
Home » Posts tagged 'Priority review' (Page 7)
Tag Archives: Priority review
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
US priority review for Eisai cancer drug lenvatinib
![]()
US priority review for Eisai cancer drug lenvatinib
Eisai has been boosted by news that regulators in the USA have agreed to a quicker review of its anticancer agent lenvatinib.
The US Food and Drug Administration has granted a priority review to Eisai’s New Drug Application for lenvatinib as a treatment for progressive radioiodine-refractory differentiated thyroid cancer. This means that the agency has assigned a Prescription Drug User Fee Act action date of April 14 next year, eight months after the NDA was submitted.
Read more at: http://www.pharmatimes.com/Article/14-10-15/US_priority_review_for_Eisai_cancer_drug_lenvatinib.aspx#ixzz3GH3iXiDU
SEE SYNTHESIS




The US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat)
13 dec 2013
Genzyme’s Cerdelga NDA receives US FDA priority review designationpharmabiz.comThe US Food and Drug Administration (FDA) has granted a six-month Priority Review designation to Genzyme’s New Drug Application (NDA) for Cerdelga (eliglustat), an investigational oral therapy for adult patients with Gaucher disease
ELIGLUSTAT TARTRATE
THERAPEUTIC CLAIM Treatment of lysosomal storage disorders
CHEMICAL NAMES
1. Octanamide, N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(1-
pyrrolidinylmethyl)ethyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)
2. bis{N-[(1R,2R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-hydroxy-1-(pyrrolidin-1-
ylmethyl)ethyl]octanamide} (2R,3R)-2,3-dihydroxybutanedioate
MOLECULAR FORMULA C23H36N2O4 . ½ C4H6O6
MOLECULAR WEIGHT 479.6
MANUFACTURER Genzyme Corp.
CODE DESIGNATION Genz-112638
CAS REGISTRY NUMBER 928659-70-5
old article cut paste
Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
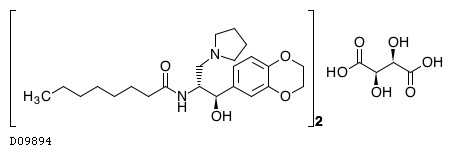
Eliglustat tartrate (USAN)
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review

Posaconazole, SCH 56592, Noxafil (Schering-Plough)
Posaconazole is a triazole antifungal drug that is used to treat invasive infections by Candida species and Aspergillus species in severely immunocompromised patients.
For prophylaxis of invasive Aspergillus and Candida infections in patients, 13 years of age and older, who are at high risk of developing these infections due to being severely immunocompromised as a result of procedures such as hematopoietic stem cell transplant (HSCT) recipients with graft-versus-host disease (GVHD), or due to hematologic malignancies with prolonged neutropenia from chemotherapy. Also for the treatment of oropharyngeal candidiasis, including oropharyngeal candidiasis refractory to itraconazole and/or fluconazole. Posaconazole is used as an alternative treatment for invasive aspergillosis, Fusarium infections, and zygomycosis in patients who are intolerant of, or whose disease is refractory to, other antifungals
Posaconazole is designated chemically as 4-[4-[4-[4-[[ (3R,5R)-5- (2,4-difluorophenyl)tetrahydro-5-(1H-1,2,4-triazol-1 -ylmethyl)-3-furanyl]methoxy]phenyl]-1 -piperazinyl]phenyl]-2-[ (1S,2S)-1 -ethyl-2- hydroxypropyl]-2,4-dihydro-3H-1,2,4-triazol-3-one with an empirical formula of C37H42F2N8O4 and a molecular weight of 700.8.
Posaconazole is used, for example, to prevent and/or treat invasive fungal infections caused by Candida species, Mucor species, Aspergillus species,Fusarium species, or Coccidioides species in immunocompromised patients and/or in patients where the disease is refractory to other antifungal agents such as amphothericin B, fluconazole, or itraconazole, and/or in patients who do not tolerate these antifungal agents.
CAS No. 171228-49-2
Posaconazole compounds have been described inU.S. Pat. Appl. No. 2003/0055067 for “Antifungal Composition with Enhanced Bioavailability,” U.S. Pat. Appl. No. 2004/0058974 for “Treating Fungal Infections,” and European Patent Publication1372394 (A1 ) for “Liquid Suspensions of Posaconazole (SCH 56592) with Enhanced Bioavailability for Treating Fungal Infections.”
| Synonyms: | Pcz;Pos;Noxafil;Sch 56592;Aids058495;Aids-058495;Posconazole;Posaconazole;Posaconazole for research;HYDROXYPROPYL]-2,4-DIHYDRO-3H-1,2,4-TRIAZOL-3-ONE |
| Molecular Formula: | C37H42F2N8O4 |
| Formula Weight: | 700.78 |
Merck’s New Drug Application for an Investigational Intravenous (IV) Formulation of NOXAFIL® (posaconazole) Receives FDA Priority Review
Marketing Authorization Application also Filed with the European Medicines Agency
WHITEHOUSE STATION, N.J., Nov. 18, 2013–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that its New Drug Application for an investigational intravenous (IV) solution formulation of the company’s antifungal agent, NOXAFIL® (posaconazole), has been accepted for priority review by the U.S. Food and Drug Administration (FDA).http://www.pharmalive.com/mercks-noxafil-nda-gets-fda-priority-review
Posaconazole (CAS Registry Number 171228-49-2; CAS Name: 2,5-anhydro-1 ,3,4-trideoxy-2- C-(2,4-difluorophenyl)-4-[[4-[4-[4-[1-[(1S,2S)-1-ethyl-2-hydroxypropyl]-1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]-1-piperazinyl]phenoxy]methyl]-1-(1 H-1 ,2,4-triazol-1-yl)-D-threo-pentitol) which is represented by the following general formula (I)

(I)
is known as an antifungal agent. It is available as an oral suspension (40 mg/ml) under the trademark NOXAFIL® from Schering Corporation, Kenilworth, NJ. WO95/17407 and WO 96/38443 disclose the compound having the general formula (I) and its use in treating fungal infections. Various pharmaceutical compositions comprising posaconazole and being adapted for oral, topical or parenteral use are described e.g. in WO 02/80678, U.S. Patent No. 5,972,381 , U.S. Patent No. 5,834,472, U.S. Patent No. 4,957,730 and WO 2005/117831. As was mentioned above, WO 95/17407 and WO 96/38443 disclose the compound having the general formula (I). However, during prosecution of the subsequently filed European patent application no. 98951994.7, now European patent EP 1 021 439 B1 , the applicant declared that the methods disclosed in these publications only lead to the compound of formula (I) as an amorphous solid.
Polymorphism is a phenomenon relating to the occurrence of different crystal forms for one molecule. There may be several different crystalline forms for the same molecule with distinct crystal structures and distinct and varying physical properties like melting point, XRPD pattern, IR-spectrum and solubility profile. These polymorphs are thus distinct solid forms which share the molecular formula of the compound from which the crystals are made up, however, they may have distinct advantageous physical properties which can have a direct effect on the ability to process and/or manufacture the drug product, like flowability, as well as physical properties such as solubility, stability and dissolution properties which can have a direct effect on drug product stability, solubility, dissolution, and bioavailability.
Three polymorphic forms of posaconazole designated as forms I, Il and III are described and characterized in WO 99/18097 (US-B-6,713,481 , US-B-6,958,337). Crystalline forms Il and III were found to be unstable under the conditions investigated, so that crystalline form I was considered to be useful in the development of a pharmaceutical product.
A. K. Saksena et al., WO 9517407; eidem, US 5661151 (1995, 1997 both to Schering);
eidem, Tetrahedron Lett. 37, 5657 (1996).
SCH-56592, a novel orally active broad spectrum antifungal agent35th Intersci Conf Antimicrob Agents Chemother (Sept 17-20, San Francisco) 1995,Abst F61
seeSaksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.-T.; Ganguly, A.K.; Bennett, F. (Schering Corp.) EP 0736030; JP 1997500658; US 5661151; US 5703079; WO 9517407
Process for the preparation of triazolonesWO 9633178
Mono N-arylation of piperazine(III): Metal-catalyzed N-arylation and its application to the novel preparations of the antifungal posaconazole and its advanced intermediateTetrahedron Lett 2002,43(18),3359
Comparative antifungal spectrum: A. Cacciapuoti et al., Antimicrob. Agents Chemother. 44, 2017 (2000).
Pharmacokinetics, safety and tolerability: R. Courtney et al., ibid. 47, 2788 (2003).
HPLC determn in serum: H. Kim et al., J. Chromatogr. B 738, 93 (2000).
Review of development: A. K. Saksena et al. inAnti-Infectives: Recent Advances in Chemistry and Structure Activity Relationships (Royal Soc. Chem., Cambridge, 1997) pp 180-199; and clinical efficacy in fungal infections: R. Herbrecht, Int. J. Clin. Pract. 58, 612-624 (2004).
synthesis 1

……………..

Synthesis of intermediate (XX): The reaction of 2-chloro-2′,4′-difluoroacetophenone (I) with sodium acetate and NaI in DMF gives 2-acetoxy-2′,4′-difluoroacetophenone (II), which by methylenation with methyltriphenylphosphonium bromide and sodium bis(trimethylsilyl)amide in THF yields 2-(2,4-difluorophenyl)-2-propen-1-ol acetate ester (III). The hydrolysis of (III) with KOH in dioxane/water affords the corresponding alcohol (IV), which is regioselectively epoxidized with titanium tetraisopropoxide and L-(+)-diethyl tartrate in dichloromethane to (S)-(-)-2-(2,4-difluorophenyl)oxirane-2-methanol (V). The reaction of (V) with 1,2,4-triazole (VI) in DMF affords (R)-2-(2,4-difluorophenyl)-3-(1,2,4-triazol-1-yl)propane-1,2-diol (VII), which is selectively mesylated with methanesulfonyl chloride and triethylamine to the monomesylate (VIII). The cyclization of (VIII) with NaH in DMF gives the oxirane (IX), which is condensed with diethyl malonate (X) by means of NaH in DMSO to yield a mixture of (5R-cis)- and (5R-trans)-5-(2,4-difluorophenyl)-2-oxo-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-carboxylic acid ethyl ester (XI). The reduction of (XI) with NaBH4 and LiCl in ethanol affords (R)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)-5-(1,2,4-triazol-1-yl) pentane-1,4-diol (XII), which is selectively tosylated with tosyl chloride and triethylamine in THF to the bistosylate (XIII). The cyclization of (XIII) by means of NaH in refluxing toluene gives (5R-cis)-5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl) tetrahydrofuran-3-methanol tosylate ester (XIV). The reaction of (XIV) with 1-(4-hydroxyphenyl)-4-(4-nitrophenyl)piperazine (XV) to obtain compound (XVI), and the following reaction sequence (XVI) to (XVII) to (XVIII) to (XIX) to (5R-cis)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(1,2,4-triazol-1-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin-1-yl]phenyl-3,4-dihydro-2H-1,2,4-triazol-3-one (XX) has been performed according to J Med Chem 1984, 27: 894-900.
………………….pat approved expiry
| United States | 5661151 | 1999-07-19 | 2019-07-19 |
| Canada | 2305803 | 2009-12-22 | 2018-10-05 |
| Canada | 2179396 | 2001-04-17 | 2014-12-20 |
| United States | 5703079 | 1994-08-26 | 2014-08-26 |
MORE INFO
| US Patent No | Patent expiry | |
|---|---|---|
| 5661151 | Jul 19, 2019 | |
| 5703079 | Aug 26, 2014 | |
| 6958337 | Oct 5, 2018 | |
| 8263600 | Apr 1, 2022 |
- Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D: Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007 Jan 25;356(4):348-59. Pubmed
- Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S: Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007 Jan 25;356(4):335-47. Pubmed
- Bhattacharya M, Rajeshwari K, Dhingra B: Posaconazole. J Postgrad Med. 2010 Apr-Jun;56(2):163-7. Pubmed
- Frampton JE, Scott LJ: Posaconazole : a review of its use in the prophylaxis of invasive fungal infections. Drugs. 2008;68(7):993-1016.Pubmed
- Schiller DS, Fung HB: Posaconazole: an extended-spectrum triazole antifungal agent. Clin Ther. 2007 Sep;29(9):1862-86. Pubmed
- Kwon DS, Mylonakis E: Posaconazole: a new broad-spectrum antifungal agent. Expert Opin Pharmacother. 2007 Jun;8(8):1167-78.Pubmed
- Groll AH, Walsh TJ: Posaconazole: clinical pharmacology and potential for management of fungal infections. Expert Rev Anti Infect Ther. 2005 Aug;3(4):467-87. Pubmed
- Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH: Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008 Oct;42(10):1429-38. Epub 2008 Aug 19. Pubmed
- Li Y, Theuretzbacher U, Clancy CJ, Nguyen MH, Derendorf H: Pharmacokinetic/pharmacodynamic profile of posaconazole. Clin Pharmacokinet. 2010 Jun;49(6):379-96. doi: 10.2165/11319340-000000000-00000. Pubmed
FDA grants priority review to Pharmacyclics drug

ibrutinib
FDA grants priority review to Pharmacyclics drug

Pharmacyclics is getting a priority review of its blood cancer treatment by federal regulators. A priority review shortens a drug evaluation by the U.S. Food and Drug Administration from 10 months to six. The acceptance of the application triggers a $75 million milestone payment to Pharmacyclics from Johnson & Johnson’s Janssen unit.

Ibrutinib (USAN[1]), also known as PCI-32765, is an experimental drug candidate for the treatment of various types of cancer. It is an orally-administered, selective and covalent inhibitor of the enzyme Bruton tyrosine kinase (Btk).[2][3][4] Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson’s Janssen Pharmaceutical division for B-cell malignancies including chronic lymphocytic leukemia, mantle cell lymphoma, diffuse large B-cell lymphoma, and multiple myeloma.[6][7][8]. Ibrutinib was first designed and synthesized at Celera Genomics by Zhengying Pan, who along with a team of chemists and biologists reported in 2007 a structure-based approach for creating a series of small molecules that inactivate BTK through covalent binding to cysteine-481 near the ATP binding domain of BTK[2]. These small molecules irreversibly inhibited BTK by using a Michael acceptor for binding to the target cysteine. In April 2006, Pharmacyclics acquired Celera’s small molecule BTK inhibitor discovery program, which included a compound, PCI-32765 (known as compound 13 in the Pan et al paper) that was subsequently chosen for further preclinical development based on the discovery of anti-lymphoma properties in vivo [5]. Since 2006, Pharmacyclics’ scientists have advanced the molecule into clinical trials and identified specific clinical indications for the drug. [2][3][4] [5] [6][7][8] It also has potential effects against autoimmune arthritis.[9]
Clinical trials
It has given good results in two phase II clinical trials.[10]
Mechanism
In preclinical studies on chronic lymphocytic leukemia (CLL) cells, ibrutinib has been reported to promote apoptosis, inhibit proliferation, and also prevent CLL cells from responding to survival stimuli provided by the microenvironment.[11] In this study, treatment of activated CLL cells with ibrutinib resulted in inhibition of Btk tyrosine phosphorylation and also effectively abrogated downstream survival pathways activated by this kinase including ERK1/2, PI3K, and NF-κB. Additionally, ibrutinib inhibited proliferation of CLL cells in vitro, effectively blocking survival signals provided externally to CLL cells from the microenvironment including soluble factors (CD40L, BAFF, IL-6, IL-4, and TNF-α), fibronectin engagement and stromal cell contact.
In early clinical studies, the activity of ibrutinib has been described to include a rapid reduction in lymphadenopathy accompanied by a transient lymphocytosis, suggesting that the drug might have direct effects on cell homing or migration to factors in tissue microenvironments.[12]
Ibrutinib has been reported to reduce CLL cell chemotaxis towards the chemokines CXCL12 and CXCL13, and inhibit cellular adhesion following stimulation at the B cell receptor.[13][14] Together, these data are consistent with a mechanistic model whereby ibrutinib blocks BCR signaling, which drives cells into apoptosis and/or disrupts cell migration and adherence to protective tumor microenvironments.
References
- ^ Statement on a Nonproprietary Name Adopted by the USAN Council
- ^ Pan, Z; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD et al. (2007). “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase”. ChemMedChem 2 (1): 58–61. doi:10.1002/cmdc.200600221. PMID 17154430.
|displayauthors=suggested (help) - ^ Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics
- ^ United States patent 7514444
- ^ Honigberg, LA; Smith, AM; Sirisawad, M; Verner, E; Loury, D; Chang, B; Li, S; Pan, Z; Thamm, DH; Miller, RA; Buggy (2010). “The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy”. Proceedings of the National Academy of Sciences of the United States of America 107 (29): 13075–80. doi:10.1073/pnas.1004594107. PMID 20615965. Unknown parameter
|firs11=ignored (help) - ^ Janssen Biotech, Inc. Announces Collaborative Development and Worldwide License Agreement for Investigational Anti-Cancer Drug, PCI-32765
- ^ Clinical trials involve PCI-32765
- ^ Clinical trials involve ibrutinib
- ^ Chang, BY; Huang, MM; Francesco, M; Chen, J; Sokolove, J; Magadala, P; Robinson, WH; Buggy, JJ (2011). “The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells”. Arthritis Research & Therapy 13 (4): R115. doi:10.1186/ar3400. PMID 21752263.
- ^ Good News Continues for Ibrutinib in CLL. 8 Dec 2012
- ^ Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy J.J., Hamdy A, Johnson AJ, Byrd JC. (2011) Bruton’s tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 117: 6287-6296
- ^ The Bruton’s tyrosine kinase (BTK) inhibitor PCI-32765 (P) in treatment-naive (TN) chronic lymphocytic leukemia (CLL) patients (pts): Interim results of a phase Ib/II study.J Clin Oncol 30, 2012 (suppl; abstr 6507)
- ^ Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N, Burger JA. (2012) The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 119: 1182-1189.
- ^ de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M. (2012) The clinically active BTK inhibitor PCI-32765 targets B-cell receptor (BCR)- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 119: 2590-2594.
External links
- BTK inhibitor PCI-32765, National Cancer Institute Drug Dictionary

FDA Grants Priority Review To New Drug Application For MNK-795
FDA Grants Priority Review To New Drug Application For MNK-795 Submitted By Depomed Licensee Mallinckrodt
Controlled Substance Analgesic Combination Product Uses Depomed’s Proprietary Acuform® Technology
NEWARK, Calif., July 29, 2013 /PRNewswire/ — Depomed, Inc. (NASDAQ:DEPO) announced today that the U. S. Food and Drug Administration (FDA) has accepted for filing a New Drug Application (NDA) from Mallinckrodt (NYSE: MNK) for MNK-795. MNK-795 is a controlled-release oral formulation of oxycodone and acetaminophen that has been studied for the management of moderate to severe acute pain where the use of an opioid analgesic is appropriate. MNK-795 is formulated with Depomed’s Acuform® drug delivery technology.
http://www.pharmalive.com/fda-grants-priority-review-to-new-drug-application-for-mnk-795
Gilead Announces U.S. FDA Priority Review Designation for Sofosbuvir for the Treatment of Hepatitis C

Sofosbuvir
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate
http://www.ama-assn.org/resources/doc/usan/sofosbuvir.pdf –for cas no

Jun. 7, 2013– Gilead Sciences, Inc. today announced that the U.S. Food and Drug Administration (FDA) has granted priority review to the company’s New Drug Application (NDA) for sofosbuvir, a once-daily oral nucleotide analogue inhibitor for the treatment of chronic hepatitis C virus (HCV) infection. The FDA grants priority review status to drug candidates that may offer major advances in treatment over existing options. Gilead filed the NDA for sofosbuvir on April 8, 2013, and FDA has set a target review date under the Prescription Drug User Fee Act (PDUFA) of December 8, 2013.

The data submitted in this NDA support the use of sofosbuvir and ribavirin (RBV) as an all-oral therapy for patients with genotype 2 and 3 HCV infection, and for sofosbuvir in combination with RBV and pegylated interferon (peg-IFN) for treatment-naïve patients with genotype 1, 4, 5 and 6 HCV infection.
Sofosbuvir is an investigational product and its safety and efficacy have not yet been established.
About Gilead Sciences
Gilead Sciences is a biopharmaceutical company that discovers, develops and commercializes innovative therapeutics in areas of unmet medical need. The company’s mission is to advance the care of patients suffering from life-threatening diseases worldwide. Headquartered in Foster City, California, Gilead has operations in North America, Europe and Asia Pacific.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]
Data from the ELECTRON trial showed that a dual interferon-free regimen of sofosbuvir plus ribavirin produced a 24-week post-treatment sustained virological response (SVR24) rate of 100% for previously untreated patients with HCV genotypes 2 or 3.[7][8]
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of sofosbuvir, ledipasvir (formerly GS-5885), and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[9]Gilead has developed a sofosbuvir + ledipasvir coformulation that is being tested with and without ribavirin.

- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S. et al. (2010). “Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus”. Journal of Medicinal Chemistry 53 (19): 7202–7218. doi:10.1021/jm100863x. PMID 20845908. edit
- “PSI-7977″. Gilead Sciences.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M. M.; Bao, D.; Chang, W.; Espiritu, C. et al. (2010). “Mechanism of Activation of PSI-7851 and Its Diastereoisomer PSI-7977″. Journal of Biological Chemistry 285 (45): 34337–34347.doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890. edit
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- Tom Murphy (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”. Bloomberg Businessweek.
- http://www.gilead.com/pr_1757156
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. Gane, E et al. New England Journal of Medicine 368:3444. January 3, 2013.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.

