
Home » Posts tagged 'Priority review' (Page 3)
Tag Archives: Priority review
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves first treatment for pediatric patients with lupus
April 26, 2019
Release
The U.S. Food and Drug Administration today approved Benlysta (belimumab) intravenous (IV) infusion for treatment of children with systemic lupus erythematosus (SLE) – often referred to as simply “lupus” – a serious chronic disease that causes inflammation and damage to various body tissues and organs. This is the first time that the FDA has approved a treatment for pediatric patients with SLE. Benlysta has been approved for use in adult patients since 2011.
“The agency expedited the review and approval of this application because Benlysta IV fulfils an unmet need for therapies, specifically in pediatric patients with SLE. While there is no cure for lupus, treatment can help our youngest patients control their disease with the hope of improving their quality of life and lowering their risk of long-term organ damage and disability,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research.
While childhood-onset SLE is rare, when diagnosed, it is generally more active in children and adolescents than adult patients, particularly in how it impacts organs such as the kidneys and central nervous system. As a result of the disease starting early in life, pediatric patients with SLE are at a higher risk for developing increased organ damage and complications from the disease as well as adverse events from the life-long treatments usually required.
The efficacy of Benlysta IV for the treatment of SLE in pediatric patients was studied over 52 weeks in 93 pediatric patients with SLE. The proportion of pediatric patients achieving the composite primary endpoint, the SLE response index (SRI-4), was higher in pediatric patients receiving Benlysta IV plus standard therapy compared to placebo plus standard therapy. Pediatric patients who received Benlysta IV plus standard therapy also had a lower risk of experiencing a severe flare, as well as longer duration of time until a severe flare (160 days versus 82 days). The drug’s safety and pharmacokinetic profiles in pediatric patients were consistent with those in adults with SLE.
Benlysta’s doctor and patient information includes a warning for mortality, serious infections, hypersensitivity and depression, based on data from the clinical studies in adults with SLE. The drug should not be administered with live vaccines. The manufacturer is required to provide a Medication Guide to inform patients of the risks associated with Benlysta.
The most common side effects in patients included nausea, diarrhea and fever. Patients also commonly experienced infusion reactions, so healthcare professionals are advised to pre-treat patients with an antihistamine.
The FDA granted this application a Priority Review designation. The FDA granted the approval of Benlysta to GlaxoSmithKline.
ブレキサノロン , Brexanolone, Allopregnanolone
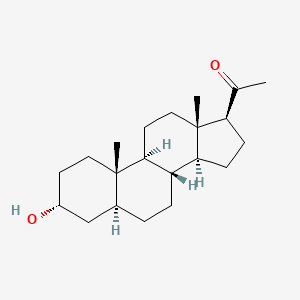

Brexanolone
318.501 g/mol, C21H34O2
CAS: 516-54-1
ブレキサノロン
The U.S. Food and Drug Administration today approved Zulresso (brexanolone) injection for intravenous (IV) use for the treatment of postpartum depression (PPD) in adult women. This is the first drug approved by the FDA specifically for PPD.
March 19, 2019
Release
The U.S. Food and Drug Administration today approved Zulresso (brexanolone) injection for intravenous (IV) use for the treatment of postpartum depression (PPD) in adult women. This is the first drug approved by the FDA specifically for PPD.
“Postpartum depression is a serious condition that, when severe, can be life-threatening. Women may experience thoughts about harming themselves or harming their child. Postpartum depression can also interfere with the maternal-infant bond. This approval marks the first time a drug has been specifically approved to treat postpartum depression, providing an important new treatment option,” said Tiffany Farchione, M.D., acting director of the Division of Psychiatry Products in the FDA’s Center for Drug Evaluation and Research. “Because of concerns about serious risks, including excessive sedation or sudden loss of consciousness during administration, Zulresso has been approved with a Risk Evaluation and Mitigation Strategy (REMS) and is only available to patients through a restricted distribution program at certified health care facilities where the health care provider can carefully monitor the patient.”
PPD is a major depressive episode that occurs following childbirth, although symptoms can start during pregnancy. As with other forms of depression, it is characterized by sadness and/or loss of interest in activities that one used to enjoy and a decreased ability to feel pleasure (anhedonia) and may present with symptoms such as cognitive impairment, feelings of worthlessness or guilt, or suicidal ideation.
Zulresso will be available only through a restricted program called the Zulresso REMS Program that requires the drug be administered by a health care provider in a certified health care facility. The REMS requires that patients be enrolled in the program prior to administration of the drug. Zulresso is administered as a continuous IV infusion over a total of 60 hours (2.5 days). Because of the risk of serious harm due to the sudden loss of consciousness, patients must be monitored for excessive sedation and sudden loss of consciousness and have continuous pulse oximetry monitoring (monitors oxygen levels in the blood). While receiving the infusion, patients must be accompanied during interactions with their child(ren). The need for these steps is addressed in a Boxed Warning in the drug’s prescribing information. Patients will be counseled on the risks of Zulresso treatment and instructed that they must be monitored for these effects at a health care facility for the entire 60 hours of infusion. Patients should not drive, operate machinery, or do other dangerous activities until feelings of sleepiness from the treatment have completely gone away.
The efficacy of Zulresso was shown in two clinical studies in participants who received a 60-hour continuous intravenous infusion of Zulresso or placebo and were then followed for four weeks. One study included patients with severe PPD and the other included patients with moderate PPD. The primary measure in the study was the mean change from baseline in depressive symptoms as measured by a depression rating scale. In both placebo controlled studies, Zulresso demonstrated superiority to placebo in improvement of depressive symptoms at the end of the first infusion. The improvement in depression was also observed at the end of the 30-day follow-up period.
The most common adverse reactions reported by patients treated with Zulresso in clinical trials include sleepiness, dry mouth, loss of consciousness and flushing. Health care providers should consider changing the therapeutic regimen, including discontinuing Zulresso in patients whose PPD becomes worse or who experience emergent suicidal thoughts and behaviors.
The FDA granted this application Priority Review and Breakthrough Therapydesignation.
Approval of Zulresso was granted to Sage Therapeutics, Inc.
Allopregnanolone, also known as 5α-pregnan-3α-ol-20-one or 3α,5α-tetrahydroprogesterone (3α,5α-THP), as well as brexanolone (USAN),[1] is an endogenous inhibitory pregnane neurosteroid[2] which has been approved by the FDA as a treatment for post-partum depression. It is synthesized from progesterone, and is a potent positive allosteric modulator of the action of γ-aminobutyric acid (GABA) at GABAA receptor.[2] Allopregnanolone has effects similar to those of other positive allosteric modulators of the GABA action at GABAA receptor such as the benzodiazepines, including anxiolytic, sedative, and anticonvulsant activity.[2][3][4] Endogenously produced allopregnanolone exerts a pivotal neurophysiological role by fine-tuning of GABAA receptor and modulating the action of several positive allosteric modulators and agonists at GABAA receptor.[5] The 21-hydroxylated derivative of this compound, tetrahydrodeoxycorticosterone (THDOC), is an endogenous inhibitory neurosteroid with similar properties to those of allopregnanolone, and the 3β-methyl analogue of allopregnanolone, ganaxolone, is under development to treat epilepsy and other conditions, including post-traumatic stress disorder (PTSD).[2]
Biochemistry
Biosynthesis
The biosynthesis of allopregnanolone in the brain starts with the conversion of progesterone into 5α-dihydroprogesterone by 5α-reductase type I. After that, 3α-hydroxysteroid dehydrogenase converts this intermediate into allopregnanolone.[2] Allopregnanolone in the brain is produced by cortical and hippocampus pyramidal neurons and pyramidal-like neurons of the basolateral amygdala.[6]
Biological activity
Allopregnanolone acts as a highly potent positive allosteric modulator of the GABAA receptor.[2] While allopregnanolone, like other inhibitory neurosteroids such as THDOC, positively modulates all GABAA receptor isoforms, those isoforms containing δ subunitsexhibit the greatest potentiation.[7] Allopregnanolone has also been found to act as a positive allosteric modulator of the GABAA-ρ receptor, though the implications of this action are unclear.[8][9] In addition to its actions on GABA receptors, allopregnanolone, like progesterone, is known to be a negative allosteric modulator of nACh receptors,[10] and also appears to act as a negative allosteric modulator of the 5-HT3 receptor.[11] Along with the other inhibitory neurosteroids, allopregnanolone appears to have little or no action at other ligand-gated ion channels, including the NMDA, AMPA, kainate, and glycine receptors.[12]
Unlike progesterone, allopregnanolone is inactive at the nuclear progesterone receptor (nPR).[12] However, allopregnanolone can be intracellularly oxidized into 5α-dihydroprogesterone, which is an agonist of the nPR, and thus/in accordance, allopregnanolone does appear to have indirect nPR-mediated progestogenic effects.[13] In addition, allopregnanolone has recently been found to be an agonist of the newly discovered membrane progesterone receptors (mPR), including mPRδ, mPRα, and mPRβ, with its activity at these receptors about a magnitude more potent than at the GABAA receptor.[14][15] The action of allopregnanolone at these receptors may be related, in part, to its neuroprotective and antigonadotropic properties.[14][16] Also like progesterone, recent evidence has shown that allopregnanolone is an activator of the pregnane X receptor.[12][17]
Similarly to many other GABAA receptor positive allosteric modulators, allopregnanolone has been found to act as an inhibitor of L-type voltage-gated calcium channels (L-VGCCs),[18] including α1 subtypes Cav1.2 and Cav1.3.[19] However, the threshold concentration of allopregnanolone to inhibit L-VGCCs was determined to be 3 μM (3,000 nM), which is far greater than the concentration of 5 nM that has been estimated to be naturally produced in the human brain.[19] Thus, inhibition of L-VGCCs is unlikely of any actual significance in the effects of endogenous allopregnanolone.[19] Also, allopregnanolone, along with several other neurosteroids, has been found to activate the G protein-coupled bile acid receptor (GPBAR1, or TGR5).[20] However, it is only able to do so at micromolar concentrations, which, similarly to the case of the L-VGCCs, are far greater than the low nanomolar concentrations of allopregnanolone estimated to be present in the brain.[20]
Biological function
Allopregnanolone possesses a wide variety of effects, including, in no particular order, antidepressant, anxiolytic, stress-reducing, rewarding,[21] prosocial,[22] antiaggressive,[23]prosexual,[22] sedative, pro-sleep,[24] cognitive, memory-impairment, analgesic,[25] anesthetic, anticonvulsant, neuroprotective, and neurogenic effects.[2] Fluctuations in the levels of allopregnanolone and the other neurosteroids seem to play an important role in the pathophysiology of mood, anxiety, premenstrual syndrome, catamenial epilepsy, and various other neuropsychiatric conditions.[26][27][28]
Increased levels of allopregnanolone can produce paradoxical effects, including negative mood, anxiety, irritability, and aggression.[29][30][31] This appears to be because allopregnanolone possesses biphasic, U-shaped actions at the GABAA receptor – moderate level increases (in the range of 1.5–2 nM/L total allopregnanolone, which are approximately equivalent to luteal phase levels) inhibit the activity of the receptor, while lower and higher concentration increases stimulate it.[29][30] This seems to be a common effect of many GABAA receptor positive allosteric modulators.[26][31] In accordance, acute administration of low doses of micronized progesterone (which reliably elevates allopregnanolone levels) has been found to have negative effects on mood, while higher doses have a neutral effect.[32]
During pregnancy, allopregnanolone and pregnanolone are involved in sedation and anesthesia of the fetus.[33][34]
Chemistry
Allopregnanolone is a pregnane (C21) steroid and is also known as 5α-pregnan-3α-ol-20-one, 3α-hydroxy-5α-pregnan-20-one, or 3α,5α-tetrahydroprogesterone (3α,5α-THP). It is very closely related structurally to 5-pregnenolone (pregn-5-en-3β-ol-20-dione), progesterone (pregn-4-ene-3,20-dione), the isomers of pregnanedione (5-dihydroprogesterone; 5-pregnane-3,20-dione), the isomers of 4-pregnenolone (3-dihydroprogesterone; pregn-4-en-3-ol-20-one), and the isomers of pregnanediol (5-pregnane-3,20-diol). In addition, allopregnanolone is one of four isomers of pregnanolone (3,5-tetrahydroprogesterone), with the other three isomers being pregnanolone (5β-pregnan-3α-ol-20-one), isopregnanolone(5α-pregnan-3β-ol-20-one), and epipregnanolone (5β-pregnan-3β-ol-20-one).
Derivatives
A variety of synthetic derivatives and analogues of allopregnanolone with similar activity and effects exist, including alfadolone (3α,21-dihydroxy-5α-pregnane-11,20-dione), alfaxolone (3α-hydroxy-5α-pregnane-11,20-dione), ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one), hydroxydione (21-hydroxy-5β-pregnane-3,20-dione), minaxolone (11α-(dimethylamino)-2β-ethoxy-3α-hydroxy-5α-pregnan-20-one), Org 20599 (21-chloro-3α-hydroxy-2β-morpholin-4-yl-5β-pregnan-20-one), Org 21465 (2β-(2,2-dimethyl-4-morpholinyl)-3α-hydroxy-11,20-dioxo-5α-pregnan-21-yl methanesulfonate), and renanolone (3α-hydroxy-5β-pregnan-11,20-dione).
Research
Allopregnanolone and the other endogenous inhibitory neurosteroids have short terminal half-lives and poor oral bioavailability, and for these reason, have not been pursued for clinical use as oral therapies, although development as a parenteral therapy for multiple indications has been carried out. However, synthetic analogs with improved pharmacokineticprofiles have been synthesized and are being investigated as potential oral therapeutic agents.
In other studies of compounds related to allopregnanolone, exogenous progesterone, such as oral micronized progesterone (OMP), elevates allopregnanolone levels in the body with good dose-to-serum level correlations.[35] Due to this, it has been suggested that OMP could be described as a prodrug of sorts for allopregnanolone.[35] As a result, there has been some interest in using OMP to treat catamenial epilepsy,[36] as well as other menstrual cycle-related and neurosteroid-associated conditions. In addition to OMP, oral pregnenolonehas also been found to act as a prodrug of allopregnanolone,[37][38][39] though also of pregnenolone sulfate.[40]
Allopregnanolone has been under development by Sage Therapeutics as an intravenously administered drug for the treatment of super-refractory status epilepticus, postpartum depression, and essential tremor.[41] As of 19 March 2019 the FDA has approved allopregnanolone for postpartum depression.
References
- ^ “ChemIDplus – 516-54-1 – AURFZBICLPNKBZ-SYBPFIFISA-N – Brexanolone [USAN] – Similar structures search, synonyms, formulas, resource links, and other chemical information”. NIH Toxnet. Retrieved 26 December 2017.
- ^ Jump up to:a b c d e f g Reddy DS (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. Progress in Brain Research. 186. pp. 113–37. doi:10.1016/B978-0-444-53630-3.00008-7. ISBN 9780444536303. PMC 3139029. PMID 21094889.
- ^ Reddy DS, Rogawski MA (2012). “Neurosteroids — Endogenous Regulators of Seizure Susceptibility and Role in the Treatment of Epilepsy”. Jasper’s Basic Mechanisms of the Epilepsies, 4th Edition: 984–1002. doi:10.1093/med/9780199746545.003.0077. ISBN 9780199746545.
- ^ T. G. Kokate, B. E. Svensson & M. A. Rogawski (September 1994). “Anticonvulsant activity of neurosteroids: correlation with γ-aminobutyric acid-evoked chloride current potentiation”. The Journal of Pharmacology and Experimental Therapeutics. 270 (3): 1223–1229. PMID 7932175.
- ^ Pinna, G; Uzunova, V; Matsumoto, K; Puia, G; Mienville, J. -M; Costa, E; Guidotti, A (2000-03-01). “Brain allopregnanolone regulates the potency of the GABAA receptor agonist muscimol”. Neuropharmacology. 39 (3): 440–448. doi:10.1016/S0028-3908(99)00149-5. PMID 10698010.
- ^ Agís-Balboa, Roberto C.; Pinna, Graziano; Zhubi, Adrian; Maloku, Ekrem; Veldic, Marin; Costa, Erminio; Guidotti, Alessandro (2006-09-26). “Characterization of brain neurons that express enzymes mediating neurosteroid biosynthesis”. Proceedings of the National Academy of Sciences. 103 (39): 14602–14607. doi:10.1073/pnas.0606544103. ISSN 0027-8424. PMC 1600006. PMID 16984997.
- ^ Mousavi Nik A, Pressly B, Singh V, Antrobus S, Hulsizer S, Rogawski MA, Wulff H, Pessah IN (2017). “Rapid Throughput Analysis of GABAA Receptor Subtype Modulators and Blockers Using DiSBAC1(3) Membrane Potential Red Dye”. Mol. Pharmacol. 92 (1): 88–99. doi:10.1124/mol.117.108563. PMC 5452057. PMID 28428226.
- ^ Morris KD, Moorefield CN, Amin J (October 1999). “Differential modulation of the gamma-aminobutyric acid type C receptor by neuroactive steroids”. Mol. Pharmacol. 56 (4): 752–9. PMID 10496958.
- ^ Li W, Jin X, Covey DF, Steinbach JH (October 2007). “Neuroactive steroids and human recombinant rho1 GABAC receptors”. J. Pharmacol. Exp. Ther. 323 (1): 236–47. doi:10.1124/jpet.107.127365. PMC 3905684. PMID 17636008.
- ^ Bullock AE, Clark AL, Grady SR, et al. (June 1997). “Neurosteroids modulate nicotinic receptor function in mouse striatal and thalamic synaptosomes”. J. Neurochem. 68 (6): 2412–23. doi:10.1046/j.1471-4159.1997.68062412.x. PMID 9166735.
- ^ Wetzel CH, Hermann B, Behl C, et al. (September 1998). “Functional antagonism of gonadal steroids at the 5-hydroxytryptamine type 3 receptor”. Mol. Endocrinol. 12 (9): 1441–51. doi:10.1210/mend.12.9.0163. PMID 9731711.
- ^ Jump up to:a b c Mellon SH (October 2007). “Neurosteroid regulation of central nervous system development”. Pharmacol. Ther. 116 (1): 107–24. doi:10.1016/j.pharmthera.2007.04.011. PMC 2386997. PMID 17651807.
- ^ Rupprecht R, Reul JM, Trapp T, et al. (September 1993). “Progesterone receptor-mediated effects of neuroactive steroids”. Neuron. 11 (3): 523–30. doi:10.1016/0896-6273(93)90156-l. PMID 8398145.
- ^ Jump up to:a b Thomas P, Pang Y (2012). “Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells”. Neuroendocrinology. 96 (2): 162–71. doi:10.1159/000339822. PMC 3489003. PMID 22687885.
- ^ Pang Y, Dong J, Thomas P (January 2013). “Characterization, neurosteroid binding and brain distribution of human membrane progesterone receptors δ and {epsilon} (mPRδ and mPR{epsilon}) and mPRδ involvement in neurosteroid inhibition of apoptosis”. Endocrinology. 154 (1): 283–95. doi:10.1210/en.2012-1772. PMC 3529379. PMID 23161870.
- ^ Sleiter N, Pang Y, Park C, et al. (August 2009). “Progesterone receptor A (PRA) and PRB-independent effects of progesterone on gonadotropin-releasing hormone release”. Endocrinology. 150 (8): 3833–44. doi:10.1210/en.2008-0774. PMC 2717864. PMID 19423765.
- ^ Lamba V, Yasuda K, Lamba JK, et al. (September 2004). “PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators”. Toxicol. Appl. Pharmacol. 199 (3): 251–65. doi:10.1016/j.taap.2003.12.027. PMID 15364541.
- ^ Hu AQ, Wang ZM, Lan DM, et al. (July 2007). “Inhibition of evoked glutamate release by neurosteroid allopregnanolone via inhibition of L-type calcium channels in rat medial prefrontal cortex”. Neuropsychopharmacology. 32 (7): 1477–89. doi:10.1038/sj.npp.1301261. PMID 17151597.
- ^ Jump up to:a b c Earl DE, Tietz EI (April 2011). “Inhibition of recombinant L-type voltage-gated calcium channels by positive allosteric modulators of GABAA receptors”. J. Pharmacol. Exp. Ther. 337 (1): 301–11. doi:10.1124/jpet.110.178244. PMC 3063747. PMID 21262851.
- ^ Jump up to:a b Keitel V, Görg B, Bidmon HJ, et al. (November 2010). “The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain”. Glia. 58 (15): 1794–805. doi:10.1002/glia.21049. PMID 20665558.
- ^ Rougé-Pont F, Mayo W, Marinelli M, Gingras M, Le Moal M, Piazza PV (July 2002). “The neurosteroid allopregnanolone increases dopamine release and dopaminergic response to morphine in the rat nucleus accumbens”. Eur. J. Neurosci. 16 (1): 169–73. doi:10.1046/j.1460-9568.2002.02084.x. PMID 12153544.
- ^ Jump up to:a b Frye CA (December 2009). “Neurosteroids’ effects and mechanisms for social, cognitive, emotional, and physical functions”. Psychoneuroendocrinology. 34 Suppl 1: S143–61. doi:10.1016/j.psyneuen.2009.07.005. PMC 2898141. PMID 19656632.
- ^ Pinna G, Costa E, Guidotti A (February 2005). “Changes in brain testosterone and allopregnanolone biosynthesis elicit aggressive behavior”. Proc. Natl. Acad. Sci. U.S.A. 102 (6): 2135–40. doi:10.1073/pnas.0409643102. PMC 548579. PMID 15677716.
- ^ Terán-Pérez G, Arana-Lechuga Y, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez Moctezuma J (October 2012). “Steroid hormones and sleep regulation”. Mini Rev Med Chem. 12 (11): 1040–8. doi:10.2174/138955712802762167. PMID 23092405.
- ^ Patte-Mensah C, Meyer L, Taleb O, Mensah-Nyagan AG (February 2014). “Potential role of allopregnanolone for a safe and effective therapy of neuropathic pain”. Prog. Neurobiol. 113: 70–8. doi:10.1016/j.pneurobio.2013.07.004. PMID 23948490.
- ^ Jump up to:a b Bäckström T, Andersson A, Andreé L, et al. (December 2003). “Pathogenesis in menstrual cycle-linked CNS disorders”. Ann. N. Y. Acad. Sci. 1007: 42–53. doi:10.1196/annals.1286.005. PMID 14993039.
- ^ Guille C, Spencer S, Cavus I, Epperson CN (July 2008). “The role of sex steroids in catamenial epilepsy and premenstrual dysphoric disorder: implications for diagnosis and treatment”. Epilepsy Behav. 13 (1): 12–24. doi:10.1016/j.yebeh.2008.02.004. PMC 4112568. PMID 18346939.
- ^ Finocchi C, Ferrari M (May 2011). “Female reproductive steroids and neuronal excitability”. Neurol. Sci. 32 Suppl 1: S31–5. doi:10.1007/s10072-011-0532-5. PMID 21533709.
- ^ Jump up to:a b Bäckström T, Haage D, Löfgren M, et al. (September 2011). “Paradoxical effects of GABA-A modulators may explain sex steroid induced negative mood symptoms in some persons”. Neuroscience. 191: 46–54. doi:10.1016/j.neuroscience.2011.03.061. PMID 21600269.
- ^ Jump up to:a b Andréen L, Nyberg S, Turkmen S, van Wingen G, Fernández G, Bäckström T (September 2009). “Sex steroid induced negative mood may be explained by the paradoxical effect mediated by GABAA modulators”. Psychoneuroendocrinology. 34 (8): 1121–32. doi:10.1016/j.psyneuen.2009.02.003. PMID 19272715.
- ^ Jump up to:a b Bäckström T, Bixo M, Johansson M, et al. (February 2014). “Allopregnanolone and mood disorders”. Prog. Neurobiol. 113: 88–94. doi:10.1016/j.pneurobio.2013.07.005. PMID 23978486.
- ^ Andréen L, Sundström-Poromaa I, Bixo M, Nyberg S, Bäckström T (August 2006). “Allopregnanolone concentration and mood–a bimodal association in postmenopausal women treated with oral progesterone”. Psychopharmacology. 187 (2): 209–21. doi:10.1007/s00213-006-0417-0. PMID 16724185.
- ^ Mellor DJ, Diesch TJ, Gunn AJ, Bennet L (2005). “The importance of ‘awareness’ for understanding fetal pain”. Brain Res. Brain Res. Rev. 49 (3): 455–71. doi:10.1016/j.brainresrev.2005.01.006. PMID 16269314.
- ^ Lagercrantz H, Changeux JP (2009). “The emergence of human consciousness: from fetal to neonatal life”. Pediatr. Res. 65 (3): 255–60. doi:10.1203/PDR.0b013e3181973b0d. PMID 19092726.
[…] the fetus is sedated by the low oxygen tension of the fetal blood and the neurosteroid anesthetics pregnanolone and the sleep-inducing prostaglandin D2 provided by the placenta (36).
- ^ Jump up to:a b Andréen L, Spigset O, Andersson A, Nyberg S, Bäckström T (June 2006). “Pharmacokinetics of progesterone and its metabolites allopregnanolone and pregnanolone after oral administration of low-dose progesterone”. Maturitas. 54 (3): 238–44. doi:10.1016/j.maturitas.2005.11.005. PMID 16406399.
- ^ Orrin Devinsky; Steven Schachter; Steven Pacia (1 January 2005). Complementary and Alternative Therapies for Epilepsy. Demos Medical Publishing. pp. 378–. ISBN 978-1-934559-08-6.
- ^ Saudan C, Desmarchelier A, Sottas PE, Mangin P, Saugy M (2005). “Urinary marker of oral pregnenolone administration”. Steroids. 70 (3): 179–83. doi:10.1016/j.steroids.2004.12.007. PMID 15763596.
- ^ Piper T, Schlug C, Mareck U, Schänzer W (2011). “Investigations on changes in ¹³C/¹²C ratios of endogenous urinary steroids after pregnenolone administration”. Drug Test Anal. 3(5): 283–90. doi:10.1002/dta.281. PMID 21538944.
- ^ Sripada RK, Marx CE, King AP, Rampton JC, Ho SS, Liberzon I (2013). “Allopregnanolone elevations following pregnenolone administration are associated with enhanced activation of emotion regulation neurocircuits”. Biol. Psychiatry. 73 (11): 1045–53. doi:10.1016/j.biopsych.2012.12.008. PMC 3648625. PMID 23348009.
- ^ Ducharme N, Banks WA, Morley JE, Robinson SM, Niehoff ML, Mattern C, Farr SA (2010). “Brain distribution and behavioral effects of progesterone and pregnenolone after intranasal or intravenous administration”. Eur. J. Pharmacol. 641 (2–3): 128–34. doi:10.1016/j.ejphar.2010.05.033. PMC 3008321. PMID 20570588.
- ^ “Brexanolone – Sage Therapeutics”. AdisInsight.
Further reading
- Herd, MB; Belelli, D; Lambert, JJ (2007). “Neurosteroid modulation of synaptic and extrasynaptic GABA(A) receptors”. Pharmacology & Therapeutics. 116 (1): 20–34. doi:10.1016/j.pharmthera.2007.03.007. PMID 17531325.
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
1-(3-Hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethanone
|
|
| Other names
ALLO; Allo; ALLOP; AlloP; Brexanolone; 5α-Pregnan-3α-ol-20-one; 3α-Hydroxy-5α-pregnan-20-one; 3α,5α-Tetrahydroprogesterone; 3α,5α-THP; Zulresso
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
|
PubChemCID
|
|
| UNII | |
| Properties | |
| C21H34O2 | |
| Molar mass | 318.501 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
//////////Brexanolone, Priority Review, Breakthrough Therapy designation, Zulresso, Sage Therapeutics Inc, FDA 2019, ブレキサノロン , Brexanolone, Allopregnanolone
CC(=O)C1CCC2C1(CCC3C2CCC4C3(CCC(C4)O)C)C
Caplacizumab-yhdp, カプラシズマブ
FDA approves first therapy Cablivi (caplacizumab-yhdp) カプラシズマブ , for the treatment of adult patients with a rare blood clotting disorder
FDA
February 6, 2019
The U.S. Food and Drug Administration today approved Cablivi (caplacizumab-yhdp) injection, the first therapy specifically indicated, in combination with plasma exchange and immunosuppressive therapy, for the treatment of adult patients with acquired thrombotic thrombocytopenic purpura (aTTP), a rare and life-threatening disorder that causes blood clotting.
“Patients with aTTP endure hours of treatment with daily plasma exchange, which requires being attached to a machine that takes blood out of the body and mixes it with donated plasma and then returns it to the body. Even after days or weeks of this treatment, as well as taking drugs that suppress the immune system, many patients will have a recurrence of aTTP,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Cablivi is the first targeted treatment that inhibits the formation of blood clots. It provides a new treatment option for patients that may reduce recurrences.”
Patients with aTTP develop extensive blood clots in the small blood vessels throughout the body. These clots can cut off oxygen and blood supply to the major organs and cause strokes and heart attacks that may lead to brain damage or death. Patients can develop aTTP because of conditions such as cancer, HIV, pregnancy, lupus or infections, or after having surgery, bone marrow transplantation or chemotherapy.
The efficacy of Cablivi was studied in a clinical trial of 145 patients who were randomized to receive either Cablivi or a placebo. Patients in both groups received the current standard of care of plasma exchange and immunosuppressive therapy. The results of the trial demonstrated that platelet counts improved faster among patients treated with Cablivi, compared to placebo. Treatment with Cablivi also resulted in a lower total number of patients with either aTTP-related death and recurrence of aTTP during the treatment period, or at least one treatment-emergent major thrombotic event (where blood clots form inside a blood vessel and may then break free to travel throughout the body).The proportion of patients with a recurrence of aTTP in the overall study period (the drug treatment period plus a 28-day follow-up period after discontinuation of drug treatment) was lower in the Cablivi group (13 percent) compared to the placebo group (38 percent), a finding that was statistically significant.
Common side effects of Cablivi reported by patients in clinical trials were bleeding of the nose or gums and headache. The prescribing information for Cablivi includes a warning to advise health care providers and patients about the risk of severe bleeding.
Health care providers are advised to monitor patients closely for bleeding when administering Cablivi to patients who currently take anticoagulants.
The FDA granted this application Priority Review designation. Cablivi also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Cablivi to Ablynx.
EU
Cablivi is the first therapeutic approved in Europe, for the treatment of a rare blood-clotting disorder
On September 03, 2018, the European Commission has granted marketing authorization for Cablivi™ (caplacizumab) for the treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP), a rare blood-clotting disorder. Cablivi is the first therapeutic specifically indicated for the treatment of aTTP 1. Cablivi was designated an ‘orphan medicine’ (a medicine used in rare diseases) on April 30, 2009. The approval of Cablivi in the EU is based on the Phase II TITAN and Phase III HERCULES studies in 220 adult patients with aTTP. The efficacy and safety of caplacizumab in addition to standard-of-care treatment, daily PEX and immunosuppression, were demonstrated in these studies. In the HERCULES study, treatment with caplacizumab in addition to standard-of-care resulted in a significantly shorter time to platelet count response (p<0.01), the study’s primary endpoint; a significant reduction in aTTP-related death, recurrence of aTTP, or at least one major thromboembolic event during study drug treatment (p<0.0001); and a significantly lower number of aTTP recurrences in the overall study period (p<0.001). Importantly, treatment with caplacizumab resulted in a clinically meaningful reduction in the use of PEX and length of stay in the intensive care unit (ICU) and the hospital, compared to the placebo group. Cablivi was developed by Ablynx, a Sanofi company. Sanofi Genzyme, the specialty care global business unit of Sanofi, will work with relevant local authorities to make Cablivi available to patients in need in countries across Europe.
About aTTP aTTP is a life-threatening, autoimmune blood clotting disorder characterized by extensive clot formation in small blood vessels throughout the body, leading to severe thrombocytopenia (very low platelet count), microangiopathic hemolytic anemia (loss of red blood cells through destruction), ischemia (restricted blood supply to parts of the body) and widespread organ damage especially in the brain and heart. About Cablivi Caplacizumab blocks the interaction of ultra-large von Willebrand Factor (vWF) multimers with platelets and, therefore, has an immediate effect on platelet adhesion and the ensuing formation and accumulation of the micro-clots that cause the severe thrombocytopenia, tissue ischemia and organ dysfunction in aTTP 2.
Note – Caplacizumab is a bivalent anti-vWF Nanobody that received Orphan Drug Designation in Europe and the United States in 2009, in Switzerland in 2017 and in Japan in 2018. The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application for caplacizumab for treatment of adults experiencing an episode of aTTP. The target action date for the FDA decision is February 6, 2019
1 http://hugin.info/152918/R/2213684/863478.pdf
![]()
More………….
EVQLVESGGG LVQPGGSLRL SCAASGRTFS YNPMGWFRQA PGKGRELVAA ISRTGGSTYY
PDSVEGRFTI SRDNAKRMVY LQMNSLRAED TAVYYCAAAG VRAEDGRVRT LPSEYTFWGQ
GTQVTVSSAA AEVQLVESGG GLVQPGGSLR LSCAASGRTF SYNPMGWFRQ APGKGRELVA
AISRTGGSTY YPDSVEGRFT ISRDNAKRMV YLQMNSLRAE DTAVYYCAAA GVRAEDGRVR
TLPSEYTFWG QGTQVTVSS
(disulfide bridge: 22-96, 153-227)
Sequence:
EU 2018/8/31 APPROVED, Cablivi
Treatment of thrombotic thrombocytopenic purpura, thrombosis
Other Names
- 1: PN: WO2011067160 SEQID: 1 claimed protein
- 98: PN: WO2006122825 SEQID: 98 claimed protein
- ALX 0081
- ALX 0681
- Caplacizumab
| FORMULA |
C1213H1891N357O380S10
|
|---|---|
| CAS |
915810-67-2
|
| MOL WEIGHT |
27875.8075
|
Caplacizumab (ALX-0081) (INN) is a bivalent VHH designed for the treatment of thrombotic thrombocytopenic purpura and thrombosis.[1][2]
This drug was developed by Ablynx NV.[3] On 31 August 2018 it was approved in the European Union for the “treatment of adults experiencing an episode of acquired thrombotic thrombocytopenic purpura (aTTP), in conjunction with plasma exchange and immunosuppression”.[4]
It is an anti-von Willebrand factor humanized immunoglobulin.[5] It acts by blocking platelet aggregation to reduce organ injury due to ischemia.[5] Results of the phase II TITAN trial have been reported.[5]
In February 2019, caplacizumab-yhdp (CABLIVI, Ablynx NV) has been approved by the Food and Drug Administration for treatment of adult patients with acquired thrombotic thrombocytopenic purpura (aTTP). The drug is used in combination with plasma exchange and immunosuppressive therapy. [6]
PATENTS
WO 2006122825
WO 2009115614
WO 2011067160
WO 2011098518
WO 2011162831
WO 2013013228
WO 2014109927
WO 2016012285
WO 2016138034
WO 2016176089
WO 2017180587
WO 2017186928
WO 2018067987
| Monoclonal antibody | |
|---|---|
| Type | Single domain antibody |
| Source | Humanized |
| Target | VWF |
| Clinical data | |
| Synonyms | ALX-0081 |
| ATC code | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C1213H1891N357O380S10 |
| Molar mass | 27.88 kg/mol |
CLIP
https://www.tandfonline.com/doi/full/10.1080/19420862.2016.1269580
Caplacizumab (ALX-0081) is a humanized single-variable-domain immunoglobulin (Nanobody) that targets von Willebrand factor, and thereby inhibits the interaction between von Willebrand factor multimers and platelets. In a Phase 2 study (NCT01151423) of 75 patients with acquired thrombotic thrombocytopenic purpura who received SC caplacizumab (10 mg daily) or placebo during plasma exchange and for 30 d afterward, the time to a response was significantly reduced with caplacizumab compared with placebo (39% reduction in median time, P = 0.005).39 The double-blind, placebo-controlled, randomized Phase 3 HERCULES study (NCT02553317) study will evaluate the efficacy and safety of caplacizumab treatment in more rapidly curtailing ongoing microvascular thrombosis when administered in addition to standard of care treatment in subjects with an acute episode of acquired thrombotic thrombocytopenic purpura. Patients will receive an initial IV dose of either caplacizumab or placebo followed by daily SC injections for a maximum period of 6 months. The primary outcome measure is the time to platelet count response. The estimated enrollment is 92 patients, and the estimated primary completion date of the study is October 2017. A Phase 3 follow-up study (NCT02878603) for patients who completed the HERCULES study is planned.
References
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Caplacizumab, American Medical Association.
- ^ World Health Organization (2011). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 106”(PDF). WHO Drug Information. 25 (4).
- ^ A Trial With Caplacizumab in Patients With Acquired Thrombotic Thrombocytopenic Purpura (HERCULES)
- ^ European Medicines Agency. “An overview of Cablivi and why it is authorised in the EU” (PDF). Retrieved 1 October 2018.
- ^ Jump up to:a b c Immune Drug Tackles Microvascular Thrombosis Disorder. Feb 2016
- ^ “FDA approved caplacizumab-yhdp”. 2019-02-07.
///////////////caplacizumab, Cablivi, Ablynx, Priority Review, Orphan Drug designation, fda 2019, eu 2018, Caplacizumab, nti-vWF Nanobody, Orphan Drug Designation, aTTP, Cablivi, Ablynx, Sanofi , ALX-0081, カプラシズマブ , PEPTIDE, ALX 0081
FDA approves first treatment Firdapse (amifampridine) for Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
FDA approves first treatment Firdapse (amifampridine) for Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
The U.S. Food and Drug Administration today approved Firdapse (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults. LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. This is the first FDA approval of a treatment for LEMS.
November 28, 2018
Release
The U.S. Food and Drug Administration today approved Firdapse (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults. LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. This is the first FDA approval of a treatment for LEMS.
“There has been a long-standing need for a treatment for this rare disorder,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “Patients with LEMS have significant weakness and fatigue that can often cause great difficulties with daily activities.”
In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with other autoimmune diseases, but more commonly occurs in patients with cancer such as small cell lung cancer, where its onset precedes or coincides with the diagnosis of cancer. The prevalence of LEMS is estimated to be three per million individuals worldwide.
The efficacy of Firdapse was studied in two clinical trials that together included 64 adult patients who received Firdapse or placebo. The studies measured the Quantitative Myasthenia Gravis score (a 13-item physician-rated categorical scale assessing muscle weakness) and the Subject Global Impression (a seven-point scale on which patients rated their overall impression of the effects of the study treatment on their physical well-being). For both measures, the patients receiving Firdapse experienced a greater benefit than those on placebo.
The most common side effects experienced by patients in the clinical trials were burning or prickling sensation (paresthesia), upper respiratory tract infection, abdominal pain, nausea, diarrhea, headache, elevated liver enzymes, back pain, hypertension and muscle spasms. Seizures have been observed in patients without a history of seizures. Patients should inform their health care provider immediately if they have signs of hypersensitivity reactions such as rash, hives, itching, fever, swelling or trouble breathing.
The FDA granted this application Priority Review and Breakthrough Therapydesignations. Firdapse also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Firdapse to Catalyst Pharmaceuticals, Inc.
///////////Priority Review, Breakthrough Therapy, Firdapse, Orphan Drug designation, fda 2018, amifampridine
FDA approves new treatment for patients with acute myeloid leukemia
|
||
November 21, 2018
Release
The U.S. Food and Drug Administration today approved Daurismo (glasdegib) tablets to be used in combination with low-dose cytarabine (LDAC), a type of chemotherapy, for the treatment of newly-diagnosed acute myeloid leukemia (AML) in adults who are 75 years of age or older or who have other chronic health conditions or diseases (comorbidities) that may preclude the use of intensive chemotherapy.
“Intensive chemotherapy is usually used to control AML, but many adults with AML are unable to have intensive chemotherapy because of its toxicities. Today’s approval gives health care providers another tool to use in the treatment of AML patients with various, unique needs. Clinical trials showed that overall survival was improved using Daurismo in combination with LDAC compared to LDAC alone for patients who would not tolerate intensive chemotherapy,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that in 2018, approximately 19,520 people will be diagnosed with AML and approximately 10,670 patients with AML will die of the disease. Almost half of the adults diagnosed with AML are not treated with intensive chemotherapy because of comorbidities and chemotherapy related toxicities.
The efficacy of Daurismo was studied in a randomized clinical trial in which 111 adult patients with newly diagnosed AML were treated with either Daurismo in combination with LDAC or LDAC alone. The trial measured overall survival (OS) from the date of randomization to death from any cause. Results demonstrated a significant improvement in OS in patients treated with Daurismo. The median OS was 8.3 months for patients treated with Daurismo plus LDAC compared with 4.3 months for patients treated with LDAC only.
Common side effects reported by patients receiving Daurismo in clinical trials include low red blood cell count (anemia), tiredness (fatigue), bleeding (hemorrhage), fever with low white blood cell count (febrile neutropenia), muscle pain, nausea, swelling of the arms or legs (edema), low platelet counts (thrombocytopenia), shortness of breath (dyspnea), decreased appetite, distorted taste (dysgeusia), pain or sores in the mouth or throat (mucositis), constipation and rash.
The prescribing information for Daurismo includes a Boxed Warning to advise health care professionals and patients about the risk of embryo-fetal death or severe birth defects. Daurismo should not be used during pregnancy or while breastfeeding. Pregnancy testing should be conducted in females of reproductive age prior to initiation of Daurismo treatment and effective contraception should be used during treatment and for at least 30 days after the last dose. The Boxed Warning also advises male patients of the potential risk of drug exposure through semen and to use condoms with a pregnant partner or a female partner that could become pregnant both during treatment and for at least 30 days after the last dose. Daurismo must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. Patients should also be advised not to donate blood or blood products during treatment. Health care providers should also monitor patients for changes in the electrical activity of the heart, called QT prolongation.
The FDA granted this application Priority Review designation. Daurismo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Daurismo to Pfizer.
//////////////Daurismo, glasdegib, fda 2018, Priority Review, Orphan Drug
FDA approves new drug Aemcolo (rifamycin), to treat travelers’ diarrhea
November 16, 2018
Release
The U.S. Food and Drug Administration today approved Aemcolo (rifamycin), an antibacterial drug indicated for the treatment of adult patients with travelers’ diarrhea caused by noninvasive strains of Escherichia coli (E. coli), not complicated by fever or blood in the stool.
“Travelers’ diarrhea affects millions of people each year and having treatment options for this condition can help reduce symptoms of the condition,” said Edward Cox, M.D., M.P.H., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Travelers’ diarrhea is the most common travel-related illness, affecting an estimated 10 to 40 percent of travelers worldwide each year. Travelers’ diarrhea is defined by having three or more unformed stools in 24 hours, in a person who is traveling. It is caused by a variety of pathogens, but most commonly bacteria found in food and water. The highest-risk destinations are in most of Asia as well as the Middle East, Africa, Mexico, and Central and South America.
The efficacy of Aemcolo was demonstrated in a randomized, placebo-controlled clinical trial in 264 adults with travelers’ diarrhea in Guatemala and Mexico. It showed that Aemcolo significantly reduced symptoms of travelers’ diarrhea compared to the placebo.
The safety of Aemcolo, taken orally over three or four days, was evaluated in 619 adults with travelers’ diarrhea in two controlled clinical trials. The most common adverse reactions with Aemcolo were headache and constipation.
Aemcolo was not shown to be effective in patients with diarrhea complicated by fever and/or bloody stool or diarrhea due to pathogens other than noninvasive strains of E. coli and is not recommended for use in such patients. Aemcolo should not be used in patients with a known hypersensitivity to rifamycin, any of the other rifamycin class antimicrobial agents (e.g. rifaximin), or any of the components in Aemcolo.
The FDA granted Aemcolo a Qualified Infectious Disease Product (QIDP)designation. QIDP designation is given to antibacterial and antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of QIDP designation, the Aemcolo marketing application was granted Priority Review under which the FDA’s goal is to take action on an application within an expedited time frame.
The FDA granted approval of Aemcolo to Cosmo Technologies, Ltd.
USFDA approval to Lumoxiti (moxetumomab pasudotoxtdfk) a new treatment for hairy cell leukemia
![]()
USFDA approval to Lumoxiti is a new treatment for hairy cell leukemia
On September 13, 2018, the U.S. Food and Drug Administration approved Lumoxiti (moxetumomab pasudotoxtdfk) injection for intravenous use for the treatment of adult patients with relapsed or refractory Hairy Cell Leukemia (HCL) who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog 1. Lumoxiti is a CD22-directed cytotoxin and is the first of this type of treatment for patients with HCL. The efficacy of Lumoxiti was studied in a single-arm, open-label clinical trial of 80 patients who had received prior treatment for HCL with at least two systemic therapies, including a purine nucleoside analog. The trial measured durable complete response (CR), defined as maintenance of hematologic remission for more than 180 days after achievement of CR. Thirty percent of patients in the trial achieved durable CR, and the overall response rate (number of patients with partial or complete response to therapy) was 75 percent. The FDA granted this application Fast Track and Priority Review designations. Lumoxiti also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Lumoxiti to AstraZeneca Pharmaceuticals. About Hairy Cell Leukemia HCL is a rare, slow-growing cancer of the blood in which the bone marrow makes too many B cells (lymphocytes), a type of white blood cells that fight infection. HCL is named after these extra B cells which look “hairy” when viewed under a microscope. As the number of leukemia cells increases, fewer healthy white blood cells, red blood cells and platelets are produced.
About Lumoxiti2 Lumoxiti (moxetumomab pasudotox) is a CD22-directed cytotoxin and a first-in-class treatment in the US for adult patients with relapsed or refractory hairy cell leukaemia (HCL) who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog. Lumoxiti is not recommended in patients with severe renal impairment (CrCl ≤ 29 mL/min). It comprises the CD22 binding portion of an antibody fused to a truncated bacterial toxin; the toxin inhibits protein synthesis and ultimately triggers apoptotic cell death.
September 13, 2018
Release
The U.S. Food and Drug Administration today approved Lumoxiti (moxetumomab pasudotox-tdfk) injection for intravenous use for the treatment of adult patients with relapsed or refractory hairy cell leukemia (HCL) who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog. Lumoxiti is a CD22-directed cytotoxin and is the first of this type of treatment for patients with HCL.
“Lumoxiti fills an unmet need for patients with hairy cell leukemia whose disease has progressed after trying other FDA-approved therapies,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This therapy is the result of important research conducted by the National Cancer Institute that led to the development and clinical trials of this new type of treatment for patients with this rare blood cancer.”
HCL is a rare, slow-growing cancer of the blood in which the bone marrow makes too many B cells (lymphocytes), a type of white blood cell that fights infection. HCL is named after these extra B cells which look “hairy” when viewed under a microscope. As the number of leukemia cells increases, fewer healthy white blood cells, red blood cells and platelets are produced.
The efficacy of Lumoxiti was studied in a single-arm, open-label clinical trial of 80 patients who had received prior treatment for HCL with at least two systemic therapies, including a purine nucleoside analog. The trial measured durable complete response (CR), defined as maintenance of hematologic remission for more than 180 days after achievement of CR. Thirty percent of patients in the trial achieved durable CR, and the overall response rate (number of patients with partial or complete response to therapy) was 75 percent.
Common side effects of Lumoxiti include infusion-related reactions, swelling caused by excess fluid in body tissue (edema), nausea, fatigue, headache, fever (pyrexia), constipation, anemia and diarrhea.
The prescribing information for Lumoxiti includes a Boxed Warning to advise health care professionals and patients about the risk of developing capillary leak syndrome, a condition in which fluid and proteins leak out of tiny blood vessels into surrounding tissues. Symptoms of capillary leak syndrome include difficulty breathing, weight gain, hypotension, or swelling of arms, legs and/or face. The Boxed Warning also notes the risk of hemolytic uremic syndrome, a condition caused by the abnormal destruction of red blood cells. Patients should be made aware of the importance of maintaining adequate fluid intake, and blood chemistry values should be monitored frequently. Other serious warnings include: decreased renal function, infusion-related reactions and electrolyte abnormalities. Women who are breastfeeding should not be given Lumoxiti.
The FDA granted this application Fast Track and Priority Review designations. Lumoxiti also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Lumoxiti to AstraZeneca Pharmaceuticals.
1 https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm620448.htm
/////////// Lumoxiti, moxetumomab pasudotoxtdfk, FDA 2018, Fast Track, Priority Review , Orphan Drug, AstraZeneca
FDA approves first treatment Libtayo (cemiplimab-rwlc) for advanced form of the second most common skin cancer
FDA approves first treatment for advanced form of the second most common skin cancer
New drug targets PD-1 pathway
September 28, 2018
Release
The U.S. Food and Drug Administration today approved Libtayo (cemiplimab-rwlc) injection for intravenous use for the treatment of patients with metastatic cutaneous squamous cell carcinoma (CSCC) or locally advanced CSCC who are not candidates for curative surgery or curative radiation. This is the first FDA approval of a drug specifically for advanced CSCC.
Libtayo works by targeting the cellular pathway known as PD-1 (protein found on the body’s immune cells and some cancer cells). By blocking this pathway, the drug may help the body’s immune system fight the cancer cells.
“We’re continuing to see a shift in oncology toward identifying and developing drugs aimed at a specific molecular target. With the Libtayo approval, the FDA has approved six immune checkpoint inhibitors targeting the the PD-1 / PD-L1 pathway for treating a variety of tumors, from bladder to head and neck cancer, and now advanced CSCC,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This type of cancer can be difficult to treat effectively when it is advanced and it is important that we continue to bring new treatment options to patients.”
CSCC is the second most common human cancer in the United States with an estimated annual incidence of approximately 700,000 cases. The most common form of skin cancer is basal cell cancer. Squamous cells are thin, flat cells that look like fish scales and are found in the tissue that forms the surface of the skin. CSCC usually develops in skin areas that have been regularly exposed to the sun or other forms of ultraviolet radiation. While the majority of patients with CSCC are cured with surgical resection, a small percentage of patients will develop advanced disease that no longer responds to local treatments including surgery and radiation. Advanced CSCC may cause disfigurement at the site of the tumor and local complications such as bleeding or infection, or it may spread (metastasize) to local lymph nodes, distant tissues and organs and become life-threatening.
The safety and efficacy of Libtayo was studied in two open label clinical trials. A total of 108 patients (75 with metastatic disease and 33 with locally-advanced disease) were included in the efficacy evaluation. The study’s primary endpoint was objective response rate, or the percentage of patients who experienced partial shrinkage or complete disappearance of their tumor(s) after treatment. Results showed that 47.2 percent of all patients treated with Libtayo had their tumors shrink or disappear. The majority of these patients had ongoing responses at the time of data analysis.
Common side effects of Libtayo include fatigue, rash and diarrhea. Libtayo must be dispensed with a patient Medication Guide that describes uses of the drug and its serious warnings. Libtayo can cause the immune system to attack normal organs and tissues in any area of the body and can affect the way they work. These reactions can sometimes become severe or life-threatening and can lead to death. These reactions include the risk of immune-mediated adverse reactions including lung problems (pneumonitis), intestinal problems (colitis), liver problems (hepatitis), hormone gland problems (endocrinopathies), skin (dermatologic) problems and kidney problems. Patients should also be monitored for infusion-related reactions.
Libtayo can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted this application Breakthrough Therapy and Priority Reviewdesignations.
The FDA granted the approval of Libtayo to Regeneron Pharmaceuticals, Inc.
Cenegermin


| Cenegermin sequence: | |
| SSSHPIFHRGEFSVCDSVSVWVGDKTTATDIKGKEVMVLGEVNIN | |
| NSVFKQYFFETKCRDPNPVDSGCRGIDSKHWNSYCTTTHTFVKAL | |
| TMDGKQAAWRFIRIDTACVCVLSRKAVR |
- OriginatorAnabasis Pharma
- DeveloperDompe Farmaceutici; Ospedale San Raffaele
- ClassEye disorder therapies; Nerve growth factors; Neuroprotectants; Proteins
- Mechanism of ActionNerve growth factor receptor agonists; Neuron stimulants
- Orphan Drug StatusYes – Keratitis; Retinitis pigmentosa
- Highest Development Phases
- RegisteredKeratitis
- Phase II Dry eyes; Glaucoma; Retinitis pigmentosa
- APPROVED FDA AUG 2018
Most Recent Events
- 28 Jul 2018No recent reports of development identified for phase-I development in Glaucoma in Italy (Ophthalmic, Drops)
- 29 May 2018Phase-II clinical trials in Glaucoma (Ophthalmic) (http://www.dompe.com/RnD-Pipeline/)
- 01 May 2018Dompé Farmaceutici completes a phase I trial in Glaucoma in USA (Ophthalmic) (NCT02855450)

August 22, 2018
Release
The U.S. Food and Drug Administration today approved the first drug, Oxervate (cenegermin), for the treatment of neurotrophic keratitis, a rare disease affecting the cornea (the clear layer that covers the colored portion of the front of the eye).
“While the prevalence of neurotrophic keratitis is low, the impact of this serious condition on an individual patient can be devastating,” said Wiley Chambers, M.D., an ophthalmologist in the FDA’s Center for Drug Evaluation and Research. “In the past, it has often been necessary to turn to surgical interventions; these treatments are usually only palliative in this disease. Today’s approval provides a novel topical treatment and a major advance that offers complete corneal healing for many of these patients.”
Neurotrophic keratitis is a degenerative disease resulting from a loss of corneal sensation. The loss of corneal sensation impairs corneal health causing progressive damage to the top layer of the cornea, including corneal thinning, ulceration, and perforation in severe cases. The prevalence of neurotrophic keratitis has been estimated to be less than five in 10,000 individuals.
The safety and efficacy of Oxervate, a topical eye drop containing cenegermin, was studied in a total of 151 patients with neurotrophic keratitis in two, eight-week, randomized controlled multi-center, double-masked studies. In the first study, patients were randomized into three different groups. One group received Oxervate, a second group received an eye drop with a different concentration of cenegermin, and the third group received an eye drop without cenegermin. In the second study, patients were randomized into two groups. One group was treated with Oxervate eye drops and the other group was treated with an eye drop without cenegermin. All eye drops in both studies were given six times daily in the affected eye(s) for eight weeks. In the first study, only patients with the disease in one eye were enrolled, while in the second study, patients with the disease in both eyes were treated in both eyes (bilaterally). Across both studies, complete corneal healing in eight weeks was demonstrated in 70 percent of patients treated with Oxervate compared to 28 percent of patients treated without cenegermin (the active ingredient in Oxervate).
The most common adverse reactions in patients taking Oxervate are eye pain, ocular hyperemia (enlarged blood vessels in the white of the eyes), eye inflammation and increased lacrimation (watery eyes).
Oxervate was granted Priority Review designation, under which the FDA’s goal is to take action on an application within six months of application filing where the agency determines that the drug, if approved, would provide a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious condition. Oxervate also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Oxervate to Dompé farmaceutici SpA.
| Clinical data | |
|---|---|
| Trade names | Oxervate, Sentinel |
| Synonyms | Recombinant human nerve growth factor; rhNGF; human beta-nerve growth factor (beta-NGF)-(1-118) peptide (non-covalent dimer) produced in Escherichia coli[1] |
| Routes of administration |
Eye drops |
| ATC code | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C583H908N166O173S8 |
| Molar mass | 13266.94 g/mol |
References
- ^ Jump up to:a bhttp://www.who.int/medicines/publications/druginformation/issues/77_INN_Recommended_List.pdf
- Jump up^ “Authorisation details”. European Medicines Agency. Retrieved 19 February 2018.
- ^ Jump up to:a b c d http://adisinsight.springer.com/drugs/800035751

{kind=link}