

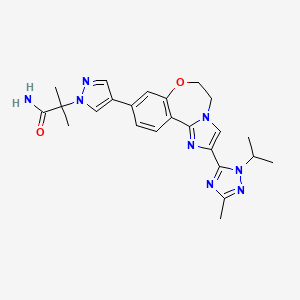
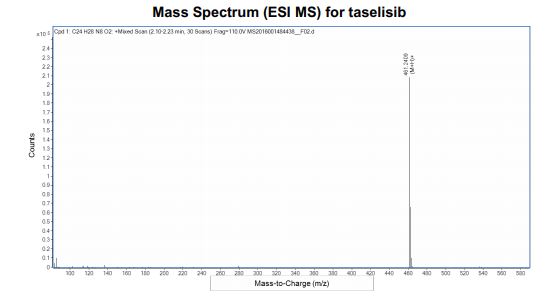
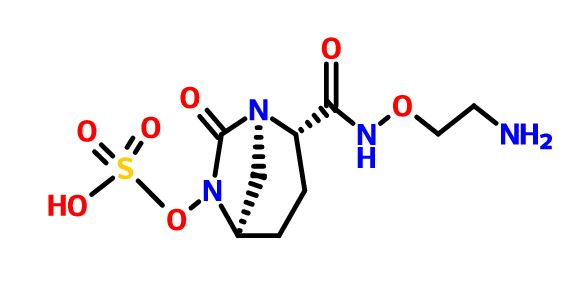
- Molecular FormulaC24H28N8O2
- Average mass460.531 Da
RG7604,Taselisib
GDC-0032, GDC0032;GDC 0032, RO5537381
1H-Pyrazole-1-acetamide, 4-[5,6-dihydro-2-[3-methyl-1-(1-methylethyl)-1H-1,2,4-triazol-5-yl]imidazo[1,2-d][1,4]benzoxazepin-9-yl]-α,α-dimethyl-
UNII:L08J2O299M
10.1021/jm4003632
2-(4-(2-(1-isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide
2-{3-[2-(1-Isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide
POLYMORPHS almost A to Z, US9266903
Taselisib (GDC-0032) is an experimental cancer drug in development by Roche. Molecule is a complex heterocycle with no chiral centres, hazardous materials are used in synthesis, preparation of impurities is a challenge. Taselisib is in phase III with Roche , clinical trials for treatment of metastatic breast cancer and non-small cell lung cancer
Taselisib (GDC-0032) is an experimental cancer drug in development by Roche. It is a small molecule inhibitor targeting phosphoinositide 3-kinase subtype PIK3CA.[1]
Taselisib is in phase III with Roche , clinical trials for treatment of metastatic breast cancer and non-small cell lung cancer.[2]
Taselisib is a phosphatidylinositol 3-kinase (PI3Kalpha) inhibitor in phase III clinical studies at Roche for the treatment of postmenopausal women with histologically or cytologically confirmed locally advanced or metastatic estrogen-receptor positive (ER+) breast cancer.
Taselisib is an orally bioavailable inhibitor of the class I phosphatidylinositol 3-kinase (PI3K) alpha isoform (PIK3CA), with potential antineoplastic activity. Taselisib selectively inhibits PIK3CA and its mutant forms in the PI3K/Akt/mTOR pathway, which may result in tumor cell apoptosis and growth inhibition in PIK3CA-expressing tumor cells. By specifically targeting class I PI3K alpha, this agent may be more efficacious and less toxic than pan PI3K inhibitors. Dysregulation of the PI3K/Akt/mTOR pathway is frequently found in solid tumors and causes increased tumor cell growth, survival, and resistance to both chemotherapy and radiotherapy. PIK3CA, which encodes the p110-alpha catalytic subunit of the class I PI3K, is mutated in a variety of cancer cell types and plays a key role in cancer cell growth and invasion.

PRODUCT PATENT
WO 2011036280
| Inventors |
Nicole Blaquiere, Steven Do, Danette Dudley, Adrian J. Folkes, Robert Heald, Timothy Heffron, Mark Jones, Aleksandr Kolesnikov, Chudi Ndubaku, Alan G. Olivero, Stephen Price, Steven Staben, Lan Wang, Less « |
| Applicant |
F. Hoffmann-La Roche Ag |
https://encrypted.google.com/patents/WO2011036280A1?cl=en
Discovery of 2-(3-(2-(1-Isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo(f)imidazo(1,2-d)(1,4)oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide (GDC-0032): A -sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity
J Med Chem 2013, 56(11): 4597

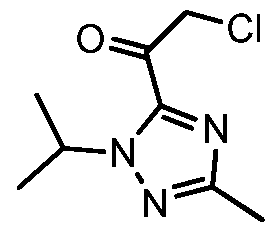
Condensation of 4-bromo-2-hydroxybenzaldehyde with glyoxal in the presence of NH3 in MeOH gives 5-bromo-2-(1H-imidazol-2-yl)phenol
Which upon annulation with 1,2-dibromoethane in the presence of Cs2CO3 in DMF at 90 °C yields 9-bromo-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepine .
Iodination of oxazepine with NIS in DMF provides 9-bromo-2,3-diiodo-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepine,
Which upon mono-deiodination by means of EtMgBr in THF at -15 °C affords 9-bromo-2-iodo-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepine .
Amidation of iodide with CO in the presence of PdCl2(PPh3)2 and HMDS in DMF at 70 °C produces the intermediate,
Which upon reaction with N,N-dimethylacetamide dimethyl acetal in the presence of DME at 65 °C furnishes intermediate . Intramolecular cyclization of this compound with isopropylamine hydrochloride in AcOH generates triazole derivative,
Which upon Suzuki coupling with dioxaborolane derivative in the presence of Pd(PPh3)4 and KOAc in CH3CN/H2O at 120 °C yields the target compound Taselisib.

Taselisib has been used in trials studying the treatment and basic science of LYMPHOMA, Breast Cancer, Ovarian Cancer, Solid Neoplasm, and HER2/Neu Negative, among others.
Solubility (25°C)
| In vitro |
DMSO |
70 mg/mL warmed (151.99 mM) |
| Water |
Insoluble |
| Ethanol |
Insoluble warmed |
Biological Activity
| Description |
Taselisib (GDC 0032) is a potent, next-generation β isoform-sparing PI3K inhibitor targeting PI3Kα/δ/γ with Ki of 0.29 nM/0.12 nM/0.97nM, >10 fold selective over PI3Kβ. |
| Features |
A beta isoform-sparing PI3K inhibitor. |
| Targets |
PI3Kδ [1]
(Cell-free assay) |
PI3Kα [1]
(Cell-free assay) |
PI3Kγ [1]
(Cell-free assay) |
PI3Kβ [1]
(Cell-free assay) |
C2β [1]
(Cell-free assay) |
View More |
| 0.12 nM(Ki) |
0.29 nM(Ki) |
0.97 nM(Ki) |
9.1 nM(Ki) |
292 nM |
|
| In vitro |
GDC-0032 is an orally bioavailable, potent, and selective inhibitor of Class I PI3Kα, δ, and γ isoforms, with 30 fold less inhibition of the PI3K β isoform relative to the PI3Kα isoform. Preclinical data show that GDC-0032 has increased activity against PI3Kα isoform (PIK3CA) mutant and HER2-amplified cancer cell lines. GDC-0032 inhibits MCF7-neo/HER2 cells proliferation with IC50 of 2.5 nM. [1] |
| Cell Data |
|
|
| Cell Lines |
Assay Type |
Concentration |
Incubation Time |
Formulation |
Activity Description |
PMID |
| human MOLM16 cells |
Proliferation assay |
|
72 h |
|
Antiproliferative activity against human MOLM16 cells after 72 hrs by Cell Titer-Blue assay |
22727640 |
|
|
| In vivo |
GDC-0032 pharmacokinetics is approximately dose proportional and time independent with a mean t1/2 of 40 hours. The combination of GDC-0032 enhances activity of fulvestrant resulting in tumor regressions and tumor growth delay (91% tumor growth inhibition (TGI)). In addition, the combination of GDC-0032 with tamoxifen enhances the efficacy of tamoxifen in vivo (102%TGI for GDC-0032). [1] |
PATENT
WO 2014140073
The invention relates to methods of making the PI3K inhibitor I (GDC-0032), named as 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanamide, having the structure:
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof.
Another aspect of the invention includes novel intermediates useful for preparing GDC- 0032 and having the structures:


The following Schemes 1-15 illustrate the chemical reactions, processes, methodology for the synthesis of GDC-0032, Formula I, and certain intermediates and reagents. Scheme 1:
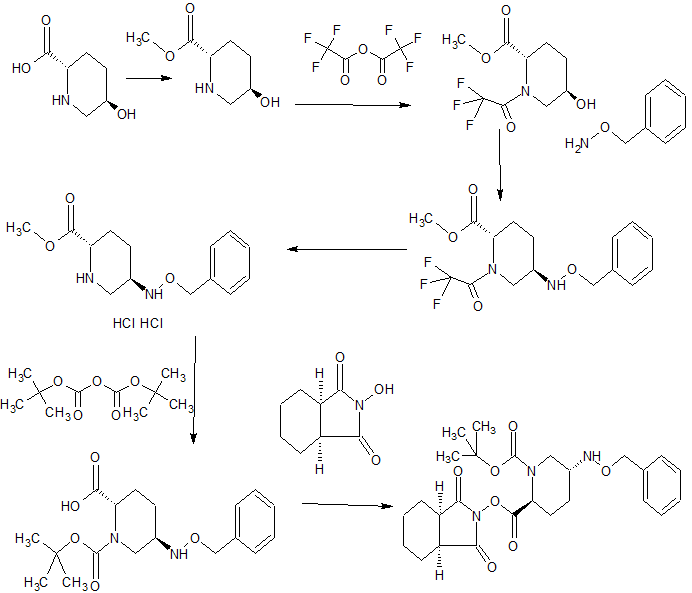
Scheme 1 shows the synthesis of intermediate isopropylhydrazine hydrochloride 4 from Boc-hydrazine 1. Condensation of 1 with acetone and magnesium sulfate gave Boc-hydrazone, tert-butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2 (Example 1). Palladium-catalyzed hydrogenation of 2 in acetic acid and methanol gave Boc-isopropyl-hydrazine 3 (Example 2) which was treated in situ with hydrogen chloride gas to give 4 (Example 3).
Alternatively, the double bond of 2 can be reduced with a hydride reagent such as sodium cyanoborohydride (Example 2).
Scheme 2:
Scheme 2 shows the synthesis of l-isopropyl-3-methyl-lH-l,2,4-triazole 7 from methyl acetimidate hydrochloride 5 and isopropylhydrazine hydrochloride 4. Reaction of 5 and 4 in triethylamine and methanol followed by cyclization of condensation product, N’- isopropylacetohydrazonamide 6 (Example 4) with triethyl orthoformate (triethoxymethane) gave 7 (Example 5). Alternatively, 4 and acetamidine can be reacted to give 6.
Or, 4 can be reacted with acetonitrile and an acid to form the corresponding salt of 6. Scheme 3:
0 K2C03, H20, MTBE w
Scheme 3 shows the synthesis of intermediate, 2-chloro-N-methoxy-N-methylacetamide 10. Reaction of 2-chloroacetyl chloride 8 and Ν,Ο-dimethylhydroxylamine hydrochloride 9 in aqueous potassium carbonate and methyl, tert-butyl ether (MTBE) gave 10 (Example 6).
Scheme 4:
Scheme 4 shows the synthesis of intermediate 4-bromo-2-fluorobenzimidamide hydrochloride 12 formed by reaction of 4-bromo-2-fluorobenzonitrile 11 with lithium hexamethyldisilazide (LiHMDS) in tetrahydrofuran (Example 7). Alternatively, 11 is treated with hydrogen chloride in an alcohol, such as ethanol, to form the imidate, ethyl 4-bromo-2- fluorobenzimidate hydrochloride, followed by ammonia in an alcohol, such as ethanol, to form 12 (Example 7).
Scheme 5:
Scheme 5 shows the synthesis of 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l,2,4-triazole V from l-isopropyl-3-methyl-lH-l,2,4-triazole 7.
Deprotonation of 7 with n-butyllithium and acylation with 2-chloro-N-methoxy-N- methylacetamide 10 gave intermediate 2-chloro-l-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)ethanone 13 (Example 8). Cyclization of 13 with 4-bromo-2-fluorobenzimidamide hydrochloride 12 and potassium hydrogen carbonate in water and THF (tetrahydrofuran) formed the imidazole V (Example 9).
Scheme 6:
Scheme 6 shows the synthesis of 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III from V. Alkylation of the imidazole nitrogen of V with a 2-hydroxyethylation reagent such as, l,3-dioxolan-2-one, gave 2-(2-(4- bromo-2-fluorophenyl)-4-( 1 -isopropyl-3-methyl- 1 H- 1 ,2,4-triazol-5 -yl)- 1 H-imidazol- 1 – yl)ethanol 14 (Example 10). Cyclization of 14 with an aqueous basic reagent, such as methyltributylammonium chloride in aqueous potassium hydroxide, gave III, which can be cystallized from ethanol and water (Example 11). Scheme 7:
IV
Scheme 7 shows the synthesis of ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2-methylpropanoate IV starting from 2-bromo-2-methylpropanoic acid 15. Alkylation of pyrazole with 15 gave 2- methyl-2-(lH-pyrazol-l-yl)propanoic acid 16 (Example 12). Esterification of 16 with sulfuric acid in ethanol gave ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17 (Example 13).
Regiospecific bromination of 17 with N-bromosuccinimide (NBS) gave IV (Example 14). Alternatively, 16 was treated in situ with a brominating reagent such as l,3-dibromo-5,5- dimethylhydantoin (DBDMH) to give 2-(4-bromo-lH-pyrazol-l-yl)-2-methylpropanoic acid which was esterified to give IV, where R is ethyl. Other esters can also be prepared, such as methyl, iso-propyl, or any alkyl, benzyl or aryl ester.
Scheme 8:
Scheme 8 shows an alternative synthesis of ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2- methylpropanoate IV starting from ethyl 2-bromo-2-methylpropanoate 18. Alkylation of pyrazole with 18 in the presence of a base such as sodium tert-butyloxide or cesium carbonate gave a mixture of ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17 and ethyl 2-methyl-3-(lH- pyrazol-l-yl)propanoate 19. Bromination of the mixture with l,3-dibromo-5,5- dimethylimidazolidine-2,4-dione (DBDMH) gave a mixture containing IV, ethyl 3-(4-bromo- lH-pyrazol-l-yl)-2-methylpropanoate 20, and 4-bromo-lH-pyrazole 21 which was treated with a strong base under anhydrous conditions, such as lithium hexamethyldisilazide in tetrahydrofuran. Acidification with hydrochloric acid gave IV.
Scheme 9:
Pd(O) catalyst
KOAc, EtOH

Scheme 9 shows the synthesis of 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)- 5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanamide, GDC-0032, 1 from ethyl 2-(4-bromo- 1 H-pyrazol- 1 -yl)-2-methylpropanoate IV (CAS Registry Number: 1040377-17-0, WO 2008/088881) and 9-bromo-2-(l-isopropyl-3-methyl-lH- 1,2,4- triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III (CAS Registry Number: 1282514-63-9, US 2012/0245144, US 8242104). Other esters besides ethyl can also be used which can be hydrolyzed with aqueous base, such as methyl, iso-propyl, or any alkyl, benzyl or aryl ester. In a one-pot Miyaura Borylation /Suzuki, Buchwald system, ethyl 2-(4-bromo-lH- pyrazol-l-yl)-2-methylpropanoate IV is reacted with 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(l,3,2- dioxaborolane), CAS Reg. No. 73183-34-3, also referred to as B2Pin2, and a palladium catalyst such as XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, CAS Reg. No. 564483- 18-7), with a salt such as potassium acetate, in a solvent such as ethanol, at about 75 °C to form the intermediate ethyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol- l-yl)propanoate 22 (Example 15, CAS Registry Number: 1201657-32-0, US 8242104, US 8263633, WO 2009/150240).

XPhos ligandIntermediate 22 can be isolated or reacted in situ (one pot) with III to form 23.
A variety of low valent, Pd(II) and Pd(0) palladium catalysts can be used during the Suzuki coupling step to form 23 (Example 16) from 22 and III, including PdCl2(PPh3)2, Pd(t- Bu)3, PdCl2 dppf CH2C12, Pd(PPh3)4, Pd(OAc)/PPh3, Cl2Pd[(Pet3)]2, Pd(DIPHOS)2, Cl2Pd(Bipy), [PdCl(Ph2PCH2PPh2)]2, Cl2Pd[P(o-tol)3]2, Pd2(dba)3/P(o-tol)3, Pd2(dba)/P(furyl)3,
Cl2Pd[P(furyl)3]2, Cl2Pd(PMePh2)2, Cl2Pd[P(4-F-Ph)3]2, Cl2Pd[P(C6F6)3]2, Cl2Pd[P(2-COOH- Ph)(Ph)2]2, Cl2Pd[P(4-COOH-Ph)(Ph)2]2, and encapsulated catalysts Pd EnCat™ 30, Pd EnCat™ TPP30, and Pd(II)EnCat™ BINAP30 (US 2004/0254066).
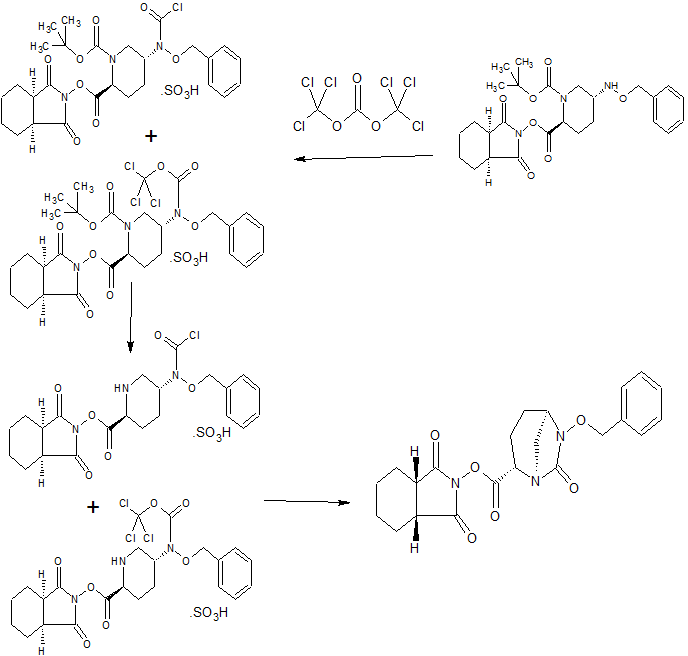
The ester group of 23 is saponified with an aqueous basic reagent such as lithium hydroxide, to give 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- IH-pyrazol- 1 -yl)-2-methylpropanoic acid II (Example 17). Intermediate 23 can be isolated or further reacted in situ with the aqueous basic reagent to form II. The carboxylic acid group of II is activated with an acyl activating reagent such as di(lH-imidazol-l-yl)methanone (carbonyl diimidazole, CDI) or Ν,Ν,Ν’,Ν’-tetramethyl- 0-(7-azabenzotriazol-l-yl)uronium hexafluorophosphate (HATU), and then reacted with an alcoholic ammonia reagent, such as ammonia dissolved in methanol, ethanol, or isopropanol, aqueous ammonium hydroxide, aqueous ammonium chloride, or ammonia dissolved in THF, to give I (Example 18).
A variety of solid adsorbent palladium scavengers can be used to remove palladium after the Suzuki coupling step to form compound I. Exemplary embodiments of palladium scavengers include FLORISIL®, SILIABOND®Thiol, and SILIABOND® Thiourea. Other palladium scavengers include silica gel, controlled-pore glass (TosoHaas), and derivatized low crosslinked polystyrene QUADRAPURE™ AEA, QUADRAPURE™ IMDAZ, QUADRAPURE™ MPA, QUADRAPURE™ TU (Reaxa Ltd., Sigma-Aldrich Chemical Co.).
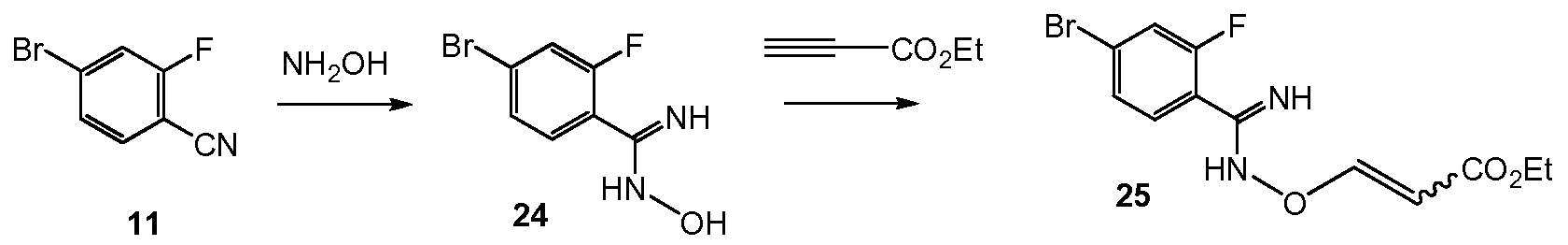


Scheme 10 shows the synthesis of 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III from 4-bromo-2-fluorobenzonitrile 11. Addition of hydroxylamine to the nitrile of 11 gave 4-bromo-2-fluoro-N-hydroxybenzimidamide 24. Michael addition of 24 to ethyl propiolate gave ethyl 3-(4-bromo-2- fluorobenzimidamidooxy)acrylate 25. Heating 25 in a high-boiling solvent such as toluene, xylene, ethylbenzene, or diphenyl oxide gave cyclized imidazole, ethyl 2-(4-bromo-2- fluorophenyl)-lH-imidazole-4-carboxylate 26, along with by-product pyrimidine, 2-(4-bromo-2- fluorophenyl)pyrimidin-4-ol. Alternatively, 25 can be cyclized to 26 with catalytic Lewis acids such as Cu(I) or Cu(II) salts. Alkylation of 26 with a 2-hydroxyethylation reagent, such as 1,3- dioxolan-2-one, in a base, such as N-methylimidazole or cesium carbonate, gave ethyl 2-(4- bromo-2-fluorophenyl)-l-(2-hydroxyethyl)-lH-imidazole-4-carboxylate 27. Ring-cyclization of 27 with an aqueous basic reagent, such as potassium hydroxide, lithium hydroxide, and methyl tributylammonium hydrochloride, gave 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepine-2-carboxylic acid 28. Addition of acetamidine to 28 with triphenylphosphine gave 9-bromo-N-(l-iminoethyl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2- carboxamide 29. Ring-cyclization of 29 with isopropylhydrazine hydrochloride 4 in acetic acid gave 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepine III.
Alternatively, 28 can be reacted with N’-isopropylacetohydrazonamide 6 to give III (Scheme 12).
Scheme 11 :
Scheme 11 shows the synthesis of 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l ,2,4-triazole V from 4-bromo-2-fluorobenzimidamide hydrochloride 12. 3-Chloro-2-oxopropanoic acid and 12 are reacted with base to give 2-(4-bromo-2-fluorophenyl)- lH-imidazole-4-carboxylic acid 30. Alternatively, 3-bromo-2-oxopropanoic acid can be reacted with 12 to give 30. Reaction of 30 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU (N,N,N’,N’-tetramethyl-0-(lH-benzotriazol-l-yl)uronium hexafluorophosphate, O- (Benzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate, CAS Ref. No. 94790- 37-1) in DMF gives intermediate, 2-(4-bromo-2-fluorophenyl)-N-(l-(2- isopropylhydrazinyl)ethylidene)-lH-imidazole-4-carboxamide 31 which need not be isolated and cyclizes upon heating to give V.
Alternatively, 5-(2-(4-chloro-2-fluorophenyl)-lH-imidazol-4-yl)-l-isopropyl-3-methyl- lH-l,2,4-triazole 44, the chloro version of V, can be prepared from 4-chloro-2-fluorobenzonitrile 38 (Scheme 15) Scheme 12:
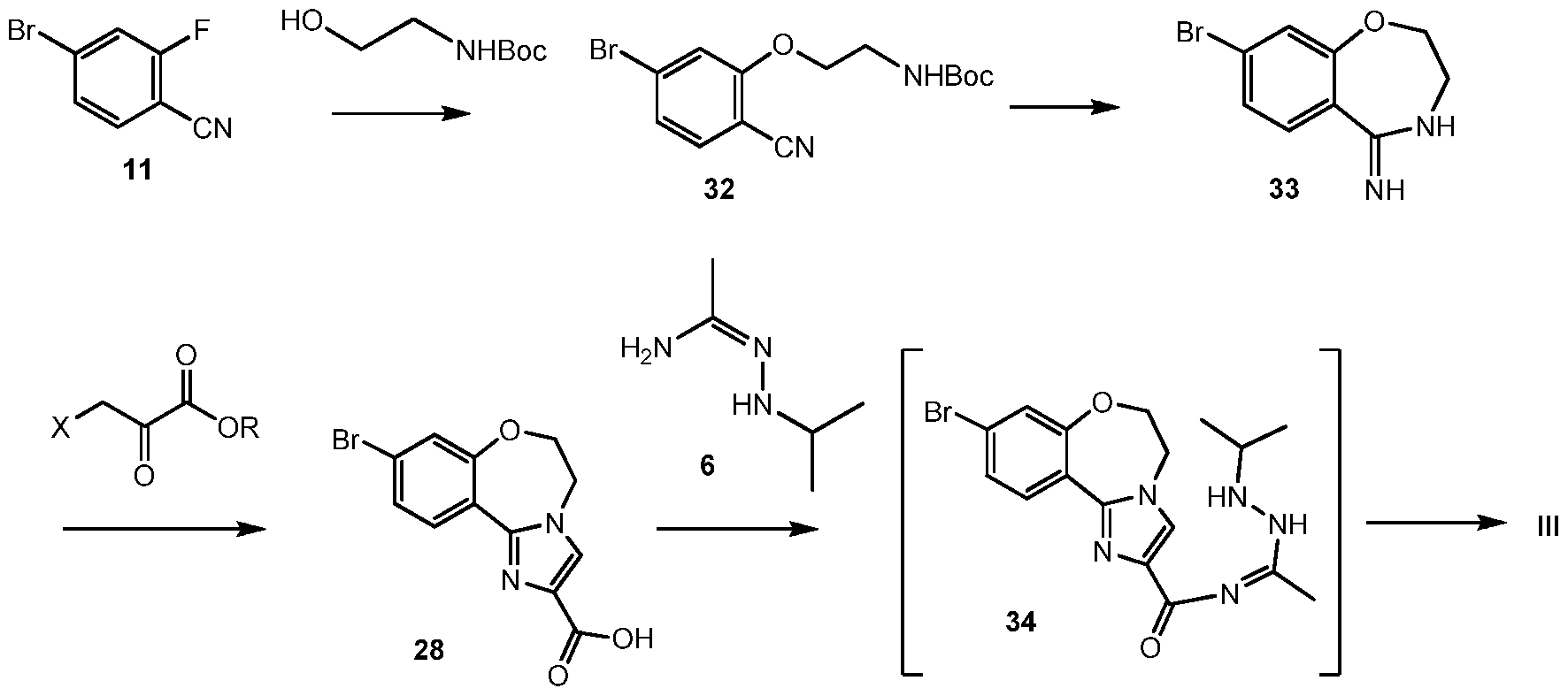
Scheme 12 shows an alternative synthesis of 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4- triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III from 4-bromo-2- fluorobenzonitrile 11. Alkylation of 11 with tert-butyl 2-hydroxyethylcarbamate gives tert-butyl 2-(5-bromo-2-cyanophenoxy)ethylcarbamate 32. Cyclization of 32 under acidic conditions, such as hydrochloric acid in ethanol, gives 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33. It will be noted that 33 has an alternative tautomeric form where the double bond is inside the oxazepine ring. Formation of the imidazole ring occurs by reaction of 3-bromo-2- oxopropanoic acid (X = Br, R = OH), or other 3-halo-2-oxopropanoic acid or ester (R = alkyl), and 33 to give 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28. Coupling of 28 with N’-isopropylacetohydrazonamide 6 and a coupling reagent such as HBTU, HATU or CDI in DMF gives intermediate, 9-bromo-N-(l-(2-isopropylhydrazinyl)ethylidene)- 5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxamide 34, which need not be isolated and forms 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III upon heating.
Alternatively, N’-isopropylacetohydrazonamide 6 is used as monohydrochloride salt, which has to be set free under the reaction conditions with an appropriate base, such as K2CO3. Scheme 13:
Scheme 13 shows an alternative synthesis of 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin- 5(2H)-imine 33 from 4-bromo-2-fluorobenzonitrile 11. Reaction of 11 with sodium methoxide in methanol gives methyl 4-bromo-2-fluorobenzimidate 35. Alkylation of 35 with 2- aminoethanol gives 4-bromo-2-fluoro-N-(2-hydroxyethyl)benzimidamide 36, followed by cyclization to 33.
Scheme 14:
37
11
Scheme 14 shows another alternative synthesis of 8-bromo-3,4- dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33 from 4-bromo-2-fluorobenzonitrile 11. Reaction of 11 with 2-aminoethanol and potassium tert-butoxide displaces fluorine to give 2-(2- aminoethoxy)-4-bromobenzonitrile hydrochloride 37. Ring closure of 37 with
trimethylaluminum gave 33. Alternatively, other trialkylaluminum reagents can be used, or magnesium alkoxide reagents such as magnesium ethoxide (magnesium bisethoxide, CAS Reg. No. 2414-98-4) to cyclize 37 to 33.
Scheme 15 shows the synthesis of 5-(2-(4-chloro-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l,2,4-triazole 44 from 4-chloro-2-fluorobenzonitrile 38. Addition of hydroxylamine to the nitrile of 38 gave 4-chloro-2-fluoro-N-hydroxybenzimidamide 39.
Michael addition of 39 to ethyl propiolate gave ethyl 3-(4-chloro-2- fluorobenzimidamidooxy)acrylate 40. Heating 40 in diphenyl oxide gave cyclized imidazole, ethyl 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylate 41. Saponification of the ester of 41 with aqueous sodium hydroxide in tetrahydrofuran gave 2-(4-chloro-2-fluorophenyl)-lH- imidazole-4-carboxylic acid 42. Reaction of 42 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU in DMF gives intermediate, 2-(4-chloro-2-fluorophenyl)-N-(l-(2- isopropylhydrazinyl)ethylidene)-lH-imidazole-4-carboxamide 43 which cyclizes upon heating to give 44.
EXAMPLES
Example 1 tert-butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2
To a solution of tert-butyl hydrazinecarboxylate 1 (CAS Reg. No. 870-46-2) (25.1 g, 0.190 mol) in acetone (185 mL) was added the magnesium sulfate (6 g) and 12 drops acetic acid (Wu et al (2012) Jour. Med. Chem. 55(6):2724-2736; WO 2007/056170; Zawadzki et al (2003) Polish Jour. Chem. 77(3):315-319). The mixture was heated to reflux for 2.5 h and cooled to rt and filtered. The filtrate was concentrated to give tert-butyl 2-(propan-2- ylidene)hydrazinecarboxylate 2 (CAS Reg. No. 16689-34-2) as an off-white solid (32 g, 98%) (used in the next step without further purification). LC-MS [M+H]+ = 172.9, RT = 2.11 min. 1H NMR 300 MHz (CDC13) d 7.35 (br s, 1H, NH), 2.04 (s, 3H), 1.82 (s, 3H), 1.54 (s, 9H); 13C NMR 300 MHz (CDC13) d 152.9, 149.7, 80.7, 28.1, 25.3, 15.9. Example 2 tert-butyl 2-isopropylhydrazinecarboxylate 3
tert-Butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2 was reduced with palladium catalyst on carbon with hydrogen gas in acetic acid and methanol to give tert-butyl 2- isopropylhydrazinecarboxylate 3 (CAS Reg. No. 16689-35-3).
Alternatively, tert-Butyl 2-(propan-2-ylidene)hydrazinecarboxylate 2 (0.51 g, 3.0 mmol) was dissolved in 20 mL of THF, treated with NaB¾CN (0.19 g, 3.0 mmol) and a few mg of bromocresol green, followed by a solution of p-toluenesulfonic acid (0.57 g, 3.0 mmol) in 1.5 mL of THF which was added dropwise over approximately 1 h to maintain the reaction pH between 3.5-5.0. After stirring at room temperature for an additional hour, the solvent was removed by rotary evaporation, and the residue was partitioned between EtOAc (30 mL) and brine. The organic phase was extracted with sat. NaHCC>3, 20 mL and brine, evaporated to a residue and dissolved in 10 mL of ethanol. The ethanolic solution was treated with 3.6 mL of 1M NaOH solution (3.6 mmol) and left to stir at rt for 30 min. The solvent was removed by rotary evaporation and the residue was taken up into ethyl acetate and extracted with water. The organic layer was evaporated under reduced pressure and the residue was purified by column chromatography using 5 % MeOH in DCM as eluent to collect tert-butyl 2- isopropylhydrazinecarboxylate 3 (0.4 g, 77 % yield): mp = 47-49 °C; Rf = 0.44 (5 % MeOH in DCM); IH NMR 300 MHz (CDC13) d 6.03 (s, N-H, IH), 3.92 (s, N-H, IH), 3.14 (m, IH), 1.46 (s, 9H), 1.02 (d, 6H, J = 6 Hz); 13C NMR 300 MHz (CDC13) d 157.2, 80.8, 51.2, 28.7, 21.0.
Example 3 isopropylhydrazine hydrochloride 4
tert-butyl 2-isopropylhydrazinecarboxylate 3 was treated with hydrochloric acid to remove the Boc protecting group and give 4 (CAS Reg. No. 16726-41-3).
Example 4 N’-isopropylacetohydrazonamide 6
Methyl acetimidate hydrochloride 5 (CAS Reg. No. 14777-27-6), isopropylhydrazine hydrochloride 4, and triethylamine were reacted in methanol to give 6 (CAS Reg. No. 73479-06- 8).
Example 5 l-isopropyl-3 -methyl- lH-l,2,4-triazole 7
N’-isopropylacetohydrazonamide 6 was treated with triethylorthoformate in ethanol, followed by triethylamine and tetrahydrofuran to give 7 (CAS Reg. No. 1401305-30-3). Example 6 2-chloro-N-methoxy-N-methylacetamide 10
To a solution of 21.2 kg potassium carbonate K2CO3 (153.7 mol, 3.0 eq) in 30 L H20 was added, Ν,Ο-dimethylhydroxylamine 9 (CAS Reg. No. 1117-97-1) (5.0 kg, 51.3 mol, 1.0 eq) at 15-20 °C. The reaction was stirred at rt for 30min and 30 L methyl tert-butyl ether (TBME) was added. After stirred for 30min, the mixture was cooled to 5°C, and 11.6 kg of 2-Chloroacetyl chloride 8 (CAS Reg. No. 79-04-9 (102.7 mol, 2.0 eq) were added slowly. The reaction was stirred at rt overnight. Organics were separated from aqueous, and aqueous was extracted with TBME (30 L). The combined organics were washed with H20 (50 L), brine (50 L) and dried over Na2S04. Filtered and concentrated under vacuum afforded 5.1 kg of 2-chloro-N-methoxy- N-methylacetamide 10 (CAS Reg. No. 67442-07-3) as a white solid.
Example 7 4-bromo-2-fluorobenzimidamide hydrochloride 12
To 35.0 L of lithium hexamethyldisilazide LiHMDS (35.0 mol, 1.4 eq, 1.0 M in THF) under N2 was added a THF solution of 4-Bromo-2-fluorobenzonitrile 11 (CAS Reg. No. 105942- 08-3) (5.0 kg in 10 L THF) at 10 °C, the mixture was stirred at rt for 3h. Cooled to -20°C and 8.3 L of HCl-EtOH (6.6 M) were added. The mixture was stirred at -10 °C for additional lh, filtered. The wet cake was washed with EA (10 L) and H20 (6 L). Drying in vacuo yielded 5.8 kg 4- bromo-2-fluorobenzimidamide hydrochloride 12 (CAS Reg. No. 1187927-25-8) as an off-white solid.
Alternatively, to a 200-L vessel was charged 4-bromo-2-fluorobenzonitrile 11 (10 kg, 50.00 mol, 1.00 equiv) and ethanol (100 L) followed by purging 40 kg Hydrogen chloride (g) at – 10 °C with stirring (Scheme 4). The resulting solution was allowed to react for an additional 36 h at 10 °C. The reaction progress was monitored by TLC until 11 was consumed completely. The resulting mixture was concentrated under vacuum while maintaining the temperature below 60 °C. The volume was concentrated to 10-15 L before 60 L MTBE was added to precipitate the product. The precipitates were collected by filtration to afford in 12 k g of ethyl 4-bromo-2- fluorobenzimidate hydrochloride 12 as a white solid. (Yield: 85%). 1H NMR δ 7.88-7.67 (m), 4.89 (br s), 4.68 (q), 3.33 (m), 1.61 (t). MS M+l: 245.9, 248.0.
To a 200L vessel, was charged ethyl 4-bromo-2-fluorobenzimidate hydrochloride (12.5 k g, 44mol, 1.00 equiv, 99%) and ethanol (125 L) followed by purging NH3 (g) at -5 °C for 12 h. The resulting solution was stirred at 30 °C for an additional 24 h. The reaction progress was monitored by TLC until SM was consumed completely. The precipitates were filtered and the filtrate was concentrated under vacuum. The product was precipitated and collected by filtration to afford 6.1 kg (54.5%) of 4-bromo-2-fluorobenzamidine hydrochloride 12 as a white solid. 1H NMR δ 9.60 (br), 7.91-7.64 (m), 3.40 (s), 2.50 (m). MS M+l: 216.9, 219.9.
Example 8 2-chloro-l-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)ethanone 13
To a 10L four necked flask was charged l-Isopropyl-3-methyl-lH-l,2,4-triazole 7 (400 g) in THF (2.5 L). The resulting solution was cooled to -40 °C and 2.5 M n-butyllithium BuLi in n- hexanes (1.41 L) was added while keeping the internal temp, below -20°C. The resulting yellow suspension was stirred at -40°C for 1 hour before being transferred. To a 20L flask was charged 2-chloro-N-methoxy-N-methylacetamide 10 (485 g) in THF (4 L). The resulting solution was cooled to -40 °C at which point a white suspension was obtained, and to this was added the solution of lithiated triazole 7 keeping the internal temp, below -20°C. At this point a yellow orange solution was obtained which was stirred at – 30°C for lhour. Propionic acid (520 mL) was added keeping the internal temp, below -20°C. The resulting off-white to yellowish suspension was warmed to -5 °C over 30 minutes. Citric acid (200 g) in water (0.8 L) was added and after stirring for 5 minutes a clear biphasic mixture was obtained. At this point stirring was stopped and the bottom aqueous layer was removed. The organic phase was washed with 20w% K3PO4 solution (1 L), 20w% K2HP04 solution (2 L), and 20w% NaCl solution (1 L). The organics was reduced to ca 4L via distillation under vacuum to afford 2-chloro-l-(l-isopropyl-3- methyl-lH-l,2,4-triazol-5-yl)ethanone 13 as a dark amber liquid which was used “as is” in the next step.
Example 9 5-(2-(4-bromo-2-fluorophenyl)- lH-imidazol-4-yl)-l -isopropyl-3-methyl- lH-l,2,4-triazole V
To a 10 L four- neck flask were charged with THF (5.6 L), 4-bromo-2- fluorobenzimidamide hydrochloride 12 (567 g), KHCO3 (567 g) and water (1.15 L). The resulting white suspension was heated to 60°C over 2 hours. At this point a hazy solution was obtained to which was added a solution of 2-Chloro-l-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5- yl)ethanone 13 in THF (2 L). This solution was stirred at 60-65 °C for 24 hours. Then the aqueous bottom layer was removed. The organic layer was concentrated under vacuum. The residue was slurried in a mixture of MIBK (1.25 L) and toluene (0.7 L), and the precipitated product was filtered giving 552 g of 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l- isopropyl-3 -methyl- lH-l,2,4-triazole V (98.0% purity, 254 nm) as a brown solid Example 10 2-(2-(4-bromo-2-fluorophenyl)-4-(l-isopropyl-3-methyl-lH-l,2,4-triazol- 5-yl)- 1 H-imidazol- 1 -yl)ethanol 14
5-(2-(4-Bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l-isopropyl-3-methyl-lH- 1,2,4- triazole V (2.75 kg, 7.55 mol) was added to a solution of 3-dioxolan-2-one (ethylene carbonate, 3.99 kg, 45.3 mol) inN-methylimidazole (12 L) at 50 °C. The suspension was heated at 80°C for 7 h until the reaction was judged complete by HPLC. The solution of 14 was cooled to 35 °C and used directly in the subsequent cyclization.
Example 11 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepine III
To a solution of 2-(2-(4-Bromo-2-fluorophenyl)-4-(l-isopropyl-3-methyl-lH-l,2,4- triazol-5-yl)-l H-imidazol- l-yl)ethanol (7.55 mmol) 14 inN-methylimidazole(12 L) at 35 °C was added methyl tributylammonium chloride (115 g, 0.453 mol), toluene (27.5 L) and 35% potassium hydroxide solution (10.6 kg, 25 mol in 22 L of water). The biphasic solution was stirred vigorously at 65 °C for 18 h when it was judged complete by HPLC. Stirring was stopped but heating was continued and the bottom aqueous layer was removed. Added isopropyl acetate (13.8 L) and the organic phase was washed twice with water (13.8 L and 27.5 L). The solvent was removed via vacuum distillation and after 30 L had been removed, isopropanol (67.6 L) was added. Vacuum distillation was resumed until an additional 30 L of solvent had been removed. Added additional isopropanol (28.8 L) and continued vacuum distillation until the volume was reduced by 42 L. Added isopropanol (4L) and the temperature was increased to >50 °C. Added water (28 L) such that the internal temperature was maintained above 50 °C, then heated to 75 °C to obtain a clear solution. The mixture was allowed to cool slowly and the product crystallized out of solution. The resulting suspension was cooled to 0 °C, held for 1 h then filtered and the cake was washed with water (5.5 L). The cake was dried at 45 °C under a nitrogen sweep to give III as a tan solid (3.30 kg, 71.6 wt %, 80.6% yield).
Example 12 2-methyl-2-(lH-pyrazol-l-yl)propanoic acid 16
2-Bromo-2-methylpropanoic acid 15 and pyrazole were reacted in triethylamine and 2- methyltetrahydrofuran to give 16.
Example 13 ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17
2-Methyl-2-(lH-pyrazol-l-yl)propanoic acid 16 was treated with sulfuric acid in ethanol to give 17. Alternatively, pyrazole (10 g, 147 mmol, 1.0 eq.) was dissolved in DMF (500 ml) at room temperature (Scheme 8). 2-Bromoisobutyrate 18 (22 ml, 147 mmol, 1.0 eq.), cesium carbonate CS2CO3 (53 g, 162 mmol, 1.1 eq) and catalytic sodium iodide Nal (2.2 g, 15 mmol, 0.1, eq) were added to the mixture that was then heated to 60 °C for 24 hr. Reaction was followed by 1H NMR and pyrazole was not detected after 24 hr. The reaction mixture was quenched with a saturated solution of NaHCC>3 (200 ml) and ethyl acetate EtOAc (150 ml) was added and organics were separated from aqueous. Organics were dried over Na2S04, filtered and concentrated under vacuum to afford an oil which was purified by flash chromatography to give 17.
Example 14 Ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2-methylpropanoate IV
Method A: Ethyl 2-methyl-2-(lH-pyrazol-l-yl)propanoate 17 was reacted with N- bromosuccinimide (NBS) in 2-methyltetrahydrofuran to give IV (CAS Reg. No. 1040377-17-0).
Method B: Ethyl 2-bromo-2-methylpropanoate 18 and pyrazole were reacted with sodium tert-butoxide in dimethylformamide (DMF) to give a mixture of ethyl 2-methyl-2-(lH- pyrazol-l-yl)propanoate 17 and ethyl 2-methyl-3-(lH-pyrazol-l-yl)propanoate 19 which was treated with l,3-dibromo-5,5-dimethylimidazolidine-2,4-dione to give a mixture of IV, ethyl 3- (4-bromo-lH-pyrazol-l-yl)-2-methylpropanoate 20, and 4-bromo-lH-pyrazole 21. The mixture was treated with a catalytic amount of lithium hexamethyldisilazide in tetrahydrofuran followed by acidification with hydrochloric acid to give IV.
Example 15 ethyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazol-l-yl)propanoate 22
To a 50 L glass reactor was charged ethyl 2-(4-bromo-lH-pyrazol-l-yl)-2- methylpropanoate IV (1.00 kg, 3.85 mol, 1.00 equiv), potassium acetate, KOAc (0.47 kg, 4.79 mol 1.25 equiv), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane),
bis(pinacolato)diboron, B2Pin2 (1.22 kg, 4.79 mol, 1.25 equiv) and ethanol (10 L, 10 vol) and the mixture was stirred until a clear solution was obtained. The solution was vacuum/degassed 3x with nitrogen. To this mixture was charged XPhos ligand (0.023 kg, 0.048 mol, 1.0 mol ) and the Pd precatalyst (0.018 kg, 0.022 mol, 0.5 mol ) resulting in a homogeneous orange solution. The solution was vacuum/degassed once with nitrogen. The internal temperature of the reaction was set to 75 °C and the reaction was sampled every 30 min once the set temperature was reached and was monitored by LC (IPC method: XTerra MS Boronic). After 5 h, conversion to 22 (CAS Reg. No. 1201657-32-0) was almost complete, with 1.3% IV remaining. Example 16 ethyl 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- IH-pyrazol- 1 -yl)-2-methylpropanoate 23
Ethyl 2-methyl-2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propanoate 22 and 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III were reacted under Suzuki conditions with palladium catalyst, in isopropanol and aqueous phosphate buffer to give 23.
A 1M solution of K3PO4 (1.60 kg in 7.6 L of water, 7.54 mol, 2.00 equiv) was charged to the above reaction mixture from Example 15, followed by the addition of a solution of III in THF (1.33 kg in 5.0 L, 3.43 mol, 0.90 equiv) over 2 min. The reaction mixture was warmed to 75 °C (internal temperature) over 45 min and stirred for 13 h at 75 °C, then analyzed by HPLC (III not detected) showing the formation of 23.
Example 17 2-(4-(2-( 1 -isopropyl-3-methyl- 1 H- 1 ,2,4-triazol-5-yl)-5 ,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- IH-pyrazol- 1 -yl)-2-methylpropanoic acid II
Ethyl 2-(4-(2-(l -isopropyl-3 -methyl- 1 H- 1 ,2,4-triazol-5-yl)-5 ,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanoate 23 was treated with aqueous lithium hydroxide to give II.
The ester saponification reaction was initiated with the addition of 3.5 M aqueous LiOH (0.74 kg in 5.0 L, 17.64 mol, 5 equiv) to the reaction mixture from Example 16 and allowed to warm to 75 °C. The mixture was sampled every 30 min (IPC method: XTerra MS Boronic) and the saponification was complete after 4.5 h (with less than 0.3% 23 remaining). The reaction mixture was concentrated via distillation to approximately half volume (starting vol = 37 L; final vol = 19 L) to remove EtOH and THF, resulting in tan-brown slurry. Water (5 L, 5 vol) was charged to the mixture and then distilled (starting vol = 25 L; final vol = 21 L). The temperature was set at 60 °C (jacket control) and then charged with isopropyl acetate, IP Ac (4 L, 4 vol). The biphasic mixture was stirred a minimum of 5 min and then the layers allowed to separate for a minimum of 5 min. The bottom aqueous layer was removed into a clean carboy and the organics were collected into a second carboy. The extraction process was repeated a total of four times, until the organic layer was visibly clear. The aqueous mixture was transferred back to the reactor and then cooled to 15 °C. A 6 M solution of HC1 (6.4 L, 38.40 mol, 10 equiv) was charged slowly until a final pH = 1 was obtained. The heterogeneous mixture was then filtered. The resulting solids were washed twice with 5 L (2 x 5 vol) of water. The filter was then heated to 80 °C and the vacuum set to -10 Psi (with nitrogen bleed) and the solids were dried for 24 h (KF = 2.0 % H20) to give 1.54 kg (95% corrected yield) of II as a white solid; 98% wt, 97.3 % pure.
Example 18 2-(4-(2-( 1 -isopropyl-3-methyl- 1 H- 1 ,2,4-triazol-5-yl)-5 ,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- lH-pyrazol- 1 -yl)-2-methylpropanamide I (GDC-0032)
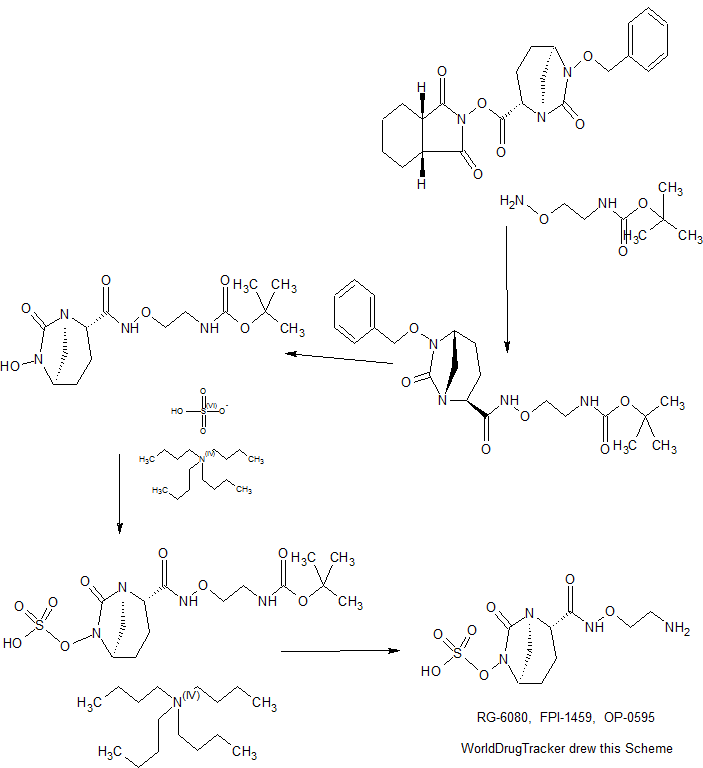
2-(4-(2-(l-Isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[l,2- d][l,4]oxazepin-9-yl)-lH-pyrazol-l-yl)-2-methylpropanoic acid II was treated with di(lH- imidazol-l-yl)methanone (carbonyldiimidazole, CDI) in tetrahydrofuran followed by methanolic ammonia to give crude I.
Solid II (1.44 kg, 3.12 mol, 1.00 equiv) was transferred into a 20 L bottle and then THF
(10 L, 7 vol) was charged. The slurry was transferred under reduced pressure into a second 50 L reactor and additional THF (5 L, 3 vol) was added for the rinse. The internal temperature of the slurry was set to 22 °C and Γ1 -carbonyldiimidazole, CDI (0.76 Kg, 5.12 mol, 1.50 equiv) was charged to the mixture and a clear solution was observed after 5 min. The reaction mixture was sampled every 30 min and analyzed by HPLC (IPC: XTerra MS Boronic method) which showed almost complete conversion to the acyl-imidazole intermediate and 1.2% remaining II after 30 min. An additional portion of CDI (0.07 kg, 0.15 mol, 0.14 equiv) was added, and the reaction mixture was stirred for 1 h and then analyzed by HPLC (IPC: XTerra MS Boronic method) which showed 0.8% remaining II.
Into a second 50-L reactor, was added NH3/MeOH (1.5 L, 10.5 mol, 3.37 equiv) and THF
(5 L, 3 vol). The acyl-imidazole intermediate was transferred to a second reactor under reduced pressure (transfer time -10 min). The internal temperature was then set to 45 °C and the volume of solvent was distilled down from 35 L to 12 L. Water (6 L, 4 vol) was then added to the mixture that was further distilled from 18 L to 11 L. Finally, another portion of water (6 L, 4 vol) was added and the solvents were distilled one last time from 17 L to 14 L, until no more THF was coming out. The reaction was then cooled down to 10 °C (internal temperature). The white slurry was filtered and the filter cake was washed with water (2 x 6 L, 2 x 4 vol). The solids were then dried at 80 °C (jacket temp) in the Aurora filter for 24 h (KF = 1.5 % H20) under vacuum to give 1.25 kg crude I, GDC-0032 (84% corrected yield, 96% wt, 97.3 % pure by HPLC) as a white solid.
A slurry of crude I (1.15 kg, 2.50 moles) in MeOH (6 L, 5 vol) was prepared and then charged to a 50 L glass reactor. Additional MeOH (24 L, 21 vol) was added to the mixture, which was then heated to 65 °C. A homogenous mixture was obtained. Si-thiol (Silicycle, Inc., 0.23 kg, 20% wt) was added to the solution via the addition port and the mixture was stirred for 3 hours. It was then filtered warm via the Aurora filter (jacket temperature = 60 °C, polish filtered and transferred directly into a second 50 L reactor with reduced pressure. The solution was then heated back to 65 °C internal temperature (IT). The homogeneous solution was cooled down to 54 °C and I seeds (12 g, 1 % wt) in MeOH (50 mL) were added with reduced pressure applied to the reactor. The mixture was then cooled down to 20 °C over 16 hours. The solids were then filtered via the Aurora filter and dried at 80 °C for 72 hours to give 921 g, 80% yield of I as a methanoate solvate (form A by XRPD,) and transferred to a pre-weighed charge-point bag.
In an isolator, the solids were slurred in IP Ac (8 L, 7 vol) and transferred to a clean 10 L reactor. The mixture was stirred for 1 h at 60 °C (IT). The solids were then filtered via the Aurora system and dried at 80 °C (jacket) for 96 h. A sample of I was removed and analyzed by GC (IP Ac = 1 %). To attempt more efficient drying, the API was transferred to two glass trays in an isolator and sealed with a drying bag before being dried in a vacuum oven set at 100 °C for 16 h. GC (IPC: Q12690V2) showed 1 % solvent was still present. The process afforded 760 g (68% corrected yield, 68% wt, 99.9 % purity by LC) of a white solid (form B by XRPD).
Crude I (340.7 g) was charged to a 2-1 . 1 1 DPI · bottle and slurried with 0.81 ,
isoamylalcohol (I A A). The slurry was transferred to a 20 L reactor and diluted with 6.7 L round- bottom flask (22 vol total). The white slurry was heated until a solution was observed (internal temperature rose to 118 °C and then cooled to 109 °C). The solution was polish filtered (0.2 μ .Μ filter). A flask was equipped with overhead stirring and the filtrate was slurried in isoamyl alcohol (344 ml ., 21 vol). The mixture was warmed to 95 °C (internal) until the solids dissolved. A slurry of charcoal (10 wt%, 0.16g) and silicycle thiol (10 wt%, 0.16g) in isoamyl alcohol (1 vol, 1 6 ml . ) was charged and the mixture was stirred at 90-95 °C for 1 h and then filtered (over Celite® pad). The clear amber colored solution was cooled to 73 °C (seeding temp range = 70 ±5 °C) and a GDC-0032 I seed (10 wt%, 0.16g) was added. The temperature of the heating mantle was turned off and the mixture was allowed to cool to room temperature overnight with stirrin (200 rpni). After 17 hr, the white solids were filtered starting with slow gravity filtration and then vacuum was applied. The solids were suction dried for 20 min with mixing until a free flowing powder was obtained. ( “rude weight prior to oven drying = 16 g. The solids were oven- dried at 100 °C for 24 h and then sampled for testing. Drying continued at 100 °C for another 24 hr. I l l NMR (DMSO d6) δ 8.38 (t), 8.01 (s), 7.87 (s), 7.44, 7.46 (d), 7.36 (s), 7.18 (br s), 6.81
(br s), 5.82 (m), 3.99 (s), 2.50 (s), 2.26 (s), 1.75 (s), 1.48, 1.46 (d).
Purified 2-(4-(2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepin-9-yl)- lH-pyrazol- 1 -yl)-2-methylpropanamide I (GDC-0032) was dry granulation formulated in tablet form by the roller compaction method (He et al (2007) Jour, of Pharm. Sci., 96(5):1342-1355) with excipients including lactose, microcrystalline cellulose (AVICEL® PH 01, FMC BioPolymer, 50 μΜ particle),
croscarmellose sodium (Ac-Di-Sol®, FMC BioPolymer), and magnesium stearate.
Example 19 4-bromo-2-fluoro-N-hydroxybenzimidamide 24
To a solution of 4-Bromo-2-fluorobenzonitrile 11 (800 g, 4 mol, 1 eq), hydroxylamine hydrochloride (695 g, 10 mol, 2.5 eq) in MeOH (2 L, 2.5 vol) was added Et3N (485 g, 4.8 mol, 1.2 eq), then the mixture was stirred at 60 °C for 40 min and checked by HPLC (no nitrile remaining). Reaction was then quenched by H20 (30 L), and lots of off-white solid was separated out, and then filtered, the filter cake was washed with water (10 L x 2) and 1350 g wet 4-bromo-2-fluoro-N-hydroxybenzimidamide 24 was obtained with 96% purity.
Example 20 ethyl 3-(4-bromo-2-fluorobenzimidamidooxy)acrylate 25
To a solution of 4-Bromo-2-fluoro-N-hydroxybenzimidamide 24 (800 g, 3.43 mol, 1 eq) and Amberlyst® A21 (20 wt%, 160 g) in PhMe (12 L, 15 vol) was added ethyl propiolate (471 g, 4.8 mol, 1.4 eq) at 10 °C. The reaction was stirred at 50 °C overnight and checked by LC-MS (ca 14A% of starting material 24 was left). Reaction was then filtered and the filtrate was concentrated under vacuum, and 1015 g ethyl 3-(4-bromo-2-fluorobenzimidamidooxy)acrylate 25 was obtained as a yellow oil with 84.9% LC purity (yield: 89%).
Example 21 ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylate 26
A solution of ethyl 3-(4-bromo-2-fluorobenzimidamidooxy)acrylate 25 (300 g, 0.91 mol, 1 eq) in diphenyl oxide (900 mL, 3 vol) was stirred at 190 °C under N2 for 1 h and checked by LC-MS (no 25 remaining). Cooled the mixture to rt and TBME (600 mL, 2 vol of 25) was added, and then PE (1.8 L, 6 vol of 25) was dropwise added to separate out solids. The mixture was stirred at rt for 20 min, and filtered to give 160 g wet cake. The wet cake was washed with PE (1 L) and dried to afford 120 g ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylate 26 with 92% LC purity as brown solids. Example 22 ethyl 2-(4-bromo-2-fluorophenyl)- 1 -(2-hydroxyethyl)- 1 H-imidazole-4- carboxylate 27
Ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylate 26 and l,3-dioxolan-2- one and N-methylimidazole were reacted to give 27.
Example 23 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28
Ethyl 2-(4-bromo-2-fluorophenyl)-l -(2-hydroxyethyl)- lH-imidazole-4-carboxylate 27, potassium hydroxide and methyl tributylammonium hydrochloride were reacted at 65 °C, cooled, and concentrated. The mixture was dissolved in ethanol and water to crystallize 28.
Example 24 9-bromo-N-(l-iminoethyl)-5,6-dihydrobenzo[f]imidazo[l,2- d] [ 1 ,4]oxazepine-2-carboxamide 29
9-Bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28, triphenylphosphine, and acetamidine were reacted to give 29.
Example 25 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine III
9-Bromo-N-( 1 -iminoethyl)-5 ,6-dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepine-2- carboxamide 29 was reacted with isopropylhydrazine hydrochloride 4 in acetic acid to give III.
Example 26 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 30
3-Chloro-2-oxopropanoic acid and 4-bromo-2-fluorobenzimidamide hydrochloride 12 are reacted with base to give 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 30.
Alternatively, to a solution of ethyl 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4- carboxylate 26 (1350 g, 4.3 mol) in THF (8.1 L, 6 vol) and H20 (4 L, 3 vol) was added NaOH (520 g, 13 mol, 3 eq), and the reaction was stirred at 65 °C for 48 h till it completed (checked by LC-MS). Adjust the mixture with 2 M HC1 to pH = 5, and product was separated out as a yellow solid, filtered to give 2.2 kg wet cake, the wet cake was washed with H20 (1.5 L), DCM (1.5 L x 3), PE (1 L), and dried to afford 970 g pure 2-(4-bromo-2-fluorophenyl)-lH-imidazole-4- carboxylic acid 30 (Scheme 10). Example 27 5-(2-(4-bromo-2-fluorophenyl)-lH-imidazol-4-yl)-l-isopropyl-3-methyl- lH-l,2,4-triazole V
Reaction of 30 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU in DMF gives intermediate, 2-(4-bromo-2-fluorophenyl)-N-(l-(2-isopropylhydrazinyl)ethylidene)- lH-imidazole-4-carboxamide 31 which cyclizes upon heating to give V.
Example 28 tert-butyl 2-hydroxyethylcarbamate gives tert-butyl 2-(5-bromo-2- cyanophenoxy)ethylcarbamate 32
Alkylation of 4-bromo-2-fluorobenzonitrile 11 with tert-butyl 2-hydroxyethylcarbamate gives 32.
Example 29 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33
Cyclization of tert-butyl 2-hydroxyethylcarbamate gives tert-butyl 2-(5-bromo-2- cyanophenoxy)ethylcarbamate 32 under acidic conditions, such as hydrochloric acid in ethanol, gives 33.
Example 30 9-bromo-5,6-dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxylic acid 28
Reaction of 3-bromo-2-oxopropanoic acid and 8-bromo-3,4- dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33 gives 28 (CAS Reg. No. 1282516-74-8).
Example 31 9-bromo-2-(l-isopropyl-3-methyl-lH-l,2,4-triazol-5-yl)-5,6- dihydrobenzo[f]imidazo[ 1 ,2-d] [ 1 ,4]oxazepine III
Coupling of 28 with N’-isopropylacetohydrazonamide 6 and coupling reagent HBTU in
DMF gives intermediate, 9-bromo-N-(l-(2-isopropylhydrazinyl)ethylidene)-5,6- dihydrobenzo[f]imidazo[l,2-d][l,4]oxazepine-2-carboxamide 34, which forms III upon heating.
Example 32 methyl 4-bromo-2-fluorobenzimidate 35
Reaction of 4-bromo-2-fluorobenzonitrile 11 with sodium methoxide in methanol gives 35.
Example 33 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33
Alkylation of methyl 4-bromo-2-fluorobenzimidate 35 with 2-aminoethanol gives 4- bromo-2-fluoro-N-(2-hydroxyethyl)benzimidamide 36, followed by cyclization to 33 (Scheme 13). Alternatively, reaction of 11 with 2- aminoethanol and potassium tert-butoxide displaces fluorine to give 2-(2-aminoethoxy)-4-bromobenzonitrile hydrochloride 37. Ring closure of 37 with trimethylaluminum gave 33 (Scheme 14). A solution of 11 (10 g, 50 mmol) and 2- aminoethanol (3.1 mL, 50.8 mmol) in 2-methyltetrahydrofuran (80 mL) was cooled to 0 °C and a solution of 1M potassium tert-butoxide in tetrahydrofuran (55 mL, 55 mmol) was slowly added while maintaining the solution temperature below 5 °C. The reaction was stirred at 0 °C for 30 min until judged complete by HPLC at which point it was warmed to 25 °C. A solution of 0.5M HC1 in isopropanol (100 mL, 50 mmol) was added and the desired HC1 salt 3 crystallized directly from the solution. The solid was collected by filtration and dried under vacuum with a nitrogen bleed to give 2-(2-aminoethoxy)-4-bromobenzonitrile hydrochloride 37 as a white solid. (12.1 g, 87 % yield).
To a flask was charged 37 (9.00 g, 32.4 mmol) and toluene (90.0 ml). The suspension was cooled to 0 °C and was added trimethylaluminum (1.8 equiv., 58.4 mmol, 2M in toluene) drop-wise over 30 minutes. The suspension was then stirred at room temperature for 1 h and then warmed to 100 °C. After 5 h, the solution was cooled to 0 °C and quenched with aqueous NaOH (2N, 90.0 ml). The suspension was extracted with EtOAc (4 x 90 ml) and the combined extracts were dried over then filtered through Celite®. The solution was concentrated and the residue triturated with EtOAc to afford 8-bromo-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-imine 33 (6.26 g, 26.0 mmol, 80% yield) as white crystalline solid.
Example 34 4-chloro-2-fluoro-N-hydroxybenzimidamide 39
To a solution of 4-chloro-2-fluorobenzonitrile 38 (400 g, 2.58 mol, 1.0 eq),
hydroxylamine hydrochloride (448 g, 6.45 mol, 2.5 eq) in MeOH (1 L, 2.5 vol) was added Et3N (313 g, 3.1 mol, 1.2 eq), then the mixture was stirred at 60 °C for 40 min and checked by HPLC (no nitrile remaining). Reaction was then quenched by H20 (10 L), and lots of off-white solid was separated out, and then filtered, the filter cake was washed with water (10 L x 2) and 378 g 4-chloro-2-fluoro-N-hydroxybenzimidamide 39 was obtained with 93% purity (Scheme 15).
Example 35 ethyl 3-(4-chloro-2-fluorobenzimidamidooxy)acrylate 40
To a solution of 4-chloro-2-fluoro-N-hydroxybenzimidamide 39 (378 g, 2 mol, 1.0 eq) and Amberlyst® A21 (20 wt%, 75.6 g) in toluene PhMe (5.6 L, 15 vol) was added ethyl propiolate (275 g, 2.8 mol, 1.4 eq) at 30 °C. The reaction was stirred at 30 °C overnight and checked by LC-MS. Reaction was then filtered and the filtrate was concentrated under vacuum, and 550 g ethyl 3-(4-chloro-2-fluorobenzimidamidooxy)acrylate 40 was obtained as a yellow oil with 83% LC purity (Scheme 15).
Example 36 ethyl 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylate 41
A solution of ethyl 3-(4-chloro-2-fluorobenzimidamidooxy)acrylate 40 (550 g, 1.9 mol, 1.0 eq, 83% LC purity) in diphenyl oxide (1.65 L, 3 vol) was stirred at 190 °C under N2 for 1 h and checked by LC-MS (no 40 remaining). Cooled the mixture to rt and PE (10 L) was added dropwise. The mixture was stirred at rt for 20 min, and filtered to give 400 g wet cake, after purified by chromatography on silica gel (PE / EA=1 / 5) to get 175 g pure ethyl 2-(4-chloro-2- fluorophenyl)-lH-imidazole-4-carboxylate 41 with 98% LC purity (Scheme 15).
Example 37 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 42
To a solution of ethyl 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylate 41 (175 g, 4.3 mol) in THF (1 L, 6 vol) and H20 (500 mL, 3 vol) was added NaOH (78 g, 1.95 mol, 3.0 eq), and the reaction was stirred at 65 °C for 48 h till it completed (checked by LC-MS). Adjust the mixture with 2 N HC1 to pH = 5, and product was separated out as a yellow solid, filtered to give 210 g wet cake, the wet cake was washed with H20 (300 mL), DCM (3 x 300 mL), PE (500 mL), and dried to afford 110 g pure 2-(4-chloro-2-fluorophenyl)-lH-imidazole-4-carboxylic acid 42 (CAS Reg. No. 1260649-87-3) (Scheme 15 ). I l l NMR (DMSO d6) δ: 12.8 (br s), 8.0, 7.9 (br s), 7.46, 7.4 (m).
PATENT
US 2014275523
SYNTHESIS
Taselisib_药物数据_原料药API_CCIS-CHEM化学平台科研物资一站式采购平台 …

CLIP
http://www.ccis-chem.com/goods.php?id=194272
Taselisib
Taselisib是罗氏集团及其下属公司Genentech和Chugai研发,目前治疗绝经后妇女雌激素受体阳性(ER +)乳腺癌和非小细胞肺癌(NSCLC)的三期临床研究均在进行中。
- 药品名称:
- Taselisib
- 研发代码:
- GDC-0032; RG-7604
- 商品名称:
- 作用机制:
- PI3K inhibitor; Cytochrome P450 CYP3A4 Inhibitors
- 适应症:
- 乳腺癌,非小细胞肺癌
- 研发阶段:
- 临床三期 (进行中)
- 研发公司:
- 罗氏 (原研)
| 分子量 |
460.53 |
| 分子式 |
C24H28N8O2 |
| CAS号 |
1282512-48-4 (Taselisib); |
| 化学名称 |
1H-Pyrazole-1-acetamide, 4-[5,6-dihydro-2-[3-methyl-1-(1-methylethyl)-1H-1,2,4-triazol-5-yl]imidazo[1,2-d][1,4]benzoxazepin-9-yl]-a,a-dimethyl- |
| Fudosteine药品(游离态)参数: |
| MW |
HD |
HA |
FRB* |
PSA* |
cLogP* |
| 460.53 |
2 |
10 |
5 |
119 |
2.548±1.034 |
|
化学合成路线及相关杂质更新时间:2015-12-15
参考文献:J. Med. Chem. 2013, 56, 4597−4610

PAPER
J Med Chem 2013, 56(11): 4597
Discovery of 2-{3-[2-(1-Isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in Vivo Antitumor Activity
Chudi O. Ndubaku*†, Timothy P. Heffron*†, Steven T. Staben†, Matthew Baumgardner‡, Nicole Blaquiere†, Erin Bradley†, Richard Bull⊥, Steven Do†, Jennafer Dotson†, Danette Dudley†, Kyle A. Edgar§, Lori S. Friedman§, Richard Goldsmith†, Robert A. Heald⊥, Aleksandr Kolesnikov†, Leslie Lee§, Cristina Lewis∥, Michelle Nannini§, Jim Nonomiya∥, Jodie Pang‡, Steve Price⊥, Wei Wei Prior§, Laurent Salphati‡, Steve Sideris∥, Jeffery J. Wallin§, Lan Wang†, BinQing Wei†, Deepak Sampath§, and Alan G. Olivero†
†Departments of Discovery Chemistry, ‡Drug Metabolism and Pharmacokinetics, §Translational Oncology, and ∥Biochemical Pharmacology, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
⊥ Argenta Discovery, 8-9 Spire Green Centre, Flex Meadow, Harlow, Essex, CM19 5TR, United Kingdom
J. Med. Chem., 2013, 56 (11), pp 4597–4610
DOI: 10.1021/jm4003632
Dysfunctional signaling through the phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway leads to uncontrolled tumor proliferation. In the course of the discovery of novel benzoxepin PI3K inhibitors, we observed a strong dependency of in vivo antitumor activity on the free-drug exposure. By lowering the intrinsic clearance, we derived a set of imidazobenzoxazepin compounds that showed improved unbound drug exposure and effectively suppressed growth of tumors in a mouse xenograft model at low drug dose levels. One of these compounds, GDC-0032 (11l), was progressed to clinical trials and is currently under phase I evaluation as a potential treatment for human malignancies.
2-{3-[2-(1-Isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (11l)
1H NMR (500 MHz, DMSO) δ 8.42 (s, 1H), 8.37 (d, J = 8.3 Hz, 1H), 8.02 (s, 1H), 7.89 (s, 1H), 7.46 (dd, J = 8.3, 1.8 Hz, 1H), 7.36 (d, J = 1.8 Hz, 1H), 7.22 (s, 1H), 6.87 (s, 1H), 5.90–5.73 (m, 1H), 4.62–4.42 (m, 4H), 2.50 (dt, J = 3.6, 1.7 Hz, 5H), 2.26 (s, 3H), 1.74 (s, 6H), 1.47 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, DMSO) δ 173.78, 158.24, 155.88, 147.31, 143.94, 136.64, 134.60, 130.26, 129.88, 126.42, 123.62, 120.32, 119.31, 116.17, 115.26, 68.31, 64.48, 49.89, 49.70, 40.06, 39.97, 39.89, 39.80, 39.72, 39.63, 39.56, 39.47, 39.30, 39.13, 38.96, 25.47, 22.34, 13.82. HRMS (ESI+): m/z (M + H+) calcd: 461.2413, found: 461.2427. Melting point: 259 °C.
POLYMORPHS almost A to Z
GDC-0032, also known as taselisib, RG7604, or the IUPAC name: 2-(4-(2-(1-isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide, has potent PI3K activity (Ndubaku et al (2013) J. Med. Chem. 56(11):4597-4610; WO 2013/182668; WO 2011/036280; U.S. Pat. No. 8,242,104; U.S. Pat. No. 8,343,955) and is being studied in patients with locally advanced or metastatic solid tumors (Juric et al “GDC-0032, a beta isoform-sparing PI3K inhibitor: Results of a first-in-human phase Ia dose escalation study”, 2013 (Apr. 7) Abs LB-64 American Association for Cancer Research Annual Meeting).
the invention relates to polymorph forms of the PI3K inhibitor I (taselisib, GDC-0032, RG7604, CAS Reg. No. 1282512-48-4, Genentech, Inc.), named as 2-(4-(2-(1-isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl)-1H-pyrazol-1-yl)-2-methylpropanamide, having the structure:
and stereoisomers, geometric isomers, tautomers, and pharmaceutically acceptable salts thereof.
Publication numberPriority datePublication dateAssigneeTitle
WO2011036280A12009-09-282011-03-31F. Hoffmann-La Roche AgBenzoxazepin pi3k inhibitor compounds and methods of use
WO2014140073A12013-03-132014-09-18F. Hoffmann-La Roche AgProcess for making benzoxazepin compounds
References
Taselisib
|
| Clinical data |
| ATC code |
|
| Identifiers |
|
|
| CAS Number |
|
| PubChem CID |
|
| ChemSpider |
|
| UNII |
|
| ChEMBL |
|
| Chemical and physical data |
| Formula |
C24H28N8O2 |
| Molar mass |
460.54 g·mol−1 |
| 3D model (JSmol) |
|
-
2-{4-[2-(1-Isopropyl-3-methyl-1H-1,2,4-triazol-5-yl)-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide
|
-
InChI=1S/C24H28N8O2/c1-14(2)32-22(27-15(3)29-32)19-13-30-8-9-34-20-10-16(6-7-18(20)21(30)28-19)17-11-26-31(12-17)24(4,5)23(25)33/h6-7,10-14H,8-9H2,1-5H3,(H2,25,33)
-
Key:BEUQXVWXFDOSAQ-UHFFFAOYSA-N
|
Taselisib
(GDC-0032, RG7604)
This compound and its uses are investigational and have not been approved by the US Food and Drug Administration. Efficacy and safety have not been established. The information presented should not be construed as a recommendation for use. The relevance of findings in preclinical studies to humans is currently being evaluated.
Taselisib, a PI3K inhibitor
Taselisib, an investigational PI3K inhibitor, is currently in clinical development based on its potential selectivity for the PI3Kα isoform.1,2 Preclinical data have shown that taselisib induced growth inhibition in PI3Kα-mutant cell lines.1 Taselisib continues to be investigated in ongoing clinical studies.
1Taselisib is an investigational PI3K inhibitor currently being studied for its potential to selectively inhibit the PI3Kα isoform.1,2
2Taselisib is designed to bind to the ATP-binding pocket of PI3Kα to potentially prevent subsequent downstream signaling.1
3In preclinical studies, taselisib induced growth inhibition in PI3Kα-mutant xenograft mouse models.1 Taselisib continues to be investigated in ongoing clinical studies.
References
- Lopez S, Schwab CL, Cocco E, et al. Taselisib, a selective inhibitor of PIK3CA, is highly effective on PIK3CA-mutated and HER2/neu amplified uterine serous carcinoma in vitro and in vivo. Gynecol Oncol.2014;135:312-317. PMID: 25172762
- Ndubaku CO, Heffron TP, Staben ST, et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem. 2013;56:4597-4610. PMID: 23662903
//////////////////RG7604, Taselisib, PHASE 3, metastatic breast cancer, non-small cell lung cancer, RO5537381, Roche
CC1=NN(C(=N1)C2=CN3CCOC4=C(C3=N2)C=CC(=C4)C5=CN(N=C5)C(C)(C)C(=O)N)C(C)C
CLIP
Manufacture of the PI3K β-Sparing Inhibitor Taselisib. Part 2: Development of a Highly Efficient and Regioselective Late-Stage Process
Frédéric St-Jean*†  , Travis Remarchuk†, Rémy Angelaud*†, Diane E. Carrera†, Danial Beaudry†, Sushant Malhotra†, Andrew McClory†, Archana Kumar†, Gerd Ohlenbusch‡, Andreas M. Schuster‡, and Francis Gosselin†
, Travis Remarchuk†, Rémy Angelaud*†, Diane E. Carrera†, Danial Beaudry†, Sushant Malhotra†, Andrew McClory†, Archana Kumar†, Gerd Ohlenbusch‡, Andreas M. Schuster‡, and Francis Gosselin†
† Department of Small Molecule Process Chemistry, Genentech Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡ Small Molecules Technical Development PTDC-C, F. Hoffmann-La Roche Ltd., Grenzacherstrasse 124, 4070 Basel, Switzerland
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.9b00050
A highly efficient and regioselective manufacturing route for the phosphoinositide 3-kinase β-sparing inhibitor taselisib was developed. Highlights of the synthesis include: (1) magnesium-mediated formation of a challenging cyclic amidine; (2) regioselective imidazole construction via alkylation/condensation with bromopyruvic acid; and (3) triazole formation with 2-isopropyl acetamidrazone to generate the key bromobenzoxazepine core intermediate. Subsequent highly efficient one-pot palladium-catalyzed Miyaura borylation/Suzuki cross-coupling/saponification, followed by a 1,1′-carbonyldiimidazole-mediated coupling with ammonia, led to the pentacyclic taselisib. This new synthetic approach provides a more efficient route to taselisib with a significant decrease in process mass intensity compared to the previous early development routes to the bromobenzoxazepine core. Finally, implementation of a controlled crystallization provided the active pharmaceutical ingredient with the desired polymorphic form.
Taselisib was obtained in 90% yield (16.2 kg) as a white to off-white solid (>99.9 wt %; >99.9 A%) as Form B crystal.
mp (DSC): 257–258 °C.
HRMS (ESI-CID) m/z Calcd for [M + H]+ C24H29N8O2: 461.2408; found: 461.2409.
1H NMR (600 MHz, DMSO-d6) δ ppm 8.41 (s, 1H), 8.37 (d, J = 8.4, 1H), 8.02 (s, 1H), 7.88 (m, 1H), 7.45 (dd, J = 8.4, 1.7 Hz, 1H), 7.36 (d, J = 1.7 Hz, 1H), 7.20 (br s, 1H), 6.84 (br s, 1 H), 5.83 (sept, J = 6.6 Hz, 1H), 4.53–4.51 (m, 2H), 4.52 (m, 2H), 2.26 (s, 3H), 1.75 (s, 6H), 1.47 (d, J = 6.7 Hz, 6H).
13C NMR (DMSO-d6, 151 MHz) δ = 173.8, 158.3, 155.9, 147.3, 144.0, 136.6, 134.6, 130.3, 129.9, 126.4, 123.6, 120.4, 119.3, 116.2, 115.3, 68.3, 64.5, 49.9, 49.7, 25.5, 25.5, 22.3, 22.3, 13.8.
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00050
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.9b00050/suppl_file/op9b00050_si_001.pdf
Ultraviolet/Visible Spectroscopy
Apparatus: Perkin Elmer, UV-6190, Lambda 25
Solvent: Acetonitrile/Water 1:1 (v/v)












////////////////

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














































































 67
67 66
66









 .
.









![[1860-5397-9-265-7]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-7.png)
![[1860-5397-9-265-i29]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i29.png)
![[1860-5397-9-265-8]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-8.png)
![[1860-5397-9-265-i30]](https://i0.wp.com/www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i30.png)































{kind=link}