PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
The most common side effects include neutropenia (low levels of neutrophils, a type of white blood cell), infusion reactions, pneumonia (infection of the lungs), upper respiratory tract infection (such as nose and throat infections), diarrhoea and bronchitis (inflammation of the airways in the lungs).[3]
In the European Union it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with relapsed and refractory multiple myeloma (MM) who have received at least two prior therapies including lenalidomide and a proteasome inhibitor (PI) and have demonstrated disease progression on the last therapy.[3]
Researchers started a Phase I study with isatuximab in combination with pomalidomide and dexamethasone for the treatment of patients with multiple myeloma (MM). The results during the Phase I trial showed that 26 out of the 45 patients discontinued the treatment due to progression of the disease. The patients had already been heavily pretreated. The latter lead to a manageable safety profile where the dose of isatuximab in combination with pomalidomide and dexamethasone would be capped to the maximum of 10 mg/kg weekly every two weeks for future studies.[12]
Based on the remarkable findings during the Phase I trial, a Phase II trial was launched where researchers investigated isatuximab as a single agent in patients with MM. The heavily pretreated patients reacted well to the single administration of isatuximab during Phase II of the trial.[13]
A Phase III combination trial for plasma cell myeloma is comparing pomalidomide and dexamethasone with and without isatuximab is in progress with an estimated completion date of 2021.[medical citation needed]
Additionally, two Phase III trials were added in 2017. The first trial highlights whether there is an added value in the combination of isatuximab with bortezomib, lenalidomide and dexamethasone. The latter will be tested in patients with newly diagnosed MM who are not qualified for a transplant (IMROZ trial). The second trial evaluates the combinations of isatuximab with carfilzomib and dexamethasone compared to carfilzomib with dexamethasone. The second trial was designed for patients who were previously treated with one to three prior lines (IKEMA). There is currently[when?] no treatment for MM, however promising improvements have been made and the study is still ongoing.[14][15]
In March 2020, it was approved for medical use in the United States.[8][9][10]
The U.S. Food and Drug Administration (FDA) approved isatuximab-irfc in March 2020, based on evidence from a clinical trial (NCT02990338) of 307 subjects with previously treated multiple myeloma.[10] The trial was conducted at 102 sites in Europe, North America, Asia, Australia and New Zealand.[10]
The trial evaluated the efficacy and side effects of isatuximab-irfc in subjects with previously treated multiple myeloma.[10] Subjects were randomly assigned to receive either isatuximab-irfc (in combination with pomalidomide and low-dose dexamethasone) or active comparator (pomalidomide and low-dose dexamethasone).[10] Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity.[10] Both subjects and health care providers knew which treatment was given.[10] The trial measured the time patients lived without the cancer growing (progression-free survival or PFS).[10]
It was approved for medical use in the European Union in May 2020.[3]
Structure and reactivity
The structure of isatuximab consists of two identical immunoglobulin kappa light chains and also two equal immunoglobulin gamma heavy chains. Chemically, isatuximab is similar to the structure and reactivity of daratumumab, hence both drugs show the same CD38 targeting. However, isatuximab shows a more potent inhibition of its ectozyme function. The latter gives potential for some non-cross reactivity. Isatuximab shows action of an allosteric antagonist with the inhibition of the CD38 enzymatic activity. Additionally, isatuximab shows potential where it can induce apoptosis without cross linking.[16] Lastly, Isatuximab reveals direct killing activity when a larger increase in apoptosis is detected in CD38 expressing cancer cells. Furthermore, isatuximab demonstrated a dose dependent inhibition of CD38 enzymatic activity. However, daratumumab with the same experimental conditions shows a more limited inhibition without a dose response.[17]
Reactions
Isatuximab binds uniquely to an epitope on the CD38 receptor and is the only CD38 antibody which can start apoptosis directly.[18] Isatuximab binds to a different CD38 epitopeamino-acid sequence than does the anti-CD38 monoclonal antibody daratumumab.[19] The binding with the CD38 receptor is mainly via the gamma heavy chains and are more potent than other CD38 antibodies such as daratumumab which can inhibit the enzymatic activity of CD38. Moreover, isatuximab inhibits the hydrolase activity of CD38.[medical citation needed]
The antibodies show signs of improving antitumor immunity by eliminating regulatory T cells, B cells and myeloid-derived suppressor cells. The difference in binding between isatuximab and daratumumab is in the recognition of the different amino acid groups. Isatuximab identifies 23 amino acids of CD38 to the contrary with daratumumab who has 27. The residue of Glu233 has a flexible sidechain and faces the N-terminal of Asp1 residue in the isatuximab light chain. The latter light chain of isatuximab is also flexible which makes the interaction between CD38/Glu233 and the Asp1 weaker than the other interactions between CD38 and isatuximab. The caspase-dependent apoptotic pathway and the lysosomal mediated cell death pathway in MM cells is induced by isatuximab. The MM cell death follows the downstream reactions of the lysosomal activation. The latter also activates the production of reactive oxygen species.[20]
Available forms
Isatuximab or isatuximab-irfc is available as a drug in an intravenous infusion form. Injection doses are 100 mg/5 mL (20 mg/mL) solution in single-dose vial or 500 mg/25 mL (20 mg/mL) solution in single-dose vial.[4]
Mechanism of action
Cancer of the blood that is distinguished by an overproduction of malignant plasma cells in the bone marrow is called multiple myeloma. The myeloma cells are marked with uniformed overexpression of CD38 surface glycoproteins. Although these proteins are also expressed on other myeloid and lymphoid cells, the extent is relatively minor compared to myeloma cells. The fact that CD38 glycoproteins carry out various important cellular functions, and that they are plentiful on the surface of myeloma cells, has made them an appealing target for multiple myeloma treatment.[21] CD38 was first described as an activation marker, but later the molecule displayed functions in adhesion to endothelial CD31 proteins, e.g. as an aiding component of the synapse complex, as well as an ectoenzyme implicated in the metabolism of extracellular NAD+ and cytoplasmic NADP. The tumour cells can evade the immune system, possibly due to adenosine, an immunosuppressive molecule that arises as a product of the ectoenzymatic activity of CD38.[22]
Isatuximab-irfc is an IgG1-derived monoclonal antibody that selectively binds to the CD38 that exists on the exterior of hematopoietic and multiple myeloma cells (as well as other tumor cells). This drug induces apoptosis of tumor cells and activates immune effector mechanisms such as complement dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC). Isatuximab-irfc is able to stimulate natural killer (NK) cells in the absence of CD38-positive target tumor cells and blocks CD38-positive T-regulatory cells.[4] Furthermore, the NADase activity of CD38 is adjusted by isatuximab, similarly to other CD38 antibodies. Contrarily to daratumumab however, isatuximab can incite apoptosis directly without cross-linking, and in its binding epitope.[23] According to the FDA, isatuximab-irfc alone has reduced ADCC and direct tumor cell killing activity in vitro in comparison to when it is combined with pomalidomide. As well as increased anti-tumor activity as opposed to isatuximab-irfc or pomalidomide only in a human multiple myeloma xenograft model.[4]
Metabolism and toxicity
Metabolism
Isatuximab-irfc is likely to be metabolized through catabolic pathways into smaller peptides. When isatuximab is at a constant state it is expected that the ≥99% elimination will occur approximately two months after the last dose was administered. The clearance percentage diminished when the dosages were increased over time, as well as when multiple doses were administered. However, the elimination of isatuximab-irfc did not differ when applied as a single agent or as a combination therapy.[4]
Toxicity
A dose-limiting toxicity (DLT) has characterized been characterized as the development of any of the following: grade ≥ 3 non-hematologic toxicity; grade 4 neutropenia or grade 4 thrombocytopenia lasting more than 5 days; grade ≥ 2 allergic reactions or hypersensitivity (i.e., infusion reactions); or any other toxicity considered to be dose-limiting by the investigators or sponsor. Grade ≤ 2 infusion reactions were excluded from the DLT definition, because, with suitable care, patients that suffered a grade 2 infusion reaction prior to completion of the infusion were able to finalize isatuximab administration.[23]
There is no recommended reduced dose of isatuximab-irfc. In the eventuality of hematological toxicity it may be necessary to delay administration so that the blood count may be recovered.[4] Although there is no counteracting agent for isatuximab, clinical experience with overdoses is seemingly nonexistent as well. Overdose symptoms will probably be in line with the side effects attached to isatuximab. Therefore, infusion reactions, gastrointestinal disturbances and an elevated risk of infections may occur. It is necessary to carefully monitor the patient in case of an overdose and to employ clinically indicated symptomatic and supportive procedures.[21]
No studies have been conducted with isatuximab concerning carcinogenicity, genotoxicity or fertility.[4]
Pregnancy
When given to pregnant women isatuximab-irfc can cause fetal injury, due to the mechanism of action. It can precipitate depletion of immune cells as well as decreased bone density in the fetus. Pregnant women are therefore notified of the potential risks to a fetus, and women that are able to reproduce are advised to use effective contraceptives during treatment and at least five months subsequent to the last dose of isatuximab-irfc.
Furthermore, it is not recommended to combine isatuximab-irfc with pomalidomide in women that are carrying a child, because pomalidomide may cause birth defects and death of the unborn child.[4]
Indications
Isatuximab is indicated as a CD38-directed cytolytic antibody. By inhibiting the enzymatic activity of CD38.
The binding of isatuximab to CD38 on multiple myeloma (MM) cells leads to a trigger to several mechanisms leading to direct apoptosis of target cancer cells. The triggered pathways are the caspase-dependent apoptotic and the lysosome-mediated cell death pathway in MM cells.[24]
It is used in a combination with dexamethasone and pomalidomide. The drug is thus to treat patients with multiple myeloma. Restrictions for the use of isatuximab is that the patients have to be adults who have at least received two previous treatments with lenalidomide and a proteasome inhibitor.[4]
Isatuximab is currently[when?] also being tested in a Phase II trial as a monotherapy against refractory/recurrent systemic light-chain amyloidosis.[24]
Efficacy and side effects
Efficacy
A Phase III study of patients with refractory and relapsed MM, who were resistant to lenalidomide and a proteasome inhibitor, and could not have received daratumumab, another anti-CD38 monoclonal antibody was published in 2019 (ICARIA-MM). The addition of isatuximab to pomalidomide and dexamethasone improved progression free survival to 11.5 months compared to 6.5 months, with an overall response rate of 63%.[25]
Side effects
Adverse reactions to isatuximab-irfc may include neutropenia, infusion-related reactions and/or secondary primary malignancies.[4] Of these three the most commonly occurring ones are the infusion-related reactions.[24] Examples of the most frequent symptoms of infusion-related reactions are dyspnea, cough, chills, and nausea, while the severest signs and symptoms included hypertension and dyspnea.[4]
Effects on animals
The activity of isatuximab has been researched in mouse tumor models. It has been proven that isatuximab leads to antitumor activity in MM cells. Furthermore, the combination of isatuximab and pomalidomide will lead to an extra enhanced antitumor activity in MM cells. Thus, pomalidomide in vivo and in vitro leads to an increase of the activity of isatuximab.[24]
Animal studies in reproduction toxicity have not yet been carried out. So, the risks of birth defects and miscarriage risks are unknown.[4]
^ Orlowski RZ, Goldschmidt H, Cavo M, Martin TG, Paux G, Oprea C, Facon T (20 May 2018). “Phase III (IMROZ) study design: Isatuximab plus bortezomib (V), lenalidomide (R), and dexamethasone (d) vs VRd in transplant-ineligible patients (pts) with newly diagnosed multiple myeloma (NDMM)”. Journal of Clinical Oncology. 36 (15_suppl): TPS8055. doi:10.1200/JCO.2018.36.15_suppl.TPS8055.
“Isatuximab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02990338 for “Multinational Clinical Study Comparing Isatuximab, Pomalidomide, and Dexamethasone to Pomalidomide and Dexamethasone in Refractory or Relapsed and Refractory Multiple Myeloma Patients (ICARIA-MM)” at ClinicalTrials.gov
The U.S. Food and Drug Administration today approved Cablivi (caplacizumab-yhdp) injection, the first therapy specifically indicated, in combination with plasma exchange and immunosuppressive therapy, for the treatment of adult patients with acquired thrombotic thrombocytopenic purpura (aTTP), a rare and life-threatening disorder that causes blood clotting.
“Patients with aTTP endure hours of treatment with daily plasma exchange, which requires being attached to a machine that takes blood out of the body and mixes it with donated plasma and then returns it to the body. Even after days or weeks of this treatment, as well as taking drugs that suppress the immune system, many patients will have a recurrence of aTTP,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Cablivi is the first targeted treatment that inhibits the formation of blood clots. It provides a new treatment option for patients that may reduce recurrences.”
Patients with aTTP develop extensive blood clots in the small blood vessels throughout the body. These clots can cut off oxygen and blood supply to the major organs and cause strokes and heart attacks that may lead to brain damage or death. Patients can develop aTTP because of conditions such as cancer, HIV, pregnancy, lupus or infections, or after having surgery, bone marrow transplantation or chemotherapy.
The efficacy of Cablivi was studied in a clinical trial of 145 patients who were randomized to receive either Cablivi or a placebo. Patients in both groups received the current standard of care of plasma exchange and immunosuppressive therapy. The results of the trial demonstrated that platelet counts improved faster among patients treated with Cablivi, compared to placebo. Treatment with Cablivi also resulted in a lower total number of patients with either aTTP-related death and recurrence of aTTP during the treatment period, or at least one treatment-emergent major thrombotic event (where blood clots form inside a blood vessel and may then break free to travel throughout the body).The proportion of patients with a recurrence of aTTP in the overall study period (the drug treatment period plus a 28-day follow-up period after discontinuation of drug treatment) was lower in the Cablivi group (13 percent) compared to the placebo group (38 percent), a finding that was statistically significant.
Common side effects of Cablivi reported by patients in clinical trials were bleeding of the nose or gums and headache. The prescribing information for Cablivi includes a warning to advise health care providers and patients about the risk of severe bleeding.
Health care providers are advised to monitor patients closely for bleeding when administering Cablivi to patients who currently take anticoagulants.
The FDA granted this application Priority Review designation. Cablivi also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Cablivi to Ablynx.
EU
Cablivi is the first therapeutic approved in Europe, for the treatment of a rare blood-clotting disorder
On September 03, 2018, the European Commission has granted marketing authorization for Cablivi™ (caplacizumab) for the treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP), a rare blood-clotting disorder. Cablivi is the first therapeutic specifically indicated for the treatment of aTTP 1. Cablivi was designated an ‘orphan medicine’ (a medicine used in rare diseases) on April 30, 2009. The approval of Cablivi in the EU is based on the Phase II TITAN and Phase III HERCULES studies in 220 adult patients with aTTP. The efficacy and safety of caplacizumab in addition to standard-of-care treatment, daily PEX and immunosuppression, were demonstrated in these studies. In the HERCULES study, treatment with caplacizumab in addition to standard-of-care resulted in a significantly shorter time to platelet count response (p<0.01), the study’s primary endpoint; a significant reduction in aTTP-related death, recurrence of aTTP, or at least one major thromboembolic event during study drug treatment (p<0.0001); and a significantly lower number of aTTP recurrences in the overall study period (p<0.001). Importantly, treatment with caplacizumab resulted in a clinically meaningful reduction in the use of PEX and length of stay in the intensive care unit (ICU) and the hospital, compared to the placebo group. Cablivi was developed by Ablynx, a Sanofi company. Sanofi Genzyme, the specialty care global business unit of Sanofi, will work with relevant local authorities to make Cablivi available to patients in need in countries across Europe.
About aTTP aTTP is a life-threatening, autoimmune blood clotting disorder characterized by extensive clot formation in small blood vessels throughout the body, leading to severe thrombocytopenia (very low platelet count), microangiopathic hemolytic anemia (loss of red blood cells through destruction), ischemia (restricted blood supply to parts of the body) and widespread organ damage especially in the brain and heart. About Cablivi Caplacizumab blocks the interaction of ultra-large von Willebrand Factor (vWF) multimers with platelets and, therefore, has an immediate effect on platelet adhesion and the ensuing formation and accumulation of the micro-clots that cause the severe thrombocytopenia, tissue ischemia and organ dysfunction in aTTP 2.
Note – Caplacizumab is a bivalent anti-vWF Nanobody that received Orphan Drug Designation in Europe and the United States in 2009, in Switzerland in 2017 and in Japan in 2018. The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application for caplacizumab for treatment of adults experiencing an episode of aTTP. The target action date for the FDA decision is February 6, 2019
This drug was developed by Ablynx NV.[3] On 31 August 2018 it was approved in the European Union for the “treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP), in conjunction with plasma exchange and immunosuppression”.[4]
It is an anti-von Willebrand factor humanized immunoglobulin.[5] It acts by blocking platelet aggregation to reduce organ injury due to ischemia.[5] Results of the phase II TITAN trial have been reported.[5]
In February 2019, caplacizumab-yhdp (CABLIVI, Ablynx NV) has been approved by the Food and Drug Administration for treatment of adult patients with acquired thrombotic thrombocytopenic purpura (aTTP). The drug is used in combination with plasma exchange and immunosuppressive therapy. [6]
Caplacizumab (ALX-0081) is a humanized single-variable-domain immunoglobulin (Nanobody) that targets von Willebrand factor, and thereby inhibits the interaction between von Willebrand factor multimers and platelets. In a Phase 2 study (NCT01151423) of 75 patients with acquired thrombotic thrombocytopenic purpura who received SC caplacizumab (10 mg daily) or placebo during plasma exchange and for 30 d afterward, the time to a response was significantly reduced with caplacizumab compared with placebo (39% reduction in median time, P = 0.005).39Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knöbl P, Wu H, Artoni A, Westwood JP, Mansouri Taleghani M, Jilma B, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 2016; 374(6):511–22; PMID:26863353; http://dx.doi.org/10.1056/NEJMoa1505533[Crossref], [PubMed], [Web of Science ®], , [Google Scholar] The double-blind, placebo-controlled, randomized Phase 3 HERCULES study (NCT02553317) study will evaluate the efficacy and safety of caplacizumab treatment in more rapidly curtailing ongoing microvascular thrombosis when administered in addition to standard of care treatment in subjects with an acute episode of acquired thrombotic thrombocytopenic purpura. Patients will receive an initial IV dose of either caplacizumab or placebo followed by daily SC injections for a maximum period of 6 months. The primary outcome measure is the time to platelet count response. The estimated enrollment is 92 patients, and the estimated primary completion date of the study is October 2017. A Phase 3 follow-up study (NCT02878603) for patients who completed the HERCULES study is planned.
Gemigliptin L-tartrate sesquihydrate was approved by Korean food and Drug Administration on June 27, 2012. It was developed and marketed as Zemiglo® by LG Life Sciences in KR.
Gemigliptin L-tartrate sesquihydrate is a dipeptidyl peptidase-4 inhibitor indicated for the treatment of type 2 diabetes mellitus.
Zemiglo® is available as tablet for oral use, containing 50 mg of free Gemigliptin. The recommended dose is 50 mg once daily taken regardless of meals.
Originator LG Life Sciences
Developer LG Chem; Sanofi
Class Antihyperglycaemics; Piperidines; Pyrimidines; Small molecules
Mechanism of Action CD26 antigen inhibitors
Marketed Type 2 diabetes mellitus
Phase II/III Acute kidney injury
24 Jun 2018 Biomarkers information updated
05 Apr 2018 LG Chem initiates enrolment in a phase I trial for Type-2 diabetes mellitus (Combination therapy) in South Korea (NCT03565458)
08 Feb 2018 LG Life Sciences completes the phase III ZEUS II trial in Type-2 diabetes mellitus (Adjunctive treatment) in South Korea (PO) (NCT02831361)
Research Code:LC-15-0444
Trade Name:Zemiglo®
MOA:Dipeptidyl peptidase-4 (DPP-4) inhibitor
Indication:Type 2 diabetes
Status:Approved 2012-06-27 korea
Company:LG Life Sciences (Originator)
Sales:ATC Code:A10BH06
Mechanism of Action
● Gemigliptin is a selective DPP-4 inhibitor[4-7].
● DPP-4 inhibition IC50=16 nM.
● Selectivity compared against DPP8/9 >3000 fold.
● Gemigliptin bound to DPP-4 enzyme with a Ki=15.2 nM.
In Vivo Efficacy
Minimum effective dose of gemigliptin in animal models:
● HbA1c reduce: DIO mice: 3 mg/kg/day for 4 weeks.
Absorption
● The oral bioavailability of gemigliptin in the rats, dogs and monkeys are species-dependent with the values of 94 %, 73 %, and 26 %, respectively.
● Gemigliptin is rapidly absorbed after single oral dose administration, with Tmax occurring 0.3 to 0.5 hr postdose in rats and dogs.
● Half-life of gemigliptin is moderate in rats, dogs, monkeys (3.6 – 5.8 hrs) and long in humans (30.8 hrs), clearance of gemigliptin ranged from 0.6 L/hr/kg (32 % of liver blood flow) in dogs to 4.1 L/hr/kg (123 % of liver blood flow).
● Volume of distribution of gemigliptin is greater than body water volume, occurring 3.7 to 14 L/kg, which suggested extensive extravascular distribution.
Distribution
● [14C] Gemigliptin and metabolites were not extensively bound to plasma proteins.
● Following oral administration of gemigliptin, [14C] gemigliptin-derived radioactivity was widely distributed to most tissues and organs in rats.
● In most tissues and organs, the total radioactivity decreased with time and was almost entirely eliminated 24 hrs after administration.
● The highest concentrations were observed in the tissues of the alimentary (the small intestine and large intestine) and the excretory or metabolic (the kidney, pancreas and liver) systems.
● Accumulation of the drug was not observed in most of the tissues or organs tested but the time course of radioactivity in testis showed some possibility of drug accumulation.
Metabolism
● Following oral administration of 10 mg/kg [14C] gemigliptin to male rats: The major circulating metabolites were LC15-0516 (dehydrated), LC15-0635 and LC15-0636 (hydroxylated), but the majority of the radioactivity in plasma was associated with the parent compound. The metabolite profile in urine was similar to that in plasma, but the profile in bile was somewhat different from that in urine or plasma. The major metabolic pathway was hydroxylation.
● Following oral administration of 50 mg [14C] gemigliptin to healthy male subjects: Gemigliptin was the most abundant component accounting for 67.2%-100% of plasma radioactivity. Unchanged gemigliptin accounted for 44.8%-67.2% of urinary radioactivity and 27.7%-51.8% of fecal radioactivity. LC15-0636 was the most abundant metabolite in plasma, accounting for 9.1%–17.5 % of plasma radioactivity. LC15-0516 and LC15-0635 were not detected in plasma samples. LC15-0636 was the only human metabolite with systemic exposure more than 10% of total drug-related exposure.
● CYP3A4 was identified as the dominant CYP isozyme converting gemigliptin to LC15-0636 in recombinant CYP/FMO enzymes.
Gemigliptin was initially developed solely by LG Life Sciences. In 2010, Double-Crane Pharmaceutical Co. (DCPC) joined with LGLS to co-develop the final compound and collaborate on the marketing of the drug in China. LGLS also announced in November 2010 that NOBEL Ilac has been granted rights to develop and commercialize gemigliptin in Turkey.
A New Drug Application (NDA) for gemigliptin in the treatment of type 2 diabetes was submitted to the Korea Food & Drug Administration (KFDA) in July 2011. Then on June 27, 2012, the KFDA has approved the manufacture and distribution of LG Life Sciences’ diabetes treatment, Zemiglo, the main substance of which is gemigliptin. LG Life Sciences signed a licensing agreement with multinational pharmaceutical companies such as Sanofi (Paris, France) and Stendhal (Mexico City, Mexico) for 104 countries. Currently, gemigliptin has been approved in 11 countries such as India, Columbia, Costa Rica, Panama, and Ecuador, and several clinical studies are in progress in Russia, Mexico, and Thailand.
History
The NDA for gemigliptin was submitted to KFDA in July, 2011 and it was approved on June 27, 2012. By the end of 2012, gemigliptin will be marketed in Korea as Zemiglo which is the fifth new DPP-4 inhibitor diabetes treatment in the world. Sanofi-Synthelabo India Private Limited announced the launch of drug for type 2 diabetes patients in India: Zemiglo (gemigliptin) on Juty 19, 2016. Zemiglo is a once daily, oral tablet. As per the International Diabetes Federation Diabetes Atlas 2015, India is home to the second largest number of adults living with diabetes worldwide, after China, with 69.1 million patients and expected to rise to 1401 million in 2040. India is the largest contributor to South East Asia regional mortality, with 1 million deaths attributable to diabetes. These statistics reveal how diabetes is fast gaining the status of a potential epidemic in India and establishes the need for treatment compliance and effective control through diet, exercise and drugs for long-term positive effects in disease management.
Mechanism of action
DPP-4 is a serine protease located on the cell surfaces throughout the body. In plasma, DPP-4 enzyme rapidly inactivates incretins including GLP-1 and GIP which are produced in the intestine depending on the blood glucose level and contribute to the physiological regulation of glucose homeostatis. Active GLP-1 and GIP increase the production and release of insulin by pancreatinc beta cells. GLP-1 also reduces the secretion of glucacon by pancreatic alpha cells, thereby resulting in a decreased hepatic glucose production. However these incretins are rapidly cleaved by DPP-4 and their effects last only for a few minutes. DPP-4 inhibitors block the cleavage of the gliptins and thus lead to an increasee insulin level and a reduced glucagon level in a glucose-dependent way. This results in a decrease of fasting and postprandial glycemia, as well as HbA1c levels.[2]
Preclinical studies
Gemigliptin is a competitive, reversible DPP-4 inhibitor (Ki = 7.25 ± 0.67 nM) with excellent selectivity over other critical human proteases such as DPP-2, DPP-8, DPP-9, elastase, trypsin, urokinase and cathepsin G. The kinetics of DPP-4 inhibition by gemigliptin was characterized by a fast association and a slow dissociation rate compared to sitagliptin (fast on and fast off rate) or vildagliptin (slow on and slow off rate). Gemigliptin was rapidly absorbed after single oral dosing and the compound was eliminated with a half-life of 3.6 h, 5.2 h, and 5.4 h in the rat, dog, and monkey, respectively.
The bioavailability of gemigliptin in the rat, dog, and monkey was species-dependent with the values of 94%, 73%, and 26%, respectively. Following the oral administration of gemigliptin in the rat, dog and monkey, about 80% inhibition of plasma DPP-4 activity were observed at the plasma levels of 18 nM, 14 nM and 4 nM, respectively.
In a diet-induced obesity model, gemigliptin reduced glucose excursion during OGTT in a dose dependent manner with the minimum effective dose of 0.3 mg/kg and enhanced glucose-stimulated plasma GLP-1 increase in a dose dependent manner reaching the maximum effect at the dose of 1 mg/kg.
Following 4 week oral repeat dosing in the DIO mice, gemigliptin reduced significantly HbA1c with the minimum effective dose of 3 mg/kg. In the beagle dog, gemigliptin significantly enhanced active GLP-1, decreased glucagon, and reduced glucose excursion during OGTT following a single dosing.
Studies on animals suggest its positive effect on hepatic and renal fibrosis .[3][4] Data on human patients are still inconclusive .[5]
Clinical studies
Monotherapy
The efficacy and safety of gemigliptin monotherapy were evaluated in two double-blind placebo controlled studies and one double-blind active-controlled study. A phase II study (study identifier: LG-DPCL002) of gemigliptin was conducted in a randomized, double-blind, placebo-controlled, parallel group design with three doses of 50, 100, and 200 mg qd for the purpose of finding a dose responsiveness and an optimal dose in patients with T2DM. The mean changes of HbA1c at week 12 from the baseline were –0.98%, –0.74%, –0.78% (when adjusted with placebo data, –0.92%, –0.68%, and –0.72%) at 50, 100, and 200 mg, respectively. Among the effective doses obtained from the phase II study in patients with T2DM, the 50 mg dose showed a similar efficacy as the 100 and 200 mg doses, within the maximum safety margin. Similar findings were reported from two phase III studies. Patients were randomized to receive gemigliptin, either a 50 mg qd (n=90) or a placebo (n=92) for 24 weeks (study identifier: LG-DPCL005; ClinicalTrials.gov registration number: NCT01601990). The placebo-subtracted changes from baseline in HbA1c were reported to be −0.71% (95% confidence interval [CI], −1.04 to −0.37) with gemigliptin 50 mg. In addition, a 28-week open-label extension study was designed to evaluate the long-term safety and efficacy of gemigliptin. Among 165 patients who consented to participate in the extension period of study LG-DPCL005, 158 patients (96%) completed their treatments for 52 weeks. All patients were switched to or continued their treatments only with gemigliptin 50 mg qd during the extension period. A further decrease in HbA1c was observed in the continued treatment with gemigliptin 50 mg in this extension period, and the mean change from baseline at 52 weeks (–0.87%) was still clinically and statistically significant (full analysis set analysis, P<0.0001). In another double-blind, active-controlled, phase III trial (study identifier: LG-DPCL011), eligible patients with HbA1c greater than 7.5% were randomized to receive gemigliptin 50 mg qd with metformin slow release (SR) qd (n=141), gemigliptin 50 mg qd (n=142), or metformin SR qd (n=150) for 24 weeks. After 24 weeks, the reduction from the baseline in HbA1c was –1.24% for gemigliptin monotherapy.
Initial combination therapy with metformin
In this randomized, double-blind, active-controlled, phase III trial (study identifier: LG-DPCL011, INICOM study; ClinicalTrials.gov registration number: NCT01787396), eligible patients with an HbA1c greater than 7.5% were randomized to gemigliptin 50 mg qd+metformin SR qd (n=141), gemigliptin 50 mg qd (n=142), or metformin SR qd (n=150). From weeks 2 to 6, metformin SR was uptitrated incrementally from 500 to 2,000 mg/day maximum in the gemigliptin/metformin and metformin groups. The mean daily doses of metformin at week 24 were 1,699 and 1,868 mg for the gemigliptin/metformin group and the metformin group, respectively. Mean change in HbA1c from baseline was –2.06% for gemigliptin/metformin group versus –1.24% for the gemigliptin group and –1.47% for the metformin group, respectively (P<0.0001 for all comparisons of combination therapy vs. monotherapy). The differences in proportions achieving an HbA1c <7% or <6.5% were also statistically significant (P<0.0001) between the combination therapy and the respective monotherapy groups.
Add-on to metformin
A 24-week, multinational, randomized, double-blind, active-controlled study (study identifier: LG-DPCL006; ClinicalTrials.gov registration number: NCT01602003) was designed to assess the efficacy and safety of gemigliptin 50 mg compared to the active control (sitagliptin) added to ongoing metformin therapy in patients with T2DM inadequately controlled with metformin alone (HbA1c, 7% to 11%). After 24 weeks, the reduction from baseline for HbA1c was 0.81% for gemigliptin 25 mg twice a day (bid) and 0.77% for gemigliptin 50 mg qd, and the differences in the least square mean changes from baseline between groups (each group of gemigliptin-sitagliptin group) were −0.011% in gemigliptin 25 mg bid and 0.004% in gemigliptin 50 mg qd. The proportion of patients achieving an HbA1c <7% at week 24 (gemigliptin 25 mg bid group, 50%; gemigliptin 50 mg qd group, 54.07%) was comparable to the results with sitagliptin 100 mg qd (48.87%). The efficacy of lowering HbA1c in the gemigliptin group was generally consistent across the subgroups based on age (<65 or ≥65 years), gender, duration of T2DM (5, >5 to 10, or >10 years), and baseline body mass index (BMI, <25 or ≥25 kg/m2). In addition, gemigliptin groups led to a significantly greater inhibition of plasma DPP-4 compared to sitagliptin. This study was extended by 28 weeks in order to evaluate the long-term efficacy and safety of gemigliptin. All treatment groups showed clinically and statistically (P<0.0001) significant improvement in glycemic control from baseline after 52 weeks. The reduction from the baseline in HbA1c was –1.06 (95% CI, –1.28 to –0.85) in the patients who continued to receive gemigliptin 50 mg qd.
Add-on to metformin and glimepiride
In this multicenter, randomized, double-blind, phase III study (study identifier: LG-DPCL010, TROICA study; ClinicalTrials.gov registration number: NCT01990469), eligible patients with inadequate glycemic control (7%≤HbA1c≤11%) were randomized to gemigliptin 50 mg qd (n=109) or placebo (n= 110). The baseline demographics were similar between groups (age, 60.9 years; BMI, 24.9 kg/m2; duration of T2DM, 12.9 years), with mean±standard deviation (SD) baseline HbA1c of 8.12%± 0.82% in the gemigliptin group and 8.15%±0.89% in the placebo group. At week 24, the adjusted mean±standard error change for HbA1c with gemigliptin was –0.88%±0.17% (change with placebo –0.01%±0.18%; difference –0.87%±0.12%; 95% CI, –1.09 to –0.64; P<0.0001).
Add-on therapy in patients with renal impairment
RI in T2DM limits the usable medications for lowering glucose level and requires frequent monitoring of renal function. Gemigliptin has balanced elimination between urinary/fecal excretion and hepatic metabolism; therefore, it does not require dose adjustment in patient with moderate to severe RI. This study evaluated the efficacy and safety of gemigliptin in T2DM patients with moderate to severe RI. This randomized, double-blind, parallel group, phase IIIb study (study identifier: LG-DPCL015, GUARD study; ClinicalTrials.gov registration number: NCT01968044) was composed of a 12-week, placebo controlled period, followed by a 40-week, double-blind active controlled extension period (placebo switched to linagliptin). A total of 132 patients with moderate or severe RI were randomized to receive gemigliptin (n=66) or placebo (n=66). Insulin was used as predominant background therapy (63.1%). At week 12, the placebo-adjusted mean change in HbA1c from the baseline was –1.20% (95% CI, –1.53 to –0.87; P<0.0001). A similar profile was also observed in other glycemic control parameters (fasting plasma glucose, glycated albumin, and fructosamine).
Effects on glycemic variability
Glycemic variability and chronic sustained hyperglycemia are the main components of dysglycemia in diabetes. The previous studies suggested that different pharmacodynamic profiles between DPP-4 inhibitors have been associated with the different effects on glycemic variability. In this study, a multicenter, randomized, active-controlled, parallel group, open-label, exploratory study was designed to evaluate the efficacy on glycemic variability and safety of initial combination therapy of gemigliptin 50 mg qd versus sitagliptin 100 mg qd, or glimepiride 2 mg qd with metformin in patients with T2DM (study identifier: LG-DPCL012, STABLE study; ClinicalTrials.gov registration number: NCT01890629). The mean amplitude of glycemic excursions (MAGE) and SD of glucose were used for assessing glucose fluctuations from the baseline after 12 weeks of treatment. At 12 weeks, MAGE was significantly lower in the DPP-4 inhibitor groups (gemigliptin and sitagliptin) than in the glimepiride group (–43.1, –38.3, and –21.7 mg/dL, respectively). Furthermore, the SD of mean glucose was significantly lower in patients with gemigliptin when compared with sitagliptin (P=0.023) and glimepiride (P=0.0058).
Ongoing studies
Several clinical studies in LG Life Sciences are actively underway to assess the efficacy and safety as an add-on combination therapy with insulin (with or without metformin) (ClinicalTrials.gov registration number: NCT02831361), to evaluate the efficacy and safety of gemigliptin-rosuvastatin fixed-dose combination in patients with T2DM and dyslipidemia in phase III clinical trials (ClinicalTrials.gov registration number: NCT02126358), and to evaluate the efficacy and safety of gemigliptin compared with vildagliptin in Russian patients with T2DM (ClinicalTrials.gov registration number: NCT02343926).
Key Characteristics
·Gemigliptin is a reversible, potent, selective, competitive, and long-acting inhibitor of DPP-4.
·Gemigliptin is orally administered 50 mg once daily either as monotherapy or in combination with other drugs. It can be taken with or without food.
·No dose adjustment is recommended for patients with renal or hepatic impairment.
·Gemigliptin shows a low propensity of drug interactions with metformin, pioglitazone, glimepiride, CYP3A4 inhibitors, rosuvastatin, or irbesartan, and dose adjustment of gemigliptin is not required for the patients who are concomitantly receiving these drugs.
·Gemigliptin decreases the mean level of HbA1c from baseline by 1.24% in monotherapy and 0.8% in add-on therapy with metformin. For gemigliptin as an initial combination with metformin, the mean reduction from baseline in HbA1c was 2.8%. In head-to-head comparisons, the mean reduction from baseline in HbA1c was 0.8% for gemigliptin with metformin and 0.8% for sitagliptin with metformin, hence the efficacy of gemigliptin is found to be comparable to sitagliptin.
·Gemigliptin was shown to be more effective in reduction of glycemic variability than glimepiride and sitagliptin with metformin as an initial combination therapy for drug naïve patients with T2DM.
·Gemigliptin is generally well tolerated in controlled clinical studies as monotherapy and as part of combination therapy. The incidences of AEs are generally similar to those of placebo and active control groups.
The present invention relates to the following formula 1- {(2S) -2- amino-I shown 4- [2,4-bis (trifluoromethyl) _5,8_ dihydro-pyrido [3,4-d ] pyrimidin -7 (6H) – yl] -4-oxo – 1.5 hydrate butyl} -5,5-difluoropiperidin-2-one (hereinafter, referred to as “compound I”) and its tartrate Preparation.
Preparation of Hydrate (Form I) of Example 1 I tartrate salt of the compound of embodiment [0072]
[0073]
[0074] The compound 2 was dissolved in about 1.87Kg 9L of ethanol.Was added 0.94Kg at O~10 ° C SOCl2, and then stirred while maintaining a low temperature.After concentration under reduced pressure, the concentrate was dissolved in MTBE 11.2L (MTBE), and the resulting mixture was adjusted to pH7~8 with ION NaOH solution.After the layers were separated, and the aqueous layer was extracted with MTBE about 3.7L and 3.7L extracted twice with MTBE, and then concentrated under reduced pressure.The resulting brown turbid solution was dissolved in 12L of ethanol, to which was added 0.47kg of dissolved water of about 1.5L of L- tartaric acid, and then stirred for I hour.The resulting crystalline slurry was filtered, washed with water and ethanol: washing (18), and then dried to obtain the title compound 1.13kg (yield 97.5%) of.
2.99 (m, 2Η), 3.11 (bt, 1H), 3.21 (bt, 1Η), 3.50 – 3.55 (m, 1Η), 3.72 – 3.91 (m, 5Η), 3.98 (t, J = 5.2Hz, 1Η) , 4.38 (s, 2Η), 4.97 -. 5.00 (m, 2Η) [0076] Example 2 compound I tartrate embodiment 1.5 hydrate (Form I) was recrystallized from water
[0077] obtained from Example 1 50g of compound I was added 250~500ml tartrate dissolved in water and water, while with ION NaOH solution was adjusted to pH6~7.Was dissolved in 23.5ml of water was added 11.7g of L- tartaric acid, and shown in Table I below, with changes in temperature, stirring speed and stirring time to obtain crystals.Then, the crystals were filtered and dried to obtain Form I.Stirring rate of change in 50~400rpm range, the temperature change in the range of 5~32 ° C.The volume of water for the recrystallization, stirring rate, temperature and mixing time shown in Table I below.
Preparation of 1.5 hydrate of tartrate salt of Compound 1 (crystal form I)
1.87 kg of the compound 2 was dissolved in about 9 L of ethanol. 0.94 kg of SOCl2was added at 0~10℃ and then stirred while maintaining low temperature. After concentrating under reduced pressure, the concentrate was dissolved in 11.2 L of MTBE (methyl t-butyl ether), and the resulting mixture was adjusted with 10 N NaOH solution to pH 7~8. After separating the layers, the aqueous layer was extracted with about 3.7 L of MTBE and twice with 3.7 L of MTBE, and then concentrated under reduced pressure. The resulting brown turbid solution was dissolved in 12 L of ethanol, 0.47 kg of L-tartaric acid dissolved in about 1.5 L of water was added thereto, and then stirred for 1 hour. The resulting crystalline slurry was filtered, washed with water and ethanol (1:8), and then dried to obtain 1.13 kg (yield 97.5%) of the title compound.
Recrystallization of 1.5 hydrate of tartrate salt of Compound 1 (crystal form I) from water
50 g of tartrate salt of Compound 1 obtained from Example 1 was added to 250~500 ml of water, and dissolved in water while adjusting the solution with 10 N NaOH to pH 6~7. 11.7 g of L-tartaric acid dissolved in 23.5 ml of water was added, and crystals were obtained with varying the temperature, stirring rate and stirring time as shown in the following Table 1. Then, the crystals were filtered and dried to obtain the crystal form I. The stirring rate was varied in the range of 50~400 rpm, and the temperature was varied in the range of 5~32℃. The volume of water used for recrystallization, the stirring rate, temperature and stirring time are represented in the following Table 1.
Table 1
Conditions for HPLC analysis
Column: Atlantis dC18 (4.6 mm I.D x 250 mm L, Particle Size 5㎛, Waters)
Column Temperature: 10℃
Mobile phase:
Mobile phase A: MeCN/TFA = 100/0.1 (v/v)
Mobile phase B: H2O/TFA = 100/0.1 (v/v)
Gradient condition:
Flow rate: 0.7 ml/min.
Detection: 256 nm, UV
Injection volume: 10㎕
Total analysis time: 55 min.
The results of the stability for the crystal form I and the crystal form II are shown in the following Table 4.
Table 4
As shown in Table 4, it could be confirmed that upon keeping the crystal form I and the crystal form II at 40±2℃, 75±5% RH or 60±2℃, 5±5% RH they exhibit a superior stability up to 8 weeks. However, according to the result of XRD analysis the crystal form I did not show any change up to 8 weeks, but the crystal form II was converted into the crystal form I at 8 week under the condition of 40℃/75% RH (see Figure 16).
Gemigliptin, a novel dipeptidyl peptidase 4 inhibitor: first new anti-diabetic drug in the history of Korean pharmaceutical industry
Abstract
Gemigliptin, a potent, selective and long-acting DPP 4 inhibitor was developed by LG Life Sciences and approved for use in patients with type 2 diabetes mellitus by the Korean Food and Drug Administration in June 2012 under the trade name Zemiglo®. Clinical pharmacokinetic and pharmacodynamic data suggest the efficacy and once daily dosing of gemigliptin. In clinical phase III studies, gemigliptin was efficacious as either monotherapy or combination therapy (add-on to metformin) and well tolerated in patients with type 2 diabetes. Further development of combination therapy is on-going.
The discovery of a series of non-peptide factor Xa (FXa) inhibitors incorporating 3-(S)-amino-2-pyrrolidinone as a central template is described. After identifying compound 4, improvements in in vitro potency involved modifications of the liphophilic group and optimizing the angle of presentation of the amidine group to the S1 pocket of FXa. These studies ultimately led to compound RPR120844, a potent inhibitor of FXa (Ki = 7 nM) which shows selectivity for FXa over trypsin, thrombin, and several fibrinolytic serine proteinases. RPR120844 is an effective anticoagulant in both the rat model of FeCl2-induced carotid artery thrombosis and the rabbit model of jugular vein thrombus formation.
-difluoro-2-oxpiperidin-l-yl)methyl]-3-oxpropyl}carbamate obtained in
PREPARATION 143 was used. [1962] 1K NMR (CD3OD) δ 5.05-4.92 (2H, m), 3.98-3.91 (2H, m), 3.85-3.79 (2H, m),
3.70-3.59 (2H, m), 3.54-3.48 (IH, m), 3.36-3.33 (2H, m), 3.24 (IH, bra), 3.14 (IH, bra), 2.83-2.76 (IH, m), 2.72-2.53 (3H, m), 2.43-2.34 (2H, m) [1963] Mass (m/e) 490 (M+l)
[1966] 14 mg of the title compound was obtained in a yield of 17% at the same manner as in PREPARATION 45, except that 43.7 mg (0.138 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[2(S)-2-methyl-5-oxomoφholin-4-yl]-butanoic acid obtained in PREPARATION 55 and 42.5 mg (0.138 mmol) of 2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine hydrochloric acid salt (product of PREPARATION 127) were used.
[1967] 1K NMR (CDCl3) δ 5.85-5.83 (IH, m), 5.09-4.92 (IH, m), 4.95-4.78 (IH, m),
4.23-4.08 (3H, m), 4.04-3.76 (3H, m), 3.73-3.66 (IH, m), 3.46-3.38 (IH, m), 3.36-3.21 (2H, m), 3.18-3.10 (2H, m), 2.96-2.81 (IH, m), 2.61-2.50 (IH, m), 1.43-1.41 (9H, m), 1.28-1.24 (3H, m)
PREPARATION 1: Synthesis of diethyl 2,2-difluoropentanedioate
To a solution of ethyl bromodifluoroacetate (33.2 g) in tetrahydrofuran (94.0 g) was added ethyl acrylate (8.2 g) and copper powder (10.9 g). After heating to 50℃, TMEDA (9.5 g) was added dropwise and the reaction mixture was then stirred for 3 hours at the same temperature. Upon disappearance of ethyl acrylate as the starting material, to the reaction solution was added methyl t-butyl ether (MTBE, 73.7 g) followed by addition of 10% aqueous ammonium chloride solution (49.8 g) dropwise, and the mixture was then stirred for 30 minutes. The remaining copper residue was removed by filtration through a celite, and methyl t-butyl ether (MTBE, 66.3 g) was added to separate the layers. The separated organic layer was washed successively with 10% aqueous NH4Cl solution (66.3 g) and 3 N aqueous hydrochloric acid solution (99.6 g) in order and then distilled under reduced pressure to obtain 55.0 g of the desired title compound.
PREPARATION 2: Synthesis of ethyl 4,4-difluoro-5-hydroxypentanoate
14.8 g of the compound obtained from the above Preparation 1 was diluted with ethanol (20.4 g) and tetrahydrofuran (69.1 g) and then cooled to 0℃. To this solution was slowly added sodium borohydride (NaBH4, 3.5 g) stepwise while keeping the internal temperature below 30℃. After confirming completion of the reaction by 1H NMR, the reaction solution was cooled to the temperature of 10℃ and 10% aqueous ammonium chloride solution (77.7 g) was slowly added. The remaining boron compound was filtered through celite, and the filtrate was distilled under reduced pressure to remove tetrahydrofuran. Then, ethyl acetate (105.2 g) was added to separate the layers, and the organic layer was distilled under reduced pressure to obtain 10.8 g of the title compound.
EXAMPLE 1: Synthesis of ethyl 4,4-difluoro-5-{[(trifluoromethyl)sulfonyl]oxy}- pentanoate
To the solution of 10.8 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (100.2 g) was added pyridine (7.0 g), and then the mixture was cooled to -5.0℃. After completion of cooling, trifluoromethane sulfonic acid anhydride (20.1 g) was slowly added dropwise while keeping the reaction temperature below 6.3℃. After stirring the reaction solution for 30 minutes, 1.5 N hydrochloric acid solution was added dropwise at 0℃ to separate the layers. The aqueous layer as separated was back-extracted twice with dichloromethane (33.4 g), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 19.7 g of the title compound as a yellow oil.
EXAMPLE 2-1: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 100.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (300.0 ml) was added pyridine (65.7 g), and the mixture was then cooled to -10.0℃. After completion of cooling, nonafluorobutanesulfonic anhydride (477.4 g) was slowly added dropwise. After stirring the reaction solution for 3 hours, 1.0 N hydrochloric acid solution (300.0 ml) was added dropwise to separate the layers. The aqueous layer as separated was back extracted once with dichloromethane (500.0 ml), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 177.5 g of the title compound.
EXAMPLE 2-2: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 500.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (1000.0 ml) was added triethylamine (389.0 g), and the mixture was then cooled to 0℃. After completion of cooling, perfluorobutanesulfonyl chloride (948.80 g) was slowly added dropwise. The reaction solution was stirred for 3 hours at room temperature, distilled under reduced pressure, dissolved in methyl t-butyl ether (MTBE, 3000.0 ml) and then washed three times with water. The organic layer thus obtained was dehydrated with magnesium sulfate, filtered through a celite and then distilled under reduced pressure to obtain 960.0 g of the title compound.
EXAMPLE 3: Synthesis of methyl (2S)-2-[(tert-butoxycarbonyl)amino]-4-oxo- pentanoate
To 25.0 g of the starting material, (3S)-3-[(t-butoxycarbonyl)amino]-4-oxo- pentanoic acid, was added t-butanol (96.9 g) followed by the addition of Boc2O (25.4 g) and dimethylaminopyridine (DMAP, 62.0 g, 0.5 mol%) at room temperature, and the reaction mixture was then stirred for 23 hours at 40℃. Upon completion of the reaction, ethylene dichloride (62.3 g) in t-butanol was added, and the mixture was then distilled under reduced pressure to obtain 30.7 g of the title compound.
EXAMPLE 4: Synthesis of tert-butyl (3S)-3-[(tert-butoxycarbonyl)amino]-4-hydroxy- butanoate
30.7 g of the compound obtained from the above Example 3 was dissolved in ethanol (112.3 g) and, after lowering the internal temperature to 10.5℃ sodium borohydride (NaBH4, 5.7 g) was slowly added dropwise. This reaction solution was stirred while maintaining the temperature below 22℃. After confirming completion of the reaction by 1H NMR and TLC, to the reaction solution was slowly added 3.0 N hydrochloric acid solution (30.7 g) dropwise at the internal temperature of 10℃ followed by addition of diluted 0.2% hydrochloric acid solution (100.0 g). The reaction solution was adjusted to pH 3~4 with addition of 9.0% aqueous hydrochloric acid solution, and then back-extracted twice with ethyl acetate (100.0 g) and toluene (44.0 g). The organic layer thus obtained was distilled under reduced pressure to obtain 25.1 g of the title compound.
EXAMPLE 5: tert-butyl (3S)-[(tert-butoxycarbonyl)amino]-4-[(methylsulfonyl)oxy]- butanoate
To 25.1 g of the compound obtained from the above Example 4 was added dichloromethane (133.0 g) and triethylamine (148.0 g), and the mixture was then cooled to 0℃. To this reaction solution was slowly added methanesulfonyl chloride (11.8 g) diluted with dichloromethane (39.9 g) dropwise for 50 minutes while maintaining the internal temperature below 12℃. After completion of the reaction, the reaction solution was washed with 0.5 N aqueous hydrochloric acid solution (120.0 g) and water (100.4 g), and then distilled under reduced pressure to obtain 31.5 g of the title compound.
EXAMPLE 6: Synthesis of tert-butyl (3S)-4-azido-3-[(tert-butoxycarbonyl)amino]- butanoate
Sodium azide (NaN3, 11.6 g) was diluted with dimethylacetamide (DMAc, 260.0 g). After elevating the internal temperature to 80℃, a solution of 31.5 g of the compound, as obtained from the above Example 5, diluted with dimethylacetamide (DMAc, 45.0 g) was added thereto. The reaction proceeded at 80℃ for 2 hours. To the reaction solution were added toluene (251.0 g) and water (320.0 g) to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 24.0 g of the title compound.
EXAMPLE 7: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate
To 21.0 g of the compound obtained from the above Example 6 was added tetrahydrofuran (93.3 g) followed by the addition of triphenylphosphine (PPh3, 21.0 g) at 40℃, the mixture was stirred for 2 hours at the same temperature, and water (3.8 g) was then added thereto. The reaction solution was distilled under reduced pressure, and the resulting triphenylphosphine oxide solid was diluted with toluene (26.0 g) and n-hexane (41.0 g), and then filtered off. The filtrate was adjusted to pH 2~3 with 1.0 N aqueous hydrochloric acid solution (110.0 g) and then subjected to separation of the layers. To remove any residual triphenylphosphine oxide solid, the aqueous layer obtained above was washed with dichloromethane (100.0 g) and then adjusted to pH 8~9 with 28% aqueous ammonia solution (7.6 g). The aqueous solution thus obtained was extracted with dichloromethane (100.0 g) and distilled under reduced pressure to obtain 8.5 g of the title compound as a white solid.
EXAMPLE 8: Synthesis of N,N-dibenzyl-L-N(Boc)-aspartamide 4-tert-butyl ester
N-Boc-L-aspartic acid 4-t-butyl ester (29.0 g, 0.10 mol) was added to THF (200 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (13.0 ml, 0.10 mol) followed by addition of N-methyl morpholine (12.0 ml, 0.10 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise dibenzylamine (21.1 ml, 0.11 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (300.0 mL) and 1 N hydrochloric acid to separate the layers, and distilled under reduced pressure to precipitate a solid. The solid was filtered and washed with ethyl acetate (100 ml), and then the washings were concentrated by distillation again under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (41.7 g, 0.89 mol).
Mass (ESI, m/z): 491 (M+Na), 469 (M+H), 413 (M-55).
EXAMPLE 9: Synthesis of N, N-diallyl-L-N(Boc)-aspartamide 4-tert-butyl ester
L-N(Boc)-aspartic acid 4-t-butyl ester (5.00 g, 17.3 mol) was added to THF (50 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (2.26 ml, 17.3 mol) followed by addition of N-methyl morpholine (1.90 ml, 17.3 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise diallylamine (2.35 ml, 19.0 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (60 ml) and 1 N hydrochloric acid and, after separating the layers, concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (6.0 g, 16.3 mol).
Mass (ESI, m/z): 391 (M+Na), 369 (M+H), 313 (M-55).
EXAMPLE 10: Synthesis of N,N-dibenzyl-4-amino-3(S)-N(Boc)-aminobutanoic acid 4-tert-butyl ester
10.0 g of the compound obtained from the above Example 8, Ru3(CO)12 (136 mg, 1mol%), and diphenylsilane (19.7 ml, 106.7 mmol) were added to tetrahydrofuran (50 ml), and the reaction solution was stirred under reflux for over 40 hours. The reaction solution was extracted with ethyl acetate (200 ml) and concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (4.7 g, 10.5 mmol).
EXAMPLE 11: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- 4-oxobutanoate
360.0 g of the starting material, N-Boc-Asp(O-t-Bu)OH, together with Boc2O (353.0 g) and ammonium bicarbonate (NH4HCO3, 123.9 g) was added to dimethylformamide (1174.6 g), and pyridine (61.0 g) was added dropwise thereto at room temperature, and the reaction mixture was then stirred for about 3 hours. Upon completion of the reaction, water (1440 ml) and toluene (1800 ml) were added to the reaction solution and stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to remove t-butanol and toluene to obtain the title compound, which was directly used in the next reaction.
EXAMPLE 12: Synthesis of (S)-tert-butyl 3-(tert-butoxycarbonylamino)-3-cyanopropanoate
To the compound obtained from Example 11 was added dimethylformamide (1019.5 g) followed by addition of cyanuric chloride (112.0 g) dropwise for 1.5 hours at temperature below 25℃. The reaction solution was stirred for one hour at room temperature, and then 0.1 N aqueous sodium hydroxide solution (1850.0 g) and toluene (1860 ml) were added thereto to separate the layers. The organic layer thus obtained was washed once again with water (700 ml) and then distilled under reduced pressure to obtain 318.3 g of the title compound.
EXAMPLE 13: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate
To 212.1 g of the compound obtained from the above Example 12 was added acetic acid (4000 ml) followed by addition of 20 wt% Pd(OH)2 (1.1 g) at 40℃. The mixture was stirred for 8 hours while keeping the internal temperature below 45℃ and 3 atmospheric pressure of hydrogen. Upon completion of the reaction, the reaction solution was distilled under reduced pressure to remove acetic acid, diluted with toluene (640 L) and then filtered through a celite. To the filtrate was added 0.25 N aqueous hydrochloric acid solution (1060 ml) to separate the layers. The aqueous layer thus obtained was basified with aqueous ammonia solution (543.1 g) and then extracted with methyl t-butyl ether (MTBE, 1000 ml). The organic layer thus obtained was distilled under reduced pressure to obtain 185.0 g of the title compound.
EXAMPLE 14: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid t-butyl ester
Triethylamine (13.2 g) was added to 16.0 g of the compound obtained from the above Example 1 or 2-1 or 2-2, and 14.1 g of the compound obtained from the above Example 7 or 13, and the mixture was then stirred for 21 hours at 40℃. Then, dichloromethane (154.8 g) and acetic acid (18.3 g) were added, and the mixture was stirred for 5 hours at room temperature. To the resulting reaction solution was added 0.5 N aqueous hydrochloric acid solution (116.8 g) and then, the mixture was stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 23.6 g of the title compound.
EXAMPLE 15: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid
23.6 g of the compound obtained from the above Example 14 was added to dichloromethane (20.0 g) followed by addition of H3PO4 (30.0 g), and the mixture was stirred for 16 hours at room temperature. After confirming the detachment of all of t-butyl group and t-butyloxycarbonyl group, the reaction solution was adjusted to pH 7.0~8.0 with 10 N aqueous hydrogen peroxide, and Boc2O (16.0 g) was added thereto. After completion of the addition, 10 N aqueous hydrogen peroxide was used to maintain the pH of the reaction solution at 8.0~9.0. After stirring for 3 hours, the resulting sodium phosphate was filtered off, and the filtrate was then adjusted to pH 2.0~3.0 with 3.0 N aqueous hydrochloric acid solution. The resulting solid was filtered and dried under nitrogen to obtain 14.5 g of the title compound.
For the title compound resulting from the above, its enantiomeric isomers―i.e. S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess of the enantiomeric isomers (S vs. R form) (enantiomeric excess; ee) was then calculated as being ee > 99%. On the other hand, in case of the Comparative Example prepared according to the prior method based on WO 06/104356, as described below, the excess (ee) of enantiomeric isomers (S vs. R form) was 80%. From this, it can be identified that the compound of formula (2) having an optically high purity could be obtained according to the method of the present invention.
COMPARATIVE EXAMPLE 1: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
COMPARATIVE EXAMPLE 1-1: Synthesis of methyl 5-amino-4,4-difluoro- pentanoate HCl
To 10.0 g of the compound obtained from Example 1 was added 40 ml of anhydrous ammonia solution (7 M solution in methanol), and the mixture was stirred for 3 hours. The reaction solution was distilled and 30 ml of hydrochloric acid solution saturated with methanol was added dropwise thereto. The reaction mixture was stirred at room temperature and then distilled to obtain 7.2 g of the title compound as a white solid.
COMPARATIVE EXAMPLE 1-2: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
To the solution of the compound (1.93 g), as obtained from the above Example 4, dissolved in dichloromethane (20.0 g) and H2O (4.0 g) were added NaBr (0.8 g) and TEMPO (11 mg, 1 mol%). To this reaction solution was slowly added a solution of 5% NaOCl (11.5 g) and NaHCO3 (1.7 g) dissolved in H2O (12.0 g) dropwise for about 2 hours while maintaining the temperature below 5℃. Upon completion of dropwise addition, the reaction solution was stirred for 30 minutes to separate the layers. To the organic layer thus obtained was added the compound (1.6 g) obtained from the above Comparative Example 1-1. After stirring for 15 minutes at room temperature, NaBH(OAc)3 (2.23 g) was added to the reaction solution. After stirring for about 19 hours, 10% aqueous NaHCO3 solution (20.0 g) and 0.5 N aqueous hydrochloric acid solution (20.0 g) were added dropwise to the reaction solution to separate the layers. The organic layer thus obtained was dehydrated under anhydrous MgSO4 to obtain 2.0 g (yield 73%) of the same title compound as Example 14, as a yellow solid. For the title compound resulting from the above, its enantiomeric isomers―i.e., S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess (ee) of the enantiomeric isomers (S vs. R form) was then calculated as being ee = 80%.
PAPER
Gemigliptin is a prolyl-specific dipeptidyl aminopeptidase IV (DPP IV, DPP-4, CD26) inhibitor
approved for the treatment of type 2 diabetes mellitus by the Korean Food and Drug Administration in
2012. Gemigliptin was discovered and developed by LG Life Sciences81 and is now the sixth DPP-4
inhibitor approved for the treatment of type 2 diabetes.82 At the time this review was prepared, there
were no publications describing the discovery strategy and preclinical data that led to the advancement
of gemigliptin to the clinic. Additionally, the synthesis of the drug has only been described in the patent
literature.83-85
The molecule was prepared via a convergent route and the synthesis of the dihydropyridopyrimidine
fragment is described in Scheme 11.85 Commercial N-Boc-3-piperidone (71) was treated with lithium
hexamethyldisilazane (LHMDS) followed by ethyl trifluoroacetate to effect a Claisen condensation,
producing diketone 72 in 81% yield. Cyclization of 72 with 2,2,2-trifluoroacetamide (73) gave bistrifluoromethyl
dihydropyridopyrimidine 74 in 23% yield. Removal of the Boc protecting group
efficiently provided amine 75 in 96% yield.
SCHEME 11
The synthesis of the carbon skeleton of the difluoropyridone fragment 80 is described in Scheme
12.84 1,4-Addition of ethyl bromodifluoroacetate (76) to ethyl acrylate (77) in the presence of copper powder and tetramethylethylenediamine (TMEDA) gave diester 78, which was selectively reduced with
sodium borohydride (NaBH4) to give alcohol 79 in 90% overall yield for the two-step procedure.
Alcohol 79 was then treated with perfluorobutanesulfonyl chloride and triethylamine to give activated
alcohol 80 in 75% yield.
87 in 51% yield. Removal of the Boc group with thionyl chloride in ethanol followed by neutralization
with aqueous sodium hydroxide and salt formation with L-tartaric acid provided gemigliptin L-tartrate
hydrate (X) in 97.5% yield.83
The completion of the synthesis of gemigliptin is described in Scheme 13.83, 84 Boc-L-aspartic acid
4-tert-butyl ester (81) was treated with ammonium bicarbonate and pyridine in the presence of di-tertbutyl
dicarbonate to give formamide 82. Dehydration of 82 to give nitrile 83 was accomplished through
reaction with cyanuric chloride in 95% overall yield for the two-step sequence. Hydrogenation of 83 in
the presence of Pearlman’s catalyst provided butyl amine 84. Alkylation of 84 with activated alcohol 80
in triethylamine followed by cyclization in acetic acid afforded difluoropyridone 85. Acidic hydrolysis
of the ester proceeded with concomitant removal of the Boc protecting group, and was followed by
reprotection of the amine with di-tert-butyl dicarbonate to give acid 86 in 84% overall yield for the
three-step procedure in >97% ee. Coupling of 86 with fragment 75 in the presence of
hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) gave amide
81. Kim, S.-H.; Lee, S.-H.; Yim, H.-J. Arch. Pharmacal Res. 2013, 36, 1185. 82. Juillerat-Jeanneret, L. J. Med. Chem. 2014, 57, 2197. 83. Park, K. S.; Yun, J. M.; Kim, B. C.; Kim, K. Y.; Lee, J. H. WO Patent 2012060590A2, 2012. 84. Kim, B. C.; Kim, K. Y.; Lee, H. B.; An, J. E.; Lee, K. W. WO Patent 2012030106A2, 2012. 85. Lee, C.-S.; Koh, J. S.; Koo, K. D.; Kim, G. T.; Kim, K.-H.; Hong, S. Y.; Kim, S.; Kim, M.-J.; Yim, H. J.; Lim, D.; Kim, H. J.; Han, H. O.; Bu, S. C.; Kwon, O. H.; Kim, S. H.; Hur, G.-C.; Kim, J. Y.; Yeom, Z.-H.; Yeo, D.-J. WO Patent 2006104356A1, 2006.
PAPER
Gemigliptin, (LC15-0444, LG Life Sciences)
Gemigliptin is a sitagliptin analogue discovered by LG Life sciences Ltd, Korea via the derivatization of the compounds. It is potent and long acting DPP-IV inhibitor with high selectivity profile (3000-fold) against isoenzymes. The binding mode of gemigliptin is not reported, but expected as sitagliptin due to structural similarity. It inhibited more than 80% of DPP-IV activity and exhibited the bioavailability of 94% in rats. It also showed the lowering of
blood glucose and elevating of GLP-1 levels in dose-dependent manner in the diet-induced obese mice. Gemigliptin displayed a noteworthy lowering in HbA1c level (0.77%) at a dose of 3.0 mg/kg.[57] It is approved by Korean FDA in June 2012 for the treatment of T2DM.[58]
Synthesis of gemigliptin involved the preparation of two key intermediates dihydropyrido[3,4-d]pyrimidine moiety 88 and β-amino acid moiety 92. Compound 86 was prepared by generating enolate from compound 85 using LHMDS and adding trifluoroacetate.
Compound 86 gave the 87 in reflux condition which after Boc deprotection afforded key amine intermediate 88. The β-amino derivative 91 was synthesized by cyclization reaction between 89 and 90, which on benzyl deprotection using Pd/C gave desired β-amino intermediate 92.
Coupling of this intermediate with 88 using EDC/HOBt followed by Boc deprotection offered
gemigliptin 94 via 93 (Scheme 13).[59,60]
[57] S. J. Yang, K. W. Min, S. K. Gupta, J. Y. Park, V. K. Shivane, S. U. Pitale, P. K.
Agarwal, A. Sosale, P. Gandhi, M. Dharmalingam, V. Mohan, U. Mahesh, D. M. Kim, Y.
S. Kim, J. A. Kim, P. K. Kim, and S. H. Baik, Diabetes, Obes. Metab., 2013, 15, 410–
416.
[58] S. H. Kim, S. H. Lee, and H. J. Yim, Arch. Pharm. Res., 2013, 36 (10), 1185-1188. [59] C. S. Lee, J. S. Koh, K. D. Koo, G. T. Kim, K. H. Kim, S. Y. Hong, S. Kim, M. J. Kim, H. J. Yim, D. Lim, H. J. Kim, H. O. Han, S. C. Bu, O. H. Kwon, S. H. Kim, G. C. Hur, J. Y. Kim, Z. H. Yeom, D. J. Yeo, WO 2006/104356 A1, 2006. [60] K. S. Park, J. M. Yun, B. C. Kim, Y. U. Kim, J. H. Lee, WO 2012/060590, 2012.
CN101151265A *2005-04-012008-03-26株式会社Lg生命科学Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent
WO2004007468A1 *2002-07-152004-01-22Merck & Co., Inc.Piperidino pyrimidine dipeptidyl peptidase inhibitors for the treatment of diabetes
WO2004069162A3 *2003-01-312005-05-19Wallace T Ashton3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes
Reference:
[1]. J. Med. Chem., Ahead of Print.
[2]. Clinical therapeutics2008, 30, 1817-1830.
[3]. Int. J. Res. Dev. Pharm. Life Sci. 2013, 2, 602-610, 609 pp.
[4]. Xenobiotica; the fate of foreign compounds in biological systems 2014, 44, 627-634.
[5]. Poster presented at the annual meeting of American Diabetes Association, 2008, San Francisco: CA.
^Lim KS, Kim JR, Choi YJ, Shin KH, Kim KP, Hong JH, Cho JY, Shin HS, Yu KS, Shin SG, Kwon OH, Hwang DM, Kim JA, Jang IJ (October 2008). “Pharmacokinetics, pharmacodynamics, and tolerability of the dipeptidyl peptidase IV inhibitor LC15-0444 in healthy Korean men: a dose-block-randomized, double-blind, placebo-controlled, ascending single-dose, Phase I study”. Clin Ther. 30 (10): 1817–30. doi:10.1016/j.clinthera.2008.10.013. PMID19014837.
^Kaji K (Mar 2014). “Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats”. J Gastroenterol.. 49 (3): 481–91. doi:10.1007/s00535-013-0783-4. PMID23475323.
^Min HS (Jun 2014). “Dipeptidyl peptidase IV inhibitor protects against renal interstitial fibrosis in a mouse model of ureteral obstruction”. Lab. Invest. 94 (5): 598–607. doi:10.1038/labinvest.2014.50. PMID24687121.
Rhee EJ, Lee WY, Yoon KH, Yoo SJ, Lee IK, Baik SH, Kim YK, Lee MK, Park KS, Park JY, Cha BS, Lee HW, Min KW, Bae HY, Kim MJ, Kim JA, Kim DK, Kim SW (December 2010). “A multicenter, randomized, placebo-controlled, double-blind phase II trial evaluating the optimal dose, efficacy and safety of LC 15-0444 in patients with type 2 diabetes”. Diabetes Obes Metab. 12 (12): 1113–1119. doi:10.1111/j.1463-1326.2010.01303.x. PMID20977584.
////////////////gemigliptin, LC15-0444, KOREA 2012, Gemigliptin tartrate sesquihydrate, GLIPTIN, ゲミグリプチン , LG Chem, Sanofi DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
Cablivi is the first therapeutic approved in Europe, for the treatment of a rare blood-clotting disorder
On September 03, 2018, the European Commission has granted marketing authorization for Cablivi™ (caplacizumab) for the treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP), a rare blood-clotting disorder. Cablivi is the first therapeutic specifically indicated for the treatment of aTTP 1. Cablivi was designated an ‘orphan medicine’ (a medicine used in rare diseases) on April 30, 2009. The approval of Cablivi in the EU is based on the Phase II TITAN and Phase III HERCULES studies in 220 adult patients with aTTP. The efficacy and safety of caplacizumab in addition to standard-of-care treatment, daily PEX and immunosuppression, were demonstrated in these studies. In the HERCULES study, treatment with caplacizumab in addition to standard-of-care resulted in a significantly shorter time to platelet count response (p<0.01), the study’s primary endpoint; a significant reduction in aTTP-related death, recurrence of aTTP, or at least one major thromboembolic event during study drug treatment (p<0.0001); and a significantly lower number of aTTP recurrences in the overall study period (p<0.001). Importantly, treatment with caplacizumab resulted in a clinically meaningful reduction in the use of PEX and length of stay in the intensive care unit (ICU) and the hospital, compared to the placebo group. Cablivi was developed by Ablynx, a Sanofi company. Sanofi Genzyme, the specialty care global business unit of Sanofi, will work with relevant local authorities to make Cablivi available to patients in need in countries across Europe.
About aTTP aTTP is a life-threatening, autoimmune blood clotting disorder characterized by extensive clot formation in small blood vessels throughout the body, leading to severe thrombocytopenia (very low platelet count), microangiopathic hemolytic anemia (loss of red blood cells through destruction), ischemia (restricted blood supply to parts of the body) and widespread organ damage especially in the brain and heart. About Cablivi Caplacizumab blocks the interaction of ultra-large von Willebrand Factor (vWF) multimers with platelets and, therefore, has an immediate effect on platelet adhesion and the ensuing formation and accumulation of the micro-clots that cause the severe thrombocytopenia, tissue ischemia and organ dysfunction in aTTP 2.
Note – Caplacizumab is a bivalent anti-vWF Nanobody that received Orphan Drug Designation in Europe and the United States in 2009, in Switzerland in 2017 and in Japan in 2018. The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application for caplacizumab for treatment of adults experiencing an episode of aTTP. The target action date for the FDA decision is February 6, 2019
This drug was developed by Ablynx NV.[3] On 31 August 2018 it was approved in the European Union for the “treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP), in conjunction with plasma exchange and immunosuppression”.[4]
It is an anti-von Willebrand factor humanized immunoglobulin.[5] It acts by blocking platelet aggregation to reduce organ injury due to ischemia.[5] Results of the phase II TITAN trial have been reported.[5]
Fedratinib had been in phase III clincial trials by Sanofi for the treatment of myelofibrosis.
However, Sanofi had discontinued this research because of the safety issues. Orphan drug designation was assigned in the U.S. and in Japan for this indication. In 2017, the clinical hold was lifted in the U.S. by Impact Biomedicines.
MOLECULAR FORMULA C27H36N6O3S
MOLECULAR WEIGHT 524.7
SPONSOR Sanofi
CODE DESIGNATIONS SAR302503; TG101348
CAS REGISTRY NUMBER……….936091-26-8
WHO 9707
TG-101348 , a dual-acting JAK2/FLT3 small molecule kinase inhibitor, has been evaluated in phase III clinical development at Sanofi (formerly known as sanofi-aventis) for the oral treatment of intermediate-2 or high risk primary myelofibrosis, post-polycythemia vera myelofibrosis or post-essential thrombocythemia myelofibrosis with splenomegaly. However, development of the compound has been discontinued due to safety issues.
In preclinical models of myeloproliferative diseases, TG-101348, administered orally, was shown to reduce V617F-expressing cell populations in a dose-dependent manner without adversely impacting normal hematopoiesis. The reduction of V617F- expressing cell populations correlated with improved survival and reduced morbidity. Orphan drug designation was assigned in the U.S. and in Japan for the treatment of secondary and primary myelofibrosis. In July 2010, TargeGen was acquired by Sanofi. In 2013, orphan drug designation was assigned by the FDA for the treatment of polycythemia vera.
Fedratinib is an orally bioavailable, small-molecule, ATP-competitive inhibitor of Janus-associated kinase 2 (JAK2) with potential antineoplastic activity. Fedratinib competes with JAK2 as well as the mutated form AK2V617F for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and the induction of tumor cell apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders (MPDs); the mutated form JAK2V617F has a valine-to-phenylalanine modification at position 617 and plays a key role in tumor cell proliferation and survival.
Fedratinib has been used in trials studying the treatment and basic science of Solid Tumor, Myelofibrosis, Renal Impairment, Neoplasm Malignant, and Hepatic Impairment, among others.
Fedratinib (TG101348; SAR302503) is an orally available inhibitor of Janus kinase 2 (JAK-2) developed for the treatment of patients with myeloproliferative diseases including myelofibrosis. Fedratinib acts as a competitive inhibitor of protein kinase JAK-2 with IC50=6 nM; related kinases FLT3 and RET are also sensitive, with IC50=25 nM and IC50=17 nM, respectively. Significantly less activity was observed against other tyrosine kinases including JAK3 (IC50=169 nM).[1] In treated cells the inhibitor blocks downstream cellular signalling (JAK-STAT) leading to suppression of proliferation and induction of apoptosis.
Myelofibrosis is a myeloid malignancy associated with anemia, splenomegaly, and constitutional symptoms. Patients with myelofibrosis frequently harbor JAK-STAT activating mutations that are sensitive to TG101348. Phase I trial results focused on safety and efficacy of Fedratinib in patients with high- or intermediate-risk primary or post–polycythemia vera/essential thrombocythemia myelofibrosis have been published in 2011.[2]
Fedratinib was originally discovered at TargeGen. In 2010, Sanofi-Aventis acquired TargeGen and continued development of fedratinib until 2013. In 2016, Impact Biomedicines acquired the rights to fedratinib from Sanofi and continued its development for the treatment of myelofibrosis and polycythemia vera. In January 2018, Celgene acquired Impact Biomedicines.[3]
Condensation of 3-bromo-N-tertbutylbenzylsulfonamide with 2-chloro-5-methyl-pyrimidin-4-ylamine in the presence of Pd2(dba)3, Xantphos, Cs2CO3 in refluxing dioxane gives sulfonamide derivative , which is coupled with 4-[2-pyrrolidin-1-yl-ethoxy]phenylamine in AcOH at 150°C to provide the title compound
EXAMPLE 90. 7V-fe^-Butyl-3-{5-methyl-2-14-(2-pyrrolidm-l-yl-ethoxy)-phenylaminol- pyrimidin-4-ylaminol-benzenesuIfonamide (Compound LVII)
LVII
[0203] A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-l-yl- ethoxy)-phenylamine (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150 °C for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum atnount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
The compound and the pharmaceutical compositions described herein can be used for treating or delaying development of myelofibrosis in a subject. N-teft-Butyl-3-[(5-methyl-2-{ [4- (2-pyrrolidin-l-ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide has the following chemical structure:
Example 4. Synthesis of TG101348
Example 4.1 N-fer^-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)-benzenesulfonamide
(Intermediate)
Example 4.1(a)
1 2 Intermediate
[0162] A mixture of 2-chloro-5-methyl-pyrimidin-4-ylamine (1) (0.4 g, 2.8 mmol), 3-bromo-N- teft-butyl-benzenesulfonamide (2) (1.0 g, 3.4 mmol), Pd2(dba¾ (0.17 g, 0.19 mmol), Xantphos (0.2 g, 3.5 mmol) and cesium carbonate (2.0 g, 6.1 mmol) was suspended in dioxane (25 mL) and heated at reflux under the argon atmosphere for 3 h. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and the filtrate
concentrated in vacuo. The residue was dissolved in EtOAc and hexanes added until solid precipitated. After filtration, the title compound (1.2 g, 98%) was obtained as a light brown solid. It was used in the next step without purification. MS (ES+): m/z 355 (M+H)+.
Example 4.1(b)
SM2 Intermediate[0163] The Intermediate was synthesized from 2,4-dichloro-5-methylpyrimidine (SMI) and N-t- butyl-3-aminobenzenesulfonamide (SM2) in the following steps: (1) Mix MeOH (6.7UOa) and SMI (Combi Blocks) (UOa); (2) Add SM2 (1.15UOa, 082eq) and H20 (8.5UOa); (3) Heat 45°C, 20h, N2, IPC CPL SM2<2%; (4) Cool 20°C; (5) Centrifuge, N2; (6) Wash H20 (2.1UOa) + MeOH (1.7UOa); (7) Mix solid in H20 (4.3UOa) + MeOH (3.4UOa); (8) Centrifuge, N2; (9) Wash H20 (2.1UOa) + MeOH (1.7UOa); and (10) Dry 45°C, vacuum, 15h. Obtained
Intermediate, mass 49.6kg (UOb); Yield 79%; OP: 99.6%.
Example 4.2 N-½ri-Butyl-3-[(5-methyl-2-{ [4-(2-pyrrolidin-l- ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide
Intermediate TG101348
Example 4.2(a)
[0164] A mixture of N-ieri-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)- benzenesulfonamide (Intermediate) (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-l-yl-ethoxy)- phenylamine (3) (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150 °C for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCC^ solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2S04 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%). ]H NMR (500 MHz, DMSO-d6): δ 1.12 (s, 9H), 1.65-1.70 (m, 4H), 2.12 (s, 3H), 2.45-2.55 (m, 4H), 2.76 (t, /=5.8 Hz, 2H), 3.99 (t, 7=6.0 Hz, 2H), 6.79 (d, 7=9.0 Hz, 2H), 7.46-7.53 (m, 4H), 7.56 (s, 1H), 7.90 (s, 1H), 8.10-8.15 (m, 2H), 8.53 (s, 1H), 8.77 (s, 1H). MS (ES+): m/z 525 (M+H)+.
Example 4.2(b)
[0165] N-½ri-Butyl-3-[(5-methyl-2-{ [4-(2-pyrrolidin-l-ylethoxy)phenyl]amino}pyrimidin-4- yl)amino]benzenesulfonamide dihydrochloride monohydrate was prepared from 4-[2-(l- pyrrolidinyl)ethoxy] aniline dihydrochloride (SM3) and Intermediate following steps (A) and (B).
[0166] Step (A), preparation of free base of SM3 (3) from SM3, comprised steps (1) – (9): (1) Solubilize NaOH (0.42UOb) in H20 (9UOb); (2) Cool <20°C, N2; (3) Add TBME (6UOb) then SM3 (Malladi Drugs) (1.06UOb); (4) Mix >20mn then stop; (5) Drain Aq Ph then extract by TBME (3UOb); (6) Combine Or Ph; (7) Concentrate, vacuum, T<40°C, to an Oil; (8) Solubilize in IPA (2.5UOb); and (9) Calculate dry extract 23%.
[0167] Step (B) comprised the steps (1) – (6): (1) Mix IPA (10.5UOb) and Intermediate (UOb); (2) Add free base of SM3 (0.75UOb, 1.33eq/ interm); (3) add HC1 cone (0.413UOb); (4) Heat 70°C, 20h, N2, IPC CPL Interm<2%; (5) Cool <20°C; (2) Centrifuge, N2; (3) Wash IPA (3UOb); (4) Dry 50°C, vacuum, 26h; (5) De-lump in Fitzmill; and (6) polybag (x2) / poly drum. Obtained TG101348 dihydrochloride monohydrate, mass 83.8kg; Yield 98%; OP: 99.5%. Example 5 Capsule Form of TG101348 and Process of Making TG101348
Example 90 N-tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benzenesulfonamide (Compound LVII)
A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150° C. for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2×30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
Example 76 N-tert-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)-benzenesulfonamide (Intermediate 33)
A mixture of 2-chloro-5-methyl-pyrimidin-4-ylamine (0.4 g, 2.8 mmol), 3-bromo-N-tert-butyl-benzenesulfonamide (1.0 g, 3.4 mmol), Pd2(dba)3 (0.17 g, 0.19 mmol), Xantphos (0.2 g, 3.5 mmol) and cesium carbonate (2.0 g, 6.1 mmol) was suspended in dioxane (25 mL) and heated at reflux under the argon atmosphere for 3 h. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and the filtrate concentrated in vacuo. The residue was dissolved in EtOAc and hexanes added until solid precipitated. After filtration, the title compound (1.2 g, 98%) was obtained as a light brown solid. It was used in the next step without purification. MS (ES+): m/z 355 (M+H)+.
Example 90N-tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benenesulfonamide (Compound LVII)
[0308]
[0309]
A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.10 g, 0.49 mmol) in aeetie acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150° C. for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaIICO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2×30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
Methods for the treatment and management of myeloproliferative diseases using 4-(amino)-2-(2,6-dioxo(3-piperidyl)-isoindoline-1,3-dione in combination with other therapies
METHODS FOR TREATING TYROSINE-KINASE-INHIBITOR-RESISTANT MALIGNANCIES IN PATIENTS WITH GENETIC POLYMORPHISMS OR AHI1 DYSREGULATIONS OR MUTATIONS EMPLOYING DIANHYDROGALACTITOL, DIACETYLDIANHYDROGALACTITOL, DIBROMODULCITOL, OR ANALOGS OR DERIVATIVES THEREOF
Metopimazine is an established antiemetic that has been approved and marketed for many years in Europe for the treatment of acute conditions. The compound does not cross the blood-brain-barrier, and is therefore free from central side effects, and is not associated with cardiovascular side effects
In May 2016, preclinical data were presented at the 2016 DDW in San Diego, CA. In rats, po NG-101 and domperidone did not penetrate the brain at therapeutically relevant concentrations, unlike metoclopramide. In dogs, the amplitude and frequency of antral contractions were increased by NG-101, whereas in rats, po metopimazine resulted in an increase in gastric emptying of solid foods. The blood-brain barrier was not readily crossed and there was no interaction with 5-HT3 or 5-HT4 receptors by NG-101 unlike metoclopramide and domperidone, respectively
Neurogastrx is investigating repurposed metopimazine (NG-101), a selective and peripherally restricted dopamine D2/D3 receptor antagonist, for the potential oral treatment of gastroparesis. By July 2014, preclinical studies were underway . SE BELOW REF
WO-2014105655: Methods for treating GI tract disorders
In May 2016, preclinical data were presented [SEE BELOW].
2016 May 24Abs 1079 NG101: A Potent and Selective Dopamine D2 Receptor Antagonist as a Potential Alternative to Metoclopramide and Domperidone for the Treatment of Gastroparesis
Digestive Disease Week
Cyril De Colle, Marieke van der Hart, Jiande Chen, Arash Rassoulpour, Pankaj J Pasricha
In July 2014, preclinical data were published. Metopimazine at 1mg/kg increased gastric motility in hound dogs. In studies in rodents, metopimazine at 3 and 10 mg/kg increased gastric emptying by 18 and 40%, respectively, compared with vehicle control
There is an increasing demand for antiemetic agents because of the most troublesome adverse effects of chemotherapy-induced nausea and emesis during cancer treatment.
However, the objective of complete prevention of emesis in all patients remains elusive. Therefore, there is a great demand for both development of (i) new antiemetic agents and (ii) new manufacturing processes for existing antiemetic agents. Metopimazine is an existing dopamine D2-receptor antagonist with potent antiemetic properties. It is chemically known as l-(3-[2-(methylsulfonyl)-10H-phenothiazin-10-yl]propyl)-4-piperidinecarboxamide , which belongs to nitrogen- and sulfur-containing tricyclic compounds (phenothiazine class of drugs) with interesting biological and pharmacological activities.
Recently, it has been found that Metopimazine plays a key role as an alternative to Ondansetron in the prevention of delayed chemotherapy-induced nausea and vomiting (CINV) in patients receiving moderate to high emetogenic noncisplatin-based chemotherapy.It has been used in France for many years for the prevention and treatment of nausea and vomiting under the brand name of Vogalene
In 1959, the first synthesis and manufacture process of Metopimazine was reported by Jacob et al.The synthesis starts from the protection of 2-(Methylsulfanyl)-10H-phenothiazine..Jacob, R. M.; Robert, J. G. German Patent No. DE1092476, 1959.
Later, in 1990, Sindelar et al. reported a modified process , which starts from synthesis of 4-(2-fluorophenylthio)-3-nitrophenylmethylsulfone..Sindelar, K.; Holubek, J.; Koruna, I.; Hrubantova, M.; Protiva, M.Collect. Czech. Chem. Commun.1990, 55, 1586– 1601, DOI: 10.1135/cccc19901586
In 2010, Satyanarayana Reddy et al. reported a modified synthetic route which starts from either N-protection using acetyl chloride or N-alkylation using dihalopropane of 2-(methylsulfanyl)-10H-phenothiazine ..Satyanarayana Reddy, M.; Eswaraiah, S.; Satyanarayana, K. Indian Patent No. 360/CHE/2010 A, Aug 19, 2011.
Synthesis of 1-(3-[2-(methylsulfonyl)-10H-phenothiazin-10-yl]propyl)piperidine-4- carboxamide (1)-Metopimazine: Pale yellow color solid, yield. 65% (82 g), DSC 189 °C.
Chemical Research Division, API R&D Centre, Micro Labs Ltd., Plot No.43-45, KIADB Industrial Area, Fourth Phase, Bommasandra-Jigani Link Road, Bommasandra, Bangalore, Karnataka 560 105, India
An efficient, practical, and commercially viable manufacturing process was developed with ≥99.7% purity and 31% overall yield (including four chemical reactions and one recrystallization) for an active pharmaceutical ingredient, called Metopimazine (1), an antiemetic drug used to prevent emesis during chemotherapy. The development of two in situ, one-pot methods in the present synthetic route helped to improve the overall yield of 1 (31%) compared with earlier reports (<15%). For the first time, characterization data of API (1), intermediates, and also possible impurities are presented. The key process issues and challenges were addressed effectively and achieved successfully.
Synthesis of 1-(3-[2-(Methylsulfonyl)-10H-phenothiazin-10-yl]propyl)-4-piperidinecarboxamide (1), Metopimazine
In ………………….. The obtained compound (1) was dried in a hot air oven at 50 °C.
WO-2014105655: Methods for treating GI tract disorders
2016 May 24Abs 1079 NG101: A Potent and Selective Dopamine D2 Receptor Antagonist as a Potential Alternative to Metoclopramide and Domperidone for the Treatment of Gastroparesis
Digestive Disease Week
Cyril De Colle, Marieke van der Hart, Jiande Chen, Arash Rassoulpour, Pankaj J Pasricha
Melting point: 220 ° C. (decomposition). EP0748807 [α] = -16.9 ° (c = 1, H 2 O).
[α]D = -17.9 (C = 0.75, DMSO, t = 23°C) at 589 nm. DOI: 10.1021/acs.oprd.6b00262
5-HT3 and 5-HT4 inhibitor that was potentially useful for the treatment of neurological disorders.
Innovators-sanofi
Hoechst Marion Roussel (Sanofi) my organisation 1993-1997 Process development at Mulund, Mumbai, India.
CENTRE IS DR RALPH STAPEL, HEAD PROCESS DEVELOPMENT, SANOFI
The 5-HT4 receptor is a G-protein coupled receptor (GPCR) which belongs to the serotonin receptor family. The role of the 5-HT4 receptor in the modulation of many diseases is well described in the literature.(1)
During the last decades, an impressive body of evidence suggested that selective stimulation of neuronal 5-HT4 receptor subtypes could be beneficial in the symptomatic treatment of memory disorders, including many antidepressants, antipsychotics, anorectics, antiemetics, gastroprokinetic agents, antimigraine agents, hallucinogens, and antactogens.(2)
Within effort to discover treatments of memory dysfunction, SL65.0102-10, a selective 5-HT4 partial agonist (Ki 6.6 μM), was discovered as promising agent for the treatment of cognition impairment. Serotonin receptors are the target of a variety of pharmaceutical drugs; SL65.0102-10 emerged as a promising 5-HT3 and 5-HT4 inhibitor that was potentially useful for the treatment of neurological disorders.(3)
Samir Jegham
Lead Generation Senior Advisor for Asia Pacific Research Hub at Sanofi
“DRUG APPROVALS INT” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
SYNTHESIS
SL65.0102-10
CONTD…………..
Synthesis
PATENT
(EP0748807) Derivatives of N- (1,4-diazabicyclo (2.2.2) -oct-2-yl) methyl benzamide, their preparation and their therapeutic use
The procedure described in Example 4.1, but from (+) – 2,2-dimethyl-1,3-dioxolane-4-methanol.
5.2. (-) – 2 – [(2,2-Dimethyl-1,3-dioxolan-4-yl) methyl] -1 H -isoindole-1,3 (2 H ) -dione.
The procedure described in Example 4.2, from methane sulfonate (+) – (2,2-dimethyl-1,3-dioxolan-4-yl) methyl.
Melting point: 81.2-81.3 ° C.
[α]= -34.9 ° (c = 1, CH 2 Cl 2 ).
5.3. (-) – 2- (2,3-dihydroxypropyl) -1 H -isoindole-1,3 (2 H ) -dione.
The procedure described in Example 4.3, from (-) – 2 – [(2,2-dimethyl-1,3-dioxolan-4-yl) methyl] -1 H -isoindole-1, 3 (2 H ) -dione.
Melting point: 122.8-122.9 ° C.
[α]= -48.8 ° (c = 1, CH 3 OH).
5.4. (-) – 2 – [(2-Phenyl-1,3-dioxolan-4-yl) methyl] -1 H -isoindole-1,3 (2 H ) -dione.
The procedure described in Example 4.4, from (-) – 2- (2,3-dihydroxypropyl) -1 H -isoindole-1,3 (2 H ) -dione.
Melting point: 84 ° C.
[α]= -59 ° (c = 1, CH 2 Cl 2 ).
5.5. Benzoate (-) – 2-bromomethyl-1- (1,3-dihydro-1,3-dioxo-2 H -isoindol-2-yl) ethyl.
The procedure described in Example 4.5, from (-) – 2 – [(2-phenyl-1,3-dioxolan-4-yl) methyl] -1 H -isoindole-1,3 ( 2 H ) -dione.
Melting point: 118.4-118.6 ° C.
[α]= -58.2 ° (c = 1, CH 2 Cl 2 ).
5.6. (+) – 2- (oxiranylmethyl) -1 H -isoindole-1,3 (2 H ) -dione. Fusion point :
The procedure described in Example 4.6, from benzoate (-) – 2-bromomethyl-1- (1,3-dihydro-1,3-dioxo-2 H -isoindol-2-yl) ethyl.
Melting point: 100.4-100.5 ° C.
[α]= + 45.5 ° (c = 1, CHCl 3 ).
The procedure described in Example 4.7, from (+) – 2- (oxiranylmethyl) -1 H -isoindole-1,3 (2 H ) -dione. Melting point: 220 ° C. (decomposition). [α] = -16.9 ° (c = 1, H 2 O).
Paper
The process development and improvements for route selection, adapted to large scale for the pilot-scale preparation of SL65.0102-10, an N-diazabicyclo[2.2.2]-octylmethyl benzamide, a 5-HT3and 5-HT4 receptor active ligand for the treatment of neurological disorders such as cognition impairment, are described in this article. Notable steps and enhancements are compared to the original route, including the improvement of a chiral epoxide synthesis by shortening the number of chemical steps, the deprotection of a quaternary ammonium salt, and the redesign of the final amidification coupling to avoid chromatography.
[α]D = -17.9 (C = 0.75, DMSO, t = 23°C) at 589 nm.
Elementary analysis: found C 43.0660%, H 5.5150%, N 12.4792%, calculated C 43.31%, H 5.68%, N 12.63%
1H AND 13C NMR PREDICT
References
(a) Hoyer, D.; Clarke, D. E.; Fozard, J. R.; Hartig, P. R.; Martin, G. R.; Mylecharane, E. J.; Saxena, P. R.;Humphrey, P. P.Pharmacol. Rev.1994, 46 ( 2) 157– 203
(b) Frazer, A.; Hensler, J. G.Chapter 13: Serotonin Receptors. In Siegel, G. J.; Agranoff, B. W.; Albers, R. W.; Fisher, S. K.; Uhler, M. D., Eds.; Basic Neurochemistry: Molecular, Cellular, and Medical Aspects; Lippincott-Raven, Philadelphia,1999; pp 263– 292.
Jegham, S.; Koenig, J. J.; Lochead, A.; Nedelec, A.; Guminski, Y.N-[(1,4-diazabicyclo[2.2.2]oct-2-yl)methyl] benzamide derivatives, their preparations and their application in therapeutics.
(a) FR 2756563 06/13/1995 9506951, 1995.
(b) US 5663173, 1997; Washington, DC: U.S. Patent and Trademark Office.
“DRUG APPROVALS INT” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
The U.S. Food and Drug Administration approved Adlyxin (lixisenatide), a once-daily injection to improve glycemic control (blood sugar levels), along with diet and exercise, in adults with type 2 diabetes.
The U.S. Food and Drug Administration approved Adlyxin (lixisenatide), a once-daily injection to improve glycemic control (blood sugar levels), along with diet and exercise, in adults with type 2 diabetes.
“The FDA continues to support the development of new drug therapies for diabetes management,” said Mary Thanh Hai Parks, M.D., deputy director, Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research. “Adlyxin will add to the available treatment options to control blood sugar levels for those with type 2.”
Type 2 diabetes affects more than 29 million people and accounts for more than 90 percent of diabetes cases diagnosed in the United States. Over time, high blood sugar levels can increase the risk for serious complications, including heart disease, blindness and nerve and kidney damage.
Adlyxin is a glucagon-like peptide-1 (GLP-1) receptor agonist, a hormone that helps normalize blood sugar levels. The drug’s safety and effectiveness were evaluated in 10 clinical trials that enrolled 5,400 patients with type 2 diabetes. In these trials, Adlyxin was evaluated both as a standalone therapy and in combination with other FDA-approved diabetic medications, including metformin, sulfonylureas, pioglitazone and basal insulin. Use of Adlyxin improved hemoglobin A1c levels (a measure of blood sugar levels) in these trials.
In addition, more than 6,000 patients with type 2 diabetes at risk for atherosclerotic cardiovascular disease were treated with either Adlyxin or a placebo in a cardiovascular outcomes trial. Use of Adlyxin did not increase the risk of cardiovascular adverse events in these patients.
Adlyxin should not be used to treat people with type 1 diabetes or patients with increased ketones in their blood or urine (diabetic ketoacidosis).
The most common side effects associated with Adlyxin are nausea, vomiting, headache, diarrhea and dizziness. Hypoglycemia in patients treated with both Adlyxin and other antidiabetic drugs such as sulfonylurea and/or basal insulin is another common side effect. In addition, severe hypersensitivity reactions, including anaphylaxis, were reported in clinical trials of Adlyxin.
The FDA is requiring the following post-marketing studies for Adlyxin:
Clinical studies to evaluate dosing, efficacy and safety in pediatric patients.
A study evaluating the immunogenicity of lixisenatide.
Adlyxin is manufactured by Sanofi-Aventis U.S. LLC, of Bridgewater, New Jersey.
Sanofi (formerly sanofi-aventis, formerly Aventis), under license from Zealand Pharma, has developed and launched lixisenatide
Lixisenatide (trade name Lyxumia) is a once-daily injectable GLP-1 receptor agonist for the treatment of diabetes, discovered by Zealand Pharma A/S of Denmark and licensed and developed by Sanofi.[1] Lixisenatide was accepted for review by the US FDA on February 19, 2013, and approved by the European Commission on February 1, 2013.[2] On September 12, 2013, Sanofi delayed the approval process in the US, citing internal data from a cardiovascular risk study. The drug will likely be resubmitted for approval in 2015.
Lixisenatide is a once-daily injectable GLP-1 receptor agonist discovered by Zealand Pharma A/S of Denmark and licensed and developed by Sanofi. As of September 2010 it is in clinical trials for diabetes. Lixisenatide was accepted for review by the US FDA on February 19, 2013, and approved by the European Commission on February 1, 2013. The drug will likely be resubmitted for approval in 2015.
Mechanism of action
GLP-1 is a naturally-occurring peptide that is released within minutes of eating a meal. It is known to suppress glucagon secretion from pancreatic alpha cells and stimulate insulin secretion by pancreatic beta cells. GLP-1 receptor agonists are used as an add-on treatment for type 2 diabetes and their use is endorsed by the European Association for the Study of Diabetes, the American Diabetes Association, the American Association of Clinical Endocrinologists and the American College of Endocrinology.
Physical and chemical properties
Lixisenatixe has been described as “des-38-proline-exendin-4 (Heloderma suspectum)-(1–39)-peptidylpenta-L-lysyl-L-lysinamide”, meaning it is derived from the first 39 amino acids in the sequence of the peptide exendin-4, found in the Gila monster (Heloderma suspectum), omitting proline at position 38 and adding six lysine residues. Its complete sequence is:[3]
The title method comprises the steps of: (1) coupling Fmoc-Lys(Boc)-OH and resin to obtain Fmoc-Lys(Boc)-resin, (2) protecting amino acid with Fmoc, conducting solid-phase synthesis to obtain lixisenatide wholly protected 20-44-peptide resin, (3) conducting solid-phase synthesis to obtain wholly protected 15-19-peptide resin, (4) coupling the wholly protected 20-44-peptide resin and wholly protected 15-19-peptide resin, (5) coupling other amino acids till solid-phase synthesis finishes, (6) cracking lixisenatide peptide resin to obtain crude peptide, and (7) purifying through RP-HPLC. The method improves crude peptide purity and purifn. yield.
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
利西拉, the English name: Lixisenatide, is a polypeptide containing 44 amino acids, the structural formula is as follows: peptide sequence as follows:
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu -Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pr O-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH 2 Li Xila to (Lixisenatide ) by Sanofi-Aventis developed once a day subcutaneously with glucagon-like peptide -I (GLP-I) receptor agonists, for the treatment of type II diabetes, on February 1, 2013 Sanofi Lee Division -Aventis of exenatide is approved EMEA, for the adjuvant treatment of poorly stable dose of basal insulin (or metformin) in the treatment of type II diabetes to improve HbAlc and postprandial blood glucose levels.
CN201210030151. 2 used in a pure solid phase sequential coupling method synthetic peptides.The method amino resin as the carrier, using conventional coupling sequence, the final cut to give Li Xila.
US6528486 patent for the compound, synthetic methods mentioned it to phase condensation method Fmoc / tBu strategy.
The [0005] W02005058954 synthesis method including the gradual condensation process Fmoc / tBu strategy, Boc strategy of gradual condensation methods and genetic engineering.
The W02001004156 synthesis method for the gradual condensation process Fmoc / tBu strategy.
Since Li Xila abroad mostly used to synthesize Fmoc solid phase synthesis method, a gradual shrinking gradually synthesis step more, resulting in more types of product impurities, US 20130284912 Special Report polypeptide impurity: Di-Ser33- Leisy pull and Di-Ala35- Li Xila come, Di-Ser 33- Li Xila come and Di-Ala35- Li Xila to atmosphere amino acid sequence as follows: Di-Ser33- Li Xila to the amino acid sequence: H-His -Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu-Trp- Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Ser-Gly-Ala-Pr 〇-Pr〇-Ser-Lys_Lys_Lys_Lys_Lys_LyS-NH2 Di-Ala35- Li Xila to the amino acid sequence: H-His-Gly- Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu-Trp-Leu-Lys -Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ala-Pr 〇-Pr〇-Ser-Lys_Lys_Lys_Lys_Lys_LyS-NH2 toxicity of these impurities are impurities larger, and very difficult to separate from the main peak , the presence of the impurities seriously affect 利西拉 to content and the use of safety.Hence the need to find an effective way to remove it and to reach the high standard level of 0.1% or less.The present inventors have found that this impurity is difficult to remove by means of the prior art, although there are ways to remove part of, but removal is not ideal, it is difficult to achieve high quality standards is likely to cause 利西拉 level while reducing their yield.
In summary, the existing Li Xila to the solid phase synthesis, low yield of the synthesis, impurities, in particular, are not well controlled impurity Di-Ser 33- Li Xila come and Di-Ala35 – Li Xila to, does not apply to industrial production
Example i ^ a: Preparation 利西拉 to fine peptide acetate Weigh 利西拉 above 44. 70g to 45L crude peptide was dissolved in water, purified by C18 column, the first purification conditions: mobile phase: A phase: 0 I% TFA; B phase: acetonitrile; gradient program was: 15% B, 60 minutes to 60% B; detection wavelength 220 nm; peak fraction collection purposes.The second purification conditions: mobile phase was: A phase: 0 3% HAC; B phase: acetonitrile; gradient program was: 10% B, 60 minutes to 60% B; detection wavelength 220 nm; peak fraction collection purposes.Desalting conditions: Mobile phase: A phase: an aqueous solution of 20 mmol / L ammonium acetate: acetonitrile = 95: 5; B phase: water: acetonitrile = 95: 5; C phase: 0.03% aqueous solution of acetic acid: acetonitrile = 95 : 5; D phase: 0.03% aqueous solution of acetic acid: acetonitrile = 50: 50; gradient program: mobile phase A isocratic for 15 minutes, convert isocratic mobile phase B for 10 minutes, is converted into the flow Phase C isocratic 10 minutes, converted into a mobile phase D isocratic 25 minutes; detection wavelength 220 nm; peak fraction collection purposes; rotary evaporation concentrated and lyophilized to give Li Xila acetate fine peptide 22. 65g which HPLC spectrum shown in Figure 5, HPLC purity of 99.75% (area normalization method), Di-Ser33- Li Xila come to 0.03% (area normalization method), Di-Ala35- Li Xila to the content of 0.05% (area normalization method).Purification total yield of 51%, total yield 41%.Its mass spectrum as shown in Figure 6, [M + H] + = 4858. 691, 利西拉 precise molecular weight to the theoretical: 4857.53, the sample mass is consistent with the theoretical molecular weight.
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
Example 2: Preparation 利西拉 to crude peptide
利西拉 [0116] Example 24 was prepared to be placed 125.4g peptide resin cleavage reaction to 10ml / g resin ratio added lysis reagent (TFA: thioanisole: EDT: TIS: water = 86: 5 : 5: 3: 1 (V / V)), stirred at room temperature 2.5h.The reaction was purified by frit funnel filtration, the filtrate was collected, the resin was washed 3 times and then a small amount of TFA, the combined filtrates concentrated under reduced pressure.Frozen precipitation in anhydrous ether was added, washed three times with anhydrous diethyl ether, and dried in vacuo to give a white solid powder, i.e. Li Xila to crude peptide 47.lg, by weight of the crude peptide yield 97.2%, HPLC purity 63.8% 0
利西拉 to crude peptide preparation: 27 patients [0117] Example
利西拉 [0118] The Example 25 was prepared to be placed 123.7g peptide resin cleavage reaction to 10ml / g resin ratio added lysis reagent (TFA: thioanisole: EDT: TIS: water = 86: 5 : 5: 3: 1 (V / V)), stirred at room temperature 2.5h.The reaction was purified by frit funnel filtration, the filtrate was collected, the resin was washed 3 times and then a small amount of TFA, the combined filtrates concentrated under reduced pressure.Frozen precipitation in anhydrous ether was added, washed three times with anhydrous diethyl ether, and dried in vacuo to give a white solid powder, i.e. Li Xila to crude peptide 46.9g, yield the crude peptide by weight 96.5%, HPLC purity 64.2% 0
28 Example 2: Preparation 利西拉 to fine peptide acetate
Example weighed 26 to 27 after 利西拉 to any 30.0g crude peptide was dissolved in 3000ml of water using Waters2545RP-HPLC system, wavelength 230nm, 50 X 250mm column of reverse phase C18 column, 0.2% TFA conventional / acetonitrile mobile phase were fractionated peaks of fractions, refined peptide purity greater than 98.5%.The fine peptide solution using Waters2545RP-HPLC system, 50 X 250mm column was C18 reverse phase column, 0.1% acetic acid / acetonitrile mobile phase transfer salt, the purpose of peak fractions were collected, concentrated by rotary evaporation and lyophilized to give Li Xila acetate fine salt peptide> 9.0g, RP-HPLC purity ≥98.5%.Purification Yield ≥30%, total yield ≥29.0%.
PATENT
CN 102875663
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
[0239] The crude peptide Li Xila to 4000g (including Li Xila to 1139g) was dissolved with purified water 100L, collected by filtration and the filtrate set aside.
[0240] purification chromatographic conditions:
[0241] HPLC Model: Novasep LC450
Column: 450X250mm, built-phenyl silane bonded silica gel as stationary phase filler, the filler particle size of 10 μ m0
flow rate: 5000ml / min.
The detection wavelength: 280nm.
Mobile phase A phase: 10% 30mM D- 30mM sodium tartrate and disodium hydrogenphosphate in methanol / 90% aqueous (v / v), adjusted to pH 2.5 with phosphoric acid.
[0246] Mobile phase A phase preparation process: Weigh 1280g 2070g D- sodium tartrate and disodium hydrogenphosphate, after an appropriate amount of purified water was dissolved through 0.45 μ m membrane filter, the filtrate collected all 300L tank, added 30L chromatographically pure After methanol was added to the 300L scale purification of water, adjusted to pH 2.5 with phosphoric acid.Repeat preparation run.
[0247] The mobile phase B phase: HPLC grade acetonitrile.
[0249] sample volume: 250.0g (6250ml).
[0250] Purification: column equilibration the sample so that after 5 minutes, run a gradient purification, monitoring and staging purposes peak fractions were collected.The collected fractions (chromatographic conditions purity testing to the same conditions as above 利西拉 determination to area normalization method measured) purity test, the purity of greater than or equal to 98% of the fractions after removing most of the acetonitrile in turn salt; purity of 70% or more less than 98% of the fraction recovered after removal of most of the acetonitrile and the purification procedure is repeated, again collected purity greater than or equal to 98% of the fraction after removal of most of the acetonitrile are also used to turn salt; purity of less than 70 % of fractions by waste disposal.
[0251] points and 16 injections, repeat the above operation.
[0252] turn salt chromatographic conditions:
[0253] HPLC Model: Novasep LC450
[0254] Column: 450 X 250mm, built-C8 reversed-phase chromatography packing, the particle size of the filler is 10 μ m.
[0255] flow rate: 5000ml / min.
[0256] The detection wavelength: 280nm.
[0257] Mobile phase A phase: 0.2% acetic acid (v / v) solution.
[0258] The mobile phase B phase: HPLC grade acetonitrile.
[0259] gradient
[0260] sample volume: 2500ml.
[0261] Purification: The column equilibration the sample for 5 minutes, run a gradient purification, monitoring and collecting the target peak fractions.The purpose of the peak fractions were concentrated by rotary evaporation under reduced pressure to 9000ml after lyophilization.
[0262] After the freeze-dried to give a white powder refined peptide 704g.Purity of 98.39%, the impurity content of less than 0.5%.Purification yield 61.8% (in crude Li Xila to content), total yield of 17.6%.
[0096] To the resulting Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHAResin mouth of a 20% strength piperidine / DMF solution for 10 minutes, the reaction was drained, washed with DMF Resin 6 (50ml * 6).Weigh Fmoc-Lys (Boc) -〇H3.52g, H0Bt1.01g, HBTU2.84g, TMP1.98ml, DMF50ml added to dissolve slowly with stirring under ice-cooling for 3 minutes, at room temperature for 2 hours, the reaction Ninhydrin detection method completed, pumping off the reaction solution, DMF the resin was washed twice (50mlX2), DCM the resin was washed twice (50mlX2), to give Fmoc-Lys (B oc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHAResin.As used in the above operation Fmoc-Lys (Boc) -OH: HOBt: HBTU: TMP ratio is 1: 1: 1: 2, wherein Fmoc-Lys (Boc) -OH is the number of moles of Fmoc-RinkAmide-MBHAResin number of moles 3 times.
[0097] Li Xila fully protected side chain was prepared to -Rink Amide-MBHA Resin:
[0098] To the resulting Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHA Resin added 20% piperidine / DMF solution for 10 minutes, drained reaction solution, washed 6 times with DMF.Weigh Jie 111〇 (3-1 ^ 8 billion (3) -0 13.528, 1 (»Shu 1.018,01 (:!! 1.391111 added 50,111,101 ^ dissolve slowly stirring for 3 minutes in an ice bath, poured into the solid phase resin is mixed with the reaction column, at room temperature for 2 hours, the reaction Ninhydrin detection method is completed, the reaction solution was deprived, DMF the resin was washed twice (50ml X 2), DCM the resin was washed twice (50ml X 2), to give Fmoc-Lys ( Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Rink Amide-MBHAResin above operation used by the Fmoc-Lys (Boc) -〇H:. HOBt: DIC ratio is 1: 1: L2, which Fmoc-Lys (Boc) is three times the number of moles -〇H Fmoc-Rink Amide-MBHA Resin moles of repeat after the coupling step, followed by the completion of the 39 lysine to first. connecting protected amino acids histidine, followed by addition of 20% piperidine / DMF solution for 10 minutes, the reaction was drained, DMF the resin was washed six times (50ml X 6), DCM the resin was washed six times (50ml X 6 ), MeOH contraction of the resin three times with MeOH 50ml, each contraction 5min. After the resin was dried in vacuo to give a full side-chain protected peptide resin to the Li Xila 27. 5g, weight resin 17. 5g.
[0099] Li Xila to crude peptide preparation:
[0100] Weigh side chains fully protected Li Xila to -Rink Amide-MBHA Resin 27. 5 grams, into a round bottom flask.Configuration 275 ml lysis buffer, wherein trifluoroacetic acid: thioanisole: ethanedithiol: anisole, phenol = 93: 4: 1: 1.5: 2 (volume ratio).Lysate in the refrigerator after the pre-freeze 1 hour before Sheng Youli put to Silas to -Rink Amide-MBHA Resin round bottom flask, stirred at room temperature for 2 hours.The reaction mixture was filtered, the resin was washed with 20ml TFA and the combined filtrate.
[0101] The volume of the filtrate was slowly poured into 2,750 ml of diethyl ether frozen (frozen advance ether), a white precipitate appears, at 3000 rpm / centrifuged 5 minutes, the resulting solid was washed twice with ether, then the solid was dried under vacuum to give Li Xila trifluoroacetate crude peptide to 15. 3g.
[0102] Li Xila to large scale production of fine peptide:
[0103] Sample Preparation: The crude peptide was dissolved in water, the sample was completely dissolved by membrane filtration, the filtrate was collected for use.
[0104] Purification conditions: Column: octadecyl silane bonded silica gel as stationary phase column, the column diameter and length: 300_X250mm.Mobile phase: A phase: 35mm〇l / L phosphoric acid solution adjusted with triethylamine to pH 6. 7; B phase: acetonitrile, flow rate: 2200ml / min, Gradient: B%: 12% ~32%, detection wavelength: 280nm .The injection volume was 75g.Purification process: the column with 50% acetonitrile rinse clean after balance sample, sample amount is 75g.Linear gradient 120min, the purpose of collecting peaks will be collected 利西拉 solution was concentrated by rotary evaporation under reduced pressure to about 80mg / ml and reserve the water temperature exceeds 40 ° C without conditions.
[0105] turn salt: turn salt conditions: Column: octadecyl silane bonded silica gel as stationary phase column, the column diameter and length: 300mmX250mm.Mobile phase: A phase: mass concentration of 0.2% aqueous acetic acid; B phase: HPLC grade acetonitrile, flow rate: 2200ml / min, detection wavelength: 280nm.Gradient: B%: 6% ~36%.The injection volume was 48-60g.Salt transfer process: the column with 50% acetonitrile rinse clean after the sample, the sample volume is 1600ml sample solution.Linear gradient 90min, the purpose of collecting peaks collected Li Xila to solutions were concentrated by rotary evaporation to about 80ml / g after go to the appropriate size vials, then freeze-dried to obtain the purity of greater than 99.5% The Li Xila come.
Sanofi Provides Update on Lixisenatide New Drug Application in U.S.
Paris, France – September 12, 2013 – Sanofi (EURONEXT: SAN and NYSE: SNY) announced today its decision to withdraw the lixisenatide New Drug Application (NDA) in the U.S., which included early interim results from the ongoing ELIXA cardiovascular (CV) outcomes study. The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
The decision to withdraw the lixisenatide application follows discussions with the U.S. Food and Drug Administration (FDA) regarding its proposed process for the review of interim data. Sanofi believes that potential public disclosure of early interim data, even with safeguards, could potentially compromise the integrity of the ongoing ELIXA study. Sanofi’s decision is not related to safety issues or deficiencies in the NDA………………………read all at
Christensen, M; Knop, FK; Holst, JJ; Vilsboll, T (2009). “Lixisenatide, a novel GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus”. IDrugs : the investigational drugs journal12 (8): 503–13. PMID19629885.
576.49306 g/mol
A guanylate cyclase activator potentially for the treatment of aortic valve stenosis.
CAS No. 254877-67-3
Originator sanofi-aventis
Developer Mayo Clinic; National Center for Advancing Translational Sciences; Sanofi; sanofi-aventis
Class Anthranilic acids; Benzamides; Cardiovascular therapies; Chlorobenzenes; Morpholines; Small molecules; Sulfonamides; Thiophenes
Mechanism of Action Guanylate cyclase stimulants
30 Jun 2015 Mayo Clinic plans a phase II trial for Aortic valve stenosis in USA (NCT02481258)
29 Jan 2014 Phase-I clinical trials in Aortic valve stenosis in USA (PO)
01 Jan 2010 Discontinued – Phase-II for Peripheral arterial occlusive disorders in Austria, Canada, France, Germany, Italy, Poland, Portugal, Russia, South Africa and USA (PO) prior to 2010
SYNTHESIS
The Intermediates hown above is used in next step shown below
The Ru(II)-catalyzed intermolecular o-C–H amidation of arenes in N-benzoylated sulfoximine with sulfonyl azides is demonstrated. The reaction proceeds with broad substrate scope and tolerates various functional groups. Base hydrolysis of the amidation product provides the anthranilic acid derivatives and methylphenyl sulfoximine (MPS) directing group. This method is successfully employed for the synthesis of HMR 1766.
Mechanism of Action:selective oral inhibitor of PI3K and mTOR
Indication:Cancer Treatment
Stage of Development: phase ll study in chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL). A phase I/II trial is assessing SAR245409 in combination with letrozole in ER/PR+ HER2- breast cancer.
SAR245409 (XL765)
SAR245409 (XL765) is an orally available inhibitor of PI3K and the mammalian target of rapamycin (mTOR), which are frequently activated in human tumors and play central roles in tumor cell proliferation. Exelixis discovered SAR245409 internally and out-licensed the compound to Sanofi. SAR245409 is being evaluated by Sanofi as a single agent and in multiple combination regimens in a variety of cancer indications. Clinical trials have included a single agent phase 2 trial in Non-Hodgkin’s lymphoma, combination phase 1b/2 trials with temozolomide in patients with glioblastoma, with letrozole in hormone receptor positive breast cancer, with bendamustine and/or rituximab in lymphoma or leukemia, and a phase 1 trial in combination with a MEK inhibitor.
SAR-245409 is an investigational drug originated by Exelixis that dually inhibits mammalian target of rapamycin (mTOR) and phosphatidylinositol 3-kinase (PI3K).
Sanofi is also evaluating the compound in phase I/II clinical trials for the treatment of malignant neoplasm as monotherpay or in combination regimen. It has also completed phase I clinical trials as an oral treatment for brain cancer.
In 2009, the drug candidate was licensed to Sanofi (formerly known as sanofi-aventis) by Exelixis worldwide for the treatment of solid tumors.
XL765 (Voxtalisib, SAR245409, Sanofi)*, a PYRIDOPYRIMIDINONE-derivative, is a highly selective, potent and reversible ATP-competitive inhibitor of pan-Class I PI3K (α, β, γ, and δ) and mTORC1/mTORC2. It is orally active, highly selective over 130 other protein kinases. In cellular assays, XL765 inhibits the formation of PIP3 in the membrane, and inhibits phosphorylation of AKT, p70S6K, and S6 phosphorylation in multiple tumor cell lines with different genetic alterations affecting the PI3K pathway.
In mouse xenograft models, oral administration of XL-765 results in dose-dependent inhibition of phosphorylation of AKT, p70S6K, and S6 with a duration of action of approximately 24 hours. Repeat dose administration of XL765 results in significant tumor growth inhibition in multiple human xenograft models in nude mice that is associated with antiproliferative, antiangiogenic, and proapoptotic effects
Compound (1) can be synthesized as described in WO 07/044813, which is hereby incorporated in its entirety.
Briefly, a base and an intermediate, compound (a), are added to solution of commercially available 2-metfiyl-2-thiopseudourea sulfate in a solvent such as water and stirred overnight at room temperature. After neutralization, compound (b) is collected by filtration and dried under vacuum. Treatment of compound (b) with POCI3 and heating at reflux for 2 hours yields compound (c) which can be concentrated under vacuum to dryness. Compound (c) can be used directly in the following reaction with ethylamine carried out in a solvent such as water with heating to give compound (d). Compound (d) is then treated with iodine monochloride in a solvent such as methanol to form compound (e). Compound (e) is then dissolved in DMA, to which ethyl acrylate, Pd(OAc)2 and a base are added. This reaction mixture is heated and reacted overnight until completion of the reaction to give compound (f), which can be purified via column chromatography.
Compound (f) is then be treated with DBU in the presence of a base, such as DIEA, and heated at reflux for 15 hours. Upon completion of the reaction, the solvent is evaporated and the residue triturated with acetone to yield compound (g). Bromination of compound (g) can be achieved through drop-wise addition of Br2 to compound (g) in CH2C12, followed by stirring overnight at room temperature. Next, filtration is carried out, and triethylamine is added so that, upon washing and drying, the product, compound (h) is obtained. A Suzuki coupling between compound (h) and lH-pyrazol-5-yl boronic acid is carried out using a Pd- catalyst such as [1,1 -bis(diphenylphosphino)ferrocene]dichloropalladium(II) in the presence of a base to yield compound (i). Finally, compound (i) can be converted to compound (1) of the instant invention through 1) oxidation of the methylthio group with m-CPBA, carried out at room temperature with stirring and 2) treatment of the resulting product dissolved in dioxane, with liquid ammonia. Stirring at room temperature overnight followed by purification by column chromatography gives the desired product, 2-amino-8-ethyl-4-methyl- 6-(lH-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one, compound (1).
Example 1 2-amino-8-ethyl-4-methyl-6-(lJΪ-pyrazol-5-yl)pyrido[2,3-</]pyrimidin-7(8J?)-one
To a solution of 2-methyl-2-thiopseudourea sulfate (Aldrich, 58.74 g, 0.422 mol) in water (1000 mL) were added sodium carbonate (81.44 g, 0.768 mol) and ethyl acetoacetate (50 g, 0.384 mol) at room temperature. The reaction mixture was stirred overnight. After neutralizing to pH = 8, the solid was collected through filtration followed by drying under vacuum overnight to afford 6-methyl-2-(methylthio)pyrimidin-4(3H)-one (57.2 g, 95% yield) of product. 1H NMR (400 MHz, DMSO-d6): δ 12.47 (bs, IH), 5.96 (bs, lH), 2.47(s, 3H), 2.17 (s, 3H).
To the round bottom flask containing 6-methyl-2-(methylthio)pyrimidin-4(3H)- one (19 g, 121.6 mmol) was added POCl3 (30 mL). The reaction mixture was heated to reflux for 2 h and then concentrated on a rotary evaporator to dryness. The crude 4-chloro- 6-methyl-2-(methylthio)pyrimidine was used directly in the next reaction without further purification.
To the 4-chloro-6-methyl-2-(methylthio)pyrimidine from above was added 30 mL of a solution of 70% ethylamine in water. The reaction mixture was heated to 50 0C for 3 h. After completion, excess ethylamine was evaporated on rotary evaporator under vacuum. The solid was filtered and dried under vacuum to afford 7V-ethyl-6-methyl-2- (methylthio)pyrimidin-4-amine (20 g, 90% yield).
To the solution of N-emyl-6-methyl-2-(methylthio)pyrimidin-4-amine (20 g, 121.6 mmol) in methanol was added iodine monochloride (26.58 g, 163.7 mmol) in small portions at 0 °C. Then the reaction mixture was stirred overnight. After evaporation of solvent, the residue was triturated with acetone. The product iV-ethyl-5-iodo-6-methyl-2- (methylthio)pyrimin-4-amine (25.2 g, 75% yield) was collected by filtration. 1H NMR (400 MHz, CDCl3): δ 5.37 (bs, IH), 3.52 (q, J = 7.2 Hz, IH), 2.50 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H).
To the solution of N-ethyl-5-iodo-6-methyl-2-(methylthio)pyrimin-4-amine (25.2 g, 81.48 mmol) in DMA (260 mL) were added ethyl acrylate (12.23 g, 122.2 mmol), Pd(OAc)2 (3.65 g, 16.25 mmol), (+)BINAP and triethyl amine (24.68 g, 244.4 mmol). Then the reaction mixture was heated to 100 0C and reacted overnight. After evaporation of solvent, the residue was diluted with water and the aqueous layer was extracted with ethyl acetate. The product (E)-ethyl-3-(4-(ethylamino)-6-methyl-2-(methylthio)pyrimidin-5- yl)acrylate (16.8 g, 73% yield) was isolated by silica gel column chromatography with 6-8% ethyl acetate in hexane as eluent. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 16.4Hz, IH), 6.20 (d, J = 16.4Hz, IH), 5.15 (bs, IH), 4.28(q, J = 7.2 Hz, 2H), 3.54 (q, J = 7.2 Hz, 2H), 2.53 (s, 3H), 2.37 (s, 3H), 1.35 (t, J = 7.2 Hz, 3H), 1.24 (t, J = 7.2 Hz, 3H).
To a solution of (E)-ethyl-3-(4-(ethylamino)-6-methyl-2-(methylthio)pyrimidin- 5-yl)acrylate (16.8 g, 59.8 mmol) in DIPEA was added l,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 18.21 g, 119.6 mmol) at room temperature. Then the reaction mixture was heated to reflux and reacted for 15 h. After evaporation of solvent, the residue was triturated with acetone. The product 8-ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (10.77 g, 77% yield) was collected by filtration. 1H NMR (400 MHz, CDCl3): δ 7.78 (d, J = 9.6 Hz, IH), 6.63 (d, J = 9.6 Hz5 IH), 4.5(q, J = 7.2 Hz, 2H), 2.67 (s, 3H), 2.62 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H).
[00187] To a solution of 8-ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)- one (6.31 g, 26.84 mmol) in DCM was added Br2 (4.79 g, 29.52 mmol) dropwise at room temperature. Then the reaction mixture was stirred at room temperature overnight. After filtration the solid was suspended in DCM (100 mL), and triethylamine (20 mL) was added. The mixture was washed with water and dried with Na2SO4, and the product 6-bromo-8- ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (6.96 g, 83 % yield) was obtained after evaporation of DCM. 1H NMR (400 MHz, CDCl3): δ 8.22 (s, IH), 4.56 (q, J = 7.2 Hz, 2H), 2.68 (s, 3H), 2.62 (s, 3H), 1.34 (t, J = 7.2Hz, 3H).
To a solution of 6-bromo-8-ethyl-4-methyl-2-(methylthio)ρyrido[2,3- d]pyrimidin-7(8H)-one (0.765 g, 2.43 mmol) in DME-H2O (10:1 11 mL) was added IH- pyrazol-5-ylboronic acid (Frontier, 0.408 g, 3.65 mmol), [1,1′- bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with CH2Cl2 (Pd(dρρρf),0.198 g, 0.243 mmol) and triethylamine (0.736 g, 7.29 mmol) at room temperature. Then the reaction mixture was heated to reflux and reacted for 4 h. After cooling down to room temperature, the reaction mixture was partitioned with water and ethyl acetate. After separation, the. organic layer was dried with Na2SO4, and the product 8- ethyl-4-methyl-2-(methylthio)-6-(lH-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one (0.567 g, 77% yield) was obtained by silica gel column chromatography. 1H NMR (400 MHz, CDCl3): δ 13.3 (bs, IH), 8.54 (s, IH), 7.82-7.07 (m, 2H), 4.45 (q, J = 7.2 Hz, 2H), 2.71 (s, 3H), 2.60 (s, 3H), 1.26 (t, J = 7.2Hz, 3H).
To the solution of 8-ethyl-4-methyl-2-(methylthio)-6-(lH-pyrazol-5- yl)pyrido[2,3-d]pyrimidin-7(8H)-one (0.123 g, 0.41mmol) in DCM (2 mL) was added MCPBA (0.176 g, 77%, 0.785 mmol) in a small portion at room temperature. Then the reaction mixture was stirred for 4 h. After evaporation of DCM, dioxane (1 mL) and liquid ammonia (1 mL) were introduced. The reaction was stirred at room temperature overnight. The product 2-amino-8-ethyl-4-methyl-6-(lH-pyrazol-5-yl)pyrido[2,3-(/lpyrimidin-7(8H)- one (50.4 mg) was obtained by silica gel column chromatography. 1H NMR (400 MHz, CD3OD): δ 8.41 (s, IH), 7.62 (d, J – 2.0 Hz, IH), 6.96 (d, J = 2.0Hz5 IH), 4.51 (q, J = 7.2Hz, 2H), 2.64 (s, 3H), 1.29 (t, J = 7.2Hz, 3H); MS (EI) for C13H14N6O: 271.3 (MH+)
References:
1. P. W. Yu, et al., Characterization of the Activity of the PI3K/mTOR Inhibitor XL765 (SAR245409) in Tumor Models with Diverse Genetic Alterations Affecting the PI3K Pathway, Mol Cancer Ther, May 2014 13; 1078-91
2. K. P. Papadopoulos, et al., Phase I Safety, Pharmacokinetic, and Pharmacodynamic Study of SAR245409 (XL765), a Novel, Orally Administered PI3K/mTOR Inhibitor in Patients with Advanced Solid Tumors, Clin Cancer Res, May 1, 2014 20; 2445

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











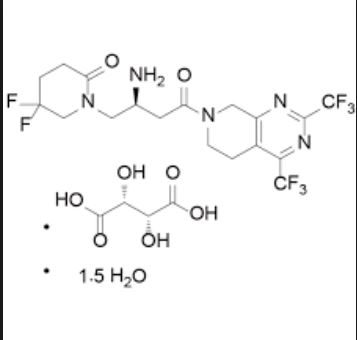








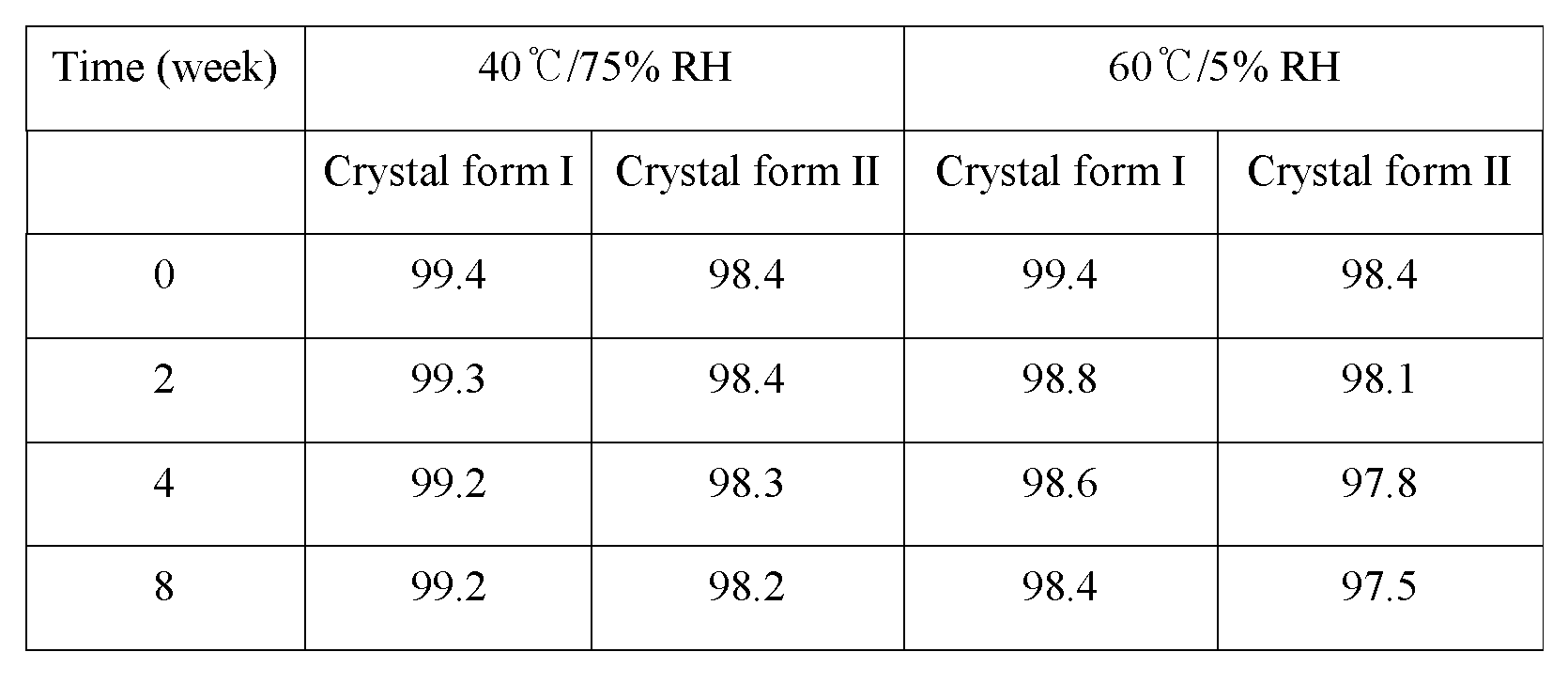





















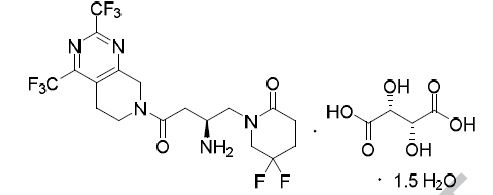




.svg)



 LIONEL MY SON
LIONEL MY SON
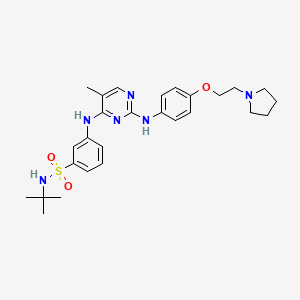
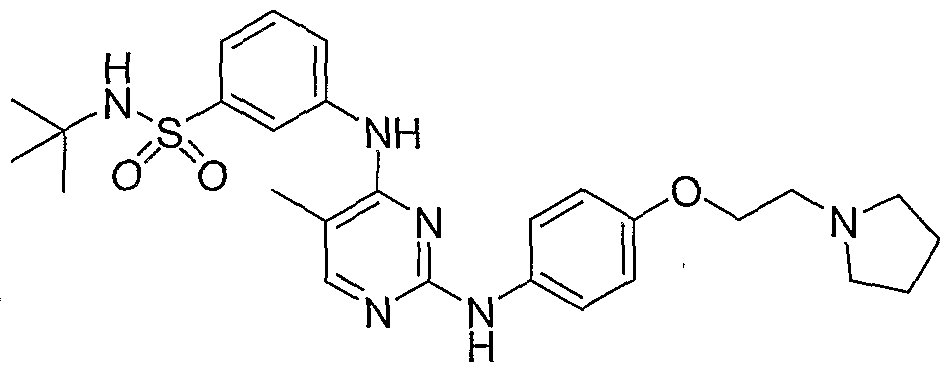












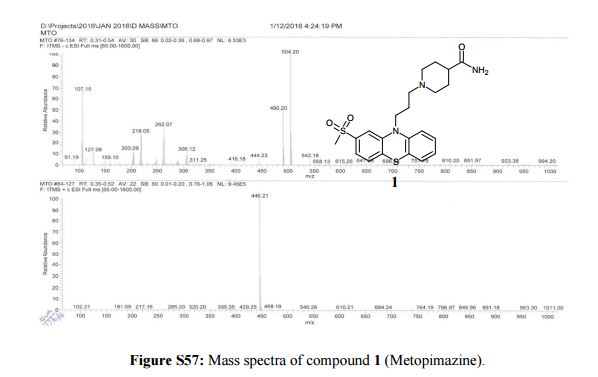
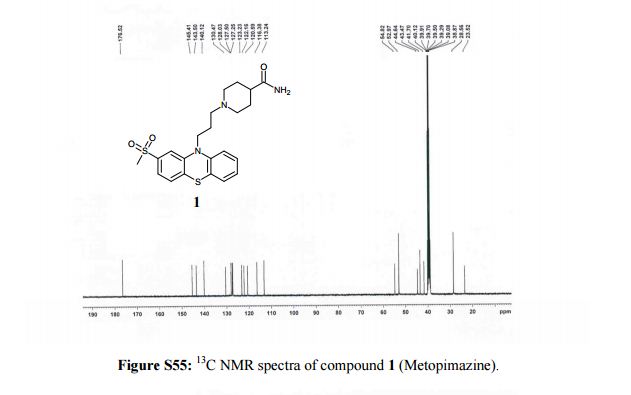
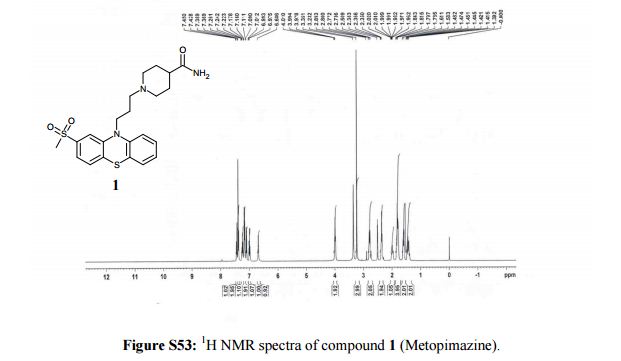












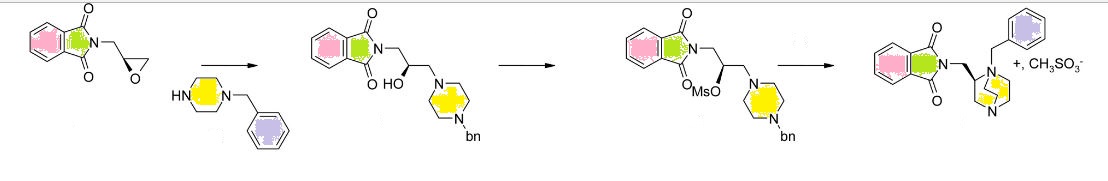







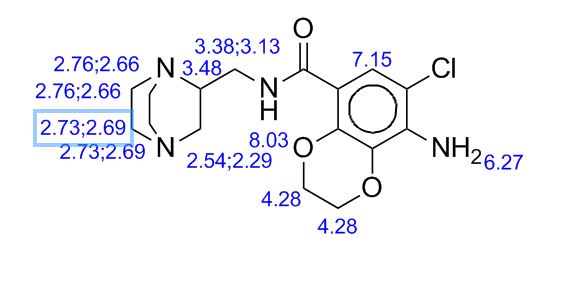
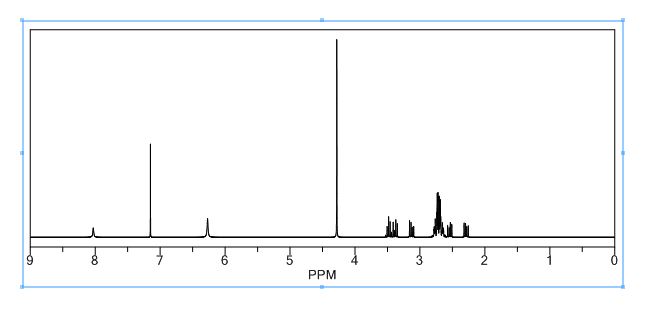
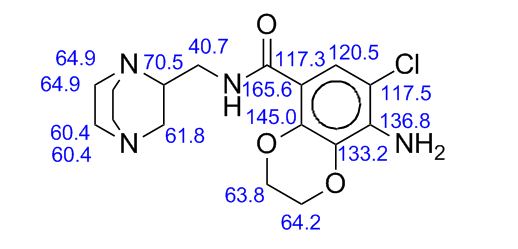
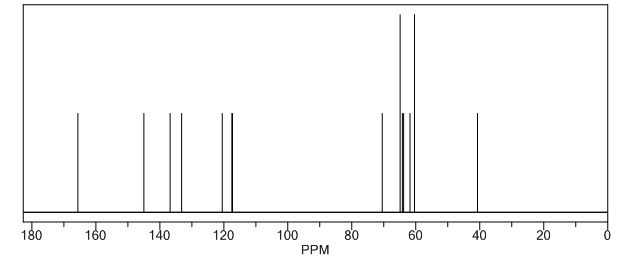



































{kind=link}
{kind=link}