution (1 x 200 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 1/1 hexanes/ethyl acetate) afforded 4-(methanesulfonyl)phenyl acetic acid methyl ester (45.42 g, 98%) as a yellow oil which solidified to a cream colored solid upon sitting over time at 25°C mp 78-80°C, EI-HRMS m/e calcd for Cι0H12O4S (M+) 228 0456, found 228 0451.
A mechanical stiπer was used for this reaction A solution of dnsopropylamme (29.2 mL, 0.21 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro- 2(lH)-pyπmιdιnone (62 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (83 4 mL, 0.21 mol) The yellow-orange reaction mixture was stiπed at -78°C for 35 min and then slowly treated with a solution of 4- (methanesulfonyl)phenyl acetic acid methyl ester (45.35 g, 0.20 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdmone (62 mL) The reaction mixture turned dark in color. The reaction mixture was then stiπed at -78°C for 50 mm, at which time, a solution of lodomethylcyclopentane (50.08 g, 0.24 mol) in a small amount of dry tetrahydrofuran was added slowly. The reaction mixture was then stiπed at -78°C for 50 mm, and then allowed to warm to 25°C, where it was stirred for 36 h. The reaction mixture was quenched with water (100 mL), and the resulting reaction mixture was concentrated in vacuo to remove tetrahydrofuran The remaining residue was diluted with ethyl acetate (1.5 L). The organic phase was washed with a saturated aqueous sodium chloπde solution (1 x 500 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid methyl ester (41.79 g, 68%) as a yellow viscous oil EI-HRMS m/e calcd for Cι6H22O4S (M+) 310.1239. found 310.1230.
A solution of 3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid methyl ester (50 96 g, 0.16 mol) in methanol (410 mL) was treated with a IN aqueous sodium hydroxide solution (345 mL, 0.35 mol). The reaction mixture was stirred at 25°C for 24 h. The reaction mixture was concentrated in vacuo to remove methanol. The resulting aqueous residue was acidified to pH = 2 with concentrated hydrochlonc acid and then extracted with ethyl acetate (5 x 200 mL) The combined organic layers were dned over sodium sulfate, filtered, and concentrated in vacuo to afford pure 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (43 61 g, 90%) as a white solid which was used without further puπfication. mp 152-154°C, EI-HRMS m e calcd for C15H20O4S (M+) 296.1082, found 296.1080.
Two separate reactions were setup in parallel: (1) A solution of (R)-(+)-4-benzyl-2- oxazohdmone (3.67 g, 20.73 mmol) m dry tetrahydrofuran (35 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (7.9 mL, 19.86 mmol). The resulting reaction mixture was stiπed at -78°C for 30 mm and then allowed to warm to 25°C, where it was stirred for 1.5 h (2) A solution of racemic 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (5.12 g, 17.27 mmol) in dry tetrahydrofuran (35 mL) was cooled to 0°C and then treated with tnethylamme (2.8 mL, 19.86 mmol). The reaction mixture was stiπed at 0°C for 10 nun and then treated dropwise with tπmethylacetyl chlonde (2.6 mL, 20.73 mmol). The resulting reaction mixture was stiπed at 0°C for 2 h and then cooled to -78°C for the addition of the freshly prepared chiral oxazolidmone. The reaction mixture containing the oxazolidmone was then added to the cooled (-78°C) mixed anhydπde solution The resulting reaction mixture was stiπed as -78°C for 1 h and allowed to gradually warm to 25°C. The reaction mixture was then stiπed at 25°C for 3 d. The resulting reaction mixture was quenched with water (100 mL) and then concentrated in vacuo to remove tetrahydrofuran. The resulting aqueous residue was diluted with ethyl acetate (600 mL). The organic layer was washed with a saturated aqueous sodium chloπde solution (1 x 300 mL), dπed over sodium sulfate, filtered, and concentrated in vacuo Thin layer chromatography using 13/7 hexanes/ethyl acetate as the developing solvent indicated the presence of two products The higher moving product had a Rf =0.32 and the lower moving product had a Rf = 0.19. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 9/1 then 13/7 hexanes/ethyl acetate) afforded two products: (1) The higher Rf product (4R, 2’S)-4-benzyl-3-[3- cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazohdm-2-one (2.12 g, 54%) as a white foam- mp 62-64°C; [c.]23 589 = +6.3° (c=0.24, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M+) 455.1766, found 455.1757. (2) The lower Rf product (4R, 2R)-4- benzyl-3-[3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.88 g, 99%) as a white foam: mp 59-61°C; [α]23 589 = -98.3° (c=0.35, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M +) 455.1766, found 455.1753. The combined mass recovery from the two products was 6.00 g, providing a 76% conversion yield for the reaction
An aqueous solution of lithium hydroperoxide was freshly prepared from mixing a solution of anhydrous lithium hydroxide powder (707.3 mg, 16.86 mmol) m 5.27 mL of water with a 30% aqueous hydrogen peroxide solution (3.44 mL, 33.71 mmol). This freshly prepared aqueous lithium hydroperoxide solution was cooled to 0°C and then slowly added to a cooled (0°C) solution of (4R, 2’R)-4-benzyl-3-[3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.84 g, 8.43 mmol) in tetrahydrofuran (33 mL) and water (11 mL). The reaction mixture was stiπed 0°C for 1.5 h The reaction mixture was then quenched with a 1.5N aqueous sodium sulfite solution (25 mL) The reaction mixture was further diluted with water (300 mL) The resulting aqueous layer was continuously extracted with diethyl ether until thm layer chromatography indicated the absence of the recovered chiral oxazolidmone in the aqueous layer The aqueous layer was then acidified to pH = 2 with a 10% aqueous hydrochlonc acid solution and extracted with ethyl acetate (300 mL) The organic extract was dned over sodium sulfate, filtered, and concentrated in vacuo to afford (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white solid (2.23 g, 89%) which was used without further puπfication Flash chromatography (Merck Silica gel 60, 70-230 mesh, 30/1 methylene chlonde/methanol then 10/1 methylene chlonde/methanol) was used to obtain a punfied sample for analytical data and afforded pure (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white foam- mp 62-64°C (foam to gel), [α]23 589 = -50.0° (c=0.02, chloroform), EI-HRMS m/e calcd for C15H20O4S (M+) 296 1082, found 296 1080
A solution of tnphenylphosphme (3.35 g, 12.79 mmol) m methylene chloπde (19 mL) was cooled to 0°C and then slowly treated with N-bromosuccmimide (2.28 g, 12.79 mmol) in small portions. The reaction mixture was stiπed at 0°C for 30 mm, and dunng this time penod, the color of the reaction mixture changed from light yellow to a darker yellow then to a purple color. The cooled purple reaction mixture was then treated with the (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid (2.23 g, 7.52 mmol) The resulting reaction mixture was then allowed to warm to 25°C over 45 mm, at which time, the reaction mixture was then treated with 2-amιnothιazole (1.88 g, 18.81 mmol) The resulting reaction mixture was stiπed at 25°C for 12 h. The reaction mixture was then concentrated in vacuo to remove methylene chloπde The remaining black residue was diluted with ethyl acetate (300 mL) and then washed well with a 10% aqueous hydrochlonc acid solution (2 x 100 mL), a 5% aqueous sodium bicarbonate solution (3 x 100 mL), and a saturated aqueous sodium chloride solution (1 x 200 mL). The organic layer was then dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl- propionamide (2.10 g, 74%) as a white foam: mp 78-80°C (foam to gel); [α]23 589 = -70.4° (c=0.027, chloroform); EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1081.
REFERENCES
Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
http://www.nature.com/nrd/journal/v8/n5/fig_tab/nrd2850_T2.html
NMR…..http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-NMR-HY-10595-13569-2014.pdf
http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-Lcms_Ms-HY-10595-13569-2014.pdf
///////////RO-28-1675, Ro 0281675
O=C(Nc1nccs1)[C@H](CC2CCCC2)c3ccc(cc3)S(C)(=O)=O

Chemical structures of Roche’s glucokinase activators (GKAs) RO-28-1675 and piragliatin, as well as the related GKA 1.
Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.

Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
Zydus Cadila’s new 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds in pipeline for diabetes type 2
List of compounds as DPP-IV inhibitors


Watch out on this post as I get to correct structure………..
![]()

2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds

One Example of 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds
CAS 1601479-87-1
(2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[5-(methylsulfonyl)-3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]tetrahydro-2H-pyran-3-amine
MW 399.45, C18 H23 F2 N3 O3 S
INTRODUCTION
Dipeptidyl peptidase IV , CD26; DPP-IV; DP-IV inhibitors acting as glucose lowering agents reported to be useful for the treatment of type 2 diabetes. compound inhibited human DPP-IV enzyme activity (IC50 < 10 nM) in fluorescence based assays.
It lowered glucose levels (with -49.10% glucose change) when administered to C57BL/6J mice at 0.3 mg/kg p.o. in oral glucose tolerance test (OGTT).
Compound displayed the following pharmacokinetic parameters in Wistar rats at 2 mg/kg p.o.: Cmax = 459.04 ng/ml, t1/2 = 59.48 h and AUC = 4751.59 h·ng/ml.
Dipeptidyl peptidase 4 (DPP-IV) inhibitor that inhibited human DPP-IV enzyme activity with an IC50 of < 10 nM in a fluorescence based assay.
Watch out on this post as I get to correct structure………..![]()

PATENT
http://www.google.com/patents/WO2014061031A1?cl=en
Compound 8: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz): 7.32-7.28 (m, IH), 7.26-7.23 (m, 2H), 4.77 (d, IH, J= 10Hz), 4.32(dd, IH, J,= 2.0Hz, J2= 10.8Hz), 4.19 (s, 4H), 3.89-3.83 (m, 4H), 3.70- 3.65 (m, IH), 3.61 (t, IH, J= 11.6Hz), 3.53-3.46 (m, IH), 3.04 (s, 3H), 2.65-2.62 (dd, IH, Ji= 1.2Hz, J2= 12Hz), 1.84 (q, IH, J = 12 Hz); ESI-MS: (+ve mode) 400.0 (M+H)+ (100 %); HPLC: 99.4 %.
Compound 4: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(hexahydropyrrolo[3, 4-c Jpyrrol- 2(lH)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz):

.23-7.20 (m, 2H), 4.64 (d, IH, J= 10.4 Hz), 4.38-4.35 (dd, IH, J,= 2.4Hz, J2= 10.4Hz), 3.69 (t, IH, J= 11Hz), 3.57-3.53 (m, 4H), 3.34-3.30 (m, 8H), 2.68-2.65 (m, IH), 2.04 (q, IH, J = 1 1.6 Hz); ESI-MS: (+ve mode) 323.9 (M+H)+ (100 %), 345.9 (M+Na)+ (20%); HPLC: 98.6 %

PATENT
IN 2012MU03030
“NOVEL DPP-IV INHIBITORS”
3030/MUM/2012
Abstract:
The present invention relates to novel compounds of the general formula (I) their tautomeric forms, their enantiomers, their diastereoisomers, their pharmaceutically accepted salts, or pro-drugs thereof, which are useful for the treatment or prevention of diabetes mellitus (DM), obesity and other metabolic disorders. The invention also relates to process for the manufacture of said compounds, and pharmaceutical compositions containing them and their use.

Pankaj R. Patel (right), Chairman and Managing Director,
////////////2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds, DPP-IV inhibitors
ZYD 1/ZYDPLA 1 From Zydus Cadila, a New NCE in Gliptin class of Antidiabetic agents.

GENERAL STRUCTURE

3-[4-(5-methyl-1,3,4-oxadiazol-2-yl)phenoxy]-5-[[(3R)-1-methyl-2-oxo-3-pyrrolidinyl]oxy]-N-2-thiazolyl- Benzamide
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide……S CONF…..WO2011013141A2
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide…..R CONF…..WO2011013141A2
CAS 1263402-84-1 R CONF
CAS 1263402-76-1 S CONF
ZYD 1/ZYDPLA 1……….Probable Representative structure only, I will modify it as per available info
Watch out on this post as I get to correct structure………..![]()

ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1.
By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
In October 2013, Zydus received IND approval from the US FDA to initiate a phase I trial in type II diabetes
Clinical trials..Type 2 Diabetes Mellitus
NCT01972893; ZYD1/1001;
CTRI/2011/04/001684;
ZYD1
ZYD1/1001
ZYD1 is a novel GLP-1 receptor agonist. The ZYD1 exhibits increased stability to proteolytic cleavage, especially against dipeptidyl peptidase-4 (DPP-IV).ZYD1 is a potent antidiabetic agent without gastrointestinal side-effects. A first in human (FIH) Phase I study intends to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ZYD1 in normal healthy adult volunteers……..https://clinicaltrials.gov/show/NCT01972893
A randomized, double blind, placebo controlled Phase I clinical study to evaluate the safety, tolerability and pharmacokinetics of ZYD1, a selective GLP-1 agonist, following the subcutaneous administrations in healthy volunteers …………http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
Some clippings I found

ONE MORE……………

Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The abstract of Poster Number: LB-PP02-4 can also be viewed on the ENDO web program at https://endo.confex.com/endo/2014endo/webprogram/authora.html. The Poster Preview is scheduled on Sunday, June 22, 2014 at McCormick Place West.
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
PATENT
http://www.google.com/patents/WO2011013141A2?cl=en
Rajendra Kharul, Mukul R. Jain, Pankaj R. Patel
Substituted benzamide derivatives as glucokinase (gk) activators

Scheme 2:

Scheme 3:

Scheme 4A:


Scheme 4B.
] Scheme 5 A:

Scheme 5B:

Scheme 6:

Example 1
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
4-(Dimethylamino)pyridine (DMAP) (0.149 g), N-(3-Dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDCI.HC1) (0.524 g) were added to a solution of 3-
( 1 -Methoxypropan-2-yloxy)-5-(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl) phenoxy) benzoic acid (0.5 g) (Intermediate 1) in dry DCM under nitrogen at 0-5 0C. 2-Aminothiazole (0.134 g) was added and the mixture was stirred for 16 h at room temperature. It was diluted with commercially available DCM. Organic phase was washed with dil HCl, saturated solution of NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude residue. The residue was chromatographed using silica gel as stationary phase and MeOH: CHCl3 gradient as mobile phase up to yield the product (0.3 g) as a white solid.
1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.92-2.01 (m, 1 H), 2.59 (s, 3 H), 2.60-2.65 (m,
I H), 2.79 (s, 3 H), 3.31-3.34 (m, 1 H), 3.36-3.44 (m ,1 H), 5.15 (t, J = 7.6 Hz, 1 H),
7.08 (s, 1 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.27-7.29 (m, 1 H), 7.40 (s, 1 H), 7.54 (s, 1 H),
7.62 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H), 12.60 (bs, 1 H); ESI-MS mix (relative intensities): 492.03 (M+H)+ (100 %), 514.02 (M+Na)+(15 %); UPLC Purity: 93.59 %, Rettime: 3.59 min.
Intermediate 1: 3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxo pyrrolidin -3-yloxy)benzoic acid
A solution of Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl- 2-oxopyrrolidin-3-yloxy)benzoate (7 g) (Intermediate 2) in a mixture of THF and methanol (1 :1 ratio) was treated with a solution of sodium hydroxide (2 g) in water and the reaction mixture was stirred for 1 h at room temperature. The resulting solution was concentrated under vacuum to remove THF and methanol, diluted with water, and washed with EtOAc. The aqueous phase was cooled and acidified with 0.1 N HCl and extracted with DCM, combined organic extracts washed with brine, dried over Na2SO4 and concentrated in vacuo to give the product (3.5 g) as white solid.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.20-2.27 (m, 1 H), 2.59-2.67 (m, 1 H), 2.77 (s, 3 H), 2.95 (s, 3 H), 3.38-3.44 (m, 1 H), 3.49-3.54 (m, 1 H), 4.96 (t, J = 7.2 Hz, 1 H), 6.93-6.95 (m, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.32-7.34 (m, 1 H), 7.52 (d, J= 8.8 Hz, 2 H), 9.96-9.98 (m, 2 H); ESI-MS (relative intensities): 431.9 (M+ Na)+ (70%).
Intermediate 2: Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2- oxo- pyrrolidin-3-yloxy)benzoate
To a stirred mixture of Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy) benzoate (15 g) (Intermediate 3), N,N-dimethylglycine hydrochloride (2.3 g), copper (II) iodide (1 g) in dry 1,4-dioxane was added 2-(4-iodophenyl)-5 -methyl- 1,3,4- oxadiazole (15.4 g) (Intermediate 4) under nitrogen. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled, quenched with water and extracted with DCM. Combined organic washings were washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude product. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether (9:1) as mobile phase to give the product (7 g) as thick liquid. 1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.91-1.98 (m, 1 H), 2.49-2.54 (m, 1 H), 2.56 (s, 3 H), 2.77 (s, 3 H), 3.34-3.41 (m, 2 H), 3.81 (s, 3 H), 5.12 (t, J= 7.6 Hz, 1 H), 7.13- 7.15 (m, 2 H), 7.22 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.97 (d, J = 8.8 Hz, 2 H); ESI- MS (relative intensities): 423.9 (M+H)+ (100%), 446.2 (M+ Na)+ (30%).
Intermediate 3: Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy)benzoate
To a stirred solution of Methyl 3, 5-dihydroxybenzoate (20 g) [CAS No. 2150- 44-9] in dry DMF was added potassium carbonate (48 g) and the suspension stirred at ambient temperature under nitrogen. To this 3-Bromo-l-methyl-pyrrolidin-2-one (4Og) (Intermediate 5) [J. Med. Chem., 1987, 30, 1995-98] was added in three equal portions in 4 h intervals at room temperature and stirred overnight at ambient temperature. It was then quenched with water. The aqueous suspension was extracted with DCM. The combined extracts were washed with water, brine, dried over Na2SO4, and filtered, concentrated under reduced pressure to get the thick liquid residue. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether as a mobile phase to yield the product as white solid (15 g).1H NMR (CDCl3, 400 MHz) δ ppm: 2.08-2.10 (m, 1 H), 2.60-2.67 (m, 1 H), 3.04 (s, 3 H), 3.40-
3.43 (m, 1 H), 3.48-3.51 (m, 1 H), 3.87 (s, 3 H), 4.91 (t, J = 7.2 Hz, 1 H), 6.59- 6.61 (m, 1 H), 7.07-7.09 (m, 1 H), 7.09-7.13 (m, 1 H), 8.02 (s, 1 H); ESI-MS (relative intensities): 287.9 (M+ Na)+ (30%).
Example 68…. S CONFIGURATION
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of S-(-)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5- [(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (3.5 g) (Intermediate 13) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, 4- (dimethylamino)pyridine (2.24 g) followed by N-(3-Dimethy lam inopropy I)-N5– ethylcarbodiimide hydrochloride (EDCI. HCl) (3.3 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.94 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (200 mL), washed with dil HCl (20 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (3.5 g). The crude brown solid was purified by solvent trituration.
1H ΝMR (CDCl3, 400 MHz) δ ppm: 2.13-2.22 (m, 1 H), 2.62 (s, 3 H), 2.56-2.64 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.92 (t, J= 7.2 Hz, 1 H), 7.01 (s, 1 H), 7.04 (t, J= 2 Hz, 1 H), 7.21 (d, J = 8.8 Hz, 2 H), 7.26 (s, 1 H), 7.36 (s, 1 H), 7.44 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 513.8 (M+Νa)+ (10 %); UPLC Purity: 98.13 %, Ret. time: 3.577 min. Chiral Purity by HPLC: 97.31 %, Ret. time: 22.93 min. % ee: 94.62 %
Intermediate 13: S-(-)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2- oxo-pyrro- lidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 1.5 g) was added to a stirring mixture of (.S)-(-)-Methyl 3- [4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy] benzoate (5.3g) (Intermediate 14) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (3.5 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.17-2.22 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.99 (t, J= 7.2 Hz, 1 H), 6.89 (t, J = 2.4 Hz, 1 H), 7.07 (d, J = 8.8 Hz, 2 H), 7.28 (s, 1 H), 7.53 (s, 1 H), 7.95 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410 (M+H)+ (100 %); UPLC Purity: 97.85 %, Ret. time: 3.136 min. Chiral Purity by HPLC: 99.59 %, Ret. Time: 57.46 min. % ee: 99.18 %
Intermediate 14: (S) -(-) -Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate
Sodium hydride suspension (0.71 g, 50 %) was added to a stirring solution of (£)-(-)- methyl 3 -(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (5.5 g) (Intermediate 15) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.91 mL) was added and stirred till the reaction completion. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vaccuo to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product (5.3 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.14-2.21 (m, 1 H), 2.58-2.63 (m, 1 H), 2.64 (s, 3 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m , 1 H), 3.89 (s, 3 H), 4.99 (t, J = 7.2 Hz, 1 H), 6.99 (t, J = 2 Hz, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.35 (s, 1 H), 7.53 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 424.1 (M+H)+ (100 %); UPLC Purity: 96.1 1 %, Ret. time: 3.68 min. Chiral Purity by HPLC: 92.05 %, Ret. Time: 39.33 min.
Intermediate 15: (S) -(-) -Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxo pyrrolidin-3-yl)oxy) benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (7 g) (Intermediate 7) and (/?)-(+)-3-hydroxy-2-pyrrolidinone (Intermediate 16) (2.4g) in dry THF (200 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (1 1.3 g) was added. Diisopropyl azodicarboxylate (DIAD) (6.2 mL) in dry THF (10 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (6 g). 1H NMR (CDCl3, 400 MHz) δ ppm: 2.26-2.33 (m, 1 H), 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.47 (m, 1 H), 3.51-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.07 (bs, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (100 %); UPLC Purity: 98.35 %, Ret. time: 3.47 min. Chiral Purity by HPLC: 95.31 %, Ret. Time: 47.97 min. ee: 90.62 %.
Intermediate 16: (R)-(+)-3-Hydroxy-2-pyrrolidinone
To a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (5)-(-)-3-hydroxy-2- pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added. To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was concentrated under vacuum to obtain residue. Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was diluted with CHCl3 and filtered through celite. Celite bed was successively washed with 1 % MeOH:CHCl3. The filtrates were combined and concentrated to dryness to remove solvents. The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min. Layers were separated, aq. layer was washed with EtOAc thrice until all organic impurities were washed out. The aq. Layer was concentrated to dryness to remove the water and solid residues were obtained. The residues obtained were washed with 1-2 % MeOH: CHCl3 (3 x 100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.
1U NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.46-2.54 (m, 1 H), 3.28-3.35 (m, IH), 3.38-3.48 (m, 1 H), 4.50 (t, J = 8.4 Hz, 1 H), 4.55 (bs, 1 H), 7.02 (bs, 1 H); [α]D25: + 68, c = l, CHCl3
Intermediate 17: (S)-(-)-3-hydroxy-2-pyrrolidinone
Cone. H2SO4 (14.8 g, 8 mL) was added drop wise over 5 min to the stirring solution of (5)-(-)-4-Amino-2-hydroxybutyric acid (15 g) [CAS No. 40371-51-5] in MeOH (95 rnL) under dry conditions using anhydrous CaCl2 guard tube. After refluxing for 4 h, the reaction mixture was allowed to cool to room temperature and diluted with water (15 mL). Potassium carbonate (24 g) was added in portions to the reaction mixture and stirred overnight (20 h). Reaction mixture was diluted with CHCl3, filtered through celite. Celite bed was thoroughly washed with 1 % MeOHiCHCl3. The filtrates were combined and evaporated to dryness to obtain thick liquid residue. The residue was subjected to aging using 1-2 % MeOHiCHCl3 and then filtered. Organic layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated to obtain the white solid. (1 1.8 g)
1H NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.48-2.55 (m, 1 H), 3.30-3.35
(m, IH), 3.36-3.50 (m, 1 H), 4.34 (t, J = 8.4 Hz, 1 H), 6.51 (bs, 1 H); [α]D25: + 98, c =
1, CHCl3
Following examples (Example 70-76) were prepared by using similar procedure as that of example lwith suitable modifications as are well within the scope of a skilled person
Example 77 R CONFIGURATION
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide
CORRECTED AS (R)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of (/?j-(+)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-
[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (0.2 g) (Intermediate 18) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, N.ΛP-dimethylamino pyridine (0.060 g) followed by EDCI. HCl (0.23 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.054 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (20 mL), washed with dil HCl (5 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (0.080 g). The crude brown solid was purified by solvent trituration.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.15-2.20 (m, 1 H), 2.55-2.60 (m, 1 H), 2.62 (s, 3 H), 2.93 (s, 3 H), 3.38-3.43 (m, 1 H), 3.47-3.53 (m, 1 H), 4.91 (t, J= 6.8 Hz, 1 H), 6.99 (d, J= 8.8 Hz, 2 H), 7.10-7.14 (m, 2 H), 7.23-7.26 (m, 1 H), 7.36 (s, 1 H), 7.43 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H), 10.75 (bs, 1 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 514.0 (M+Na)+ (20 %); UPLC Purity: 95.25 %, Ret.time: 3.578 min. Chiral Purity by HPLC: 95.93 %, Ret.time: 14.17min. % ee: 91.86 %
Intermediate 18: (R)-(+)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl- 2-oxo- pyrrolidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 0.35 g) was added To a stirring mixture of (/?)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate (1.1 g) (Intermediate 19) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (0.76 g).
1H NMR (DMSO-J6, 400 MHz) δ ppm: 1.92-1.99 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 3.31 (s, 3 H), 3.32-3.40 (m, 2 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.08 (s, 1 H), 7.14 (s, 1 H), 7.23 (d, J= 8.8 Hz, 2 H), 7.40 (s, 1 H), 7.99 (d, J= 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (65 %), 410.1 (M+H)+ (100 %); UPLC Purity: 96.95 %, Ret. time: 3.12 min. Chiral Purity by HPLC: 89.04 %, Ret. Time: 48.15 min. Intermediate 19: (R)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate:
Sodium hydride suspension (0.16 g, 50 %) was added to a stirring solution of (R)- (+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (1.5 g) (Intermediate 20) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.20 mL) was added and stirred till the reaction completed. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vacuum to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product
(1.2 g).
1U NMR (DMSO-J6, 400 MHz) δ ppm: 1.95-1.98 (m, 1 H), 2.51-2.55 (m, 1 H), 2.56 (s, 3 H), 2.88 (s, 3 H), 3.29-3.34 (m, 1 H), 3.37-3.40 (m ,1 H), 3.81 (s, 3 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.13-7.17 (m, 2 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.41 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 423.9 (M+H)+ (100 %); UPLC Purity: 90.38 %, Ret. time: 3.68 min.
Intermediate 20: (R)-(+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxopyrrolidin -3-yl)oxy)benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (2.5 g) (Intermediate 7) and (5)-(-)-3-hydroxy-2-pyrrolidinone (Intermediate 17) (0.8 g) in dry THF (70 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (3.77 g) was added. Diisopropyl azodicarboxylate (DIAD) (2.1 mL) in dry THF (2 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (2 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.23-2.30 (m, 1 H); 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.46 (m, 1 H), 3.50-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (45 %); UPLC Purity: 96.40 %, Ret. time: 3.48 min. Chiral Purity by HPLC: 90.92 %, Ret. Time: 48.36 min.


http://zyduscadila.com/wp-content/uploads/2015/09/ZYDPLA1-a-Novel-LongActing-DPP-4-Inhibitor.pdf
http://zyduscadila.com/wp-content/uploads/2015/05/PressNote23-10-13.pdf
http://zyduscadila.com/wp-content/uploads/2015/07/annual_report_14-15.pdf
http://pharmaxchange.info/press/2012/08/glucokinase-activators-gkas-in-diabetes-management/
LB-PP02-4 ZYDPLA1, a novel long-acting DPP-4 inhibitor
Jt Int Congr Endocrinol Annu Meet Endocr Soc (ICE/ENDO) (June 21-24, Chicago) 2014, Abst LBSU-1075
LB-PP02-4 ZYDPLA1, a Novel Long-Acting DPP-4 Inhibitor
Session: LBSU 1074-1087-Diabetes & Obesity
Translational
Disclosure: MRJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. AAJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. RB: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. HP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. SK: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. KP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VKR: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PRP: Chairman, Cadila Healthcare Limited, Ahmedabad, India. RD: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India.

////////Dipeptidyl Peptidase IV, CD26, DPP-IV, DP-IV, Inhibitors
MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan

MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan
KENILWORTH, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) has approved MARIZEV® (omarigliptin) 25 mg and 12.5 mg tablets, an oral, once-weekly DPP-4 inhibitor indicated for the treatment of adults with type 2 diabetes. Japan is the first country to have approved omarigliptin……….http://www.mercknewsroom.com/news-release/prescription-medicine-news/marizev-omarigliptin-mercks-once-weekly-dpp-4-inhibitor-type
syn…….https://newdrugapprovals.org/2014/04/18/omarigliptin-mk-3102-in-phase-3-for-type-2-diabetes/

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
/////////////MARIZEV, (Omarigliptin), Merck’s, Once-Weekly, DPP-4 Inhibitor, Type 2 Diabetes, Approved, Japan
TENELIGLIPTIN

TENELIGLIPTIN
| Teneligliptin; 760937-92-6; UNII-28ZHI4CF9C; Teneligliptin (INN); 28ZHI4CF9C | |
| MF | C22H30N6OS |
|---|---|
| MW | 426.5782 g/mol |
Teneligliptin (INN; trade name Tenelia) is a pharmaceutical drug for the treatment of type 2 diabetes mellitus. It is approved for use in Japan.[1] It belongs to the class of anti-diabetic drugs known as dipeptidyl peptidase-4 inhibitors or “gliptins”.[2] {(2S,4S)-4-[4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)-1-piperazinyl]-2-pyrrolidinyl}(1,3-thiazolidin-3-yl)methanone
Teneligliptin was launched in Japan in 2012 by Mitsubishi Pharma and Daiichi Sankyo for the treatment of type 2 diabetes mellitus. In 2013, the indication was partially changed to include it as a combination therapy with existing oral hypoglycemic agents, such as biganides, alpha-glucosidaseinhibitors, rapid-acting insulin secretagogues, and insulin preparations, as well as sulfonylureas and thiazolidines that had been approved for the combination.
In 2014, the product was registered in KR for the treatment of type 2 diabetes mellitus.
In 2013, Mitsubishi Tanabe Pharma filed for approval in Japan for use of the compound as combination therapy for the treatment of diabetes type 2.
| CAS | 760937-92-6 |
|---|
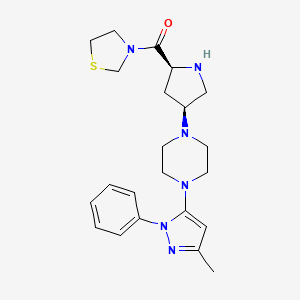
3-{(2S,4S)-4-[4-(3-methyl-l -phenyl- 1 H- pyrazol-5-yl)- l-piperazinyl]-2-pyrrolidinylcarbonyl}-l , 3-thiazolidine is represented structurally by a compound of formula (I):

Teneligliptin (CAS 760937-92-6) is a novel, potent and long-lasting dipeptidyl peptidase-4 inhibitor in treatment of type 2 diabetes. Dipeptidyl-peptidase-4 (DPP- 4) inhibitor has been demonstrated to improve glycemic control, in particular postparandial hyperglycemic control.
Despite of their common mechanism of action, DPP-4 inhibitors show marked structural heterogeneity. DPP-4 inhibitors may be classified into peptidomimetic (i.e. sitagliptin, vildagliptin, saxagliptin, and anagliptin) and non-peptidomimetic (i.e. alogliptin and linagliptin) subtypes.
Teneligliptin, is chemically known as a 3- {((2S,4S)-4-(4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-yl 25 carbonyl}thiazolidine hemipentahydrobromide hydrate and is peptidomimetic with the molecular formula of C22H30N6OS.2½HBr.xH2O and molecular weight of 642.88 g/mol for hemipentahydrobromide. The hydrate can be from mono to dihydrate.
U.S. Patent No. 7,074,794 B2 (the US ‘794) discloses teneligliptin as L-proline derivative and its pharmaceutically acceptable salts which exhibits a Dipeptidyl 5 peptidase IV (DPP-IV) inhibitory activity, which is useful for the treatment or prophylaxis of diabetes, obesity, HIV infection, cancer metastasis, dermopathy, prostatic hyperplasia, periodontitis, autoimmune diseases and the like.
The example-222 of the US ‘794 discloses the process for the preparation of teneligliptin as trihydrochloride salt U.S. Patent No. 8,003,790 B2 (the US ‘790) discloses salts of proline derivative, solvate thereof and production method thereof. In particular, the US ‘790 discloses 2.0 hydrochloride or 2.5 hydrochloride; 2.0 hydrobromide or 2.5 hydrobromide, and hydrates thereof teneligliptin.
The US ‘790 B2 further discloses different salts 15 of teneligliptin which are incorporated herein as reference in their entirety U.S. PG-Pub. No. 2011/0282058 A1 discloses salts of 3-{((2S,4S)-4-(4-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-ylcarbonyl}thiazolidine with mono-, di- and tri-basic acids or a solvate thereof. 20 International (PCT) publication No. WO 2012/165547 A1 discloses a process for preparation of teneligliptin and pharmaceutically acceptable salts thereof.
International (PCT) publication No. WO 2007/127635 A2 (the WO ‘635 A2) discloses a process for the preparation of diketo-piperazine and piperidine 25 derivatives. In particular, the WO ‘635 A2 discloses the process for preparation of 4-oxo-2-(thiazolidine-3-carbonyl)-pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (III)] by reacting piperazine with aryl halide.
International (PCT) publication No. WO 2012/099915 A1 (the WO ‘915 A1) 5 discloses the process for the preparation of deuterated thiazolidine derivatives. The WO ‘915 A1 also discloses the process for the preparation of 1-(3-methyl-1- phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by condensation of 5- chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by deprotection of Boc-protected 1-(3-methyl-1-phenyl-1Hpyrazol-5-yl)piperazine with triflouroacetic acid.
U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine by condensation of 5-chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine in presence of n-BuLi in tetrahydrofuran. Bioorganic & Medicinal Chemistry, 14(11), 3662-3671 (2006),
Bioorganic & Medicinal Chemistry, 20(16), 5033-5041 (2012) and U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate by reacting (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid with 25 thiazolidine in presence of HOBT and EDC.HCl in dimethylformamide solvent.
Bioorganic & Medicinal Chemistry, 15(2), 641-655 (2007) discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3- carbonyl)pyrrolidine-1-carboxylate by treating (2S,4S)-tert-butyl 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate with tetrabutylammonium fluoride in tetrahydrofuran.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the 5 process for the preparation of herein compound (II) after by reacting 1-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) with (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate in presence of sodium triacetoxyborohydride. There is provided different alternative processes for the preparation of teneligliptin and intermediates thereof.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) also discloses the process for the preparation of 4-[4-(5-methyl-2-phenyl-2H-pyrazol-3-yl)-piperazin- 1-yl]-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (II)] after by reacting 1-(3-methyl-1-phenyl-1H-pyrazol-5- 15 yl)piperazine [herein compound (V)] with (2S,4S)-tert-butyl 4-[[(1,1- dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate in presence of trifluoromethylsulfonic anhydride and diisopropylethylamine. 3 – [[(2S, 4S) -4- [4- (3- methyl-1-phenyl–1H- pyrazol-5-yl) -1-piperazinyl ] -2-pyrrolidinyl] carbamoyl] thiazolidine, having the formula below, is a very novel DPP-4 inhibitor potential.

World Patent Application No. W02012099915 for Ge Lieting discloses a process for the preparation route is as follows:

Journal B10rganic & Medicinal Chemistry, 2012, 20, 5705-5719 also discloses a preparation method for Ge Lieting, the route is as follows:

[0009] 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine, was prepared for the Ge Lieting key intermediate. Journals B10rganic & Medicinal Chemistry, 2012,20,5705-5719 reported the preparation of the intermediates prepared route is as follows:

[0011] The preparative route after the N-Boc-N- acetoacetyl piperazine phenylhydrazine and methanesulfonic acid in an ethanol solution of the reaction at room temperature 14h, concentrated under reduced pressure after addition of pyridine.Was added phosphorus oxychloride in pyridine, 20h post treatment reaction at room temperature the reaction system. The compound obtained above was then added trifluoroacetic acid was dissolved in methylene chloride after, after treatment at room temperature for 1.5h to give 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine.
The reaction process requires mesylate mesylate flammable, easy-absorbent deliquescence, and has a strong corrosive and irritating, easy to cause the body burns; phosphorus oxychloride, a highly toxic substance, water violent hair in the air smoke, hydrolyzed into phosphoric acid and hydrogen chloride, is very unstable, to operate a lot of trouble; trifluoroacetic acid is highly corrosive and irritant, can cause the body burns; low yield of the reaction (10%). Seeking a simple operation, high reaction yield, low cost and suitable for industrial production production process 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine has a very important role in the field of medicine.
…………………………………….


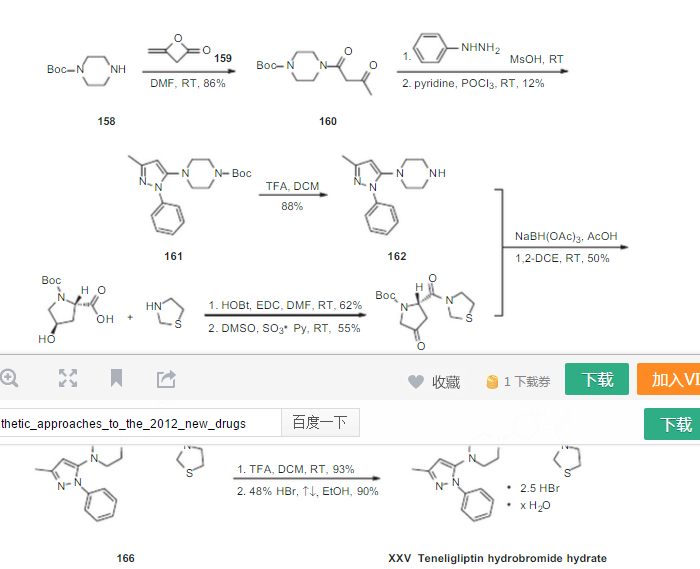
since the capture is staggered, compd 165 is not clear in above pic see below
…………
if above section iis not clear see at ……..http://www.allfordrugs.com/2015/07/03/teneligliptin/
…………………….
reaction scheme in http://www.google.com/patents/CN104177295A?cl=en

Description: LR as Lawesson reagent (Lawesson Reagent), is a sulfur oxygen exchange reagent. The present invention provides a method for preparing key intermediates Ge Lieting method, comprising the steps of: (I) N-Boc-N- acetoacetyl piperazine Lawesson’s reagent in the presence of an organic solvent, with a phenylhydrazine of the formula occurs ⑴ reaction shown:

(2) the step (1) The product was dissolved in an organic solvent, the following formula (II) in concentrated hydrochloric acid to deprotected shown:

Volume 20, Issue 19, 1 October 2012, Pages 5705–5719


………………………..
http://www.google.co.in/patents/WO2015019238A1?cl=en
Example 5: Preparation of {(2^,.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -vHpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
Activated carbon (10 g) was added to a solution of the residue (obtained in Example 4) in isopropyl alcohol (1000 mL) at 30°C to 35°C. The reaction mixture was filtered through a Hyflo® bed. The filtrate was heated to a temperature of 70°C to 75°C. Hydrobromic acid (48%; 168 g) was slowly added to the filtrate at 70°C to 75°C over a period of 10 minutes to 15 minutes. The reaction mixture was stirred for 2.5 hours at 70°C to 77°C. The progress of the reaction was monitored by HPLC. After completion of the reaction, the reaction mixture was cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes. The reaction mixture was filtered to obtain a solid. The solid obtained was washed with isopropyl alcohol (2 x 200 mL), and dried at 50°C under reduced pressure for 15 hours to obtain crude {(25*,45)-4-[4-(3-methyl-l-phenyl-lH- pyrazol-5 -yl)piperazin- 1 -yl]pyrrolidin-2-yl} ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate.
Yield: 90%
Example 6: Purification of {(2^’.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -yllpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
A reaction mixture containing {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5- yl)piperazin- 1 -yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate (100 g; prepared according to the process of Example 5) in ethanol (700 mL) was heated at 70°C to 75°C to obtain a solution. The solution was filtered at the same temperature. The filtrate was allowed to cool to a temperature of 65 °C to 68°C, and deionized water (10 mL) was added at the same temperature. The solution was cooled to a temperature of 55°C to 60°C, and stirred at the same temperature for 2 hours. The solution was further cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes to obtain a solid. The solid was filtered, washed with ethanol (100 mL), and dried at 45°C to 50°C under reduced pressure for 18 hours to 20 hours to obtain pure {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin-l- yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone hemipentahydrobromide hydrate .
Yield: 90%
HPLC Purity: 99.93%
| WO2012099915A1 * | 18 Jan 2012 | 26 Jul 2012 | Hongwen Zhu | Thiazolidine derivatives and their therapeutic use |
| WO2012165547A1 * | 31 May 2012 | 6 Dec 2012 | Mitsubishi Tanabe Pharma Corporation | Method for manufacturing pyrazole derivative |
| WO2014041560A2 * | 28 Aug 2013 | 20 Mar 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of teneligliptin |
| US7074794 | 10 Aug 2001 | 11 Jul 2006 | Mitsubishi Pharma Corporation | Proline derivatives and the use thereof as drugs |
| US8003790 | 17 Feb 2006 | 23 Aug 2011 | Mitsubishi Tanabe Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| US20050256310 * | 12 May 2005 | 17 Nov 2005 | Pfizer Inc | Therapeutic compounds |
| EP1854795A1 * | 17 Feb 2006 | 14 Nov 2007 | Mitsubishi Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| EP1894567A1 * | 2 Jun 2006 | 5 Mar 2008 | Mitsubishi Tanabe Pharma Corporation | Concomitant pharmaceutical agents and use thereof |
| US20040106655 * | 10 Aug 2001 | 3 Jun 2004 | Hiroshi Kitajima | Proline derivatives and the use thereof as drugs |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015019238A1 * | 28 Jul 2014 | 12 Feb 2015 | Ranbaxy Laboratories Limited | Process for the preparation of n-protected (5s)-5-(1,3-thiazolidin-3-ylcarbonyl)pyrrolidin-3-one |
| Patent | Submitted | Granted |
|---|---|---|
| Proline derivatives and use thereof as drugs [US7060722] | 2005-11-03 | 2006-06-13 |
| Proline derivatives and the use thereof as drugs [US7074794] | 2004-06-03 | 2006-07-11 |
| Proline derivatives and use thereof as drugs [US2006173056] | 2006-08-03 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US8003790] | 2009-08-27 | 2011-08-23 |
| METHOD OF TREATING ABNORMAL LIPID METABOLISM [US2010305139] | 2010-12-02 | |
| COMBINED USE OF DIPEPTIDYL PEPTIDASE 4 INHIBITOR AND SWEETENER [US2010113382] | 2010-05-06 | |
| CONCOMITANT PHARMACEUTICAL AGENTS AND USE THEREOF [US2009082256] | 2009-03-26 | |
| PROPHYLACTIC/THERAPEUTIC AGENT FOR ABNORMALITIES OF SUGAR/LIPID METABOLISM [US2009088442] | 2009-04-02 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US2011282058] | 2011-11-17 |
- Joanne Bronson, Amelia Black, T. G. Murali Dhar, Bruce A. Ellsworth, and J. Robert Merritt. “Teneligliptin (Antidiabetic)”. Annual Reports in Medicinal Chemistry 48: 523–524. doi:10.1016/b978-0-12-417150-3.00028-4.
- Kishimoto, M (2013). “Teneligliptin: A DPP-4 inhibitor for the treatment of type 2 diabetes”. Diabetes, metabolic syndrome and obesity : targets and therapy 6: 187–95. doi:10.2147/DMSO.S35682. PMC 3650886. PMID 23671395.
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
EU approves Lilly diabetes drug Trulicity, dulaglutide
Regulators in Europe have given the green light to Eli Lilly’s Trulicity, its once-weekly glucagon-like peptide-1 receptor agonist for type 2 diabetes.
Read more at: http://www.pharmatimes.com/Article/14-11-25/EU_approves_Lilly_diabetes_drug_Trulicity.aspx
Dulaglutide is a glucagon-like peptide 1 receptor agonist (GLP-1 agonist) for the treatment of type 2 diabetes that can be used once weekly.[1][2]GLP-1 is a hormone that is involved in the normalization of level of glucose in blood (glycemia). The FDA approved dulaglutide for use in the United States in September 2014.[3] The drug is manufactured by Eli Lilly under the brand name Trulicity.[3]

Mechanism of action
Dulaglutide binding to glucagon-like peptide 1 receptor, slows gastric emptying and increases insulin secretion by beta cells in the pancreas. Simultaneously the compound reduces the elevated glucagon secretion by alpha cells of the pancreas, which is known to be inappropriate in the diabetic patient. GLP-1 is normally secreted by L cells of the gastrointestinal mucosa in response to a meal.[4]
Medical uses[
The compound is indicated for adults with type 2 diabetes mellitus as an adjunct to diet and exercise to improve glycemic control. Dulaglutide is not indicated in the treatment of subjects with type 1 diabetes mellitus or patients with diabetic ketoacidosis. Dulaglutide can be used either stand-alone or in combination with other medicines for type 2 diabetes, in particular metformin, sulfonylureas, thiazolidinediones, and insulin taken concomitantly with meals.[5]
Side effects
The most common side effects include gastrointestinal disorders, such as dyspepsia, decreased appetite, nausea, vomiting, abdominal pain, diarrhea.[6] Some patients may experience serious adverse reactions: acute pancreatitis (symptoms include persistent severe abdominal pain, sometimes radiating to the back and accompanied by vomiting),hypoglycemia, renal impairment (which may sometimes require hemodialysis). The risk of hypoglycemia is increased if the drug is used in combination with sulfonylureas orinsulin.[7][8]
Contraindications
The compound is contraindicated in subjects with hypersensitivity to active principle or any of the product’s components. As a precautionary measure patients with a personal or family history of medullary thyroid carcinoma or affected by multiple endocrine neoplasia syndrome type 2 should not take dulaglutide, because for now it is unclear whether the compound can increase the risk of these cancers.[9]
![]()
References
- JCourtney Aavang Tibble, Tricia Santos Cavaiola, Robert R Henry (2013). “Longer Acting GLP-1 Receptor Agonists and the Potential for Improved Cardiovascular Outcomes: A Review of Current Literature”. Expert Rev Endocrinol Metab 8 (3): 247–259.doi:10.1586/eem.13.20.
- “Lilly’s Once-Weekly Dulaglutide Shows Non-Inferiority to Liraglutide in Head-to-Head Phase III Trial for Type 2 Diabetes”. Eli Lilly. Feb 25, 2014.
- “FDA approves Trulicity to treat type 2 diabetes” (Press release). FDA. Sep 18, 2014.
- Nadkarni P, Chepurny OG, Holz GG (2014). “Regulation of glucose homeostasis by GLP-1”. Prog Mol Biol Transl Sci 121: 23–65. doi:10.1016/B978-0-12-800101-1.00002-8.PMC 4159612. PMID 24373234. Retrieved 2014-09-29.
- Terauchi Y, Satoi Y, Takeuchi M, Imaoka T (July 2014). “Monotherapy with the once weekly GLP-1 receptor agonist dulaglutide for 12 weeks in Japanese patients with type 2 diabetes: dose-dependent effects on glycaemic control in a randomised, double-blind, placebo-controlled study”. Endocr. J. PMID 25029955. Retrieved 2014-09-29.
- Nauck M, Weinstock RS, Umpierrez GE, Guerci B, Skrivanek Z, Milicevic Z (August 2014). “Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5)”. Diabetes Care 37 (8): 2149–58.doi:10.2337/dc13-2761. PMID 24742660.
- Amblee A (April 2014). “Dulaglutide for the treatment of type 2 diabetes”. Drugs Today50 (4): 277–89. doi:10.1358/dot.2014.50.4.2132740. PMID 24918645.
- Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E (February 2014). “Glucagon-like peptide-1 receptor agonists and pancreatitis: a meta-analysis of randomized clinical trials”. Diabetes Res. Clin. Pract. 103 (2): 269–75. doi:10.1016/j.diabres.2014.01.010.PMID 24485345.
- Samson SL, Garber A (April 2013). “GLP-1R agonist therapy for diabetes: benefits and potential risks”. Curr Opin Endocrinol Diabetes Obes 20 (2): 87–97.doi:10.1097/MED.0b013e32835edb32. PMID 23403741. Retrieved 2014-09-30.
| Identifiers | |
|---|---|
| CAS number | 923950-08-7 |
| ATC code | None |
| Chemical data | |
| Formula | C2646H4044N704O836S18 |
| Mol. mass | 59669.81 g/mol |
