
Home » Posts tagged 'teva' (Page 2)
Tag Archives: teva
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Teva: FDA Approves of Generic Tobi in US

TOBRAMYCIN
JERUSALEM–(BUSINESS WIRE)–Oct. 14, 2013– Teva Pharmaceutical Industries Ltd. announces today that the U.S. Food and Drug Administration has granted approval of the generic equivalent to Tobi® (Tobramycin Inhalation Solution USP) in the United States. Pursuant to an agreement with Novartis on this product, Teva expects to launch this product in late November. Marketed by Novartis, Tobi had annual sales of approximately $350 million in the United States, according to IMS data as of June 30, 2013. READ ALL AT………..
http://www.drugs.com/news/teva-fda-approves-tobi-us-48168.html

Tobramycin is an aminoglycoside antibiotic derived from Streptomyces tenebrarius and used to treat various types of bacteria infections, particularly Gram-negative infections. It is especially effective against species of Pseudomonas.[1]

Tobramycin works by binding to a site on the bacterial 30S and 50S ribosome, preventing formation of the 70S complex. As a result, mRNA cannot be translated into protein and cell death ensues. Tobramycin is preferred over gentamicin for Pseudomonas aeruginosapneumonia due to better lung penetration.


Like all aminoglycosides, tobramycin does not pass the gastro-intestinal tract, so forsystemic use it can only be given intravenously or intramuscularly. Ophthalmic (tobramycin only, Tobrex, or combined with dexamethasone, sold as TobraDex) and nebulised formulations both have low systemic absorption. The formulation for injection is branded Nebcin. The nebulised formulation (brand name Tobi) is indicated in the treatment of exacerbations of chronic infection with Pseudomonas aeruginosa in patients diagnosed with cystic fibrosis. A proprietary formulation of micronized, nebulized tobramycin has been tested as a treatment for bacterial sinusitis.[2] Tobrex is a 0.3% tobramycin sterile ophthalmic solution is produced by Bausch & Lomb Pharmaceuticals. Benzalkonium chloride 0.01% is added as a preservative. It is available by prescription only in the United States and Canada. In certain countries, such as Italy, it is available over the counter. Tobrex and TobraDex are indicated in the treatment of superficial infections of the eye, such as bacterial conjunctivitis. Tobramycin (injection) is also indicated for various severe or life-threatening gram-negative infections : meningitis in neonates, brucellosis, pelvic inflammatory disease, Yersinia pestis infection (plague).

Like other aminoglycosides, tobramycin is ototoxic: it can cause hearing loss, or a loss ofequilibrioception, or both in genetically susceptible individuals. These individuals carry a normally harmless genetic mutation that allows aminoglycosides such as tobramycin to affect cochlear cells. Aminoglycoside-induced ototoxicity is generally irreversible.

As with all aminoglycosides, tobramycin is also nephrotoxic, meaning it is toxic to thekidneys. This effect can be particularly worrisome when multiple doses accumulate over the course of a treatment or when the kidney concentrates urine by increasing tubular reabsorption during sleep. Adequate hydration may help prevent excess nephrotoxicity and subsequent loss of renal function. For these reasons parenteral tobramycin needs to be carefully dosed by body weight, and its serum concentration monitored. Tobramycin is thus said to be a drug with a narrow therapeutic index.
|
Mass-spectrum of tobramycin |
|
|

- “Tobramycin” (pdf). Toku-E. 2010-01-12. Retrieved 2012-06-11.
- “Nebulized Tobramycin in treating bacterial Sinusitis” (Press release). July 22, 2008. Retrieved 2009-12-06.

A widely accepted therapy for treating respiratory infections caused by Gram- negative bacteria involves intravenous administration of a single antibiotic or combinations of antibiotics. Gibson et al., 2003 Am. J. Respir. Crit. Care. Med.
168(8):918-951 ; Ramsey, 1996 N. Engl. J. Med. 335(3): 179-188. However, this method of treatment has several significant limitations including: (1 ) narrow spectrum of activity of existing antibiotics, (2) insufficient concentrations of antibiotic reaching the respiratory tract to ensure rapid onset and high rates of bacterial killing, and (3) development of adverse side effects due to high systemic concentrations of drug.
Aerosol administration of antibiotics (Conway, 2005 Chronic Repir. Dis. 2:35- 41 ; O’Riordan, 2000 Respir. Care 45(7):836-845) addresses several of the limitations of parenteral administration (Flume and Klepser, 2002 Pharmacotherapy 22(3 Pt 2):71 S-79S; Kuhn, 2001 Chest. 120:94S-98S). It enables topical delivery of high concentrations of drug to the endobronchial spaces and reduces side effects by lowering systemic exposure to antibiotic. However, patients having chronic respiratory conditions, such as chronic obstructive pulmonary disease, cystic fibrosis and bronchiestasis, may receive prolonged and repeated antibiotic therapies over the entire duration of their adult lives. Gibson et al., 2003 Am. J. Respir. Crit. Care. Med. 168(8):918-951 ; Ramsey, 1996 N. Engi. J. Med. 335(3): 179-188. Therefore, cumulative antibiotic toxicity and development of resistance remains a significant problem. Chronic obstructive pulmonary disease (COPD), a smoking-related condition characterized by progressive and poorly reversible airflow obstruction and airway inflammation, is the fourth most common cause of death in developed countries. COPD is projected to be the third leading cause of global deaths in 2020 and is the only one of the four most common causes of death with an increasing mortality rate. In 2008 in the United States, there were an estimated 10 million patients diagnosed with chronic obstructive pulmonary disease (COPD). SDI COPD Claims Analysis, May 2009. Murray et at., 1997 Lancer 349: 1269-76. Approximately 7 million U.S. patients receive treatment for COPD. Mannino et al; The Epidemiology and
Economics of COPD, Proc Am Tho e Soc 2007. In the US, direct COPD costs in 2002 were approximately $18.0 billion. Statistics from National Center for Health Statistics, National Health Interview Survey: Research for the 1995-2004 redesign, Hyattsville, Maryland: U.S. Department of Health and Human Services, CDC, NCHS. Vital and Health Stat 2( 26), 1999.
The clinical course of COPD is characterized by chronic disability, with intermittent, acute exacerbations which may be triggered by a variety of stimuli including exposure to pathogens, inhaled irritants (e.g., cigarette smoke), allergens, or pollutants. “Acute exacerbation” refers to worsening of a patient’s COPD symptoms from his or her usual state that is beyond normal day-to-day variations, and is acute in onset. See, Rabe et al., 2007 Am J Res Crit Care Med, 176: 532- 555. Acute exacerbations of COPD greatly affect the health and quality of life of patients with COPD. Bathoorn, E, Int J Chron Obstruct Pulmon Dis. 2008 3(2):217- 229. Acute exacerbation of COPD is a key driver of the associated substantial socioeconomic costs of the disease. Approximately 73% ($13 billion) of direct COPD costs in 2002 were due to hospitalizations related to acute exacerbations of COPD. Investigators from the Burden of Obstructive Lung Disease (BOLD) Initiative have estimated the cumulative discounted cost of COPD care in the US to be $880 billion by 2020 – an average of more than $44 billion per year over two decades. Lee et al., 2006 ATS Proceedings, 3:A598. Multiple studies have also shown that prior exacerbation is an independent risk factor for future hospitalization for COPD.
Garcia-Aymerich et al., 2003, Thorax, 58:100-105. Hospitalization consumes roughly 70% of COPD healthcare expenditure in the US. McGhan et al., 2007, Chest, 132(6): 1748-1755. Accordingly, for a new drug therapy to significantly reduce the health and economic costs of COPD, it must address acute exacerbations of COPD.
It is clear that there is a continued need for an improved method of treatment for acute and chronic respiratory infections caused by Gram-negative and Gram- positive bacteria, particularly multidrug resistant bacteria, such as P. aeruginosa. This is particularly evident in patients having chronic respiratory conditions where current therapies are limited by problems with development of resistance and toxicity. Such method of treatment would preferably comprise inhalation of an aerosolized antibiotic composition that delivers a therapeutically effective amount of the active pharmaceutical ingredients directly to the endobronchial space of the airways or to the nasal passages. Such treatment would ideally be efficacious, reduce the frequency of drug resistance, and improve safety.
PCT Publication No. WO2005/1 0022 to Gilead Sciences, Inc. (formerly Corus Pharma) discloses a fosfomycin plus tobramycin combination formulation for delivery by aerosolization. The concentrated fosfomycin/tobramycin combination formulation containing an efficacious amount of fosfomycin and tobramycin is able to inhibit susceptible bacteria. Fosfomycin and tobramycin are formulated in solution such that when reconstituted, the pH is between 4.5 and 8.0 or as a dry powder. Also disclosed is a method for treatment of respiratory tract infections by a formulation delivered as an aerosol having mass medium aerodynamic diameter predominantly from 1 to 5 microns, produced by a jet or ultrasonic nebulizer (or equivalent) or dry powder inhaler.
tobramycin solution for inhalation of various formulations are described in the prior art. 例如,美国专利第5,508,269号公开了一种制剂,该制剂包括在Iml的盐水中40_100mg (毫克)的氨基糖苷被稀释成四分之一生理盐水强度,该制剂的PH值在5. 5到6. 5之间,其中该溶液以5ml浓缩形式气雾给药。 For example, U.S. Patent No. 5,508,269 discloses a preparation which is included in the brine Iml 40_100mg (mg) was diluted into a quarter normal saline aminoglycoside strength, the PH value of the preparation of 5 . 5 to 6.5 between, wherein the solution in concentrated form 5ml aerosol administration. [0011] 美国专利6987094公开了一种气雾制剂,其包括75mg/ml妥布霉素的水性溶液,该溶液包括O. 45%w/v (质量/体积百分比)的氯化钠,该制剂的pH值在4. O到5. 5之间,而渗透压在250到450m0sm/l (毫渗透克分子/毫升)。[0011] U.S. Patent 6,987,094 discloses an aerosol formulation which comprises an aqueous solution of 75mg/ml tobramycin, the solution comprises O. 45% w / v (mass / volume) of sodium chloride, the preparation the pH value of 4. O to 5.5, between the osmotic pressure of 250 to 450m0sm / l (mg penetration mol / ml). [0012] 美国专利申请2007/0116649公开了一种气雾制剂,其包括约100mg/ml到200mg/ ml的抗革兰阴性抗生素。 [0012] U.S. Patent Application 2007/0116649 discloses an aerosol formulation comprising about 100mg/ml to 200mg / ml of antibiotics against gram-negative. 提到了妥布霉素制剂,但是没有公开有妥布霉素的实验。Tobramycin formulations mentioned, but there is no disclosure tobramycin experiment. [0013] 美国专利申请2007/0071686公开了一种妥布霉素组合物,其包括约80mg/ml到120mg/ml的妥布霉素、一酸性辅助剂和低浓度的氯化钠。[0013] U.S. Patent Application 2007/0071686 discloses a tobramycin composition comprises about 80mg/ml to 120mg/ml of tobramycin, an acidic auxiliary agents and a low concentration of sodium chloride. 所述酸性辅助剂可以是硫酸钠或磷酸钠。 The acidic auxiliary agent may be sodium or sodium phosphate. 根据US2007/0071686,活性剂的浓度不超过120mg/ml,这是因为据说妥布霉素的浓度因粘度原因对雾化有负面影响。According to US2007/0071686, the concentration of the active agent is not more than 120mg/ml, this is because the concentration of tobramycin said reasons due to the viscosity of a negative impact on atomization. 而且,根据US2007/0071686的妥布霉素组合物利用喷雾器给药给患者,即活性成分通过潮式呼吸吸入。Moreover, the composition according to US2007/0071686 of tobramycin administered to patients using the spray, that the active ingredient through the tidal breathing inhalation. [0014] 欧洲专利2186508尤其公开了一种少于4ml溶液的组合物,其包括的60 – 200mg/ ml的、在生理上可接受的载体上的氨基糖苷抗生素。 [0014] In particular, in European Patent 2,186,508 discloses a composition of the solution is less than 4ml, comprising of 60 – 200mg / ml, and in a physiologically acceptable carrier aminoglycoside antibiotics. EP2186508中的实验显示,包括120mg/ ml妥布霉素的组合物使用PARI LC PLUS®牌喷雾器(德国Starnberg的Pari Boy N压缩器公司)给药需要约10分钟。EP2186508 The experiments showed that including 120mg / ml tobramycin compositions using PARI LC PLUS ® brand spray (Starnberg, Germany The Pari Boy N compressor company) to about 10 minutes of administration. 尽管这少于商业上可获得的TOBI®给药所需的时间,但从患者配合度和患者友好角度考虑,所需的时间还是太长。 Although this is less than the commercially available TOBI ® dosing time required, but with the degree of the patient, and patient-friendly point of view, the time is too long. EP2186508提到,使用呼吸致动吸入装置对于前面提到的商业上可获得的系统可以获得更快的给药时间。 EP2186508 mentioned breath actuated inhalation device used for the previously mentioned commercially available systems can be obtained faster delivery time. 但是,EP2186508 中使用呼吸致动吸入装置(AcroDose™)所获得的给药时间的实验仅限于低浓度的妥布霉素(60mg/ml)。However, EP2186508 using the breath actuated inhaler device (AcroDose ™) obtained experimental delivery time is limited to low concentrations of tobramycin (60mg/ml). 其还指出,使用AcroDose™系统的60mg/ml组合物给药,还必须给药第二可分量。 It also noted that the use AcroDose ™ system 60mg/ml composition administered may also be administered a second component. 从患者友好和配合角度来看,需要加入和给药第二可分量代表一种缺点。 Friendship and cooperation from the patient’s point of view, and the administration need to add a second component represents a drawback can be. [0015] 妥布霉素溶液也以局部给药出名,例如治疗角膜炎,参见Davis等人在“Canad. J Ophtal. ”(加拿大眼科杂志,1978年第13期273页)的文章,Davis等人在“Arch Opthalmol” (眼科杂志,1978年第96卷123-125页)的文章和Unter man等人在“ J. Cataract Refract. Surg. ”(白内障手术杂志,1988年第14卷500-504页)的文章。[0015] tobramycin solution is also famous for topical administration, such as treatment keratitis, see Davis et al in “Canad. J Ophtal.” (Canadian Journal of Ophthalmology, 1978 13 273) of the article, Davis, etc. in “Arch Opthalmol” (Ophthalmology 1978 Volume 96 pages 123-125) of articles and Unter man and others in “J. Cataract Refract. Surg.” (cataract surgery magazine, Volume 14, 500-504, 1988 pages) of the article. [0016] 现有给药手段和治疗方案的公知缺点是给药所需的时间,尤其影响患者的配合度和患者的生活质量。 Existing methods of administration and treatment programs known

FDA OKs Teva’s Injectable Treanda

FDA OKs Teva’s Injectable Treanda
FDA Approves Teva’s Injectable Treanda

bendamustine
Sept. 17, 2013 (GLOBES)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA; TASE: TEVA) has announced that the US Food and Drug Administration (FDA) has approved a new injectable version Treanda for treatment of indolent B-cell non-Hodgkin lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen, and chronic lymphocytic leukemia. read all at
http://www.pharmalive.com/fda-oks-tevas-injectable-treanda
Bendamustine (INN, trade names Treakisym, Ribomustin, Levact and Treanda; also known as SDX-105) is a nitrogen mustard used in the treatment of chronic lymphocytic leukemia[1] and lymphomas. It belongs to the family of drugs called alkylating agents. It is also being studied for the treatment of sarcoma.[2]
History
Bendamustine was first synthesized in 1963 by Ozegowski and Krebs in East Germany(the former German Democratic Republic). Until 1990 it was available only in East Germany. East German investigators found that it was useful for treating chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myelomaand lung cancer.
Bendamustine received its first marketing approval in Germany, where it is marketed under the tradename Ribomustin, by Astellas Pharma GmbH’s licensee, Mundipharma International Corporation Limited. It is indicated as a single-agent or in combination with other anti-cancer agents for indolent non-Hodgkin’s lymphoma, multiple myeloma, and chronic lymphocytic leukemia. SymBio Pharmaceuticals Ltd holds exclusive rights to develop and market bendamustine HCl in Japan and selected Asia Pacific Rim countries.
In March 2008, Cephalon received approval from the United States Food and Drug Administration to market bendamustine in the US, where it is sold under the tradename Treanda, for treatment of chronic lymphocytic leukemia.[3]
In October 2008, the FDA granted further approval to market Treanda for the treatment of indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. [4]
Bendamustine, 4-{5-[Bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric acid:

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there under the tradename Cytostasan®. See, e.g., W. Ozegowski and D. Krebs, IMET 3393 γ-[1-methyl-5-bis-(β-chloroethyl)-aminobenzimidazolo-(2)]-butyryl chloride, a new cytostatic agent of the group of benzimidazole nitrogen mustards. Zbl. Pharm. 110, (1971) Heft 10, 1013-1019, describing the synthesis of bendamustine hydrochloride monohydrate. Since that time, it has been marketed in Germany under the tradename Ribomustin®. Bendamustine is an alkylating agent that has been shown to have therapeutic utility in treating diseases such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to technical difficulties in its preparation and administration. Researchers, therefore, have investigated methods of improving the preparation and stability of bendamustine and its formulations. For example, German (GDR) Patent No. 159877 discloses a method for preparing bendamustine free base by reaction of the bis-hydroxyl precursor with thionyl chloride followed by recrystallization from water.
German (GDR) Patent No. 34727 discloses a method of preparing derivatives of bendamustine. The described derivatives differ from bendamustine in the substitution at the 1-position.
German (GDR) Patent No. 80967 discloses an injectable preparation of bendamustine hydrochloride monohydrate, ascorbic acid, and water. GDR 80967 describes that lyophilization of compounds such as bendamustine is only possible if the compound is of sufficient stability that it can withstand the processing conditions. The preparation described in GDR 80967 is not lyophilized.
German (GDR) Patent No. 159289 discloses a ready-to use, injectable solution of bendamustine hydrochloride that avoids lyophilization. GDR 159289 describes an anhydrous solution of bendamustine hydrochloride in 1,2-propylene glycol or ethanol.
U.S. application Ser. No. 11/330,868, filed Jan. 12, 2006, assigned to Cephalon, Inc., Frazer, P A, discloses methods of preparing lyophilized pharmaceutical compositions comprising bendamustine hydrochloride.
Chemotherapeutic uses
Bendamustine has been used both as sole therapy and in combination with other agents including etoposide, fludarabine, mitoxantrone,methotrexate, prednisone, rituximab, vincristine and 90Y-ibritumomab tiuxetan.
One combination for stage III/IV relapsed or refractory indolent lymphomas and mantle cell lymphoma (MCL), with or without prior rituximab-containing chemoimmunotherapy treatment, is bendamustine with mitoxantrone and rituximab.[5] In Germany in 2012 it has become the first line treatment of choice for indolent lymphoma.[6] after Trial results released in June 2012 showed that it more than doubled disease progression-free survival when given along with rituximab. The combination also left patients with fewer side effects than the older R-CHOP treatment.[7]
Common adverse reactions are typical for the class of nitrogen mustards, and include nausea, fatigue, vomiting, diarrhea, fever, constipation, loss of appetite, cough, headache, unintentional weight loss, difficulty breathing, rashes, and stomatitis, as well as immunosuppression, anemia, and low platelet counts. Notably, this drug has a low incidence of hair loss (alopecia) unlike most other chemotherapy drugs.[8]
References
- Kath R, Blumenstengel K, Fricke HJ, Höffken K (January 2001). “Bendamustine monotherapy in advanced and refractory chronic lymphocytic leukemia”. J. Cancer Res. Clin. Oncol. 127 (1): 48–54. doi:10.1007/s004320000180. PMID 11206271.
- Bagchi S (August 2007). “Bendamustine for advanced sarcoma”. Lancet Oncol. 8 (8): 674. doi:10.1016/S1470-2045(07)70225-5.PMID 17726779.
- “Cephalon press release – Cephalon Receives FDA Approval for TREANDA, a Novel Chemotherapy for Chronic Lymphocytic Leukemia”. Retrieved 2008-03-23.
- “Cephalon press release -Cephalon Receives FDA Approval for TREANDA to Treat Patients with Relapsed Indolent Non-Hodgkin’s Lymphoma”. Retrieved 2008-11-03.
- Weide R, Hess G, Köppler H, et al. (2007). “High anti–lymphoma activity of bendamustine/mitoxantrone/rituximab in rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A muticenter phase II study of the German Low Grade Lymphoma Study Group (GLSG)”. Leuk. Lymphoma. 48 (7): 1299–1306. doi:10.1080/10428190701361828. PMID 17613757.
- New Combo Replaces CHOP for Lymphoma. Dec 2012
- “‘Rediscovered’ Lymphoma Drug Helps Double Survival: Study”. June 3, 2012.
- Tageja, Nishant; Nagi, Jasdeepa; “Bendamustine: something old, something new”; Cancer Chemotherapy and Pharmacology, 2010 Aug;66(3):413-23. doi: 10.1007/s00280-010-1317-x.
External links
- Manufacturer’s official website intended for US patients
more info
Bendamustine hydrochloride, 4-{5-[Bis(2-chloroethyl) amino]- l-methyl-2- benzimidazolyl} butyric acid hydrochloride, of the formula (VI) :

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there, as the hydrochloride salt, under the trade name Cytostasan®. Since that time, it has been marketed in Germany under the trade name Ribomustin®. Bendamustine Hydrochloride as injection is available in the United States under the tradename Treanda®. Bendamustine hydrochloride is an alkylating agent that is approved for the treatment of non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia.
Bendamustine hydrochloride is a benzimidazole analog. While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to formation of non-bendamustine products (i.e. “degradation impurities”) which leads to technical difficulties in its preparation and administration. In light of its instability in aqueous solution, bendamustine is supplied as a lyophilized cake of bendamustine hydrochloride salt. US2006/159713, US 2006/128777 and WO2010/036702 disclose various impurities of Bendamustine hydrochloride which are as follows:

PC-1 PC-2
Jena et al. were the first to disclose the synthesis of Bendamustine hydrochloride in German (GDR) Patent No. 34727. Krueger et al. in German (GDR) Patent No. 159877 recite a method as summarized in scheme-1, for the synthesis of bendamustine hydrochloride comprising the reaction of the 4-[l-methyl-5-bis-(2- hydroxyethyl)-benzimidazolyl-2]butyric acid ethyl ester (4) (or the corresponding methyl, propyl or butyl ester) with thionyl chloride in chloroform at 0-5°C to form 4-[l- methyl-5-bis-(2-chloroethyl)-benzimidazolyl-2]butyric acid ethyl ester (5). Excess of thionyl chloride is destroyed by stirring the reaction mixture in aqueous HCl. Finally chloroform is distilled off and stirred at 95°C for 3 hours. The reaction mixture is partially concentrated and the residue is diluted with water and stirred upto crystallization. Further purification is done by recrystallization from water.
Scheme-1: Method disclosed by Krueger et al. in DD159877 for the synthesis of Bendamustine hydrochloride

Bendamustine hydrochloride (6)
Ozegowski et al in Zentralblatt fuer Pharmazie, Pharmakotherapie und Laboratoriumsdiagnostik 1 10 (10), 1013-1019 (1971) discloses a process for the preparation of bendamustine hydrochloride monohydrate. The Chinese journal “Chinese journal of New Drugs “, 2007, No. 23, Vol. 16, 1960-61 and J. Prakt. Chem. 20, 178-186 (1963) disclose another method for the synthesis of Bendamustine hydrochloride monohydrate starting from 2,4-dinitrochlorobenzene as summarized in scheme-2.

The crucial conversions are reaction of l-methyl-2-(4′-ethyl butyrate)-5- amino]-lH-benzimidazole 6 with ethylene oxide in the presence of water, sodium acetate and acetic acid, by maintaining at 5°C for 5 hours and overnight at 20°C to give 4-{5-[bis-(2-hydroxy-ethyl)-amino]-l-methyl-lH-benzimidazol-2-yl}-butyric acid ethyl ester (dihydroxy ester) 7 as a jelly mass, which on chlorination using thionyl chloride in chloroform and subsequent in situ hydrolysis with concentrated HCI gave bendamustine hydrochloride. It also discloses a process for the recrystallization of bendamustine hydrochloride from water and the product obtained is a monohydrate with a melting point of 148-151°C.
IP.com Journal 2009, 9(7B), 21 discloses another process as shown below for the preparation of ethyl-4-[5-[bis(2-hydroxyethyl) amino]- l-methylbenzimidazol-2- yl]butanoate (III) wherein ethyl-4-(5 -amino- 1 -methyl- lH-benzo[d]imidazol-2-yl) butanoate (II) is reacted with 2-halo ethanol in the presence of an inorganic base selected from the group consisting potassium carbonate, potassium bicarbonate, sodium

The PCT application WO 2010/042568 assigned to Cephalon discloses the synthesis of Bendamustine hydrochloride as summarized in schem-3 starting from 2,4- dintroaniline in six steps. The crucial step is reductive alkylation of Il-a, using borane- tetrahydrofuran and chloroacetic acid at ambient temperature, producing compound of formula I-a. Acid mediated hydrolysis of I-a using concentrated hydrochloric acid at reflux produced bendamustine hydrochloride which has a purity of 99.1%. The above PCT Patent application also discloses a method of purification of Bendamustine hydrochloride by agitating the Bendamustine hydrochloride in a mixture of DMF and THF at 75°C for about 30 minutes followed by cooling to ambient temperature and isolating the solid by filtration.
Scheme-3:


iil-a


Bemdamuatine hydrochloride
The PCT application WO 2011/079193 assigned to Dr. Reddy’s Laboratories discloses the synthesis of Bendamustine hydrochloride as summarized in schem-4 starting from compound of formula (II). The crucial step is alkylation of compound of formula II with 2-haloethanol in the presence of an organic base to give a compound of formula (III) which on chlorination with a chlorinating agent affords a compound of formula (IV). Compound of formula (IV) on hydrolysis in acidic medium gives bendamustine hydrochloride. It further discloses purification of bendamustine hydrochloride using aqueous hydrochloric acid and acetonitrile.
Scheme-4:

Bendamustine hydrochloride (Pure)
The most of the prior art processes described above involve
• The use of ethylene oxide for the preparation of bendamustine hydrochloride, which is often not suitable for industrial scale processes due to difficulty in handling ethylene oxide, since it is shipped as a refrigerated liquid.
• Further, the known processes involve the use of strongly acidic conditions and high temperatures for the hydrolysis of ethyl ester of bendamustine and subsequent in-situ formation of bendamustine hydrochloride, thereby resulting in increased levels of various process-related impurities IMP. -A (RRT-0.46), IMP. -B (RRT-1.27) and IMP. -C (RRT-1.31) whose removal is quite difficult and make the process less economically viable.

IMP.-B
International Application Publication No. WO 2009/120386 describes various solid forms of bendamustine hydrochloride designated as bendamustine hydrochloride Form 1, bendamustine hydrochloride Form 2, bendamustine hydrochloride Form 3, bendamustine hydrochloride Form 4, amorphous bendamustine hydrochloride or a mixture thereof, processes for their preparation and lyophilized composition comprising the solid forms. According to the disclosure, monohydrate of bendamustine hydrochloride has been prepared previously. The monohydrate has a reported melting point of 152-156°C which is similar to that of the observed melting point of bendamustine hydrochloride Form 2.
It is known that synthetic compounds can contain extraneous compounds or impurities resulting from their synthesis or degradation. The impurities can be unreacted starting materials, by-products of the reaction, products of side reactions, or degradation products. Generally, impurities in an active pharmaceutical ingredient (API) may arise from degradation of the API itself, or during the preparation of the API. Impurities in Bendamustine hydrochloride or any active pharmaceutical ingredient (API) are undesirable and might be harmful.
Regulatory authorities worldwide require that drug manufacturers isolate, identify and characterize the impurities in their products. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurity is present in the lowest possible levels, even if structural determination is not possible. The product mixture of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. Side products and byproducts of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product mixture. At certain stages during processing of the active pharmaceutical ingredient, the product is analyzed for purity, typically, by HPLC, TLC. or GC analysis, to determine if it is suitable for continued processing and, ultimately, for use in a pharmaceutical product. Purity standards are set with the intention of ensuring that an API is as free of impurities as possible, and, thus, are as safe as possible for clinical use. The United States Food and Drug Administration guidelines recommend that the amounts of some impurities are limited to less than 0.1 percent.
Generally, impurities are identified spectroscopically and by other physical methods, and then the impurities are associated with a peak position in a chromatogram (or a spot on a TLC plate). Thereafter, the impurity can be identified by its position in the chromatogram, which is conventionally measured in minutes between injection of the sample on the column and elution of the particular component through the detector, known as the “retention time” (“RT”). This time period varies daily based upon the condition of the instrumentation and many other factors. To mitigate the effect that such variations have upon accurate identification of an impurity, practitioners use “relative retention time” (“RRT”) to identify impurities. The RRT of an impurity is its retention time divided by the retention time of a reference marker.
It is known by those skilled in the art, the management of process impurities is greatly enhanced by understanding their chemical structures and synthetic pathways, and by identifying the parameters that influence the amount of impurities in the final product.
Therefore, there remains a need for improved process for the preparation of bendamustine hydrochloride, producing high yield and purity, and well-suited for use on an industrial scale. Despite the existence of various polymorphic forms of bendamustine hydrochloride, there exists a need for a simple process for the preparation of the stable form of bendamustine hydrochloride which is amenable to scale up and results in high yield and purity.
Bendamustine, (4-{5-[bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric

Bendamustine
is an atypical structure with a benzimidazole ring, which structure includes an active nitrogen mustard. Bendamustine was initially synthesized in 1963 in the German Democratic Republic and was available from 1971 to 1992 in that location under the name Cytostasan®. Since that time, it has been marketed in Germany under the tradename Ribomustin®. It is currently available for use in the United States under the tradename Treanda® (Cephalon, Inc., Frazer, Pa.). It has been widely used to treat chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
Like other nitrogen mustards, bendamustine hydrolyzes in aqueous solution, with the major degradant being the primary alcohol HP1 (See U.S. application Ser. No. 11/330,868, the entirety of which is incorporated herein):
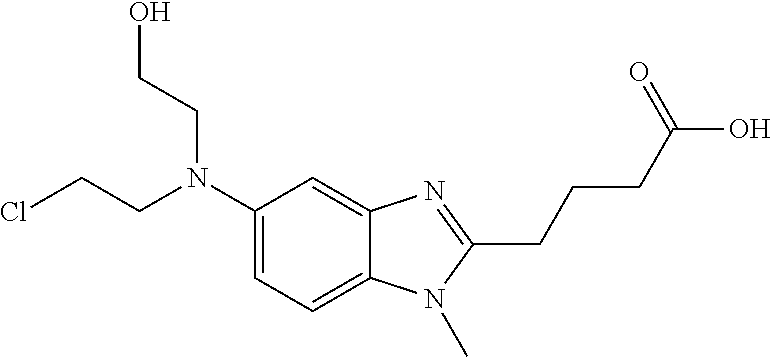
In light of its instability in aqueous solution, bendamustine is currently supplied as a lyophilized powder for injection. Just prior to its infusion, the medical practitioner reconstitutes the powder with Sterile Water for Injection. Reconstitution should yield a clear, colorless to pale yellow solution and the powder should completely dissolve in about 5 minutes. If particulate matter is observed, the reconstituted product should not be used and should be discarded. The reconstituted product is then transferred to a 0.9% Sodium Chloride Injection infusion bag within 30 minutes of reconstitution. This admixture should be a clear and colorless to slightly yellow solution. If the admixture comprises particulate matter or is discolored, it should be discarded and a fresh sample prepared.
The salt bendamustine hydrochloride is an alkylating agent, originally synthesized in 1963 at the Institute for Microbiology & Experimental Therapy in Jena, German Democratic Republic, with the intent to produce an agent with both alkylating and antimetabolite properties. Jenapharm (now Schering AG) formerly marketed it in Germany under the trade name Cytostasan from 1971 to 1992. Cytostasan was a lyophilised powder for solution for injection (vials) conatining 25 mg of Bendamustine HCI. It was widely used but never studied systematically in patients until the 1990s, then German investigators demonstrated its clinical activity in a number of malignancies. Since 1993, Ribosepharm was marketing bendamustine in Germany under the brand name Ribomustin RBO. Ribomustin is available as a lyophilized powder for injection, containing 100 mg of drug in each 50 ml_ vial, or 25 mg of drug in each 20 ml_ vial, also comprising mannitol, and indicated for the treatment of chronic lymphocytic leukemia. The lyophilized powder is reconstituted as close to the time of patient administration as possible with 40 ml_ (for a 100 mg product) or 10 mL (for a 25 mg product) of sterile water for injection. The reconstituted product then is further diluted to 500 mL with 0.9% sodium chloride for injection. The route of administration is by intravenous infusion over 30 to 60 minutes.
Another bendamustine product is sold in the United States by Cephalon, Inc. as TREANDA® for Injection, a lyophilized powder in a single-use vial indicated for the treatment of patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin’s lymphoma. A 25 mg dose vial contains 25 mg of bendamustine hydrochloride and 42.5 mg of mannitol, and a 100 mg dose vial contains 100 mg of bendamustine hydrochloride and 170 mg of mannitol.
TREANDA is intended for intravenous infusion only after reconstitution with Sterile Water for Injection USP, and then further dilution with either 0.9% Sodium
Chloride Inj.ection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Inj.ection, USP. The pH of the reconstituted solution is 2.5-3.5. TREANDA is supplied as a sterile non-pyrogenic white to off-white lyophilized powder, in a single-use vial.
Bendamustine hydrochloride is very unstable in an aqueous solution. The bis-2-chlorethylamino bond is hydrolyzed in weak acid, neutral, or alkaline solution. Monohydroxybendamustine [HP-1 ] is formed rapidly in the presence of water. Bendamustine ethyl ester [BM1 EE] is formed when bendamustine reacts with ethyl alcohol. BM1 EE can be formed during drug substance manufacturing, e.g., during recrystalization and/or purification processes. BM1 EE is a more potent cytotoxic drug than bendamustine.
Teva and Alexza Announce Teva’s License to Market ADASUVE® in the U.S.

Loxapine (Loxapac, Loxitane)
is a typical antipsychotic medication, used primarily in the treatment of schizophrenia. It is a member of the dibenzoxazepine class and structurally related to clozapine (which belongs to the chemically akin class ofdibenzodiazepines). Several researchers have argued that Loxapine may behave as anatypical antipsychotic.
Loxapine may be metabolized by N-demethylation to amoxapine, a tetracyclic antidepressant.
Adasuve® (loxapine) inhalation powder is approved in the U.S. for the acute treatment of agitation associated with schizophrenia and bipolar disorder
May 8, 2013
Teva Pharmaceuticals USA, Inc., a subsidiary of Teva Pharmaceutical Industries Ltd (NYSE: TEVA), and Alexza Pharmaceuticals, Inc. (NASDAQ: ALXA) announced today that the companies have entered into an exclusive U.S. license and supply agreement for ADASUVE (loxapine) inhalation powder 10 mg for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. Teva will be responsible for all U.S. commercial and clinical activities for ADASUVE, including U.S. post-approval clinical studies, and has gained rights to conduct additional clinical trials of ADASUVE for potential new indications in neurological disorders. Alexza will be responsible for manufacturing and supplying ADASUVE to Teva for commercial sales and clinical trials.
About Alexza Pharmaceuticals, Inc.
Alexza Pharmaceuticals is focused on the research, development and commercialization of novel, proprietary products for the acute treatment of central nervous system conditions, including agitation, acute repetitive seizures and insomnia. Alexza’s products are based on the Staccato®system, a hand-held inhaler that is designed to deliver a drug aerosol to the deep lung, providing rapid systemic delivery and therapeutic onset, in a simple, non-invasive manner.
FDA Approves New Drug Application (NDA) for Teva’s Quartette (levonorgestrel/ethinyl estradiol and ethinyl estradiol) Tablets for the Prevention of Pregnancy

levonorgestrel

ethinyl estradiol
Quartette™ Represents the Next Generation of Extended Regimen Oral Contraceptives
JERUSALEM 30 MAR 2013
Teva Pharmaceutical Industries Ltd. today announced that the U.S. Food and Drug Administration (FDA) has approved Quartette™ (levonorgestrel/ethinyl estradiol and ethinyl estradiol) tablets for the prevention of pregnancy. Quartette™ represents the next generation of extended regimen oral contraceptives to be approved by the FDA, and was designed to minimize breakthrough bleeding (BTB) between scheduled periods. The approval of Quartette™ demonstrates Teva’s continued commitment to the development and production of an innovative range of pharmaceutical products that support the health of women around the world.
“Breakthrough bleeding can be experienced with any birth control pill, especially during the first few months, and is one of the reasons a large number of women discontinue extended regimens,” said Dr. James A. Simon, clinical professor of Obstetrics and Gynecology at the George Washington University School of Medicine. “The estrogen in Quartette™ increases at specific points and provides four short light periods a year. Breakthrough bleeding decreases over time, which might help encourage patient adherence.”
The approval was based on a development program that included results from Phase I, Phase II and Phase III clinical trials designed to evaluate the safety and efficacy of Quartette™. The Phase III clinical trial, which involved more than 3,000 women, found that Quartette™ was 97 percent effective at preventing pregnancy. Data further demonstrated that the most common adverse reactions (≥2%) in the Phase III clinical trial were headaches, heavy/irregular vaginal bleeding, nausea/vomiting, acne, dysmenorrhea, weight increased, mood changes, anxiety/panic attack, breast pain and migraines. The primary clinical trial that evaluated the efficacy of Quartette™ also assessed BTB. BTB and unscheduled spotting decreased over successive 91 day cycles.1
Quartette™ features a unique 91-day oral regimen, whereby the dose of estrogen increases at three distinct points over the first 84 days and the amount of progestin remains consistent; this is followed by seven days of 10 mcg of ethinyl estradiol.
“Teva is the leader in the pharmaceutical industry in the marketing and development of extended regimen oral contraceptives, and Quartette™ represents the next generation of these contraceptives. It is a uniquely differentiated product and is based on Teva’s research into when breakthrough bleeding is most likely to occur with these regimens,” said Jill DeSimone, senior vice president & general manager, Global Teva Women’s Health. “Quartette™ is the newest product in our global women’s health franchise and is an example of our dedication to providing a variety of contraceptive and family planning options that fit women’s lifestyles.”
Nippon and Teva receive approval for biosimilar G-CSF (fligrastim) in Japan

As with the original drug, acts on neutrophil precursor cells, to promote the proliferation, differentiation and its biosimilar filgrastim of (recombinant) promotes the release of neutrophils from the bone marrow, enhances its function. In the field of cancer treatment, it is used for chemotherapy-induced neutropenia mainly cancer.
Filgrastim is a granulocyte colony-stimulating factor (G-CSF) analog used to stimulate the proliferation and differentiation of granulocytes.[1] It is produced by recombinant DNA technology. The gene for human granulocyte colony-stimulating factor is inserted into the genetic material of Escherichia coli. The G-CSF then produced by E. coli is different from G-CSF naturally made in humans.
It is marketed by Amgen under the brand name Neupogen, in India it is also marketed by Abbott Healthcare under the brand name Imumax, Dr. Reddy’s Laboratories under the brand name Grafeel, In Pakistan CCL Pharmaceuticals (Pvt) Ltd under the brand name Grastin, Zenotech Laboratories Limited under the brand name Nugraf, Raichem lifesciences under the brand name Shilgrast, Intas Biopharmaceuticals under the brand name Neukine, Emcure biopharmaceuticals under the brand name Emgrast, Reliance Life Sciences under the brand name Religrast and Sandoz under the name Zarzio.
Apricus Biosciences is currently developing and testing a product (under the brand name Nupen) which can deliver filgrastim through the skin to improve post-chemotherapy recovery of neutrophil counts.
Phase III Study of Oral Laquinimod for Relapsing-Remitting Multiple Sclerosis

Laquinimod
5-chloro-N-ethyl-4-hydroxy-1-methyl-2-oxo-
N-phenyl-1,2-dihydroquinoline-3-carboxamide
Laquinimod is an experimental immunomodulator developed by Active Biotech and Teva. It is currently being investigated as an oral treatment for multiple sclerosis (MS).
Laquinimod is the successor of Active Biotech’s failed experimental immunomodulator linomide.[1]
The compound has been investigated in two Phase II trials using successive magnetic resonance scans (MRI). Laquinimod seems to be able to reduce the MS disease activity on MRI.[2][3] However, the response to a given dose was discrepant between both studies.[4]
Phase III studies for MS started in December 2007.[5] In 2011, Teva announced its clinical trials involving laquinimod had failed, being unable to significantly reduce relapses into MS among patients beyond a placebo.[6] However, the final results of above mentioned phase III trial proved oral laquinimod administered once daily slowed the progression of disability and reduced the rate of relapse in patients with relapsing–remitting multiple sclerosis [7]
Mar 6, 2013 –
CONCERTO Study Enrolling Patients Globally to Evaluate Impact of Laquinimod on Disability Progression
Teva Pharmaceutical Industries Ltd. and Active Biotech announced today enrollment of the first patient in the CONCERTO study – the third Phase III placebo-controlled study designed to evaluate the efficacy, safety and tolerability of once-daily oral laquinimod in patients with relapsing-remitting multiple sclerosis (RRMS). The primary outcome measure of CONCERTO will be confirmed disability progression as measured by the Expanded Disability Status Scale (EDSS).
“Previous Phase III studies in more than 2,400 people with RRMS suggest a unique profile of laquinimod, directly affecting the neurodegenerative processes that lead to disability progression, the main concern in the treatment of RRMS,” said CONCERTO principal investigator, Dr. Timothy Vollmer, Professor of Neurology, University of Colorado Denver, Medical Director of the Rocky Mountain Multiple Sclerosis Center, and Co-Director of the RMMSC at Anschutz. “We are currently enrolling patients in this third pivotal study to further examine the clinical benefits of laquinimod on disability progression, the primary endpoint of the CONCERTO trial, and brain atrophy, at both the previously studied 0.6 mg dose, and now a higher 1.2 mg dose.”
The multinational, randomized, double blind placebo-controlled study will aim to enroll approximately 1,800 patients at more than 300 sites globally (http://clinicaltrials.gov/show/NCT01707992). Along with the primary endpoint of time to confirmed disability progression, the study will also examine the impact of laquinimod on endpoints such as percent change in brain volume and other clinical and MRI markers of disease activity.
“For nearly 30 years, Teva has been focused on improving the lives of people with multiple sclerosis by delivering innovative treatment options that address this complex disease,” said Dr. Michael Hayden, President of Global R&D and Chief Scientific Officer at Teva Pharmaceutical Industries Ltd. “The CONCERTO study demonstrates our commitment to collaborating with MS communities worldwide to further develop laquinimod and address unmet patient needs.”
ABOUT LAQUINIMOD
Laquinimod is an oral, once-daily CNS-active immunomodulator with a novel mechanism of action being developed for the treatment of MS. In animal models laquinimod crosses the blood brain barrier to potentially have a direct effect on resident CNS inflammation and neurodegeneration. The global Phase III clinical development program evaluating oral laquinimod in MS includes two pivotal studies, ALLEGRO and BRAVO.
In addition to the MS clinical studies, laquinimod is currently in clinical development for Crohn’s disease and Lupus.
ABOUT CONCERTO
CONCERTO is a multinational, multicenter, randomized, double-blind, parallel-group, placebo-controlled study followed by an active treatment phase, to evaluate the efficacy, safety and tolerability of two doses of oral administration of laquinimod 0.6 mg/day or 1.2 mg/day in subjects with RRMS. This third Phase III laquinimod study will evaluate laquinimod in approximately 1,800 patients for up to 24 months, after which patients will continue to an active treatment period with laquinimod for an additional 24 months. The primary outcome measure will be time to confirmed disability progression as measured by the Expanded Disability Status Scale (EDSS). The study will also examine the impact of laquinimod on endpoints such as percent change in brain volume, as well as other clinical and MRI markers of disease activity.
ABOUT MULTIPLE SCLEROSIS
MS is the leading cause of neurological disability in young adults. It is estimated that more than 400,000 people in the United States are affected by the disease and that two million people may be affected worldwide. Multiple sclerosis is a degenerative disease of the central nervous system in which inflammation and axonal damage and loss result in the development of progressive disability.
ABOUT TEVA
Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) is a leading global pharmaceutical company, committed to increasing access to high-quality healthcare by developing, producing and marketing affordable generic drugs as well as innovative and specialty pharmaceuticals and active pharmaceutical ingredients. Headquartered in Israel, Teva is the world’s leading generic drug maker, with a global product portfolio of more than 1,000 molecules and a direct presence in about 60 countries. Teva’s branded businesses focus on CNS, oncology, pain, respiratory and women’s health therapeutic areas as well as biologics. Teva currently employs approximately 46,000 people around the world and reached $20.3 billion in net revenues in 2012.
ABOUT ACTIVE BIOTECH
Active Biotech AB is a biotechnology company with focus on autoimmune/inflammatory diseases and cancer. Projects in or entering pivotal phase are laquinimod, an orally administered small molecule with unique immunomodulatory properties for the treatment of multiple sclerosis, TASQ for prostate cancer as well as ANYARA for use in cancer targeted therapy, primarily of renal cell cancer. In addition, laquinimod is in Phase II development for Crohn’s and Lupus. Further projects in clinical development comprise the two orally administered compounds, 57-57 for SLE & Systemic Sclerosis and RhuDex(TM) for RA. Please visit http://www.activebiotech.com for more information.
- Tan IL, Lycklama à Nijeholt GJ, Polman CH et al. (April 2000). “Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trials”. Mult Scler 6 (2): 99–104. PMID 10773855.
- Comi G, Pulizzi A, Rovaris M et al. (June 2008). “Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study”. Lancet 371 (9630): 2085–2092. doi:10.1016/S0140-6736(08)60918-6. PMID 18572078.
- Polman C, Barkhof F, Sandberg-Wollheim M et al. (March 2005). “Treatment with laquinimod reduces development of active MRI lesions in relapsing MS”. Neurology 64 (6): 987–91. doi:10.1212/01.WNL.0000154520.48391.69. PMID 15781813.
- Keegan BM, Weinshenker BG (June 2008). “Laquinimod, a new oral drug for multiple sclerosis”. Lancet 371 (9630): 2059–2060. doi:10.1016/S0140-6736(08)60894-6. PMID 18572062.
- ClinicalTrials.gov NCT00509145 Safety and Efficacy of Orally Administered Laquinimod Versus Placebo for Treatment of Relapsing Remitting Multiple Sclerosis (RRMS) (ALLEGRO)
- Kresege, Naomi (1 August 2011). “Teva’s Copaxone Successor Fails in Latest Clinical Trial”. Bloomberg. http://www.bloomberg.com/news/2011-08-01/teva-s-oral-multiple-sclerosis-drug-fails-to-meet-goal-of-clinical-trial.html. Retrieved 2 August 2011. “Teva Pharmaceutical Industries Ltd. (TEVA)’s experimental multiple sclerosis pill failed to reduce relapses more than placebo in a clinical trial, dealing a blow to the company’s effort to find a successor to an older drug.”
- (Comi et al. N Engl J Med 2012;366:1000).

EP 1073639; JP 2002513006; US 6077851; WO 9955678
5-Chloroisatoic anhydride (I) is alkylated with iodomethane and NaH to afford (II). Subsequent condensation of anhydride (II) with the malonic monoamide (III) in the presence of NaH in hot DMA furnishes the target quinoline carboxamide.
…

Reaction of 2-amino-6-chlorobenzoic acid (I) with phosgene and NaHCO3 in dioxane gives 5-chloroisatoic anhydride (II), which is methylated by means of iodomethane and NaH in DMF to yield 5-chloro-1-methylisatoic anhydride (III). Finally, anhydride (III) is condensed with the malonic monoamide (IV) by means of NaH in hot dimethylacetamide. Alternatively, condensation of anhydride (III) with ethoxy malonyl chloride (V) by means of NaOMe and triethylamine in dichloromethane affords 5-chloro-4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3- carboxylic acid ethyl ester (VI), which is finally condensed with N-ethylaniline (VII) in refluxing toluene. Alternatively, ester (VI) is hydrolyzed by means of concentrated HCl in hot Ac2O to give the carboxylic acid (VIII), which is finally condensed with N-ethylaniline (VII) by means of SOCl2 and TEA in dichloromethane
Phase III Study of Teva’s Milprosa (Progesterone) Vaginal Ring Published in Fertility and Sterility

Progesterone
Data Demonstrated Once-Weekly Milprosa™ Provides Similar Pregnancy Rates to Daily 8 Percent Progesterone Vaginal Gel
Mar 4, 2013 –
Teva Pharmaceutical Industries Ltd. today announced the publication of results of the Phase III clinical trial of Milprosa™ (progesterone) vaginal ring in Fertility and Sterility. The study compared the efficacy and safety of once-weekly Milprosa™ to daily 8 percent progesterone vaginal gel for luteal phase support in in vitro fertilization (IVF) and found that clinical pregnancy rates per retrieval at eight and 12 weeks were comparable between patient groups. Adverse event (AE) profiles were similar between the two treatment groups and consistent with known AEs associated with progesterone.
“The study results demonstrate that Milprosa™ may be an effective and safe option for progesterone supplementation during the luteal phase among women undergoing IVF,” said Laurel Stadtmauer, M.D., Ph.D., professor of Obstetrics and Gynecology at Jones Institute for Reproductive Medicine at Eastern Virginia Medical School and study author. “Since normal luteal function may be compromised among women undergoing IVF, progesterone supplementation is essential and the more options patients have, the better. If approved, the once-weekly dosing of Milprosa™ may offer convenience for patients.”
The Phase III randomized, single-blinded, multicenter, noninferiority study was conducted at 22 clinical sites in the U.S. and included 1,297 patients between the ages of 18 and 42. Of enrolled patients, 646 were randomized to Milprosa™ and 651 to the 8 percent progesterone vaginal gel.
“The Fertility and Sterility publication of the Milprosa™ Phase III data is a significant milestone for Teva, especially because fertility is a meaningful new area of specialization for the company and one in which significant unmet need exists,” said Jill DeSimone, senior vice president & general manager, Global Teva Women’s Health. “We look forward to continuing to share important updates about Milprosa™ and demonstrating our investment in and commitment to women’s health.”
About the Study
The Phase III study randomized patients into two treatment groups: one group received once-weekly Milprosa™ and the other received daily 8 percent progesterone vaginal gel. Milprosa™ and the vaginal gel were initiated on the day following egg retrieval and continued through 12 weeks’ gestation. Efficacy was evaluated by comparing clinical pregnancy rates of patients at eight and 12 weeks gestation.
- At week eight, clinical pregnancy rates per retrieval were 48.0 percent for the Milprosa™ group and 47.2 percent for the vaginal gel group (between-group difference, 0.8%; 95% CI, -4.6%, 6.3%).
- At week 12, clinical pregnancy rates per retrieval for Milprosa™ and the vaginal gel were 46.4 percent and 45.2 percent respectively (between-group difference, 1.3%; 95% CI, -4.1%, 6.7%).
A secondary efficacy endpoint was the rate of live birth.
- The overall live birth rate per retrieval for women using Milprosa™ was 45.2 percent; among women using the vaginal gel, the rate was 43.3 percent.
- The majority of patients pregnant at week 12, when progesterone treatment ended, went on to have a live birth: 97.4 percent for the Milprosa™ group and 96.5 percent for the vaginal gel group.
The most commonly reported adverse events (those greater than or equal to 10% in the Milprosa™ treatment group) were nausea, headache, abdominal pain, post-procedural discomfort, abdominal distension, back pain, fatigue, vomiting and constipation. Serious adverse events (SAEs) occurred in approximately 12 percent of all patients, with no significant difference in the rate between treatment groups. The majority of SAEs that occurred were mild to moderate in severity and not related to treatment. Rates of discontinuation of treatment due to AEs were low and similar between both groups (approximately 6%).
About Milprosa™ (Progesterone) Vaginal Ring
Milprosa™ is an investigational, once-weekly progesterone ring inserted in the vagina. It is flexible and designed to continuously release a steady dose of micronized progesterone. Milprosa™ is in development to support embryo transplantation and early pregnancy (up to 10 weeks post-embryo transfer) by supplementation of corpus luteal function as part of an Assisted Reproductive Technology (ART) treatment program for infertile women.
About Supplementation of Corpus Luteal Function
The corpus luteum is a temporary endocrine gland that develops during the luteal phase of a woman’s menstrual cycle. It is an important contributor of progesterone and is critical for the maintenance of early pregnancy. During in vitro fertilization, progesterone supplementation is needed because natural levels of the hormone may be insufficient. This supplementation improves implantation rates and thus pregnancy rates. Additionally, progesterone supplementation supports early pregnancy.
About Teva
Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) is a leading global pharmaceutical company, committed to increasing access to high-quality healthcare by developing, producing and marketing affordable generic drugs as well as innovative and specialty pharmaceuticals and active pharmaceutical ingredients. Headquartered in Israel, Teva is the world’s leading generic drug maker, with a global product portfolio of more than 1,000 molecules and a direct presence in about 60 countries. Teva’s branded businesses focus on CNS, oncology, pain, respiratory and women’s health therapeutic areas as well as biologics. Teva currently employs approximately 46,000 people around the world and reached $20.3 billion in net revenues in 2012.
